

Abstract
This study elucidates the mechanism of CCL25 and CCR9 in rheumatoid arthritis. RA synovial fluid (SF) expresses elevated levels of CCL25 compared to OA SF and plasma from RA and normal. CCL25 was released into RA SF by fibroblasts (FLS) and macrophages (MΦs) stimulated with IL-1β and IL-6. CCR9 is also presented on IL-1β and IL-6 activated RA FLS and differentiated MΦs. Conversely, in RA PBMCs neither CCL25 nor CCR9 are impacted by 3-month longitudinal TNF inhibitor therapy. CCL25 amplifies RA FLS and monocyte infiltration via p38 and ERK phosphorylation. CCL25-stimulated RA FLS secrete potentiated levels of IL-8 which is disrupted by p38 and ERK inhibitors. CCL25 polarizes RA monocytes into non-traditional M1 MΦs that produce IL-8 and CCL2. Activation of p38 and ERK cascades are also responsible for the CCL25-induced M1 MΦ development. Unexpectedly, CCL25 was unable to polarize RA PBMCs into effector Th1/Th17 cells. Consistently, lymphokine like RANKL was uninvolved in CCL25-induced osteoclastogenesis; however, this manifestation was regulated by osteoclastic factors such as RANK, cathepsin K (CTSK) and TNFα. In short, we reveal that CCL25/CCR9 manipulates RA FLS and MΦ migration and inflammatory phenotype in addition to osteoclast formation via p38 and ERK activation.
Keywords: RA, CCL25, CCR9, macrophages, fibroblasts
Graphical Abstract
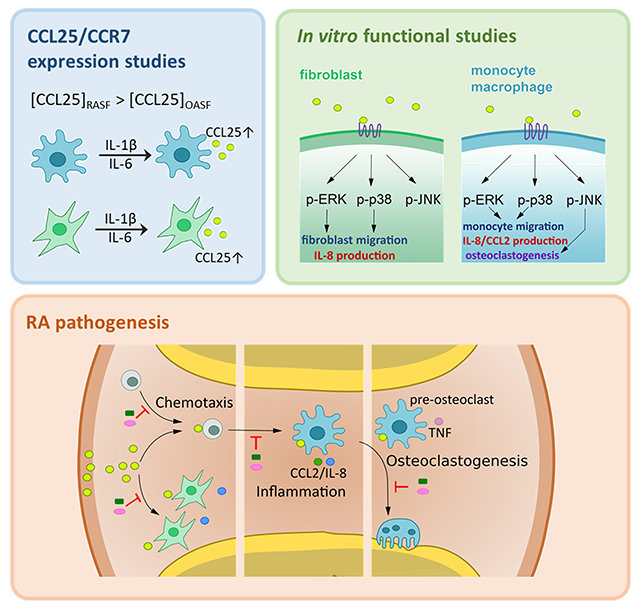
INTRODUCTION
Rheumatoid Arthritis (RA) is a chronic autoimmune disease in which synovial tissue (ST) macrophages (MΦs) and fibroblasts (FLS) play a critical role in pannus formation [1]. Notably, joint monokines and fibrokines attract cells that express their corresponding receptors to potentiate RA inflammatory and erosive phenotypes [2]. In this study, we investigated the expression and the mechanism by which ligation of CCL25 to CCR9 amplifies RA pathology.
CCR9 belongs to the β-chemokine receptor family that binds to its non-promiscuous ligand CCL25, also known as thymus expressed chemokine (TECK) [3, 4]. In physiological condition, CCL25 is expressed in the thymus or the small intestine and recruits CCR9 positive CD4+ and CD8+ T cells and IgA secreting B cells [5–7]. Additionally, the number of lymphocytes is only mildly reduced in CCR9−/− murine intestines under homeostatic conditions, in part due to compensatory action modulated by the CXCR3 function [5]. Extending these observations, in colitis patients, upregulation of CCL25 contributes to 90% of the colon CD4+T cells expressing CCR9, compared to 10% of CD3+CCR9+ colon cells detected in normal (NL) individuals [8].
Nevertheless, the impact of CCL25 and CCR9 on effector T cell differentiation is rather conflicting. In TNFSF15 transgenic mice, the frequency of CCR9+ cells was markedly elevated in mesenteric lymph nodes (MLNs) which led to potentiated Th1 and Th17 cell differentiation [9]. Similarly, TLR4−/− mice were resistant to experimental autoimmune encephalomyelitis (EAE), due to reduced levels of CCL25 and decreased the number of CCR9+CD4+ T cells in the spinal cord that contributed to diminished Th17 cell development [10]. In contrast, others have shown that CCR9−/− mice are highly susceptible to colitis, in part because T cells from MLNs secrete significantly higher levels of IFNγ and IL-17 compared to wild type (WT) mice [11]. Yet, the number of circulating CCR9+ T cells is elevated in patients with small bowel inflammation; these cells are also responsible for enhanced IFNγ and IL-17 production from MLNs of individuals with Crohn disease compared to normal controls [12, 13].
Intriguingly, expression of murine CCL25 is enriched during wound healing in oral mucosa and antigen presentation to facilitate leukocyte recruitment and retention [14, 15]. Consistently, in labial salivary glands of Sjogren’s Syndrome (SS) patients, the enhanced CCL25 concentration was directly connected to B cell hyperactivity. It was also determined that the circulating CCR9+ T cells attracted by CCL25 in SS patients exhibited a specific phenotype [16]. Compared to CXCR5+ T cells, expression levels of IL-7R, ICOS, PD1, IL-21, IL-10, IFNγ and IL-17 were highly upregulated in SS CCR9+ T cells [16]. Moreover, CCR9 deficiency or blockade ameliorated collagen-induced arthritis (CIA) disease activity [17].
The expression of CCR9 is also implicated in melanoma, ovarian, lung, breast and colon cancer [18–22]. Because of the broad expression pattern and functional importance of CCR9 in cancer, this receptor is considered as a tumor biomarker [20]. CCL25 interaction with CCR9 fosters tumor chemoresistance and metastasis via diverse signaling pathways and strategies. Due to the pivotal role of CCL25 and CCR9 in cancer biology, various therapeutic approaches have been developed to disrupt their action [22].
In this study, we sought to elucidate the expression pattern, regulatory factors and functional consequence of CCL25 and CCR9 in RA synovitis. We found that CCL25 and CCR9 are highly co-expressed on RA relative to NL synovial tissue (ST) FLS, MΦ and vasculature. Interestingly, the elevated release of CCL25 from RA synovial fluid (SF) instigates RA FLS and monocytes migration through activation of ERK and p38 pathways. In RA FLS, expression of CCL25 and CCR9 is modulated by IL-1β and IL-6 stimulation. In contrast, while the expression of CCR9 in RA MΦs was unaffected by inflammatory stimuli, differentiation of monocytes to MΦs, markedly increased its cell surface levels. We found that ligation of CCL25 to CCR9 in RA FLS and MΦs was accompanied by secretion of proangiogenic factors such as IL-8 and/or CCL2. Surprisingly, joint CCL25 was unable to amplify RA Th1 or Th17 cell polarization. Later in the disease, transcriptional upregulation of RANK and cathepsin K (CTSK) advances CCL25-induced osteoclastogenesis; while induction of TNFα stabilizes this manifestation. Finally, since TNF inhibitor (i) therapy was inconsequential on CCL25 and CCR9 expression, targeted therapy against this chemokine and receptor pair may provide a novel strategy for these RA patients.
RESULTS
CCL25 and CCR9 are mainly expressed on RA ST FLS and MΦs
To determine the significance of CCL25 and CCR9 in RA, their expression was characterized in ST and/or SF. Co-expression of CCL25 and CCR9 was markedly higher in RA compared to NL ST lining (x2), sublining cells (x2-4) and vasculature (x3-8), while their presentation in RA was comparable to OA STs (Figs. 1A, 1F). Moreover, CCL25 concentration was higher in RA SF compared to OA SF and plasma from NL and RA individuals (Fig. 1B). We substantiated that in RA ST lining and sublining, CCL25 is primarily expressed in vimentin+ RA FLS and CD68+ MΦs (Supplementary 1; Fig. S5) and its expression was further amplified by IL-1β, IL-6 and/or LPS (x2) stimulation, while TNF had no impact on this function (Figs. 1C–D). In contrast, inflammatory stimuli were unable to potentiate CCL25 transcription in endothelial cells; despite its higher expression levels in RA compared to NL ST vasculature (Figs 1A, 1E). In line with morphological studies, Western blot and FACS analyses displayed that CCR9 frequency was 25-48% increased on RA FLS (Figs. 1F–H). It was also shown that MΦs differentiated from RA monocytes, expressed higher cell surface levels of CCR9 (9-20x) (Figs. 1I). Consistent with CCL25, CCR9 cell surface levels were unaffected by inflammatory signals in endothelial cells (Fig. 1J). Taken together, RA FLS and MΦs, by secreting CCL25 and expressing cell-surface CCR9, are both producers and responders of this pathway.
