

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease with an estimated heritability between 40 and 50%. DNA methylation patterns can serve as proxies of (past) exposures and disease progression, as well as providing a potential mechanism that mediates genetic or environmental risk. Here, we present a blood-based epigenome-wide association study meta-analysis in 9706 samples passing stringent quality control (6763 patients, 2943 controls). We identified a total of 45 differentially methylated positions (DMPs) annotated to 42 genes, which are enriched for pathways and traits related to metabolism, cholesterol biosynthesis, and immunity. We then tested 39 DNA methylation–based proxies of putative ALS risk factors and found that high-density lipoprotein cholesterol, body mass index, white blood cell proportions, and alcohol intake were independently associated with ALS. Integration of these results with our latest genome-wide association study showed that cholesterol biosynthesis was potentially causally related to ALS. Last, DNA methylation at several DMPs and blood cell proportion estimates derived from DNA methylation data were associated with survival rate in patients, suggesting that they might represent indicators of underlying disease processes potentially amenable to therapeutic interventions.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by progressive degeneration of motor neurons in the brain and spinal cord (1). The disease affects about 1 in 350 people, with death typically occurring within 2 to 5 years after onset. The heritability of ALS is estimated to be around 50% (2), showing that a considerable portion of the risk could be conferred by environmental and lifestyle risk factors. However, the identification of these factors has proven difficult because of several challenges such as recall and measurement bias, resulting in a large body of literature with conflicting results and only a few established factors related to ALS risk or patient survival (3–6). Epigenetic patterns, which act at the interface between genes and environment, can serve as proxies of (past) exposures, therefore enabling the study of these exposures and putative risk factors. Moreover, the identification of ALS-associated epigenetic factors could provide insights into disease etiology and disease processes.
DNA methylation is one of the best characterized and most stable epigenetic modifications and plays an important role in gene regulation, genomic stability, and genomic imprinting (7–9). The development of standardized assays for quantifying DNA methylation has enabled the systematic analysis of associations between methylomic variation and a wide range of human diseases, including cancer, schizophrenia, and various neurodegenerative diseases (10, 11). DNA methylation in whole blood captures a wide range of putative ALS risk factors at a molecular level, including smoking, alcohol intake, body mass index (BMI), biological age, and various metabolic and inflammatory proteins (12–18). Leveraging DNA methylation as proxies for these risk factors offers several advantages because it is (i) not prone to recall bias (relevant for smoking and alcohol), (ii) may capture information not (accurately) captured by the self-report (such as passive and past smoking) and provides a quantifiable measure (19), and (iii) is relatively stable in the short term [especially relevant for immunological proteins (18)]. Moreover, many risk factor studies have been conducted in small samples (3, 6), whereas our large DNA methylation study can provide a well-powered alternative that jointly considers the molecular correlates of many risk factors. We, therefore, performed a blood-based DNA methylation study of ALS incorporating 9706 samples that passed stringent quality control.
RESULTS
Epigenome-wide association study meta-analysis of ALS identifies 45 DMPs
We quantified genome-wide DNA methylation in whole blood from 10,462 individuals using the Illumina HumanMethylation450 (450 k) array (6275 samples) and the Illumina MethylationEPIC (EPIC) array (4187 samples). We merged individual-level DNA methylation array data from 14 countries into four strata (MinE 450 K, MinE EPIC, AUS1, and AUS2; see Materials and Methods and fig. S1). A total of 6763 patients with ALS and 2943 control individuals passed our stringent quality control, which was followed by normalization of signal intensities in each stratum (Table 1, data file S1, and tables S1 to S5). Samples excluded from our analyses did not show different demographic or clinical characteristics compared to the subset selected for analyses (data file S2).
Table 1. Demographic and clinical characteristics of study population.
Shown are numbers (and percentages) of samples that passed quality control.
| Project MinE | External | |||
|---|---|---|---|---|
| MinE 450 k | MinE EPIC | AUS1* | AUS2* | |
| (N = 4474) | (N = 3897) | (N = 1088) | (N = 247) | |
| Diagnosis | ||||
| Control | 1436 (32%) | 915 (23%) | 493 (45%) | 99 (40%) |
| Case | 3038 (68%) | 2982 (77%) | 595 (55%) | 148 (60%) |
| Sex at birth | ||||
| Female | 1863 (42%) | 1700 (44%) | 487 (45%) | 124 (50%) |
| Male | 2611 (58%) | 2197 (56%) | 601 (55%) | 123 (50%) |
| Age (years) | ||||
| Mean (SD) | 63 (± 11) | 61 (± 13) | 70 (± 12) | |
| Missing | 438 (9.8%) | 949 (24.4%) | 77 (7.1%) | |
| Site of onset † | ||||
| Bulbar | 861 (28%) | 739 (25%) | 173 (29%) | 36 (24%) |
| Generalized | 98 (3%) | 112 (4%) | 0 (0%) | 0 (0%) |
| Spinal | 2023 (67%) | 2060 (69%) | 0 (0%) | 0 (0%) |
| Thoracic | 10 (0%) | 5 (0%) | 0 (0%) | 0 (0%) |
| Missing | 46 (1.5%) | 66 (2.2%) | 422 (70.9%) | 112 (75.7%) |
| Survival status † | ||||
| Alive | 437 (14%) | 1112 (37%) | 516 (87%) | 43 (29%) |
| Dead | 2564 (84%) | 1845 (62%) | 79 (13%) | 87 (59%) |
| Missing | 37 (1.2%) | 25 (0.8%) | 0 (0%) | 18 (12.2%) |
| Survival (months) † | ||||
| Median (Q1-Q3) | 31.4 (31.4–48.9) | 31.3 (21.1–47.1) | 31.5 (23.6–44.4) | 38.3 (25.4–66.5) |
| Missing | 17 (0.7%) | 9 (0.5%) | 2 (2.5%) | 1 (1.1%) |
| C9orf72 status † ‡ | ||||
| Expanded (≥30) | 200 (7%) | 155 (5%) | ||
| Normal | 2809 (92%) | 2780 (93%) | ||
| Missing | 29 (1.0%) | 47 (1.6%) | ||
Data only included in case/control analyses.
Case only.
Dead only.
We performed an epigenome-wide association study (EWAS) in each of the four strata using two methods to adjust for known and unknown confounders. First, we used a linear model adjusting for known confounders and a calibrated number of principal components (PCs) to adjust for unknown confounding factors (fig. S2), followed by correction for residual bias and inflation in test statistics using bacon (hereafter referred to as the LB model) (20). Second, we used MOA (mixed linear model–based omic association) as implemented in the OSCA software in which the random effect of total genome-wide DNA methylation captures the correlation structure between probes and directly controls for the genomic inflation (21). The MOA algorithm did not converge for the AUS2 stratum, resulting in a total sample size of 9459 for the MOA results. Test statistics across strata were combined using an inverse variance-weighted (IVW) fixed-effects meta-analysis (22). Inflation of the test statistics was well controlled in both the LB (λ = 1.046; Fig. 1) and the MOA results, respectively (λ = 0.984; Fig. 1), and we observed little heterogeneity between strata (figs. S3 to S5). Various sensitivity analyses indicated that the results were robust to changes in analysis strategy, including adjustment for population stratification (10 genetic PCs), using M values instead of β values, using functional normalization (23) instead of dasen (24), and excluding specific strata or experimental batches (figs. S6 to S8). Last, application of a method that we recently described (25) led to the removal of likely cross-hybridizing probes, including four probes that showed high homology to the C9orf72 repeat locus (fig. S9). In total, 724,712 positions passed quality control and were included in the meta-analysis. Of these, 332,066 were specific to the EPIC array, and 26,367 were specific to the 450 k array, respectively.
Fig. 1. EWAS meta-analysis.
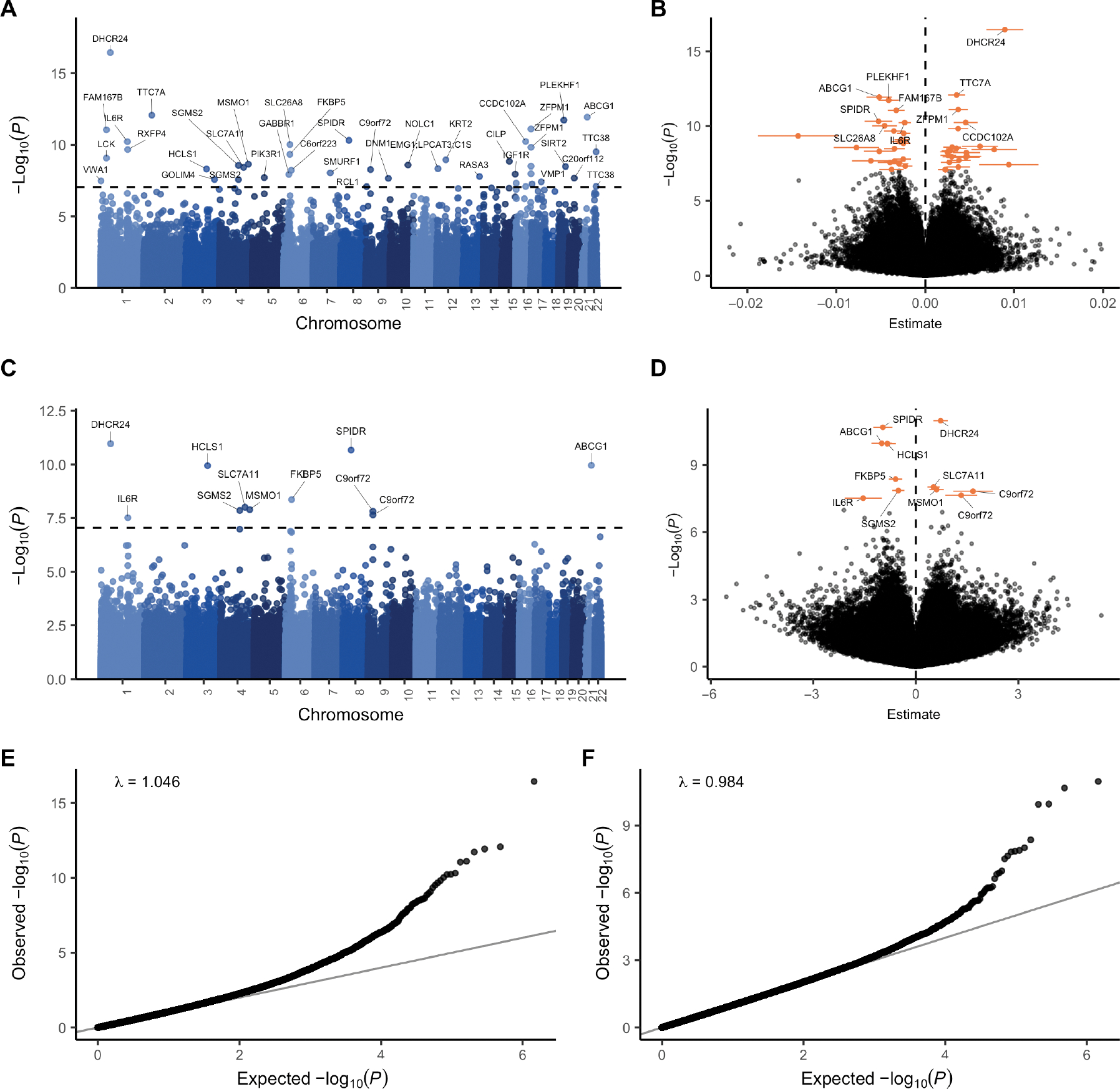
EWAS on 6763 patients and 2943 controls. (A and C) Manhattan plot comparing (A) LB (linear model + bacon) and (C) OSCA MOA association P values [−log10(P), y axis] and genomic location (x axis). The dashed line indicates the genome-wide significance threshold (9 × 10−8). Sites were annotated with the nearest protein-coding gene in ensembl [some gene labels in (A) could not be clearly displayed; all labels are presented in fig. S10]. (B and D) Volcano plots showing (B) LB and (D) OSCA MOA estimated effect sizes (x axis) and association P values [−log10(P), y axis]. Ninety-five percent confidence intervals are shown for DMPs, and the nearest genes are shown for the top 10 DMPs identified with the LB algorithm and for all DMPs identified with the MOA algorithm. (E and F) Quantile-quantile plot showing observed (E) LB and (F) OSCA MOA P values [−log10(P), y axis] against the expected distribution under the null (x axis).
