

Abstract
Adiponectin has been demonstrated to be a mediator of insulin sensitivity; however, the underlined mechanisms remain unclear. SESN2 is a stress-inducible protein that phosphorylates AMPK in different tissues. In this study, we aimed to validate the amelioration of insulin resistance by globular adiponectin (gAd) and to reveal the role of SESN2 in the improvement of glucose metabolism by gAd. We used a high-fat diet-induced wild-type and SESN2−/− C57BL/6J insulin resistance mice model to study the effects of six-week aerobic exercise or gAd administration on insulin resistance. In vitro study, C2C12 myotubes were used to determine the potential mechanism by overexpressing or inhibiting SESN2. Similar to exercise, six-week gAd administration decreased fasting glucose, triglyceride and insulin levels, reduced lipid deposition in skeletal muscle and reversed whole-body insulin resistance in mice fed on a high-fat diet. Moreover, gAd enhanced skeletal muscle glucose uptake by activating insulin signaling. However, these effects were diminished in SESN2−/− mice. We found that gAd administration increased the expression of SESN2 and Liver kinase B1 (LKB1) and increased AMPK-T172 phosphorylation in skeletal muscle of wild-type mice, while in SESN2−/− mice, LKB1 expression was also increased but the pAMPK-T172 was unchanged. At the cellular level, gAd increased cellular SESN2 and pAMPK-T172 expression. Immunoprecipitation experiment suggested that SESN2 promoted the formation of complexes of AMPK and LKB1 and hence phosphorylated AMPK. In conclusion, our results revealed that SESN2 played a critical role in gAd-induced AMPK phosphorylation, activation of insulin signaling and skeletal muscle insulin sensitization in mice with insulin resistance.
Keywords: Adiponectin, Sestrins, AMP-Activated protein kinase, Exercise training, Glucose homeostasis
Graphical abstract

Abbreviation
- Sestrin2
SESN2
- AMP-activated protein kinase
AMPK
- Globular adiponectin
gAd
- Liver kinase B1
LKB1
- Type 2 diabetes mellitus
T2DM
- Adiponectin
Acrp30
- Adiponectin receptor1
AdipoR1
- Threonine protein kinase 11
STK11
- Wild type
WT
- Normal chow
NC
- High-fat diet
HFD
- HFD control
HC
- HFD exercise group
HE
- HFD gAd inject group
HC + gAd
- Dulbecco's modified Eagle's medium
DMEM
- Poly vinylidene difluoride
PVDF
- Oral Glucose Tolerance Test
OGTT
- Area under curve
AUC
- Horseradish peroxidase
HRP
- Total cholesterol
TC
- Triglycerides
TG
- GlVucose transporter 4
GLUT4
- GLUT4 Storage Vesicles
GSV
- Control
Con
- siRNA -SESN2 group
si-SESN2
Introduction
Insulin resistance is a decrease in insulin sensitivity of target cells and in the ability of tissue cells to take up and utilize glucose, which plays a typical role in the pathogenesis of Type 2 diabetes mellitus (T2DM). Over the past 40 years, the incidence of diabetes has increased significantly worldwide. As of 2017, the prevalence of diabetes among Chinese adults was 10.9% and the number of people with diabetes was 114 million.1,2 Currently, there are many studies dedicated to improving high-fat diet-induced insulin resistance with the aim of becoming a breakthrough in the treatment of T2DM. The adipokine adiponectin has emerged as insulin sensitizing adipokine and anti-inflammatory, insulin sensitizing factor.3, 4, 5Adiponectin is present in human plasma as full-length adiponectin (Acrp30) and as a C-terminal globular fragment (gAcrp30/gAd).6,7 The C-terminal globular fragment is produced by proteolytic cleavage and is thought to be the pharmacologically active moiety.6 The physiological actions of adiponectin are initiated by binding with its specific transmembrane receptor, mainly adiponectin receptor1 (adipoR1), in skeletal muscle. A wide variety of explanations for adiponectin's glucose lowering and insulin sensitizing properties has been proposed, which have been derived predominantly from in vitro and ex vivo studies, including: suppression of gluconeogenesis,8,9 increased AMPK/ACC-dependent fatty acid oxidation in liver and skeletal muscle.3,7,8,10 However, consistent model for adiponectin's action in vivo is lacking, and the mechanisms by which adiponectin ameliorates insulin resistance are a matter of active debate.
Exercise is likewise considered to be one of the most important approaches to ameliorating insulin resistance since it results in numerous adaptations in multiple tissues and organ systems.11 Studies have shown that exercise-associated signals can directly activate exercise-responsive signaling molecules, such as AMPK, to propagate the exercise signal. Aerobic exercise induces alterations in cellular energy balance that can result in exercise intensity-dependent activation of AMPK in skeletal muscle and promote energy metabolism and participate in the glucose homeostasis regulation.12, 13, 14 It has been demonstrated that aerobic exercise typically restores plasma adiponectin, moreover, increases the protein expression of AdipoR1 in skeletal muscle, which is closely related to adiponectin signaling.15, 16, 17
Sestrins (Sestrin1, 2, and 3, gene name: Sesn) are a classical family of stress-inducible proteins that regulate metabolism through sensing nutrient levels and redox status in cells, tissues and organs.18 The SESN2 knockout has been associated with insulin resistance.19 Long-term treadmill exercise in mice increases Sestrin2 protein expression in quadriceps, and acute treadmill exercise also increases Sestrin2 protein expression that associates with their effector AMPKα.20 It is well established that SESN2 are involved in the AMPK signaling pathway in different tissues or cell lines.12,18,21 Liver kinase B1 (LKB1), also called Serine/Threonine protein kinase 11 (STK11), phosphorylates and activates the AMPK subfamily kinases and plays a crucial role in regulating glucose homeostasis and energy metabolism in skeletal muscle.22 Some studies have reported that sestrin2 can directly associate with AMPK and promote AMPK phosphorylation by the upstream kinase LKB1 by acting as a “scaffold” between them.20,23,24
It has been reported that two-week globular adiponectin treatment ameliorates lipid-induced insulin resistance and increases absolute rates of fatty acid oxidation and glucose oxidation in muscle.25 The mechanism involved may be related to that gAd induces the translocation of LKB1 from the nucleus to the cytosol in L6 myoblasts,26 as well as in C2C12 myoblasts,27 suggesting that gAd works by regulating LKB1. And AMPK as a direct target of LKB1 and its intimate relationship with SESN2 led us to speculate that gAd may exert its effect of enhancing insulin sensitivity through them. Interestingly, a new study shows that gAd treatment induced a marked increase in SESN2 protein expression in MCF-7 cells and SESN2 knockdown led to the prevention of gAd-stimulated AMPK phosphorylation, suggesting that SESN2 mediates gAd-induced AMPK phosphorylation.28
Given that prior studies have demonstrated that aerobic exercise or globular adiponectin can improve insulin resistance in mice,11,25,29,30 we hypothesized that the insulin-sensitizing properties of adiponectin might be due to the activation of LKB1-AMPK pathway in skeletal muscle, similar to exercise. To address this hypothesis, we performed a comprehensive series of studies to assess the effects of six-week gAd treatment or aerobic exercise on skeletal muscle, in a high-fat diet mouse model of insulin resistance. Here, we demonstrate that six-week gAd treatment or aerobic exercise improves whole-body insulin sensitivity in insulin resistance mice via LKB1-SESN2-AMPK in skeletal muscle in which SESN2 contributes to an essential role, leading to increased insulin signaling. Taken together, these results provide a referable perspective on the mechanisms by which gAd or aerobic exercise reverses insulin resistance.
