

Abstract
Tissue-resident memory T cells (TRM cells) provide protective immunity, but the contributions of specific tissue environments to TRM cell differentiation and homeostasis are not well understood. In the present study, the diversity of gene expression and genome accessibility by mouse CD8+ TRM cells from distinct organs that responded to viral infection revealed both shared and tissue-specific transcriptional and epigenetic signatures. TRM cells in the intestine and salivary glands expressed transforming growth factor (TGF)-β-induced genes and were maintained by ongoing TGF-β signaling, whereas those in the fat, kidney and liver were not. Constructing transcriptional–regulatory networks identified the transcriptional repressor Hic1 as a critical regulator of TRM cell differentiation in the small intestine and showed that Hic1 overexpression enhanced TRM cell differentiation and protection from infection. Provision of a framework for understanding how CD8+ TRM cells adapt to distinct tissue environments, and identification of tissue-specific transcriptional regulators mediating these adaptations, inform strategies to boost protective memory responses at sites most vulnerable to infection.
Upon infection, naive CD8+ T cells become activated and subsequently undergo proliferation and differentiate to effector cells that produce inflammatory cytokines and secrete cytolytic granules. Adhesion molecules and chemokines recruit activated T cells into infected tissues. As the infection is cleared, the pathogen-specific T cell population contracts and a small number of memory T cells are retained in tissues and provide long-lived, localized immunity from reinfection (TRM cells)1–4. The influence of unique tissue environments on TRM cell differentiation and function remains unclear.
Based on chromatin accessibility and gene expression, CD8+ TRM precursor cells within tissues can be distinguished from both circulating effector and memory-precursor CD8+ T cells within the first week of infection5–7. The upregulation of molecules that prevent tissue egress, CD69 and CD103, and the downregulation of lymphoid-homing molecules, including S1PR1, CCR7 and CD62L, are key for TRM cell formation2,8. TRM cells share characteristics with circulating effector and memory CD8+ T cells, including expression of inflammatory cytokines and cytolytic molecules, sustained lifespan and functional protection from reinfection7,9. Accordingly, TRM cell differentiation requires transcription factors with well-established roles in effector (Blimp1 (ref. 10), Notch2 (ref. 11) and Egr2 (ref. 12)) and circulating memory (Runx3 (ref. 5) and Nr4a1 (ref. 13)) CD8+ T cells. However, transcription factors that support differentiation of effector (T-bet) and circulating memory (Eomes) CD8+ T cells can suppress TRM cell differentiation14. TGF-β regulates a critical nexus in these processes because it enhances tissue entry, mediates upregulation of adhesion molecules, including CD103, and contributes to downregulation of T-bet cells and Eomes6,14–16.
Transcriptional networks required for tissue residency of CD8+ T cells are also important for the maintenance of other immune cell populations in nonlymphoid tissues, including innate lymphoid cells (ILCs), natural killer (NK) cells and macrophages5,10,17,18. Thus, TRM cells may use common transcriptional modules shared among leukocytes to establish and maintain residency in nonlymphoid tissues. Elucidating a ‘core’ tissue-residency program has relied on identifying common features of TRM cells that are distinct from circulatory T cells, but by necessity this neglects divergent gene expression arising in different nonlymphoid tissues. T cells in nonlymphoid tissues encounter varying nutrient availability, pH, oxygen tension and cytokine milieus19, and genes that vary in expression among TRM cells in different tissues5,9,10,20,21 and mediate functional tissue adaptations have been identified. For example, skin, adipose and intestinal TRM cells differentially express and depend on specific fatty acid-binding protein isoforms20,22, and expression of the prototypical TRM cell markers CD69 and CD103 varies across tissues. Despite these observations, tissue-specific gene-expression programs have not been comprehensively characterized for CD8+ TRM cells.
To identify gene-expression and genome-accessibility changes that arise in CD8+ T cell populations responding to systemic viral infection in distinct tissues environments, we used RNA-sequencing (RNA-seq) and assay for transposase-accessible chromatin with high throughput (ATAC-seq) of cells from small intestine (SI) intraepithelial lymphocytes (IEL), kidney, liver, salivary glands (SG) and adipose tissue, as well as the spleen and blood. Single-cell (sc)RNA-seq was used to differentiate ubiquitous tissue-specific changes to the TRM cell population from differences in the abundance of shared, heterogeneous TRM cell populations. These results allowed prediction of tissue-specific transcriptional regulators of TRM cell populations. We found that CD8+ TRM cells from each tissue displayed unique transcriptional modules and functional activities, highlighting the critical idea that broad features of TRM cells may not always be extrapolated from studies of an individual tissue and establishing a framework for understanding organ-specific transcriptional regulation of TRM cell differentiation.
Results
TRM cells show shared and tissue-specific gene-expression programs.
To understand the relationship between idiosyncratic features of TRM cells and the tissue environment, we infected mice with lymphocytic choriomeningitis virus (LCMV) Armstrong, which generates CD8+ TRM cells in a broad range of tissues1. CD8+ P14 T cells were transferred into CD45 congenic recipient mice 1 d before infection to allow for the systematic phenotyping and purification of TRM cells. P14 CD8+ T cells were isolated from blood, spleen, IEL, kidney, SG, liver and fat between day 30 and day 40 post-infection. Cells in the tissue were distinguished from those in the vasculature by intravenous (IV) administration of antibodies to CD8α before sacrifice23. IV-negative (IV−) P14 cells in tissues other than the spleen and blood were defined as TRM cells (used as such hereafter, unless otherwise specified; Extended Data Fig. 1). We observed considerable variation in the expression of CD69 and CD103 on P14 cells isolated from tissues at days 30–40 (Fig. 1a,b). Of the tissues assessed, only IEL and SG TRM cells expressed substantial levels of CD103, although the frequency of CD103+ cells was considerably higher in the IEL (Fig. 1a,b). Varying frequencies of CD69+ P14 cells were observed across all of the tissues, ranging from >90% in the IEL to approximately 15% in the liver (Fig. 1a,b). Similar results were observed in mice infected intravenously with Listeria monocytogenes that expresses the GP33 peptide (LM-GP33) (Extended Data Fig. 2a,b), indicating common differentiation and adaptation pathways associated with TRM cells.
Fig. 1 |. TRM cells in distinct tissue microenvironments possess unique transcriptional programs.
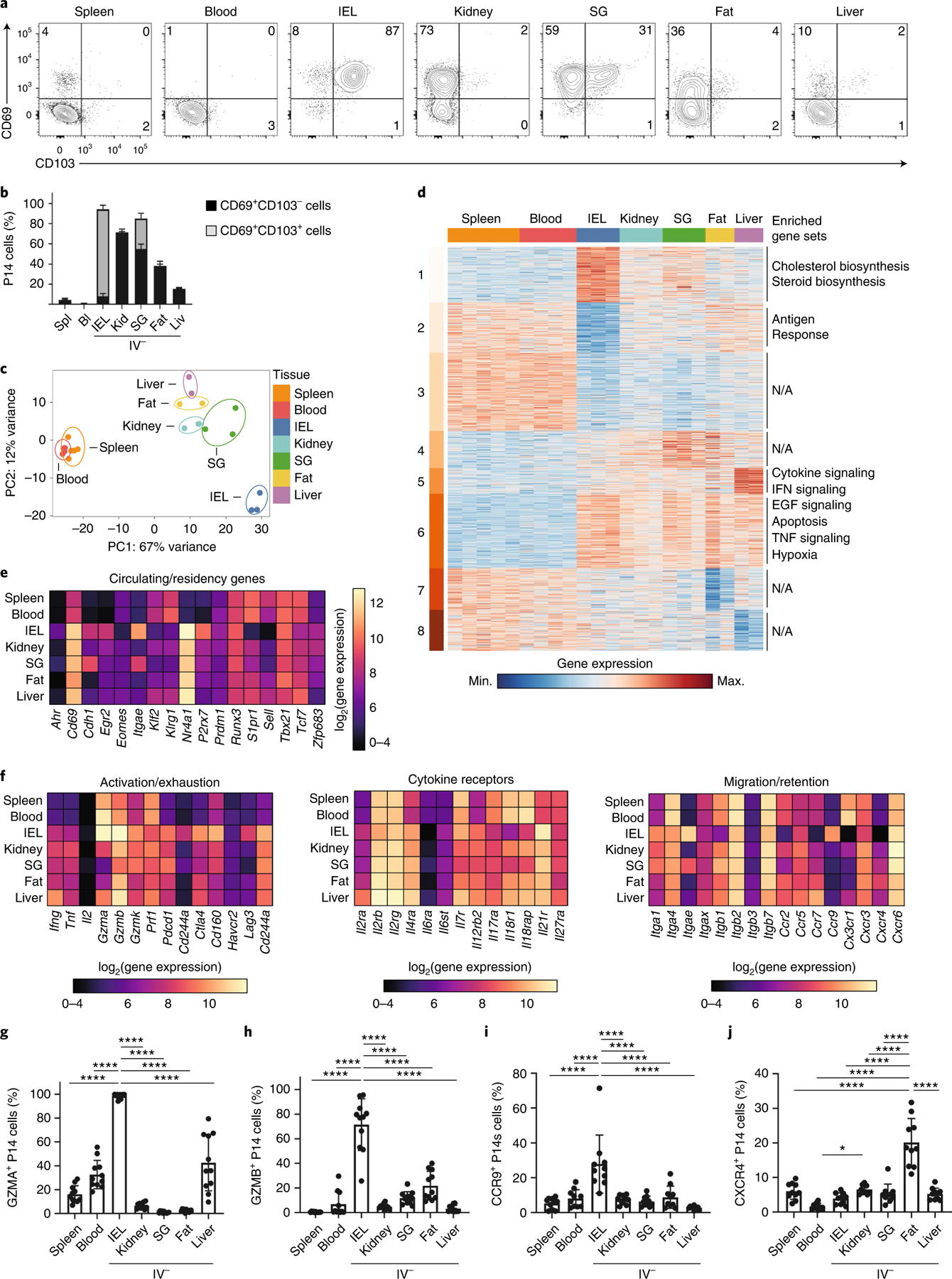
a,b, Representative flow cytometry plots (a) and quantification (b) of CD69 and CD103 expression in CD8+ TRM cells isolated from IEL, kidney, SG, fat and liver. c, PCA analysis of RNA-seq of CD8+ TRM cells isolated from IEL, kidney, SG, fat and liver, as well as memory CD8+ cells from spleen and blood. Two to three experimental replicates were used per sequenced tissue sample, generated by pooling tissues from multiple mice. d, Heatmap of 2,820 DEGs from RNA-seq dataset in c, clustered with k-means = 8. Enriched gene sets are indicated on the right. EGF, Epidermal growth factor; N/A, not available. e, The log2(expression) values for select genes previously associated with circulating or tissue-resident CD8+ T cells from the RNA-seq dataset in c. f, The log2(expression) values for select genes associated with activation/exhaustion (left), cytokine receptors (center) and migration/retention (right) from the RNA-seq dataset in c. g–j, Percentage of GZMA+ (g), GZMB+ (h), CCR9+ (i) and CXCR4+ (j) P14 cells isolated from the indicated tissues as assessed by flow cytometry. Quantification of flow cytometry data in b and g–j displays the mean ± s.d. for 12 (b), 11 (g and h) or 10 (i and j) mice from 3 experimental replicates. The significance was calculated using a one-way ANOVA and corrected for multiple comparisons using Tukey’s test. ****P < 0.0001.
To define the transcriptional adaptations of TRM cells to distinct tissue environments, we compared gene expression in P14 T cells sorted from the spleen and blood and IV− P14 cells sorted from the IEL, kidney, liver, SG and fat at days 30–40 post-infection. Principal component analysis (PCA) of these data showed that all TRM cell populations were separated distinctly from circulating P14 cells in the spleen and blood in principal component (PC)1 (Fig. 1c)5,6,10. PC2 separated TRM cells by tissue: liver TRM cells were on one end, IEL TRM cells on the opposite end and all other TRM cells were distributed in between (Fig. 1c). The distance of IEL TRM cells from other tissue populations indicated that they were the most transcriptionally distinct compared with the other TRM cell populations. By comparing splenic P14 cells with each of the other P14 populations, we identified 2,820 differentially expressed genes (DEGs) (Fig. 1d and Supplementary Table 1). Using k-means clustering (k = 8), we identified shared and tissue-specific clusters of gene expression. Clusters 3 and 6 comprised the adaptions of P14 cells to tissue residence, because these genes were differentially regulated in all TRM cell populations compared with circulating P14 cells in the spleen and blood (Fig. 1d). In contrast, clusters 1, 5, 7 and 8 possessed tissue-specific, gene-expression patterns (Fig. 1d). Cluster 1 comprised genes upregulated in IEL TRM cells compared with all other sequenced populations, and was enriched for genes with a role in cholesterol and steroid biosynthesis (Fig. 1d).
We next compared the expression of genes previously shown to be important for the establishment and maintenance of circulating and TRM cells. TRM cells in the IEL had the lowest expression of genes associated with circulating memory cells, including Eomes, Tcf7, Klf2, S1pr1 and Sell, whereas TRM cells in other tissues showed intermediate expression (Fig. 1e). In addition, we compared the expression of select genes in three functional categories: genes associated with activation or exhaustion, genes encoding cytokine receptors and genes important for migration or retention (Fig. 1f). Under homeostatic conditions, we observed constitutive expression of Gzma in the IEL, liver, spleen and blood memory P14 CD8+ T cells (Fig. 1g), with higher expression in the IEL than the liver (Fig. 1g). Granzyme B expression was high in the IEL TRM cells compared with the other TRM cell populations (Fig. 1h). Similar results were seen after infection with LM-GP33 (Extended Data Fig. 2c,d). IEL TRM cells had high expression of Il7r and low expression of Il2rb, a component of the interleukin (IL)-2 and IL-15 receptor, compared with TRM cells in the kidney, SG and fat (Fig. 1f), consistent with previous observations that kidney and SG TRM cells depend on IL-15 signaling for survival, whereas IEL TRM cells do not24.
