

Abstract
Neurodegenerative disorders are characterized by neuronal loss, usually in late life. But recently, abnormalities of proteins implicated in neurodegenerative disorders have been identified in disorders of childhood, raising the possibility that clues to susceptibility to and prevention of neurodegenerative disorders may be identifiable before symptoms of disease arise. This review leverages these new and evolving findings to test our hypothesis, first proposed in 2010, that proteins implicated in neurodegenerative disorders play important roles in brain development by examining evidence in the peer-reviewed literature published in the past five years for the relevance of these proteins in normal and disease-associated brain development.
Keywords: Disease mechanisms, Genetic disorders, Proteins, Biomarkers
Introduction
The biomedical community often views development and senescence as two completely different processes. But nervous system malfunction is often either a long-term consequence of a lifelong condition or an acquired, late-life change in the quantity, structure, or function of a protein or pathway of developmental significance. Understanding the relationships between nervous system development and degeneration may afford early detection of susceptibility to and prevention of neurodegenerative disease. In 2010, we proposed the hypothesis that proteins implicated in Alzheimer and Parkinson diseases are important for nervous system development.1 Since then, support for this hypothesis and identification of developmentally important molecules involved in these and other late-life neurodegenerative disorders, including frontotemporal dementia and Huntington disease, have vastly advanced, raising the promise of identifying early biomarkers and therapeutic targets for these conditions.2,3
We performed a systematic literature review to examine the expression and roles of proteins implicated in neurodegenerative disorders during early brain development (Table 1). Identification of neuro-embryologically important molecules and pathways that are involved in late-life neurodegenerative disorders holds the promise of identifying biomarkers; predictive, pathogenic, and etiologic mechanisms; and therapeutic targets for these common and devastating diseases.1 Our findings of protein and pathway coincidences between genesis and degeneration in the nervous system are consistent with the hypothesis that proteins implicated in neurodegeneration play roles in nervous system development in prenatal and early postnatal life and underscore the promise of identifying susceptible individuals before neurodegeneration begins.
TABLE 1.
Examples of Proteins Implicated in Normal Nervous System Development and Late-Life Neurodegenerative Disease
| Disease | Protein | Putative Function in Normal Nervous System Development | Putative Role in Disease Pathogenesis and/or Progression |
|---|---|---|---|
| Alzheimer disease | Aβ | Proliferation and differentiation of neural stem cells; neuronal migration, neurite outgrowth, and synaptogenesis; neuronal plasticity and learning; cell signaling receptor; binding partner of N-methyl-D-aspartate receptor | Activates Wnt signaling and thereby enhances proliferation of neural stem cells and inhibits their differentiation; generates intracellular C-terminal domain which translocates to the nucleus and transcriptionally represses the canonical, β-catenin‐dependent branch of the Wnt signaling pathway; modulates Notch signaling; inhibits Shh signaling |
| Rac1 | Dendritogenesis | Cytoplasmic translocation of SET, decreased activity of protein phosphatase 2A, and consequent accumulation of hyperphosphorylated tau | |
| MAPT/tau | Maturational predominant localization to and dephosphorylation in axons | Persistence of diffuse cellular localization and phosphorylation with loss of dendritic spines | |
| Frontotemporal dementia | Progranulin | Plays role in lysosomal protein trafficking | Homozygous or compound heterozygous mutations lead to neuronal ceroid lipofuscinosis (CLN11 type); hemizygous mutations lead to frontotemporal dementia |
| Huntington disease | Huntingtin | Unknown (knockout or trinucleotide repeat = embryonic lethal) | Degeneration of medium spiny neurons and gliosis in striatum and cerebral cortex |
| Parkinson disease | Glucocerebrosidase | Lysosomal glycoside hydrolase that cleaves the glycolipid glucosylceramide | Homozygous or compound heterozygous mutations lead to the lysosomal storage disorder, Gaucher disease; hemizygous mutations predispose to Parkinson disease and Lewy body dementia, mechanism unknown; possibly related to generalized decrease in lysosomal function with consequent accumulation of α-synuclein; impaired mitochondrial autophagy; misfolded glucocerebrosidase with consequent impairment of Parkin ubiquitin lyase function and accumulation of protein aggregates and damaged mitochondria |
| PINK, Parkin | PINK: mitochondrial serine/threonine kinase important for oxidative phosphorylation; Parkin: ubiquitin lyase that earmarks damaged mitochondria for destruction | Mutations of PINK or uncoupling of oxidative phosphorylation inactivate PINK preventing it from “attracting” Parkin and causing it to accumulate in the mitochondrial membrane; mutations of Parkin cause damaged mitochondria to accumulate; Parkin mutations are seen in juvenile Parkinson disease |
Literature search
In November 2020, we conducted a search of peer-reviewed articles published in the past five years listed in the NCBI PubMed database using the implicated protein names, respectively, for each of the four neurodegenerative diseases under consideration (Table 2). Each protein name in turn was joined with the Boolean operator “AND” first with the relevant disease name and then with, in turn, “brain development,” “brain embryogenesis,” and, in the case of progranulin and glucocerebrosidase, “neuronal ceroid lipofuscinosis” and “Gaucher disease,” respectively. Of 77 peer-reviewed articles identified, 73 were deemed by both authors and a colleague not directly involved in the study (N.B.; Acknowledgments section) to be primary research relevant to the possible roles of these proteins in brain development and are cited in this review.
TABLE 2.
Protein Names Used for Literature Search Relevant to Each Neurodegenerative Disease
| Neurodegenerative Disease | Protein |
|---|---|
| Alzheimer disease | Amyloid-beta (Aβ) |
| ApoE | |
| Neurofilaments | |
| Presenilin | |
| Sirtuins | |
| Tau | |
| Frontotemporal dementia | Progranulin |
| Huntington disease | Huntingtin |
| Sirtuins | |
| Parkinson disease | α-Synuclein |
| β-Synuclein | |
| Glucocerebrosidase | |
| Parkin | |
| Sirtuins | |
| Tau |
Alzheimer disease
The clinical, pathological, and epidemiological features of Alzheimer disease4 are summarized in Box 1.
BOX 1. Alzheimer Disease.
Alzheimer disease is the most common primary cause of dementia.
Clinically, Alzheimer disease is characterized by progressive loss of cognitive function, including memory, praxis, language function, and complex perception.
Aging, genetic predisposition, obesity and metabolic syndrome, cerebrovascular disease, neuroinflammation, head trauma, and environmental exposures have all been hypothesized to play etiologic roles in Alzheimer disease.4
Pathologically, it is characterized by the accumulation of extracellular senile plaques, consisting primarily of beta-amyloid protein, and intracellular neurofibrillary tangles, consisting primarily of hyperphosphorylated tau protein, in the cerebral cortex. It remains unclear whether these findings represent causal abnormalities, final pathological common pathways, or epiphenomena in the disease.
By 40 years of age, most people with Down syndrome have β-amyloid plaques in their brains. Approximately half of all people with Down syndrome will develop Alzheimer disease as they age.
Protein markers
Typical processing of amyloid precursor protein (APP) involves sequential cleavage by α- and γ-secretases. In contrast, in Alzheimer disease, APP is processed by sequential cleavage by β- and γ-secretases (Fig). This aberrant cleavage leads to generation of Aβ and subsequent translocation of the Aβ intracellular C-terminal domain from the cytoplasm to the nucleus of the cell, inducing Notch signaling and maintenance of the undifferentiated state in neural stem cells. This differentiation arrest may undermine compensation for degeneration of mature neurons.5
FIGURE.
