

Summary
Dysregulated maternal fatty acid metabolism increases the risk of congenital heart disease (CHD) in offspring with an unknown mechanism, and the effect of folic acid fortification in preventing CHD is controversial. Using gas chromatography coupled to either a flame ionization detector or mass spectrometer (GC-FID/MS) analysis, we find that the palmitic acid (PA) concentration increases significantly in serum samples of pregnant women bearing children with CHD. Feeding pregnant mice with PA increased CHD risk in offspring and cannot be rescued by folic acid supplementation. We further find that PA promotes methionyl-tRNA synthetase (MARS) expression and protein lysine homocysteinylation (K-Hcy) of GATA4 and results in GATA4 inhibition and abnormal heart development. Targeting K-Hcy modification by either genetic ablation of Mars or using N-acetyl-L-cysteine (NAC) decreases CHD onset in high-PA-diet-fed mice. In summary, our work links maternal malnutrition and MARS/K-Hcy with the onset of CHD and provides a potential strategy in preventing CHD by targeting K-Hcy other than folic acid supplementation.
Keywords: congenital heart disease, maternal fatty acids, palmitic acid, methionyl-tRNA synthetase, protein lysine homocysteinylation, GATA4
Graphical abstract
Highlights
-
•
Maternal high palmitic acid is associated with increased risk of CHD in offspring
-
•
Palmitic acid promotes MARS and K-Hcy modification through activating NF-κB
-
•
GATA4 transcriptional acitivity is inhibited by K-Hcy modification
-
•
Inhibition of K-Hcy rescued PA-induced CHD occurrence in mice
Zhao et al. report that elevated maternal serum palmitic acid (PA) is associated with the risk of congenital heart disease (CHD) in offspring. PA induces CHD phenotypes in mice via promoting K-Hcy of GATA4, therefore inhibiting its transcriptional activity. Inhibition of K-Hcy rescued PA-induced CHD occurrence.
Introduction
Congenital heart disease (CHD), the most common congenital human birth defect, affects 9.1 per 1,000 live births worldwide.1 CHD onset is influenced by both genetic and nutritional factors.2 The periconceptional administration of folic acid, which is a synthetic form of folate, is the most effective method for CHD prevention. Epidemiological studies have shown that folic acid fortification reduces the risk of CHD in newborns,3,4,5 whereas exposure to folic acid antagonists during pregnancy increases the risk of CHD in offspring.6
Our previous genetic and biochemical studies revealed that folic acid supplementation may mainly reduce homocysteine levels in cells, the elevation of which causes CHD. Genetic noncoding variations in homocysteine removal genes, including methionine synthase (MTR), MTR reductase (MTRR), and cystathionine β-synthase (CBS), decrease the expression of these genes and lead to the accumulation of homocysteine, thereby increasing the risk of CHD.7,8,9 Moreover, increased proteasome activity-mediated folate transmembrane transport decreases the risk of CHD by reducing homocysteine levels.10 With regards to the mechanisms underlying the teratogenic effects of homocysteine, we found that increased homocysteine levels could be sensed by methionyl-tRNA synthetase (MARS), leading to the lysine homocysteinylation (K-Hcy) modification of proteins and generation of K-Hcy signals.11 Increased K-Hcy levels inhibit developmental signals and lead to birth defects. In human genetic studies, we observed increased copy numbers of MARS and/or MARS2 in patients with CHD, validating the pathological effects of K-Hcy.12 These findings suggest that teratogenic K-Hcy levels are determined by both the substrate homocysteine and enzyme MARS.13 Although K-Hcy could be alleviated by lowering homocysteine levels using folic acid supplementation, which is performed globally, the incidence of CHD has increased significantly in recent years due to unknown reasons.1,9
In addition to folate-homocysteine balance, dysregulated maternal lipid metabolism contributes to the risk of CHD in offspring. For example, maternal obesity is associated with numerous pregnancy-related adverse outcomes, including an increased risk of congenital septal anomalies and neural tube defects.2,4,14,15,16,17 In addition, high maternal triglyceride (TG) levels during early pregnancy are associated with an increased risk of CHD in offspring.18 A high-fat diet during pregnancy also increases this risk, as shown by human cohort19 and animal model studies.20,21 Considering that fatty acids play a central role in embryonic development,22,23 these findings suggest that increased maternal circulating fatty acids may increase the risk of CHD in offspring by changing the developmental environment of the embryo. However, the mechanisms by which increased fatty acids generate teratogenic signals are unknown.
We previously found that a high-fat diet activates MARS expression in several organs of mice, suggesting a potential relationship between fatty acids and K-Hcy signals.24 Considering that a high-fat diet is correlated with dysregulated MARS and K-Hcy levels and that free fatty acids (FFAs) have a signaling role in cells, we hypothesized that imbalanced maternal fatty acids may affect embryonic MARS expression, amplify the signal from homocysteine independent of folate or folic acid effects, and increase the risk of CHD in offspring. In this study, we aimed to identify CHD-related FFAs in serum samples obtained from pregnant women. In addition, we used in vitro assays, cell cultures, and mouse models to investigate how imbalanced FFA levels increase the risk of CHD by attenuating the CHD-protective effect of folate.
Results
Increased serum palmitic acid levels during early pregnancy are associated with increased CHD risk in offspring
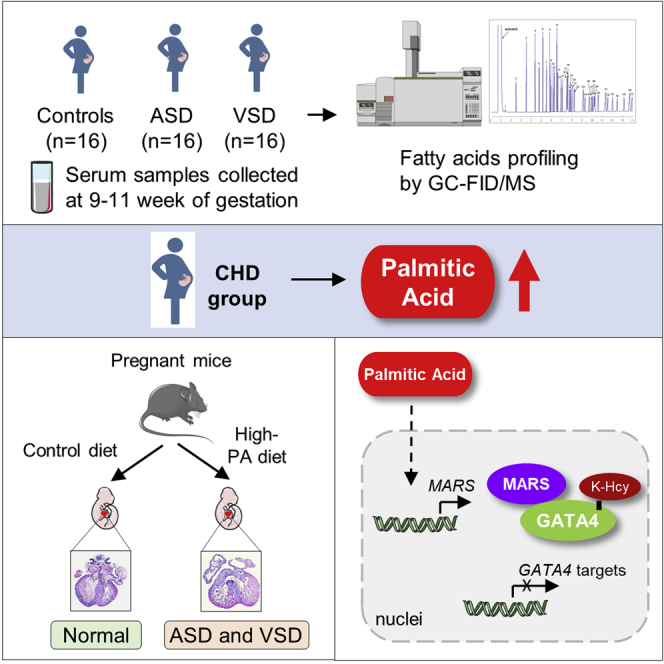
To investigate whether FFAs were associated with the risk of CHD, we screened FFA levels in the serum samples of 32 pregnant women bearing children with CHD and 16 pregnant women bearing healthy children (Figure S1A). The screening was performed using gas chromatography coupled to either a flame ionization detector or mass spectrometer (GC-FID/MS). In total, 11 types of FFAs and total FFA levels were successfully determined (Table S1; Figure S1B). In addition, the levels of 13 nutrients were measured using conventional clinical methods, including established CHD-related maternal nutrition factors, such as TGs, folate, vitamin B12, and homocysteine (Table S2). We compared the serum concentrations of maternal metabolites between pregnant women bearing children with CHD and those bearing healthy children and found that the levels of FFA types and total FFA increased significantly in pregnant women bearing children with CHD (Table S1; Figures S1C–S1M).
Two statistical models, including the two-tailed t test and ANOVA F test, were used to analyze 25 types of serum nutrients, including 11 types of FFAs, total FFA levels, and 13 nutrients measured using conventional clinical methods, to identify significant features (Figure 1A). The association was assumed to be significant after Bonferroni correction at p < 2.0 × 10−3 (0.05/25). C16:0, C18:0, C18:1n9, C18:2n6, C20:3n6, and FFAs were significant after t tests with Bonferroni corrections. All FFAs showed strong correlations with each other (Pearson’s correlation coefficient > 0.8; Figure S1N). After combining the F tests and performing Bonferroni corrections, maternal palmitic acid (PA; C16:0), oleic acid (C18:1n9), and total FFA levels were significant. A permutation test performed by randomly shuffling the values based on the random forest model indicated that PA, total FFA, and eicosatrienoic acid (C20:3n6) were the top three important fatty acid types (Figure 1B). Maternal PA and total FFA showed a strong correlation (Pearson’s correlation coefficient = 0.99, p = 1.2 × 10−40; Figure 1C) and were the two most promising fatty acid types associated with CHD risk.
Figure 1.

Increased serum PA levels during early pregnancy are associated with increased CHD risk in offspring
(A) FFA evidence of association analysis for CHD in offspring using t tests and ANOVA F tests. The x axis shows the different FFA levels, and the y axis represents the −log10 p values using the two different models. The dashed horizontal red line indicates the 5% Bonferroni threshold.
(B) Permutation importance showed PA (C16:0) as the most important variable in the random forest model.
(C) Pearson’s correlation coefficient was used to correlate the total FFA (y) and C16:0 (x). The Pearson’s correlation coefficient was r = 0.97.
Elevated PA levels in pregnant mice induce the onset of CHD
We explored whether increased levels of maternal PA indeed caused, and were not just correlated with, CHD in offspring. We assessed the teratogenic effects of PA in a mouse model fed with a high-PA diet. Eight-week-old C57BL/6J female mice were fed high-PA chow prepared by supplementing standard chow diet (CD) with either 0.5% or 1% PA. The ratio of PA to normal chow was optimized to ensure that increased circulating PA levels were comparable to those observed in clinical patients. CD supplemented with 1% PA (10 g/kg added to the standard CD) induced a 50% increase in PA concentration in mouse serum, a rise that mimicked the PA levels observed in clinical patients with CHD compared with the control group (Figure S2A). We also established mouse models with high levels of other fatty acids, which were also increased in the pregnant women bearing children with CHD. These included oleic acid (C18:1n9; Figure S2B), linoleic acid (C18:2n6; Figure S2C), stearic acid (C18:0; Figure S2D), and eicosatrienoic acid (C20:3n6; Figure S2E). Pregnant mice were administered high-fatty-acid chow or normal chow from embryonic day 0.5 (E0.5) to E13.5. Next, the cardiac phenotypes of the embryos at E14.5 were examined using histological analyses. During the feeding period, the high-PA chow did not affect the food intake, water intake, body weight, blood pressure, pulse, blood glucose, glucose tolerance, and fat mass of pregnant mice (Figures S2F–S2N). Moreover, no differences were observed in the total number of fetuses and incidence of absorbed fetuses between the different groups of pregnant mice (Figures S2O and S2P). However, ventricular septal defect (VSD) and atrial septal defect (ASD) phenotypes were observed in the hearts of embryos obtained from high-PA-diet-fed female mice (Figure 2A). The proportion of pregnant mice with offspring having CHD increased from 25% (3 out of 12) in the control group to 100% (12 out of 12) in the high-PA-feeding group (Figure 2B). The CHD occurrence ratio for all offspring significantly increased from 2.97% in the control group (3 in 101) to 31.96% (31 in 97) in the high-PA-diet-fed group, indicating a statistically significant difference between these groups (p = 6.4 × 10−8, relative risk [RR] = 10.78, 95% confidence interval [CI] = 3.40–34.04; Figure 2C). We also evaluated the gender of the embryos and found no differences in the embryos with CHD (Figure S2Q). In contrast, in the mice administered other kinds of high-fatty-acid chow, such as oleic acid, stearic acid, eicosatrienoic acid, or linoleic acid, the incidence of CHD in the offspring did not increase (Figures 2B and 2C). These results suggest that increased maternal PA levels affect heart development and increase the risk of CHD in mouse embryos.