Figure 1. The expression of CCL25 and CCR9 is accentuated in RA compared to NL specimens.
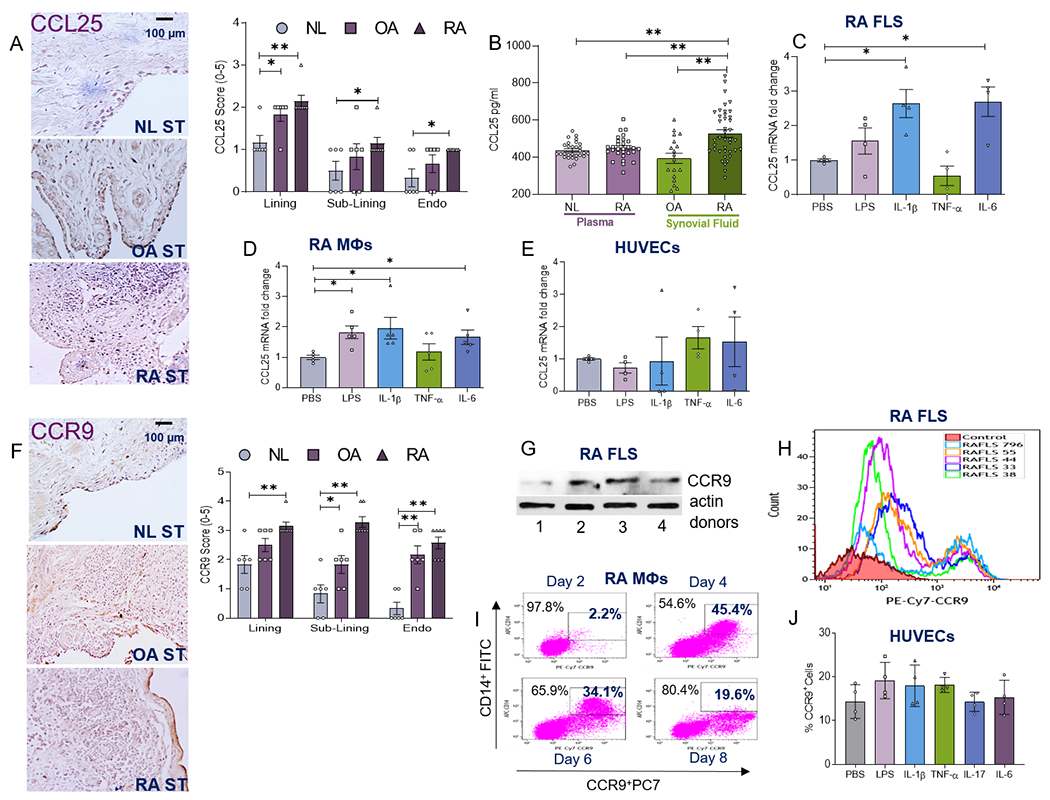
NL (n=6), OA (n=7) and RA (n=7) STs were immunostained with CCL25 Ab (1:50) (A) or CCR9 Ab (1:250) (F) and staining was scored on a 0-5 scale (scale bar= 100μm; original magnification×400), n=6–7 different tissues from 6 to 7 different NL, OA and RA donors in 3 different experiments. B. The protein concentration of CCL25 was quantified in NL plasma (n=29 different donors), RA plasma (n=29 different donors), OA SF (n=18 different donors) and RA SF (n=40 different donors) using R&D Systems Duoset ELISA in 3 different experiments. RA FLS (C), in vitro differentiated RA MΦs (D) or HUVECs (E) were untreated (PBS) or treated with 100ng/ml of LPS, IL-1β, TNFα, IL-6 for 6h before quantifying the expression of CCL25 by real-time RT-PCR and normalizing to GAPDH, (n=4–5 independent experiments) using 4 different RA FLS donors, MΦs obtained from 5 different RA patients and 4 independent experiments using HUVECs. G. CCR9 protein levels (1:500) were assessed in 4 different RA FLS donors compared to equal loading (actin) by Western blot analysis. H. Frequency of CCR9 (PE-Cy7 labeled CCR9 Ab, 1:200) positive cells were determined in 5 different RA FLS donors (796, 55, 44, 33 and 38) in 3 independent experiments. I. One representative of 3 different RA patients is shown as % CD14+CCR9+RA PB MΦs from days 2, 4, 6 & 8 in vitro differentiation by flow cytometry analysis, n=3 different RA MΦs were used in 3 independent experiments. J. HUVECs were untreated (PBS) or treated with 100ng/ml of LPS, IL-1β, TNFα or IL-6 for 24h prior to quantifying the expression of CCR9 by flow cytometry analysis, n=4 independent experiments. All the flow cytometry stainings were normalized to unstained viable cells. The data are shown as mean ± SEM, * represents p<0.05 and ** denotes p<0.01 using one-way ANOVA followed by Tukey’s multiple comparisons or Bonferroni test (Fig. A and F).
CCL25 attracts CCR9+ RA FLS via MAPK signaling
Given that CCR9 expression was expanded on RA FLS, we asked if the inflammatory factors present in RA SF could impact its frequency on these cells. Interestingly, several monokines including IL-1β, TNFα, IL-6 amplified the CCR9 cell surface expression on RA FLS by 4-6 fold (Fig. 2A). Next, the significance of CCL25 was examined in a model system relevant to FLS migration and its contribution to pannus formation. Using a scratch assay, CCL25 dose-dependently (250-500ng/ml) potentiated RA FLS migration by 10-20 fold (Figs. 2B–C). In contrast, RA FLS proliferation was unaffected by CCL25 stimulation, despite this process being amplified by bFGF treatment at 72h and 96h (Fig. 2D). Subsequently, the mechanism of CCL25-induced RA FLS migration was examined. While p38, ERK and JNK pathways were phosphorylated by CCL25 stimulation in RA FLS (Fig. 2E); activation of AKT1 and STAT3 was not implicated in this feature. In spite of, p38i and ERKi efficiency in obstructing CCL25-induced RA FLS migration (4-6x); JNKi was ineffective on this manifestation (Figs. 2F–G). In RA FLS, CCL25 inflammatory response was mediated by IL-8 (3.5x) secretion; however, levels of IL-6, CCL2, CCL5, MMP1 and MMP3 were unchanged (Fig. 2H). Consistent with FLS migration, only p38 and ERK pathways were involved in IL-8 production accentuated by CCL25 stimulation (Fig. 2I). Collectively, our findings indicate that phosphorylation of p38 and ERK by CCL25 escalates CCR9+ RA FLS recruitment and joint IL-8 secretion.
Figure 2. CCL25 promotes RA FLS recruitment via scratch assay and inflammatory response through p38 and ERK activation.
