

Abstract
Clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) protein gene editing is poised to transform the treatment of genetic diseases. However, limited progress has been made toward precise editing of DNA via homology-directed repair (HDR) that requires careful orchestration of complex steps. Herein, dendrimer-based lipid nanoparticles (dLNPs) are engineered to co-encapsulate and deliver multiple components for in vivo HDR correction. BFP/GFP switchable HEK293 cells with a single Y66H amino acid mutation are employed to assess HDR-mediated gene editing following simultaneous, one-pot delivery of Cas9 mRNA, single-guide RNA, and donor DNA. Molar ratios of individual LNP components and weight ratios of the three nucleic acids are systematically optimized to increase HDR efficiency. Using flow cytometry, fluorescence imaging, and DNA sequencing to quantify editing, optimized 4A3-SC8 dLNPs edit >91% of all cells with 56% HDR efficiency in vitro and >20% HDR efficiency in xenograft tumors in vivo. Due to the all-in-one simplicity and high efficacy, the developed dLNPs offer a promising route toward the gene correction of disease-causing mutations.
Keywords: CRISPR/Cas, gene editing, mRNA delivery, nanoparticles, nucleic acid delivery
In the short time since its discovery in 2013,[1] CRISPR/Cas (clustered regularly interspaced short palindromic repeat/CRISPR-associated (Cas) protein) has been quickly applied to the treatment of diseases such as thalassemia, sickle cell disease, familial hypercholesterolemia, Duchenne muscular dystrophy, cancer, and Leber congenital amaurosis.[2] However, progress to date for in vivo editing has largely been limited to gene knockouts via an error-prone DNA repair mechanism known as non-homologous end joining (NHEJ). True correction of genetic disease and cancer mutations will require homology-directed repair (HDR), an approach currently hindered by the lack of carriers that can mediate this complex DNA repair pathway. Here we overcome this challenge by reporting a non-viral, all-in-one approach using dendrimer-based lipid nanoparticles (dLNPs) for precise gene correction by HDR in vitro and in vivo.
CRISPR offers several highly desirable traits for gene editing including sequence-dependent target specificity and editing permanence in non-dividing cells.[1d,e,2e] These attributes could, in theory, allow many genetic diseases to be cured by a single treatment. Most in vivo editing efforts so far have utilized NHEJ, whereby insertions and/or deletions (indels) can occur at the site of the Cas9 and single-guide RNA (sgRNA) induced double-stranded break (DSB) in the DNA. The introduction of indels into the target site typically results in mutations that subsequently render the protein non-functional or truncated.[1b,3] In diseases where knockout of a target is beneficial, the NHEJ mechanism is highly useful. However, in most genetic diseases and cancer applications, HDR correction of the sequence is required for therapeutic benefit.[2e,4] Additionally, in many genetic diseases wherein the mutated genomic sequence codes for a partially active protein that still retains some productive activity, NHEJ could be quite detrimental as it could eliminate previously existing activity.
In contrast, through utilization of HDR, cells can accurately correct the mutated genetic sequence of interest to a precisely fixed sequence. Rather than forming indels at the cut site via NHEJ, the DSB can be repaired when in close proximity to a strand of DNA containing the correct amino acid sequence flanked by 5′ and 3′ regions of overlapping homology to the endogenous DNA.[4a,c,5] In order to utilize this repair process, there are three required components that must work in tandem with one another: Cas9 protein, sgRNA, and a single-stranded DNA (ssDNA) template. Thus, the requirements for HDR correction are much greater than those for NHEJ gene knockouts, which has limited advancement of gene correction efforts.
Due to the difficult challenge of delivering these multiple cargoes, discovery, and engineering of delivery systems is an important goal.[2e,4] While viral vectors are effective for gene delivery, they present several shortcomings for inducing HDR including restrictive packaging limits, risk of random deleterious integration into the genome, and potential immune response.[6] Among non-viral options for deploying Cas enzymes, delivery of Cas9 mRNA may yield more protein than direct delivery of Cas9 protein on a mass basis and may be safer than delivery of pDNA encoding for Cas9 that could possibly integrate into the host genome.[1f,2e–g,7] To the best of our knowledge, a non-viral delivery system for achieving HDR in vivo using a fully nucleic acid-mediated approach has not been reported. Herein, we report an all-in-one non-viral dLNP system[8] capable of inducing HDR in vivo.
