

Summary
Pediatric acute myeloid leukemia (pAML) is typified by high relapse rates and a relative paucity of somatic DNA mutations. Although seminal studies show that splicing factor mutations and mis-splicing fuel therapy-resistant leukemia stem cell (LSC) generation in adults, splicing deregulation has not been extensively studied in pAML. Herein, we describe single-cell proteogenomics analyses, transcriptome-wide analyses of FACS-purified hematopoietic stem and progenitor cells followed by differential splicing analyses, dual-fluorescence lentiviral splicing reporter assays, and the potential of a selective splicing modulator, Rebecsinib, in pAML. Using these methods, we discover transcriptomic splicing deregulation typified by differential exon usage. In addition, we discover downregulation of splicing regulator RBFOX2 and CD47 splice isoform upregulation. Importantly, splicing deregulation in pAML induces a therapeutic vulnerability to Rebecsinib in survival, self-renewal, and lentiviral splicing reporter assays. Taken together, the detection and targeting of splicing deregulation represent a potentially clinically tractable strategy for pAML therapy.
Keywords: pediatric AML, hematopoietic stem cells, pre-mRNA splicing, splicing modulation, SF3B1, RBFOX2, CD47, embryonic stem cells
Graphical abstract
Highlights
-
•
Pediatric acute myeloid leukemia stem cells harbor increased exon skipping events
-
•
Decreased RBFOX2 induces embryonic splicing and CD47 splice isoform upregulation
-
•
Lentiviral splicing reporter studies show exon skipping reversal with Rebecsinib
-
•
Rebecsinib inhibits pediatric acute myeloid leukemia stem cell propagation
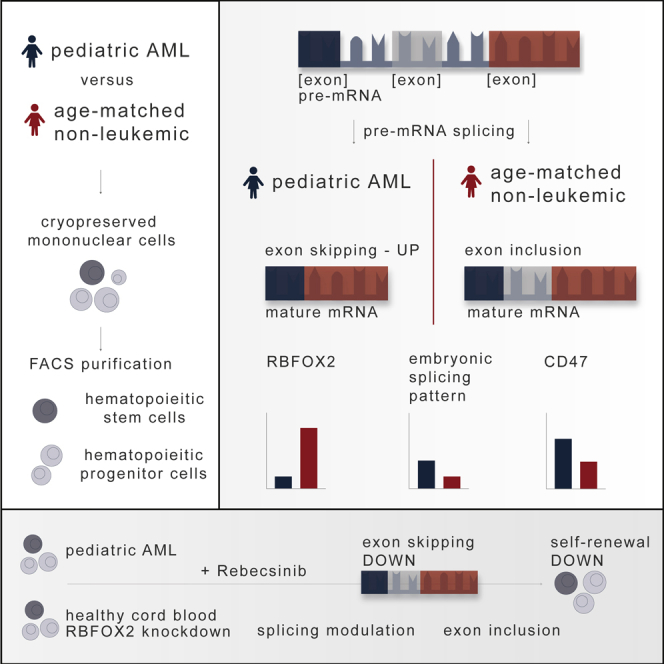
Pediatric acute myeloid leukemia stem cells (LSCs) harbor increased exon skipping events and decreased RBFOX2 expression, which is linked to embryonic splicing patterns and CD47 splice isoform upregulation. van der Werf et al. show that the reversal of malignant exon skipping with Rebecsinib, a selective splicing modulator, prevents pediatric LSC propagation.
Introduction
Alternative pre-mRNA splicing involves tightly regulated intron removal and exon joining, which is crucial for converting pre-mRNAs into mRNAs that are subsequently translated into functional proteins.1,2 The expression of different splice isoforms can have wide-ranging effects on stem cell functional properties, including differentiation, self-renewal, dormancy, and homing. Deregulation of alternative splicing has been linked to somatic mutations in splicing regulatory genes, such as SRSF2, U2AF1, and SF3B1, and confers a poor prognosis in human adult myeloid malignancies, such as acute myeloid leukemia (AML).3,4,5,6,7 Moreover, mis-splicing can occur in stem cell regulatory transcripts,8 in some cases, as a response to inflammatory cytokine-inducible RNA editing by ADAR1, which introduces new splice acceptor sites through adenosine-to-inosine (A-to-I) RNA editing.4,9,10,11,12
Previously, we identified splice isoform signatures that distinguished therapy-resistant acute myeloid leukemia stem cells (LSCs) from healthy age-matched hematopoietic stem cells (HSCs) and hematopoietic progenitor cells (HPCs).4 In addition, we pre-clinically sensitized adult LSCs to a splicing modulator, 17S-FD-895 (Rebecsinib) in stromal co-cultures and humanized mouse models.4 This discovery of splicing deregulation in therapy-resistant myeloid malignancies, in the absence of recurrent splicing factor mutations, fueled our investigation of splicing deregulation in children diagnosed with AML. Although pediatric AML (pAML) is frequently therapeutically recalcitrant, precise delineation of the mechanisms of therapeutic resistance has been hindered by its relative rarity. Recent research has revealed a relative paucity of somatic mutations in pAML compared with adult AML.13,14 This finding suggests that pAML propagation might be driven by epigenomic alterations and splicing deregulation rather than solely by somatic DNA mutations.13,14,15,16
To elucidate the relative importance of somatic DNA drivers and splicing deregulation in pAML, we performed whole exome sequencing and multiplexed single-cell proteogenomics analyses, purified HSC and HPC RNA sequencing (RNA-seq) analyses with splicing-specific computational bioinformatics pipelines, single-cell biosensing dual-fluorescence lentiviral splicing reporter assays, and lentiviral shRNA knockdown assays. Using these methods, we discovered that downregulation of RBFOX2, a repressor of embryonic alternative pre-mRNA splicing, was associated with CD47 splice isoforms upregulation, which has been reported to prevent programmed cell removal by macrophages and fuel LSC propagation in adult AML.17,18 Moreover, in vitro assays and humanized AML mouse model studies revealed selective sensitivity of pAML to splicing modulation with Rebecsinib, thereby setting the stage for the development of clinical splicing modulator approaches aimed at eradicating LSCs in refractory AML in children.
Results
Whole-exome sequencing detection of somatic mutations in pAML
To examine recurrent somatic mutations, we performed targeted whole-exome sequencing analyses of 124 gene mutations on pAML (n = 11) and adult de novo AML (dnAML; n = 3) peripheral blood and bone marrow mononuclear cell samples (Figures 1A, S1A, and S1B, Table S1). While bulk samples lacked detectable mutations in splicing factor genes, such as SF3B1, SRSF2, U2AF1, U2AF2, or ZRSR2 (Figure 1B), we could not exclude the existence of splicing factor mutations in rare cell populations.
Figure 1.

Comparative targeted whole-exome sequencing analysis of pediatric and adult acute myeloid leukemia samples
(A) Study design schematic.
(B) Targeted whole-exome (next-generation) sequencing analysis of 124 hematopoietic malignancy-associated genes was performed on DNA extracted from peripheral blood or bone marrow mononuclear cells isolated from pediatric AML (pAML, n = 11) patients and adult de novo AML (dnAML, n = 3) patients. See also Figure S1.
Single-cell proteogenomics detection of clonal heterogeneity in pAML
To rule out recurrent splicing factor mutations in relatively rare cell populations, we analyzed three pAML patients (266, 451, and 678) using multiplexed single-cell proteogenomics analyses (Figure 2).19,20 To isolate single cells for DNA genotypes and cell surface protein expression, we used a droplet microfluidics platform (Tapestri)19,20 followed by immunoprecipitation with antibody-oligo conjugates to detect cell surface receptors (CD3, CD7, CD11b, CD34, CD38, CD45, CD56, and ROR1), and we queried DNA mutations in 45 myeloid malignancy-associated genes, covering 312 amplicons. With this myeloid malignancy proteogenomics platform, 8,325, 7,658, and 5,440 single cells were analyzed, respectively, for each of the three pAML samples as shown in the uniform manifold approximation and projection (UMAP) plots (Figures 2A, 2B, 2C (left) and S2A, S2B, S2C). To correlate immunophenotype with DNA mutation status, we mapped detectable single-nucleotide variants (SNVs) with the immunophenotypes (Figures 2A, 2B, 2C (right) and S2A, S2B, S2C). Next, we correlated the DNA mutation with cell immunophenotype. Patient 266 (an 11-year-old girl) had multiple mutations in TP53, KIT, ASXL1, DNMT3A, ETV6, KDM6A, SETBP1, EZH2, PHF6, BCOR, and SF3B1 (Figure 2A). Patient 451 (a 9-year-old boy) had mutations in RAD21, FLT3, WT1, RUNX1, DNMT3A, NF1, BRAF, RUNX1, NF1, EZH2, and SF3B1 (Figure 2B). Patient 678 (a 1-year-old boy) had mutations in KRAS, SETBP1, FLT3, TET2, KIT, BCOR, RAD21, EZH2, and WT1 (Figure 2C). In all three pAML patients studied by single-cell proteogenomic sequencing, there was no clear evidence of immunophenotypic segregation. In addition, different clones harbored only minor differences in somatic mutations. In two out of three patients (451 and 266), we identified an identical mutation in a non-coding region of the splicing factor, SF3B1. While intriguing and included as part of the standard Tapestri myeloid mutation panel, the clinical implications of this SF3B1 mutation are not known and will require detailed epidemiologic studies to elucidate in the context of myeloid malignancies in both pediatric and adult patients.
Figure 2.

Single-cell proteogenomic sequencing analysis of clonal heterogeneity in pAML
Single-cell proteogenomic profiling was performed using the MissionBio Tapestri system to identify clonal somatic mutations and proteomic alterations. Uniform manifold approximation and projection (UMAP) plots (left) are shown from three separate pAML patients and were generated using eight antibody-oligo conjugates (CD3, CD7, CD11b, CD34, CD38, CD45, CD56, and ROR1) and a DNA mutation panel covering 45 myeloid genes with 312 amplicons.
(A) pAML 266; correspondence of immunophenotyping and single nucleotide variant (SNV) analysis with ROR1, CD7, CD38, and CD34 cell surface protein expression is shown in a heatmap with each row associated with a single cell (right).
(B) pAML 451; correspondence of immunophenotyping and SNV analysis with CD34, CD38, CD7, and ROR1 cell surface protein expression is shown in a heatmap with each row associated with a single cell (right).
(C) pAML 678; correspondence of immunophenotyping and SNV analysis with CD7, CD38, CD34, and ROR1 cell surface protein expression is depicted (right). See also Figure S2.