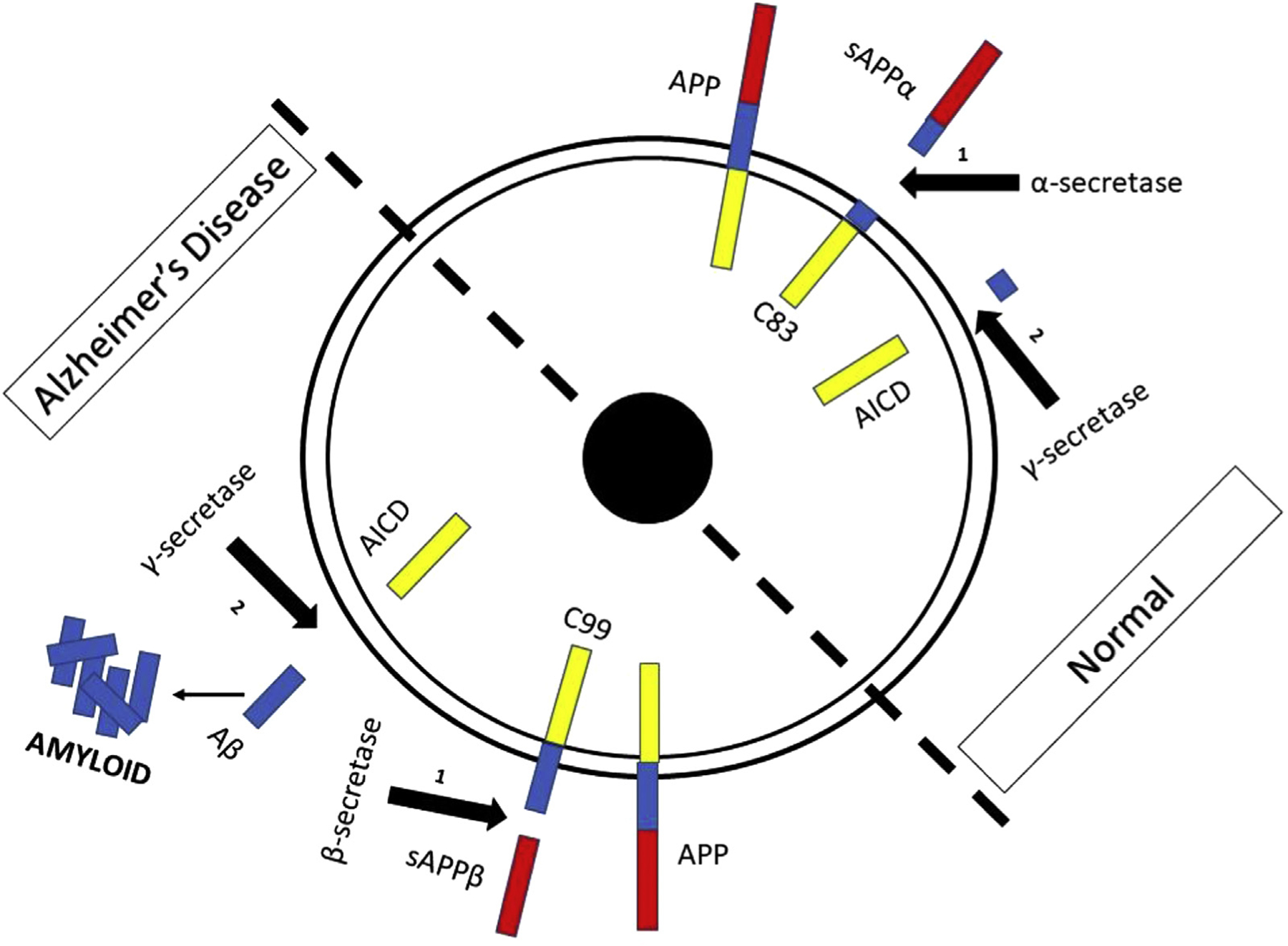
Processing of amyloid precursor protein under normal circumstances and in Alzheimer disease. APP, amyloid precursor protein; sAPPα,β, soluble amyloid precursor protein α,β; C83 & C99, C-terminal APP fragments; AICD, amyloid intracellular domain. The color version of this figure is available in the online edition.
In addition, Alzheimer diseaseeassociated nuclear translocation of the intracellular C-terminal domain of the APP transcriptionally represses the canonical β-catenin–dependent branch of the Wnt signaling pathway and, as occurs with Aβ40, ensures maintenance of proliferative potential in neural stem cells. Similarly, high concentrations of Aβ42, another cleavage product, inhibit signaling through the sonic hedgehog (Shh) pathway and enhance levels of glycogen synthase kinase 3β, thereby inhibiting proliferation, neuronal differentiation, and axon and synapse formation in adult neural stem and precursor cells.5
Recent evidence demonstrates that adult neurogenesis depends on synthesis of cyclic AMP (cAMP) and phosphorylation of cyclic AMP response element binding protein (CREB) and suggests that inhibition of the degradation of cAMP reverses mitophagy, enhances neurogenesis, and restores memory function in Aβ-neuroblastoma cell culture and murine models of Alzheimer disease.6 Furthermore, pharmacologic agents that enhance activation of or reactivate the brain-derived neurotrophic factor (BDNF), TrkA, and TrkB pathways and phosphorylation of Akt and CREB during adulthood have shown evidence of activating neurogenesis and reversing cognitive decline in murine models of Alzheimer disease.7
In Alzheimer disease, maintenance and redirection of dendrites are deficient. Dendrites decrease in number and complexity in the forebrain and hippocampus. Underlying this late-life aberration of dendrite integrity is aberrancy of the distribution of Rac1 and consequently aberrant translocation of the protein SET from the nucleus to cytoplasm. This results in decreased activity of protein phosphatase 2A, a phosphatase that normally would dephosphorylate tau protein.8 Hyperphosphorylated tau and related accumulation of Aβ are hallmarks and biomarkers for the severity of Alzheimer disease.9
Both Alzheimer disease and treatment of mature neurons in culture with Ab are associated with redistribution out of axons and accumulation in dendrites of the microtubule-associated protein, MAPT/tau, and hyperphosphorylation of MAPT/tau in the dendritic pattern. While the mechanism of this redistribution and accumulation is not definitively known, Aβ induces an increase in local production of MAPT/tau in dendrites. In addition, inhibition of either proteosomal or autophagic protein degradation in the dendritosomatic compartment of neurons also causes accumulation of MAPT/tau in dendrites. Accumulation of MAPT/tau results in loss of dendritic spines similar to that seen in Alzheimer disease.10
How accumulation of tau proteins results in the loss of dendritic spines and destruction of neurons is not clear. There is some evidence that developmental, C1q-dependent synaptic pruning by microglia is reactivated by accumulation of phospho-tau.11
Neurodevelopmental roles of protein markers
APP appears to be important for proliferation and differentiation of neural stem cells, neuronal migration, neurite outgrowth, and synaptogenesis during embryogenesis and development, and neuronal plasticity and learning throughout life. It has been hypothesized to be a receptor important for cell signaling and a binding partner of the N-methyl-D-aspartate receptor. Its intracellular C-terminal domain has been proposed to be a transcriptional regulator.5
The role of APP and its cleavage products in signal transduction is critically important for brain development. Under normal circumstances, after α-secretase cleavage, APP liberates sAPPα, an activator of Notch signaling and inducer of glial differentiation of neural precursor cells.5
sAPPα is also an agonistic ligand for insulin/insulin-like growth factor receptor in the developing cerebral cortex.12 Activation of this receptor is required for development of the embryonic hippocampus and cingulate cortex and facilitates axon outgrowth in cortical neurons in culture.13
Wnt signaling is also critically important during brain development and is associated with the maintenance of self-renewal, differentiation, directionally specific migration, and maturation of neural stem cells.5
Activation of the Shh pathway is essential for brain development. Activation of Shh results in its binding to the Patched (Ptch) receptor and lifts its inhibition of Smoothened (Smo) which, in turn, activates the transcription factor, Gli. Gli activation plays an important role in proliferation, differentiation, axonal outgrowth, and synapse elaboration in neural stem cells.5
Each of the pathways engaged by normal processing of APP is aborted or diverted by Alzheimer disease–type processing.5 Thus, the processing route of APP functions as an arbiter of the “choice” between development and stagnation or dysgenesis in the embryonic brain.
