

Summary
Accumulation of senescent cells contributes to age-related diseases including idiopathic pulmonary fibrosis (IPF). Insulin-like growth factor binding proteins (IGFBPs) regulate many biological processes; however, the functional contributions of IGFBP2 in lung fibrosis remain largely unclear. Here, we report that intranasal delivery of recombinant IGFBP2 protects aged mice from weight loss and demonstrated antifibrotic effects after bleomycin lung injury. Notably, aged human-Igfbp2 transgenic mice reveal reduced senescence and senescent-associated secretory phenotype factors in alveolar epithelial type 2 (AEC2) cells and they ameliorated bleomycin-induced lung fibrosis. Finally, we demonstrate that IGFBP2 expression is significantly suppressed in AEC2 cells isolated from fibrotic lung regions of patients with IPF and/or pulmonary hypertension compared with patients with hypersensitivity pneumonitis and/or chronic obstructive pulmonary disease. Altogether, our study provides insights into how IGFBP2 regulates AEC2-cell-specific senescence and that restoring IGFBP2 levels in fibrotic lungs can prove effective for patients with IPF.
Keywords: aging, fibrosis, lung, IGFBP2, P21, γ-H2AX, PPAR-α, senescence, SASP
Graphical abstract
Highlights
-
•
IGFBP2 expression in AEC2 cells is downregulated during lung fibrosis
-
•
Igfbp2 deficiency increases P21, a senescence marker, following lung injury
-
•
Intranasal delivery of recombinant IGFBP2 protects aged mice following lung injury
-
•
Aged human Igfbp2 transgenic mice blunts bleomycin-induced lung fibrosis
Chin et al. report that AEC2-cell-derived IGFBP2 inhibits P21 and negatively correlates with lung fibrosis. Using preclinical models, they reveal that AEC2-cell-derived IGFBP2 ameliorates lung fibrosis by suppressing senescence and SASP factors. Further, they show diminished IGFBP2 expression specifically in AEC2 cells of patients with IPF and IPF-PAH.
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common and severe form of interstitial lung disease, affecting more than 5 million individuals worldwide.1 IPF is a heterogeneous disease often occurring in the elderly, with a median survival rate of less than 3 years.2 Furthermore, development of pulmonary arterial hypertension (PAH) secondary to lung fibrosis is a major factor of all-cause mortality in patients with IPF. PAH may greatly reduce life expectancy in patients with IPF to less than 1 year, and currently, no therapies are approved for this disease.3,4 Advanced age is a significant risk factor for IPF, and yet very little is known about how age contributes to the pathogenesis of IPF.5 Although much of the pathogenesis of IPF remains unclear, alveolar epithelium has emerged as a principal site of injury in IPF. This disease is typically characterized by aberrant alveolar epithelium that triggers exaggerated myofibroblast activation and excessive extracellular matrix deposition.6 More specifically, chronic injury to alveolar epithelial type 2 (AEC2) cells has been linked to the pathogenesis of IPF.7
Cellular senescence has recently been implicated as a critical underpinning of age-related fibroproliferative diseases including IPF.8 Cellular senescence, a stress response associated with stable cell-cycle arrest, is accompanied by robust secretion of pro-inflammatory mediators leading to senescence-associated secretory phenotype (SASP).9 Mounting evidence suggests that P21 and phosphorylation of H2AX are two key molecular markers in the senescence pathway10,11,12 and that accumulation of P21 is typically observed during the process of senescence development.13 Recent studies demonstrate that selective removal of senescent cells can ameliorate a number of age-related diseases.14,15 However, the underlying mechanisms of cell senescence in alveolar epithelium remains poorly understood.
The insulin-like growth factor (IGF) system consists of 6 IGF-binding proteins (IGFBPs) with high affinity binding to IGF-I and IGF-II but not insulin.16,17,18 IGFBPs are ubiquitously expressed in most tissues, and their actions are complex and depend upon the tissue and their ability to interact with proteins in the extracellular matrix.19 Although the actions of IGFs are modulated by IGFBPs, several studies suggest that cellular functions can also be affected via IGF-independent mechanisms.20,21 Growing evidence suggests that IGFBPs play an important role in senescence and aging.22,23,24 In particular, IGFBP2 may play an important role in lung function as IGFBP2 levels can predict the risk of a rapid evolution of systemic sclerosis-induced interstitial lung disease.25 More recent studies demonstrate that IGFBP2 undergoes epigenetic alterations in a tissue-specific manner, contributing to the development of chronic diseases.26,27
In the current study, we investigated whether IGFBP2 regulates cellular senescence and secretion of senescent factors specifically in AEC2 cells in the development of pulmonary fibrosis. To this end, using several in vitro and in vivo models, we explored the presence and functional contributions of IGFBP2 in senescence-mediated lung fibrosis. Utilizing gene silencing and overexpression approaches, we show that IGFBP2 inhibits P21 and γ-H2AX in murine lung epithelial cells under fibrotic stimuli. We demonstrate that recombinant IGFBP2 delivered locally reduced lung fibrosis and inhibited senescence after bleomycin injury in aged mice. Importantly, we report that transgenic expression of human Igfbp2 specifically in AEC2 cells significantly reduced senescence, pro-inflammatory mediators, and extracellular matrix markers in response to low-dose bleomycin-induced pulmonary fibrosis in aged mice. Finally, we found significantly reduced expression of PPARA and IGFBP2 in AEC2 cells of patients with IPF and/or IPF-PAH. These findings may offer critical insights for future therapeutic approaches that could prove beneficial for patients with IPF and IPF-PAH.
Results
Selective loss of IGFBP2 in AEC2 cells in non-resolving lung fibrosis of aged mice model
Intratracheal administration of bleomycin is the most widely used model for studying pulmonary fibrosis in mice.28 However, several studies have utilized young mice in an attempt to recapitulate features of IPF. Since IPF is an age-associated chronic disease, we intratracheally administered single low-dose bleomycin (1 U/kg body weight) to aged mice (78–82 weeks old) after intubation. Body weight was measured every week and was significantly lower in bleomycin-treated mice compared with normal saline at 7, 14, and 21 days (Figure 1A). To detect fibrosis, lungs were evaluated for histological features. Both trichrome and Sirius red staining increased after 28 and 50 days of bleomycin exposure compared with relative controls (Figures 1B and S1A). Total lung collagen content was significantly increased in lungs at 28 days compared with saline controls, and that increase was also significant in lungs at 50 days compared with 28 days of bleomycin injury, indicating non-resolving lung fibrosis (Figures 1C and S1B). The protein levels of collagen-I, fibronectin, and vimentin were increased in the lung tissues of aged mice subjected to low-dose intratracheal bleomycin administration. In the total lung, bleomycin treatment decreased Igfbp2 expression at both mRNA and protein levels but increased P21 protein levels (senescence marker), concomitant with the activation of extracellular matrix (ECM) proteins (Figures 1D, 1E, and S1C). For the detection of IGFBP2 levels in circulation, ELISA analysis revealed no significant difference in the aged mice after 14 days of bleomycin injury (Figure 1F). AEC2 cells are major facultative progenitor cells that play a critical role in the progression of lung fibrosis.29 In line with this, gene expression analysis revealed significantly decreased Igfbp2 mRNA levels in primary AEC2 cells after 14 days of bleomycin injury (Figures 1G and S2). Consistent with the fibrotic changes, multicolor immunohistological analyses revealed elevated P21 and γ-H2AX staining, molecular markers for cellular senescence, specifically in AEC2 cells after 28 and 50 days of bleomycin challenge (Figures 1H–1J). Taken together, these data strongly indicate that reduced expression of IGFBP2, specifically in AEC2 cells, is associated with cellular senescence in aged murine model of bleomycin-induced persistent lung fibrosis.
Figure 1.

Low-dose bleomycin induces irreversible pulmonary fibrosis in aged mice
(A) Line plot showing body weights of aged wild-type (WT; C57BL/6J) mice 7, 14, and 21 days after intratracheal administration of normal saline (equal volume) or bleomycin (1 U/kg body weight) (n = 8 WT saline; n = 8 WT bleomycin). ∗∗∗p < 0.001, two-way ANOVA with Tukey’s post-hoc test.
(B) Representative images of Mason’s trichrome-stained lung sections of aged mice 28 or 50 days after intratracheal administration of bleomycin. Scale bars, 50 μm (top) and 1 mm (bottom) (n = 8 WT saline; n = 8 WT bleomycin).
(C) Hydroxyproline content (μg per mg of lung tissue) in the lungs of aged mice 28 or 50 days after intratracheal administration of bleomycin (1 U/kg body weight). ∗∗∗p < 0.001, ∗p < 0.05, one-way ANOVA with Tukey’s post-hoc test.
(D) Western blot for expression of IGFBP2, P21, collagen-1, fibronectin, and vimentin in the lung homogenates of aged mice 14 days after low-dose bleomycin challenge. β-actin served as the internal control. Note that same samples were run on different gels (n = 6 WT saline; n = 8 WT bleomycin).
(E) Igfbp2 mRNA expression in the lung homogenates of aged mice subjected to low-dose bleomycin challenge after 14 days.
(F) IGFBP2 protein expression was determined by ELISA in the serum samples of aged mice 14 days after bleomycin injury (n = 5 WT saline; n = 6 WT bleomycin).
(G) Igfbp2 mRNA expression in the primary AEC2 cells isolated from aged mice subjected to low-dose bleomycin challenge after 14 days. Each sample is obtained from 4 mice lungs (n = 3 WT saline; n = 3 WT bleomycin). NS, not significant, ∗∗∗∗p < 0.0001, ∗∗p < 0.01, Student’s unpaired two-tailed t test.
(H) Representative multicolor immunohistochemistry of lung sections from aged WT mice subjected to low-dose bleomycin after 28 and 50 days. Green color indicates surfactant protein C (SPC) expression; brown color indicates P21 (top) and phospho-H2AX (bottom) expression. Insets: zoom-in images to show green and brown color localization. Black arrowheads highlight the double-positive AEC2 cells. Scale bars, 10 μm.
(I and J) Quantification of double-positive cells for P21 (left) and phospho-H2AX (right) expression in SPC + cells in the lungs of aged WT mice subjected to low-dose bleomycin after 28 and 50 days, respectively. Each data point represents per field image of a sample. Data are mean ± SEM. ∗∗∗∗p < 0.0001, ∗∗p < 0.01, ∗p < 0.05, one-way ANOVA with Tukey’s post-hoc test.