Figure 2.

Elevated PA levels in pregnant mice induced CHD onset
(A) CHD phenotypes in embryos from mice with high levels of PA. Representative histological staining results are shown (scale bar, 500 μm).
(B) Proportion of pregnant mice bearing CHD offspring after being fed normal chow and fatty-acid-rich chow (n = 12 mice in the normal and PA groups; n = 10 mice in other groups).
(C) Incidence of CHD in embryos when pregnant mice were fed normal chow or fatty-acid-rich chow. Significance was calculated using a one-way ANOVA.
(D) MARS expression and K-Hcy levels in the heart tissues of mouse embryos were detected by western blotting (n = 6 per group).
(E) Homocysteine levels in the heart tissues of mouse embryos did not change in the high-PA-diet group (n = 7 per group).
(F) HTL levels in the heart tissues of mouse embryos were elevated in the high-PA-diet group (n = 7 per group).
The data are expressed as mean ± SEM. ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05, non-significant (ns)p > 0.05 using unpaired Student’s t tests.
See also Figure S1.
PA levels are associated with increased MARS expression and protein K-Hcy
We explored how increased maternal PA levels led to the occurrence of CHD in offspring. High-fat diets are known to increase the risk of CHD in offspring.21,25 The findings of our previous study showed that a high-fat diet activated MARS expression and amplified the signal from homocysteine to cause protein K-Hcy modification.24 Therefore, we hypothesized that PA alone was sufficient to activate MARS, as PA is the predominant form of fatty acids in the high-fat diet (https://researchdiets.com/). By comparing homocysteine, MARS, and K-Hcy levels in mouse heart tissues between embryos from maternal high-PA-diet-fed mice and maternal normal CD-fed mice, we found that MARS expression and K-Hcy levels (Figure 2D), but not homocysteine levels (Figure 2E), were notably increased in the heart tissues of embryos from maternal high-PA-diet-fed mice. As the active form of Hcy in generating K-Hcy modification catalyzed by MARS, homocysteine thiolactone (HTL) levels were significantly increased in embryonic hearts of maternal high-PA-diet-fed mice (Figure 2F). Collected results suggested that PA may activate MARS to generate HTL and promote pan-K-Hcy modification.
Thereafter, we confirmed whether PA activated MARS expression by conducting experiments using cultured cells. We found that, in the cultured mouse cardiac muscle cell line HL-1, rat myoblast cell line H9C2, human embryonic kidney-derived HEK293T cells, and primary neonatal rat ventricular myocytes (NRVMs), elevated PA levels strongly activated both the mRNA (Figure 3A) and protein expression (Figure 3B) of MARS. However, other common FFAs did not cause a significant increase in MARS expression (Figures 3A and 3B). Moreover, elevated PA levels led to dose- and time-dependent increases in MARS expression and pan-K-Hcy levels in HL-1, H9C2, HEK293T, and NRVM cells (Figures 3C and 3D). Folic acid supplementation in the culture media did not reduce pan-K-Hcy levels in PA-treated cells (Figure 3E). Instead, supplementation with N-acetyl-L-cysteine (NAC), a well-known antioxidant that also decreased HTL formation (Figure S3A), and MARS enzymatic inhibitor acetyl Hcy thioether (AHT)12 decreased pan-K-Hcy levels and blocked the effects of PA in promoting K-Hcy (Figures 3E and 3F). These results indicated that PA increased K-Hcy levels by activating MARS expression. To examine whether PA abrogated folate effects by regulating folate metabolism, we measured expression of key folate metabolism enzymes by qRT-PCR in both mice models and HL-1 cells, and it was found that neither Rfc1, Dhfr, Mtr, or Mthfr was changed after PA treatment, which conclude the possibility that PA regulates folate metabolism (Figures S3B and S3C). In addition, supplementation with cycloheximide, an RNA synthesis inhibitor, blocked the effects of PA on increasing the MARS protein levels (Figure 3G). Considering that the changing MARS mRNA patterns were in accordance with its protein levels (see Figures 3A and 3B), these results indicate that PA increases the transcription levels of MARS.
Figure 3.

PA increased MARS expression and protein lysine homocysteinylation
(A and B) PA, but not other fatty acids, strongly activated MARS mRNA expression (A) and protein expression (B) in HL-1, H9C2, HEK293T, and NRVM cells. Cells were treated with fatty acids for 9 h before harvesting (n = 4 biological replicates per group).
(C) Elevated PA levels led to a dose-dependent increase in MARS expression in HL-1, H9C2, HEK293T, and NRVM cells. Cells were treated with PA for 9 h before harvesting. CBB, Coomassie brilliant blue.
(D) PA led to time-dependent increases in MARS expression in HL-1, H9C2, HEK293T, and NRVM cells. Cells were treated with PA (0.5 mM) for the indicated times before harvesting. CBB, Coomassie brilliant blue.
(E) NAC, but not folic acid, decreased pan-K-Hcy levels in PA-treated cells. The cells were treated with 0.5 mM PA, 1 mM NAC, and 100 nM folic acid for 9 h before harvesting. CBB, Coomassie brilliant blue.
(F) AHT decreased pan-K-Hcy levels in PA-treated cells. The cells were treated with 0.5 mM PA and 50 μM AHT for 9 h before harvesting.
(G) Cycloheximide (CHX) treatment blocked the effect of PA on MARS expression. Cells were treated with 200 μg/mL CHX and 0.5 mM PA for 9 h before harvesting.
PA increases MARS transcription by activating the NF-κB pathway
To explore how PA activates MARS transcription, we predicted potential transcriptional factors in the MARS promoter regions in humans, mice, and rats using the online Jaspar database (http://jaspar.genereg.net). We found that RELA, MXI1, MEIS2, TCF4, KLF2, and CEBPA have potential binding sites in the promoter region of MARS (Figure S6). The knockdown of RELA, but not of the other transcription factors, led to decreased mRNA levels of MARS in cultured HL-1 and HEK293T cells (Figure S5). Considering that RELA, also known as the nuclear factor κB (NF-κB) p65 subunit, was reported to be activated by PA through Toll-like receptor 4,26 these observations suggest that PA activates MARS expression through RELA. Upon NF-κB activation, phosphorylated RELA translocated to the nucleus and bound to the consensus sequence 5′-GGRNNYYCC-3′ (R = A or G, Y = T or C) located in the promoters or enhancers of the target genes. We first confirmed that PA activated the NF-κB pathway in cultured HEK293T and HL-1 cells (Figure 4A). The knockdown of RELA decreased both the mRNA and protein levels of MARS and blocked the effect of PA in activating MARS expression (Figures 4B and 4C). By conducting in vitro electrophoretic mobility shift assays (EMSAs; Figure 4D) and luciferase assays (Figure 4E), we confirmed that RELA binds to the promoter region of MARS. Moreover, we validated that RELA bound to the MARS promoters in human and mouse cell lines using chromatin immunoprecipitation (Figure 4F). Supplementation of PA in the culture media led to an increased binding affinity between RELA and MARS promoters as more immunoprecipitated promoter fragments were amplified (Figure 4F). These results indicate that PA may activate MARS through the NF-κB pathway. The NF-κB pathway was also activated in the heart tissue of embryos from pregnant mice fed a high-PA diet (Figure 4G). We also performed immunohistochemistry (IHC) analysis for heart sections and confirmed that the NF-κB pathway was activated and K-Hcy was elevated in heart tissue of embryos from pregnant mice fed a high-PA diet (Figure 4H). Taken together, these results suggest that increased PA levels enhance MARS transcription by activating the NF-κB pathway.
Figure 4.

PA increases MARS transcription by activating the NF-κB pathway
(A) PA activated the NF-κB pathway in cultured HEK293T and HL-1 cells. Cells were treated with PA for 4 h before harvesting.
(B) Knockdown of RELA blocked the effect of PA on MARS mRNA expression (n = 5 biological replicates per group).
(C) Knockdown of RELA blocked the effect of PA on activating MARS protein expression.
(D) EMSAs showed RELA bound to the MARS promoter region in three species.
(E) Effects of p65 on the transcription activities of the human MARS promoter in HEK293T cells and mouse Mars promoter in HL-1 cells, determined using dual-luciferase reporter assays (n = 3 biological replicates per group).
(F) Chromatin immunoprecipitation (ChIP)-PCR (top) and ChIP-qPCR (bottom) assays were performed using p65 antibody in both HL-1 and HEK293T cells untreated or treated with PA. Rabbit immunoglobulin G (IgG) antibody was used as the control (n = 4 per group).
(G) The NF-κB pathway was activated in the embryonic heart tissue of maternal high-PA-chow-fed mice, as detected by western blotting (n = 6 mice per group).
(H) IHC staining of pIKKa/β and K-Hcy for embryonic heart sections of mice model. Scale bar, 500 μm (n = 3 mice for chow-diet group, n = 5 mice for high-PA group).
The data are expressed as mean ± SEM. ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05, nsp > 0.05 using unpaired Student’s t tests.
MARS promotes the K-Hcy of GATA4
We investigated the mechanisms underlying the teratogenic effects of K-Hcy on heart development. In our previous studies,12,24 we systematically identified MARS-interacting proteins and K-Hcy-modified protein substrates. However, no transcription factors modified by K-Hcy were identified. We presumed that this was because of their low expression levels. Considering that most of the PA-correlated phenotypes in both clinical patients and the mouse models were septal defects, which were reported to be associated with genetic mutations in transcription factors belonging to families, such as GATA, TBX, and NKX, we tested the interactions between MARS and GATA4, TBX5, NKX2-5, MEF2A, and SRF.
First, we validated that MARS was located in both the cytosol and nucleus (Figure 5A). Next, we screened the potential interactions between MARS and the aforementioned transcription factors using co-immunoprecipitation assays with exogenous proteins. We found that GATA4 (Figure 5B), but not the other CHD-related transcription factors (Figure S6A), interacted with MARS. The interaction between GATA4 and MARS was confirmed by co-immunoprecipitation assays using endogenous GATA4 and MARS in cultured HL-1 cells (Figure 5C) and human ventricular septum samples from patients with CHD who had undergone surgery (Figure 5D). Moreover, the interaction between GATA4 and MARS was validated by the results of tandem affinity purification performed using GATA4 as the bait (Table S3). Our results suggest that changes in the signaling of GATA4, which is involved in septal defect pathogenesis, may contribute to the pathology associated with K-Hcy. Therefore, we used liquid chromatography followed by tandem mass spectrometry (LC-MS/MS) to screen for K-Hcy-modified sites in GATA4. We found that the lysine 300 residue of GATA4 (K300) (K299 in mice and rats) was homocysteinylated in HEK293T cells (Figure 5E). GATA4 K300 was also homocysteinylated in the cardiac tissues of embryos from high-PA-diet-fed mice (Figure S6B).
