

Summary
Hyperpolarizing GABAAR currents, the unitary events that underlie synaptic inhibition, are dependent upon efficient Cl− extrusion, a process that is facilitated by the neuronal specific K+/Cl− co-transporter KCC2. Its activity is also a determinant of the anticonvulsant efficacy of the canonical GABAAR-positive allosteric: benzodiazepines (BDZs). Compromised KCC2 activity is implicated in the pathophysiology of status epilepticus (SE), a medical emergency that rapidly becomes refractory to BDZ (BDZ-RSE). Here, we have identified small molecules that directly bind to and activate KCC2, which leads to reduced neuronal Cl− accumulation and excitability. KCC2 activation does not induce any overt effects on behavior but prevents the development of and terminates ongoing BDZ-RSE. In addition, KCC2 activation reduces neuronal cell death following BDZ-RSE. Collectively, these findings demonstrate that KCC2 activation is a promising strategy to terminate BDZ-resistant seizures and limit the associated neuronal injury.
Keywords: epilepsy, benzodiazepine, refractory status epilepticus, KCC2, neuronal Cl− extrusion, synaptic inhibition, neuronal death, neuroprotection, EEG
Graphical abstract
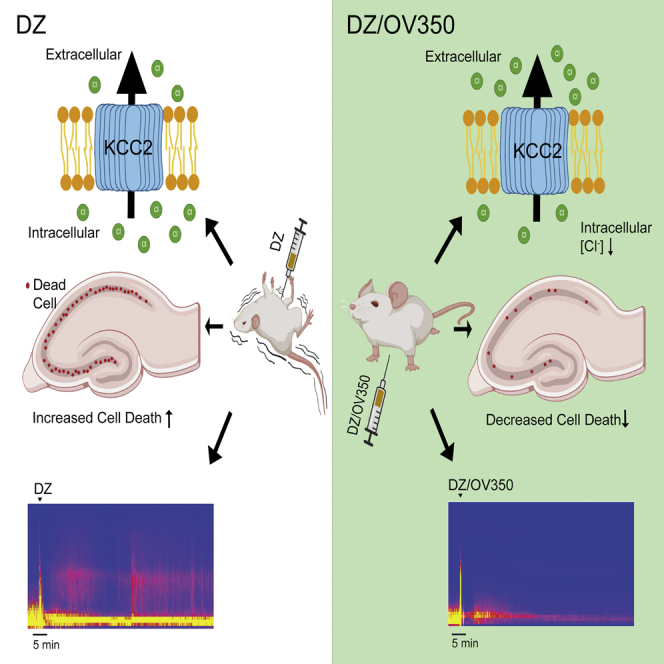
Highlights
-
•
Identification of small molecules that bind to and activate KCC2
-
•
Activation of KCC2 reduces neuronal Cl− accumulation
-
•
KCC2 activation restores the efficacy of benzodiazepine to arrest seizures
-
•
KCC2 activation reduces neuronal death following seizures
Impairments in KCC2-dependent neuronal Cl− extrusion are believed to contribute to the gross increases in excitability that lead to benzodiazepine-resistant seizures. Jarvis et al. demonstrate that pharmacological activation of KCC2 reduces neuronal excitability, restores the anticonvulsant efficacy of benzodiazepines, and reduces seizure-induced neuronal cell death.
Introduction
Synaptic inhibition in the adult brain is principally mediated by γ-aminobutyric acid receptors (GABAARs), receptors that act to limit neuronal excitability via Cl−-dependent neuronal hyperpolarization.1 These receptors are also the sites of action for barbiturates, benzodiazepines (BDZs) and neurosteroids, all of which exert their therapeutic efficacy as GABAAR positive allosteric modulators (PAMs). In contrast, GABAAR activation leads to depolarization of the neuronal membrane potential in embryonic and post-natal neurons. This shift from depolarizing to hyperpolarizing GABAAR currents reflects elevations in Cl− extrusion by mature neurons, a phenomenon due in part to increased activity of the electroneutral neuron-specific K+/Cl− co-transporter KCC2, or SLC12A5.2,3,4,5 Consistent with its role in supporting fast synaptic inhibition, mutations in KCC2 identified in humans result in severe epilepsy, developmental delay, and premature death.6,7,8,9 Likewise, deficits in KCC2 expression levels seen in epileptic foci resected from patients with treatment resistant epilepsy correlate with depolarizing GABAAR currents.8,10
Studies in mice have further illustrated the essential role that KCC2 plays in brain function as global knockout of the SLC12A5 gene leads to death shortly after birth,11 while global knockout of the KCC2b isoform is lethal before post-natal day 21 (P21). Conditional inactivation of KCC2 in the hippocampus leads to neuronal Cl− accumulation and depolarizing GABAAR currents. These deficits in inhibition parallel the development of spontaneous seizures, neuronal apoptosis, and reactive astrocytosis.12 Studies in rodents have further suggested that deficits in KCC2 activity contribute to onset of status epilepticus (SE) and the subsequent development of BDZ resistance (BDZ-RSE).13,14,15,16,17,18 Consistent with this, indirectly increasing KCC2 by decreasing its phosphorylation slows the onset of SE and prevents the subsequent development of BDZ-RSE.13,17 In addition, globally increasing KCC2 expression levels has been shown to enhance the efficacy of BDZ to alleviate SE.19 Despite the attraction of KCC2 as a therapeutic target, molecules that directly bind to and activate this transporter have not been described.
Here, we describe a family of small molecules that bind to and activate KCC2. We demonstrate that KCC2 activation reduces neuronal Cl− accumulation and excitability. KCC2 activation prevents the development of BDZ-RSE and limits the subsequent neuronal injury. Together, our results suggest that potentiating KCC2 may be an efficacious strategy to arrest BDZ-resistant seizures and to limit the associated neuronal injury.
Results
Identification of small molecules that activate and directly bind to KCC2
To identify chemical entities capable of directly activating KCC2, we used a multi-tiered drug screening cascade (Figure 1A). We first measured the effect on potassium flux of 1.3 million compounds from the AstraZeneca screening collection, tested at 30 μM using an established thallium TI influx assay in HEK-293 cells overexpressing KCC2.17,20,21 Compounds that exhibited >30% potentiation of KCC2 activity with EC50 of <30 μM were selected for further characterization. To control for compounds that may non-specifically modulate KCC2 activity, we tested active compounds in analogous Tl influx assays in HEK-293 cells overexpressing KCC3, KCC4, or NKCC1 as well as untransfected HEK-293 cells. Only those compounds selectively modulating potassium flux in KCC2-transfected cells were progressed. To eliminate compounds that may indirectly modulate KCC2 activity by modifying its phosphorylation, we examined the effect of compounds on a panel of 348 protein kinases (SelectScreen Kinase Profiling; Thermo Fisher Scientific; Table S1) that included multiple PKC isoforms, SPAK(STK39), and WNK kinases, all of which phosphorylate KCC2 on the key regulatory residues S940 and T1007 to modulate its activity and membrane trafficking.13,18,22,23 By eliminating compounds that exhibited significant activity (IC50 < 10 μM) against any of these kinases, we identified a series of fused aminopyrimidine compounds typified by compound 1 (Cmp1; Figure S1). Cmp1 was able to increase KCC2 activity with an EC50 of 2.01 μM, a concentration that did not modify the activity of KCC3, KCC4, or NKCC1, other solute carriers that contribute to neuronal Cl− homeostasis (Figure 1B).
Figure 1.

Identification and optimization of small-molecule potentiators of KCC2
(A) Drug screening workflow illustrates the steps taken to identify KCC2 direct activators, a multi-tiered program starting from a compound library of 1.3 million compounds.
(B) The EC50 of Cmp1 for KCC2, KCC3, KCC4, and NKCC1 was measured using a Tl influx FLIPR assay in expressing HEK-293 cells.
(C) HEK-293 cells expressing KCC2 were exposed to vehicle (V) or 30 μM Cmp1 for 90 min and then heated to 37°C–58°C. Following centrifugation, soluble fractions were immunoblotted with KCC2 and GAPDH antibodies. The migration of molecular mass standards is indicated by the arrows on the left of the gels.
(D) The level of remaining soluble native KCC2 at each temperature was normalized to that seen at 37°C (Ix/I37°C), and these data were then used to construct a thermal melt curve for KCC2 under control conditions and in the presence of Cmp1 (n = 4 transfections).
(E) An isothermal dose-response curve was constructed for destabilization of KCC2 by Cmp1 (n = 4 transfections).
We went on to determine whether Cmp1 interacts directly with KCC2 using the cellular thermal shift assay (CETSA), a label-free method to assess target engagement under physiological conditions. The assay relies on the principle that ligand binding results in a change in thermal stability (either stabilization or destabilization) of the bound protein. CETSA measures the amount of soluble (or non-denatured) protein remaining following heating at various temperatures in the absence or presence of a ligand.24,25
To examine the effects of Cmp1 on the thermal stability of KCC2, the transporter was transiently expressed in HEK-293 cells.20,21,26 48 h after transfection, live expressing cells were exposed to Cmp1 or vehicle and heated from 37°C to 58°C. Cells were immediately lysed in 0.2% NP40 and then centrifuged at 12,000 × g to remove aggregated and denatured proteins along with cell debris. The levels of soluble KCC2 at each temperature were measured via immunoblotting with KCC2 antibodies. We first examined the molecular mass of KCC2 in control cell lysates produced under these conditions, and the majority of KCC2 immunoreactivity migrated as major bands of approximately 130 kDa species on SDS-PAGE, the predicted molecular mass of monomeric KCC2. However, in some lysates, higher-molecular-mass species greater than 250 kDa were observed (Figure S2).23,26,27,28 These larger species are SDS-resistant denatured aggregates that are commonly seen upon solubilization of hydrophobic integral membrane proteins including KCC2.29 Since CETSA measure the temperature at which proteins denature, these higher-molecule-mass species were excluded from our analysis.