LSC signature identification in pAML
The relative paucity of somatic mutational drivers in pAML samples suggests that other molecular mechanisms, including pre-mRNA splicing alterations, may fuel therapeutic resistance and LSC generation.4 While malignant pre-mRNA splicing deregulation has been shown to be an essential driver of therapy-resistant LSC generation in adults, it has not been reported in pAML. To investigate the role of malignant pre-mRNA splicing in pAML, we collected bone marrow or peripheral blood from children diagnosed with AML (pAML; 1 to 14 years of age) and age-matched non-leukemic individuals (pNL) followed by differential gene expression and splice isoform RNA-seq analyses. Also, we analyzed adult dnAML patients (34 to 83 years of age) and adult secondary AML (sAML; 59 to 82 years of age) patients who were previously shown to harbor distinct splicing patterns (Figures 1A, S1A, and S1B, Table S1).4
Several adult dnAML and sAML studies show that LSCs can be enriched in the CD34+CD38−Lineage– HSC or the CD34+CD38+Lineage− HPC compartment and can be distinguished from normal blood cells based on gene expression and splicing signatures as well as the capacity to self-renew in immunocompromised mouse models.21,22 Similar to adults with AML, the frequency of LSCs can distinguish children with AML that are more likely to become resistant to treatment regimens aimed at eliminating dividing cells.23,24 Accordingly, characterization and elimination of LSCs appear to be critical. To investigate the cell type and context-specific role of malignant pre-mRNA splicing in pAML pathogenesis, we performed RNA-seq on FACS-purified CD34+CD38−Lineage– cells (HSCs) and CD34+CD38+Lineage– cells (HPCs).
In sorted HSCs, differential gene expression analyses detected 178 genes that were significantly differentially expressed in pAML versus age-matched non-leukemic controls (Table S2; adjusted p value < 0.05). Initially, we evaluated the expression of leukemia-associated markers that have previously been used to discriminate LSCs from normal HSCs. These phenotypic markers include CD47, CLL-1 (also known as CLEC12a), TIM3, CD7, CD123, CD96, CD33, and CD44.25,26 Using this RNA-seq analysis platform, we found significantly increased expression levels for each of these LSC markers in HSCs derived from pAML compared with non-leukemic (pNL) samples (Figure 3A).21,27 In contrast to pNL HSCs, LSC gene expression patterns were similar between pAML and adult dnAML HSCs (Figure 3B).28 Because of its role in evasion of programmed cell removal by macrophages, CD47 upregulation in pAML HSCs is particularly notable as a potential driver of therapeutic resistance.
Figure 3.

Whole-transcriptome detection of splicing deregulation in pAML compared with pediatric non-leukemic (pNL) hematopoietic stem cells
(A and B) Transcript expression levels (log counts per million; CPM) of LSC-associated phenotypic markers in pAML (n = 9) versus pNL (n = 3; A) and adult dnAML (n = 3; B) derived hematopoietic stem cells (HSCs; moderated t statistic with Benjamini-Hochberg correction method for multiple testing; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005).
(C) Schematic of alternative splicing event detection.
(D) Quantification of differential splicing events, including exon skipping (ES), intron retention (IR), alternative 3′ splice site usage (A3SS), and alternative 5′ splice site usage (A5SS) in pAML HSCs compared with pNL HSCs using multivariate analysis of transcript splicing (rMATS) bioinformatics analysis.
(E) Percentage of exon skipping in pAML HSCs compared with pNL HSCs.
(F) Heatmap of top 200 distinct splice isoforms in pAML HSCs versus pNL HSCs.
(G) Quantification of differential splicing events (ES, IR, A3SS, A5SS) in pAML HSCs versus adult dnAML HSCs.
(H) Functional enrichment analysis of pAML HSCs versus adult dnAML HSCs. See also Figure S3.
In addition, we compared several gene expression signatures that have been reported in the literature in HSCs and HPCs (Figure S3A).29,30,31,32 As expected, sorted HSCs derived from adult dnAML cannot be distinguished from pAML-derived HSCs as both cell populations include malignant cells (Figure S3B). In contrast, pAML-derived HSCs can be distinguished from non-leukemic HSCs derived from age-related bone marrow aspirates (Figures S3C–S3E). Similar results were obtained with FACS-purified HPCs derived from pAML, pNL, and adult dnAML aspirates. Together, these data suggest an enrichment of LSC expression signatures in both the CD34+CD38−Lineage– HSC and the CD34+CD38+Lineage− HPC compartment in pAML.
Detection of pre-mRNA splicing disruption in LSCs
By employing multivariate analysis of transcript splicing, rMATS, we detected alternative splicing alterations in hg38-aligned whole-transcriptome RNA sequencing data derived from pAML and non-leukemic HSCs and HPCs FACS-purified from age-matched bone marrow aspirates (Figure 3).33 In these splicing analyses, we detected 2,987 differential splicing events involving 1,982 genes (false discovery rate, FDR < 0.05; percent spliced in, PSI > 0.1) in pAML-derived HSCs compared with age-matched controls. These alternative splicing events included exon skipping (ES), intron retention (IR), and alternative 5′ splice site (A5SS) as well as alternative 3′ splice site (A3SS) usage (Figure 3C, Table S3).34 ES was the most frequent event in our analysis with more than 2,000 differential events identified (Figure 3D). Interestingly, the cassette exons identified were skipped more frequently in pAML HSCs compared with non-leukemic controls (Figure 3E). In addition, we identified distinct differences in splice isoforms expressed by pAML compared with non-leukemic HSCs and HPCs (Figures 3F and S3F). Prominent splicing differences between pAML and age-matched normal HSCs and HPCs suggest that a reasonable therapeutic index may be achieved with selective small molecule splicing modulatory strategies in pAML.
Next, comparative RNA-seq was used to distinguish splicing patterns between pAML and adult dnAML. Notably, we found 3,335 differential splicing events involving 2,071 genes in pAML HSCs compared with adult dnAML HSCs (FDR < 0.05; PSI > 0.1; Figures 3G and S3G). To identify genes that were differentially spliced in pAML only, we investigated the overlap between differential splicing events in pAML compared with adult dnAML, and pAML compared with pNL. Subsequently, splicing events unique to pediatric HSCs were utilized in enrichment analysis to identify associated pathways and functions. Functional enrichment analysis revealed widespread effects of genes involved in mRNA processing, including several members of the serine/arginine-rich gene family and numerous heterogeneous human ribonucleoproteins (Figure 3H). Similarly, differentially splicing events and the associated genes in pAML-derived HSCs compared with adult sAML-derived HSCs showed a comparable distribution of event types and overlapping categories in the enrichment analysis results (Figure S3H). These results underscore fundamental splicing differences between pAML and adult AML. Collectively, these data emphasize the importance of studying the cell type and context-specific pathogenesis of pAML.
Live lentiviral splicing biosensor reporter assays quantify leukemia propagation
To further analyze alternative splicing events in pAML cells, we re-engineered a lentiviral EF1alpha (EF1a) promoter-driven MAPT-mini-cassette-containing dual-fluorescence GFP/RFP reporter capable of detecting ES (GFP) and exon inclusion (RFP) at the single-stem-cell level (Figures 4A and 4B).35 Specifically, we transduced immunomagnetic bead-selected CD34+ cells derived from pAML bone marrow aspirates as well as pAML cell lines with the lentiviral dual-fluorescence splicing reporter. Using this lentiviral splicing reporter assay, we identified a strong GFP signal indicative of ES in pAML CD34+ cells (Figure 4C). Moreover, we found that CD34+ cells from pAML patients harbored significantly increased levels of GFP when compared with CD34+ cells derived from healthy umbilical cord blood (Figure 4D). In addition to validating RNA-seq-based data from pAML-derived HSCs versus age-matched controls, this innovative lentiviral dual-fluorescence splicing reporter system enables real-time in vitro and in vivo quantification of splicing alterations that will facilitate therapeutic splicing modulator development.
Figure 4.

Lentiviral splicing reporter and RNA sequencing-based detection of splicing alterations in pAML
(A) Schematic of lentiviral dual-fluorescence MAPT splicing reporter.
(B) Representative image of Kasumi-1, a pAML cell line, stably transduced with the pCDH-EF1-IRES-Puro MAPT splicing reporter lentiviral vector. Left: confocal fluorescence microscopic image of lentiviral splicing reporter GFP expression (scale bar, 50 μm). Right: confocal fluorescence microscopic image of lentiviral splicing reporter RFP expression (scale bar, 50 μm).
(C) FACS analysis of GFP and RFP mean fluorescence intensity (MFI) in the live cell population of CD34+ selected pAML patient samples (n = 4 biological replicates).
(D) FACS analysis of GFP MFI in the live cell population of CD34+ selected pAML samples versus CD34+ cord blood (CB) samples (Student’s t test, p = 0.029; n = 4 biological replicates).
(E) Whole-transcriptome (RNA-seq)-based quantification of CD44 splice isoforms in pAML (n = 9, blue) versus pNL (n = 3; red) HSCs (Student's t test, p < 0.05).
(F) RNA-seq-based quantification of CD47 splice isoform expression in pAML (blue) versus pNL (red) HSCs (Student's t test, CD47-201 p = 0.3766; CD47-202 p = 0.0019; CD47-204 p = 0.0167; CD47-205 p = 0.0134). See also Figure S4.
Identification of splicing deregulation biomarkers of splicing deregulation in pAML stem cells
While we identified global splicing differences between pAML and age-matched non-leukemic cells, we also identified specific changes in pro-survival splice isoform expression.4,36 Specifically, we found that the pro-survival splice isoform MCL1-L, a member of the BCL2 gene family, was highly expressed in pAML HPCs when compared with non-leukemic HPCs (Figures S4A and S4B). Along with MCL1-L, we detected upregulation of pro-survival BCL2-L transcript levels in pAML HSCs and HPCs (Figure S4C). In addition, we identified differential splicing of the A-to-I RNA editing enzyme, ADAR1. Specifically, the ratio of the interferon-inducible splice isoform of ADAR1, ADAR1 p150, was significantly increased compared with the non-interferon-responsive isoform, ADAR1 p110, in pAML compared with pNL HSC and HPC samples (Figure S4D). In accordance with an increased ratio of ADAR1 p150 to ADAR1 p110, we identified widespread editing events in pAML compared with pNL HSCs (Figure S4E). Concurrently, we identified RNA editing and concomitant differential splicing of PTPN6, a protein tyrosine phosphatase that serves as a driver of oncogenic transformation (Figures S4F and S4G).37 Since A-to-I RNA editing can remove the 3′ splice acceptor adenosine, future studies will explore whether ADAR1 activity can be linked or even cause differential exon usage in pAML.38
Interestingly, LSC-related genes, including CD44 and CD47, were differentially spliced. Specifically, we identified significantly increased levels of CD44-008 (also known as CD44v6), an important driver of cancer stem cell self-renewal (Figures 4E and S4H).28,36 In addition, we found differential splicing of CD47, the malignant “don’t eat me signal” that enables evasion of programmed cell removal by macrophages (Figures 4F and S4I).17,18 Previously, we discovered that downregulation of muscleblind-like splicing regulator 3 (MBNL3) was associated with activation of human embryonic stem cell (ESC) alternative splicing involved in expression and splicing of CD44 as a driver of blast crisis LSC generation in chronic myeloid leukemia.36 More recently, it was shown that splicing of CD47 is regulated by RNA binding fox-1 homolog 2 (RBFOX2), a master regulator of alternative splicing in embryonic stem cells.39,40,41 Thus, we investigated the relative roles of MBNL genes and RBFOX2 in pAML splicing deregulation.
