

Summary
The origin, composition, and significance of the distal male urethral microbiome are unclear, but vaginal microbiome dysbiosis is linked to new sex partners and several urogynecological syndromes. We characterized 110 urethral specimens from men without urethral symptoms, infections, or inflammation using shotgun metagenomics. Most urethral specimens contain characteristic lactic acid bacteria and Corynebacterium spp. In contrast, several bacteria associated with vaginal dysbiosis were present only in specimens from men who reported vaginal intercourse. Sexual behavior, but not other evaluated behavioral, demographic, or clinical variables, strongly associated with inter-specimen variance in urethral microbiome composition. Thus, the male urethra supports a simple core microbiome that is established independent of sexual exposures but can be re-shaped by vaginal sex. Overall, the results suggest that urogenital microbiology and sexual behavior are inexorably intertwined, and show that the male urethra harbors female urogenital pathobionts.
Keywords: urobiome, urethra, urethritis, sexually transmitted infection, microbiome, microbiota, development, idiopathic
Graphical abstract
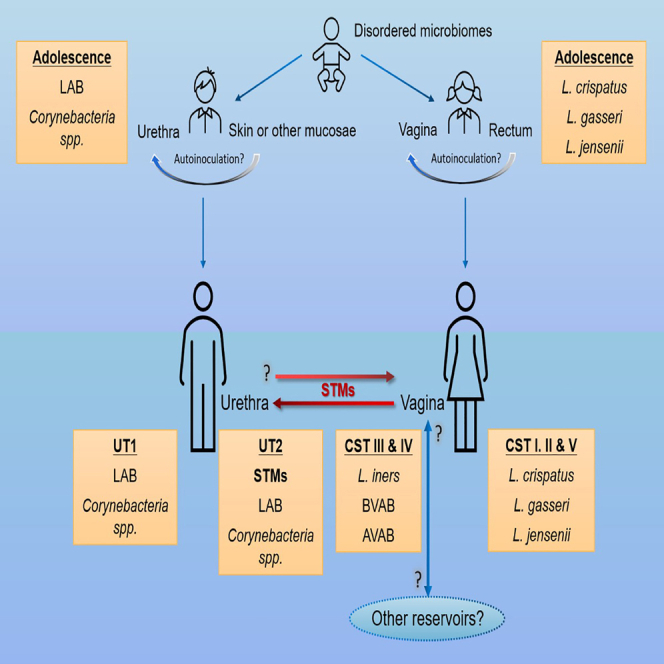
Model of the development and succession of urogenital microbiome composition depicting how vaginal sex could reshape the male urethral microbiome by introduction of sexually transmitted microorganisms (STMs).
Highlights
-
•
The adult male urethra usually supports a characteristic core microbiome
-
•
Bacteria associated with vaginal dysbiosis in women colonize some men
-
•
Vaginal dysbiosis-associated bacteria are only detected in men who have vaginal sex
-
•
Sexual behavior is an important determinant of microbiome composition
Toh et al. characterize the urethral microbial communities in men who lack urethral symptoms and inflammation and discover that a few common bacteria are present in most men. However, several bacteria associated with reproductive tract disease in women only colonize men who have vaginal sex with women.
Introduction
The male urinary and reproductive tracts merge at the post-prostatic urethra, and microorganisms that exit or enter the male urogenital tract traverse the penile urethra (PU). Similar to other mucosae, the PU is richly endowed with innate and adaptive immune cells that can detect and respond to microorganisms.1 Nonetheless, the PU can be infected by and transmit a broad array of sexually transmitted bacterial, viral, and eukaryotic pathogens.2 Communities of commensal microorganisms (microbiomes) associated with a healthy gastrointestinal tract and female reproductive tract (FRT) protect these organs against infection and promote health.3,4 It is unclear if the healthy PU supports a characteristic microbiome, or microbiomes, that contribute to PU health or disease.
Several factors have impeded characterization of the PU microbiome. PU sampling is painful and is rarely indicated in healthy individuals; hence the PU microbiome has mostly been studied in men with sexually transmitted infections (STIs). Some PU microorganisms can be detected in non-invasive specimens, such as urine, but these approaches may oversample the upper urogenital tract and proximal urethra and under-sample the PU epithelium.5,6 The utility of PCR-based cultivation-independent microbial identification approaches, including 16S rRNA gene sequencing, has also been constrained by low microbial biomass in urogenital specimens, inability of these approaches to detect all the diverse types of microorganisms that have been documented in urogenital specimens (bacteria, viruses, fungi, protists, parasites), and signal to noise issues.7,8
There is increasing evidence that microorganisms do colonize the healthy PU. In a nongonococcal urethritis (NGU) case-control study in men, Bowie and colleagues cultivated streptococci, lactobacilli, and a broader variety of anaerobic bacteria from 91% of the urethral specimens from the controls.9 Similar bacteria were detected in urine and urethral swabs of asymptomatic men using 16S rRNA gene sequencing in two other studies, which additionally showed that many of these corresponded to bacterial vaginosis associated bacteria (BVAB).5,10 Other studies of urine, primarily from control men enrolled in studies of urogenital disease, have replicated these findings.11,12,13,14,15,16 Although stability of the adult PU microbiome has not been evaluated, some PU bacteria that have been observed in adults were detected in consecutive monthly urine specimens collected from adolescents over a 3-month interval.17 Conversely, some prevalent bacteria in adults were not detected in adolescents.17
Existing data suggest that the healthy PU microbiome is simple, stable, and may be linked to environmental and sexual exposures17,18,19; however, the taxonomic resolution of most studies has been low, so the identities of many PU microorganisms are unclear. The relationship between specific sexual behaviors and PU microbiome composition is also unknown.
Here, we use stringent inclusion criteria informed by clinical examination, STI testing, measurements of urethral inflammation, and behavioral surveys to identify healthy men with no signs or symptoms of urethral inflammation or disease to identify correlates of PU microbiome composition. PU specimens from 110 men and 24 vaginal specimens from a separate validation cohort were characterized using a shotgun metagenomic sequencing approach, allowing us to perform a detailed characterization of the PU microbiome. Roles of STI risk factors and sexual behaviors in PU microbiome composition were also evaluated. We show that the healthy PU supports a core microbiome that may be re-shaped by penile-vaginal (vaginal) sex. We also show that silent carriage of a wide array of FRT pathogens and pathobionts, but not protective Lactobacillus spp., is common in men who lack urethral disease.
Results
Participant characteristics and enrollment strategy
We screened 164 volunteers who visited a public health clinic in Indianapolis for STI screening, to identify healthy adult cis-gender men without signs and symptoms of urethral inflammation, infection, or disease as part of the Idiopathic Urethritis Men’s Project.20,21,22 Fifty-four men were excluded for positive STI or urethral inflammation tests, antibiotic use in the past month, history of urogenital or systemic disease, other findings of urogenital abnormality including urogenital surgery, or incomplete survey results (Figure 1). The mean age of the remaining 110 men was 28.7 ± 10.7 years, 35% (38 of 110) were Black, 53% (58 of 110) were White, 13% (14 of 110) were other (Asian and more than one race), and 89% (98 of 110) identified as non-Hispanic or Latino. Seventy-five (68%) self-identified as heterosexual, 22 (20%) as homosexual, and 13 (12%) as bisexual/other, and most were sexually active in the prior year (108 of 110) (Tables S1 and S3).
Figure 1.

Study participant inclusion and exclusion flowchart
Participants were excluded from this analysis if they had urethral discharge, tested positive for C. trachomatis, M. genitalium, N. gonorrhoeae, T. vaginalis, or urethrotropic Neisseria meningitidis strain US_NmUC, exhibited >1 polymorphonuclear leukocytes per high-power field (PMN/HPF) on their urethral Gram stain smear, reported antibiotic use in the past month, had genitourinary tract symptoms, or genital skin conditions.
PU can be colonized by FRT bacteria, but the microbiomes of the PU and FRT differ
The PU swabs were sequenced to an average depth of 31,303,028 ± 9,466,191 reads, and the sequences were annotated using MetaPhlAn3.23 A total of 117 different bacterial and 26 viruses were detected (Table S4). Bacteria were detected in 92 specimens and these sequences accounted for 95.0% of the total microorganism sequences, on average per specimen (Figures 2A–2C). Viral sequences were less prevalent (41 of 92) (44.6%) and abundant (5.0%). Streptococcus mitis, a lactic acid bacterium (LAB) (bacterium in the order Lactobacillales that produce lactic acid) that has been cultured from male urogenital specimens previously,24,25 accounted for 24.2% of the sequences. Many sequences corresponded to BVAB (e.g., Gardnerella vaginalis, Atopobium vaginae [recently renamed Fannyhessea vaginae26], Prevotella amnii) or aerobic vaginitis associated bacteria (AVAB) (Streptococcus agalactiae and Streptococcus anginosis).27,28,29 Bacteriophages (phages) accounted for 62% of the viral sequences. Streptococcus phages were detected in 11 specimens, but no individual phages were prevalent. Sexually transmitted viruses that can silently colonize the PU such as Alphapapilloma viruses30 and adenovirus31 were found in two and one specimens, respectively. Systemic viruses that are commonly detected in male urogenital specimens, including cytomegalovirus32 and Epstein-Barr virus,33 were each found in two specimens. Human herpesvirus 6 was detected in six specimens.
Figure 2.

Pie charts depicting percent composition of the top 10 most abundant PU microorganisms based on relative abundance
“Other” includes all remaining taxa detected excluding the top 10 most abundant. Healthy controls: (A) overall microbial composition, (B) viruses only, (C) bacteria only; Vaginal specimens: (D) overall microbial composition, (E) viruses only, (F) bacteria only.
Eighteen PU specimens yielded few or no microorganism sequences. This suggested that the urethral microbiome is sparse or absent in some men, or that our approach failed to detect or annotate sequences from microorganisms that were present. Separately, although sequences from FRT pathobionts were prevalent in the urethral specimens, sequences from several FRT health-associated Lactobacillus spp. were uncommon or absent. Therefore, we evaluated if our microbial annotation approach was too stringent using vaginal specimens collected in another study as positive controls. We obtained vaginal swabs from four women who enrolled in a prior study of incident bacterial vaginosis (iBV)34 that were collected every other day before (−2 days), during, and up to 8 days before development of iBV (six specimens from each woman). These specimens were sequenced and annotated identically to the PU specimens. Lactobacillus crispatus, Lactobacillus colehominis, Lactobacillus gasseri, Lactobacillus jensenii and other less-common protective Lactobacillus spp. were abundant in the pre-BV specimens whereas a broad range of BVAB such as G. vaginalis, A. vaginae, and Prevotella bivia, were detected in the iBV specimens, confirming that our approach could detect sequences from a wide range of FRT bacteria (Figures 2D–2F) (Table S4). S. mitis was not detected in the FRT specimens, consistent with this microorganism being an infrequent FRT colonizer.35 Re-annotation of the PU sequences using a less stringent K-mer based approach (Kraken2)36 (Table S5) yielded similar species-level identifications. This suggested that some PU specimens contain few or no microorganisms, and that our primary annotation approach captured most of the microbial diversity present. Subsequent analyses were performed using the species-level MetaPhlAn3 annotations because Kraken2 also identified many invariant taxa, possibly due to human contamination in reference microbial genome sequences.37 Thus, the healthy male genital tract may harbor FRT pathobionts, but the microbiomes of the PU and FRT differ.