To quantify the effects of Cmp1 on KCC2 thermal stability, the level of the remaining 130 kDa species seen at increasing temperatures was normalized to that at 37°C (100%). Using these criteria, Cmp1 induced a leftward shift in the melting temperature of KCC2 compared with vehicle. The respective IC50 values were 50.9°C ± 0.7°C and 48.3°C ± 0.3°C for vehicle and Cmp1, respectively. This thermal destabilization is consistent with direct binding of Cmp1 to KCC2 (Figures 1C and 1D). The interaction was further characterized by generating an isothermal dose response fingerprint at 50°C, revealing Cmp1 destabilized KCC2 stability with an IC50 value of 1.1 ± 0.7 μM (Figure 1E).
Cmp1 was used as the starting point for a program of medicinal chemistry optimization. Using FLIPR assays together with a suite of in vitro drug absorption, distribution, metabolism, and excretion assays (ADME) and predictive tools, a series of compounds were created with the aim of optimizing for potency, lipophilicity, solubility, metabolic stability, and brain penetration. One of these derivatives, OV350 (350; Figure S1), exhibited an EC50 for KCC2 of 261.4 ± 22.2 nM and a maximum potentiation of 90% at a saturating concentration of 3 μM using 1% β-cyclodextrin (BCD) as a vehicle during TI uptake (Figure 2A). To gain mechanistic insights into our compounds, we evaluated the effects of 350 (10 μM) on the initial rates of TI uptake by HEK-293 cells expressing KCC2. The initial rate of TI uptake relative to vehicle-treated controls was increased by 301.2% ± 25.2% by 350 (p = 0.0023; Figures 2B and 2C) in cells expressing KCC2. Given that these measurements are made at saturating substrate concentrations,17,20,21 this result suggests that 350 acts to increase KCC2 maximal velocity. Thus, we have identified small molecules that directly bind to KCC2 and increase its maximal velocity.
Figure 2.

Small-molecule activators increase KCC2 activity without modifying its plasma membrane accumulation or phosphorylation
(A) The effects of increasing amounts of 350 formulated in 1% BCD on KCC2 activity expressed in HEK-293 cells using TI influx (n = 3 transfections).
(B) HEK-293 cells expressing KCC2 or untransfected (UT) cells were incubated with V or 10 μM 350 for 30 min. Representative traces of TI influx for each experimental group measured over 150 s are shown.
(C) After subtraction for values seen in UT cells, the initial rate of TI uptake was determined, and data were normalized to rates seen in KCC2-expressing cells exposed to V (100%) (n = 5 transfections). The green arrow indicates the addition of stimulus buffer.
(D) HEK-293 cells expressing GlyRα1 and KCC2 neurons were subjected to gramicidin perforated patch-clamp recordings in the presence of bumetanide (10 μM). After attaining perforation, cultures were exposed to 350 or V (1% BCD) for 15 min. EGly was then determined using voltage ramps to determine the polarity of 50 μM glycine-induced currents, and representative current-voltage (I-V) plots are shown for HEK-293 cells at 0 (blue) and 15 (red) min treatment for V and 350.
(E) Individual shifts in EGly are show for cells incubated with 350 (0.3 and 3 μM) or V for 15min.
(F) Mean shifts in EGly were determined for cells treated with V, 0.3 μM 350, and 3 μM 350 (n = 5 transfections).
(G) 350-induced changes in [Cl−] were calculated from EGABA using the Nernst equation for V, 0.3 μM 350, and 3 μM 350 (n = 5 transfections).
(H) HEK-293 cells (UT) or those expressing KCC2 were exposed to V, 0.3 μM, or 3 μM 350 for 15 min and biotinylated with NHS-biotin. Surface and total extracts were subsequently immunoblotted with KCC2 and actin antibodies. The ratio of surface/total KCC2 immunoreactivity was determined and normalized to V (100%) (n = 3 transfections).
(I) HEK-293 cells were treated with V (−) or 3 μM 350 (+) for 15 min. Cell lysates were immunoblotted with KCC2, pS940, pT1007, and actin antibodies. The ratios of pS940/KCC2 and pT1007/KCC2 were then compared with V (100%) (n = 3 transfections).
In all panels, p values were determined using t tests, ∗p < 0.05.
KCC2 activators do not modify transporter accumulation on the plasma membrane accumulation or its phosphorylation
To confirm the validity of our measurements using TI flux, we expressed KCC2 in HEK-293 cells with the ɑ1 subunit of the glycine receptor (GlyRɑ1), a condition that forms homomeric glycine-activated ion channels when expressed in this system. We then used the gramicidin perforated patch-clamp technique to measure the reversal potential for GlyRɑ1-mediated currents (EGly) in cells exposed to 350. The use of this technique, while limited in throughput, facilitates high-resolution single-cell recordings without disturbing endogenous intracellular Cl− levels.20,21
To calculate EGly values, cells were exposed to 50 μM Gly, and the polarity of Gly-induced currents was determined at different holding potentials (Figures 2D and 2E). 15 min incubation with either 0.3 or 3 μM 350 induced shifts of −15.6 ± 4.5 (∗p = 0.022) and −32.2 ± 9.5 (∗p = 0.001) mV in EGly, respectively, while vehicle was without effect (−0.8 ± 0.5 mV; p = 0.512) (Figure 2F). As determined using the Nernst equation, 0.3 and 3 μM 350 reduced intracellular Cl− levels by 12.5 ± 4.5 (p = 0.0013) and 15.2 ± 6.2 (p = 0.0215) mM, respectively, while vehicle was without effect (1.8 ± 0.9 mM; p = 0.234) (Figure 2G).
The effects of 350 on the plasma membrane stability of KCC2 expression was assessed using biotinylation, as described previously.20,21 Compared with vehicle, neither 0.3 or 3 μM 350 modified the level of KCC2 on the plasma membrane (Figure 2H; 94.5% ± 8.4% [p = 0.745] and 104.7% ± 8.4% of control [p = 0.736], respectively). The reliability of our biotinylation procedure was assessed via immunoblotting with antibodies against the cytosolic protein actin. Actin was present in the total lysates but was absent from the surface fractions (Figure 2H).
The activity of KCC2 is subject to dynamic modulation via phosphorylation of critical intracellular residues within its C-terminal cytoplasmic domain. Central to this process are serine 940 (S940) and threonine 1007 (T1007), which activate and inhibit KCC2 activity, respectively.30 We thus examined the effects of 350 on the phosphorylation of these residues using immunoblotting with characterized phospho-specific antibodies pS940 and pT1007, respectively. 350 (3 μM) did not significantly modify phosphorylation of S940 (95.3% ± 10.2% of control; p = 0.764) or T1007 (108% ± 10.2% of control; p = 0.649) (Figure 2I).
Our studies suggest that 350 potentiates KCC2 activity without modifying either its plasma membrane accumulation or the phosphorylation of the key regulatory residues S940 or T1007.
KCC2 activation in neurons reduces neuronal Cl− levels
To test if 350 modifies neuronal Cl− levels, we used gramicidin-perforated patch-clamp recording to measure the reversal potentials of GABAAR-mediated currents (EGABA) in 18–20 days in vitro (DIV) hippocampal neurons.13,28 At this developmental stage, GABAAR activation leads to hyperpolarization, a phenomenon critically dependent upon KCC2.3 Our measurements were performed in the presence of bumetanide (10 μM) and tetrodotoxin (TTX; 300 nM) to limit the contributions of NKCC1 and activity-dependent shifts in Cl− levels, respectively.28 Cultures were exposed to 350 or vehicle for 15 min, and voltage ramps were used to determine changes in the reversal potential of GABAAR currents over time (Figure 3A). These data were then used to determine shifts in [Cl−] levels. At 15 min, 300 nM 350 (∼EC50) significantly reduced EGABA from −75 ± 3.1 to −85.1 ± 4.2 mV (Figure 3B; ∗p = 0.0104), a net −9.1 ± 2.2 mV negative shift. This shift reflected a significant decrease of intracellular Cl− from 7.7 ± 0.4 to 5.1 ± 0.4 mM following incubation with 350 (Figure 3C; ∗p = 0.0153). In contrast, vehicle did not significantly modify EGABA (Figure 3B; 0 = −77.2 ± 3.3 and 15 = −78.0 ± 4.3 mV, respectively; p = 0.580) or chloride levels (Figure 3C; 0 = 7.6 ± 0.5 and 15 = 7.4 ± 0.8 mM; p = 0.1646).
Figure 3.

KCC2 activation reduces neuronal Cl− accumulation and slows the development of seizure-like events in brain slices
(A) 18–21 DIV hippocampal neurons were subjected to gramicidin perforated patch-clamp recordings in the presence of bumetanide (10 μM) and TTX (300 nM). After perforation, cultures were exposed to 300 nM 350 or V (1% BCD) for 15 min. EGABA was then determined using voltage ramps to determine the polarity of muscimol-induced currents, and representative I-V plots are shown for neurons at 0 (blue) and 15 (red) min treatment for V and 350.
(B) EGABA values were measured at 0 and 15 min following treatment with V or 300 nM 350 (n = 5 cultures).
(C) [Cl−] values were calculated from EGABA values for V- or 350-treated neurons (n = 5 cultures).
(D) 18–21 DIV hippocampal neurons were incubated with 300 nM 350 or V for 1 h. Neurons were subjected to whole-cell recording using an electrolyte containing 30 mM Cl−. 5 min later, basal EGABA, and [Cl−] were determined and compared between treatments (n = 5 cultures).
(E) Neurons treated as above were exposed to 11K (10 μM), and the shifts in EGABA (11KΔEGABA) and in [Cl−] (11KΔCl−) were compared (n = 5 cultures).