In order to achieve correction of a mutated genetic sequence via HDR, there are challenging barriers that must be overcome (Figure 1). Here we pursued an approach of an all nucleic acid CRISPR/Cas system consisting of Cas9 mRNA, sgRNA, and donor ssDNA encapsulated in dLNPs to enable cytoplasmic delivery.[9] After Cas9 mRNA translation, the ribonucleoprotein complex (RNP)[1a,4c,10] is formed, consisting of the Cas9 nuclease and sgRNA, which has been shown to further bind ssDNA[11] prior to nuclear localization driven by the nuclear localization signal. RNPs locate the target sequence in the genomic DNA wherein the sgRNA will bind in an anti-parallel complementary fashion upstream of the protospacer adjacent motif sequence.[4c,12] Once bound, the Cas9 nuclease will cleave the genomic DNA, resulting in a DSB,[4c,13] wherein the ssDNA HDR donor template can be copied and incorporated into the genomic DNA (Figure 1).[4a,c,5a–c,f,g] Due to these barriers, we used an engineering optimization approach to identify formulations that can overcome these challenges with HDR efficiency as the paramount goal. As a starting point, we employed a library of degradable, ionizable dendrimer-based lipids[8a] with the ability to be positively charged at low pH to bind RNAs during self-assembly, uncharged at neutral pH to reduce toxicity, and positively charged again at the maturing endosome pH to facilitate endosomal release. Prior work exploring ionizable dendrimer lipids to deliver short siRNAs/miRNAs (18–22 bp)[8a–c,e] or long messenger RNAs (>1000 nt)[8d,f] have revealed that creation of new lipid designs[14] and formulation reengineering[8d,f] are useful approaches to develop carriers for new opportunities. Indeed, it has been shown that optimal carriers for short RNAs are not always effective for long RNAs.[8g,14a,15] Such efforts on LNP engineering have not yet been directed toward HDR-mediated genome correction.
Figure 1.
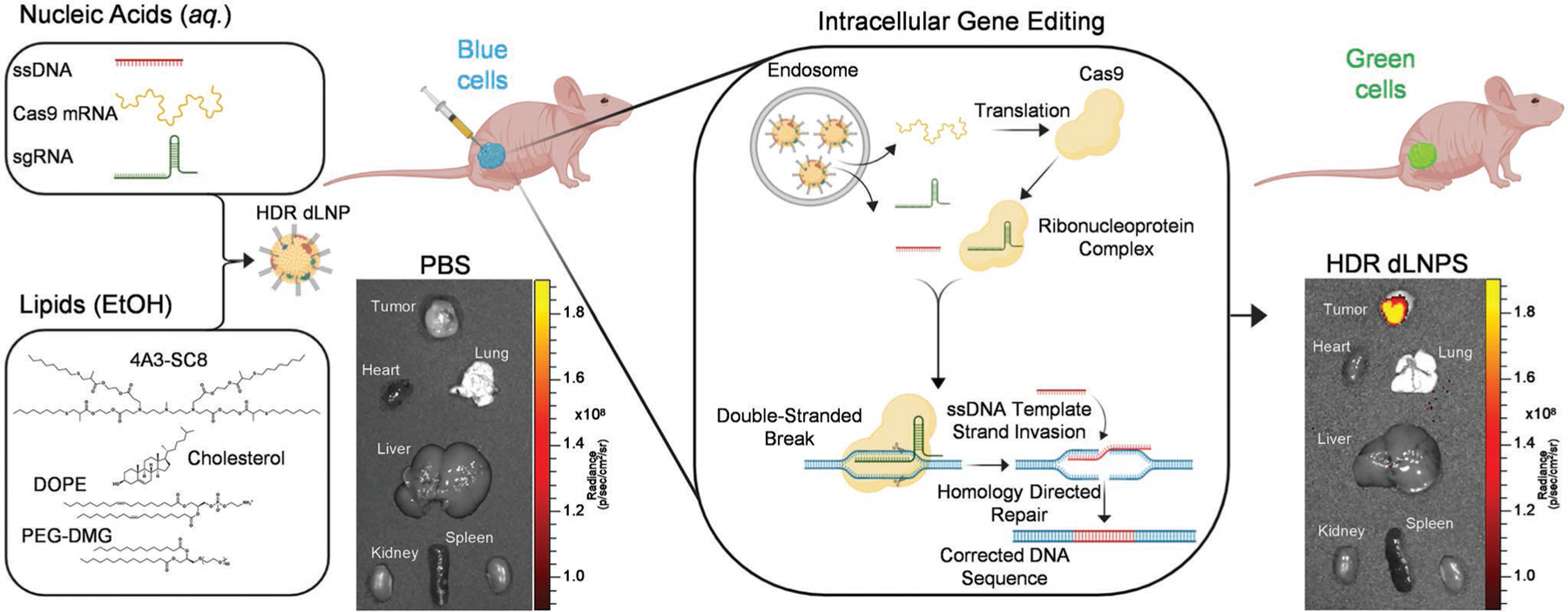