Embryonic neurogenesis depends critically on synthesis of cAMP and phosphorylation of CREB.7,14 Activation of the BDNF, TrkA, and TrkB pathways and phosphorylation of Akt and CREB enable embryonic neurogenesis.
Rho family GTPases, including Rac1 and Rac3, have a critical role in dendritogenesis during brain development.15 The distribution of Rac1 in the cell controls translocation of the protein SET from the nucleus to cytoplasm and activation of protein phosphatase 2A, a phosphatase that dephosphorylates tau protein.8 Hyperphosphorylated tau and related accumulation of Aβ are hallmarks of the early-onset dementia associated with Down syndrome.9
The microtubule-associated protein, MAPT/tau, is developmentally regulated in a cyto-regional fashion. In young neurons, MAPT/tau is evenly distributed through the dendrites, cell body, and axon. As the cell matures and its processes become more obviously polarized, MAPT/tau is redistributed in graded fashion over developmental time. In mature neurons, MAPT/tau is seen predominantly in axons, with low levels in dendrites. Moreover, differential patterns of phosphorylation of MAPT/tau are seen in axons and dendrites, with the axonal pattern predominating in mature neurons.10
It is of interest that the placentas of fetuses with Down syndrome overexpress APP and not many of the other proteins encoded by genes on chromosome 21. This overexpression is accompanied by increased apoptosis and decreased trophoblastic syncytialization of the placenta.16 Furthermore, the APP cleavage product, Aβ, inhibits the transition of mesenchymal stem cells to pericytes through activation of the ERK 1/2-MAPK pathway,17 effectively inhibiting the vascular investment of organs such as the placenta. Given the vascular contributions to Alzheimer disease pathogenesis, it would be interesting to know if APP or Aβ over-production alters vascular biology in the brains of individuals with Down syndrome in a way that predisposes them to early-onset Alzheimer disease.
It is also of interest that late-onset generalized myoclonic epilepsy is prevalent among adults with Down syndrome older than 50 years (46%) and with nonsyndromic Alzheimer disease (13%).18–20 The reasons for the occurrence of myoclonic epilepsy in and the difference in its incidence between these populations are not clear. Notably, the occurrence of epilepsy in children with Down syndrome has been hypothetically attributed to postnatal progressive reduction in the number and length of dendritic spines on specific neurons,21 and the consequent change in excitatory/inhibitory ratio in the brain.22 It would be interesting to determine whether, in addition to the deposition of amyloid, the excitatory-to-inhibitory ratio continues to change into older adulthood.
Many of the proteins found in the postsynaptic density, aberrancy of which is associated with neurodevelopmental disorders including epilepsy, autism, and schizophrenia, feature prominently on the list of proteins decreased in abundance in the brains of tau overexpressing mice, a model for neurodegenerative diseases including Alzheimer disease, relative to wild-type mice.23
Life course aspects of protein markers
Many studies have sought to discover early-life environmental and genetic factors that cause or predispose people to Alzheimer disease.24 For example, clearance by vascular pericytes of Aβ from the nervous system is dependent throughout life on low-density lipoprotein receptor–related protein 1A and is variably robust depending on apolipoprotein E (ApoE) subtype; genetic variation in production and structure of these species alters risk for Alzheimer disease.25 A meta-analysis of studies that examined the inverse correlation and dose response between exposure to air pollution and cognitive function identified very small particulate matter as the component of air pollution and the ApoE4 allele as the genetic factor most consistently associated with lower cognitive function relative to control populations and the youngest and oldest subjects as the most consistently affected by the impact of these determinants. Very small particulate matter exposure and the ApoE4 allele appear to have additive effects. The effect on cognitive function in childhood appears to be operative even in utero. It is tempting to hypothesize, but difficult to determine, whether early exposure to air pollution predisposes to cognitive loss or neurodegenerative disorders in late adulthood.26 It is also interesting to hypothesize that early enactment of the amyloidogenic, Aβ-generating cleavage of APP does not result in neuronal damage until later in life because substances or cellular elements produced in early life protect neurons from such damage. Indeed, there is evidence that proteins secreted by umbilical cord blood mesenchymal stem cells protect murine hippocampal neurons from the effects of Aβ and prevent its accumulation in mice.27,28 Mesenchymal stem cells enhance clearance of Aβ through autophagy and upregulation of enzymes that degrade this peptide.16,29
In addition, given the findings regarding APP- and Aβ-induced changes in the placental vasculature of fetuses with Down syndrome,16,17 it would be interesting to know whether disordered regulation of APP occurs in the placentas of those destined to develop sporadic, non–Down syndrome–associated Alzheimer disease.
Finally, it should be noted that while amyloidogenic aggregation of Aβ is pathogenic, a similar process involving other proteins is critical for normal embryogenesis. For example, localization specificity of phosphorylase activity and the resulting definition of cell type polarity in the developing Drosophila gastrula involve amyloid-like aggregation of phosphorylases.30
Frontotemporal dementia
The clinical, pathological, and epidemiological features of frontotemporal dementia31–34 are summarized in Box 2.
BOX 2. Frontotemporal Dementia.
Frontotemporal dementia is characterized by personality changes, apathy, and deterioration in and paucity of the use of language. Personality changes can include loss of empathy and sympathy; inability to recognize affective, emotional, and sarcastic or ironic nuances in communication with others is also common. Compulsive and perseverative behaviors and hyperorality, particularly around food and eating, are seen in many patients.31
Frontotemporal dementia most frequently affects individuals younger in age than those affected by Alzheimer disease.
Pathological changes in the frontal and temporal lobes of patients with frontotemporal dementia include gliosis, lipofuscinosis, and accumulation of lysosomal proteins.32
In 60% of patients with frontotemporal dementia, there is no family history of the disease. Of the remaining patients, 60% have familial mutations in MAPT, C9orf72, or GRN. Mutations in other genes have been described.33
The association of frontotemporal dementia with amyotrophic lateral sclerosis occurs primarily in patients with mutations of C9orf72, but can occur in patients with mutations of other genes, including FUS and OPTN.34
Protein markers
In 5–10% of all patients with frontotemporal dementia who constitute a subset of those with autosomal dominant disease, frontotemporal dementia is caused by haploinsufficiency of progranulin, encoded by the GRN gene, because of mutation of one GRN allele.32,35 Most of the relevant GRN mutations involve a premature stop codon and consequent nonsense-mediated decay of the resultant progranulin fragment, impairment of progranulin secretion, or absence of a functionally important cysteine residue in the protein.35
Other patients with familial frontotemporal dementia have etiologic mutations of the MAPT gene that encodes tau protein. Mice in which MAPT has been engineered to overexpress a repressible form of the human mutant tau protein develop progressive locomotor hyperactivity, the severity of which is age dependent. The mutant tau aggregates and forms deposits in the frontal lobes of the mice. Enhancing the prevalence of the N-ace-tylglucosamine adduct or decreasing phosphorylation of the mutant tau decreases tau aggregation and deposition in the frontal lobes and prevents the hyperactive behavior. It has been conjectured that the murine hyperactivity is the equivalent of the wandering behavior prevalent in people with frontotemporal dementia.11,36
Neurodevelopmental roles of protein markers
Frontotemporal dementia shares many of its pathological features with a neurodegenerative disorder with onset in adolescence, the variety of neuronal ceroid lipofuscinosis (NCL) due to mutations of the GRN gene which encodes the protein progranulin. Unlike genetic cases of frontotemporal dementia, NCL is caused by homozygous or compound heterozygous mutations in GRN. NCL due to GRN mutations is an autosomal recessive disorder that differs clinically from frontotemporal dementia. It results in seizures, myoclonus, retinitis pigmentosa, cataracts, cerebellar ataxia, and dementia.37 The divergence of age of onset and disease manifestations between NCL and frontotemporal dementia reflects one of a handful of situations of gene dose dependence of clinical phenotype.