Decreased RBFOX2 expression in pAML
Recently, expression of RBFOX2 (also known as RBM9) was detected in the human T cell line JURKAT.42 Subsequently, a role for RBFOX2 was identified in B cell non-Hodgkin lymphoma and in the human pAML cell line Kasumi-1. This pAML cell line harbors a RUNX1/RUNX1T1 fusion oncogene, which is typified by a 2-fold reduction in RBFOX2 expression.43,44 In addition, the splicing regulator RBFOX2 promotes ESC survival. Together with MBNL proteins, as well as pyrimidine tract binding (PTBP) proteins, RBFOX2 is a pre-mRNA splicing regulator that is normally expressed at lower levels in ESCs compared with differentiated cell types.40,45
Strikingly, both pAML-derived HSCs and HPCs showed significant downregulation of RBFOX2 (Figures 5A and S5A). Similar to the pattern of reduced RBFOX2 expression in pAML and ESCs, pAML HSCs showed decreased expression of MBNL1 (Student’s t test, p = 0.022) and MBNL2 (Student’s t test, p = 0.018) (Figure 5B, Table S2).42 Also, we identified significantly increased expression of a CELF2, a repressor of both RBFOX2 and MBNL genes, in HPCs (Student’s t test, p = 0.035) (Table S2).40 Since RBFOX2 was the only significantly differentially expressed gene related to splicing in both HSC and HPC cell types, we explored the contribution of RBFOX2 to splicing deregulation in pAML.
Figure 5.

RBFOX2 downregulation and reversion to an embryonic stem cell splicing program in pAML
(A) RBFOX2 protein expression levels were quantified in pAML (n = 5 biological replicates) and cord-blood-derived HSCs (n = 3 biological replicates) and hematopoietic progenitor cells (HPCs; pAML, n = 5; pNL, n = 4 biological replicates) by FACS analysis as shown by MFI (Student’s t test HSC [left] p = 0.04; HPC [right] p = 0.06).
(B) RNA-seq-based cytoscape network analysis of RBFOX2 and embryonic stem cell (ESC) specific alternative splicing program-related transcripts. Nodes are colored according to log fold change.
(C) Volcano plot highlighting RBFOX2 target genes that show differential exon usage in pAML (false discovery rate, FDR < 0.05; percent spliced in, PSI > 0.1). A negative PSI reflects exon inclusion in pAML, while a positive PSI reflects ES in pAML versus pNL cells.
(D) Heatmap of exon expression levels after unsupervised clustering of RBFOX2 target genes in pAML HSCs and HPCs (n = 9 and n = 8, respectively; blue) compared with pNL (n = 3 and n = 6, respectively; red).
(E) Cytoscape network analysis of differentially spliced RBFOX2 target genes implicated in “Pathways in Cancer” in pAML HPCs. The top 200 ES events in this comparison were used as seeds for network propagation on the STRING high-confidence interactome. Differential expression log fold change is mapped to node color. Triangles depict seed list genes, and circles represent associated genes. See also Figure S5.
Subsequently, we performed motif enrichment analysis using RNA mapping analysis and plotting server (rMAPS).46 RBFOX2 is known to promote ES when binding upstream of an exon, and exon inclusion when binding downstream of an exon. In our analyses, HPCs showed significant enrichment for the RBFOX2 binding motif upstream of the alternatively spliced exon (Figure S5B). Next, we identified RBFOX2 target genes based on the oRNAment database, which uses an algorithm to predict genes with target binding sites, and an eCLIP study.39,41 Genes present in both studies were used for subsequent analysis of RBFOX2 target genes (Figure 5C). In this analysis, we focused on genes that have been shown to have differential exon usage in ESCs.40 Consistent with the behavior in ESCs, these exons were found to be highly expressed in pAML (Figure 5D). Thus, both pAML HSCs and HPCs express exons linked to RBFOX2 repression in ESCs. These data are indicative of reversion to an embryonic splicing pattern in pAML. Thus, we performed network propagation analysis on the STRING high confidence interactome using the top differentially spliced RBFOX2 target genes as a seed list.47 Together, these findings implicate that decreased RBFOX2 expression drives widespread splicing deregulation of several processes key for leukemia survival (Figures 5E and S5C). Notably, this RBFOX2 differentially spliced target gene analysis revealed upregulation of “the don’t eat me signal,” CD47 (Figure 5E), thereby underscoring the potential importance of immune evasion in pAML pathogenesis. Moreover, differential expression of RBFOX2 and differential splicing of RBFOX2 target genes also seems important when comparing pAML and adult dnAML (Figures S5D and S5E).
Embryonic splicing leads to splicing modulator sensitivity in pAML
Next, we examined the direct contribution of RBFOX2 to self-renewal, as measured in hematopoietic colony replating assays. First, we examined self-renewal upon lentiviral shRNA knockdown of RBFOX2 in cord-blood-derived hematopoietic stem and progenitor cells (HSPCs; Figure S6A). In this study, shRNA knockdown of RBFOX2 significantly increased the replating capacity of cord-blood-derived HSPCs (Figure 6A).
Figure 6.

Rebecsinib inhibition of pAML LSC survival and self-renewal
(A) Colony assay of CD34+ cord blood cells in long-term stromal co-cultures following lentiviral shRNA control (shControl, n = 7 biological replicates) or shRNA knockdown of RBFOX2 (shRBFOX2, n = 7 biological replicates; Student’s t test, p = 0.03). Data represent mean ± SD.
(B) Percent colony survival and self-renewal (replating) of CD34+ cord blood samples after treatment with 100 or 1,000 nM of Rebecsinib normalized to vehicle-treated controls. Mean colony percentages ± SD are shown for each treatment condition (n = 7 biological replicates). Statistical analysis was performed by one-way ANOVA (survival [left] 100 nM Rebecsinib shControl versus shRBFOX2 p = 0.003, 1,000 nM Rebecsinib shControl versus shRBFOX2 p < 0.001; self-renewal [right] 100 nM Rebecsinib shControl versus shRBFOX2 p < 0.001, 1,000 nM Rebecsinib shControl versus shRBFOX2 p < 0.001).
(C) Survival and self-renewal of CD34+ cells derived from pAML (n = 6 biological replicates) or cord blood (CB) (n = 7 biological replicates) in long-term stromal co-cultures. Data represent mean ± SD for each treatment condition. Statistical analysis was performed by one-way ANOVA (survival [left] 0.1 μM Rebecsinib pAML versus CB p = 0.001, 1 μM Rebecsinib pAML versus CB p = 0.001, 10 μM Rebecsinib pAML versus CB p = 0.001; self-renewal [right] 0.1 μM Rebecsinib pAML versus CB p = 0.001, 1 μM Rebecsinib pAML versus CB p = 0.001, 10 μM Rebecsinib pAML versus CB p = 0.001).
(D) Self-renewal of CD34+ pAML (n = 6 biological replicates) versus adult dnAML (n = 9 biological replicates) treated with Rebecsinib. Data represent mean ± SD for each treatment condition. Statistical analysis was performed by one-way ANOVA (pAML versus adult dnAML p < 0.001).
(E) Representative fluorescence microscopic imaging analysis of the pAML cell line Kasumi-1 cells stably transduced with the lentiviral pCDH-EF1-IRES-Puro MAPT dual-fluorescence splicing reporter treated with (left) DMSO (scale bar, 200 μm) and (middle) 100 nM Rebecsinib (scale bar, 200 μm). Right: the ratio of RFP/GFP was quantified using Volocity software (Quorum Technologies) following Rebecsinib treatment and normalized to DMSO vehicle controls (n = 1).
(F) FACS analysis of RFP/GFP MFI in the live cell population of CD34+ selected pAML patient samples treated with 100 nM Rebecsinib for 72 h (n = 6 biological replicates, Student's t test, p = 0.016).
(G) FACS analysis of RFP/GFP MFI in MOLM13 (a leukemia cell line derived from a 20-year-old male) cells stably transduced with the lentiviral pCDH-EF1a-IRES-Puro MAPT dual-fluorescence splicing reporter and treated in vitro with Rebecsinib (100 nM) versus DMSO (n = 3 technical replicates, data represent mean ± SEM, Student's t test, p < 0.0001).
(H) MOLM13-MAPT splicing reporter stably transduced cells were engrafted into irradiated NSG-SGM mice, treated with vehicle (n = 3 biological replicates) or Rebecsinib (10 or 20 mg/kg; n = 2, n = 3 technical replicates), and sacrificed 24 h post-treatment. The ratio of RFP/GFP MFI in live, human CD45+ cells in bone marrow, spleen, and peripheral blood was quantified by FACS analysis using a MACS Quant instrument and FlowJo software. Data represent mean ± SEM for each treatment condition. Statistical analyses were performed using unpaired, two-tailed Student’s t test (bone marrow [left] vehicle versus 20 mg/kg p = 0.0013, peripheral blood [middle] vehicle versus 20 mg/kg p = 0.0488, spleen [right] vehicle versus 20 mg/kg p < 0.0001). See also Figure S6.
In parallel, we examined the response to splicing modulation using the small molecule inhibitor Rebecsinib, previously known as 17S-FD-895.4 Rebecsinib, which binds within the spliceosome core adjacent to SF3B1, SF3B3, and PHF5A, induces alternative intron and exon usage and reduces self-renewal and survival of LSCs in adult sAML.4 Strikingly, the knockdown of RBFOX2 in cord-blood-derived HSPCs significantly increased sensitivity to Rebecsinib in both colony survival and colony replating assays (Figure 6B). Next, we performed functional hematopoietic colony survival and self-renewal (replating) assays in HSPCs isolated from pAML bone marrow aspirates. In these Rebecsinib sensitivity experiments, we detected a dose-dependent reduction in clonogenicity and replating (Figure 6C). This effect was detected at doses as low as 100 nM in pAML, whereas cord-blood-derived HSPCs appeared to be unaffected (Figure 6C). Notably, pAML samples were significantly more sensitive to splicing modulation than adult dnAML and similar in sensitivity to sAML (Figures 6D and S6B). Interestingly, splicing modulation with Rebecsinib induced MCL1 exon 2 skipping, thereby eliciting pro-apoptotic MCL1-S isoform expression rather than pro-survival MCL1-L transcripts expression (Figure S6C and Table S4). Moreover, we established the relative potency of Rebecsinib using the same leukemic cell line, MOLM13, which had been previously used to assess the potency of other splicing modulators, including E7107 and H3B-8800 (Figure S6D).48,49
In addition, we treated Kasumi-1 leukemic cells transduced with the lentiviral splicing reporter with 100 nM Rebecsinib and performed time-lapse fluorescence microscopic imaging analysis (Figure 6E). Throughout the time course, there was an increase in the ratio of RFP to GFP (normalized to DMSO-treated cells), which was indicative of splicing modulation following Rebecsinib treatment (Figure 6E) In addition, we transduced the lentiviral splicing reporter and treated HSPCs isolated from pAML peripheral blood samples and bone marrow aspirates. Again, we found an increase in the ratio of RFP to GFP indicative of splicing modulation following Rebecsinib (Figure 6F). These results suggest that global ES in pAML can be corrected by splicing modulation treatment using Rebecsinib.
