

Abstract
Down syndrome (DS) is the most common genetic disorder leading to developmental disability. The phenotypes associated with DS are complex and vary between affected individuals. Placental abnormalities in DS include differences in cytotrophoblast fusion that affect subsequent conversion to syncytiotrophoblast, atypical oxidative stress/antioxidant balance, and increased expression of genes that are also upregulated in the brains of individuals with Alzheimer’s disease. Placentas in DS are prematurely senescent, showing atypical evidence of mineralization. Fetuses with DS are especially susceptible to adverse obstetric outcomes, including early in utero demise, stillbirth and growth restriction, all of which are related to placental function. The placenta, therefore, may provide key insights towards understanding the phenotypic variability observed in individuals with DS and aid in identifying biomarkers that can be used to evaluate phenotypic severity and prenatal treatments in real time. To address these issues, many different mouse models of DS have been generated to identify the mechanisms underlying developmental changes in many organ systems. Little is known, however, regarding placental development in the currently available mouse models of DS. Based upon the relative paucity of data on placental development in preclinical mouse models of DS, we recommend that future evaluation of new and existing models routinely include histologic and functional assessments of the placenta. In this paper we summarize studies performed in the placentas of both humans and mouse models with DS, highlighting gaps in knowledge and suggesting directions for future research.
Keywords: Down syndrome, Trisomy 21, Placenta, Mouse models, Biomarkers
1. Introduction
Down syndrome (DS) or trisomy 21 (T21) affects 1 in 700–1200 live births and is the most common genetic disorder leading to developmental disability [1,2]. This syndrome results from abnormalities in both gene dosage imbalance as well as globally dysregulated gene expression [3,4]. The phenotypes associated with DS are complex and vary significantly between affected individuals [3]. For example, intellectual disability is present in all individuals with DS, however IQ varies significantly [5]. Other phenotypic features, such as cardiac defects, are only seen in 40–50% of individuals with DS [6].
The placenta has many functions to ensure an optimal environment for offspring survival. Abnormalities of placental function are widely recognized as impacting pregnancy outcome and the long-term health of the child [7,8]. Multiple processes are dysregulated in the placentas of fetuses with DS [9–14]. Fetuses with DS are especially susceptible to adverse obstetric outcomes, including in utero demise, stillbirth and growth restriction, all of which can be a consequence of poor placental function [15–17]. The presence of aneuploid cells confined to the placenta is also associated with poor pregnancy outcomes [18–20].
During human pregnancies, the placenta is difficult to evaluate in real time due to the potential risks to the pregnant woman and her fetus. Also, given that there is generally only one fetus in each pregnancy and there is a relatively long gestational period, it is difficult to generate longitudinal data and control for natural genetic variation. Given what is known about the impact of placental function on human development, this organ may provide key insights towards understanding the phenotypic variability in individuals with DS. To address these issues, many different mouse models of DS have been generated to identify the mechanisms underlying developmental changes in many organ systems [4,21–23]. Little is known, however, regarding placental development in currently available mouse models of DS. This review will summarize what is currently known regarding the human placenta in DS, as well as in mouse models of DS, identifying knowledge gaps and future directions for research.
2. Comparisons between the typically developing human and murine placentas
Typical embryonic development is dependent upon having enough oxygen, nutrient and waste exchange through the placenta. This process, however, can vary among species [24]. The fetal portion of the placenta is derived from two basic elements: an outer epithelium derived from the trophoblast cell lineage and an underlying vascular network and stroma that are derived from embryonic mesoderm [25]. The other key portion of the placenta includes the maternal decidua and vasculature. Concerted interaction between the fetal and maternal elements are required for successful implantation [25]. Considerable differentiation in the trophoblast lineage produces different cell subtypes with specialized endocrine, vascular, immunological and transport functions [24]. Although human and rodent placentas have different organizational structure, both species have discoid hemochorial placentas and undergo the same fundamental steps during development (Supplemental Fig. 1). These include trophoblastic invasion, vascularization of the trophoblast to establish and maintain feto-placental vasculature and maternal vascular remodeling to establish utero-placental circulation [24]. Detailed anatomical comparisons of the mouse and human placentas have been published elsewhere [25–35] and are summarized in Table 1.
Table 1.
Comparison of human and murine placentas.
| Feature | Euploid Human | T21 Human | Wild-type Murine | DS Mouse Models |
|---|---|---|---|---|
| Gross Shape | Discoid | Unchanged | Discoid | Unchanged |
| Transport Barrier | Hemochorial | Unchanged | Hemochorial | Unchanged |
| Trophoblast layers | Monochorial (1 STB layer) | Abnormal (2 layers due to defective cell fusion) | Trichorial (2 STB layers and 1 TGC layer) | Unknown |
| Site of Fetal-Maternal Exchange | Chorionic villi (via STB) | Abnormal (abnormal cell fusion and signaling) | Labyrinth (via STB) | Abnormal in T16 model |
| Syncytiotropho-blast Fusion | Syncytin 1 and 2 | Abnormal (delayed or absent) | Syncytin A and B | Unknown |
| Hormone synthesis | STB | Abnormal | TGC, SpTb | Unknown |
| Decidual/Endo-vascular Invasion | Extravillous trophoblast (interstitial and endovascular) | Abnormal (increased apoptosis, abnormal cell signaling and decreased invasion) | Primary TGC (implantation), Glycogen cells (only interstitial invasion), Secondary TGC (endovascular invasion) | Decreased glycogen and giant cells in T16 model |
| Endometrial Decidualization | Day 14 of menstrual cycle | Unchanged | Begins at fertilization | Unknown |
| Maternal uterine natural killer cells | Secretion of VEGF and NO to promote invasion | Unknown | Secretion of VEGF and NO to promote invasion | Unknown |
| Selective MHC class I expression by trophoblast | HLA-C, -G, E | Abnormal (Decreased expression of HLA-G) | H2–K | Increased expression in T16 model |
| Oxidative stress | Increases throughout gestation | Abnormally elevated | Increases throughout gestation | Unknown |
| Gestational length | 40 weeks | Unchanged | 3 weeks | Unchanged |
| Embryos | 1 | Unchanged | 4–12 | Decreased |
| Vascular invasion | Myometrial involvement (First 1/3) | Abnormal | Limited to the decidua | Unknown |
| Regulators of angiogenesis | VEGF, PlGF, sFlt-1 | Abnormal (Decreased levels of PlGF) | VEGF, Proliferin, sFlt-1 | Unknown |
| Placental Hormones | hGH/hCS 5-member gene cluster, Prolactin, Adipokines, hCG, Progesterone | Abnormal (Abnormal hCG production and secretion) | Prl 22-member gene cluster, growth hormone, placental lactogens, Progesterone, Adipokines | Unknown |
| Spiral arteries | 100–150 | Unknown | 5–30 | Unknown |
| Transformation of Spiral Arteries | Trophoblast dependent (with uterine natural killer cell assistance) | Abnormal | Uterine natural killer cell dependent | Unknown |
| Choriovitelline Placenta | Transient role in early pregnancy | Unchanged | Functions throughout pregnancy | Unchanged |
| Stem cell differentiation | Mediated by low oxygen tension | Abnormal (Increased oxidative stress) | Mediated by low oxygen tension | Unknown |
| Progesterone Production | Corpus luteum until, then STB | Defective | Corpus luteum throughout gestation | Unknown |
| Development of the choriovitelline placenta (yolk sac) | Day 8 | Unknown | Day 8 | Unknown |
| Early nutrition | Histiotrophic from endometrial glandular secretions | Unchanged | Histiotrophic from endometrial glandular secretions | Unchanged |
| Establishment of chorioallantoic circulation | 12 weeks | Unknown | Day 10.5 | Unknown |
| Temporal relationship | First trimester | Unchanged | Mid trimester | Unchanged |
TGC = Trophoblast giant cell, STB = syncytiotrophoblast, SpTb = spongiotrophoblast, VEGF = vascular endothelial growth factor, NO = nitric oxide, hGH = human growth factor, hCS = human chorionic sommatotropin, hCG = human chorionic gonadotropin Prl = prolactin, sFlt-1 = soluble fms-like tyrosine kinase.
While the mouse placenta is not identical to its human counterpart many studies have shown that many cell lineages are largely conserved and similar genes direct placental development in both species (Table 2 and Supplemental Table 1) [30,31]. Also, in both humans and mice there is differential expression of genes throughout gestation [30,33].
Table 2.
