

Abstract
Amyotrophic Lateral Sclerosis (ALS) is an incurable neurodegenerative disease which often occurs concurrently with frontotemporal dementia (FTD), another disorder involving progressive neuronal loss. ALS and FTD form a neurodegenerative continuum and share pathological and genetic features. Mutations in a multitude of genes have been linked to ALS/FTD, including FUS. The FUS protein aggregates and forms inclusions within affected neurons. However, the precise mechanisms connecting protein aggregation to neurotoxicity remain under intense investigation. Recent evidence points to the contribution of epigenetics to ALS/FTD. A main epigenetic mechanism involves the post-translational modification (PTM) of histone proteins. We have previously characterized the histone PTM landscape in a FUS ALS/FTD yeast model finding decreased acetylation on lysine residues 14 and 56 of Histone H3. Here, we describe the first report of amelioration of disease phenotypes by controlling histone acetylation on specific modification sites. We show that inhibiting histone deacetylases (HDACs), via treatment with Trichostatin A (TSA), suppresses the toxicity associated with FUS overexpression in yeast by preserving the levels of H3K56ac and H3K14ac without affecting FUS expression or its aggregation. Our data raises the novel hypothesis that the toxic effect of protein aggregation in neurodegeneration is related to its association with altered histone marks. Altogether, we demonstrate the ability to counter the repercussions of protein aggregation on cell survival by preventing specific histone modification changes. Our findings launch off a novel mechanistic framework that will enable alternative therapeutic approaches for ALS/FTD and other neurodegenerative diseases.
Keywords: FUS, Amyotrophic Lateral Sclerosis, Frontotemporal Dementia, Histone Acetylation, Trichostatin A
Amyotrophic Lateral Sclerosis (ALS) is a relentless, fatal neurodegenerative disease characterized by the loss of motor neurons in the spinal cord, brain, and brainstem leading to progressive paralysis.(1) ALS shares genetic and pathological features with frontotemporal dementia (FTD). FTD primarily affects the frontal and temporal lobes of the brain causing deficits in the ability to reason and control movement.(2) ALS and FTD lie on two ends of a disease continuum.(2) Prognosis for ALS/FTD patients is poor. There is no cure, and no therapy is able to stop or modify disease progression.
Mutations in a large number of genes have been associated with ALS/FTD. The protein products of these mutant genes aggregate and form inclusions within affected neurons, leading to their demise. Among these, Superoxide Dismutase 1 (SOD1), TAR DNA-binding protein 43 (TDP-43) and Fused in Sarcoma (FUS) have been thoroughly studied. FUS is a RNA-binding protein involved in various processes, including splicing and DNA damage repair.(3) Despite intense investigation, the molecular channels connecting protein aggregation to neurotoxicity remain elusive.
Epigenetics refers to heritable alterations in gene expression occurring without modification to the genome. A key epigenetic mechanism is the post-translational modification (PTM) of histone proteins.(4) The N-terminal tails of histones are heavily modified with numerous chemical moieties, including methylation, acetylation and phosphorylation.(5) Histone PTMs affect gene expression by controlling the transcription machinery’s access to DNA and by recruitment of transcription factors.(6) These modifications comprise a ‘histone code’ that other proteins can ‘write,’ ‘erase’ and ‘read.’(7) In the case of histone acetylation, histone acetyltransferases (HATs) are responsible for installing these groups while histone deacetylases (HDACs) remove them.(8)
HDAC inhibition has arisen as a potential strategy for treating ALS/FTD and other neurodegenerative diseases. For instance, the HDAC inhibitor Scriptaid can clear SOD1 aggregates in vitro.(9) Moreover, pan-HDAC inhibitors are neuroprotective in TDP-43 ALS models.(10) Similarly, phenylbutyrate, a general HDAC inhibitor, improves clinical phenotypes in an SOD1 ALS mouse model.(11) Trichostatin A (TSA), a pan-class I HDAC inhibitor, decreases motor neuron death in the same mouse model.(12)
Yeast provides an expedient, cost-efficient model to study certain aspects of neurodegeneration. FUS overexpression in yeast recapitulates several features of ALS/FTD pathology including FUS mislocalization and aggregation.(13) Furthermore, in contrast to mammalian cell models, yeast FUS ALS/FTD models display overt cellular demise allowing for convenient exploration of chemical tools to prevent cell death. Recent evidence has tied FUS proteinopathy to the epigenome. FUS overexpression in yeast is associated with alterations to the histone PTM landscape.(8,14) In particular, FUS overexpression is connected to decreases in the levels of acetylation on Lysines 14 and 56 of Histone H3.(14) Histone PTMs are accessible pharmacological targets, and thus we speculated that chemical interventions aimed at modulating relevant epigenetic changes might lead to improved cell survival. Treatment with an HDAC inhibitor, such as TSA, could preserve histone acetylation levels by preventing the removal of acetyl groups from histone tails.