Subsequently, we treated MOLM13 cells stably transduced with the lentiviral splicing reporter (MOLM13-MAPT) with 100 nM Rebecsinib and found a similar increase in the ratio of RFP to GFP by flow cytometry after treatment (Figure 6G). Then, we engrafted MOLM13-MAPT intravenously into sublethally irradiated (150 cGy) NSG-SGM mice and treated them with vehicle or Rebecsinib (10 mg/kg or 20 mg/kg) and sacrificed the mice 24 h post-treatment. The ratio of RFP to GFP was calculated in live, human CD45+ cells retrieved from bone marrow, spleen, and peripheral blood by flow cytometry (Figure 6H) and in vivo fluorescent imaging (Figure S6E). These in vivo data show a significant increase in the RFP-to-GFP ratio between the vehicle and 20 mg/kg Rebecsinib group in all tissues and recapitulate the dose-dependent increase in the RFP-to-GFP ratio seen in in vitro assays. Taken together, these data suggest that splicing deregulation induces a dose-dependent therapeutic vulnerability to pharmacologic splicing modulation.
Discussion
Compared with adult dnAML, pAML is associated with a relatively low mutational burden. This likely stems from the relative dearth of DNA-damaging environmental exposures and age-related clonal hematopoiesis in children.13,16,50,51 Hence, we posited that transcriptomic and epitranscriptomic (post-transcriptional) rather than genomic alterations may prevail as drivers of LSC generation in children.13
To investigate this hypothesis, we utilized (1) whole-exome and single-cell proteogenomics analyses focused on myeloid mutations, (2) transcriptome-wide analyses of FACS-purified HSCs and HPCs followed by differential splicing analyses, (3) dual-fluorescence lentiviral single-stem-cell splicing reporter assays, (4) HSC and HPC survival and replating assays, and (5) humanized pAML mouse models treated with a selective splicing modulator, Rebecsinib. Using this combinatorial approach, we identified splicing deregulation typified by increased ES levels in pAML HSCs compared with age-matched non-leukemic HSC samples.52 In addition, we identified fundamental differences between pAML and adult dnAML, which emphasize the importance of studying differential splicing in pAML specifically. Beyond classes of genes known to be associated with cancer, such as in cell cycle and metabolic pathways, the process of pre-mRNA splicing was frequently disrupted in pediatric samples.
Intriguingly, in two out of three patients studied by single-cell proteogenomics analyses, we identified an identical mutation in a non-coding region of the splicing factor SF3B1, which is included in the standard Tapestri myeloid mutation panel. However, the clinical significance of this mutation has not been elucidated. Moreover, there was no identifiable correlation between clonal somatic mutation profiles and immunophenotype suggesting that epigenetic or epitranscriptomic (post-transcriptional) alterations, rather than splicing factor mutations, contribute to splicing deregulation in pAML. As a potential driver of splicing deregulation, we found significant downregulation of RBFOX2, a repressor of embryonic splicing, in pAML. Moreover, decreased RBFOX2 expression correlated with differential exon usage and upregulation of LSC survival, self-renewal, and immune evasion splice isoforms. Specifically, CD47 was one of the few genes found to be both differentially spliced and expressed in pAML. Increased expression of CD47 indicates that LSCs in pAML may not be cleared by endogenous macrophages.17 Interestingly, CD47 blockade using an anti-CD47 antibody, now known as magrolimab, was recently shown to be effective in eliminating AML stem cells in adults.53 Future studies will focus on uncovering the potential of this immune checkpoint inhibitor in pAML.
In addition, we detected MCL1 and BCL2 pro-survival isoforms as well as ADAR1p150 self-renewal isoform upregulation in pAML HSC. Further studies demonstrated that splicing deregulation in pAML induced a therapeutic vulnerability to a small molecule splicing modulator, Rebecsinib (17S-FD-895), in lentiviral splicing reporter assays in vitro and in humanized pAML mouse models.4,49,54 Together, these data indicate that enhanced pAML LSC survival and self-renewal is fueled by alternative splicing and potentially through RNA editing. In the future, we will focus on pAML serial LSC transplantation experiments with Rebecsinib as well as ADAR1 reporter studies that may further define a link between splicing deregulation, RNA editing, and LSC propagation. In addition, we will focus our studies on biomarkers of potential drug response.
In summary, we have identified splicing deregulation, in the setting of RBFOX2 downregulation, which induces a therapeutic vulnerability to a small molecule splicing modulator, Rebecsinib. This approach may be utilized to detect and target therapy-resistant LSCs in pAML and cancer stem cells in other malignancies that become therapeutically recalcitrant as a consequence of splicing deregulation.
Limitations of the study
The insights garnered regarding pAML stem cell splicing deregulation in this study underscore the importance of interpatient variability as well as the cell type and context-specific changes in splicing and responses to targeted splicing modulation. While a non-coding SF3B1 splicing mutation was detected in two of the patients by single-cell proteogenomic sequences, the clinical significance of this finding still needs to be elucidated. The paucity of patient samples for this rare disease and the study of rare stem cell populations was also challenging with regard to RNA sequencing-based alternative splicing analyses. However, these challenges provided the impetus for developing lentiviral dual-fluorescence splicing reporter systems that are more quantitative and can be evaluated in real time both in vitro and in vivo. In future investigational new drug (IND) enabling studies, RNA sequencing-based biomarker development will be performed to further elucidate the selectivity of Rebecsinib as well as the therapeutic index prior to initiating a phase 1 clinical trial. While increases in ADAR1-mediated RNA editing and splicing deregulation suggest that transcript editing and splicing may change protein levels, further studies of both protein isoforms and expression levels will need to be incorporated as an important IND enabling future direction to conform with the pediatric research equity act.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD45 BB515 (Clone HI30) | BD | Cat# 564586; RRID: AB_2869588 |
| CD38 PE-Cy7 | BD | Cat# 335790; RRID: AB_399969 |
| CD45 APC | Invitrogen | Cat# MHCD4505; RRID: AB_10372216 |
| CD34 BV421 (Clone 581) | BD | Cat# 562577; RRID: AB_2687922 |
| CD8-PE-Cy 5.5 | BD | Cat# 555368; RRID: AB_395771 |
| CD56− PE-Cy 5.5 | BD | Cat# 555517; RRID: AB_395907 |
| CD4− PE-Cy 5.5 | BD | Cat# 555348; RRID: AB_395753 |
| CD3− PE-Cy 5.5 | BD | Cat# 555334; RRID: AB_395741 |
| CD19− PE-Cy 5.5 | BD | Cat# 555414; RRID: AB_395814 |
| CD2− PE-Cy 5.5 | BD | Cat# 555328; RRID: AB_395735 |
| CD14− PerCP-Cy5.5 | BD | Cat# 550787; RRID: AB_393884 |
| CD8-PE-Cy 5.5 | BD | Cat# 555368; RRID: AB_395771 |
| RBFOX2 (anti-Fox2/RBM9) | Abcam | Cat# ab57154; RRID: AB_2285090 |
| Goat anti-mouse IgG2a H&L (PE) | Abcam | Cat# ab74490; RRID: AB_1310345 |
| Bacterial and virus strains | ||
| 2ND and 3rd Generation HIV Lentivirus | Core facility at UC San Diego | N/A |
| Biological samples | ||
| CD34+ Cord Blood | AllCells or Lonza | https://allcells.com; Cat# 2C-101 |
| Adult AML patient samples | Obtained from patients in clinical trials of the Dutch-Belgian Cooperation Trial Group for Hematology-Oncology (HOVON) according to METc-VUmc | N/A |
| Adult secondary AML patient samples | Obtained through patients consented at UC San Diego Health according to Institutional Review Board-approved protocols | For patient characteristics see Crews et al.4 |
| Pediatric AML and pediatric non-leukemic patient samples | Obtained from patients through a written informed consent from the Dutch Childhood Oncology Group (DCOG) according to METc-VUmc | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Rebecsinib (17S-FD-895) | Dr. Michael Burkart (Villa et al.55) | N/A |
| Critical commercial assays | ||
| RNeasy Micro Kit | Qiagen | Cat# 74004 |
| Super-Script III | ThermoFisher Scientific | Cat# 11752250 |
| SYBR GreenER | ThermoFisher Scientific | Cat# 11761500 |
| Live/Dead Fixable NearIR Dead Cell Stain Kit | ThermoFisher Scientific | Cat# L10119 |
| Live/Dead Fixable Aqua Dead Cell Stain Kit | ThermoFisher Scientific | Cat# L34957 |
| CD34 Micro-Bead Kit, Ultra-Pure, human | Miltenyi | Cat# 130-100-453 |
| MyeloCult™ H5100 | Stem Cell Technologies | Cat# 05150 |
| MethoCult™ H4330 | Stem Cell Technologies | Cat# 04330 |
| Mission Bio Tapestri Single Cell (Myeloid DNA Core Ambient Kit Version 2) | Mission Bio | https://missionbio.com |
| Deposited data | ||
| Analyzed data and code | This paper | https://github.com/ucsd-ccbb/pAML_StemCell_Methods |
| Generated dataset | This paper | dbGaP: http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs003196.v1.p1; accession code: phs003196.v1.p1 |
| Experimental models: Cell lines | ||
| Kasumi-1 | ATCC | Cat# CRL-2724; RRID:CVCL_0589 |
| THP-1 | ATCC | Cat# TIB-202; RRID:CVCL_0006 |
| KG1a | ATCC | Cat# CCL-246.1; RRID:CVCL_1824 |
| MOLM-13 | DMSZ | Cat# ACC-554; RRID:CVCL_2119 |
| SI/SI (IL-3, CSF) (SL) | StemCell Technologies Ltd. | Cat# 00302 |
| M2-10B4 (IL-3, G-CSF) (M2) | StemCell Technologies Ltd. | Cat# 00301 |
| Experimental models: Organisms/strains | ||
| Mouse: NOD.Cg-PrkdcscidIl2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ (NSGS-SGM) | Jackson Labs | N/A |
| Oligonucleotides | ||
| Human HPRT primers | Crews et al.4 | N/A |
| Human MCL1-L primers | Goff et al.56 | N/A |
| Human MCL1-S primers | Goff et al.56 | N/A |
| Recombinant DNA | ||
| MAPT Reporter | Stoilov et al.35 | N/A |
| pCDH-EF1a-IRES-Puro MAPT | This paper | See Figure 4 |
| pSMART hEF1a ShRBFOX2 | Horizon Discovery (Waterbeach, UK) | https://horizondiscovery.com/en/gene-modulation/knockdown/shrna/products/smartvector-lentiviral-shrna?nodeid=entrezgene-23543 |
| pSMART hEF1a ShControl | Horizon Discovery (Waterbeach, UK) | https://horizondiscovery.com/en/gene-modulation/knockdown/controls/products/smartvector-lentiviral-controls |
| Software and algorithms | ||
| Bcl2fastq (v2.17) | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html | |
| FastQC | Wingett and Andrews57 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| STAR | Dobin et al.58 | https://github.com/alexdobin/STAR |
| RSEM | Li and Dewey59 | https://deweylab.github.io/RSEM/ |
| GENCODE | https://www.gencodegenes.org/human/release_19.html | |
| EdgeR | Robinson et al.60 | http://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Limma | Ritchie et al.61 | https://www.bioconductor.org/packages/release/bioc/html/limma.html |
| Limma-voom | Law et al.62 | https://www.bioconductor.org/packages/release/bioc/html/limma.html |
| webGestalt | Liao et al.63 | http://www.webgestalt.org/ |
| GSVA | Hänzelmann et al.64 | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| gProfiler | Reimand et al.65 | https://biit.cs.ut.ee/gprofiler/page/r |
| rMATS turbo | Shen et al.66 | https://github.com/Xinglab/rmats-turbo |
| rmats2sashimiplot | https://github.com/Xinglab/rmats2sashimiplot/ | |
| rMAPS2 Motifi Map | Hwang et al.46 | http://rmaps.cecsresearch.org/MTool/ |
| Mosaic | https://github.com/MissionBio/mosaic-jupyter | |
| STRING high confidence interactome | Szklarczyk et al.47 | 9606.protein.links.v11.0.txt |
| Cytoscape | Shannon et al.67 | https://cytoscape.org/ |
| VisJS2jupyter | Rosenthal et al.68 | https://github.com/ucsd-ccbb/visJS2jupyter |
| network_bio_toolkit | https://github.com/ucsd-ccbb/network_bio_toolkit | |
| PicardTools | https://broadinstitute.github.io/picard/ | |
| Genome Analysis Toolkit (GATK) | McKenna et al.69 | https://gatk.broadinstitute.org/hc/en-us |
| RepeatMasker | https://www.repeatmasker.org/ | |
| FlowJo | FLOW JO LLC | https://www.flowjo.com/ |
| GraphPad Prism | GraphPad Software Inc. | https://www.graphpad.com/scientific-software/prism/ |
| Microsoft Excel | Microsoft | |
| GATK Resource Bundle | https://gatk.broadinstitute.org/hc/en-us/articles/360035890811-Resource-bundle | |
| Gnomad | https://gnomad.broadinstitute.org/ | |
| Other | ||
| Spliceosome Protein Annotations | Cvitkovic and Jurica70 | http://spliceosomedb.ucsc.edu/ |
| ExAC | https://gnomad.broadinstitute.org/downloads#exac-variants | |
| RNA Binding Protein Targets by eCLIP | Van Nostrand et al.39 | N/A |
| oRNAment | Benoit et al.41 | N/A |
| REDIportal.80 | Lo Giudice et al.71 | N/A |
| dbSNP | Sherry et al.72 | N/A |
Resource availability
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Catriona Jamieson (cjamieson@health.ucsd.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact (C.H.M.J) with a completed Materials Transfer Agreement.