4A3-SC8 HDR dLNPs effectively orchestrate HDR-mediated gene editing in vitro and in vivo. All-in-one dLNPs containing three nucleic acids (Cas9 mRNA, sgRNA, and ssDNA) are able to escape the endosome and release nucleic acid cargoes into the cytoplasm. Cas9 mRNAs are then translated into Cas9 proteins which associate with sgRNAs to form ribonucleoprotein complexes (RNPs). RNPs/ssDNA template traverses the nuclear membrane and locate their target site in the genomic DNA creating a double-stranded break (DSB) wherein the ssDNA template containing a corrected sequence is incorporated into the genomic DNA.
Here, we specifically focused on co-encapsulation of three components (Cas9 mRNA, sgRNA, and donor DNA), which presented a mixture of nucleic acids of different lengths and chemical compositions. We reasoned that the overall physical properties of the combined nucleic acid cargoes would exhibit hydrophobic and electrostatic characteristics more similar to long RNAs than short RNAs.[8a] Given previous data demonstrating that alterations to the molar ratios of the four core lipid components (ionizable amino dendrimer lipid, amphipathic phospholipid, cholesterol, PEG2000-DMG) within dLNPs can drastically alter their ability to effectively deliver short (siRNA) to long (mRNA) cargoes,[8d] we initially focused on dendrimers and formulation parameters (38.5:30:30:1.5; Dendrimer:Cholesterol:DOPE:PEG-DMG) with the most flexibility across nucleic acid types (Figure S1, Supporting Information). The screening library of dendrimers that we selected for analysis consisted of four distinct amine cores (3A3, 3A5, 4A1, 4A3) and nine peripheries with different alkyl chain lengths (SC5, SC6, SC7, SC8, SC9, SC10, SC11, SC12, SC14) totaling 36 individual dendrimer lipids (Figure 2A,B).[8a] Each of the dendrimers was then combined with cholesterol, DOPE, and PEG-DMG in ethanol at the optimized internal molar ratios and rapidly mixed with luciferase (Luc) mRNA in an acidic aqueous buffer to form dLNPs.
Figure 2.