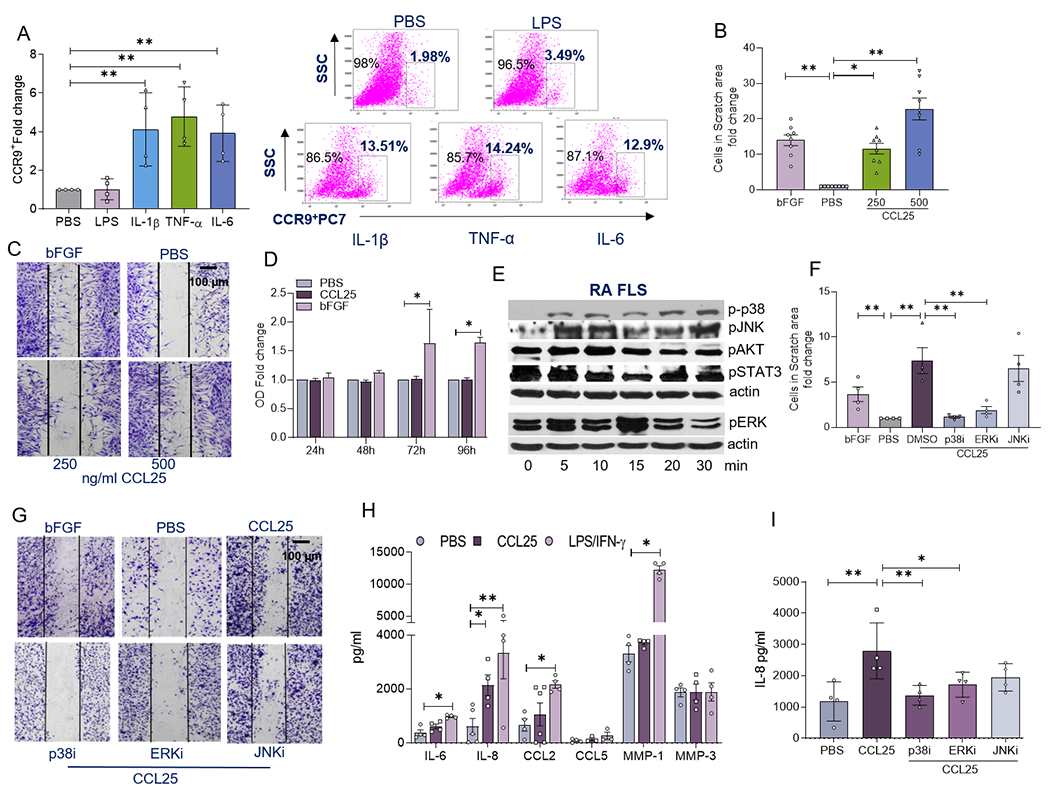
A. RA FLS were untreated (PBS) or treated with 100ng/ml of LPS, IL-1β, TNFα or IL-6 for 24h before quantifying % CCR9+ cells by flow cytometry analysis, n=4 independent experiments were performed using 4 different RA FLS lines. B and C. Representative images are shown for RA FLS scratch area assay where cells were untreated (PBS) or were treated with bFGF (+Ctl; 100ng/ml) or CCL25 (250–500ng/ml) for 24h and the number of cells was normalized to PBS in 4 different RA FLS patients used in 4 independent experiments. D. RA FLS were either untreated (PBS) or treated with CCL25 (500 ng/ml) or bFGF (+ control; 100ng/ml) for 24h, 48h, 72h or 96h prior to performing the MTT assay, using 4 different RA FLS donors in 2 independent experiments. E. RA FLS was stimulated with 250ng/ml of CCL25 from 0-30 min prior to detecting phosphorylation of p38, JNK, AKT, STAT3 and ERK in addition to equal actin loading by Western blot analysis, n=3-4 independent experiments were performed using 3 different RA FLS donors. Note that p-p38, pJNK, pAKT and pSTAT3 have the same actin; while pERK was detected on a different gel and has a different equal loading. F and G. RA FLS were untreated (PBS) or treated with bFGF (+control; 100ng/ml) and the experimental group consisted of CCL25 (250ng/ml) with DMSO or combined with 10μM of p38i (SB203580), ERKi (PD98059) or JNKi (SP600125) (pretreated for 18h before stimulation) for 24h, prior to determining the number of cells in the scratch area, n=4 independent experiments were performed using 4 different RA FLS. Data are shown as fold increase relative to the PBS control. H. RA FLS was untreated or treated with 100ng/ml of LPS/IFNγ or 250ng/ml of CCL25 for 24h before quantifying supernatant IL-6, IL-8, CCL2, CCL5, MMP1, MMP3 protein levels by ELISA (R&D Systems), n=3-6 independent experiments were performed using 3 to 4 different RA FLS. I. RA FLS was untreated or stimulated with CCL25 (250ng/ml) with DMSO or combined with 10μM of p38i (SB203580), ERKi (PD98059) or JNKi (SP600125) for 24h, before measuring IL-8 levels by ELISA, n=4 independent experiments were performed using 4 different RA FLS cell lines. The data are shown as mean ± SEM, * represents p<0.05 and ** denotes p<0.01 using one-way ANOVA followed by Tukey’s multiple comparisons.
CCL25 is a monocyte chemoattractant that cultivates a narrow range of M1 monokines
Since CCL25 and CCR9 are co-localized in RA MΦs, studies were conducted to elucidate their mechanism of action. Differentiation of RA monocytes into MΦs upregulates CCR9 expression (Fig. 3A–B). Conversely, CCR9 cell surface expression was not influenced by inflammatory factors or RA SF stimulation in RA MΦs (Fig. 3C–D, Supplementary 1; Fig. S6). Next, we asked whether TNFi therapy would impact CCR9 and CCL25 function in RA blood cells. For this purpose, transcription levels of CCR9 and CCL25 were compared in RA patients pre- and post-TNFi therapy. We found that CCR9 and CCL25 transcription was unchanged in RA PBMCs (+/− DMARDs) prior to and following TNFi therapy (Figs. 3E–F). Corroborating our findings in RA FLS, monocyte trafficking was dose-responsively expanded by CCL25 stimulation (Fig. 3G). Interestingly like RA FLS, phosphorylation of p38, ERK and JNK were enhanced by CCL25 signaling in RA MΦs without affecting AKT and STAT3 activation (Fig. 3H). However, unlike RA FLS, inhibition of all MAPK pathways was implicated in CCL25-driven monocyte infiltration (Fig. 3I). IL-8 and CCL2 (2x) production was accentuated in CCL25-stimulated RA MΦs, while expression of TNFα, IL-6, CCL5, MMP1 and MMP9 was unaffected (Fig. 3J). In RA MΦs, CCL25-modulated IL-8 and CCL2 secretion were dependent on p38 and ERK phosphorylation, however, JNK signaling was not involved in this process (Fig. 3K–L). Interestingly, neither IL-8 nor CCL2 protein levels were potentiated by CCL25-stimulation in RA FLS and MΦ coculture compared to PBS treatment (Supplementary 1; Figs. S1 & S2). Moreover, zymosan-induced RA MΦ phagocytosis was suppressed by CCL25 treatment (Fig. 3M). In short, our results indicate that the standard of care, TNFi therapy, does not affect the CCL25/CCR9 pathway and that this chemokine and receptor pair manipulates the infiltrating RA monocytes to advance inflammation and repress phagocytosis.
Figure 3. Activation of p38 and ERK pathways fosters CCL21-induced RA monocyte infiltration and differentiation into a specific M1 MΦ profile.
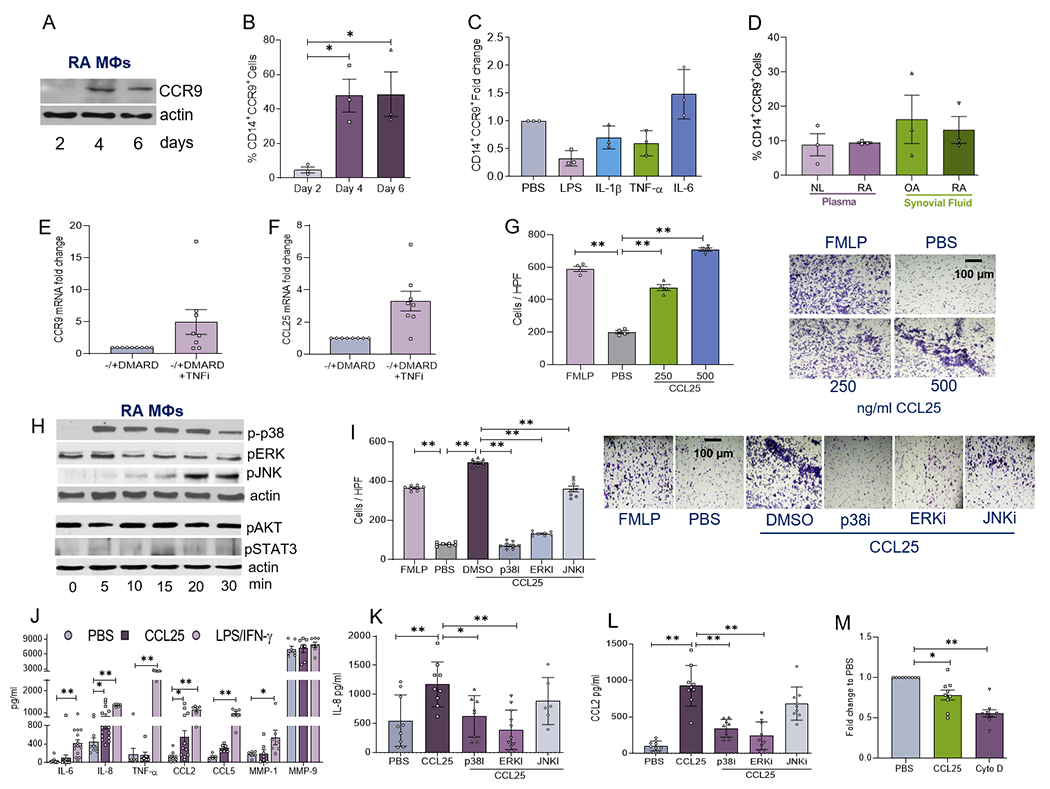