Experimental model and subject details
Animal experiments
All mouse studies were completed in the Sanford Consortium vivarium in accordance with the University Laboratory Animal Resources and Institutional Animal Care and Use Committee of the University of California regulations (IACUC). Four-week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ (NSGS-SGM) mice were purchased from Jackson Laboratories. Both sexes were used in the study. After an acclimation period of one week, animals were irradiated with 150cGy. 24 h later, animals were injected with approximately 5 × 104 MOLM13 cells stably transduced with a lentiviral splicing reporter (MOLM13-MAPT) intravenously. Animals were housed in groups. Mice were randomly assigned to experimental groups. Experimental animals were maintained in the Sanford Consortium vivarium according to IACUC regulations (https://blink.ucsd.edu/_files/sponsortab/iacuc/Policy%206%20Breeding.pdf). Transplanted animals were monitored regularly for health assessment. Peripheral blood screening for human CD45+ was regularly performed to detect disease.
Human subjects
Cryopreserved mononuclear cells isolated from adult AML diagnostic bone marrow samples were obtained from patients in clinical trials of the Dutch-Belgian Cooperation Trial Group for Hematology-Oncology (HOVON) in accordance with the VU Amsterdam (Vrije Universiteit) research ethics review committee (ETCO), METc-VUmc, and the declaration of Helsinki. Pediatric AML and pediatric non-leukemic samples, obtained from lymphoma patients without bone marrow involvement, were collected after obtaining written informed consent from patients treated on the Dutch Childhood Oncology Group (DCOG), which has been approved by METc-VUmc. Based on CD34 positivity and age (pediatric versus adult), samples were included. CD34 negative samples were excluded from this study. Primary adult acute myeloid leukemia samples were obtained from consenting patients at the University of California in accordance with a UC San Diego human research protections program Institutional Review Board approved protocol (#131550). The IRB reviewed this protocol and found that it meets the requirements as stated in 45 CFR 46.404 and 21 CFR 50.51. Human cord blood samples were purchased as purified CD34+ cells from AllCells Inc or Lonza Bioscience. (See Table S1 for patient characteristics).
Cell lines
Human cell lines Kasumi-1 and MOLM13 were obtained from the American Type Culture Collection (ATCC, Manassas, VA) or the German Collection of Microorganisms and Cell Cultures (DMSZ) and cultured in RPMI-1640 supplemented with 20% fetal bovine serum. Murine cell lines SL and M2 were obtained from Stem Cell Technologies Ltd. and cultured in DMEM, and RPMI-1640, respectively, supplemented with 10% fetal bovine serum, 1% L-glutamine, and penicillin-streptomycin.
Method details
Next generation sequencing (NGS) analysis
For NGS (targeted whole exome sequencing) analyses, DNA was extracted from white blood cells isolated from blood or bone marrow specimens. Solution-based hybrid capture was used to select the mutational hotspot regions or (for a select group of genes) the entire coding region (all exons) of a panel of 124 genes. The resulting DNA libraries were sequenced using a sequencing-by-synthesis technology. The data were aligned to the Genome Reference Consortium Human Build 37 (GRCh37) reference sequence. Bioinformatics analysis was used to identify variants (single nucleotide changes, small insertions, or deletions) in the captured regions of the 124 genes at greater than 5% allele frequency and represented by a minimum of 100 reads (100x coverage). This test was adopted and its performance characteristics were determined by the UC San Diego Clinical Genomics Laboratory.
Single cell proteogenomics analysis
Patient samples were thawed, washed with 1x DPBS, quantified by hemacytometer and checked for viability by trypan blue staining (cell viability >95%). The cells were resuspended in 1xDPBS at a concentration of 8,000–10,000 cells/μL and incubated with TruStain FcX plus Staining Buffer (MissionBio), dextran sulfate (100 g/mL) for 3 min at room temperature. A pool of eight oligo-conjugated antibodies (CD3, CD7, CD11b, CD34, CD38, CD45, CD56, ROR1) (MissionBio) were added to the cells and incubated for 30 min at room temperature. The cells were then washed 3x with DPBS plus 5% fetal bovine serum followed by re-quantification and resuspended at 4,000 cells/μL in Tapestri Cell Buffer (MissionBio). The cell suspension was loaded into the Tapestri microfluidics cartridge (MissionBio) following the manufacturer’s procedure. Following successful encapsulation including the Myeloid Panel reverse DNA primer (30μM) (MissionBio), the cells were lysed, digested and barcoded with the Myeloid Panel forward DNA primer (30μM) (MissionBio) for the DNA and barcoded for protein with an antibody Tag primer (30μM). The barcoded cells were UV treated, subjected to PCR amplification per the manufacturer’s procedure. The DNA PCR products were isolated from individual droplets followed by purification with Ampure XP Beads (Beckman Coulter). The protein PCR products were obtained from the Ampure XP bead supernatant followed by DNA indexing primer. The protein-labeled PCR products were incubated with a biotin tag oligo (5μM) at 95°C for 5 min followed by incubation on ice for 5 min. The protein PCR products were purified using Streptavidin C1 beads (Thermo Fisher) and the beads were used as a PCR template for tagmentation with i5/i7 Illumina indices followed by a purification with Ampure XP beads. The DNA and protein libraries were quantified by Qubit fluorometry (Thermo Fisher) and the Agilent 2100 Tapestation (Agilent). The DNA and protein libraries were pooled and sequenced using the NextSeq 550 Mid-Output system by Scripps Sequencing Core. Following sequencing, the FASTQ single cell DNA and protein files were analyzed using the Python notebook Tapestri Mosaic pipeline (MissionBio).
Patient sample processing and preparation for whole transcriptome sequencing
Whole transcriptome sequencing (RNA-seq) was performed on pediatric (6 females and 10 males; age 1–14) and adult (1 female; 4 males; age 32–61) patients across two cell types (See Table S1 for patient characteristics). Peripheral blood or bone marrow derived mononuclear cells were isolated by Ficoll-paque density centrifugation, transferred, and stored in liquid nitrogen. CD34+ cells were selected from peripheral blood mononuclear cells from all patients by magnetic bead separation (MACS; Miltenyi, Bergisch Gladbach, Germany; CD34 Micro-Bead Kit, Ultra-Pure, human, Milteny).10 The CD34+ enriched fractions were stained with fluorescent antibodies against human CD45, CD34, CD38, Lineage markers (BD Pharmingen; CD2 PE-Cy5, 1:20, cat 555328; CD3 PE-Cy5, 1:20 cat 555334; CD4 PE-Cy5, 1:10, cat 555348; CD8 PE-Cy5, 1:50, cat 555368; CD14 PerCP-Cy5.5, 3:100, cat 550787; CD19 PE-Cy5, 1:50, cat 555414; CD20 PE-Cy5, 1:20, cat 555624; CD56 PE-Cy5, 1:10, cat 555517; CD45 APC, 1:50, cat 335790; CD34 BV421, 1:100, cat 562577; CD38 PE-Cy7, 1:50, cat 335790), and propidium iodide. Cells were FACS-purified using a FACS Aria II (Sanford Consortium Stem Cell Core Facility) into hematopoietic stem cell (Lin-CD45+CD34+CD38−) and progenitor (Lin-CD45+CD34+CD38+) populations directly into RLT lysis buffer (Qiagen) for RNA extraction (RNeasy Micro Kit, Qiagen) followed by RNA-Seq (The Scripps Research Institute Next Generation Sequencing Core).