IGFBP2 downregulation in adenoviral TGF-β1-induced lung fibrosis in aged mice model
Next, we investigated the importance of IGFBP2 in an additional model of lung fibrosis in aged mice. For the development of lung fibrogenesis, we performed intratracheal delivery of adenovirus (5 × 108 PFU) expressing transforming growth factor β1 (TGF-β1) to aged (78–82 weeks older) mice. In this setting, aged mice who received a single dose of adenovirus expressing TGF-β1 developed lung fibrosis with increased Sirius red positivity and trichrome staining compared with mice who received empty or null-adenovirus (Figure 2A). Total lung hydroxyproline content was elevated in adenoviral TGF-β1-treated mice at 28 days (Figure 2B). Furthermore, multicolor immunohistological analyses revealed significantly increased P21 and γ-H2AX staining in association with reduced IGFBP2 protein expression specifically in AEC2 cells after 28 days of adenoviral TGF-β1-mediated lung injury (Figures 2C and 2D). Together, these results imply that decreased expression of IGFBP2 is associated with markers of cellular senescence—P21 and γ-H2AX, selectively in AEC2 cells in the pathogenesis of adenoviral TGF-β1-induced lung fibrosis.
Figure 2.

IGFBP2 downregulation in adenoviral TGF-β1-induced pulmonary fibrosis in aged mice
(A) Sirius red (top)- or Mason’s trichrome (bottom)-stained lung sections of aged (78–82 weeks old) WT mice 28 days after intratracheal administration of Ad-null or Ad-TGF-β1 virus (5 × 108 PFU). Scale bars, 50 μm (n = 6 Ad-null; n = 6 Ad-TGF-β1).
(B) Hydroxyproline content (μg per mg of lung) in the lungs of 18-month-old mice 28 days after intratracheal administration of Ad-null or Ad-TGF-β1 virus (n = 6 Ad-null; n = 6 Ad-TGF-β1).
(C) Representative double-color immunohistochemistry lung images of aged WT mice challenged with Ad-Null or Ad-TGF-β1 virus. Green color indicates SPC expression; brown color indicates IGFBP2 or P21 or phospho-H2AX expression. Black arrowheads indicate the double-positive AEC2 cells. Scale bars, 10 μm (n = 6 Ad-null; n = 6 Ad-TGF-β1).
(D) Quantification of percentages of double-positive cells for IGFBP2, P21, and phospho-H2AX expression in SPC + cells, respectively. Data are mean ± SEM ∗∗p < 0.01, and ∗∗∗p < 0.001, Student’s unpaired two-tailed t test.
IGFBP2 deficiency elevates senescent markers under pro-fibrotic stimuli
To investigate the biological relevance of IGFBP2, mouse lung epithelial (MLE-12) cells were exposed to chronic or acute fibrotic stimuli—atazanavir (20 μg/mL) + 0.1% hypoxia, cigarette smoke extract (100 μg/mL), and bleomycin (10 μg/mL) as independent models of alveolar epithelial cell injury. To reliably induce senescence and to evaluate the chronic effects of IGFBP2, MLE-12 cells were exposed (long term) to atazanavir (senescence inducer) and 0.1% hypoxia or a two-hit model of bleomycin treatment. Both chronic in vitro models revealed decreased protein expression of IGFBP2 coupled with increased protein expression of P21 and phospho-H2AX compared with relative controls (Figures 3A, 3B, S3A, and S3B). These findings suggest that reduced IGFBP2 is associated with increased expression of P21 and phospho-H2AX under chronic pro-fibrotic stimuli.
Figure 3.

IGFBP2 deficiency increases P21 expression in response to fibrotic stimuli in vitro
(A) Western blot for the expression of IGFBP2, P21, and phospho-H2AX in MLE-12 cells pretreated with atazanavir (ATZ; 20 μg/mL) for 1 h and exposed to hypoxia treatment for 72 h.
(B) Western blot for the expression of IGFBP2, P21, and phospho-H2AX in MLE-12 cells exposed to absence or presence of chronic exposure to bleomycin (two-hit model; 10 μg/mL).
(C) Western blot for the expression of IGFBP2 and P21 in MLE-12 cells exposed to absence or presence of hypoxia treatment at 4 h. β-Actin served as an internal control.
(D) Western blot for the expression of IGFBP2 and P21 in the cytosolic and nuclear fractions of MLE-12 cells that were exposed to absence or presence of hypoxia treatment. α-Tubulin and histone-3 served as internal controls.
(E) Western blot for the expression of IGFBP2 and P21 in MLE-12 cells exposed to absence or presence of cigarette smoke treatment (100 μg/mL).
(F) Non-targeting or Igfbp2 siRNA-transduced MLE-12 cells were exposed to absence or presence of hypoxia treatment at 4 h. Western blot for the expression of IGFBP2 and P21.
(G) Non-targeting or Igfbp2 siRNA-transduced MLE-12 cells were challenged with absence or presence of bleomycin exposure (10 μg/mL). Western blot for the expression of IGFBP2, P21, and phospho-H2AX in MLE-12 cells subjected to bleomycin exposure at 4 h. β-Actin served as an internal control. Data are representative of minimum of 3 independent experiments.
Repetitive acute injuries are believed to cause chronic disease, and therefore we evaluated the acute effects of IGFBP2 in vitro. The protein levels of P21 were increased and IGFBP2 protein levels were decreased in MLE-12 cells exposed (short term) to 0.1% hypoxia challenge (Figures 3C and S3C). Furthermore, IGFBP2 protein levels were decreased in the nuclear fractions of MLE-12 cells challenged with hypoxia compared with normoxia (Figures 3D and S3D). To analyze IGFBP2 expression in additional fibrosis-relevant systems, we next exposed (short term) MLE-12 cells to cigarette smoke insult. To this end, we observed reduced expression of IGFBP2, but P21 expression was not significantly altered after cigarette smoke injury (Figures 3E and S3E).
Next, to evaluate the function of IGFBP2, we performed small interfering RNA (siRNA) transfection in MLE-12 cells challenged with either hypoxia or bleomycin insults. We showed increased P21 protein levels in MLE-12 cells treated with Igfbp2 siRNA compared with non-targeting siRNA, and that increase was also observed after hypoxia or bleomycin treatments (Figures 3F, 3G, S3F, and S3G). Surprisingly, expression levels of phospho-H2AX were not altered by Igfbp2 knockdown, indicating that IGFBP2 deficiency does not directly control phosphorylation of H2AX following bleomycin challenge (Figures 3G and S3G). Together, these observations indicate that IGFBP2 regulates P21 accumulation in response to acute pro-fibrotic stimuli.
Lentiviral expression of IGFBP2 reduces senescent markers and β-galactosidase activity
To determine the action of IGFBP2, we undertook a lentiviral transduction approach in MLE-12 cells exposed to fibrotic stimuli. Consistent with the results from Igfbp2 silencing, lentivirus-mediated expression of Igfbp2 decreased P21 levels in MLE-12 cells in response to hypoxia treatment when compared with mock-virus-treated MLE-12 cells exposed to hypoxia (Figures 4A and S4A). Furthermore, P21 tend to show decreased levels in both cytosolic and nuclear fractions in Igfbp2 lentivirus-transduced MLE-12 cells in response to hypoxia treatment (Figures 4B and S4B). To further validate our findings, we analyzed the role of IGFBP2 in response to cigarette smoke and bleomycin stimuli. Conversely, lentiviral expression of Igfbp2 did not reduce P21 expression in MLE-12 cells under cigarette smoke conditions (Figures 4C and S4C). Interestingly, lentiviral expression of Igfbp2 decreased not only P21 expression but also phospho-H2AX in MLE-12 cells exposed to bleomycin treatment, indicating an indirect regulation between IGFBP2 and phospho-H2AX (Figures 4D and S4D). Since senescence-associated (SA) β-galactosidase (β-gal) activity is a widely used marker for senescence, we evaluated its association on the endogenous function of IGFBP2 in MLE-12 cells exposed to atazanavir (protease inhibitor) and hypoxia as well as bleomycin treatments. Biochemical analysis showed that SA β-gal activity significantly increased in MLE-12 cells after treatment with atazanavir and hypoxia (4 days) or bleomycin (2 days). Importantly, Igfbp2 expressed through lentivirus transduction significantly decreased SA β-gal activity in MLE-12 cells after treatment with atazanavir and hypoxia or bleomycin at 96 or 48 h, respectively (Figures 4E and 4F). Collectively, our results indicate that elevated expression of IGFBP2 reduces β-gal activity in mouse lung epithelial cells following hypoxia and bleomycin pro-fibrotic stimuli.
Figure 4.

Stable transduction with Igfbp2 lentivirus vector decreased P21 expression and β-galactosidase activity in vitro
(A) Mock-virus- and Igfbp2 lentivirus-transduced MLE-12 cells in the absence or presence of hypoxia treatment at 4 h. Western blot for the expression of IGFBP2 and P21. β-Actin served as internal control.
(B) Western blot for the expression of IGFBP2 and P21 in the cytosolic and nuclear fractions of Igfbp2 lentivirus-transduced MLE-12 cells in the absence or presence of hypoxia treatment at 4 h. α-Tubulin and histone-3 served as internal controls.
(C) Western blot for the expression of IGFBP2, P21, and phosph-H2AX in Igfbp2 lentivirus-transduced MLE-12 cells in the absence or presence of cigarette smoke treatment (100 μg/mL).
(D) Western blot for the expression of IGFBP2, P21, and phospho-H2AX in Igfbp2 lentivirus-transduced MLE-12 cells in the absence or presence of bleomycin (10 μg/mL). β-Actin served as internal control.
(E) Bar graph showing the β-galactosidase activity of MLE-12 cells pretreated with ATZ for 1 h and subjected to hypoxia for 96 h.
(F) Bar graph showing the β-galactosidase activity of MLE-12 cells treated with bleomycin for 48 h. Data are representative of minimum of 3 independent experiments. Data are mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001, one-way ANOVA with Tukey’s post-hoc test.
PPARα regulates IGFBP2 expression in lung fibrosis
Peroxisome proliferator-activated receptor α (PPARα) plays a vital role in cellular metabolism and functions as a transcription factor for Igfbp2, indicating its direct target.30,31 We therefore examined whether Ppara activation is involved in the pathogenesis of pulmonary fibrosis through induction of Igfbp2. Consistent with previous studies, we demonstrated decreased PPARα expression in MLE-12 cells exposed to chronic bleomycin challenge and also in primary murine AEC2 cells after 14 days of bleomycin injury. To evaluate the regulatory relationship between PPARα and IGFBP2, we performed siRNA transfection in MLE-12 cells challenged with bleomycin. We showed decreased Igfbp2 expression in Ppara siRNA-transfected MLE-12 cells compared with non-targeting siRNA, and that decrease was also observed after bleomycin injury (Figures 5A–5C and S5A–S5C). Using immunohistological staining, we found that PPARα expression was observed in AEC2 cells and that expression was significantly lower in bleomycin-treated lungs compared with saline-treated lungs (Figures 5D and 5E). Notably, gene expression analysis revealed that mRNA levels of Ppara were significantly lower in primary AEC2 cells of patients with IPF compared with patients with chronic obstructive pulmonary disease (COPD) but not in patients with hypersensitivity pneumonitis (HP) (Figure 5F; Table S1). Using multicolor immunohistochemical staining, we confirmed that AEC2 cells in IPF lung sections had lower PPARα protein expression compared with AEC2 cells from healthy donors (Figures 5G and 5H; Table S2). Together, these data demonstrate that PPARα regulates IGFBP2 expression in mouse lung epithelial cells after bleomycin injury and is significantly reduced in AEC2 cells of patients with IPF.