Figure 5.

MARS promotes lysine-homocysteinylation of GATA4
(A) MARS is located in both the cytosol and nuclei of HL-1, HEK239T, and NRVM cells.
(B) Co-immunoprecipitation assays showed that exogenous MARS interacted with exogenous GATA4 in cultured HL-1 cells.
(C and D) Co-immunoprecipitation assays showed that endogenous MARS interacted with endogenous GATA4 in cultured HL-1 cells (C) and human heart tissue (D).
(E) GATA4 K300 was homocysteinylated, as determined by LC-MS/MS. The MS/MS spectrum of the peptide is shown.
(F) MARS overexpression enhanced the K-Hcy modification of GATA4 in HL-1 cells.
(G) PA did not increase the K-Hcy-modified level of GATA4 in MARS-knockdown HL-1 cells.
The overexpression of MARS increased the K-Hcy-modified level of GATA4 in cultured HL-1 cells (Figure 5F). However, PA did not increase the K-Hcy-modified level of GATA4 in Mars-knockdown HL-1 cells (Figure 5G). Moreover, the K-Hcy-modified level of GATA4 was increased in the embryos from high-PA-diet-fed mice compared with that in embryos from normal-diet-fed mice (Figure 2E). Taken together, these results confirm that PA activates MARS and leads to the increased K-Hcy modification of GATA4.
K-Hcy inactivates GATA4 and impedes its binding to promoters
We explored whether K-Hcy modification affects GATA4 function. The K300 site is evolutionarily conserved across species, from Danio rerio to Homo sapiens (Figure S6C), suggesting that K-Hcy potentially affects GATA4 function. The increased K-Hcy modification of GATA4, induced by increased PA levels, did not alter GATA4 expression at either the mRNA or protein levels (Figures S6D and S6E) and did not affect the nuclear localization of GATA4, as shown by the western blotting analysis (Figure S6F). To better evaluate GATA4 activity under different treatments, we detected the mRNA expression of well-characterized GATA4 transcription-regulated targets, including Nppa, Myh7, Tnni3, and Myl1.27,28,29 The mRNA expression of these targets was significantly decreased in PA-treated HL-1 and H9C2 cells but not in Mars-knockdown HL-1 and H9C2 cells (Figure 6A). The deletion of GATA4 has been reported to inhibit cell proliferation,30 and the downregulation of GATA4 causes cardiomyocyte apoptosis.31 These effects are parallel with the anti-proliferative and pro-apoptotic roles of PA32; therefore, the effects of PA and MARS on cell proliferation and apoptosis were evaluated. Since PA, NF-κB, and GATA4 have all been reported to be associated with cell proliferation and apoptosis, we monitored proliferation and apoptosis in HL-1 and H9C2 cells. PA treatment decreased cell proliferation and promoted apoptosis in HL-1 and H9C2 cells, whereas the knockdown of Mars partially alleviated these effects (Figures S6G and S6H). Proliferation and apoptosis also accounted for the teratogenic role of PA and MARS. Moreover, in Gata4-knockdown HL-1 and H9C2 cells, the expressions of Nppa, Myh7, Tnni3, and Myl1 decreased significantly and were no longer responsive to PA treatment (Figure 6B). In addition to Nppa, Myh7, Tnni3, and Myl1, other Gata4 downstream targets, such as Myl3, Tnnc1, and Nppb,33 were generally regulated by PA in normal, but not Gata4-knockdown, HL-1 and H9C2 cells (Figure S6I). These results indicate that K-Hcy modification inhibits GATA4 activity.
Figure 6.

K-Hcy inactivates GATA4 and impedes its binding to the promoter
(A) PA treatment decreased the mRNA levels of GATA4 downstream targets (Nppa, Myh7, Tnni3, and Myl1) in HL-1 (top) and H9C2 cells (bottom), and this effect was abolished in Mars-knockdown cells (n = 4 biological replicates per group).
(B) PA did not decrease the mRNA levels of GATA4 downstream targets in Gata4-knockdown HL-1 (top) and H9C2 (bottom) cells (n = 4 biological replicates per group).
(C) EMSAs showed that HTL decreased the binding affinity of GATA4 for the Nppa probe. The sequence of the FAM-labeled probe was obtained from the Mus Musculus Nppa promoter.
(D) ChIP-PCR (top) and ChIP-qPCR (bottom) assays showed that PA decreased the binding of GATA4 to the Mus musculus Nppa and Myh7 promoters (n = 5 biological replicates per group).
(E) K-Hcy levels in exogenous wild-type GATA4 and GATA4 K300W mutants.
(F) The EMSA showed that the GATA4 K300W mutant exhibited a lower binding affinity for the Nppa probe than wild-type GATA4.
(G) Overexpression of wild-type GATA4, but not K300W, rescued the effect of PA-mediated cardiac gene suppression in shGATA4 HL-1 and H9C2 cells (n = 4 biological replicates per group).
The data are expressed as mean ± SEM. ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05, nsp > 0.05 using unpaired Student’s t tests.
The K300 site is located close to the GATA4 zinc-finger DNA-binding domain (amino acids 271–295), suggesting that K-Hcy modification impairs the DNA-binding affinity of GATA4 to gene promoters. By using in vitro EMSAs, we confirmed that GATA4 bound to the Nppa promoter region consisting of the GATA4-binding motif “HGATAR” (Figure 6C). However, when we increased the K-Hcy level of GATA4 by adding HTL during EMSA, the binding affinity of GATA4 for the Nppa promoter decreased significantly (Figure 6C). Moreover, we found that although GATA4 bound to the promoter regions of Nppa and Myh7, elevated K-Hcy levels induced by the increased PA levels in the culture media abrogated the binding of GATA4 to these promoter regions in HL-1 cells as revealed by chromatin immunoprecipitation analyses (Figure 6D). Moreover, to mimic the bulky side-chain effects of K-Hcy, we created mutant constructs of GATA4 in which the modifiable lysine was mutated to tryptophan (GATA4 K300W mutant). GATA4 K300W showed diminished K-Hcy levels (Figure 6E). By using immunofluorescence staining in HL-1 cells that stably overexpressed wild-type GATA4 or the GATA4 K300W mutant, we found that the K300W mutant did not exhibit altered nuclear localization (Figure S6J). The EMSAs validated that the binding affinity of the GATA4 K300W mutant to the gene promoter was weaker than that of the wild-type GATA4 (Figure 6F). This notion was further confirmed by the lower expression levels of the downstream targets of GATA4 when overexpressing the GATA4 K300W mutant compared with overexpressing wild-type GATA4 (Figure 6G). By rescuing wild-type GATA4 and GATA4 K300W in shGATA4 HL-1 and H9C2 cells combined with PA treatment, we found that PA suppressed cardiac gene expression in shGATA4+GATA4 wild-type cells. PA treatment did not suppress cardiac gene expression in shGATA4+GATA4 K300W cells, suggesting that K300 was essential for PA-mediated cardiac gene suppression (Figure 6G). These results confirmed that the K-Hcy modification of GATA4 inhibited GATA4 transcriptional activity.
Reduced GATA4 function has previously been reported to be associated with a bias toward endothelial cell fate.34 Therefore, we tested the expressions of the core transcription regulators of endothelial cells (SOX17) and ETS factors (ETS1) in heart tissues from the embryos of high-PA-diet-fed mice. The expressions of SOX17 and ETS1 were increased (Figure 2E), suggesting that endothelial/endocardial programs were upregulated. These phenomena could also provide an explanation for PA-induced CHDs.
Blocking K-Hcy rescues GATA4 function and decreases CHD prevalence in mice with high levels of PA
To further confirm that gestational PA induces CHD in offspring by activating MARS and K-Hcy, we investigated whether depleting MARS could block the teratogenic effects of PA. Eight-week-old wild-type female mice were mated with 8-week-old Mars heterozygous knockout male mice. Pregnant mice were administered high-PA chow from E0.5–E13.5. The cardiac phenotypes of the embryos at E14.5 were examined using histological analyses, and their genotypes were tested using PCR. We found that although high-PA chow induced the occurrence of CHD phenotypes, the incidence of CHD was lower in Mars heterozygous knockout embryos than in wild-type embryos (Figure 7A). The results show that the cardiac K-Hcy of GATA4 decreased; the cardiac expression levels of the downstream targets of GATA4 increased; and the endothelial/endocardial regulators decreased in Mars heterozygous knockout embryos compared with those in wild-type embryos (Figure 7B). Blocking the activation of MARS therefore rescued the teratogenic effects of PA. These findings indicate a potential strategy for preventing high-PA-induced CHD occurrence when MARS is overactivated.
Figure 7.

Blocking K-Hcy rescues GATA4 function and decreases CHD prevalence in mice with high PA levels
(A) Mars heterozygous knockout embryos exhibited decreased CHD incidence compared with wild-type embryos (n = 15 per group).
(B) K-Hcy levels of endogenous GATA4, protein levels of GATA4 downstream targets (NPPA, TNNI3, and MYL1), and endocardial/endothelial factors (ETS1 and SOX17) in the heart tissues of wild-type and Mars+/− embryonic mice. The quantification of blots is shown on the right (n = 6 mice per group).
(C) NAC, but not folic acid, decreased the levels of endogenous GATA4 in HL-1 and HEK293T cells. Cells were treated with 0.5 mM PA, 1 mM NAC, and 100nM folic acid for 9 h before harvesting.
(D) NAC increased the mRNA levels of GATA4 downstream targets in PA-treated HL-1 and HEK293T cells (n = 5 per group). Cells were treated with 0.5 mM PA and 1 mM NAC for 9 h before harvesting.
(E) NAC, but not folic acid, decreased PA-induced CHD incidence (n = 9 mice per group).
(F) Incidence of CHD in embryos from the indicated groups of pregnant mice. Significance was calculated using a one-way ANOVA.
(G) K-Hcy levels of endogenous GATA4, protein levels of GATA4 downstream targets (NPPA, TNNI3, and MYL1), and endocardial/endothelial factors (ETS1 and SOX17) in the heart tissues of embryonic mice with the indicated treatment. The quantification of blots is shown on the right (n = 3 mice per group).
The data are expressed as mean ± SEM. ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05, nsp > 0.05 using unpaired Student’s t tests.
