

Abstract
The blood–brain barrier (BBB) is a physiological barrier maintaining a specialized brain micromilieu that is necessary for proper neuronal function. Endothelial tight junctions and specific transcellular/efflux transport systems provide a protective barrier against toxins, pathogens, and immune cells. The barrier function is critically supported by other cell types of the neurovascular unit, including pericytes, astrocytes, microglia, and interneurons. The dysfunctionality of the BBB is a hallmark of neurological diseases, such as ischemia, brain tumors, neurodegenerative diseases, infections, and autoimmune neuroinflammatory disorders. Moreover, BBB dysfunction is critically involved in epilepsy, a brain disorder characterized by spontaneously occurring seizures because of abnormally synchronized neuronal activity. While resistance to antiseizure drugs that aim to reduce neuronal hyperexcitability remains a clinical challenge, drugs targeting the neurovasculature in epilepsy patients have not been explored. The use of novel imaging techniques permits early detection of BBB leakage in epilepsy; however, the detailed mechanistic understanding of causes and consequences of BBB compromise remains unknown. Here, we discuss the current knowledge of BBB involvement in temporal lobe epilepsy with the emphasis on the neurovasculature as a therapeutic target.
Keywords: angiopoietin/Tie2, blood–brain barrier, cerebral edema, epilepsy animal models, in vitro BBB models, neurovascular unit, temporal lobe epilepsy, VEGF/VEGFR, Wnt/β‐catenin
Targeting vascular dysfunction at the neurovascular unit in epilepsy and neurological diseases.
1. EPILEPSY—CLINICAL FEATURES, DIAGNOSIS, AND THE FUNCTION OF THE BBB
Epilepsy is one of the most frequent chronic neurological diseases worldwide, affecting about 65 million people in all age groups [1]. The clinical hallmarks of epilepsy are spontaneous recurrent seizures, with distinct semiology based on the function of the cortical areas involved in seizure‐associated hyperexcitation [2]. Epilepsy is a major burden for patients, caregivers, health care and economic systems [3]. Patients with epilepsy suffer from decreased quality of life, adverse medication effects, cognitive impairment, and educational, vocational and social consequences. Increased mortality is associated with epilepsy because of seizure related injuries, accidents, increased risk of psychiatric comorbidities and sudden unexpected death in epilepsy patients (SUDEP) [4, 5]. The International League Against Epilepsy (ILAE) classifies epilepsy into four main types: (1) focal, (2) generalized, (3) combined generalized and focal, and (4) unknown. Etiologies of epilepsies are diverse, for example, structural, genetic, infectious, metabolic, immune and unknown causes [6]. In acquired epilepsies, such as temporal lobe (TLE) epilepsy, these etiologies lead to the transformation of “normal” brain tissue into a hyperexcitable neuronal network, a process termed epileptogenesis. The mechanisms of epileptogenesis and epilepsy progression are poorly understood and involve a variety of pathophysiological processes, including dysfunction of the BBB [7, 8].
The first line of treatment and mainstay in epilepsy therapy are antiseizure drugs (ASD) [4]. Despite the current medical treatment with multiple well‐tolerated ASD, one‐third of patients suffer from refractory epilepsy (RE) [9]. In the case of RE, the chance of becoming seizure‐free is only 5%–10% with additional ASD and <5%–10% with further ASD application. Therefore, other therapeutic strategies, such as curative or palliative epilepsy surgery, neurostimulation or ketogenic diet should be considered in these cases [4, 10].
Despite diagnostic and therapeutic advancements, many patients suffering from RE cannot be treated adequately, given that ASD offer only symptomatic (seizure‐suppressing) relief but no curative therapy [10]. Hence, understanding the pathophysiology of epileptogenesis is a key requirement for the development of causal epilepsy therapy. One important pathomechanism of epileptogenesis is a dysfunctional BBB, resulting in the subsequent leakage of albumin and immune cells into the brain parenchyma [11, 12]. Repetitive seizures further lead to BBB malfunction, possibly contributing to a vicious circle that results in treatment failure.
Structurally, the BBB separates the brain parenchyma from the systemic circulation and maintains a permissive micromilieu necessary for proper neuronal function (Figure 1) [13, 14, 15]. During development, endothelial cells receive specific cues that induce BBB characteristics [15]. Their functional interaction with other cells of the central nervous system (CNS) such as neuroblasts, pericytes, perivascular fibroblast, astrocytes, microglia and nerve endings, collectively known as the neurovascular unit (NVU), is essential for BBB maturation [13, 15]. Impairment of these interactions may have fatal consequences. For example, pericyte deficiency is associated with BBB breakdown during development and aging [16, 17, 18] and has been implicated in the pathogenesis of epilepsy [19].
FIGURE 1.
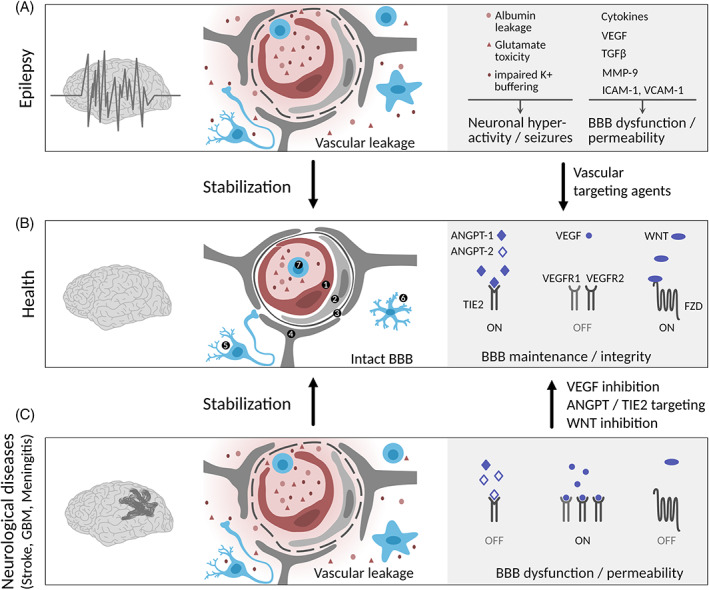
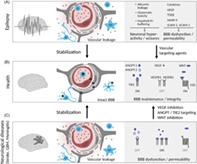
Vascular dysfunction at the neurovascular unit in epilepsy. Schematic drawing of the cellular and molecular composition of the NVU in health, epilepsy, and neurological diseases with BBB dysfunction (stroke, GBM, meningitis). (A) In epilepsy, BBB disruption leads to leakage of blood components (albumin, neurotransmitter (e.g., glutamate), K+ ions), leukocyte infiltration, aberrant transport and clearance of molecules, neuronal hyperactivity, and BBB damage. Microglia and astrocytes release cytokines and growth factors that further promote EC activation and BBB dysfunction. (B) An intact BBB protects the brain parenchyma from factors present in the systemic circulation and maintains a highly regulated brain micromilieu required for brain homeostasis and proper neuronal functioning. (C) BBB integrity can be re‐established upon vascular‐targeting therapies in neurological diseases associated with BBB dysfunction and cerebral edema. Endothelial cell (1), pericyte (2), basal lamina (3), astrocyte (4), neuron (5), microglia/activated microglia (6), leukocyte (7). Illustration: Visual Science Communication
BBB properties are induced by Wnt/β‐catenin signaling, and mainly driven by Wnt7a/Wnt7b and the non‐Wnt‐related Norrie disease protein, which are master regulators of endothelial barrier properties in the brain and in the retina [20, 21, 22, 23]. Endothelial‐specificity of Wnt7 is guaranteed by a receptor complex that is formed by frizzled 4, low‐density lipoprotein receptor‐related protein 5/6, G‐protein coupled receptor 124 and the reversion‐inducing‐cysteine‐rich protein with kazal motifs [23, 24, 25]. Wnt/β‐catenin signaling is strongly active during brain development, and at low level in the adult to maintain BBB properties [14, 20, 25]. Furthermore, vascular endothelial growth factor (VEGF) is crucial for brain angiogenesis and BBB development [26]. VEGF evokes immature, leaky vessels [27] and the downregulation of endothelial claudin‐5 and occludin, thereby promoting BBB breakdown in vivo [28].
In addition to inductive cues, brain vessel maturation requires pericyte‐secreted angiopoietin(Ang)‐1 that signals endothelial Tie2 to maintain BBB characteristics and reduce permeability [13, 29]. The endothelial barrier is established by inter‐endothelial junctions with the absence of fenestrations, low level vesicular transport, and expression of the ATP‐binding cassette (ABC) transporters Abcb1a (P‐gp, MDR1) and Abcg2 (breast cancer resistance protein, BCRP), forming a metabolic barrier for blood–borne toxic compounds and drugs [14, 30]. Claudin‐5 is a major constituent of tight junctions which are intermingled with adherens junctions, systems that are both linked to the cytoskeleton [13, 14, 31]. Proteins that bind to the VE‐cadherin/β‐catenin complex include cerebral cavernous malformations (CCM) proteins 1, 2, and 3. Loss‐of‐function mutations in either CCM1, 2, or 3 lead to vascular instability specifically in the CNS, which often result in neurological symptoms such as seizures and epilepsy, as well as hemorrhagic strokes [32]. Both, rare genetic and more frequent sporadic forms of CCM have defects in endothelial‐polarization, aberrant TGFβ and β‐catenin signaling, as well as a mesenchymal/stem cell‐like signature of clonal endothelial cells [33, 34]. Interestingly, CCM‐targeting approaches with Propanolol, a betablocker known to reduce BBB leakage and inflammation, are currently pursued in clinical trials [35] (see Table 1 for all therapeutic approaches mentioned throughout the article).
TABLE 1.
Experimental and clinical approaches with BBB‐restoring efficacy in epilepsy and neurological disorders with a dysfunctional BBB.
| Drug/therapeutic | Target/mode of action | Preclinical model | Clinical approach | Reference(s) |
|---|---|---|---|---|
| Propanolol | β‐adrenergic receptor antagonist, anti‐angiogenic, anti‐inflammatory | Repurposing in rare vascular diseases, e.g., CCM | [35] | |
| RepSox | TGF‐β‐RI/ALK5 inhibitor, regulator of claudin‐5 expression | Kainic acid model of TLE | [160] | |
| Ketogenic diet | GLUT1 targeting | Childhood epilepsy, recurrent glioblastoma |
[10] [169] |
|
|
Sunitinib, Anti‐VEGF antibodies / VEGFR2 pathway inhibitors |
Tyrosine kinase inhibitor, VEGF inhibition, prevention of angiogenesis |
Rat TLE model, Hippocampal cultures |
[54] [8] [55] |
|
| Tie2 activators, Angiopoietin‐2 inhibitors | Angiopietin‐Tie2 signaling, vascular stabilization, edema prevention | Preclinical models for glioma, stroke, retinal diseases | Clinical trials for macula edema |
[56] [57] |
| Everolimus, Rapamycin | mTOR antagonist | Rodent epilepsy models | Tuberous sclerosis complex |
[46] |
| Dexamethasone | Glucocorticoid, stabilization of endothelial‐cell junctions, edema prevention | Rodent epilepsy models | Temporal lobe epilepsy, Glioblastoma |
[10] [163] |
| Engineered Wnt ligands | Wnt/β‐catenin signaling | Preclinical glioma and stroke models | [62] | |
| Losartan | Angiotensin‐type 1 receptor antagonist, TGFβ signaling inhibitor | Rodent epilepsy models |
[80] [81] [82] [46] |
|
| Tariquidar | P‐gp inhibitor | Rodent epilepsy models |
[70] [12] |
|
| IPR‐179 | MMP2/9 inhibitor | Rodent epilepsy models |
[87] [86] |
|
| Imatinib | Tyrosine kinase and PDGFR inhibitor | Hippocampal cultures | Chronic myeloid leukemia and glioblastoma‐associated epilepsy |
[91] [46] |
|
Natalizumab, Anti‐VCAM‐1 antibodies |
α4 integrin, blockade of immune cell infiltration | Pilocarpine model of SE | Adjunctive therapy for patients with drug resistant epilepsy/Clinical phase II |
[94] [95] |
| Verapamil | Calcium channel blocker, inhibitor of P‐gp | Adjuvant treatment in drug resistant epilepsy | [157, 158] |
Abbreviations: BBB, blood–brain barrier; CCM, cerebral cavernous malformation; GLUT1, glucose transporter protein type 1; MMP, matrix metalloproteinase; mTOR, mammalian target of rapamycin; PDGFRβ, platelet‐derived growth factor receptor beta; P‐gp, P‐glycoprotein; SE, status epilepticus; TLE, temporal lobe epilepsy; TGF‐β‐RI/ALK5, transforming growth factor‐beta receptor I/activin like kinase 5; VCAM‐1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor.