Figure 5.

Reduced PPARA expression in the AEC2 cells from fibrotic lung regions of patients with IPF
(A) Western blot for the expression of PPARα and β-actin in MLE-12 cells exposed to absence or presence of bleomycin at 4 h.
(B) Non-targeting or Ppara siRNA-transduced MLE-12 cells were exposed to absence or presence of bleomycin treatment at 4 h. Western blot for the expression of PPARα and IGFBP2. β-Actin served as internal control. Data are representative of minimum of 3 independent experiments.
(C) Ppara mRNA expression in the primary AEC2 cells isolated from aged mice subjected to low-dose bleomycin challenge after 14 days. Eukaryotic 18S rRNA was used as an endogenous control (n = 5 WT saline; n = 5 WT bleomycin).
(D) Representative multicolor color immunohistochemistry of lung sections from aged WT mice 28 days after bleomycin injury. Green color indicates SPC expression; brown color indicates PPARα expression. Scale bars, 10 μm (n = 5 WT saline; n = 5 WT bleomycin).
(E) Quantification of percentages of double-positive cells for SPC and PPARα in the lungs of aged WT mice subjected to low-dose bleomycin after 28 days.
(F) Ppara mRNA expression was determined by qPCR in the primary AEC2 cells of patients with IPF (n = 21) compared with HP (n = 5) or COPD (n = 9).
(G) Representative multicolor immunohistological staining of PPARA and SPC. Arrows indicate examples of SPC-positive and PPARA-positive cells. Staining was performed with lung sections from 2 healthy controls and 2 patients with IPF.
(H) Quantification of percentages of double-positive cells for SPC and PPARA in the fibrotic lung regions of patients with IPF and donor (healthy) controls. Data are mean ± SEM. NS, not significant; ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, one-way ANOVA with Tukey’s post-hoc test (for multiple group comparisons) or Student’s unpaired two-tailed t test (for two group comparisons).
Recombinant IGFBP2 exhibits antifibrotic effects after bleomycin lung injury
We next tested the antifibrotic potential of IGFBP2 in the context of bleomycin-induced lung fibrosis. Current FDA-approved drugs highlight the need for more effective treatments. Therefore, we focused on IGFBP2 as a potential therapeutic target for lung fibrosis. Aged mice were treated with intranasal delivery of recombinant IGFBP2 protein (25 μg/kgwt), containing surfactant (50 mg/kgwt), once every 3 days after bleomycin challenge, and lung tissues were harvested after 14 and 28 days. Body weight was assessed every 7 days, and weight loss was significantly improved in the IGFBP2-treated group when compared with the bleomycin and surfactant group (Figures 6A and 6B). Histological staining revealed that collagen deposition was lower in the lungs of IGFBP2-treated mice after bleomycin injury relative to control mice (Figure 6C). In addition, hydroxyproline content was significantly reduced in IGFBP2-treated mice compared with vehicle-treated mice after 28 days of bleomycin lung injury (Figure 6D). Interestingly, IGFBP2-treated mice showed improved overall survival (Figure 6E). Using senescence RT2 Profiler PCR array, we found that important fibrosis related genes—Tgfb1, Vim, Nox4, and igf1—and the senescence gene Cdkn1b were downregulated in AEC2 cells of IGFBP2-treated mice after 14 days of bleomycin lung injury (Figures S6A and S6B). Immunohistological analyses revealed decreased levels of the ECM marker fibronectin in IGFBP2-treated mice compared with vehicle-treated mice after 28 days of bleomycin lung injury (Figure 6F). Furthermore, the protein levels of senescence markers—P21 and phospho-H2AX—were decreased in the lung tissues of IGFBP2-treated mice compared with vehicle-treated mice after 14 days of bleomycin injury (Figures 6G and S6C). To provide more direct visualization, we performed multicolor immunohistological analyses and found that P21 levels, specifically in AEC2 cells, were significantly reduced in IGFBP2-treated mice compared with vehicle-treated mice after 14 days of bleomycin lung injury (Figures 6H and 6I). Overall, these results suggest the therapeutic potential of IGFBP2 in the development of lung fibrosis.
Figure 6.

Intranasal treatment of recombinant IGFBP2 alleviates bleomycin-induced pulmonary fibrosis in aged mice
(A) Schematic representation of the experimental approach. Aged WT mice were exposed to saline or bleomycin treated with or without recombinant IGFBP2 protein (25 μg/kgwt), containing Curosurf (50 mg/kgwt), by intranasal instillation and euthanized 14 and 28 days later.
(B) Body weights of IGFBP2-treated and vehicle-treated mice were measured and represented as bar graph (n = 8 per group). ∗∗∗p < 0.001 and ∗p < 0.05, two-way ANOVA.
(C) Representative images of Sirius red (top) and Mason’s trichrome (middle) staining from lung sections at day 28. Representative whole lung images of Mason’s trichrome (below) staining from lung sections at day 28. Scale bars, 50 μm (top and middle) and 1 mm (bottom).
(D) Total lung hydroxyproline content was quantitated 28 days after bleomycin treatment and represented as bar graph (n = 4 per group). ∗p < 0.05, one-way ANOVA with Tukey’s post-hoc test.
(E) Survival of mice 28 days after intranasal treatment of IGFBP2 presented as line graph (n = 10 mice per group). Animal survival was assessed by the Kaplan-Meier analysis using log-rank test.
(F) Representative immunohistochemically stained lung images of fibronectin from aged WT mice 28 days after bleomycin injury (n = 6 per group). Scale bars, 50 μm (top and middle) and 1 mm (below).
(G) Western blot for the expression of P21 and fibronectin 14 days after bleomycin injury (n = 4 per group). β-Actin served as internal control.
(H) Representative multicolor immunohistochemistry-stained lung images of SPC (red) and P21 (brown) from aged WT mice 14 days after bleomycin injury (n = 8 per group). Black arrowheads indicate the double-positive AEC2 cells. Scale bars, 50 μm.
(I) Bar graph showing the percentages of double-positive P21 and SPC AEC2 cells that were quantified. Data are mean ± SEM. ∗p < 0.05, ∗∗∗∗p < 0.001, one way ANOVA with Tukey’s post-hoc test.
Senescence RT2 Profiler PCR array in AEC2 cells of human Igfbp2 transgenic mice
To address whether IGFBP2 regulates cellular senescence in the development of pulmonary fibrosis, we performed senescence RT2 Profiler PCR array specifically in primary AEC2 cells in the pathogenesis of bleomycin-induced lung fibrosis. To achieve physiological relevance, we utilized human Igfbp2-inducible transgenic (Igfbp2Tg) aged mice in which the human Igfbp2 gene was genetically activated in AEC2 cells by combining human Igfbp2 floxed alleles with the tamoxifen-inducible SPC-CreERT2 knockin allele (Figures S7A and S7B). Utilizing RT2 Profiler PCR array, first we examined the expression of senescence genes in lung homogenates and in AEC2 cells of aged (36 weeks old) Igfbp2fl/fl littermates exposed to bleomycin injury compared with normal saline. We identified significant upregulation of important senescence genes—Cdkn1a (P21), Cdkn2b, Pai-1, Igfbp7, and Sparc—along with 11 other senescent-related genes (Figures S8A, S8B, S9A, and S9B). Next, we assessed the expression of senescence genes specifically in AEC2 cells of aged (36 weeks old) Igfbp2Tg mice after 14 days of bleomycin injury. qPCR array data analysis revealed 31 upregulated and 21 downregulated genes relevant to the cellular senescence pathway. Primary AEC2 cells isolated from aged Igfbp2Tg mice showed downregulation of important senescence genes, namely Cdkn1a (P21), Pai-1, Irf-5, Irf-7, and Tp53bp1 as well as fibronectin compared with aged Igfbp2fl/fl littermates exposed to bleomycin injury (Figures S9C and S9D). Collectively, these data indicate that transgenic expression of Igfbp2 downregulates Cdkn1a (P21) and other senescence-related genes specifically in AEC2 cells in the pathogenesis of bleomycin-induced lung fibrosis in aged mice.
Transgenic expression of human Igfbp2 reduces ECM deposition, senescence-associated secretory phenotype, and senescence
We next examined the in vivo effect of low-dose bleomycin exposure in human Igfbp2 inducible transgenic (Igfbp2Tg) aged (36 weeks old) mice. The Igfbp2Tg mice were first assessed for weight loss at 7-day intervals after intratracheal instillation of bleomycin injury. The Igfbp2Tg mice were significantly protected from weight loss at days 7, 14, and 21 relative to aged Igfbp2fl/fl littermates after bleomycin injury (Figure 7A). Both Sirius red and trichrome staining and hydroxyproline contents were significantly reduced in Igfbp2Tg mice relative to aged Igfbp2fl/fl littermates after 28 days of bleomycin treatment (Figures 7B and 7C). Similarly, compared with aged Igfbp2fl/fl littermates, Igfbp2Tg mice showed decreased ECM markers—collagen-1, fibronectin, and vimentin—together with decreased P21 expression in the lungs at 14 days after bleomycin injury (Figures 7D and S10). Furthermore, quantitative RT-PCR analysis of AEC2 cells from aged Igfbp2Tg mice showed downregulation of SASP factors, namely Il-1β, Tnf-α, Mcp-1, Stat6, and Il-4, compared with aged Igfbp2fl/fl littermates challenged with bleomycin treatment, indicating abrogation of pulmonary fibrosis (Figure 7E). Multicolor histological examination of aged Igfbp2Tg mice revealed significantly decreased γ-H2AX staining in AEC2 cells after bleomycin injury (Figures 7F and 7G). Overall, these data strongly suggest that senescence markers, SASP factors, and ECM markers were substantially ameliorated in Igfbp2Tg mice and demonstrate the functional importance of IGFBP2 in the context of bleomycin-induced lung fibrosis.
Figure 7.

Effects of aged human Igfbp2 transgenic mice challenged with bleomycin treatment
(A) Line plot showing the change in body weights of aged (36 weeks) WT and human Igfbp2 transgenic (Tg) mice subjected to intratracheal administration of bleomycin treatment (0.75 U/kg bodyweight) (n = 7 Igfbp2 fx/fx; n = 7 Igfbp2 Tg). ∗∗∗p < 0.001 and ∗∗p < 0.01, two-way ANOVA.