(F) Freshly prepared 350 μm brain slices from C57Bl/6 mice were incubated in ACSF supplemented with V or 350 (1 μM) for 1 h at 32°C and then with ACSF deficient in Mg+2 (0-Mg) (black arrowhead). Field recordings were then performed within the entorhinal cortex as a means of monitoring neuronal excitability. The first SLE and entrance into LRD development are indicated.
(G) The time to the first SLE and to LRDs were compared between treatments (n = 7 mice).
(H) The interictal interval was compared between the first and second SLE using slices treated with V or 350 (∗p < 0.05; n = 7 mice).
In all panels, p values were determined using t tests, ∗p < 0.05.
To measure the effects of the compound on KCC2 activity under more dynamic conditions, we incubated cultures in 350 for 1 h and subjected them to whole-cell patch-clamp recording to artificially impose a 32 mM Cl− load on neurons via the patch pipette.13,17 5 min after break-in, an initial EGABA value was determined, and the effects of a subsequent 5 min incubation with the selective KCC2 inhibitor VU0463271 (11K; 10 μM) were examined. 350-treated cells exhibited lower basal EGABA values compared with controls (Figure 3D; −59.3 ± 2.1 and −47.4 ± 2.3 mV, respectively; ∗p = 0.025), reflecting decreased neuronal Cl− levels (Figure 3D; 21.2 ± 3.5 and 14.9 ± 4.5 mM Cl−, respectively; ∗p = 0.040). The magnitude of the 11K-induced shift in EGABA (Δ11KEGABA) was significantly larger for neurons exposed to 350 compared with vehicle (Figure 3E; 18.5 ± 4.6 and 7.9 ± 4.2 mV, respectively; ∗p = 0.009), Moreover, the 11K-induced increases in Cl− (Δ11K [Cl−]) were also higher for neurons treated with 350 compared with vehicle (Figure 3E; 16.2 ± 2.3 and 9.5 ± 3.7 mM, respectively; ∗p = 0.021). Collectively, these results demonstrate that 350 increases KCC2 activity in neurons and reduces Cl− accumulation.
KCC2 activation limits neuronal hyperexcitability
We went on to assess the effects of 350 on the development of “seizure-like events” (SLEs) in brain slices exposed to artificial cerebrospinal fluid (ACSF) deficient in Mg+2 (0-Mg), a method used widely to increase neuronal excitability.31 To do so, acute slices were incubated at 32°C with 1 μM 350 or vehicle for 1 h and then continually perfused with 0-Mg. Field recordings within the entorhinal cortex were then employed to monitor the development of SLEs and their transition to late recurrent discharges (LRDs), which are believed to be analogous to the development of SE.13,32 350 did not modify the appearance of the first SLE relative to control (Figures 3F and 3G; 350 = 11.3 ± 3.3 min and vehicle = 10.5 ± 2.6 min, respectively; p = 0.256). However, 350 did slow the development of LRDs (Figures 3F and 3G; 350 = 28.4 ± 6.4 min, vehicle = 12.4 ± 4.1 min; ∗p = 0.010). We also examined the effects on the interictal interval by comparing the duration between the first and second SLE. 350 significantly increased the interictal interval (Figure 3H; 350 = 4.2 ± 0.5 min, vehicle = 2.6 ± 0.3 min; ∗p = 0.005), and activity returned to baseline following termination of the initial SLE. In contrast, no significant differences in SLE duration were evident in vehicle-treated slices (350 = 49 ± 15 s, min, vehicle = 34 ± 9 s, p = 0.371). 350 evidently slows the development of LRDs and increases the interictal interval, suggesting that KCC2 activation limits neuronal hyperexcitability.
KCC2 activation protects against pentylenetetrazol-induced motor convulsions
We examined distribution of drug in the brain following subcutaneous (s.c.) injection (50 mg/kg). 350 was detected at 30 min and reached a maximal concentration of 675.75 ± 80.5 nM at 4 h, a level that was maintained at 8 h (Figure 4A). Given the ability of 350 to partition into the brain, we examined whether it induces any gross effects on animal behavior. To do so, mice were injected with 350 (50 mg/kg) or vehicle s.c., and the animals’ behavior was compared in the open field.33,34 350 did not modify the total distance traveled during the trial (Figure 4B; 350 = 61.3 ± 2.3 and vehicle = 64.5 ± 2.1 m, respectively; p = 0.253). Likewise, the time spent in the center zone was comparable (Figure 4B; 350 = 18.4% ± 2.1%, vehicle = 16.6% ± 2.5%; p = 0.658).
Figure 4.

KCC2 activation does not induce gross modifications in mouse behavior but does protect against PTZ-induced seizures
(A) Mice were injected s.c. with 50 mg/kg 350 in 5% BCD. Drug accumulation was then measured over a time course of 8 h via liquid chromatography tandem mass spectrometry (LC-MS/MS) (n = 3–4 mice).
(B) Mice were dosed with 350 50 mg/kg s.c., and 2–3 h later, they were placed in the center of a 60 × 60 cm open field and allowed to explore for 40 min. The total distance traveled and time in the center of the area were then quantified and compared (n = 9 mice).
(C) Mice were injected with 25 mg/kg 350 (i.v.) or V. 1 h later, mice were subjected to continuous video monitoring and injected with increasing amounts of PTZ to a maximum of 200 mg/kg. The doses of PTZ to induce hindlimb clonus were compared (n = 14 mice).
In all panels, p values were determined using t tests, ∗p < 0.05.
To test whether 350 exerts anticonvulsant efficacy, we determined its effects on motor seizures induced by the convulsant pentylenetetrazole (PTZ), a GABAAR antagonist, as measured using the modified Racine scale (stage 1–5 seizures).35,36 Mice were injected intravenously (i.v.) with 25 mg/kg 350 or vehicle and subjected to continuous video monitoring.35 1 h later, mice were infused with increasing amounts of PTZ, and the dose to induce hindlimb clonus (Racine scale = 4) was determined. The dose of PTZ required to induce hindlimb clonus was significantly increased by 350 (Figure 4C; control = 84.5 ± 5.1 mg/kg and 350 = 137.4 ± 7.5 mg/kg, respectively; ∗p = 0.001). 30 min following i.v. injection at 25 mg/kg, 350 was detected in plasma and brain at 7.6 ± 4.4 and 41.5 ± 11.5 μM, respectively (Figure S3).
Collectively, these results suggest that 350 freely distributes in the brain and that, while it does not lead to any stereotyped effects on behavior, it does protect against late-stage seizures induced by PTZ, a pharmacological screening tool widely used to identify anticonvulsants.
KCC2 activation prevents the development of BDZ refractory seizures
To further explore the anticonvulsant properties of 350, its effects on the development of kainic acid (KA)-induced SE in mice was examined using electroencephalographic (EEG) recording.18 This model was chosen because of the similarities with patients undergoing SE, as KA-induced seizures become refractory to BDZs within minutes.17,37,38,39,40
Mice implanted with EEG/electromyography (EMG) electrodes were first injected s.c. with 50 mg/kg 350 or vehicle and then 2 h later with KA (20 mg/kg intraperitoneally [i.p.]). Seizures were allowed to proceed for a further 2 h before administration of a saturating concentration of diazepam (DZ; 5 mg/kg i.p.), after which recordings were extended for a further 60 min (Figures 5A–5C). The effects of 350 on the development of SE were first examined, revealing increased latency to first seizure (Figure 5D; vehicle = 10.2 ± 2.5 min, 350 = 14.4 ± 2.9 min; ∗p = 0.0329) and to SE compared with vehicle (Figure 5D; vehicle = 38.3 ± 3.4 min, 350 = 54.5 ± 6.9 min; ∗p = 0.0230). In addition, the time spent in epileptiform activity was lower in mice pretreated with 350 (Figure 5D; vehicle = 85% ± 5.4%, 350 = 62.5% ± 8.4%; p = 0.0230). To provide quantitative insights into the effects of KA, recordings were subjected to fast Fourier transformation (FFT), used to transform the EEG signals from the time domain into the frequency domain to form a power spectral density plot for frequencies between 0 and 100 Hz 2 h after KA injection.17 In mice pretreated with 350, total EEG power was significantly reduced compared with vehicle (Figure 5E; 4.2 ± 2.4 × 10−6 and 6.9 ± 3.2 × 10−7 V2 for vehicle and 350, respectively; ∗p = 0.0024).
Figure 5.

Pretreatment of mice with KCC2 activators prevents the development of BDZ-RSE
(A) Line diagram outlining the experimental design.
(B) Exemplar EEG traces are shown for mice injected with V (s.c.) 2 h prior to dosing i.p. with 20 mg/kg KA. 2 h following KA injection, mice were dosed i.p. with 5 mg/kg DZ, and EEG recordings were extended for a further 1 h. The lower trace represents an enlargement of the green bar indicated in the upper trace.
(C) Exemplar EEG traces are shown for mice injected with 50 mg/kg 2 h prior to dosing i.p. with 20 mg/kg KA. 2 h following KA injection, mice were dosed i.p. with 5 mg/kg DZ, and EEG recordings were extended for a further 1 h. The lower trace represents an enlargement of the green bar indicated in the upper trace.
(D) The time to the first seizure, the onset of SE, and the percentage of total time in epileptiform activity were compared between treatment groups (n = 9 mice).
(E) EEG recordings were subjected to FFT, and spectral plots are shown for frequencies between 1 and 100 Hz for mice treated with V or 350 2 h after KA injection. Total EEG power was then compared between treatments (n = 9 mice).
(F) EEG recordings from mice pretreated with V 2 h following KA injection and 10 min after dosing with DZ were subjected to FFT, and spectral plots are shown for frequencies between 1 and 100 Hz. Total EEG power was then compared (n = 9 mice).
(G) EEG recordings from mice pretreated with 350 2 h following KA injection and 10 min after dosing with DZ were subjected to FFT, and spectral plots are shown for frequencies between 1 and 100 Hz. Total EEG power was then compared between treatments (n = 9 mice).