Two distinct microbiomes are associated with the PU
Clustering has been used to identify characteristic microbiome community state types or enterotypes in the vagina and gut,38,39 respectively, and assess if specific organs support core microbiomes, defined by Shade and Handelsman as “the suite of members shared among microbial consortia from similar habitats.”40 Clustering performed with compositional datasets, like relative abundance, has limitations,41 so the MetaPhlAn3 taxa counts were transformed from Simplex space into Euclidian space using two approaches, a default centered-log ratio (CLR) transformation and a custom additive log ratio (ALR) transformation that used human sequences as the invariant taxon.42,43 To validate the ALR approach, the numbers of G. vaginalis genomes, measured by quantitative PCR, and the ratio of G. vaginalis genomes to human whole-genome sequences were compared. These were strongly positively correlated (Spearman correlation 0.82, p = 1.2 × 10−23) across several orders of magnitude (Figure S1), showing ALR abundance is a reasonable proxy for bacterial genome counts.44 Clustering based on Euclidian distance, using ALR or CLR transformed data, sorted the specimens into clusters we called urethrotypes (UTs) (Figures 3A and S2A). Calinski-Harabasz (CH) and Silhouette index analyses determined that two clusters were the optimal number based on the maximum index values45 using either the ALR or the CLR transformed data (Figures 3B, 3C, and S2B). Similar clusters were reproduced using bacterial and viral taxa or only the bacterial taxa (Figures 3A and S3A). Finally, similar clusters were also found when principal-component analysis separated the taxa using the ALR abundance of bacterial or all taxa (Figures 3Dand S3C). Thus, the clustering results were primarily driven by bacteria. Clusters from the ALR data were designated UT1 and UT2, and contained 66 and 26 specimens, respectively.
Figure 3.

Clustering results based on Euclidian distance using ALR-transformed data reveals two urethrotype clusters
(A–D) (A) Heatmap of ALR-transformed proportions of the top 50 most abundant microbial taxa found in the penile urethral specimens of 92 men reveals two urethrotype clusters, UT1 and UT2. Metadata at the top of the heatmap include type of sexual activity (none, rectal only, vaginal only, vaginal and rectal sex) conducted at specific time intervals (last 60 days, last 1 year, lifetime) and urethrotypes (UT1 = pink, UT2 = blue). The bar graph depicts the absolute abundance of microbial sequences on a log scale. The colored bar indicates the relative abundance of a given species. As the color bar becomes redder, the relative abundance of the microorganism increases, (B) CH index analysis and (C) Silhouette analysis was used to determine the optimal number of UT clusters. (D) Relationships among communities visualized by principal-component analysis based on bacterial ALR abundance.
UT1 is dominated by simple communities of Streptococcus and Corynebacterium spp
One hundred total bacterial species were detected in the UT1 specimens, but the richness of the microbiomes in the individual specimens was low (Chao1: 6.13 ± 6.71; Ace: 6.12 ± 9.13) (Figure 4C). Only 16 of these bacteria were detected in more than 10% of UT1 specimens, while the other 84 bacteria only accounted for 19% of UT1 bacterial sequences (Table S4) (Figure 4A), indicating that UT1 harbors a simple microbiome. S. mitis was especially prevalent (54 of 66) and accounted for 35.5% of UT1 sequences.
Figure 4.

Species diversity (richness) of UT1 and UT2
(A) Alpha diversity between UT1 and UT2 measured by various indices. Significant p values are indicated (∗∗p < 0.01, ∗∗∗p < 0.001).
(B and C) (B) Top 20 most abundant bacterial species in UT1, (C) Top 20 most abundant bacterial species in UT2. Error bars indicate standard error.
(D) Heatmap with hierarchal clustering by Euclidean distance depicting the percentage of unique Gardnerella genomospecies-specific reads in participants in whom Gardnerella sequences were detected by MetaPhlAn3
S. mitis was the only taxon whose ALR abundance was significantly higher in UT1 than UT2 after application of Wilcoxon’s signed rank test and a Benjamini-Hochberg (WBH) multiple test correction (p = 0.0024) (Table S6). Lower proportions of one or more of 15, primarily aerobic, Corynebacterium spp. were detected in 61% (40 of 66) of the specimens and accounted for 8.7% of UT1 bacterial sequences (Table S4). Other LAB including various viridans streptococci (Streptococcus pseudopneumoniae, Streptococcus milleri), Lactobacillus iners, and AVAB (S. agalactiae and S. anginosis) were detected in 92% (11 of 12) of the S. mitis-negative specimens, and 50% (27 of 54) of the S. mitis-positive specimens and accounted for 22.4% of UT1 sequences. All these Streptococcus,24 and most of these Corynebacterium spp.46 have been cultured from male urogenital specimens previously. Thus, like in adolescents,17 core communities of LAB and Corynebacterium spp. colonize the PU in most adults.
UT2 specimens are dominated by BVAB
Eighty-three different bacteria were detected in the 26 UT2 specimens, 66 of which were also detected in UT1 specimens. The ALR abundance of 56 of these bacteria did not significantly differ between UT1 and UT2, although the prevalence and abundance of many of these was low (Table S6). Like in UT1, Corynebacterium and LAB spp. were prevalent (81% [52 of 66] and 88% [23 of 26], respectively) in UT2 specimens, suggesting that these organisms constitute a core PU microbiome (Table S4) (Figure 4B). In contrast, the richness of UT2 specimens (Chao1: 9.39 ± 4.26; Ace: 10.4 ± 5.48) was higher than UT1 specimens (p = 0.00001 for Chao1; p = 0.00008 for Ace, Wilcoxon’s signed rank test) (Figure 4C). In addition, the ALR abundance of nine bacteria (Aerococcus christensenii, G. vaginalis, A. vaginae, Veillonella montpellierensis, P. amnii, Dialister micraerophilus, Sneathia amnii (recently renamed Sneathia vaginalis47), Mageeibacillus indolicus, and L. iners) was higher in UT2 than in UT1 specimens (p < 0.05 WBH) (Table S6) (Figure 5). All these bacteria were prevalent (range from 100% for G. vaginalis to 54% for M. indolicus) and collectively accounted for 85.9% of UT2 sequences (Table S4). All these bacteria are associated with BV, AV, or other non-optimal vaginal community state types,27,38,48,49,50 and many can form inter-species biofilms with the keystone species G. vaginalis.51
Figure 5.

Violin plots showing the difference in ALR abundance of the 12 significant taxa between UT1 and UT2
The white bar represents the interquartile range, and the black bar represents the median value. Taxa with p value <0.05 were considered significantly different. The Wilcoxon Rank-Sum Test was used to generate the reported unadjusted p values. The violin plot outlines represent kernel probability density (the width of the shaded area represents the proportion of the data located there).
Phenotypically diverse clinical isolates originally grouped into G. vaginalis52 correspond to at least four named Gardnerella spp. and multiple additional unnamed genomospecies (GS).53,54 Some GS are associated with specific BV phenotypes, and co-infection with multiple GS is associated with incident BV.55 We applied a modification of the approach developed by Potter et al.,54 to determine which GS were present in PU specimens. Sequences unique to all nine GS defined by Potter et al. were detected, and sequences from more than one GS (range 2–9, median 5) were detected in 93% of the G. vaginalis-positive specimens (Figure 4D) (Table S7). Sequences from GS03, associated with recurrent BV, were the most prevalent and abundant, and sequences from GS04, associated with metronidazole treatment-refractory BV, were detected in several specimens.55 Thus, phylogenetically distinct G. vaginalis strains can colonize the PU.
BVAB and core urethral bacteria may inhabit different PU niches
Given that the absolute number of bacteria in vaginal specimens from women with BV is higher compared with healthy women, and the reverse is true for women with AV,56 the relationship between PU microbiome composition and ALR abundance was investigated. When all taxa were considered, their combined ALR abundance was significantly higher in UT2 compared with UT1 (p = 3.8 × 10−8, Wilcoxon’s signed rank test). In contrast, the ALR abundance of core taxa was more similar (p = 0.084, Wilcoxon’s signed rank test).
Competitive exclusion dictates that competition between sympatric species eventually leads to extinction of the less fit species.57 Two observations suggested that core bacteria and BVAB might inhabit different niches. First, many BVAB are obligate anaerobes, whereas most of the core bacteria that we detected are not.58 Second, ALR abundance of core bacteria was similar in UT1 and UT2, suggesting that core organisms do not compete with BVAB. The taxa count data were analyzed using network analysis (SPIEC-EASI) (Figures 6A and 6B).59 All the UT2 BVAB we identified were positively associated with, and many were connected to, one another by multiple edges in networks generated using either covariance selection (Glasso) or neighborhood selection (MB) approaches (Figures 6A and 6B) (Table S8). No edges were detected between L. iners, S. mitis, or other core LAB and other bacteria, indicating no strong ecological interactions among those taxa. In addition, gene ontology (GO) analysis determined that several genes and pathways that mediate anaerobic growth (e.g., fumarate reductase) and utilization of alternate urinary tract carbon and nitrogen sources (e.g., allantoin) were significantly enriched in the UT2 compared with the UT1 metagenome (Figures 6C–6E) (Table S8). Overall, these observations are consistent with the hypothesis that BVAB and core bacteria inhabit different niches in the urethra.
Figure 6.