Genes involved in placental development.
| Gene Name | Function | Cell/Tissue type in Human | Cell/Tissue type in Mouse |
|---|---|---|---|
| ID2/Id2 | Stem cell marker | TSC | TSC |
| ASH2/Mash2 | Transcription factor required for spongiotrophoblast development and proliferation | TSC | Chorion, ectoplacental cone, SpTb |
| GCM1/Gcm1 | Transcription factor required for initiation and morphogenesis and syncytiotrophoblast differentiation | CTB, STB | CTB, STB |
| HAND1/Hand1 | Transcription factor required for giant cell differentiation | Trophectoderm | Trophectoderm, TGC |
| TEAD3/Tead3 | Transcription factor involved in regulation of placental lactogen expression | STB | STB |
| MMP9/Mmp9 | Matrix degrading enzyme | EVT | TGC |
| Integrin α1β1 | Cell adhesion molecule | EVT | TGC |
| BHLHE40/Stra13 | Transcription factor | EVT | TGC |
| GLUT1 (SLC2A1)/Glut1 (Slc2a1) | Glucose transport | Mesenchyme or endothelium, CTB | Mesenchyme or endothelium, CTB |
| MEST | Exact role unknown. Abnormal imprinting associated with pregnancy loss. | Mesenchyme or endothelium | Mesenchyme or endothelium |
| PLAC1/Plac1 | Trophoblast differentiation, FGF7 signaling | CTB, STB | All trophoblast cell types |
| IGF2/IgF2 | Promotion of fetal and placental growth, as well as nutrient transfer. Imprinted. | EVT | CTB, Glycogen cells |
| PECAM 1(CD31)/Pecam1 (Cd31) | Adhesion molecule involved in angiogenesis | EVT | EVT, TGC |
| UPA (PLAU)/Upa (Plau) | Role in trophoblast invasion | EVT | TGC |
| HGF/Hgf | Role in angiogenesis and villous development | Mesenchyme or endothelium | Mesenchyme or endothelium |
| VEGF/Vegf | Regulation of angiogenesis | CTB, STB | TGC |
| VEGFR-2/FlK1 | VEGF receptor | Mesenchyme or endothelium | Mesenchyme or endothelium |
| VEGFR1/Flt-1 | VEGF receptor | CTB, STB, EVT, mesenchyme or endothelium | Glycogen cells, mesenchyme or endothelium |
| FGFR2/Fgfr2 | Role in mitogenesis and differentiation | TSC | TSC |
| EOMES/Eomes | Transcription factor | TSC | TSC |
| TFAP2C/Ap-2gamma | Transcription Factor | TSC | TSC |
| NODAL/Nodal | Required for maintenance of human embryonic stem cell pluripotency and may play a role in human placental development | TSC | TSC |
ID2 = inhibitor of DNA binding 2, ASH2 = Achaete-scute family bHLH transcription factor 2, GCM1/Gcm1 = glial cells missing, HAND1 = Heart and neural crest derivatives expressed 1, TEAD3 = TEA domain transcription factor 3, MMP9 = Matrix metalloproteinase 9, BHLHE40 = Basic helix-loop-helix family member e40, GLUT-1/Glut-1 = Glucose transporter 1, MEST = Mesoderm specific transcript, PLAC1 = Placental specific protein 1, IGF2 = Insulin-like growth factor 2, PECAM = platelet endothelial cell adhesion molecule-1, PLAU-plasminogen activator urokinase, HGF/Hgf = Hepatocyte growth factor, VEFGF/R = Vascular endothelial growth factor/Receptor s-Flt1 = soluble fms-like tyrosine kinase 1, FGFR2 = Fibroblast growth factor receptor 2, TFAP2C = Transcription factor AP-2 gamma, TGC = Trophoblast giant cell, STB = syncytiotrophoblast, SpTb = spongiotrophoblast TSC = Trophoblast stem cells, CTB = cytotrophblast
3. The human placenta in trisomy 21
Compared to euploid pregnancies, studies conducted on placental specimens and cell cultures from pregnancies affected by T21 have shown differences in histology, function and gene expression.
3.1. Histological appearance
Limited reports exist regarding the histological appearance of placentas from pregnancies affected by T21. Examination of 14 aneuploid placentas (including three cases of trisomy 21) found that villous mineralization was more common in placentas from aneuploid compared to euploid stillbirths [34]. In euploid pregnancies, as gestational age advances, basal plate calcification is common, but villous mineralization is not. This can be a sign of cessation of fetal artery perfusion and intrauterine cardiac failure and hydrops [34].
A pathological analysis of placentas from 10 cases of trisomy 21 (four liveborn, six stillborn) delivered between 16- and 40-weeks’ gestation, demonstrated hemorrhagic endovasculitis in approximately 80% of T21 placentas compared to only 5% in euploid placentas [35]. Hemorrhagic endovasculitis is associated with stillbirth and fetal growth restriction. Coexisting lesions are often present, including feto-placental vessel thrombi, and villous fibrosis. This may indicate that there is primary placental dysfunction, abnormal vascular development or an abnormal feto-maternal interface [36].
3.2. Villous morphology
Histological analysis of placental biopsies and amniocytes in unaffected and affected pregnancies demonstrated that there was an increase in invasive cytotrophoblast apoptosis, vascular abnormalities, inflammation, villous hypoplasia and intervillous fibrin deposition during the second and third trimester [9–12]. Roberts et al. examined cross sections of placental villi obtained at 10–14 weeks of gestation. Compared to euploid, T21 placentas had an increased percentage of double layer trophoblast and an increased proportion of villous capillaries with nucleated red cells and basophilic stippling, indicating delayed maturation, differentiation and abnormal erythropoiesis [37]. Another study of second and third trimester placentas showed varying amounts of large chorionic villi interspersed with normal appearing villi. Small muscular arteries and arterioles were also absent from some large villi, giving the villi a hypovascular appearance. As seen in previous studies, there was also an increased percentage of two-layer trophoblast observed in T21 placentas [12].
3.3. Cytotrophoblast function
Two differentiation pathways exist for cytotrophoblasts (CTB); formation of the syncytiotrophoblast (STB) and formation of invasive extravillous trophoblasts (EVT). Both pathways show striking differences between T21 and euploid placentas (Fig. 1). Studies conducted using CTB derived from first, second and third trimester euploid placentas found that there was in vitro aggregation and fusion within 48–72 h of forming the STB. However, in T21, aggregation occurred, but there was little or no fusion [9,14]. Interestingly, abnormal trophoblast fusion and differentiation in T21 placentas is reversible in vitro with the addition of exogenous activin-A, a paracrine factor involved in crosstalk between trophoblasts and mesenchymal cells [38,39]. In euploid placentas it is highly secreted by mesenchymal cells but is secreted at a significantly lower level in T21.
Fig. 1.
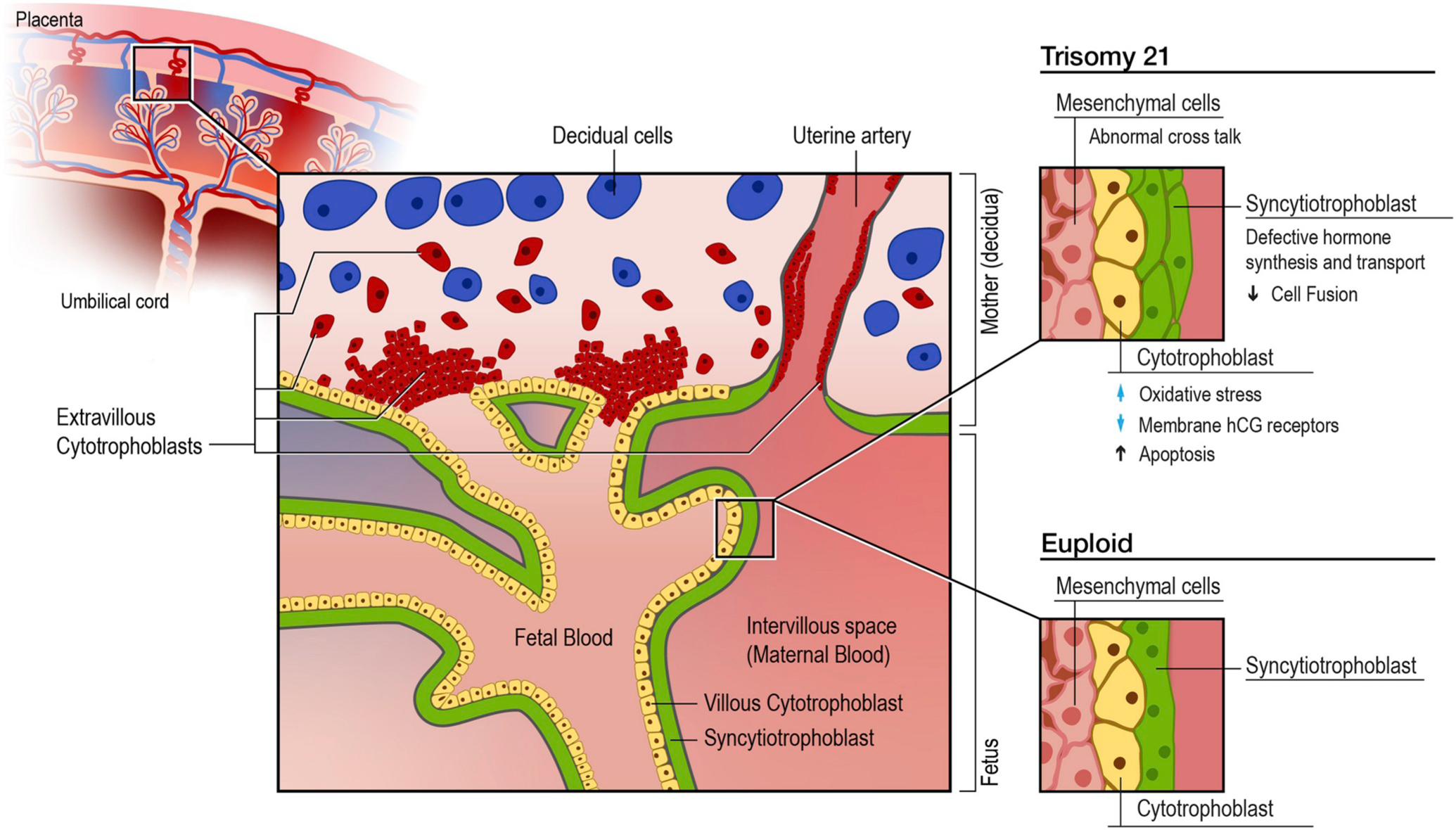
Cell types and functional abnormalities in T21 and euploid placentas.