(B) Sirius red (top)- or Mason’s trichrome (middle)-stained lung sections and whole-lung images (trichrome; below) of aged Igfbp2 fx/fx and human Igfbp2 Tg mice 28 days after intratracheal administration of bleomycin treatment. Scale bars, 50 μm (top and middle) and 1 mm (below) (n = 8 Igfbp2 fx/fx; n = 8 Igfbp2 Tg).
(C) Total lung collagen content measured by hydroxyproline assay in aged Igfbp2 fx/fx and human Igfbp2 Tg mice 28 days after intratracheal administration of bleomycin treatment (n = 4 Igfbp2 fx/fx; n = 8 Igfbp2 Tg). ∗∗p < 0.01 and ∗p < 0.05, one way ANOVA with Tukey’s post-hoc test.
(D) Western blot for the expression of IGFBP2, P21, collagen-I, fibronectin, and vimentin (n = 6 Igfbp2 fx/fx; n = 8 Igfbp2 Tg).
(E) qPCR analysis for mRNA expression of tumor necrosis factor α (TNF-α), IL-1β, MCP-1, IL-6, STAT3, STAT6, and IL-4 in aged WT and human Igfbp2 Tg mice 14 days after intratracheal administration of bleomycin. Each sample is obtained from 4 mice lungs (n = 6 Igfbp2 fx/fx; n = 6 Igfbp2 Tg). ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, and ∗p < 0.05. Student’s unpaired two-tailed t test.
(F) Representative double-color immunohistochemistry-stained lung images of SPC (green) and phospho-H2AX (brown) expression from aged Igfbp2 fx/fx and Igfbp2 Tg mice 28 days after bleomycin injury. Black arrowheads indicate the double-positive AEC2 cells. Scale bars, 10 μm.
(G) Bar graph showing the percentages of double-positive p-H2AX and SPC AEC2 cells that were quantified. Data are mean ± SEM. NS, not significant; ∗∗∗∗p < 0.001, one way ANOVA with Tukey’s post-hoc test.
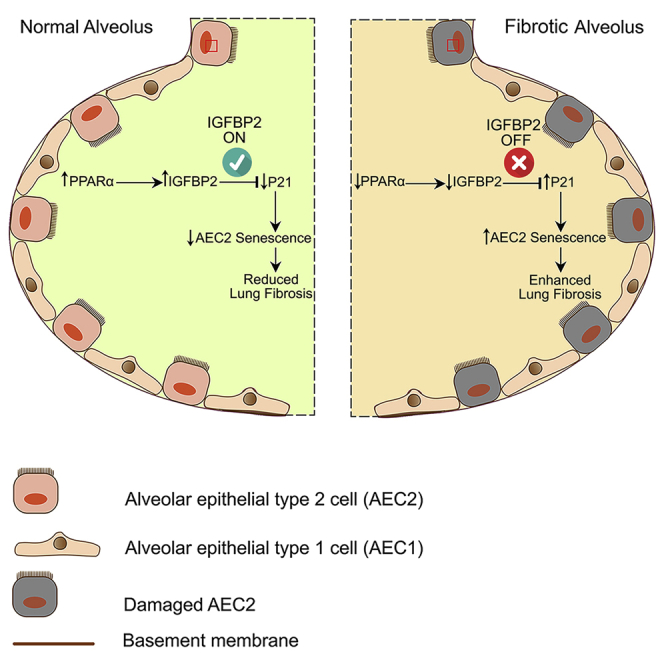
(H) Schema represents molecular regulation of IGFBP2 signaling involving senescence in the AEC2 cells of the aged lung.
Alveolar IGFBP2 expression was diminished in patients with IPF and IPF-PAH
To decipher the role of IGFBP2 in the pathogenesis of IPF and related subtypes, we performed transcriptomic expression studies in primary AEC2 cells isolated from fibrotic lung regions of patients with IPF and non-IPF (COPD or HP) patients (Table S1). In this cohort (n = 43), the median (interquartile range) age of patients with IPF was 66 years (ranged from 60 to 71.8 years), patients with COPD was 66 years (62.5–70.5 years), and patients with HP was 60.5 years (57–71 years), respectively. We found statistically significant differences of IGFBP2 expression in AEC2 cells between non-IPF (COPD or HP) patients and patients with IPF (p < 0.01). Further analysis showed that IGFBP2 expression is significantly lower in patients with IPF compared with either patients with COPD (p < 0.05) or HP (p < 0.01) (Figure 8A). Compared with patients with IPF with non-smoking history, IGFBP2 mRNA expression was not significantly altered in patients with IPF with smoking history (Figure 8B). Pulmonary hypertension and type 2 diabetes are common and significant comorbidities associated with IPF.32 Therefore, we examined the alveolar IGFBP2 relationship between patients with IPF and in combination with type 2 diabetes or PAH. No statistical significance was observed in alveolar IGFBP2 expression between patients with IPF alone compared with those with IPF and coexisting with patients with type 2 diabetes (Figure 8C). In contrast, IGFBP2 mRNA expression in AEC2 cells was significantly lowered in patients with IPF in combination with PAH (IPF-PAH) compared with IPF alone (Figures 8D and S11). Consistent with the mRNA levels of IGFBP2 expression in AEC2 cells, multicolor immunohistochemical analyses confirmed significantly decreased protein expression of IGFBP2 in AEC2 cells of fibrotic lung regions in patients with IPF compared with healthy controls (Figures 8E and 8F; Table S2). Overall, these results imply that the loss of IGFBP2 in AEC2 cells of patients with IPF compared with patients with COPD or HP is associated with pulmonary hypertension.
Figure 8.

IGFBP2 expression was suppressed in the primary AEC2 cells of fibrotic lungs obtained from patients with IPF
(A) IGFBP2 mRNA expression was determined by qPCR in the primary AEC2 cells isolated from fibrotic lung regions of patients with IPF (n = 27) compared with patients with COPD (n = 9) or HP (n = 5). ∗p < 0.05 and ∗∗p < 0.01, one-way ANOVA with Tukey’s post-hoc test.
(B) IGFBP2 mRNA expression in primary AEC2 cells obtained from patients with IPF with smoking history (n = 19) compared with patients with IPF with non-smoking history (n = 6).
(C) IGFBP2 mRNA expression in primary AEC2 cells obtained from patients with IPF with type 2 diabetes (n = 4) compared with patients with IPF with no type 2 diabetes (n = 7).
(D) IGFBP2 mRNA expression determined by qPCR in the primary AEC2 cells obtained from patients with IPF with pulmonary hypertension (MPAP ≥ 25 mmHg) (n = 13) compared with patients with IPF with no pulmonary hypertension (n = 14). MPAP, mean pulmonary artery pressure.
(E) Representative multicolor immunohistological staining of SPC and IGFBP2. Arrows indicate examples of SPC-positive and IGFBP2-positive cells. Staining was performed with lung sections from 2 healthy controls and 2 patients with IPF.
(F) Quantification of percentages of double-positive cells for SPC and IGFBP2 in the fibrotic lung regions of patients with IPF and donor (healthy) controls. Data are expressed as mean ± SEM. NS, not significant; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.0001, Student’s unpaired two-tailed t test.
Discussion
Enhanced alveolar epithelial progenitor cell senescence has been linked to age-related and end-stage IPF. However, the mechanisms driving AEC2 senescence remain unclear. In this study, we demonstrated that loss of IGFBP2 specifically in AEC2 cells leads to P21 and phospho-H2AX-mediated senescence and thus promotes persistent lung fibrosis. Our findings provide evidence that inactivation of transcription factor PPARα results in the loss of IGFBP2 expression in aged mice after low-dose bleomycin lung injury. Moreover, intranasal treatment with IGFBP2 recombinant protein attenuated bleomycin-induced pulmonary fibrosis in aged mice. Transgenic aged mice that express human Igfbp2 specifically in AEC2 cells showed reduced senescence, SASP factors, and collagen deposition and improved pulmonary fibrosis after bleomycin lung injury. Notably, low transcript levels of IGFBP2 were found in the AEC2 cells obtained from fibrotic lung lesions of patients with IPF and IPF-PAH compared with non-IPF (COPD or HP) patients.
Dysregulated AEC2 cells have been intimately linked to the pathogenesis of IPF.33 In a paracrine manner, injured AEC2 cells are the prime signaling partners for the activation of fibroblasts.34 In addition, recent studies suggest a critical link between the IGF system and pulmonary fibrosis.35 We and others have previously highlighted the crucial role of IGFBP5 in the pathogenesis of IPF.36,37 Present study findings demonstrate suppressed levels of IGFBP2 expression in AEC2 cells both in aged mice and in humans with lung fibrosis. Decreased expression of IGFBP2 specifically in AEC2 cells of aged fibrotic lungs may seem counterintuitive to a recent report showing higher levels of circulating IGFBP1 and IGFBP2 in the serum of patients with IPF.38 However, the elevated serum levels of IGFBP2 in patients with IPF may be reasoned due to aberrant secretion by immune cells eliciting an immune response or other comorbid factors such as lung to liver interactions involving a systemic disease. A recent single-cell RNA sequencing dataset from IPF cell atlas shows higher transcript levels of IGFBP2 in AEC2 cells of patients with IPF or interstitial lung disease (ILD). This discrepancy might be due to AEC2 heterogeneity in distinct stages of fibrotic lungs and limitations of single-cell RNA (scRNA) sequencing including bias of transcript coverage.39,40 Alternatively, a recent study demonstrating upregulation of IGF1 in the fibrotic lung tissue of patients with IPF41 suggests that lower levels of IGFBPs may be regulating IGF-dependent and -independent effects and is a subject of further investigation.
Fibrosis has been strongly linked to senescence in the lung.8 Primarily, cellular senescence is an evolutionarily selected process that counteracts early-life cancer.13 In the first instance, senescence may act as a protective response by limiting proliferation, but studies have shown that accumulation of senescence is the key pathogenic mechanism that drives the development of pulmonary fibrosis.8,42 Advanced age is the predominant risk factor for age-related pathologies and, importantly, for the development of pulmonary fibrosis.43 Recent studies utilize aged mice as an improved model for IPF.44 In line with this, we confirm that challenging aged mice with low-dose bleomycin or Ad-TGF-β1 injury resulted in non-resolving lung fibrosis.
Alveolar epithelial cell senescence is evident in IPF and in a variety of experimental models of lung fibrosis.45 Development of senescence biomarkers and senolytic drugs is an active area of research for age-related diseases. New therapeutic targets directed toward AEC2 senescence are therefore urgently needed. In this study, we show that silencing Igfbp2 in mouse lung epithelial cells increased P21 levels in response to multiple fibrotic stimuli. Similarly, lentiviral-mediated expression of Igfbp2 decreased both P21 and γ-H2AX levels and reduced SA β-gal activity in response to both hypoxia and bleomycin stimuli. Consistently, recent reports demonstrated upregulation of P21 expression in human lungs with IPF compared with donor lungs.45,46 In this study, we speculate that IGFBP2 controlling P21 expression may involve a network of genes that include Wnt/β-catenin signaling.47,48 Other studies show that IGFBP2 is detected in the nuclei and that such nuclear translocation is mediated by a functional nuclear localization signal sequence.49 In agreement with this, we show that upregulated IGFBP2 expression in the nuclear fraction is relative to the cytosolic fraction in mouse lung epithelial cells and that IGFBP2 regulates P21 expression after hypoxia and bleomycin stimuli.