The LB meta-analysis resulted in 44 differentially methylated positions (DMPs) (P < 9 × 10−8; Fig. 1, A and B, Table 2, fig. S10, and data file S3), and the MOA meta-analysis resulted in 11 significant DMPs (P < 9 × 10−8; Fig. 1, C and D, and data file S4) (26). The MOA DMPs comprised a subset of the LB DMPs, with the exception of cg01589155, which is annotated to the C9orf72 locus; this site was significant in MOA (P = 1.51 × 10−8) and just below the significance threshold in the LB results (P = 2.59 × 10−7) (fig. S11). Effect sizes were generally small, and we observed both hypermethylated (51%) and hypomethylated (49%) DMPs associated with ALS (Fig. 1, B and D). On the basis of the nearest gene mapping, these DMPs were annotated to 42 unique genes. In addition, we annotated each site with cis-eQTMs (cis expression quantitative trait methylations) in blood calculated in an external dataset [six Dutch biobanks included in Biobanking and BioMolecular Resources Research Infrastructure (BBMRI) (27)]. This revealed that DNA methylation at 18 sites was significantly associated with the expression of at least one nearby gene [false discovery rate (FDR) < 0.05], which included the nearest gene in 14 of 18 sites (Table 2 and data file S5). The DMPs included multiple colocalized positions (<250 kb), including four DMPs in ZFPM1, two DMPs in C9orf72, two DMPs in SGSM2, two DMPs in TTC38, two DMPs near LCK, and two DMPs in and near GPR97. Most of the colocalized DMPs were highly correlated (|r| > 0.25), and we also found several distant DMPs to be highly correlated (figs. S12 and S13).
Table 2. Top 10 DMPs.
Details of the 10 most strongly associated sites identified with the LB algorithm. Position, Chromosome:bp (GRCh37); Nearest gene, nearest gene based on GRCh37 (Ensembl release 75); cis-eQTM, the top cis-eQTM for the respective probe; cis-eQTM FDR, P value corresponding to the top cis-eQTM, FDR-corrected for the number of tests for the respective probe; B, regression coefficient (representing the mean change in β values) and their 95% confidence intervals; P value, P value from the LB algorithm; PMS indicates that the probe is part of the respective PMS (polymethylation score); Trait, overlap with enriched traits from the MRC-IEU and NGDC EWAS databases (showing a maximum of five traits). HGF, hepatocyte growth factor; N.CDase, neutral ceramidase; FGF.21, fibroblast growth factor 21; CI, confidence interval.
| Probe | Position | Nearest gene | cis-eQTM (direction) | cis-eQTM FDR | B (95% CI) | P value | PMS | Traits |
|---|---|---|---|---|---|---|---|---|
| cg17901584 | 1:55353706 | DHCR24 | DHCR24 (−) | 2.9 × 10−62 | 0.0090 (0.0069–0.011) | 3.6 × 10−17 | BMI, HDL-c, and HGF | Hepatic fat, BMI, metabolic trait, and (serum) triglycerides |
| cg06528816 | 2: 47242277 | TTC7A | TTC7A (−) | 0.13 | 0.0035 (0.0026–0.0045) | 8.5 × 10−13 | Allergic sensitization and total serum IgE | |
| cg06500161 | 21: 43656587 | ABCG1 | ABCG1 (−) | 1.6 × 10−25 | −0.0052 (−0.0066 to −0.0038) | 1.2 × 10−12 | BMI, HDL-c, N.CDase, and FGF.21 | Hepatic fat, BMI, metabolic trait, and (serum) triglycerides |
| cg14945937 | 19: 30162771 | PLEKHF1 | PLEKHF1 (−) | 0.02 | −0.0041 (−0.0053 to −0.003) | 1.9 × 10−12 | ||
| cg08940169 | 16: 88540241 | ZFPM1 | PIEZO1* (−) | 0.08 | 0.0037 (0.0026–0.0048) | 7.8 × 10−12 | Allergic sensitization, total serum IgE, childhood asthma, and schizophrenia | |
| cg07571745 | 1:32715428 | FAM167B | CCDC28B* (−) | 0.26 | −0.0033 (−0.0042 to −0.0023) | 8.9 × 10−12 | ||
| cg14195992 | 8: 48265917 | SPIDR | SPIDR (−) | 0.0059 | −0.0053 (−0.0068 to −0.0037) | 4.8 × 10−11 | Birth weight | |
| cg08851837 | 16: 57558820 | CCDC102A | GPR56* (+) | 0.84 | 0.0045 (0.0032–0.0059) | 5.8 × 10−11 | ||
| cg09257526 | 1:154379696 | IL6R | ATP8B2* (−) | 0.0031 | −0.0023 (−0.003 to −0.0016) | 5.9 × 10−11 | Alcohol consumption per day | |
| cg15782984 | 6: 35993792 | SLC26A8 | SLC26A8 (−) | 0.007 | −0.0046 (−0.0059 to −0.0032) | 9.5 × 10−11 |
The association between DNA methylation and the nearest gene was not significant (FDR > 0.05)
Sensitivity analyses indicate that ALS-associated differential methylation is not driven by genetic variation in cis or trans, riluzole use, or C9orf72 status
We performed sensitivity analyses to evaluate whether our results were driven by known biological factors associated with ALS or by genetic variation. First, we examined the effects of the C9orf72 repeat expansion by performing an EWAS meta-analysis excluding 371 carriers of this mutation. Overall, the results were highly correlated (fig. S14), except for cg01589155 and cg23074747 (located within the C9orf72 repeat and in a CpG island just upstream of the repeat, respectively), which were strongly driven by C9orf72 carrier status. Second, to delineate whether DMPs were influenced by riluzole use, we performed an EWAS on riluzole use in patients with ALS (N users = 1803, N nonusers = 451), finding no evidence of shared signals between the ALS EWAS and the riluzole EWAS (fig. S15). Last, we investigated whether results were driven by genetic variation. For each DMP, we iteratively adjusted for all genetic variants in cis (<250 kb, including variants overlapping the CpG site or probe) as detected in our overlapping whole-genome sequencing (WGS) data (28) (NALS = 5755; Ncontrols = 2184) and blood trans methylation quantitative trait loci (trans-mQTLs)s as reported in the Genetics of DNA Methylation Consortium (GoDMC) database (http://mqtldb.godmc.org.uk). We found no evidence that the DMPs were driven by either genetic variants in cis or in trans (fig. S16).
Enrichment analyses of genes annotated to ALS-associated differential DNA methylation implicate metabolic, inflammatory, and cholesterol pathways
Gene set analysis
To characterize the EWAS results, we performed gene set enrichment analyses based on both nearest genes and cis-eQTMs annotated to each tested position (29, 30). We considered both the default threshold used in the methylGSA package (P < 0.001) and the stringent genome-wide significance threshold (9 × 10−8) to select DMPs for enrichment analyses.
We identified two main categories of enriched pathways: First, in both the LB and MOA results, we identified cholesterol/steroid biosynthesis–related pathways. These included the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway steroid biosynthesis and the gene ontology (GO) pathway cholesterol biosynthetic process, sterol biosynthetic process, organic hydroxy compound biosynthetic process, and secondary alcohol biosynthetic process, which were enriched among the MOA results (Table 3). In addition, we found that these and related pathways were enriched among annotated cis-eQTMs in both the LB and MOA results (table S5). The enrichments were mainly driven by four DMPs: three covarying DMPs in DHCR24 (cg17901584), MSMO1 (cg05119988), and ABCG1 (cg06500161) (figs. S12 and S13) and a DMP in SLC7A11 (cg06690548). Of these, cg17901584, cg05119988, and cg06500161 were strongly associated with the expression of the nearest gene in blood (DHCR24, MSMO1, and ABCG1, respectively; Table 2 and data file S5).
Table 3. Gene set enrichments.
Details of the gene sets that were significantly enriched (FDR < 0.05) among the MOA and LB results based on nearest genes annotated to each site. Method, EWAS method and P value cutoff applied to the respective EWAS test statistics resulting in the input probes for the shown enrichment analyses; N overlap, number of genes that overlap with genes in the respective pathway; N genes, total number of genes in the pathway; FDR, FDR-controlled P values (Wallenius’ noncentral hypergeometric distribution).
| Method | Database | Pathway | N overlap | N genes | FDR |
|---|---|---|---|---|---|
| LB (P < 0.001) | KEGG | Cytokine-cytokine receptor interaction | 36 | 262 | 0.0012 |
| KEGG | NK cell-mediated cytotoxicity | 22 | 108 | 0.036 | |
| MOA (P < 0.001) | – | – | – | – | – |
| LB (P < 9 × 10−8) | – | – | – | – | – |
| MOA (P < 9 × 10−8) | KEGG | Steroid biosynthesis | 2 | 18 | 0.015 |
| GO BP | Cholesterol biosynthetic process | 3 | 71 | 0.021 | |
| GO BP | Sterol biosynthetic process | 3 | 77 | 0.021 | |
| GO BP | Organic hydroxy compound biosynthetic process | 4 | 251 | 0.021 | |
| GO BP | Secondary alcohol biosynthetic process | 3 | 71 | 0.021 |
Second, the immune-related KEGG pathways cytokine–cytokine receptor interaction and natural killer (NK) cell–mediated cytotoxicity were enriched in the LB results (at P < 0.001) but not in the MOA results (Table 3).
EWAS database enrichments
To further characterize the results, we assessed whether the DMPs overlapped with trait-associated positions reported in publicly available EWAS databases (31, 32). For the LB results, we found a significant overlap (FDR < 0.05) with 23 traits in the MRC Integrative Epidemiology Unit (IEU) database (Fig. 2A and Table 4) and 20 traits in the National Genomics Data Center (NGDC) database (fig. S17 and data files S6 and S7), with a total of 23 of 44 DMPs overlapping with one or more enriched traits. For the MOA results, we found a significant overlap (FDR < 0.05) with 20 traits in the MRC-IEU database (fig. S18) and 14 traits in the NGDC database (fig. S19), with a total of 8 of 11 DMPs overlapping with one or more of the enriched traits.
Fig. 2. EWAS database enrichments.