Illustration shows placental layers and cell types. Boxes shows magnified view of the cell types in the chorionic villi. The cytotrophoblasts (yellow) are precursors to the syncytiotrophoblast (green), which remains double layered secondary to poor fusion and cell differentiation. The box labeled euploid development shows the syncytiotrophoblast as a multinucleated monolayer surround the underlying cytotrophoblast layer.
In one study, analysis of CTB isolated from euploid and trisomic placentas showed defective STB formation and function leading to abnormally hyperglycosylated beta human chorionic gonadotropin (βhCG). This abnormal and weakly bioactive βhCG molecule cannot correctly stimulate CTB differentiation. In vitro function can be restored by treatment with non-hyperglycosylated βhCG [40]. Another cell culture and RNA expression study performed on placental samples at 7–12 weeks of gestation demonstrated a significant increase in the Luteinizing Hormone/Chorionic Gonadotropin Receptor (LHCGR) mRNA transcription in T21 placentas compared to controls. Despite this, the synthesis of high molecular weight mature LHCGR proteins was significantly reduced in T21 compared to unaffected pregnancies, suggesting a lack of utilization of circulating βhCG [41,42].
To identify the membrane proteins required for human trophoblast fusion, one study found that in euploid placentas cell fusion proteins are highly expressed. For example, syncytin-1 is expressed at elevated levels in the EVT and syncytin-2 is expressed in villous CTB. Syncytin-2 and zona occludens-1 (ZO-1, tight junction protein) are also highly expressed in isolated CTB. Production of these proteins rapidly decreases during STB formation. Syncytin-1 and connexin 43 (Cx43, gap junction protein) increase with cell aggregation and fusion. The expected time-dependent variations in expression of these membrane proteins are not observed in T21 placentas [39]. Additionally, a more recent study found that interferon induced transmembrane proteins (IFITMs) impair STB formation and inhibit syncytin activity, which may explain these findings given that there increased interferon activity seen in Down syndrome [43,44]. In cell cultures from placentas at 18–22 weeks, T21 CTBs were more likely to undergo apoptosis and have abnormal expression of cell surface markers compared to euploid. Specific findings included decreased expression of VE-cadherin (cell-cell adhesion glycoprotein) and upregulation of matrix metallopeptidase 9 (MMP-9, extracellular matrix breakdown). These results imply that in T21, there are abnormalities in the differentiation pathways that lead to CTB invasion of the uterus, and that these cells may be eliminated by programmed cell death from the maternal-fetal interface. In addition, up-regulation of MMP may be a compensatory mechanism for poor uterine invasion [13]. CTBs are also involved in activating the MHC class Ib molecule HLA-G, which is thought to play a role in maternal tolerance of the fetal hemiallograft, which was found to have decreased expression in T21 placentas [13,45, 46].
Lastly, in trophoblasts the keratin cytoskeleton appears to be important in maintaining structural integrity. Keratin 8 (KRT8) also functions in signal transduction and potentially in apoptosis. Klugman et al. demonstrated that in trophoblasts from T21 placentas, TNF-related Apoptosis-I inducing ligand (TRAIL) was significantly overexpressed compared to euploid. There was also an inverse relationship between KRT8 and TRAIL in T21 [47].
3.4. Oxidative stress
STBs have high synthetic and metabolic activities and are therefore vulnerable to oxidative stress [7]. In euploid pregnancies, STBs can adapt to minimal increases in reactive oxidation species (ROS) by restoring the oxidant/antioxidant balance. If this response is inadequate it can lead to hypoxia and chronic oxidative stress, which increases the vascularity of the villi and inhibits the differentiation of CTB to STB [48].
SOD-1, the gene for intra-cellular Cu, Zn-superoxide dismutase (SOD), maps to human chromosome 21. In T21, there are three copies of this gene. A study examining the impact of oxidative stress on placental development showed that overexpression of SOD-1 impairs STB formation [49,50]. It has been shown that ROS, overexpression of SOD-1 and abnormal oxidative status, demonstrated by a significant increase in catalase activity, are present in T21 CTB. In turn, T21 trophoblasts cannot compensate for the oxidative imbalance [49,50]. Excess SOD in T21 placentas prevents differentiation of CTB to STB, thereby down-regulating STB-specific hCG, placental growth factor secretions and synthesis of syncytin [51].
3.5. Placental hormone expression
Prenatal screening for aneuploidy can be performed by measurement of placental derived cell-free DNA or biochemical analytes in maternal serum during the first and/or second trimester (Table 3). In the first trimester, maternal serum free βhCG or total βhCG, and placental derived pregnancy-associated plasma protein-A (PAPP-A) are measured. In the second trimester, alpha-fetoprotein (AFP), unconjugated estriol (uE3), inhibin-A (DIA), and βhCG are measured. Cell-free fetal DNA can be measured any time after 10 weeks’ gestation [52]. Serum analyte trends in pregnancies affected with Down syndrome show significantly decreased PAPP-A, mild decrease in AFP and uE3, and increased DIA, βhCG and cell-free DNA [53–55]. These abnormalities are thought to be secondary to abnormal trophoblast invasion, increased trophoblast apoptosis and delayed maturation of the feto-placental unit [56,57].
Table 3.
Markers used for prenatal screening in trisomy 21.
| Hormone | Typical Expression | First Trimester in T21 | Second Trimester in T21 |
|---|---|---|---|
| βhCG | - Detectable at 4 weeks | ↑↑ | ↑↑ |
| - Peaks at 10 weeks | |||
| - Declines until 20 weeks | |||
| - Constant until term | |||
| PAPP-A | - Detectable at 8 weeks | ↓↓ | ↔ |
| - Steady increase until term | |||
| AFP | - Detectable at 12 weeks | ↓ | ↓ |
| - Peaks at 32 weeks | |||
| - Declines until term | |||
| uE3 | - Detectable pre-implantation | ↓ | ↓ |
| - Steady increase until term | |||
| DIA | - Detectable at 5 weeks | ↑ | ↑↑ |
| - Peaks at 8 weeks | |||
| - Steady decrease from 8 to 16 weeks | |||
| - Steady increase until 36 weeks | |||
| Activin-A | - Detectable at 8 weeks | ↔ | ↔ |
| - Constant level throughout 1st and 2nd trimester | |||
| - Steady increase from 24 weeks to term | |||
| PlGF | - Increases steadily throughout gestation | ↓ | ↓ |
| - Peaks at 30 weeks | |||
| - Steady decrease until term | |||
| Cell-free DNA | - Increases throughout gestation | ↑ | ↑ |
| – 0.1% increase/week from 10 to 20 weeks | |||
| – 1% increase/week from 21 weeks to term |
↓ or ↑ = 25% difference, ↓↓ or ↑↑ = 50% difference, ↔ distribution overlaps with normal population.