Transcription factor PPARα was reported to upregulate the promoter activity of Igfbp2 in metformin-treated primary hepatocytes.30 In the current study, we demonstrated that PPARα was not only associated with but also transcriptionally regulates Igfbp2 expression in the context of lung injury. Moreover, reduced levels of Ppara were confirmed in AEC2 cells of patients with IPF. It is possible that transcription factors other than PPARα may be involved in the regulation of IGFBP2 expression and awaits further investigation. Given the pleiotropic effects and diverse roles of PPARα, its therapeutic potential needs to be elucidated in future studies.
Previous studies have shown that AEC2 cells undergo senescence and secrete pro-fibrotic mediators in bleomycin-induced pulmonary fibrosis.50 In this study, we showed that intranasal delivery of IGFBP2 suppressed important fibrotic genes—Tgfb1 and Nox4—and the senescence gene Cdkn1b, encoding P27 in AEC2 cells of aged mice after bleomycin lung injury. These findings indicate potential regulation between IGFBP2, TGF-β1, and NOX4 in lung fibrosis. We further observed downregulation of senescent markers—Cdkn1a, encoding P21, Pai-1, Irf-5, Irf-7, and Tp53bp1—in aged human Igfbp2 transgenic mice compared with aged wild-type mice challenged with bleomycin treatment. Interestingly, a recent report showed that interferon-regulated members—IRF-5 and IRF-7—induce senescence in immortal fibroblasts.51 Another report showed increased P53BP1-positive foci in the development of radiation-induced lung injury.52 Importantly, emerging evidence suggests that DNA damage-induced P21 expression increasing to stable levels allows the cell to permanently exit the cell cycle and undergo senescence or apoptosis.11,53 Cellular senescence is also accompanied by a pro-inflammatory phenotype, termed SASP. Specifically, we show significant downregulation of established SASP components—Tnf-α, Il-1β, Mcp-1, and Stat6—in AEC2 cells expressing human Igfbp2 in response to bleomycin injury. Although interleukin-4 (IL-4) was upregulated, recent reports suggest that IL-4 plays a dichotomous role in pulmonary fibrosis.54,55 These data support the concept that IGFBP2 inhibits P21 and γ-H2AX-mediated senescence and SASP factors in AEC2 cells and thereby prevents lung fibrogenesis (Figure 7H). In addition, the ability of recombinant IGFBP2 to reduce bleomycin-induced lung fibrosis in aged mice provides important insights about the therapeutic potency of IGFBP2 for patients with IPF.
To translate our in vitro and in vivo findings to human lung disease, we determined that IGFBP2 mRNA levels were significantly lower in AEC2 cells from patients with IPF compared with patients with either COPD or HP. Furthermore, IGFBP2 mRNA expression was significantly lowered in AEC2 cells from patients with both IPF and PAH compared with patients with IPF alone. There are some limitations to this study. Patients were studied from a single center with end-stage IPF. This limited our ability to comprehend the earliest levels and functions of IGFBP2 in the pathogenesis of IPF.
In conclusion, we identified an antifibrogenic role of IGFBP2 and demonstrated that the localized presence of IGFBP2 regulates P21 and γ-H2AX-mediated senescence in an AEC2-cell-specific manner with implications for the treatment of IPF.
Limitations of the study
Our study demonstrated that loss of IGFBP2 specifically in AEC2 cells mediates senescence through PPARα and the P21 axis and promotes persistent lung fibrosis. Furthermore, intranasal delivery of recombinant IGFBP2 protein attenuated lung fibrosis after bleomycin injury in aged mice. However, there are limitations in this study that readers should keep in mind. First, IGFBP2 as a therapeutic agent to delineate late or established lung fibrosis in an aged mice setup has not been addressed. Similarly, the senescence super array used was a targeted array that might not have covered all the relevant genes contributing to cellular senescence, but this approach provides higher sensitivity and reproducibility within the pathway of genes examined. Second, patient samples used in our study were from a single center cohort with end-stage IPF. Future studies need to preferentially explore cellular interactions mediated by prolonged senescence and its regulation by IGF signaling members in the pathogenesis of lung fibrosis.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat-anti-IGFBP2 | R&D systems | Cat# MAB7971; RRID:AB_2264598 |
| Rabbit anti-IGFBP2 for IHC | Abcam | Cat# ab188200 |
| Rabbit anti-P21 | Abcam | Cat# ab188224; RRID:AB_2734729 |
| Rabbit anti-Collagen 1 | EMD Millipore | Cat# AB765P; RRID:AB_92259 |
| Rabbit anti-PPARα | LSBio | Cat# LS-C312574 |
| Rabbit anti-Fibronectin | Abcam | Cat# ab2413; RRID:AB_2262874 |
| Rabbit anti-Vimentin | CST | Cat# 5741S; RRID:AB_10695459 |
| HRP conjugated-β actin | Santa Cruz | Cat# sc-47778HRP; RRID:AB_2714189 |
| Mouse anti-α tubulin | CST | Cat# 3873; RRID:AB_1904178 |
| Rabbit anti-H3 Histone | CST | Cat# 4499; RRID:AB_10544537 |
| Rabbit anti-phospho-Histone 2AX | CST | Cat# 2577; RRID:AB_2118010 |
| Rabbit anti-Prosurfactant Protein C | Abcam | Cat# ab211326; RRID:AB_2927746 |
| Bacterial and virus strains | ||
| Ad-Null | Vector Biolabs | Cat#1240 |
| Ad-m-TGFβ1 | Vector Biolabs | Cat#ADV-274099 |
| mouse Igfbp2 lentiviral particle | Origene | Cat#: MR204287L3V |
| Lenticontrol particles | Origene | Cat# PS100092V |
| Chemicals, peptides, and recombinant proteins | ||
| human IGFBP2 recombinant protein | R&D Systems | Cat# 797-B2-025 |
| Bleomycin | EMD Millipore | Cat# 203401 |
| DMSO | Sigma- Aldrich | Cat# D8418 |
| 4% PFA | Santa Cruz | Cat# sc-281692 |
| DMEM/F12 | Gibco | Cat# 11-320-033 |
| Plasmocin | invivogen | Cat# ant-mpp |
| Collagenase type 1 | Worthington Biochemical | Cat# LS004197 |
| Dispase | Corning | Cat# 354235 |
| Green chromogen | Leica | Cat# DC9913 |
| Bond polymer refine system | Leica | Cat# DS9800 |
| BondTM Primary antibody diluent | Leica | Cat# AR9352 |
| BOND Epitope Retrieval Solution 1 | Leica | Cat# AR9961 |
| BOND Epitope Retrieval Solution 2 | Leica | Cat# AR9640 |
| Wash solution 10X concentration | Leica | Cat# AR9590 |
| BOND Universal Covertile | Leica | Cat# S21.4611 |
| Tween-20 | Sigma- Aldrich | Cat# P9416 |
| PVDF membrane | EMD Millipore | Cat# IPVH00010 |
| SuperSignalTM Pico or Femto substrate | Thermo Fisher Scientific | Cat# 34095 |
| RIPA buffer | Thermo Fisher scientific | Cat# 89901 |
| Igfbp2 | Life science technology | Hs01040719_m1 |
| 18S rRNA | Life science technology | Hs99999901_s1 |
| Pparα | Life science technology | Mm00440939_m1 and Hs00231882_m1 |
| Igfbp2 siRNA | Horizon Discovery | L-062198-01-0005 |
| Pparα siRNA | Santa Cruz | sc-36308 |
| Critical commercial assays | ||
| High-Capacity RNA-to-cDNA™ Kit | Thermo Fisher scientific | Cat# 4387406 |
| TaqMan™ Fast Advanced Master Mix | Thermo Fisher scientific | Cat# 44-445-56 |
| Hydroxyproline assay | Cell Biolab | Cat# STA-675 |
| Βeta-galactosidase activity assay | Biovision | Cat# K821- 100 |
| RT2 profiler PCR assay | Qiagen | Cat# PAMM-050ZC-6 |
| IGFBP2 ELSA assay | R&D systems | Cat# DY797 |
| BCA protein assay | Thermo Fisher Scientific | Cat# 23227 |
| Experimental models: Cell lines | ||
| MLE-12 | ATCC | CRL-2110 |
| Experimental models: Organisms/strains | ||
| Igfbp2flx/flxtransgenic mouse | Leeds University | |
| Sftpc-CreERT2 | The Jackson Laboratory | RRID#IMSR_JAX:028054 |
| C57BL6/J | The Jackson Laboratory | RRID#IMSR_JAX:000664 |
| Oligonucleotides | ||
| IL-1βCCH | PrimerBank ID 6680415a1 | 5′-GCAACTGTTCCTGAACTCAACT-3′ and 5′-ATCTTTTGGGGTCCGTCAACT-3′ |
| IL-4 | PrimerBank ID 10946584a1 | 5′- GGTCTCAACCCCCAGCTAGT-3′ 5′- GCCGATGATCTCTCTCAAGTGAT-3′ |
| IL-6 | PrimerBank ID 13624311a1 | 5′-TAGTCCTTCC-3′ 5′-TACCCCAATTTCC-3′ |
| MCP-1 | PrimerBank ID 6755430a1 | 5′- TTAAAAACCTGGATCGGAACCAA-3′ 5′- GCATTAGCTTCAGATTTACGGGT-3′ |
| STAT-3 | PrimerBank ID 13277852a1 | 5′- CAATACCATTGACCTGCCGAT-3′ 5′- GAGCGACTCAAACTGCCCT-3′ |
| STAT6 | PrimerBank ID 6678155a1 | 5′- CTCTGTGGGGCCTAATTTCCA-3′ 5′- CATCTGAACCGACCAGGAACT-3′ |
| TNF-α | PrimerBank ID 7305585a1 | 5′- CCCTCACACTCAGATCATCTTCT-3′ 5′- GCTACGACGTGGGCTACAG-3′ |
| Software and algorithms | ||
| ImageJ 1.53t | FIJI | https://imagej.nih.gov/ij/ |
| Graphpad Prism 9.0 | Graphpad software LLC | https://www.graphpad.com |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Angara Sureshbabu (suresh.angara@nortonthoracic.org).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Human lung tissues
The study protocol was approved by the local ethics committee under the St. Joseph’s Hospital and Medical Center Institutional Review Board. All enrolled patients have provided informed consent (IRB# PHX-21-500-138-73-18) and approval was obtained from the respective local ethical committees for medical research.