RNA-sequencing analyses
RNA-Seq was performed on Illumina’s NextSeq 500 sequencer with 150bp paired-end reads. Sequencing data were de-multiplexed and produced as fastq files using Illumina’s bcl2fastq (v2.17). Quality control of the raw fastq files was performed using the software tool FastQC.57 Sequencing reads were aligned to the human genome (hg19) using the STAR v2.5.1a aligner.58 Read and transcript quantification was performed with RSEM v1.3.0 and GENCODE annotation (genocode.v19.annotation.gtf).59 The R BioConductor packages edgeR and Limma were used to implement the limma-voom method for differential expression analysis at both the gene and transcript levels.60,61,62 The experimental design was modeled upon disease in each tissue type (∼0 + disease). Genes or transcripts with an adjusted p value of <0.05 (based on the moderated t-statistic using the Benjamini-Hochberg (BH) method for multiple testing correction) were considered significantly differentially expressed (DE).73 Functional enrichment of the differentially expressed genes/transcripts was performed using the Bioconductor package webGesalt (v0.3.0) and Gene Set Enrichment Analysis using the Bioconductor package GSVA (v1.22.4) with the msigdb hallmark (H) and C2 ontologies (https://www.gsea-msigdb.org/gsea/msigdb/).64,74
Differential splicing analyses
To quantify splicing changes in all comparisons, rMATS turbo (version 4.1) was used to calculate Inclusion Level Differences for all detectable splicing changes and assign false discovery rate (FDR) adjusted p values.66 Sorted BAM files generated with STAR (v2.5.3a) were used as input along with read length (75) and the GRCh38 v82 gtf file (for transcript definitions).58 The rMATS software produces a list of annotated differential splicing events for 5 individual types: Alternative 5′ Splice Site (A5′SS), Alternative 3′ Splice Site (A3′SS), Mutually Exclusive Exon (MXE), Retained Intron (RI), and Skipped Exon (SE). Each type is defined by a combination of Inclusion Junctions (IJ) and Skipping Junctions (SJ), i.e. SE events incorporate two IJ’s that span the introns from the upstream exon to the included exon and from the included exon to the downstream exon, with one SJ that connects the upstream exon to the downstream exon. The normalized counts for the IJ’s and SJ’s are used to calculate Inclusion Levels for each sample, which are used for calculating Inclusion Level Difference between the two groups and the statistical determination of significance. Splicing Events with an FDR <0.05 were considered for downstream analysis.
After assigning significant differential splicing events to genes, enrichment analysis was performed with WebGestalt (http://www.webgestalt.org/).63 These results include enrichment of Gene Ontology categories (Biological Process, Molecular Function, and Cellular Compartment) and Pathways (KEGG, WikiPathway, Reactome). Visualization of specific differential splicing events was performed with rmats2sashimiplot (https://github.com/Xinglab/rmats2sashimiplot/) which generates plots of read and junction coverage surrounding inclusion/skipping events. Motif enrichment analysis was performed using the rMAPS2 Motif Map tool (http://rmaps.cecsresearch.org/MTool/) which takes rMATS output files as input and generates plots of the alternatively spliced exon and intron regions containing enriched canonical RNA-binding protein binding sequences along with associated statistics for the enrichment.46
Network analysis of differentially spliced genes
Significantly differentially spliced genes were used as seeds for network propagation on the STRING high confidence interactome (https://stringdb-static.org/download/protein.links.v11.0.txt.gz).47,75 The most proximal genes to the seed set were identified using a network propagation method, using degree-matched sampling to generate proximity z-scores for each gene in the network. Genes with a Z score >2 were retained in the network and used for visualization and downstream analysis. A graph-based modularity maximization clustering algorithm was used to identify groups of genes within the most proximal genes which were highly interconnected. Genes in the entire network and within each of these clusters were annotated with associated pathways identified by functional enrichment analysis, with the g:Profiler tool using the proximal gene set as the background gene list for enrichment of the clusters and the STRING interactome genes as the background for the entire network enrichment.65
Network visualization and propagation was performed using Cytoscape and VisJS2jupyter.67,68 The subgraph composed of the most proximal genes was visualized using a modified spring-embedded layout algorithm, modified by cluster membership, so that genes belonging to the same cluster are separated from other clusters. Differential expression log fold change was mapped to the node color, for the significantly differentially expressed genes (FDR<0.05) within the subgraph.
RNA editing analyses
RNA editing analysis was performed as described previously.35 Briefly, RNA reads were aligned with STAR (v2.5.2b) and deduplicated with Picard MarkDuplicates. Next, alignments were processed sequentially according to GATK best practices v3.8 for calling RNA-Seq variants with tools SplitNCigarReads, RealignerTargetCreator, IndelRealigner, BaseRecalibrator, PrintReads.69 Variants were called with HaplotypeCaller and filtered with VariantFiltration and mismatches in the first 6 base pairs were discarded. Non-Alu variants were further processed after Alu removal by RepeatMasker. Sites were retained if not within 4bp of splicing junctions, not in homopolymer runs, minimum of three reads with the alternative allele, ten total reads, and a minimum allele frequency of 0.10. Known RNA editing sites were identified according to REDIportal.71 To identify ADAR-specific RNA edits, A to G variants in genes on the positive strand and T to C variants on the negative strand were retained. The final list of RNA edits was annotated with VEP and sites that exist in ExAC, gnomad, and dbSNP removed.72
Gene list acquisition
All human genes were retrieved from the Spliceosome database to acquire a list of alternative splicing related genes (http://spliceosomedb.ucsc.edu/).70 This list was limited to relevant genes by filtering for genes in the categories: hnRNP, SR protein, U1 snRNP, U5 snRNP, U11/U12 snRNP, 17S U2 snRNP, 17S U2 snRNP associated, pre-mRNA/mRNA binding proteins, alter-native splicing factor. This resulted in 691 unique genes.
Lentiviral RBFOX2 shRNA knockdown
CD34+ primary samples were transduced with pSMART hEF1a shCTRL and shRBFOX2 (MOI: 200) for 48h. Plasmids were purchased from Horizon Discovery (Waterbeach, UK). Upon treatment, RNA was extracted using RNeasy micro extraction kits (QIAGEN, Germantown, MD) following the manufacturer’s protocol including a DNase incubation step to digest any trace genomic DNA present. RNA was subjected to cDNA synthesis using the Super-Script III (ThermoFisher Scientific) kit followed by qRT-PCR using SYBR GreenER (ThermoFisher Scientific) master mix according to the manufacturer’s recommended procedures. qRT-PCR was performed using SYBR GreenER Super Mix (Life Technologies) on BioRad iQ5, C1000 Touch, or BioRad CFX384 instruments.45 In addition, cells were stained with fluorescent antibodies against human CD45, CD34, CD38, RBFOX2 (CD45 APC, 1:50, cat 335790; CD34 BV421, 1:100, cat 562577; CD38 PE-Cy7, 1:50, cat 335790; RBFOX2 unconjugated, 1:25, cat ab74490) and NearIR (Invitrogen).
Chemical synthesis and preparation of splicing modulatory drugs
Synthesis of Rebecsinib compound has been previously described.4,55 Meanwhile, synthesis was optimized for a completed pre-investigational new drug application (pre-IND) study (PIND 153126).
MTT growth inhibition assay
Growth inhibitory effects of Rebecsinib were determined using the MTT dye reduction assay. Upon 96 h drug exposure, MTT salt (5 mg/mL) was added for 6 h. The crystals were dissolved with 100 μL acidified isopropanol, and formazan production was quantitated using a spectrophotometer at 562 nm.76 For this assay, Rebecsinib (MW: 552.71 g/mol) was prepared at a concentration of 10 mg/mL.
Lentiviral splicing reporter
For the generation of the lentiviral dual fluorescence splicing reporter, a previously described construct35 was cloned into a lentiviral pCDH-EF1a-MCS-T2A backbone (System Biosciences). For the evaluation of in vitro splicing activity using a two-color fluorescent splicing reporter system, CD34+ primary samples, Kasumi-1 and MOLM13 cells were cultured in RPMI-1640 medium containing 20% fetal bovine serum. Cells were stably transduced with a lentiviral splicing reporter and sorted for GFP+ (Kasumi-1-MAPT or MOLM13-MAPT).35 MOLM13-MAPT Cells were exposed to doses of Rebecsinib ranging from 0.03μM to 3μM for 24 h. Samples were stained with DAPI or NearIR (Invitrogen) and fluorescent antibodies against human CD45, CD34, and CD38 (CD45 APC, 1:50, cat 335790; CD34 BV421, 1:100, cat 562577; CD38 PE-Cy7, 1:50, cat 335790). Subsequently, we ran samples on the BD LSRFortessa. Mean fluorescence intensity (MFI) was calculated with FlowJo software based on live cells (DAPI negative or NearIR negative). Similarly stably transduced Kausmi1-MAPT cells were stained with DAPI and run on the BD LSRFortessa to assess baseline levels of GFP and RFP. MFI was calculated with FlowJo software based on live cells (DAPI negative). Primary patient samples were CD34+ selected by magnetic bead separation (MACS; Miltenyi, Bergisch Gladbach, Germany) and transduced with the lentiviral splicing reporter (MOI:15) for 48 h in StemPro and cytokines (Thermo Fischer Scientific). CD34+ cells were treated with 100nM Rebecsinib for 72 h in a stromal co-culture (described under In vitro stromal co-culture assays). Cells were stained with LIVE/DEAD Fixable Aqua (cat L34957, 1:100). Subsequently, samples were run on the BD LSRFortesssax20. MFI was calculated with FlowJo software based on live cells (Aqua negative).
In addition, 600,000 stably transduced Kasumi-1 MAPT cells were plated in 35mm glass bottom culture dishes (MatTrek Life Sciences) and treated with DMSO control. Cells were allowed to settle for 30 min prior to imaging. Cells were imaged every 10 min for 21 h using the Fluoview FV10i Olympus confocal microscope with 5% CO2 (Tokai Hit, Japan). On day two, 600,000 Kasumi-1 MAPT cells were plated in 35mm glass bottom culture dishes (MatTrek Life Sciences) and treated with 100nM Rebecsinib. Cells were allowed to settle for 30 min prior to imaging. Cells were imaged every 10 min for 24 h using the Fluoview FV10i Olympus confocal microscope with 5% CO2 (Tokai Hit, Japan). Images were analyzed using Volocity software (Quorum Technologies Inc.) to determine the mean intensity of RFP and GFP.
In vivo lentiviral splicing reporter assays
Four-week-old NSGS-SGM mice were purchased from Jackson Laboratories. Chow was switched to low-fluorescent chow two days after arrival. After acclimation, animals were irradiated with 150cGy, and transplanted with 50K Molm13 MAPT intravenously within 24 h. Two to three weeks after transplant, animals were given one dose of vehicle (n = 3), 10 mg/kg (n = 2), or 20 mg/kg (n = 3) of Rebecsinib intravenously. Animals were sacrificed ∼24 h after dose, wherein bone marrow, spleen, and peripheral blood were harvested. Tissues were processed for single cell suspension and stained with Live/Dead NearIR Stain (1:1000), CD45 BV421 (1:100), and CD38 PE-Cy7 (1:50). FlowJo software was used to calculate the RFP/GFP MFI Ratio. Subsequently, an unpaired t-test between vehicle and 20 mg/kg Rebecsinib was performed.
In vitro stromal co-culture assays
Stromal co-culture experiments were performed as previously described.56 Briefly, mouse bone marrow cells lines SL (expressing human SCF and IL3) and M2 (expressing human IL3 and G-CSF) were irradiated, mixed, and incubated overnight to facilitate attachment. CD34+ were selected from pediatric AML (n = 6), adult de novo AML (n = 9) and adult sAML (n = 9) primary samples. CD34+ cord blood (CB) (n = 7) (All Cells Inc.) cells were utilized as a control. 10,000–15,000 CD34+ cells were added to SLM2 stroma in 1mL of Myelocult H5100 (Stem Cell Technologies, Vancouver). Rebecsinib or vehicle (DMSO) was added at the initiation of co-culture at indicated concentrations. After 1 week both attached to stroma and floating cells were collected, resuspended in fresh media, and plated in methylcellulose (MC) (Stem Cell Technologies, MethoCult, H4330) in triplicates. After 2 weeks, primary colonies (>40 cells) were counted and some individual colonies were plucked, and cells were resuspended and re-plated again in fresh MC. Secondary colonies were counted after an additional 14 days. Basal colony formation of untreated cells was considered to be 100% and results are presented as % of change. Data (means) for summarized AML or CB samples are presented. Error bars indicate the SD Statistical analysis included Student’s t test and one-way ANOVA, including All Pair-wise Multiple Comparison Procedures (Holm-Sidak method).
