

Abstract
Metabolic alterations are a key hallmark of cancer cells, and the augmented synthesis and use of nucleotide triphosphates is a critical and universal metabolic dependency of cancer cells across different cancer types and genetic backgrounds. Many of the aggressive behaviours of cancer cells, including uncontrolled proliferation, chemotherapy resistance, immune evasion and metastasis, rely heavily on augmented nucleotide metabolism. Furthermore, most of the known oncogenic drivers upregulate nucleotide biosynthetic capacity, suggesting that this phenotype is a prerequisite for cancer initiation and progression. Despite the wealth of data demonstrating the efficacy of nucleotide synthesis inhibitors in preclinical cancer models and the well-established clinical use of these drugs in certain cancer settings, the full potential of these agents remains unrealized. In this Review, we discuss recent studies that have generated mechanistic insights into the diverse biological roles of hyperactive cancer cell nucleotide metabolism. We explore opportunities for combination therapies that are highlighted by these recent advances and detail key questions that remain to be answered, with the goal of informing urgently warranted future studies.
Subject terms: Cancer metabolism, Cancer metabolism
Overactive nucleotide synthesis is a hallmark of cancers and inhibitors of nucleotide synthesis pathways have shown promise in some cancer types. In this Review, Mullen and Singh give an overview of the role of aberrant nucleotide synthesis in supporting cancer cell growth, immune evasion, metastasis and resistance to cancer therapies, with a focus on identifying opportunities for the use of combination therapies to target these pathways more effectively.
Introduction
The hyperactive synthesis and use of nucleotide triphosphates (NTPs) and their deoxy counterparts (dNTPs) is a universal feature of cancer cells that is highly druggable. The supraphysiological abundance of intracellular nucleotides contributes to many aspects of cancer cell behaviour, including uncontrolled proliferation, immune evasion, metastasis and therapy resistance. Furthermore, many oncogenic drivers have been shown to upregulate nucleotide biosynthesis, suggesting that this phenotype is critical for cancer initiation and progression downstream of oncogene activation. The reliance of cancer cells on hyperactive nucleotide synthesis reflects the fact that (d)NTPs are rate-limiting for several essential biological processes that are themselves hyperactive in cancer cells, including DNA replication and repair, transcription, ribosome biogenesis and post-translational protein glycosylation.
The synthesis of purine and pyrimidine nucleotides occurs by two distinct pathways: the de novo pathway, which involves the incorporation of small precursors into nucleotides in an energy-intensive, multistep series of enzymatic reactions, and the nucleoside/nucleobase salvage pathway, in which a nucleoside or nucleobase is converted to its cognate nucleoside monophosphate (NMP) in a single phosphorylation or phosphoribosyltransferase reaction, respectively. There are major differences in the de novo and salvage synthesis of pyrimidines and purines, with important implications regarding which substrates can be salvaged to bypass the blockade of de novo synthesis (Fig. 1).
Fig. 1. Biosynthetic pathways for pyrimidine and purine nucleotides, relevant inhibitors and oncogenic regulators.

The biosynthetic pathways leading to pyrimidine (part a) and purine (part b) nucleotides are shown. For both pyrimidines and purines, de novo synthesis entails a complex series of steps that transform amino acids and phosphoribosyl pyrophosphate (PRPP) into uridine monophosphate (UMP) or inosine monophosphate (IMP), respectively; salvage pathways recycle nucleosides and nucleobases to form nucleoside monophosphates (NMPs) or deoxy NMPs in a single step using ATP or PRPP, respectively. The de novo synthesis pathways are shown by red arrows and enzymes, and nucleotide salvage pathways are shown by blue arrows and enzymes. Inhibitors of these pathways are shown in purple boxes. Selected oncogenic regulators that influence these pathways are shown in yellow. In addition to the oncogenic regulators shown, the pentose phosphate pathway (PPP) can also be upregulated by the oncogenic factors hypoxia-inducible factor 1α (HIF1α), mucin 1 (MUC1), MYC and SREBP1. Dashed inhibitory arrows in part b indicate that the inhibitors pemetrexed (Pem) and methotrexate (MTX) inhibit GART and ATIC indirectly by disruption of the folic acid cycle. 5,10-MTHF, 5,10-methylenetetrahydrofolate; 5-FU, 5-fluorouracil; ADA, adenosine deaminase; ADSL, adenylosuccinate lyase; ADSS, adenylosuccinate synthetase; AICAR, 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside; AMPD, AMP deaminase; APRT, adenine phosphoribosyltransferase; BQ, brequinar; CAD, carbamoyl phosphate synthetase II, aspartate transcarbamoylase and dihydroorotase; CDA, cytidine deaminase; CMP, cytidine monophosphate; CTP, cytidine triphosphate; dA, deoxyadenosine; dC, deoxycytidine; dCDP, deoxycytidine phosphate; DCK, deoxycytidine kinase; dCMP, deoxycytidine monophosphate; DCTD, deoxycytidylate deaminase; dCTP, deoxycytidine triphosphate; DHF, dihydrofolate; DHFR, DHF reductase; DHO, dihydroorotate; DHODH, DHO dehydrogenase; dT, thymidine; dTTP, deoxythymidine triphosphate; dU, deoxyuridine; dUMP, deoxyuridine monophosphate; FGAR, phosphoribosyl-N-formylglycineamide; FAICAR, 5-formamidoimidazole-4-carboxamide ribotide; GAR, glycineamide ribonucleotide; GMPS, GMP synthase; GOF-mut-p53, gain-of-function mutant p53; HGPRT, hypoxanthine–guanine phosphoribosyltransferase; HU, hydroxyurea; IMPDH1/2, IMP dehydrogenases 1 and 2; Lef, leflunomide; MAPK, mitogen-activated protein kinase; MPA, mycophenolic acid; mTOR, mammalian target of rapamycin; PNP, purine nucleoside phosphorylase; RNR, ribonucleotide reductase; THF, tetrahydrofolate; TK1/2, thymidine kinases 1 and 2; TS, thymidylate synthase; UCK1/2, uridine–cytidine kinases 1 and 2; UDP, uridine diphosphate; UMPS, UMP synthase; UTP, uridine triphosphate; XMP, xanthine monophosphate.
Nucleotide synthesis inhibitors were among the first antineoplastic agents discovered and are still the backbone of therapy for many cancer indications. Recently, nucleotide synthesis has been ‘rediscovered’ many times over as a crucial cancer vulnerability in various large-scale, unbiased functional genomic1–3 and chemical4–6 screening approaches as well as in metabolomics-based investigations of particular cancers7–11. This has renewed interest in the field and sparked high-impact preclinical studies that have served as a rationale for several ongoing clinical trials and drug development campaigns. Given the vital nature of (d)NTPs, one might expect systemic treatment with nucleotide synthesis inhibitors to be prohibitively toxic. However, drugs in this class are generally clinically well tolerated with a manageable toxicity profile, likely due to the existence of multiple independent enzymatic pathways to generate (d)NTPs. Based on the clinical toxicity profiles of inhibitors of de novo nucleotide synthesis, it appears that cells in most terminally differentiated tissues can generate sufficient (d)NTPs through salvage pathways to maintain homeostasis. However, proliferating cells that must regenerate cell populations with high turnover (such as haematopoietic cells in the bone marrow, which maintain adequate blood cell populations, and progenitor cells in the gastrointestinal tract that maintain the gut epithelium) require the de novo pathways to support continuous proliferation12–15. In addition to explaining how de novo pathway inhibitors are tolerated, salvage pathways also provide a potential mechanism of adaptive resistance to these drugs (discussed later).
There are several nucleotide metabolism enzymes for which clinically approved or clinical-grade inhibitors are available (Fig. 1). In this Review, we provide a brief overview of nucleotide metabolism and discuss clinically relevant inhibitors of nucleotide synthesis enzymes, focusing on several enzymes for which clinically approved inhibitors exist: dihydroorotate dehydrogenase (DHODH), inosine monophosphate dehydrogenases 1 and 2 (IMPDH1/2), thymidylate synthase (TS), dihydrofolate reductase (DHFR) and ribonucleotide reductase (RNR). Inhibition of each of these enzymes causes a distinct profile of (d)NTP depletion, leading to important differences in downstream consequences for the cell. In the following sections, we discuss recent insights into the cellular consequences of targeting nucleotide metabolism enzymes, with a focus on emerging avenues for combination therapy strategies (Table 1). We delineate how these agents disrupt vital cancer cell activities, including cell growth and proliferation, immune evasion, metastasis and resistance to therapy. Finally, we explore how cancer cells might have intrinsic or acquired resistance to these nucleotide synthesis inhibitors as well as the potential for future preclinical and clinical studies involving these agents.
Table 1.
Proposed combination therapy strategies with nucleotide synthesis inhibitors
| Combination agent (target) | Mechanism | Cancer type | Validation level |
|---|---|---|---|
| Dihydroorotate dehydrogenase | |||
| Gemcitabine (ribonucleotide reductase inhibitor and DNA chain terminator) | Decreased molecular competition by endogenous dCTP | PDAC11 | Animal models |
| Doxorubicin (topoisomerase II inhibitor) | Impaired DNA repair | TNBC9 | Animal models |
| Floxuridine (5-FU prodrug, TS inhibitor) | Unknown | KRAS-mutant PDAC5 | Cell culture |
| Cisplatin/etoposide (alkylating agent/topoisomerase II inhibitor) | Unknown | SCLC2 | Animal models |
| Azacytidine (DNMT1 inhibitor) | Decreased molecular competition by endogenous dCTP | MDS131 | Animal models |
| Dipyridamole (ENT1/2 inhibitor) | Enhanced (d)NTP depletion | CRC144, neuroblastoma145, AML146 | Cell culture, animal models |
| ENT1 knockout | Enhanced (d)NTP depletion | PDAC149 | Animal models |
| GPX4 inhibitor (ferroptosis inducer) | Increased mitochondrial lipid peroxidation | Multiple139 | Animal models |
| Deoxycytidylate deaminase knockout | Combinatorial dUMP depletion and dTTP depletion | SCLC2 | Cell culture |
| TRAIL (apoptosis inducer) | Decreased mitochondrial membrane potential | Multiple141 | Cell culture |
| Buparlisib (PI3K inhibitor) | Combinatorial de novo pathway blockade | GBM with PTEN deletion7 | Animal models |
| Lapatinib (EGFR inhibitor) | Combinatorial de novo pathway blockade | GBM with EGFR amplification7 | Animal models |
| Prednisolone (glucocorticoid receptor agonist) | Unknown | KRAS-mutant PDAC5 | Cell culture |
| Thapsigargin (ER stress inducer) | Depleted deoxyuridine, leading to amplified ER stress and ROS | PDAC78 | Cell culture |
| TS | |||
| Carboplatin (alkylating agent) | Unknown | NSCLC112 | FDA approved |
| Pembrolizumab (PD1 blockade) | Immunogenic cell death, enhanced T cell OXPHOS | NSCLC111,112 | FDA approved |
| Inosine monophosphate dehydrogenases 1 and 2 | |||
| Radiation therapy | Inhibited radiation therapy-induced DNA double-strand break repair | GBM136 | Animal models |
| Temozolomide (alkylating agent) | Increased salvage of methylated O6-MG nucleobases leading to increased O6-MG incorporation into DNA | GBM134 | Animal models |
| Anti-PD1 antibody | Increased cancer cell presentation of immunogenic antigens via MHC class I | TNBC119 | Animal models |
| Ribonucleotide reductase | |||
| DI-39/VE-822 (DCK inhibitor/ATR inhibitor) | Combined dCTP depletion and replication stress | B-ALL60,156 | Animal models |
5-FU, 5-fluorouracil; AML, acute myeloid leukaemia; B-ALL, B cell acute lymphoblastic leukaemia; CRC, colorectal cancer; DCK, deoxycytidine kinase; dCTP, deoxycytidine triphosphate; (d)NTP, (deoxy)nucleotide triphosphate; dTTP, deoxythymidine triphosphate; dUMP, deoxyuridine monophosphate; ENT1/2, equilibrative nucleoside transporters 1 and 2; ER, endoplasmic reticulum; GBM, glioblastoma multiforme; MDS, myelodysplastic syndrome; NSCLC, non-small-cell lung cancer; O6-MG, O6-methylguanine; OXPHOS, oxidative phosphorylation; PDAC, pancreatic ductal adenocarcinoma; ROS, reactive oxygen species; SCLC, small-cell lung cancer; TNBC, triple-negative breast cancer; TRAIL, TNF-related apoptosis-inducing ligand; TS, thymidylate synthase.
Overview of nucleotide metabolism
Here, we briefly explain the biosynthetic pathways for pyrimidine and purine nucleotides, discuss how nucleotide levels are regulated during cell division and highlight common examples of oncogenic dysregulation of nucleotide metabolism. For further details on nucleotide synthesis pathways, we refer readers to excellent reviews16–18.
Synthesis of pyrimidines and purines
The de novo and salvage pathways of pyrimidines versus purines occur via fundamentally different logic (Fig. 1). The pyrimidine de novo pathway first builds the aromatic base (orotate) and only then adds a ribose 5-phosphate moiety in a phosphoribosyl pyrophosphate (PRPP)-dependent reaction, whereas the purine de novo pathway begins with PRPP and builds the aromatic base onto the ribose skeleton in a stepwise fashion. However, both de novo pathways require ATP, glutamine-derived nitrogen and aspartate. The pyrimidine nucleoside salvage pathway proceeds by the phosphorylation of (deoxy)nucleosides to form (d)NMPs in a single ATP-dependent step catalysed by uridine–cytidine kinase 1 (UCK1) and UCK2, deoxycytidine kinase (DCK) and thymidine kinase 1 (TK1) and TK2. In mammalian cells, pyrimidine nucleobases — uracil, cytosine, thymine — cannot be salvaged by PRPP-dependent phosphoribosyltransferase reactions19 or be converted to orotate to enter the de novo pathway, and therefore free pyrimidine nucleobases are not efficiently salvaged20. Purine ribonucleosides — adenosine, guanosine and inosine — cannot be converted to their cognate NMPs and must first be catabolized to their cognate nucleobases (adenine, guanine and hypoxanthine) by purine nucleoside phosphorylase (PNP) and then converted to NMPs in a PRPP-dependent phosphoribosyltransferase reaction catalysed by adenine phosphoribosyltransferase (APRT) or hypoxanthine–guanine phosphoribosyltransferase (HGPRT). However, the purine deoxynucleosides deoxyadenosine and deoxyguanosine can be phosphorylated by DCK to form dAMP and dGMP, respectively — this set of reactions is considered the purine nucleoside–nucleobase salvage pathway (Fig. 1).