Within this dataset, expression of the chemokine receptors CCR9, which has a role in the recruitment of CD8+ T cells to the intestine25,26, and CXCR4, which mediates CD8+ T cell homing to the bone marrow through interactions with CXCL12 (ref. 27), had the highest expression in TRM cells isolated from the IEL and fat, respectively (Fig. 1f). We found that 25–30% of SI-resident TRM cells maintained the expression of CCR9 once established in the tissue (Fig. 1i). Adipocytes can secrete CXCL12, which recruits macrophages to the tissue28. TRM cells isolated from adipose tissue had higher expression of CXCR4 compared with memory T cells in circulation or TRM cells in other tissues (Fig. 1j), suggesting that CXCR4 may play a role in their recruitment and/or retention within adipose tissue. Analysis of published datasets10,20 that characterized TRM cells in other experimental systems identified similar patterns of tissue-specific gene expression (Extended Data Fig. 2e,f). Compared with P14 cells in the circulation, TRM cells displayed higher expression of messenger RNA for a number of genes encoding inhibitory receptors (including Pdcd1, Lag3 and Ctla4), with IEL TRM cells having the highest mRNA expression of many of these receptors (Fig. 1f). However, programmed cell death protein 1 (PD-1) or Lag3 protein was not detected on the TRM cell surface directly ex vivo (Extended Data Fig. 2g), suggesting that TRM cells in these tissues may be poised to upregulate expression of these proteins.
Genes upregulated across TRM cells in multiple tissues, such as Fos, Jun and Nr4a1, are upregulated by tissue digestion29,30. To understand how tissue digestion impacted gene expression, we compared digestion with collagenase at 37 °C with digestion with cold active protease (CAP) at 4 °C. CAP digestion yielded lower numbers of TRM cells than collagenase digestion and did not affect expression levels of CD69 and CD103, but cleaved CD8α and CD62L (Extended Data Fig. 3a,b). To determine how tissue digestion impacted the transcriptional profile, we performed RNA-seq on P14 cells isolated from the spleen and TRM cells from the kidney with collagenase, CAP or dithioerythritol (DTE). We identified 620 DEGs (Extended Data Fig. 3c and Supplementary Table 2). PCA analysis showed that both digestion method and tissue defined the variation observed between samples (Extended Data Fig. 3d). Although CD69 expression was increased in samples digested at 37 °C, its expression was still elevated in CAP-digested kidney TRM cells compared with splenic memory T cells (Extended Data Fig. 3e). Analysis of previously published core TRM cell signatures5,10 indicated that some, though not all, of the genes among these signatures were additionally upregulated by digestion at 37 °C (Extended Data Fig. 3f,g). Analysis of our TRM cell RNA-seq dataset after removing the digestion-associated genes indicated that the TRM cells in each tissue remained distinct from circulating cells, as well as each other (Extended Data Fig. 3h). Collectively, these data demonstrate that TRM cells responding to the same infection possess both shared and tissue-specific, gene-expression programs and provide a basis for investigating the unique, tissue-dependent requirements for establishing this key protective population.
TRM cells show inter- and intra-tissue heterogeneity.
To address intra-tissue heterogeneity, we used scRNA-seq to profile circulating and TRM cell populations. Spleen and blood circulating CD8+ T cells were separated from TRM cells on the UMAP (Uniform Manifold Approximation and Projection) dimensional reduction plot and TRM cells from different tissues clustered largely separate from each other (Fig. 2a). Top genes identified as enriched in each tissue by bulk RNA-seq displayed similar patterns of gene expression in the scRNA-seq data (Extended Data Fig. 4a). In addition, the removal of digestion-associated genes from the scRNA-seq dataset did not influence the enrichment of the core TRM cell signature upregulated in T cells isolated from tissues (Extended Data Fig. 5). Unbiased clustering identified 12 distinct clusters (Fig. 2b). As expected, memory CD8+ T cells from spleen and blood separated into two clusters, representing cells that are more similar to effector memory T cells (TEM cells) and central memory T cells (TCM cells) (Fig. 2b–e). We observed analogous subsets of cells enriched for expression of effector- versus memory-associated genes among tissue-isolated TRM cells to varying degrees (Fig. 2f,g and Supplementary Table 3)21. Cluster 3 corresponded to P14 cells in the liver that expressed memory T cell-associated genes such as Il7r, Tcf7 and Ifng. Correspondingly, cluster 7 included liver cells that expressed effector-cell-associated genes, such as Klrg1, Gzma and Gzmb (Fig. 2f). Consistent with differential protein expression of CD69 and CD103 within tissues, the scRNA-seq data showed a gradient of expression for Cd69 and Itgae (encoding CD103) and the tissue-resident gene signature (Extended Data Figs. 5a and 6a). In the IEL, kidney and SG, heterogeneity within a tissue correlated with additional markers of tissue residency, including the corresponding decreased expression of Tcf7 (ref. 31), Il18r1 (ref. 32) and Ly6c2 (ref. 32) (Extended Data Fig. 6a,b–d). Among the most highly variable genes within each tissue, a number of genes (Odc1, Rgs1 and Dusp2) encode for molecules with an as yet unknown function in TRM cells (Extended Data Fig. 6a). Notably, in spite of the low frequency of CD69+ cells among the IV− P14 cells in the liver, the cells clustered together (Fig. 2a), arguing that the CD69− cells were not simply recirculating cells captured in the tissue. Similarly, expression of CD103 in TRM cells in SG and IEL did not result in their co-clustering (Fig. 2a). Thus, the tissue origin was the most important factor in gene expression by TRM cells.
Fig. 2 |. ScRNA-seq identifies distinct tissue and function-specific transcriptional programs in TRM cells.
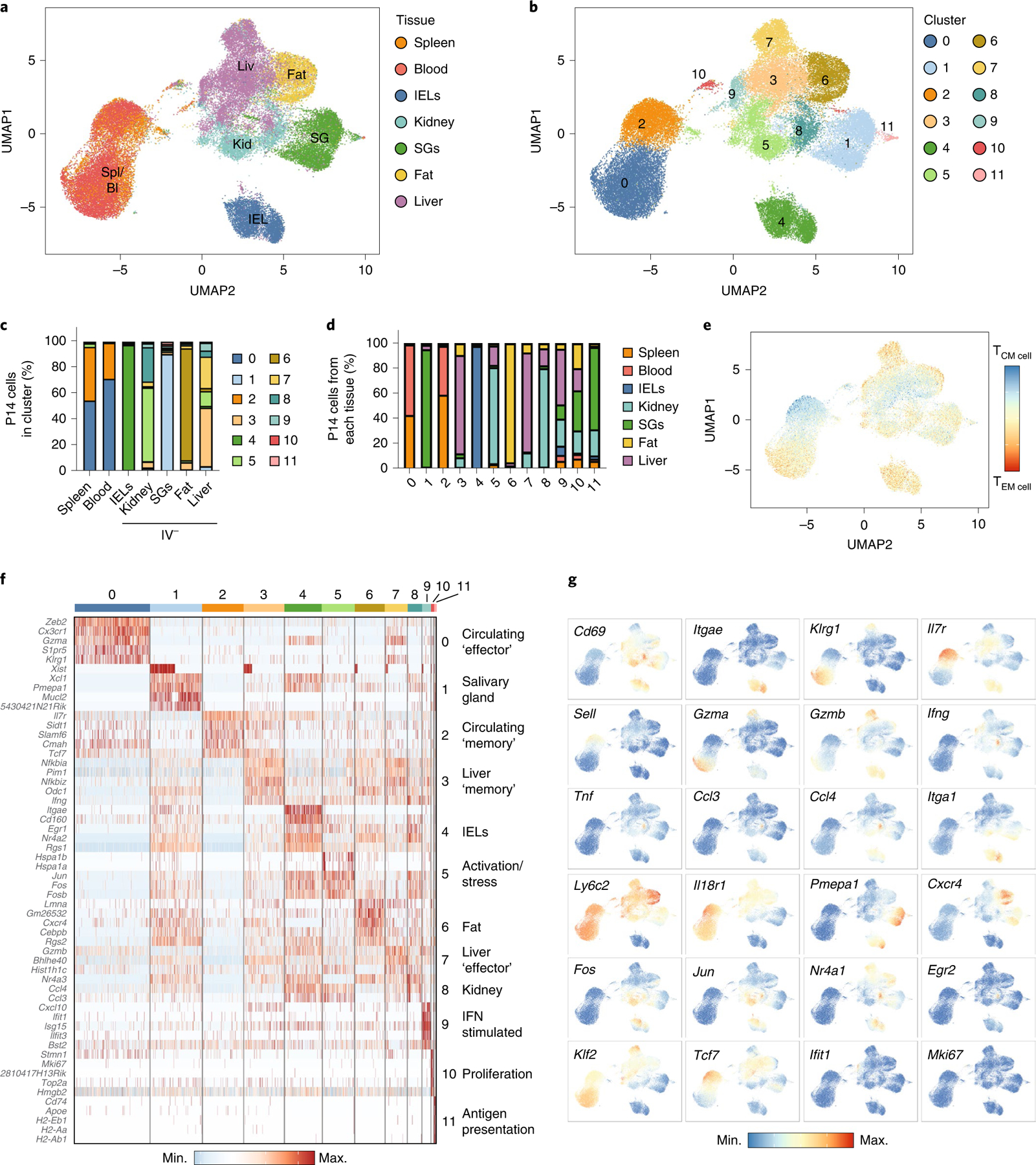
a,b, UMAP dimensional reduction colored by tissue (a) and cluster (b) of scRNA-seq of circulating and resident P14 cells isolated from the indicated tissues harvested 32 d after initial infection with LCMV. c, Percentage of P14 cells assigned to each cluster for each tissue for the scRNA-seq dataset in a. d, Percentage of P14 cells from each tissue in each cluster. e, UMAP dimensional reduction showing the difference in the TCM cell or TEM cell gene score for each cell. f, Heatmap showing the top five genes from each cluster. g, UMAP dimensional reduction colored by gene expression. The sequenced cells comprising each tissue in this dataset were derived from at least three experimental replicates and comprise pooled tissue samples from multiple mice.
Multiple genes in cluster 1 (SG), including Pmepa1, act as negative regulators of TGF-β signaling, whereas the upregulation of Pmepa1 and Xcl1 was also observed in IEL TRM cells (Fig. 2f). TRM cells isolated from the fat upregulated Cxcr4 compared with TRM cells isolated from other tissues (Fig. 2f,g), whereas IEL TRM cells displayed elevated expression of Itgae, activation genes such as Nr4a2 and Egr1 and inhibitory receptors such as Cd160 (Fig. 2f,g). Cluster 9 included genes upregulated during interferon (IFN) stimulation (Fig. 2f,g). Although bulk RNA-seq indicated that P14 cells in the liver uniquely upregulated IFN- and inflammation-induced genes, scRNA-seq showed that this signature arose in only a small subset of the total cells isolated from the liver (Fig. 2f,g). Thus, scRNA-seq showed that, in spite of clear ‘on and off ’ expression or heterogeneity of intracellular molecules and surface receptors, such as CD103, CD69 and IL-18 receptor (IL-18R), TRM cells from a given tissue were relatively similar in overall gene expression.
Ongoing TGF-β signaling is required for SI and SG TRM cells.
TGF-β is important for the formation of TRM cells within diverse tissues such as the skin, SI, SG and kidney, and also plays a direct role in the upregulation of CD103 (refs. 6,15,16,33). To gain insight into the range of tissues where TGF-β may shape TRM cell gene expression during homeostasis, we looked for enrichment of TGF-β signaling using a published TGF-β gene-expression signature based on the in vitro treatment of CD8+ T cells with TGF-β34. Cells with a high ‘TGF-β score’ were observed primarily within the IEL and SG (Fig. 3a,b), the two tissues with significant CD103 expression. To test whether sustained TGF-β signaling was important for TRM cell homeostasis in a tissue-specific context, we deleted Tgfbr2 in established TRM cell populations. Tgfbr2fl/flR26-CreERT2−/− or Tgfbr2+/+R26-CreERT2+/− P14 (hereafter wild-type (WT)) and Tgfbr2fl/flR26-CreERT2+/− P14 cells (hereafter TGFβR2 KO) were transferred at a 1:1 ratio into congenically distinct mice that were then infected with LCMV. Tamoxifen was administered daily from day 14 to day 18 post-infection to induce deletion of Tgfbr2 after formation of TRM cells and the relative ratio WT:TGFβR2 KO cells was assessed at day 40 post-infection. We observed a twofold decrease in the relative frequency of TGFβR2 KO cells in the IEL and SG TRM cell compartment compared with WT cells, with no significant loss of TGFβR2 KO CD8+ P14 cells in spleen, blood, kidney, fat and liver (Fig. 3c,d). We also observed a significant decrease in the frequency of CD103+ TGFβR2 KO cells compared with WT cells isolated from the IEL and SG (Fig. 3e,f). There was no decrease in the frequency of CD69+ cells isolated from any of the tissues or the circulation (Fig. 3e,f). These results indicate that constitutive signaling through TGFβR2 was specifically required to maintain CD103+ cells in the IEL and SG and suggests that the signals promoting TRM cell survival during homeostasis were unique to each tissue.
Fig. 3 |. Sustained TGF-β signaling is required for the maintenance of TRM cells in IEL and SG.