Materials and methods
Animals
All animal protocols were approved by the Tianjin Medical University Animal Care. SESN2-knockout (KO) mice, which are characterized by a generalized lack of SESN2, were maintained on a C57BL/6J background. Four-week-old male WT and KO mice were housed at a constant temperature (22 °C–24 °C) with a fixed 12-h light, 12-h dark cycle and free access to food and water. After 1 week of acclimation, the mice were randomly divided into an NC group (n = 7, only WT mice) and an HFD group (n = 38, WT and KO mice), fed on normal chow (NC) and high-fat diet (HFD) (60% calories from fat, Research Diets D12492), respectively, for up to six weeks. Then, the HFD group randomized into HFD control (HC), HFD exercise group (HE) and HFD gAd inject group (HC + gAd), and these three groups continually fed an HFD continually for up to 12 weeks. At the end of the intervention, n = 6,6,5,5,6,4 for WT-HC, WT-HE, WT-HC + gAd, SESN2−/−-HC, SESN2−/−-HE, SESN2−/−-HC + gAd, respectively. The mice from the exercise group were exercised on a motor-driven rodent treadmill 5 days a week for a total of 6 weeks. The mice initially ran at the intensity of 50% O2max for 30 min/day during the first week; thereafter, the running intensity and time were increased to 75% O2max (12 m/min) for 60 min/day. For adiponectin treatment, mice were given intraperitoneal injections of globular adiponectin (5 μg/kg of body weight, ProSpec) for six weeks. After 48 h of the last bout of exercise, the mice were fasted for 12–16 h then anesthetized with isoflurane and sacrificed and the quadriceps muscle was dissected.
Oral Glucose Tolerance Test (OGTT), fasting serum insulin, and metabolic parameters
Mice were fasted overnight prior to administration of OGTT. The test started at 8 a.m. d-glucose (Sigma, USA) was dissolved in ddH2O and administered orally to the fasted mice (2 g/kg of body weight) using a 20-gauge stainless steel gavage feeding needle. Samples of whole blood (10–20 μl each) were collected from a tail clip bleed immediately before and 15, 30, 60 and, 120 min after administration of glucose. Blood glucose levels were measured using the Blood Glucose Monitoring System (One Touch, Life Scan, USA). The blood samples were taken from vena caudalis, insulin level was measured by insulin ELISA kit (MLbio, China). Total cholesterol (TC) and triglycerides (TG) were determined by enzymatic kits (Applygen, China).
Cell culture and treatments
C2C12 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and maintained at 37 °C in a humidified atmosphere with 5% CO2. Cells were seeded in a six-well plate and differentiated in DMEM with 2% horse serum for six days. For SESN2 knockdown experiments, C2C12 cells was transfected with negative control or siRNA against SESN2 (GenePharma, Shanghai, China) using Lipofectamine RNAiMAX (Invitrogen). For adenoviruses treatment, C2C12 myotubes were infected with Ad-SESN2, using a multiplicity of infection (MOI) of 100 and harvested 48 h post-infection on day 6 of differentiation. For gAd treatment, gAd (0.5 μg/ml) was added to the cells for 8 h at 37 °C. Media lacking gAd served as a vehicle control. All experiments were run in triplicate.
Immunoblotting and immunoprecipitation
Protein extracts from C2C12 cell and skeletal muscle were prepared with NP-40 lysis buffer (150 mM sodium chloride, 1.0% NP-40, 50 mM Tris, pH 8.0, protease inhibitor cocktail, and phosphatase inhibitor cocktail). The extract was diluted in SDS loading buffer and heated at 75 °C for 10 min. Equal amounts of protein were subjected to electrophoresis in 10% SDS/PAGE at 110 V and transferred onto 0.45 μm poly vinylidene difluoride (PVDF) membranes at 330 mA for 1.5 h. The membranes were blocked by 5% nonfat milk for 1 h at room temperature and incubated overnight at 4 °C with the primary antibody for SESN2 (Abcam, USA), pAMPK-Thr172, AMPKα, AKT, pAKT-Ser473, AS160, pAS160-Thr642, GLUT4 (Cell Signaling Technology, USA), AdipoR1 (Santa, USA), LKB1 (Proteintech, China), β-tubulin (Utibody, China). The immunoreactive proteins were detected with a chemiluminescent horseradish peroxidase (HRP) substrate (Millipore, USA), analyzed with a scanner and digitalized using the image analysis software (Quantity One, Hercules, CA, USA). All results are representative of three independent experiments. Anti-LKB1 primary antibody was cross-linked to Dynabeads® Protein A (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Tissue lysates were precleared with IgG Dynabeads® Protein A for 10 min and then incubated with LKB1-Dynabeads. AMPKα2 immunoprecipitated complexes were washed five times with washing buffer. Proteins were eluted by boiling in loading buffer and then processed for Western blot analysis.
Statistical analysis
All data are expressed as means ± standard error of the mean (SEM). Significant differences were assessed by one-way or two way ANOVA followed by the Student–Newman–Keuls test. The p < 0.05 was considered statistically significant.
Results
Globular adiponectin improved insulin sensitivity in mice fed on a high-fat diet
As presented in Fig. 1A–D, after six weeks of HFD, C57BL/6J mice developed notably higher fasting glucose and serum insulin levels and caused impaired glucose tolerance, which was indicative of increased insulin resistance compared with the NC group. Aerobic exercise or globular adiponectin intervention reduced fasting glucose and serum insulin levels in insulin-resistant mice (Fig. 1E and F). Consistent with published reports, we observed significantly improved glucose tolerance in wild-type mice after 6-week aerobic exercise compared with mice in HC group (Fig. 1H). Meanwhile, six-week globular adiponectin treatment exerted a similar effect. In SESN2−/− mice, neither exercise nor gAd treatment produced significant changes to the fasting blood glucose. Both interventions reduced serum insulin levels but were still significantly increased compared to their corresponding WT groups. Further, in the absence of SESN2, the improvement of glucose tolerance by exercise was reduced. In addition, a high-fat diet impaired glucose tolerance in the gAd treatment group was significantly reduced compared with the HC group mice (Fig. 1I). However, unlike in WT mice, the ameliorative effect of gAd on SESN2−/− mice was relatively feeble, as evidenced by the significant difference in blood glucose levels between the two groups at 30-min points, showing a prolonged glycemic plateau. Taken together, these results suggest that gAd treatment improves insulin resistance in the high-fat diet mice similarly to aerobic exercise, and more notably that SESN2 may be important in it.