Physiological control of nucleotide levels
To undergo cell division, resting cells must dramatically increase their dNTP and NTP pools by 5–10-fold (ref. 21) and by at least 10-fold (ref. 16), respectively. Although de novo nucleotide synthesis is far more expensive than nucleoside salvage in terms of carbon and energy, it is required to rapidly expand nucleotide pools to meet these demands. NTP concentrations in proliferating cells are on the order of 100 µM to several millimolar, whereas dNTP concentrations are typically on the order of 10–100 µM, with relative dNTP concentrations generally being balanced within a given cell type22. De novo pathway activity is generally very low or absent in non-proliferating cells and must be induced to generate sufficient (d)NTPs for proliferation (which is thought to largely explain the low toxicity of systemic inhibition of the de novo pathway in most tissues, as discussed later)16. Many canonical drivers of cell proliferation, such as mammalian target of rapamycin (mTOR) signalling, increase the expression and activity of key de novo pathway enzymes, including the carbamoyl phosphate synthetase 2 (CPSII), aspartate transcarbamoylase (ATC) and dihydroorotase (DHOase) complex (CAD)23. To ensure a balance in the relative abundance of various (d)NTPs, key de novo pathway enzymes, such as RNR and the CAD complex components, are allosterically downregulated by their (d)NTP end products in negative feedback loops16. Additionally, nucleotide synthesis is balanced by nucleotide catabolism, which is initiated by nucleotide phosphorylases24. This complex interplay allows cells to dynamically fine-tune their nucleotide pools to meet the demands of cell division.
Oncogenic activation of nucleotide synthesis
Although the precise intracellular concentrations of (d)NTPs vary widely based on cell type, cellular context and culture conditions, a review of over 600 published values estimates that cancer cells have, on average, 6–11-fold greater dNTP and 1.25–5-fold greater NTP concentrations than non-malignant proliferating cells22. Numerous well-known oncogenes have been shown to enforce hyperactive (d)NTP synthesis; for example, mutant KRAS8,25, PI3K26 and MYC27,28, which are all frequent drivers of human cancer, promote the activity and expression of key de novo pathway enzymes and indirectly support de novo pathway flux by increasing cellular uptake of glucose and other nutrients to provide the required ATP, ribose and amino acids. Conversely, loss-of-function mutations or silencing of nucleotide catabolism enzymes, such as SAM domain and HD domain-containing 1 (SAMDH1), which converts dNTPs to deoxynucleosides, have been reported to increase nucleotide pools by preventing nucleotide degradation29,30. Given the ubiquitous upregulation of nucleotide levels in human cancer22 and the crucial role that this plays in various malignant cell behaviours (detailed below) as well as the fact that de novo pathway activity is dispensable for the homeostasis of most mammalian tissues16, de novo pathway enzymes are rational targets for cancer therapy.
Targeting nucleotide synthesis
Nucleotide synthesis enzymes have proven to be highly tractable drug targets. In this section, we briefly discuss prominent clinically druggable enzymes, their inhibitors and their mechanisms of action. A representative (but not exhaustive) description of clinically relevant nucleotide synthesis inhibitors is provided in Table 2.
Table 2.
Structure and mechanism of clinically approved or investigational nucleotide synthesis inhibitors
| Target enzyme | Compound | Structure | Mechanism |
|---|---|---|---|
| Dihydroorotate dehydrogenase | Teriflunomide (active metabolite of leflunomide)40 | 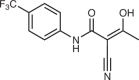 |
Non-competitive with DHO, reversible |
| Brequinar40 | 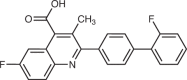 |
Non-competitive with DHO, reversible | |
| Thymidylate synthase | 5-Fluorouracil37 | 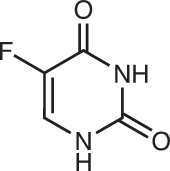 |
Competitive with dUMP, reversible |
| Pemetrexed38 | 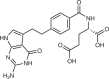 |
Competitive with 5,10-MTHF, reversible | |
| DHF reductase | Methotrexate32 | 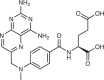 |
Competitive with DHF, reversible |
| IMPDH1/2 | Mycophenolic acid41 | 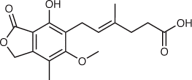 |
Uncompetitive with IMP, reversible |
| IMPDH1/2 and GMP synthase | Mizoribine42 | 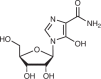 |
Competitive with IMP, reversible |
| Ribonucleotide reductase | Hydroxyurea46 | 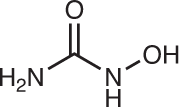 |
Non-competitive with NDPs, irreversible |
| Gemcitabine47,48 | 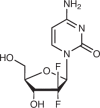 |
Competitive with NDPs, reversible |
5,10-MTHF, 5,10-methylenetetrahydrofolate; DHF, dihydrofolate; DHO, dihydroorotate; dUMP, deoxyuridine monophosphate; IMP, inosine monophosphate; IMPDH1/2, inosine monophosphate dehydrogenases 1 and 2; NDP, ribonucleotide diphosphate.
Inhibitors of DHFR
Following seminal observations by Farber and Diamond in the 1940s describing the efficacy of antifolate compounds against childhood leukaemia31, agents targeting folic acid metabolism and downstream thymidine synthesis have become the backbone of therapies for many cancers. Methotrexate (MTX), a competitive inhibitor of DHFR, is commonly used for the treatment of various cancers and autoimmune conditions32. DHFR inhibition causes depletion of folic acid cycle intermediates, which are methyl and formyl donors for de novo thymidine and purine synthesis, respectively (Fig. 1). It is thought that the anticancer efficacy of MTX is primarily mediated by indirect TS inhibition through the depletion of tetrahydrofolate and the subsequent depletion of deoxythymidine triphosphate (dTTP), although other mechanisms are likely to play important roles in MTX activity against rheumatoid arthritis33,34 and in certain cancer contexts35,36.
Inhibitors of TS
The importance of dTTP is illustrated by the anticancer effectiveness of direct TS inhibitors such as 5-fluorouracil (5-FU) and the folate analogue pemetrexed. 5-FU is metabolized to 5-fluoro-deoxyuridine monophosphate and competitively inhibits TS by displacing its endogenous substrate, deoxyuridine monophosphate (dUMP)37. One drawback of this mechanism is that 5-FU induces dUMP accumulation, thereby reducing its own effectiveness as it must out-compete endogenous dUMP. Pemetrexed avoids this drawback as it competitively inhibits TS at its folate binding site and its inhibitory activity is therefore unaffected by dUMP accumulation38. Although pemetrexed also inhibits DHFR and multiple enzymes in the purine de novo synthesis pathway, it is thought that dTTP depletion is its main mechanism of cytotoxicity39. Regardless of their precise mechanisms, which remain incompletely understood, MTX, 5-FU and pemetrexed have proven clinical efficacy and have become staple treatments for many cancers.
Inhibitors of DHODH
DHODH is a key enzyme in the pyrimidine de novo pathway that is strictly required for the synthesis of uridine monophosphate (UMP) from aspartate, and its inhibition causes depletion of all pyrimidine (d)NTPs as they are all derived from UMP (Fig. 1a). DHODH is the only mitochondrial enzyme in the pyrimidine de novo pathway and resides on the outer surface of the inner mitochondrial membrane; its activity is obligately coupled to the electron transport chain as it donates electrons from the oxidation of dihydroorotate to coenzyme Q, which in turn are passed to complex III and can fuel oxidative phosphorylation (OXPHOS).
Leflunomide and its active metabolite teriflunomide are the only DHODH inhibitors to have gained FDA approval to date and are indicated in certain autoimmune syndromes (discussed in detail below). Owing to the relatively poor potency of leflunomide and its known off-target interactions, more potent and selective DHODH inhibitors have been developed and are currently in clinical trials; the most well-characterized of these is brequinar. The precise mechanisms of the various DHODH inhibitors are not completely defined, but it has been shown that brequinar and teriflunomide both bind the hydrophobic ubiquinone-binding pocket of DHODH and are non-competitive with dihydroorotate40. Although DHODH inhibition has shown impressive efficacy in many preclinical studies, it is still awaiting clinical validation for the treatment of patients with cancer.
Inhibitors of IMPDH
Inosine monophosphate (IMP), an intermediate in the purine de novo pathway, can be converted either to AMP or GMP (Fig. 1b). The two-step GMP-producing arm of this pathway is the target of several approved drugs. Mycophenolic acid (MPA) is a non-competitive inhibitor of IMPDH1 and IMPDH2, which convert IMP to XMP before GMP synthesis41. Mizoribine, a nucleoside analogue that is converted to mizoribine monophosphate, is a competitive inhibitor of IMPDH1, IMPDH2 and GMPS, the enzyme that catalyses the conversion of XMP to GMP42. Both MPA and mizoribine cause selective depletion of (d)GTP. As is the case for DHODH, inhibitors of (d)GTP synthesis have gained approval for autoimmune conditions but have not yet shown clinical efficacy in patients with cancer despite robust preclinical activity in animal models of cancer43.
Inhibitors of RNR
RNR is required for the de novo synthesis of all dNTPs as it reduces the 2′ carbon of the ribose moiety of NDPs to generate dNDPs (Fig. 1). RNR plays a critical role in balancing the levels of dNTPs as it is allosterically regulated by negative feedback loops that ensure that no single dNTP species is overabundant or deficient, and its expression and stability are also regulated at several other levels44,45.
Hydroxyurea is an approved RNR inhibitor that is indicated for the treatment of sickle-cell anaemia, myeloproliferative disorders and other diseases. Hydroxyurea is converted to a nitroxide free radical, which in turn quenches a tyrosyl free radical in the active site of RR, resulting in irreversible RNR inhibition46. Gemcitabine is another RNR-targeting agent used to treat various malignancies, competitively inhibiting the RRM1 subunit of RNR47,48 in addition to other cytotoxic mechanisms49.
Effects of inhibiting (d)NTP synthesis on growth and proliferation
Nucleotides and deoxynucleotides are essential substrates for many anabolic processes that are critical for cell growth and proliferation. Therefore, (d)NTP depletion using de novo pathway inhibitors impairs several processes that promote tumour growth, including DNA replication/repair, ribosome biogenesis and protein translation, oncogenic mRNA transcription, post-translational protein glycosylation and maintenance of homeostatic cellular redox balance. In this section, we discuss the cellular consequences of nucleotide depletion downstream of nucleotide synthesis inhibitors and the potential utility of these agents as components of combination therapies.
Effects of (d)NTP synthesis inhibition on DNA replication and repair
During DNA replication, the relative abundance of all four dNTPs is tightly controlled by RNR and SAMDH1, and an imbalance in dNTP pools causes misincorporation of bases, leading to mutation or DNA damage. Inhibition of TS by 5-FU, pemetrexed or MTX results in a large excess of deoxyuridine triphosphate (dUTP) over dTTP (Fig. 2a). This causes misincorporation of uracil instead of thymine into DNA, which is sensed by the cell and repaired by base excision repair mechanisms. The massive accumulation of such DNA lesions ultimately results in ‘thymineless death’50; the precise mechanism of this process is not completely understood but might involve FAS–FASL signalling51 or the cyclic futile repair of uracil misincorporation, leading to progressively widening single-stranded gaps in DNA52.
Fig. 2. Nucleotides fuel cancer cell growth and proliferation.

a, Deoxythymidine triphosphate (dTTP) is required for the synthesis of DNA. Depletion of dTTP by the inhibition of thymidine synthase (TS) results in the accumulation of deoxyuridine triphosphate (dUTP) and an increase in the dUTP-to-dTTP ratio. As DNA polymerases cannot distinguish dUTP from dTTP, this leads to widespread misincorporation of uracil and a massive DNA damage response, ultimately resulting in thymineless death. b, Nucleotide triphosphates (NTPs) are required for the synthesis of RNA; inhibition of dihydroorotate dehydrogenase (DHODH) or inosine monophosphate dehydrogenase (IMPDH) depletes NTPs and disrupts RNA transcription. DHODH inhibition by brequinar (BQ) or leflunomide (Lef) impairs the productive elongation of RNA polymerase II (Pol II) by an unknown mechanism and thereby inhibits oncogenic transcription. Pol II pause release is normally triggered by positive transcription elongation factor B (P-TEFB), which phosphorylates DSIF and NELF; this causes DSIF to promote elongation and results in the dissociation of NELF (left). Pyrimidine depletion using BQ or Lef or purine depletion using mycophenolic acid (MPA) disrupt ribosomal RNA (rRNA) synthesis by starving RNA Pol I of NTP substrates and thereby hinder ribogenesis and the translation of oncogenic proteins (right). c, Uridine triphosphate (UTP) is required for the synthesis of uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), the substrate for O-GlcNAcylation of proteins by O-GlcNAc transferase (OGT). O-GlcNAcylation of various proteins, including MYC, hypoxia-inducible factor 1α (HIF1α) and phosphofructokinase 1 (PFK1), promotes cancer progression by diverse mechanisms. d, Generation of mitochondrial reactive oxygen species (mitoROS) requires high-energy electrons, either derived from α-ketoglutarate (αKG) via the TCA cycle or from DHODH via reduced coenzyme Q. DHODH-dependent mitoROS generation thus frees up αKG to produce cytosolic NADPH and reduced glutathione (GSH) via a pathway involving malate dehydrogenase 1 (MDH1) and malic enzyme 1 (ME1), allowing for control of cytosolic oxidative stress. Deoxyuridine (dU) produced downstream of DHODH serves as another cytosolic reactive oxygen species (ROS) quenching agent. Blockade of dU synthesis by inhibition of DHODH or cytidine deaminase (CDA) by the CDA inhibitor tetrahydrouridine (THU) accentuates oxidative and endoplasmic reticulum (ER) stress upon treatment with thapsigargin. 5,10-MTHF, 5,10-methylenetetrahydrofolate; 5-FU, 5-fluorouracil; CIII, complex III; CIV, complex IV; CTP, cytidine triphosphate; dC, deoxycytidine; DHF, dihydrofolate; DHFR, dihydrofolate reductase; DHO, dihydroorotate; dTMP, deoxythymidine monophosphate; dUMP, deoxyuridine monophosphate; GSSG, oxidized glutathione; MTX, methotrexate; MUC1, mucin 1; OAA, oxaloacetate; Pem, pemetrexed; PyrDNP, pyrimidine de novo pathway; rC, cytidine; rU, uridine; THF, tetrahydrofolate; UDP, uridine diphosphate; UGGP, UDP-glucose/galactose pyrophosphorylase; UMP, uridine monophosphate.
dNTP insufficiency caused by de novo pathway inhibition results in the accumulation of stalled DNA replication forks, which is known as replication stress. Replication stress activates the DNA damage checkpoint kinases ATM and ATR, which in turn pause cell cycle progression and promote replication stress resolution53. Failure of this checkpoint system can result in catastrophic chromosomal damage and cell death. The replication stress response has emerged as a crucial regulator of dNTP pools during pharmacological nucleotide depletion as well as during unperturbed DNA replication54. In both contexts, ATR directly upregulates dNTP biosynthetic capacity by phosphorylating and activating DCK55 and inducing the expression of the RNR subunit RRM2 (ref. 56). Additionally, ATM has been shown to stabilize the RNR subunit RRM2B (also known as p53R2) by phosphorylation57, and fibroblasts isolated from individuals with germline homozygous loss-of-function ATM mutations display reduced expression of all three RNR subunits relative to the wild-type counterparts58. Thus, the replication stress response is a fundamental mechanism by which cells sense dNTP deficiency and adaptively increase dNTP pools.