SPIEC-EASI network visualizations were generated by using two inference methods to construct a microbiome association network from all the bacterial operational taxonomic units (OTUs) (117)
Each node diameter is proportional to the mean of that OTU’s relative abundance. Nodes are colored based on the urethrotype (UT1 = pink; UT2 = blue) in which the taxon is most abundant. Topology network using (A) graphical least absolute shrinkage and selection operator (Glasso), and (B) Meinshausen-Buhlmann’s neighborhood selection (MB). Edges indicate the two nodes are connected. Taxa definitions: Ac = Aerococcus christensenii; Al = Anaerococcus lactolyticus; Ama = Actinobaculum massiliense; Ao = Alloscardovia omnicolens; Ar = Actinomyces radingae; Atu = Actinomyces turicensis; Aul = Actinotignum urinale; Av = Atopobium vaginae; Cac = Cutibacterium acnes; Cgl = Corynebacterium glucuronolyticum; Ch = Corynebacterium hadale; Cpg = Corynebacterium pseudogenitalium; Cpp = Corynebacterium pyruviciproducens; Csm = Corynebacterium simulans; Csg = Corynebacterium singulare; C1 = Corynebacterium sp. NML140438; Ef = Enterococcus faecalis; Fh = Facklamia hominis; Ge = Gemella haemolysans; Gv = Gardnerella vaginalis; Hh = Haemophilus haemolyticus; Hq = Haemophilus quentini; Li = Lactobacillus iners; Mi = Mageeibacillus indolicus; Ml = Micrococcus luteus; Pam = Prevotella amnii; Pbv = Prevotella bivia; Pbc = Prevotella buccalis; Pds = Prevotella disiens; Pl = Propionimicrobium lymphophilum; Pm = Prevotella melaninogenica; P1 = Prevotella sp. oral taxon 299; P2 = Prevotella sp. S7 18; Pt = Prevotella timonensis; Sag = Streptococcus agalactiae; Sam = Sneathia amnii; San = Streptococcus anginosis group; Sho = Staphylococcus hominis; Sml = Streptococcus milleri; Smi = Streptococcus mitis; Spm = Streptococcus pneumoniae; Spp = Streptococcus pseudopneumoniae; S1 = Streptococcus sp. HMSC034E03; Up = Ureaplasma parvum; Uu = Ureaplasma urealyticum; Va = Veillonella atypica; V1 = Veillonellaceae bacterium DNF00626; Vm = Veillonella montpellierensis.
(C–E) Relative abundance of top 20 significant gene ontology (GO) terms in UT1 and UT2 generated with HUMAnN 3.0 (all p values <0.001). GO terms were mapped from the gene families in the output, and the same GO terms of different taxa were combined. Wilcoxon signed rank test was used to identify the differentially abundant GO terms between UT1 and UT2, and the GO terms were ordered by p value. GO definitions: BP = Biological process; CC = Cellular component; MF = Molecular function.
UT2, but not UT1, is associated with vaginal sex
Streptococcus spp. were among the most stable and abundant bacteria detected in the in urine of sexually inexperienced adolescents in one study,17 whereas FRT bacteria were the most prevalent bacteria detected in urogenital specimens in another study of adult male STI clinic attendees.5,10
Since the participants all completed detailed surveys, we tested if membership in UT1 or UT2 was associated with any demographic or STI risk factors, but failed to identify any significant associations (Table S9). However, use of self-reported sexual orientation to infer current and past patterns of sexual behavior has limitations60 and we observed several instances where men who self-identified as heterosexual reported current or past same-sex behaviors as well as the reverse scenario (Table S3). Therefore, we tested if UT1 or UT2 associated with specific sexual behaviors in specific time intervals. UT2 was significantly associated with vaginal sex in the past 60 days (odds ratio [OR] = 6.01; 95% confidence interval [CI] 1.64–22.00), and in the past year (OR = 6.86; 95% CI 1.49–31.58), but not in an individual’s lifetime (OR = 14.64; 95% CI 0.84–255.00) (Table S9). In contrast, insertive penile-anal (anal sex) and insertive penile-oral sex (oral sex) were not associated with UT1 or UT2 in these intervals (Table S9).
Specific bacteria are significantly associated with vaginal sex
Given that UT2 was associated with vaginal sex, we evaluated associations between specific bacteria, individual sexual behaviors, and combinations of sexual behaviors in three intervals (past 60 days, past year, ever) using participant survey data (Table S3). The odds of detecting several bacteria were elevated in men who reported vaginal sex (Table S9). For example, the odds of detecting G. vaginalis were higher in men who reported vaginal sex in the past 60 days (OR = 6.97; 95% CI 2.49–19.51), past year (OR = 11.79; 95% CI 3.21–43.30), or ever (OR = 35.5352; 95% CI 2.05–616.68) compared with the men who did not. Notably, in the 14 men who had never had vaginal sex, we did not detect A. vaginae, P. amnii, L. iners, Streptococcus anginosus, M. indolicus, G. vaginalis, V. montpellierensis, or S. amnii. Most of the associations we identified above remained significant when combinations of sexual behaviors were considered (Table S10). For example, the OR of detecting G. vaginalis in men who reported vaginal and rectal sex in the past year, compared with men who only reported rectal sex, was 12.09 (95% CI 2.31–63.42). Some bacteria were more prevalent in men who did not report vaginal sex. The OR of detecting Corynebacterium glucuronolyticum (OR = 0.27; 95% CI 0.08–0.91) was lower in men who reported vaginal sex in the past year compared with men who did not. A few bacteria were associated with rectal and oral sex, but these associations were comparatively weak and involved less prevalent bacteria (Table S10). All associations mentioned above remained significant when covariates including age, race, and urethral STI history were considered in multivariate analyses, and no associations were identified with non-sexual covariates (Table S11). Thus, colonization of the PU by many FRT bacteria may be contingent upon vaginal sex.
The effects of vaginal sex can be detected after extended intervals
To test if the associations with vaginal sex we observed were transient, we evaluated if any bacteria were more prevalent in men who had reported vaginal sex within 1 year, but not in the past 60 days (n = 6), compared with men who did not have vaginal sex in the past year (n = 26). Despite the small sample size, two FRT bacteria, G. vaginalis (OR = 7.67; 95% CI 1.04–56.77) and Veillonellaceae bacterium DNF00626 (OR = 29.44; 95% CI 1.20–719.88), were still associated with vaginal sex after 60 days, and several other BVAB were trending toward but did not reach significance (Tables S2 and S10). This indicated that bacteria remained associated with vaginal sex for at least 60 days after exposure. To assess if colonization with FRT bacteria persisted beyond a year, we tested if these organisms were enriched in men who reported vaginal sex in their lifetimes, but not the past year (n = 12), compared with men who never had vaginal sex (n = 14). FRT bacteria were detected in a few of the men who reported vaginal sex more than a year ago, but this comparison did not reach significance (Table S10).
Sexual behaviors significantly associate with variance in PU microbiome composition
Since specific bacteria were associated with vaginal sex, we investigated the contribution of specific sexual behaviors, relative to other variables we captured, on the variance in PU microbiome composition. Univariate PERMANOVA regression analysis was performed using the survey questions as independent variables. Except for sexual behavior, no other variables including age, race, and STI history were significant (data not shown). Next, sexual behaviors performed at different intervals (i.e., past 60 days, past year, ever, respectively) were treated as a polytomous variable (i.e., with the category of oral, rectal, vaginal sex, and their combinations). Polytomous sexual behavior in the past 60 days and year significantly contributed to the variance, and the effect of lifetime sexual behavior was trending toward significant (p < 0.01 within 60 days, p < 0.02 within 1 year, p = 0.066 ever), explaining 10.25%, 10.08%, and 4.74% of the variance of the PU microbiome composition, respectively (Table S11). Since sexual behavior was the key factor associated with PU composition, the effects of each individual sexual behavior were dissected using a multivariate PERMANOVA regression analysis where the behaviors were treated as separate independent variables (Table 1). Vaginal sex was the only independent variable associated significantly with variation in PU microbiome composition (p value <0.002 within 60 days, p value <0.001 within 1 year, and p value <0.005 ever) and explained 4.26%, 4.14%, and 3.37% of the variance in the past 60 days, past year, and ever, respectively.
Table 1.
PERMANOVA regression analysis of effects of sexual behavior
| R2 | p value | |
| A. Time of Behavior | ||
| Past 60 days | 0.1025 | 0.0060 |
| Past 1 year | 0.1008 | 0.0130 |
| Lifetime | 0.0474 | 0.0660 |
| B. Time and Type of Behavior | ||
| Oral 60 days | 0.0096 | 0.5115 |
| Vaginal 60 days | 0.0426 | 0.0020 |
| Rectal 60 days | 0.0141 | 0.1588 |
| Oral 1 year | 0.0067 | 0.8432 |
| Vaginal 1 year | 0.0414 | 0.0010 |
| Rectal 1 year | 0.0182 | 0.0569 |
| Vaginal lifetime | 0.0337 | 0.0050 |
| Rectal lifetime | 0.0070 | 0.8821 |
A. Univariate effects at three time intervals (past 60 days, past year, lifetime). B. Univariate effects on all combinations of specific sexual behaviors (oral, vaginal, rectal) at three time intervals (past 60 days, past year, lifetime). R2 depicts the proportion of variation in the data explained by the group being tested. P value indicates whether this result was a result of chance.
Discussion
PU microbiology is highly relevant to human health because the urethra can transmit STIs, and cryptic pathogens have been implicated in a variety of idiopathic urogenital syndromes.11,61,62,63,64 Overall, our findings establish a baseline for studying this microbiome in male urogenital tract health and disease.
Consortia of LAB and Corynebacterium spp. similar to those we observed here have also been detected in adolescents17 and adults,65 so these may be resilient microbial communities. Alternately, these observations could reflect continuous seeding of the PU with microorganisms from other body surfaces without colonization.17,18,66 In either case, the apparent lack of interactions between BVAB and core bacteria, similar loads of core bacteria in UT1 and UT2, numerous differences observed in the metagenomes of UT1 and UT2, and predicted oxygen requirements of BVAB and core bacteria all suggest that these groups of bacteria inhabit different urethral niches. We speculate that core bacteria colonize the urethral meatus where oxygen availability may be higher, whereas BVAB colonize the mucin-rich penile urethra.67 Determining if the PU microbiota contributes to urethral health is a key area for future study. If PU core bacteria contribute to STI colonization resistance, like vaginal lactobacilli,68 additional risks of the increasingly common use of broad-spectrum antimicrobials in STI prophylaxis need to be considered.69 In contrast, if the PU microbiota is dispensable, broader application of prophylaxis may be warranted to eliminate male reservoirs of female urogenital pathogens.
Our observations suggest that PU colonization by FRT bacteria is contingent upon vaginal exposures and are consistent with prior reports that have documented suspected female to male transmission of undifferentiated G. vaginalis strains and BV biofilms.70,71 Testing this and the reverse, if men can transmit these bacteria to women, directly will be difficult because this would ideally require sampling of sexual dyads before and after first partnered sexual activity, because it would be expected that existing dyads would already share readily transmissible microorganisms. Alternately, it might be possible to address these questions using a prospective dyad study design that incorporates interval antibiotic treatment of one or both partners or periods of voluntary abstinence. Nonetheless, incident BV is strongly associated with new male sex partners,72 but attempts to prevent BV by treating male partners with antibiotics have been mostly unsuccessful.73,74,75,76,77,78 Notably, none of these treatment trials assessed if all the BVAB we observed in men here were eliminated. Thus, improved understanding of how antibiotics impact the PU microbiota could inform future BV partner-treatment trials and provide insights into the role of sexual exposures in BV and other genitourinary syndromes.