Fig. 1.

Globular adiponectin improves insulin sensitivity in mice fed on high-fat diet.
Fasting glucose (A), Oral Glucose Tolerance Test (OGTT) (B) and Area under curve (AUC) below baseline blood glucose (C) were measured after 6-week normal chow (NC) or high-fat diet (HFD) in wild-type mice (n = 7/per group). Additionally, serum insulin (D) was also measured using ELISA Kit. After exercise or gAd intervention completed, Fasting glucose (E) and serum insulin (F) were measured. (n = 6,6,5,5,6,4 for WT-HC, WT-HE, WT-HC + gAd, SESN2−/−-HC, SESN2−/−-HE, SESN2−/−-HC + gAd, respectively). Meanwhile, OGTT showed exercise or gAd impact on blood glucose (G–I). a, p < 0.05 WT-HC vs WT-HE; b, p < 0.05 WT-HC vs WT-HC + gAd; c, p < 0.05 SESN2−/−-HC vs SESN2−/−-HE; d, p < 0.05 WT-HE vs SESN2−/−-HE; e, p < 0.05 SESN2−/−-HC vs SESN2−/−-HC + gAd; f, p < 0.05 WT-HC + gAd vs SESN2−/−-HC + gAd. Data are shown as means ± standard deviation (SD). NC - normal chow, HFD - high-fat diet, HC - HFD control, HE - HFD exercise group, HC + gAd - HFD gAd inject group, WT - wild type, SESN2 - Sestrin2.
Effects of exercise or globular adiponectin on energy metabolism in mice fed on a high-fat diet
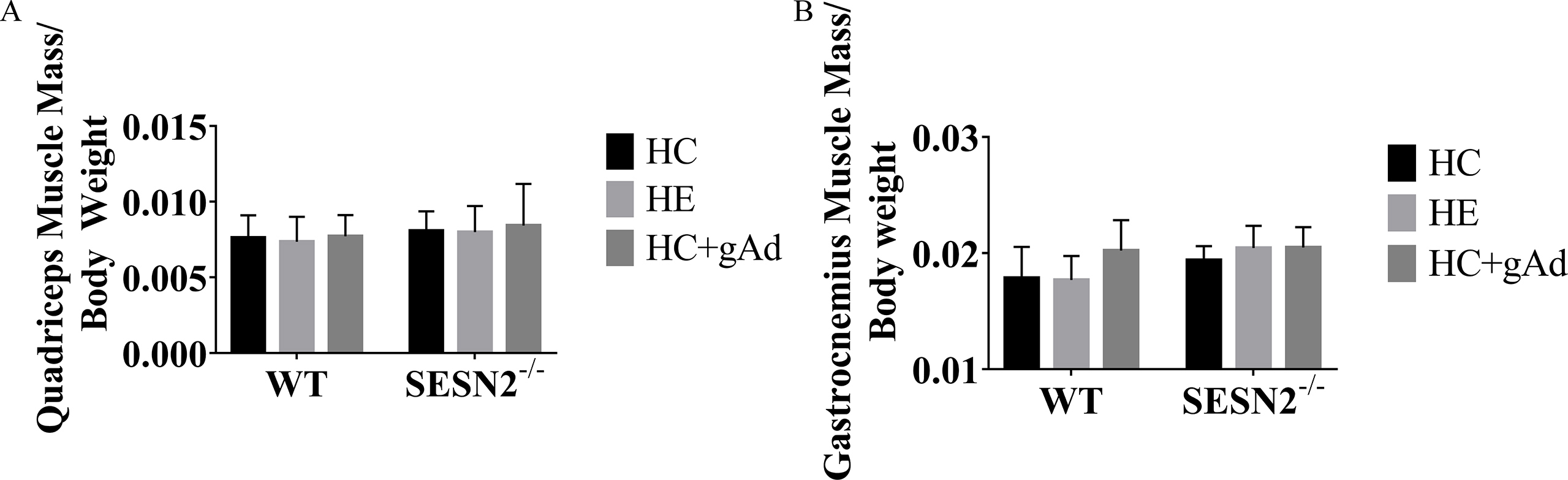
Aerobic exercise or gAd intervention did not modify the food intake and energy intake in the two groups of wild-type and SESN2−/− mice (Fig. 2A and B). While two interventions significantly reduced body weight in mice fed a high-fat diet, mainly contributed by decreases in ratio of fat mass rather than changes in the ratio of lean body mass (Fig. 2C–E). The body weight of SESN2−/− mice was generally lower than that of wild-type mice, which might be due to their lower initial body weight. Exercise did not decrease the body weight of SESN2−/− mice, but gAd still significantly induced weight loss in SESN2−/− mice. Besides, the fat-reducing effect of both interventions remained. The serum was collected and circulating TC and TG were measured with the ELISA kit. As shown in Fig. 2F and G, aerobic exercise or gAd intervention significantly reduced the serum triglyceride and total cholesterol levels in wild-type mice. However, in SESN2−/− mice, aerobic exercise or gAd intervention did not affect serum TG, but serum TG was significantly increased in the SESN2−/−-HE group than in the WT-HE group, suggesting that SESN2 may be involved in the process of lowering serum TG levels by aerobic exercise. In contrast, TC levels in the SESN2−/−-HC + gAd group were significantly higher than those in the WT-HC + gAd group, suggesting that SESN2 may be involved in the regulation of serum TC levels by gAd. Oil Red O staining of each group showed exercise or gAd treatment led to decreased lipid deposition in quadriceps. However, the SESN2 ablation prevented the protective effect of both interventions on ectopic lipid deposition in skeletal muscle (Fig. 2H, S1).
Fig. 2.

Effects of exercise or globular adiponectin on energy metabolism in mice fed on high-fat diet.
Wild-type and SESN2−/− mice were randomly allocated to sedentary or exercise group (75% O2max intensity, 12 m/min, 1 h/day, 5 days/week) or gAd injection group (5 μg/kg body weight, 7 days/week), feeding with 60% high-fat diet. Average food intake and average energy intake were determined. Additionally, final body weight (C), fat mass/body weight (D) and lean mass/body weight (E) were measured. Serum total cholesterol (TC) (F) and triglycerides TG (G) were measured with ELISA kits. Representative images of oil red staining within muscle fibers of each group (H). (n = 6,6,5,5,6,4 for WT-HC, WT-HE, WT-HC + gAd, SESN2−/−-HC, SESN2−/−-HE, SESN2−/−-HC + gAd, respectively). Data are shown as mean ± standard deviation (SD). ∗, p < 0.05. HC - HFD control, HE - HFD exercise group, HC + gAd - HFD gAd inject group, WT - wild type, SESN2 - Sestrin2.