CCR9 protein expression was analyzed by Western blotting and normalized to actin (A) or % CD14+CCR9+cells were evaluated by flow cytometry (B) in RA MΦ progenitor cells differentiated over 2-6 days, n=3 independent experiments from 5 different RA patients. RA PB in vitro differentiated MΦs were untreated (PBS) or treated with 100ng/ml of LPS, IL-1β, TNFα or IL-6 (C) or 1:10 of NL plasma, RA plasma, OA SF or RA SF (D) for 24h before quantifying % of CD14+CCR9+ by flow cytometry analysis. Data are shown as fold increase above PBS control, n=3 independent experiments from 3 different RA patients. Expression of CCR9 (E) and CCL25 (F) was analyzed by qPCR and normalized to GAPDH in PBMCs from RA patients on ±DMARDS and following 3-month of TNFi therapy, n=8 different RA patients in 3 experiments (please see M&M). G. Chemotaxis was performed using NL PB monocytes in response to PBS, FMLP (+Ctl, 50nM), CCL25 (250-500ng/ml) for 2h and cells were counted in 3 HPF/condition, n=4 independent experiments from 4 different RA patients. H. RA PB in vitro differentiated MΦs were activated with 250ng/ml of CCL25 from 0-30 min before detecting phosphorylation of p38, ERK, JNK, AKT and STAT3 in addition to equal actin loading by Western blot analysis, n=4 independent experiments using 4 different donors. Note that p-p38, pERK and pJNK have the same actin; while pAKT and pSTAT3 were detected on a different gel and have a different equal loading. I. Chemotaxis was performed using NL PB monocytes in response to PBS or bFGF (+control; 100ng/ml) and the experimental group c2onsisted of CCL25 (250ng/ml) with DMSO or combined with 10μM of p38i (SB203580), ERKi (PD98059) or JNKi (SP600125) (pretreated 1h before the assay) for 24h, prior to determining the number of cells in 3 HPF, n=8 independent experiments with 1 donor per experiment. J. RA PB in vitro MΦs was untreated, stimulated with 100ng/ml of LPS/IFNγ or 250ng/ml of CCL25 for 24h before quantifying the supernatant levels of IL-6, IL-8, TNFα, CCL2, CCL5, MMP1, MMP9 by ELISA (R&D Systems), n=10-15 independent experiments from 10 to 15 different patients. RA PB in vitro MΦs was untreated, stimulated with 250ng/ml of CCL25 with DMSO or combined with 10μM of p38i (SB203580), ERKi (PD98059) or JNKi (SP600125) for 24h, prior to measuring IL-8 (K) or CCL2 (L) levels by ELISA, n=8-10 independent experiments from 8 to 10 different patients. M. Phagocytosis was assayed in RA PB in vitro MΦs by measuring the engulfment of Zymosan in absence of treatment (PBS) or presence of CCL25 (250ng/ml) or Cytochalasin D (-Ctl, 2μM) for 6h following the manufacturer’s instruction, n=9 independent experiments from 9 different RA patients. The data are shown as mean ± SEM, * represents p<0.05 and ** denotes p<0.01 using one-way ANOVA followed by Tukey’s multiple comparisons.
CCL25 ligation transforms CCR9+ myeloid progenitor cells into mature osteoclasts
CCL25 and CCR9 play an important role in T effector cell function, thus experiments were performed to test their influence on RA Th1/Th17 cell differentiation. While the positive controls IL-12 and IL-1β+IL-6+TGFβ enhanced polarization of RA Th1 and Th17 cells, this component was unaffected by CCL25 stimulation (Figs. 4A–C). Supporting this notion, the lymphoid nuclear factor of activated T-cells (NFATc) was not induced in CCL25-activated osteoclast progenitor cells (Fig. 4D). On the contrary, transcriptional upregulation of myeloid receptor activator of nuclear factor κ B (RANK) and Cathepsin K (CTSK) initiates CCL25-mediated osteoclast formation (Fig. 4D). Due to potentiated IL-8 and CCL2 production and MAPK activation in CCL25 stimulated RA myeloid cells, we characterized the impact of these monokines and signaling pathways on RA osteoclastogenesis. Surprisingly, while Abs against CCL2 and IL-8 did not impair CCL25-induced osteoclast maturation, this function was abrogated by MAPK inhibitors (Fig. 4E). It was also noted that in addition to RANK and CTSK, the expression of TNFα (2x) was elevated by CCL25 once RA osteoclasts were fully differentiated (Fig. 4F). Lack of NFATc transcription in CCL25-driven myeloid progenitor cells (Fig. 4D) was consistent with the absence of RANKL induction in osteoclasts formed by CCL25 (Fig. 4F). These findings highlight the importance of myeloid cells and lack of T cell involvement in CCL25-induced RA osteoclast formation.
Figure 4. CCL25-mediated osteoclastogenesis is mainly regulated by myeloid cells.
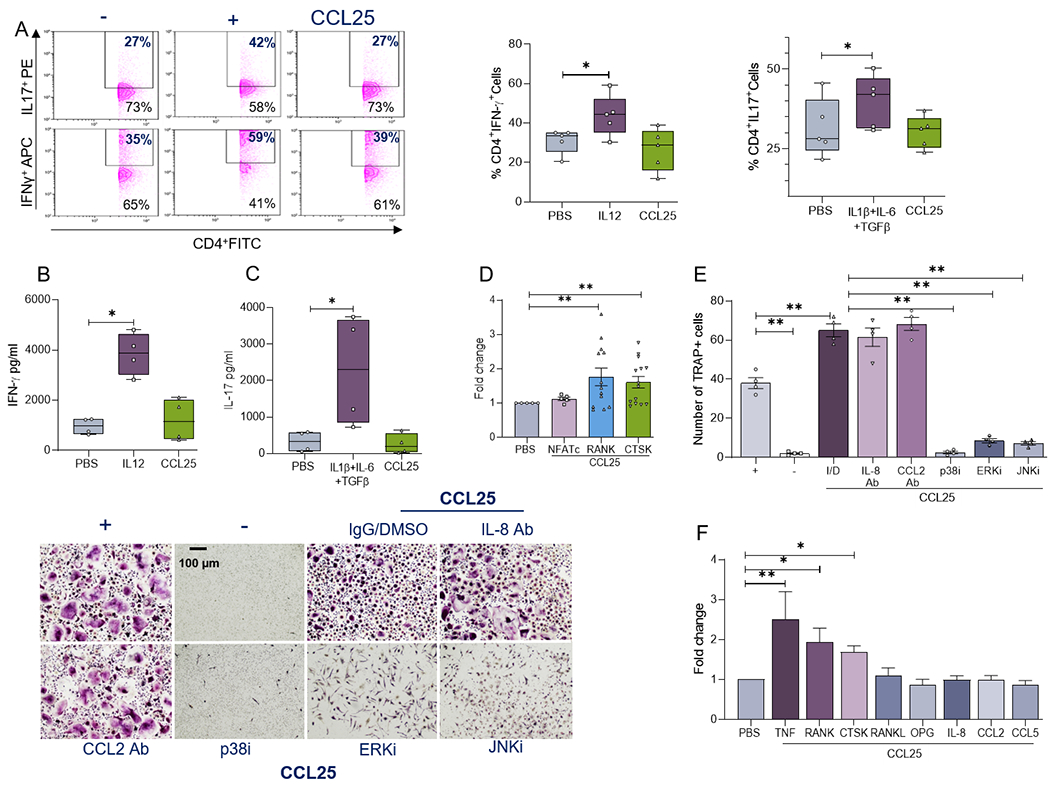
RA PBMCs were cultured in presence of CD3 Ab plus CD28 Ab (0.25μg/ml each) which were untreated (PBS) or treated with IL-12 (10ng/ml; Th1 +Ctl), IL-1β+IL-6 (20ng/ml)+TGFβ (4ng/ml; Th17+Ctl) or CCL25 (250ng/ml). Following 4-5 days of treatment, frequency of CD4+IFNγ+(Th1) and CD4+IL-17+(Th17) cells was determined by flow cytometry, n=5 independent experiments from 5 different RA patients (A) and secretion of IFNγ (B) or IL-17 (C) was measured in supernatants by ELISA, n=4 independent experiments from 4 different RA patients. D. RA osteoclast progenitor cells were cultured in suboptimal condition (10ng/ml M-CSF+RANKL) for 7 days prior to PBS or CCL25 (250ng/ml) stimulation for 6h. Levels of NFATc, RANK and CTSK were determined by real-time RT-PCR and normalized to GAPDH, n=5-15 independent experiments from 5 to 15 RA patients. E. RA osteoclast progenitor cells cultured in suboptimal condition was treated with CCL25 (250ng/ml) in presence of IgG/DMSO (I/D), 10μg/ml of IL-8 Ab or CCL2 Ab (R&D Systems) as well as 10μM of p38i (SB203580), ERKi (PD98059) or JNKi (SP600125) for 14-21 days prior to TRAP staining, n=4 independent experiments from 4 different RA patients. F. Mature RA osteoclasts were treated with PBS or CCL25 (250ng/ml) for 6h prior to quantifying transcription of TNF, RANK, CTK, RANKL, OPG, IL-8, CCL2 and CCL5 by real-time RT-PCR and normalized to GAPDH, n=10-12 independent experiments from 10 to 15 different RA patients. The data are shown as mean ± SEM, * represents p<0.05 and ** denotes p<0.01 using one-way ANOVA followed by Tukey’s multiple comparisons.