Given the importance of the mentioned pathways in development, vascular endothelial growth factor (VEGF), angiopoietin/Tie2 and Wnt/β‐catenin signaling also interfere with barrier properties in glioblastoma [36, 37, 38], brain ischemia [39] and bacterial meningitis [40]. These pathways are key regulators of BBB dysfunction in CNS disorders, and thus could also play a role in epilepsy (Figure 1).
Broadening our current knowledge and understanding of BBB dysfunction will open new avenues for the development of disease modifying therapies and possibly preventive, antiepileptogenic treatment strategies in the future. The aim of this review is to summarize and discuss the implications of BBB dysfunction for epilepsy with special emphasis on the neurovasculature as a novel therapeutic target. As BBB‐targeting therapy has not yet been attempted in epilepsy, here we discuss potential targets that have been successfully applied in other CNS disorders associated with BBB dysfunction.
2. IMPAIRMENT OF THE BBB AS A CONSEQUENCE OF ICTOGENIC AND EPILEPTOGENIC BRAIN LESIONS
Although epilepsy emerged as a consequence of neuronal hyperexcitability, BBB disruption is increasingly recognized as a critical factor for seizure development (Figure 1, [11, 12]). While there are many excellent reviews that describe the BBB involvement in epilepsy, we here aim to highlight if the restoration of the BBB could be used as a potential therapeutic target for seizure management and epilepsy therapy. Different components of the NVU can be affected by various pathomechanisms of epileptogenesis that compromise the BBB, including leakage of serum proteins, excessive glutamate release, glucose transporter dysfunction, angiogenesis and neuroinflammation [11, 41, 42]. While novel imaging techniques permit early detection of BBB leakage in epilepsy patients [11, 43, 44], basic proof of concept that BBB modulation can change seizure pathology has not been shown preclinically or clinically. This is however an important and clinically relevant topic, and thus methods for diagnosis and treatments in improving the vascular integrity are under development [45, 46].
Although pursued for some time [12, 42], interests in understanding the mechanisms underlying BBB dysfunction in epilepsy emerged in the 1980s with ultrastructural analyses of human epilepsy tissue, which revealed increased pinocytosis, alterations of tight junctions and of the capillary basement membrane [47, 48, 49]. Additional studies indicated a role for deficient glucose transport and brain ion homeostasis. Under normal conditions the glucose transporter (GLUT1) transports plasma glucose across the BBB, whilst potassium (K+) homeostasis is controlled by voltage‐dependent ion channels expressed by glia. Evidence obtained from epileptic patients undergoing cortical resection indicated the alteration of GLUT1 expression, and positron emission tomography studies showed decreased brain glucose uptake and hypometabolism in the epileptogenic region [50, 51] (clinically targeted by ketogenic diet [10]).
A crucial role for angiogenesis in the progression of TLE was demonstrated by Rigau et al. who described angiogenesis, aberrant vascularization, and BBB alterations in temporal lobe epilepsy (TLE) [52], which is in line with the overexpression of angiogenic factors. Rigau and colleagues have shown a correlation between an increased density of the microvascular network with seizure frequency, loss of tight junctions, immunoglobulin (IgG) leakage and accumulation in neurons of hippocampal resections from TLE patients [52]. Furthermore, excessive angiogenesis was evident in the acute and chronic phase of epilepsy in a rat TLE model [52]. Gene expression analysis of epileptic tissue obtained from patients undergoing epilepsy surgery further identified VEGF signaling as a crucial mediator of pharmacoresistant TLE [8, 53], suggesting the inhibition of the VEGF/VEGFR2 pathway might offer a therapeutic strategy to restore BBB function for seizure prevention and progression. Indeed, inhibitors of VEGF/VEGFR2 signaling, such as Sunitinib, prevented angiogenesis, hippocampal atrophy and seizures in rat TLE [54]. In hippocampal cultures, VEGF neutralization prevented seizure‐like event induced vascular remodeling and the downregulation of ZO‐1 [8, 55].
In addition, evidence for a role of angiopoietin/Tie2 signaling in TLE has been suggested as the Tie2 receptor tyrosine kinase was upregulated at disease onset and in chronic epilepsy [52]. Therefore, targeting Tie2 signaling for the restoration of BBB function, which is being pursued in neurological diseases associated with vascular leakage syndromes, might offer new therapeutic possibilities for BBB/neurovascular targeting in epilepsy [27, 56, 57]. BBB‐restoring drugs that alleviate leakage and inflammation include mTOR (mammalian target of rapamycin) inhibitors, that is, Rapamycin/Everolimus, and corticosteroids that are currently in clinical use [10, 45, 46]. Patients with mTOR‐dependent malformations of cortical development (MCD) associated with seizures display vessel abnormalities in the affected, dysmorphic cortical tissue [45, 58]. In MCD animal models, the hypervascularization is sensitive to Rapamycin, that is approved for epilepsy treatment in tuberous sclerosis complex [58]. Mutations in mTOR pathway genes, such as the GATOR1 complex (DEPDC5, NPRL2, NPRL3) could serve as genetic‐/biomarkers for BBB restoring therapies and patient stratification [10].
A common gene signature for the dysregulation of BBB was found between different neurovascular disorders [14, 59]. Daneman and colleagues demonstrated that CNS endothelial cells responded similarly to stroke, traumatic brain injury, neuroinflammation and seizures with a “BBB dysfunction signature” that resembled endothelial gene expression in peripheral organs, [59]. These findings are further supported by an unbiased omics expression analysis of microvessels isolated from brain tumor [37], stroke [60, 61] and meningitis models [40]. These reports suggest that vascular or BBB‐targeted therapies might show efficacy across a range of brain malignancies as described by Martin and colleagues targeting BBB dysfunction in brain tumors and stroke with engineered Wnt ligands [62].
In epilepsy, the parenchymal accumulation of IgG and albumin was demonstrated to be the cause of BBB dysfunction in patients and animal models [52, 63, 64, 65]. Seminal work, which is basis for studying BBB disruption in seizure pathologies identified increased BBB permeability in acute seizures [41, 42, 52, 63, 64, 65], which also persisted in chronic epilepsy [52, 65]. This increased BBB permeability is associated with several downstream effects, such that neuronal activity is directly affected. Ivens et al. provided initial mechanistic insight of albumin‐mediated BBB changes [65] and were further confirmed in subsequent studies [42, 66, 67]. Extravasated albumin enters astrocytes via TGFβ receptors, leading to a Smad2‐mediated downregulation of inward rectifying potassium (Kir4.1) channels on astrocytes that ensure clearance of excess extracellular K+ [42, 65, 66, 67]. Vascular injury‐mediated brain exposure to serum albumin induced transcriptional changes that can be blocked by TGFβ inhibitors [67], thus suggesting the TGFβ pathway as a therapeutic target [45, 46]. Additionally, the expression of glutamate transporters by astrocytes is decreased by brain interstitial albumin [42]. These events lead to the reduction of extracellular potassium and glutamate buffering, resulting in neuronal hyperexcitability, and epileptogenesis (Figure 1). Albumin‐induced astrocyte activation further results in the release of proinflammatory cytokines and chemokines [42]. Moreover, albumin can also be taken up by neurons, which can strongly increase the synthesis and release of glutamate, leading to additional hyperexcitability and epileptogenesis [42, 52, 64]. These events are accompanied by dysregulated expression of uptake transporters for the uptake of essential compounds like glucose and monocarboxylates at the BBB, together with enhanced expression and activity of efflux transporters such as P‐gp and drug‐metabolizing enzymes of the cytochrome P450 family [68]. The latter processes, known as the “transporter hypothesis,” have been implicated in pharmacoresistance to ASD since they have been speculated to restrict ASDs from entering the brain [12, 41, 69]. Furthermore, increased P‐gp expression is associated with higher seizure frequency in both animal models and TLE patients [41]. Conversely, in experimental epilepsy tariquidar, a P‐gp inhibitor, improved responses to ASD [70].
It is still not clear if BBB breakdown is the cause or consequence of seizures. The first scenario is supported by a study from Marchi et al. [63], in which they showed transient, osmotic BBB opening is sufficient to cause seizures in the absence of CNS pathologies. Further crucial work established a direct connection of seizures and BBB breakdown via drainage of serum albumin [41, 42, 45]. This view is supported by several studies that demonstrated the disruption of BBB by bile salts, albumin or mannitol [41, 42, 64, 65, 71, 72]. Additional work from Van Vliet and colleagues indicated that continuous BBB opening by mannitol in the chronic epileptic phase increased seizure frequency and further promoted BBB damage [64]. In a rare, monogenetic form of epilepsy named incontinentia pigmenti, caused by a mutation in the NF‐κB essential modulator (NEMO), loss of brain vessels and BBB function in patients was shown to be causative for disease onset [73]. On the contrary, Prager et al. showed recurrent seizures promoted microvascular injury which led to neurovascular decoupling and BBB dysfunction, thus supporting the second scenario of BBB disruption is a resultant of seizures [74]. Similarly, Vazana et al. provided evidence that repetitive seizures triggered excess glutamate release and BBB leakage [75]. Work by Rüber and colleagues demonstrated seizure‐induced changes in BBB function in patients with epilepsy [44].
However, despite proven BBB disruption by means of fluorescein tracers [76], several studies provided evidence that BBB opening does not always promote spontaneous seizures, suggesting that seizures gradually develop over time [42, 64, 71, 72, 77].
Friedman and colleagues demonstrated a causal link between BBB disruption and subclinical, seizure activity [78]. The authors identified paroxysmal slow cortical activity in EEG as an indicator of subclinical seizure activity in Alzheimer's disease and epilepsy patients. Moreover, intraventricular injection of serum albumin provoked nonconvulsive seizure activity in rodents (PSWE, paroxysmal slow wave events), suggesting neurovasculature involvement in the development of epilepsy pathology also provides an interesting prognostic indicator and potential therapeutic target [78]. Further, studies by the Kaufer laboratory [79] indicated that albumin leakage, and downstream activation of TGFβ signaling in astrocytes can also lead to PSWE and seizure vulnerability, which was inhibited by TGFβ receptor inhibitors. Inhibitors of TGFβ signaling (e.g., Losartan, FDA‐approved) have been shown to interfere with BBB dysfunction, ictogenesis and epileptogenesis [46, 80, 81, 82].
At the BBB, matrix metalloproteinases (MMP) play an important role in extracellular matrix (ECM) remodeling. Digestion of ECM surrounding brain capillaries and degradation of tight junction molecules by MMPs can affect barrier permeability [83]. In line with these findings increased MMP‐9 immunoreactivity has been reported in epileptogenic lesions resected from patients [84, 85]. In a pilocarpine‐induced rat model of epilepsy, MMP‐2 and MMP‐9 activity decreased tight junction expression, and increased barrier leakage [86]. Moreover, the exposure of rat capillaries to glutamate led to similar results, and this leakage was reverted with the blockade of cytosolic phospholipase A2 (cPLA2), suggesting that cPLA2 can be a target for barrier improvement [86]. Importantly, treatment with the MMP2/9 inhibitor IPR‐179 has been shown to have antiseizure and antiepileptogenic effects in rodent epilepsy models [87].