Six-week exercise or gAd treatment modulated the insulin signaling pathway in skeletal muscle
As Fig. 3A, C, D shown, exercise or gAd administration induced increased adiponectin and adipoR1 protein levels in skeletal muscle in wild-type mice, which is consistent with the previous studies.17,31,32 After SESN2 deletion, exercise did not increase the adipoR1 expression like it did in WT mice. Although gAd still upregulated the expression of adipoR1, the protein content of adipoR1 in SESN2−/−-HC + gAd remained lower than that in WT-HC + gAd. To investigate the effects of exercise or globular adiponectin treatment on the skeletal muscle insulin signaling pathway, we examine pAKT-Ser473, AKT, pAS160-Thr642, AS160 and glucose transporter 4 (GLUT4) protein content (Fig. 3B). AKT has an important function in the insulin signaling pathway, and the level of pAKT-Ser473 could represent the activation of this pathway. The most abundant glucose transporter aiding in skeletal muscle glucose uptake is GLUT4.33 Moreover, AS160 serves as a critical signaling junction in facilitating GLUT4 trafficking to the plasma membrane.34 In wild-type mice, after six-week exercise or globular adiponectin treatment, phosphorylation of AKT was increased in comparison with control, whereas expression of total AKT did not change (Fig. 3E). Two interventions inhibited the GAP activity of AS160 via its phosphorylation at Thr642 (Fig. 3F). This allowed GTP loading of Rabs that aid in vesicular trafficking of GLUT4 to the plasma membrane and thereby increased glucose uptake. Moreover, the expression of GLUT4 were elevated in skeletal muscle after exercise or gAd treatment (Fig. 3G). While in SESN2−/− mice, aerobic exercise or gAd treatment did not alter the levels of either AKT or AS160 phosphorylation. More remarkably, the protein level of GLUT4 was also not significantly changed. Taken together, our findings suggest that the six-week exercise or globular adiponectin treatment activates the insulin signaling pathway in skeletal muscle and SESN2 plays an essential role in this process.
Fig. 3.

Six-week exercise or gAd treatment modulated the insulin signaling pathway in skeletal muscle.
Representative Western blot (A–B) and densitometric quantification of adiponectin (C), adiponectin receptor1 (AdipoR1) (D), AKT, pAKT-Ser473 (E), AS160, pAS160-Thr642 (F) and glucose transporter 4 (GLUT4) (G) in quadriceps of male WT and SESN2−/− mice following six-week exercise or gAd treatment. β-tubulin was used as the internal control (n = 3 per group). Data were presented as means ± standard error of the mean (SEM); ∗, p < 0.05. HC - HFD control, HE - HFD exercise group, HC + gAd - HFD gAd inject group, WT - wild type, SESN2 - Sestrin2.
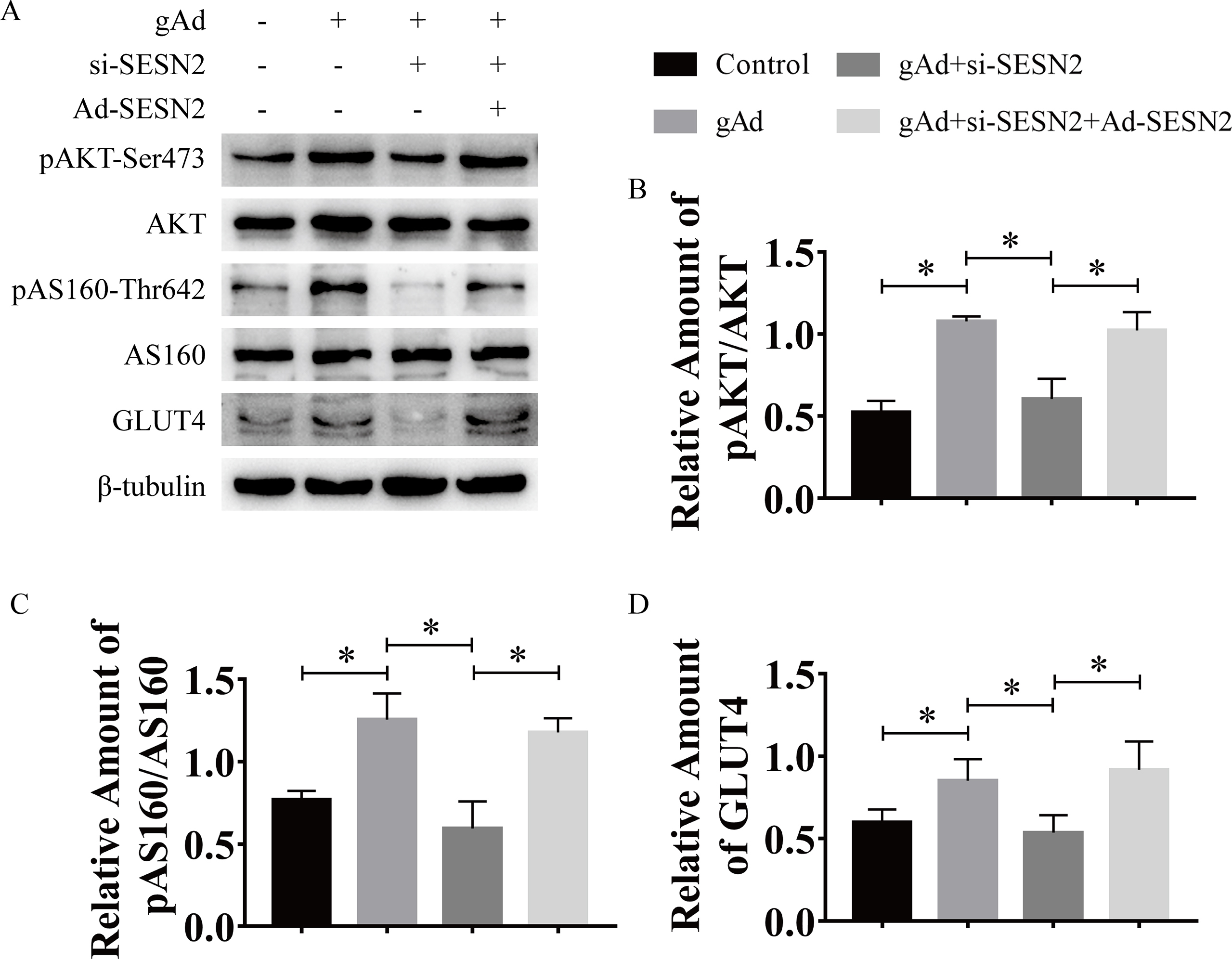
SESN2 deletion affected the activation of the LKB1/AMPK signaling pathway by gAd in skeletal muscle
Considering the vital role of AMPK and SESN2 in skeletal muscle metabolism, we hypothesized that AMPK and SESN2 might be important in the improvement of insulin sensitivity by aerobic exercise or gAd. Therefore, we examined the protein expression of AMPK and its upstream kinase LKB1 and SESN2 in skeletal muscle (Fig. 4A). We found that aerobic exercise or gAd significantly increased the content of LKB1 in skeletal muscle of wild-type mice. However, ablation of SESN2 prevented the increased LKB1 protein expression by aerobic exercise, indicating that SESN2 is involved in the regulation of LKB1 in skeletal muscle by aerobic exercise. Meanwhile, gAd tended to increase LKB1 protein content (p = 0.056) in SESN2 mice and no significant change compared with its corresponding wild-type group (Fig. 4B), suggesting that LKB1 is regulated by gAd independently of SESN2 in skeletal muscle. The expression of SESN2 was elevated by exercise or gAd (Fig. 4C). Moreover, SESN2 expression was absent in SESN2−/− mice, confirming that the SESN2 knockout mice model was successfully created. Consistent with previous studies, gAd increased the AMPK phosphorylation in skeletal muscle, resembled aerobic exercise, while knockout of SESN2 impaired the phosphorylation of AMPK induced by both interventions.