The combined inhibition of the replication stress response and nucleotide synthesis has shown synthetic lethality against p53-deficient solid tumours59, B cell acute lymphoblastic leukaemia (B-ALL)60, acute myeloid leukaemia (AML)61 and phosphatase and tensin homologue (PTEN)-deficient breast cancer10. It has also been suggested that the enforced differentiation of AML blasts by DHODH inhibition4,62 is caused by ATR–CHK1 activation63. Future studies are needed to further characterize the role of ATM–ATR in mediating resistance to nucleotide starvation.
Effects of (d)NTP synthesis inhibition on ribosome biogenesis and protein translation
While ribosomal RNA (rRNA) makes up the vast majority of RNA mass in all cells, increased rRNA mass and nucleolar hypertrophy are frequently observed in cancer cells, which facilitates hyperactive protein translation. Whereas dNTPs are required for DNA synthesis, NTPs are necessary for RNA transcription and their depletion using inhibitors that block both dNTP and NTP formation, such as inhibitors of DHODH or IMPDH, would be expected to limit rRNA transcription and translational capacity (Fig. 2b). Indeed, Kofuji et al. recently reported that the genetic deletion of both IMPDH1 and IMPDH2 or their pharmacological inhibition using MPA is needed to inhibit rRNA and tRNA transcription enough to normalize nucleolar mass and morphology and inhibit tumour growth in glioblastoma multiforme (GBM) tumour xenografts64. These findings demonstrate that inhibition of multiple redundant enzymes (for example, IMPDH1 and IMPDH2) can be required to deplete end product metabolites from cells. Fortunately, MPA inhibits both enzymes. Similarly, DHODH inhibition was recently reported to limit rRNA transcription in GBM models and thereby inhibit ribosome biogenesis and GBM progression in vivo65. Thus, it appears that either purine or pyrimidine NTPs can become limiting for rRNA transcription. This has important implications for combination therapy strategies as hyperactive protein translation is a known mechanism of resistance to chemotherapy agents such as cisplatin66 and paclitaxel67.
Effects of (d)NTP synthesis inhibition on mRNA transcription and control of cancer cell differentiation
Nucleotide starvation dramatically influences the transcriptomic profile of cancer cells. In an elegant study using a zebrafish model of melanoma initiation6, White et al. found that the effects of DHODH inhibition were similar to those caused by the genetic deletion of Spt5, an RNA polymerase II (Pol II)-associated factor critical for the transition of Pol II from the promoter-proximal paused state to productive elongation68 (Fig. 2b). Depletion of pyrimidine nucleotides with leflunomide globally disrupted Pol II elongation and suppressed the expression of key melanoma driver genes in vitro, and treatment of mice with leflunomide inhibited the growth of melanoma xenograft tumours6. These findings remain to be extended to other malignancies.
Interestingly, whereas DHODH inhibition suppresses neural crest lineage formation in melanoma6, Sykes et al. discovered that the DHODH inhibitor brequinar enforces the differentiation of leukaemic blasts in various animal models of AML, resulting in disease control and enhanced survival4. Longitudinal assessment of syngeneic AML blasts in mice demonstrated that brequinar treatment induces a transcriptomic signature consistent with normal neutrophil differentiation. Thus, the transcriptomic consequences of DHODH inhibition appear to be highly dependent on the context. Regardless, ongoing clinical trials are evaluating the efficacy of DHODH inhibitors against AML and other haematological malignancies (NCT04609826, NCT02509052 and NCT05246384).
In addition to DHODH4,62, targeting of IMPDH1 and IMPDH2 (ref. 43), DHFR69, TS70 and RNR71 have all been shown to promote cancer cell differentiation in various contexts. It has been postulated that this effect may result from the disruption of DNA replication, imbalance in (d)NTP pools, downregulation of critical differentiation-opposing transcription factors such as MYC, or some combination thereof. Interestingly, pharmacological upregulation of RNR (using nelarabine) or genetic knockdown of SAMDH1 were recently shown to induce AML differentiation, despite both perturbations increasing total (d)NTP pools, demonstrating that nucleotide imbalance — rather than starvation — induces differentiation in this context72. The molecular links between (d)NTP level perturbation and cellular differentiation in both cancer and normal physiology remain to be fully elucidated.
Effects of (d)NTP synthesis inhibition on post-translational protein glycosylation
Uridine triphosphate or cytidine triphosphate (CTP) are required for the activation of sugar moieties for glycosylation reactions. As a result, the depletion of pyrimidine NTPs might impair glycosylation reactions. Although the global effects on protein (and lipid) glycosylation downstream of pyrimidine depletion remain to be characterized, this relationship has been validated for glycosylation with O-linked N-acetylglucosamine (O-GlcNAc) (Fig. 2c). O-GlcNAc modification occurs on serine and threonine residues, often in direct competition with phosphorylation modifications, and has been shown to regulate the function and stability of proteins that directly regulate cancer progression such as MYC73. DHODH inhibition in AML blasts causes a decrease in global protein O-GlcNAcylation4 and MYC protein abundance74; therefore, it has been postulated that decreased MYC stability resulting from impaired O-GlcNAcylation could be responsible for DHODH inhibitor-mediated AML differentiation4,62,74. Given the global upregulation of O-GlcNAcylation in cancer and the resulting regulation of key metabolic enzymes, transcription factors and epigenetic modifiers73,75, impaired cancer cell O-GlcNAcylation downstream of pyrimidine depletion likely has pleiotropic consequences. Global proteomics studies are needed to reveal novel oncogenic proteins whose activity is enhanced by pyrimidine-dependent glycosylation and to delineate the contribution of impaired glycosylation to the loss of cell viability upon pyrimidine starvation.
Nucleotide metabolism alteration regulates ROS homeostasis
Cancer cells must manage oxidative stress by maintaining reactive oxygen species (ROS) levels within a narrow range76. Disruption of cancer cell ROS homeostasis can occur downstream of many different metabolic perturbations (such as hyperglycaemia)77 or genotoxic perturbations (such as gemcitabine)78 or via changes in the microenvironment (such as acidosis)79, commonly by depletion of the cellular antioxidants NADPH and reduced glutathione (GSH). Interestingly, the pyrimidine nucleosides uridine80,81 and deoxyuridine78 have emerged as powerful relievers of oxidative stress. Indeed, it was recently shown that the production of deoxyuridine by cytidine deaminase (CDA) attenuates ROS accumulation and mitigates endoplasmic reticulum (ER) stress in pancreatic ductal adenocarcinoma (PDAC) cells treated with thapsigargin or gemcitabine78. Inhibition of CDA (by tetrahydrouridine) or of DHODH (by leflunomide) depleted intracellular deoxyuridine and accentuated thapsigargin-mediated cell death, whereas deoxyuridine supplementation rescued cells from thapsigargin toxicity by attenuating oxidative and ER stress78 (Fig. 2d). These findings suggest that the reprogramming of nucleoside pools is an important adaptive mechanism to survive therapy-induced oxidative and ER stress.
DHODH is unique among nucleotide synthesis enzymes in that it localizes to the inner mitochondrial membrane and is obligately coupled to the electron transport chain. Accordingly, DHODH knockdown has been shown to disrupt complex III activity, cause ROS accumulation and decrease mitochondrial membrane potential82. KRAS-driven tumorigenesis requires mitochondrial ROS to attenuate ERK activity and maintain optimal ERK signalling intensity83, while cytosolic regeneration of NADPH by malic enzyme 1 (ME1) is required to maintain adequate cytosolic GSH levels to handle oxidative stress84. Since both of the aforementioned pathways require α-ketoglutarate, it has been postulated5 that DHODH-dependent mitochondrial ROS generation frees up α-ketoglutarate to enter NADPH-producing pathways in the cytosol and thereby facilitates redox homeostasis (Fig. 2d).
Altered nucleotide metabolism drives cancer cell immune evasion
Mounting preclinical evidence has demonstrated that deranged cancer cell metabolism facilitates immune evasion by altering the metabolic landscape of the tumour microenvironment (TME)85. For example, enhanced cancer cell uptake of glucose and glutamine can deprive immune effector cells of these nutrients and thereby inhibit their anticancer activity86. Conversely, cancer cell secretion of lactate87 or kynurenine88 can directly suppress immune cell effector function. Evidence is emerging that altered nucleotide handling by cancer cells facilitates their immune escape by various mechanisms.
The role of purinergic signalling in immunity and autoimmunity
In the 1970s, it was discovered that germline loss-of-function mutations in adenosine deaminase (ADA) and PNP cause severe combined immunodeficiency syndrome, characterized by a complete absence of T and B lymphocyte-mediated immunity and debilitating predisposition to infection89,90. Paradoxically, loss of the protein fatty acid metabolism–immunity nexus (FAMIN, also known as LACC1), which was recently discovered to be a multifunctional enzyme with four distinct purine metabolism activities, including ADA and PNP activities91, causes Still disease, a severe juvenile-onset autoimmune syndrome92. These data show that purine metabolism plays a crucial and complex role in human immunity, and so it is perhaps unsurprising that cancer hijacks these pathways to avoid immune destruction.
The purine nucleoside adenosine is a suppressor of anticancer immunity. Adenosine signalling through cell surface adenosine receptors dampens the anticancer activity of T cells93, macrophages94, innate immune cells95 and dendritic cells96 in the TME (Fig. 3a). Adenosine production is an attractive therapeutic target as adenosine accumulation in the TME is observed across various solid tumours and is correlated with poor patient outcomes97,98. Accumulation of adenosine can occur through the breakdown of extracellular ATP by the sequential action of the 5′-ectonucleotidase enzymes CD39, which converts ATP or ADP to AMP, and CD73, which converts AMP to adenosine99; these enzymes are frequently upregulated in human cancers and are correlated with poor prognosis100,101. Anti-CD73 antibodies have been shown to enhance anticancer immunity and restrain tumour growth in various preclinical mouse models101–103, leading to ongoing clinical trials involving anti-CD73 antibodies (NCT05173792, NCT05143970, NCT05431270, NCT05174585, NCT04989387 and NCT04668300) and CD73 inhibitors (NCT05227144 and NCT04104672) for various cancer indications.
Fig. 3. Altered nucleoside handling facilitates cancer cell immune evasion.

a, Cancer cell-directed adenosine build-up in the tumour microenvironment dampens anticancer immunity by signalling to various immune cell subsets. Extracellular ATP, which is a powerful immunostimulatory molecule, is cleared by CD39 and CD73, which are overexpressed in cancer cells. This process yields adenosine, which is immunosuppressive. Adenosine can also be directly exported from cancer cells through equilibrative nucleoside transporters 1 and 2 (ENT1/2). Extracellular adenosine dampens the antitumour activity of T cells, natural killer (NK) cells and dendritic cells (DCs); it also promotes the immunosuppressive activity of tumour-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), thereby facilitating cancer cell immune evasion. b, The multifunctional purine catabolism enzyme fatty acid metabolism–immunity nexus (FAMIN) serves as a biochemical immune checkpoint by dampening DC-mediated T cell priming. Genetic inactivation of FAMIN in DCs accelerates antigen presentation downstream of cytosolic acidification resulting from increased NADH production by inosine monophosphate (IMP) dehydrogenases 1 and 2 (IMPDH1/2). FAMIN inactivation also abolishes DC secretion of inosine, which normally signals through adenosine receptors on T cells to depress T cell activation during priming. A2AR, adenosine A2A receptor; ADSL, adenylosuccinate lyase; ADSS, adenylosuccinate synthetase; AMPD, AMP deaminase; APRT, adenine phosphoribosyltransferase; GMPR, GMP reductase; GMPS, GMP synthase; HGPRT, hypoxanthine–guanine phosphoribosyltransferase; MHC I, MHC class I; PNP, purine nucleoside phosphorylase; PRPP, phosphoribosyl pyrophosphate; PurDNP, purine de novo pathway; XMP, xanthine monophosphate.
Extracellular ATP is itself a strong pro-inflammatory signal104 that is often present at extremely high levels in the TME105,106. High concentrations of extracellular ATP normally indicate cell lysis, as ATP can only enter the extracellular milieu in large quantities upon necrosis or other forms of immunogenic cell death. Apoptotic regulatory T cells have been shown to release abundant amounts of ATP and then convert it to adenosine through their own cell-surface CD39 and CD73, thereby causing immunosuppression by their own cell death107. These findings show that, in addition to producing extracellular adenosine, CD39 and CD73 suppress immunity by clearing ATP from the TME.