Quantification and statistical analysis
Statistical analyses
For splicing reporter assays, qRT-PCR analyses, and flow cytometry, data were analyzed using Microsoft Excel and plotted for graph preparation and statistical analyses in Prism GraphPad (San Diego, CA). Differences were assessed by unpaired or paired Student’s t-tests, as indicated in the figure legends, and considered statistically significant for p values of <0.05. For stromal co-culture assays and multiple group comparisons, data (means) were calculated and graphed. Error bars indicate the SD or SEM, as indicated in individual figure legends. Student’s t test and one-way ANOVA statistical analyses were performed using Prism GraphPad and comparisons described in each figure legend.
Acknowledgments
We would like to thank our funding agencies for their vital support, including Padres Pedal the Cause, NIH/NCI R01CA205944, NIH/NIDDK R01DK114468-01, NIH/NCI 2P30CA023100-28, CIRM TRAN1-10540, MPN Research Foundation, LLS Blood Cancer Discoveries, NIH/NCATS UL1TR001442, and NASA NRA NNJ13ZBG001N. Also, we would like to thank the Moores Family Foundation, the Koman Family Foundation, the Sanford Stem Cell Institute, and the UC San Diego Moores Cancer Center for their generous support. I.v.d.W. was supported by the Diamond Program of the Graduate Oncology School Amsterdam, and C.H.M.J. is supported by the Koman Family Presidential Endowed Chair for Cancer Research.
Author contributions
I.v.d.W., P.K.M., S.K.S., L.L., C.N.M., and C.H.M.J. designed research, performed experiments, and analyzed the data. R.H.D. engineered the lentiviral dual-fluorescence splicing reporter. J.C., G.J.L.K., P.W., and L.A.C. supported study design. T.C.W. performed all computational analyses related to RNA-seq differential expression, differential splicing, network generation, and single-cell data. T.C.W. and A.M. performed the RNA editing analysis of the RNA-seq data. K.M.F. supervised the bioinformatic analysis. W.C.C., J.J.L.C., and M.D.B. synthesized and prepared splicing modulatory drugs. M.E.D. designed and performed the single-cell immunophenotyping. C.H.M.J. designed the study and supervised the analysis. I.v.d.W., M.E.D., S.K.S., and C.H.M.J. wrote the manuscript.
Declaration of interests
M.D.B. is a co-founder of Aspera Biomedicines. C.H.M.J. is a co-founder of Aspera Biomedicines and Impact Biomedicines and has received royalties for intellectual property licensed by Forty Seven Inc.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: March 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.100962.
Supplemental information
Data and code availability
Data: The datasets generated during this study are available through dbGaP/GEO Sequence Read Archive (accession codes are provided in the Key resources table).
Code: All code and processed data can be accessed via GitHub: https://github.com/ucsd-ccbb/pAML_StemCell_Methods.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Chua B.A., Van Der Werf I., Jamieson C., Signer R.A.J. Post-transcriptional regulation of homeostatic, stressed, and malignant stem cells. Cell Stem Cell. 2020;26:138–159. doi: 10.1016/j.stem.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rossi D.J., Jamieson C.H.M., Weissman I.L. Stems cells and the pathways to aging and cancer. Cell. 2008;132:681–696. doi: 10.1016/j.cell.2008.01.036. [DOI] [PubMed] [Google Scholar]
- 3.Adamia S., Haibe-Kains B., Pilarski P.M., Bar-Natan M., Pevzner S., Avet-Loiseau H., Lode L., Verselis S., Fox E.A., Burke J., et al. A genome-wide aberrant RNA splicing in patients with acute myeloid leukemia identifies novel potential disease markers and therapeutic targets. Clin. Cancer Res. 2014;20:1135–1145. doi: 10.1158/1078-0432.CCR-13-0956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crews L.A., Balaian L., Delos Santos N.P., Leu H.S., Court A.C., Lazzari E., Sadarangani A., Zipeto M.A., La Clair J.J., Villa R., et al. RNA splicing modulation selectively impairs leukemia stem cell maintenance in secondary human AML. Cell Stem Cell. 2016;19:599–612. doi: 10.1016/j.stem.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogawa S. Splicing factor mutations in AML. Blood. 2014;123:3216–3217. doi: 10.1182/blood-2014-04-566752. [DOI] [PubMed] [Google Scholar]
- 6.Yoshida K., Ogawa S. Splicing factor mutations and cancer. Wiley Interdiscip. Rev. RNA. 2014;5:445–459. doi: 10.1002/wrna.1222. [DOI] [PubMed] [Google Scholar]
- 7.van der Werf I., Wojtuszkiewicz A., Meggendorfer M., Hutter S., Baer C., Heymans M., Valk P.J.M., Kern W., Haferlach C., Janssen J.J.W.M., et al. Splicing factor gene mutations in acute myeloid leukemia offer additive value if incorporated in current risk classification. Blood Adv. 2021;5:3254–3265. doi: 10.1182/bloodadvances.2021004556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abrahamsson A.E., Geron I., Gotlib J., Dao K.-H.T., Barroga C.F., Newton I.G., Giles F.J., Durocher J., Creusot R.S., Karimi M., et al. Glycogen synthase kinase 3β missplicing contributes to leukemia stem cell generation. Proc. Natl. Acad. Sci. USA. 2009;106:3925–3929. doi: 10.1073/pnas.0900189106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zipeto M.A., Court A.C., Sadarangani A., Delos Santos N.P., Balaian L., Chun H.J., Pineda G., Morris S.R., Mason C.N., Geron I., et al. ADAR1 activation drives leukemia stem cell self-renewal by impairing let-7 biogenesis. Cell Stem Cell. 2016;19:177–191. doi: 10.1016/j.stem.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang Q., Crews L.A., Barrett C.L., Chun H.-J., Court A.C., Isquith J.M., Zipeto M.A., Goff D.J., Minden M., Sadarangani A., et al. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA. 2013;110:1041–1046. doi: 10.1073/pnas.1213021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang Q., Isquith J., Zipeto M.A., Diep R.H., Pham J., Delos Santos N., Reynoso E., Chau J., Leu H., Lazzari E., et al. Hyper-editing of cell-cycle regulatory and tumor suppressor RNA promotes malignant progenitor propagation. Cancer Cell. 2019;35:81–94.e7. doi: 10.1016/j.ccell.2018.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang Q., Isquith J., Ladel L., Mark A., Holm F., Mason C., He Y., Mondala P., Oliver I., Pham J., et al. Inflammation-driven deaminase deregulation fuels human pre-leukemia stem cell evolution. Cell Rep. 2021;34:108670. doi: 10.1016/j.celrep.2020.108670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolouri H., Farrar J.E., Triche T., Ries R.E., Lim E.L., Alonzo T.A., Ma Y., Moore R., Mungall A.J., Marra M.A., et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018;24:103–112. doi: 10.1038/nm.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brunner A.M., Graubert T.A. Genomics in childhood acute myeloid leukemia comes of age. Nat. Med. 2018;24:7–9. doi: 10.1038/nm.4469. [DOI] [PubMed] [Google Scholar]
- 15.Reedijk A.M.J., Klein K., Coebergh J.W.W., Kremer L.C., Dinmohamed A.G., de Haas V., Versluijs A.B., Ossenkoppele G.J., Beverloo H.B., Pieters R., et al. Improved survival for children and young adolescents with acute myeloid leukemia: a Dutch study on incidence, survival and mortality. Leukemia. 2019;33:1349–1359. doi: 10.1038/s41375-018-0314-7. [DOI] [PubMed] [Google Scholar]
- 16.Brandsma A.M., Bertrums E.J.M., van Roosmalen M.J., Hofman D.A., Oka R., Verheul M., Manders F., Ubels J., Belderbos M.E., van Boxtel R. Mutation signatures of pediatric acute myeloid leukemia and normal blood progenitors associated with differential patient outcomes. Blood Cancer Discov. 2021;2:484–499. doi: 10.1158/2643-3230.BCD-21-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaiswal S., Jamieson C.H.M., Pang W.W., Park C.Y., Chao M.P., Majeti R., Traver D., van Rooijen N., Weissman I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Advani R., Flinn I., Popplewell L., Forero A., Bartlett N.L., Ghosh N., Kline J., Roschewski M., LaCasce A., Collins G.P., et al. CD47 blockade by Hu5F9-G4 and Rituximab in non-hodgkin's lymphoma. N. Engl. J. Med. 2018;379:1711–1721. doi: 10.1056/NEJMoa1807315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miles L.A., Bowman R.L., Merlinsky T.R., Csete I.S., Ooi A.T., Durruthy-Durruthy R., Bowman M., Famulare C., Patel M.A., Mendez P., et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature. 2020;587:477–482. doi: 10.1038/s41586-020-2864-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morita K., Wang F., Jahn K., Hu T., Tanaka T., Sasaki Y., Kuipers J., Loghavi S., Wang S.A., Yan Y., et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020;11:5327. doi: 10.1038/s41467-020-19119-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jan M., Chao M.P., Cha A.C., Alizadeh A.A., Gentles A.J., Weissman I.L., Majeti R. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc. Natl. Acad. Sci. USA. 2011;108:5009–5014. doi: 10.1073/pnas.1100551108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarry J.E., Murphy K., Perry R., Sanchez P.V., Secreto A., Keefer C., Swider C.R., Strzelecki A.C., Cavelier C., Récher C., et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J. Clin. Invest. 2011;121:384–395. doi: 10.1172/jci41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanekamp D., Denys B., Kaspers G.J.L., te Marvelde J.G., Schuurhuis G.J., De Haas V., De Moerloose B., de Bont E.S., Zwaan C.M., de Jong A., et al. Leukaemic stem cell load at diagnosis predicts the development of relapse in young acute myeloid leukaemia patients. Br. J. Haematol. 2018;183:512–516. doi: 10.1111/bjh.14991. [DOI] [PubMed] [Google Scholar]
- 24.Zeijlemaker W., Grob T., Meijer R., Hanekamp D., Kelder A., Carbaat-Ham J.C., Oussoren-Brockhoff Y.J.M., Snel A.N., Veldhuizen D., Scholten W.J., et al. CD34(+)CD38(-) leukemic stem cell frequency to predict outcome in acute myeloid leukemia. Leukemia. 2019;33:1102–1112. doi: 10.1038/s41375-018-0326-3. [DOI] [PubMed] [Google Scholar]
- 25.Taussig D.C., Pearce D.J., Simpson C., Rohatiner A.Z., Lister T.A., Kelly G., Luongo J.L., Danet-Desnoyers G.A.H., Bonnet D. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106:4086–4092. doi: 10.1182/blood-2005-03-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Rhenen A., Moshaver B., Kelder A., Feller N., Nieuwint A.W.M., Zweegman S., Ossenkoppele G.J., Schuurhuis G.J. Aberrant marker expression patterns on the CD34+CD38− stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia. 2007;21:1700–1707. doi: 10.1038/sj.leu.2404754. [DOI] [PubMed] [Google Scholar]