Fig. 4.

SESN2 deletion affected the activation of LKB1/AMPK signaling pathway by exercise or gAd in skeletal muscle.
Representative Western blot (A) and densitometric quantification of LKB1 (B), SESN2 (C), AMPK, pAMPK-Thr172 (D) in quadriceps of male WT and SESN2−/− mice following six-week exercise or gAd treatment (n = 3/per group). β-tubulin was used as the internal control. Data were presented as means ± standard error of the mean (SEM); ∗, p < 0.05. HC - HFD control, HE - HFD exercise group, HC + gAd - HFD gAd inject group, WT - wild type, SESN2 - Sestrin2, AMPK - AMP-activated protein kinase, LKB1 - Liver kinase B1.
SESN2 promoted the binding of LKB1 to AMPK in response to gAd treatment in myotubes
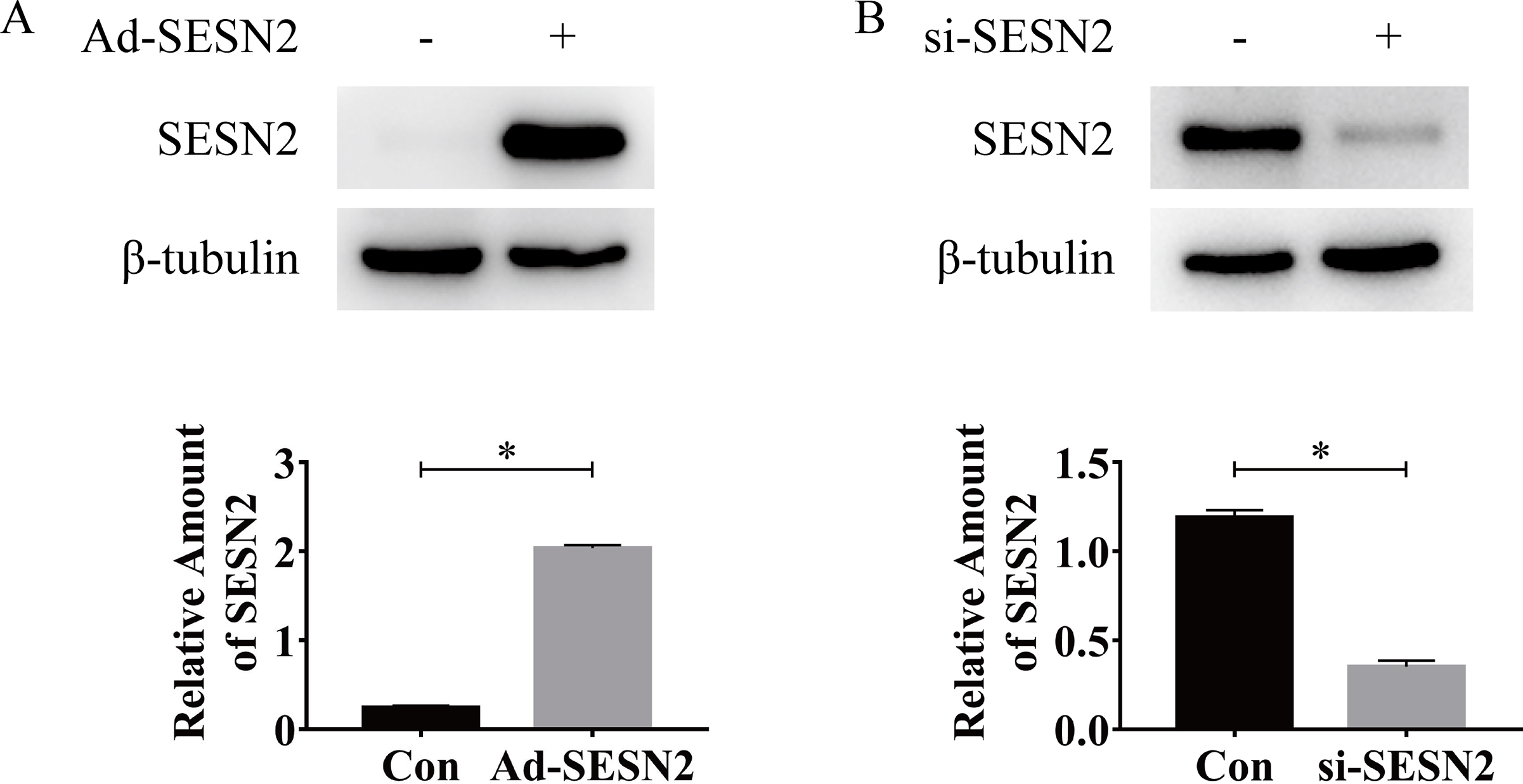
To clarify the signaling mechanisms by which gAd activates AMPK in C2C12 myotubes, we examined the role of SESN2 signaling. As shown in Fig. 5A, gAd treatment induced a marked increase in SESN2 protein expression and reached the peak at 8 h (Fig. 5A). Then we generated Ad-SESN2 and siRNA-SESN2. Ad-SESN2 elevated SESN2 expression and siRNA-SESN2 decreased the SESN2 content in myotubes (Sup Fig. 1). Additionally, the content of LKB1 was significantly increased and was independent of the expression of SESN2 in gAd treated myotubes (Fig. 5 B, C). Consistent with the previous studies, gAd treatment significantly increased the phosphorylation level of AMPK, however, sestrin2 siRNAs fully reversed the effect (Fig. 5 B, D). To further verify the relationship between SESN2, LKB2 and AMPK, we conducted immunoprecipitation experiments. LKB1 immune complexes revealed a significant increase in LKB1-AMPK interactions after gAd treatment compared to the control group, however, the absence of SESN2 prevented the binding between LKB1 and AMPK. Then after overexpression of SESN2 in the rescue group, we found that the binding of LKB1 and AMPK was increased again. All these results suggest that SESN2 promotes the formation of complexes of AMPK and LKB1, which causes subsequent phosphorylation of AMPK. In subsequent experiments verifying the functional role of SESN2 in the activation of the insulin signaling pathway by gAd, increased phosphorylation of AKT and AS160 and protein amount of GLUT4 by gAd were markedly prevention by gene silencing of SESN2 in myotubes. Moreover, overexpression of SESN2 rescued the activation of the insulin signaling pathway (Fig. S2). Collectively, these results reveal that SESN2 plays a critical role in gAd-induced AMPK phosphorylation, activation of insulin signaling pathway and insulin sensitization.