To test if TSA can ameliorate FUS proteinopathy, we measured the growth of yeast overexpressing FUS or a vector control in various concentrations of the drug. DMSO was used as a vehicle control. Serial yeast dilutions were grown on selective media supplemented with either glucose or galactose. Glucose suppresses FUS expression, while galactose activates it. Both FUS and vector control strains grew well on glucose, regardless of drug condition (Figure 1). Hence, TSA does not appear to impact yeast growth at the concentrations used. As previously reported, (13,14) FUS overexpression leads to a dramatic decrease in cell growth compared to a vector control (Figure 1). Excitingly, TSA ameliorated growth suppression in FUS yeast. TSA-treated FUS yeast grow better than untreated and vehicle control (Figure 1, black arrows). To semi-quantitatively measure the effect of TSA on yeast growth in the context of FUS overexpression, we assessed the image density of the middle spot from each growth condition and compared it to the corresponding spot in the vector control. Yeast overexpressing FUS treated with 2.50 μM TSA grew significantly better than untreated or DMSO-treated yeast (Figure 1). Our results establish that interfering with HDAC activity via TSA treatment ameliorates the growth suppression associated with FUS overexpression in yeast.
Figure 1. Trichostatin A relieves growth suppression in a FUS ALS/FTD yeast model.
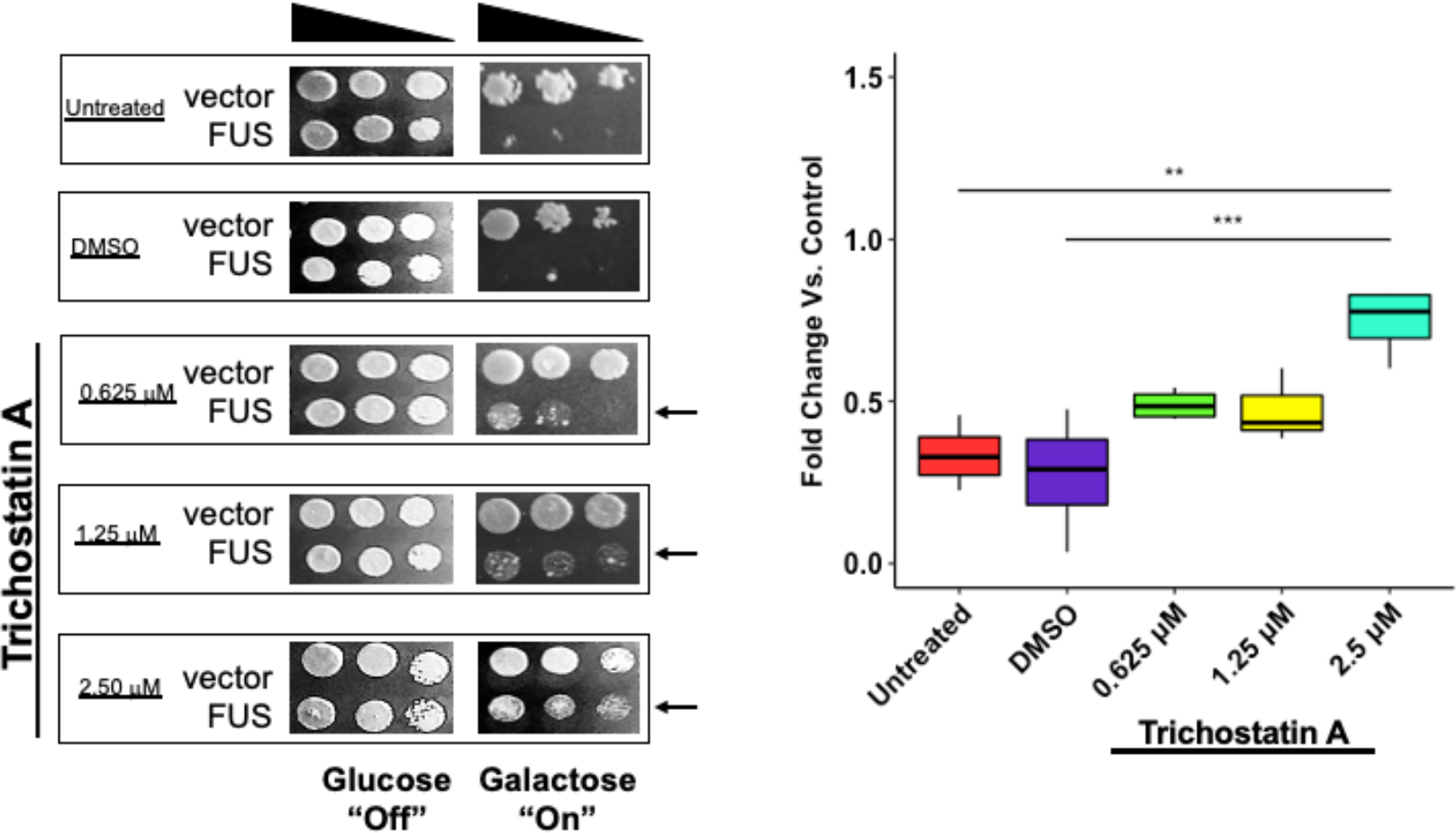
Yeast expressing FUS or an empty vector were serially diluted 5-fold and spotted on glucose (off) or galactose (on) medium in the absence (untreated, DMSO) or presence of TSA at varying concentrations. Densitometric measurement of cell density compared to the untreated control is depicted. Box plot whiskers represent upper and lower quartiles. *** = p < 0.001, ** = p < 0.01. n = 4.