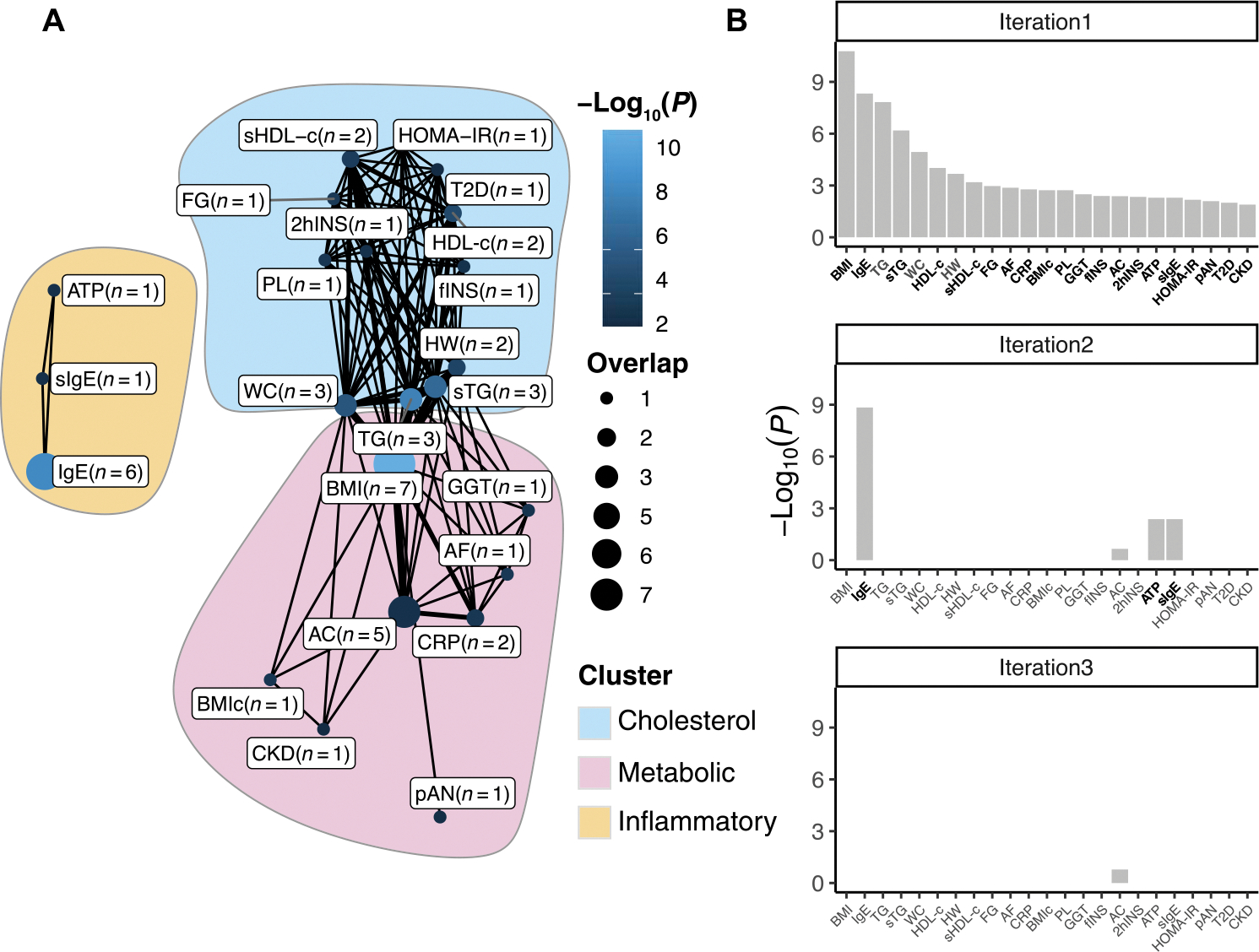
Significant overlap (Fisher’s exact test, FDR < 0.05) between traits included in the MRC-IEU EWAS database and ALS-associated positions identified using the LB model. (A) Network showing the traits that significantly (FDR < 0.05) overlap with the ALS-associated positions. Nodes indicate the overlap between ALS-associated positions and positions associated with indicated traits, with larger nodes indicating more overlap, and lighter shades of blue indicating stronger associations. Edges indicate probe overlap between the traits, with thicker lines indicating more overlapping probes. Colored surfaces indicate the clusters (cholesterol, metabolic, and inflammatory) identified using the Louvain clustering algorithm. (B) Identification of independent clusters of traits. The first iteration shows the traits that significantly overlap with the ALS-associated probes at FDR < 0.05. In subsequent iterations, the probes belonging to the trait with the lowest-enrichment P value were excluded, and enrichment tests were performed using the remaining traits. This algorithm was repeated, retaining traits that were nominally significant (P < 0.05, indicated in bold), until at most one trait remained nominally significant. At the third iteration, no traits remained nominally significant (P < 0.05), showing that both BMI and related traits (including triglycerides and HDL-c) and IgE and related traits (atopy) show independent overlap with the ALS-associated positions. IgE, total serum IgE; TG, triglycerides; sTG, serum triglycerides; WC, waist circumference; sHDL-c, serum HDL-c: HW, hypertriglyceridemic waist; FG, fasting glucose; AF, atrial fibrillation; BMIc, BMI change; PL, postprandial lipemia; GGT, gamma-glutamyl transferase; fINS, fasting insulin; AC, alcohol consumption per day; 2hINS, 2-hour insulin; ATP, atopy; sIgE, high serum IgE; pAN, plasma adiponectin; T2D, type 2 diabetes; CKD, chronic kidney disease; HOMA-IR, homeostatic Model Assessment of Insulin Resistance.
Table 4. EWAS database enrichments.
Ten strongest enrichments within the MRC-IEU EWAS database. FDR is the FDR-corrected P values from a Fisher’s exact test. Effect directions indicate whether the ALS EWAS and trait EWAS effect sizes share the same direction of effect (for example, an opposite direction of effect for BMI indicates that DNA methylation changes at overlapping positions associated with a lower BMI are also associated with a higher ALS risk). EWAS method indicates whether DMPs identified with respective method were enriched for the given trait.
| Trait | FDR | Effect directions | EWAS method |
|---|---|---|---|
| BMI | 1.36 × 10−9 | Opposite | LB and MOA |
| Total serum IgE | 1.93 × 10−7 | Opposite | LB |
| Triglycerides | 4.02 × 10−7 | Opposite | LB and MOA |
| Serum triglycerides* | 1.32 × 10−5 | Opposite | LB and MOA |
| Waist circumference | 1.85 × 10−4 | Opposite | LB and MOA |
| HDL-c | 0.0013 | Equal | LB and MOA |
| Hypertriglyceridemic waist | 0.0024 | Equal | LB and MOA |
| Serum HDL-c* | 0.0066 | Equal | LB and MOA |
| Fasting glucose | 0.0097 | Opposite | LB and MOA |
| Atrial fibrillation | 0.011 | Opposite | LB and MOA |
Note that we adhered to the trait descriptions as provided in the database: serum, plasma, and whole-blood measurements are included as distinct traits (“triglycerides” and “high-density lipoprotein cholesterol” refer to whole-blood measurements).
Among the strongest enrichments in the MRC-IEU database (all results shown in data files S6 and S7) were BMI, total serum immunoglubulin E (IgE) (only enriched among the LB results), (serum) triglycerides, waist circumference, and high-density lipoprotein cholesterol (HDL-c), of which all showed effect directions opposite to those found for ALS, except for HDL-c (Table 4). Using the Louvain clustering algorithm (33), we found that the overlapping traits clustered into two (MOA) to three (LB) clusters. These included two connected cholesterol-related (including HDL-c and triglycerides) and metabolism-related (including BMI and alcohol consumption) clusters, which were identified in the results from both EWAS methods. In addition, in the LB results, we identified an inflammation-related trait cluster that included traits such as total serum IgE and atopy. We found that this inflammation-related cluster was independent of the other clusters, as indicated by iterative analyses presented in Fig. 2B, showing that only the immune-related traits remained significant (P < 0.05) after excluding BMI-related probes (figs. S17 to S19).
Polymethylation scores for BMI, HDL-c, alcohol intake, and white blood cell proportions are associated with ALS
To gain further insight into potential intermediate phenotypes associated with ALS, we used 39 published polymethylation scores (PMSs) as proxies for various traits and exposures, including BMI, HDL-c, low-density lipoprotein cholesterol (LDL-c), total cholesterol, alcohol consumption, smoking, white blood cell (WBC) proportions [CD4T, CD8T, monocytes, granulocytes, and NK cells], biological age, and a collection of immunological and neurological proteins (12–18, 34, 35).
First, we performed a validation analysis for each of the PMSs for which we had relevant clinical/exposure data available (see Materials and Methods and table S4). We selected PMSs with an explained variance of ≥5%, as indicated by an incremental R2 between the null model (including known covariates and control probe PCs) and the model including the respective PMS (Fig. 3A). Two PMSs that were included in the validation analysis did not meet the implemented threshold of ≥5% (LDL-c and total cholesterol).
Fig. 3. Polymethylation score analyses on disease risk and patient survival.
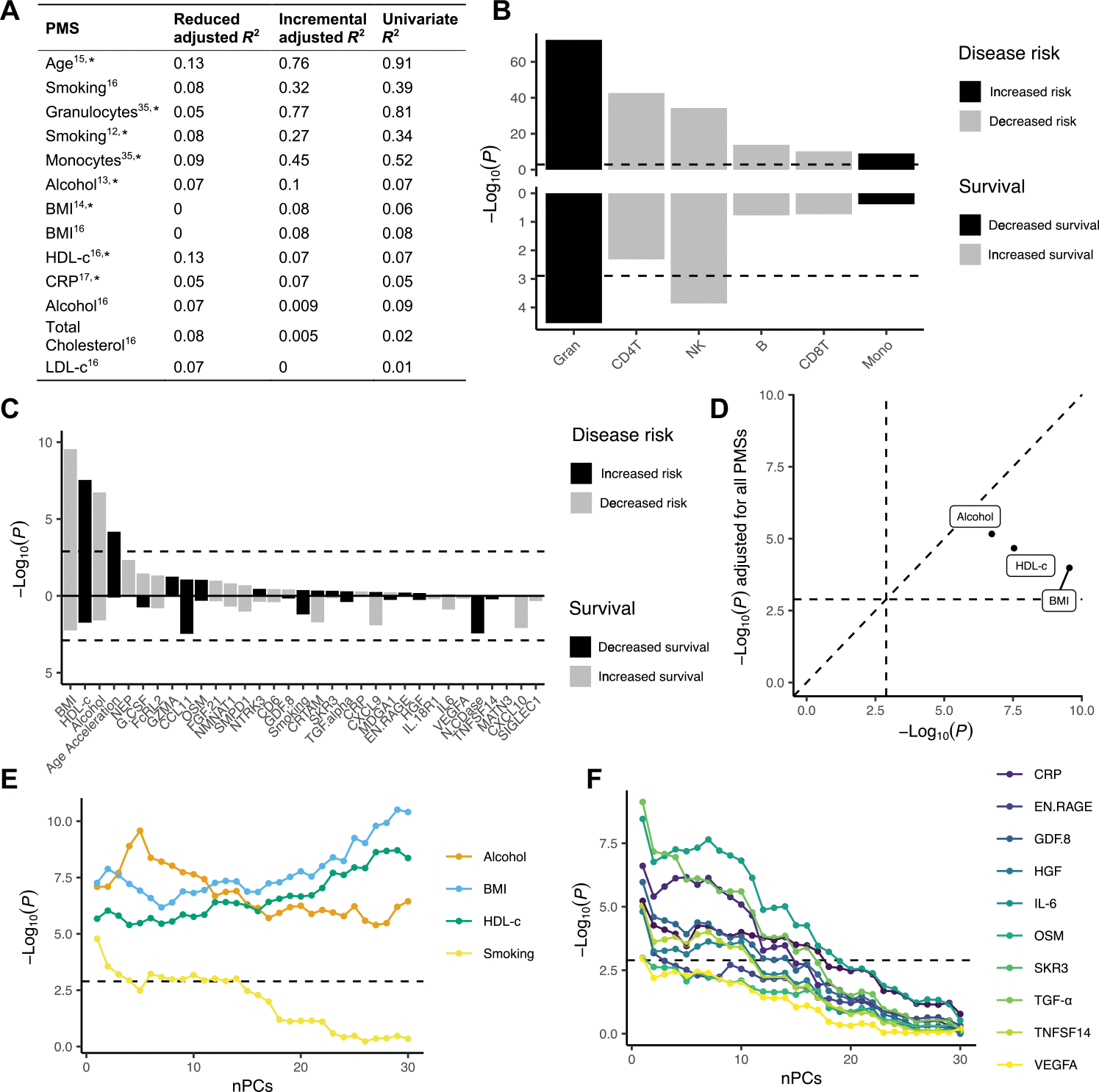
Polymethylation scores (PMSs) were determined as proxies for various traits, exposures, proteins, and WBC proportions, calculated as weighted sums based on probes and weights derived from published papers, respectively. Case-control association analyses were performed on 6763 patients and 2943 controls; survival analyses were performed within 5162 patients. (A) Explained variance of PMSs calculated in samples for which both DNA methylation data and biomarker/clinical data were available (N = 800 of 2000). Reduced R2 represents the variance explained by the null model, whereas the incremental R2 represents the additional variance explained by the PMS over the null model. Last, the explained variance of the univariate model of the respective PMS is displayed (see Materials and Methods). The asterisk indicates that the PMS was used in the association tests. (B and C) The top panel shows association P values from logistic regression [−log10(P), y axis] for each PMS (x axis). (B) WBC proportions and (C) various traits and exposures, colored by whether a higher score is associated with increased (black) or decreased (gray) disease risk. The bottom panel shows the Cox PH P values [−log10(P), y axis] for each PMS (x axis), colored by whether a higher score is associated with decreased (black) or increased (gray) survival, respectively. The dashed line indicates the significance threshold (1.3 × 10−3). (D) Original P values [−log10(P), x axis] compared to P values after including all PMSs as fixed covariates in the logistic regression model [−log10(P), y axis] for the ALS-associated traits/exposures. (E and F) Association P values [−log10(P), y axis] upon incrementally adding principal components (PCs) as fixed covariates in the logistic regression model. HGF, hepatocyte growth factor; EN.RAGE, extracellular newly identified RAGE-binding protein; GDF8, growth/differentiation factor 8; OSM, Oncostatin-M, SKR3, Serine/threonine-protein kinase receptor R3; TNFSF14, tumor necrosis factor ligand superfamily member 14; VEGFA, vascular endothelial growth factor A; nPCs, number of principal components.