Fig. 5.

SESN2 promoted the binding of LKB1 to AMPK in response to gAd treatment in myotubes.
C2C12 myotubes were treated with gAd (0.5 μg/ml) for the indicated time periods. The expression level of SESN2 (A) was determined by Western blot analysis. Representative Western blot and densitometric quantification of LKB1, pAMPK-Thr172 and AMPK in gAd treated myotubes (B–D). After transfection with SESN2 siRNA and Ad-SESN2, AMPK associated with LKB1 (E) was measured by immunoprecipitation assay in gAd treated myotubes (n = 3 per group). β-tubulin was used as the internal control. Data were presented as means ± standard error of the mean (SEM); ∗, p < 0.05. SESN2 - Sestrin2, AMPK - AMP-activated protein kinase, LKB1 - Liver kinase B1. si-SESN2 - siRNA -SESN2 group.
Discussion
Exercise is likely to exert beneficial effects on obesity-related diseases, such as T2DM, and may contribute to healthy longevity.35,36 Strikingly, our study showed that gAd intervention led to the activation of the insulin pathway, with the extent of their recovery found to be similar to that in high-fat diet fed mice subjected to exercise, suggesting that gAd may represent “exercise-mimetic”. Nonetheless, it is likely that the physiological change linking exercise to adiponectin expression may or may not occur, but exercise and adiponectin can exert positive metabolic and muscular effects independent of each other.
In terms of improving systemic glucose metabolism, consistent with previous studies, we found that gAd significantly reduced blood glucose levels and improved impaired glucose tolerance in high-fat diet mice, which is in agreement with the improvement of glucose metabolism by aerobic exercise.37,38 Additionally, muscle lipid deposition is considered to be an important determinant of insulin resistance in skeletal muscle,39,40 since increases in intramyocellular lipid and intermyocellular adipose tissue are associated with impaired skeletal muscle insulin signaling. Our study found that either exercise or gAd intervention reduced lipid ectopic deposition in the quadriceps muscle of mice and that SESN2 was involved.
Any impairment in skeletal muscles’ AKT-AS160-GLUT4 axis leads to dire consequences in terms of whole-body glucose homeostasis.34 In basal condition, AKT are present in the cytoplasm. AS160, via its N-terminal binds to GSVs (GLUT4 Storage Vesicles) and tethers GLUT4. AS160 has a functional GAP domain in its C-terminal against several Rab GTPases that mediate vesicular traffic. This does not allow GDP-loaded Rabs to mediate GLUT4 exocytosis under basal conditions. In an insulin-stimulated condition, PI3K dependent increase in the pool of PIP3 attracts AKT by its PH domain to the plasma membrane. An activated AKT phosphorylates AS160, hence inactivating its GAP activity and promoting dissociation from GSVs. This allows GTP loading of Rabs that aid in vesicular trafficking of GLUT4 to the plasma membrane. Moreover, there was a study limited to AKT-Ser473 based on results in human skeletal muscle indicating that insulin-stimulated AKT-Ser473 phosphorylation (as opposed to AKT-Thr308) was closely related to AS160 phosphorylation and glucose uptake.41
Because a high-fat diet is associated with abnormalities in glucose metabolism, we next tested how gAd influences glucose homeostasis. We observed herein that gAd successfully improved glucose metabolism in mice fed on a high-fat diet, similarly to exercise, but this should be carefully interpreted as a high-fat diet is not fully corresponding to diabetes-mediated changes in glucose metabolism. Furthermore, based on the fact that skeletal muscle contributes largely to glucose homeostasis by facilitating glucose uptake as well as the critical role of activation of the AKT/AMPK pathway in improving glucose metabolism,42 we evaluated the effect of exercise or gAd intervention on AKT and its downstream molecules together with AMPK. In this study, given the role of AS160 on GLUT4 translocation mentioned above, gAd effectively stimulated phosphorylation of AKT and AS160, thereby enhancing glucose uptake and GLUT4 translocation. Indeed, AMPK and AKT are generally accepted to respond to exercise/muscle contraction and insulin/PI3K, respectively.43, 44, 45 In conclusion, our results demonstrate that gAd is effective in the improvement of glucose metabolism in skeletal muscle cells in vitro and in vivo.
In the models of myocardial ischemia, SESN2 has been shown to act as a “scaffold” between AMPK and LKB1, promoting phosphorylation of the AMPK-Thr172 site by enhancing the binding of AMPK to LKB1.24,46 A recent study found that gAd induced a significant increase in SESN2 protein expression and promoted the formation of SESN2-AMPK and SESN2-LKB1 complexes in MCF-7 (human breast cancer) cells, while SESN2 knockdown prevented gAd-induced AMPK phosphorylation in MCF-7 cells.28 Several studies have described that gAd induced the translocation of LKB1 from the nucleus to the cytosol in L6 myoblasts and in C2C12 myoblasts.26,27 However, no study has revealed the relationship between the SESN2/LKB1/AMPK triad in the process of insulin sensitization by gAd in skeletal muscle. In this study, we found that gAd administration increased the protein expression of SESN2 and LKB1 and activated AMPK in an animal model, while in SESN2−/− mice, LKB1 protein content was also increased without activating AMPK. At a cellular level, gAd spontaneously induced increased cellular SESN2 protein expression and AMPK phosphorylation, along with detectable activation of the AKT pathway. Importantly, the silencing of SESN2 prevented AMPK phosphorylation by reducing the binding between LKB1 and AMPK. Furthermore, overexpression of SESN2 rescued the diminished phosphorylation of AMPK due to the absence of SESN2 in myotubes treated with gAd. We conclude that SESN2 increases AMPK phosphorylation at Thr172 by promoting the binding between LKB1 and AMPK in C2C12 myotubes treated with gAd.