We verified that the reduction of growth suppression did not result from changes in FUS levels or aggregation. There was no significant difference in FUS expression in any of the treatment conditions (Figure S1a). We confirmed that TSA did not affect FUS aggregation (Figure S1b and Figure S1c). Additionally, we established that TSA did not impact FUS aggregates or their cellular distribution (Figure S2). TSA and other HDAC inhibitors have been previously shown to trigger expression of Hsp70 which helps clear FUS aggregates and reduce proteotoxic stress.(15,16) However, as we see no reduction in FUS aggregation, we suspect the toxicity mitigation we observe does not involve proteostasis enhancement.
To clarify if the rescue elicited by TSA is FUS-specific and not generally applicable to misfolded proteins, we treated yeast overexpressing TDP-43 with TSA. As shown previously, (14,17) TDP-43 overexpression is very toxic (Figure S3, left panel). However, in contrast to FUS, TDP-43 proteinopathy is not linked to histone H3 acetylation disturbances.14 TDP-43 yeast treated with 2.50 μM TSA did not display growth improvement (Figure S3, right panel) suggesting that the effect of TSA is specific to FUS proteinopathy. We verified TSA did not impact TDP-43 expression levels (Figure S4a).
To probe whether detoxification occurs through epigenetic mechanisms, we asked if TSA restored levels of H3K14ac and H3K56ac. As previously reported,(14) untreated and DMSO-treated yeast displayed significantly decreased H3K14ac and H3K56ac compared to untreated vector control. Remarkably, TSA-treated FUS yeast showed a significant increase in H3K14ac levels compared to untreated and DMSO-treated FUS yeast (Figure 2a). Surprisingly, TSA-treated yeast also showed significantly higher H3K56ac levels (Figure 2b). H3K56 is deacetylated by Hst3 and Hst4, which are insensitive to TSA.(18,19) However, acetylation of H3K56 is mediated by the lysine acetyltransferase Rtt109 in concert with the chaperones Vsp75 and Asf1.(20,21) Interestingly, H3K14ac drives Rtt109 to acetylate H3K56ac via Asf1 binding.(21) Through this crosstalk mechanism, it is possible that H3K14ac recovery is promoting the rise in H3K56ac levels.
Figure 2. Trichostatin A restores acetylation on Lysines 14 and 56 of Histone H3 in yeast FUS ALS/FTD models.