a,b, Violin plot showing TGF-β signature score by tissue in scRNA-seq analysis of resident and circulating P14 cells from Fig. 2 (a) and UMAP dimensional reduction colored by TGF-β score (main) and tissue (inset) (b). c,d, Representative flow cytometry plots (WT CD45.1, TGFβR2 KO CD45.1.2) (c) and quantification (d) comparing relative numbers of WT and TGFβR2 KO P14 cells. e,f, Representative flow cytometry plots (e) and quantification (f) comparing CD69 and CD103 expression in WT and TGFβR2 KO P14 cells. Quantification of flow cytometry data in d and f displays the mean ± s.d. for ten mice, three experimental replicates for all tissues. The boxplot in a shows the median. The lower and upper hinges correspond to the first and third quartiles, and the upper whisker extends from the hinge to the largest value no further than 1.5 × interquartile range from the hinge. The significance in d is calculated with a one-way ANOVA and corrected for multiple comparisons using Tukey’s test. The significance in f is calculated using a two-way ANOVA and corrected for multiple comparison’s using Sidak’s multiple comparison test. NS, not significant. ****P < 0.0001. *P < 0.05.
TRM cells acquire tissue-specific chromatin accessibility changes.
To gain insight into the transcription factors directing gene expression changes associated with TRM cell differentiation in specific tissues, we next assessed the genome accessibility in distinct tissue environments. We performed ATAC-seq on P14 cells isolated from the spleen and IV− P14 TRM cells from the IEL, kidney, SG, fat and liver after infection with LCMV. The similarity across samples was assessed using Spearman’s correlation, and samples from the IEL and spleen clustered by replicate, whereas kidney, SG, fat and liver samples clustered among themselves (Extended Data Fig. 7a). We identified 7,150 peaks that were differentially accessible between P14 cells in the spleen and each of the other tissues (Fig. 4a, Extended Data Fig. 7b–d and Supplementary Table 4). Peaks in differentially accessible regions (DARs) were enriched within intergenic and intronic regions compared with all peaks (Extended Data Fig. 7b). We identified clusters of accessible genomic regions with tissue-specific, tissue-shared, broadly circulating and broadly resident expression profiles (Fig. 4a). To understand the relationship between DARs and DEGs, we assigned each DAR to the nearest gene, and then we assigned any differentially accessible genes to the corresponding DAR cluster. Many of these genes followed the same general pattern as the DAR with which they are associated (Fig. 4a).
Fig. 4 |. TRM cells in distinct tissue microenvironments possess unique epigenetic programs.
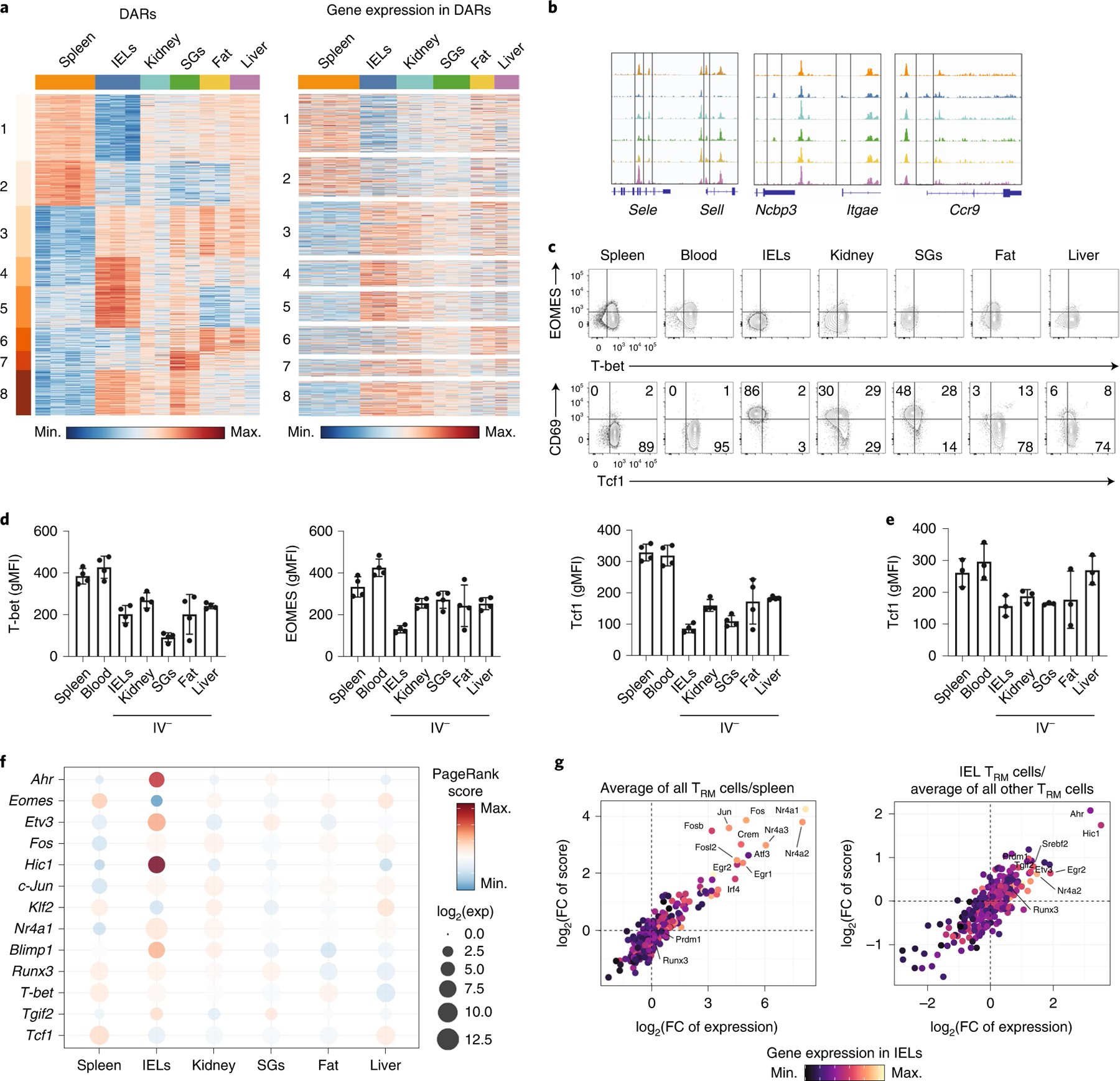
a, Heatmap showing 7,150 DARs clustered with k-means = 8 (left) from ATAC-seq of P14 cells in the spleen and IV− P14 cells isolated from the IEL, kidney, SG, fat and liver, and gene expression for the gene nearest to each DAR (right). Only DEGs are shown. Each sequenced tissue possesses two to four experimental replicates, generated by pooling tissues from multiple mice. b, ATAC tracks of DARs from select genes. c,d, T-bet, EOMES and Tcf1 expression assessed in P14 cells isolated from mice 30–40 d after initial infection with LCMV. Flow cytometry plots (c) and quantification (d) from one representative experiment out of three total experiments with a total of eleven mice. gMFI, geometric mean of median fluorescence intensity. e, Tcf1 expression assessed in P14 cells isolated from mice 30–34 d after initial infection with LM-GP33. Quantification is from one representative experiment out of three experiments with a total of ten mice. f, PageRank score and gene expression for select TFs displayed by individual tissues. g, Average PageRank score and gene expression of transcription factors displayed as the average in all TRM cells over P14 cells in the spleen (left) PageRank score and gene expression in IEL TRM cells over the average of all other TRM cells (right). FC, fold-change.
Consistent with their expression patterns, the genes of canonical markers of circulation (Sell) and tissue residency (Itgae and Ccr9) displayed alterations in accessibility (Fig. 4b). An accessible peak at the transcription start site (TSS) of CD62L (encoded by Sell) had the greatest accessibility in the P14 splenocytes. Additional accessible regions specific to both IEL and SG TRM cells were identified at the TSS of Itgae (Fig. 4b). A uniquely accessible region in the TSS of Ccr9 was observed in IEL TRM cells (Fig. 4b), consistent with observed protein expression. In addition to the epigenetic profiling of TRM cells across tissues, we assessed the expression of the transcription factors (TFs) T-bet, Eomes and Tcf1, which suppress TRM cell differentiation14,31. Although T-bet was downregulated in TRM cells in all tissues compared with those in the circulation, Eomes was more strongly downregulated in IEL TRM cells than in TRM cells isolated from other sites (Fig. 4c,d). Tcf1 was downregulated in all tissue TRM cells compared with circulatory memory cells, with the most prominent downregulation in the IEL, followed by the SG (Fig. 4c,d). Similarly, Tcf1 downregulation by the IEL, kidney, SG and fat TRM cells compared with circulating memory cells were observed in the response to LM-GP33 infection (Fig. 4e).
To predict key TFs that may play a role in mediating tissue-specific transcriptional programs, we used the published Taiji pipeline that had identified TFs important for the differentiation of both circulating and tissue-resident T cell populations5,35,36. This approach generates a network of DARs and DEGs and predicts the TFs most likely to be driving these differences, based on the TF motif enrichment. IEL TRM cells had the highest PageRank score for TFs associated with residency, including Blimp1, and a negative correlation with genes known to promote recirculation or inhibit residency, including Klf2, Eomes and Tcf1 (Fig. 4f and Supplementary Table 5)10. In contrast, circulating memory cells in the spleen and liver TRM cells were enriched for TFs associated with memory formation, including Tcf1 and Eomes (Fig. 4f). In the present study, we observed that the PageRank score indicated Runx3 activity in IEL, kidney, SG and spleen (Fig. 4f), consistent with reports on its role in both TRM cell differentiation and circulating memory T cells5,37. An analysis of TFs enriched across TRM cells from multiple tissues compared with P14 cells from the spleen identified Nr4a1 and Jun (Fig. 4g), which have established functional roles in the formation of TRM cells in vivo7,13.
In the SI TRM cells, PageRank identified Blimp1, which has a critical role in the formation of TRM cells10. Fewer Gzmb-Cre+/−Prdm1fl/fl P14 T cells were detected in the IEL than in the kidney at day 60 post-infection with LCMV (Extended Data Fig. 8). Ahr, which is critical for maintenance of skin CD8+ TRM and liver-resident NK cells38,39, and Hic1, a ZBTB trans-repressor expressed in SI, but not spleen or blood leukocytes40, were predicted to be the strongest gut-specific transcription factors by PageRank (Fig. 4g). Thus, at the epigenetic level, TRM cells possessed unique and overlapping adaptations to distinct tissue environments.
Transcriptional repressor Hic1 regulates IEL TRM cell formation.
Hic1 is important for the accumulation of CD4+ and CD8+ T cells in the SI under homeostatic conditions40. We observed that Hic1 was expressed primarily by IEL TRM cells (Fig. 5a). In addition, a published scRNA-seq dataset7 indicated that Hic1 was upregulated at early timepoints (day 5) post-infection with LCMV and maintained until at least 90 d post-infection in SI CD8+ TRM cells (Fig. 5b). Further analysis of published datasets indicated that Hic1 was upregulated in SI-resident CD4+ T cells41, macrophages17 and type 2 innate lymphocytes (ILC2s)42 compared with these cell types in other tissues (Extended Data Fig. 9a), suggesting common adaptation by diverse cell types to SI residency.
Fig. 5 |. Loss of Hic1 prevents the formation of SI TRM cells.