NAC and AHT can inhibit MARS activity and decrease K-Hcy levels in cultured cells. We found that supplementation with NAC and AHT, but not folic acid, decreased the K-Hcy-modified level of GATA4 and abrogated the effect of PA treatment in cultured HL-1 and HEK293T cells (Figures 7C and S7A). In addition, NAC or AHT supplementation increased the expressions of the downstream targets of GATA4 in PA-treated HL-1 and HEK293T cells (Figures 7D and S7B). NAC also alleviated PA-induced apoptosis and the inhibition of proliferation (Figures S7C and S7D). In the high-PA-diet-fed mice, a daily dose of 300 mg/kg NAC, administered from E0.5 to the end of the experiment, significantly decreased the high-PA-diet-induced incidence of CHD. The percentage of pregnant mice bearing offspring with CHD decreased significantly (Figures 7E and 7F), whereas the incidence of CHD in embryos decreased from 29.33% (22 in 75) to 8.64% (7 in 81; Figure 7F). Notably, NAC decreased the levels of GATA4 and activated GATA4 in the embryonic hearts of offspring obtained from high-PA-diet-fed mice (Figure 7G). In contrast, folic acid supplementation during pregnancy did not rescue the above phenotypes caused by high-PA-chow feeding, including the increased K-Hcy modification of GATA4, decreased expression levels of GATA4-regulated targets, increased expression levels of endothelial/endocardial regulators, and increased incidence of CHD in embryos (Figures 7E and 7F). These findings suggest that PA promotes K-Hcy by activating MARS expression and blocking the response to folic acid. This indicates that the inhibition of MARS-mediated signaling under increased circulating PA levels can be used to prevent CHD (Figure 7G).
Discussion
Using clinical samples and in vitro and in vivo models, we showed that increased maternal PA levels amplified teratogenic K-Hcy signals by activating MARS transcription. Increased K-Hcy inhibited embryonic GATA4 signaling and increased the risk of CHD in the offspring. Although we did not determine the levels of PA in the earlier stages of pregnancy, the teratogenic effects of PA were evident in our mouse model. This suggested that increased maternal circulating PA levels from E0.5–E13.5, which included the entire period required for embryonic heart development, led to an elevated risk of CHD in the offspring. We also found that increased maternal PA levels did not induce the onset of CHD in Mars-knockout mice, confirming that PA exerted its teratogenic effects via MARS-mediated K-Hcy signaling.
The K-Hcy modification of proteins, which was determined by both homocysteine concentrations and MARS protein levels, caused the onset of CHD. Our findings indicate that PA activates MARS transcription, suggesting that high maternal levels of PA may abrogate the protective effect of folic acid, as the PA-induced elevation of K-Hcy does not require high levels of homocysteine. That is, under normal PA levels, MARS proceeds normally in translation, and the K-Hcy level is determined by the intracellular homocysteine concentration; hence, both K-Hcy and homocysteine respond to folate or folic acid levels. However, when PA levels are high, the expression of MARS increases, thereby markedly increasing the efficiency of K-Hcy formation. Under these conditions, supplementation with folic acid did not reduce protein K-Hcy. Accordingly, in our high-PA-diet-fed mouse model, the inhibition of MARS, but not supplementation of folic acid, could rescue all the pathological effects caused by PA (see Figure 7).
We have previously conducted a cell-wide proteomics survey and identified K-Hcy-modified proteins.12 However, no transcription factors associated with the K-Hcy modification of proteins have been identified, possibly because of the low protein density of transcription factors in cells. Because defects in transcription factors are known to be the principal cause of CHD, we hypothesized that CHD-related transcription factors are affected by K-Hcy. We found that GATA4 was heavily modified by K-Hcy. GATA4 plays a crucial role in the regulation of heart development, which has been shown in several studies, as best evidenced by the fact that GATA4 mutations are associated with numerous congenital septal defects in humans.34,35,36,37,38,39 GATA4 has the ability to bind through its zinc-finger DNA-binding domain and sequence-specific DNA motifs and regulate the transcription of target genes. We found a homocysteinylated lysine residue located close to the GATA4 zinc-finger DNA-binding domain and found that K-Hcy modification abrogated the DNA-binding affinity of GATA4. These results indicated that, in addition to genetic mutations, post-translational modifications also led to the inactivation of GATA4 and the onset of CHD. As we did not obtain proteome-wide K-Hcy-modified transcription factors, it is possible that K-Hcy modifies and affects proteins other than GATA4 that are involved in septal defects. Elevated oxidative stress is also an important factor involved in PA and K-Hcy-induced CHD onset, and reactive oxygen species is widely acknowledged as a risk factor for multiple developmental defects, including incorrect implantation of embryos, miscarriages, premature births, low birth weight, and malformations.40,41 In this study, we found that K-Hcy regulates GATA4 activity, which further demonstrates how K-Hcy specifically contributes to cardiac dysplasia and septal defects.
Our study found that increased maternal PA levels caused VSD and ASD phenotypes in mice, which was in accordance with the maternal hyperhomocysteine-induced and GATA4 mutation-induced CHD phenotypes in humans.42,43,44,45,46 These phenomena suggest that PA signals have a crosstalk with homocysteine signals and synergistically regulate GATA4 signals. Besides septal defects, GATA4 mutations have also been reported to be associated with complex CHDs. A study has further revealed that GATA4 is a dose-sensitive regulator of cardiac morphogenesis.47 Embryos expressing 70% less GATA4 protein had a common atrioventricular canal (CAVC), double outlet right ventricle (DORV), hypoplastic ventricular myocardium, and normal coronary vasculature and died between embryonic day E13.5 and E16.5, whereas embryos expressing 50% less GATA4 protein survived normally. Besides regulating embryonic heart development, GATA4 plays a key role in postnatal cardiac structural remodeling, as loss-of-function mutations of GATA4 were reported to be responsible for familial dilated cardiomyopathy,48 sporadic dilated cardiomyopathy,49 familial atrial fibrillation,50 and lone atrial fibrillation.51 It will be interesting to explore the connection between nutrient signals and GATA4 function in cardiac remodeling in the future. In this study, we did not observe additional CHD phenotypes, such as CAVC or DORV, as PA only partially inhibited GATA4 activity. As for the relationship between fatty acids and CHD, a previous study showed that maternal type 2 diabetes, which was induced by a 15-week high-fat diet, significantly induced septal defects and persistent truncus arteriosus in the development heart of the offspring, along with increased oxidative stress and apoptosis in the embryonic heart.21 However, few mutations in the genes necessary for fatty acid metabolism have been found in patients with CHD. In a case report,52 a patient with a multiple acyl-coenzyme A (CoA) dehydrogenase deficiency (glutaric aciduria type II [GAII]), which is a deficiency in the electron transfer flavoprotein and leads to the accumulation of FFAs in the plasma, was born with ASD and VSD. These results indicate that exogenous FFA signals from the diet may be much more important than inborn fatty acid metabolic abnormalities.
In conclusion, we found that increased levels of maternal PA increased the risk of CHD in offspring by inhibiting GATA4 signaling in embryos through the NF-κB-MARS-K-Hcy pathway. The inhibition of MARS prevented the PA-induced elevation of K-Hcy in the embryonic heart and decreased the risk of CHD in the offspring, highlighting the potential use of targeting this pathway for the prevention and treatment of CHD.
Limitations of the study
When analyzing fatty acids levels from clinical samples, we did not collect cord blood, which is a direct link between mother and offspring, and detecting metabolic changes in cord blood may better interpret the influence to offspring from mother. K-Hcy modification affects numerous substrates such as histones that will influence embryonic development.53 In this study, we found that PA also increased total K-Hcy levels of histones in fetus heart tissue (Figure S7E), which contributed to the pathogenic role of PA/K-Hcy-induced CHD. The functions of K-Hcy on histones need to be further studied. AHT was previously reported12 to inhibit CHD incidence induced by all-trans retinoic acid in mice and chicken embryos by inhibiting K-Hcy, and if AHT could rescue PA-induced CHD warrants further validation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| MARS | Abcam | Cat# ab50793, RRID: AB_881492 |
| NF-κB p65 | Cell Signaling Technology | Cat# 8242, RRID: AB_10859369 |
| NF-κB p65 (phospho S536) | Cell Signaling Technology | Cat# 3033, RRID: AB_331284 |
| IKK alpha | Cell Signaling Technology | Cat# 2682, RRID: AB_331626 |
| Phospho-IKKα/β (Ser176/180) | Cell Signaling Technology | Cat# 2697, RRID: AB_2079382 |
| GATA4 Monoclonal Antibody | Thermo Fisher Scientific | Cat# MA5-15532, RRID: AB_10989032 |
| NPPA | Abclonal | Cat# A14755, RRID: AB_2761631 |
| TNNI3 | Abclonal | Cat# A0152, RRID: AB_2756984 |
| MYL1 | Abclonal | Cat# A8438, RRID: AB_2770495 |
| SOX17 | Abclonal | Cat# A18858, RRID: AB_2862485 |
| ETS1 | Abcam | Cat# ab225868 |
| Flag | Abmart | Cat# M20008; RRID: AB_2713960 |
| HA | Abmart | Cat# M20003 |
| Actin | Genscript | Cat# A00702-100, RRID: AB_914102 |
| Goat anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 488 | Thermo Fisher Scientific | Cat# A32731, RRID: AB_2633280 |
| Anti-mouse secondary antibody | Genscript | Cat# A00160, RRID: AB_1968937 |
| Anti-rabbit secondary antibody | Genscript | Cat# A00098, RRID: AB_1968815 |
| K-Hcy antibody | This Study | N/A |
| Biological samples | ||
| Serum Samples from pregnant women as 9–11 weeks of gestation | This Study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine 3000 | Invitrogen | Cat# L3000015 |
| Cycloheximide | MedChemExpress | Cat# HY-12320 |
| Palmitic acid | Sigma Aldrich | Cat# P0500 |
| Oleic acid | Sigma Aldrich | Cat# O1008 |
| Stearic acid | Sigma Aldrich | Cat# 85679 |
| Linoleic acid | Sigma Aldrich | Cat# L1376 |
| Eicosatrienoic acid | Yuanye BioTechnology | Cat# Y28446 |
| N-acetyl-L-cysteine | MedChemExpress | Cat# HY-B0215 |
| L-Homocysteine | Sigma Aldrich | Cat# 44925 |
| L-Homocysteine Thiolactone hydrochloride | Sigma Aldrich | Cat# H6503 |
| anti-FLAG M2 affinity gel | Sigma Aldrich | Cat# A2220 |
| anti-FLAG M2 magnetic beads | Sigma Aldrich | Cat# M8823 |
| Folic acid | MedChemExpress | Cat# HY-16637 |
| AHT | N/A | 10.15252/emmm.201809469 |
| RNA isolater | Vazyme | Cat# R401-01 |
| Penicillin-Streptomycin | Invitrogen | Cat# 15070063 |
| DAPI | Sigma-Aldrich | Cat# D8417 |
| Critical commercial assays | ||
| Dual-Luciferase Reporter Assay System | Promega | Cat# E1910 |
| Chromatin Immunoprecipitation (ChIP) Assay Kit | Merck | Cat# 17-295 |
| Elecsys Folate III | Roche | Cat# 07027290190 |
| Homocysteine Enzyme Immunoassay Kit | Axis Shield | Cat# FHCY100 |
| Cell Counting Kit-8 | Dojindo | Cat# CK04 |
| FITC Annexin V Apoptosis Detection Kit I | BD Pharmingen | Cat# 556547 |
| DAB Substrate Kit | Beyotime | Cat# P0203 |
| Pierce™ ECL Plus Western Blotting Substrate | Thermo Fisher Scientific | Cat# 32132 |
| HiScript III 1st Strand cDNA Synthesis Kit | Vazyme | Cat# R312-01 |
| ClonExpress MultiS One Step Cloning Kit | Vazyme | Cat# C113-02 |
| ChamQ SYBR Color qPCR Master Mix | Vazyme | Cat# Q411-02 |
| Deposited data | ||
| GC-FID/MS data | MetaboLights | http://www.ebi.ac.uk/metabolights/MTBLS6982 |
| Experimental models: Cell lines | ||
| HL-1 | Merck | Cat# SCC065 RRID: CVCL_0303 |
| H9C2 | ATCC | Cat# CRL-1446 |
| HEK293T | ATCC | Cat# CRL-11268 |
| Experimental models: Organisms/strains | ||
| C57 BL/6J | Shanghai SLAC Laboratory Animal Co., Ltd. | N/A |
| Mars heterozygous knockout mice | Gempharmatech | N/A |
| Sprauge-Dawley rats | Shanghai SLAC Laboratory Animal Co., Ltd. | N/A |
| Oligonucleotides | ||
| siRNA Oligonucleotides | This Study | Table S5 |
| qRT-PCR primers | This Study | Table S5 |
| ChIP primers | This Study | Table S5 |
| Recombinant DNA | ||
| Mammalian Expression Plasmids | This Study | Table S5 |
| Software and algorithms | ||
| ImageJ | ImageJ | RRID:SCR_003070 |
| Graphpad Prism 8 | Graphpad software |
https://www.graphpad.com/ RRID:SCR_002798 |
| Proteome Discoverer 2.4 | Thermo Fisher Scientific | RRID:SCR_014477 |
| Mascot 2.7.0 | Matrix Science |
http://www.matrixscience.com RRID:SCR_014322 |
| R 2.17 | R studio | https://www.r-project.org/ |
| JASPAR database | JASPAR |
http://jaspar.genereg.net RRID:SCR_003030 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jian-Yuan Zhao (zhaojy@vip.163.com).