We found that PMSs for HDL-c, monocyte cell proportion, and granulocyte cell proportion were positively associated with ALS (P < 1.3 × 10−3; Fig. 3, B and C, and data file S8), and the PMSs for alcohol intake, BMI, and the other WBC proportions (CD4T, CD8T, NK, and B cells) were negatively associated with ALS, a result that reflects the nature of proportion data given the positive associations of other cell types (P < 1.3 × 10−3; Fig. 3, B and C, and data file S8). Although we did find a significant association for epigenetic age acceleration [P = 6.7 × 10−5; clock of Zhang et al. (15) adjusted for chronological age], there was significant heterogeneity between strata (Cochran’s Q test P < 0.1/39; data file S8), which led us to exclude age acceleration for further consideration. In addition, we considered the multitissue clock of Horvath (36) and the clock of Hannum et al. (37), but the associations for both did not pass the multiple testing threshold (P = 1.8 × 10−3 and P = 0.23, respectively, at a multiple testing threshold of 1.3 × 10−3; fig. S20).
Conditional analyses showed that PMSs HDL-c, BMI, and alcohol were independently associated with ALS, although the HDL-c and BMI associations were attenuated after mutual adjustment (Fig. 3D and fig. S21). The WBC associations also remained significant (P < 1.3 × 10−3) after mutual adjustment for the other PMSs, except for a subset of immunological proteins that attenuated the associations (fig. S22). Adjustment for DMPs showed that signal is shared between several DMPs and ALS-associated PMSs (fig. S23); most notably, the alcohol intake association became not significant (P > 1.3 × 10−3) upon adjustment for two covarying DMPs in SLC7A11 (cg06690548) and C6orf223 (cg18120259) (figs. S12 and S13). The HDL-c association became not significant (P > 1.3 × 10−3) upon adjustment for two covarying DMPs in DHCR24 (cg17901584) and ABCG1 (cg06500161) (figs. S12 and S13). We assessed whether the associations were primarily driven by carriers of the C9orf72 repeat expansion but found no evidence that this was the case (fig. S24) nor were the PMS associations primarily driven by specific strata or experimental batches as evidenced by leave-one-out analyses (figs. S25 to S28).
Last, in addition to the PC-adjusted models, we also evaluated less stringent models, showing that various immunological and neurological proteins such as C-reactive protein (CRP), interleukin-6 (IL-6), transforming growth factor–α (TGF-α), and chemokine eotaxin-1 (CCL11) as well as smoking were significantly associated with ALS when PCs were excluded (P < 1.3 × 10−3; Fig 3, E and F, and fig. S29).
Survival analyses indicate that WBC proportions and DNA methylation at five ALS-associated DMPs are associated with disease progression
A total of 5138 patients met the inclusion criteria for the survival analyses (see the “Survival analyses” section in Materials and Methods). Comparison of included and excluded patients (data file S2) shows that both exhibit characteristics that match population-based studies (38). This indicates that we have included a representative sample of patients with the entire spectrum of disease characteristics.
We performed multivariate Cox proportional hazards (PHs) meta-analyses on the 45 DMPs identified using the MOA and LB models. A total of five DMPs showed a significant association with survival after correcting for known confounders and PCs (0.05/45 = p < 1.11 × 10−3) and cross-validation between three sensitivity analyses. Effect sizes were moderate and showed both shorter and longer survival time between DNA methylation and overall survival (data file S9).
All reported positions were not affected by the addition of time-varying effects in the Cox PH model or by applying a restricted cubic spline with varying complexity to model the baseline log cumulative hazard (fig. S30). Moreover, after adjusting for C9orf72 carrier status in the multivariate Cox PH model, the positions (besides the C9orf72 mapped probe) remained significantly associated with survival (P < 1.11 × 10−3; fig. S30). Four positions showed a significant (FDR < 0.05) cis-eQTM effect with FKBP5, ATP8B2, SPIDR, and DHCR24 (Table 5).
Table 5.
DMPs associated with survival. Details of the positions significantly associated with survival (P < 1.11 × 10−3). Position, Chromosome:bp (GRCh37); Nearest gene, nearest gene based on Ensembl GRCh37 (75); cis-eQTM, the top cis-eQTM for the respective probe; cis-eQTM FDR, P value corresponding to the top cis-eQTM, FDR-corrected for the number of tests for the respective probe; PMS, probe is part of the respective PMS (polymethylation score); HR, hazard ratio; Trait, overlap with significantly enriched traits (FDR < 0.05) from the MRC-IEU and NGDC EWAS databases (showing a maximum of five traits). HGF, hepatocyte growth factor. Survival analyses were performed within 5162 patients.
| Probe | Position | Nearest gene | cis-eQTM (direction) | cis-eQTM FDR | HR (95% CI) | P value | PMS | Traits |
|---|---|---|---|---|---|---|---|---|
| cg14195992 | 8:48265917 | SPIDR | SPIDR (−) | 0.0059 | 0.07 (0.025–0.2) | 4.7 × 10−7 | ||
| cg03546163 | 6:35654363 | FKBP5 | FKBP5 (−) | 0.016 | 0.19 (0.087–0.41) | 2.7 × 10−5 | HDL-c | BMI, waist circumference, alcohol consumption per day, and chronic kidney disease |
| cg09257526 | 1:154379696 | IL6R | ATP8B2* (−) | 0.0031 | 0.0049 (0.00045–0.053) | 1.3 × 10−5 | Alcohol consumption per day | |
| cg17901584 | 1:55353706 | DHCR24 | DHCR24 (−) | 2.9 × 10−62 | 4.6 (2.2–9.8) | 1.0 × 10−5 | BMI, HDL-c, HGF | Hepatic fat, BMI, metabolic trait, and (serum) triglycerides |
| cg01589155 | 9:27573532 | C9orf72 | 47 (6.2–360) | 2.0 × 10−4 |
The association between DNA methylation and the nearest gene was not significant (FDR > 0.05).
We also assessed whether the PMSs were associated with survival, finding that a higher proportion of granulocytes was significantly associated with decreased survival and a higher proportion of NK cells was associated with increased survival (P < 1.3 × 10−3; Fig. 3, B and C, bottom; and data file S10). These associations were robust in sensitivity analyses (figs. S31 and S32) and persisted upon adjustment for C9orf72 carrier status (fig. S33).
DISCUSSION
In this study, we present genome-wide DNA methylation data on more than 10,000 individuals, with extensive clinical data and WGS data available for most of the samples. After thorough quality control and extensive sensitivity analyses, we identified a total of 45 DMPs at which variable DNA methylation is robustly associated with ALS (p < 9 × 10−8). By using enrichment analyses, PMSs, and survival analyses, we highlight a role for metabolic, inflammatory, and cholesterol pathways and identify WBC proportions and several DMPs as potential disease modifiers in ALS.
Genes annotated to DMPs were enriched for pathways related to cholesterol biosynthesis. The main drivers of these enrichments include cg17901584 (DHCR24), cg06500161 (ABCG1), cg05119988 (MSMO1), and cg06690548 (SLC7A11), with DNA methylation at the first three positions being associated with expression of their annotated genes in blood. These genes are all involved in cholesterol biosynthesis and lipid transport, and DNA methylation at these positions has been robustly linked to HDL- and total cholesterol, triglyceride concentration, and BMI-related traits such as diabetes and hepatic fat content (31, 32). Both cg17901584 (DHCR24) and cg06500161 (ABCG1) are included in the HDL-cholesterol PMS and explain a considerable part of the association that we found between elevated HDL cholesterol and ALS. Moreover, we identified two covarying probes in SGMS2 (sphingomyelin synthase 2), which is of interest given that altered sphingolipid synthesis has recently been linked to ALS (39).
cg06690548 (annotated to SLC7A11) has also been previously associated with alcohol intake and related factors such as gamma-glutamyl transferase (GGT) and phosphatidylethanol (13, 31, 32), and the association between the alcohol PMS and ALS was primarily driven by this DMP. Alcohol has been extensively studied in previous epidemiological studies of risk factors for ALS with varying results, but a recent review suggests that alcohol has a risk-decreasing effect, which is in line with our current results (40). Previous work showed that increased DNA methylation at cg06690548 is associated with down-regulation of SLC7A11 in brain tissue (41). SLC7A11 encodes xCT, a cystine-glutamate antiporter that imports cystine while exporting glutamate, the former being an essential precursor of glutathione, the major antioxidant in the brain. It is possible, therefore, that the association found in SLC7A11—and by extension alcohol as risk factor for ALS—is related to two well-established pathologic processes in ALS: glutamate excitotoxicity and/or oxidative stress.
Both the EWAS trait enrichments and PMS analyses indicate that lower BMI is associated with ALS. The BMI association persisted after adjustment for other PMSs, including those for HDL cholesterol and alcohol intake, although these PMSs are not perfect proxies of the respective covariates. Lowered BMI throughout the course of the disease (42), as well as various other systemic metabolic alterations, including hypermetabolism and hyperlipidemia (39, 43), have been reported in patients with ALS and mouse models of the disease. Several pathophysiological mechanisms underlying alterations in (lipid) metabolism have been implicated, although it is not clear whether these represent a cause or consequence of the disease. For example, metabolism may be altered because of mitochondrial defects, uncontrolled fasciculations, or increased respiratory effort (43). These findings may be connected as patients with ALS may compensate for hypermetabolism by increasing energy intake that could in turn lead to hyperlipidemia (43). In addition, the immune alterations that we found may be related to these findings, because it has been shown that metabolism and the immune system are connected (44). However, the metabolic- and cholesterol-related findings were statistically independent of the immune-related findings and thus did not support a shared mechanistic pathway. The finding of disrupted metabolic pathways may be a potential avenue for therapeutic intervention, because diet represents a modifiable factor and previous studies in patients with ALS and animals suggest that dietary intervention could benefit disease prognosis, for example, by compensating for defects in lipid metabolism or compensating for increased energy demand or lower BMI (45).
It is important to note that our analyses can say little about causality, and we need to be cautious in concluding that these factors represent major risks for the disease. However, Mendelian randomization (MR) analyses in our latest genome-wide association study (GWAS) (28) indicate that blood cholesterol is causally related to ALS, whereas no causal evidence for (among others) BMI, triglycerides, blood pressure, and other metabolic traits was found. This shows that the causal role for cholesterol in ALS might be independent of other metabolic traits. Although these MR analyses assessed blood cholesterol, neurons are thought to use similar molecular mechanisms (46), and the shared genetic susceptibility between cholesterol and ALS risk could therefore indicate that cholesterol is also raised in the spinal cord and brain. Cholesterol is involved in many crucial processes in the central and peripheral nervous system, including membrane fluidity, synapse formation facilitation, neurite growth, and long-term potentiation (39). Alternatively, it has been suggested that the energetic needs of large motor neurons make it selectively vulnerable for alterations in metabolism or could be the source of oxidative stress (47). Moreover, lipid concentration in the blood and autophagy are related (48), as illustrated by a recent study showing that high cholesterol leads to increased protein aggregation through autophagy impairment in mouse models of Alzheimer’s disease (49).