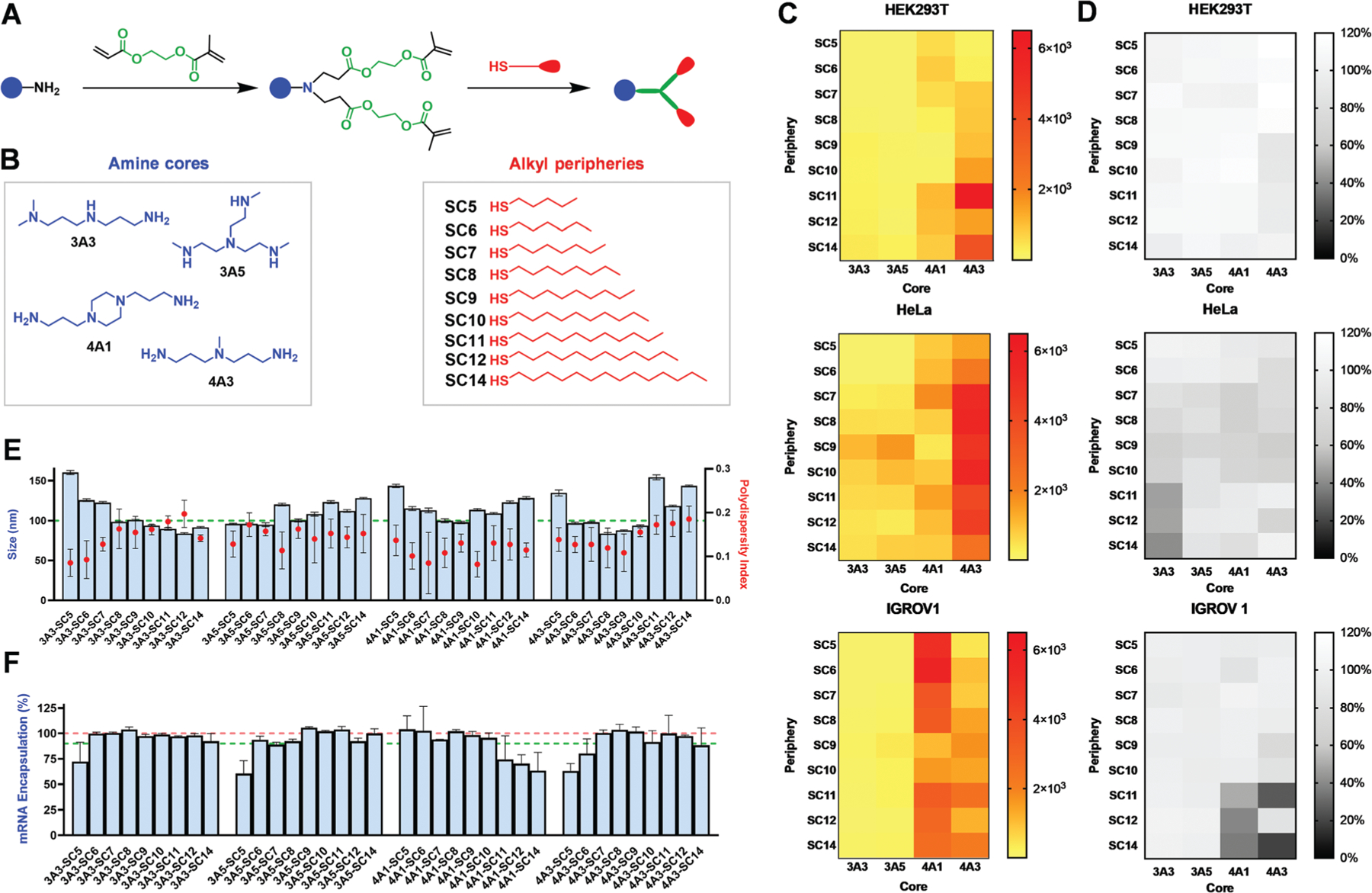
The chemical identity of dendrimers in dLNPs influences luciferase mRNA delivery efficacy across cell types. A) Modular degradable dendrimers were synthesized containing an ionizable amine core, ester linkages, and alkyl-thiol peripheries via sequential Michael addition. B) Four different amine cores and nine alkyl peripheries were selected to form a dendrimer library consisting of 36 distinct structures for efficacy assessment. C) HEK293T, HeLa, and IGROV1 cells were transfected with dLNPs containing firefly luciferase mRNA at a dose of 12.5 ng (6.672 × 10−3 m) and analyzed for fold increase in bioluminescence after normalization to background and viability (N = 4). D) All cells were assessed for viability following transfection with each of the dLNP formulations and exhibited minimal to no cytotoxicity (N = 4). E) Most dLNPs were ≈100 nm in diameter and uniform (PDI <0.2; N = 5). F) mRNA encapsulation did not vary significantly between dLNP formulations (N = 4).