A provocative report by Saveljeva et al. recently elucidated a complex mechanism by which FAMIN restrains antigen-specific immunity and prevents deleterious autoinflammation upon influenza infection108. Genetic inactivation of FAMIN in dendritic cells enhanced their uptake, MHC class II-mediated presentation and MHC class I-mediated cross-presentation of antigens, resulting in amplified T cell priming. Mechanistically, FAMIN inactivation in dendritic cells had two main effects: first, it increased flux through IMPDH1 and IMPDH2 (possibly by limiting purine nucleobase salvage by preventing FAMIN-mediated PNP activity), thereby increasing the cytosolic NADH-to-NAD+ ratio; this, in turn, caused cytoplasmic acidification that resulted in accelerated vesicle trafficking. Second, it decreased the synthesis and secretion of inosine, which normally dampens T cell activation during dendritic cell priming by signalling through the T cell adenosine A2A receptor in a manner analogous to adenosine (Fig. 3b). Mice with germline dendritic cell-specific FAMIN inactivation displayed enhanced anticancer immunity relative to FAMIN-competent counterparts and rejected syngeneic tumour grafts, establishing FAMIN as a bona fide biochemical immune checkpoint and target for cancer immunotherapy108.
Effects of (d)NTP synthesis inhibition on the nucleoside activation of TLRs
The ribonucleosides guanosine and uridine promote pro-inflammatory signalling through binding to Toll-like receptor 7 (TLR7) and TLR8, respectively109,110. TLRs activate innate immunity pathways upon binding pathogen-associated molecular patterns and damage-associated molecular patterns. Several studies have shown that TLR7 and TLR8, which recognize pathogen-associated molecular patterns and damage-associated molecular patterns in the lysosomal compartment, require guanosine or uridine binding for full activation109,110. Although the effects of nucleotide synthesis inhibitors on TLR7 and TLR8 signalling remain uncharacterized, it is possible that depletion of uridine or guanosine might dampen the activity of TLR7 and TLR8 and suppress the pro-inflammatory responses of dendritic cells or monocytes. This mechanism might partially underlie the immunomodulatory activity of DHODH inhibitors or IMPDH1/2 inhibitors, which could potentially deplete lysosomal uridine or guanosine, respectively.
TS and IMPDH1/2 inhibitors improve response to immune-checkpoint blockade therapy
Some of the most widely used anticancer agents target TS, including 5-FU, pemetrexed and MTX. In 2018, pemetrexed, carboplatin and the anti-PD1 antibody pembrolizumab became the first combination of chemotherapy and immunotherapy to gain FDA approval, based on improved progression-free survival and overall response rate over chemotherapy alone in patients with advanced non-small-cell lung cancer111. In animal models, pemetrexed enhances anticancer immunity and anti-PDL1 antibody efficacy by inducing the immunogenic cell death of cancer cells, promoting the infiltration of favourable immune subsets (such as activated CD8+ T cells) into the tumour and increasing the capacity of T cells to perform OXPHOS, thereby enhancing their effector function112. The mechanistic underpinnings of these observations remain to be fully characterized.
In a functional genomic screen for regulators of antigen presentation in diffuse large B cell lymphoma cells, TS was implicated as a potent negative regulator of MHC class I cell surface expression, and pemetrexed was shown to increase diffuse large B cell lymphoma MHC class I cell surface abundance113. Inducing MHC class I expression in cancer cells promotes anticancer immunity in several preclinical models114–118, but the in vivo relevance of this mechanism for TS inhibitors remains to be validated.
The IMPDH and GMPS inhibitor mizoribine was recently shown to enhance cancer cell immunogenicity by increasing the expression of immunoproteasome subunits and driving MHC class I presentation of cancer-associated neoantigens. In this study, pretreating 4T1 breast cancer cells with mizoribine before engraftment into syngeneic murine hosts resulted in an improved response of mice to anti-PD1 antibody treatment compared to mice with non-pretreated grafts119. However, the study did not address the potential immunosuppressive effects of mizoribine on the anti-PD1 response as mizoribine was not administered to tumour-bearing mice. Therefore, it is unclear whether systemic administration of GTP-depleting agents, such as MPA or mizoribine, can enhance immunotherapy efficacy against pre-existing tumours, and this warrants direct testing.
Dual effects of nucleotide synthesis inhibitors on autoimmunity and cancer
The efficacy of nucleotide synthesis inhibitors in combination with immune-checkpoint blockade appears paradoxical as these agents are also commonly used to treat autoimmune syndromes. For example, MTX, leflunomide and teriflunomide are each approved for the treatment of rheumatoid arthritis and multiple sclerosis, while MTX and MPA are used to prevent organ transplant rejection and graft versus host disease. In autoimmune indications, these therapies are thought to work at least in part by inhibiting autoreactive T cell-mediated tissue destruction, and this is supported by in vitro mechanistic evidence as well as animal and human studies120,121.
The above data raise an obvious question: if these drugs suppress T cell attack on host tissues in autoimmune disease settings, why would they not also impair T cell attack on cancer cells? Although a complete answer to this question is far from clear, multiple human studies have provided insight. In a randomized, double-blind, placebo-controlled study of healthy volunteers, daily teriflunomide treatment for a period of 30 days did not affect the delayed-type hypersensitivity response to recall antigens from Myobacterium tuberculosis and Candida albicans and, although teriflunomide did decrease antibody titre in response to vaccination with a rabies neoantigen, the antibody titre observed in the treatment cohort was well above the minimum threshold required for rabies seroprotection122. This suggests that prolonged teriflunomide treatment does not meaningfully impair the adaptive immune response against certain known pathogen-associated antigens. For MTX, the data is more mixed and suggests that, while MTX does not impair responsiveness to influenza vaccination, it might do so for pneumococcal vaccines123. However, the above data suggest that T cell-dependent immunity is largely preserved during treatment with antifolates or DHODH inhibitors. Further studies are needed to determine if these observations hold true for patients with cancer, who may have systemic immunosuppression from their disease or concurrent therapies.
The recent Teri-DYNAMIC trial (NCT01863888), which examined T cell activity and clonal diversity in patients with multiple sclerosis treated with teriflunomide, suggested that the degree of teriflunomide-mediated T cell functional impairment depends on the affinity of the T cell receptor (TCR) to its cognate antigen–MHC complex for each T cell clonal population124. The authors argue that high-affinity T cell clones, including those that drive autoimmune disease, are more reliant on mitochondrial OXPHOS than their medium-affinity and low-affinity counterparts and are therefore more markedly affected by OXPHOS impairment upon teriflunomide treatment. This was supported by animal models of T cell activation with known varying TCR–antigen affinities, which confirmed that the effector function of T cells with a high TCR–antigen affinity was more markedly inhibited by teriflunomide than that of T cells with lower TCR affinity for the same antigen. Therefore, the metabolic requirements for effector T cell response might vary depending on the T cell affinity for different antigens associated with the host, the cancer or the pathogen.
For these reasons, when considering the addition of nucleotide synthesis inhibitors to immunotherapy regimens, their ability to promote cancer cell immunogenicity must be weighed against their potential inhibitory effects on host immunity, which appear highly context specific and antigen dependent. The clinical use of DHODH and IMPDH inhibitors as therapies for autoimmune syndromes does not preclude their inclusion in combination strategies with immunotherapy, especially given clinical experience with pemetrexed, and the effect of these agents on anticancer immunity and immunotherapy efficacy must be directly tested in immunocompetent animal models to address this open question.
Nucleotide metabolism in metastasis
Metastasis is a key cause of most cancer-related deaths in patients with solid tumors. Metastatic colonization is a multistep process that requires cancer cells to escape from the primary tumour site, survive harsh conditions in the systemic circulation and subsequently establish a new niche in a distal organ. The transcriptional signatures associated with metastasis have been well studied; however, the role of cancer cell metabolism, and of nucleotide metabolism in particular, is still emerging. We highlight recent advances in this area below.
GTP activates prometastatic GTPases
Monomeric GTPase proteins — most prominently RHO GTPase family members — play a crucial role in several processes necessary for cancer cell metastasis, including migration, invasion and extracellular matrix degradation125. These proteins are active only in their GTP-bound ‘on’ state, so one might expect that their activation is dependent on sufficient GTP pools to power guanine nucleotide exchange factors. Indeed, Wawrzyniak et al. showed that depleting GTP pools by overexpressing GMP reductase (GMPR) or by inhibiting IMPDH impairs melanoma migration and invasion in vitro and impairs metastasis in tumour xenograft models by preventing the activation of RHO GTPase family members RHO, RAC and CDC42 (ref. 126) (Fig. 4). Human metastatic melanoma lesions frequently show GMPR downregulation and IMPDH2 upregulation relative to primary tumour tissue, suggesting that GTP pool augmentation drives melanoma metastasis126. Concordantly, Kollareddy et al. showed that oncogenic gain-of-function mutations of p53, which are frequently observed across several cancer types127, promote metastasis by upregulating the expression of de novo GTP synthesis genes128. Mutant (but not wild-type) p53 binds the GMP synthase (GMPS) promoter and drives its transcription, resulting in increased GTP pools. Consistent with these findings, short hairpin RNA-mediated knockdown of GMPS abolished brain metastasis of highly CNS-metastatic human breast cancer xenografts, which correlated with a reduction in the proportion of GTP-bound RAS, RAC1 and CDC42 upon GMPS knockdown in vitro128. These studies highlight the importance of augmented GTP pools in promoting metastasis across diverse cancer types, establishing GTP synthesis as a druggable target to block metastasis.
Fig. 4. Supraphysiological GTP abundance promotes metastasis through the activation of RHO-family GTPases.
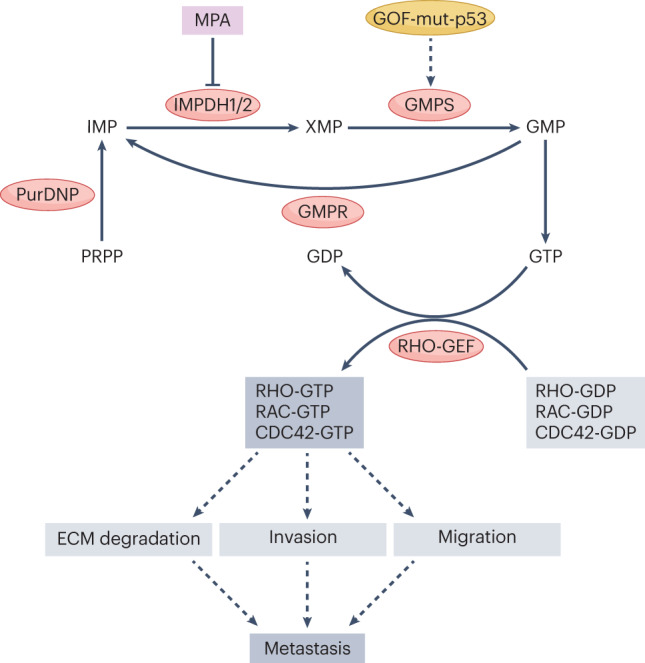
Small GTPase proteins of the RHO family support multiple behaviours necessary for metastasis, including extracellular matrix (ECM) degradation, basement membrane invasion and cell migration. These proteins are active in their GTP-bound state and inactivated by hydrolysis of bound GTP to GDP. Reactivation is catalysed by guanine nucleotide exchange factors (GEFs), which replace bound GDP with a new molecule of GTP. Depletion of GTP by genetic or pharmacological inhibition of inosine monophosphate (IMP) dehydrogenases 1 and 2 (IMPDH1/2) or GMP synthase (GMPS), or by overexpression of GMP reductase (GMPR), inhibits RHO-family GTPase activity by decreasing the GTP-bound fraction and blocks cancer metastasis in various animal models. GOF-mut-p53, gain-of-function mutant p53; MPA, mycophenolic acid; PRPP, phosphoribosyl pyrophosphate; PurDNP, purine de novo pathway; XMP, xanthine monophosphate.
DHODH as a metastatic driver
Inhibition of the pyrimidine de novo pathway was recently shown to abolish liver metastasis in murine models of colorectal cancer129; in this study, Yamaguchi et al. generated patient-derived xenografts (PDXs) from patients with colorectal cancer and compared the metabolic profile of PDX tissues that efficiently metastasized to the mouse liver against those that did not. Pyrimidine de novo pathway intermediates were heavily enriched in metastatic PDX tissues, and inhibition of DHODH using leflunomide treatment consistently diminished liver metastasis in this system, an effect the authors attributed to the disruption of proliferation of liver metastatic colonies. However, it is worth noting that partial loss-of-function germline mutations in DHODH are the cause of Miller syndrome, a genetic disorder characterized by craniofacial malformations due to failure of normal pharyngeal arch migration during embryonic development130. Given its role in cell migration during development, DHODH might have important roles in cell migration during cancer metastasis, which warrants further study.
Nucleotide metabolism and therapy resistance
Cancer therapy resistance, defined as the failure of medical interventions to eradicate a tumour or control tumour growth, can be viewed as the cause of most cancer deaths. Nucleotide metabolism has been implicated in resistance to numerous cancer therapies, including genotoxic chemotherapy, radiotherapy and targeted agents (as well as immunotherapy, as discussed above); we discuss this below.
Molecular competition of endogenous nucleotides with nucleoside analogue chemotherapy drugs
Gemcitabine, or 2′,2′-difluorodeoxycytidine, is a deoxynucleoside analogue prodrug that must pass through the pyrimidine nucleoside salvage pathway for activation. After uptake by nucleoside transporters, gemcitabine is phosphorylated by DCK to form gemcitabine monophosphate and then further phosphorylated to form gemcitabine diphosphate and gemcitabine triphosphate, which exert their toxicity by inhibiting RNR or incorporating into DNA and causing chain termination, respectively49.