βhCG and its receptor are also important regulators of embryo implantation and pregnancy maintenance. The beta subunit is placenta specific and is primarily produced by the STB. In addition to its early role in the maintenance of progesterone levels, βhCG facilitates trophoblast differentiation, endometrial decidualization, invasion of maternal spiral arteries and angiogenesis. It also plays a small role in stimulating the thyroid to produce thyroid hormone, which is essential for brain and nervous system development [41,58].
Elevated levels of βhCG in mid-pregnancy are associated with pre-eclampsia, fetal growth restriction and T21 [41,42]. The etiology of maternal serum βhCG elevation in T21 is unclear. However, it has been proposed that it could be due to up-regulation of the βhCG gene, increased half-life of the hyperglycosylated βhCG hormone, or reduced availability of the LHCGR [41].
PAPP-A is a metalloproteinase that increases the bioavailability of insulin-like growth factor binding proteins. It is expressed by the STB, as well as in many other human tissues, but at lower levels than in the placenta [59]. PAPP-A levels are also low in serum from pregnant women carrying fetuses with T21. Extremely low levels are a risk factor for impaired fetal growth, preterm delivery, preeclampsia and stillbirth [60, 61]. A role for this hormone in fetal growth has also been shown in knockout mice [62].
Maternal serum AFP is decreased in the first and second trimester in women carrying fetuses with T21 [63,64]. Although not a placental product, AFP levels are significantly increased in placental tissue while in the fetal liver, AFP levels are only slightly lower than euploid controls. This indicates that there may be a transport defect leading to placental accumulation [57]. In contrast to T21, elevated maternal serum levels of AFP are associated with growth restriction (<5%), preterm delivery and stillbirth, indicating normal placental function, but abnormal fetal production [61].
Estriol (UE3) is a cholesterol derived hormone that regulates utero-placental blood flow, development of placental vascularization and maternal cardiovascular remodeling. It is synthesized by STB from the precursor dehydroepiandrosterone (DHEAS), which originates in the fetal adrenal gland. Estrogen levels remain low in the first trimester of pregnancy and progressively increase, remaining elevated until the onset of labor [58]. UE3 is low in T21, however it is unknown whether this is secondary to fetal production or placental dysfunction [65].
Another placental hormone impacted by T21 is inhibin A. βhCG secretion is in part controlled by inhibin A (inhibition) and activin-A (secretion). Maternal serum inhibin A is elevated in T21 secondary to increased placental production. The exact mechanism for this increased production is unclear, however, it has been postulated that increased activin A production leads to increased βhCG, which in turn leads to increased inhibin A [57]. Conflicting data exist regarding the relationship between βhCG levels and both inhibin A and activin A in T21, given that levels of these hormones are low in both amniotic fluid and mesenchymal cells [66]. Finally, placental growth factor (PlGF) is a member of the vascular endothelial growth factor (VEGF) family. It is an important local mediator of angiogenesis and regulator of maternal Insulin derived growth factor 1 (IGF-1). It is produced by villous CTB, STB and EVT. Although its utility for screening in T21 is currently unclear, decreased levels of PlGF are associated with fetal growth restriction. Compared to euploid placentas, PlGF levels are reduced during the first and second trimester in T21 [57,67–69].
3.6. Placental senescence
The placenta ages as the pregnancy advances, but premature placental aging is associated with adverse pregnancy outcomes. It is also known that DS has been associated with early onset and higher incidence of age-related conditions such as Alzheimer’s disease [3]. A few studies in T21 placentas have found evidence of premature aging, which could be due to increased oxidative stress, mitochondrial dysfunction or telomere shortening that is associated with aneuploidy [10,13,70]. For example, USP16 is overexpressed in DS. It has been shown that upregulation of USP16 in the Ts65Dn mouse model and primary human fibroblasts results in downregulation of the Wnt pathway and reduces stem cell renewal. Although it the role of USP16 in placental development has not been fully elucidated it has been shown that Cdkn2a knockout mice have abnormal placental development [71]. USP 16 regulates activation of Cdkn2a, which in turn acts as a negative regulator of the Wnt pathway. This is relevant since Wnt signaling pathways plays a crucial role in stem cell maintenance, aging and cellular senescence in various tissues including the placenta [72,73].
Additionally, in a study conducted by Wong et al. using samples from first, second and third trimester placentas with T21, a 2.3-fold increase in amyloid precursor protein (APP) expression was found. Results also showed that increased expression of APP was associated with increased apoptosis, which in turn, decreased cell growth and suppressed differentiation of the STB [74].
Rozovski et al. also showed that several key genes that were overexpressed in T21 trophoblast had similar expression patterns to those found in the brains of people with Alzheimer’s disease (Supplemental Table 1). For example, APP was increased by two-fold and LOX was over expressed by three-fold [75].
3.7. Placental gene expression and epigenetic modifications
Hsa21 is the smallest human chromosome, representing ~1% of the human genome [76]. Although small, there is significant enrichment for genes located in cytoskeletal structures compared to the rest of the genome. Importantly, these cytoskeletal proteins play an important role in the development of the placenta and nervous system. Hsa21 also contains 31 transcription factors [76]. Overexpression of some of these transcription factors has been seen in multiple studies (Supplemental Table 1) and may lead to altered transcription of non-Hsa21 targets [3]. However, further studies are needed to confirm these hypotheses.
Kipp et al. evaluated placental samples from second and third trimester pregnancies with T21. They demonstrated that in T21 samples there was a trend towards higher levels of inhibin alpha (INHA) mRNA levels and decreased levels of Wilms Tumor 1 (WT1) mRNA levels, a repressor of INHA. The pathological basis of this alteration is not completely understood [77].
Bianco et al. studied gene expression profiles of second trimester placental samples from pregnancies affected by trisomies 13, 18 and 21. T21 was associated with the highest number of dysregulated genes. Greater than 90% of the dysregulated genes did not map to chromosome 21. T21 also had more pleiotropic effects and affected a larger number of signaling pathways than T13 or T18. These signaling pathways, however, were less crucial for development. In T21 there was also differential expression of 160 genes when compared to euploid placentas (145 up-regulated and 15 down-regulated). Differential expression was most notable for genes encoded by chromosome 21 (ABCG, ITSN1, ADAMTS1 and PLAC4), as well as HGF, EGFR, S100A and RAC1 (Supplemental Table 1) [78].
Using microarrays, Rozovski et al. performed gene expression analyses of chorionic villus samples (CVS) from T21 and euploid controls. They found genes that mapped to chromosome 21 were over-represented by 4.5-fold, but most of the differentially expressed genes did not map to chromosome 21. Some had greater than 10-fold over-expression. The most significant difference was seen in MEST, which was increased 13.7-fold in T21. Another interesting finding in this study was the two-fold increase of ITM2B [75].
Gene expression can also be impacted by DNA methylation. Studies regarding the methylation status of DS placentas present conflicting data, which may be secondary to differences in gestational ages. Lim et al. examined first trimester CVS samples and found differential gene expression in trisomic placentas compared to euploid controls. They demonstrated that 47 genes on Hsa21 had increased expression in T21 placentas; these genes were globally hypomethylated [79]. Another study by Jin et al. analyzed CVS samples from the first and second trimesters and found global DNA hypermethylation. More specifically, genes involved in epigenetic regulation (Ten-eleven translocation methylcytosine dioxygenase 1 and 2; TET1, TET2 and RE1-Silencing Transcription factor; REST) were hypermethylated (Supplemental Table 1) [80].
Recently, investigators have become interested in miRNA expression and function due to their involvement in post-transcriptional regulation of gene expression and its potential impact on development, including in cases of T21. An analysis of miRNAs identified 34 that were significantly differentially expressed (16 upregulated, 18 downregulated) in T21 compared to controls, and 76 possible gene targets located on chromosome 21. Differences were not seen in chromosome 21 derived miRNA. Target genes on chromosome 21 were associated with T21-related complications such as intellectual disability, neurobehavioral manifestations, and congenital abnormalities [81].
3.8. Placental proteome dysregulation
The underlying mechanisms of phenotypic variability in T21 are poorly understood. To date, no prenatal biomarkers that predict phenotype in T21-have been identified. In this context, the placental proteome offers a unique opportunity to identify biomarkers associated with disease severity and to potentially monitor treatment response and predict outcomes [82,83]. Several studies have looked for protein biomarkers using maternal serum. Few however show significant placental expression (Supplemental Table 1) [84–91].