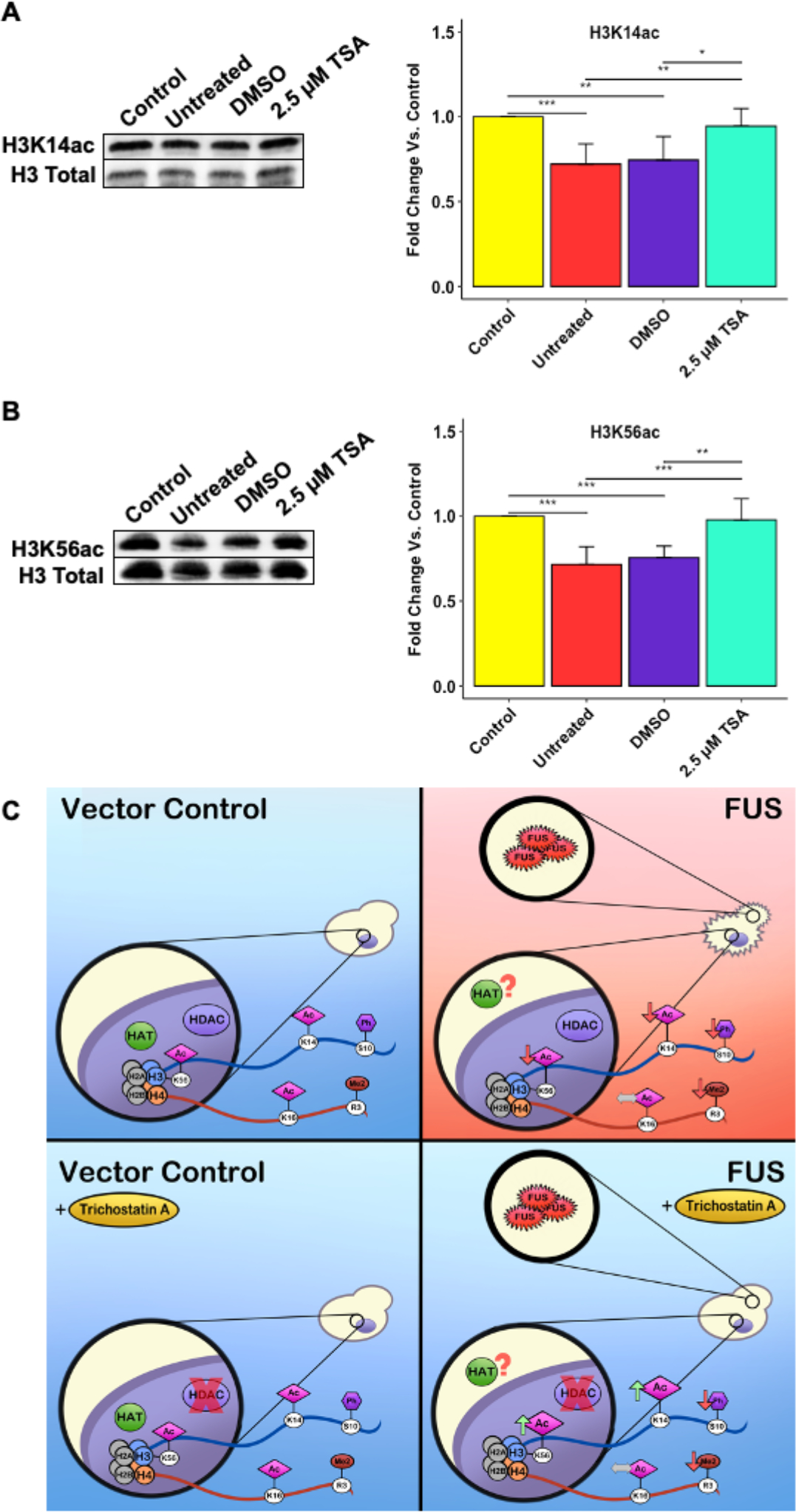
Representative Western blots displaying the levels of H3K14ac (A, n = 6) and H3K56ac (B, n = 6) are shown. Histograms compiling multiple biological replicates are shown. (C) Putative mechanism for TSA effects in FUS ALS/FTD yeast models. Error bars represent +SD. * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
TSA increases global acetylation levels in yeast, but at concentrations higher than those used in this study. (22,23) To verify that 2.50 μM TSA did not affect acetylation at modification sites uncoupled from FUS proteinopathy, we measured H4K16ac levels (Figure S5). TSA did not increase H4K16ac, suggesting that 2.50 μM TSA restores acetylation only for those modification sites originally impacted by FUS overexpression. To further investigate if 2.50 μM TSA raised acetylation levels outside of the context of FUS proteinopathy, we probed for H3K14ac and H3K56ac in vector control yeast, where the levels of these PTMs are not decreased. TSA has no significant effect on H3K14ac or H3K56ac levels in vector control yeast (Figure S6). We also verified TSA did not impact H3K14ac or H3K56ac levels in TDP-43 yeast (Figure S4b and S4c).