Life course aspects of protein markers
Progranulin is important throughout the life cycle for lysosomal protein trafficking. Localization of the lysosomal proteins glucocerebrosidase, hexosaminidase A, and prosaposin to the lysosomes is dependent on progranulin.38 It is not clear why two completely different diseases, with different time course and phenotype, arise from haploinsufficiency and total insufficiency, respectively, of progranulin. But progranulin deficiency in neurons increases cellular autophagy and causes abnormal enlargement of lysosomes. Enhancement of progranulin levels restores the lower incidence of autophagy and returns lysosome size to control levels.39
Progranulin is expressed in the placenta, epidermis, microvasculature, and brain during murine development.40 GRN−/− mice have decreased angiogenesis in the labyrinth section of the placenta.41 GRN expression was upregulated in the villous tropho-blasts of women who had preeclampsia (PE) during their pregnancies.42
Huntington disease
The clinical, pathological, and epidemiological features of Huntington disease43 are summarized in Box 3.
BOX 3. Huntington Disease.
Huntington disease is a progressive lethal autosomal dominant disorder caused by excessive trinucleotide repeats in the HTT gene that encodes the protein huntingtin.
The wild-type HTT gene contains fewer than 20 CAG repeats; patients with Huntington disease have an HTT gene with more than 36 repeats. Repeat number correlates inversely with age at onset of symptoms of any given severity.
These symptoms most often include chorea and cognitive and psychiatric dysfunction. The latter two can precede motor dysfunction by many years. The cognitive dysfunction involves attention, memory, and speech; the psychiatric dysfunction results in anxiety, apathy, irritability, and, in some cases, aggressive behavior. The motor dysfunction can include dystonia, bradykinesia, and rigidity in addition to chorea.43
Pathologically, Huntington disease is characterized by a loss of striatal volume, primarily due to apoptosis of medium spiny neurons. Cerebral cortical volume loss is common as well. Eventually, other regions of the brain are involved, and patients with advanced Huntington disease have brains that often weigh 70% of normal control brains.43
Protein markers
Huntington disease is caused by expanded CAG repeats of the HTT gene, which codes for the protein huntingtin. Because most people who carry an expanded HTT allele are initially asymptomatic and then slowly become increasingly symptomatic over time, it was deemed critical to develop an animal model in which the early course and evolution of the disease could be studied. Many of the pathological findings in the brains of some mice engineered to express mutant huntingtin are also seen as differences in comparative studies of postmortem brain tissue from control subjects and patients with Huntington disease, respectively. Other murine models, however, do not exhibit neurodegeneration or exhibit it in a distribution different from that seen in human Huntington disease. One such model, R6/2 mice, exhibits increased vascular branching and pericyte activation in the striatum followed by the cortex, much the same as that seen in postmortem brains of patients with Huntington disease.44,45 This mouse line is transgenic for the 5’ end of the human HTT gene, carrying over 100 CAG repeats.46
Mouse models of Huntington disease have revealed that synaptic dysfunction results from expression of the mutant huntingtin. Recent studies have suggested that Huntington disease is a circuit disorder affecting the corticobasal ganglia-thalamocortical loop and rendering corticostriatal neurons injurious to their targets. Dysgenesis of corticostriatal connections, including those with non-neuronal cells, may underlie this aberrant interaction.47
Recent human pathological studies have suggested that hyperphosphorylation and aggregation of tau protein contribute secondarily to the cognitive dysfunction of Huntington disease. The mechanistic relationship between huntingtin and tau is not clear, but pharmacologic modification of tau has been suggested as symptomatic treatment for Huntington disease.48
Neurodevelopmental roles of protein markers
Huntingtin is expressed in preimplantation embryos, and knockout of huntingtin expression is lethal in embryos.49 Huntingtin plays a scaffolding role in vesicular and BDNF transport, cell division, and ciliogenesis.47 Expression of trinucleotide repeat–expanded huntingtin results in behavioral, cognitive, brain morphological, cellular, and metabolic abnormalities that are detectible as early as 4 weeks of age in rats and 14 days of age in mice.49
Huntingtin is not expressed in the placenta. A meta-analysis of raw microarray data deposited in the Gene Expression Omnibus (GEO) database compared peripheral blood samples from people with cardiovascular disease (CVD), placentas from pregnant women who had PE, and pregnant women with uncomplicated pregnancies and found 22 commonly expressed genes in CVD and PE. Further gene set enrichment analyses identified dysregulated pathways that overlapped with those seen in individuals with Huntington disease.50
The creation of neuruloids that mimic the ontogeny of the caudal forebrain and diencephalon has afforded in vitro study of the effects of knockout and trinucleotide repeat amplification of the gene for huntingtin on brain embryogenesis.51 Several findings from this study speak to the role of proteins that are important in late-life neurodegenerative disease during embryogenesis. First, knockout and CAG copy number amplification of HTT have similar effects on morphogenesis and the single-cell transcriptomes of neuruloids, suggesting that trinucleotide repeat amplification in Huntington disease results in loss of function. Second, both CAG expansion and knockout of HTT lead to a decrease in the lumen size of neuruloids and extension of its PAX6 domain, which condenses over time in wild-type neuruloids. Third, changes in the transcriptomes of neuruloid cells with either knockout or CAG expansion of HTT suggest blocked polarization of neuroepithelial cells.
Studies of the striatum in R6/2 mice and homozygous HTT mutant rats demonstrated dysregulation of expression of genes related to neural transmission and signaling in the dopaminergic and glutamatergic pathways in early postnatal life. Furthermore, examination of differentiative capacity of subventricular zone precursor and stem cells in both models demonstrated decreased neuronal differentiation with both increased oligodendroglial differentiation and apoptotic cell death in evidence. Behaviorally, animals that expressed the trinucleotide repeat expanded HTT demonstrated increased risk-taking and less anxious behavior traits. Of potential therapeutic and mechanistic interest, treatment with vorinostat, a histone deacetylase inhibitor, reversed or prevented the behavioral, biochemical, and cellular accompaniments of expression of mutant huntingtin.49
Life course aspects of protein markers
Questions about the relevance of rodent models to the roles of huntingtin in developing humans and to Huntington disease have led to an effort to develop and validate models of Huntington disease in larger mammals. Initial efforts to produce a porcine model were problematic because insertion of the human HTT gene results in embryonic lethality. Subsequently, a porcine model was produced by CRISPR-Cas9-facilitated replacement of the pig exon 1 with human exon 1 containing 150 CAG repeats in the endogenous porcine HTT gene. Unlike rodent models, this porcine model of Huntington disease exhibits neurodegeneration in the striatum and cerebral cortex, but not in the cerebellum. The striatal degeneration predominantly involved medium spiny neurons and was accompanied by gliosis, as is the case in human Huntington disease.52 It will be important to examine these pigs with Huntington disease prenatally and in early life to discern developmental changes in the brain related to aberrant function of huntingtin.
Parkinson disease
The clinical, pathological, and epidemiological features of Parkinson disease53 are summarized in Box 4.
BOX 4. Parkinson Disease.
Parkinson disease is a progressive movement disorder that affects 1–2% of the population older than 65 years.
Clinically, it is characterized by bradykinesia, tremor, rigidity, and loss of postural control.