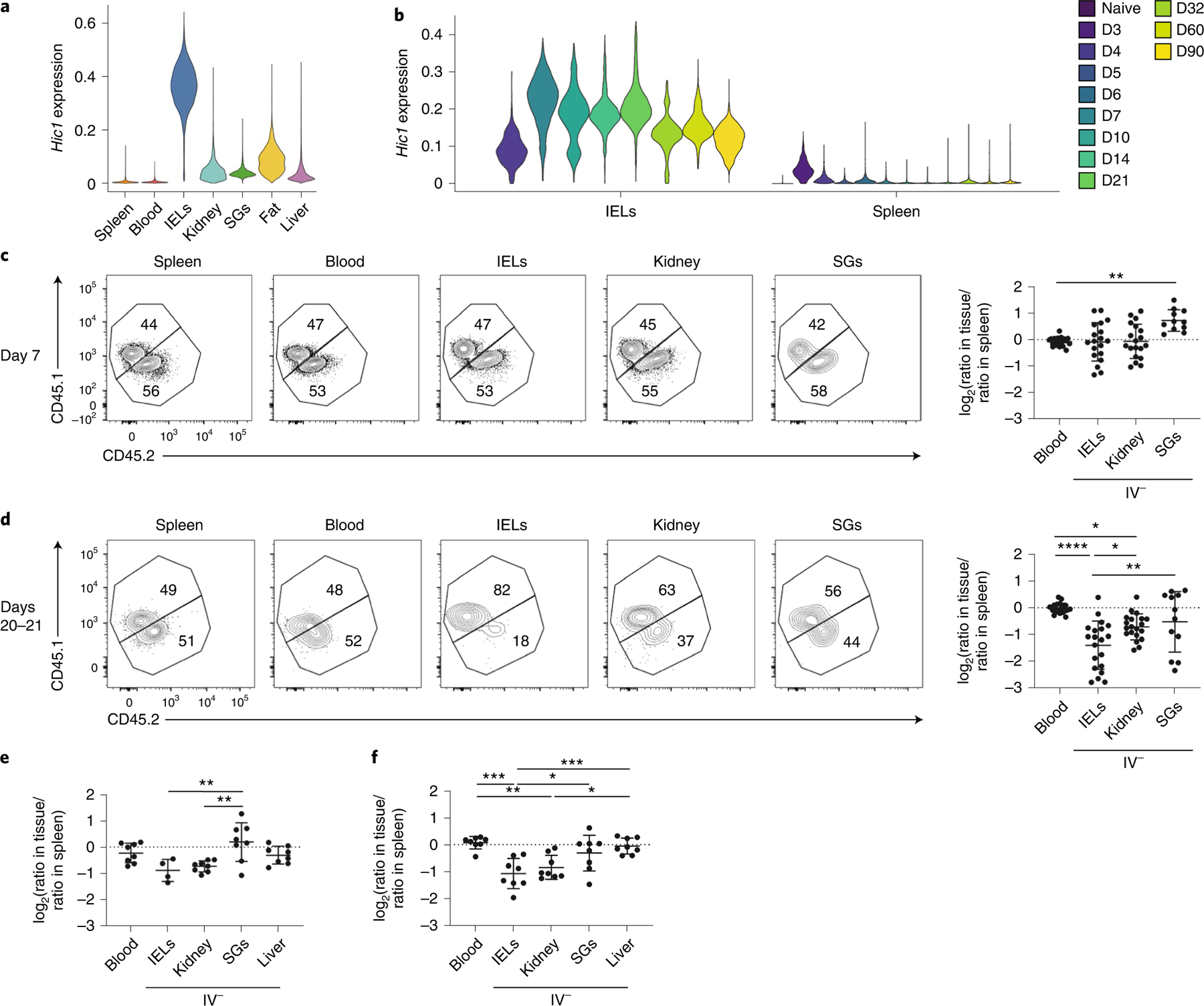
a, Violin plots displaying Hic1 expression after MAGIC imputation in scRNA-seq data from Fig. 2. b, Hic1 expression in scRNA-seq dataset from Kurd et al.7. The violin plot is colored by time after infection with LCMV. D, Day. c–f, A 1:1 mixed transfer of P14 cells transduced with a control shRNA or a Hic1-targeting shRNA before infection with LCMV or LM-GP33. Representative flow cytometry plot (Ctrl CD45.1, Hic1 KD CD45.1.2) (left) and quantification (right) of the relative ratios between Hic1-targeting and control shRNAs are normalized to the spleen on days 7–8 (c) or 20–21 (d) after initial infection with LCMV. e,f, Quantification of the relative ratios between Hic1-targeting and control shRNAs normalized to the spleen on day 7 (e) or day 20 (f) after initial infection with LM-GP33. Graphs in c and d display the mean ± s.d. for 19 mice from 5 separate experiments for blood, spleen, IEL and kidney, and 11 mice from 3 experiments for SG. Graphs in e and f display the mean ± s.d. for eight mice from two separate experiments. The significance was calculated using a one-way ANOVA and corrected for multiple comparisons using Tukey’s test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To determine whether Hic1 had a specific role in the formation of SI TRM cells, activated P14 CD8+ T cells transduced with a retroviral vector encoding CD19–short hairpin (sh)RNA as control (Ctrl) or Hic1–shRNA (Hic1 KD) were mixed at a 1:1 ratio and adoptively transferred into recipient mice 1 h before infection with LCMV. At days 7–8 post-infection, the percentage of Hic1 KD cells was increased relative to Ctrl cells in the SG, whereas their frequencies were similar in all the other tissues (Fig. 5c). At days 20–21 post-infection we observed a decrease in the fraction of Hic1 KD cells in the kidney and an even greater decrease in the SI compared with Ctrl cells (Fig. 5d). Similarly, the frequency of Hic1 KD cells was increased in the SG compared with Ctrl cells at day 7 post-infection with LM-GP33 (Fig. 5e), whereas Hic1 KD cells were reduced in the IEL and kidney at day 20 (Fig. 5e,f). The percentage of CD69+CD103− and CD69+CD103+ Hic1 KD and Ctrl cells were similar at days 7–8 and days 20–21 post-LCMV infection in the IEL and kidney (Extended Data Fig. 9b,c), whereas the percentage of CD69+CD103− cells in the kidney and the percentage of CD69+CD103+ cells in the IEL were reduced among Hic1 KD compared with Ctrl cells at day 20 post-infection with LM-GP33 (Extended Data Fig. 9d). There was no change in the relative fraction of Hic1 KD KLRG1+CD127− terminal effector (TE), KLRG1−CD127+ memory precursor (MP), CD127−CD62L− t-TEM, CD127+CD62L− TEM and CD127+CD62L+ TCM cells post-LCMV infection at day 7 and days 20–21, whereas there was a small, but significant, decrease in Hic1 KD TEM cells and a significant small increase in Hic1 KD TCM cells compared with Ctrl cells at day 20 post-LM-GP33 infection (Extended Data Fig. 9e–h), suggesting that memory T cell formation in the absence of Hic1 might be influenced by the kinetics or type of infection.
Next, P14 cells transduced with either an empty retroviral vector (Ctrl) or a retroviral vector encoding Hic1 cDNA (Hic1 OE) were mixed at a 1:1 ratio and adoptively transferred into recipient mice 1 h before infection with LCMV. At day 7 post-infection, the frequency of Hic1 OE cells in the IEL was approximately 11-fold higher than in Hic1 OE cells in the spleen (Fig. 6a,b). At day 20 post-infection, the frequency of Hic1 OE cells was higher compared with Ctrl cells in the spleen, blood, kidney and IEL, whereas Hic1 OE cells were greatly reduced in the SG (Fig. 6b). A similar increase in the percentage of Hic1 OE IEL cells and a decrease in the percentage of Hic1 OE SG cells compared with Ctrl cells were observed at days 7 and 20 post-infection with LM-GP33 (Fig. 6c,d). We detected a greater frequency of CD69+CD103+ Hic1 OE cells compared with Ctrl cells in the IEL at day 7, but not day 21 post-LCMV infection (Extended Data Fig. 9i,j). We also detected increased frequency of Hic1 OE MP cells and decreased frequency of Hic1 OE TE cells at day 7 post-infection (Extended Data Fig. 9k)43, and increased frequency of Hic1 OE TEM cells and decreased frequency of Hic1 OE TCM cells at day 21 (Extended Data Fig. 9l).
Fig. 6 |. Hic1 overexpression enhances the formation of SI TRM. cells.
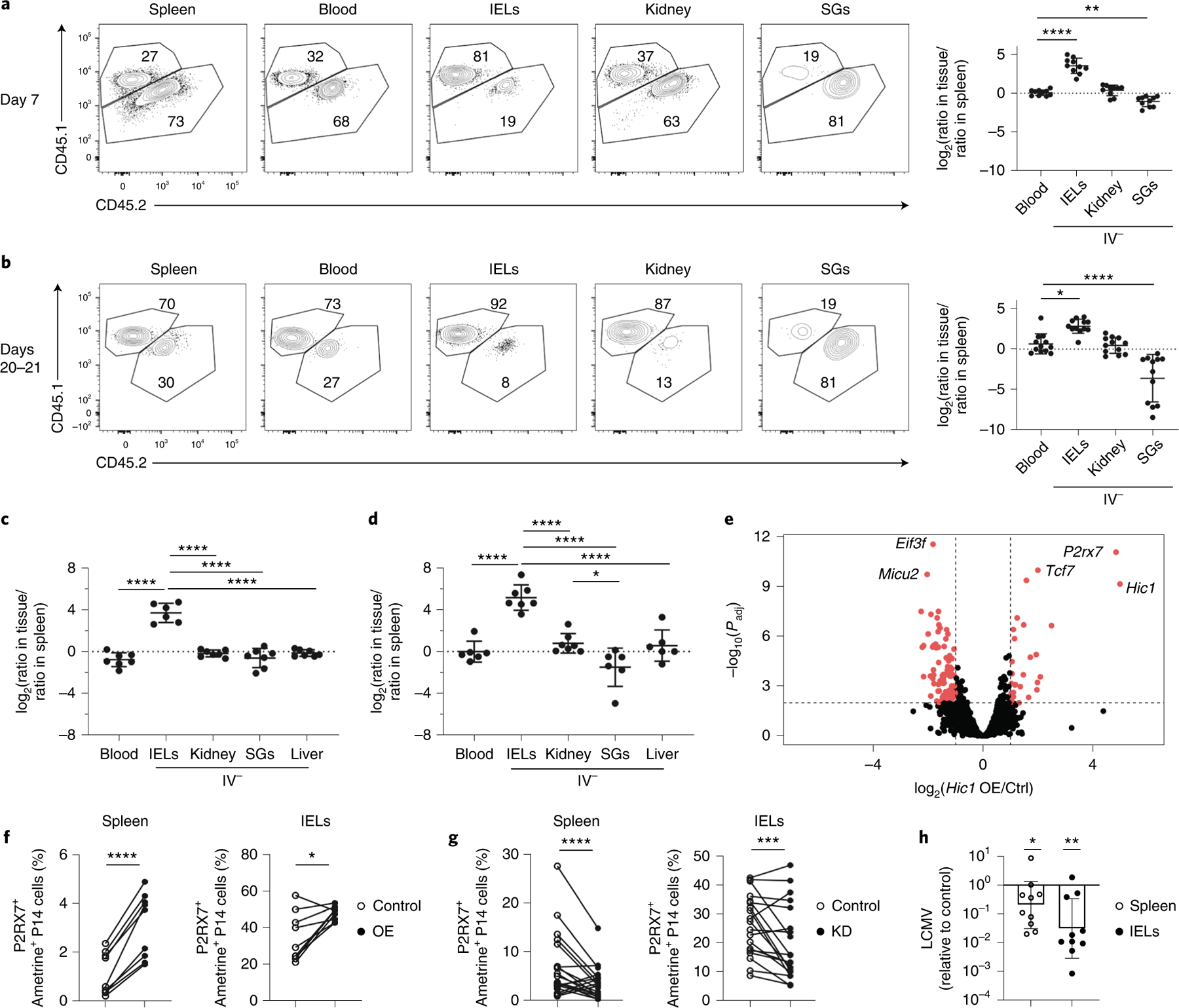
a–d, A 1:1 mixed transfer of P14 cells transduced with a control or a Hic1-overexpression construct before infection with LCMV or LM-GP33. Representative flow cytometry plot (Ctrl CD45.1.2, Hic1 KD CD45.1) (left) and quantification (right) of the relative ratios between Hic1 and control-transduced P14 cells are normalized to the spleen on days 7–8 (a) or days 20–21 (b) after initial infection with LCMV. Quantification of the relative ratios between Hic1 and control-transduced P14 cells was normalized to the spleen on day 7 (c) or day 20 (d) after initial infection with LM-GP33. e, RNA-seq of control and Hic1-overexpressing P14 cells isolated from the spleen at day 7 post-infection with LCMV. f,g, Percentage of P2RX7+ametrine+ P14 cells isolated from the spleen (left) or IEL (right) in Hic1-overexpression at day 7 post-infection (f) and in Hic1 knockdown at day 7 post-infection (g). h, Bar plot showing LCMV expression in mice that received Hic1-overexpressing P14 cells normalized to mice receiving control P14 cells. LCMV titers were assessed by qPCR relative to HPRT. Graphs in a and b display the mean ± s.d. from 10 mice (a) and 12 mice (b) from 3 separate experiments. Graphs in c and d display the mean ± s.d. for six mice (c) and seven mice (d) from two separate experiments. Significance in a–d was calculated using a one-way ANOVA and corrected for multiple comparisons using Tukey’s test. Graphs in f display three individual experiments with three mice each. Graphs in g display mean ± s.d. for 18 mice from 4 separate experiments. Significance in f and g was calculated using a two-sided, paired Student’s t-test and connecting lines indicate that the Ctrl and Hic1 KD/OE cells were isolated from the same mouse. The graph in h displays the geometric mean ± s.d. from two different experiments with five mice each. The significance was calculated using an unpaired, two-sided Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To assess how Hic1 mediated these effects, we performed RNA-seq on Hic1 OE cells and Ctrl cells sorted from the spleen at day 7 post-LCMV infection. We observed elevated expression of P2rx7, an ATP receptor important for memory T cell differentiation and TRM cell homeostasis43,44, in Hic1 OE cells (Fig. 6e and Supplementary Table 6). A higher percentage of Hic1 OE cells in the spleen and IEL was P2RX7+ compared with Ctrl cells in the same tissues (Fig. 6f), whereas both splenic and IEL Hic1 KD cells had decreased expression of P2RX7 (Fig. 6g). Expression of P2rx7 was highest in SI CD8+ TRM cells, CD4+ TRM cells, macrophages and ILC2s compared with the corresponding resident populations in other sites (Extended Data Fig. 9m).
To assess the functional capacity of P14 cells that overexpressed Hic1, Ctrl cells or Hic1 OE cells were adoptively transferred into distinct recipients 1 h before intravenous infection with LM-GP33, followed by LCMV infection on day 20 post-LM-GP33 infection. The quantitative (q)PCR assessment of viral load at day 3 post-LCMV infection indicated a reduction in LCMV titers in the spleen, and even more in the SI, in mice that received Hic1 OE cells compared with those receiving Ctrl cells (Fig. 6h). Of note, analysis of gene expression in a human scRNA-seq dataset showed enrichment of the core TRM cells and TGF-β-induced gene-expression signatures45, as well as elevated Hic1 expression in T cells isolated from the intestine and rectum compared with peripheral blood mononuclear cells (Extended Data Fig. 10). These observations indicated that Hic1 was critical for establishing a mature T cell population in the SI, and that its expression could be detrimental to the formation of TRM cells in other tissues. Thus, parallel transcriptional pathways may support both mouse and human TRM cell populations and favor seeding as well as maintenance of resident T cell populations in a particular tissue.
Discussion
In the present study, we showed that TRM cells in distinct tissues possessed transcriptional and epigenetic programs that comprised both broadly shared tissue-resident and tissue-specific signatures. We observed variances in TRM cell populations that included the differential expression of and dependence on genes known to be important for the generation and function of TRM cells, and we identified previously unknown tissue-specific transcriptional regulators. Our observations highlighted the broad functional, transcriptional and epigenetic adaptations of TRM cells with the same antigen specificity to a range of tissue environments, thus establishing a framework for identifying targets that influence TRM cell populations within specific organs to enhance therapeutic strategies.
Using RNA-seq, scRNA-seq and ATAC-seq assays, we observed that TRM cells from each tissue were more similar to each other than to circulating memory T cells of the same pathogen specificity, with TRM cells from the IEL being most distinct. ScRNA-seq revealed substantial heterogeneity in the expression of numerous genes among TRM cells within a tissue, consistent with previous reports of intra-tissue functional heterogeneity7,9,46. Ultimately, the systematic comparison of gene expression and chromatin accessibility across TRM cell populations emphasized the idea that statements about the generation, function and homeostasis of TRM cell populations need to consider the tissue-specific context in each case.