As we observed evidence of histone PTM crosstalk, we wondered if other non-acetyl histone modifications connected to FUS proteinopathy would recover in response to TSA. FUS yeast, regardless of treatment conditions, retained reduced H3S10ph and H4R3me2asym levels (Figure S7). This suggests that the decreases in these PTMs are independent of histone acetylation. Furthermore, it establishes that the amelioration of growth suppression elicited by TSA is not related to changes in the levels of H3S10ph or H3Rme2asym. Akin to histone acetylation, TSA did not affect neither H3S10ph nor H4R3me2asym in vector control yeast (Figure S8).
Collectively, we show that TSA relieves growth suppression in yeast overexpressing FUS. This mitigation of toxicity is accompanied by preservation of H3K14ac and H3K56ac levels, but not H3S10ph or H4R3me2asym levels. We did not observe any changes in FUS expression or aggregation with TSA treatment. To the best of our knowledge, this is the first report of its kind tying specific histone acetylation sites to improvements in ALS/FTD cell demise. Pan-histone acetylation has been shown to increase after HDAC inhibition, but specific residues were not implicated.(12,24) Furthermore, our data raises the novel hypothesis that the toxic effect of protein aggregation in neurodegeneration is at least in part related to its association with altered histone marks, a notion that is not well characterized in ALS/FTD. We hypothesize reduction of toxicity occurs through modulation of epigenetic pathways. Very recently, pathogenic FUS has been found to interact with nucleoporins and impede nucleocytoplasmic transport.(25) It is possible that defects in nucleocytoplasmic transport result in mislocalization of HATs, resulting in impaired H3K14ac and H3K56ac which could then lead to aberrant gene expression and ultimately cell death (Figure 2c). However, further investigation is needed to definitively establish such a mechanism.
In conclusion, our results highlight that changes in histone PTMs are closely associated with the cell demise elicited by FUS proteinopathy. We illustrate the ability to counter the detrimental effects of protein aggregation on cell survival by modulating specific histone modification changes via chemical treatment. Our results allude to novel mechanistic networks involving epigenetics that will enable new treatments for ALS/FTD and other neurodegenerative diseases.
Supplementary Material
ACKNOWLEDGMENTS
We thank Prof. James Shorter (University of Pennsylvania) for kindly sharing reagents. We also thank Royena Tanaz for technical support.
Funding Sources
Brooklyn College, CUNY and NIH (K22NS091314) supported M.P.T. The Graduate Center, CUNY and Brooklyn College supported S.A.B and S.N.C.
ABBREVIATIONS
- ALS
Amyotrophic Lateral Sclerosis
- FTD
Frontotemporal Dementia
- FUS
Fused in Sarcoma
- HAT
Histone Acetyltransferase
- HDAC
Histone Deacetylase
- PTM
Post-translational Modification
- TSA
Trichostatin A
Footnotes
ASSOCIATED CONTENT
Supporting Information
Supplementary materials outlining detailed Materials and Methods information, Supplementary Figures presenting Trichostatin A’s effect on FUS expression and aggregation, its effect on TDP-43 proteinopathy models and its impact on other histone modification levels, as well as Supplementary References are available free of charge. (PDF)
REFERENCES
- [1].Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D, Orozco G, and Chinea A (2015) A comprehensive review of amyotrophic lateral sclerosis, Surgical Neurology International 6, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nolan M, Talbot K, and Ansorge O (2016) Pathogenesis of FUS-associated ALS and FTD: insights from rodent models, Acta Neuropathologica Communications 4, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shang Y, and Huang EJ (2016) Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis, Brain Research 1647, 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Allis CD, and Jenuwein T (2016) The molecular hallmarks of epigenetic control, Nature Reviews Genetics 17, 487. [DOI] [PubMed] [Google Scholar]
- [5].Luger K, and Mader AW (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. (Cover story), Nature 389, 251. [DOI] [PubMed] [Google Scholar]
- [6].Mazzio EA, and Soliman KF (2012) Basic concepts of epigenetics: impact of environmental signals on gene expression, Epigenetics 7, 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Strahl BD, and Allis CD (2000) The language of covalent histone modifications, Nature 403, 41–45. [DOI] [PubMed] [Google Scholar]