Materials availability
All reagents generated in this study are available from the lead contact without restriction.
Experimental model and subject details
Study participants
Human serum samples were obtained from pregnant women who visited the Obstetrics and Gynecology Hospital of Fudan University between January 2018 and December 2019. The study design and conduct were approved and supervised by the Ethics Committee of the Obstetrics and Gynecology Hospital of Fudan University through Ethics Vote 2015-17-C1 in accordance with the criteria established by the Declaration of Helsinki. Written informed consent was obtained from all patients and controls before the start of the study. Serum samples were obtained from women in their first trimester at weeks 9–11 of gestation. The case group included 16 pregnant women bearing children with VSDs and 16 pregnant women bearing children with ASDs. The control group consisted of 32 pregnant women bearing healthy children. CHD phenotypes were first identified by cardiac screening examinations during week 22 of gestation and confirmed after birth using neonatal echocardiography. Participants presenting clinical features of developmental syndromes, multiple major developmental anomalies, known chromosomal abnormalities, or a family history of CHD in a first-degree relative (parent, sibling, or child) were excluded. All participants were unrelated to the ethnic Han Chinese. The demographic characteristics of the pregnant women bearing children with or without CHD are shown in Table S2.
Animal models
C57BL/6 mice were obtained from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China), housed in polycarbonate cages, and provided food and water at a 12-h light:dark cycle. Male and female mice were paired (1:2 ratio) overnight. Mating was verified by vaginal plug observations the following morning, which was considered E0.5. Pregnant females were randomly assigned to the control and experimental groups (n ≥ 8 per group).
A high-fatty acid diet-fed pregnant mouse model was established by changing the normal diet to a fatty acid-rich diet from E0.5 to E14.5. The control group was fed with a standard chow diet. The diet composition is shown in Table S4. The rescue groups received N-acetyl-L-cysteine (NAC) at 300 mg/kg or folic acid at 10 mg/kg body weight per day by tail vein injection from E0.5 to E14.5. Embryos were dissected from the decidual capsules in uteri at E14.5 for the analysis of heart defects.
Mars heterozygous knockout mice were generated using the CRISPR-Cas9-mediated genome editing system on a C57BL/6 background by GemPharmatech Co., Ltd. (Jiangsu Province, China). The genotypes of the offspring were identified using PCR. The primers used are shown in Table S5.
All experimental procedures involving animals were approved by the Fudan University Institutional Animal Care and Use Committee and conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Cell lines
Rat myoblast cells (H9C2), mouse cardiac cells (HL-1), and human embryonic kidney cells (HEK293T) were cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (HyClone, South Logan, UT, USA) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA), 100 units/mL penicillin (Invitrogen), and 100 μg/mL streptomycin (Invitrogen). The cells were incubated in 5% CO2 at 37°C. Cell transfection was performed using Lipofectamine 3000 (Invitrogen) according to manufacturer’s instruction. For treatment of fatty acids, 2.5 mM stock solution was prepared, as described previously.55 Each fatty acid was added to the medium for 9 h before harvesting.
Neonatal rat ventricular myocytes culture
Neonatal rat ventricular myocytes (NRVMs) were isolated from 1-day-old Sprauge-Dawley rats. The hearts from 2-day-old rats were aseptically removed and immediately placed in cold PBS buffer. The ventricles were dissected, minced and digested with 0.15% trypsin for 10 min at 37°C and the supernatant was then transferred to a centrifuge tube containing DMEM supplemented with 10% FBS. The steps for digestion were repeated ∼10 times to maximize yield. Following centrifugation at 50 x g for 10 min, the cell pellet was resuspended in cultured medium. The suspended cells were plated and incubated at 37°C for 1 h. Thereafter, the culture medium containing non-adherent cells were collected and these enriched cardiomyocytes were seeded in a 24-well plate at a concentration of 1 x 105 cells per well for use in further experiments. 0.1 mM bromodeoxyuridine (BrdU) was added to inhibit fibroblast proliferation.
Method details
Physiological index evaluation of the experimental animals
Pregnant mice were housed individually in cages. Food intake and weight of each pregnant mouse were recorded daily at 9 a.m. from E0.5 to E13.5. Blood pressure, pulse, and random blood glucose of each pregnant mouse were assessed simultaneously every 3 days from E0.5 to E13.5 by using a non-invasive animal blood-pressure meter (Visitech Systems; BP-2000, Apex, New York, USA) and glucose meter (Accu-chek; Roche, Basel, Switzerland). For glucose tolerance test (GTT), mice were starved for 6 h and injected with glucose (1 g/kg glucose per mouse). Blood glucose level was assessed at 0, 15, 30, 60, and 120 min after injection using a glucose meter.
Mouse embryo heart isolation and histological analysis
Embryonic hearts were dissected at E14.5 and fixed in 4% paraformaldehyde. The hearts were dehydrated using graded ethanol series, followed by vitrification with dimethylbenzene. The heart tissues were embedded in paraffin and sectioned at 5 μm. The sections were then de-paraffinized, rehydrated in graded alcohol series, and stained with hematoxylin–eosin. The stained heart sections were imaged using Olympus IX73 inverted microscope.
Immunohistochemistry
Paraffin embedded heart tissue sections were de-paraffinized twice with xylene and rehydrated. Antigen retrieval was performed in citric acid (pH 6.0) for 10 min. Endogenous peroxidase was blocked with 3% H2O2 in methanol. For detection of K-Hcy, sections were incubated with 10mM iodoacetamide resolved in 50 mM Tris-HCl (pH 8.0) for 1 h at room temperature. Samples were then incubated with anti-p-IKKα/β (1: 500 dilutions in 10% goat serum) and K-Hcy antibodies (1: 500 dilutions in 10% goat serum) overnight at 4°C, and then the appropriate secondary antibody was applied and incubated at 37°C for 1 h. Sections were developed using a DAB substrate kit (#P0203, Beyotime), and the reaction was stopped with water. The stained heart sections were imaged using Olympus IX73 inverted microscope.
Quantification of metabolites in the plasma
EDTA-treated plasma samples were obtained, centrifuged immediately, and stored in a −80°C freezer until analysis for folate, homocysteine, and triglyceride content. Folate concentration was determined using the Elecsys Folate III competitive chemiluminescent immunoassay (Roche) by using a Cobas E411 Analyzer (Roche). Homocysteine concentration was quantified using the Axis Homocysteine Enzyme Immunoassay Kit (Axis-Shield; Norton, MA). TG, total cholesterol, HDL-C, LDL-C, apolipoprotein A1, apolipoprotein B, FFA, fasting blood glucose, and glycated albumin were analyzed using an automatic biochemical analyzer (Hitachi 7180; WAKO).
Gas chromatography coupled to either a flame ionization detector or mass spectrometer analysis of fatty acids
Gas chromatography coupled to either a flame ionization detector or mass spectrometer (GC-FID/MS) analysis of fatty acids was performed as previously described.54 In brief, 50 μL of serum samples were added to 600 μL of precooled methanol. Supernatants were collected after 10 min of centrifugation (12,000 × g, 4°C). Next, 20 μL of internal standards in hexane (1 mg/mL methyl heptadecanoate, 0.5 mg/mL methyl tricosanoate, and 28 mg/mL butylated hydroxytoluene) were added to a Pyrex tube, followed by the addition of 100 μL of the above supernatant and 1 mL of methanol–hexane mixture (4:1, v/v). The tubes were cooled in liquid nitrogen for 15 min. Next, 100 μL of precooled acetyl chloride was added, and the mixture was flushed briefly with nitrogen gas. The tubes were screw-capped and maintained at 25°C in the dark for 24 h. The tubes were then cooled in an ice bath for 10 min, followed by gradual addition of 2.5 mL of 6% K2CO3 solution (with shaking) for neutralization. After the tubes were left to stand for 30 min, 200 μL of hexane was added to extract the methylated fatty acids. The mixture was left to stand for 10 min, and then the upper layer was transferred to a glass sample vial. This extraction process was repeated twice, and the supernatants were combined and evaporated to dryness. The residues were dissolved in 100 μL of hexane and subjected to GC-FID/MS analysis. For tissues, approximately 10 mg of sample was homogenized in 500 μL of methanol by using a TissueLyser at 20 Hz for 90 s. Next, 100 μL of homogenate mixture was transferred to a Pyrex tube for methylation, as described above.
Methylated fatty acids were measured on a Shimadzu GCMS-QP2010Plus spectrometer (Shimadzu Scientific Instruments, USA) equipped with a mass spectrometer with an electron impact (EI) ion source and flame ionization detector (FID). One microliter of the sample was injected into an Agilent DB-225 capillary GC column (10 m, 0.1 mm ID, 0.1 μm film thickness) equipped with a splitter (1:60). Helium gas was used as the carrier and makeup gas. The injection port and detector temperatures were set at 230°C. The column temperature was set at 55°C for 1 min, increased to 205°C at a rate of 30°C/min, maintained at 205°C for 3 min, and increased to 230°C (5°C/min). The MS spectra were acquired using an EI voltage of 70 eV and an m/z range of 45–450. Methylated fatty acids were identified by comparing with a chromatogram obtained from a mixture of 37 known standards and confirmed on the basis of mass spectral data. Each fatty acid was quantified using FID data from the signal integrals and internal standards.