Our results also point toward a role for the immune system in ALS. The EWAS results were enriched for immune-related traits including IgE and allergic sensitization; these results were independent of predicted WBC proportions. DMPs driving these enrichments included, among others, cg06528816 (annotated to TTC7A) and a cluster of three covarying DMPs in the ZFPM1 gene, both implicated in immune-related traits such as IgE, asthma, and allergic sensitization (31, 32). Our PMS analyses corroborate the role of immunity in ALS because we found that WBC proportions were altered in ALS, with a higher ratio of granulocytes and a lower ratio of lymphocytes in patients with ALS (CD4T, CD8T, and NK cells). We further found that increased granulocyte proportions are associated with worse prognosis, whereas NK cell proportions are associated with better prognosis, indicating that WBC proportions might have prognostic value. The role of immunity is further supported by our observation that various PMSs for various inflammatory proteins including CRP, IL-6, TGF-α, and CCL11 were elevated in patients with ALS; although these differences remained after adjustment for WBC proportions, they disappeared upon adjustment for principal components. Our findings are in line with previous studies that identified higher ratios of neutrophils and/or granulocytes to lymphocytes in patients with ALS, elevated inflammatory proteins, and an association between higher neutrophil proportions and worse prognosis (50, 51). Although immune alterations could be part of a systemic aspect of ALS, there is evidence that suggests that the peripheral immune system contributes to neuroinflammation, the latter being an established phenomenon in ALS as well as other neurodegenerative diseases (50). Especially interesting in this regard are recent analyses showing that mast cells infiltrate skeletal muscles at the neuromuscular junction and degranulate to help recruit neutrophils (50), which prevent reinnervation capacity and may thus be a potential mechanism causing worse prognosis. In line with this, we identified an enrichment for IgE (and related traits such as allergy and atopy), which activate mast cells, and found that increased proportions of granulocytes were associated with ALS and patient survival. Thus, these findings could be of interest for new treatments, especially given that mast cell activity can be influenced therapeutically (50).
We do not replicate the recently reported association between epigenetic age acceleration and survival (52). In our analyses, we adjusted for sampling age, because it has been shown to be crucial when studying epigenetic age acceleration (53), especially given that age of onset affects disease progression in ALS (1). As we have shown, both survival and age of onset were associated with age acceleration when sampling age was not accounted for, but the associations disappeared upon adjustment. In addition, in our case/control analysis, we observed substantial heterogeneity among strata; hence, our results do not support an unambiguous role for age acceleration in ALS.
We must acknowledge the limitations of our study. First, our cross-sectional design hinders inferences about causality. MR analyses presented in our recent GWAS (28) did not find evidence for a causal role of the DMPs identified in this study; although this may indicate a lack of power, it could also indicate that the results reflect the consequences of disease processes rather than causal mechanisms. In that case, the value of the identified DNA methylation changes would lie primarily in revealing underlying disease processes in ALS. Furthermore, the identified ALS- and survival-associated DNA methylation patterns could be of interest as potential starting points for new disease-modifying treatments.
Second, we note that we collected DNA from whole blood rather than from brain tissue. Although some blood DNA methylation patterns reflect those in brain tissue more closely than others—as previously shown for the DMPs that we identified in the C9orf72 locus (54)—DNA methylation is often tissue specific (55). However, in contrast to brain tissue, blood DNA methylation is accessible, allowing for sampling close to disease onset and in large numbers. Leveraging the large body of literature available on blood DNA methylation allowed us to uncover risk factors and pathways related to ALS.
Last, the stringent adjustment for confounding that we applied by using PCs and random effects models [OSCA (21)] may have obscured biological signals of interest. For example, our results indicate that the additional DMPs identified using the LB algorithm are enriched for inflammatory pathways and traits, which corroborates previous findings that suggest that uncaptured variation can be explained by cell type heterogeneity and related immune processes (56). Similarly, we show that the associations found for various immunological proteins such as CRP and IL-6 disappeared upon PC adjustment. This relates to the discussion on whether to treat variables such as cell type proportions as nuisance variables in an EWAS or view them as variables that provide valuable information in themselves (57). In this study, we therefore struck a balance by opting for a two-way approach, combining a stringently corrected EWAS with a more targeted approach where we studied “confounders” such as WBC proportions, smoking, and BMI as outcomes of interest, assessing them with both stringent (including PCs) and more lenient models.
MATERIALS AND METHODS
Study design
This study aimed to identify differential DNA methylation in patients diagnosed with definite, probable, and probable laboratory-supported ALS according to the revised El Escorial Criteria (58). First, we implemented a comprehensive pipeline tailored to large-scale epigenome-wide studies to identify individually methylated positions in 6763 patients with ALS and 2943 controls without motor neuron diseases. We explored the biological meaning of the results by performing gene set enrichment analyses and by overlapping our results with trait-associated positions reported in publicly available EWAS databases. Power analysis calculated with the EPIC array online tool (26) showed that for 96.6% of sites, we had >80% power to detect a mean DNA methylation difference of 1% using the default significance threshold (P < 9 × 10−8). Second, we applied 39 DNA methylation–based proxies of putative ALS risk factors. Last, we leveraged clinical data to perform survival analysis and reveal indicators of disease progression.
Samples were collected across 14 countries (2:1 case/control ratio). Population-based controls were matched for age, sex, and geographical region in a 1:2 ratio and not screened for (subclinical) signs of ALS. Experimental batches were processed in the same laboratory and sequenced in the same series depicting the origin of each DNA sample, resulting in 44 independent batches after quality control. Strata, for analyses, were defined as samples within the Project MinE sequencing consortium stratified by array technology (MinE 450 k and MinE EPIC), and the external Australian data were stratified into two strata based on differences in signal intensities (AUS1 and AUS2) (see Supplementary Materials and Methods “QC and normalization” section, fig. S1, and QC figure 50 for more details).
DNA methylation was quantified using Illumina 450 k and EPIC arrays. We applied extensive quality control leading to the exclusion of 756 (7.2%) samples (based on several technical metrics, relatedness, genotype concordance, and sex concordance) and 175,134 (24%) probes (based on technical metrics, cross-reactivity, and overlap with common single-nucleotide polymorphisms). For further details on cohorts and QC, see Supplementary Materials and Methods. The investigators were not blinded to the experimental conditions during experiments and the analyses.
Statistical analysis
Epigenome-wide association study
Two approaches were used to perform EWAS analyses:
Linear regression was performed at each site, testing for an association between DNA methylation β values and case-control status, adjusting for the following fixed covariates: sex, experimental batch, predicted age, estimated WBC, 30 control probe PCs, and m array-wide residual PCs (see Supplementary Materials and Methods). The number of array-wide PCs (m) was optimized in each stratum by evaluating the sample-size normalized inflation factors (λ1000). The number of PCs (m) were chosen so that for each stratum, λ1000 ≤ 1.15 (m is 30, 15, 25, and 30 for the MinE 450 k, MinE EPIC, AUS1, and AUS2 strata, respectively). We then corrected for remaining inflation and/or bias in test statistics of each stratum using the bacon algorithm (20). Hereafter, we refer to this model as the LB model.
Mixed linear model analyses were performed using the MOA algorithm implemented in the OSCA software (v0.45) (21). This method tests for an association between case-control status and DNA methylation at a given position, adjusting for both fixed effects (we included predicted age, sex, and experimental batch) and a random genome-wide DNA methylation factor per person with variance-covariance matrix between individuals built from genome-wide DNA methylation sites (11, 21, 59).
For both the linear model and MOA results, test statistics across strata were combined using an IVW fixed-effects meta-analysis (22). Positions with a two-tailed P value of <9 × 10−8 were considered genome-wide significant and termed DMPs (26). DMPs were considered significantly heterogeneous when Cochran’s Q P values < 0.1 (corrected for the number of DMPs).
Cis-eQTM analyses
For each position, we tested for an association between DNA methylation and gene expression of genes in cis (<250 kb) using linear regression, adjusting for age, sex, strata, WBC composition, and 20 PCs as fixed effects (10 PCs derived from gene expression data and 10 PCs derived from the DNA methylation data) (27). We corrected the test statistics for bias and inflation (estimated on the basis of the association between DNA methylation and expression of all genes using the bacon algorithm). For each site, two-tailed P values were corrected for the number of tested genes using FDR correction.
Correlation analyses
β values were first adjusted for the covariates used in the LB algorithm; pairwise correlations were calculated among the residuals of this regression using Pearson’s correlation coefficient. Correlations were calculated per stratum (within ALS cases) and combined in an IVW meta-analysis of Fisher’s z-transformed correlation values (22).
Enrichment analyses
Gene set analyses were performed using the Wallenius’ noncentral hypergeometric distribution (29, 30). This method takes into account that the number of CpGs assigned to each gene differs by accounting for the probability of a gene being selected using Wallenius’ noncentral hypergeometric distribution. Two-tailed Fisher’s exact tests were used for trait enrichment analyses. Resulting P values from the enrichment analyses were corrected for multiple testing using FDR correction. The filtering procedure for gene sets and traits and the backgrounds used are described in Supplementary Materials and Methods.
PMS analyses
Incremental R2 estimates from linear regression were used to determine whether the PMS increased the predictive ability above and beyond that of the null model that included the phenotype measure as the dependent variable and case-control status, predicted age, sex, experimental batch, WBC, and 30 control probe PCs as independent variables. For each stratum, we tested for an association between the PMS and case/control status using logistic regression. Sex, predicted age, WBC, experimental batch, 30 control probe PCs, and m array-wide PCs (see the “Epigenome-wide association study” section) were included as fixed covariates for all PMSs except for DNA methylation age and WBC proportions. For DNA methylation age, we additionally adjusted for chronological age (representing age acceleration). For the WBC PMSs, we did not adjust for array-wide PCs because these essentially represent WBC proportions (56). Strata test statistics were combined using an IVW fixed-effects meta-analysis (22). We corrected for the number of PMSs tested using the Bonferroni correction [two-tailed P value of <1.3 × 10−3 (0.05/39)]. PMSs were considered significantly heterogeneous when Cochran’s Q P values < 2.3 × 10−3 (0.01/39).
Survival analyses
We used a multivariate Cox PH regression model to test for an association between survival and DMPs and PMSs, adjusting for predicted age, sex, experimental batch, WBC, 30 control probe PCs, and m array-wide PCs (see the “Epigenome-wide association study” section). The PH assumption of the Cox model was checked using Schoenfeld and martingale residuals. In addition, the Royston-Parmar spline model was performed using the flexsurvspline function from the R package flexsurv. Model complexity was assessed by the addition of up to five knots compared to one single knot. Test statistics were combined using IVW fixed-effects meta-analysis. We corrected for the number of tests using the Bonferroni correction (two-tailed P < 1.11 × 10−3 for DMPs and two-tailed P < 1.3 × 10−3 for PMSs). Positions were considered significantly heterogeneous when Cochran’s Q P values < 0.1 (corrected for the number of tests performed).
Supplementary Material
Acknowledgments:
We acknowledge the Older Australian Twins Study (OATS) research team: https://cheba.unsw.edu.au/project/older-australian-twins-study. We thank the participants and their informants for their time and generosity in contributing to this research.