To our knowledge, only two studies have investigated global proteomic dysregulation in T21 placentas using two-dimensional gel electrophoresis coupled with tandem mass spectroscopy (Supplemental Table 1). In the first, Sun et al. compared the proteomic profiles of five T21 and five euploid second and third trimester placentas. They identified seven significantly up-regulated (ANXA2, ERp29, SOD1, PSMA2, PPIA and FGB) and three down-regulated proteins (ECHS1, PRDX6 and PDIA3) in T21 placentas [90]. In the second, Chen et al. found 80 unique dysregulated proteins in T21 placentas between 16 and 18 weeks of gestation. In addition to a few proteins in maternal plasma that have been described in other studies (A1AT, VIM), this study reported novel placental proteins (ATXN3, XRCC2, CSH1, GBP2 and SPRED2) that can be used in combination with other T21-specific biomarkers [92].
4. The placenta in mouse models of down syndrome
In the human, functional understanding of the human placenta is limited to postpartum studies and some in vitro systems. Because of this, animal models are critical for investigation of basic placental biology. There are, however, limitations to using mouse models, given that no single mouse model completely recapitulates the full phenotype observed in people with T21 [92]. There are also many developmental processes that occur during intrauterine human life that are postnatal events in mice. Another issue is that significant differences exist in the lengths of murine and human pregnancies. On the other hand, changes in the amount of oxidative stress within the placenta throughout gestation are similar in both species [30]. Lastly in mice, the persistent yolk sac provides a secondary circulation, which may protect against environmental insults. Recent data, however, indicate that the human yolk sac may play more than just a vestigial role [93].
To better understand the pathophysiology of DS, many mouse models of DS have been generated [23]. The Ts16 mouse was created in 1985 but was shown to be lethal during the embryonic period. This model harbors a full trisomy of Mmu16, including 119 protein-coding genes that are orthologous to human chromosome 21 (Hsa21). Due to its early lethality, this mouse is not useful for studying DS phenotypes. Information on its placental development is, however, available and summarized below [94]. Subsequently, efforts have focused on generating models with segmental Mmu10, Mmu16 and Mmu17 trisomies that carry three copies of Hsa21 orthologous genes. The Ts1Cje, Ts65Dn and Dp(16)1/Yey mice are the most commonly used models in current studies of DS. While all three have large segments of triplicated Hsa21-orthologous genes on Mmu16, each was engineered using a distinct methodology, resulting in different cytogenetic profiles, numbers of triplicated genes, and prenatal and postnatal phenotypes [21]. Currently only limited data on the placentas from Ts1Cje mice exist, and there are no data on placentas from Ts65Dn or Dp(16)1/Yey mice.
4.1. Trisomy 16 (T16) mouse model
In one study, App expression was analyzed in T16 embryos and placentas at days E15 and E17. Although localization of App mRNA and protein in the brain and placenta was unchanged, there was a marked increase in App expression in the placenta that did not correlate with obvious differences in morphology [94].
In another study, histological analysis at days E15 and E17 found that there were decreased numbers of glycogen cells, giant cells and spongiotrophoblast cells. This study also demonstrated labyrinth and placental weights to be smaller than in controls [95].
Lastly, a study by Kornguth et al. showed that T16 placentas had a less extensively developed labyrinth, causing decreased nutrient supplies. Additionally, the junctional zone showed no significant signs of normal differentiation throughout gestation. When comparing placental cells and neurons, there was an altered pattern of proteins and increased levels of class I MHC H-2Kk on their cell surfaces, potentially affecting cell-cell interactions [96].
4.2. Ts1Cje mouse model
The Ts1Cje mouse demonstrates mild to moderate phenotypic abnormalities. Pennings et al. performed a gene expression study that compared fetal livers and placentas of Ts1Cje and wild-type embryonic mice at day E15.5. Increased expression of genes in the trisomic segment was shown. Forty-eight differentially expressed genes were present in the placentas of affected mice. Most were ascribed to the gene dosage effects of the trisomic locus at Mmu16. Interestingly, the fetal liver of Ts1Cje embryos exhibited more differentially expressed genes (109 genes) with very little overlap with the placenta outside the Mmu16 trisomic genes [97].
5. Future research and conclusions
In this review we have shown that compared to euploid placentas, there are multiple differences in the placentas of human fetuses with T21. By contrast, scant attention has been paid to placental development in preclinical mouse models of DS. This is a lost opportunity for understanding clues to the mechanisms underlying atypical development in DS and associated disease.
A central issue is whether the abnormalities seen in human placentas in T21 have downstream consequences regarding fetal or infant survival, growth restriction, and brain or another organ development. For instance, cardiac anomalies are seen in 40% of individuals with DS [6]. Although the mechanisms between the placenta, fetal heart development and later cardiac dysfunction have not been fully elucidated, it is hypothesized that there are links in common genetic developmental pathways such as Wnt/beta catenin signaling, angiogenesis and cell adhesion. Other theories include placental insufficiency leading to growth restriction, poor nutrient transfer and oxidative stress [98].
Placental abnormalities in DS include differences in CTB fusion that affect subsequent conversion to STB, atypical oxidative stress/antioxidant balance, and increased expression of genes that are also upregulated in the brains of individuals with Alzheimer’s disease. There is also a suggestion that placentas in human T21 are prematurely senescent, showing atypical evidence of mineralization. Is there a connection between the premature aging of the placenta and the premature aging of people with DS?
Initiatives such as the Human Placenta Project, created to address the need for better real-time assessment of human placental structure and function, and the Placental Atlas Tool were developed to create a shared research platform for placental data. These resources will be important in establishing normal parameters for placental development and creating data repositories to distinguish normal from abnormally developing placentas [99,100].
Based upon the relative paucity of data on placental development in preclinical mouse models of DS, we recommend that future evaluation of new and existing models routinely include histologic and functional assessments of the placenta. It is crucial to understand the role of this important organ in subsequent development of brain and other structural and functional abnormalities.
Supplementary Material
{kind=link}
Acknowledgments
This research was supported [in part] by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health. Grant# HG200399.
Footnotes
Declaration of competing interest
The authors declared no conflict of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.placenta.2019.10.002.
References
- [1].de Graaf G, Buckley F, Dever J, Skotko BG, Estimation of live birth and population prevalence of Down syndrome in nine U.S. states, Am. J. Med. Genet 173 (10) (2017) 2710–2719, 10.1002/ajmg.a.38402. [DOI] [PubMed] [Google Scholar]
- [2].Data and Statistics on Down Syndrome, Division of Birth Defects and Developmental Disabilities, 2017. NCBDDD, Centers for Disease Control and Prevention Web site, https://www.cdc.gov/ncbddd/birthdefects/downsyndrome/data.html.