DISCUSSION
The current study describes a hitherto undefined role for CCL25 and CCR9 in RA pannus formation. Expression of CCL25 and CCR9 was potentiated in RA compared to NL ST FLS and MΦs. Intriguingly, CCL25 and CCR9 levels were highly responsive to inflammatory stimuli in RA FLS, and both were mutually amplified by IL-1β and IL-6 stimulation. On the contrary, in RA MΦs, cell surface expression of CCR9 was unresponsive to inflammatory stimuli, while its levels were upregulated by the differentiation. Nevertheless, certain monokines such as IL-1β and IL-6, but not TNF, upregulated CCL25 transcription in RA MΦs. In line with these observations, the expression of CCL25 and CCR9 in RA PBMCs was unaffected by longitudinal TNFi therapy. We exhibited that CCL25-induced p38 and ERK phosphorylation was responsible for joint infiltration of RA FLS and monocytes in addition to the secretion of proangiogenic factors, IL-8 and CCL2, from these cells. Furthermore, activation of MAPK and induction of RANK, CTK and TNF were implicated in CCL25-induced osteoclastogenesis (Fig. 5).
Figure 5. Ligation of CCL25 to CCR9+ RA FLS and MΦ can potentially advance pannus formation.
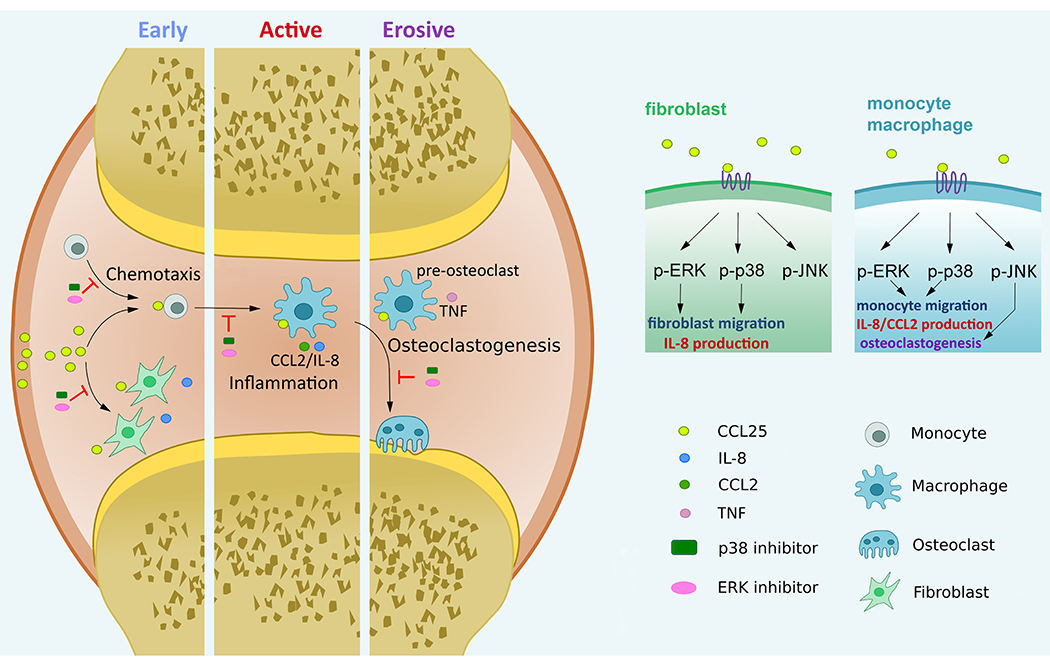
CCL25 induces RA FLS and monocyte migration and contributes to an inflammatory phenotype orchestrated by RA FLS and MΦ crosstalk. RA osteoclast formation driven by CCL25 is instigated through transcriptional upregulation of RANK, CTSK and TNF, which is accompanied by activation of p38 and ERK pathways.
Earlier studies have observed that CCL25 and CCR9 concentrations were elevated in RA compared to OA specimen [23]; however, their cellular expression and mechanism of function was undefined. Expression of CCL25 & CCR9 was detected in the small intestine; where they contribute to dendritic & T cell infiltration as well as promoting T cell homing to the thymus and spleen [24]. Moreover, CCR9 is present in melanoma skin lesions and on lymphoblastic leukemia cells and its ligation to CCL25 facilitates cancer cell polarization, microvilli absorption and tumor metastasis [25, 26].
Interestingly, elevated expression of CCL25 in cutaneous basal cell carcinoma patients is in part responsible for fibroblast influx and tissue matrix remodeling [27]. Others have shown that thioglycollate induced inflammation, escalates the frequency of CD11b+F480+CCR9+ macrophages in the peritoneal cavity [28]. CCR9+ MΦs played an integral role in liver inflammation instigated in preclinical hepatitis [29]. Extending these observations, elevated CCL25 levels in RA SF, attracted CCR9+ MΦs and RA FLS into the joints. In contrast to earlier findings in colitis [30, 31] or cancer [32], where CCR9 is primarily expressed on intestinal T cells or has a lymphocytic origin, neither CCL25 nor CCR9 was detected in RA or OA joint T cells. Mechanistically, retinoic acid increases CCR9 expression on mouse naive CD4+ T cells and this function is dependent on NFATC2 and retinoic acid receptors [33]. On the contrary, IL-2 and IL-4 treatment downregulate the expression of CCR9 expression on CD4+ T cells [34]. Although the mechanism of CCL25/CCR9 function has been mainly focused on T cells; in RA STs it is MΦs and FLS that are the targets cells for this pathway.
Stimulation with LPS, IL-1β and IL-6, markedly enhanced CCL25 expression on RA MΦs. In opposition to previous studies [35], expression of CCR9 or CCL25 was not impacted by TNFα treatment in RA MΦs. Corroborating these results, transcription levels of CCR9 and CCL25 were unchanged in RA PBMCs following 3-month of TNFi therapy. While inflammatory stimuli were ineffective on myeloid CCR9 cell surface expression, these levels were upregulated when RA monocytes differentiated into mature MΦs. Notably, in RA FLS, IL-1β and IL-6 were the overlapping factors that amplified both CCR9 and CCL25 levels. Although CCL25 and CCR9 have not been implicated in the RA FLS function, it has been shown that CCR9+ MΦs advanced liver fibrosis through the expression of collagen1α1, transforming growth factor (TGF)β1 and tissue inhibitor of metalloproteinase (TIMP)1 [36]. In agreement with this notion, MΦ-modulated liver inflammation was mitigated in CCR9−/− compared to WT mice [29]. These studies indicate that CCL25/CCR9 activation contributes to crosstalk between MΦs and fibroblasts.
We demonstrate that joint CCL25 facilitates RA FLS and monocyte migration and triggers IL-8 and/or CCL2 production, via phosphorylation of ERK and p38 pathways. Nonetheless, in RA FLS and MΦs, AKT and STAT3 signaling pathways were not activated by CCL25 signaling. Conversely, others have shown that CCL25-induced AKT phosphorylation triggers NF-κB and mTOR activation; leading to an anti-apoptotic mechanism in lung cancer [37] and chemoresistance in prostate cancer [38]. However, in an experimental model of myocardial infarction (MI), cardiac remodeling in WT tissues exhibited greater ERK and p38 phosphorylation compared to healthy or MI tissues harvested from CCR9−/− mice [39]. These findings indicate that the differential signaling pathways activated by CCL25 in diseased tissues are primarily dependent on the cell types that express CCR9 and respond to CCL25 stimulation.
Contrasting RA FLS migration, the JNK pathway is uniquely involved in CCL25-induced monocyte migration. Supporting our findings, the elevation of cardiac CD68+ MΦs was detected in WT compared to CCR9−/− mice induced with MI [39]. Earlier studies have shown that NL monocytes stimulated with CCL25 produce increased TNF and IL-6 levels [17]. Opposing these findings, induction of sepsis in CCR9−/− mice resulted in greater disease severity and higher peritoneal production of IL-6, TNF and CXCL10 relative to WT mice [28]. RA MΦs polarized by CCL25 exhibit a non-traditional M1 profile that expresses IL-8 and CCL2; while IL-6, TNFα, CCL5, MMP1 and MMP9 production levels remained unchanged. We found that zymosan ligation to myeloid TLR2 and TLR6 and stimulation of phagocytosis [40] was suppressed by CCL25. We postulate that the suppressed antigen presentation observed in CCL25-activated RA MΦs may be the consequence of its potentiated inflammatory response. Much like CCL25’s incapability to potentiate phagocytosis, RA CCR9+ PBMCs were not differentiated into effector Th1/Th17 cells by CCL25 stimulation. Our results suggest that CCL25/CCR9-induced pathology is differentially regulated in RA compared to SS [16] or EAE [10]. In SS patients, IL-7R elevation in CCR9+ T cells and response to IL-7 and antigen were responsible for Th17 cell polarization [16]. In the EAE model, TLR4 induced upregulation of CCL25 and CCR9 was involved in Th17 cell infiltration [10]. We suspect that the lack of Th1/Th17 master regulators (IL-12, IL-6, IL-1β) is accountable for CCL25’s inability to polarize RA naïve cells into Th1/Th17 cells.