Plasmid constructs
Whole-length GATA4 and p65 were amplified from HEK293T or HL-1 cDNA and cloned into the Xho I and EcoR I restriction sites of the pcDNA3.1-Flag vector. Human and mouse MARS promoter were amplified from HEK293T or HL-1 genome and cloned into the Xho I and Hind III restriction sites of the pGL3-Basic vector. Plasmids were constructed using ClonExpress MultiS One Step Cloning Kit (#C113-02; Vazyme). The GATA4 mutant was generated by site-directed mutagenesis by using the Mut Express MultiS Fast Mutagenesis kit (#C215-01; Vazyme), according to manufacturer’s instructions. The primer sequences were shown in Table S5.
Nuclei and histone isolation
Cells or heart tissues were washed twice with PBS and homogenized in 200 μL of lysis buffer (10 mM HEPES (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 0.1% Triton X-100) with protease inhibitor mixture (#4693116001, Sigma-Aldrich). After incubation for 5 min on ice, the nuclei were collected in the pellet by low speed centrifugation (1500 × g, 4 min, 4°C). The nuclei were washed once with the lysis buffer without 0.1% Triton X-100 and then lysed in 200 μL of nuclei lysis buffer (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, and protease inhibitor mixture). After 10 min incubation on ice, soluble histones were separated from chromatin by centrifugation (2000 × g, 4 min). The supernatants were then added with 10 mM iodoacetamide and reacted for 1h at 4°C before subject to western blotting for detecting homocysteinylated histones.
Immunoprecipitation and western blotting
Cultured cells or cells extracted from the heart tissues were homogenized with 0.5% NP-40 buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5% Nonidet P-40, and a mixture of protease inhibitors. After centrifugation at 12,000 x g and 4°C for 15 min, the supernatant of the lysates was collected for western blotting. For immunoprecipitation, cell or tissue lysates were incubated with anti-Flag M2 affinity gel (#A2220, Sigma Aldrich) or specific primary antibodies for 3 h at 4°C. The beads were washed three times with NP-40 buffer before boiling with SDS loading buffer, followed by the standard western blotting procedures. For detection of K-Hcy in cultured cells or heart tissues, 10 mM iodoacetamide was added to the lysates and reacted for 1 h at 4°C before western blotting or immunoprecipitation. Antibodies used are shown in key resources table. Detection was performed by measuring chemiluminescence on a Typhoon FLA 9500 (GE Healthcare, Little Chalfont, UK) using Pierce ECL Plus Western Blotting Substrate (#32132, Thermo Fisher Scientific). The intensity of the blots was qualified using ImageJ.
Preparation of K-Hcy antibody
The pan K-Hcy was generated in our previous study.24 Briefly, 10 mg chicken egg albumin55 was homocysteinylated by incubating with 1 mM HTL in 0.1 M Na2CO3 (pH 8.0) with 1:10 (v/v) pyridine at 25°C for 14 h. Next, 15 mM iodoacetamide was added and reacted in the dark at room temperature for 1 h. The modified proteins were purified by passing reaction mixtures through a Sephadex G-25 gel filtration column with 50 mM Tris buffer as the mobile phase in an AKTA-FPLC system (GE Healthcare, Chicago, IL, USA) to remove organic reagents. The sample was desalted using a G25 column and dried with a desiccator to obtain a powder as a homocysteine-modified antigen. The antigen was sent to Abmart (Abmart Shanghai Co.,Ltd) to produce K-Hcy antibodies. The specificity of the antibody was validated in our previous study.24
RNA extraction and quantitative real-time PCR
Total RNA was isolated from cultured cells or heart tissues by using RNA isolater (Vazyme) and then converted to cDNA by using HiScript III first Strand cDNA Synthesis Kit (#R312, Vazyme). The Mars, Nppa, Tnni3, Myh7, Myl1, Rfc1, Mtr, Dhfr and Mthfr mRNA levels were measured by quantitative real-time PCR using ChamQ SYBR Color qPCR Master Mix (#Q411, Vazyme) on the ABI Prism 7900 sequence detection system (Applied Biosystems, Foster City, CA, USA), with actin as an internal reference gene. Primers used for qRT-PCR were shown in Table S5.
Dual-luciferase reporter assay
For the MARS promoter luciferase reporter assay, HEK293T and HL-1 cells were seeded in a 24-well plate. The cells were transfected with 1 μg of pGL3-MARS-promoter plasmid and 20 ng of pRL-TK vector (Promega); half of the cells were additionally co-transfected with 50 ng of the pcDNA3.1-p65 expression plasmid or empty pcDNA3.1 vector by using Lipofectamine 3000. The transfection efficiency was monitored using the Renilla luciferase pRL-TK vector as an internal control. Two days after transfection, cell lysates were collected and subjected to luciferase assay by using the Dual-Luciferase Reporter Assay System (#E1910, Promega), according to manufacturer’s protocol. Three independent transfection experiments were performed, and each luciferase assay was performed in triplicate. Normalized data were calculated as the ratio of the Firefly/Renilla luciferase activities.
Electrophoretic mobility shift assay
EMSA was conducted as previously described.56 Three pairs of 6-carboxy-fluorescein (FAM)-labeled double-stranded DNA probes containing a putative p65-binding site (human, mouse and rat) were generated by annealing their respective complementary oligonucleotides. Probe sequences were shown in Table S5.
HEK293T cells were transfected with pcDNA3.1-p65-Flag or pcDNA3.1-GATA4-Flag plasmids. After 36 h of transfection, cells were harvested and lysed in 0.5% NP-40 buffer, and Flag-tagged proteins were enriched using anti-FLAG M2 magnetic beads (Thermo Fisher Scientific). The FAM-labeled probe (1 pmol) and 20 μg of immunoprecipitated protein were incubated in reaction buffer containing 5 mM MgCl2, 2 mM EDTA, 50 ng/μL poly (dI-dC), 2.5% glycerol, and 0.5 mg/mL BSA for 20 min at 25°C. The reaction mixture without the immunoprecipitated protein was served as a negative control. For the cold competition assay, 50 pmol and 100 pmol unlabelled probes were added into the reaction. The samples were subjected to 10% non-denaturing PAGE and analyzed using a Typhoon FLA 9500 scanner.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was carried out using the Chromatin Immunoprecipitation (ChIP) Assay Kit (Merck). Briefly, cells were cross-linked by 1% (v/v) formaldehyde for 10 min at 37°C before harvesting. DNA was then sonicated to generate 200- to 500-bp DNA fragments. The sheared chromatin was immunoprecipitated by incubation with p65 or GATA4 antibody or normal rabbit IgG overnight at 4°C. The precipitated DNA fragments were then identified by PCR and quantified by qRT-PCR. Primers are listed in Table S5.
RNAi
Synthetic oligos were used for siRNA-mediated silencing of MARS, RELA, CEBPA, MEIS2, KLF2, MXI1 and TCF4, and scramble siRNA was used as a control. Cells were transfected with siRNAs by using Lipofectamine 3000, according to manufacturer’s instructions. Knockdown efficiency was verified using western blotting and qRT-PCR. The siRNA sequences were shown in Table S5.
Sample preparation for LC-MS/MS analysis
For detecting GATA4-interacting proteins and homocysteinylation sites on GATA4, HEK293T cells were transfected with the GATA4-Flag vector and treated with 0.2 mM palmitic acid for 16 h before harvesting. Cells were lysed in 0.5% NP-40 buffer supplemented with 5 mM iodoacetamide (#I1149, Sigma Aldrich) and protease inhibitor (#4693116001, Sigma Aldrich), and the supernatants were then immunoprecipitated with anti-FLAG M2 magnetic beads (M8823-1mL; Sigma Aldrich) for 3 h at 4°C. For detecting K-Hcy sites of GATA4 from heart tissues, heart tissues were homogenized in 0.5% NP-40 buffer supplemented with 5 mM iodoacetamide and protease inhibitor, and the supernatants were then immunoprecipitated with anti-GATA4 antibody. For detecting K-Hcy sites, the precipitates were washed twice with 0.1% NP-40 buffer, twice with ddH2O, and three times with 50 mM NH4HCO3, after which on-bead tryptic digestion was performed at 37°C overnight. The peptides in the supernatants were collected through centrifugation and dried in a speed vacuum (Eppendorf). The obtained peptides were stored at −80°C until LC-MS/MS analysis.
LC-MS/MS analysis
Samples were analyzed on a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific, Rockford, IL, USA) coupled with high-performance liquid chromatography (EASY-nLC 1200 System; Thermo Fisher Scientific). Dried peptide samples were re-dissolved in Solvent A (0.1% formic acid in water) and loaded onto a trap column (100 μm × 2 cm, home-made; particle size, 3 μm; pore size, 120 Å; SunChrom, USA) with a maximum pressure of 280 bar by using Solvent A, and then separated on an in-house 150 μm × 12 cm silica microcolumn (particle size, 1.9 μm; pore size, 120 Å; SunChrom, USA) with a gradient of 5%–35% mobile phase B (acetonitrile and 0.1% formic acid) at a flow rate of 600 nL/min for 75 min. The MS analysis for Q Exactive HF-X was performed using one full scan (300–1,400 m/z, R = 60,000 at 200 m/z) at automatic gain control target of 3e6 ions, followed by up to 20 data-dependent MS/MS scans with higher-energy collision dissociation (target, 2 × 103 ions; maximum injection time, 40 ms; isolation window, 1.6 m/z; normalized collision energy, 27%). Detection was performed using Orbitrap (R = 15,000 at 200 m/z). Data were acquired using Xcalibur software (Thermo Fischer Scientific).
K-Hcy site identification
RAW files were processed with UniProt Homo Sapiens (UP000005640) or Mus musculus (UP000000589) protein database and using Proteome Discoverer (version 2.4, Thermo Fisher Scientific) with Mascot (version 2.7.0, Matrix Science). The ptmRS node was used in K-Hcy sites analysis workflow. The mass tolerances were 10 ppm for precursor and fragment Mass Tolerance 0.05 Da. Up to two missed cleavages were allowed. The acetylation on the protein N-terminal, oxidation on methionine and carbamidomethylation on cysteine were set as variable modifications. For searching for K-Hcy peptides, carbamidomethylated Hcy on lysine [C(6)H(10)N(2)O(2)S, 174.046 Da] was set as variable modification. False discovery rate (FDR) thresholds for peptide were set to 0.01.
Hcy and HTL quantification
Heart tissues were homogenized on ice-cold PBS (PBS) and centrifuged (10,000 x g) at 4°C for 15 min, and the supernatants were collected for Hcy quantification. Hcy concentration was determined using an Axis Homocysteine Enzyme Immunoassay Kit (Axis-Shield). HTL was assayed as the following: heart tissues were harvested by PBS washing followed by denaturing by pre-chilled 80% methanol (dissolved in ddH2O, precooled in −80°C). The lysate was centrifuged (10,000 x g) at 4°C for 10 min. The supernatant was vacuum dried, then re-dissolved in ddH2O and subjected to ultra-filtration on a polyvinylidene fluoride low protein binding membrane (Millex-GV4 and Millex-HV4, Millipore). The collected metabolites were extracted and the HTL was analyzed using liquid chromatography-mass spectrometry (LC-MS) as previously described.57
Cell proliferation and apoptosis assay
Cell viability was assessed using the Cell Counting Kit-8 (Dojindo) according to the manufacturer’s guidelines. Briefly, a total of approximately 5 × 103 cells were seeded in 96-well plates, 10 μL CCK-8 solution was added to each well and cultivated for 1 h. The absorbance of the reaction system was measured spectrophotometrically at 450 nm. Cell apoptosis was detected using an Annexin V-FITC Apoptosis Detection Kit (BD Pharmingen) according to the manufacturer’s guidelines. Data were collected on an Accuri C6 flow cytometer (BD Biosciences).