In all panels, p values were determined using t tests, ∗p < 0.05.
2 h after KA injection, mice were injected with DZ, and its ability to suppress EEG power was compared between treatment groups 10 min before and 10 min after dosing. Consistent with previously published studies using the KA model,1,17,37 DZ did not modify EEG power in mice pretreated with vehicle (Figure 5F; 4.2 ± 2.4 × 10−6 and 2.9 ± 2.7 × 10−6 V2 pre- and post-DZ, respectively; p = 0.105). In contrast, in mice pretreated with 350, DZ significantly reduced EEG power (Figure 5G; 6.9 ± 3.2 × 10−7 and 2.7 ± 3.2 × 10−7 V2, pre- and post-DZ, respectively; ∗p = 0.004).
To confirm that 350 acts centrally to mediate its effects on seizure activity, we directly injected 350 (20 μM; in 1% BCD) or vehicle into the hippocampus of mice implanted with EEG/EMG electrodes.17 30 min later, mice were injected with KA and a further 2 h later with DZ, as detailed above. Relative to vehicle, 350 increased the latency to the first seizure and to SE and reduced the percentage of time in epileptiform activity (Figure S4; ∗p = 0.039, 0.005, and 0.002, respectively). 2 h after KA injection, the latency to seizure reduction by DZ (∗p = 0.04), percentage of epileptiform activity (p = 0.018), and percentage of DZ insensitivity were significantly reduced in mice pretreated with 350 compared with vehicle-treated controls (Figure S4).
KCC2 activation arrests ongoing BDZ-RSE
To evaluate the potential of KCC2 activators to terminate ongoing SE, we again employed the KA model. Mice were injected with KA (20 mg/kg i.p.). 2 h later, mice were dosed with DZ alone or DZ/350 (i.p.; DZ = 5 mg/kg; 350 = 50 mg/kg), and recordings were extended for a further 60 min (Figures 6A–6C). Spectral data were then subjected to FFT, and the total EEG power between 1 and 100 Hz of the 2 h following KA treatment (pre-drug) and 30 min following their injection (post-drug) were analyzed (Figure 6D). First, we compared pre-drug EEG between treatment groups. These values were not significantly different (Figure 6E; DZ alone = 1.1 ± 0.2 × 10−6 V2 and DZ/350 = 9.2 ± 1 × 10−7 V2; p = 0.3732). In addition, the latency to the first seizure and to SE and the percentage of time in epileptiform activity were also comparable between treatment groups (Figure S5).
Figure 6.

KCC2 activation restores the efficacy of DZ to terminate ongoing BDZ-RSE
(A) Line diagram outlining the experimental design utilized.
(B) Exemplar EEG traces are shown for mice injected i.p. with 20 mg/kg KA, followed 2 h later by 5 mg/kg DZ. The lower trace represents an enlargement of the region in the upper trace indicated by the green bar.
(C) Exemplar EEG traces are shown for mice injected i.p. with 20 mg/kg KA, followed 2 h later by DZ and 350 (5 and 50 mg/kg, respectively). The lower trace represents an enlargement of the region in the upper trace indicated by the green bar.
(D) EEG recordings 2 h after KA injection and 30 min following DZ or DZ/350 treatment were subjected to FFT, and spectral plots are shown for frequencies between 1 and 100 Hz. EEG δ, θ, α, β, and low- and high- γ power frequencies are indicated.
(E) Total EEG power was compared for treatment groups 2 h following KA injection (DZ: n = 13, DZ/350, n = 15 mice).
(F) Total EEG power 30 min after drug treatment was normalized to values seen after KA injection (100%) (DZ: n = 13, DZ/350 n = 15 mice).
(G) The percentage of DZ-insensitive mice were compared between treatment groups (DZ: n = 13, DZ/350: n = 15 mice).
(H) EEG power frequencies 30 min after injection with DZ/350 were compared with those seen with DZ alone (100%) (DZ: n = 13, DZ/350: n = 15 mice).
In all panels, p values were determined using t tests, ∗p < 0.05.
To examine the effects of drugs on EEG power, total power was compared in 2 min epochs prior to and 30 min after injection. In mice treated with DZ alone, variable but non-significant effects of DZ on EEG power were evident (Figure 6E; 67.8% ± 9.4% of control; p = 0.214). In contrast, EEG power was reduced to 21.7% ± 1.5% of control in mice treated with DZ and 350 (Figure 6F; ∗p = 0.007). We also compared DZ insensitivity between treatment groups, which was defined as the number of animals that did not show a reduction of >50% in EEG power within 20 min of treatment. While 62% of mice exhibited DZ insensitivity, this was abolished by 350 (Figure 6F).
To assess possible effects on the EEG frequency bands, δ (1–4 Hz), θ (4–8 Hz), α (8–13 Hz), β (13–30 Hz), low γ (30–50 Hz), and high γ (50–100 Hz) were assessed by normalization to values seen in mice treated with DZ. 30 min after injection, θ (38.7% ± 7.4% of control; ∗p = 0.0158), α (41.8% ± 7.6% of control; ∗p = 0.0490), low-γ (52.6% ± 14.7% of control; ∗p = 0.0447) and high-γ (48.9% ± 13.4% of control; ∗p = 0.0315) power were significantly reduced, while δ (37.1% ± 7.3% of control; p = 0.097) and β (54.3% ± 11.5% of control; p = 0.142) power were not (Figure 6H).
Finally, we measured 350 accumulation in the brain and plasma for up to 30 h following a single i.p. dose of 50 mg/kg. 350 was detected at 4.2 ± 1.3 and 5.4 ± 1.7 μM in the brain and plasma, respectively, 30 min following injection, consistent with its efficacy to arrest BDZ-RSE. 350 reached maximum levels of 6.6 ± 1.3 and 4.7 ± 0.9 μM in the brain and plasma, respectively, at 1 h. 350 was detected in the brain at 372 ± 17.7 and 33 ± 8.0 nM 15 and 30 h after injection (Figure S6). Based on an EC50 of approximately 260 nM, 350 is present in the brain at levels enough to activate KCC2 for at least 15 h following i.p. dosing at 50 mg/kg.
Collectively, these results demonstrate that activation of KCC2 is sufficient to arrest ongoing BDZ-RSE.
KCC2 activation limits neuronal cell death following BDZ-RSE
KA-induced seizures lead to neuronal cell death in many brain regions 48–72 h after treatment.41 To determine if KCC2 activators have an impact on the extent of neuronal injury, mice were maintained for 48 h after KA treatment, and no mortality was seen in either experimental group following i.p. drug administration. Following cardiac perfusion, 30 μm brain sections were subjected to terminal deoxynucleotidyl transferase dUTP nick end (TUNEL) staining to visualize dead cells and counterstained with DAPI (Figure 7). The number of TUNEL+ cells in the CA1 region of the hippocampus was then compared between treatment groups. Consistent with published studies, high levels of TUNEL+ cells were found in mice injected with DZ alone. To quantify this result, we first compared the number of dead cells per slice. DZ/350 treatment reduced the number of TUNEL+ CA1 cells compared with mice treated with DZ alone (Figure 7B; DZ = 43.3 ± 2.7, DZ/350 = 32.3 ± 2.1; p = 0.0034). Likewise, the number of dead cells/mouse was also reduced (Figure 7C; DZ = 45.2 ± 2.0, 31.3 ± 3.6; p = 0.0228). Thus, 350 limits neuronal cell death following BDZ-RSE.
Figure 7.

KCC2 activation reduces neuronal cell death 48 h after BDZ-RSE
(A) Mice that survived for 48 h following KA-induced seizures were sacrificed, and brain sections were subjected to TUNEL/DAPI staining followed by confocal microscopy. The righthand panels (scale bar: 50 μm) are enlargements of the boxed areas in the lefthand panels (scale bar: 1 mm).
(B) The number of TUNEL+ neurons was quantified in hippocampal slices from both treatment groups (n = 18 and 22 slices for DZ and DZ/350, respectively).
(C) The number TUNEL+ neurons was quantified in mice from both treatment groups (n = 4 and 5 mice for DZ and DZ/350, respectively).
In all panels, p values were determined using t tests, ∗p < 0.05.
Discussion
Accumulating evidence suggests that deficits in the activity of KCC2 activity contribute to the pathophysiology of epilepsy. To further address the therapeutic efficacy of KCC2 activation, we have identified a family of small molecules that potently and selectively activate this transporter. Our screen exploited the ability of KCC2 activity to be visualized using TI imaging via an FLIPR assay, which allowed us to identify activators from a proprietary library of 1.3 million distinct chemical entities. Compared with our simple scalable assays, other screens using Cl− imaging were performed at much lower scales with libraries composed of between 20,000 and 90,000 compounds.21,42,43
Direct interaction of Cmp1 with KCC2 was observed using CETSA, and subsequent optimization resulted in the identification of 350. 350 potently increases KCC2 activity, as measured by patch-clamp recording and TI flux, without modifying its plasma membrane stability or the phosphorylation of the key regulatory sites S940 and T1007 within KCC2 expressed in HEK-293 cells.22,30 In addition to these well-characterized sites, KCC2 is phosphorylated on up to 9 other amino acids in the brain.23 The significance of these residues for transporter activity remains unknown as is their role in mediating the efficacy of 350.
Collectively, our results suggest that the compounds identified in our studies act as direct activators of KCC2. This is in contrast to other indirect potentiators of this transporter, which have been shown to increase expression levels or to modify its phosphorylation.17,42,43,44 Consistent with the role of KCC2 in limiting neuronal excitability, 350 reduced neuronal Cl− accumulation and the development of LRDs in acute brain slices exposed to 0-Mg. 350 rapidly distributed to the brain and did not appear to induce any gross effects on mouse behavior in the open field but acted to protect against PTZ-induced motor seizures. We assessed whether 350 impacts development of SE in mice induced by KA. The onset of SE was slowed in mice pre-dosed with 350, and the power of KA-induced seizures was also reduced. Collectively, these results suggest that 350 exhibits anticonvulsant activity.