TGF-β is a pleiotropic cytokine known to affect transcriptional programs at barrier sites and is important for the formation of TRM cells in diverse tissues, including the skin, SI, kidney and SG6,8,14–16,34. The TGF-β-induced gene expression signature in IEL and SG TRM cells persisted long after TRM cell formation and viral clearance, indicating that ongoing TGF-β signaling is important for TRM cell maintenance in a tissue-specific manner. We found that the loss of TGFβR2 resulted in decreased numbers of TRM cells in the IEL and SG, but not the kidney or liver, consistent with reports that ongoing TGF-β signaling is also required for maintenance of TRM cells within the epidermis but not the liver47. These findings further highlight the previously underestimated differences across TRM cell populations from their residing tissues.
Leveraging gene expression and chromatin accessibility data, the PageRank algorithm identified known TRM cell regulators, such as Ahr, Blimp1 and Nr4a1, and the transcriptional repressor Hic1 as one of the top predicted regulators of differential gene expression in SI TRM cells. We found that Hic1 expression regulated TRM cell formation, particularly in the SI. Loss of Hic1 did not prevent T cell access to the SI, but resulted in defective TRM cell persistence in the SI and a partial loss of kidney TRM cells, whereas overexpression of Hic1 led to increased accumulation of TRM cells in the SI. Hic1 overexpression led to a significant decrease in established TRM cells in the SG, indicating that adaptation to one tissue may impair TRM cell homeostasis in another environment. Hic1 mediated changes in expression of P2RX7, a sensor of damage-associated molecular patterns shown to play an important role in TRM cell formation43,44,48. Hic1-overexpressing CD8+ TRM cells had higher expression of P2RX7 than controls and Hic1 knockdown resulted in lower expression of P2RX7, suggesting that Hic1 may regulate adaptation to the SI environment. Both Hic1 and P2RX7 are induced by retinoic acid, which is produced by the intestinal epithelium and promotes the differentiation of gut-homing immune cells and collaborates with TGF-β in promoting mucosal immunity40,49–51. Thus, the tissue milieu may provide additional remodeling of transcriptional networks that promote adaptation to that specific tissue.
In addition to the CD8+ TRM cells, immune cells such as ILCs, macrophages, NK cells and CD4+ T cells have permanent residence in many tissues5,10,17,18. Blimp1 and Hobit collaborate to promote tissue residency of CD8+ TRM, NK and NKT cells by repressing genes associated with circulation and tissue egress10. Comparing expression of Hic1 across CD8+ T cells, CD4+ T cells, ILC2s, NK cells and macrophages indicated the tissue-specific upregulation of Hic1 by cells in the SI compared with immune cells in other tissues, supporting the idea of a broad role for Hic1 in establishing the resident immune program in the SI17,41,42,52. Similarly, organ-specific expression of fatty acid-binding protein isoforms in CD8+ TRM cells is driven by secreted factors derived from the tissues and this expression is mirrored by other resident immune cell types within each organ17. Thus, in addition to providing an atlas of distinct and overlapping TRM cell features in diverse tissue environments, these findings collectively raise the possibility of ‘programming’ tissue-tailored immune responses, where immune cells that promote or regulate inflammation could be transcriptionally engineered for trafficking to, retention in and function within a particular tissue.
Online content
Any methods, additional references, Nature Research reporting summaries, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41590-022-01229-8.
Methods
Mice.
All mouse strains were bred and housed in specific pathogen-free conditions in accordance with the Institutional Animal Care and Use Guidelines of the University of California, San Diego (UCSD) at a temperature between 18 °C and 23 °C with 40–60% humidity. Male and female mice were both used in the present study. All mice used were on a C57BL/6J background. P14, Tgfbr2fl/fl mice (stock no. 012603, Jackson Laboratory), R26Cre-ERT2 (stock no. 008463, Jackson Laboratory), Thy1.1 and CD45.1 congenic mice were bred in house. Prdm1fl/fl (stock no. 008100, Jackson Laboratory) and Gzmb-cre (stock no. 003734, Jackson Laboratory) spleens were a gift from the laboratory of S. Kaech. To delete floxed alleles using Cre-ERT2, we administered 1 mg of tamoxifen (Cayman Chemical Company) emulsified in 100 μl of sunflower seed oil (Sigma-Aldrich) via daily intraperitoneal injections on days 14–18 of infection. All animal studies were approved by the Institutional Animal Care and Use Committees of UCSD and performed in accordance with UC guidelines.
Cell culture.
PLAT-E cells were cultured in Dulbecco’s modified Eagle’s medium + d-glucose supplemented with 10% bovine growth serum, 100 U ml−1 of penicillin, 100 μg ml−1 of streptomycin, 292 μg ml−1 of l-glutamine, 10 mM Hepes and 55 μM 2-mercaptoethanol. Enriched CD8+ T cells were maintained in RPMI + l-glutamine supplemented with 10% fetal bovine serum (FBS), 100 U ml−1 of penicillin, 100 μg ml−1 of streptomycin, 292 μg ml−1 of l-glutamine, 10 mM Hepes and 55 μM 2-mercaptoethanol.
Infection studies.
C57BL/6J P14 CD8+ T cells congenic for CD45 or Thy1 were adoptively transferred at 5 × 104 cells per recipient mouse by intravenous (i.v.) injection. Donor mice were sex and age matched to recipients or female donors were transferred into male recipients. For cotransfers, Tgfbr2fl/flER-Cre+ and the corresponding control P14 CD8+ T cells were mixed in a 1:1 ratio and adoptively transferred by i.v. injection into CD45 or Thy1 congenic recipients. Mice were then infected with 2 × 105 plaque-forming units (p.f.u.) of LCMV Armstrong by intraperitoneal (i.p.) injection or with 5 × 103 colony-forming units (c.f.u.) of LM-gp33 by i.v. injection 24 h after transfer. For cotransfers of transduced cells, P14 cells were mixed at a 1:1 ratio of ametrine+ cells and a total of 5 × 105 P14 cells was transferred by i.v. injection into CD45 or Thy1 congenic recipients. Then, 1 h after transfer, recipient mice were infected with either 2 × 105 p.f.u. of LCMV by i.p. injection or 5 × 103 c.f.u. of LM-gp33 by i.v. injection.
Preparation of single-cell suspensions.
To identify CD8+ T cells in the vasculature of nonlymphoid tissues (SI, kidney, SG, fat and liver), 3 μg of CD8α (53–6.7) conjugated to APC-eFluor 780 was injected intravenously into mice 3 min before sacrifice, as has been previously described21. Cells labeled with low to no CD8α antibody were considered to be outside the vasculature. Single-cell suspensions of splenocytes were prepared by mechanical disaggregation followed by treatment with ACK (ammonium–chloride–potassium) lysing buffer. Blood samples were treated with ACK lysing buffer. SI IEL were prepared through the removal of Peyer’s patches and the luminal contents. The SI was cut longitudinally and into 1-cm pieces, then incubated at 37 °C for 30 min in Hanks’ balanced salt solution with 2.1 mgml−1 of sodium bicarbonate, 2.4 mgml−1 of Hepes, 8% bovine growth serum and 0.154 mgml−1 of DTE (EMD Millipore). The kidneys, SG, fat and liver were minced into small pieces and then incubated in RPMI with 1.2 mgml−1 of Hepes, 292 μg ml−1 of l-glutamine, 1 mM MgCl , 1 mM CaCl , 5% FBS and 100 U ml−1 of collagenase (Worthington) at 37 °C for 30 min. Lymphocytes from the SI, kidney, SG and liver were separated on a 44%/67% Percoll density gradient. For digestion with cold active protease, the kidney and SG were minced into small pieces and then shaken at 4 °C for 30 min in phosphate-buffered saline (PBS) with 10 mgml−1 of protease from Bacillus sp. (Sigma-Aldrich), 0.5 mM EDTA and 125 U ml−1 of DNase (Sigma-Aldrich). Digestion was quenched with an equal volume of PBS containing 20% bovine growth serum. Lymphocytes were separated using a 44%/67% Percoll density gradient.
Generation of retroviral supernatant and CD8+ T cell transduction.
PLAT-E cells were plated in a 10-cm tissue culture dish 1 d before transfection. The next day, each plate was transfected with 5 μg of pCL-Eco and 10 μg of the plasmid of interest using TransIT-LT1 (Mirus). Retroviral supernatant was collected 48 and 72 h after transfection. CD8+ T cells were isolated from the spleen and lymph nodes and negatively enriched, as previously described53, then 2 × 105 P14 cells were plated in a 6-well dish coated with goat anti-hamster immunoglobulin (Ig) G (H (heavy) + L (light); Thermo Fisher Scientific), anti-CD3 (catalog no. 145–2C11, eBioscience) and anti-CD28 (catalog no. 37.51, eBioscience). Then, 18 h after plating, T cell culture medium was removed and replaced with retroviral supernatant supplemented with 50 μM 2-mercaptoethanol and 8 μg ml−1 of polybrene (Millipore). CD8+ T cells were spinfected for 60 min at 800g and 37 °C; 2 h after spinfection, the retroviral supernatant was removed and replaced with T cell culture medium. Then 24 h after transduction, all ametrine+ cells as assessed by flow cytometry were considered to be transduced.
Flow cytometry and cell sorting.
Cells were incubated with the indicated antibodies for 20 min at 4 °C in PBS supplemented with 2% bovine growth serum and 0.01% sodium azide. Intracellular staining was completed using the FoxP3 transcription factor staining kit (eBioscience). For assays with CD8+ T cell stimulation, P14 cells from each tissue were incubated for 3 h in T cell culture medium at 37 °C with 10 nM GP33–41 peptide and Protein Transport Inhibitor (eBioscience) (see Supplementary Table 7 for a list of antibodies used in the present study). Stained cells were analyzed using LSRFortessa or LSRFortessa X-20 cytometers (BD), FACSDiva software and Flowjo software (TreeStar). All sorting was performed on BD FACSAria Fusion instruments.
Bulk RNA-seq.
For each replicate, cells from 10–15 mice were pooled and then sorted for bulk RNA-seq. Then, 32 d after initial infection with LCMV, 5 × 103 P14 cells were sorted from the spleen and blood and 5 × 103 IV− P14 cells were sorted from the IEL, kidney, SG, fat and liver into PBS + 5% bovine serum albumin (BSA). Cells, 1 × 103, were then resorted into 1× TCL lysis buffer + 1% 2-mercaptoethanol. Library preparation for ultra-low-input RNA-seq was performed as described online (https://www.immgen.org/img/Protocols/ImmGenULI_RNAseq_methods.pdf).
Trimmomatic was used to remove adapters and trim low-quality reads (NexteraPE-PE.fa:2:30:10:1:TRUE LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:25)54. Trimmed reads were then aligned to the gencode M25 annotation of the mm10 genome using STAR with the default conditions55. Aligned reads were then quantified with featureCounts56 (-t exon -g gene_id -p -B) and DEGs were identified using DEseq2 (ref. 57). The list of all DEGs was generated by combining the contrasts of each tissue compared with the spleen and including all genes where the log2(fold-change) > 1 and Padj < 0.05. PCA analysis in Fig. 1 was generated with the plotPCA function in DEseq2 (ref. 57). Clustering in Fig. 1c was performed using pheatmap with a k-mean = 8 and all heatmaps were generated using the pheatmap package. Gene set variation analysis (GSVA) was performed using the GSVA package in R58. Raw expression counts were used as input and kcdf was set to Poisson. The TGF-β gene list was obtained from Nath et al.34.
10× Genomics library preparation and sequencing.
Cells, 1 × 104, were sorted into T cell culture medium as described above. Samples were spun down at 500 relative centrifugal force (r.c.f.) for 5 min and then resuspended in PBS + 0.04% (w:v) BSA. Samples were then loaded into Chromium Chip B (10× Genomics) and partitioned into Gel Bead In-Emulsions (GEMs) in a chromium controller (10× Genomics). ScRNA libraries were generated according to the Chromium Single Cell 3′ Reagent Kits v.3 User Guide and sequenced on a HiSeq 4000.
Reads were aligned to the mm10 genome using cellranger count59. The resulting counts matrix was then processed using Seurat60 and cells with <500 or >2,500 detected genes or a mitochondrial read percentage >10 were discarded. For analysis of P14 cells across all tissues, all samples were combined into a counts matrix using the merge function in Seurat. Data were log(normalized) and scaled using NormalizeData and ScaleData. The top 2,000 most variable genes were calculated using FindVariableGenes and then used in the PCA calculation with RunPCA. The top 20 PCs were used to calculate a UMAP dimensional reduction using the RunUMAP function. Louvain clustering was performed with Seurat’s FindClusters based on the top 20 PCs with the resolution set to 0.35. For visualizing the intra-tissue heterogeneity, each tissue dataset was normalized separately using sctransform in Seurat. The top ten genes, as ranked by residual_variance after running sctransform for each tissue, were plotted. In addition, data imputation was performed using MAGIC61 with the log(normalized expression) values and the default settings and the exact solver. Seurat’s AddModuleScore with default settings function was used to calculate scores for the TGF-β gene list (Supplementary Table 8).
The human single-cell data were normalized using scuttle’s logNormCounts after running quickCluster with the patient ID as a blocking factor. Cell type was annotated using SingleR62 with the MonacoImmuneData as a reference. Subsequently the dataset was filtered on T cells and patients with ulcerative colitis were excluded. The TGF-β and TRM signature score were calculated using AUCell63.
ATAC-seq.