Sexual behavior has been ignored in most human microbiome studies. Thus, our observation that sexual behavior explains more than 10% of variance in PU microbiota composition in specific intervals is even more striking when additional context is considered. Many participants in our study reported few, infrequent, or no vaginal exposures, and the prevalence of many FRT bacteria we identified is low in the overall adult female population. Much smaller effect sizes have been attributed to what are believed to be key drivers of the composition of other human microbiomes. For example, a recent study of more than 4,000 adults that considered hundreds of covariates was only able to account for 16% of gut microbiome variance.79 However, the apparent powerful influence of vaginal sex on the PU microbiome may reflect that this site is less likely to be exposed to other sources of microorganisms than the skin and mucosal surfaces of the gut and lungs.
The associations between vaginal sex and BVAB that we observed may not be surprising in the context of what is known about the role of vaginal sex in the dispersal of sexually transmitted pathogens. The rarity of protective vaginal Lactobacillus spp. observed in our study specimens is consistent with some prior observations,10,11,17 and may reflect that these bacteria persist in and seed the vagina from a gastrointestinal reservoir.80,81 Our failure to detect microorganisms associated with oral and anal sex may not be due to differences in sample size; oral sex was more prevalent than vaginal sex in all intervals when partner gender was not considered (Table S3). We also failed to detect several known and putative sexually transmitted and urinary pathogens that have been observed in prior studies of men with urogenital disease or men in whom urethral inflammation was not evaluated.11,12,13,14,15,16 Some of the differences might be explained by the more sensitive PCR approaches used in prior studies. Alternately, adaptations that permit specific BVAB to colonize the PU without eliciting inflammation may not be widely distributed, and we strictly excluded men with urethral/inflammation or disease. Natural history studies of the urethral microbiome in controlled settings seem warranted to differentiate these possibilities, considering their high public health significance.
We propose a model that integrates our findings with prior observations regarding the composition, development, and succession of urogenital microbiomes. Culture and metagenomic approaches have identified a broad array of bacterial taxa in pediatric vaginal and male urine specimens.82,83,84,85,86,87 However, few common patterns have emerged across these studies, and there seems to be general agreement that pediatric urogenital microbiomes are disorganized and sparse. By adolescence, unknown behavioral and/or developmental changes in male individuals permit prolonged colonization of the distal PU by core microorganisms.17 We hypothesize that vaginal sex might then promote colonization of the deeper PU by sexually transmitted microorganisms without replacement of the core PU microorganisms. In contrast, protective Lactobacillus spp., possibly from the rectum,81 begin to stably colonize the vaginal mucosa,88 concomitant with an increase in glycogen levels proceeding menarche in adolescent females.89 Sexually transmitted microorganisms may then be introduced by vaginal sex71 or other types of sexual exposures.90
Limitations of the study
Our observations suggest that PU colonization by FRT bacteria is contingent upon vaginal exposures and are consistent with prior reports that documented suspected female to male transmission of undifferentiated G. vaginalis strains and BV biofilms.70,71 Testing this and the reverse, if men can transmit these bacteria to women, could be difficult because this would ideally involve sampling of sexual dyads before and after first partnered sexual activity, as existing dyads might already share readily transmissible microorganisms. Alternately, it might be possible to address these questions using a prospective dyad study design that incorporates interval antibiotic treatment of one of the partners and/or periods of voluntary abstinence.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human-derived urethral swabs | This paper | |
| Human-derived vaginal swabs | Muzny et al. 201834 | |
| Chemicals, peptides, and recombinant proteins | ||
| Lysozyme | Millipore Sigma | SAE0152 |
| AMPure XP Reagent Beads | Beckman Coulter | A63881 |
| Critical commercial assays | ||
| DNeasy Blood & Tissue Kit | Qiagen | Cat. No. 69506 |
| NexteraXT DNA Library Prep Kit | Illumina | FC-131-1096 |
| NexteraXT Index Kit Set A | Illumina | FC-131-2001 |
| Qubit 1X dsDNA HS Assay Kit | ThermoFisher | Q32854 |
| Deposited data | ||
| Raw metagenomic sequencing data of urethral swabs (human reads removed) | This paper | NCBI Bio Project Accession number: PRJNA785561 |
| Raw metagenomic sequencing data of vaginal swabs (human reads removed) | This paper | NCBI Accession number: PRJNA707585 |
| Sequences deposited | This paper | NCBI SRA Bio Sample accessions from SAMN23566502 to SAMN23566611 |
| Oligonucleotides | ||
| Gardnerella vaginalis_cpn60_Forward Primer- CGCATCTGCTAAGGATGTTG | Menard et al. 200891 | AF240579.3 |
| Gardnerella vaginalis_cpn60_Reverse Primer- CAGCAATCTTTTCGCCAACT | Menard et al. 200891 | AF240579.3 |
| Gardnerella vaginalis_cpn60_Probe- FAM-TGCAACTATTTCTGCAGCAGATCC-TAMRA | Menard et al. 200891 | AF240579.3 |
| Recombinant DNA | ||
| plasmid pGEMT-easy(cpn60) | This paper | |
| Mock community B, Even, Low Concentration v5.1L | BEI Resources, NIAID, NIH | HM782D |
| Software and algorithms | ||
| Custom scripts for data analysis | This paper | https://github.com/qunfengdong/HealthyMaleUrethralMicrobiome |
| MetaPhlAn3 | Beghini et al. 202123 | https://github.com/biobakery/biobakery/wiki/metaphlan3 |
| Kraken2 | Wood et al. 201936 | https://github.com/DerrickWood/kraken2 |
| R | R Core Team 202292 | https://www.R-project.org/ |
| R package "MASS" | Venables et al. 200293 | https://cran.r-project.org/web/packages/MASS/index.html |
| R package "microbiome" | Lahti, Shetty 201794 | https://www.bioconductor.org/packages/release/bioc/html/microbiome.html |
| R package "cluster" | Maechler et al. 202295 | https://cran.r-project.org/web/packages/cluster/index.html |
| R package "clusterSim" | Walesiak, Dudek 202096 | https://cran.r-project.org/web/packages/clusterSim/index.html |
| R package "circlize" | Gu et al. 201497 | https://cran.r-project.org/web/packages/circlize/index.html |
| R package "ComplexHeatmap" | Gu et al. 201698; Gu 202299 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| R package ”vegan” | https://github.com/vegandevs/vegan | https://cran.r-project.org/web/packages/vegan/index.html |
| R package "phyloseq" | McMurdie, Holmes 2013100 | https://www.bioconductor.org/packages/release/bioc/html/phyloseq.html |
| R package ”taxize” | Chamberlain, Szocs 2013101 | https://cran.r-project.org/web/packages/taxize/index.html |
| R package "ggplot2" | Wickham 2016102 | https://ggplot2.tidyverse.org |
| R package "ggbiplot" | https://github.com/vqv/ggbiplot | https://github.com/vqv/ggbiplot |
| R package "ggpubr" | https://rpkgs.datanovia.com/ggpubr/ | https://rpkgs.datanovia.com/ggpubr/ |
| Sparse InverseE Covariance Estimation for Ecological Association INference (SPIEC-EASI) | Kurtz et al. 201559 | https://github.com/zdk123/SpiecEasi |
| Cytoscape | Shannon et al. 2003103 | https://cytoscape.org |
| Gardnerella genomospecies (GS) identification | Potter et al. 201954 | |
| HUMAnN 3.0 | Beghini et al. 202123 | https://github.com/biobakery/humann |
| R package ”epiR” | https://cran.r-project.org/web/packages/epiR/epiR.pdf | |
| Microsoft Excel | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| HiSeq4000 | Illumina | |
| NovaSeq6000 | Illumina | |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David E. Nelson (nelsonde@indiana.edu).
Materials availability
All primers, probe, and plasmid generated for this study are available from the lead contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Study population and cross-sectional design
This is an analysis of asymptomatic cis-gender men who enrolled as healthy controls in a case-control research study named the Idiopathic Urethritis Men’s Project (IUMP), while undergoing STI screening, at the Bell Flower Clinic in Indianapolis, Indiana, between August 4th, 2016 and December 11th, 2019. Participants provided written informed consent for the collection of two distal urethral swabs, followed by a first-catch urine specimen, and completed a detailed computer-assisted self-interviewing health and behavioral questionnaire (Methods S1, Table S1). A genital examination and a urethral Gram stain were performed to evaluate for urethral discharge, Gram-negative intracellular diplococci, and the number of polymorphonuclear leukocytes per high-power field (PMN/HPF). Participants were excluded if they tested positive for Chlamydia trachomatis, Mycoplasma genitalium, Neisseria gonorrhoeae, Trichomonas vaginalis, or urethritis clade Neisseria meningitidis US_NmUC strains,104 had urethral discharge, exhibited >1 PMN/HPF on their Gram stain smear, reported antibiotic use in the last month, or reported urethral symptoms. This study was approved by the Indiana University-Purdue University-Indianapolis (IUPUI) Institutional Review Board and the Marion County Public Health Department.