Pathologically, Parkinson disease results in intraneuronal eosinophilic inclusions consisting primarily of the protein α-synuclein (the so-called Lewy bodies) and neuronal loss in the pars compacta of the substantia nigra.
While most Parkinson disease is sporadic, 10% of cases cluster in families with autosomal dominant or recessive inheritance. This has led to the hypothesis that both genetic and environmental factors contribute to the pathogenesis of the disease.53
Protein markers
Mutations of the GBA1 gene, which codes for the enzyme glucocerebrosidase, are associated with susceptibility to Parkinson disease and Lewy body dementia. However, the relationship is both complex and mechanistically cryptic. Carriers of mutations of the GBA1 gene, only some of which are the same mutations that, in their homozygous state, cause Gaucher disease, are at increased risk for Parkinson disease and Lewy body dementia. The GBA1 gene is of variable penetrance, and it appears that its penetrance is influenced by age and by the presence of variants of two other genes, SCNA and CTSB.54,55 This is of particular interest because the lysosomal cysteine protease, cathepsin B, encoded by the CTSB gene, is thought to enhance the aggregation of α-synuclein, a key contributor to the pathogenesis of Parkinson disease.56 In addition, the specific GBA1 mutation in a carrier influences the age at onset of symptoms in Parkinson disease.55
One theory regarding the relationship between GBA1 mutations and susceptibility to Parkinson disease relates to the finding that decreased glucocerebrosidase activity results in accumulation of glucosylceramide and glucosylsphingosine, which, in turn, decreases lysosomal function and, consequently, α-synuclein degradation, and enhances α-synuclein aggregation.57
But the relationship between GBA1 carrier status and susceptibility to Parkinson disease is not as simple as the degree in reduction of enzyme activity. While homozygotes for GBA1 mutations, who have neuropathic Gaucher disease (see the next section), are at greater risk than the general population to develop Parkinson disease, 90% of patients with neuropathic Gaucher disease do not develop Parkinson disease and mutations of GBA1 are found in patients with Parkinson disease with and without Gaucher disease.57 Studies of siblings concordant for Gaucher disease have found they are sometimes discordant for Parkinson disease and have not yet identified etiologic factors underlying this discordance.58
Glucocerebrosidase synthesis and organellar disposition require protein synthesis in the endoplasmic reticulum, cleavage of its signal peptide, glycosylation in the Golgi, packaging in endocytic vesicles, and trafficking to the lysosome. Mutations that do not result in reduction of enzyme activity have been shown to impair mitochondrial autophagy and lysosomal trafficking and to enhance and perpetuate aggregation of α-synuclein. Misfolding of the enzyme is also thought to play a role in this pathologic process.59 Misfolded proteins are disposed of by the cell in a process that requires activity of the endoplasmic reticulum and the ubiquitin-proteasome system, including the Parkinson disease–associated ubiquitin E3 lyase, parkin. It has been hypothesized that misfolded mutant glucocerebrosidase produces a toxic gain of function that overwhelms or damages the ability of the endoplasmic reticulum to dispose of not only glucocerebrosidase but also all misfolded or ubiquitinated proteins ordinarily earmarked for destruction. In addition, endoplasmic reticulum stress leads to cellular inrush of calcium and consequent mitochondrial dysfunction and oxidative injury.57 It has been proposed that regulation of mitochondrial number and autophagy of oxidatively damaged mitochondria are disturbed in mutant GBA1 heterozygous but not homozygous neuroblasts.60
In view of these proposed mechanisms, several approaches to therapy for GBA1-related Parkinson disease have been suggested. These include glucocerebrosidase replacement therapy potentially with direct delivery into the central nervous system, glucoceramide and glucosphingosine reduction therapy, and chaperonin therapy aimed at reducing the incidence of protein misfolding.57 Studies to date suggest that none of these proposed therapies prevent the development of Parkinson disease in patients with Gaucher disease.58
Recent studies using induced pluripotent stem cells derived from patients with simultaneous mutations in the Parkinson disease–associated genes LRRK2 and GBA1 and differentiated to dopaminergic neurons indicate that LRRK2 kinase regulates glucocerebrosidase activity through the GTPase, Rab10. Parkinson disease–associated mutant LRRK2 kinase has increased kinase activity and decreased glucocerebrosidase activity. CRISPR-Cas correction of the LRRK2 mutation or pharmacologic inhibition of LRRK2 kinase results in restoration of glucocerebrosidase activity in these iPSC-derived dopaminergic neurons. Furthermore, these “repaired” cells do not demonstrate the accumulation of oxidation products seen in the double mutant cells.61
PTEN-induced kinase (PINK) is a mitochondrial serine/threonine kinase that is inhibited when mitochondria are damaged and when oxidative phosphorylation is uncoupled. Under these pathological conditions, PINK accumulates in the mitochondrial membrane and attracts the ubiquitin protein ligase, parkin, the activity of which results in mitochondrial degradation, a process known as mitophagy. Mitophagy involving PINK and parkin is associated with autosomal recessive, juvenile Parkinson disease62 and a host of other conditions in which mitochondria are damaged and earmarked for destruction. For example, PE results in placental mitophagy63; conversely, cancer cells, with their relative preference for anaerobic metabolism, underutilize mitophagy and accumulate aberrant mitochondria.64
Neurodevelopmental roles of protein markers
While neither GBA1 nor the PRKN genes are expressed in the placenta, mesenchymal stem cells derived from the human placenta spontaneously produce neurotrophic factors that provide neuroprotection as well as immunosuppression when used as a cell-based therapy for Parkinson disease in a rat model.65
Homozygous and compound heterozygous mutations of GBA1 result in the metabolic disorder, Gaucher disease, a disorder with onset often in childhood that includes both neuropathic and non-neuropathic variants. Classically, Gaucher disease has been divided into three subtypes: Type 1, non-neuropathic; Type 2, infantile, acute neuropathic; and Type 3, chronic neuropathic. More recently, this classification has been called into question, and the notion of a “spectrum” of nervous system involvement and rapidity of progression with disease has been proposed. Neurological manifestations include prominent brainstem and, particularly, cranial nerve dysfunction with swallowing difficulty, stridor, and oculomotor palsy, rigidity and hypotonia evolving to hypertonia, and, in infantile acute disease, opisthotonos. Seizures are a late manifestation. Pathologically, in the central nervous system, there is an accumulation of tubular inclusions consisting primarily of glucocerebroside in the cytoplasm of neurons.66
It has been shown that particular mutations in GBA1 are highly associated with more severe neuropathic, classically Type 2 Gaucher disease, while other mutations are more likely to cause non-neuropathic Type 1 Gaucher disease. An almost 13-year study of 2304 patients with Parkinson disease who had undergone GBA1 sequencing demonstrated that patients with Parkinson disease who carried a GBA1 allele associated with severe neuropathic Gaucher disease were three times more likely to have aggressive, global cognitive decline associated with their Parkinson disease as those who carried a non-neuropathic allele or who were non-carriers of a GBA1 mutant.67
Life course aspects of protein markers
Patients with neuropathic Gaucher disease are at higher risk than the general public for development in later life of Parkinson disease, and heterozygotes with GBA1 mutations that are associated with more aggressive neuropathic Gaucher disease are most likely, if they develop Parkinson disease, to have associated aggressive cognitive decline.67 In a similar vein, heterozygosity for mutations in NPC1 and NPC2, the genes that, if mutated in both copies, would result in Niemann Pick type C disease, another metabolic disorder that can affect the nervous system, has been shown to correlate with higher incidence of a variety of neurodegenerative diseases, including Parkinson disease, progressive supranuclear palsy, and corticobasal degeneration and other dementias.68 This raises the hypothesis that these predisposing mutations decrease endogenous resistance to insults, endogenous or exogenous, that are the proximate cause of neurodegenerative disorders such as Parkinson disease. Suspected insults in this regard have included oxidative stress, mitochondrial damage, and misfolding and consequent aggregation of cellular proteins.53
Summary and future questions
New information that has emerged since we first proposed that proteins implicated in Alzheimer and Parkinson diseases play major roles in brain development1 supports and extends this hypothesis. The threads that now seem to tie these diseases together are abnormalities of protein processing, trafficking, and aggregation; mitochondrial dysfunction; and oxidative stress.9,10,62,69,70 The developmental roles of proteins implicated in the pathogenesis of neurodegenerative diseases and the association of genes that mediate susceptibility to these disorders with diseases of childhood suggest that the origins of neurodegeneration may be there in prenatal life. However, it remains probable that, in some cases, different modifications of the same protein are pathogenic in different ways at different epochs in the life cycle and are not pathogenically related to one another. Model systems facilitate longitudinal examination of the structure and function of proteins and protein networks from embryogenesis through senescence and collectively mimic many of the characteristics of neurodevelopmental and neurodegenerative diseases (Table 3). In addition, new insights into the complex roles of gene dose and epigenetic context in determination of phenotypic features and age at onset of disease have emerged.26–28,54,55,66,67 Further study of protein processing and trafficking, mitochondrial function, and resistance to oxidative stress in the developing nervous system may shed light on susceptibility to late-life neurodegeneration and identify therapeutic and preventive targets for neurodegenerative diseases.