- 27.Hosen N., Park C.Y., Tatsumi N., Oji Y., Sugiyama H., Gramatzki M., Krensky A.M., Weissman I.L. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc. Natl. Acad. Sci. USA. 2007;104:11008–11013. doi: 10.1073/pnas.0704271104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin L., Hope K.J., Zhai Q., Smadja-Joffe F., Dick J.E. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 29.van Galen P., Hovestadt V., Wadsworth Ii M.H., Hughes T.K., Griffin G.K., Battaglia S., Verga J.A., Stephansky J., Pastika T.J., Lombardi Story J., et al. Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell. 2019;176:1265–1281.e24. doi: 10.1016/j.cell.2019.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ng S.W.K., Mitchell A., Kennedy J.A., Chen W.C., McLeod J., Ibrahimova N., Arruda A., Popescu A., Gupta V., Schimmer A.D., et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540:433–437. doi: 10.1038/nature20598. [DOI] [PubMed] [Google Scholar]
- 31.Huang B.J., Smith J.L., Farrar J.E., Wang Y.-C., Umeda M., Ries R.E., Leonti A.R., Crowgey E., Furlan S.N., Tarlock K., et al. Integrated stem cell signature and cytomolecular risk determination in pediatric acute myeloid leukemia. Nat. Commun. 2022;13:5487. doi: 10.1038/s41467-022-33244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eppert K., Takenaka K., Lechman E.R., Waldron L., Nilsson B., van Galen P., Metzeler K.H., Poeppl A., Ling V., Beyene J., et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011;17:1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 33.Shen S., Park J.W., Lu Z.X., Lin L., Henry M.D., Wu Y.N., Zhou Q., Xing Y. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA. 2014;111:E5593–E5601. doi: 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greenberg D.S., Soreq H. In: Brenner's Encyclopedia of Genetics. Second Edition. Maloy S., Hughes K., editors. Academic Press; 2013. Alternative splicing; pp. 97–98. [DOI] [Google Scholar]
- 35.Stoilov P., Lin C.-H., Damoiseaux R., Nikolic J., Black D.L. A high-throughput screening strategy identifies cardiotonic steroids as alternative splicing modulators. Proc. Natl. Acad. Sci. USA. 2008;105:11218–11223. doi: 10.1073/pnas.0801661105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holm F., Hellqvist E., Mason C.N., Ali S.A., Delos-Santos N., Barrett C.L., Chun H.-J., Minden M.D., Moore R.A., Marra M.A., et al. Reversion to an embryonic alternative splicing program enhances leukemia stem cell self-renewal. Proc. Natl. Acad. Sci. USA. 2015;112:15444–15449. doi: 10.1073/pnas.1506943112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beghini A., Ripamonti C.B., Peterlongo P., Roversi G., Cairoli R., Morra E., Larizza L. RNA hyperediting and alternative splicing of hematopoietic cell phosphatase (PTPN6) gene in acute myeloid leukemia. Hum. Mol. Genet. 2000;9:2297–2304. doi: 10.1093/oxfordjournals.hmg.a018921. [DOI] [PubMed] [Google Scholar]
- 38.Kapoor U., Licht K., Amman F., Jakobi T., Martin D., Dieterich C., Jantsch M.F. ADAR-deficiency perturbs the global splicing landscape in mouse tissues. Genome Res. 2020;30:1107–1118. doi: 10.1101/gr.256933.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Nostrand E.L., Freese P., Pratt G.A., Wang X., Wei X., Xiao R., Blue S.M., Chen J.-Y., Cody N.A.L., Dominguez D., et al. A large-scale binding and functional map of human RNA-binding proteins. Nature. 2020;583:711–719. doi: 10.1038/s41586-020-2077-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han H., Irimia M., Ross P.J., Sung H.K., Alipanahi B., David L., Golipour A., Gabut M., Michael I.P., Nachman E.N., et al. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature. 2013;498:241–245. doi: 10.1038/nature12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benoit Bouvrette L.P., Bovaird S., Blanchette M., Lécuyer E. oRNAment: a database of putative RNA binding protein target sites in the transcriptomes of model species. Nucleic Acids Res. 2020;48 doi: 10.1093/nar/gkz986. D166–d173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gazzara M.R., Mallory M.J., Roytenberg R., Lindberg J.P., Jha A., Lynch K.W., Barash Y. Ancient antagonism between CELF and RBFOX families tunes mRNA splicing outcomes. Genome Res. 2017;27:1360–1370. doi: 10.1101/gr.220517.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quentmeier H., Pommerenke C., Bernhart S.H., Dirks W.G., Hauer V., Hoffmann S., Nagel S., Siebert R., Uphoff C.C., Zaborski M., et al. RBFOX2 and alternative splicing in B-cell lymphoma. Blood Cancer J. 2018;8:77. doi: 10.1038/s41408-018-0114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grinev V.V., Barneh F., Ilyushonak I.M., Nakjang S., Smink J., van Oort A., Clough R., Seyani M., McNeill H., Reza M., et al. RUNX1/RUNX1T1 mediates alternative splicing and reorganises the transcriptional landscape in leukemia. Nat. Commun. 2021;12:520. doi: 10.1038/s41467-020-20848-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Venables J.P., Lapasset L., Gadea G., Fort P., Klinck R., Irimia M., Vignal E., Thibault P., Prinos P., Chabot B., et al. MBNL1 and RBFOX2 cooperate to establish a splicing programme involved in pluripotent stem cell differentiation. Nat. Commun. 2013;4:2480. doi: 10.1038/ncomms3480. [DOI] [PubMed] [Google Scholar]
- 46.Hwang J.Y., Jung S., Kook T.L., Rouchka E.C., Bok J., Park J.W. rMAPS2: an update of the RNA map analysis and plotting server for alternative splicing regulation. Nucleic Acids Res. 2020;48 doi: 10.1093/nar/gkaa237. W300–w306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K.P., et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van der Werf I., Wojtuszkiewicz A., Yao H., Sciarrillo R., Meggendorfer M., Hutter S., Walter W., Janssen J., Kern W., Haferlach C., et al. SF3B1 as therapeutic target in FLT3/ITD positive acute myeloid leukemia. Leukemia. 2021;35:2698–2702. doi: 10.1038/s41375-021-01273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steensma D.P., Wermke M., Klimek V.M., Greenberg P.L., Font P., Komrokji R.S., Yang J., Brunner A.M., Carraway H.E., Ades L., et al. Results of a clinical trial of H3B-8800, a splicing modulator, in patients with myelodysplastic syndromes (MDS), acute myeloid leukemia (AML) or chronic myelomonocytic leukemia (CMML) Blood. 2019;134:673. doi: 10.1182/blood-2019-123854. [DOI] [Google Scholar]
- 50.Thomas D., Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood. 2017;129:1577–1585. doi: 10.1182/blood-2016-10-696054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaiswal S., Ebert B.L. Clonal hematopoiesis in human aging and disease. Science. 2019;366:eaan4673. doi: 10.1126/science.aan4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Black D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 53.Sikic B.I., Lakhani N., Patnaik A., Shah S.A., Chandana S.R., Rasco D., Colevas A.D., O'Rourke T., Narayanan S., Papadopoulos K., et al. First-in-Human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol. 2019;37:946–953. doi: 10.1200/jco.18.02018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seiler M., Yoshimi A., Darman R., Chan B., Keaney G., Thomas M., Agrawal A.A., Caleb B., Csibi A., Sean E., et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018;24:497–504. doi: 10.1038/nm.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Villa R., Mandel A.L., Jones B.D., La Clair J.J., Burkart M.D. Structure of FD-895 revealed through total synthesis. Org. Lett. 2012;14:5396–5399. doi: 10.1021/ol3023006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goff D.J., Court Recart A., Sadarangani A., Chun H.J., Barrett C.L., Krajewska M., Leu H., Low-Marchelli J., Ma W., Shih A.Y., et al. A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell. 2013;12:316–328. doi: 10.1016/j.stem.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wingett S.W., Andrews S. FastQ Screen: a tool for multi-genome mapping and quality control. F1000Res. 2018;7:1338. doi: 10.12688/f1000research.15931.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Law C.W., Chen Y., Shi W., Smyth G.K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liao Y., Wang J., Jaehnig E.J., Shi Z., Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47 doi: 10.1093/nar/gkz401. W199–w205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reimand J., Kull M., Peterson H., Hansen J., Vilo J. g:Profiler--a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35:W193–W200. doi: 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shen S., Park J.W., Huang J., Dittmar K.A., Lu Z.X., Zhou Q., Carstens R.P., Xing Y. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res. 2012;40:e61. doi: 10.1093/nar/gkr1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rosenthal S.B., Len J., Webster M., Gary A., Birmingham A., Fisch K.M. Interactive network visualization in Jupyter notebooks: visJS2jupyter. Bioinformatics. 2018;34:126–128. doi: 10.1093/bioinformatics/btx581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cvitkovic I., Jurica M.S. Spliceosome database: a tool for tracking components of the spliceosome. Nucleic Acids Res. 2013;41:D132–D141. doi: 10.1093/nar/gks999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lo Giudice C., Tangaro M.A., Pesole G., Picardi E. Investigating RNA editing in deep transcriptome datasets with REDItools and REDIportal. Nat. Protoc. 2020;15:1098–1131. doi: 10.1038/s41596-019-0279-7. [DOI] [PubMed] [Google Scholar]
- 72.Sherry S.T., Ward M., Sirotkin K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999;9:677–679. [PubMed] [Google Scholar]
- 73.Benjamini Y., Drai D., Elmer G., Kafkafi N., Golani I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001;125:279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- 74.Tarca A.L., Draghici S., Khatri P., Hassan S.S., Mittal P., Kim J.S., Kim C.J., Kusanovic J.P., Romero R. A novel signaling pathway impact analysis. Bioinformatics. 2009;25:75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cowen L., Ideker T., Raphael B.J., Sharan R. Network propagation: a universal amplifier of genetic associations. Nat. Rev. Genet. 2017;18:551–562. doi: 10.1038/nrg.2017.38. [DOI] [PubMed] [Google Scholar]
- 76.van Meerloo J., Kaspers G.J.L., Cloos J. Cell sensitivity assays: the MTT assay. Methods Mol. Biol. 2011;731:237–245. doi: 10.1007/978-1-61779-080-5_20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data: The datasets generated during this study are available through dbGaP/GEO Sequence Read Archive (accession codes are provided in the Key resources table).
Code: All code and processed data can be accessed via GitHub: https://github.com/ucsd-ccbb/pAML_StemCell_Methods.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.