- [3].Antonarakis SE, Down syndrome and the complexity of genome dosage imbalance, Nat. Rev. Genet 18 (3) (2017) 147–163, 10.1038/nrg.2016.154. [DOI] [PubMed] [Google Scholar]
- [4].Letourneau A, Antonarakis SE, Genomic determinants in the phenotypic variability of Down syndrome, Prog. Brain Res 197 (2012) 15–28, 10.1016/B978-0-444-54299-1.00002-9. [DOI] [PubMed] [Google Scholar]
- [5].Delabar JM, Aflalo-Rattenbac R, Creau N, Developmental defects in trisomy 21 and mouse models, Sci. World J 6 (2006) 1945–1964, 10.1100/tsw.2006.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Vis JC, Duffels MG, Winter MM, et al. , Down syndrome: a cardiovascular perspective, J. Intellect. Disabil. Res 53 (5) (2009) 419–425, 10.1111/j.1365-2788.2009.01158.x. [DOI] [PubMed] [Google Scholar]
- [7].Burton GJ, Fowden AL, Thornburg KL, Placental origins of chronic disease, Physiol. Rev 96 (4) (2016) 1509–1565, 10.1152/physrev.00029.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].von Beckerath AK, Kollmann M, Rotky-Fast C, Karpf E, Lang U, Klaritsch P, Perinatal complications and long-term neurodevelopmental outcome of infants with intrauterine growth restriction, Am. J. Obstet. Gynecol 208 (2) (2013), 10.1016/j.ajog.2012.11.014, 130.e1–130.e6. [DOI] [PubMed] [Google Scholar]
- [9].Frendo JL, Vidaud M, Guibourdenche J, et al. , Defect of villous cytotrophoblast differentiation into syncytiotrophoblast in Down’s syndrome, J. Clin. Endocrinol. Metab 85 (10) (2000) 3700–3707, 10.1210/jcem.85.10.6915. [DOI] [PubMed] [Google Scholar]
- [10].Amiel A, Fejgin MD, Liberman M, Sharon Y, Kidron D, Biron-Shental T, Senescence in amniocytes and placentas from trisomy 21 pregnancies, J. Matern. Fetal Neonatal Med 26 (11) (2013) 1086–1089, 10.3109/14767058.2013.768982. [DOI] [PubMed] [Google Scholar]
- [11].Pidoux G, Guibourdenche J, Frendo JL, et al. , Impact of trisomy 21 on human trophoblast behaviour and hormonal function, Placenta 25 (Suppl) (2004) S79–S84, 10.1016/j.placenta.2004.01.007. A. [DOI] [PubMed] [Google Scholar]
- [12].Qureshi F, Jacques SM, Johnson MP, et al. , Trisomy 21 placentas: histopathological and immunohistochemical findings using proliferating cell nuclear antigen, Fetal Diagn. Ther 12 (4) (1997) 210–215, 10.1159/000264470. [DOI] [PubMed] [Google Scholar]
- [13].Wright A, Zhou Y, Weier JF, et al. , Trisomy 21 is associated with variable defects in cytotrophoblast differentiation along the invasive pathway, Am. J. Med. Genet 130A (4) (2004) 354–364, 10.1002/ajmg.a.30254. [DOI] [PubMed] [Google Scholar]
- [14].Massin N, Frendo JL, Guibourdenche J, et al. , Defect of syncytiotrophoblast formation and human chorionic gonadotropin expression in Down’s syndrome, Placenta 22 (Suppl A) (2001) S93–S97, 10.1053/plac.2001.0658. [DOI] [PubMed] [Google Scholar]
- [15].Guseh SH, Little SE, Bennett K, Silva V, Wilkins-Haug LE, Antepartum management and obstetric outcomes among pregnancies with Down syndrome from diagnosis to delivery, Prenat. Diagn 37 (7) (2017) 640–646, 10.1002/pd.5054. [DOI] [PubMed] [Google Scholar]
- [16].Wessels MW, Los FJ, Frohn-Mulder IM, Niermeijer MF, Willems PJ, Wladimiroff JW, Poor outcome in Down syndrome fetuses with cardiac anomalies or growth retardation, Am. J. Med. Genet 116A (2) (2003) 147–151, 10.1002/ajmg.a.10823 [doi]. [DOI] [PubMed] [Google Scholar]
- [17].Sparks TN, Griffin E, Page J, Pilliod R, Shaffer BL, Caughey AB, Down syndrome: perinatal mortality risks with each additional week of expectant management, Prenat. Diagn 36 (4) (2016) 368–374, 10.1002/pd.4792 [doi]. [DOI] [PubMed] [Google Scholar]
- [18].Toutain J, Goutte-Gattat D, Horovitz J, Saura R, Confined placental mosaicism revisited: impact on pregnancy characteristics and outcome, PLoS One 13 (4) (2018), 10.1371/journal.pone.0195905 e0195905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sparks TN, Thao K, Norton ME, Mosaic trisomy 16: what are the obstetric and long-term childhood outcomes? Genet. Med 19 (10) (2017) 1164–1170, 10.1038/gim.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wan J, Li R, Zhang Y, et al. , Pregnancy outcome of autosomal aneuploidies other than common trisomies detected by noninvasive prenatal testing in routine clinical practice, Prenat. Diagn 38 (11) (2018) 849–857, 10.1002/pd.5340. [DOI] [PubMed] [Google Scholar]
- [21].Guedj F, Pennings JL, Massingham LJ, et al. , An integrated human/murine transcriptome and pathway approach to identify prenatal treatments for Down syndrome, Sci. Rep 6 (2016) 32353, 10.1038/srep32353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ferres MA, Bianchi DW, Siegel AE, Bronson RT, Huggins GS, Guedj F, Perinatal natural history of the Ts1Cje mouse model of Down syndrome: growth restriction, early mortality, heart defects, and delayed development, PLoS One 11 (12) (2016), 10.1371/journal.pone.0168009 e0168009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Herault Y, Delabar JM, Fisher EMC, Tybulewicz VLJ, Yu E, Brault V, Rodent models in Down syndrome research: impact and future opportunities, Dis. Model Mech 10 (10) (2017) 1165–1186, 10.1242/dmm.029728 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Grigsby PL, Animal models to study placental development and function throughout normal and dysfunctional human pregnancy, Semin. Reprod. Med 34 (1) (2016) 11–16, 10.1055/s-0035-1570031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cross JC, Genetic insights into trophoblast differentiation and placental morphogenesis, Semin. Cell Dev. Biol 11 (2) (2000) 105–113, 10.1006/scdb.2000.0156. [DOI] [PubMed] [Google Scholar]
- [26].Enders AC, Carter AM, Comparative placentation: some interesting modifications for histotrophic nutrition – a review, Placenta 27 (Suppl A) (2006) S11–S16, 10.1016/j.placenta.2005.10.013. [DOI] [PubMed] [Google Scholar]
- [27].Rawn SM, Cross JC, The evolution, regulation, and function of placenta-specific genes, Annu. Rev. Cell Dev. Biol 24 (2008) 159–181, 10.1146/annurev.cellbio.24.110707.175418. [DOI] [PubMed] [Google Scholar]
- [28].Cross JC, Baczyk D, Dobric N, et al. , Genes, development and evolution of the placenta, Placenta 24 (2–3) (2003) 123–130, doi: S0143–4004(02)90887–8. [DOI] [PubMed] [Google Scholar]
- [29].Burdon C, Mann C, Cindrova-Davies T, Ferguson-Smith AC, Burton GJ, Oxidative stress and the induction of cyclooxygenase enzymes and apoptosis in the murine placenta, Placenta 28 (7) (2007) 724–733, doi: S0143–4004(06)00288–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gheorghe C, Mohan S, Longo LD, Gene expression patterns in the developing murine placenta, J. Soc. Gynecol. Investig 13 (4) (2006) 256–262, 10.1016/j.jsgi.2006.02.007. [DOI] [PubMed] [Google Scholar]
- [31].Cox B, Kotlyar M, Evangelou AI, et al. , Comparative systems biology of human and mouse as a tool to guide the modeling of human placental pathology, Mol. Syst. Biol 5 (1) (2009) 279, 10.1038/msb.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Silva JF, Serakides R, Intrauterine trophoblast migration: a comparative view of humans and rodents, Cell Adhes. Migrat 10 (1–2) (2016) 88–110, 10.1080/19336918.2015.1120397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sitras V, Fenton C, Paulssen R, Vartun A, Acharya G, Differences in gene expression between first and third trimester human placenta: a microarray study, PLoS One 7 (3) (2012), 10.1371/journal.pone.0033294 e33294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pierce BT, Martin LS, Hume RF Jr., Calhoun BC, Muir-Padilla J, Salafia CM, Relationship between the extent of histologic villous mineralization and stillbirth in aneuploid and euploid fetuses, J. Soc. Gynecol. Investig 9 (5) (2002) 290–293. S1071557602001673. [DOI] [PubMed] [Google Scholar]
- [35].Salafia CM, Burns JP, Wiley PS, The incidence of hemorrhagic endovasculitis is increased in cases of both liveborn and stillborn trisomy 21, Am. J. Med. Genet. Suppl 45 (4) (1989) A90. [Google Scholar]
- [36].Sander CM, Gilliland D, Richardson A, Foley KM, Fredericks J, Stillbirths with placental hemorrhagic endovasculitis: a morphologic assessment with clinical implications, Arch. Pathol. Lab Med 129 (5) (2005) 632–638. OA4193. [DOI] [PubMed] [Google Scholar]