TABLE 3.
Developmental Model Systems Used to Study Neurodegenerative Diseases
| Developmental Model System | Neurodegenerative Disease(S) Studied | Advantage | Limitation |
|---|---|---|---|
| Human embryonic stem cells | Familial Alzheimer disease76 | Ability to manipulate genome by CRISPR/Cas9; uniform genetic background | Need to know specific genetic aberration responsible |
| Human induced pluripotent stem cells and adult neural stem cells | Alzheimer disease77,78 Parkinson disease79 Huntington disease80 |
Can study cells from patients with disease without genetic manipulation | Genetic background of cells from different patients is different |
| Murine embryonic fibroblasts | Parkinson disease81 | Easy to obtain and grow | Question of relevance as model of human neural cells and systems |
| Zebrafish | Alzheimer disease82 Parkinson disease83,84 |
Easily bred and genetically manipulated; transparent skin affords visibility of organs | Question of relevance as model of human nervous system function |
| Mutant or genetically engineered mice and rats | Alzheimer disease12,85 Parkinsons disease86,87 |
Easily bred and genetically manipulated; uniform genetic background | Question of relevance as model of human nervous system function |
The present study largely examined proteins important in late-life disease and queried their functions during normal development. Approaching this topic from another vantage point – specifically, examining these proteins from the standpoint of their role in neurodevelopmental disorders – is similarly fruitful. Many proteins implicated in Alzheimer disease are also implicated in severe autism.71 Dysregulation of secretion of sAPPα has been described in autism and fragile X syndrome. Of note, fragile X mental retardation protein represses transcription of the APP gene. Activation of the metabotropic glutamate receptor (mGluR5) releases this repression, enhancing production of APP and secretion of sAPPα.71,72 In addition, sAPPα has been demonstrated to have anabolic properties and may be responsible for the increase in brain size seen in both some people with autism and most people with fragile X syndrome.73
While examples of abnormalities of protein sequence or post-translational modification can lead to neurodevelopmental and neurodegenerative dysfunction, abnormalities of protein concentration, subcellular or extracellular localization, or temporal expression during the life cycle and proteomic abnormalities that represent aberrant changes in coexpression during the life cycle are also sources of disorder. The effects of maturational delay or precocity of the anatomic and proteomic components of neural networks on propensity for late-life degeneration or disorganization of these networks are yet to be fully discerned.74,75 Novel approaches, including use of bioinformatic techniques, to represent and characterize protein coexpression changes over time and space in health and disease will afford a more holistic view of neurodevelopmental clues to neurodegeneration.75
Acknowledgments
The authors are grateful to Nancy Bowen, M.P.H., Dr.P.H. for assistance in retrieving and evaluating references for this review.
Footnotes
Conflict of interest and source of funding statement: The authors declare no conflict of interest or financial disclosures concerning the materials or methods used in this study or the findings specified in this article.
References
- 1.Rogers D, Schor NF. The child is father to the man: developmental roles for proteins of importance for neurodegenerative disease. Ann Neurol. 2010;67: 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strosznajder AK, Wójtowicz S, Jeżyna MJ, et al. Recent insights on the role of PPAR-β/δ in neuroinflammation and neurodegeneration, and its potential target for therapy. Neuromolecular Med. 2021;23:86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Boer EMJ, Orie VK, Williams T, et al. TDP-43 proteinopathies: a new wave of neurodegenerative diseases. J Neurol Neurosurg Psychiatry. 2020;92:86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breijyeh Z, Karaman R. Comprehensive review on Alzheimer disease: causes and treatment. Molecules. 2020;25:5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coronel R, Palmer C, Bernabeu-Zornoza A, et al. Physiological effects of amyloid precursor protein and its derivatives on neural stem cell biology and signaling pathways involved. Neural Regen Res. 2019;14:1661–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartolome F, de la Cueva M, Pascual C, et al. Amyloid β-induced impairments on mitochondrial dynamics, hippocampal neurogenesis, and memory are restored by phosphodiesterase 7 inhibition. Alzheimers Res Ther. 2018;10:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lei H, Zhang Y, Huang L, et al. L-3-n-Butylphthalide regulates proliferation, migration, and differentiation of neural stem cell in vitro and promotes neurogenesis in APP/PS1 mouse model by regulating BDNF/TrkB/CREB/Akt pathway. Neurotox Res. 2018;34:477–488. [DOI] [PubMed] [Google Scholar]
- 8.Borin M, Saraceno C, Catania M, et al. Rac1 activation links tau hyperphosphorylation and Aβ dysmetabolism in Alzheimer disease. Acta Neuropathol Commun. 2018;6:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee CW, Stankowski JN, Chew J, et al. The lysosomal protein cathepsin L is a progranulin protease. Mol Neurodegener. 2017;12:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balaji V, Kaniyappan S, Mandelkow E, et al. Pathological missorting of endogenous MAPT/Tau in neurons caused by failure of protein degradation systems. Autophagy. 2018;14:2139–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X, Smith K, Pearson M, et al. Early intervention of tau pathology prevents behavioral changes in the rTg4510 mouse model of tauopathy. PLoS One. 2018;13:e0195486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aulston BD, Schapansky J, Huang Y, et al. Secreted amyloid precursor protein alpha activates neuronal insulin receptors and prevents diabetes-induced encephalopathy. Exp Neurol. 2018;303:29–37. [DOI] [PubMed] [Google Scholar]
- 13.Jin J, Ravindran P, Di Meo D, Püschel AW. Igf1R/InsR function is required for axon extension and corpus callosum formation. PLoS One. 2019;14:e0219362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ortega-Martínez S A new perspective on the role of the CREB family of transcription factors in memory consolidation via adult hippocampal neurogenesis. Front Mol Neurosci. 2015;8:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corbetta S, Gualdoni S, Ciceri G, et al. Essential role of Rac1 and Rac3 GTPases in neuronal development. FASEB J. 2009;23:1347–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong OGW, Cheung CLY, Ip PPC, et al. Amyloid precursor protein overexpression in Down syndrome trophoblast reduces cell invasiveness and interferes with syncytialization. Am J Pathol. 2018;188:2307–2317. [DOI] [PubMed] [Google Scholar]