In the normal brain, the neurovascular coupling maintains cerebral flow according to the demand of neuronal activity. It has been shown that pericytes, which are integral for BBB function [14], regulate capillary diameter in response to neuronal activity and thus play a key role in the maintenance of neurovascular coupling [88]. Seizures are initially accompanied by reversible capillary vasodilation and pericyte elongation, indicating a vasoregulatory role of pericytes [74]. However, after subsequent seizures, the vasodilatory responses decrease significantly, and recurrent seizures result in irreversible constriction of pericytes and capillaries, thus rendering the vascular system unresponsive [74]. These findings suggest that pericyte injury may underlie neurovascular decoupling and suggest pericytic injury as an inducer for vascular dysfunction in epilepsy. However, this view has been challenged by the fact that pericytes are located at brain capillaries whereas neurovascular coupling takes place at arterioles that are covered by smooth muscle cells [89]. Moreover, neurovascular coupling depends upon the expression of caveolin‐1 and endothelial nitric oxide synthase in the arteriolar region of the vasculature at which pericytes are not present [90]. Klement et al. demonstrated pericyte‐glia scarring at leaky capillaries in experimental epilepsy and identified PDGFRβ modulation (by Imatinib) as a possible strategy to remedy BBB dysfunction [91].
Taken together, current evidence suggests a bidirectional interplay between impaired BBB function and epilepsy [12, 41, 42]. BBB disruption and leakage promote seizures which consequently potentiate further leakage and BBB dysfunction, ultimately leading to additional seizures, inflammation and damage of NVU cells [41, 42]. Therefore, BBB breakdown can induce epilepsy, and conversely, seizure‐induced BBB breakdown may lead to epilepsy progression. Because of the importance of BBB in determining the long‐term outcome of epilepsy, the restoration of BBB function holds promise as a target for therapy and should be further pursued in future therapeutic approaches.
3. IMPAIRMENT OF THE BBB AS A CONSEQUENCE OF INFLAMMATORY BRAIN LESION FORMATIONS
Primary ictogenic and epileptogenic brain lesions may cause seizures and epilepsy, with subsequent disruption of the BBB and infiltration of distinct subsets of immune cells [92, 93]. In addition, primary adhesion, transmigration and invasion of the brain parenchyma by immune cells can lead to BBB disruption and cortical inflammation, promoting seizure development and epilepsy. Moreover, the induction and recurrence of seizures were inhibited by antibodies directed against α4 integrin or vascular cell adhesion molecule‐1 (VCAM‐1) in experimental epilepsy [94]. Targeting of α4 integrin by Natalizumab as an adjunctive therapy for patients with drug‐resistant epilepsy was pursued in a clinical phase II study (NCT03283371) [95]. However, selection of patients with focus on active neuroinflammation and BBB dysfunction using imaging‐associated markers was suggested for future studies as the endpoint was not met [95].
Further work demonstrated adaptive and innate autoimmune inflammation affected the allocortex of the amygdala and hippocampus as a major cause of temporal lobe seizures (TLS) and TLE in humans [96, 97, 98]. In some cases of autoimmune TLS and TLE, specific autoantibodies (AABs) that bind to intracellular or plasma membrane antigens, expressed in both, neurons and astrocytes, illustrated the presence of an adaptive neural autoimmune reaction. In patients with AABs to intracellular neural antigens cytotoxic T cells may play a major pathogenic role in the development of neuronal dysfunction and cell death, as intracellular antigens are not initially accessible to antibodies. The release of intracellular antigens may initiate B cell responses and autoantibody production [99]. In patients with AABs directed against extracellular domains of neural plasma membrane proteins, such as ionotropic and metabotropic neurotransmitter receptors and synaptic adhesion molecules, those AABs have been shown to exert direct effects on the function and cellular localization of these antigens, rendering these AABs directly pathogenic [99]. Nevertheless, a pathogenic role of cytotoxic T cells has also been suggested [99, 100]. Cytotoxic T cells are deemed pathogenic in autoimmune TLS and TLE because of their strong clonal expansions, parenchymal localization in close spatial proximity, granular expression and the release of cytotoxic effector molecules towards target cells [100, 101, 102, 103]. However, their cognate antigen(s) remain elusive so far.
For pharmacoresistant TLE with hippocampus sclerosis of unknown origin, infiltration of cytotoxic T cells and increased expression of intercellular adhesion molecule‐1 (ICAM‐1) on endothelial cells have been described as predominantly localized in the hippocampal CA1 sector [92, 93, 104, 105, 106, 107, 108]. These infiltrates correlated with the extent of hippocampal neuronal loss, suggesting a role for cytotoxic T cells in driving epileptogenic neurodegeneration [106, 107, 108]. This is further supported by the observation that neuronal antigen‐reactive cytotoxic T cells cause BBB disruption, accompanied by acute temporomesial adaptive and innate inflammation and TLS. This is followed by hippocampal damage with microglia activation, astrogliosis, and TLE development in mice [109].
CNS invasion of leukocytes in inflammatory disorders of the brain has been shown to be a multistep process including (i) rolling and adhesion to the vascular endothelium via the binding of integrin α4β1 (very late antigen‐4; VLA‐4) to VCAM‐1, (ii) paracellular penetration of the endothelial monolayer and the endothelial basement membrane (BM) following interactions with chemokines/chemokine receptors and lymphocyte function‐associated antigen 1 (LFA‐1) with ICAM‐1, (iii) accumulation of leukocytes within the perivascular space defined by the outer endothelial BM and the inner parenchymal BM, (iv) penetration of the parenchymal BM and glia limitans and invasion of the brain parenchyma [110].
In experimental autoimmune encephalomyelitis (EAE) and humans with multiple sclerosis endothelial ion channels, in particular the TWIK‐related potassium channel‐1, regulates the expression levels of endothelial adhesion molecules (ICAM‐1, VCAM‐1, etc.) and thereby regulates trafficking of lymphocytes across the endothelial barrier (steps i and ii) [111, 112]. Moreover, lymphocyte‐derived MMP‐9 and MMP‐2 are involved in lymphocyte trafficking across the parenchymal barrier and invasion into the brain parenchyma (step iv) [113, 114, 115]. It is not determined if these mechanisms are also relevant for cytotoxic T cells driving autoimmune TLS and TLE. Of note, a recent study investigated the immune microenvironment/immunotranscriptome in drug resistant epilepsy patient biopsies using single cell transcriptomics which may provide a resource for the design and development of future drugs [116].
4. IMAGING OF BBB DYSFUNCTION IN THE EPILEPTIC BRAIN
Imaging of BBB compromise has been demonstrated in epilepsy patients [117, 118] and animal models of epilepsy [75, 119, 120]. In neuroimaging, it is crucial to distinguish the three categories of BBB dysfunction in epilepsy patients and to acknowledge that they may occur simultaneously: (i) BBB dysfunction of pathological entities underlying structural epilepsies, for example, tumors, (ii) more discrete BBB dysfunction interictally as a permanent correlate of epilepsy, that is, repeated uniform seizures, and (iii) postictally as transient microstructural sequelae of a single epileptic seizure. In the clinical routines, the structural status of the BBB is assessed by means of computer tomography, Single Photon Emission Tomography (SPECT), and Magnetic Resonance Imaging (MRI), though contrast‐enhanced MRI sequences are most commonly applied [121]. Gadolinium is administered as a contrast agent, which decreases longitudinal relaxation time (T1) and, thus, increases the signal on T1‐weighted images when it accumulates in the extravascular compartment. Of the established protocols, dynamic contrast‐enhanced MRI (DCE‐MRI), which consists of the intravenous administration of Gadolinium before the rapid acquisition of several T1‐weighted volumes, is most sensitive for BBB integrity assessment [122]. However, albeit not yet established, contrast‐enhanced quantitative MRI has been shown to have a higher diagnostic value in the assessment of BBB integrity, since it directly measures the relaxation time instead of signal intensities of conventional MRI [123]. While most pathological entities in structural epilepsy are defined by abnormal BBB functions (not addressed in this review), there are growing interests related to BBB dysfunction in small vessels that are temporally and anatomically associated with epileptic seizures (ii, iii). The imaging evidence for a permanent BBB dysfunction of epilepsy (ii) is sparse and is only drawn from animal studies investigating epileptogenesis [82, 124]. However, these studies cannot answer the most crucial question: whether the BBB dysfunction is a cause or a consequence of epilepsy or both [125].
While it is difficult to perform ictal imaging in animals, it is almost impossible to design a study involving ictal acquisition of brain images from patients. Most of the studies are hence geared towards describing the neurostructural sequelae of epileptic seizures, which does not allow for an estimation of the onset of the dysfunction or its regression. Permeability is visualized by measuring the exchange of blood components in the extravascular compartment. Vasogenic edema may cause alterations of diffusivity, which can be measured by diffusion weighted imaging (DWI, Figure 2, I). Zhong et al. demonstrated a decline in the apparent diffusion coefficient after a bicuculline‐induced status epilepticus in rodents, which reached a plateau within 30 min after drug administration [126]. This effect was also observed in epilepsy patients [127]. Horowitz and colleagues were the first to demonstrate temporomesial parenchymal contrast enhancement of Gadolinium on post‐contrast MRI after a complex partial seizure [117]. The use of DCE‐MRI and quantitative T1 may unmask even subtle and focal postictal enhancements of Gadolinium‐based contrast agent in the patient brain on an individual level (Figure 2, II) [44, 128]. Besides the evidence for peri‐ictal BBB dysfunction, ictal hyperperfusion is known to occur, which can be measured by SPECT [129] (Figure 2, III), that subsequently turns into postictal hypoperfusion, which may be measured by Arterial Spin Labeling (ASL) [130]. As links have been established between brain perfusion and neurovascular permeability in epilepsy [131], peri‐ictal alterations in brain perfusion may be of interest. In this context, the transverse relaxation time (T2) of the ASL signal magnetization is being put in the focus of MR‐physicists [132, 133]. Differing T2 times of water protons in the vessel and gray matter compartments enable the identification of protons in the respective compartments, which can then be used to infer information on the microstructural status of the BBB separating these two compartments. One study has further suggested that ictally extravasated iron may be measured within the seizure‐onset zone using quantitative susceptibility mapping [134]. High expectations are placed on the ultra‐high field scanner: While gray matter T1 values increase with field strength but Gadolinium relaxation time is roughly the same, the net effect is a higher relative contrast enhancement in the ultra‐high field, allowing for the detection of more subtle BBB compromise [135].
FIGURE 2.
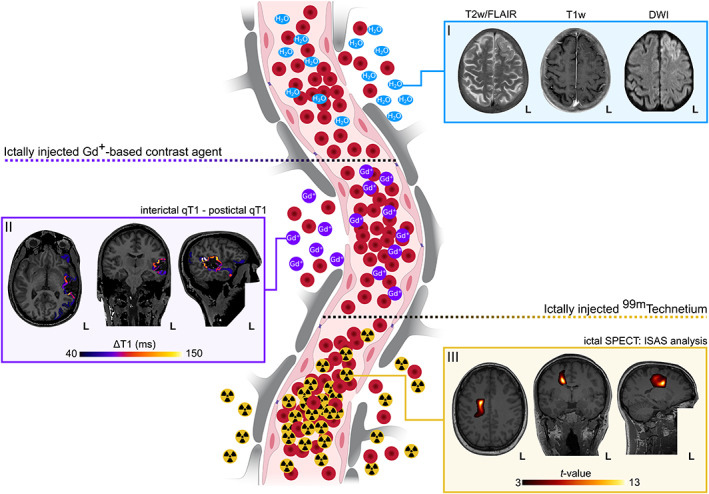
Schematic of the physiological underpinnings of postictal imaging findings and presentation of exemplary cases. Patient I: 52 years old, male, no lesion in MRI, focal seizure with impaired awareness and motor onset, ictal EEG onset in left fronto‐temporal electrodes. Postictal imaging finding: Left frontal cortical T2 hyperintensity, hypointense thickened cortex in T1, hyperintense diffusion signal. Patient II: 34 years old, male, resected left‐temporal cavernoma, generalized tonic–clonic seizure, ictal EEG onset in left centro‐temporal electrodes. Postictal imaging finding: Postictally reduced qT1 as compared to the interictal volume indicative of Gd+‐accumulation. Patient III: 27 years old, female, no lesion in MRI, focal seizure with impaired awareness and motor onset, no EEG correlate. Postictal imaging finding: Postictal hyperperfusion in right precuneus as comparted to interictal SPECT volume. DWI, diffusion weighted image; Gd+, gadolinium; ISAS: ictal‐interictal SPECT analyzed by SPM; qT1, quantitative T1; T2w, T2 weighted image; T1w, T1 weighted image; SPECT, single photon emission computed tomography; ΔT1, interictal qT1—postictal qT1
5. IN VITRO BBB MODELS IN EPILEPSY
Despite the abundance of in vitro BBB models that simulate the physiological in vivo BBB architecture and function [136, 137], models mimicking the dysfunctional BBB are sparse. Since BBB dysfunction can be either cause or consequence of epilepsy, in vitro BBB models could be utilized in studying mechanisms of BBB damage, or to screen potential drugs to tighten the neurovasculature. While in vivo experimental epilepsy models in rodents do not accurately reflect complex seizure‐like events emanating from neural cells in human epilepsy because of inter‐species differences [138], human hippocampus/cortical slices in co‐culture with human brain endothelial cells could serve as a novel epilepsy model in vitro. In comparison to physiological in vitro BBB models, we suggest to utilizing primary human brain microvascular endothelial cells (HBMEC) isolated from surgically resected brain tissue of epilepsy patients as shown in Figure 3A (adapted from [139]). HBMEC obtained from surgical resections displayed BBB characteristics as shown by their expression of VE‐cadherin and claudin‐5, and functionally with high transendothelial electrical resistance (TEER) and low capacitance (Ccl) values when cultured in transwell inserts (Figure 3B,C). Monoculture models for assessing BBB dysfunction [39, 136, 140] show that isolated HBMEC are pure as they express endothelial junction marker and display the typical spindle morphology of endothelial cells. Furthermore, the TEER values obtained for HBMEC are much higher than that of mouse brain endothelial cells (around 25 Ω cm2) [136, 140]. This indicates the feasibility of a tight functional barrier with freshly isolated HBMEC from patient material.