KA-induced SE in rodents develops resistance to termination by BDZs, like that seen in patients with SE.45 We established that pretreatment of mice with 350 prevented the development of BDZ-RSE 4 h later. Critically, at this time point, 350 was present in the brain at a concentration sufficient to robustly potentiate KCC2 activity based on an EC50 of 260 nM. In addition to pretreatment, we further established that 350 was efficacious in restoring the efficacy of BDZs to terminate ongoing BDZ-RSE. Evidence accrued from patients and animal models suggest that multiple factors, ranging from modifications in membrane trafficking and subunit composition of GABAARs to deficits in GABA release, contribute to BDZ-RSE.46 However, increased intracellular accumulation of Cl− leading to depolarizing GABAARs is also widely believed to be of significance.47 The ability of 350 to prevent and arrest BDZ-RSE suggest that deficits in KCC2 activity are central to the pathophysiology of this trauma.
Prolonged seizure activity in humans and animal models leads to neuronal cell death. Cell death is evident within 48–72 h in mice that survive KA-induced SE.48,49,50 We observed that neuronal cell death in the hippocampus of mice treated with 350/DZ was significantly reduced compared with controls treated with DZ alone. This effect may be due to the ability of 350 to limit hyperexcitability, but previous in vitro and in vivo studies have shown that reducing or transiently inhibiting KCC2 expression levels is sufficient to induce neuronal apoptosis.3,4 Regardless of the precise mechanism, our study provides evidence that KCC2 activation arrests BDZ-RSE and limits the extent of the subsequent neuronal injury.
In summary, we have developed brain penetrant small-molecule activators of KCC2 that act directly on the target, reduce neuronal excitability, and show efficacy in treating BDZ-RSE. Collectively, our results suggest that KCC2 activation may be a viable strategy to treat refractory epilepsies in humans and limit the associated brain injury.
Limitations of the study
Our data prove that molecules that bind to an active KCC2 reduce neuronal excitability and are efficacious in arresting BDZ-RSE. However, there are several limitations of our study that warrant further discussion. The efficacy of 350 to arrest BDZ-RSE was tested at a single dose, which resulted in high levels of drug accumulation in the brain (approximately 4 μM) relative to its EC50 for KCC2 (260 nM). Thus, further studies are required to determine the relationship between drug concentration in the brain and suppression of EEG power during RSE. Such pharmaco-EEG measurement data will identify the effective dose of the drug that suppresses seizures and minimizes side effects. An additional limitation of our study is that we only studied SE induced by KA. To ensure our findings are not model specific, testing the efficacy of KCC2 activators to arrest BDZ-RSE caused by other agents will confirm the utility of this mechanism to treat refractory seizures. RSE leads to neuronal death and reactive gliosis, processes that underlie epileptogenesis and the development of recurrent seizures. Our study demonstrates that KCC2 activation reduces neuronal death 48 h after RSE. Studies using sustained exposure will determine if this neuroprotective efficacy of KCC2 activation impacts the development of spontaneous seizures and recurrent seizures following RSE. Finally, our study demonstrates that KCC2 activators restore or augment the anticonvulsant efficacy of BDZs. Thus, it will be essential to assess if similar synergy is seen on the anxiolytic, amnesic, hypnotic, and sedative efficacies of BDZs.
Data sharing
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy, described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse KCC2 | Neuromab | N1/12 |
| Mouse Actin | Sigma | MAB1501 |
| Rabbit GAPDH | Cell Signaling Technology | 2118 |
| Rabbit KCC2 pS940 | Phosphosolutions | P1551-940 |
| Rabbit KCC2 pT1007 | Phosphosolutions | P1551-1007 |
| Bacterial and virus strains | ||
| E. coli JM109 | Promega | P9751 |
| Chemicals, peptides, and recombinant proteins | ||
| Diazepam | Tocris | 2805/50 |
| Tetrodotoxin | Tocris | 1078/1 |
| Bumetanide | Tocris | 3108/50 |
| Muscimol | Tocris | 0289/1 |
| Glycine | Tocris | 0219/1G |
| VU0463271 | Tocris | 3888/10 |
| Gramicidin A | Sigma | 50845 |
| Ouabain | Sigma | O3125 |
| Premium Grade Sulfo-NHS-LC-Biotin | Thermofisher | PG82075 |
| Monomeric Avidin agarose | Thermofisher | 20228 |
| Kainic acid | Cayman | 58002-62-3 |
| Captisol (SBE-β-CD) | Ligand Pharmaceuticals | RC-0C7-020 |
| β-cyclodextrin | Selleckchem | E0046 |
| Pentylenetetrazole | Tocris | 2687/1G |
| Critical commercial assays | ||
| FluxOR K+ assay | Molecular Devices | R8223 |
| Lipofectamine 2000 | Thermofisher | STEM00001 |
| PlasmidPlusMega Kit | QIAGEN | 12981 |
| BCA protein assay | Thermofisher | 23227 |
| Click-iT™ Plus TUNEL | Thermofisher | C10618 |
| ProLong™ Gold Antifade Mountant with DAPI | Thermofisher | P36941 |
| FluxOR Dye | Thermofisher | F10016 |
| BackDrop™ | Thermofisher | B10512 |
| SelectScreen Kinase Profiling | Thermofisher | https://www.thermofisher.com/us/en/home/products-and-services/services/custom-services/screening-and-profiling-services/selectscreen-profiling-service/selectscreen-kinase-profiling-service.html |
| Deposited data | ||
| Compound synthesis and structures | WO2021180952A1 | https://worldwide.espacenet.com |
| Experimental models: Organisms/strains | ||
| HEK-293 | ATCC | CRL-1573 |
| Mice C57BL/6J | Jackson | 000,664 |
| Recombinant DNA | ||
| Human KCC2b | Addgene | 79,711 |
| GFP | Origene | PS100010 |
| Human Glycine receptor α1 subunit | Origene | RG210390 |
| Software and algorithms | ||
| ImageJ | Schneider et al. | https://imagej.nih.gov/ij/ |
| GraphPad | Prism | https://www.prismsoftware.com |
| LabChart 8.1 | ADInstruments | https://www.adinstruments.com |
| Clampex 10 | Molecular devices | https://www.moleculardevices.com/ |
| FUJIFILM X RAW STUDIO | Fuji | https://app.fujifilm.com |
| Other | ||
| EEG/EMG headmounts | Pinnacle Technology | 8201 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Stephen J. Moss (stephen.moss@tufts.edu).
Materials availability
Requests for materials should be addressed to Dr. Aaron Goldman (agoldman@ovidrx.com) or Dr. Rebecca Jarvis (rebecca.jarvis@astrazeneca.com). Compound requests will require an MTA given their potential for commercial development. Requests for KCC2 expression plasmids should be addressed to Addgene. The antibodies described in the paper are available from NeuroMab (anti-KCC2) or Phosphosolutions (pS940, and pT1007).
Experimental model and subject details
C57BL/6J (000,664) mice were purchased from Jackson Labs (https://www.jax.org). Male mice between 10 and 12 weeks of age were used for all experimentation and were allowed to recover for 7 days after surgery, prior to experimentation. Forebrain neurons were prepared from E18, or P1 C57BL/6J mice. All animal experimentation was approved by the Tufts University IACUC committee (https://viceprovost.tufts.edu/about-iacuc).
Method details
Antibodies and expression constructs. Mouse anti-KCC2 was purchased from NeuroMab and used at 1/500. Mouse anti actin was from Sigma and used at 1/1000, while rabbit anti GAPDH was from Cell Signaling and used at 1/1000. Rabbit anti-pS940 and pT1007 were from Phospho-solutions and used at 1/1000, and 1/750 respectively.28 The KCC2b construct used in these studies was obtained from Origene (SC304801) and contains human SLC12A5 cDNA cloned into the pCMV6-XL5 vector. The GFP (PS100010) and human GlyRα1(RG210390) cDNAs were also obtained from Origene and expressed using pCMV6-XL5.
Drug screening using Tl flux
HEK-293 cells were transiently transfected with a cDNA encoding human KCC2 using Lipofectamine 2000 reagent and seeded into 384-well PDL coated black-clear plates. 48 h after transfection KCC2 activity was measured using the Tl+ FLIPR (Fluorometric Imaging Plate Reader) assay was performed using the FluxOR™ Potassium Ion Channel Assay. Transfected cells and untransfected controls were initially washed in HBBS using a BioTek Elx 405 plate washer. TI sensitive FluxOR dye was added in HBSS supplemented with 20 mM HEPES, pH 7.3, in addition to BackDrop background suppressor. Test compounds (1.3 million) were added to a final concentration of 30μM (1% DMSO) and incubated for 1h. Stimulus buffer was added containing 5 mM K+/0.5 mM TL and cells were imaged for 120 s using an FLIPR TETRA workstation. Total TI uptake at 120 s was then compared to that seen at 0 time. The final concentrations of K+ and Cl− in the assay were 5 and 30 mM respectively, sufficient for maximal active KCC220,21,51 Song et al., 2002). Data were expressed as relative fluorescence units (RFU). The EC50 values and potency for initial hits and subsequent derivatives were determined using a maximal concentration of 100 μM drug and serial dilutions down to 10 nM.
Compound synthesis
All compounds were synthesized in house using up using multistep synthetic schemes resulting in the creation of a subseries of 114 members. Compounds were purified by HPLC (<98%) and their final structure was confirmed using NMR. In depth synthesis of all subseries members are outlined by Jarvis and Burli 2021; “Fused pyrimidine compounds as KCC2 activators” Patent WO2021180952A1, (https://worldwide.espacenet.com).