Taken together, these results suggest that long-term injection of gAd improves insulin resistance in HFD-fed mice, similar to aerobic exercise. On the one hand, gAd activates the insulin signaling pathway to increase glucose uptake in skeletal muscle. On the other hand, gAd activates AMPK by promoting the binding of LKB1 and AMPK, which in turn regulates energy metabolism. In contrast, SESN2 ablation affected the insulin-sensitive effects of gAd by blocking the binding of LKB1 and AMPK. Most importantly, our study provides a novel insight into the mechanism by which gAd reverses skeletal muscle insulin resistance in mice.
Submission statement
All authors have read and agree with manuscript content. In this section is a statement regarding that while this manuscript is being reviewed for this journal, the manuscript will not be submitted elsewhere for review and publication.
Ethical approval statement
Animals were housed at 22 °C–24 °C under 12 h dark and 12 h light cycle. Food and water were provided ad libitum during experimental period. All procedures were performed in accordance with the guidelines established by Animal Care and Committee Guidelines of Tianjin Medical University Animal Care and Use Committee (approval number: SYXK-2019-0004).
Authors' contribution
XML performed the experiments, analyzed and interpreted the data, and drafted the manuscript. YY, HS, and SJL performed part of the experiments and analyzed the data; YMN analyzed data; LF critically revised the manuscript and was responsible for important intellectual content. All authors approved the final version for publication.
Conflict of interest
The authors declare that there is no duality of interest associated with this manuscript.
Acknowledge statement
This study was supported by grants from the Natural Science Foundation of China (NSFC) 31871206, 32171135(to LF), and 31671237(to YMN). The authors are grateful to Chunxia Yu, Lu Wang, Xiao Han and Yating Huang for their technical assistance.
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.smhs.2022.08.001.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Li Y, Teng D, Shi X, et al. Prevalence of diabetes recorded in mainland China using 2018 diagnostic criteria from the American Diabetes Association: national cross sectional study. BMJ. 2020;369:m997. doi: 10.1136/bmj.m997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang T, Lu J, Shi L, et al. Association of insulin resistance and β-cell dysfunction with incident diabetes among adults in China: a nationwide, population-based, prospective cohort study. Lancet Diabetes Endocrinol. 2020;8(2):115–124. doi: 10.1016/s2213-8587(19)30425-5. [DOI] [PubMed] [Google Scholar]
- 3.Iwabu M, Yamauchi T, Okada-Iwabu M, et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature. 2010;464(7293):1313–1319. doi: 10.1038/nature08991. [DOI] [PubMed] [Google Scholar]
- 4.Christiansen T, Paulsen SK, Bruun JM, Ploug T, Pedersen SB, Richelsen B. Diet-induced weight loss and exercise alone and in combination enhance the expression of adiponectin receptors in adipose tissue and skeletal muscle, but only diet-induced weight loss enhanced circulating adiponectin. J Clin Endocrinol Metab. 2010;95(2):911–919. doi: 10.1210/jc.2008-2505. [DOI] [PubMed] [Google Scholar]
- 5.Taylor EB. The complex role of adipokines in obesity, inflammation, and autoimmunity. Clin Sci (Lond) 2021;135(6):731–752. doi: 10.1042/CS20200895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fruebis J, Tsao TS, Javorschi S, et al. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci U S A. 2001;98(4):2005–2010. doi: 10.1073/pnas.98.4.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomas E, Tsao T, Saha A, et al. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc Natl Acad Sci U S A. 2002;99(25):16309–16313. doi: 10.1073/pnas.222657499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8(11):1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 9.Miller RA, Chu Q, Le Lay J, et al. Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. J Clin Invest. 2011;121(6):2518–2528. doi: 10.1172/JCI45942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamauchi T, Kamon J, Waki H, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7(8):941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 11.Mcgee SL, Hargreaves M. Exercise adaptations: molecular mechanisms and potential targets for therapeutic benefit. Nat Rev Endocrinol. 2020;16(9):495–505. doi: 10.1038/s41574-020-0377-1. [DOI] [PubMed] [Google Scholar]
- 12.Wang T, Niu Y, Liu S, Yuan H, Liu X, Fu L. Exercise improves glucose uptake in murine myotubes through the AMPKalpha2-mediated induction of Sestrins. Biochim Biophys Acta, Mol Basis Dis. 2018;1864(10):3368–3377. doi: 10.1016/j.bbadis.2018.07.023. [DOI] [PubMed] [Google Scholar]
- 13.Roberts CK, Hevener AL, Barnard RJ. Metabolic syndrome and insulin resistance: underlying causes and modification by exercise training. Compr Physiol. 2013;3(1):1–58. doi: 10.1002/cphy.c110062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Meo S, Iossa S, Venditti P. Improvement of obesity-linked skeletal muscle insulin resistance by strength and endurance training. J Endocrinol. 2017;234(3):R159–R181. doi: 10.1530/joe-17-0186. [DOI] [PubMed] [Google Scholar]
- 15.Krause MP, Milne KJ, Hawke TJ. Adiponectin-consideration for its role in skeletal muscle health. Int J Mol Sci. 2019;20(7):1528. doi: 10.3390/ijms20071528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoue A, Cheng XW, Huang Z, et al. Exercise restores muscle stem cell mobilization, regenerative capacity and muscle metabolic alterations via adiponectin/AdipoR1 activation in SAMP10 mice. J Cachexia Sarcopenia Muscle. 2017;8(3):370–385. doi: 10.1002/jcsm.12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farias JM, Maggi RM, Tromm CB, et al. Exercise training performed simultaneously to a high-fat diet reduces the degree of insulin resistance and improves adipoR1-2/APPL1 protein levels in mice. Lipids Health Dis. 2012;11:134. doi: 10.1186/1476-511x-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ro SH, Fay J, Cyuzuzo CI, et al. SESTRINs: emerging dynamic stress-sensors in metabolic and environmental health. Front Cell Dev Biol. 2020;8 doi: 10.3389/fcell.2020.603421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JH, Budanov AV, Talukdar S, et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metabol. 2012;16(3):311–321. doi: 10.1016/j.cmet.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Niu Y, Yuan H, Huang J, Fu L. AMPK binds to Sestrins and mediates the effect of exercise to increase insulin-sensitivity through autophagy. Metabolism. 2015;64(6):658–665. doi: 10.1016/j.metabol.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 21.Quan N, Wang L, Chen X, et al. Sestrin2 prevents age-related intolerance to post myocardial infarction via AMPK/PGC-1α pathway. J Mol Cell Cardiol. 2018;115:170–178. doi: 10.1016/j.yjmcc.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shan T, Xu Z, Liu J, Wu W, Wang Y. Lkb1 regulation of skeletal muscle development, metabolism and muscle progenitor cell homeostasis. J Cell Physiol. 2017;232(10):2653–2656. doi: 10.1002/jcp.25786. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Liu S, Yuan H, Niu Y, Fu L. Sestrin 2 induces autophagy and attenuates insulin resistance by regulating AMPK signaling in C2C12 myotubes. Exp Cell Res. 2017;354(1):18–24. doi: 10.1016/j.yexcr.2017.03.023. [DOI] [PubMed] [Google Scholar]