FIGURE 3.
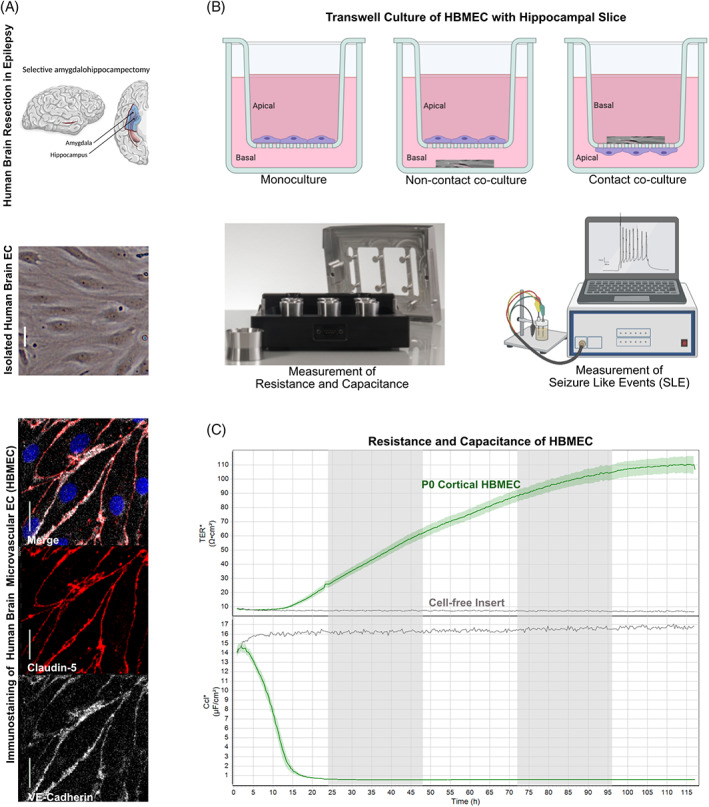
In vitro BBB co‐culture model of hippocampal slices with brain endothelial cells. (A) Brain endothelial cells (EC) isolated from the temporal cortex surgically resected from TLE patients by selective amygdalahippocampectomy (top panel; adapted from [139]) exhibit the classic spindle shape of brain ECs (middle panel) and express the BBB markers VE‐cadherin (white) and claudin‐5 (red). Scale bars 10 mm. (B) The isolated brain ECs cultured on transwell inserts represent a BBB in vitro monoculture model. The co‐culture of the cortical ECs on the transwell insert membrane (apical) with the hippocampal tissue slice obtained from TLE patient surgery in the bottom chamber (basal) represent the non‐contact co‐culture model for the BBB in vitro. The co‐culture of the hippocampal slice on top of the transwell insert membrane (basal) with the cortical ECs on the bottom of the insert membrane (apical) represents a contact co‐culture BBB in vitro epilepsy model. Impedance measurements performed on these transwell inserts for the in vitro BBB model in a cellZscope (nanoAnalytics) device provide resistance and capacitance values reflecting BBB function. Electrophysiological recordings from the hippocampal slice either from the insert or from the bottom chamber provide measurements of seizure‐like events from the epileptic tissue. (C) Representative transendothelial electrical resistance (TEER) and capacitance (Ccl) values from a monolayer of cortical brain ECs (green line) isolated from a TLE patient show high resistance and low capacitance values indicating a functional monoculture BBB model in vitro (as in A) compared to the cell‐free insert (black line).
The combination of primary endothelial cells isolated from healthy‐appearing cortical tissue with hippocampal slices of epileptic foci, resected during epilepsy surgery could be utilized for establishing an in vitro BBB model to mimic human epilepsy. Cortical slices can also be utilized followed by treatment with epilepsy inducing agents, such as kainic acid [141, 142, 143]. Three possible scenarios are displayed in Figure 3B: (1) HBMEC monocultures to study BBB function or dysfunction, HBMEC co‐cultures with epileptic brain slices in a (2) contact or (3) non‐contact manner to study a bidirectional interaction of the neurovasculature with epileptic tissue. The HBMEC monoculture model is useful for testing BBB effects of known or novel pathways in epilepsy pathogenesis, or for testing drug candidates targeting these pathways. The co‐culture models provide the possibility to monitor in vitro, the seizure‐like events in epileptic brain slices as described [144, 145], and assess the effect of epileptic changes on BBB function. Alternatively, seizure‐like events can be measured in slices treated with ASD [145, 146] using the transwell set‐up. The improvement of seizure‐like events in the basal only application (Figure 3B contact or non‐co‐culture) would reveal drug efficacy as the drug is directly accessible by the epileptic cells of the slice located on the basal side. However, no improvement upon apical application of drugs in this co‐culture setting would indicate the BBB as a transport barrier for the drug. Thus, the model system not only allows for ASD screening but also testing the permeability of the BBB. The contact and non‐contact aspects of this model would aid in studying the physical interaction of epileptic cells with the intact vasculature and can be used to obtain insights into the role of other NVU cell‐types in epilepsy.
As seizure recordings in vitro are performed on slices cultured on transwell membranes [147, 148, 149, 150], a combination with BBB functional measurements on brain endothelial cells [39, 136, 140] appears to be an option to further investigate the mechanisms of BBB dysfunction in human epilepsy. In addition, the in vitro BBB model would be crucial in elucidating mechanisms of drug resistance in epilepsy [151]. Theories have been proposed to explain the ASD resistance: (a) a lack of transport of the drug across the BBB and (b) as a result from an upregulation of multidrug resistance proteins such as P‐gp at the BBB upon treatment with ASD [68, 151]. The Janigro group showed the inhibition of P‐gp in vitro led to an increased permeability of phenytoin in endothelial cell cultures from drug resistant patients [151]. Thus, in vitro models could be utilized for the measurement of drugs reaching the basal chamber of the transwell filter system across the in vitro BBB when applied apically. The cells of the BBB could also be analyzed for expression of genes and proteins related to drug resistant pathways [136, 140]. The direct effect of ASD on BBB function can also be investigated, and repeated ASD administrations can be used to study drug resistance [9]. Furthermore, in vitro BBB models comprised of patient material, can be valuable for assessing BBB function from drug resistant versus responding patients [151, 152]. While epilepsy animal models provide insights into mechanisms of epileptogenesis, complementary analyses in human epilepsy surgery specimen allow to investigate the dysregulation of genes and pathways, for example, mTOR signaling, in individual patients with the potential to prevent the occurrence of chronic seizures by means of personalized, targeted epilepsy therapy [10, 153, 154].
6. THE ROLE OF THE BBB IN PHARMACORESISTANCE
Pharmacoresistance (PR) to anticonvulsive drugs is a major treatment obstacle for patients with focal epilepsies, and thus novel therapy strategies to interfere with underlying pathomechanisms are needed. PR is defined by a lack of seizure control in an epilepsy patient despite the application of two appropriately chosen and used antiseizure medications (ASM), and this affects approximately one third of epilepsy patients [9]. Whereas multidrug‐resistance (MDR) is defined as the lack of seizure control despite receiving more than two ASM, which are directed against different drug targets, for example, ion channels or receptors [155]. One established cause for MDR is the presence of molecules in the BBB actively transporting substances, including ASM, out of the brain to maintain it toxin‐free. These so‐called ATP‐binding cassette transporters (ABC‐t), such as P‐glycoprotein (P‐gp) or multidrug resistance‐associated protein (MRP‐1), play a critical role in the pharmacokinetics of different drugs classes including ASM. This transporter hypothesis can explain PR and MDR to a broad range of ASM, even when administered simultaneously. Since drug transporter expression can be induced by several factors, such as hypoxia, inflammation, or even seizures themselves, a high frequency of uncontrolled seizures in epilepsy patients can thus increase the likelihood of drug resistance [155]. Furthermore, many of the established causes of epilepsy can contribute to PR or MDR at the same time. A continuous dysfunctional leaky BBB can result in repeated epileptogenic stimuli associated with epilepsy progression and PR [156]. Both BBB related mechanisms of PR in epilepsy, (i) the activation of drug transporters and (ii) the repetitive leakage of albumin and other substances through the BBB, are potential treatment targets in PR. Inhibitors of P‐gp, such as the repurposed Verapamil, have already been studied in refractory epilepsy patients in pilot studies with reported success in a relevant proportion of treated patients [157, 158]. However, randomized prospective controlled trials are still pending.
Regarding the second BBB related mechanism of PR, it has been proposed that drugs stabilizing the BBB or preventing pro‐epileptic sequelae of continual BBB dysfunction could represent a new generation of agents to prevent seizure activity in epilepsy patients [159, 160]. One study showed the tight junction protein claudin‐5 is enriched in brain endothelial cells and was significantly diminished in surgically resected brain tissue from patients with treatment‐resistant epilepsy. Concomitantly, dynamic contrast‐enhanced MRI in these patients showed widespread BBB disruption, and the administration of anti‐claudin‐5 antibody induced convulsions in a non‐human primate [161]. A knockdown of claudin‐5 in mice leads to spontaneous recurrent seizures, severe neuroinflammation, and mortality, whilst RepSox, a regulator of claudin‐5 expression, could prevent seizures in experimental epilepsy [160]. Several other mechanisms of BBB dysfunction described in this review are potential targets for epilepsy therapy and reducing the seizure frequency and duration could also reduce potential PR, as these processes are interlinked. BBB homeostasis could be achieved by the targeting of angiogenic growth factors, Wnt/β‐catenin signaling (further discussed below) and established drugs such as Rapamycin/Everolimus and the angiotensin‐type 1 receptor antagonist losartan [10, 153, 159].
7. THE BBB AS A POTENTIAL THERAPEUTIC TARGET IN EPILEPSY—APPROACHES FOR BBB STABILIZATION AND PREVENTION OF SEIZURES AND EPILEPSY
BBB dysfunction has been identified as a therapeutic target in several neurological disorders associated with vascular leakage. Although the exact characteristic of BBB dysfunction in epilepsy is not completely resolved, current evidence suggests a peri‐ictal BBB dysfunction in both, animal models and epilepsy patients. While many experimental therapeutic approaches have been tested in experimental seizure models, there are currently no therapies directly targeting the neurovasculature in epileptogenesis and ictogenesis. At present, novel therapeutic approaches are aimed at indirect restoration of the BBB, through the inhibition of inflammation caused by NVU injuries [45, 46]. Consequently, drugs that tighten the BBB may be considered as therapy or prevention of epileptic seizures and further development of epilepsy (Figure 1). While the identification of patients at risk for seizures and epilepsy that might respond to BBB‐tightening therapeutics represents a major clinical challenge, drugs that are already employed for vascular leakage syndromes might be considered for treatment of patients where—based on clinical and radiological data—a leaky BBB is considered instrumental in seizure development and sustainment.