For each replicate, cells from 10–15 mice were pooled and then 2 × 104 cells were sorted into PBS + 5% BSA and spun down at 500g for 20 min at 4 °C. The cell pellet was resuspended in 25 μl of lysis buffer and then spun down at 600g for 30 min at 4 °C. The nuclei pellet was resuspended in 25 μl of transposition reaction mixture containing Tn5 transposase from Nextera DNA Sample Perp Kit (Illumina) and incubated at 37 °C for 30 min. The transposase-associated DNA was then purified using the Zymo DNA clean-up kit. To amplify the library, the DNA was first amplified for five cycles using indexing primers from the Nextera kit and NEBNext High-Fidelity 2× PCR master mix. To reduce the PCR amplification bias, after the first 5 cycles, 5 μl of amplified DNA was used to perform qPCR to determine the number of cycles for the second round of PCR. The total amplified DNA was then size selected to fragments <800 bp using gel purification. The size of the pooled library was examined by tapestation. The final library was then sequenced on a HiSeq 4000 to get at least 10 million reads. Sequencing results were initially analyzed and processed using the ENCODE ATAC-seq pipeline, including read trimming, quality filtering, alignment and peak calling64,65.We performed each ATAC-seq experiment at least twice and used the irreproducibility discovery rate framework to identify the reproducible peaks. DARs were identified using diffbind66, filtering out regions with <20 reads in any sample or less than a fourfold difference in the number of reads between the spleen and any other tissue. Heatmaps were generated using pheatmap. PageRank analysis was performed as previously described35–37. As outlined above, the normalized counts table generated for the RNA-seq data, the alignment files generated by the ATAC-seq pipeline and the optimal peaks list were used as the inputs for this analysis.
LCMV titers by qPCR.
Tissues were homogenized and RNA was extracted. Complementary DNA was synthesized using the superscript IV transcriptase. The following primers were used for Hprt (forward: TGAAGAGCTACTGTAA TGATCAGTCAAC; reverse: AGCAAGCTTGCAACCTTAACCA) and LCMV gp (glycoprotein) (forward: CATTCACCTGGACTTTGTCAGACTC; reverse: CATTCACCTGGACTTTGTCAGACTC).
Statistical methods.
Statistical tests were performed using Prism (7.0/9.0; Graphpad) and R (v.4.1). Two-tailed, paired or unpaired Student’s t-test, or one- or two-way analysis of variance (ANOVA) was used for comparisons between groups. P values <0.05 were considered significant.
Data availability
All bulk RNA-seq, ATAC-seq and scRNA-seq datasets have been uploaded to the Gene Expression Omnibus repository (accession no. GSE182276). The following published datasets were used in addition: accession nos. GSE125527 (ref. 45), GSE70813 (ref. 10), GSE131847 (ref. 7), PRJNA414132 (ref. 20), GSE117568 (ref. 42), GSE63340 (ref. 17) and GSE128197 (ref. 41). The mouse reference genome mm10 has been used for RNA-seq, ATAC-seq and scRNA-seq analysis.
Extended Data
Extended Data Fig. 1 |. Gating strategy.

a, Gating strategy used to identify indicated IV− TRM populations.
Extended Data Fig. 2 |. Phenotypic characterization of TRM after LM-gp33 infection and expression of select genes in TRM from other published datasets.

a-b, CD69 and CD103 expression by CD8+ TRM isolated from tissues 30–40 days after infection with LM-GP33. Representative flow cytometry plots (a) and quantification (b). c-d, Percent of GZMA+ (c) and GZMB+ (d) P14 cells isolated from the indicated tissues 30–40 days after infection with LM-GP33 as assessed by flow cytometry. Datasets are from (e) Mackay et al, Science 2016. (f) Han et al, Immunity 2017. (g) ex vivo PD1 and Lag3 expression in P14 cells isolated from the indicated tissues. Quantification of flow cytometry data in b, c and d displays the mean ± SD for 10 mice from 3 experimental replicates. Data in g shows a representative experiment with 3 mice from a total of 3 experiments with 10 mice. Significance was calculated using a one-way ANOVA and corrected for multiple comparisons using Tukey’s test. ****p < 0.0001.
Extended Data Fig. 3 |. Collagenase digestion induces upregulation of a subset of genes also associated with tissue residency.
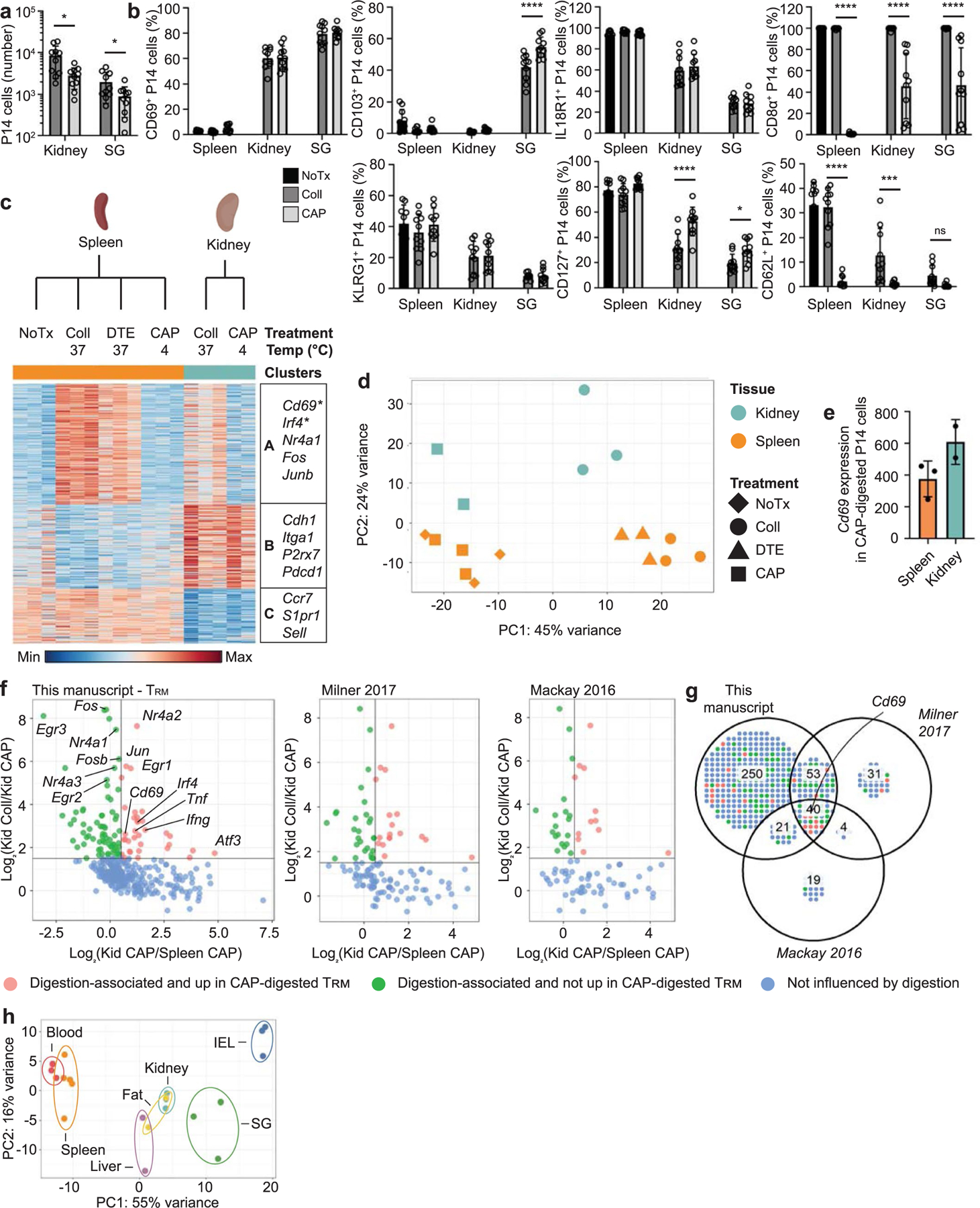
a-d, P14 cells were adoptively transferred into CD45 congenic hosts one day prior to infection with LCMV. 30–40 days after initial infection, P14 cells were isolated from tissues using no additional treatment (NoTx), collagenase (Coll), or a cold active protease (CAP). a, Quantification of the number of P14 cells recovered from each tissue using the indicated digestion methods. b, Percent of P14 cells expressing CD69 (top left), CD103 (top center left), IL-18R1 (top center right), CD8a (top right), KLRG1 (bottom left), CD127 (bottom center), or CD62L (bottom right) assessed by flow cytometry. c-e, RNA-sequencing of P14 cells isolated from the spleen or kidney using NoTx, Coll, dithioerythritol (DTE), or CAP. c, Differentially expressed genes (348) were clustered with k-means = 3. Select genes in each cluster displayed on the right. Genes that were upregulated in CAP-treated tissues compared to CAP-treated spleens indicated with an asterisk. d, Principal Component Analysis. e, Cd69 expression by P14 cells isolated from the spleen or kidney with CAP. f,g, Genes included in the TRM signatures from this paper (left), Milner et al, Nature 2017 (center) and Mackay et al, Science 2016 (right) were selected. f, Corresponding expression values for collagenase-digested kidney, CAP-digested kidney, and CAP-digested spleen samples were plotted. Each gene in the corresponding TRM signature is represented by a single point and colored by influence of digestion on expression. g, Venn diagram of the preceding data. h, Principal component analysis of RNA-sequencing data from Fig. 1 with all digestion-associated genes removed. Genes were considered digestion-associated if they were expressed above a minimum threshold and at >1.5 fold in collagenase-digested kidney compared to CAP-digested kidney samples. Graphs in a and b display the mean ± SD for 10 mice from 3 experimental replicates. RNA-seq data displayed in c-f contains 2–3 experimental replicates for each sample, and tissues from multiple mice were pooled. Graph in e displays the mean ± SD. Significance calculated using a two-way ANOVA and correcting for multiple comparisons using Dunnett’s test. *p < 0.05, ***p < 0.001, ****p < 0.0001.
Extended Data Fig. 4 |. Top enriched genes identified in bulk RNA-sequencing of TRM are also found in scRNA-sequencing.

a, The top 5 genes enriched in bulk RNA-sequening samples for TRM isolated from the blood, IEL, SG, fat, and liver are shown on a UMAP dimensional reduction plot.
Extended Data Fig. 5 |. Removal of digestion-associated gene signature from the TRM gene signature does not alter the enrichment of tissue signature.

a,b, scRNA-sequencing data described in Fig. 2. Each cell was scored based on the enrichment of genes included in the indicated signatures. Cells were colored by score on a UMAP dimensional reduction (a) and separated by cluster and ordered based on score (b).
Extended Data Fig. 6 |. TRM differentiation programs are a source of intra-tissue heterogeneity.
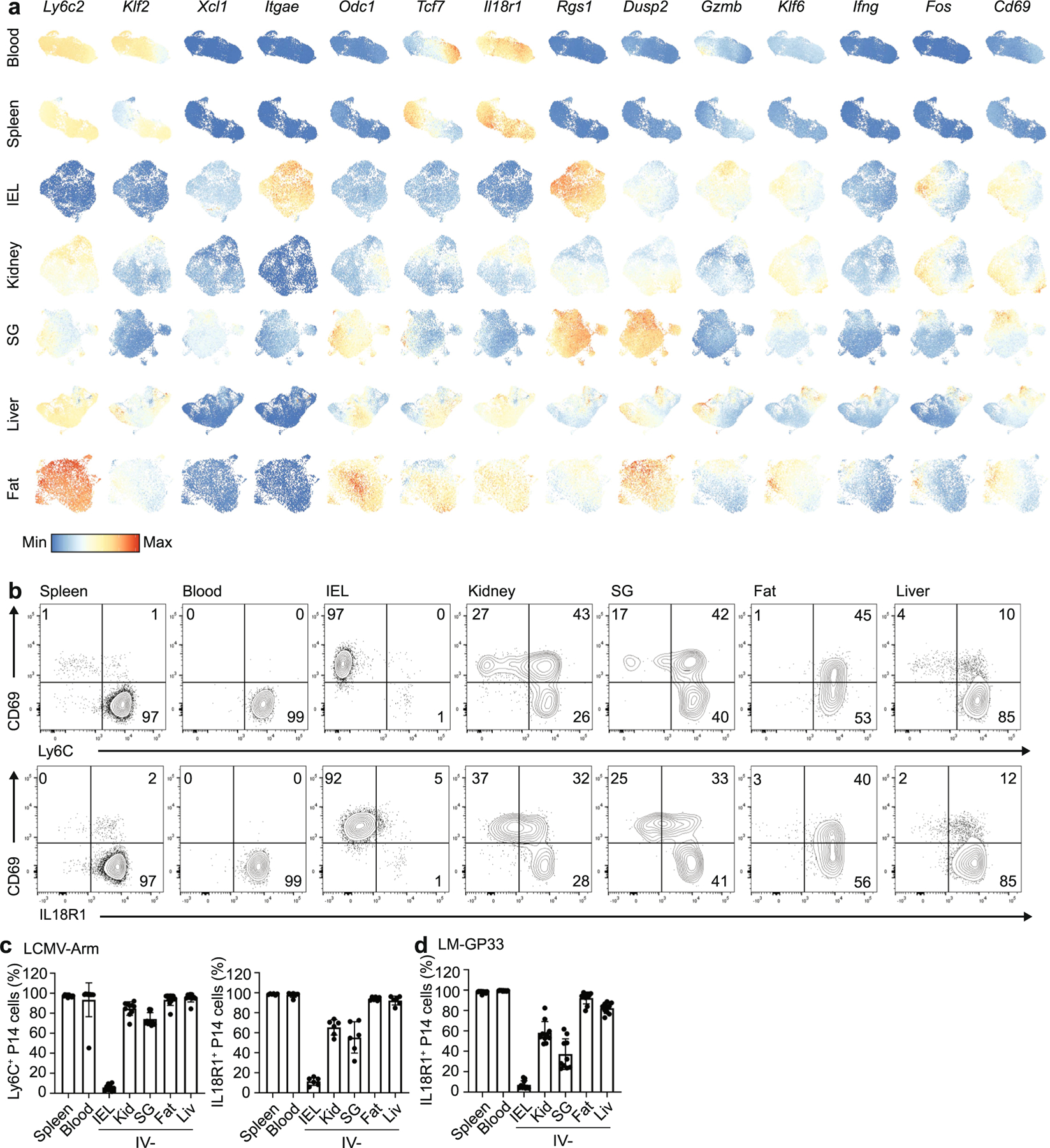
a, UMAP dimensional reduction of scRNA-sequencing of TRM separated by tissue. Cells were colored by the expression of the indicated genes. Scales are consistent across tissues to allow for comparison within and among tissues. b-c, Expression of CD69, Ly6C, IL18R1 on P14 cells harvested 30–40 days after initial infection with LCMV. Representative flow cytometry plots (b) and quantification (c). d, Quantification of IL18R1 expression on P14 cells harvested from the indicated tissues 30–40 days after initial infection with LM-GP33. Quantification of flow cytometry data in c and d displays the mean ± SD for 6 (c) 10 (d) mice from 2 experimental replicates.