Individuals with IPF, ILD, COPD or HP were enrolled for lung transplantation procedure. The explant lung tissues were obtained during the above surgery procedure. All individuals who were enrolled for lung transplant surgery have respiratory illness and majority of them were undertaking generic medication for hypertension, diabetes or gastroesophageal reflex disease. Healthy control tissues were obtained from excess donor lungs. For middle age purposes, individuals ≤ 45 years of age were excluded from this study. The details of patient demographics and clinical characteristics in each group were provided in Tables S1 and S2.
Cell culture
MLE-12 (mouse lung epithelial) cell line was obtained from A.T.C.C. (American Type Culture Collection, Rockville, Maryland, USA), and cultured in DMEM/F12 medium (Gibco) enriched with 2% fetal bovine serum (Gibco) and 50 μg/ml plasmocin (Invivogen) in a humidified atmosphere with 5% CO2 at 37 °C.
A corresponding Sftpc-cre mouse line was obtained from Jackson Laboratories (RRID#IMSR_JAX:028054). To induce homologous recombination by CreERT2, 5 consecutive intraperitoneal tamoxifen injections (0.45 mg/kg body weight) were given at 36 weeks old male or female mice.
Animals
Aged 18-month-old (78 weeks old or greater) C57BL/6J (RRID#IMSR_JAX:000664) mice were obtained from the Jackson Laboratories (Bar Harbor, Maine, USA). Human-Igfbp2 tamoxifen inducible transgenic mice were generated by Drs. Kearney and Wheatcroft (Leeds University, United Kingdom), and were purchased from Genoway (Lyon, France). Animals were housed under pathogen-free conditions with food and water ad libitum. All experiments and procedures were approved by the Institutional Animal Care and Use Committee at St. Joseph’s Hospital and Medical Center (Phoenix, Arizona).
Method details
Generation of SFTPC-cre-ERT2-human-Igfbp2 transgenic mice
Surfactant protein C (Sftpc)-cre hIgfbp2flox (human Igfbp2 knocked into the ROSA26 locus flanked by a floxed STOP codon) was generated by crossing a mouse line expressing tamoxifen-inducible cre under control of the AEC2 cell-specific promoter Sftpc with human-Igfbp2flox. The primer sequences for genotyping the Igfbp2Cre transgene were 5′- ACACCGGCCTTATTCCAAG-3′, 5′- TGCTTCACAGGGTCGGTAG-3′, 5′-TGCTTCACAGGGTCGGTAG-3′. The band was observed at 210 bp for the Sftpc-CreERT2 and 327 bp band for the wild type. Igfbp2LoxP allele was detected by amplifying a 245 bp for the homozygous Igfbp2 and a 778 bp band for the wild type (Figures S7A and S7B). The following primer sequences used for Igfbp2Loxp were 5′-CTCCCAAAGTCGCTCTGAGTTGTTATCA-3′, 5′-CGATTTGTGGTGTATGTAACTAATCTGTCTGG-3′ and 5′- GCAGTGAGAAGAGTACCACCATGAGTCC-3′ respectively.
Bleomycin-induced pulmonary fibrosis
Aged (78–82 weeks older) male and female C57BL/6J mice received intratracheal bleomycin (1 U/kg body weight) (Catalog # 203401; EMD Millipore, Burlington, Massachusetts, USA) or normal saline as previously described.56 To study the molecular signaling of IGFBP2, aged (36 weeks or greater) human-Igfbp2 transgenic mice and corresponding littermates (flox) mice received intratracheal instillation of bleomycin (0.75 U/kg body weight) or saline. After the instillation, the anesthetized mice were kept on a warm bed for recovery. The experimental animals were monitored daily for adverse clinical signs, including body weight, appearance, hydration status, and any behavioral changes.
Adenoviral TGFβ1 induced pulmonary fibrosis
Aged (78–82 weeks older) male and female C57BL/6J mice received intratracheal null adenovirus (Catalog # 1240; Vector Biolabs) or adenovirus-TGFβ1 (Catalog # ADV-274099; Vector Biolabs) as previously described.57 Mice received 5.0 × 108 pfu of adenovirus-TGFβ1 or adenovirus-null and were sacrificed by day 28 under ketamine/xylazine anesthesia.
Intranasal instillation of recombinant IGFBP2 protein, in vivo
Single dose of normal saline or bleomycin was administered intratracheally to each group of aged mice. Following bleomycin injury, recombinant IGFBP2 protein (Catalog# 797-B2-025; R&D Systems), containing surfactant (Curosurf®; Chiesi Farmaceutici, Italy) was delivered intranasally every 3 days and euthanized 14 and 28 days later. Curosurf was used as a delivery vehicle to AEC2 cells in the context of lung injury.58
Isolation of primary murine or human AEC2 cells
AEC2 cells were isolated from murine or human lungs as previously described with slight modifications.59 Briefly, Sftpc-Cre-ERT2-human-IGFBP2flox transgenic mice and corresponding littermate’s mice were sacrificed after 14 days of bleomycin treatment, and the lung was perfused with 30 ml of saline to remove the blood. Either murine or human lungs were digested using mixture of 1 mg/ml of collagenase I and 5 U/ml of Dispase at 37°C for 25 minutes. Lung cell suspensions were passed through 400 μm filters (Pluriselect, USA) and neutralized by equal volume of 20% FBS/DMEM (Invitrogen, USA) containing 200 U/mL DNase (Catalog # D-4527, Sigma-Aldrich). First, suspended cells were separated using CD45 MACS cell separation magnetic beads (Catalog # 130-052-301, Miltenyi Biotec). Subsequent cell suspensions were sequentially filtered through 100-, 40-, 20 μm filters (Pluriselect, USA). After filtration, cells were in MACS buffer and separated by EpCAM magnetic beads (catalog #130-105-958, Miltenyi Biotec). The resulting CD45(-) EpCAM(+) population was enriched for AEC2 cells followed by flow cytometry and quantitative PCR analysis.
Short-term exposures of hypoxia and cigarette smoke treatment
Approximately 16 h before the exposure to hypoxia or cigarette smoke (CS) extract, 1 × 106 MLE-12 were seeded on to the 10 cm dish. Subsequently, MLE-12 cells were transferred to the incubation chamber with 0.1% O2 and 5% of CO2 or cigarette smoke (CS) extract (100 μg/ml) at regular incubation chamber for indicated time point exposures. After hypoxia or CS treatments, cells were harvested for downstream experiments.
Long-term exposures of atazanavir and hypoxia treatment
Briefly, 1 × 106 MLE-12 were seeded on to the 10 cm dish. MLE-12 cells were pre-treated with Atazanavir (20 μM) for 1 h and transferred to incubation chamber with 0.1% O2 and 5% of CO2 for 72 h.
Short-term or long-term exposures of bleomycin treatment
Briefly, 1 × 106 MLE-12 cells were incubated with short (4 h) or chronic (6 day; 2 hit) bleomycin (10 μg/ml) exposures as previously described.60 In chronic bleomycin exposure, cells were challenged to two, 24h exposures of low-dose bleomycin on days 1 and 4, followed by 48 hr post-exposure incubation periods (6 days, two 24 h exposures on days 1 and 4). After short or chronic exposures, MLE-12 cells were harvested for downstream experiments.
Cytosolic/ nuclear protein fractionation
1.0 × 107 MLE-12 cells in the 10 cm dish were exposed to the hypoxia condition for 4 and 24 hours, respectively. Cells were harvested, and resuspended with 1x cytosolic extraction buffer (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.05% NP40 pH 7.9). Then, cells were incubated on ice for 10 mins, and lysed with 25 μl Thermo Scientific™ RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific, catalog #89901) with protease and phosphatase inhibitor cocktails (Fisher Scientific, catalog #78445). The supernatant was collected after centrifugation for 10 mins at 800xg. The pellet was washed twice with 1x cytosolic extraction buffer. The pellet was lysed with 50 μl Thermo Scientific™ RIPA Lysis and Extraction Buffer and incubated on ice for 30 minutes by vortexing for 5-minute intervals. The nuclear fraction was collected from the supernatant after the centrifugation for 30 minutes at 14,000xg. The cytosolic/nuclear fraction was detected by immunoblotting assay.
β-galactosidase activity assay
MLE-12 cells were seeded at 8×105 cells per 10 cm dish at 37°C for overnight, and 20μM atazanavir (Sigma-Aldrich, catalog #SML1796-5MG) was added two times at 24 h intervals. Simultaneously, MLE-12 cells were incubated in hypoxia (0.1%) for 96 h. Endogenous β-galactosidase activity was measured by the β-Gal Activity Assay Kit (BioVision, catalog# K821-100). Briefly, MLE-12 cells were lysed using 100 μl ice cold β-Gal assay buffer for 15 min after atazanavir and hypoxia treatments for 96 h. Supernatant was collected after centrifugation at 10,000 X g for 10 min at 4°C. About 10 μl of supernatant was used along with β-Gal substrate and fluorescence was measured using Spectramax i3 fluorometer (Molecular Devices, Inc.) in kinetic mode for 5 – 60 min at 37 oC.
Lentivirus transduction
Briefly, 1x104 cells were mixed with mock or Igfbp2 lentivirus (catalog # MR204287L3V; OriGene Technologies) at MOI=80 in the 500 ml of 10 μg/ml polybrene/DMEM-F12. The mixture was plated on the 24-well plate and centrifuged at 900g for 2 hours at 25°C. After spinfection, the extra 500 μL of 2%FBS/DMEM-F12 were added dropwise onto the cells. The plate was incubated for 48 h at 37°C, and replaced the medium to 2% FBS/DMEM-F12. 0.5 μg/ml puromycin was used for the selection. The cells were lysed and the IGFBP2 level was estimated by immunoblotting.
Senescence gene expression profiling
Cellular senescence gene expression profiling was performed using RT2 Profiler PCR Array (catalog #330231, Qiagen). AEC2 cells were isolated from the lungs of normal saline or bleomycin treated mice. RNA was extracted as per the manufacturer instructions described in RT2 first strand kit (catalog #330401, Qiagen). cDNA was combined with RT2 SYBR Green qPCR Master Mix (catalog #330520, Qiagen), and ΔΔCT was measured by StepOnePlus™ Real-Time PCR System (catalog #4376600, Thermo Fisher Scientific). The further analysis and fold regulation were calculated based on the ΔΔCT method using the GeneGlobe data analysis web portal (www.qiagen.com/geneglobe) (Figures S8A, S8B, and S9A–S9D). The primers for senescence associated secretory phenotype (SASP) genes were obtained from RealTime Primers (# OS1, realtime primers.com). The list of primer sequences for the quantitative PCR for the SASP genes are listed in key resource table.