Of interest, albeit currently theoretical with regard to epilepsy therapy, considerable progress has been achieved in the treatment of age‐related macular degeneration (AMD) and diabetic macular edema (DME), both characterized by an impaired, leaky blood–retina barrier (BRB), [27, 56, 162]. Since anatomical structures and physiological functions of BRB and BBB overlap, the same class of drugs could potentially be applied for disorders where BRB and BBB impairment are instrumental in the pathophysiology of the diseases.
Examples of such drugs include the synthetic glucocorticoid dexamethasone, VEGF inhibitors, and, more recently, Tie2‐activating substances. Dexamethasone (Dex) has been successfully applied for DME and is widely used for vasogenic cerebral edema resolution. Dex is currently the gold‐standard for the treatment of peritumoral edema in glioblastoma patients, despite the observation that its application is associated with immunosuppression and limited survival [163].
Dex is also applied for epilepsy therapy where it was shown to reduce inflammation, ictogenesis, and epileptogenesis [45, 46]. Mechanistically, Dex preserved junctional integrity [46]. Further studies also demonstrated a BBB‐tightening effect using porcine brain microvascular endothelial cells, where Dex was able to rescue VEGF‐mediated permeability [163]. Interestingly, Dex has been shown to regulate the expression of Ang‐1 and VEGF, providing mechanistic insight in the BBB stabilizing role [164]. However, Dex treatment induces profound undesired effects, thus limiting its potential long‐term application in epilepsy [45, 46, 163].
VEGF inhibitors (Ranibizumab, Aflibercept) are approved for the treatment of DME and AMD, and adjuvant therapy in solid cancers (Bevacizumab) [27, 162]. In glioblastoma, bevacizumab led to the extension of progression‐free survival, potentially associated with an anti‐edema and corticoid‐sparing effect [27, 163]. However, VEGF also directly affects the central and peripheral nervous system, and described neuroprotective effects could prevent durable usage of VEGF inhibitors in epilepsy patients [165]. In epilepsy models, Sunitinib has shown efficacy for seizure prevention [8].
Comparable to VEGF signaling, the Wnt/β‐catenin pathway mitigates vascular leakage in models of brain pathologies [38, 166], but in contrast to VEGF approved therapeutic options for patients are currently missing. However, the brain endothelial‐specific receptor complex for Wnt7a/Wnt7b may provide an interesting future target for BBB modulation [167]. Indeed, in stroke and glioma preclinical models, specific targeting of the vasculature by adeno‐associated virus (AAV) coding for a dominant active form of Wnt7, leading to Wnt/β‐catenin activation in brain endothelial cells, ameliorated BBB failure and disease progression [62]. However, whether this therapeutic intervention might also be suitable for the treatment of epilepsy, further investigation is required.
Noteworthy in this context is that activators of Tie2 signaling, such as the Ang‐2 inhibitor Trebananib (AMG386), and inhibitors of the vascular endothelial protein tyrosine phosphatase (VE‐PTP), can prevent cerebrovascular leakage in animal models of stroke and glioma [36, 39, 163]. The involvement of angiopoietin/Tie2 signaling in epilepsy was emphasized by Rigau et al. [52], suggesting therapeutic targeting of this pathway for BBB restoration and epilepsy therapy.
The VE‐PTP inhibitor Razuprotafib, a brain‐penetrating small molecule Tie2 activator has been investigated in clinical trials for patients with SARS‐CoV‐2‐associated pulmonary vascular leakage syndrome (NCT04511650, NCT04488081). In DME and AMD the therapeutic efficacy of razuprotafib was demonstrated and appeared to be associated with limited systemic toxicity, suggesting the possibility of repurposing the drug for cerebrovascular leakage syndromes treatments [27, 56]. Furthermore, Faricimab, an investigational bispecific anti‐VEGF/Ang‐2 antibody that inhibits VEGF and activates Tie2, demonstrated efficacy across two global phase III studies for DME (NCT03622580, NCT03622593) and AMD (NCT03823287, NCT03823300) [57], suggesting these novel classes of vascular‐stabilizing drugs are able to tighten the BRB and BBB, and therefore could be applicable in patients with cerebrovascular leakage syndromes associated with epilepsy and other neurological disorders.
8. CONCLUSION AND FUTURE DIRECTIONS
A need for the development of novel epilepsy drugs remains unchanged as drug resistance continues to be a burden in one third of epilepsy patients. The BBB involvement in epilepsy—in general and specifically in drug resistant forms of epilepsy—hereby represents an important topic that is an underappreciated aspect of seizure management and should be pursued in future therapeutic approaches. It is well accepted that a dysfunctional BBB and subsequent leakage of blood contents into the brain parenchyma are important pathomechanisms of epileptogenesis. Currently, antiseizure approaches to remedy BBB malfunction act indirectly on NVU and/or parenchymal cells (i.e., to reduce inflammation) and do not directly target the BBB [45, 46].
Interestingly, as discussed in the previous sections, basic proof of concept that BBB modulation may change seizure pathology has recently been provided by Greene et al. who demonstrated that restoring BBB integrity prevented seizures [160]. The authors demonstrated that restoration of claudin‐5 levels attenuated seizures and neuroinflammation, and provided evidence for a direct correlation between human epilepsy and claudin‐5 levels at the BBB. Thus, studies modulating the BBB to alter seizure pathology preclinically or clinically are at initial stages. Methods for diagnosis, and treatment for improving BBB function are under development. Novel imaging techniques for the identification of BBB dysfunction in epilepsy patients might further evolve as a biomarker in future clinical routine.
Along with above mentioned neurovascular signaling pathways that have been targeted in other CNS pathologies for the restoration of BBB function, the current review emphasized the exploration of novel therapeutic approaches for targeting the BBB in epilepsy therapy as opposed to parenchymal targets that have met with pharmacoresistance [168].
In the light of the points discussed above, below are some important issues/questions that will need to be addressed in the future.
BBB and NVU dysfunctions have emerged as a hallmarks of epilepsy, however, detailed mechanistic understanding on the role of the neurovasculature as a potential target in epilepsy therapy is still needed.
Will targeting of the neurovasculature provide a therapeutic strategy in refractory epilepsy therapy, and as a remedy for BBB dysfunction provide a future therapeutic strategy in refractory epilepsy therapy?
Will diagnostic approaches, such as imaging of BBB dysfunction in epilepsy patients, be applicable in identifying individuals at risk for seizure development?
AUTHOR CONTRIBUTIONS
All authors equally wrote, reviewed and edited the manuscript. All authors have approved the final version of this manuscript.
CONFLICT OF INTEREST
The authors declare no competing interest.
ACKNOWLEDGEMENTS
This work was supported by the LOEWE Center for Personalized Translational Epilepsy Research (CePTER; YR, SB, KD, EF, SL, FR, TR, LMW, KHP), the LOEWE Frankfurt Cancer Institute (FCI; YR, KHP), the German Research Foundation (LI 911/7‐1; SL), the Excellence Cluster Cardio‐Pulmonary Institute (SL), the European Union HORIZON 2020 ITN “BtRAIN” (SL), the Uniscientia foundation (190‐2022; KHP) and the German Centre for Heart and Circulation Research (DZHK; SL, YR, KHP). We would kindly like to acknowledge editing of language and grammar by Jennifer H. Lun. Apologies to all authors, whose work could not be cited because of space limitations. Open Access funding enabled and organized by Projekt DEAL. [Correction added on 12 January 2023, after first online publication: Projekt DEAL funding statement has been added.]
Reiss Y, Bauer S, David B, Devraj K, Fidan E, Hattingen E, et al. The neurovasculature as a target in temporal lobe epilepsy. Brain Pathology. 2023;33(2):e13147. 10.1111/bpa.13147
Funding information European Commission, Grant/Award Number: HORIZON 2020 ITN “BtRAIN”; Excellence Cluster Cardio‐Pulmonary Institute; German Centre for Heart and Circulation Research (DZHK); German Research Foundation (DFG), Grant/Award Number: LI 911/7‐1; LOEWE Center for Personalized Translational Epilepsy Research (CePTER); LOEWE Frankfurt Cancer Institute (FCI); Uniscientia Foundation, Grant/Award Number: 190‐2022
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Thijs RD, Surges R, O'Brien TJ, Sander JW. Epilepsy in adults. Lancet. 2019;393(10172):689–701. [DOI] [PubMed] [Google Scholar]
- 2. Rosenow F, Lüders H. Presurgical evaluation of epilepsy. Brain. 2001;124(9):1683–700. [DOI] [PubMed] [Google Scholar]
- 3. Willems LM, Hamer HM, Knake S, Rosenow F, Reese JP, Strzelczyk A. General trends in prices and prescription patterns of anticonvulsants in Germany between 2000 and 2017: analysis of national and cohort‐based data. Appl Health Econ Health Policy. 2019;17(5):707–22. [DOI] [PubMed] [Google Scholar]
- 4. Tang F, Hartz AMS, Bauer B. Drug‐resistant epilepsy: multiple hypotheses, few answers. Front Neurol. 2017;8:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Strzelczyk A, Griebel C, Lux W, Rosenow F, Reese JP. The burden of severely drug‐refractory epilepsy: a comparative longitudinal evaluation of mortality, morbidity, resource use, and cost using German health insurance data. Front Neurol. 2017;8:712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klein P, Dingledine R, Aronica E, Bernard C, Blümcke I, Boison D, et al. Commonalities in epileptogenic processes from different acute brain insults: do they translate? Epilepsia. 2018;59(1):37–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lanen RHV, Melchers S, Hoogland G, Schijns OE, Zandvoort MAV, Haeren RH, et al. Microvascular changes associated with epilepsy: a narrative review. J Cereb Blood Flow Metab. 2021;41(10):2492–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Hauser WA, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia. 2010;51(6):1069–77. [DOI] [PubMed] [Google Scholar]
- 10. Rosenow F, Alphen NV, Becker A, Chiocchetti A, Deichmann R, Deller T, et al. Personalized translational epilepsy research—novel approaches and future perspectives part I: clinical and network analysis approaches. Epilepsy Behav. 2017;76:13–8. [DOI] [PubMed] [Google Scholar]
- 11. Gorter JA, Aronica E, van Vliet EA. The roof is leaking and a storm is raging: repairing the blood–brain barrier in the fight against epilepsy. Epilepsy Curr. 2019;19(3):177–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Löscher W, Friedman A. Structural, molecular, and functional alterations of the blood–brain barrier during epileptogenesis and epilepsy: a cause, consequence, or both? Int J Mol Sci. 2020;21(2):591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liebner S, Dijkhuizen RM, Reiss Y, Plate KH, Agalliu D, Constantin G. Functional morphology of the blood–brain barrier in health and disease. Acta Neuropathol. 2018;135(3):311–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Profaci CP, Munji RN, Pulido RS, Daneman R. The blood–brain barrier in health and disease: important unanswered questions. J Exp Med. 2020;217(4):e20190062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ben‐Zvi A, Liebner S. Developmental regulation of barrier‐ and non‐barrier blood vessels in the CNS. J Intern Med. 2021;292:31–46. [DOI] [PubMed] [Google Scholar]