Lack of T cell participation is also evident in CCL25-driven osteoclast formation, as the maturation of the progenitor cells is accentuated by RANK and CTSK but not RANKL or NFATc. It is also notable that TNF is expressed in osteoclast matured by CCL25, while its expression was undetectable in CCL25-differentiated MΦs. Taken together, the CCL25/CCR9 pathway interconnects RA MΦ and FLS function to potentially advance pannus formation and neovascularization.
MATERIALS AND METHODS
Ethical Approval:
Patients were recruited from the practices of orthopedic surgeons or rheumatologists in the group practice of the academic physicians of the University of Illinois at Chicago. STs and fibroblasts extracted from the synovium were obtained from individuals undergoing total joint replacement or synovectomy. RA patients were diagnosed according to the 1987 revised criteria from the ACR [41]. All studies were approved by the University of Illinois at Chicago Institutional Review Board and all donors provided written informed consent. Data recorded at the time of the ST collection are the date of collection and patient diagnosis. RA ST or SF samples submitted to our research required no special handling.
Immunohistochemistry:
STs from RA, OA and NLs were de-identified and were formalin-fixed, paraffin-embedded and sectioned. Briefly, slides were deparaffinized in xylene and antigens were unmasked by incubating slides in Proteinase K digestion buffer (Dako, CA). STs were stained with CCL25 (TECK, 1:50; Santa Cruz ), CCR9 (1:250, R&D Systems) or control IgG Abs and staining was scored on a 0-5 scale [42] by two blinded observers. Where 0= normal appearance, 1= minimal changes, 2= mixed appearance, 3= moderate changes, 4= marked changes and 5= severe changes [43].
Normal and RA cell isolation:
Studies were approved by the University of Illinois at Chicago (UIC) IRB board and all donors gave informed written consent. RA patients were diagnosed according to the 1987 revised criteria from the ACR [41]. RA peripheral blood was drawn into tubes containing citrate phosphate dextrose solution. Mononuclear cells were separated by histopaque gradient centrifugation and monocytes were isolated from NL or RA peripheral blood (PB) using a negative selection kit without CD16 depletion (StemCell Technology, Cat#19058) according to the manufacturer’s instruction [42, 43]. RA PB MΦs were differentiated in vitro in the media containing 20% FBS.
RA patient population:
Peripheral blood was obtained from patients with RA, diagnosed as aforementioned [41]. Blood was obtained from 6 women and 2 men with an average age of 60.4 years. At baseline, patients were treated with methotrexate (n=1), no therapy (n=1), corticosteroid (n=2), methotrexate plus corticosteroid (n=2), leflunomide, hydroxychloroquine plus corticosteroid (n=1), and methotrexate, sulfasalazine, hydroxychloroquine plus corticosteroid (n=1). Following the 3-month of TNFi therapy, blood was procured from the same RA patients (n=8). These studies were approved by the UIC IRB board and all donors gave informed written consent.
ELISA:
Human CCL25, IL-8, IL-6, TNFα, CCL2, CCL5, MMP1 and MMP9 protein levels were quantified by ELISA according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN, USA).
Western blot analysis:
To determine CCL25 signaling, RA FLS or MΦs were untreated or stimulated with CCL25 (250ng/ml) for 0-30 min and phosphorylation of p38, ERK, JNK, Akt, STAT3 (Cell Signaling Technology, 1:1000) was determined by Western blotting. In a different experiment, RA MΦ progenitor cells in culture were harvested on days 2, 4, 6 and 8 following differentiation. The collected lysates were probed for CCR9 (R&D Systems, 1:500) and actin (1:3000).
MTT assay:
To determine the impact of CCL25 on RA ST fibroblast proliferation, MTT assay was performed. Cells were plated to 60–70% confluency in a 96 well plate and were either untreated (PBS) or treated with CCL25 (500 ng/ml) or bFGF (+ control; 100 ng/ml). After 24, 48, 72 or 96h of treatment, MTT solution (5mg/ml) was added into each well for 3 h. Thereafter, the MTT reaction was stopped and the OD was determined at 570nm [44]. The data in each time point are shown as fold increase above the counterpart control.
Flow cytometry analysis:
Human Th1 and Th17 cell frequency was quantified in RA PBMCs cultured in 0.25μg/ml of CD3 Ab and CD28 Ab (Biolegend) and cells were untreated (PBS) or treated with IL-12 (20ng/ml; +control for Th1), (4ng/ml) TGFβ+IL-6+IL-1β (R&D, 20ng/ml each; +control for Th17) or CCL25 (250ng/ml) for 4-5 days. Prior to staining, cells were treated with BFA (5μg/ml), PMA (50ng/ml) and ionomycin (1μg/ml) for 4h. Next, FITC-conjugated CD4 Ab (Biolegend) staining, cells were fixed and permeabilized using cyto-fastfix/perm buffer set (Biolegend cat#426803) and RA PBMCs were stained with APC-conjugated IFNγ or PE-labeled IL-17 Ab (Invitrogen). In a different experiment, RA FLS, HUVECs or RA MΦs were untreated or treated with 100ng/ml of LPS, IL-1β, TNFα or IL-6 for 24h prior to quantifying the frequency of viable CCR9+ cells (Biolegend, 1:200) or CD14+ (Biolegend, 1:1000) CCR9+ cells. For extracellular staining, DAPI or 7AAD negative cells were regarded as viable and for intracellular staining zombie violet negative cells (before fixation and permeabilization) were considered as viable. All the stainings were normalized to viable unstained cells. Data are presented at % CCR9+ cells in RA FLS, HUVECs or RA MΦs. Gating strategy for CD14+ MΦs, RA FLS, HUVECs and CD4+ T cells are shown in supplementary 2–4.
RA ST fibroblast scratch assay:
Fibroblasts from fresh RA ST were isolated by mincing and digestion in a solution of Dispase, collagenase, and DNase. Cells were used between passages 3-9 [42–45]. A scratch was created in the middle of the wells that contained confluent RA FLS [46]. Thereafter, RA FLS were untreated (PBS) or treated with bFGF (+control; 100ng/ml) and the experimental group consisted of CCL25 (250 or 500ng/ml) with DMSO or combined with 10μM of p38i (SB203580), ERKi (PD98059) or JNKi (SP600125) for 24h. Subsequently, cells were fixed for 1h with 10% formalin and stained for 1h with 0.05% crystal violet before imaging with NIS-Elements Software from Nikon. The cell number in the scratch area was counted and compared to the untreated control [44, 46].
Real-time RT-PCR:
RNA isolated using TRIzol (Life Technologies) was reverse transcribed to cDNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems) for subsequent qRT-PCR analysis. We used Taqman primer/probe mixes predesigned by Integrated DNA Technologies and Taqman gene expression master mix (Applied Biosystems) to perform qRT-PCR. Data are presented as fold changes in RNA levels compared to control treatment, calculated following the 2–ΔΔCt method. The expression level of each gene was normalized to GAPDH.
HUVECs:
Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza and used at passages 3-6. Confluent HUVECs were untreated (PBS) or treated with 100ng/ml of LPS, IL-1β, TNFα or IL-6 for 6h or 24h before quantifying the expression of CCL25 and CCR9 by real-time RT-PCR or flow cytometry analysis.
Transwell monocyte migration assay:
Chemotaxis was assayed in 24-well transwell. Briefly, blood monocytes were negatively selected and loaded onto inserts at 1×106 cells. PBS (-Ctl), FMLP (+Ctl, 50nM), CCL25 (250 or 500ng/ml) with DMSO or combined with 10μM of p38i (SB 203580), ERKi (PD98059) or JNKi (SP600125) (inhibitors were pre-incubated 1h prior to initiation) was placed in the lower compartment. Following 2h of incubation, cells on the inserts were detached and cells migrated to the lower surface were fixed with 10% formalin for 1h. Thereafter, the migrated cells were stained with 0.05% crystal violet, imaged and counted.
Phagocytosis:
Phagocytosis was assayed in RA MΦs by measuring the engulfment of Zymosan in absence of treatment (PBS) or presence of CCL25 (250ng/ml) or Cytochalasin D (-Ctl, 2μM) for 6h following the manufacturer’s instruction (Cell Biolabs).