Funding:
The research reported in this publication was supported by grants from The Dutch Research Council (NWO) (VENI scheme grant 09150161810018 to W.v.R.) and Prinses Beatrix Spierfond (neuromuscular fellowship grant W.F19–03 to W.v.R.), The Prinses Beatrix Spierfonds (W.OR20–08 to J.J.F.A.v.V. and J.H.V.), The Canadian Institutes of Health Research (FRN 159279 to J.P.R.), The Dutch Research Council (NWO) (VIDI grant 91719350 to K.P.K.), The European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 772376-EScORIAL to J.H.V.), the Swedish Brain Foundation (grant nos. 2012–0262, 2012–0305, 2013–0279, 2016–0303, 2018–0310, and 2020–0353 to P.M.A.), the Swedish Research Council (grant nos. 2012–3167 and 2017–03100 to P.M.A.), the Knut and Alice Wallenberg Foundation (grant nos. 2012.0091, 2014.0305, and 2020.0232 to P.M.A.), the Ulla-Carin Lindquist Foundation and the Västerbotten County Council (grant no. 56103–7002829 to P.M.A.), and King Gustaf V’s and Queen Victoria’s Freemason’s Foundation. This is an EU Joint Programme–Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organizations under the aegis of JPND (www.jpnd.eu) [United Kingdom, Medical Research Council (MR/L501529/1; MR/R024804/1) and Economic and Social Research Council (ES/L008238/1)] and through the Motor Neurone Disease Association (MNDA). This study represents independent research part funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. A.A.-C. is supported by an NIHR Senior Investigator Award. Samples used in this research were entirely/in part obtained from the U.K. National DNA Bank for MND Research, funded by the MND Association and the Wellcome Trust. We would like to thank people with MND and their families for their participation in this project. We acknowledge sample management undertaken by Biobanking Solutions funded by the Medical Research Council at the Centre for Integrated Genomic Medical Research, University of Manchester. R.J.P. is funded through the Gravitation program of the Dutch Ministry of Education, Culture, and Science and the Netherlands Organization for Scientific Research (BRAINSCAPES). G.L.S. was supported by a PhD studentship from the Alzheimer’s Society. S.T.N. acknowledges support through a FightMND Mid-Career Fellowship. V.S. is supported by the Italian Ministry of Health, AriSLA, and E-Rare Joint Transnational Call. A.A.K. is funded by the MNDA and NIHR Maudsley Biomedical Research Centre. D.B., E.T., and H.R. are employees of Biogen. L.H.v.d.B. reports grants from the Netherlands ALS Foundation, grants from The Netherlands Organization for Health Research and Development (Vici scheme), grants from The European Community’s Health Seventh Framework Programme [grant agreement no. 259867 (EuroMOTOR) to L.H.v.d.B.], and grants from The Netherlands Organization for Health Research and Development (the STRENGTH project, funded through the EU Joint Programme–Neurodegenerative Disease Research, JPND), during the conduct of the study. Project MinE Belgium was supported by a grant from IWT (no. 140935), the ALS Liga België, the National Lottery of Belgium, and the KU Leuven Opening the Future Fund. P.V.D. holds a senior clinical investigatorship of FWO-Vlaanderen and is supported by the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders, the ALS Liga België, and the KU Leuven funds “Een Hart voor ALS”, “Laeversfonds voor ALS Onderzoek”, and the “Valéry Perrier Race against ALS Fund”. This work was supported by the Italian Ministry of Health (Ministero della Salute, Ricerca Sanitaria Finalizzata, grant RF-2016–02362405 to A. Chiò), the Progetti di Rilevante Interesse Nazionale program of the Ministry of Education, University and Research (grant 2017SNW5MB to A. Chiò); the European Commission’s Health Seventh Framework Programme (FP7/2007–2013 under grant agreement 259867 to A. Chiò), and the Joint Programme–Neurodegenerative Disease Research (Strength, ALS-Care and Brain-Mend projects), granted by Italian Ministry of Education, University, and Research. This study was performed under the Department of Excellence grant of the Italian Ministry of Education, University and Research to the “Rita Levi Montalcini” Department of Neuroscience, University of Torino, Italy. We acknowledge funding from the Australian National Health and Medical Research (NHMRC) Council: 1151854, 1083187, 1173790, 1078901, 1113400, 1095215, and 1176913 Enabling Grant #402703 to N.R.W. Additional funding was provided by the Motor Neurone Disease Research Institute of Australia Ice Bucket Challenge grant for the SALSA-SGC consortium. The OATS (used for controls) was facilitated through Twins Research Australia, a national resource in part supported by a Centre for Research Excellence from the Australian NHMRC Council (NHMRC 1079102 to N.R.W.). Funding for this study was awarded by the (NHMRC)/Australian Research Council Strategic Award (grant 401162 to N.R.W.) and NHMRC grants (1405325, 1024224, 1025243, 1045325, 1085606, 568969, and 1093083 to N.R.W.). The collaboration project is cofunded by the PPP Allowance made available by Health~Holland, Top Sector Life Sciences & Health, to stimulate public-private partnerships. This study was supported by the ALS Foundation Netherlands. This work was sponsored by NWO Domain Science for the use of the national computer facilities. A.N.B. is grateful to the Suna and Inan Kirac Foundation and Koc University for the excellent research environment created and for financial support. G.A.R. is supported by the Canadian Institutes of Health. Several authors of this publication are members of the Netherlands Neuromuscular Center (NL-NMD) and the European Reference Network for rare neuromuscular diseases EURO-NMD. French ALS patients of the Pitié-Salpêtrière hospital (Paris) have been collected with ARSla funding support.
Footnotes
Competing interests: J.H.V. has sponsored research agreements with Biogen. L.H.v.d.B. receives personal fees from Cytokinetics, outside the submitted work. A.A.-C. has served on scientific advisory boards for Mitsubishi Tanabe Pharma, OrionPharma, Biogen Idec, Lilly, GSK, Apellis, Amylyx, and Wave Therapeutics. A.C. serves on scientific advisory boards for Mitsubishi Tanabe, Roche, Biogen, Denali, and Cytokinetics. P.V.D. reports grants from CSL Behring (paid to institution and participated in advisory board meetings of Biogen, Alexion Pharmaceuticals, Ferrer, QurAlis, argenx, UCB, and Augustine Therapeutics (paid to institution). P.M.A. works as a consultant to Biogen, Roche, Avrion, Regeneron, and Orphazyme; as a clinical trial site investigator for Biogen, Alexion, Sanofi, AL-S Pharma, Amylyx, and Orphazyme; as, since 1993, Director of the ALS genetic laboratory at Umeå University Hospital that performs genetic testing for SOD1 mutation; and as a member of the ClinGen ALS Gene Curation Expert panel.
Consortia
BIOS (Biobank-based Integrative Omics Study) Consortium
Management team Bastiaan T. Heijmans61 (chair), Peter A.C. t Hoen62, Joyce van Meurs63, Rick Jansen64, Lude Franke65
Cohort collection Dorret I. Boomsma66, René Pool66, Jenny van Dongen66, Joukje J. Hottenga66 (Netherlands Twin Register); Marleen M.J. van Greevenbroek67, Coen D.A. Stehouwer67, Carla J.H. van der Kallen67, Casper G. Schalkwijk67 (Cohort study on Diabetes and Atherosclerosis Maastricht); Cisca Wijmenga65, Lude Franke65, Sasha Zhernakova65, Ettje F. Tigchelaar65 (LifeLines Deep); P. Eline Slagboom61, Marian Beekman61, Joris Deelen61, Diana van Heemst68 (Leiden Longevity Study); Jan H. Veldink1, Leonard H. van den Berg1 (Prospective ALS Study Netherlands); Cornelia M. van Duijn69, Bert A. Hofman70, Aaron Isaacs69, André G. Uitterlinden63 (Rotterdam Study)
Data generation Joyce van Meurs63 (chair), P. Mila Jhamai63, Michael Verbiest63, H. Eka D. Suchiman61, Marijn Verkerk63, Ruud van der Breggen61, Jeroen van Rooij63, Nico Lakenberg61
Data management and computational infrastructure Hailiang Mei71 (chair), Maarten van Iterson61, Michiel van Galen62, Jan Bot72, Dasha V. Zhernakova65, Rick Jansen64, Peter van ‘t Hof71, Patrick Deelen65, Irene Nooren72, Peter A.C. t Hoen62, Bastiaan T. Heijmans61, Matthijs Moed61
Data analysis group Lude Franke65 (co-chair), Martijn Vermaat62, Dasha V. Zhernakova65, René Luijk61, Marc Jan Bonder65, Maarten van Iterson61, Patrick Deelen65, Freerk van Dijk 73, Michiel van Galen62, Wibowo Arindrarto71, Szymon M. Kielbasa74, Morris A. Swertz73, Erik W. van Zwet74, Rick Jansen64, Peter A.C. t Hoen62 (co-chair), Bastiaan T. Heijmans61 (co-chair)
Affiliations
61Molecular Epidemiology, Department of Biomedical Data Sciences, Leiden University Medical Center, Leiden 2300 RC, Netherlands. 62Department of Human Genetics, Leiden University Medical Center, Leiden 2300 RC, Netherlands. 63Department of Internal Medicine, ErasmusMC, Rotterdam 3015 GD, Netherlands. 64Department of Psychiatry, VU University Medical Center, Neuroscience Campus Amsterdam, Amsterdam 1081 HV, Netherlands. 65Department of Genetics, University of Groningen, University Medical Centre Groningen, Groningen 9700 RB, Netherlands. 66Department of Biological Psychology, VU University Amsterdam, Neuroscience Campus Amsterdam, Amsterdam 1081 HV, Netherlands. 67Department of Internal Medicine and School for Cardiovascular Diseases (CARIM), Maastricht University Medical Center, Maastricht 6211 LK, Netherlands. 68Department of Gerontology and Geriatrics, Leiden University Medical Center, Leiden 2300 RC, Netherlands. 69Department of Genetic Epidemiology, ErasmusMC, Rotterdam 3015 GD, Netherlands. 70Department of Epidemiology, ErasmusMC, Rotterdam 3015 GD, Netherlands. 71Sequence Analysis Support Core, Department of Biomedical Data Sciences, Leiden University Medical Center, Leiden 2300 RC, Netherlands. 72SURFsara, Amsterdam 1098 XG, Netherlands. 73Genomics Coordination Center, University Medical Center Groningen, University of Groningen, Groningen 9700 RB, Netherlands. 74Medical Statistics, Department of Biomedical Data Sciences, Leiden University Medical Center, Leiden 2300 RC, Netherlands.
BRAIN MEND Consortium (Biological Resource Analysis to Identify New MEchanisms and phenotypes in Neurodegenerative Diseases Consortium)
Ammar Al-Chalabi9,60, Naomi R. Wray3,44, Gilbert Bensimon75,44, Orla Hardiman59, Adriano Chiò18, Jan H. Veldink1, George Davey Smith76,77, Jonathan Mill2
Affiliations
75Département de Pharmacologie Clinique, Hôpital de la Pitié-Salpêtrière, UPMC Pharmacologie, AP-HP, Paris 75013, France. 76MRC Integrative Epidemiology Unit, University of Bristol, Bristol BS8 1TH, UK. 77Population Health Science, Bristol Medical School, Bristol, Bristol BS8 1TH, UK.
Data and materials availability:
All summary statistics are available as supplementary data. Individual-level DNA methylation data from Project MinE are available upon request in the European Genome-phenome Archive (EGAS00001004587). The DNA methylation data for the Australian cohorts are deposited on dbGAP and available under accession number phs002068.v1.p1. Code is available at https://github.com/pjhop/EWAS_MinE.