- 24.Quan N, Sun W, Wang L, et al. Sestrin2 prevents age-related intolerance to ischemia and reperfusion injury by modulating substrate metabolism. Faseb J. 2017;31(9):4153–4167. doi: 10.1096/fj.201700063R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X, Zhang D, Vatner DF, et al. Mechanisms by which adiponectin reverses high fat diet-induced insulin resistance in mice. Proc Natl Acad Sci U S A. 2020;117(51):32584–32593. doi: 10.1073/pnas.1922169117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vu V, Bui P, Eguchi M, Xu A, Sweeney G. Globular adiponectin induces LKB1/AMPK-dependent glucose uptake via actin cytoskeleton remodeling. J Mol Endocrinol. 2013;51(1):155–165. doi: 10.1530/JME-13-0059. [DOI] [PubMed] [Google Scholar]
- 27.Deepa SS, Zhou L, Ryu J, et al. APPL1 mediates adiponectin-induced LKB1 cytosolic localization through the PP2A-pkcζ signaling pathway. Mol Endocrinol. 2011;25(10):1773–1785. doi: 10.1210/me.2011-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pham DV, Raut PK, Pandit M, et al. Globular adiponectin inhibits breast cancer cell growth through modulation of inflammasome activation: critical role of Sestrin2 and AMPK signaling. Cancers. 2020;12(3):613. doi: 10.3390/cancers12030613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang X, Zhang Y, Hu W, et al. Different effects of leucine supplementation and/or exercise on systemic insulin sensitivity in mice. Front Endocrinol. 2021;12:651303. doi: 10.3389/fendo.2021.651303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, Liu Y, Liu B, et al. Mechanisms of aerobic exercise upregulating the expression of hippocampal synaptic plasticity-associated proteins in diabetic rats. Neural Plast. 2019;2019:7920540. doi: 10.1155/2019/7920540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang SP, Chen YH, Chang WC, Liu IM, Cheng JT. Increase of adiponectin receptor gene expression by physical exercise in soleus muscle of obese Zucker rats. Eur J Appl Physiol. 2006;97(2):189–195. doi: 10.1007/s00421-006-0163-3. [DOI] [PubMed] [Google Scholar]
- 32.Martinez-Huenchullan SF, Tam CS, Ban LA, Ehrenfeld-Slater P, Mclennan SV, Twigg SM. Skeletal muscle adiponectin induction in obesity and exercise. Metabolism. 2020;102:154008. doi: 10.1016/j.metabol.2019.154008. [DOI] [PubMed] [Google Scholar]
- 33.Deshmukh AS, Murgia M, Nagaraj N, Treebak JT, Cox J, Mann M. Deep proteomics of mouse skeletal muscle enables quantitation of protein isoforms, metabolic pathways, and transcription factors. Mol Cell Proteomics. 2015;14(4):841–853. doi: 10.1074/mcp.M114.044222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma M, Dey CS. AKT ISOFORMS-AS160-GLUT4: the defining axis of insulin resistance. Rev Endocr Metab Disord. 2021;22(4):973–986. doi: 10.1007/s11154-021-09652-2. [DOI] [PubMed] [Google Scholar]
- 35.Paffenbarger RS, Jr., Hyde RT, Wing AL, Lee IM, Jung DL, Kampert JB. The association of changes in physical-activity level and other lifestyle characteristics with mortality among men. N Engl J Med. 1993;328(8):538–545. doi: 10.1056/nejm199302253280804. [DOI] [PubMed] [Google Scholar]
- 36.Hawley JA, Hargreaves M, Joyner MJ, Zierath JR. Integrative biology of exercise. Cell. 2014;159(4):738–749. doi: 10.1016/j.cell.2014.10.029. [DOI] [PubMed] [Google Scholar]
- 37.Mardare C, Krüger K, Liebisch G, et al. Endurance and resistance training affect high fat diet-induced increase of ceramides, inflammasome expression, and systemic inflammation in mice. J Diabetes Res. 2016;2016:4536470. doi: 10.1155/2016/4536470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Genders AJ, Kuang J, Marin EC, et al. Exercise does not improve insulin resistance and mitochondrial characteristics together. J Endocrinol. 2021;252(2):91–105. doi: 10.1530/joe-21-0242. [DOI] [PubMed] [Google Scholar]
- 39.Kiefer LS, Fabian J, Rospleszcz S, et al. Distribution patterns of intramyocellular and extramyocellular fat by magnetic resonance imaging in subjects with diabetes, prediabetes and normoglycaemic controls. Diabetes Obes Metabol. 2021;23(8):1868–1878. doi: 10.1111/dom.14413. [DOI] [PubMed] [Google Scholar]
- 40.Klein S, Gastaldelli A, Yki-Järvinen H, Scherer PE. Why does obesity cause diabetes? Cell Metabol. 2022;34(1):11–20. doi: 10.1016/j.cmet.2021.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bouzakri K, Zachrisson A, Al-Khalili L, et al. siRNA-based gene silencing reveals specialized roles of IRS-1/Akt2 and IRS-2/Akt1 in glucose and lipid metabolism in human skeletal muscle. Cell Metabol. 2006;4(1):89–96. doi: 10.1016/j.cmet.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 42.Richter EA, Hargreaves M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol Rev. 2013;93(3):993–1017. doi: 10.1152/physrev.00038.2012. [DOI] [PubMed] [Google Scholar]
- 43.Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA, Kiens B. Isoform-specific and exercise intensity-dependent activation of 5’-AMP-activated protein kinase in human skeletal muscle. J Physiol. 2000;528 Pt 1(Pt 1):221–226. doi: 10.1111/j.1469-7793.2000.t01-1-00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang X, Liu G, Guo J, Su Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci. 2018;14(11):1483–1496. doi: 10.7150/ijbs.27173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chung I, Kim SA, Kim S, et al. Biglycan reduces body weight by regulating food intake in mice and improves glucose metabolism through AMPK/AKT dual pathways in skeletal muscle. Faseb J. 2021;35(8):e21794. doi: 10.1096/fj.202002039RR. [DOI] [PubMed] [Google Scholar]
- 46.Morrison A, Chen L, Wang J, et al. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. Faseb J. 2015;29(2):408–417. doi: 10.1096/fj.14-258814. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.