Method details
DNA extraction
DNA was extracted from urethral swabs using the DNeasy Blood and Tissue Kit (Qiagen, USA), and eluted using Tris-EDTA buffer. DNA was also extracted from molecular grade water spiked with a pure culture of Thermus thermophilus cells (an environmental hyperthermophile unlikely to be present in reagents or specimens) (reagent contamination control) or a mock bacterial community of known composition listed in the table below (HM782-D, BEI resources, NIAID, NIH) (Mock community B, Even, Low Concentration v5.1L) (extraction/annotation control). The mock bacterial community contains a pool of approximately 100 ng of the bacterial genomic DNA mixture (see table below) suspended in 25 μL TE buffer (10 mM Tris-HCl and 1 mM EDTA, pH ∼ 7.4).
| Organism | NCBI Reference Sequence |
|---|---|
| Acinetobacter baumannii, strain 5377 | NC_009085 |
| Actinomyces odontolyticus, strain 1A.21 | NZ_AAYI02000000 |
| Bacillus cereus, strain NRS 248 | NC_003909 |
| Bacteroides vulgatus, strain ATCC® 8482™ | NC_009614 |
| Clostridium beijerinckii, strain NCIMB 8052 | NC_009617 |
| Deinococcus radiodurans, strain R1 (smooth) | NC_001263, NC_001264 |
| Enterococcus faecalis, strain OG1RF | NC_17316 |
| Escherichia coli, strain K12, sub-strain MG1655 | NC_000913 |
| Helicobacter pylori, strain 26695 | NC_000915 |
| Lactobacillus gasseri, strain 63 AM | NC_008530 |
| Listeria monocytogenes, strain EGDe | NC_003210 |
| Neisseria meningitidis, strain MC58 | NC_003112 |
| Propionibacterium acnes, strain KPA171202 | NC_006085 |
| Pseudomonas aeruginosa, strain PAO1-LAC | NC_002516 |
| Rhodobacter sphaeroides, strain ATH 2.4.1 | NC_007493, NC_007494 |
| Staphylococcus aureus, strain TCH1516 | NC_010079 |
| Staphylococcus epidermidis, FDA strain PCI 1200 | NC_004461 |
| Streptococcus agalactiae, strain 2603 V/R | NC_004116 |
| Streptococcus mutans, strain UA159 | NC_004350 |
| Streptococcus pneumoniae, strain TIGR4 | NC_003028 |
Vaginal swab DNA from a separate cohort of African American women enrolled in a prior incident bacterial vaginosis (iBV) study was also sequenced to test if our approaches could detect common FRT bacteria.34
Metagenomic shotgun sequencing
Dual-indexed sequencing libraries were prepared from 1 ng total swab or control DNA using the Nextera XT Library Preparation kit (Illumina Inc., USA), pooled, and pair end (2x150 b) sequenced on the Illumina HiSeq4000 or NovaSeq platforms at the Indiana University Center for Medical Genomics.
Data preprocessing
Most sequences from the 110 participants were human 27,092,065 ± 8,140,893 (86.6%), and 461,885 ± 1,411,457 (1.5%) were annotated to specific microorganisms. 18 participants with low or no bacteria reads (MetaPhlAn3 Bacteria/Human reads <= 0.01%) were subsequently removed from the downstream analyses. For the remaining 92 participants, the number of human and microbial sequences were 27,275,575 ± 8,461,033 (85.8%), and 551,858 ± 1,528,493 (1.7%) respectively.
Data analysis
MetaPhlAn3 taxa counts were subject to additive log transformation (ALR) using human sequences (the log of microbial counts plus 0.1 divided by human reads), and a default centered-log ratio (CLR) transformation by R package "microbiome". Urethrotype (UT) classification was performed with the partitioning around medoids clustering algorithm (pam by R package “cluster”) based on Euclidean distances of both ALR (Figures 3 and S3) and CLR (Figure S2) transformed microbial taxon counts. Calinski-Harabasz (CH) and Silhouette index using the transformed ALR and CLR dataset were used to identify the optimal number of clusters,105,45 which were designated as urethrotypes in this study (Figures 3, S2, and S3). In addition to pam, other R packages (clusterSim, circlize, ComplexHeatmap, vegan, phyloseq and taxize), and custom R scripts were used to generate the heatmaps for the ALR and CLR transformed data. Principal component analysis was performed and plotted by R using ALR transformed data.41 Wilcoxon signed-rank test was used for detecting differential abundance in ALR transformed data and Fisher’s exact test was used to compare the prevalence of taxa between urethrotypes. R package vegan was used to calculate Chao I and Ace alpha diversity indices. Spearman correlations were calculated using Gardnerella vaginalis qPCR genome counts and ALR transformed sequence counts.
Taxonomic profiling analyses
Raw sequences were demultiplexed and annotated using MetaPhlAn3 and Kraken2 (Tables S2 and S3).23,36 Human sequences were counted using Kraken2.106 Only T. thermophilus sequences were detected in the reagent contamination control (Table S2). All microorganisms in the mock community were detected, confirming that our extraction approach could isolate genomic DNA from diverse bacteria, and our annotation approaches could detect these bacteria (Table S2).
Quantitative PCR
Gardnerella vaginalis-specific qPCR targeting the cpn60 gene was performed using an Eppendorf Realplex4 cycler to quantify organism load, compared with a plasmid standard curve (pGEMT-easy(cpn60)), as previously described with modifications:91 each reaction contained 0.25 nmol of probe, 0.5 nmol of gene-specific primers, 1 μl of extracted DNA, and 5 μl of 2XFastStart TaqMan Probe Master mix (Roche, USA) in a total of 10 μl. Results were expressed as copies of microorganism per sample.
Network visualization of positive associations
Raw taxa counts were further analyzed using a microbiome network analysis method, Sparse InversE Covariance Estimation for Ecological Association INference (SPIEC-EASI),59 by applying either covariance selection (Glasso) or neighborhood selection (MB). The network was visualized using Cytoscape.103
Gardnerella genomospecies identification
PU sequences that matched nine Gardnerella genomospecies (GS) were identified using a modification of an approach that was developed to define these GS and identify GS-specific sequences (Potter et al., 2019). After human sequences were removed, Gardnerella sequences from the PU specimens were compared with the nine GS using NCBI BLAST+2.12.0. (Sayers et al., 2022). First, unique matches that mapped to only one GS were identified. Perfect matches that contained no mismatches or gaps were then identified from the unique matches. Those perfect matches were then blasted against the NCBI nt nucleotide database to check if they also matched perfectly to other non Gardnerella bacteria; if so, they were removed from the subsequent analysis. The numbers and proportions of the remaining GS-specific sequences for each subject were calculated.
Functional metabolic profiling
Raw PU sequences were also used to determine the abundance of microbial genes corresponding to specific metabolic pathways using HUMAnN 3.0 (Beghini et al., 2021). Gene Ontology (GO) terms were mapped from the corresponding genes in the HUMAnN output. The same GO terms from different taxa were then combined, and Wilcoxon signed rank test was used to identify differentially abundant GO terms between UT1 and UT2.
Quantification and statistical analysis
Statistical analysis was performed using R statistical programing, GraphPad Prism 9, and Microsoft Excel.
Chi-square tests were used to examine if there were associations between UTs and any of the individual items in the questionnaire, and p-values were adjusted by Benjamini-Hochberg correction.107 Confusion matrices (with 0.5 added to each cell if at least 1 cell was 0) were then used to calculate odds ratios (ORs) with 95% confidence intervals (CI) between UTs and sexual behaviors (vaginal, rectal, and oral sex in the past 60 days, past year or lifetime) using R package “epiR”.
Permutational multivariate analyses of variance (PERMANOVA) regressions were performed on the Euclidean distance matrix by R package vegan, derived from the ALR transformed taxa, to identify factors associated with variation in microbial composition.108 Questionnaire covariates were individually tested in the univariate PERMANOVA regression, and p-values were adjusted by Benjamini-Hochberg correction.107 Sexual behaviors at different intervals (past 60 days, past year, lifetime), were treated as a polytomous variable (i.e., multiple categories: none, vaginal-only, rectal-only, oral-only, oral-vaginal, oral-rectal, vaginal-rectal, and oral-rectal-vaginal) in the univariate PERMANOVA regression models. Finally, individual sexual behaviors were treated as separate independent variables in a multivariate PERMANOVA regression model.
Custom R scripts and confusion matrices (with 0.5 added to each cell if at least 1 cell was 0) for each taxon and sexual behavior (vaginal, rectal, oral, vaginal and rectal, vaginal and oral, rectal and oral, within the same time periods as above) were used to calculate ORs to test for association of individual taxa with specific sexual behaviors. The same taxa (response variable) and sexual behaviors (explanatory variables) were used in logistic regression models with covariates (age, race, STI diagnosis in the past 60 days and lifetime, history of self-reported chlamydia, gonorrhea, trichomoniasis, syphilis or NGU) to test if any of those covariates were significantly associated with the taxa. To investigate whether any taxa were associated with vaginal sex in certain time intervals, we also calculated ORs for 1) vaginal sex between the last 60 days and 1 year versus no vaginal sex in the last year, and 2) vaginal sex ever but not in the last year versus no vaginal sex ever.
Acknowledgments
This manuscript is dedicated in loving memory of our colleague Dr. Byron Batteiger.
The authors thank the study participants and the Marion County Public Health Department’s Bellflower STD clinic staff, Xiaoli Zhang for banking specimens, and are grateful to Drs. Stanley Spinola, Frank Yang, Ted Bae, and Brad Griesenauer for critical review of the manuscript. The mock community reagent was obtained through BEI Resources, National Institute of Allergy and Infectious Diseases, National Institutes of Health, as part of the Human Microbiome Project: Genomic DNA from Microbial Mock Community B (Even, Low Concentration), v5.1L, for 16S rRNA Gene Sequencing, HM-782D.
Funding: This work was supported by grant R01AI116706 from the National Institute of Allergy and Infectious Diseases to D.E.N. Grant K23AI106957-01A1 from the National Institute of Allergy and Infectious Diseases supported the efforts of C.A.M.
Author contributions
Conceptualization, D.E.N., B.E.B., B.V.D.P., and Q.D.; Methodology, E.T., Y.X., X.G, Q.D., and D.E.N.; Software, Y.X., and Q.D.; Formal Analysis, Y.X., X.G., and Q.D.; Investigation, E.T., T.A.B., J.A.W., N.G., L.J.F., and Y.X.,; Writing – Original Draft, E.T. and D.E.N.; Writing – Review & Editing, E.T., Y.X., S.J.J., Q.D., C.A.M., J.D.F., and D.E.N.; Funding Acquisition, D.E.N. and Q.D.; Resources, D.E.N. and C.A.M.; Supervision, D.E.N. and Q.D.
Declaration of interests
Y.X., X.G., T.A.B., B.V.D.P., N.G., L.J.F., J.D.F., and Q.D. have no conflicts of interest. S.J.J. has received honoraria and consulting fees from Hologic, Inc. E.T., J.A.W., and. D.E.N. retain the patent for the US_NmUC diagnostic assay used in this manuscript. C.A.M. is a consultant for Lupin Pharmaceuticals, BioFire Diagnostics, Cepheid, and PhagoMed. She has also received research funding support from Lupin Pharmaceuticals, Abbott Molecular, and Gilead as well as speaker honoraria from Abbott Molecular, Cepheid, Roche Diagnostics, and Becton Dickinson.
Published: March 21, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.100981.
Contributor Information
Qunfeng Dong, Email: qdong@luc.edu.
David E. Nelson, Email: nelsonde@indiana.edu.
Supplemental information
Gene Ontology (GO) terms from different taxa were mapped from the gene families, combined, and used to identify differential GO terms whose abundance differed between UT1 and UT2.
Each row includes the odds ratio and its 95% confidence interval for the specific levels of comparison for types of sexual behavior (oral, rectal, vaginal), in specific time intervals (1 year, 60 days, lifetime), and covariates used for each taxon.