Gemcitabine metabolites compete with their endogenous deoxycytidylate counterparts (Fig. 5a) and the toxicity of gemcitabine is related to the ratio of gemcitabine-derived metabolites to endogenous dNTPs in target cells. Shukla et al. showed that acquired resistance to gemcitabine in PDAC cells is caused by increased deoxyCTP (dCTP) pools downstream of mucin 1 (MUC1) and hypoxia-inducible factor 1α (HIF1α), which funnel glucose through the non-oxidative pentose phosphate pathway to provide PRPP for de novo pyrimidine synthesis and enhance CTP synthetase activity to increase dCTP pools (Fig. 5a). In murine models using gemcitabine-resistant PDAC cell lines or gemcitabine-naive PDXs, inhibition of DHODH with leflunomide treatment or inhibition of HIF1α by digoxin treatment restored gemcitabine sensitivity in PDAC tumours by depleting dCTP in cancer cells11. These results have prompted a phase II clinical trial testing the addition of digoxin to standard-of-care chemotherapy in patients with resectable PDAC (NCT04141995).
Fig. 5. Hyperactive nucleotide synthesis confers resistance to a range of therapeutic interventions.

a, Gemcitabine, or 2′,2′-difluorodeoxycytidine (dFdC), is in molecular competition with endogenous deoxycytidylate species at every stage of its metabolism, from uptake to phosphorylation to incorporation into elongating nascent DNA. The efficacy of gemcitabine can therefore be enhanced by depleting cellular deoxycytidylate nucleotides through either dihydroorotate dehydrogenase (DHODH) inhibition or disruption of oncogenic signalling through mucin 1 (MUC1) and hypoxia-inducible factor 1α (HIF1α) as this increases the proportion of gemcitabine-derived nucleotides relative to their endogenous counterparts. b, Various cytotoxic anticancer therapies operate by causing double-strand breaks (DSBs) in DNA. Adaptive resistance to these therapies emerges downstream of hyperactive DSB repair, which requires large amounts of deoxynucleotide triphosphates (dNTPs). Inhibition of dNTP synthesis therefore enhances the efficacy of these therapies in many contexts. c, Aside from its role in pyrimidine synthesis, DHODH also opposes ferroptosis by neutralizing mitochondrial lipid peroxide radicals through the generation of reduced ubiquinol and opposes apoptosis by contributing to mitochondrial membrane potential. DHODH inhibition therefore sensitizes cancer cells to ferroptosis and apoptosis-inducing agents such as GPX4 inhibitors and TNF-related apoptosis-inducing ligand (TRAIL), respectively. ΔΨm, mitochondrial membrane potential; 5-FU, 5-fluorouracil; BQ, brequinar; CAD, carbamoyl phosphate synthetase II, aspartate transcarbamoylase, dihydroorotase; CAF, cancer-associated fibroblast; CMPK, cytidine monophosphate kinase; CTPS, cytidine triphosphate synthase; dC, deoxycytidine; dCDP, deoxycytidine phosphate; DCK, deoxycytidine kinase; dCMP, deoxycytidine monophosphate; dCTP, deoxycytidine triphosphate; dFdCDP, gemcitabine diphosphate; dFdCMP, gemcitabine monophosphate; dFdCTP, gemcitabine triphosphate; DHO, dihydroorotate; dTTP, deoxythymidine triphosphate; ENT1/2, equilibrative nucleoside transporters 1 and 2; G6P, glucose 6-phosphate; GSH, reduced glutathione; GSSG, oxidized glutathione; IMM, inner mitochondrial membrane; Lef, leflunomide; MPA, mycophenolic acid; NDPK, nucleoside-diphosphate kinase; NonOx PPP, non-oxidative pentose phosphate pathway; Pem, pemetrexed; PRPP, phosphoribosyl pyrophosphate; RNR, ribonucleotide reductase; TAM, tumour-associated macrophage; UMP, uridine monophosphate; UMPS, UMP synthase; UTP, uridine triphosphate.
The same principle of molecular competition of endogenous nucleotides with nucleoside analogue therapies was more recently observed for decitabine (5-aza-2′-deoxycytidine) and azacytidine (5-azacytidine), which are used to treat AML and myelodysplastic syndrome. Like gemcitabine, decitabine and azacytidine require activation by the pyrimidine nucleoside salvage pathway and ultimately compete with endogenous dCTP for incorporation into DNA. Using primary blood and bone marrow samples from patients with myelodysplastic syndrome engrafted in immunodeficient mice, Kayamori et al. showed that combination treatment with decitabine and the DHODH inhibitor PTC299 lowered disease burden and prolonged mouse survival compared to treatment with either agent alone131. A phase I trial testing the combination of the DHODH inhibitor JNJ-74856665 and azacytidine in myeloid malignancies is ongoing (NCT04609826).
Gemcitabine competes with deoxycytidine for phosphorylation by DCK. In orthotopic murine models of PDAC, cancer cells induce tumour-associated macrophages to secrete deoxycytidine, which diminishes cancer cell uptake and phosphorylation of gemcitabine by direct competition, thus conferring resistance to gemcitabine132 (Fig. 5a). Similarly, in PDAC cell culture and organoid models, cancer-associated fibroblasts have also been shown to secrete deoxycytidine and promote gemcitabine resistance133, and this mechanism likely contributes to gemcitabine resistance in human PDAC. In a PDAC mouse model, systemic elimination of myeloid cells restored gemcitabine sensitivity, implicating the exchange of deoxycytidine from myeloid cells to cancer cells in the TME as a key mediator of gemcitabine resistance in PDAC132.
The alkylating agent temozolomide is a standard-of-care chemotherapy agent for GBM that exerts its toxicity by alkylating guanine residues in DNA and RNA to form O6-methylguanine (O6-MG). Following the catabolism of DNA and RNA, O6-MG can be salvaged by HGPRT, ultimately forming (d)O6-methylguanosine triphosphate that can be reincorporated into DNA and RNA, causing further damage. Thus, O6-MG species are in molecular competition with their endogenous guanylate counterparts for incorporation into DNA and RNA. Shireman et al. recently showed that, upon temozolomide treatment, GBM cells increase their use of the purine de novo pathway and dampen their use of the purine nucleobase salvage pathway by epigenetically downregulating the expression of salvage pathway enzymes134. This causes decreased O6-MG salvage and thereby mitigates temozolomide-mediated DNA damage. Consistent with this mechanism, combination treatment with the IMPDH inhibitor MPA and temozolomide resulted in greater DNA damage in GBM cells in vitro and prolonged survival in GBM PDX models compared to temozolomide monotherapy134.
Pyrimidine dNTP depletion impairs DNA repair in response to genotoxic chemotherapy
DHODH inhibition can improve cancer cell sensitivity to genotoxic agents even if they are not nucleoside analogues that show direct molecular competition with (d)NTPs. For example, triple-negative breast cancer cells upregulate flux through the pyrimidine de novo pathway in response to doxorubicin or cisplatin9 in vitro, resulting in increased dCTP and dTTP pools that facilitate DNA repair under genotoxic stress. Consistently, combination treatment with leflunomide and doxorubicin outperformed either single agent in a triple-negative breast cancer xenograft model9. Independent studies reported similar benefits for brequinar and doxorubicin treatment in a melanoma xenograft model135. DHODH inhibitors have also been shown to improve the efficacy of other genotoxic agents, including the combination of cisplatin plus etoposide in murine models of small-cell lung cancer2 and oxaliplatin in cell culture models of PDAC5, suggesting that pyrimidine dNTP upregulation is crucial for handling genotoxic stress across multiple cancers.
(d)NTPs promote resistance to radiation therapy
Radiation therapy is an effective treatment for many cancers, although for some aggressive cancers, such as GBM, relapse and subsequent disease progression are common following treatment. Metabolic profiling of a large panel of GBM cell lines showed that purine nucleotide abundance, and particularly the abundance of (d)GTP, correlates strongly with resistance to radiation therapy136. Accordingly, depletion of (d)GTP by MPA sensitized resistant GBM cell lines to radiation therapy by preventing the repair of radiation-induced DNA double-strand breaks (DSBs); conversely, supplementation of purines conferred resistance to radiation therapy by accelerating DSB repair. The radiosensitizing activity of MPA was robustly observed across flank and orthotopic PDX models of GBM136; these results, along with other studies, have justified an ongoing phase I trial testing the addition of MPA to standard-of-care radiation therapy (with optional addition of temozolomide) in patients with GBM or gliosarcoma (NCT04477200).
Radiation therapy is also used for the treatment of borderline-resectable PDAC tumours to make them resectable. In cultured pancreatic cancer cells, increased cellular nucleotide pools downstream of MUC1 overexpression impart resistance to radiation, and supplementing exogenous nucleosides diminishes radiation-induced DNA damage137.
Conceptually, the combination of nucleotide depletion with genotoxic chemotherapy or radiation therapy differs from combining multiple genotoxic agents or multiple genotoxic agents plus radiation therapy, as nucleotide depletion deprives cells of the dNTPs necessary for DNA repair (Fig. 5b). It might be more rational to combine DSB-inducing agents with nucleotide synthesis inhibitors than with other DSB-inducing agents as it appears that resistance to these interventions is not caused by a lack of DSB formation but is instead due to hyperactive DSB repair, which requires increased dNTP pools136.
(d)NTP synthesis inhibitors synergise with inhibitors of oncogenic signalling kinases
As mentioned above, flux through the pyrimidine de novo pathway is increased downstream of various oncogenic signalling pathways8,18,26,28. Downstream of PI3K or MAPK pathway activation, CAD activity is accelerated by its phosphorylation by S6K (on Ser1869)23 or ERK (on Thr456)138, respectively. Accordingly, GBM tumours with PTEN loss or EGFR amplification show increased CADS1869 or CADT456 phosphorylation, respectively. Combination treatment with leflunomide and either a PI3K or EGFR inhibitor (based on the genetic background) led to enhanced pyrimidine depletion in vitro and prolonged survival in a GBM xenograft model, showing that redundant targeting of pyrimidine de novo synthesis (that is, inhibition of both DHODH and upstream oncogenic signalling that upregulates flux through the pathway) is efficacious in certain settings7.
DHODH inhibitors have also shown synergy with kinase inhibitors, such as everolimus and palbociclib, in KRAS-mutant solid tumours5, although the precise mechanism for these results remains to be determined. It is likely that nucleotide depletion can cooperate with disruption of oncogenic signalling in a variety of genetic contexts; the full landscape of these relationships remains to be characterized.
(d)NTP synthesis inhibitors synergise with cell death inducers
Mao et al. recently reported that DHODH inhibitors sensitize cancer cells to several ferroptosis-inducing agents through a mechanism independent of pyrimidine nucleotide depletion, as restoration of pyrimidine pools with uridine supplementation did not rescue sensitization to ferroptosis139. DHODH supplies reducing power to the inner mitochondrial membrane in the form of reduced ubiquinol, an obligate DHODH reaction product, and therefore limits the peroxidation of lipids in the inner mitochondrial membrane — a key step in ferroptosis (Fig. 5c). Consequently, a combination of GPX4 inhibitors and brequinar resulted in profound induction of cancer cell ferroptosis in vitro in a manner dependent on mitochondrial lipid peroxidation, and a combination of brequinar and sulfasalazine (a clinically approved drug with ferroptosis-inducing activity) synergistically limited tumour growth in various tumour xenograft systems139.
TNF-related apoptosis-inducing ligand (TRAIL) has been investigated as an anticancer therapy based on its ability to induce cancer cell apoptosis140. Inhibition or knockdown of DHODH sensitizes cancer cells to TRAIL-induced apoptosis in vitro in a manner that is rescuable by uridine supplementation, suggesting a mechanism dependent on pyrimidine nucleotide depletion141. DHODH inhibitors have been reported to decrease mitochondrial membrane potential5,82, the maintenance of which plays a crucial role in opposing apoptosis142. Therefore, it is possible that DHODH inhibitors could cooperate with apoptosis-inducing agents such as venetoclax and other BCL-2 antagonists (Fig. 5c). A phase I trial investigating the combination of the DHODH inhibitor JNJ-74856665 and venetoclax for myeloid malignancies is ongoing (NCT04609826).
Resistance to nucleotide synthesis inhibitors
There are three possible mechanisms that could explain the resistance of cancer cells to nucleotide synthesis inhibitors. First, cancer cells might execute oncogenic growth despite severe (d)NTP shortage. Second, cancer cells might acquire sufficient (d)NTPs to enable disease progression despite inhibition of key de novo pathway enzymes. Finally, the degree and timing of nucleotide depletion in vivo might be insufficient to cause cancer cell nucleotide starvation, either because of insufficient target engagement or a suboptimal dose regimen. The first possibility can be ruled out as there is no existing data to support this hypothesis, and (d)NTPs are strictly required for the DNA and RNA synthesis necessary for a cell to duplicate. The third possibility, for which there are plausible examples (discussed below), represents a problem of pharmacokinetics rather than cell-autonomous resistance. Therefore, we first focus on the second possibility and highlight evidence for this category of resistance mechanism.
Nucleoside salvage and resistance
Because nucleoside salvage pathways can supply nucleotides independently of de novo synthesis pathways, salvage pathways might be expected to confer resistance to de novo pathway inhibitors. Pyrimidine nucleoside salvage requires far less ATP and no additional input of aspartate or glutamine as compared to the de novo synthesis. Therefore, it might be favourable for cancer cells to maximize their salvage pathway utilization to conserve energy and biomass for other anabolic processes. Indeed, isotopic labelling experiments have shown that cancer cells preferentially rely on pyrimidine nucleoside salvage for dNTP synthesis, as the contribution of nucleoside salvage to pyrimidine residues in DNA is generally greater than 50% and can be as high as 80% (ref. 143). Supplementation of cells with supraphysiological uridine concentrations completely rescues the proliferation rate under DHODH inhibition, even for cell lines with relatively low basal pyridine nucleoside salvage activity143, showing that enforced flux through the pyrimidine nucleoside salvage pathway by mass action can rescue cell growth under ablation of the de novo pathway; however, the extent to which this occurs under conditions of physiological extracellular nucleoside abundance, especially in vivo, was not explored in this study.
The ability of nucleoside salvage to compensate for blockade of de novo synthesis depends on which node is targeted. For example, RNR inhibition can be bypassed by DCK-mediated and TK1/2-mediated salvage of deoxynucleosides but not by salvage of ribonucleosides or nucleobases. TS inhibition can only be bypassed by TK1 and TK2. The bypass strategies for various metabolites under DHODH or IMPDH inhibition are complex and can be inferred from Fig. 1.