Since non-viral nanoparticles can exhibit some degree of cell-type specificity,[16] the library of selected dendrimer compounds was screened for both effective delivery (as quantified by luciferase expression) and toxicity across three different cell lines (HEK293T, HeLa, and IGROV-1) to identify the most active formulation across multiple cell types (Figure 2C,D). Additionally, to rule out any discrepancies in delivery that may be due to ineffective nucleic acid binding or distortions during particle formation, RNA encapsulation efficiency and particle size and uniformity were assessed using the Ribogreen assay and dynamic light scattering (DLS), respectively. As expected, there were no major differences observed with respect to dLNP size, as most averaged roughly 100 nm in diameter; all nanoparticles were uniform (PDI <0.2) (Figure 2E); and most dLNPs effectively encapsulated >92% of mRNA at the tested ratios (Figure 2F). In contrast to the lack of variance observed with respect to these aspects of the different dLNPs, notable differences in delivery efficacy were illuminated between dendrimer compounds. With the goal set to maximize HDR across cell lines, we noted the highest delivery for formulations containing dendrimers with 4A1 and 4A3 amine cores. Increased alkyl chain length dependence was observed in IGROV1 and HEK293T cells. In consideration of the data as a whole, 4A3 emerged as the lead amine core for long nucleic acid delivery due to its activity across all three cell lines, including marked efficacy in HeLa cells.
To further define the role of alkyl chain length in delivery efficiency and toxicity, a dose-response assay was conducted using the top amine core candidate (4A3) and all previously examined alkyl peripheries (SC5-SC14) across the same three cell lines (Figures S2–S4, Supporting Information). A slight trend emerged between increasing alkyl chain length and delivery efficacy. However, some mild increases in cytotoxicity were also observed with increasing alkyl chain length (Figure S5, Supporting Information). In consideration of these two factors, it was concluded that 4A3-SC8 dLNPs demonstrated an optimal balance of pronounced delivery efficacy and minimal toxicity (Figure 2).
Accomplishing non-viral HDR-mediated gene editing requires synthetic carriers to deliver nucleic acids with very large differences in size: Cas9 mRNA (≈4500 nt), modified sgRNA (≈120 nt), and ssDNA HDR template (127 nt) (Figure 1). This challenge is further compounded by the fact that Cas9 mRNA must first be translated into Cas9 protein before it can accomplish guided gene editing. To address these factors using the optimized 4A3-SC8 dLNPs, we systematically analyzed both the kinetics of cargo delivery with respect to protein expression and the ability of 4A3-SC8 dLNPs to encapsulate multiple nucleic acids inside of a single nanoparticle. HEK293 cells expressing a GFP sequence with a single Y66H amino acid mutation (CAT in place of TAC) were employed to quantify NHEJ and HDR events.[17] With this mutated sequence (CAT), the cells fluoresce blue instead of green; however, when the mutation is corrected to TAC, the cells regain normal green fluorescence (Figure S6, Supporting Information). As such, insertion of the correct amino acid sequence into this position restores GFP function. If indels are present in the sequence, (indicative of NHEJ), the cells lose their fluorescence (Figure 3A,B).[17]
Figure 3.