RA osteoclast differentiation:
To generate mature osteoclasts, RA monocytes cultured in 0% FBS RPMI media were allowed to attach for 1h. Thereafter, cells were cultured in αMEM (10% FBS) and were treated with 20ng/ml of human M-CSF & RANKL (+control) for 14-21 days and the culturing media was replaced every 3-4 days. In addition, cells in suboptimal conditions (10ng/ml of M-CSF+RANKL) were cultured in the presence of the test reagents to determine their ability to form mature osteoclasts for 14-21 days. To quantify osteoclast formation, TRAP staining was performed according to the manufacturer’s instruction (Sigma-Aldrich). In vitro experiments were performed in triplicates and the total number of osteoclasts was quantified by counting TRAP+ multinuclear (> 3 nuclei) cells in each well [47–49].
Statistical Analysis:
For comparison among multiple groups, one-way ANOVA followed by Tukey’s multiple comparisons or Bonferroni test was employed, using Graph Pad Prism 8 software. The data were also analyzed using a two-tailed Student’s t-test for paired or unpaired comparisons between two groups. Values of p < 0.05 were considered significant.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by awards from the Department of Veteran’s Affairs MERIT Award BX002286, the National Institutes of Health NIH AI147697, AR056099 and AR065778 and the National Psoriasis Foundation (NPF), Pfizer Investigator-Initiated Research (IIR) Program and Chicago Biomedical Consortium (CBC) Accelerator Award.
Footnotes
CONFLICT OF INTEREST
The authors have declared that no commercial or financial conflict of interest exists.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are presented in this paper. No shared databases were used or created.
REFERENCES
- 1.Brennan FM and McInnes IB, Evidence that cytokines play a role in rheumatoid arthritis. Journal of Clinical Investigation 2008. 118: 3537–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McInnes IB and Schett G, The pathogenesis of rheumatoid arthritis. N Engl J Med 2011. 365: 2205–2219. [DOI] [PubMed] [Google Scholar]
- 3.Papadakis KA, Prehn J, Nelson V, Cheng L, Binder SW, Ponath PD, Andrew DP and Targan SR, The role of thymus-expressed chemokine and its receptor CCR9 on lymphocytes in the regional specialization of the mucosal immune system. J Immunol 2000. 165: 5069–5076. [DOI] [PubMed] [Google Scholar]
- 4.Kunkel EJ, Campbell DJ and Butcher EC, Chemokines in lymphocyte trafficking and intestinal immunity. Microcirculation 2003. 10: 313–323. [DOI] [PubMed] [Google Scholar]
- 5.Stenstad H, Svensson M, Cucak H, Kotarsky K and Agace WW, Differential homing mechanisms regulate regionalized effector CD8alphabeta+ T cell accumulation within the small intestine. Proc Natl Acad Sci U S A 2007. 104: 10122–10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Svensson M, Marsal J, Ericsson A, Carramolino L, Broden T, Marquez G and Agace WW, CCL25 mediates the localization of recently activated CD8alphabeta(+) lymphocytes to the small-intestinal mucosa. J Clin Invest 2002. 110: 1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wendland M, Czeloth N, Mach N, Malissen B, Kremmer E, Pabst O and Forster R, CCR9 is a homing receptor for plasmacytoid dendritic cells to the small intestine. Proc Natl Acad Sci U S A 2007. 104: 6347–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trivedi PJ, Bruns T, Ward S, Mai M, Schmidt C, Hirschfield GM, Weston CJ and Adams DH, Intestinal CCL25 expression is increased in colitis and correlates with inflammatory activity. J Autoimmun 2016. 68: 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng L, Zhang X, Chen J, Ichikawa R, Wallace K, Pothoulakis C, Koon HW, Targan SR and Shih DQ, Sustained Tl1a (Tnfsf15) Expression on Both Lymphoid and Myeloid Cells Leads to Mild Spontaneous Intestinal Inflammation and Fibrosis. Eur J Microbiol Immunol (Bp) 2013. 3: 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Han J, Wu M, Xu L, Wang Y, Yuan W, Hua F, Fan H, Dong F, Qu X and Yao R, Toll-Like Receptor 4 Promotes Th17 Lymphocyte Infiltration Via CCL25/CCR9 in Pathogenesis of Experimental Autoimmune Encephalomyelitis. J Neuroimmune Pharmacol 2019. 14: 493–502. [DOI] [PubMed] [Google Scholar]
- 11.Wurbel MA, McIntire MG, Dwyer P and Fiebiger E, CCL25/CCR9 interactions regulate large intestinal inflammation in a murine model of acute colitis. PLoS One 2011. 6: e16442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papadakis KA, Prehn J, Moreno ST, Cheng L, Kouroumalis EA, Deem R, Breaverman T, Ponath PD, Andrew DP, Green PH, Hodge MR, Binder SW and Targan SR, CCR9-positive lymphocytes and thymus-expressed chemokine distinguish small bowel from colonic Crohn’s disease. Gastroenterology 2001. 121: 246–254. [DOI] [PubMed] [Google Scholar]
- 13.Saruta M, Yu QT, Avanesyan A, Fleshner PR, Targan SR and Papadakis KA, Phenotype and effector function of CC chemokine receptor 9-expressing lymphocytes in small intestinal Crohn’s disease. J Immunol 2007. 178: 3293–3300. [DOI] [PubMed] [Google Scholar]
- 14.McGrory K, Flaitz CM and Klein JR, Chemokine changes during oral wound healing. Biochem Biophys Res Commun 2004. 324: 317–320. [DOI] [PubMed] [Google Scholar]
- 15.Otten K, Dragoo J, Wang HC and Klein JR, Antigen-induced chemokine activation in mouse buccal epithelium. Biochem Biophys Res Commun 2003. 304: 36–40. [DOI] [PubMed] [Google Scholar]
- 16.Blokland SLM, Hillen MR, Kruize AA, Meller S, Homey B, Smithson GM, Radstake T and van Roon JAG, Increased CCL25 and T Helper Cells Expressing CCR9 in the Salivary Glands of Patients With Primary Sjogren’s Syndrome: Potential New Axis in Lymphoid Neogenesis. Arthritis Rheumatol 2017. 69: 2038–2051. [DOI] [PubMed] [Google Scholar]
- 17.Yokoyama W, Kohsaka H, Kaneko K, Walters M, Takayasu A, Fukuda S, Miyabe C, Miyabe Y, Love PE, Nakamoto N, Kanai T, Watanabe-Imai K, Charvat TT, Penfold ME, Jaen J, Schall TJ, Harigai M, Miyasaka N and Nanki T, Abrogation of CC chemokine receptor 9 ameliorates collagen-induced arthritis of mice. Arthritis Res Ther 2014. 16: 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amersi FF, Terando AM, Goto Y, Scolyer RA, Thompson JF, Tran AN, Faries MB, Morton DL and Hoon DS, Activation of CCR9/CCL25 in cutaneous melanoma mediates preferential metastasis to the small intestine. Clin Cancer Res 2008. 14: 638–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh R, Stockard CR, Grizzle WE, Lillard JW Jr. and Singh S, Expression and histopathological correlation of CCR9 and CCL25 in ovarian cancer. Int J Oncol 2011. 39: 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tu Z, Xiao R, Xiong J, Tembo KM, Deng X, Xiong M, Liu P, Wang M and Zhang Q, CCR9 in cancer: oncogenic role and therapeutic targeting. J Hematol Oncol 2016. 9: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson-Holiday C, Singh R, Johnson EL, Grizzle WE, Lillard JW Jr. and Singh S, CCR9-CCL25 interactions promote cisplatin resistance in breast cancer cell through Akt activation in a PI3K-dependent and FAK-independent fashion. World J Surg Oncol 2011. 9: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen HJ, Edwards R, Tucci S, Bu P, Milsom J, Lee S, Edelmann W, Gumus ZH, Shen X and Lipkin S, Chemokine 25-induced signaling suppresses colon cancer invasion and metastasis. J Clin Invest 2012. 122: 3184–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Endres M, Andreas K, Kalwitz G, Freymann U, Neumann K, Ringe J, Sittinger M, Haupl T and Kaps C, Chemokine profile of synovial fluid from normal, osteoarthritis and rheumatoid arthritis patients: CCL25, CXCL10 and XCL1 recruit human subchondral mesenchymal progenitor cells. Osteoarthritis Cartilage 2010. 18: 1458–1466. [DOI] [PubMed] [Google Scholar]