Hydroxyproline assay
The collagen content was measured by hydroxyproline assay kit (catalog #STA-675, Cell Biolabs), and the experiment was performed according to the manufacturer’s instructions. Briefly, mouse lung tissues were dried at 60°C overnight, and the dry weights were recorded. The samples were hydrolysed by mixing the 6 N hydrochloric acid at 95°C for 24 hours. Then, a Chloramine T mixture reagent is added to each sample and incubated for 30 minutes at room temperature. Finally, Ehrlich’s reagent is added to each sample and the colorimetric 4-hydroxyproline absorbance was measured at 560 nm using BioTek plate reader (Agilent, United States). Hydroxyproline content in each sample was calculated as ‘μg per mg lung tissue’.
Histology and multicolor immunohistochemistry
Murine lung samples were collected and fixed by using the standard protocols as previously described.61 Briefly, the paraffin-embedded section was cut at 3μm thickness and both histological and immunohistochemical staining was performed by the Leica Bond-Rx automated system (Leica, IL). The following antibodies, IGFBP2 (abcam ab188200, 1:2000), P21 (abcam ab188224, 1:200), p-H2AX (CST 2577, 1:200), PPARα (LSBio LS-C312574, 1: 200) and proSPC (abcam ab211326, 1:2000) were used for immunohistochemistry staining. The green chromogen (Leica DS9913) was used for prosurfactant protein C, while the bond polymer refine system (Leica DS9800) was used for IGFBP2, P21, p-H2AX and PPARα protein expression. Images were acquired by the Nikon Eclipse microscope coupled with NIS-Element imaging software.
Histological digital analyses
Fiji open source image processing software package v1.53c (http://fiji.sc) was used for the quantification of collagen (Sirius red and Mason’s trichrome) measurements. Quantification of lung sections were performed in 10 different fields in a random fashion manner.
Immunoblotting
Total mouse lungs or MLE-12 Cells were lysed by RIPA Lysis Extraction Buffer (catalog #89901; Thermo Fisher Scientific) along with protease and phosphatase inhibitors cocktail (catalog #78445; Thermo Fisher Scientific). Total protein concentration was determined by Pierce BCA protein assay reagent kit (catalog #23227; Thermo Fisher Scientific) according to the manufacturer’s protocol. 40 μg of total protein was loaded and separated by SDS-polyacrylamide gel electrophoresis. The gel was transferred using a Mini Trans-Blot cell (Biorad) to PVDF membrane (catalog #IPVH00010; EMD Millipore). Proteins were detected by mouse anti-IGFBP2 (R&D Systems, catalog #MAB7971), anti-P21 (Abcam, catalog #ab188224), anti-β-actin (catalog #sc-47778HRP; Santa Cruz Biotechnology) and anti-histone H3 (catalog #4499; Cell Signaling Technology). Immunoblots were incubated with SuperSignal™ West Pico or Femto Maximum Sensitivity Substrate (catalog #34095; Thermo Fisher Scientific).
Quantification and statistical analysis
All statistical tests were analyzed using SPSS version 25.0 (IBM Corporation) or GraphPad Prism version 9.0 (GraphPad Software). Statistical analysis was performed using 2-way ANOVA or 1-way ANOVA followed by Tukey’s post-hoc test for multi-group comparisons. Student’s unpaired two-tailed t-test was used to compare two groups. P value of less than 0.05 was considered statistically significant.
Acknowledgments
The authors like to thank Babita Bisht, University of Iowa, for inputs on statistical analyses; Jesse Canez, Norton Thoracic Institute, for helping with genotyping experiments; Valentina Pujadas, Norton Thoracic Institute, for helping with image acquisition; and Kristine Nally for proofreading the manuscript.
Author contributions
A.S. conceived the study. C.C. performed the majority of experiments. A.S. and R.R. performed and contributed to experiments. A.S., C.C., R.R., and K.S. contributed to human data analyses. S.B.W. and M.T.K provided mouse strains. A.S. wrote the manuscript, oversaw analysis, and supervised the entire project. All authors critically revised the manuscript and gave final approval of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: February 13, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.100945.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Meltzer E.B., Noble P.W. Idiopathic pulmonary fibrosis. Orphanet J. Rare Dis. 2008;3:8. doi: 10.1186/1750-1172-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ley B., Collard H.R., King T.E. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011;183:431–440. doi: 10.1164/rccm.201006-0894CI. [DOI] [PubMed] [Google Scholar]
- 3.Nadrous H.F., Pellikka P.A., Krowka M.J., Swanson K.L., Chaowalit N., Decker P.A., Ryu J.H. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest. 2005;128:2393–2399. doi: 10.1378/chest.128.4.2393. [DOI] [PubMed] [Google Scholar]
- 4.Yasui K., Yuda S., Abe K., Muranaka A., Otsuka M., Ohnishi H., Hashimoto A., Takahashi H., Tsuchihashi K., Takahashi H., et al. Pulmonary vascular resistance estimated by Doppler echocardiography predicts mortality in patients with interstitial lung disease. J. Cardiol. 2016;68:300–307. doi: 10.1016/j.jjcc.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 5.Janssens J.P., Pache J.C., Nicod L.P. Physiological changes in respiratory function associated with ageing. Eur. Respir. J. 1999;13:197–205. doi: 10.1034/j.1399-3003.1999.13a36.x. [DOI] [PubMed] [Google Scholar]
- 6.Katzenstein A.L.A. Pathogenesis of “fibrosis” in interstitial pneumonia: an electron microscopic study. Hum. Pathol. 1985;16:1015–1024. doi: 10.1016/s0046-8177(85)80279-3. [DOI] [PubMed] [Google Scholar]
- 7.Winters N.I., Burman A., Kropski J.A., Blackwell T.S. Epithelial injury and dysfunction in the pathogenesis of idiopathic PulmonaryFibrosis. Am. J. Med. Sci. 2019;357:374–378. doi: 10.1016/j.amjms.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schafer M.J., White T.A., Iijima K., Haak A.J., Ligresti G., Atkinson E.J., Oberg A.L., Birch J., Salmonowicz H., Zhu Y., et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017;8:14532. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He S., Sharpless N.E. Senescence in health and disease. Cell. 2017;169:1000–1011. doi: 10.1016/j.cell.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fragkos M., Jurvansuu J., Beard P. H2AX is required for cell cycle arrest via the p53/p21 pathway. Mol. Cell Biol. 2009;29:2828–2840. doi: 10.1128/MCB.01830-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galanos P., Vougas K., Walter D., Polyzos A., Maya-Mendoza A., Haagensen E.J., Kokkalis A., Roumelioti F.M., Gagos S., Tzetis M., et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016;18:777–789. doi: 10.1038/ncb3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yosef R., Pilpel N., Papismadov N., Gal H., Ovadya Y., Vadai E., Miller S., Porat Z., Ben-Dor S., Krizhanovsky V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017;36:2280–2295. doi: 10.15252/embj.201695553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campisi J., d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 14.Luo C., Zhou S., Zhou Z., Liu Y., Yang L., Liu J., Zhang Y., Li H., Liu Y., Hou F.F., Zhou L. Wnt9a promotes renal fibrosis by accelerating cellular senescence in tubular epithelial cells. J. Am. Soc. Nephrol. 2018;29:1238–1256. doi: 10.1681/ASN.2017050574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogrodnik M., Miwa S., Tchkonia T., Tiniakos D., Wilson C.L., Lahat A., Day C.P., Burt A., Palmer A., Anstee Q.M., et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017;8:15691. doi: 10.1038/ncomms15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allard J.B., Duan C. IGF-binding proteins: why do they exist and why are there so many? Front. Endocrinol. 2018;9:117. doi: 10.3389/fendo.2018.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ewald C.Y., Landis J.N., Porter Abate J., Murphy C.T., Blackwell T.K. Dauer-independent insulin/IGF-1-signalling implicates collagen remodelling in longevity. Nature. 2015;519:97–101. doi: 10.1038/nature14021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sureshbabu A., Tonner E., Flint D.J. Insulin-like growth factor binding proteins and mammary gland development. Int. J. Dev. Biol. 2011;55:781–789. doi: 10.1387/ijdb.113364as. [DOI] [PubMed] [Google Scholar]
- 19.Bach L.A. IGF-binding proteins. J. Mol. Endocrinol. 2018;61:T11–T28. doi: 10.1530/JME-17-0254. [DOI] [PubMed] [Google Scholar]
- 20.Frommer K.W., Reichenmiller K., Schutt B.S., Hoeflich A., Ranke M.B., Dodt G., Elmlinger M.W. IGF-independent effects of IGFBP-2 on the human breast cancer cell line Hs578T. J. Mol. Endocrinol. 2006;37:13–23. doi: 10.1677/jme.1.01955. [DOI] [PubMed] [Google Scholar]
- 21.Sureshbabu A., Okajima H., Yamanaka D., Tonner E., Shastri S., Maycock J., Szymanowska M., Shand J., Takahashi S.I., Beattie J., et al. IGFBP5 induces cell adhesion, increases cell survival and inhibits cell migration in MCF-7 human breast cancer cells. J. Cell Sci. 2012;125:1693–1705. doi: 10.1242/jcs.092882. [DOI] [PubMed] [Google Scholar]
- 22.Byun H.O., Lee Y.K., Kim J.M., Yoon G. From cell senescence to age-related diseases: differential mechanisms of action of senescence-associated secretory phenotypes. BMB Rep. 2015;48:549–558. doi: 10.5483/bmbrep.2015.48.10.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim K.S., Kim M.S., Seu Y.B., Chung H.Y., Kim J.H., Kim J.R. Regulation of replicative senescence by insulin-like growth factor-binding protein 3 in human umbilical vein endothelial cells. Aging Cell. 2007;6:535–545. doi: 10.1111/j.1474-9726.2007.00315.x. [DOI] [PubMed] [Google Scholar]
- 24.Kim K.S., Seu Y.B., Baek S.H., Kim M.J., Kim K.J., Kim J.H., Kim J.R. Induction of cellular senescence by insulin-like growth factor binding protein-5 through a p53-dependent mechanism. Mol. Biol. Cell. 2007;18:4543–4552. doi: 10.1091/mbc.e07-03-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gester F., Henket M., Deseny D., Moermans C., André B., Malaise M., Louis R., Guiot J. IGFBP-2: a new pathway in systemic sclerosis associated interstitial lung disease. Eur. Respir. J. 2019;54 doi: 10.1183/13993003.congress-2019.OA3597. [DOI] [Google Scholar]