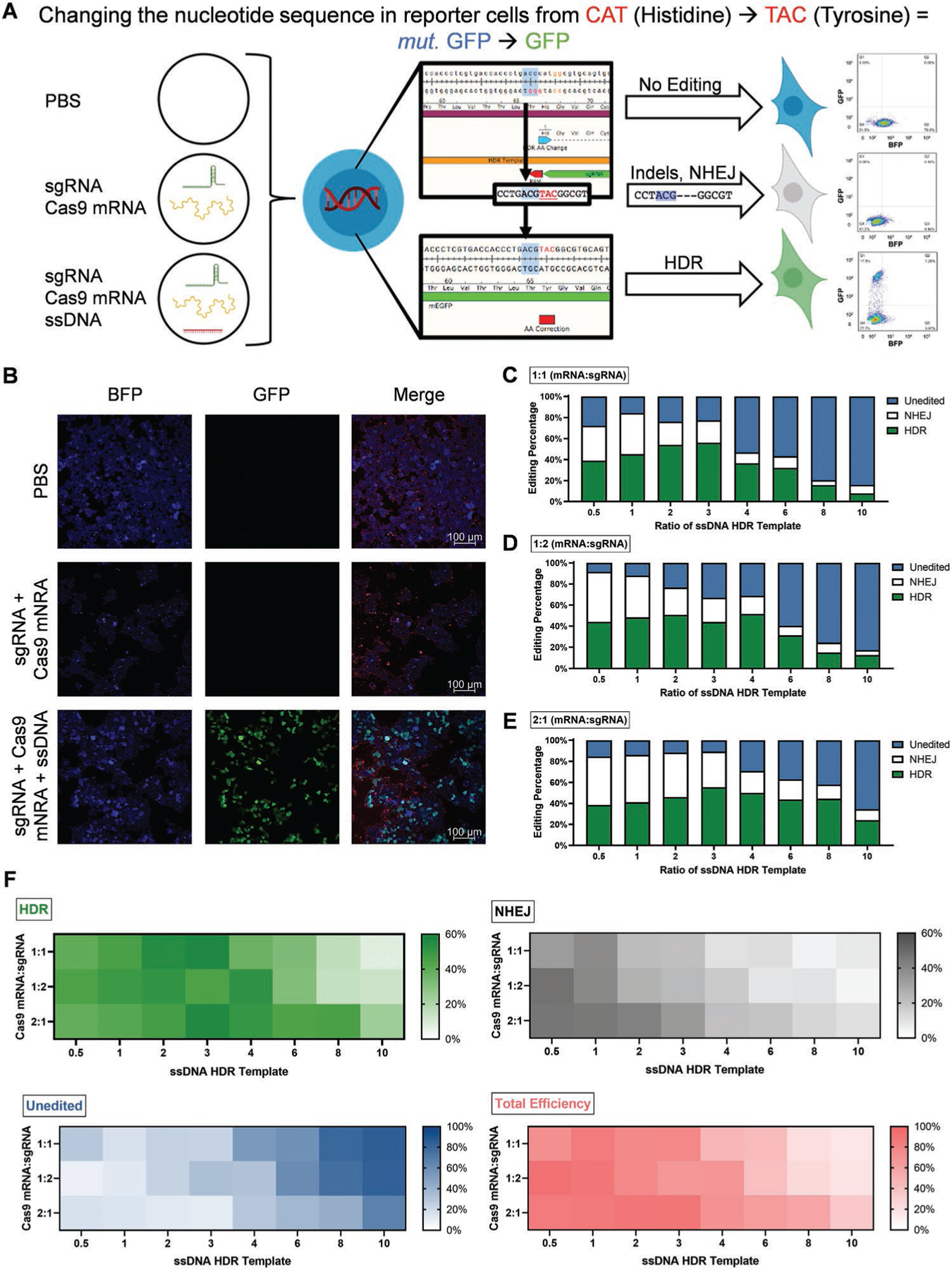
4A3-SC8 dLNPs successfully induced HDR in HEK293 cells containing a GFP sequencing with a Y66H mutation via one-pot delivery of Cas9 mRNA, sgRNA, and a corrected ssDNA template. A) HEK293 cells contain a single amino acid mutation in their GFP sequence (Y66H) that alters their fluorescence. Depending on the gene editing technique employed, the fluorescence can be eliminated (NHEJ) or restored to native GFP (HDR). B) HEK293 (Y66H) cells were transfected with dLNPs containing 1000 ng of only Cas9 mRNA and sgRNA at a 2:1 ratio or dLNPs containing Cas9 mRNA, sgRNA, and ssDNA template at a 2:1:1 ratio and imaged using confocal microscopy for mutated GFP (blue) and GFP (green) signal. CellMask Orange was used to stain plasma membranes in merged images. C–E) The amount and type of gene editing induced were assessed using TIDER to analyze DNA sequencing following transfection with HDR dLNPs containing Cas9 mRNA:sgRNA ratios fixed at 1:1 (C), 1:2 (D), and 2:1 (E) (N = 3). F) Across all three fixed ratios of Cas9 mRNA:sgRNA similar trends in the amount of HDR, NHEJ, total efficacy, and unedited cells were observed, with the 2:1 Cas9 mRNA:sgRNA exhibiting the highest degree of all forms of editing (N = 3).