Cellular thermal shift assay
HEK-293 cells expressing KCC2 were exposed to Cmp1 or vehicle for 90 min while heated to 37-58°C and then rapidly cooled on ice. Cultures were subsequently lysed with 0.2% NP40, centrifuged at 12,000 x g to remove aggregated and denatured proteins in addition to cell debris, at 4°C. The supernatants were subjected to SDS-PAGE and immunoblotted with KCC2 and GAPDH antibodies. The levels of soluble KCC2 at increasing temperature were normalized to levels seen at 37°C (100%). The respective lx/l37°C values were then used to construct melting curves for KCC2 at each temperature. The IC50 values were then compared for KCC2 exposed to vehicle and Cmp1. The Isothermal dose analysis was performed at 50°C using increasing concentrations of Cmp1 for 1-10,0000 nM. The level of remaining soluble KCC2 at increasing amounts of Cmp1 were normalized to that seen at 3 nM (100%).24,25
Kinetic analysis of TI uptake
To measure the effects of drugs on the kinetics of TI uptake transfected HEK-293 were seed in 96 well plates. 48 h later cells were incubated with 10 μM 350, in 1 mg/mL 2-Hydroxypropyl-β-cyclodextrin (BCD), or vehicle, TI sensitive dye, 20 μM bumetanide, 20 μM ouabain and 2.5 mM probenecid for 1 h at room temperature. Cells were imaged using a FlexStation 3 microplate reader and following addition of stimulus buffer and TI uptake was determine every 1.5 s over a time course of 10 s.17 The initial rate of TI update was determined over this linear time course and expressed as RFU/s.
HEK-293 cell culture and biotinylation
HEK-293 cells were purchased from ATCC (CRL-1573) and transfected with human KCC2 and GFP expression plasmids using Lipofectamine 2000. 48-72 h following transfection, cultures were labeled with NHS-Biotin at 4°C for 30 min. Following cell lysis and purification on immobilized avidin, surface and total fractions were subject to SDS-PAGE and immunoblotted with KCC2 and actin antibodies as detailed previously.20,26
Determining KCC2 activity in HEK-293 cells using perforated patch-clamp recording
Briefly HEK-293 cells were transiently transfected with cDNA for GFP, GlyR1, and KCC2.20,21 48 h later recordings were conducted at room temperature in bath saline containing 140 mM NaCl, 2.5 mM KCl, 2.5 mM MgCl2, 2.5 mM CaCl2, 10 mM HEPES, 11 mM glucose, pH 7.4. Isolated GFP+ cells were visualized by epifluorescence and perforated with gramicidin (50 μg/mL) inside a patch pipette containing 140 mM KCl, 10 mM HEPES, 10μM bumetanide, pH 7.4. EGly values were obtained by application of glycine (50 μM) during positive-going voltage ramps (20 mV; 1-s duration). These measurements were made at 0 and 15 min after incubation with 350 in 1 mg/mL 2-Hydroxypropyl-β-cyclodextrin, or this vehicle. Data were acquired at 10 kHz with an Axopatch 200B amplifier and Clampex 10 software (Molecular Devices, Sunnyvale, CA). For gramicidin perforated-patch experiments, intracellular Cl− values were back-calculated using measured EGly values and the Nernst equation.17,52
Immunoblotting
Protein levels in cell lysates and tissue fractions were quantified using the BCA assay. 20-50 μg of protein was subject to SDS-PAGE and transferred to a membrane. Membranes were subsequently immunoblotted with antibodies against KCC2, pS940, pT1007, actin or GAPDH. Blots were visualized using enhanced chemiluminescene and quantified using a Fuji imager and quantified using Fuji software under linear conditions.17,26,28,53
Neuronal cultures
Hippocampal neurons were prepared from C57BL/6J P1 mice and plated at 450,000 cells per dish and maintained at 37°C in a humidified 5% CO2 incubator for up to 22 days before experimentation.20,28
Measuring KCC2 activity in neurons using patch-clamp recording
Recordings were performed on DIV 18-21 hippocampal cultures at 34°C, in saline contained the following (in mM): 140 NaCl, 2.5 KCl, 2.5 CaCl2, 2.5 MgCl2, 10 HEPES, and 11 glucose, pH 7.4 as detailed previously.28,52 For perforated-patch experiments, pipettes contained (in mM): 140 KCl and 10 HEPES, pH 7.4 with KOH and gramicidin A (50 μg/mL; MedChem Express). To measure EGABA we used 20 mV voltage-ramp protocols over 1-s periods to determine the reversal potentials of the leak-subtracted currents induced by 10 μM muscimol, a GABAAR agonists. For whole-cell experiments, pipettes contained (in mM): 115 K-meth-SO4, 30 KCl, 2 Mg-ATP, 4 Na-ATP, 0.4 Na-GTP, and 10 HEPES, pH 7.4 with KOH as outlined previously. All voltages from whole-cell experiments were corrected offline using a calculated liquid junction potential value in Clampex (Molecular Devices).54,55 All measurements were made in the presence of 300 nM Tetrodotoxin and 10 μM bumetanide. Intracellular Cl− values were back-calculated using measured EGABA values and the Nernst equation. Neurons that exhibited basal resting membrane potentials below −60 mV were considered unhealthy and excluded from analysis.
In vitro seizure assays
Horizontal/Coronal slices (350 μM) are prepared from 5-7 week old male mice and placed in a submerged chamber for a 60-min recovery period at 32°C in normal ACSF.13,32 Slices were then transferred to a Warner Instruments recording chamber and electrodes of 0.5-1 mOhm were inserted into layer III/IV of the medial entorhinal cortex, several cell layers deep. Slices were perfused for 10 min with normal ACSF before exposure to media deficient in Mg+2 supplemented containing vehicle (1 mg/mL 2-Hydroxypropyl-β-cyclodextrin), or 350. Recordings were made with a Multi-Clamp 700B amplifier with Clampex 10 acquisition software (Molecular Devices). Seizure activity was defined as a change greater that 3x the SD of the 1 min of baseline activity prior to 0-Mg2+ exposure. LRD were defined as periods of seizure activity with less interictal durations of <20 s.13,32
Measuring drug accumulation in brain and plasma exposure
Mice were injected subcutaneously (SC), IV, or IP with 350 in 5% BCD in a maximum volume of 300 μL. Plasma and brain samples were rapidly frozen on dry ice. Tissue and plasma samples were extracted in acetonitrile and the solvent phases was removed, for LC-MS/MS (Integrated Analytical Solutions, Inc, Berkeley, CA 94710; www.ianalytical.net).
Mouse behavioral analysis and PTZ-induced seizure assays
Mice were dosed SC with 50 mg/kg 350 or vehicle. 2-3 h later mice were placed in the center of the open field and the total distance traveled was monitored over a 40 min trial. The total distance traveled and the % time spent in the center zone was then determined as described previously.33,34 For PTZ induced seizures mice were injected IV with 25 mg/kg 350, or vehicle. 1 h later mice were subject to continuous video recording and injected with increasing concentration of PTZ. The dose required to induce the first hindlimb clonus was then determined and compared between treatment groups. All IV drug dosing was performed using a remote mini pump to avoid handling-induced seizures.35,36 For data analysis investigators were blind to drug and experimental conditions.
EEG recordings and data analysis
Surgeries were performed as described previously13,54,55 on C57BL/6J adult male mice (11 weeks) were anesthetized with isoflurane inhalation. Electroencephalography/electromyography (EEG/EMG) headmounts (3-channel, Pinnacle Technology #8201) were superglued to the skull aligned with lambda, and 4 screws inserted for subdural recording contacts above the frontal and parietal lobes. Silver epoxy was placed under each screw head to provide electrical connectivity between the electrodes and the headmount. Mice were then allowed to recover for 7 days before experimentation. On the day of recording, mice were acclimated to the EEG room 1 h prior to recording. For the 350 pretreatment study, after 1-h baseline recordings were obtained, mice were subcutaneously injected with 50 mg/kg 350, followed by 20 mg/kg kainate injection (IP) 2 h after. 5 mg/kg Diazepam (DZ) was administered to the animals 2 h-post KA injection and the recording continued for another hour.
To assess the efficacy of drugs to arrest ongoing SE, mice were intraperitoneally injected with 20 mg/kg kainate to induce seizures. 2 h after kainate injection, 5 mg/kg DZ or 50 mg/kg 350 and 5 mg/kg DZ was administered to the mice and the recording continued for another hour. Onset to first seizure and SE was performed by analysis of the EEG recordings. A seizure was defined as activity 2.5x the SD of the baseline of the preceding 1 min of activity that persists for at least 20 s. SE was defined as seizure activity lasting longer that 30 min, or continuous events that were separated by less than 30 s return to baseline. Percentage of epileptiform activity was determined by dividing the cumulative of all epileptiform activity over the duration of EEG recording. Fast-fourier transformation (FFT) was used to transform the EEG signals from the time domain into the frequency domain to form a power spectral density (PSD) plot using Labchart software [8K FFT size, Hann (cosine-bell), 87.5% window overlap]. EEG frequency analysis was performed by binning the total signal into the following frequencies (1-4 Hz), theta (4-8 Hz), alpha (8-13 Hz), beta (13-30 Hz), low gamma (30-50 Hz) and high gamma (50-100 Hz). The contribution of each frequency band to total power was then determined and compared between treatments.17,26,28,53 For the pretreatment study, total power 10 min before and 10 min after DZ treatment was compared. For the study where the effect of 350 on ongoing SE was investigated, total power in 2-min epochs immediately before and 30 min after injection were compared. Suppression was defined by a 50% reduction of total power. DZ insensitivity was calculated by the percentage of animals which did not show a 50% reduction of total power, within 20 min post-DZ or DZ+350 treatment. For data analysis investigators were blind to drug and experimental conditions.