- 16. Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature. 2010;468(7323):562–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood–brain barrier. Nature. 2010;468(7323):557–61. [DOI] [PubMed] [Google Scholar]
- 18. Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68(3):409–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yamanaka G, Takata F, Kataoka Y, Kanou K, Morichi S, Dohgu S, et al. The neuroinflammatory role of pericytes in epilepsy. Biomedicines. 2021;9(7):759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, et al. Wnt/β‐catenin signaling controls development of the blood–brain barrier. J Cell Biol. 2008;183(3):409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/β‐catenin signaling is required for CNS, but not non‐CNS, angiogenesis. Proc Natl Acad Sci U S A. 2009;106(2):641–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt signaling regulates organ‐specific assembly and differentiation of CNS vasculature. Science. 2008;322(5905):1247–50. [DOI] [PubMed] [Google Scholar]
- 23. Wang Y, Cho C, Williams J, Smallwood PM, Zhang C, Junge HJ, et al. Interplay of the Norrin and Wnt7a/Wnt7b signaling systems in blood–brain barrier and blood–retina barrier development and maintenance. Proc Natl Acad Sci U S A. 2018;115(50):201813217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vallon M, Yuki K, Nguyen TD, Chang J, Yuan J, Siepe D, et al. A RECK‐WNT7 receptor‐ligand interaction enables isoform‐specific regulation of Wnt bioavailability. Cell Rep. 2018;25(2):339–349.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vanhollebeke B, Stone OA, Bostaille N, Cho C, Zhou Y, Maquet E, et al. Tip cell‐specific requirement for an atypical Gpr124‐ and Reck‐dependent Wnt/β‐catenin pathway during brain angiogenesis. Elife. 2015;4:e06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wittko‐Schneider IM, Schneider FT, Plate KH. Cerebral angiogenesis, methods and protocols. Methods Mol Biol. 2014;1135:3–20. [DOI] [PubMed] [Google Scholar]
- 27. Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell. 2019;176(6):1248–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF‐mediated disruption of endothelial CLN‐5 promotes blood–brain barrier breakdown. Proc Natl Acad Sci U S A. 2009;106(6):1977–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood–brain barrier: from physiology to disease and Back. Physiol Rev. 2019;99(1):21–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miller D. Regulation of ABC transporters at the blood–brain barrier. Clin Pharmacol Ther. 2015;97(4):395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dejana E, Vestweber D. Chapter six the role of VE‐cadherin in vascular morphogenesis and permeability control. Prog Mol Biol Transl. 2013;116:119–44. [DOI] [PubMed] [Google Scholar]
- 32. Awad I, Jabbour P. Cerebral cavernous malformations and epilepsy. Neurosurg Focus. 2006;21(1):1–9. [DOI] [PubMed] [Google Scholar]
- 33. Malinverno M, Maderna C, Taha AA, Corada M, Orsenigo F, Valentino M, et al. Endothelial cell clonal expansion in the development of cerebral cavernous malformations. Nat Commun. 2019;10(1):2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bravi L, Malinverno M, Pisati F, Rudini N, Cuttano R, Pallini R, et al. Endothelial cells lining sporadic cerebral cavernous malformation cavernomas undergo endothelial‐to‐mesenchymal transition. Stroke. 2018;47(3):886–90. [DOI] [PubMed] [Google Scholar]
- 35. Cuesta AM, Gallardo‐Vara E, Casado‐Vela J, Recio‐Poveda L, Botella LM, Albiñana V. The role of propranolol as a repurposed drug in rare vascular diseases. Int J Mol Sci. 2022;23(8):4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Scholz A, Harter PN, Cremer S, Yalcin BH, Gurnik S, Yamaji M, et al. Endothelial cell‐derived angiopoietin‐2 is a therapeutic target in treatment‐naive and bevacizumab‐resistant glioblastoma. EMBO Mol Med. 2016;8(1):39–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tacchio MD, Macas J, Weissenberger J, Sommer K, Bähr O, Steinbach JP, et al. Tumor vessel normalization, immunostimulatory reprogramming, and improved survival in glioblastoma with combined inhibition of PD‐1, angiopoietin‐2, and VEGF. Cancer Immunol Res. 2019;7(12):1910–27. [DOI] [PubMed] [Google Scholar]
- 38. Reis M, Czupalla CJ, Ziegler N, Devraj K, Zinke J, Seidel S, et al. Endothelial Wnt/β‐catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF‐B expression. J Exp Med. 2012;209(9):1611–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gurnik S, Devraj K, Macas J, Yamaji M, Starke J, Scholz A, et al. Angiopoietin‐2‐induced blood–brain barrier compromise and increased stroke size are rescued by VE‐PTP‐dependent restoration of Tie2 signaling. Acta Neuropathol. 2016;131(5):753–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Devraj G, Guérit S, Seele J, Spitzer D, Macas J, Khel MI, et al. HIF‐1α is involved in blood–brain barrier dysfunction and paracellular migration of bacteria in pneumococcal meningitis. Acta Neuropathol. 2020;140(2):183–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van Vliet EA, Aronica E, Gorter JA. Role of blood–brain barrier in temporal lobe epilepsy and pharmacoresistance. Neuroscience. 2014;277:455–73. [DOI] [PubMed] [Google Scholar]
- 42. van Vliet EA, Aronica E, Gorter JA. Blood–brain barrier dysfunction, seizures and epilepsy. Semin Cell Dev Biol. 2015;38:26–34. [DOI] [PubMed] [Google Scholar]
- 43. Swissa E, Serlin Y, Vazana U, Prager O, Friedman A. Blood–brain barrier dysfunction in status epileptics: mechanisms and role in epileptogenesis. Epilepsy Behav. 2019;101:106285. [DOI] [PubMed] [Google Scholar]
- 44. Rüber T, David B, Lüchters G, Nass RD, Friedman A, Surges R, et al. Evidence for peri‐ictal blood–brain barrier dysfunction in patients with epilepsy. Brain. 2018;141(10):2952–65. [DOI] [PubMed] [Google Scholar]
- 45. Baruah J, Vasudevan A, Köhling R. Vascular integrity and signaling determining brain development, network excitability, and epileptogenesis. Front Physiol. 2020;10:1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. van Vliet EA, Marchi N. Neurovascular unit dysfunction as a mechanism of seizures and epilepsy during aging. Epilepsia. 2022;63:1297–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cornford EM, Oldendorf WH. Epilepsy and the blood–brain barrier. Adv Neurol. 1986;44:787–812. [PubMed] [Google Scholar]
- 48. Cornford EM. Epilepsy and the blood brain barrier: endothelial cell responses to seizures. Adv Neurol. 1999;79:845–62. [PubMed] [Google Scholar]
- 49. Kasantikul V, Brown WJ, Oldendorf WH, Crandall PC. Ultrastructural parameters of limbic microvasculature in human psychomotor epilepsy. Clin Neuropathol. 1983;2(4):171–8. [PubMed] [Google Scholar]
- 50. Janigro D. Blood–brain barrier, ion homeostasis and epilepsy: possible implications towards the understanding of ketogenic diet mechanisms. Epilepsy Res. 1999;37(3):223–32. [DOI] [PubMed] [Google Scholar]
- 51. Marchi N, Granata T, Ghosh C, Janigro D. Blood–brain barrier dysfunction and epilepsy: pathophysiologic role and therapeutic approaches. Epilepsia. 2012;53(11):1877–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rigau V, Morin M, Rousset MC, Bock FD, Lebrun A, Coubes P, et al. Angiogenesis is associated with blood–brain barrier permeability in temporal lobe epilepsy. Brain. 2007;130(7):1942–56. [DOI] [PubMed] [Google Scholar]
- 53. Castro‐Torres RD, Ureña‐Guerrero ME, Morales‐Chacón LM, Lorigados‐Pedre L, Estupiñan‐Díaz B, Rocha L, et al. New aspects of VEGF, GABA, and glutamate signaling in the neocortex of human temporal lobe pharmacoresistant epilepsy revealed by RT‐qPCR arrays. J Mol Neurosci. 2020;70(6):916–29. [DOI] [PubMed] [Google Scholar]
- 54. Benini R, Roth R, Khoja Z, Avoli M, Wintermark P. Does angiogenesis play a role in the establishment of mesial temporal lobe epilepsy? Int J Dev Neurosci. 2016;49(1):31–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Morin‐Brureau M, Lebrun A, Rousset MC, Fagni L, Bockaert J, Bock FD, et al. Epileptiform activity induces vascular remodeling and zonula occludens 1 downregulation in organotypic hippocampal cultures: role of VEGF signaling pathways. J Neurosci. 2011;31(29):10677–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Campochiaro PA, Peters KG. Targeting Tie2 for treatment of diabetic retinopathy and diabetic macular edema. Curr Diabetes Rep. 2016;16(12):126. [DOI] [PubMed] [Google Scholar]
- 57. Liberski S, Wichrowska M, Kocięcki J. Aflibercept versus Faricimab in the treatment of neovascular age‐related macular degeneration and diabetic macular edema: a review. Int J Mol Sci. 2022;23(16):9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang L, Huang T, Teaw S, Bordey A. Hypervascularization in mTOR‐dependent focal and global cortical malformations displays differential rapamycin sensitivity. Epilepsia. 2019;60(6):1255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Munji RN, Soung AL, Weiner GA, Sohet F, Semple BD, Trivedi A, et al. Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood–brain barrier dysfunction module. Nat Neurosci. 2019;22(11):1892–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kestner RI, Mayser F, Vutukuri R, Hansen L, Günther S, Brunkhorst R, et al. Gene expression dynamics at the neurovascular unit during early regeneration after cerebral ischemia/reperfusion injury in mice. Front Neurosci. 2020;14:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Spitzer D, Guérit S, Puetz T, Khel MI, Armbrust M, Dunst M, et al. Profiling the neurovascular unit unveils detrimental effects of osteopontin on the blood–brain barrier in acute ischemic stroke. Acta Neuropathol. 2022;144(2):305–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Martin M, Vermeiren S, Bostaille N, Eubelen M, Spitzer D, Vermeersch M, et al. Engineered Wnt ligands enable blood–brain barrier repair in neurological disorders. Science. 2022;375(6582):eabm4459. [DOI] [PubMed] [Google Scholar]
- 63. Marchi N, Angelov L, Masaryk T, Fazio V, Granata T, Hernandez N, et al. Seizure‐promoting effect of blood–brain barrier disruption. Epilepsia. 2007;48(4):732–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. van Vliet EA, Araújo SDC, Redeker S, Schaik RV, Aronica E, Gorter JA. Blood–brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain. 2007;130(2):521–34. [DOI] [PubMed] [Google Scholar]
- 65. Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, et al. TGF‐β receptor‐mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2006;130(2):535–47. [DOI] [PubMed] [Google Scholar]
- 66. David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, et al. Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci. 2009;29(34):10588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cacheaux LP, Ivens S, David Y, Lakhter AJ, Bar‐Klein G, Shapira M, et al. Transcriptome profiling reveals TGF‐signaling involvement in epileptogenesis. J Neurosci. 2009;29(28):8927–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Han H, Mann A, Ekstein D, Eyal S. Breaking bad: the structure and function of the blood–brain barrier in epilepsy. AAPS J. 2017;19(4):973–88. [DOI] [PubMed] [Google Scholar]
- 69. Tishler DM, Weinberg KI, Hinton DR, Barbaro N, Annett GM, Raffel C. MDR1 gene expression in brain of patients with medically intractable epilepsy. Epilepsia. 1995;36(1):1–6. [DOI] [PubMed] [Google Scholar]