Staged and simultaneous delivery approaches were compared to identify the most convenient and effective approach for HDR correction. Because Cas9 is delivered in the form of mRNA, it was contemplated that staged delivery using two or three separate dLNPs could aid HDR by allowing time for mRNA translation to protein. On the other hand, simultaneous co-delivery of all three nucleic acids in one nanoparticle would be more convenient and translatable to in vivo editing.
First, to test the staged two-particle approach, 4A3-SC8 dLNPs containing Cas9 mRNA were administered to HEK293 B/GFP cells followed 24h later by 4A3-SC8 dLNPs containing both modified sgRNA and ssDNA HDR template in a single nanoparticle at fixed ratios of 1:1, either by weight or by moles (Figure S7, Supporting Information). Second, to test the threeparticle approach, each set of dLNPs contained only one of the following: Cas9 mRNA, modified sgRNA, or ssDNA HDR template. Third, the approach of an all-in-one simultaneous dLNP delivery was tested wherein a single dLNP formulation contained all three nucleic acid components for HDR (Figure S8, Supporting Information). To confirm that all three nucleic acids (Cas9 mRNA, sgRNA, ssDNA) required for HDR were co-encapsulated into a single dLNP formulation, we quantified nucleic acid loading and verified encapsulation (Figure S13, Supporting Information). Encouragingly, HDR was achieved by all three approaches as evaluated by GFP signal using flow cytometry at a similar efficiency of ≈18% (Figures S7 and S8, Supporting Information). Although all three approaches were viable, we hereafter focused on all-in-one simultaneous dLNP delivery because it was the most facile and efficacy was not decreased when compared with the staged approaches. Simultaneous, one pot delivery is especially valuable when considering accomplishing non-viral HDR in vivo as it ensures all three components will be internalized into an individual target cell, rather than only one or two of the necessary components.
Building on the all-in-one nanoparticle approach, we sought to improve HDR gene correction by optimizing the ratio of nucleic acids within the dLNPs. Sets of 4A3-SC8 dLNPs were created wherein Cas9 mRNA and sgRNA were fixed at ratios of 1:1 (Figure 3C), 1:2 (Figure 3D), and 2:1 (Figure 3E), respectively, by weight. Then, with the hypothesis that with more ssDNA HDR template available, cells could achieve a higher amount of correction via HDR, the ssDNA HDR template was titrated into the nucleic acid mixtures at increasing ratios of 0.5, 1, 2, 3, 4, 6, 8, and 10 (Table S1, Supporting Information). Among all groups tested, the ratios of 1:1:3 and 2:1:3 mRNA:modified sgRNA:ssDNA HDR template were the most efficacious, both resulting in similar HDR correction rates of 56% as quantified via DNA sequencing. DNA sequencing was instituted as our primary analytical technique for detecting gene editing events because it provides an unambiguous quantification of nucleic acid modifications at single base resolution in DNA, thereby avoiding any bias that may be associated with fluorescence reporter techniques. Interestingly, the amount of HDR achieved in all groups (1:1, 1:2, and 2:1) appeared to hit a corrective maxima of ≈50% HDR when the ratio of ssDNA HDR template included in the HDR dLNPs was between 3 and 4. In the groups where Cas9 mRNA and sgRNA were fixed at ratios of 1:1 and 1:2, and the amount of ssDNA in each group was fixed at ratios of 3 and 4, respectively, the amount of HDR correction induced dramatically tapered off. However, in the group where Cas9 mRNA and sgRNA were fixed at a ratio of 2:1, the amount of HDR achieved declined as the ratio of ssDNA HDR template increased beyond 3, but not nearly as substantially nor as sharply as in the 1:1 and 1:2 groups (Figure 3E). When analyzed for the amount and type of editing achieved across the three Cas9 mRNA:sgRNA fixed groups, common trends emerged with a progressive increase in total editing efficiency, HDR, and NHEJ up until a ssDNA ratio of ≈4 wherein editing efficiency began to decline and the number of unedited cells started to increase. Notably, editing of all forms also appeared to decline more gradually after a ssDNA ratio of 4 in the 1:2 and 2:1 groups (Figure 3F). In agreement with previous adenovirus (AAV) delivery data, these results indicate that Cas9 may not be the limiting factor for inducing high levels of HDR-mediated correction.