- 17.Xu L, Li J, Luo Z, et al. Aβ inhibits mesenchymal stem cell-pericyte transition through MAPK pathway. Acta Biochim Biophys Sin (Shanghai). 2018;50: 776–781. [DOI] [PubMed] [Google Scholar]
- 18.Aller-Alvarez JS, Menéndez-González M, Ribacoba-Montero R, et al. Myoclonic epilepsy in Down syndrome and Alzheimer disease. Neurologiá. 2017;32: 69–73. [DOI] [PubMed] [Google Scholar]
- 19.Beagle AJ, Darwish SM, Ranasinghe KG, et al. Relative incidence of seizures and myoclonus in Alzheimer’s disease, dementia with Lewy bodies, and frontotemporal dementia. J Alzheimers Dis. 2017;60:211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McVicker RW, Shanks OEP, McClelland RJ. Prevalence and associated features of epilepsy in adults with Down’s syndrome. Br J Psychiatry. 1994;164: 528–532. [DOI] [PubMed] [Google Scholar]
- 21.Takashima S, Becker LE, Armstrong DL, Chan F-W. Abnormal neuronal development in the visual cortex of the human fetus and infant with Down’s syndrome, a quantitative and qualitative Golgi study. Brain Res. 1981;225:1–21. [DOI] [PubMed] [Google Scholar]
- 22.Sarnat HB. Excitatory/inhibitory synaptic ratios help explain epileptogenesis in polymicrogyria and Down syndrome. Pediatr Neurol. 2021;116:41–54. [DOI] [PubMed] [Google Scholar]
- 23.Dejanovic B, Huntley MA, De Mazière A, et al. Changes in the synaptic proteome in tauopathy and rescue of Tau-induced synapse loss by C1q antibodies. Neuron. 2018;100:1322–1336.e7. [DOI] [PubMed] [Google Scholar]
- 24.Seifan A, Schelke M, Obeng-Aduasare Y, Isaacson R. Early life epidemiology of Alzheimer disease–a critical review. Neuroepidemiology. 2015;45:237–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma Q, Zhao Z, Sagare AP, et al. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-β42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol Neurodegener. 2018;13:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kilian J, Kitazawa M. The emerging risk of exposure to air pollution on cognitive decline and Alzheimer disease - evidence from epidemiological and animal studies. Biomed J. 2018;41:141–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim DH, Lim H, Lee D, et al. Thrombospondin-1 secreted by human umbilical cord blood-derived mesenchymal stem cells rescues neurons from synaptic dysfunction in Alzheimer disease model. Sci Rep. 2018;8:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim DH, Lee D, Lim H, et al. Effect of growth differentiation factor-15 secreted by human umbilical cord blood-derived mesenchymal stem cells on amyloid beta levels in in vitro and in vivo models of Alzheimer disease. Biochem Biophys Res Commun. 2018;504:933–940. [DOI] [PubMed] [Google Scholar]
- 29.Xu Z, Nan W, Zhang X, et al. Umbilical cord mesenchymal stem cells conditioned medium promotes Aβ25–35 phagocytosis by modulating autophagy and Aβ-degrading enzymes in BV2 cells. J Mol Neurosci. 2018;65:222–233. [DOI] [PubMed] [Google Scholar]
- 30.Nil Z, Hervás R, Gerbich T, et al. Amyloid-like assembly activates a phosphatase in the developing drosophila embryo. Cell. 2019;178:1403–1420.e21. Erratum in: Cell 2019;179:801. [DOI] [PubMed] [Google Scholar]
- 31.Coraini A, Basciotta M. Frontotemporal dementia as underlying cause of newly altered mental status in a 59-year-old female: a case presentation and literature review. J Community Hosp Intern Med Perspect. 2020;10:446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arrant AE, Onyilo VC, Unger DE, Roberson ED. Progranulin gene therapy improves lysosomal dysfunction and microglial pathology associated with frontotemporal dementia and neuronal ceroid lipofuscinosis. J Neurosci. 2018;38: 2341–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gowell M, Baker I, Ansorge O, Husain M. Young-onset frontotemporal dementia with FUS pathology. Pract Neurol. 2020;21:149–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pang W, Hu F. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J Neurochem. 2021;157:334–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bartoletti-Stella A, De Pasqua S, Baiardi S, et al. Characterization of novel progranulin gene variants in Italian patients with neurodegenerative diseases. Neurobiol Aging. 2021;97, 145.e7–145.e15. [DOI] [PubMed] [Google Scholar]
- 36.Midorikawa A, Suzuki H, Hiromitsu K, Kawamura M. Wandering behavior of a severely demented patient with frontotemporal dementia. Neurocase. 2016;22:220–224. [DOI] [PubMed] [Google Scholar]
- 37.Huin V, Barbier M, Bottani A, et al. Homozygous GRN mutations: new phenotypes and new insights into pathological and molecular mechanisms. Brain. 2020;143:303–319. Erratum in: Brain 2020;143:e24. [DOI] [PubMed] [Google Scholar]
- 38.Arrant AE, Roth JR, Boyle NR, et al. Impaired β-glucocerebrosidase activity and processing in frontotemporal dementia due to progranulin mutations. Acta Neuropathol Commun. 2019;7:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elia LP, Mason AR, Alijagic A, Finkbeiner S. Genetic regulation of neuronal progranulin reveals a critical role for the autophagy-lysosome pathway. J Neurosci. 2019;39:3332–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Daniel R, Daniels E, He Z, Bateman A. Progranulin (acrogranin/PC cell-derived growth factor/granulin-epithelin precursor) is expressed in the placenta, epidermis, microvasculature, and brain during murine development. Dev Dyn. 2003;227:593–599. [DOI] [PubMed] [Google Scholar]
- 41.Xu B, Chen X, Ding Y, et al. Abnormal angiogenesis of placenta in progranulin-deficient mice. Mol Med Rep. 2020;22:3482–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stubert J, Schattenberg F, Richter DU, et al. Trophoblastic progranulin expression is upregulated in cases of fetal growth restriction and preeclampsia. J Perinat Med. 2012;40:475–481. [DOI] [PubMed] [Google Scholar]
- 43.Lahue RS. New developments in Huntington disease and other triplet repeat diseases: DNA repair turns to the dark side. Neuronal Signal. 2020;4: NS20200010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shah K, Abbruscato T. Chapter 6. The blood-brain barrier: a restricted gateway to the brain. In: Conn PM, ed. Conn’s Translational Neuroscience. Cambridge, MA: Academic Press; 2016:141–146. [Google Scholar]
- 45.Padel T, Roth M, Gaceb A, et al. Brain pericyte activation occurs early in Huntington disease. Exp Neurol. 2018;305:139–150. [DOI] [PubMed] [Google Scholar]
- 46.Jackson Laboratories. A field guide to working with mouse models of Huntington disease. Available at: https://jackson.jax.org/rs/444-BUH-304/images/Guide_Huntingtons_Disease.pdf. Accessed November 18, 2020.