Nucleoside uptake blockade can overcome resistance to de novo pathway inhibitors
Many studies have reported in vitro synergism between brequinar and dipyridamole, an inhibitor of equilibrative nucleoside transporter 1 (ENT1) and ENT2 (refs. 143–146), which are thought to be the main mediators of nucleoside influx147. However, the preclinical in vivo performance of this combination strategy has been mixed, with, at best, modest improvement over brequinar alone, although this may be influenced by the impressive efficacy of brequinar monotherapy in these model systems144,145. The poor performance of dipyridamole could be a pharmacokinetic problem as it is very rapidly cleared from the plasma148. Because of this, ongoing clinical trials involving dipyridamole for COVID-19 (NCT05166876 and NCT04391179) call for administration three or four times a day, whereas preclinical studies in cancer thus far have used daily administration, which likely results in periods when dipyridamole exposure is insufficient to effectively block ENT-mediated nucleoside uptake. Tumour-specific ENT1 knockout was recently shown to dramatically enhance DHODH inhibitor efficacy in an aggressive, brequinar-resistant mouse model of PDAC149, supporting the notion that the combined blockade of ENT1 and DHODH is efficacious and arguing that the short half-life of dipyridamole could explain its mixed performance in other studies144,145. Thus, the in vivo potential of combined pharmacological inhibition of DHODH and ENT-mediated nucleoside salvage remains to be rigorously evaluated with a long-acting ENT inhibitor.
The importance of nucleoside salvage as a resistance mechanism against inhibition of de novo synthesis is further underscored by studies demonstrating sufficient nucleoside and nucleobase levels in plasma22 and tumour interstitial fluid150 to support nucleoside salvage in cancer cells. As previously mentioned, stromal cells in the TME, including tumour-associated macrophages and cancer-associated fibroblasts, can secrete abundant nucleosides and nucleobases132,133, and this might contribute to compensatory tumour nucleoside salvage pathway flux under inhibition of de novo synthesis in vivo.
Targeting nucleoside salvage enzymes can overcome de novo pathway inhibitor resistance
Nucleoside salvage pathway enzymes have been implicated in DHODH inhibitor resistance. Li et al. argue that low expression of the enzyme deoxycytidine deaminase (DCTD), which converts deoxycytidine monophosphate (dCMP) to dUMP, explains the sensitivity of genetically engineered mouse models of small-cell lung cancer to brequinar compared to genetically engineered mouse models of PDAC and lung adenocarcinoma, which have relatively higher DCTD expression2. DCTD produces dUMP, which otherwise becomes limiting upon DHODH inhibition; accordingly, knockout of DCTD enhanced brequinar sensitivity in cell lines derived from PDAC and lung adenocarcinoma mouse models2. In a similar example, DCK is required to maintain dCTP pools upon inhibition of RNR in B-ALL cells60; the combined inhibition of RNR and DCK was shown to deplete dCTP in vitro and eradicate engrafted B-ALL in vivo when combined with ATR inhibition by inducing lethal replication stress60.
Nucleotide recycling by autophagy-mediated degradation of RNA
In addition to the de novo and salvage pathways, cells can obtain nucleotides through autophagy — specifically, the degradation of cellular nucleic acids in autophagosomes. This process yields (d)NMPs and does not depend on the uptake of extracellular nucleosides or on phosphorylation or phosphoribosylation of nucleosides or nucleobases by nucleoside salvage enzymes. It was recently discovered that the depletion of either purine or pyrimidine NTPs induces autophagy in HEK-293T cells151. Purine depletion activated autophagy by antagonizing mTOR activation, whereas pyrimidine depletion did so through an apparently mTOR-independent mechanism. These results suggest that NTP abundance has divergent roles in regulating autophagy151 and further work is needed to understand if these mechanisms are operative in cancer.
PDAC and other KRAS-driven malignancies are conditionally dependent on autophagy, especially in hypoxic and nutrient-poor regions of the TME152. However, the specific autophagy-derived substrates that enable cancer cell survival under starvation conditions are incompletely defined. Isotopic labelling pulse-chase experiments using cell lines from KRAS-driven genetically engineered mouse models of non-small-cell lung cancer have shown that autophagy-derived amino acids and four carbon units can sustain de novo purine and pyrimidine synthesis under nutrient starvation and that nucleoside supplementation rescues cell death of autophagy-incompetent cells under the same conditions153. In Caenorhabditis elegans, it has been shown that autophagic degradation of rRNA in lysosomes (termed ‘ribophagy’) by the T2 endonuclease rnst-2 (a homologue of human ribonuclease T2) is required to prevent embryonic lethality in worm embryos with loss-of-function mutations in de novo pyrimidine synthesis enzymes, suggesting that ribophagy can prevent lethal nucleotide starvation in certain contexts154. The role of autophagy and ribophagy in maintaining nucleotide pools in human cancer cells to confer resistance to purine or pyrimidine de novo pathway inhibitors remains to be explored.
Evaluating nucleotide synthesis inhibitors
Dose regimen
Inhibitors of metabolic enzymes are fundamentally different from alkylating chemotherapies such as platinum-based agents, cyclophosphamide, temozolomide and busulfan. For alkylating agents, the cumulative drug exposure over time (the area under the curve) is proportional to anticancer efficacy and patient toxicity, reflecting the fact that these drugs covalently bind DNA and other molecules and are thus physically expended by exerting their toxicity. Clinical experience has shown that infrequent administration of the maximum tolerated dose (MTD), followed by a period of recovery, is generally optimal for maximizing therapeutic effects and minimizing toxicity with these compounds. However, for nucleotide synthesis inhibitors, this logic does not apply, as there is likely a systemic exposure threshold above which the target enzyme is effectively inhibited and below which the target pathway is not blockaded at all. The cumulative time during which the target pathway is inhibited, rather than the total drug exposure per se, is likely to be the parameter that determines efficacy and toxicity. Infrequent administration of the MTD for nucleotide synthesis inhibitors likely results in far higher drug exposure than is necessary to inhibit the target initially, increasing the chance of off-target toxicity, followed by long periods during which the target pathway is uninhibited. The duration of this period is dependent on the dosing schedule and half-life of the drug.
In the early 1990s, the infrequent MTD dosing paradigm was applied in clinical trials of brequinar monotherapy in several cancer settings, which uniformly failed owing to a lack of efficacy12–15. Weekly administration of brequinar, which has a half-life of around 10–12 hours at clinically tested doses155, almost certainly resulted in subtherapeutic drug levels for several days between doses. Thus, in hindsight, the negative results of these trials must be interpreted with caution, and future trials involving nucleotide synthesis inhibitors must carefully consider pharmacokinetic parameters when designing their dosage regimen to avoid similar potential pitfalls. Encouragingly, ongoing and recently completed human trials of brequinar and other DHODH inhibitors for haematological cancers (NCT04609826, NCT02509052 and NCT05246384) and COVID-19 (NCT04575038) have supported the safety of a daily dose schedule in these settings and, as mentioned previously, prolonged daily administration of teriflunomide was shown to be safe without clinically significant immunosuppression in a randomized trial of healthy volunteers122.
Target engagement monitoring
Longitudinal quantification of plasma biomarkers for target enzyme inhibition (for example, monitoring dihydroorotate or other upstream metabolites for DHODH inhibitors) is a useful non-invasive means of ensuring sustained pharmacodynamic target engagement in vivo. When possible, a direct comparison of pre-treatment and on-treatment levels of these biomarkers would provide a powerful corroboration of target enzyme inhibition. This may be clinically possible for AML and other haematological malignancies for which serial cancer cell sampling from the peripheral blood is feasible in some patients. For preclinical animal studies, metabolomic profiling of tumour tissue harvested at necropsy is a suitable alternative. Metabolic imaging modalities that enable tissue localization of exogenous metabolites, such as PET tracing of 18F-labelled metabolites and MRI tracing of 13C-labelled metabolites, represent another powerful tool to characterize the tumour and systemic host response to nucleotide synthesis inhibitors156,157.
Conclusion
Deranged nucleotide metabolism has a substantial footprint on virtually all malignant cell activities across diverse cancer types and thus represents a pan-cancer metabolic dependency. In addition to their canonical role as substrates for nucleic acid biogenesis, nucleotides fuel a wide array of processes that are hyperactive in cancer cells. Therefore, nucleotide synthesis inhibitors have tremendous potential as elements of combination therapy strategies that has yet to be fully realized. This Review has compiled an illustrative but by no means exhaustive set of combination therapy concepts that have emerged from recent preclinical advances and await further validation (Table 1). In conjunction with hypothesis-driven investigations, the full landscape of nucleotide depletion-induced cancer cell dependencies can now be elucidated with the use of genome-wide functional genomic screening technologies, and these urgently needed studies will undoubtedly uncover additional cancer-essential functions of nucleotide metabolism reprogramming.
Ongoing and future clinical investigations involving nucleotide synthesis inhibitors will benefit from the standardization of dose regimens based on rigorous pharmacokinetic and pharmacodynamic characterization of these agents. Drug development campaigns will likely generate more potent and selective inhibitors of existing clinical drug targets, such as DHODH, TS, RNR, CD73 and ENT1/2, and will likely bring other key enzymes, such as DCK, UCK and CDA, under clinical pharmacological control. The medical impact of these developments will reverberate beyond oncology and will open new avenues for the treatment of infection, autoimmune disease, neurodegenerative syndromes and other diseases.
Acknowledgements
These studies are supported by F30CA265277 to N.J.M. and R01CA163649, R01CA270234, R01CA210439, R01CA216853, R01CA256911 and U54CA274329 to P.K.S. from the National Cancer Institute of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
N.J.M. and P.K.S. performed the literature search and developed the outline. N.J.M. developed the initial draft. P.K.S. edited the manuscript, added further content and provided mentoring and supervision.
Peer review
Peer review information
Nature Reviews Cancer thanks Natalia Tretyakova, Caius Radu, Costas Lyssiotis and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Biancur DE, et al. Functional genomics identifies metabolic vulnerabilities in pancreatic cancer. Cell Metab. 2021;33:199–210. doi: 10.1016/j.cmet.2020.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li L, et al. Identification of DHODH as a therapeutic target in small cell lung cancer. Sci. Transl Med. 2019;11:517. doi: 10.1126/scitranslmed.aaw7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu XG, et al. Functional genomics in vivo reveal metabolic dependencies of pancreatic cancer cells. Cell Metab. 2021;33:211–221. doi: 10.1016/j.cmet.2020.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sykes DB, et al. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell. 2016;167:171–186.e15. doi: 10.1016/j.cell.2016.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koundinya M, et al. Dependence on the pyrimidine biosynthetic enzyme DHODH is a synthetic lethal vulnerability in mutant KRAS-driven cancers. Cell Chem. Biol. 2018;25:705–717.e11. doi: 10.1016/j.chembiol.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 6.White RM, et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471:518–522. doi: 10.1038/nature09882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, et al. Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells. Sci. Transl. Med. 2019;11:eaau4972. doi: 10.1126/scitranslmed.aau4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santana-Codina N, et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018;9:4945. doi: 10.1038/s41467-018-07472-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown KK, Spinelli JB, Asara JM, Toker A. Adaptive reprogramming of de novo pyrimidine synthesis is a metabolic vulnerability in triple-negative breast cancer. Cancer Discov. 2017;7:391–399. doi: 10.1158/2159-8290.CD-16-0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mathur D, et al. PTEN regulates glutamine flux to pyrimidine synthesis and sensitivity to dihydroorotate dehydrogenase inhibition. Cancer Discov. 2017;7:380–390. doi: 10.1158/2159-8290.CD-16-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shukla SK, et al. MUC1 and HIF-1α signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell. 2017;32:71–87.e7. doi: 10.1016/j.ccell.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maroun J, et al. Multicenter phase II study of brequinar sodium in patients with advanced lung cancer. Cancer Chemother. Pharmacol. 1993;32:64–66. doi: 10.1007/BF00685878. [DOI] [PubMed] [Google Scholar]
- 13.Moore M, et al. Multicenter phase II study of brequinar sodium in patients with advanced gastrointestinal cancer. Invest. New Drugs. 1993;11:61–65. doi: 10.1007/BF00873913. [DOI] [PubMed] [Google Scholar]
- 14.Natale R, et al. Multicenter phase II trial of brequinar sodium in patients with advanced melanoma. Ann. Oncol. 1992;3:659–660. doi: 10.1093/oxfordjournals.annonc.a058298. [DOI] [PubMed] [Google Scholar]
- 15.Cody R, et al. Multicenter phase II study of brequinar sodium in patients with advanced breast cancer. Am. J. Clin. Oncol. 1993;16:526–528. doi: 10.1097/00000421-199312000-00014. [DOI] [PubMed] [Google Scholar]