REFERENCES AND NOTES
- 1.van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, van den Berg LH, Amyotrophic lateral sclerosis. Lancet 390, 2084–2098 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Ryan M, Heverin M, McLaughlin RL, Hardiman O, Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol. 76, 1367–1374 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Chalabi A, Hardiman O, The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 9, 617–628 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Xue A, Jiang L, Zhu Z, Wray NR, Visscher PM, Zeng J, Yang J, Genome-wide analyses of behavioural traits are subject to bias by misreports and longitudinal changes. Nat. Commun. 12, 20211 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armon C, An evidence-based medicine approach to the evaluation of the role of exogenous risk factors in sporadic amyotrophic lateral sclerosis. Neuroepidemiology 22, 217–228 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Martin S, Al Khleifat A, Al-Chalabi A, What causes amyotrophic lateral sclerosis? F1000Res. 6, 371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones PA, Takai D, The role of DNA methylation in mammalian epigenetics. Science 293, 1068–1070 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Schübeler D, Function and information content of DNA methylation. Nature 517, 321–326 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Smith ZD, Meissner A, DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 14, 204–220 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Dor Y, Cedar H, Principles of DNA methylation and their implications for biology and medicine. Lancet 392, 777–786 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Nabais MF, Laws SM, Lin T, Vallerga CL, Armstrong NJ, Blair IP, Kwok JB, Mather KA, Mellick GD, Sachdev PS, Wallace L, Henders AK, Zwamborn RAJ, Hop PJ, Lunnon K, Pishva E, Roubroeks JAY, Soininen H, Tsolaki M, Mecocci P, Lovestone S, Kłoszewska I, Vellas B, Furlong S, Garton FC, Henderson RD, Mathers S, McCombe PA, Needham M, Ngo ST, Nicholson G, Pamphlett R, Rowe DB, Steyn FJ, Williams KL, Anderson TJ, Bentley SR, Dalrymple-Alford J, Fowder J, Gratten J, Halliday G, Hickie IB, Kennedy M, Lewis SJG, Montgomery GW, Pearson J, Pitcher TL, Silburn P, Zhang F, Visscher PM, Yang J, Stevenson AJ, Hillary RF, Marioni RE, Harris SE, Deary IJ, Jones AR, Shatunov A, Iacoangeli A, van Rheenen W, van den Berg LH, Shaw PJ, Shaw CE, Morrison KE, Al-Chalabi A, Veldink JH, Hannon E, Mill J, Wray NR, McRae AF, Meta-analysis of genome-wide DNA methylation identifies shared associations across neurodegenerative disorders. Genome Biol. 22, 90 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bollepalli S, Korhonen T, Kaprio J, Anders S, Ollikainen M, EpiSmokEr: A robust classifier to determine smoking status from DNA methylation data. Epigenomics 11, 1469–1486 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Liu C, Marioni RE, Hedman ÅK, Pfeiffer L, Tsai P-C, Reynolds LM, Just AC, Duan Q, Boer CG, Tanaka T, Elks CE, Aslibekyan S, Brody JA, Kühnel B, Herder C, Almli LM, Zhi D, Wang Y, Huan T, Yao C, Mendelson MM, Joehanes R, Liang L, Love S-A, Guan W, Shah S, A. F M.c. Rae A. Kretschmer H. Prokisch K. Strauch A. Peters PM. Visscher NR. Wray X. Guo KL. Wiggins AK. Smith EB. Binder KJ. Ressler MR. Irvin DM. Absher D. Hernandez L. Ferrucci S. Bandinelli K. Lohman J. Ding L. Trevisi S. Gustafsson JH. Sandling L. Stolk AG. Uitterlinden I. Yet JE. Castillo-Fernandez TD. Spector JD. Schwartz P Vokonas L. Lind Y. Li M. Fornage DK. Arnett NJ Wareham N. Sotoodehnia KK. Ong JB. van Meurs J, Conneely KN, Baccarelli AA, Deary IJ, Bell JT, North KE, Liu Y, Waldenberger M, London SJ, Ingelsson E, Levy D, A DNA methylation biomarker of alcohol consumption. Mol. Psychiatry 23, 422–433 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamilton OKL, Zhang Q, McRae AF, Walker RM, Morris SW, Redmond P, Campbell A, Murray AD, Porteous DJ, Evans KL, McIntosh AM, Deary IJ, Marioni RE, An epigenetic score for BMI based on DNA methylation correlates with poor physical health and major disease in the Lothian Birth Cohort. Int. J. Obes. 43, 1795–1802 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Q, Vallerga CL, Walker RM, Lin T, Henders AK, Montgomery GW, He J, Fan D, Fowdar J, Kennedy M, Pitcher T, Pearson J, Halliday G, Kwok JB, Hickie I, Lewis S, Anderson T, Silburn PA, Mellick GD, Harris SE, Redmond P, Murray AD, Porteous DJ, Haley CS, Evans KL, McIntosh AM, Yang J, Gratten J, Marioni RE, Wray NR, Deary IJ, McRae AF, Visscher PM, Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 11, 54 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCartney DL, Hillary RF, Stevenson AJ, Ritchie SJ, Walker RM, Zhang Q, Morris SW, Bermingham ML, Campbell A, Murray AD, Whalley HC, Gale CR, Porteous DJ, Haley CS, McRae AF, Wray NR, Visscher PM, McIntosh AM, Evans KL, Deary IJ, Marioni RE, Epigenetic prediction of complex traits and death. Genome Biol. 19, 136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ligthart S, Marzi C, Aslibekyan S, Mendelson MM, Conneely KN, Tanaka T, Colicino E, Waite LL, Joehanes R, Guan W, Brody JA, Elks C, Marioni R, Jhun MA, Agha G, Bressler J, Ward-Caviness CK, Chen BH, Huan T, Bakulski K, Salfati EL; WHI-EMPC Investigators G Fiorito; CHARGE epigenetics of Coronary Heart Disease S. Wahl K Schramm J Sha DG Hernandez AC Just JA. Smith N. Sotoodehnia LC Pilling JS Pankow PS Tsao C Liu W. Zhao S. Guarrera VJ. Michopoulos AK. Smith MJ. Peters D. Melzer P. Vokonas M. Fornage H Prokisch JC. Bis AY. Chu C. Herder H. Grallert C. Yao S. Shah A.l. F. Mc Rae H. Lin S. Horvath D. Fallin A. Hofman NJ. Wareham KL. Wiggins AP. Feinberg JM. Starr PM. Visscher JM Murabito SLR. Kardia DM Absher EB. Binder AB. Singleton S. Bandinelli A. Peters M. Waldenberger G. Matullo JD. Schwartz EW. Demerath AG. Uitterlinden JBJ. van Meurs OH Franco Y-D. Chen I, Levy D, Turner ST, Deary IJ, Ressler KJ, Dupuis J, Ferrucci L, Ong KK, Assimes TL, Boerwinkle E, Koenig W, Arnett DK, Baccarelli AA, Benjamin EJ, Dehghan A, DNA methylation signatures of chronic low-grade inflammation are associated with complex diseases. Genome Biol. 17, 255 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gadd DA, Hillary RF, McCartney DL, Stevenson AJ, Nangle C, Flaig R, Harris SE, Walker RM, Shi L, Tucker-Drob EM, Deary IJ, Porteous DJ, Hayward C, Visscher PM, Cox SR, Evans KL, McIntosh AM, Marioni RE, DNA methylation proxies for 16 plasma proteins predict the incidence of 7 leading causes of morbidity. BioRxiv, 2020.12.01.404681 (2020). [Google Scholar]
- 19.Amador C, Zeng Y, Barber M, Walker RM, Campbell A, McIntosh AM, Evans KL, Porteous DJ, Hayward C, Wilson JF, Navarro P, Haley CS, Genome-wide methylation data improves dissection of the effect ofsmoking on body mass index. PLOS Genet. 17, e1009750 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Iterson M, van Zwet EW, Heijmans BT, Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol. 18, 19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang F, Chen W, Zhu Z, Zhang Q, Nabais MF, Qi T, Deary IJ, Wray NR, Visscher PM, McRae AF, Yang J, OSCA: A tool for omic-data-based complex trait analysis. Genome Biol. 20, 107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwarzer G, meta: An R package for meta-analysis (2019). [Google Scholar]
- 23.Fortin J-P, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, Greenwood CM, Hansen KD, Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 15, 503 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pidsley R, Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC, A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics 14, 293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hop PJ, Zwamborn RAJ, Hannon EJ, Dekker AM, van Eijk KR, Walker EM, Iacoangeli A, Jones AR, Shatunov A, Khleifat AA, Opie-Martin S, Shaw CE, Morrison KE, Shaw PJ, McLaughlin RL, Hardiman O, Al-Chalabi A, van den Berg LH, Mill J, Veldink JH, Cross-reactive probes on Illumina DNA methylation arrays: Alarge study on ALS shows that a cautionary approach is warranted in interpreting epigenome-wide association studies. NAR Genom. Bioinformatics 2, lqaa105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mansell G, Gorrie-Stone TJ, Bao Y, Kumari M, Schalkwyk LS, Mill J, Hannon E, Guidance for DNA methylation studies: Statistical insights from the Illumina EPIC array. BMC Genomics 20, 366 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hop PJ, Luijk R, Daxinger L, van Iterson M, Dekkers KF, Jansen R; BIOS Consortium JBJ van Meurs, P. A. C. ‘t Hoen, M. A. Ikram, M. M. J. Greevenbroek, D. I. Boomsma, P. E. Slagboom, J. H. Veldink, E. W. van Zwet, B. T. Heijmans, Genome-wide identification of genes regulating DNA methylation using genetic anchors for causal inference. Genome Biol. 21, 220 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Rheenen W, van der Spek RAA, Bakker MK, van Vugt JJFA, Hop PJ, Zwamborn RAJ, de Klein N, Westra H-J, Bakker OB, Deelen P, Shireby G, Hannon E, Moisse M, Baird D, Restuadi R, Dolzhenko E, Dekker AM, Gawor K, Westeneng H-J, Tazelaar GHP, van Eijk KR, Kooyman M, Byrne RP, Doherty M, Heverin M, Al Khleifat A, Iacoangeli A, Shatunov A, Ticozzi N, Cooper-Knock J, Smith BN, Gromicho M, Chandran S, Pal S, Morrison KE, Shaw PJ, Hardy J, Orrell RW, Sendtner M, Meyer T, Başak N, van der Kooi AJ, Ratti A, Fogh I, Gellera C, Lauria G, Corti S, Cereda C, Sproviero D, D’Alfonso S, Sorarù G, Siciliano G, Filosto M, Padovani A, Chiò A, Calvo A, Moglia C, Brunetti M, Canosa A, Grassano M, Beghi E, Pupillo E, Logroscino G, Nefussy B, Osmanovic A, Nordin A, Lerner Y, Zabari M, Gotkine M, Baloh RH, Bell S, Vourc’h P, Corcia P, Couratier P, Millecamps S, Meininger V, Salachas F, J. S M.o. Pardina A. Assialioui R. Rojas-García PA. Dion JP. Ross AC. Ludolph JH. Weishaupt D. Brenner A. Freischmidt G. Bensimon A. Brice A. Durr CAM. Payan S. Saker-Delye NW. Wood S. Topp R. Rademakers L. Tittmann W. Lieb A. Franke S. Ripke A. Braun J. Kraft DC. Whiteman CM. Olsen AG. Uitterlinden A. Hofman M. Rietschel S. Cichon MM. Nöthen P. Amouyel; SLALOM Consortium; PARALS Consortium; SLAGEN Consortium; SLAP Consortium BJ. Traynor AB Singleton M. Neto M.i., Cauchi RJ, Ophoff RA, Wiedau-Pazos M, Lomen-Hoerth C, van Deerlin VM, Grosskreutz J, Roediger A, Gaur N, Jörk A, Barthel T, Theele E, Ilse B, Stubendorff B, Witte OW, Steinbach R, Hübner CA, Graff C, Brylev L, Fominykh V, Demeshonok V, Ataulina A, Rogelj B, Koritnik B, Zidar J, Ravnik-Glavač M, Glavač D, Stević Z, Drory V, Povedano M, Blair IP, Kiernan MC, Benyamin B, Henderson RD, Furlong S, Mathers S, McCombe PA, Needham M, Ngo ST, Nicholson GA, Pamphlett R, Rowe DB, Steyn FJ, Williams KL, Mather KA, Sachdev PS, Henders AK, Wallace L, de Carvalho M, Pinto S, Petri S, Weber M, Rouleau GA, Silani V, Curtis CJ, Breen G, Glass JD, Brown RH, Landers JE, Shaw CE, Andersen PM, Groen EJN, van Es MA, Pasterkamp RJ, Fan D, Garton FC, McRae AF, Smith G. D.a, Gaunt TR, Eberle MA, Mill J, McLaughlin RL, Hardiman O, Kenna KP, Wray NR, Tsai E, Runz H, Franke L, Al-Chalabi A, Van Damme P, van den Berg LH, Veldink JH, Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 53, 1636–1648 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phipson B, Maksimovic J, Oshlack A, missMethyl: An R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics 32, 286–288 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Ren X, Kuan PF, methylGSA: A Bioconductor package and Shiny app for DNA methylation data length bias adjustment in gene set testing. Bioinformatics 35, 1958–1959 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Staley J, Battram T, Crawford G, Prince C, Babaei MS, Hemani G, Suderman M, Relton C, Gaunt T, EWAS Catalog: The MRC-IEU catalog of epigenome-wide association studies [accessed 6 June 2020 at http://ewascatalog.org/].