Intrahippocampal drug delivery
C57BL/6 adult male mice (12 weeks) were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine (i.p.). A lengthwise incision was made to expose the skull and a small burr hole was made over the hippocampus (−2 mm posterior to bregma and 1.5 mm lateral to the midline). A micromanipulator was used to lower a microcannula-depth electrode device, manufactured in-house from a stainless-steel cannula (Plastics One) and an EEG head mount (Pinnacle Technology, Inc.). All animals recovered for a week before experimentation. On the day of experimentation mice were dosed with 20 μM 350, or vehicle in PBS in a total volume of 100 nL directly into the hippocampus. Basal EEG activity was monitored for 30 min prior to the injection of KA (20 mg/kg; IP).17
TUNEL assay
2-day post-EEG recordings, whole mouse brains were harvested and drop-fixed in 4% paraformaldehyde (PFA) solution overnight. PFA-fixed brains were cryoprotected in 30% sucrose solution prior to OCT embedding. 30-μm coronal brain sections were obtained from a Leica CM1900 cryostat. TUNEL assay was performed on hippocampal brain sections, using Click-iT Plus TUNEL Assay Kits for In Situ Apoptosis Detection with AlexaFluor 594 and sections were mounted with Prolong Gold Antifade mountant with DAPI (Thermofisher). 40X confocal Z-stacks images of CA1 hippocampal region were acquired by Leica Falcon SP8 and maximum projections of z-stacks were processed using Fiji software. The number of TUNEL positive cells in the region of interest in CA1 pyramidal layer was quantified. For data analysis investigators were blind to drug and experimental conditions.
Quantification and statistical analysis
Statistical analysis
The normality of datasets was tested using the D’Agostino-Pearson and the Shapiro-Wilk normality tests. Student’s t test or Welch’s t-tests (paired and unpaired, as appropriate), were then used to determine significance using GraphPad (Prism). EGG data was analyzed using LabChart 8.1 (ADInstruments), while electrophysiological data were analyzed using Clampex 10 (Molecular devices). All Data represents mean ± SEM The p values and N numbers for each dataset and included in the Figure legend. The p values are also quoted in the text.
Acknowledgments
S.J.M. is supported by National Institutes of Health (NIH) – National Institute of Neurological Disorders and Stroke grants NS087662, NS081986, NS108378, NS101888, NS103865, and NS111338 and NIH – National Institute of Mental Health grant MH118263. K.A., L.C.C., and R.A.C. were fellows of and funded by the AstraZeneca Postdoctoral Program. We thank Jake Dengler for assistance with the graphical abstract, which was created with biorender.com.
Author contributions
S.F.J.N., A.J.N., R.A.C., K.A., F.W., A.E.-S., M.P.F.-C., T.Z.D., J.L.S., L.M., A.K., L.C.C., and J.L.M. performed experimentation and data curation and analyzed data. R.J., Q.W., R.W.B., N.J.B., I.P.C., A.J.G., J.L.M., and S.J.M. provided resources, developed methodology, and analyzed data. I.P.C., J.L.M., A.J.G., and S.J.M. obtained funding. R.J., S.F.J.N., and S.J.M. wrote the manuscript.
Declaration of interests
S.J.M. serves as a consultant for AstraZeneca, Ovid Therapeutics, and SAGE Therapeutics, relationships that are regulated by Tufts University. S.J.M. holds equity in SAGE Therapeutics and Ovid Therapeutics. R.J., T.J., I.K.G., A.K., L.M., and I.P.C. are employees of AstraZeneca and hold equity. A.G. is an employee of Ovid Therapeutics and holds equity. R.J. and R.W.B. hold a patent describing the KCC2 activators, WO2021180952A1 (https://worldwide.espacenet.com).
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research.
Published: March 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.100957.
Supplemental information
The effects of 1μM Cmp1 on the activity of 348 individual protein kinases was measured via ATP hydrolysis using SelectScreen kinase Profiling (Thermofischer).
Data and code availability
This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Sieghart W., Savić M.M. International union of basic and clinical pharmacology. CVI: GABAA receptor subtype- and function-selective ligands: key issues in translation to humans. Pharmacol. Rev. 2018;70:836–878. doi: 10.1124/pr.117.014449. [DOI] [PubMed] [Google Scholar]
- 2.Kaila K., Price T.J., Payne J.A., Puskarjov M., Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014;15:637–654. doi: 10.1038/nrn3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kontou G., Josephine Ng S.F., Cardarelli R.A., Howden J.H., Choi C., Ren Q., Rodriguez Santos M.A., Bope C.E., Dengler J.S., Kelley M.R., et al. KCC2 is required for the survival of mature neurons but not for their development. J. Biol. Chem. 2021;296:100364. doi: 10.1016/j.jbc.2021.100364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Virtanen M.A., Uvarov P., Mavrovic M., Poncer J.C., Kaila K. The multifaceted roles of KCC2 in cortical development. Trends Neurosci. 2021;44:378–392. doi: 10.1016/j.tins.2021.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Rivera C., Voipio J., Payne J.A., Ruusuvuori E., Lahtinen H., Lamsa K., Pirvola U., Saarma M., Kaila K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 6.Kahle K.T., Merner N.D., Friedel P., Silayeva L., Liang B., Khanna A., Shang Y., Lachance-Touchette P., Bourassa C., Levert A., et al. Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep. 2014;15:766–774. doi: 10.15252/embr.201438840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stödberg T., McTague A., Ruiz A.J., Hirata H., Zhen J., Long P., Farabella I., Meyer E., Kawahara A., Vassallo G., et al. Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat. Commun. 2015;6:8038. doi: 10.1038/ncomms9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palma E., Amici M., Sobrero F., Spinelli G., Di Angelantonio S., Ragozzino D., Mascia A., Scoppetta C., Esposito V., Miledi R., Eusebi F. Anomalous levels of Cl- transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc. Natl. Acad. Sci. USA. 2006;103:8465–8468. doi: 10.1073/pnas.0602979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saitsu H., Watanabe M., Akita T., Ohba C., Sugai K., Ong W.P., Shiraishi H., Yuasa S., Matsumoto H., Beng K.T., et al. Impaired neuronal KCC2 function by biallelic SLC12A5 mutations in migrating focal seizures and severe developmental delay. Sci. Rep. 2016;6:30072. doi: 10.1038/srep30072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huberfeld G., Wittner L., Clemenceau S., Baulac M., Kaila K., Miles R., Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hübner C.A., Stein V., Hermans-Borgmeyer I., Meyer T., Ballanyi K., Jentsch T.J. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- 12.Kelley M.R., Cardarelli R.A., Smalley J.L., Ollerhead T.A., Andrew P.M., Brandon N.J., Deeb T.Z., Moss S.J. Locally reducing KCC2 activity in the Hippocampus is sufficient to induce temporal lobe epilepsy. EBioMedicine. 2018;32:62–71. doi: 10.1016/j.ebiom.2018.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore Y.E., Deeb T.Z., Chadchankar H., Brandon N.J., Moss S.J. Potentiating KCC2 activity is sufficient to limit the onset and severity of seizures. Proc. Natl. Acad. Sci. USA. 2018;115:10166–10171. doi: 10.1073/pnas.1810134115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pathak H.R., Weissinger F., Terunuma M., Carlson G.C., Hsu F.C., Moss S.J., Coulter D.A. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J. Neurosci. 2007;27:14012–14022. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barmashenko G., Hefft S., Aertsen A., Kirschstein T., Köhling R. Positive shifts of the GABAA receptor reversal potential due to altered chloride homeostasis is widespread after status epilepticus. Epilepsia. 2011;52:1570–1578. doi: 10.1111/j.1528-1167.2011.03247.x. [DOI] [PubMed] [Google Scholar]
- 16.Deeb T.Z., Nakamura Y., Frost G.D., Davies P.A., Moss S.J. Disrupted Cl(-) homeostasis contributes to reductions in the inhibitory efficacy of diazepam during hyperexcited states. Eur. J. Neurosci. 2013;38:2453–2467. doi: 10.1111/ejn.12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee K.L., Abiraman K., Lucaj C., Ollerhead T.A., Brandon N.J., Deeb T.Z., Maguire J., Moss S.J. Inhibiting with-no-lysine kinases enhances K+/Cl- cotransporter 2 activity and limits status epilepticus. Brain. 2022;145:950–963. doi: 10.1093/brain/awab343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silayeva L., Deeb T.Z., Hines R.M., Kelley M.R., Munoz M.B., Lee H.H.C., Brandon N.J., Dunlop J., Maguire J., Davies P.A., Moss S.J. KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc. Natl. Acad. Sci. USA. 2015;112:3523–3528. doi: 10.1073/pnas.1415126112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheung D.L., Cooke M.J., Goulton C.S., Chaichim C., Cheung L.F., Khoshaba A., Nabekura J., Moorhouse A.J. Global transgenic upregulation of KCC2 confers enhanced diazepam efficacy in treating sustained seizures. Epilepsia. 2022;63:e15–e22. doi: 10.1111/epi.17097. [DOI] [PubMed] [Google Scholar]