Immunofluorescence
For detecting subcellular localization of GATA4 and K300W mutants, HL-1 cells were transfected with Flag-tagged GATA4 and K300W mutant for 36 h. Cells were harvested and washed with PBS buffer twice to remove the remaining medium. Paraformaldehyde (4%) was used to fix the cells, 0.5% Triton X-100 in PBS was added and incubated for 20 min at 25°C. Then the cells were incubated with 5% goat serum for 1 h at room temperature and followed by incubating with anti-Flag antibody (1:500 in 5% goat serum) at 4°C overnight. After washing with PBS, cells were incubated with Alexa Flour 488 donkey anti-rabbit secondary antibody for 1 h at 25°C in the dark. Cells were washed and incubated with DAPI for 5 min to display the nuclei. Cells were observed under a fluorescence microscope (Olympus FV3000, Tokyo, Japan).
Quantification and statistical analysis
Two-tailed t-tests and ANOVA F-tests were used to analyze the association of each FFA during human pregnancy with CHD risk in the offspring. The contribution of each FFA to CHD risk was further assessed by taking the permutation variable importance, which measures the contribution of an FFA by randomly shuffling values for that FFA and observing the most important FFAs in the random forest model. Pearson’s correlation coefficient was used to measure the association between different FFAs. Pooled results were expressed as the mean or SEM. A one-way ANOVA was performed for multi-group analyses, and two-tailed Student’s t-tests or t-tests with Welch’s correction were performed for two-group analyses. Differences were considered statistically significant if the p value was less than 0.05. Statistical analyses were performed using Prism software (version 8.0; GraphPad Software, Inc.), Excel (Microsoft Corp.), and R version 2.17.
Acknowledgments
This work was supported by grants from National Key R&D Program of China (nos. 2020YFA0803601 and 2019YFA0801900), the fellowship of China Postdoctoral Science Foundation (2020M670987), and the National Natural Science Foundation of China (nos. 82130015, 31871432, 81771627, 81971061, 81770312, 81970572, and 32000895). Objects in graphical abstract were acquired from Figdraw (www.figdraw.com, ID: 533420148).
Author contributions
J.-Y.Z., K.S., J.C., and X.-Y.Z. conceived of the presented idea. R.Z., L.C., W.-J.G., L.L., Z.-Z.C., and J.C. performed the experiments. J.X., Z.-Y.Z., and W.-D.Z. assisted with the experiments. R.Z., Z.-Z.C., X.-Y.Z., and J.C. performed the analysis. J.-Y.Z. and R.Z. drafted the manuscript. All authors discussed the results and commented on the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: February 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.100953.
Contributor Information
Xiang-Yu Zhou, Email: husq04@163.com.
Jing Cao, Email: caojing73@126.com.
Kun Sun, Email: sunkun@xinhuamed.com.cn.
Jian-Yuan Zhao, Email: zhaojy@vip.163.com.
Supplemental information
Data and code availability
-
•
GC-FID/MS raw data were deposited in the MetaboLights repository (www.ebi.ac.uk/metabolights/) under the accession number MTBLS6982.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.van der Linde D., Konings E.E.M., Slager M.A., Witsenburg M., Helbing W.A., Takkenberg J.J.M., Roos-Hesselink J.W. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J. Am. Coll. Cardiol. 2011;58:2241–2247. doi: 10.1016/j.jacc.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 2.Botto L.D., Correa A. Decreasing the burden of congenital heart anomalies: an epidemiologic evaluation of risk factors and survival. Prog. Pediatr. Cardiol. 2003;18:111–121. doi: 10.1016/s1058-9813(03)00084-5. [DOI] [Google Scholar]
- 3.Botto L.D., Mulinare J., Erickson J.D. Do multivitamin or folic acid supplements reduce the risk for congenital heart defects? Evidence and gaps. Am. J. Med. Genet. 2003;121a:95–101. doi: 10.1002/ajmg.a.20132. [DOI] [PubMed] [Google Scholar]
- 4.Feng Y., Wang S., Chen R., Tong X., Wu Z., Mo X. Maternal folic acid supplementation and the risk of congenital heart defects in offspring: a meta-analysis of epidemiological observational studies. Sci. Rep. 2015;5:8506. doi: 10.1038/srep08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Botto L.D., Mulinare J., Erickson J.D. Occurrence of congenital heart defects in relation to maternal multivitamin use. Am. J. Epidemiol. 2000;151:878–884. doi: 10.1093/oxfordjournals.aje.a010291. [DOI] [PubMed] [Google Scholar]
- 6.Hernández-Díaz S., Werler M.M., Walker A.M., Mitchell A.A. Folic acid antagonists during pregnancy and the risk of birth defects. N. Engl. J. Med. 2000;343:1608–1614. doi: 10.1056/NEJM200011303432204. [DOI] [PubMed] [Google Scholar]
- 7.Zhao J.Y., Yang X.Y., Gong X.H., Gu Z.Y., Duan W.Y., Wang J., Ye Z.Z., Shen H.B., Shi K.H., Hou J., et al. Functional variant in methionine synthase reductase intron-1 significantly increases the risk of congenital heart disease in the han Chinese population. Circulation. 2012;125:482–490. doi: 10.1161/CIRCULATIONAHA.111.050245. [DOI] [PubMed] [Google Scholar]
- 8.Zhao J.Y., Yang X.Y., Shi K.H., Sun S.N., Hou J., Ye Z.Z., Wang J., Duan W.Y., Qiao B., Chen Y.J., et al. A functional variant in the cystathionine beta-synthase gene promoter significantly reduces congenital heart disease susceptibility in a Han Chinese population. Cell Res. 2013;23:242–253. doi: 10.1038/cr.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao J.Y., Qiao B., Duan W.Y., Gong X.H., Peng Q.Q., Jiang S.S., Lu C.Q., Chen Y.J., Shen H.B., Huang G.Y., et al. Genetic variants reducing MTR gene expression increase the risk of congenital heart disease in Han Chinese populations. Eur. Heart J. 2014;35:733–742. doi: 10.1093/eurheartj/eht221. [DOI] [PubMed] [Google Scholar]
- 10.Wang D., Wang F., Shi K.H., Tao H., Li Y., Zhao R., Lu H., Duan W., Qiao B., Zhao S.M., et al. Lower circulating folate induced by a fidgetin intronic variant is associated with reduced congenital heart disease susceptibility. Circulation. 2017;135:1733–1748. doi: 10.1161/CIRCULATIONAHA.116.025164. [DOI] [PubMed] [Google Scholar]
- 11.Jakubowski H. Homocysteine modification in protein structure/function and human disease. Physiol. Rev. 2019;99:555–604. doi: 10.1152/physrev.00003.2018. [DOI] [PubMed] [Google Scholar]
- 12.Mei X., Qi D., Zhang T., Zhao Y., Jin L., Hou J., Wang J., Lin Y., Xue Y., Zhu P., et al. Inhibiting MARSs reduces hyperhomocysteinemia-associated neural tube and congenital heart defects. EMBO Mol. Med. 2020;12:e9469. doi: 10.15252/emmm.201809469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jakubowski H., Zhang L., Bardeguez A., Aviv A. Homocysteine thiolactone and protein homocysteinylation in human endothelial cells: implications for atherosclerosis. Circ. Res. 2000;87:45–51. doi: 10.1161/01.res.87.1.45. [DOI] [PubMed] [Google Scholar]
- 14.Correa A., Marcinkevage J. Prepregnancy obesity and the risk of birth defects: an update. Nutr. Rev. 2013;71:S68–S77. doi: 10.1111/nure.12058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stothard K.J., Tennant P.W.G., Bell R., Rankin J. Maternal overweight and obesity and the risk of congenital anomalies: a systematic review and meta-analysis. JAMA. 2009;301:636–650. doi: 10.1001/jama.2009.113. [DOI] [PubMed] [Google Scholar]
- 16.Persson M., Razaz N., Edstedt Bonamy A.K., Villamor E., Cnattingius S. Maternal overweight and obesity and risk of congenital heart defects. J. Am. Coll. Cardiol. 2019;73:44–53. doi: 10.1016/j.jacc.2018.10.050. [DOI] [PubMed] [Google Scholar]
- 17.Cai G.J., Sun X.X., Zhang L., Hong Q. Association between maternal body mass index and congenital heart defects in offspring: a systematic review. Am. J. Obstet. Gynecol. 2014;211:91–117. doi: 10.1016/j.ajog.2014.03.028. [DOI] [PubMed] [Google Scholar]
- 18.Nederlof M., de Walle H.E.K., van Poppel M.N.M., Vrijkotte T.G.M., Gademan M.G.J. Deviant early pregnancy maternal triglyceride levels and increased risk of congenital anomalies: a prospective community-based cohort study. BJOG. 2015;122:1176–1183. doi: 10.1111/1471-0528.13393. [DOI] [PubMed] [Google Scholar]
- 19.Smedts H.P.M., Rakhshandehroo M., Verkleij-Hagoort A.C., de Vries J.H.M., Ottenkamp J., Steegers E.A.P., Steegers-Theunissen R.P.M. Maternal intake of fat, riboflavin and nicotinamide and the risk of having offspring with congenital heart defects. Eur. J. Nutr. 2008;47:357–365. doi: 10.1007/s00394-008-0735-6. [DOI] [PubMed] [Google Scholar]
- 20.Mdaki K.S., Larsen T.D., Wachal A.L., Schimelpfenig M.D., Weaver L.J., Dooyema S.D.R., Louwagie E.J., Baack M.L. Maternal high-fat diet impairs cardiac function in offspring of diabetic pregnancy through metabolic stress and mitochondrial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2016;310:H681–H692. doi: 10.1152/ajpheart.00795.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Y., Reece E.A., Zhong J., Dong D., Shen W.B., Harman C.R., Yang P. Type 2 diabetes mellitus induces congenital heart defects in murine embryos by increasing oxidative stress, endoplasmic reticulum stress, and apoptosis. Am. J. Obstet. Gynecol. 2016;215:366.e1–366.e10. doi: 10.1016/j.ajog.2016.03.036. ARTN 366.e1-e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Innis S.M. Fatty acids and early human development. Early Hum. Dev. 2007;83:761–766. doi: 10.1016/j.earlhumdev.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 23.Chirala S.S., Chang H., Matzuk M., Abu-Elheiga L., Mao J., Mahon K., Finegold M., Wakil S.J. Fatty acid synthesis is essential in embryonic development: fatty acid synthase null mutants and most of the heterozygotes die in utero. Proc. Natl. Acad. Sci. USA. 2003;100:6358–6363. doi: 10.1073/pnas.0931394100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang D., Zhao R., Qu Y.Y., Mei X.Y., Zhang X., Zhou Q., Li Y., Yang S.B., Zuo Z.G., Chen Y.M., et al. Colonic lysine homocysteinylation induced by high-fat diet suppresses DNA damage repair. Cell Rep. 2018;25:398–412.e6. doi: 10.1016/j.celrep.2018.09.022. [DOI] [PubMed] [Google Scholar]