- 70. Löscher W, Potschka H, Sisodiya SM, Vezzani A. Drug resistance in epilepsy: clinical impact, potential mechanisms, and new innovative treatment options. Pharmacol Rev. 2020;72(3):606–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, et al. Lasting blood–brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004;24(36):7829–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tomkins O, Friedman O, Ivens S, Reiffurth C, Major S, Dreier JP, et al. Blood–brain barrier disruption results in delayed functional and structural alterations in the rat neocortex. Neurobiol Dis. 2007;25(2):367–77. [DOI] [PubMed] [Google Scholar]
- 73. Ridder DA, Wenzel J, Müller K, Töllner K, Tong XK, Assmann JC, et al. Brain endothelial TAK1 and NEMO safeguard the neurovascular unitTAK1 and NEMO protect the neurovascular unit. J Exp Med. 2015;212(10):1529–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Prager O, Kamintsky L, Hasam‐Henderson LA, Schoknecht K, Wuntke V, Papageorgiou I, et al. Seizure‐induced microvascular injury is associated with impaired neurovascular coupling and blood–brain barrier dysfunction. Epilepsia. 2019;60(2):322–36. [DOI] [PubMed] [Google Scholar]
- 75. Vazana U, Veksler R, Pell GS, Prager O, Fassler M, Chassidim Y, et al. Glutamate‐mediated blood–brain barrier opening: implications for neuroprotection and drug delivery. J Neurosci. 2016;36(29):7727–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. van Vliet EA, Zibell G, Pekcec A, Schlichtiger J, Edelbroek PM, Holtman L, et al. COX‐2 inhibition controls P‐glycoprotein expression and promotes brain delivery of phenytoin in chronic epileptic rats. Neuropharmacology. 2010;58(2):404–12. [DOI] [PubMed] [Google Scholar]
- 77. Frigerio F, Frasca A, Weissberg I, Parrella S, Friedman A, Vezzani A, et al. Long‐lasting pro‐ictogenic effects induced in vivo by rat brain exposure to serum albumin in the absence of concomitant pathology. Epilepsia. 2012;53(11):1887–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Milikovsky DZ, Ofer J, Senatorov VV Jr, Friedman AR, Prager O, Sheintuch L, et al. Paroxysmal slow cortical activity in Alzheimer's disease and epilepsy is associated with blood–brain barrier dysfunction. Sci Transl Med. 2019;11(521):eaaw8954. [DOI] [PubMed] [Google Scholar]
- 79. Senatorov VV Jr, Friedman AR, Milikovsky DZ, Ofer J, Saar‐Ashkenazy R, Charbash A, et al. Blood–brain barrier dysfunction in aging induces hyperactivation of TGFβ signaling and chronic yet reversible neural dysfunction. Sci Transl Med. 2019;11(521):eaaw8283. [DOI] [PubMed] [Google Scholar]
- 80. Bar‐Klein G, Cacheaux LP, Kamintsky L, Prager O, Weissberg I, Schoknecht K, et al. Losartan prevents acquired epilepsy via TGF‐β signaling suppression. Ann Neurol. 2014;75(6):864–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Weissberg I, Wood L, Kamintsky L, Vazquez O, Milikovsky DZ, Alexander A, et al. Albumin induces excitatory synaptogenesis through astrocytic TGF‐β/ALK5 signaling in a model of acquired epilepsy following blood–brain barrier dysfunction. Neurobiol Dis. 2015;78:115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bar‐Klein G, Lublinsky S, Kamintsky L, Noyman I, Veksler R, Dalipaj H, et al. Imaging blood–brain barrier dysfunction as a biomarker for epileptogenesis. Brain. 2017;140(6):1692–705. [DOI] [PubMed] [Google Scholar]
- 83. Rempe RG, Hartz AM, Bauer B. Matrix metalloproteinases in the brain and blood–brain barrier: versatile breakers and makers. J Cereb Blood Flow Metab. 2016;36(9):1481–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Konopka A, Grajkowska W, Ziemiańska K, Roszkowski M, Daszkiewicz P, Rysz A, et al. Matrix metalloproteinase‐9 (MMP‐9) in human intractable epilepsy caused by focal cortical dysplasia. Epilepsy Res. 2013;104(1–2):45–58. [DOI] [PubMed] [Google Scholar]
- 85. Li S, Yu S, Zhang C, Shu H, Liu S, An N, et al. Increased expression of matrix metalloproteinase 9 in cortical lesions from patients with focal cortical dysplasia type IIb and tuberous sclerosis complex. Brain Res. 2012;1453:46–55. [DOI] [PubMed] [Google Scholar]
- 86. Rempe RG, Hartz AMS, Soldner ELB, Sokola BS, Alluri SR, Abner EL, et al. Matrix metalloproteinase‐mediated blood–brain barrier dysfunction in epilepsy. J Neurosci. 2018;38(18):4301–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Broekaart DWM, Bertran A, Jia S, Korotkov A, Senkov O, Bongaarts A, et al. The matrix metalloproteinase inhibitor IPR‐179 has antiseizure and antiepileptogenic effects. J Clin Invest. 2020;131(1):e138332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hamilton NB, Attwell D, Hall CN. Pericyte‐mediated regulation of capillary diameter: a component of neurovascular coupling in health and disease. Front Neuroenergetics. 2010;2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hill RA, Tong L, Yuan P, Murikinati S, Gupta S, Grutzendler J. Regional blood flow in the normal and ischemic brain is controlled by arteriolar smooth muscle cell contractility and not by capillary pericytes. Neuron. 2015;87(1):95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chow BW, Nuñez V, Kaplan L, Granger AJ, Bistrong K, Zucker HL, et al. Caveolae in CNS arterioles mediate neurovascular coupling. Nature. 2020;579(7797):106–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Klement W, Blaquiere M, Zub E, de Bock F, Boux F, Barbier E, et al. A pericyte‐glia scarring develops at the leaky capillaries in the hippocampus during seizure activity. Epilepsia. 2019;60(7):1399–411. [DOI] [PubMed] [Google Scholar]
- 92. Neumann AM, Abele J, Kirschstein T, Engelmann R, Sellmann T, Köhling R, et al. Mycophenolate mofetil prevents the delayed T cell response after pilocarpine‐induced status epilepticus in mice. PLoS One. 2017;12(11):e0187330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zattoni M, Mura ML, Deprez F, Schwendener RA, Engelhardt B, Frei K, et al. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J Neurosci. 2011;31(11):4037–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Fabene PF, Mora GN, Martinello M, Rossi B, Merigo F, Ottoboni L, et al. A role for leukocyte‐endothelial adhesion mechanisms in epilepsy. Nat Med. 2008;14(12):1377–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. French JA, Cole AJ, Faught E, Theodore WH, Vezzani A, Liow K, et al. Safety and efficacy of Natalizumab as adjunctive therapy for people with drug‐resistant epilepsy. Neurology. 2021;97(18):e1757–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ravizza T, Gagliardi B, Noé F, Boer K, Aronica E, Vezzani A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29(1):142–60. [DOI] [PubMed] [Google Scholar]
- 97. Bauer J, Becker AJ, Elyaman W, Peltola J, Rüegg S, Titulaer MJ, et al. Innate and adaptive immunity in human epilepsies. Epilepsia. 2017;58(S3):57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Geis C, Planagumà J, Carreño M, Graus F, Dalmau J. Autoimmune seizures and epilepsy. J Clin Invest. 2019;129(3):926–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Golombeck KS, Bönte K, Mönig C, van Loo KM, Hartwig M, Schwindt W, et al. Evidence of a pathogenic role for CD8+ T cells in anti‐GABAB receptor limbic encephalitis. Neurology – Neuroimmunol Neuroinflammation. 2016;3(3):e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Bracher A, Alcalá C, Ferrer J, Melzer N, Hohlfeld R, Casanova B, et al. An expanded parenchymal CD8+ T cell clone in GABAA receptor encephalitis. Ann Clin Transl Neurol. 2020;7(2):239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Plonquet A, Gherardi RK, Créange A, Antoine JC, Benyahia B, Grisold W, et al. Oligoclonal T‐cells in blood and target tissues of patients with anti‐Hu syndrome. J Neuroimmunol. 2002;122(1–2):100–5. [DOI] [PubMed] [Google Scholar]
- 102. Voltz R, Dalmau J, Posner JB, Rosenfeld MR. T‐cell receptor analysis in anti‐Hu associated paraneoplastic encephalomyelitis. Neurology. 1998;51(4):1146–50. [DOI] [PubMed] [Google Scholar]
- 103. Bien CG, Vincent A, Barnett MH, Becker AJ, Blümcke I, Graus F, et al. Immunopathology of autoantibody‐associated encephalitides: clues for pathogenesis. Brain. 2012;135(5):1622–38. [DOI] [PubMed] [Google Scholar]
- 104. Tezer FI, Firat A, Tuzun E, Unal I, Soylemezoglu F, Bilginer B, et al. Immunopathology in drug resistant mesial temporal lobe epilepsy with different types of hippocampal sclerosis. Int J Neurosci. 2017;128(5):1–26. [DOI] [PubMed] [Google Scholar]
- 105. Gales JM, Prayson RA. Chronic inflammation in refractory hippocampal sclerosis‐related temporal lobe epilepsy. Ann Diagn Pathol. 2017;30:12–6. [DOI] [PubMed] [Google Scholar]
- 106. Lu JQ, Steve TA, Wheatley M, Gross DW. Immune cell infiltrates in hippocampal sclerosis: correlation with neuronal loss. J Neuropathol Exp Neurol. 2017;76(3):206–15. [DOI] [PubMed] [Google Scholar]
- 107. Nakahara H, Konishi Y, Beach TG, Yamada N, Makino S, Tooyama I. Infiltration of T lymphocytes and expression of ICAM‐1 in the hippocampus of patients with hippocampal sclerosis. Acta Histochem Cytoc. 2010;43(6):157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Volmering E, Niehusmann P, Peeva V, Grote A, Zsurka G, Altmüller J, et al. Neuropathological signs of inflammation correlate with mitochondrial DNA deletions in mesial temporal lobe epilepsy. Acta Neuropathol. 2016;132(2):277–88. [DOI] [PubMed] [Google Scholar]
- 109. Pitsch J, Loo KMJ, Gallus M, Dik A, Kamalizade D, Baumgart A, et al. CD8+ T‐lymphocyte driven limbic encephalitis results in temporal lobe epilepsy. Ann Neurol. 2021;89(4):666–85. [DOI] [PubMed] [Google Scholar]
- 110. Engelhardt B, Sorokin L. The blood–brain and the blood–cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol. 2009;31(4):497–511. [DOI] [PubMed] [Google Scholar]
- 111. Bittner S, Ruck T, Schuhmann MK, Herrmann AM, Maati HMO, Bobak N, et al. Endothelial TWIK‐related potassium channel‐1 (TREK1) regulates immune‐cell trafficking into the CNS. Nat Med. 2013;19(9):1161–5. [DOI] [PubMed] [Google Scholar]
- 112. Bittner S, Ruck T, Fernández‐Orth J, Meuth SG. TREK‐King the blood–brain‐barrier. J Neuroimmune Pharm. 2014;9(3):293–301. [DOI] [PubMed] [Google Scholar]
- 113. Agrawal S, Anderson P, Durbeej M, Rooijen NV, Ivars F, Opdenakker G, et al. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med. 2006;203(4):1007–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood–brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol. 2001;153(5):933–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Gerwien H, Hermann S, Zhang X, Korpos E, Song J, Kopka K, et al. Imaging matrix metalloproteinase activity in multiple sclerosis as a specific marker of leukocyte penetration of the blood–brain barrier. Sci Transl Med. 2016;8(364):364ra152. [DOI] [PubMed] [Google Scholar]
- 116. Kumar P, Lim A, Hazirah SN, Chua CJH, Ngoh A, Poh SL, et al. Single‐cell transcriptomics and surface epitope detection in human brain epileptic lesions identifies pro‐inflammatory signaling. Nat Neurosci. 2022;25(7):956–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Horowitz SW, Merchut M, Fine M, Azar‐Kia B. Complex partial seizure‐induced transient MR enhancement. J Comput Assist Tomogr. 1992;16(5):814–6. [DOI] [PubMed] [Google Scholar]
- 118. Tomkins O, Shelef I, Kaizerman I, Eliushin A, Afawi Z, Misk A, et al. Blood–brain barrier disruption in post‐traumatic epilepsy. J Neurol Neurosurg Psychiatry. 2008;79(7):774–7. [DOI] [PubMed] [Google Scholar]
- 119. Librizzi L, Noè F, Vezzani A, de Curtis M, Ravizza T. Seizure‐induced brain‐borne inflammation sustains seizure recurrence and blood–brain barrier damage. Ann Neurol. 2012;72(1):82–90. [DOI] [PubMed] [Google Scholar]