- 26.Kammel A., Saussenthaler S., Jähnert M., Jonas W., Stirm L., Hoeflich A., Staiger H., Fritsche A., Häring H.U., Joost H.G., et al. Early hypermethylation of hepatic Igfbp2 results in its reduced expression preceding fatty liver in mice. Hum. Mol. Genet. 2016;25:2588–2599. doi: 10.1093/hmg/ddw121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wittenbecher C., Ouni M., Kuxhaus O., Jähnert M., Gottmann P., Teichmann A., Meidtner K., Kriebel J., Grallert H., Pischon T., et al. Insulin-like growth factor binding protein 2 (IGFBP-2) and the risk of developing type 2 diabetes. Diabetes. 2019;68:188–197. doi: 10.2337/db18-0620. [DOI] [PubMed] [Google Scholar]
- 28.Rangarajan S., Bone N.B., Zmijewska A.A., Jiang S., Park D.W., Bernard K., Locy M.L., Ravi S., Deshane J., Mannon R.B., et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat. Med. 2018;24:1121–1127. doi: 10.1038/s41591-018-0087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacob A., Vedaie M., Roberts D.A., Thomas D.C., Villacorta-Martin C., Alysandratos K.D., Hawkins F., Kotton D.N. Derivation of self-renewing lung alveolar epithelial type II cells from human pluripotent stem cells. Nat. Protoc. 2019;14:3303–3332. doi: 10.1038/s41596-019-0220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang H.S., Cho H.C., Lee J.H., Oh G.T., Koo S.H., Park B.H., Lee I.K., Choi H.S., Song D.K., Im S.S. Metformin stimulates IGFBP-2 gene expression through PPARalpha in diabetic states. Sci. Rep. 2016;6:23665. doi: 10.1038/srep23665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin M., Kang H.S., Park J.H., Bae J.H., Song D.K., Im S.S. Recent insights into insulin-like growth factor binding protein 2 transcriptional regulation. Endocrinol. Metab. 2017;32:11–17. doi: 10.3803/EnM.2017.32.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raghu G., Amatto V.C., Behr J., Stowasser S. Comorbidities in idiopathic pulmonary fibrosis patients: a systematic literature review. Eur. Respir. J. 2015;46:1113–1130. doi: 10.1183/13993003.02316-2014. [DOI] [PubMed] [Google Scholar]
- 33.Yao C., Guan X., Carraro G., Parimon T., Liu X., Huang G., Mulay A., Soukiasian H.J., David G., Weigt S.S., et al. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2021;203:707–717. doi: 10.1164/rccm.202004-1274OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakai N., Tager A.M. Fibrosis of two: epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta. 2013;1832:911–921. doi: 10.1016/j.bbadis.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun W., Jing X., Yang X., Huang H., Luo Q., Xia S., Wang P., Wang N., Zhang Q., Guo J., Xu Z. Regulation of the IGF1 signaling pathway is involved in idiopathic pulmonary fibrosis induced by alveolar epithelial cell senescence and core fucosylation. Aging (Albany NY) 2021;13:18852–18869. doi: 10.18632/aging.203335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sureshbabu A., Tonner E., Allan G.J., Flint D.J. Relative roles of TGF-beta and IGFBP-5 in idiopathic pulmonary fibrosis. Pulm. Med. 2011;2011:517687. doi: 10.1155/2011/517687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yasuoka H., Zhou Z., Pilewski J.M., Oury T.D., Choi A.M.K., Feghali-Bostwick C.A. Insulin-like growth factor-binding protein-5 induces pulmonary fibrosis and triggers mononuclear cellular infiltration. Am. J. Pathol. 2006;169:1633–1642. doi: 10.2353/ajpath.2006.060501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guiot J., Bondue B., Henket M., Corhay J.L., Louis R. Raised serum levels of IGFBP-1 and IGFBP-2 in idiopathic pulmonary fibrosis. BMC Pulm. Med. 2016;16:86. doi: 10.1186/s12890-016-0249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun Y.L., Hurley K., Villacorta-Martin C., Huang J., Hinds A., Gopalan K., Caballero I.S., Russo S.J., Kitzmiller J.A., Whitsett J.A., et al. Heterogeneity in human induced pluripotent stem cell-derived alveolar epithelial type II cells revealed with ABCA3/SFTPC reporters. Am. J. Respir. Cell Mol. Biol. 2021;65:442–460. doi: 10.1165/rcmb.2020-0259OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lähnemann D., Köster J., Szczurek E., McCarthy D.J., Hicks S.C., Robinson M.D., Vallejos C.A., Campbell K.R., Beerenwinkel N., Mahfouz A., et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020;21:31. doi: 10.1186/s13059-020-1926-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hernandez D.M., Kang J.H., Choudhury M., Andrianifahanana M., Yin X., Limper A.H., Leof E.B. IPF pathogenesis is dependent upon TGFbeta induction of IGF-1. Faseb. J. 2020;34:5363–5388. doi: 10.1096/fj.201901719RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rana T., Jiang C., Liu G., Miyata T., Antony V., Thannickal V.J., Liu R.M. PAI-1 regulation of TGF-beta1-induced alveolar type II cell senescence, SASP secretion, and SASP-mediated activation of alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 2020;62:319–330. doi: 10.1165/rcmb.2019-0071OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meiners S., Eickelberg O., Königshoff M. Hallmarks of the ageing lung. Eur. Respir. J. 2015;45:807–827. doi: 10.1183/09031936.00186914. [DOI] [PubMed] [Google Scholar]
- 44.Hohmann M.S., Habiel D.M., Coelho A.L., Verri W.A., Hogaboam C.M. Quercetin enhances ligand-induced apoptosis in senescent idiopathic pulmonary fibrosis fibroblasts and reduces lung fibrosis in vivo. Am. J. Respir. Cell Mol. Biol. 2019;60:28–40. doi: 10.1165/rcmb.2017-0289OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lehmann M., Korfei M., Mutze K., Klee S., Skronska-Wasek W., Alsafadi H.N., Ota C., Costa R., Schiller H.B., Lindner M., et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur. Respir. J. 2017;50:1602367. doi: 10.1183/13993003.02367-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okuda R., Aoshiba K., Matsushima H., Ogura T., Okudela K., Ohashi K. Cellular senescence and senescence-associated secretory phenotype: comparison of idiopathic pulmonary fibrosis, connective tissue disease-associated interstitial lung disease, and chronic obstructive pulmonary disease. J. Thorac. Dis. 2019;11:857–864. doi: 10.21037/jtd.2019.02.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamei J., Toyofuku T., Hori M. Negative regulation of p21 by beta-catenin/TCF signaling: a novel mechanism by which cell adhesion molecules regulate cell proliferation. Biochem. Biophys. Res. Commun. 2003;312:380–387. doi: 10.1016/j.bbrc.2003.10.129. [DOI] [PubMed] [Google Scholar]
- 48.Verma B.K., Kondaiah P. Regulation of beta-catenin by IGFBP2 and its cytoplasmic actions in glioma. J. Neuro Oncol. 2020;149:209–217. doi: 10.1007/s11060-020-03596-4. [DOI] [PubMed] [Google Scholar]
- 49.Azar W.J., Zivkovic S., Werther G.A., Russo V.C. IGFBP-2 nuclear translocation is mediated by a functional NLS sequence and is essential for its pro-tumorigenic actions in cancer cells. Oncogene. 2014;33:578–588. doi: 10.1038/onc.2012.630. [DOI] [PubMed] [Google Scholar]
- 50.Jiang C., Liu G., Luckhardt T., Antony V., Zhou Y., Carter A.B., Thannickal V.J., Liu R.M. Serpine 1 induces alveolar type II cell senescence through activating p53-p21-Rb pathway in fibrotic lung disease. Aging Cell. 2017;16:1114–1124. doi: 10.1111/acel.12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Q., Tang L., Roberts P.C., Kraniak J.M., Fridman A.L., Kulaeva O.I., Tehrani O.S., Tainsky M.A. Interferon regulatory factors IRF5 and IRF7 inhibit growth and induce senescence in immortal Li-Fraumeni fibroblasts. Mol. Cancer Res. 2008;6:770–784. doi: 10.1158/1541-7786.MCR-07-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beach T.A., Groves A.M., Johnston C.J., Williams J.P., Finkelstein J.N. Recurrent DNA damage is associated with persistent injury in progressive radiation-induced pulmonary fibrosis. Int. J. Radiat. Biol. 2018;94:1104–1115. doi: 10.1080/09553002.2018.1516907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feringa F.M., Raaijmakers J.A., Hadders M.A., Vaarting C., Macurek L., Heitink L., Krenning L., Medema R.H. Persistent repair intermediates induce senescence. Nat. Commun. 2018;9:3923. doi: 10.1038/s41467-018-06308-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huaux F., Liu T., McGarry B., Ullenbruch M., Phan S.H. Dual roles of IL-4 in lung injury and fibrosis. J. Immunol. 2003;170:2083–2092. doi: 10.4049/jimmunol.170.4.2083. [DOI] [PubMed] [Google Scholar]
- 55.Izbicki G., Or R., Christensen T.G., Segel M.J., Fine A., Goldstein R.H., Breuer R. Bleomycin-induced lung fibrosis in IL-4-overexpressing and knockout mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2002;283:L1110–L1116. doi: 10.1152/ajplung.00107.2002. [DOI] [PubMed] [Google Scholar]
- 56.Helms M.N., Torres-Gonzalez E., Goodson P., Rojas M. Direct tracheal instillation of solutes into mouse lung. J. Vis. Exp. 2010;29:1941. doi: 10.3791/1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bellaye P.S., Shimbori C., Upagupta C., Sato S., Shi W., Gauldie J., Ask K., Kolb M. Lysyl oxidase-like 1 protein deficiency protects mice from adenoviral transforming growth factor-beta1-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2018;58:461–470. doi: 10.1165/rcmb.2017-0252OC. [DOI] [PubMed] [Google Scholar]
- 58.Das P., Curstedt T., Agarwal B., Prahaladan V.M., Ramirez J., Bhandari S., Syed M.A., Salomone F., Casiraghi C., Pelizzi N., Bhandari V. Small molecule inhibitor adjuvant surfactant therapy attenuates ventilator- and hyperoxia-induced lung injury in preterm rabbits. Front. Physiol. 2020;11:266. doi: 10.3389/fphys.2020.00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sureshbabu A., Syed M., Das P., Janér C., Pryhuber G., Rahman A., Andersson S., Homer R.J., Bhandari V. Inhibition of regulatory-associated protein of mechanistic target of rapamycin prevents hyperoxia-induced lung injury by enhancing autophagy and reducing apoptosis in neonatal mice. Am. J. Respir. Cell Mol. Biol. 2016;55:722–735. doi: 10.1165/rcmb.2015-0349OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shaghaghi H., Cuevas-Mora K., Para R., Tran C., Roque W., Robertson M.J., Rosas I.O., Summer R., Romero F. A model of the aged lung epithelium in idiopathic pulmonary fibrosis. Aging (Albany NY) 2021;13:16922–16937. doi: 10.18632/aging.203291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sureshbabu A., Patino E., Ma K.C., Laursen K., Finkelsztein E.J., Akchurin O., Muthukumar T., Ryter S.W., Gudas L., Choi A.M.K., Choi M.E. RIPK3 promotes sepsis-induced acute kidney injury via mitochondrial dysfunction. JCI Insight. 2018;3:e98411. doi: 10.1172/jci.insight.98411. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.