Extended Data Fig. 7 |. TRM in distinct tissue microenvironments possess unique epigenetic programs.
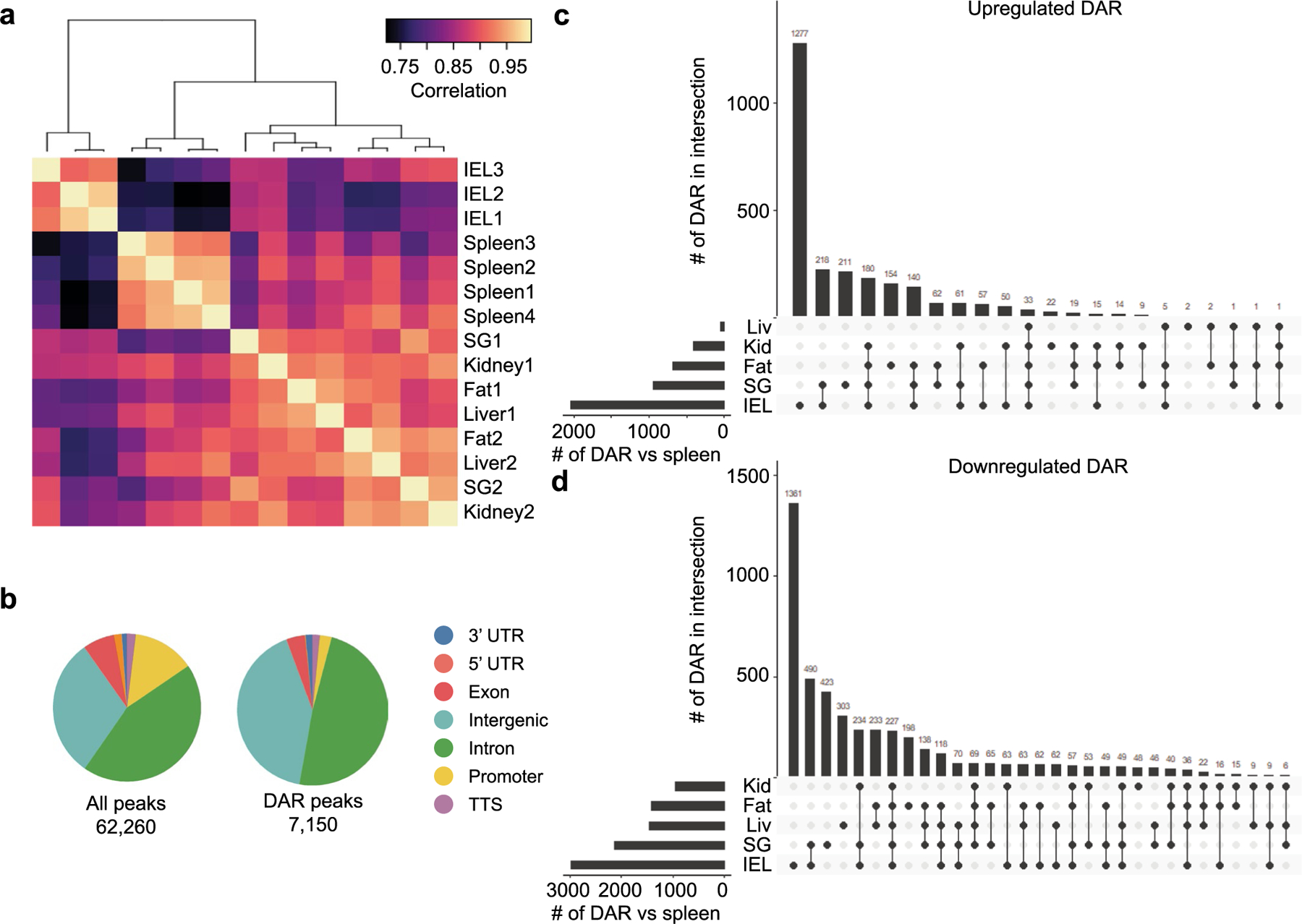
a-d, ATAC-seq of P14 CD8+ T cells in the spleen and IV− P14 CD8+ T cells isolated from the IEL, kidney, SG, fat, and liver. a, Pearson correlation for all peaks across all samples. b, Annotation of the genomic region type for all identified accessible regions (left) and DAR (right). c,d, Shared and unique upregulated DAR (c) and downregulated DAR (d) in each tissue compared to the spleen for all DAR with a p-value <0.05 and a fold change >4 using a Wald statistics.
Extended Data Fig. 8 |. Blimp1 deletion impairs TRM formation in the IEL and SG more than the kidney.
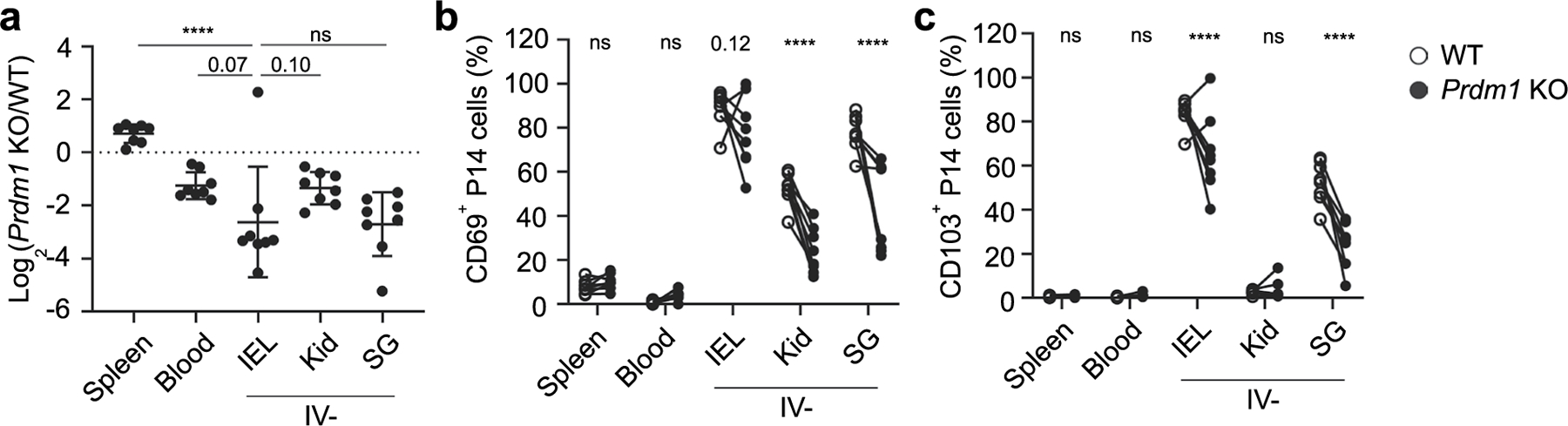
a-c, Gzmb-Cre−/−Prdm1fl/fl (WT) and Gzmb-Cre+/−Prdm1fl/fl (KO) were transferred at a 1:1 ratio into congenically distinct recipients one day prior to infection with LCMV. Tissues were harvested 60 days after initial infection. a, Ratio of KO to WT P14 cells in the indicated tissues. b-c, % of CD69+ (b) and CD103+ (c) P14 cells for WT and KO populations. Graphs display mean ± SD for a combined 2 experimental replicates, each with m = 4 mice. Significance in (a) calculated with a one-way ANOVA using Tukey’s multiple comparison test. Significance in (b-c) calculated with a two-way ANOVA using with Sidak’s multiple comparison test. ****p < 0.0001.
Extended Data Fig. 9 |. Hic1 is critical for the differentiation of small intestine TRM.
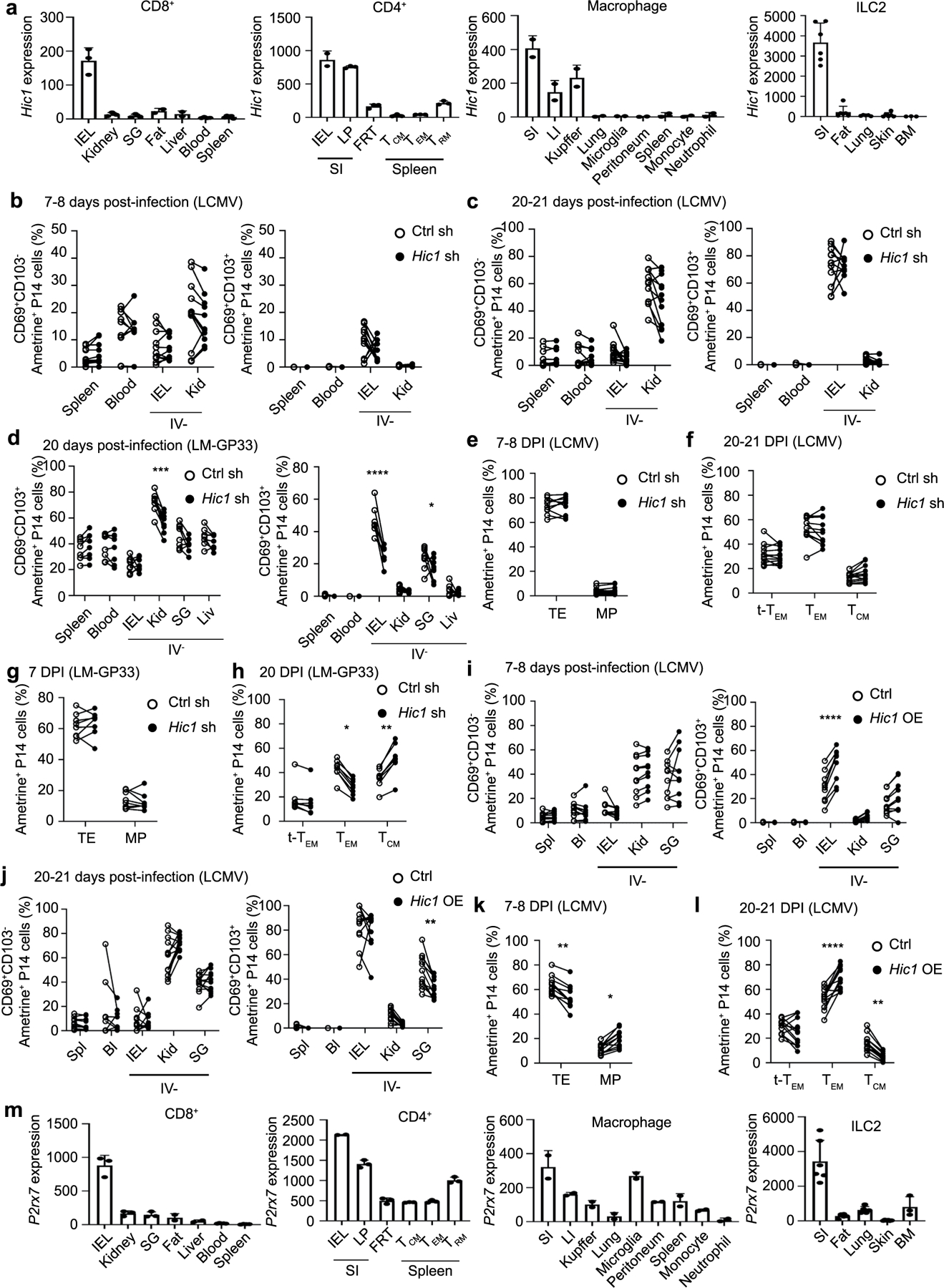
a, Hic1 expression by resident immune cell populations isolated from the indicated tissues. b-g, 1:1 mixed transfer of P14 cells transduced with a control shRNA or a Hic1-targeting shRNA. b-c, Percentage of P14 cells that are CD69+CD103− (left) or CD69+CD103+ (right) on day 7–8 (b) or day 20–21 post-infection with LCMV (c). d, Percentage of P14 cells that are CD69+CD103− (left) or CD69+CD103+ (right) on day 20 post-infection with LM-GP33. e, Percentage of P14 cells that were terminal effectors (TE, KLRG1+CD127−) or memory precursors (MP, KLRG1−CD127+) on day 7–8 post-infection with LCMV. f, Percentage of P14 cells that are terminal effector memory (tTEM, CD127-CD62L-), effector memory (TEM, CD127+CD62L-), or central memory (TCM, CD127+CD62L+) on day 20–21 post-infection with LCMV. g-h, Percentage of P14 cells that were TE or MP on day 7 (g) or day 20 (h) after infection with LM-GP33. i-l, 1:1 mixed transfer of P14 cells transduced with a control vector or a Hic1-overexpression vector. i-j, Percentage of P14 cells that are CD69+CD103− (left) or CD69+CD103+ (right) on day 7–8 (i) or day 20–21 (j) post-infection with LCMV. k, Percentage of P14 cells that were TE or MP on day 7–8 post-infection with LCMV l, Percentage of P14 cells that were tTEM, TEM, or TCM on day 20–21 post-infection with LCMV. m, P2xr7 expression by resident immune cell populations isolated from the indicated tissues. Graphs in a and m display mean ± SD for the expression values from RNA-Seq samples (22 samples for CD8+, 17 samples for CD4+, 18 samples for Macrophages, 26 samples for ILC2). Graphs in b, c, e, f, il display mean ± SD for 11 mice from 3 experimental replicates. Graphs in d, g, and h display mean ± SD for 8 mice from 2 individual experiments. Significance calculated with a two-way ANOVA using with Sidak’s multiple comparison test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Extended Data Fig. 10 |. Human TRM recapitulate phenotypes observed in murine TRM.