- 25.Siddeek B., Mauduit C., Chehade H., Blin G., Liand M., Chindamo M., Benahmed M., Simeoni U. Long-term impact of maternal high-fat diet on offspring cardiac health: role of micro-RNA biogenesis. Cell Death Dis. 2019;5:71. doi: 10.1038/s41420-019-0153-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quan J., Liu J., Gao X., Liu J., Yang H., Chen W., Li W., Li Y., Yang W., Wang B. Palmitate induces interleukin-8 expression in human aortic vascular smooth muscle cells via Toll-like receptor 4/nuclear factor-kappaB pathway (TLR4/NF-kappaB-8) J. Diabetes. 2014;6:33–41. doi: 10.1111/1753-0407.12073. [DOI] [PubMed] [Google Scholar]
- 27.Molkentin J.D. The zinc finger-containing transcription factors GATA-4, -5, and -6. Ubiquitously expressed regulators of tissue-specific gene expression. J. Biol. Chem. 2000;275:38949–38952. doi: 10.1074/jbc.R000029200. [DOI] [PubMed] [Google Scholar]
- 28.Bisping E., Ikeda S., Sedej M., Wakula P., McMullen J.R., Tarnavski O., Sedej S., Izumo S., Pu W.T., Pieske B. Transcription factor GATA4 is activated but not required for insulin-like growth factor 1 (IGF1)-induced cardiac hypertrophy. J. Biol. Chem. 2012;287:9827–9834. doi: 10.1074/jbc.M111.338749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang Q., Molkentin J.D. Divergent signaling pathways converge on GATA4 to regulate cardiac hypertrophic gene expression. J. Mol. Cell. Cardiol. 2002;34:611–616. doi: 10.1006/jmcc.2002.2011. [DOI] [PubMed] [Google Scholar]
- 30.Kohlnhofer B.M., Thompson C.A., Walker E.M., Battle M.A. GATA4 regulates epithelial cell proliferation to control intestinal growth and development in mice. Cell. Mol. Gastroenterol. Hepatol. 2016;2:189–209. doi: 10.1016/j.jcmgh.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bisping E., Ikeda S., Kong S.W., Tarnavski O., Bodyak N., McMullen J.R., Rajagopal S., Son J.K., Ma Q., Springer Z., et al. Gata4 is required for maintenance of postnatal cardiac function and protection from pressure overload-induced heart failure. Proc. Natl. Acad. Sci. USA. 2006;103:14471–14476. doi: 10.1073/pnas.0602543103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang L., Guan G., Lei L., Liu J., Cao L., Wang X. Oxidative and endoplasmic reticulum stresses are involved in palmitic acid-induced H9c2 cell apoptosis. Biosci. Rep. 2019;39 doi: 10.1042/BSR20190225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Misra C., Sachan N., McNally C.R., Koenig S.N., Nichols H.A., Guggilam A., Lucchesi P.A., Pu W.T., Srivastava D., Garg V. Congenital heart disease-causing Gata4 mutation displays functional deficits in vivo. PLoS Genet. 2012;8:e1002690. doi: 10.1371/journal.pgen.1002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ang Y.S., Rivas R.N., Ribeiro A.J.S., Srivas R., Rivera J., Stone N.R., Pratt K., Mohamed T.M.A., Fu J.D., Spencer C.I., et al. Disease model of GATA4 mutation reveals transcription factor cooperativity in human cardiogenesis. Cell. 2016;167:1734–1749.e22. doi: 10.1016/j.cell.2016.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okubo A., Miyoshi O., Baba K., Takagi M., Tsukamoto K., Kinoshita A., Yoshiura K., Kishino T., Ohta T., Niikawa N., Matsumoto N. A novel GATA4 mutation completely segregated with atrial septal defect in a large Japanese family. J. Med. Genet. 2004;41:e97. doi: 10.1136/jmg.2004.018895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarkozy A., Conti E., Neri C., D'Agostino R., Digilio M.C., Esposito G., Toscano A., Marino B., Pizzuti A., Dallapiccola B. Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J. Med. Genet. 2005;42:e16. doi: 10.1136/jmg.2004.026740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J., Fang M., Liu X.Y., Xin Y.F., Liu Z.M., Chen X.Z., Wang X.Z., Fang W.Y., Liu X., Yang Y.Q. A novel GATA4 mutation responsible for congenital ventricular septal defects. Int. J. Mol. Med. 2011;28:557–564. doi: 10.3892/ijmm.2011.715. [DOI] [PubMed] [Google Scholar]
- 38.Yang Y.Q., Wang J., Liu X.Y., Chen X.Z., Zhang W., Wang X.Z. Mutation spectrum of GATA4 associated with congenital atrial septal defects. Arch. Med. Sci. 2013;9:976–983. doi: 10.5114/aoms.2013.39788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Y.Q., Wang J., Liu X.Y., Chen X.Z., Zhang W., Wang X.Z., Liu X., Fang W.Y. Novel GATA4 mutations in patients with congenital ventricular septal defects. Med. Sci. Mon. Int. Med. J. Exp. Clin. Res. 2012;18 doi: 10.12659/msm.882877. CR344-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pietryga M., Dydowicz P., Toboła K., Napierała M., Miechowicz I., Gąsiorowska A., Brązert M., Florek E. Selected oxidative stress biomarkers in antenatal diagnosis as 11-14 gestational weeks. Free Radic. Biol. Med. 2017;108:517–523. doi: 10.1016/j.freeradbiomed.2017.04.020. [DOI] [PubMed] [Google Scholar]
- 41.Toboła-Wróbel K., Pietryga M., Dydowicz P., Napierała M., Brązert J., Florek E. Association of oxidative stress on pregnancy. Oxid. Med. Cell. Longev. 2020;2020:6398520. doi: 10.1155/2020/6398520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie H.H., Li J., Li P.Q., Zhang A.A., Li Y., Wang Y.Z., Xie D.X., Xie X.D. A genetic variant in a homocysteine metabolic gene that increases the risk of congenital cardiac septal defects in Han Chinese populations. IUBMB Life. 2017;69:700–705. doi: 10.1002/iub.1651. [DOI] [PubMed] [Google Scholar]
- 43.Malik R.A., Lone M.R., Ahmed A., Koul K.A., Malla R.R. Maternal hyperhomocysteinemia and congenital heart defects: a prospective case control study in Indian population. Indian Heart J. 2017;69:17–19. doi: 10.1016/j.ihj.2016.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosenquist T.H., Ratashak S.A., Selhub J. Homocysteine induces congenital defects of the heart and neural tube: effect of folic acid. Proc. Natl. Acad. Sci. USA. 1996;93:15227–15232. doi: 10.1073/pnas.93.26.15227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hobbs C.A., Cleves M.A., Melnyk S., Zhao W., James S.J. Congenital heart defects and abnormal maternal biomarkers of methionine and homocysteine metabolism. Am. J. Clin. Nutr. 2005;81:147–153. doi: 10.1093/ajcn/81.1.147. [DOI] [PubMed] [Google Scholar]
- 46.Kalisch-Smith J.I., Ved N., Sparrow D.B. Environmental risk factors for congenital heart disease. Cold Spring Harbor Perspect. Biol. 2020;12:a037234. doi: 10.1101/cshperspect.a037234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pu W.T., Ishiwata T., Juraszek A.L., Ma Q., Izumo S. GATA4 is a dosage-sensitive regulator of cardiac morphogenesis. Dev. Biol. 2004;275:235–244. doi: 10.1016/j.ydbio.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 48.Zhao L., Xu J.H., Xu W.J., Yu H., Wang Q., Zheng H.Z., Jiang W.F., Jiang J.F., Yang Y.Q. A novel GATA4 loss-of-function mutation responsible for familial dilated cardiomyopathy. Int. J. Mol. Med. 2014;33:654–660. doi: 10.3892/ijmm.2013.1600. [DOI] [PubMed] [Google Scholar]
- 49.Li J., Liu W.D., Yang Z.L., Yuan F., Xu L., Li R.G., Yang Y.Q. Prevalence and spectrum of GATA4 mutations associated with sporadic dilated cardiomyopathy. Gene. 2014;548:174–181. doi: 10.1016/j.gene.2014.07.022. [DOI] [PubMed] [Google Scholar]
- 50.Yang Y.Q., Wang M.Y., Zhang X.L., Tan H.W., Shi H.F., Jiang W.F., Wang X.H., Fang W.Y., Liu X. GATA4 loss-of-function mutations in familial atrial fibrillation. Clin. Chim. Acta. 2011;412:1825–1830. doi: 10.1016/j.cca.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 51.Jiang J.Q., Shen F.F., Fang W.Y., Liu X., Yang Y.Q. Novel GATA4 mutations in lone atrial fibrillation. Int. J. Mol. Med. 2011;28:1025–1032. doi: 10.3892/ijmm.2011.783. [DOI] [PubMed] [Google Scholar]
- 52.Farag E., Argalious M., Narouze S., DeBoer G.E., Tome J. The anesthetic management of ventricular septal defect (VSD) repair in a child with mitochondrial cytopathy. Can. J. Anaesth. 2002;49:958–962. doi: 10.1007/BF03016883. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Q., Bai B., Mei X., Wan C., Cao H., Dan L., Wang S., Zhang M., Wang Z., Wu J., et al. Elevated H3K79 homocysteinylation causes abnormal gene expression during neural development and subsequent neural tube defects. Nat. Commun. 2018;9:3436. doi: 10.1038/s41467-018-05451-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.An Y., Xu W., Li H., Lei H., Zhang L., Hao F., Duan Y., Yan X., Zhao Y., Wu J., et al. High-fat diet induces dynamic metabolic alterations in multiple biological matrices of rats. J. Proteome Res. 2013;12:3755–3768. doi: 10.1021/pr400398b. [DOI] [PubMed] [Google Scholar]
- 55.Daud A.I., Loo K., Pauli M.L., Sanchez-Rodriguez R., Sandoval P.M., Taravati K., Tsai K., Nosrati A., Nardo L., Alvarado M.D., et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J. Clin. Invest. 2016;126:3447–3452. doi: 10.1172/JCI87324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo X., Wang L., Li J., Ding Z., Xiao J., Yin X., He S., Shi P., Dong L., Li G., et al. Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature. 2015;517:640–644. doi: 10.1038/nature13899. [DOI] [PubMed] [Google Scholar]
- 57.Barathi S., Angayarkanni N., Pasupathi A., Natarajan S.K., Pukraj R., Dhupper M., Velpandian T., Muralidharan C., Sivashanmugham M. Homocysteinethiolactone and paraoxonase: novel markers of diabetic retinopathy. Diabetes Care. 2010;33:2031–2037. doi: 10.2337/dc10-0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
GC-FID/MS raw data were deposited in the MetaboLights repository (www.ebi.ac.uk/metabolights/) under the accession number MTBLS6982.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.