- 120. Wunder A, Schoknecht K, Stanimirovic DB, Prager O, Chassidim Y. Imaging blood–brain barrier dysfunction in animal disease models. Epilepsia. 2012;53(s6):14–21. [DOI] [PubMed] [Google Scholar]
- 121. Veksler R, Shelef I, Friedman A. Blood–brain barrier imaging in human neuropathologies. Arch Med Res. 2014;45(8):646–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Heye AK, Culling RD, Hernández MDCV, Thrippleton MJ, Wardlaw JM. Assessment of blood–brain barrier disruption using dynamic contrast‐enhanced MRI. A systematic review. Neuroimage Clin. 2014;6:262–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hattingen E, Jurcoane A, Daneshvar K, Pilatus U, Mittelbronn M, Steinbach JP, et al. Quantitative T2 mapping of recurrent glioblastoma under bevacizumab improves monitoring for non‐enhancing tumor progression and predicts overall survival. Neuro Oncol. 2013;15(10):1395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. van Vliet EA, Otte WM, Gorter JA, Dijkhuizen RM, Wadman WJ. Longitudinal assessment of blood–brain barrier leakage during epileptogenesis in rats. A quantitative MRI study. Neurobiol Dis. 2014;63:74–84. [DOI] [PubMed] [Google Scholar]
- 125. Friedman A. Blood–brain barrier dysfunction, status epilepticus, seizures, and epilepsy: a puzzle of a chicken and egg? Epilepsia. 2011;52(s8):19–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zhong J, Petroff OAC, Prichard JW, Gore JC. Changes in water diffusion and relaxation properties of rat cerebrum during status epilepticus. Magnet Reson Med. 1993;30(2):241–6. [DOI] [PubMed] [Google Scholar]
- 127. Alsop DC, Connelly A, Duncan JS, Hufnagel A, Pierpaoli C, Rugg‐Gunn FJ. Diffusion and perfusion MRI in epilepsy. Epilepsia. 2002;43(s1):69–77. [Google Scholar]
- 128. Gorter JA, van Vliet EA, Aronica E. Status epilepticus, blood–brain barrier disruption, inflammation, and epileptogenesis. Epilepsy Behav. 2015;49:13–6. [DOI] [PubMed] [Google Scholar]
- 129. von Oertzen TJ, Mormann F, Urbach H, Reichmann K, Koenig R, Clusmann H, et al. Prospective use of subtraction ictal SPECT coregistered to MRI (SISCOM) in presurgical evaluation of epilepsy. Epilepsia. 2011;52(12):2239–48. [DOI] [PubMed] [Google Scholar]
- 130. Gaxiola‐Valdez I, Singh S, Perera T, Sandy S, Li E, Federico P. Seizure onset zone localization using postictal hypoperfusion detected by arterial spin labelling MRI. Brain. 2017;140(11):2895–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Farrell JS, Colangeli R, Wolff MD, Wall AK, Phillips TJ, George A, et al. Postictal hypoperfusion/hypoxia provides the foundation for a unified theory of seizure‐induced brain abnormalities and behavioral dysfunction. Epilepsia. 2017;58(9):1493–501. [DOI] [PubMed] [Google Scholar]
- 132. Schidlowski M, Boland M, Rüber T, Stöcker T. Blood–brain barrier permeability measurement by biexponentially modeling whole‐brain arterial spin labeling data with multiple T2‐weightings. NMR Biomed. 2020;33(10):e4374. [DOI] [PubMed] [Google Scholar]
- 133. Schidlowski M, Stirnberg R, Stöcker T, Rüber T. Reliability of quantitative transverse relaxation time mapping with T2‐prepared whole brain pCASL. Sci Rep‐UK. 2020;10(1):18299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zimmer TS, David B, Broekaart DWM, Schidlowski M, Ruffolo G, Korotkov A, et al. Seizure‐mediated iron accumulation and dysregulated iron metabolism after status epilepticus and in temporal lobe epilepsy. Acta Neuropathol. 2021;142(4):729–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Canjels LPW, Jansen JFA, van den Kerkhof M, Alers R, Poser BA, Wiggins CJ, et al. 7T dynamic contrast‐enhanced MRI for the detection of subtle blood–brain barrier leakage. J Neuroimaging. 2021;31(5):902–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Czupalla CJ, Liebner S, Devraj K. Cerebral angiogenesis, methods and protocols. Methods Mol Biol. 2014;1135:415–37. [DOI] [PubMed] [Google Scholar]
- 137. Erickson MA, Wilson ML, Banks WA. In vitro modeling of blood–brain barrier and interface functions in neuroimmune communication. Fluids Barriers Cns. 2020;17(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Grainger AI, King MC, Nagel DA, Parri HR, Coleman MD, Hill EJ. In vitro models for seizure‐liability testing using induced pluripotent stem cells. Front Neurosci. 2018;12:590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Spencer D, Burchiel K. Selective amygdalohippocampectomy. Epilepsy Res Treat. 2012;2012:382095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hansen L, Lohfink N, Vutukuri R, Kestner RI, Trautmann S, Hecht M, et al. Endothelial Sphingosine‐1‐phosphate receptor 4 regulates blood–brain barrier permeability and promotes a homeostatic endothelial phenotype. J Neurosci. 2021;42(10):1908–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Smith MD, Adams AC, Saunders GW, White HS, Wilcox KS. Phenytoin‐ and carbamazepine‐resistant spontaneous bursting in rat entorhinal cortex is blocked by retigabine in vitro. Epilepsy Res. 2007;74(2–3):97–106. [DOI] [PubMed] [Google Scholar]
- 142. Sandow N, Kim S, Raue C, Päsler D, Klaft ZJ, Antonio LL, et al. Drug resistance in cortical and hippocampal slices from resected tissue of epilepsy patients: no significant impact of P‐glycoprotein and multidrug resistance‐associated proteins. Front Neurol. 2015;6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Campos G, Fortuna A, Falcão A, Alves G. In vitro and in vivo experimental models employed in the discovery and development of antiepileptic drugs for pharmacoresistant epilepsy. Epilepsy Res. 2018;146:63–86. [DOI] [PubMed] [Google Scholar]
- 144. Jones RSG, da Silva AB, Whittaker RG, Woodhall GL, Cunningham MO. Human brain slices for epilepsy research: pitfalls, solutions and future challenges. J Neurosci Methods. 2016;260:221–32. [DOI] [PubMed] [Google Scholar]
- 145. Raimondo JV, Heinemann U, Curtis M, Goodkin HP, Dulla CG, Janigro D, et al. Methodological standards for in vitro models of epilepsy and epileptic seizures. A TASK1‐WG4 report of the AES/ILAE translational TASK force of the ILAE. Epilepsia. 2017;58(S4):40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wickham J, Brödjegård NG, Vighagen R, Pinborg LH, Bengzon J, Woldbye DPD, et al. Prolonged life of human acute hippocampal slices from temporal lobe epilepsy surgery. Sci Rep‐UK. 2018;8(1):4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Heuzeroth H, Wawra M, Fidzinski P, Dag R, Holtkamp M. The 4‐aminopyridine model of acute seizures in vitro elucidates efficacy of new antiepileptic drugs. Front Neurosci. 2019;13:677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Avoli M, Jefferys JGR. Models of drug‐induced epileptiform synchronization in vitro. J Neurosci Methods. 2016;260:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lapilover EG, Lippmann K, Salar S, Maslarova A, Dreier JP, Heinemann U, et al. Peri‐infarct blood–brain barrier dysfunction facilitates induction of spreading depolarization associated with epileptiform discharges. Neurobiol Dis. 2012;48(3):495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Antonio LL, Anderson ML, Angamo EA, Gabriel S, Klaft ZJ, Liotta A, et al. In vitro seizure like events and changes in ionic concentration. J Neurosci Methods. 2016;260:33–44. [DOI] [PubMed] [Google Scholar]
- 151. Cucullo L, Hossain M, Rapp E, Manders T, Marchi N, Janigro D. Development of a humanized In vitro blood–brain barrier model to screen for brain penetration of antiepileptic drugs. Epilepsia. 2007;48(3):505–16. [DOI] [PubMed] [Google Scholar]
- 152. Grimminger T, Pernhorst K, Surges R, Niehusmann P, Priebe L, Von LM, et al. Levetiracetam resistance: synaptic signatures & corresponding promoter SNPs in epileptic hippocampi. Neurobiol Dis. 2013;60:115–25. [DOI] [PubMed] [Google Scholar]
- 153. Aronica E, Becker AJ, Spreafico R. Malformations of cortical development. Brain Pathol. 2012;22(3):380–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Becker AJ. Review: Animal models of acquired epilepsy: insights into mechanisms of human epileptogenesis. Neuropathol Appl Neurobiol. 2018;44(1):112–29. [DOI] [PubMed] [Google Scholar]
- 155. Czornyj L, Auzmendi J, Lazarowski A. Transporter hypothesis in pharmacoresistant epilepsies. Is it at the central or peripheral level? Epilepsia Open. 2022;7(Suppl 1):S34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Salar S, Maslarova A, Lippmann K, Nichtweiss J, Weissberg I, Sheintuch L, et al. Blood–brain barrier dysfunction can contribute to pharmacoresistance of seizures. Epilepsia. 2014;55(8):1255–63. [DOI] [PubMed] [Google Scholar]
- 157. Narayanan J, Frech R, Walters S, Patel V, Frigerio R, Maraganore DM. Low dose verapamil as an adjunct therapy for medically refractory epilepsy – an open label pilot study. Epilepsy Res. 2016;126:197–200. [DOI] [PubMed] [Google Scholar]
- 158. Asadi‐Pooya AA, Razavizadegan SMA, Abdi‐Ardekani A, Sperling MR. Adjunctive use of verapamil in patients with refractory temporal lobe epilepsy: a pilot study. Epilepsy Behav. 2013;29(1):150–4. [DOI] [PubMed] [Google Scholar]
- 159. Klein P, Friedman A, Hameed MQ, Kaminski RM, Bar‐Klein G, Klitgaard H, et al. Repurposed molecules for antiepileptogenesis: missing an opportunity to prevent epilepsy? Epilepsia. 2020;61(3):359–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Greene C, Hanley N, Reschke CR, Reddy A, Mäe MA, Connolly R, et al. Microvascular stabilization via blood–brain barrier regulation prevents seizure activity. Nat Commun. 2022;13(1):2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Wakayama E, Kuzu T, Tachibana K, Hirayama R, Okada Y, Kondoh M. Modifying the blood–brain barrier by targeting claudin‐5: safety and risks. Ann N Y Acad Sci. 2022;1514(1):62–9. [DOI] [PubMed] [Google Scholar]
- 162. Sadiq MA, Halim MS, Hassan M, Onghanseng N, Karaca I, Agarwal A, et al. Pharmacological agents in development for diabetic macular edema. Int J Retin Vitreous. 2020;6(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 163. Dubinski D, Hattingen E, Senft C, Seifert V, Peters KG, Reiss Y, et al. Controversial roles for dexamethasone in glioblastoma – opportunities for novel vascular targeting therapies. J Cereb Blood Flow Metab. 2019;39(8):1460–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Kim H, Lee JM, Park JS, Jo SA, Kim YO, Kim CW, et al. Dexamethasone coordinately regulates angiopoietin‐1 and VEGF: a mechanism of glucocorticoid‐induced stabilization of blood–brain barrier. Biochem Bioph Res Commun. 2008;372(1):243–8. [DOI] [PubMed] [Google Scholar]
- 165. Lange C, Storkebaum E, de Almodóvar CR, Dewerchin M, Carmeliet P. Vascular endothelial growth factor: a neurovascular target in neurological diseases. Nat Rev Neurol. 2016;12(8):439–54. [DOI] [PubMed] [Google Scholar]
- 166. Lengfeld JE, Lutz SE, Smith JR, Diaconu C, Scott C, Kofman SB, et al. Endothelial Wnt/β‐catenin signaling reduces immune cell infiltration in multiple sclerosis. Proc Natl Acad Sci U S A. 2017;114(7):E1168–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Eubelen M, Bostaille N, Cabochette P, Gauquier A, Tebabi P, Dumitru AC, et al. A molecular mechanism for Wnt ligand‐specific signaling. Science. 2018;361(6403):eaat1178. [DOI] [PubMed] [Google Scholar]
- 168. Chen Z, Brodie MJ, Liew D, Kwan P. Treatment outcomes in patients with newly diagnosed epilepsy treated with established and new antiepileptic drugs: a 30‐year longitudinal cohort study. JAMA Neurol. 2017;75(3):279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, et al. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 2008;7(6):500–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.