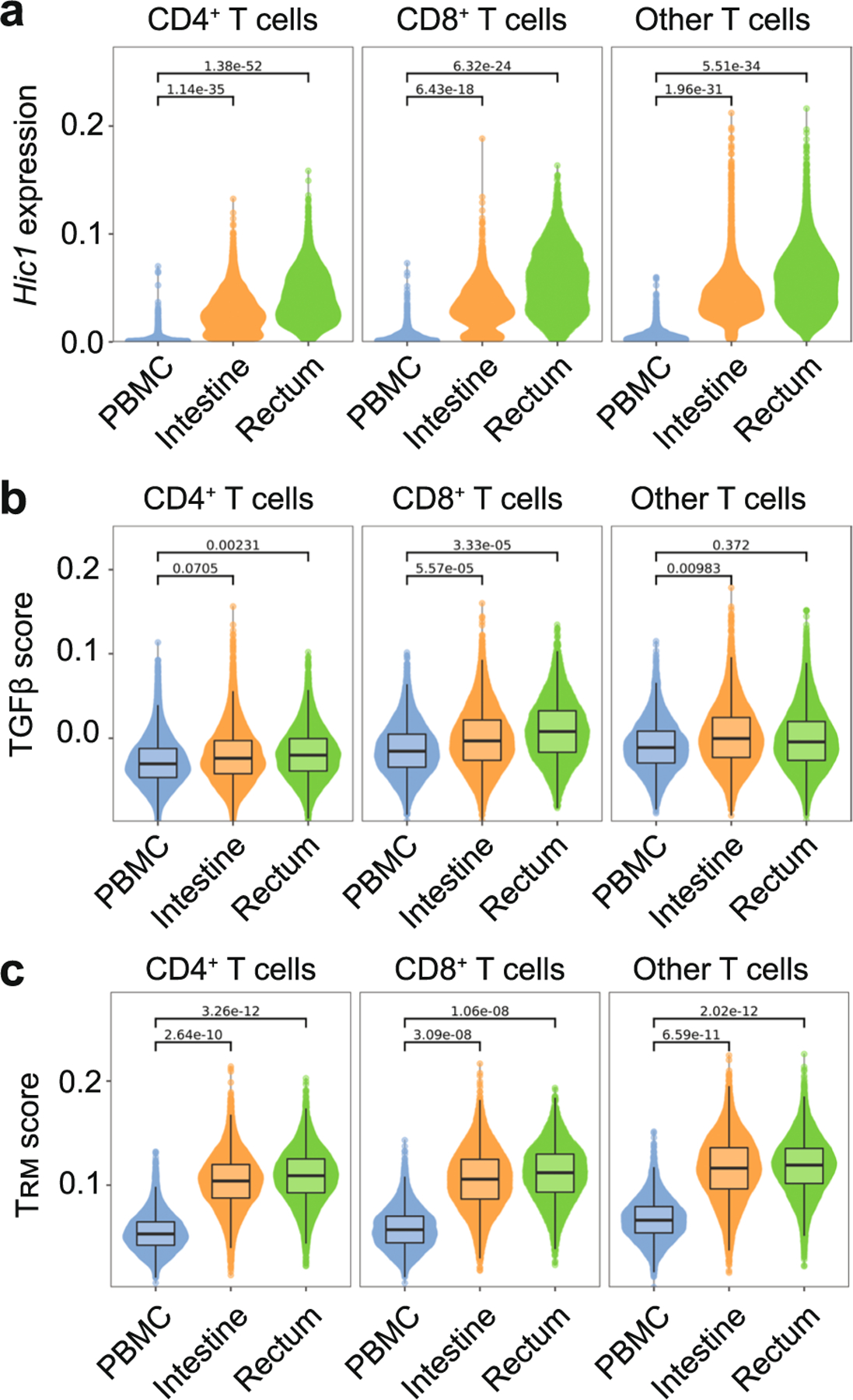
a-c, Single-cell RNA-sequencing of healthy human tissue in Boland et al, Science Immunology 2020. a, Hic1 expression after MAGIC imputation. b,c, Individual cells are scored based on enrichment for genes included in the TGFβ signature (b) and TRM signature (c). Single cell data was pooled from 13 different healthy donors for PBMC and rectum biopsies and 10 healthy donors for intestinal samples. Boxplot shows median. The lower and upper hinges correspond to the first and third quartiles. The upper whisker extends from the hinge to the largest value no further than 1.5 * IQR from the hinge. Statistics were calculated by aggregating the scRNA data to pseudo-bulk samples for each patient and cell type. A T statistics test as implemented in the R package limma was then used to calculate the P values.
Supplementary Material
Acknowledgements
This work was funded by the National Institutes of Health (grant no. AI067545 to A.W.G. and no. AI132122 to A.W.G. and J.T.Chang) and the American Cancer Society Postdoctoral Fellowship (grant no. PF-20–048-01-LIB to J.T.Crowl). ATAC-seq and scRNA-seq using the 10× Genomics platform was conducted at the IGM Genomics Center, UCSD and supported by grant nos. P30KC063491 and P30CA023100. A.W.G. is a UCSD Tata Chancellor’s Professor. M.H. was supported by the German Research Foundation fellowship (no. HE 8656/1–1). We thank H. Nguyen for assistance with measuring LCMV titers, the Goldrath laboratory members for technical advice, helpful discussion and critical reading of the manuscript and the Immunological Genome Project for reagents and sample/data processing.
Footnotes
Reporting summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Competing interests
A.W.G. is a member of the ArsenalBio scientific advisory board. J.T. Crowl is a current employee of Outpace Bio. The remaining authors declare no competing interests.
Additional information
Extended data Extended data are available for this paper at https://doi.org/10.1038/s41590-022-01229-8.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41590-022-01229-8.
Peer review information Nature Immunology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Ioana Visan in collaboration with the Nature Immunology team.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Steinert EM et al. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell 161, 737–749 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Skon CN et al. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol 14, 1285–1293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gebhardt T et al. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol 10, 524–530 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Masopust D & Soerens AG Tissue-resident T cells and other resident leukocytes. Annu. Rev. Immunol 37, 521–546 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milner JJ et al. Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mackay LK et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol 14, 1294–1301 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Kurd NS et al. Early precursors and molecular determinants of tissue-resident memory CD8+ T lymphocytes revealed by single-cell RNA sequencing. Sci. Immunol 5, eaaz6894 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casey KA et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol 188, 4866–4875 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milner JJ et al. Heterogenous populations of tissue-resident CD8+ T cells are generated in response to infection and malignancy. Immunity 52, 808–824.e807 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackay LK et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Hombrink P et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat. Immunol 17, 1467–1478 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Du N et al. EGR2 is critical for peripheral naive T-cell differentiation and the T-cell response to influenza. Proc. Natl Acad. Sci. USA 111, 16484–16489 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boddupalli CS et al. ABC transporters and NR4A1 identify a quiescent subset of tissue-resident memory T cells. J. Clin. Invest 126, 3905–3916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mackay LK et al. T-box transcription factors combine with the cytokines TGF-beta and IL-15 to control tissue-resident memory T cellfate. Immunity 43, 1101–1111 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Ma C, Mishra S, Demel EL, Liu Y & Zhang N TGF-beta controls the formation of kidney-resident T cells via promoting effector T cell extravasation. J. Immunol 198, 749–756 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang N & Bevan MJ Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39, 687–696 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lavin Y et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frizzell H et al. Organ-specific isoform selection of fatty acid-binding proteins in tissue-resident lymphocytes. Sci. Immunol 5, eaay9283 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Krausgruber T et al. Structural cells are key regulators of organ-specific immune responses. Nature 583, 296–302 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han SJ et al. White adipose tissue is a reservoir for memory T cells and promotes protective memory responses to infection. Immunity 47, 1154–1168.e1156 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milner JJ et al. Delineation of a molecularly distinct terminally differentiated memory CD8 T cell population. Proc. Natl Acad. Sci. USA 117, 25667–25678 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan Y et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252–256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson KG et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc 9, 209–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schenkel JM et al. IL-15-Independent maintenance of tissue-resident and boosted effector memory CD8 T cells. J. Immunol 196, 3920–3926 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Svensson M et al. CCL25 mediates the localization of recently activated CD8alphabeta+ lymphocytes to the small-intestinal mucosa. J. Clin. Invest 110, 1113–1121 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zabel BA et al. Human G protein-coupled receptor GPR-9–6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine-mediated chemotaxis. J. Exp. Med 190, 1241–1256 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazo IB et al. Bone marrow is a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity 22, 259–270 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Kim D et al. CXCL12 secreted from adipose tissue recruits macrophages and induces insulin resistance in mice. Diabetologia 57, 1456–1465 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Adam M, Potter AS & Potter SS Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: a molecular atlas of kidney development. Development 144, 3625–3632 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Flanagan CH et al. Dissociation of solid tumor tissues with cold active protease for single-cell RNA-seq minimizes conserved collagenase-associated stress responses. Genome Biol 20, 210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu J et al. T cell factor 1 suppresses CD103+ lung tissue-resident memory T cell development. Cell Rep 31, 107484 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Liao W et al. The downregulation of IL-18R defines bona fide kidney-resident CD8+ T cells. iScience 24, 101975 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thom JT, Weber TC, Walton SM, Torti N & Oxenius A The salivary gland acts as a sink for tissue-resident memory CD8+ T cells, facilitating protection from local cytomegalovirus infection. Cell Rep 13, 1125–1136 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Nath AP et al. Comparative analysis reveals a role for TGF-beta in shaping the residency-related transcriptional signature in tissue-resident memory CD8+ T cells. PLoS ONE 14, e0210495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu B et al. Epigenetic landscapes reveal transcription factors that regulate CD8+ T cell differentiation. Nat. Immunol 18, 573–582 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang K, Wang M, Zhao Y & Wang W Taiji: system-level identification of key transcription factors reveals transcriptional waves in mouse embryonic development. Sci. Adv 5, eaav3262 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang D et al. The transcription factor Runx3 establishes chromatin accessibility of cis-regulatory landscapes that drive memory cytotoxic T lymphocyte formation. Immunity 48, 659–674.e656 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaid A et al. Persistence of skin-resident memory T cells within an epidermal niche. Proc. Natl Acad. Sci. USA 111, 5307–5312 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khan TN, Mooster JL, Kilgore AM, Osborn JF & Nolz JC Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J. Exp. Med 213, 951–966 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burrows K et al. The transcriptional repressor HIC1 regulates intestinal immune homeostasis. Mucosal Immunol 10, 1518–1528 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Beura LK et al. CD4+ resident memory T cells dominate immunosurveillance and orchestrate local recall responses. J. Exp. Med 216, 1214–1229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ricardo-Gonzalez RR et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat. Immunol 19, 1093–1099 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borges da Silva H et al. The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8+ T cells. Nature 559, 264–268 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Borges da Silva H et al. Sensing of ATP via the purinergic receptor P2RX7 promotes CD8+ TRM cell generation by enhancing their sensitivity to the cytokine TGF-beta. Immunity 53, 158–171 e156 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boland BS et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci. Immunol 5, eabb4432 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Milner JJ & Goldrath AW Transcriptional programming of tissue-resident memory CD8+ T cells. Curr. Opin. Immunol 51, 162–169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mani V et al. Migratory DCs activate TGF-beta to precondition naive CD8+ T cells for tissue-resident memory fate. Science 366, eaav5728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stark R et al. TRM maintenance is regulated by tissue damage via P2RX7. Sci. Immunol 3, eaau1022 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Hashimoto-Hill S, Friesen L, Kim M & Kim CH Contraction of intestinal effector T cells by retinoic acid-induced purinergic receptor P2X7. Mucosal Immunol 10, 912–923 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mucida D, Park Y & Cheroutre H From the diet to the nucleus: vitamin A and TGF-beta join efforts at the mucosal interface of the intestine. Semin. Immunol 21, 14–21 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heiss K et al. High sensitivity of intestinal CD8+ T cells to nucleotides indicates P2X7 as a regulator for intestinal T cell responses. J. Immunol 181, 3861–3869 (2008). [DOI] [PubMed] [Google Scholar]
- 52.McFarland AP et al. Multi-tissue single-cell analysis deconstructs the complex programs of mouse natural killer and type 1 innate lymphoid cells in tissues and circulation. Immunity 54, 1320–1337.e1324 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen R et al. In vivo RNA interference screens identify regulators of antiviral CD4+ and CD8+ T cell differentiation. Immunity 41, 325–338 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hanzelmann S, Castelo R & Guinney J GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf 14, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng GX et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stuart T et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Dijk D et al. Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729.e727 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aran D et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol 20, 163–172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aibar S et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Consortium EP An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Davis CA et al. The encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res 46, D794–D801 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ross-Innes CS et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All bulk RNA-seq, ATAC-seq and scRNA-seq datasets have been uploaded to the Gene Expression Omnibus repository (accession no. GSE182276). The following published datasets were used in addition: accession nos. GSE125527 (ref. 45), GSE70813 (ref. 10), GSE131847 (ref. 7), PRJNA414132 (ref. 20), GSE117568 (ref. 42), GSE63340 (ref. 17) and GSE128197 (ref. 41). The mouse reference genome mm10 has been used for RNA-seq, ATAC-seq and scRNA-seq analysis.