- 24.Zabel BA, Agace WW, Campbell JJ, Heath HM, Parent D, Roberts AI, Ebert EC, Kassam N, Qin S, Zovko M, LaRosa GJ, Yang LL, Soler D, Butcher EC, Ponath PD, Parker CM and Andrew DP, Human G protein-coupled receptor GPR-9-6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine-mediated chemotaxis. J Exp Med 1999. 190: 1241–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou B, Leng J, Hu M, Zhang L, Wang Z, Liu D, Tong X, Yu B, Hu Y, Deng C, Liu Y and Zhang Q, Ezrin is a key molecule in the metastasis of MOLT4 cells induced by CCL25/CCR9. Leuk Res 2010. 34: 769–776. [DOI] [PubMed] [Google Scholar]
- 26.Heinrich EL, Arrington AK, Ko ME, Luu C, Lee W, Lu J and Kim J, Paracrine Activation of Chemokine Receptor CCR9 Enhances The Invasiveness of Pancreatic Cancer Cells. Cancer Microenviron 2013. 6: 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Omland SH, Wettergren EE, Mollerup S, Asplund M, Mourier T, Hansen AJ and Gniadecki R, Cancer associated fibroblasts (CAFs) are activated in cutaneous basal cell carcinoma and in the peritumoural skin. BMC Cancer 2017. 17: 675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizukami T, Kanai T, Mikami Y, Hayashi A, Doi T, Handa T, Matsumoto A, Jun L, Matsuoka K, Sato T, Hisamatsu T and Hibi T, CCR9+ macrophages are required for eradication of peritoneal bacterial infections and prevention of polymicrobial sepsis. Immunol Lett 2012. 147: 75–79. [DOI] [PubMed] [Google Scholar]
- 29.Nakamoto N, Ebinuma H, Kanai T, Chu PS, Ono Y, Mikami Y, Ojiro K, Lipp M, Love PE, Saito H and Hibi T, CCR9+ macrophages are required for acute liver inflammation in mouse models of hepatitis. Gastroenterology 2012. 142: 366–376. [DOI] [PubMed] [Google Scholar]
- 30.Park C, Cheung KP, Limon N, Costanzo A, Barba C, Miranda N, Gargas S, Johnson AMF, Olefsky JM and Jameson JM, Obesity Modulates Intestinal Intraepithelial T Cell Persistence, CD103 and CCR9 Expression, and Outcome in Dextran Sulfate Sodium-Induced Colitis. J Immunol 2019. 203: 3427–3435. [DOI] [PubMed] [Google Scholar]
- 31.Igaki K, Komoike Y, Nakamura Y, Watanabe T, Yamasaki M, Fleming P, Yang L, Soler D, Fedyk E and Tsuchimori N, MLN3126, an antagonist of the chemokine receptor CCR9, ameliorates inflammation in a T cell mediated mouse colitis model. Int Immunopharmacol 2018. 60: 160–169. [DOI] [PubMed] [Google Scholar]
- 32.Qiuping Z, Jei X, Youxin J, Wei J, Chun L, Jin W, Qun W, Yan L, Chunsong H, Mingzhen Y, Qingping G, Kejian Z, Zhimin S, Qun L, Junyan L and Jinquan T, CC chemokine ligand 25 enhances resistance to apoptosis in CD4+ T cells from patients with T-cell lineage acute and chronic lymphocytic leukemia by means of livin activation. Cancer Res 2004. 64: 7579–7587. [DOI] [PubMed] [Google Scholar]
- 33.Ohoka Y, Yokota A, Takeuchi H, Maeda N and Iwata M, Retinoic acid-induced CCR9 expression requires transient TCR stimulation and cooperativity between NFATc2 and the retinoic acid receptor/retinoid X receptor complex. J Immunol 2011. 186: 733–744. [DOI] [PubMed] [Google Scholar]
- 34.Qiuping Z, Qun L, Chunsong H, Xiaolian Z, Baojun H, Mingzhen Y, Chengming L, Jinshen H, Qingping G, Kejian Z, Zhimin S, Xuejun Z, Junyan L and Jinquan T, Selectively increased expression and functions of chemokine receptor CCR9 on CD4+ T cells from patients with T-cell lineage acute lymphocytic leukemia. Cancer Res 2003. 63: 6469–6477. [PubMed] [Google Scholar]
- 35.Schmutz C, Cartwright A, Williams H, Haworth O, Williams JH, Filer A, Salmon M, Buckley CD and Middleton J, Monocytes/macrophages express chemokine receptor CCR9 in rheumatoid arthritis and CCL25 stimulates their differentiation. Arthritis Res Ther 2010. 12: R161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu PS, Nakamoto N, Ebinuma H, Usui S, Saeki K, Matsumoto A, Mikami Y, Sugiyama K, Tomita K, Kanai T, Saito H and Hibi T, C-C motif chemokine receptor 9 positive macrophages activate hepatic stellate cells and promote liver fibrosis in mice. Hepatology 2013. 58: 337–350. [DOI] [PubMed] [Google Scholar]
- 37.Li B, Wang Z, Zhong Y, Lan J, Li X and Lin H, CCR9-CCL25 interaction suppresses apoptosis of lung cancer cells by activating the PI3K/Akt pathway. Med Oncol 2015. 32: 66. [DOI] [PubMed] [Google Scholar]
- 38.Sharma PK, Singh R, Novakovic KR, Eaton JW, Grizzle WE and Singh S, CCR9 mediates PI3K/AKT-dependent antiapoptotic signals in prostate cancer cells and inhibition of CCR9-CCL25 interaction enhances the cytotoxic effects of etoposide. Int J Cancer 2010. 127: 2020–2030. [DOI] [PubMed] [Google Scholar]
- 39.Huang Y, Wang D, Wang X, Zhang Y, Liu T, Chen Y, Tang Y, Wang T, Hu D and Huang C, Abrogation of CC chemokine receptor 9 ameliorates ventricular remodeling in mice after myocardial infarction. Sci Rep 2016. 6: 32660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Underhill DM, Macrophage recognition of zymosan particles. J Endotoxin Res 2003. 9: 176–180. [DOI] [PubMed] [Google Scholar]
- 41.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, Medsger TAJ, Mitchell DM, Neustadt DH, Pinals RS, Schaller JG, Sharp JT, Wilder RL and Hunder GG, The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis and Rheumatism 1988. 31: 315–324. [DOI] [PubMed] [Google Scholar]
- 42.Pickens SR, Chamberlain ND, Volin MV, Pope RM, Talarico NE, Mandelin AM 2nd, and Shahrara S, Characterization of interleukin-7 and interleukin-7 receptor in the pathogenesis of rheumatoid arthritis. Arthritis Rheum 2011. 63: 2884–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pickens SR, Chamberlain ND, Volin MV, Pope RM, Mandelin AM 2nd, and Shahrara S, Characterization of CCL19 and CCL21 in rheumatoid arthritis. Arthritis Rheum 2011. 63: 914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elshabrawy HA, Volin MV, Essani AB, Chen Z, McInnes IB, Van Raemdonck K, Palasiewicz K, Arami S, Gonzalez M, Ashour HM, Kim SJ, Zhou G, Fox DA and Shahrara S, IL-11 facilitates a novel connection between RA joint fibroblasts and endothelial cells. Angiogenesis 2018. 21: 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chamberlain ND, Vila OM, Volin MV, Volkov S, Pope RM, Swedler W, Mandelin AM 2nd, and Shahrara S, TLR5, a novel and unidentified inflammatory mediator in rheumatoid arthritis that correlates with disease activity score and joint TNF-alpha levels. J Immunol 2012. 189: 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morgan R, Endres J, Behbahani-Nejad N, Phillips K, Ruth JH, Friday SC, Edhayan G, Lanigan T, Urquhart A, Chung KC and Fox DA, Expression and function of aminopeptidase N/CD13 produced by fibroblast-like synoviocytes in rheumatoid arthritis: role of CD13 in chemotaxis of cytokine-activated T cells independent of enzymatic activity. Arthritis Rheumatol 2015. 67: 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim SJ, Chen Z, Chamberlain ND, Essani AB, Volin MV, Amin MA, Volkov S, Gravallese EM, Arami S, Swedler W, Lane NE, Mehta A, Sweiss N and Shahrara S, Ligation of TLR5 Promotes Myeloid Cell Infiltration and Differentiation into Mature Osteoclasts in Rheumatoid Arthritis and Experimental Arthritis. J Immunol 2014. 193: 3902–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim SJ, Chang HJ, Volin MV, Umar S, Van Raemdonck K, Chevalier A, Palasiewicz K, Christman JW, Volkov S, Arami S, Maz M, Mehta A, Zomorrodi RK, Fox DA, Sweiss N and Shahrara S, Macrophages are the primary effector cells in IL-7-induced arthritis. Cellular & Molecular Immunology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Raemdonck K, Umar S, Palasiewicz K, Volkov S, Volin MV, Arami S, Chang HJ, Zanotti B, Sweiss N and Shahrara S, CCL21/CCR7 signaling in macrophages promotes joint inflammation and Th17-mediated osteoclast formation in rheumatoid arthritis. Cellular and Molecular Life Sciences 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are presented in this paper. No shared databases were used or created.