Sheet “60d-1yr+ vs. 1year-” compares (1) vaginal sex in the past year, but not in the past 60 days, and (2) no vaginal sex in the past year. Sheet “1year-ever+ vs. ever-” compares (1) vaginal sex in the lifetime, but not in the past year, and (2) never had vaginal sex.
Data and code availability
Raw sequences from the PU specimens have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive under Bio Project Accession number: PRJNA785561 (http://www.ncbi.nlm.nih.gov/bioproject/785561), and the vaginal specimen sequences are deposited under Accession number: PRJNA707585 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA707585/). All the sequences deposited in the NCBI SRA database are under the Bio Sample accessions from SAMN23566502 to SAMN23566611. All data supporting the findings of this study are included in the paper and supplemental information files.
Custom scripts are archived at GitHub (https://github.com/qunfengdong/HealthyMaleUrethralMicrobiome).
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
References
- 1.Pudney J., Anderson D. Innate and acquired immunity in the human penile urethra. J. Reprod. Immunol. 2011;88:219–227. doi: 10.1016/j.jri.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moi H., Blee K., Horner P.J. Management of non-gonococcal urethritis. BMC Infect. Dis. 2015;15:294. doi: 10.1186/s12879-015-1043-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pickard J.M., Zeng M.Y., Caruso R., Núñez G. Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017;279:70–89. doi: 10.1111/imr.12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma B., Forney L.J., Ravel J. Vaginal microbiome: rethinking health and disease. Annu. Rev. Microbiol. 2012;66:371–389. doi: 10.1146/annurev-micro-092611-150157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong Q., Nelson D.E., Toh E., Diao L., Gao X., Fortenberry J.D., Van der Pol B. The microbial communities in male first catch urine are highly similar to those in paired urethral swab specimens. PLoS One. 2011;6:e19709. doi: 10.1371/journal.pone.0019709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pohl H.G., Groah S.L., Pérez-Losada M., Ljungberg I., Sprague B.M., Chandal N., Caldovic L., Hsieh M. The urine microbiome of healthy men and women differs by urine collection method. Int. Neurourol. J. 2020;24:41–51. doi: 10.5213/inj.1938244.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karstens L., Asquith M., Caruso V., Rosenbaum J.T., Fair D.A., Braun J., Gregory W.T., Nardos R., McWeeney S.K. Community profiling of the urinary microbiota: considerations for low-biomass samples. Nat. Rev. Urol. 2018;15:735–749. doi: 10.1038/s41585-018-0104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karstens L., Asquith M., Davin S., Fair D., Gregory W.T., Wolfe A.J., Braun J., McWeeney S. Controlling for contaminants in low-biomass 16S rRNA gene sequencing experiments. mSystems. 2019;4:e00290-19. doi: 10.1128/mSystems.00290-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowie W.R., Pollock H.M., Forsyth P.S., Floyd J.F., Alexander E.R., Wang S.P., Holmes K.K. Bacteriology of the urethra in normal men and men with nongonococcal urethritis. J. Clin. Microbiol. 1977;6:482–488. doi: 10.1128/jcm.6.5.482-488.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson D.E., Van Der Pol B., Dong Q., Revanna K.V., Fan B., Easwaran S., Sodergren E., Weinstock G.M., Diao L., Fortenberry J.D. Characteristic male urine microbiomes associate with asymptomatic sexually transmitted infection. PLoS One. 2010;5:e14116. doi: 10.1371/journal.pone.0014116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srinivasan S., Chambers L.C., Tapia K.A., Hoffman N.G., Munch M.M., Morgan J.L., Domogala D., Lowens M.S., Proll S., Huang M.L., et al. Urethral microbiota in men: association of Haemophilus influenzae and Mycoplasma penetrans with nongonococcal urethritis. Clin. Infect. Dis. 2020;73:e1684–e1693. doi: 10.1093/cid/ciaa1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frølund M., Wikström A., Lidbrink P., Abu Al-Soud W., Larsen N., Harder C.B., Sørensen S.J., Jensen J.S., Ahrens P. The bacterial microbiota in first-void urine from men with and without idiopathic urethritis. PLoS One. 2018;13:e0201380. doi: 10.1371/journal.pone.0201380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manhart L.E., Khosropour C.M., Liu C., Gillespie C.W., Depner K., Fiedler T., Marrazzo J.M., Fredricks D.N. Bacterial vaginosis-associated bacteria in men: association of Leptotrichia/Sneathia spp. with nongonococcal urethritis. Sex. Transm. Dis. 2013;40:944–949. doi: 10.1097/OLQ.0000000000000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis D.A., Brown R., Williams J., White P., Jacobson S.K., Marchesi J.R., Drake M.J. The human urinary microbiome; bacterial DNA in voided urine of asymptomatic adults. Front. Cell. Infect. Microbiol. 2013;3:41. doi: 10.3389/fcimb.2013.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Y., Gao H., Mihindukulasuriya K.A., La Rosa P.S., Wylie K.M., Vishnivetskaya T., Podar M., Warner B., Tarr P.I., Nelson D.E., et al. Biogeography of the ecosystems of the healthy human body. Genome Biol. 2013;14:R1. doi: 10.1186/gb-2013-14-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bajic P., Dornbier R.A., Doshi C.P., Wolfe A.J., Farooq A.V., Bresler L. Implications of the genitourinary microbiota in prostatic disease. Curr. Urol. Rep. 2019;20:34. doi: 10.1007/s11934-019-0904-6. [DOI] [PubMed] [Google Scholar]
- 17.Nelson D.E., Dong Q., Van der Pol B., Toh E., Fan B., Katz B.P., Mi D., Rong R., Weinstock G.M., Sodergren E., Fortenberry J.D. Bacterial communities of the coronal sulcus and distal urethra of adolescent males. PLoS One. 2012;7:e36298. doi: 10.1371/journal.pone.0036298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zozaya M., Ferris M.J., Siren J.D., Lillis R., Myers L., Nsuami M.J., Eren A.M., Brown J., Taylor C.M., Martin D.H. Bacterial communities in penile skin, male urethra, and vaginas of heterosexual couples with and without bacterial vaginosis. Microbiome. 2016;4:16. doi: 10.1186/s40168-016-0161-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mändar R., Punab M., Borovkova N., Lapp E., Kiiker R., Korrovits P., Metspalu A., Krjutškov K., Nõlvak H., Preem J.K., et al. Complementary seminovaginal microbiome in couples. Res. Microbiol. 2015;166:440–447. doi: 10.1016/j.resmic.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 20.Batteiger T.A., Jordan S.J., Toh E., Fortenberry L., Williams J.A., LaPradd M., Katz B., Fortenberry J.D., Dodge B., Arno J., et al. Detection of rectal Chlamydia trachomatis in heterosexual men who report cunnilingus. Sex. Transm. Dis. 2019;46:440–445. doi: 10.1097/OLQ.0000000000000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jordan S.J., Toh E., Williams J.A., Fortenberry L., LaPradd M.L., Katz B.P., Batteiger B.E., Nelson D.E., Batteiger T.A. Aetiology and prevalence of mixed-infections and mono-infections in non-gonococcal urethritis in men: a case-control study. Sex. Transm. Infect. 2020;96:306–311. doi: 10.1136/sextrans-2019-054121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jordan S.J., Toh E., Williams J.A., Fortenberry L.J., LaPradd M., Ryan J.D., Nelson D.E., Batteiger T.A. No pathogen-specific sign or symptom predicts the etiology of monomicrobial nongonococcal urethritis in men. Sex. Transm. Dis. 2020;47:329–331. doi: 10.1097/OLQ.0000000000001158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beghini F., McIver L.J., Blanco-Míguez A., Dubois L., Asnicar F., Maharjan S., Mailyan A., Manghi P., Scholz M., Thomas A.M., et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife. 2021;10:e65088. doi: 10.7554/eLife.65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruoff K.L., Fishman J.A., Calderwood S.B., Kunz L.J. Distribution and incidence of viridans streptococcal species in routine clinical specimens. Am. J. Clin. Pathol. 1983;80:854–858. doi: 10.1093/ajcp/80.6.854. [DOI] [PubMed] [Google Scholar]
- 25.Mores C.R., Price T.K., Wolff B., Halverson T., Limeira R., Brubaker L., Mueller E.R., Putonti C., Wolfe A.J. Genomic relatedness and clinical significance of Streptococcus mitis strains isolated from the urogenital tract of sexual partners. Microb. Genom. 2021;7:mgen000535. doi: 10.1099/mgen.0.000535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nouioui I., Carro L., García-López M., Meier-Kolthoff J.P., Woyke T., Kyrpides N.C., Pukall R., Klenk H.P., Goodfellow M., Göker M. Genome-based taxonomic classification of the phylum actinobacteria. Front. Microbiol. 2018;9:2007. doi: 10.3389/fmicb.2018.02007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oakley B.B., Fiedler T.L., Marrazzo J.M., Fredricks D.N. Diversity of human vaginal bacterial communities and associations with clinically defined bacterial vaginosis. Appl. Environ. Microbiol. 2008;74:4898–4909. doi: 10.1128/AEM.02884-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang C., Fan A., Li H., Yan Y., Qi W., Wang Y., Han C., Xue F. Vaginal bacterial profiles of aerobic vaginitis: a case-control study. Diagn. Microbiol. Infect. Dis. 2020;96:114981. doi: 10.1016/j.diagmicrobio.2019.114981. [DOI] [PubMed] [Google Scholar]
- 29.Donders G.G.G., Vereecken A., Bosmans E., Dekeersmaecker A., Salembier G., Spitz B. Definition of a type of abnormal vaginal flora that is distinct from bacterial vaginosis: aerobic vaginitis. BJOG. 2002;109:34–43. doi: 10.1111/j.1471-0528.2002.00432.x. [DOI] [PubMed] [Google Scholar]