- [37].Roberts L, Sebire NJ, Fowler D, Nicolaides KH, Histomorphological features of chorionic villi at 10–14 weeks of gestation in trisomic and chromosomally normal pregnancies, Placenta 21 (7) (2000) 678–683, 10.1053/plac.2000.0553. [DOI] [PubMed] [Google Scholar]
- [38].Gerbaud P, Pidoux G, Guibourdenche J, et al. , Mesenchymal activin-A overcomes defective human trisomy 21 trophoblast fusion, Endocrinology 152 (12) (2011) 5017–5028, 10.1210/en.2011-1193. [DOI] [PubMed] [Google Scholar]
- [39].Malassine A, Pidoux G, Gerbaud P, Frendo JL, Evain-Brion D, Human trophoblast in trisomy 21: a model for cell-cell fusion dynamic investigation, Adv. Exp. Med. Biol 714 (2011) 103–112, 10.1007/978-94-007-0782-5_4. [DOI] [PubMed] [Google Scholar]
- [40].Pidoux G, Gerbaud P, Cocquebert M, et al. , Review: human trophoblast fusion and differentiation: lessons from trisomy 21 placenta, Placenta 33 (Suppl) (2012) S81–S86, 10.1016/j.placenta.2011.11.007. [DOI] [PubMed] [Google Scholar]
- [41].Banerjee S, Smallwood A, Chambers AE, et al. , A link between high serum levels of human chorionic gonadotrophin and chorionic expression of its mature functional receptor (LHCGR) in Down’s syndrome pregnancies, Reprod. Biol. Endocrinol 3 (2005), 25-7827-3-25, 1477-7827-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chambers AE, Mills WE, Mercade I, et al. , The utility of circulating LHCGR as a predictor of Down’s syndrome in early pregnancy, BMC Pregnancy Childbirth 14 (2014), 10.1186/1471-2393-14-197, 197-2393-14-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Buchrieser J, Degrelle SA, Coudere T, et al. , IFITM proteins inhibit placental syncytiotrophoblast formation and promote fetal demise, Science 365 (2019) 176–180. [DOI] [PubMed] [Google Scholar]
- [44].Sullivan KD, Lewis HC, Hill AA, et al. , Trisomy 21 consistently activates the interferon response, Elife 5 (2016), 10.7554/eLife.16220 e16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Colucci F, Boulenouar S, Kieckbusch J, Moffett A, How does variability of immune system genes affect placentation? Placenta 32 (2011) 539–545, 10.1016/j.placenta.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fisher SJ, Why is placentation abnormal in preeclampsia? Am. J. Obstet. Gynecol 213 (4 Suppl) (2015) S115–S122, 10.1016/j.ajog.2015.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Klugman SD, Gross SJ, Liang J, et al. , Expression of keratin 8 and TNF-related apoptosis-I inducing ligand (TRAIL) in Down syndrome placentas, Placenta 29 (4) (2008) 382–384, 10.1016/j.placenta.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schoots MH, Gordijn SJ, Scherjon SA, van Goor H, Hillebrands JL, Oxidative stress in placental pathology, Placenta 69 (2018) 153–161. S0143–4004(18)30070–5. [DOI] [PubMed] [Google Scholar]
- [49].Malassine A, Frendo JL, Evain-Brion D, Trisomy 21-affected placentas highlight prerequisite factors for human trophoblast fusion and differentiation, Int. J. Dev. Biol 54 (2–3) (2010) 475–482, 10.1387/ijdb.082766am. [DOI] [PubMed] [Google Scholar]
- [50].Frendo JL, Therond P, Guibourdenche J, Vidaud M, Evain-Briona D, Implication of copper zinc superoxide dismutase (SOD-1) in human placenta development, Ann. N. Y. Acad. Sci 973 (2002) 297–301, 10.1111/j.1749-6632.2002.tb04654.x. [DOI] [PubMed] [Google Scholar]
- [51].Banerjee S, Smallwood A, Nargund G, Campbell S, Placental morphogenesis in pregnancies with Down’s syndrome might provide a clue to pre-eclampsia, Placenta 23 (2–3) (2002) 172–174, 10.1053/plac.2001.0767. [DOI] [PubMed] [Google Scholar]
- [52].Practice bulletin no. 163 summary: screening for fetal aneuploidy, Obstet. Gynecol 127 (5) (2016) 979–981, 10.1097/AOG.0000000000001439. [DOI] [PubMed] [Google Scholar]
- [53].Dugoff L, Society for Maternal-Fetal Medicine, First- and second-trimester maternal serum markers for aneuploidy and adverse obstetric outcomes, Obstet. Gynecol 115 (5) (2010) 1052–1061, 10.1097/AOG.0b013e3181da93da. [DOI] [PubMed] [Google Scholar]
- [54].Palomaki GE, Eklund EE, Neveux LM, Lambert Messerlian GM, Evaluating first trimester maternal serum screening combinations for down syndrome suitable for use with reflexive secondary screening via sequencing of cell free DNA: high detection with low rates of invasive procedures, Prenat. Diagn 35 (8) (2015) 789–796, 10.1002/pd.4609. [DOI] [PubMed] [Google Scholar]
- [55].Taglauer ES, Wilkins-Haug L, Bianchi DW, Review: cell-free fetal DNA in the maternal circulation as an indication of placental health and disease, Placenta 35 (Suppl) (2014) S64–S68, 10.1016/j.placenta.2013.11.014 (Suppl). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Baer RJ, Currier RJ, Norton ME, et al. , Obstetric, perinatal, and fetal outcomes in pregnancies with false-positive integrated screening results, Obstet. Gynecol 123 (3) (2014) 603–609, 10.1097/AOG.0000000000000145. [DOI] [PubMed] [Google Scholar]
- [57].Newby D, Aitken DA, Crossley JA, Howatson AG, Macri JN, Connor JM, Biochemical markers of trisomy 21 and the pathophysiology of down’s syndrome pregnancies, Prenat. Diagn 17 (10) (1997) 941–951, 10.1002/(SICI)1097-0223(199710)17:103.0.CO, 2-G. [DOI] [PubMed] [Google Scholar]
- [58].Oratz S, The hormones of the placenta, Sci. J. Lander Coll. Arts Sci 8 (1) (2014) 35. [Google Scholar]
- [59].Gyrup C, Christiansen M, Oxvig C, Quantification of proteolytically active pregnancy-associated plasma protein-A with an assay based on quenched fluorescence, Clin. Chem 53 (5) (2007) 947–954, clinchem.2006.080614. [DOI] [PubMed] [Google Scholar]
- [60].Kingdom JC, Audette MC, Hobson SR, Windrim RC, Morgen E, A placenta clinic approach to the diagnosis and management of fetal growth restriction, Am. J. Obstet. Gynecol 218 (2S) (2018) S803–S817. S0002–9378(17)32340–2. [DOI] [PubMed] [Google Scholar]
- [61].Gaccioli F, Aye ILMH, Sovio U, Charnock-Jones DS, Smith GCS, Screening for fetal growth restriction using fetal biometry combined with maternal biomarkers, Am. J. Obstet. Gynecol 218 (2S) (2018) S725–S737. S0002–9378(17)32476–6. [DOI] [PubMed] [Google Scholar]
- [62].Randhawa R, Cohen P, The role of the insulin-like growth factor system in prenatal growth, Mol. Genet. Metab 86 (1–2) (2005) 84–90, doi: S1096–7192(05)00249–0. [DOI] [PubMed] [Google Scholar]
- [63].Donalson K, Turner S, Morrison L, Liitti P, Nilsson C, Cuckle H, Maternal serum placental growth factor and alpha-fetoprotein testing in first trimester screening for down syndrome, Prenat. Diagn 33 (5) (2013) 457–461, 10.1002/pd.4087. [DOI] [PubMed] [Google Scholar]
- [64].Huang T, Dennis A, Meschino WS, Rashid S, Mak-Tam E, Cuckle H, First trimester screening for Down syndrome using nuchal translucency, maternal serum pregnancy-associated plasma protein A, free-beta human chorionic gonadotrophin, placental growth factor, and alpha-fetoprotein, Prenat. Diagn 35 (7) (2015) 709–716, 10.1002/pd.4597. [DOI] [PubMed] [Google Scholar]
- [65].Newby D, Aitken DA, Howatson AG, Connor JM, Placental synthesis of oestriol in Down’s syndrome pregnancies, Placenta 21 (2–3) (2000) 263–267, 10.1053/plac.1999.0469. [DOI] [PubMed] [Google Scholar]
- [66].Dalgliesh GL, Aitken DA, Lyall F, Howatson AG, Connor JM, Placental and maternal serum inhibin-A and activin-A levels in Down’s syndrome pregnancies, Placenta 22 (2–3) (2001) 227–234, 10.1053/plac.2000.0598. [DOI] [PubMed] [Google Scholar]
- [67].Spencer K, Liao AW, Skentou H, Ong CY, Nicolaides KH, Maternal serum levels of total activin-A in first-trimester trisomy 21 pregnancies, Prenat. Diagn 21 (4) (2001) 270–273, 10.1002/pd.53. [DOI] [PubMed] [Google Scholar]
- [68].Johnson J, Pastuck M, Metcalfe A, et al. , First-trimester Down syndrome screening using additional serum markers with and without nuchal translucency and cell-free DNA, Prenat. Diagn 33 (11) (2013) 1044–1049, 10.1002/pd.4194. [DOI] [PubMed] [Google Scholar]