- 16.Lane AN, Fan TW-M. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015;43:2466–2485. doi: 10.1093/nar/gkv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tong X, Zhao F, Thompson CB. The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr. Opin. Genet. Dev. 2009;19:32–37. doi: 10.1016/j.gde.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villa E, Ali ES, Sahu U, Ben-Sahra I. Cancer cells tune the signaling pathways to empower de novo synthesis of nucleotides. Cancers. 2019;11:688. doi: 10.3390/cancers11050688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, et al. Identification and characterization of human uracil phosphoribosyltransferase (UPRTase) J. Hum. Genet. 2007;52:415–422. doi: 10.1007/s10038-007-0129-2. [DOI] [PubMed] [Google Scholar]
- 20.Pérignon JL, Bories DM, Houllier AM, Thuillier L, Cartier PH. Metabolism of pyrimidine bases and nucleosides by pyrimidine-nucleoside phosphorylases in cultured human lymphoid cells. Biochim. Biophys. Acta. 1987;928:130–136. doi: 10.1016/0167-4889(87)90113-3. [DOI] [PubMed] [Google Scholar]
- 21.Ferraro P, Franzolin E, Pontarin G, Reichard P, Bianchi V. Quantitation of cellular deoxynucleoside triphosphates. Nucleic Acids Res. 2010;38:e85. doi: 10.1093/nar/gkp1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Traut TW. Physiological concentrations of purines and pyrimidines. Mol. Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 23.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339:1323–1328. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stillman B. Deoxynucleoside triphosphate (dNTP) synthesis and destruction regulate the replication of both cell and virus genomes. Proc. Natl Acad. Sci. USA. 2013;110:14120–14121. doi: 10.1073/pnas.1312901110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawada K, Toda K, Sakai Y. Targeting metabolic reprogramming in KRAS-driven cancers. Int. J. Clin. Oncol. 2017;22:651–659. doi: 10.1007/s10147-017-1156-4. [DOI] [PubMed] [Google Scholar]
- 26.Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer. 2020;20:74–88. doi: 10.1038/s41568-019-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong Y, Tu R, Liu H, Qing G. Regulation of cancer cell metabolism: oncogenic MYC in the driver’s seat. Signal. Transduct. Target. Ther. 2020;5:124. doi: 10.1038/s41392-020-00235-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y-C, et al. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS ONE. 2008;3:e2722. doi: 10.1371/journal.pone.0002722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldstone DC, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480:379–382. doi: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- 30.Franzolin E, et al. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc. Natl Acad. Sci. USA. 2013;110:14272–14277. doi: 10.1073/pnas.1312033110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farber S, Diamond LK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 1948;238:787–793. doi: 10.1056/NEJM194806032382301. [DOI] [PubMed] [Google Scholar]
- 32.Waltham MC, Holland JW, Robinson SC, Winzor DJ, Nixon PF. Direct experimental evidence for competitive inhibition of dihydrofolate reductase by methotrexate. Biochem. Pharmacol. 1988;37:535–539. doi: 10.1016/0006-2952(88)90225-0. [DOI] [PubMed] [Google Scholar]
- 33.Cronstein BN, Aune TM. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat. Rev. Rheumatol. 2020;16:145–154. doi: 10.1038/s41584-020-0373-9. [DOI] [PubMed] [Google Scholar]
- 34.Friedman B, Cronstein B. Methotrexate mechanism in treatment of rheumatoid arthritis. Joint Bone Spine. 2019;86:301–307. doi: 10.1016/j.jbspin.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neradil J, Pavlasova G, Veselska R. New mechanisms for an old drug; DHFR- and non-DHFR-mediated effects of methotrexate in cancer cells. Klin. Onkol. 2012;25:2S87–92. [PubMed] [Google Scholar]
- 36.Sramek M, Neradil J, Sterba J, Veselska R. Non-DHFR-mediated effects of methotrexate in osteosarcoma cell lines: epigenetic alterations and enhanced cell differentiation. Cancer Cell Int. 2016;16:14. doi: 10.1186/s12935-016-0289-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peters GJ, et al. Induction of thymidylate synthase as a 5-fluorouracil resistance mechanism. Biochim. Biophys. Acta. 2002;1587:194–205. doi: 10.1016/S0925-4439(02)00082-0. [DOI] [PubMed] [Google Scholar]
- 38.Shih C, et al. LY231514, a pyrrolo[2,3-d]pyrimidine-based antifolate that inhibits multiple folate-requiring enzymes. Cancer Res. 1997;57:1116–1123. [PubMed] [Google Scholar]
- 39.Adjei AA. Pharmacology and mechanism of action of pemetrexed. Clin. Lung Cancer. 2004;5:S51–S55. doi: 10.3816/CLC.2004.s.003. [DOI] [PubMed] [Google Scholar]
- 40.McLean JE, Neidhardt EA, Grossman TH, Hedstrom L. Multiple inhibitor analysis of the brequinar and leflunomide binding sites on human dihydroorotate dehydrogenase. Biochemistry. 2001;40:2194–2200. doi: 10.1021/bi001810q. [DOI] [PubMed] [Google Scholar]
- 41.Ransom JT. Mechanism of action of mycophenolate mofetil. Ther. Drug Monit. 1995;17:681–684. doi: 10.1097/00007691-199512000-00023. [DOI] [PubMed] [Google Scholar]
- 42.Yokota S. Mizoribine: mode of action and effects in clinical use. Pediatr. Int. 2002;44:196–198. doi: 10.1046/j.1328-8067.2002.01536.x. [DOI] [PubMed] [Google Scholar]
- 43.Naffouje R, et al. Anti-tumor potential of IMP dehydrogenase inhibitors: a century-long story. Cancers. 2019;11:1346. doi: 10.3390/cancers11091346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elledge SJ, Zhou Z, Allen JB. Ribonucleotide reductase: regulation, regulation, regulation. Trends Biochem. Sci. 1992;17:119–123. doi: 10.1016/0968-0004(92)90249-9. [DOI] [PubMed] [Google Scholar]
- 45.Guarino E, Salguero I, Kearsey SE. Cellular regulation of ribonucleotide reductase in eukaryotes. Semin. Cell Dev. Biol. 2014;30:97–103. doi: 10.1016/j.semcdb.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 46.Yarbro JW. Mechanism of action of hydroxyurea. Semin. Oncol. 1992;19:1–10. [PubMed] [Google Scholar]
- 47.Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006;17:v7–v12. doi: 10.1093/annonc/mdj941. [DOI] [PubMed] [Google Scholar]
- 48.Cerqueira NMFSA, Fernandes PA, Ramos MJ. Understanding ribonucleotide reductase inactivation by gemcitabine. Chemistry. 2007;13:8507–8515. doi: 10.1002/chem.200700260. [DOI] [PubMed] [Google Scholar]
- 49.Plunkett W, et al. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin. Oncol. 1995;22:3–10. [PubMed] [Google Scholar]
- 50.Ahmad SI, Kirk SH, Eisenstark A. Thymine metabolism and thymineless death in prokaryotes and eukaryotes. Annu. Rev. Microbiol. 1998;52:591–625. doi: 10.1146/annurev.micro.52.1.591. [DOI] [PubMed] [Google Scholar]
- 51.Houghton JA, Harwood FG, Tillman DM. Thymineless death in colon carcinoma cells is mediated via Fas signaling. Proc. Natl Acad. Sci. USA. 1997;94:8144–8149. doi: 10.1073/pnas.94.15.8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goulian M, et al. Mechanism of thymineless death: 73. Pediatr. Res. 1985;19:756–756. doi: 10.1203/00006450-198507000-00093. [DOI] [Google Scholar]
- 53.Gaillard H, García-Muse T, Aguilera A. Replication stress and cancer. Nat. Rev. Cancer. 2015;15:276–289. doi: 10.1038/nrc3916. [DOI] [PubMed] [Google Scholar]
- 54.Diehl FF, et al. Nucleotide imbalance decouples cell growth from cell proliferation. Nat. Cell Biol. 2022;24:1252–1264. doi: 10.1038/s41556-022-00965-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beyaert M, Starczewska E, Van Den Neste E, Bontemps F. A crucial role for ATR in the regulation of deoxycytidine kinase activity. Biochem. Pharmacol. 2016;100:40–50. doi: 10.1016/j.bcp.2015.11.022. [DOI] [PubMed] [Google Scholar]
- 56.Gong C, et al. ATR-CHK1-E2F3 signaling transactivates human ribonucleotide reductase small subunit M2 for DNA repair induced by the chemical carcinogen MNNG. Biochim. Biophys. Acta. 2016;1859:612–626. doi: 10.1016/j.bbagrm.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 57.Chang L, et al. ATM-mediated serine 72 phosphorylation stabilizes ribonucleotide reductase small subunit p53R2 protein against MDM2 to DNA damage. Proc. Natl Acad. Sci. USA. 2008;105:18519–18524. doi: 10.1073/pnas.0803313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eaton JS, Lin ZP, Sartorelli AC, Bonawitz ND, Shadel GS. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J. Clin. Invest. 2007;117:2723–2734. doi: 10.1172/JCI31604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hubackova S, et al. Replication and ribosomal stress induced by targeting pyrimidine synthesis and cellular checkpoints suppress p53-deficient tumors. Cell Death Dis. 2020;11:110. doi: 10.1038/s41419-020-2224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Le TM, et al. ATR inhibition facilitates targeting of leukemia dependence on convergent nucleotide biosynthetic pathways. Nat. Commun. 2017;8:241. doi: 10.1038/s41467-017-00221-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fordham SE, et al. Inhibition of ATR acutely sensitizes acute myeloid leukemia cells to nucleoside analogs that target ribonucleotide reductase. Blood Adv. 2018;2:1157–1169. doi: 10.1182/bloodadvances.2017015214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Christian S, et al. The novel dihydroorotate dehydrogenase (DHODH) inhibitor BAY 2402234 triggers differentiation and is effective in the treatment of myeloid malignancies. Leukemia. 2019;33:2403–2415. doi: 10.1038/s41375-019-0461-5. [DOI] [PubMed] [Google Scholar]
- 63.Dembitz V, et al. The ribonucleoside AICAr induces differentiation of myeloid leukemia by activating the ATR/Chk1 via pyrimidine depletion. J. Biol. Chem. 2019;294:15257–15270. doi: 10.1074/jbc.RA119.009396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kofuji S, et al. IMP dehydrogenase-2 drives aberrant nucleolar activity and promotes tumorigenesis in glioblastoma. Nat. Cell Biol. 2019;21:1003–1014. doi: 10.1038/s41556-019-0363-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lafita-Navarro MC, et al. Inhibition of the de novo pyrimidine biosynthesis pathway limits ribosomal RNA transcription causing nucleolar stress in glioblastoma cells. PLoS Genet. 2020;16:e1009117. doi: 10.1371/journal.pgen.1009117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu M, Tao Z, Wang S, Jiang Y, Qu M. Suppression of oncogenic protein translation via targeting eukaryotic translation initiation factor 4E overcomes chemo-resistance in nasopharyngeal carcinoma. Biochem. Biophys. Res. Commun. 2019;512:902–907. doi: 10.1016/j.bbrc.2019.03.118. [DOI] [PubMed] [Google Scholar]
- 67.Xi C, et al. Inhibition of eukaryotic translation initiation factor 4E is effective against chemo-resistance in colon and cervical cancer. Biochem. Biophys. Res. Commun. 2018;503:2286–2292. doi: 10.1016/j.bbrc.2018.06.150. [DOI] [PubMed] [Google Scholar]
- 68.Zhou Q, Li T, Price DH. RNA polymerase II elongation control. Annu. Rev. Biochem. 2012;81:119–143. doi: 10.1146/annurev-biochem-052610-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fawal M-A, Jungas T, Davy A. Inhibition of DHFR targets the self-renewing potential of brain tumor initiating cells. Cancer Lett. 2021;503:129–137. doi: 10.1016/j.canlet.2021.01.026. [DOI] [PubMed] [Google Scholar]
- 70.Siddiqui A, et al. Thymidylate synthase maintains the de-differentiated state of triple negative breast cancers. Cell Death Differ. 2019;26:2223–2236. doi: 10.1038/s41418-019-0289-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Makishima M, Okabe-Kado J, Honma Y. Growth inhibition and differentiation induction in human monoblastic leukaemia cells by 1alpha-hydroxyvitamin D derivatives and their enhancement by combination with hydroxyurea. Br. J. Cancer. 1998;77:33–39. doi: 10.1038/bjc.1998.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang H, et al. Disruption of dNTP homeostasis by ribonucleotide reductase hyperactivation overcomes AML differentiation blockade. Blood. 2022;139:3752–3770. doi: 10.1182/blood.2021015108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hanover JA, Chen W, Bond MR. O-GlcNAc in cancer: an oncometabolism-fueled vicious cycle. J. Bioenerg. Biomembr. 2018;50:155–173. doi: 10.1007/s10863-018-9751-2. [DOI] [PubMed] [Google Scholar]
- 74.Dang W, et al. Pharmacological inhibition of dihydroorotate dehydrogenase induces apoptosis and differentiation in acute myeloid leukemia cells. Haematologica. 2018;103:1472–1483. doi: 10.3324/haematol.2018.188185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ferrer CM, Sodi VL, Reginato MJ. O-GlcNAcylation in cancer biology: linking metabolism and signaling. J. Mol. Biol. 2016;428:3282–3294. doi: 10.1016/j.jmb.2016.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nogueira V, Hay N. Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013;19:4309–4314. doi: 10.1158/1078-0432.CCR-12-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chatterjee A, et al. MnTE-2-PyP protects fibroblast mitochondria from hyperglycemia and radiation exposure. Redox Biol. 2022;52:102301. doi: 10.1016/j.redox.2022.102301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Olou AA, King RJ, Yu F, Singh PK. MUC1 oncoprotein mitigates ER stress via CDA-mediated reprogramming of pyrimidine metabolism. Oncogene. 2020;39:3381–3395. doi: 10.1038/s41388-020-1225-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abrego J, et al. GOT1-mediated anaplerotic glutamine metabolism regulates chronic acidosis stress in pancreatic cancer cells. Cancer Lett. 2017;400:37–46. doi: 10.1016/j.canlet.2017.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lebrecht D, Vargas-Infante YA, Setzer B, Kirschner J, Walker UA. Uridine supplementation antagonizes zalcitabine-induced microvesicular steatohepatitis in mice. Hepatology. 2007;45:72–79. doi: 10.1002/hep.21490. [DOI] [PubMed] [Google Scholar]
- 81.Lebrecht D, et al. Uridine supplementation antagonizes zidovudine-induced mitochondrial myopathy and hyperlactatemia in mice. Arthritis Rheum. 2008;58:318–326. doi: 10.1002/art.23235. [DOI] [PubMed] [Google Scholar]
- 82.Fang J, et al. Dihydro-orotate dehydrogenase is physically associated with the respiratory complex and its loss leads to mitochondrial dysfunction. Biosci. Rep. 2013;33:e00021. doi: 10.1042/BSR20120097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weinberg F, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl Acad. Sci. USA. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Son J, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bader JE, Voss K, Rathmell JC. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol. Cell. 2020;78:1019–1033. doi: 10.1016/j.molcel.2020.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chang C-H, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Scott KEN, Cleveland JL. Lactate wreaks havoc on tumor-infiltrating T and NK cells. Cell Metab. 2016;24:649–650. doi: 10.1016/j.cmet.2016.10.015. [DOI] [PubMed] [Google Scholar]