- [8].Bennett SA, Tanaz R, Cobos SN, and Torrente MP (2019) Epigenetics in amyotrophic lateral sclerosis: a role for histone post-translational modifications in neurodegenerative disease, Translational Research 204, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Corcoran LJ, Mitchison TJ, and Liu Q (2004) A Novel Action of Histone Deacetylase Inhibitors in a Protein Aggresome Disease Model, Current Biology 14, 488–492. [DOI] [PubMed] [Google Scholar]
- [10].Sanna S, Esposito S, Masala A, Sini P, Nieddu G, Galioto M, Fais M, Iaccarino C, Cestra G, and Crosio C (2020) HDAC1 inhibition ameliorates TDP-43-induced cell death in vitro and in vivo, Cell Death & Disease 11, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, Dangond F, Cormier KA, Cudkowicz ME, Brown RH Jr., and Ferrante RJ (2005) Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice, J Neurochem 93, 1087–1098. [DOI] [PubMed] [Google Scholar]
- [12].Yoo Y-E, and Ko C-P (2011) Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis, Experimental Neurology 231, 147–159. [DOI] [PubMed] [Google Scholar]
- [13].Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, and Gitler AD (2011) Molecular Determinants and Genetic Modifiers of Aggregation and Toxicity for the ALS Disease Protein FUS/TLS, PLoS biology 9, e1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen K, Bennett SA, Rana N, Yousuf H, Said M, Taaseen S, Mendo N, Meltser SM, and Torrente MP (2018) Neurodegenerative Disease Proteinopathies Are Connected to Distinct Histone Post-translational Modification Landscapes, ACS Chem Neurosci 9, 838–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Park JA, Kim YE, Seok HJ, Park WY, Kwon HJ, and Lee Y (2011) Differentiation and upregulation of heat shock protein 70 induced by a subset of histone deacetylase inhibitors in mouse and human embryonic stem cells, BMB Rep 44, 176–181. [DOI] [PubMed] [Google Scholar]
- [16].Kuta R, Larochelle N, Fernandez M, Pal A, Minotti S, Tibshirani M, St. Louis K, Gentil BJ, Nalbantoglu JN, Hermann A, and Durham HD (2020) Depending on the stress, histone deacetylase inhibitors act as heat shock protein co-inducers in motor neurons and potentiate arimoclomol, exerting neuroprotection through multiple mechanisms in ALS models, Cell Stress and Chaperones 25, 173–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Johnson BS, McCaffery JM, Lindquist S, and Gitler AD (2008) A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity, Proc Natl Acad Sci U S A 105, 6439–6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hachinohe M, Hanaoka F, and Masumoto H (2011) Hst3 and Hst4 histone deacetylases regulate replicative lifespan by preventing genome instability in Saccharomyces cerevisiae, Genes to Cells 16, 467–477. [DOI] [PubMed] [Google Scholar]
- [19].Workman JJ, Chen H, and Laribee RN (2016) <em>Saccharomyces cerevisiae</em> TORC1 Controls Histone Acetylation by Signaling Through the Sit4/PP6 Phosphatase to Regulate Sirtuin Deacetylase Nuclear Accumulation, Genetics 203, 1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fillingham J, Recht J, Silva AC, Suter B, Emili A, Stagljar I, Krogan NJ, Allis CD, Keogh M-C, and Greenblatt JF (2008) Chaperone Control of the Activity and Specificity of the Histone H3 Acetyltransferase Rtt109, Molecular and Cellular Biology 28, 4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cote JM, Kuo Y-M, Henry RA, Scherman H, Krzizike DD, and Andrews AJ (2019) Two factor authentication: Asf1 mediates crosstalk between H3 K14 and K56 acetylation, Nucleic acids research 47, 7380–7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Weerasinghe SVW, Wambua M, and Pflum MKH (2010) A histone deacetylase-dependent screen in yeast, Bioorganic & Medicinal Chemistry 18, 7586–7592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bernstein BE, Tong JK, and Schreiber SL (2000) Genomewide studies of histone deacetylase function in yeast, Proceedings of the National Academy of Sciences 97, 13708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rossaert E, Pollari E, Jaspers T, Van Helleputte L, Jarpe M, Van Damme P, De Bock K, Moisse M, and Van Den Bosch L (2019) Restoration of histone acetylation ameliorates disease and metabolic abnormalities in a FUS mouse model, Acta Neuropathologica Communications 7, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lin Y-C, Kumar MS, Ramesh N, Anderson EN, Nguyen AT, Kim B, Cheung S, McDonough JA, Skarnes WC, Lopez-Gonzalez R, Landers JE, Fawzi NL, Mackenzie IRA, Lee EB, Nickerson JA, Grunwald D, Pandey UB, and Bosco DA (2021) Interactions between ALS-linked FUS and nucleoporins are associated with defects in the nucleocytoplasmic transport pathway, Nature Neuroscience 24, 1077–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.