- 20.Conway L.C., Cardarelli R.A., Moore Y.E., Jones K., McWilliams L.J., Baker D.J., Burnham M.P., Bürli R.W., Wang Q., Brandon N.J., et al. N-Ethylmaleimide increases KCC2 cotransporter activity by modulating transporter phosphorylation. J. Biol. Chem. 2017;292:21253–21263. doi: 10.1074/jbc.M117.817841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cardarelli R.A., Jones K., Pisella L.I., Wobst H.J., McWilliams L.J., Sharpe P.M., Burnham M.P., Baker D.J., Chudotvorova I., Guyot J., et al. The small molecule CLP257 does not modify activity of the K(+)-Cl(-) co-transporter KCC2 but does potentiate GABAA receptor activity. Nat. Med. 2017;23:1394–1396. doi: 10.1038/nm.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kahle K.T., Deeb T.Z., Puskarjov M., Silayeva L., Liang B., Kaila K., Moss S.J. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 2013;36:726–737. doi: 10.1016/j.tins.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smalley J.L., Kontou G., Choi C., Ren Q., Albrecht D., Abiraman K., Santos M.A.R., Bope C.E., Deeb T.Z., Davies P.A., et al. Isolation and characterization of multi-protein complexes enriched in the K-Cl Co-transporter 2 from brain plasma membranes. Front. Mol. Neurosci. 2020;13:563091. doi: 10.3389/fnmol.2020.563091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawatkar A., Schefter M., Hermansson N.O., Snijder A., Dekker N., Brown D.G., Lundbäck T., Zhang A.X., Castaldi M.P. CETSA beyond soluble targets: a broad application to multipass transmembrane proteins. ACS Chem. Biol. 2019;14:1913–1920. doi: 10.1021/acschembio.9b00399. [DOI] [PubMed] [Google Scholar]
- 25.Martinez N.J., Asawa R.R., Cyr M.G., Zakharov A., Urban D.J., Roth J.S., Wallgren E., Klumpp-Thomas C., Coussens N.P., Rai G., et al. A widely-applicable high-throughput cellular thermal shift assay (CETSA) using split Nano Luciferase. Sci. Rep. 2018;8:9472. doi: 10.1038/s41598-018-27834-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee H.H.C., Walker J.A., Williams J.R., Goodier R.J., Payne J.A., Moss S.J. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem. 2007;282:29777–29784. doi: 10.1074/jbc.M705053200. [DOI] [PubMed] [Google Scholar]
- 27.Williams J.R., Sharp J.W., Kumari V.G., Wilson M., Payne J.A. The neuron-specific K-Cl cotransporter, KCC2. Antibody development and initial characterization of the protein. J. Biol. Chem. 1999;274:12656–12664. doi: 10.1074/jbc.274.18.12656. [DOI] [PubMed] [Google Scholar]
- 28.Lee H.H.C., Deeb T.Z., Walker J.A., Davies P.A., Moss S.J. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat. Neurosci. 2011;14:736–743. doi: 10.1038/nn.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Medina I., Friedel P., Rivera C., Kahle K.T., Kourdougli N., Uvarov P., Pellegrino C. Current view on the functional regulation of the neuronal K(+)-Cl(-) cotransporter KCC2. Front. Cell. Neurosci. 2014;8:27. doi: 10.3389/fncel.2014.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moore Y.E., Kelley M.R., Brandon N.J., Deeb T.Z., Moss S.J. Seizing control of KCC2: a new therapeutic target for epilepsy. Trends Neurosci. 2017;40:555–571. doi: 10.1016/j.tins.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Anderson W.W., Lewis D.V., Swartzwelder H.S., Wilson W.A. Magnesium-free medium activates seizure-like events in the rat hippocampal slice. Brain Res. 1986;398:215–219. doi: 10.1016/0006-8993(86)91274-6. [DOI] [PubMed] [Google Scholar]
- 32.Kelley M.R., Deeb T.Z., Brandon N.J., Dunlop J., Davies P.A., Moss S.J. Compromising KCC2 transporter activity enhances the development of continuous seizure activity. Neuropharmacology. 2016;108:103–110. doi: 10.1016/j.neuropharm.2016.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tretter V., Revilla-Sanchez R., Houston C., Terunuma M., Havekes R., Florian C., Jurd R., Vithlani M., Michels G., Couve A., et al. Deficits in spatial memory correlate with modified {gamma}-aminobutyric acid type A receptor tyrosine phosphorylation in the hippocampus. Proc. Natl. Acad. Sci. USA. 2009;106:20039–20044. doi: 10.1073/pnas.0908840106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vien T.N., Ackley M.A., Doherty J.J., Moss S.J., Davies P.A. Preventing phosphorylation of the GABA A R beta3 subunit compromises the behavioral effects of neuroactive steroids. Front. Mol. Neurosci. 2022;15:817996. doi: 10.3389/fnmol.2022.817996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimada T., Yamagata K. Pentylenetetrazole-induced kindling mouse model. J. Vis. Exp. 2018:56573. doi: 10.3791/56573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Erum J., Van Dam D., De Deyn P.P. PTZ-induced seizures in mice require a revised Racine scale. Epilepsy Behav. 2019;95:51–55. doi: 10.1016/j.yebeh.2019.02.029. [DOI] [PubMed] [Google Scholar]
- 37.Reddy D.S., Kuruba R. Experimental models of status epilepticus and neuronal injury for evaluation of therapeutic interventions. Int. J. Mol. Sci. 2013;14:18284–18318. doi: 10.3390/ijms140918284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crawshaw A.A., Cock H.R. Corrigendum to “Medical management of status epilepticus: emergency room to intensive care unit” [Seizure: Eur. J. Epilep. 75 (2020) 145-152] Seizure. 2020;80:282. doi: 10.1016/j.seizure.2020.05.021. [DOI] [PubMed] [Google Scholar]
- 39.Crawshaw A.A., Cock H.R. Medical management of status epilepticus: emergency room to intensive care unit. Seizure. 2020;75:145–152. doi: 10.1016/j.seizure.2019.10.006. [DOI] [PubMed] [Google Scholar]
- 40.Betjemann J.P., Lowenstein D.H. Status epilepticus in adults. Lancet Neurol. 2015;14:615–624. doi: 10.1016/S1474-4422(15)00042-3. [DOI] [PubMed] [Google Scholar]
- 41.Ben-Ari Y., Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–587. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- 42.Gagnon M., Bergeron M.J., Lavertu G., Castonguay A., Tripathy S., Bonin R.P., Perez-Sanchez J., Boudreau D., Wang B., Dumas L., et al. Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med. 2013;19:1524–1528. doi: 10.1038/nm.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prael Iii F.J., Kim K., Du Y., Spitznagel B.D., Sulikowski G.A., Delpire E., Weaver C.D. Discovery of small molecule KCC2 potentiators which attenuate in vitro seizure-like activity in cultured neurons. Front. Cell Dev. Biol. 2022;10:912812. doi: 10.3389/fcell.2022.912812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang X., Drotar J., Li K., Clairmont C.D., Brumm A.S., Sullins A.J., Wu H., Liu X.S., Wang J., Gray N.S., et al. Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci. Transl. Med. 2019;11:eaau0164. doi: 10.1126/scitranslmed.aau0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niquet J., Baldwin R., Suchomelova L., Lumley L., Naylor D., Eavey R., Wasterlain C.G. Benzodiazepine-refractory status epilepticus: pathophysiology and principles of treatment. Ann. N. Y. Acad. Sci. 2016;1378:166–173. doi: 10.1111/nyas.13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burman R.J., Rosch R.E., Wilmshurst J.M., Sen A., Ramantani G., Akerman C.J., Raimondo J.V. Why won't it stop? The dynamics of benzodiazepine resistance in status epilepticus. Nat. Rev. Neurol. 2022;18:428–441. doi: 10.1038/s41582-022-00664-3. [DOI] [PubMed] [Google Scholar]
- 47.Burman R.J., Selfe J.S., Lee J.H., van den Berg M., Calin A., Codadu N.K., Wright R., Newey S.E., Parrish R.R., Katz A.A., et al. Excitatory GABAergic signalling is associated with benzodiazepine resistance in status epilepticus. Brain. 2019;142:3482–3501. doi: 10.1093/brain/awz283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bengzon J., Mohapel P., Ekdahl C.T., Lindvall O. Neuronal apoptosis after brief and prolonged seizures. Prog. Brain Res. 2002;135:111–119. doi: 10.1016/S0079-6123(02)35011-8. [DOI] [PubMed] [Google Scholar]
- 49.Walker M.C. Pathophysiology of status epilepticus. Neurosci. Lett. 2018;667:84–91. doi: 10.1016/j.neulet.2016.12.044. [DOI] [PubMed] [Google Scholar]
- 50.Dingledine R., Varvel N.H., Dudek F.E. When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv. Exp. Med. Biol. 2014;813:109–122. doi: 10.1007/978-94-017-8914-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Agez M., Schultz P., Medina I., Baker D.J., Burnham M.P., Cardarelli R.A., Conway L.C., Garnier K., Geschwindner S., Gunnarsson A., et al. Molecular architecture of potassium chloride co-transporter KCC2. Sci. Rep. 2017;7:16452. doi: 10.1038/s41598-017-15739-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deeb T.Z., Lee H.H.C., Walker J.A., Davies P.A., Moss S.J. Hyperpolarizing GABAergic transmission depends on KCC2 function and membrane potential. Channels. 2011;5:475–481. doi: 10.4161/chan.5.6.17952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moore Y.E., Conway L.C., Wobst H.J., Brandon N.J., Deeb T.Z., Moss S.J. Developmental regulation of KCC2 phosphorylation has long-term impacts on cognitive function. Front. Mol. Neurosci. 2019;12:173. doi: 10.3389/fnmol.2019.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nathanson A.J., Zhang Y., Smalley J.L., Ollerhead T.A., Rodriguez Santos M.A., Andrews P.M., Wobst H.J., Moore Y.E., Brandon N.J., Hines R.M., et al. Identification of a core amino acid motif within the alpha subunit of GABAARs that promotes inhibitory synaptogenesis and resilience to seizures. Cell Rep. 2019;28:670–681.e8. doi: 10.1016/j.celrep.2019.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hines R.M., Maric H.M., Hines D.J., Modgil A., Panzanelli P., Nakamura Y., Nathanson A.J., Cross A., Deeb T., Brandon N.J., et al. Developmental seizures and mortality result from reducing GABAA receptor alpha2-subunit interaction with collybistin. Nat. Commun. 2018;9:3130. doi: 10.1038/s41467-018-05481-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The effects of 1μM Cmp1 on the activity of 348 individual protein kinases was measured via ATP hydrolysis using SelectScreen kinase Profiling (Thermofischer).
Data Availability Statement
This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.