- 88.Ala M. The footprint of kynurenine pathway in every cancer: a new target for chemotherapy. Eur. J. Pharmacol. 2021;896:173921. doi: 10.1016/j.ejphar.2021.173921. [DOI] [PubMed] [Google Scholar]
- 89.Giblett E, Ammann A, Sandman R, Wara D, Diamond L. Nucleoside-phosphorylase deficiency in a child with severely defective T-cell immunity and normal B-cell immunity. Lancet. 1975;305:1010–1013. doi: 10.1016/S0140-6736(75)91950-9. [DOI] [PubMed] [Google Scholar]
- 90.Giblett E, Anderson J, Cohen F, Pollara B, Meuwissen H. Adenosine-deaminase deficiency in two patients with severely impaired cellular immunity. Lancet. 1972;300:1067–1069. doi: 10.1016/S0140-6736(72)92345-8. [DOI] [Google Scholar]
- 91.Cader MZ, et al. FAMIN is a multifunctional purine enzyme enabling the purine nucleotide cycle. Cell. 2020;180:278–295.e23. doi: 10.1016/j.cell.2019.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yasin S, Schulert GS. Systemic juvenile idiopathic arthritis and macrophage activation syndrome: update on pathogenesis and treatment. Curr. Opin. Rheumatol. 2018;30:514–520. doi: 10.1097/BOR.0000000000000526. [DOI] [PubMed] [Google Scholar]
- 93.Ohta A, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl Acad. Sci. USA. 2006;103:13132–13137. doi: 10.1073/pnas.0605251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang J, et al. Tumor-derived adenosine promotes macrophage proliferation in human hepatocellular carcinoma. J. Hepatol. 2021;74:627–637. doi: 10.1016/j.jhep.2020.10.021. [DOI] [PubMed] [Google Scholar]
- 95.Strakhova R, Cadassou O, Cros-Perrial E, Jordheim LP. Regulation of tumor infiltrated innate immune cells by adenosine. Purinergic Signal. 2020;16:289–295. doi: 10.1007/s11302-020-09701-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Novitskiy SV, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood. 2008;112:1822–1831. doi: 10.1182/blood-2008-02-136325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Antonioli L, Blandizzi C, Pacher P, Haskó G. Immunity, inflammation and cancer: a leading role for adenosine. Nat. Rev. Cancer. 2013;13:842–857. doi: 10.1038/nrc3613. [DOI] [PubMed] [Google Scholar]
- 98.Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer. 2017;17:709–724. doi: 10.1038/nrc.2017.86. [DOI] [PubMed] [Google Scholar]
- 99.Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013;19:355–367. doi: 10.1016/j.molmed.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cai XY, et al. High expression of CD39 in gastric cancer reduces patient outcome following radical resection. Oncol. Lett. 2016;12:4080–4086. doi: 10.3892/ol.2016.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.King RJ, et al. CD73 induces GM-CSF/MDSC-mediated suppression of T cells to accelerate pancreatic cancer pathogenesis. Oncogene. 2022;41:971–982. doi: 10.1038/s41388-021-02132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wurm M, et al. A novel antagonistic CD73 antibody for inhibition of the immunosuppressive adenosine pathway. Mol. Cancer Ther. 2021;20:2250–2261. doi: 10.1158/1535-7163.MCT-21-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Roh M, Wainwright DA, Wu JD, Wan Y, Zhang B. Targeting CD73 to augment cancer immunotherapy. Curr. Opin. Pharmacol. 2020;53:66–76. doi: 10.1016/j.coph.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. 2010;29:5346–5358. doi: 10.1038/onc.2010.292. [DOI] [PubMed] [Google Scholar]
- 105.Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol. Rev. 2017;276:121–144. doi: 10.1111/imr.12528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pellegatti P, et al. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS ONE. 2008;3:e2599. doi: 10.1371/journal.pone.0002599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Maj T, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017;18:1332–1341. doi: 10.1038/ni.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Saveljeva S, et al. A purine metabolic checkpoint that prevents autoimmunity and autoinflammation. Cell Metab. 2022;34:106–124.e10. doi: 10.1016/j.cmet.2021.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hu T, Suter SR, Mumbleau MM, Beal PA. TLR8 activation and inhibition by guanosine analogs in RNA: importance of functional groups and chain length. Bioorg. Med. Chem. 2018;26:77–83. doi: 10.1016/j.bmc.2017.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang Z, et al. Structural analysis reveals that Toll-like receptor 7 is a dual receptor for guanosine and single-stranded RNA. Immunity. 2016;45:737–748. doi: 10.1016/j.immuni.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 111.Borghaei H, et al. 24-Month overall survival from KEYNOTE-021 cohort G: pemetrexed and carboplatin with or without pembrolizumab as first-line therapy for advanced nonsquamous non-small cell lung cancer. J. Thorac. Oncol. 2019;14:124–129. doi: 10.1016/j.jtho.2018.08.004. [DOI] [PubMed] [Google Scholar]
- 112.Schaer DA, et al. The folate pathway inhibitor pemetrexed pleiotropically enhances effects of cancer immunotherapy. Clin. Cancer Res. 2019;25:7175. doi: 10.1158/1078-0432.CCR-19-0433. [DOI] [PubMed] [Google Scholar]
- 113.Dersh D, et al. Genome-wide screens identify lineage- and tumor-specific genes modulating MHC-I- and MHC-II-restricted immunosurveillance of human lymphomas. Immunity. 2021;54:116–131.e10. doi: 10.1016/j.immuni.2020.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yamamoto K, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. 2020;581:100–105. doi: 10.1038/s41586-020-2229-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Goel S, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548:471–475. doi: 10.1038/nature23465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gu SS, et al. Therapeutically increasing MHC-I expression potentiates immune checkpoint blockade. Cancer Discov. 2021;11:1524–1541. doi: 10.1158/2159-8290.CD-20-0812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kang SH, et al. Inhibition of MEK with trametinib enhances the efficacy of anti-PD-L1 inhibitor by regulating anti-tumor immunity in head and neck squamous cell carcinoma. Oncoimmunology. 2019;8:e1515057. doi: 10.1080/2162402X.2018.1515057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kalbasi A, et al. Uncoupling interferon signaling and antigen presentation to overcome immunotherapy resistance due to JAK1 loss in melanoma. Sci. Transl. Med. 2020;12:eabb0152. doi: 10.1126/scitranslmed.abb0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Keshet R, et al. Targeting purine synthesis in ASS1-expressing tumors enhances the response to immune checkpoint inhibitors. Nat. Cancer. 2020;1:894–908. doi: 10.1038/s43018-020-0106-7. [DOI] [PubMed] [Google Scholar]
- 120.Brown PM, Pratt AG, Isaacs JD. Mechanism of action of methotrexate in rheumatoid arthritis, and the search for biomarkers. Nat. Rev. Rheumatol. 2016;12:731–742. doi: 10.1038/nrrheum.2016.175. [DOI] [PubMed] [Google Scholar]
- 121.Genestier L, et al. Immunosuppressive properties of methotrexate: apoptosis and clonal deletion of activated peripheral T cells. J. Clin. Invest. 1998;102:322–328. doi: 10.1172/JCI2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bar-Or A, et al. Randomized study of teriflunomide effects on immune responses to neoantigen and recall antigens. Neurol. Neuroimmunol. Neuroinflamm. 2015;2:e70. doi: 10.1212/NXI.0000000000000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brezinschek HP, Hofstaetter T, Leeb BF, Haindl P, Graninger WB. Immunization of patients with rheumatoid arthritis with antitumor necrosis factorα therapy and methotrexate. Curr. Opin. Rheumatol. 2008;20:295–299. doi: 10.1097/BOR.0b013e3282ffdeca. [DOI] [PubMed] [Google Scholar]
- 124.Klotz L, et al. Teriflunomide treatment for multiple sclerosis modulates T cell mitochondrial respiration with affinity-dependent effects. Sci. Transl. Med. 2019;11:eaao5563. doi: 10.1126/scitranslmed.aao5563. [DOI] [PubMed] [Google Scholar]
- 125.Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu. Rev. Biochem. 1999;68:459–486. doi: 10.1146/annurev.biochem.68.1.459. [DOI] [PubMed] [Google Scholar]
- 126.Wawrzyniak JA, et al. A purine nucleotide biosynthesis enzyme guanosine monophosphate reductase is a suppressor of melanoma invasion. Cell Rep. 2013;5:493–507. doi: 10.1016/j.celrep.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010;2:a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kollareddy M, et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat. Commun. 2015;6:7389. doi: 10.1038/ncomms8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yamaguchi N, et al. PCK1 and DHODH drive colorectal cancer liver metastatic colonization and hypoxic growth by promoting nucleotide synthesis. eLife. 2019;8:e52135. doi: 10.7554/eLife.52135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rainger J, et al. Miller (Genee-Wiedemann) syndrome represents a clinically and biochemically distinct subgroup of postaxial acrofacial dysostosis associated with partial deficiency of DHODH. Hum. Mol. Genet. 2012;21:3969–3983. doi: 10.1093/hmg/dds218. [DOI] [PubMed] [Google Scholar]
- 131.Kayamori K, et al. DHODH inhibition synergizes with DNA-demethylating agents in the treatment of myelodysplastic syndromes. Blood Adv. 2021;5:438–450. doi: 10.1182/bloodadvances.2020001461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Halbrook CJ, et al. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 2019;29:1390–1399.e6. doi: 10.1016/j.cmet.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dalin S, et al. Deoxycytidine release from pancreatic stellate cells promotes gemcitabine resistance. Cancer Res. 2019;79:5723–5733. doi: 10.1158/0008-5472.CAN-19-0960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shireman JM, et al. De novo purine biosynthesis is a major driver of chemoresistance in glioblastoma. Brain. 2021;144:1230–1246. doi: 10.1093/brain/awab020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dorasamy MS, Ab A, Nellore K, Wong PF. Synergistic inhibition of melanoma xenografts by brequinar sodium and doxorubicin. Biomed. Pharmacother. 2019;110:29–36. doi: 10.1016/j.biopha.2018.11.010. [DOI] [PubMed] [Google Scholar]
- 136.Zhou W, et al. Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat. Commun. 2020;11:3811. doi: 10.1038/s41467-020-17512-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gunda V, et al. MUC1-mediated metabolic alterations regulate response to radiotherapy in pancreatic cancer. Clin. Cancer Res. 2017;23:5881–5891. doi: 10.1158/1078-0432.CCR-17-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Graves LM, et al. Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature. 2000;403:328–332. doi: 10.1038/35002111. [DOI] [PubMed] [Google Scholar]
- 139.Mao C, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–590. doi: 10.1038/s41586-021-03539-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Snajdauf M, et al. The TRAIL in the treatment of human cancer: an update on clinical trials. Front. Mol. Biosci. 2021;8:628332. doi: 10.3389/fmolb.2021.628332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.He T, et al. Inhibition of the mitochondrial pyrimidine biosynthesis enzyme dihydroorotate dehydrogenase by doxorubicin and brequinar sensitizes cancer cells to TRAIL-induced apoptosis. Oncogene. 2014;33:3538–3549. doi: 10.1038/onc.2013.313. [DOI] [PubMed] [Google Scholar]
- 142.Gottlieb E, Armour SM, Harris MH, Thompson CB. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003;10:709–717. doi: 10.1038/sj.cdd.4401231. [DOI] [PubMed] [Google Scholar]
- 143.Abt ER, et al. Metabolic modifier screen reveals secondary targets of protein kinase inhibitors within nucleotide metabolism. Cell Chem. Biol. 2020;27:197–205.e6. doi: 10.1016/j.chembiol.2019.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cuthbertson CR, et al. The dihydroorotate dehydrogenase inhibitor brequinar is synergistic with ENT1/2 Inhibitors. ACS Pharmacol. Transl Sci. 2020;3:1242–1252. doi: 10.1021/acsptsci.0c00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yu Y, et al. Therapeutic targeting of both dihydroorotate dehydrogenase and nucleoside transport in MYCN-amplified neuroblastoma. Cell Death Dis. 2021;12:821. doi: 10.1038/s41419-021-04120-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gaidano V, et al. The synergism between DHODH inhibitors and dipyridamole leads to metabolic lethality in acute myeloid leukemia. Cancers. 2021;13:1003. doi: 10.3390/cancers13051003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pastor-Anglada M, Pérez-Torras S. Emerging roles of nucleoside transporters. Front. Pharmacol. 2018;9:606. doi: 10.3389/fphar.2018.00606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nielsen-Kudsk F, Pedersen AK. Pharmacokinetics of dipyridamole. Acta Pharmacol. Toxicol. 1979;44:391–399. doi: 10.1111/j.1600-0773.1979.tb02350.x. [DOI] [PubMed] [Google Scholar]
- 149.Mullen NJ, et al. ENT1 blockade by CNX-774 overcomes resistance to DHODH inhibition in pancreatic cancer. Cancer Lett. 2023;552:215981. doi: 10.1016/j.canlet.2022.215981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sullivan MR, et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife. 2019;8:e44235. doi: 10.7554/eLife.44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mimura K, et al. Genome-wide CRISPR screening reveals nucleotide synthesis negatively regulates autophagy. J. Biol. Chem. 2021;296:100780. doi: 10.1016/j.jbc.2021.100780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab. 2017;25:1037–1043. doi: 10.1016/j.cmet.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Guo JY, et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016;30:1704–1717. doi: 10.1101/gad.283416.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu Y, et al. Autophagy-dependent ribosomal RNA degradation is essential for maintaining nucleotide homeostasis during C. elegans development. eLife. 2018;7:e36588. doi: 10.7554/eLife.36588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Noe DA, et al. Phase I and pharmacokinetic study of brequinar sodium (NSC 368390) Cancer Res. 1990;50:4595–4599. [PubMed] [Google Scholar]
- 156.Nathanson DA, et al. Co-targeting of convergent nucleotide biosynthetic pathways for leukemia eradication. J. Exp. Med. 2014;211:473–486. doi: 10.1084/jem.20131738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Liu Y, Zhou Q, Song S, Tang S. Integrating metabolic reprogramming and metabolic imaging to predict breast cancer therapeutic responses. Trends Endocrinol. Metab. 2021;32:762–775. doi: 10.1016/j.tem.2021.07.001. [DOI] [PubMed] [Google Scholar]