- 30.Giuliano A.R., Nielson C.M., Flores R., Dunne E.F., Abrahamsen M., Papenfuss M.R., Markowitz L.E., Smith D., Harris R.B. The optimal anatomic sites for sampling heterosexual men for human papillomavirus (HPV) detection: the HPV detection in men study. J. Infect. Dis. 2007;196:1146–1152. doi: 10.1086/521629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanaoka N., Ito S., Konagaya M., Nojiri N., Yasuda M., Fujimoto T., Deguchi T. Infectious human adenoviruses are shed in urine even after disappearance of urethral symptoms. PLoS One. 2019;14:e0212434. doi: 10.1371/journal.pone.0212434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cannon M.J., Hyde T.B., Schmid D.S. Review of cytomegalovirus shedding in bodily fluids and relevance to congenital cytomegalovirus infection. Rev. Med. Virol. 2011;21:240–255. doi: 10.1002/rmv.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Israele V., Shirley P., Sixbey J.W. Excretion of the Epstein-Barr virus from the genital tract of men. J. Infect. Dis. 1991;163:1341–1343. doi: 10.1093/infdis/163.6.1341. [DOI] [PubMed] [Google Scholar]
- 34.Muzny C.A., Blanchard E., Taylor C.M., Aaron K.J., Talluri R., Griswold M.E., Redden D.T., Luo M., Welsh D.A., Van Der Pol W.J., et al. Identification of key bacteria involved in the induction of incident bacterial vaginosis: a prospective study. J. Infect. Dis. 2018;218:966–978. doi: 10.1093/infdis/jiy243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rabe L.K., Winterscheid K.K., Hillier S.L. Association of viridans group streptococci from pregnant women with bacterial vaginosis and upper genital tract infection. J. Clin. Microbiol. 1988;26:1156–1160. doi: 10.1128/jcm.26.6.1156-1160.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wood D.E., Lu J., Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Breitwieser F.P., Pertea M., Zimin A.V., Salzberg S.L. Human contamination in bacterial genomes has created thousands of spurious proteins. Genome Res. 2019;29:954–960. doi: 10.1101/gr.245373.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ravel J., Gajer P., Abdo Z., Schneider G.M., Koenig S.S.K., McCulle S.L., Karlebach S., Gorle R., Russell J., Tacket C.O., et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA. 2011;108(Suppl 1):4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arumugam M., Raes J., Pelletier E., Le Paslier D., Yamada T., Mende D.R., Fernandes G.R., Tap J., Bruls T., Batto J.M., et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shade A., Handelsman J. Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 2012;14:4–12. doi: 10.1111/j.1462-2920.2011.02585.x. [DOI] [PubMed] [Google Scholar]
- 41.Gloor G.B., Macklaim J.M., Pawlowsky-Glahn V., Egozcue J.J. Microbiome datasets are compositional: and this is not optional. Front. Microbiol. 2017;8:2224. doi: 10.3389/fmicb.2017.02224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aitchison J. The statistical analysis of compositional data. J. Roy. Stat. Soc. B. 1982;44:139–160. doi: 10.1111/j.2517-6161.1982.tb01195.x. [DOI] [Google Scholar]
- 43.Aitchison J., Greenacre M. Biplots of compositional data. Roy. Stat. Soc. Appl. Stat. C. 2002;51:375–392. doi: 10.1111/1467-9876.00275. [DOI] [Google Scholar]
- 44.Regalado J., Lundberg D.S., Deusch O., Kersten S., Karasov T., Poersch K., Shirsekar G., Weigel D. Combining whole-genome shotgun sequencing and rRNA gene amplicon analyses to improve detection of microbe-microbe interaction networks in plant leaves. ISME J. 2020;14:2116–2130. doi: 10.1038/s41396-020-0665-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y., Li Z., Xiong H., Gao X., Wu J. IEEE International Conference on Data Mining; Sydney, NSW, Australia: 2010. ”Understanding of Internal Clustering Validation Measures,” 2010; pp. 911–916. [DOI] [Google Scholar]
- 46.Funke G., von Graevenitz A., Clarridge J.E., 3rd, Bernard K.A. Clinical microbiology of coryneform bacteria. Clin. Microbiol. Rev. 1997;10:125–159. doi: 10.1128/CMR.10.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eisenberg T., Gronow S., Falgenhauer J., Imirzalioglu C., Mühldorfer K., Rau J., Blom J., Fawzy A., Glaeser S.P., Kämpfer P. Sneathia vaginalis sp. nov. (Fusobacteriales, Leptotrichiaceae) as a replacement of the species 'Sneathia amnii' Harwich et al. 2012 and 'Leptotrichia amnionii' Shukla et al. 2002, and emended description of Sneathia Collins et al. Int. J. Syst. Evol. Microbiol. 2019;71 doi: 10.1099/ijsem.0.004663. 2001. [DOI] [PubMed] [Google Scholar]
- 48.Fredricks D.N., Fiedler T.L., Marrazzo J.M. Molecular identification of bacteria associated with bacterial vaginosis. N. Engl. J. Med. 2005;353:1899–1911. doi: 10.1056/NEJMoa043802. [DOI] [PubMed] [Google Scholar]
- 49.Tao Z., Zhang L., Zhang Q., Lv T., Chen R., Wang L., Huang Z., Hu L., Liao Q. The pathogenesis of Streptococcus anginosus in aerobic vaginitis. Infect. Drug Resist. 2019;12:3745–3754. doi: 10.2147/IDR.S227883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Austin M.N., Rabe L.K., Srinivasan S., Fredricks D.N., Wiesenfeld H.C., Hillier S.L. Mageeibacillus indolicus gen. nov., sp. nov.: a novel bacterium isolated from the female genital tract. Anaerobe. 2015;32:37–42. doi: 10.1016/j.anaerobe.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Machado A., Cerca N. Influence of biofilm formation by Gardnerella vaginalis and other anaerobes on bacterial vaginosis. J. Infect. Dis. 2015;212:1856–1861. doi: 10.1093/infdis/jiv338. [DOI] [PubMed] [Google Scholar]
- 52.Piot P., Van Dyck E., Peeters M., Hale J., Totten P.A., Holmes K.K. Biotypes of Gardnerella vaginalis. J. Clin. Microbiol. 1984;20:677–679. doi: 10.1128/jcm.20.4.677-679.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vaneechoutte M., Guschin A., Van Simaey L., Gansemans Y., Van Nieuwerburgh F., Cools P. Emended description of Gardnerella vaginalis and description of Gardnerella leopoldii sp. nov., Gardnerella piotii sp. nov. and Gardnerella swidsinskii sp. nov., with delineation of 13 genomic species within the genus Gardnerella. Int. J. Syst. Evol. Microbiol. 2019;69:679–687. doi: 10.1099/ijsem.0.003200. [DOI] [PubMed] [Google Scholar]
- 54.Potter R.F., Burnham C.A.D., Dantas G. In silico analysis of Gardnerella genomospecies detected in the setting of bacterial vaginosis. Clin. Chem. 2019;65:1375–1387. doi: 10.1373/clinchem.2019.305474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turner E., Sobel J.D., Akins R.A. Prognosis of recurrent bacterial vaginosis based on longitudinal changes in abundance of Lactobacillus and specific species of Gardnerella. PLoS One. 2021;16:e0256445. doi: 10.1371/journal.pone.0256445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oerlemans E.F.M., Wuyts S., Bellen G., Wittouck S., De Boeck I., Ruban K., Allonsius C.N., van den Broek M.F.L., Donders G.G.G., Lebeer S. The dwindling microbiota of aerobic vaginitis, an inflammatory state enriched in pathobionts with limited TLR stimulation. Diagnostics. 2020;10:879. doi: 10.3390/diagnostics10110879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hardin G. The competitive exclusion principle. Science. 1960;131:1292–1297. doi: 10.1126/science.131.3409.1292. [DOI] [PubMed] [Google Scholar]
- 58.Bergey's Manual of Systematics of Archaea and Bacteria. (2015) 10.1002/9781118960608. [DOI]
- 59.Kurtz Z.D., Müller C.L., Miraldi E.R., Littman D.R., Blaser M.J., Bonneau R.A. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput. Biol. 2015;11:e1004226. doi: 10.1371/journal.pcbi.1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Young R.M., Meyer I.H. The trouble with "MSM" and "WSW": erasure of the sexual-minority person in public health discourse. Am. J. Public Health. 2005;95:1144–1149. doi: 10.2105/AJPH.2004.046714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Javier-DesLoges J., McKay R.R., Swafford A.D., Sepich-Poore G.D., Knight R., Parsons J.K. The microbiome and prostate cancer. Prostate Cancer Prostatic Dis. 2022;25:159–164. doi: 10.1038/s41391-021-00413-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rizzo A., Santoni M., Mollica V., Fiorentino M., Brandi G., Massari F. Microbiota and prostate cancer. Semin. Cancer Biol. 2022;86:1058–1065. doi: 10.1016/j.semcancer.2021.09.007. [DOI] [PubMed] [Google Scholar]
- 63.Li W.T., Iyangar A.S., Reddy R., Chakladar J., Bhargava V., Sakamoto K., Ongkeko W.M., Rajasekaran M. The bladder microbiome is associated with epithelial-mesenchymal transition in muscle invasive urothelial Bladder carcinoma. Cancers. 2021;13:3649. doi: 10.3390/cancers13153649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee H.Y., Wang J.W., Juan Y.S., Li C.C., Liu C.J., Cho S.Y., Yeh H.C., Chueh K.S., Wu W.J., Wu D.C. The impact of urine microbiota in patients with lower urinary tract symptoms. Ann. Clin. Microbiol. Antimicrob. 2021;20:23. doi: 10.1186/s12941-021-00428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mehta S.D., Zhao D., Green S.J., Agingu W., Otieno F., Bhaumik R., Bhaumik D., Bailey R.C. The microbiome composition of a man's penis predicts incident bacterial vaginosis in his female sex partner with high accuracy. Front. Cell. Infect. Microbiol. 2020;10:433. doi: 10.3389/fcimb.2020.00433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu C.M., Hungate B.A., Tobian A.A.R., Serwadda D., Ravel J., Lester R., Kigozi G., Aziz M., Galiwango R.M., Nalugoda F., et al. Male circumcision significantly reduces prevalence and load of genital anaerobic bacteria. mBio. 2013;4:e00076. doi: 10.1128/mBio.00076-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Russo C.L., Spurr-Michaud S., Tisdale A., Pudney J., Anderson D., Gipson I.K. Mucin gene expression in human male urogenital tract epithelia. Hum. Reprod. 2006;21:2783–2793. doi: 10.1093/humrep/del164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dabee S., Passmore J.A.S., Heffron R., Jaspan H.B. The complex link between the female genital microbiota, genital infections, and inflammation. Infect. Immun. 2021;89:e00487-20. doi: 10.1128/IAI.00487-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grant J.S., Stafylis C., Celum C., Grennan T., Haire B., Kaldor J., Luetkemeyer A.F., Saunders J.M., Molina J.M., Klausner J.D. Doxycycline prophylaxis for bacterial sexually transmitted infections. Clin. Infect. Dis. 2020;70:1247–1253. doi: 10.1093/cid/ciz866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schwebke J.R., Muzny C.A., Josey W.E. Role of Gardnerella vaginalis in the pathogenesis of bacterial vaginosis: a conceptual model. J. Infect. Dis. 2014;210:338–343. doi: 10.1093/infdis/jiu089. [DOI] [PubMed] [Google Scholar]