- 32.Liu D, Zhao L, Wang Z, Zhou X, Fan X, Li Y, Xu J, Hu S, Niu M, Song X, Li Y, Zuo L, Lei C, Zhang M, Tang G, Huang M, Zhang N, Duan L, Lv H, Zhang M, Li J, Xu L, Kong F, Feng R, Jiang Y, EWASdb: Epigenome-wide association study database. Nucleic Acids Res. 47, D989–D993 (2019), [accessed 6 June 202 at https://ngdc.cncb.ac.cn/ewas/atlas]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E, Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, P10008 (2008). [Google Scholar]
- 34.Yousefi PD, Richmond R, Langdon R, Ness A, Liu C, Levy D, Relton C, Suderman M, Zuccolo L, Validation and characterisation of a DNA methylation alcohol biomarker across the life course. Clin. Epigenetics 11, 163 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teschendorff AE, Breeze CE, Zheng SC, Beck S, A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies. BMC Bioinformatics 18, 105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horvath S, DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan J-B, Gao Y, Deconde R, Chen M, Rajapakse I, Friend S, Ideker T, Zhang K, Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hardiman O, Al-Chalabi A, Brayne C, Beghi E, van den Berg LH, Chio A, Martin S, Logroscino G, Rooney J, The changing picture of amyotrophic lateral sclerosis: Lessons from European registers. J. Neurol. Neurosurg. Psychiatry 88, 557–563 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Tracey TJ, Kirk SE, Steyn FJ, Ngo ST, The role of lipids in the central nervous system and their pathological implications in amyotrophic lateral sclerosis. Semin. Cell Dev. Biol. 112, 69–81 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Peng B, Yang Q, Joshi RB, Liu Y, Akbar M, Song B-J, Zhou S, Wang X, Role of alcohol drinking in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Int. J. Mol. Sci. 21, 2316 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vallerga CL, Zhang F, Fowdar J, McRae AF, Qi T, Nabais MF, Zhang Q, Kassam I, Henders AK, Wallace L, Montgomery G, Chuang Y-H, Horvath S, Ritz B, Halliday G, Hickie I, Kwok JB, Pearson J, Pitcher T, Kennedy M, Bentley SR, Silburn PA, Yang J, Wray NR, Lewis SJG, Anderson T, Dalrymple-Alford J, Mellick GD, Visscher PM, Gratten J, Analysis of DNA methylation associates the cystine–glutamate antiporter SLC7A11 with risk of Parkinson’s disease. Nat. Commun. 11, 1238 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Westeneng H-J, van Veenhuijzen K, van der Spek RA, Peters S, Visser AE, van Rheenen W, Veldink JH, van den Berg LH, Associations between lifestyle and amyotrophic lateral sclerosis stratified by C9orf72 genotype: A longitudinal, population-based, case-control study. Lancet Neurol. 20, 373–384 (2021). [DOI] [PubMed] [Google Scholar]
- 43.Dupuis L, Pradat P-F, Ludolph AC, Loeffler J-P, Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 10, 75–82 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Tall AR, Yvan-Charvet L, Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 15, 104–116 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pape JA, Grose JH, The effects of diet and sex in amyotrophic lateral sclerosis. Rev. Neurol. (Paris) 176, 301–315 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hartmann H, Ho WY, Chang J-C, Ling S-C, Cholesterol dyshomeostasis in amyotrophic lateral sclerosis: Cause, consequence, or epiphenomenon?FEBS J. in press; (2021). [DOI] [PubMed] [Google Scholar]
- 47.Schmitt F, Hussain G, Dupuis L, Loeffler J-P, Henriques A , A plural role for lipids in motor neuron diseases: Energy, signaling and structure. Front. Cell. Neurosci. 8, 25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ, Autophagy regulates lipid metabolism. Nature 458, 1131–1135 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barbero-Camps E, Roca-Agujetas V, Bartolessis I, de Dios C, Fernández-Checa JC, Marí M, Morales A, Hartmann T, Colell A, Cholesterol impairs autophagy-mediated clearance of amyloid beta while promoting its secretion. Autophagy 14, 1129–1154 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Béland L-C, Markovinovic A, Jakovac H, De Marchi F, Bilic E, Mazzini L, Kriz J, Munitic I, Immunity in amyotrophic lateral sclerosis: Blurred lines between excessive inflammation and inefficient immune responses. Brain Commun. 2, fcaa124 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCombe PA, Lee JD, Woodruff TM, Henderson RD , The peripheral immune system and amyotrophic lateral sclerosis. Front. Neurol. 11, 279 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang M, McKeever PM, Xi Z, Moreno D, Sato C, Bergsma T, McGoldrick P, Keith J, Robertson J, Zinman L, Rogaeva E, DNA methylation age acceleration is associated with ALS age of onset and survival. Acta Neuropathol. 139, 943–946 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.El Khoury LY, Gorrie-Stone T, Smart M, Hughes A, Bao Y, Andrayas A, Burrage J, Hannon E, Kumari M, Mill J, Schalkwyk LC, Systematic underestimation of the epigenetic clock and age acceleration in older subjects. Genome Biol. 20, 283 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Russ J, Liu EY, Wu K, Neal D, Suh ER, Irwin DJ, M.c CT. Millan MB. Harms NJ. Cairns EM. Wood SX. Xie L. Elman LM. Cluskey M. Grossman VM. Van Deerlin EB. Lee, Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathol. 129, 39–52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Braun PR, Han S, Hing B, Nagahama Y, Gaul LN, Heinzman JT, Grossbach AJ, Close L, Dlouhy BJ, Howard III MA, Kawasaki H, Potash JB, Shinozaki G, Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl. Psychiatry 9, 47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farré P, Jones MJ, Meaney MJ, Emberly E, Turecki G, Kobor MS, Concordant and discordant DNA methylation signatures of aging in human blood and brain. Epigenetics Chromatin 8, 19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lappalainen T, Greally JM, Associating cellular epigenetic models with human phenotypes. Nat. Rev. Genet. 18, 441–451 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases, El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1, 293–299 (2000). [DOI] [PubMed] [Google Scholar]
- 59.Nabais MF, Lin T, Benyamin B, Williams KL, Garton FC, Vinkhuyzen AAE, Zhang F, Vallerga CL, Restuadi R, Freydenzon A, Zwamborn RAJ, Hop PJ, Robinson MR, Gratten J, Visscher PM, Hannon E, Mill J, Brown MA, Laing NG, Mather KA, Sachdev PS, Ngo ST, Steyn FJ, Wallace L, Henders AK, Needham M, Veldink JH, Mathers S, Nicholson G, Rowe DB, Henderson RD, McCombe PA, Pamphlett R, Yang J, Blair IP, McRae AF, Wray NR, Significant out-of-sample classification from methylation profile scoring for amyotrophic lateral sclerosis. Npj Genomic Med. 5, 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA, Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Iterson M, Tobi EW, Slieker RC, den Hollander W, Luijk R, Slagboom PE, Heijmans BT, MethylAid: Visual and interactive quality control of large Illumina 450k datasets. Bioinformatics 30, 3435–3437 (2014). [DOI] [PubMed] [Google Scholar]
- 62.van Iterson M, Cats D, Hop P; BIOS Consortium BT Heijmans, omicsPrint: Detection of data linkage errors in multiple omics studies. Bioinformatics 34, 2142–2143 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Lehne B, Drong AW, Loh M, Zhang W, Scott WR, Tan S-T, Afzal U, Scott J, Jarvelin M-R, Elliott P, McCarthy MI, Kooner JS, Chambers JC, A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol. 16, 37 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou W, Laird PW, Shen H, Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 45, e22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luijk R, Goeman JJ, Slagboom EP, Heijmans BT, van Zwet EW, An alternative approach to multiple testing for methylation QTL mapping reduces the proportion of falsely identified CpGs. Bioinformatics 31, 340–345 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Royston P, Parmar MKB, Flexible parametric proportional-hazards and proportional-odds models for censored survival data, with application to prognostic modelling and estimation of treatment effects. Stat. Med. 21, 2175–2197 (2002). [DOI] [PubMed] [Google Scholar]
- 67.Zhang Z, Reinikainen J, Adeleke KA, Pieterse ME, Groothuis-Oudshoorn CG, Time-varying covariates and coefficients in Cox regression models. Ann. Transl. Med. 6, 121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huisman MHB, de Jong SW, van Doormaal PTC, Weinreich SS, Schelhaas HJ, van der Kooi AJ, de Visser M, Veldink JH, van den Berg LH, Population based epidemiology of amyotrophic lateral sclerosis using capture-recapture methodology. J. Neurol. Neurosurg. Psychiatry 82, 1165–1170 (2011). [DOI] [PubMed] [Google Scholar]
- 69.Tunca C, Şeker T, Akçimen F, Coşkun C, Bayraktar E, Palvadeau R, Zor S, Koçoğlu C, Kartal E, Şen NE, Hamzeiy H, Erimiş AÖ, Norman U, Karakahya O, Olgun G, Akgün T, Durmuş H, Şahin E, Çakar A, Gürsoy EB, Yıldız GB, İşak B, Uluç K, Hanağası H, Bilgiç B, Turgut N, Aysal F, Ertaş M, Boz C, Kotan D, İdrisoğlu H, Soysal A, Adatepe NU, Akalın MA, Koç F, Tan E, Oflazer P, Deymeer F, Taştan Ö, Çiçek AE, Kavak E, Parman Y, Başak AN, Revisiting the complex architecture of ALS in Turkey: Expanding genotypes, shared phenotypes, molecular networks, and a public variant database. Hum. Mutat. 41, e7–e45 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Debray S, Race V, Crabbé V, Herdewyn S, Matthijs G, Goris A, Dubois B, Thijs V, Robberecht W, Van Damme P, Frequency of C9orf72 repeat expansions in amyotrophic lateral sclerosis: A Belgian cohort study. Neurobiol. Aging 34, 2890.e7–2890.e12 (2013). [DOI] [PubMed] [Google Scholar]
- 71.Goris A, van Setten J, Diekstra F, Ripke S, Patsopoulos NA, Sawcer SJ; International Multiple Sclerosis Genetics Consortium, M. van Es; Australia and New Zealand MS Genetics Consortium, P. M. Andersen, J. Melki, V. Meininger, O. Hardiman, J. E. Landers, R. H. Brown, A. Shatunov, N. Leigh, A. Al-Chalabi, C. E. Shaw, B. J. Traynor, A. Chio, G. Restagno, G. Mora, R. A. Ophoff, J. R. Oksenberg, P. VanDamme, A. Compston, W. Robberecht, B. Dubois, L. H. van den Berg, P. L. De Jager, J. H. Veldink, P. I. W. de Bakker, No evidence forshared genetic basis of common variants in multiple sclerosis and amyotrophic lateral sclerosis. Hum. Mol. Genet. 23, 1916–1922 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Traxinger K, Kelly C, Johnson BA, Lyles RH, Glass JD, Prognosis and epidemiology of amyotrophic lateral sclerosis. Clin. Pract. 3, 313–320 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Byrne S, Elamin M, Bede P, Hardiman O, Absence of consensus in diagnostic criteria for familial neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 83, 365–367 (2012). [DOI] [PubMed] [Google Scholar]
- 74.Sachdev PS, Lammel A, Trollor JN, Lee T, Wright MJ, Ames D, Wen W, Martin NG, Brodaty H, Schofield PR; OATS research team A comprehensive neuropsychiatric study of elderly twins: The Older Australian Twins Study. Twin Res. Hum. Genet. 12, 573–582 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All summary statistics are available as supplementary data. Individual-level DNA methylation data from Project MinE are available upon request in the European Genome-phenome Archive (EGAS00001004587). The DNA methylation data for the Australian cohorts are deposited on dbGAP and available under accession number phs002068.v1.p1. Code is available at https://github.com/pjhop/EWAS_MinE.