- [69].Cowans NJ, Kisanga MC, Spencer K, Maternal serum placental growth factor in second trimester trisomy 21 pregnancies, Prenat. Diagn 32 (2) (2012) 117–121, 10.1002/pd.2904. [DOI] [PubMed] [Google Scholar]
- [70].Sultana Z, Maiti K, Dedman L, Smith R, Is there a role for placental senescence in the genesis of obstetric complications and fetal growth restriction? Am. J. Obstet. Gynecol 218 (2S) (2018) S762–S773. S0002–9378(17)32332–3. [DOI] [PubMed] [Google Scholar]
- [71].Gal H, Lysenko M, Stroganov S, Vadai E, Youssef SA, Tzadikevitch-Geffen K, Rotkopf R, Biron-Shental T, de Bruin A, Neeman M, Krizhanovsky V, Molecular pathways of senescence regulate placental structure and function, EMBO J (2019), 10.15252/embj.2018100849 e100849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Adorno M, Sikandar S, Mitra SS, Kuo A, Nicolis Di Robilant B, Haro-Acosta V, Ouadah Y, Quarta M, Rodriguez J, Qian D, Reddy VM, Cheshier S, Garner CC, Clarke MF, Usp16 contributes to somatic stem-cell defects in Down’s syndrome, Nature 501 (2013) 380–384, 10.1038/nature12530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Adorno M, di Robilant BN, Sikandar SS, et al. , Usp16 modulates Wnt signaling in primary tissues through Cdkn2a regulation, Sci. Rep 8 (1) (2018) 17506, 10.1038/s41598-018-34562-w. Published 2018 Nov 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wong OGW, Cheung CLY, Ip PPC, Ngan HYS, Cheung ANY, Amyloid precursor protein overexpression in Down syndrome trophoblast reduces cell invasiveness and interferes with syncytialization, Am. J. Pathol 188 (10) (2018) 2307–2317, doi: S0002–9440(17)30904–5. [DOI] [PubMed] [Google Scholar]
- [75].Rozovski U, Jonish-Grossman A, Bar-Shira A, Ochshorn Y, Goldstein M, Yaron Y, Genome-wide expression analysis of cultured trophoblast with trisomy 21 karyotype, Hum. Reprod 22 (9) (2007) 2538–2545, 10.1093/humrep/dem214. [DOI] [PubMed] [Google Scholar]
- [76].Hattori M, Fujiyama A, Taylor TD, Watanabe H, et al. , Chromosome 21 mapping and sequencing consortium. The DNA sequence of human chromosome 21, Nature 405 (2000) 311–319. [DOI] [PubMed] [Google Scholar]
- [77].Kipp JL, Lambert-Messerlian G, Eklund E, Rodriguez G, Demczuk M, Gundogan F, Expression of transcription factors controlling alpha inhibin gene expression in placental tissues from pregnancies affected by fetal Down syndrome, Prenat. Diagn 32 (3) (2012) 302–305, 10.1002/pd.3826. [DOI] [PubMed] [Google Scholar]
- [78].Bianco K, Gormley M, Farrell J, et al. , Placental transcriptomes in the common aneuploidies reveal critical regions on the trisomic chromosomes and genome-wide effects, Prenat. Diagn 36 (9) (2016) 812–822, 10.1002/pd.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lim JH, Kim SY, Han JY, Kim MY, Park SY, Ryu HM, Comprehensive investigation of DNA methylation and gene expression in trisomy 21 placenta, Placenta 42 (2016) 17–24, 10.1016/j.placenta.2016.03.012. [DOI] [PubMed] [Google Scholar]
- [80].Jin S, Lee YK, Lim YC, et al. , Global DNA hypermethylation in Down syndrome placenta, PLoS Genet 9 (6) (2013), 10.1371/journal.pgen.1003515 e1003515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lim JH, Kim DJ, Lee DE, et al. , Genome-wide microRNA expression profiling in placentas of fetuses with Down syndrome, Placenta 36 (3) (2015) 322–328, 10.1016/j.placenta.2014.12.020. [DOI] [PubMed] [Google Scholar]
- [82].Hourrier S, Salomon LJ, Dreux S, Muller F, Screening for adverse pregnancy outcome at early gestational age, Clin. Chim. Acta 411 (21–22) (2010) 1547–1552, 10.1016/j.cca.2010.06.024. [DOI] [PubMed] [Google Scholar]
- [83].Goetzinger KR, Odibo AO, Screening for abnormal placentation and adverse pregnancy outcomes with maternal serum biomarkers in the second trimester, Prenat. Diagn 34 (7) (2014) 635–641, 10.1002/pd.4370. [DOI] [PubMed] [Google Scholar]
- [84].Pennings JL, Koster MP, Rodenburg W, Schielen PC, de Vries A, Discovery of novel serum biomarkers for prenatal Down syndrome screening by integrative data mining, PLoS One 4 (11) (2009), 10.1371/journal.pone.0008010 e8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kang Y, Dong X, Zhou Q, et al. , Identification of novel candidate maternal serum protein markers for Down syndrome by integrated proteomic and bioinformatic analysis, Prenat. Diagn 32 (2012) 284–292, 10.1002/pd.3829. [DOI] [PubMed] [Google Scholar]
- [86].Kolialexi A, Tsangaris GT, Papantoniou N, et al. , Application of proteomics for the identification of differentially expressed protein markers for Down syndrome in maternal plasma, Prenat. Diagn 28 (2008) 691–698, 10.1002/pd.2040. [DOI] [PubMed] [Google Scholar]
- [87].Yu B, Zhang B, Wang J, et al. , Preliminary proteomic-based identification of a novel protein for Down’s syndrome in maternal serum, Exp. Biol. Med 237 (2012) 530–539, 10.1258/ebm.2012.011312. [DOI] [PubMed] [Google Scholar]
- [88].Lopez Uriarte GA, Burciaga Flores CH, Torres de la Cruz VM, et al. , Proteomic profile of serum of pregnant women carring a fetus with Down syndrome using nano uplc Q-tof ms/ms technology, J. Matern. Fetal Neonatal Med 31 (2018) 1483–1489, 10.1080/14767058.2017.1319923. [DOI] [PubMed] [Google Scholar]
- [89].Nagalla SR, Canick JA, Jacob T, et al. , Proteomic analysis of maternal serum in Down syndrome: identification of novel protein biomarkers, J. Proteome Res 6 (2007) 1245–1257, 10.1021/pr060539h. [DOI] [PubMed] [Google Scholar]
- [90].Sun CJ, Yan LY, Wang W, Yu S, Wang X, Zhang WY, Proteomic analysis of the alteration of protein expression in the placenta of Down syndrome, Chin. Med. J 124 (22) (2011) 3738–3745. [PubMed] [Google Scholar]
- [91].Chen CP, Chen YH, Chern SR, et al. , Placenta proteome analysis from Down syndrome pregnancies for biomarker discovery, Mol. Biosyst 8 (9) (2012) 2360–2372, 10.1039/c2mb25081k. [DOI] [PubMed] [Google Scholar]
- [92].Aziz NM, Guedj F, Pennings JLA, et al. , Lifespan analysis of brain development, gene expression and behavioral phenotypes in the Ts1Cje, Ts65Dn and dp(16)1/yey mouse models of Down syndrome, Dis. Model Mech 11 (6) (2018), 10.1242/dmm.031013dmm031013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Cindrova-Davies T, Jauniaux E, Elliot MG, Gong S, Burton GJ, Charnock-Jones DS, RNA-seq reveals conservation of function among the yolk sacs of human, mouse, and chicken, Proc. Natl. Acad. Sci. U. S. A 114 (24) (2017) E4753–E4761, 10.1073/pnas.1702560114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Bersu ET, Mossman HW, Kornguth SE, Altered placental morphology associated with murine trisomy 16 and murine trisomy 19, Teratology 40 (5) (1989) 513–523, 10.1002/tera.1420400514. [DOI] [PubMed] [Google Scholar]
- [95].Holtzman DM, Bayney RM, Li YW, et al. , Dysregulation of gene expression in mouse trisomy 16, an animal model of Down syndrome, EMBO J 11 (2) (1992) 619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kornguth S, Bersu E, Anderson M, Markley J, Correlation of increased levels of class I MHC H-2Kk in the placenta of murine trisomy 16 conceptuses with structural abnormalities revealed by magnetic resonance microscopy, Teratology 45 (4) (1992) 383–391, 10.1002/tera.1420450409. [DOI] [PubMed] [Google Scholar]
- [97].Pennings JL, Rodenburg W, Imholz S, et al. , Gene expression profiling in a mouse model identifies fetal liver- and placenta-derived potential biomarkers for Down Syndrome screening, PLoS One 6 (2011), 10.1371/journal.pone.0018866 e18866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Burton GJ, Jauniaux E, Development of the human placenta and fetal heart: synergic or independent? Front. Physiol 9 (2018) 37, 10.3389/fphys.2018.00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Ilekis JV, Keller M, Shlionskaya A, et al. , The placental atlas tool (PAT): a collaborative research and discovery platform for the placental research community, Placenta (2019), 10.1016/j.placenta.2019.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Guttmacher AE, Maddox YT, Spong CY, The human placenta project: placental structure, development, and function in real time, Placenta 35 (5) (2014) 303–304, 10.1016/j.placenta.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.