- 71.Swidsinski A., Doerffel Y., Loening-Baucke V., Swidsinski S., Verstraelen H., Vaneechoutte M., Lemm V., Schilling J., Mendling W. Gardnerella biofilm involves females and males and is transmitted sexually. Gynecol. Obstet. Invest. 2010;70:256–263. doi: 10.1159/000314015. [DOI] [PubMed] [Google Scholar]
- 72.Verstraelen H., Verhelst R., Vaneechoutte M., Temmerman M. The epidemiology of bacterial vaginosis in relation to sexual behaviour. BMC Infect. Dis. 2010;10:81. doi: 10.1186/1471-2334-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Swedberg J., Steiner J.F., Deiss F., Steiner S., Driggers D.A. Comparison of single-dose vs one-week course of metronidazole for symptomatic bacterial vaginosis. JAMA. 1985;254:1046–1049. [PubMed] [Google Scholar]
- 74.Vejtorp M., Bollerup A.C., Vejtorp L., Fanøe E., Nathan E., Reiter A., Andersen M.E., Strømsholt B., Schrøder S.S. Bacterial vaginosis: a double-blind randomized trial of the effect of treatment of the sexual partner. Br. J. Obstet. Gynaecol. 1988;95:920–926. doi: 10.1111/j.1471-0528.1988.tb06581.x. [DOI] [PubMed] [Google Scholar]
- 75.Mengel M.B., Berg A.O., Weaver C.H., Herman D.J., Herman S.J., Hughes V.L., Koepsell T.D. The effectiveness of single-dose metronidazole therapy for patients and their partners with bacterial vaginosis. J. Fam. Pract. 1989;28:163–171. [PubMed] [Google Scholar]
- 76.Vutyavanich T., Pongsuthirak P., Vannareumol P., Ruangsri R.A., Luangsook P. A randomized double-blind trial of tinidazole treatment of the sexual partners of females with bacterial vaginosis. Obstet. Gynecol. 1993;82:550–554. [PubMed] [Google Scholar]
- 77.Colli E., Landoni M., Parazzini F. Treatment of male partners and recurrence of bacterial vaginosis: a randomised trial. Genitourin. Med. 1997;73:267–270. doi: 10.1136/sti.73.4.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schwebke J.R., Lensing S.Y., Lee J., Muzny C.A., Pontius A., Woznicki N., Aguin T., Sobel J.D. Treatment of male sexual partners of women with bacterial vaginosis: a randomized, double-blind, placebo-controlled trial. Clin. Infect. Dis. 2021;73:e672–e679. doi: 10.1093/cid/ciaa1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Falony G., Joossens M., Vieira-Silva S., Wang J., Darzi Y., Faust K., Kurilshikov A., Bonder M.J., Valles-Colomer M., Vandeputte D., et al. Population-level analysis of gut microbiome variation. Science. 2016;352:560–564. doi: 10.1126/science.aad3503. [DOI] [PubMed] [Google Scholar]
- 80.Duar R.M., Lin X.B., Zheng J., Martino M.E., Grenier T., Pérez-Muñoz M.E., Leulier F., Gänzle M., Walter J. Lifestyles in transition: evolution and natural history of the genus Lactobacillus. FEMS Microbiol. Rev. 2017;41:S27–S48. doi: 10.1093/femsre/fux030. [DOI] [PubMed] [Google Scholar]
- 81.Antonio M.A.D., Rabe L.K., Hillier S.L. Colonization of the rectum by Lactobacillus species and decreased risk of bacterial vaginosis. J. Infect. Dis. 2005;192:394–398. doi: 10.1086/430926. [DOI] [PubMed] [Google Scholar]
- 82.Myhre A.K., Bevanger L.S., Berntzen K., Bratlid D. Anogenital bacteriology in non-abused preschool children: a descriptive study of the aerobic genital flora and the isolation of anogenital Gardnerella vaginalis. Acta Paediatr. 2002;91:885–891. doi: 10.1080/080352502760148586. [DOI] [PubMed] [Google Scholar]
- 83.Hill G.B., St Claire K.K., Gutman L.T. Anaerobes predominate among the vaginal microflora of prepubertal girls. Clin. Infect. Dis. 1995;20(Suppl 2):S269–S270. doi: 10.1093/clinids/20.supplement_2.s269. [DOI] [PubMed] [Google Scholar]
- 84.Xiaoming W., Jing L., Yuchen P., Huili L., Miao Z., Jing S. Characteristics of the vaginal microbiomes in prepubertal girls with and without vulvovaginitis. Eur. J. Clin. Microbiol. Infect. Dis. 2021;40:1253–1261. doi: 10.1007/s10096-021-04152-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kinneman L., Zhu W., Wong W.S.W., Clemency N., Provenzano M., Vilboux T., Jane't K., Seo-Mayer P., Levorson R., Kou M., et al. Assessment of the urinary microbiome in children younger than 48 months. Pediatr. Infect. Dis. J. 2020;39:565–570. doi: 10.1097/INF.0000000000002622. [DOI] [PubMed] [Google Scholar]
- 86.Fredsgaard L., Thorsteinsson K., Bundgaard-Nielsen C., Ammitzbøll N., Leutscher P., Chai Q., Jensen A.M., Sørensen S., Pedersen L.M., Hagstrøm S., Arenholt L.T.S. Description of the voided urinary microbiota in asymptomatic prepubertal children - a pilot study. J. Pediatr. Urol. 2021;17:545.e1–545.e8. doi: 10.1016/j.jpurol.2021.03.019. [DOI] [PubMed] [Google Scholar]
- 87.Storm D.W., Copp H.L., Halverson T.M., Du J., Juhr D., Wolfe A.J. A Child's urine is not sterile: a pilot study evaluating the Pediatric Urinary Microbiome. J. Pediatr. Urol. 2022;18:383–392. doi: 10.1016/j.jpurol.2022.02.025. [DOI] [PubMed] [Google Scholar]
- 88.Hickey R.J., Zhou X., Settles M.L., Erb J., Malone K., Hansmann M.A., Shew M.L., Van Der Pol B., Fortenberry J.D., Forney L.J. Vaginal microbiota of adolescent girls prior to the onset of menarche resemble those of reproductive-age women. mBio. 2015;6:e00097-15. doi: 10.1128/mBio.00097-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nunn K.L., Ridenhour B.J., Chester E.M., Vitzthum V.J., Fortenberry J.D., Forney L.J. Vaginal glycogen, not estradiol, is associated with vaginal bacterial community composition in black adolescent women. J. Adolesc. Health. 2019;65:130–138. doi: 10.1016/j.jadohealth.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Berger B.J., Kolton S., Zenilman J.M., Cummings M.C., Feldman J., McCormack W.M. Bacterial vaginosis in lesbians: a sexually transmitted disease. Clin. Infect. Dis. 1995;21:1402–1405. doi: 10.1093/clinids/21.6.1402. [DOI] [PubMed] [Google Scholar]
- 91.Menard J.P., Fenollar F., Henry M., Bretelle F., Raoult D. Molecular quantification of Gardnerella vaginalis and Atopobium vaginae loads to predict bacterial vaginosis. Clin. Infect. Dis. 2008;47:33–43. doi: 10.1086/588661. [DOI] [PubMed] [Google Scholar]
- 92.Team R.C. 2022. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 93.Venables W.N., Ripley B.D., Venables W.N. 4th Edition. Springer; 2002. Modern Applied Statistics with S. [Google Scholar]
- 94.Lahti L., Shetty S. 2017. Tools for Microbiome Analysis in R. [Google Scholar]
- 95.Maechler M., Rousseeuw P., Struyf A., Hubert M., Hornik K. 2022. Cluster: Cluster Analysis Basics and Extensions. [Google Scholar]
- 96.Dudek A., Walesiak M. The choice of variable normalization method in cluster analysis. Education Excellence and Innovation Management: A 2025 Vision to Sustain Economic Development during Global Challenges. 2020:325–340. [Google Scholar]
- 97.Gu Z., Gu L., Eils R., Schlesner M., Brors B. Circlize Implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 98.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 99.Gu Z. Complex heatmap visualization. iMeta. 2022;1:e43. doi: 10.1002/imt2.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McMurdie P.J., Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chamberlain S.A., Szöcs E. taxize: taxonomic search and retrieval in R. F1000Res. 2013;2:191. doi: 10.12688/f1000research.2-191.v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wickham H. 2nd ed. Springer International Publishing; 2016. ggplot2 : Elegant Graphics for Data Analysis. Use R! [Google Scholar]
- 103.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Toh E., Williams J.A., Qadadri B., Ermel A., Nelson D.E. Development of a SimpleProbe real-Time PCR Assay for rapid detection and identification of the US novel urethrotropic clade of Neisseria meningitidis ST-11 (US_NmUC) PLoS One. 2020;15:e0228467. doi: 10.1371/journal.pone.0228467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Calinski T., Harabasz J. A dendrite method for cluster analysis. Comm. in Stats. - Theory & Methods. 1974;3:1–27. [Google Scholar]
- 106.Bush S.J., Connor T.R., Peto T.E.A., Crook D.W., Walker A.S. Evaluation of methods for detecting human reads in microbial sequencing datasets. Microb. Genom. 2020;6:mgen000393. doi: 10.1099/mgen.0.000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 108.Anderson M.J. Permutational multivariate analysis of variance (PERMANOVA) 2017. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene Ontology (GO) terms from different taxa were mapped from the gene families, combined, and used to identify differential GO terms whose abundance differed between UT1 and UT2.
Each row includes the odds ratio and its 95% confidence interval for the specific levels of comparison for types of sexual behavior (oral, rectal, vaginal), in specific time intervals (1 year, 60 days, lifetime), and covariates used for each taxon.
Sheet “60d-1yr+ vs. 1year-” compares (1) vaginal sex in the past year, but not in the past 60 days, and (2) no vaginal sex in the past year. Sheet “1year-ever+ vs. ever-” compares (1) vaginal sex in the lifetime, but not in the past year, and (2) never had vaginal sex.
Data Availability Statement
Raw sequences from the PU specimens have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive under Bio Project Accession number: PRJNA785561 (http://www.ncbi.nlm.nih.gov/bioproject/785561), and the vaginal specimen sequences are deposited under Accession number: PRJNA707585 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA707585/). All the sequences deposited in the NCBI SRA database are under the Bio Sample accessions from SAMN23566502 to SAMN23566611. All data supporting the findings of this study are included in the paper and supplemental information files.
Custom scripts are archived at GitHub (https://github.com/qunfengdong/HealthyMaleUrethralMicrobiome).
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.