- 47.Cepeda C, Levine MS. Synaptic dysfunction in Huntington disease: lessons from genetic animal models. Neuroscientist. 2020;1073858420972662. 10.1177/1073858420972662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masnata M, Salem S, de Rus Jacquet A, et al. Targeting tau to treat clinical features of Huntington disease. Front Neurol. 2020;11:580732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siebzehnrubl FA, Raber KA, Urbach YK, et al. Early postnatal behavioral, cellular, and molecular changes in models of Huntington disease are reversible by HDAC inhibition. Proc Natl Acad Sci U S A. 2018;115:E8765–E8774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sitras V, Fenton C, Acharya G. Gene expression profile in cardiovascular disease and preeclampsia: a meta-analysis of the transcriptome based on raw data from human studies deposited in Gene Expression Omnibus. Placenta. 2015;36:170–178. [DOI] [PubMed] [Google Scholar]
- 51.Haremaki T, Metzger JJ, Rito T, et al. Self-organizing neuruloids model developmental aspects of Huntington disease in the ectodermal compartment. Nat Biotechnol. 2019;37:1198–1208. [DOI] [PubMed] [Google Scholar]
- 52.Yan S, Tu Z, Liu Z, et al. A huntingtin knockin pig model recapitulates features of selective neurodegeneration in Huntington disease. Cell. 2018;173: 989–1002.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Minakaki G, Krainc D, Burbulla LF. The convergence of alpha-synuclein, mitochondrial, and lysosomal pathways in vulnerability of midbrain dopaminergic neurons in Parkinson disease. Front Cell Dev Biol. 2020;8:580634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Do J, McKinney C, Sharma P, Sidransky E. Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegener. 2019;14:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blauwendraat C, Reed X, Krohn L, et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson disease and Lewy body dementia. Brain. 2020;143:234–248. Erratum in: Brain 2020;143:e33 Erratum in: Brain 2020;143:e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsujimura A, Taguchi K, Watanabe Y, et al. Lysosomal enzyme cathepsin B enhances the aggregate forming activity of exogenous α-synuclein fibrils. Neurobiol Dis. 2015;73:244–253. [DOI] [PubMed] [Google Scholar]
- 57.Johnson PH, Weinreb NJ, Cloyd JC, et al. GBA1 mutations: prospects for exosomal biomarkers in α-synuclein pathologies. Mol Genet Metab. 2020;129: 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lopez G, Steward A, Ryan E, et al. Clinical evaluation of sibling pairs with Gaucher disease discordant for parkinsonism. Mov Disord. 2020;35:359–365. [DOI] [PubMed] [Google Scholar]
- 59.Indellicato R, Trinchera M. The link between Gaucher disease and Parkinson disease sheds light on old and novel disorders of sphingolipid metabolism. Int J Mol Sci. 2019;20:3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morén C, Juárez-Flores DL, Chau KY, et al. GBA mutation promotes early mitochondrial dysfunction in 3D neurosphere models. Aging (Albany NY). 2019;11:10338–10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ysselstein D, Nguyen M, Young TJ, et al. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson disease patients. Nat Commun. 2019;10:5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson disease. Neuron. 2015;85:257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ausman J, Abbade J, Ermini L, et al. Ceramide-induced BOK promotes mitochondrial fission in preeclampsia. Cell Death Dis. 2018;9:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yan C, Gong L, Chen L, et al. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy. 2020;16: 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Conese M, Cassano R, Gavini E, et al. Harnessing stem cells and neurotrophic factors with novel technologies in the treatment of Parkinson disease. Curr Stem Cell Res Ther. 2019;14:549–569. [DOI] [PubMed] [Google Scholar]
- 66.Alaei MR, Tabrizi A, Jafari N, Mozafari H. Gaucher disease: new expanded classification emphasizing neurological features. Iran J Child Neurol. 2019;13: 7–24. [PMC free article] [PubMed] [Google Scholar]
- 67.Liu G, Boot B, Locascio JJ, et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson. Ann Neurol. 2016;80:674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schneider SA, Tahirovic S, Hardy J, et al. Do heterozygous mutations of Niemann-Pick type C predispose to late-onset neurodegeneration: a review of the literature. J Neurol. 2021;268:2055–2064. [DOI] [PubMed] [Google Scholar]
- 69.Paushter DH, Du H, Feng T, Hu F. The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol. 2018;136:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Naia L, Carmo C, Campesan S, et al. Mitochondrial SIRT3 confers neuroprotection in Huntington disease by regulation of oxidative challenges and mitochondrial dynamics. Free Radic Biol Med. 2021;163:163–179. [DOI] [PubMed] [Google Scholar]
- 71.Sokol DK, Maloney B, Long JM, Ray B, Lahiri DK. Autism, Alzheimer disease, and fragile X. Neurology. 2011;76:1344–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McLane RD, Schmitt LM, Pedapati EV, et al. Peripheral amyloid precursor protein derivative expression in fragile X syndrome. Front Integr Neurosci. 2019;13:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ray B, Long JM, Sokol DK, Lahiri DK. Increased secreted amyloid precursor protein (Sappα) in severe autism: proposal of a specific anabolic pathway and putative biomarker. PLoS One. 2011;6:e20405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sarnat HB. Immunocytochemical markers of neuronal maturation in human diagnostic neuropathology. Cell Tissue Res. 2015;359:279–294. [DOI] [PubMed] [Google Scholar]
- 75.Jia P, Manuel AM, Fernandes BS, Dai Y, Zhao Z. Distinct effect of prenatal and postnatal brain expression across 20 brain disorders and anthropometric social traits: a systematic study of spatiotemporal modularity. Brief Bioinform. 2021: bbab214. 10.1093/bib/bbab214 (online ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ferreccio A, Mathieu J, Detraux D, et al. Inducible CRISPR genome editing platform in naive human embryonic stem cells reveals JARID2 function in self-renewal. Cell Cycle. 2018;17:535–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barreau K, Montero-Menei C, Eyer J. The neurofilament derived-peptide NFL-TBS.40–63 enters in-vitro in human neural stem cells and increases their differentiation. PLoS One. 2018;13:e0201578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Amponsah AE, Guo R, Kong D, et al. Patient-derived iPSCs, a reliable in vitro model for the investigation of Alzheimer disease. Rev Neurosci. 2021;32:379–402. [DOI] [PubMed] [Google Scholar]
- 79.Trombetta-Lima M, Sabogal-Guáqueta AM, Dolga AM. Mitochondrial dysfunction in neurodegenerative diseases: a focus on iPSC-derived neuronal models. Cell Calcium. 2021;94:102362. [DOI] [PubMed] [Google Scholar]
- 80.Li B, Ye F, Chen L, et al. Generation of two induced pluripotent stem cell lines SPPHi001-A and SPPHi002-A from a Huntington disease family of South-western China. Stem Cell Res. 2021;50:102149. [DOI] [PubMed] [Google Scholar]
- 81.Geng K, Fu N, Yang X, Xia W. Adjudin delays cellular senescence through Sirt3 mediated attenuation of ROS production. Int J Mol Med. 2018;42:3522–3529. [DOI] [PubMed] [Google Scholar]
- 82.Jiang H, Newman M, Lardelli M. The zebrafish orthologue of familial Alzheimer disease gene PRESENILIN 2 is required for normal adult melanotic skin pigmentation. PLoS One. 2018;13:e0206155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hu Z, Zhang J, Zhang Q. Expression pattern and functions of autophagy-related gene atg5 in zebrafish organogenesis. Autophagy. 2011;7:1514–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sánchez E, Azcona LJ, Paisán-Ruiz C. Pla2g6 deficiency in zebrafish leads to dopaminergic cell death, axonal degeneration, increased β-synuclein expression, and defects in brain functions and pathways. Mol Neurobiol. 2018;55: 6734–6754. [DOI] [PubMed] [Google Scholar]
- 85.Aghajanov M, Chavushyan V, Matinyan S, et al. Alzheimer disease-like pathology-triggered oxidative stress, alterations in monoamines levels, and structural damage of locus coeruleus neurons are partially recovered by a mix of proteoglycans of embryonic genesis. Neurochem Int. 2019;131: 104531. [DOI] [PubMed] [Google Scholar]
- 86.Barkhuizen M, van Dijck FJP, Jellema RK, et al. The influence of anesthetics on substantia nigra tyrosine hydroxylase expression and tau phosphorylation in the hypoxic-ischemic near-term lamb. Pediatr Res. 2018;83: 1190–1199. [DOI] [PubMed] [Google Scholar]
- 87.Wnuk A, Rzemieniec J, Staroń J, et al. Prenatal exposure to benzophenone-3 impairs autophagy, disrupts RXRs/PPARγ signaling, and alters epigenetic and post-translational statuses in brain neurons. Mol Neurobiol. 2019;56: 4820–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]