[2c] Rather, the availability of the ssDNA template proximal to the cut site may be more important for increasing HDR, wherein an optimal ratio led to the highest HDR balancing the total amount of all three components (Cas9 mRNA, sgRNA, ssDNA). For all ratios, HDR dLNP size and polydispersity were measured to identify whether any correlation existed between HDR efficiency and either of these characteristics. Despite a slight inverted bell curve trend in particle diameter, no other notable differences existed between formulations with all HDR dLNPs exhibiting uniformity and averaging ≈150 nm in diameter (Figure S9, Supporting Information).
To confirm the results of DNA sequencing, HDR correction was also measured using flow cytometry. The amount of HDR achieved in the groups where Cas9 mRNA and sgRNA were fixed at ratios of 1:1 and 1:2 appeared to be somewhat independent of the amount of ssDNA HDR template included in the dLNPs with conditions inducing HDR at a rate of 30–35%, which could be related to the random viral integration of B/GFP DNA into the model HEK cells, although more work will be needed to study this further. In contrast, when Cas9 mRNA and modified sgRNA were fixed at ratios of 2:1, there was a nearly linear increase in the amount of HDR achieved in concordance with an increase in ssDNA HDR template. Notably, although there was not a large variance in the amount of HDR achieved in the 1:1 and 1:2 Cas9 mRNA:sgRNA groups, many of the formulations appeared to hit a similar asymptote that mirrored the maxima of ≈36% HDR achieved in the 2:1 Cas9 mRNA:sgRNA group with respect to amount of HDR induced (Figure S10, Supporting Information). To further confirm correction of the amino acid sequence and visualize the ability of the 4A3-SC8 dLNPs to induce HDR, nanoparticles were loaded Cas9 mRNA, modified sgRNA, and ssDNA HDR template at a ratio of 2:1:1 and cells were imaged using confocal microscopy (Figure 3B). As expected, corrected cells were abundant and expressing bright GFP signal which was notably absent in the PBS control.
With an established system for accomplishing high levels of HDR in vitro, we next evaluated the ability of 4A3-SC8 dLNPs containing nucleic acid ratios of either 1:1:8, 1:1:3, or 2:1:3 Cas9 mRNA:sgRNA:ssDNA, respectively, to induce HDR in vivo in a proof-of-principle experiment to aid future translatable disease correction. Xenograft tumors were generated utilizing a 1:1 mixture of the HEK293 B/GFP cells and Matrigel, which was then injected subcutaneously in the right hind legs of athymic nude Foxn1nu mice. Once the tumors were 25 mm3 in size, each set of 4A3-SC8 dLNPs containing one of the three ratios (1:1:8, 1:1:3, 2:1:3) of Cas9 mRNA, modified sgRNA, and ssDNA HDR template were formulated and injected intratumorally at a dose of 0.5 mg kg−1 total nucleic acids. After 5 days, the tumors along with internal organs were resected and imaged using IVIS for GFP signal (Figure 4A). Bright green GFP signal, indicative of gene correction via HDR, was readily apparent in the tumors treated with 4A3-SC8 dLNPs containing HDR machinery, compared with no detectable GFP signal in the PBS control (Figure 4B,C). In addition to IVIS imaging, tumors were sectioned and imaged using confocal microscopy. As expected, the tumors again demonstrated bright GFP signal indicating that HDR had been achieved (Figure 4D).
Figure 4.
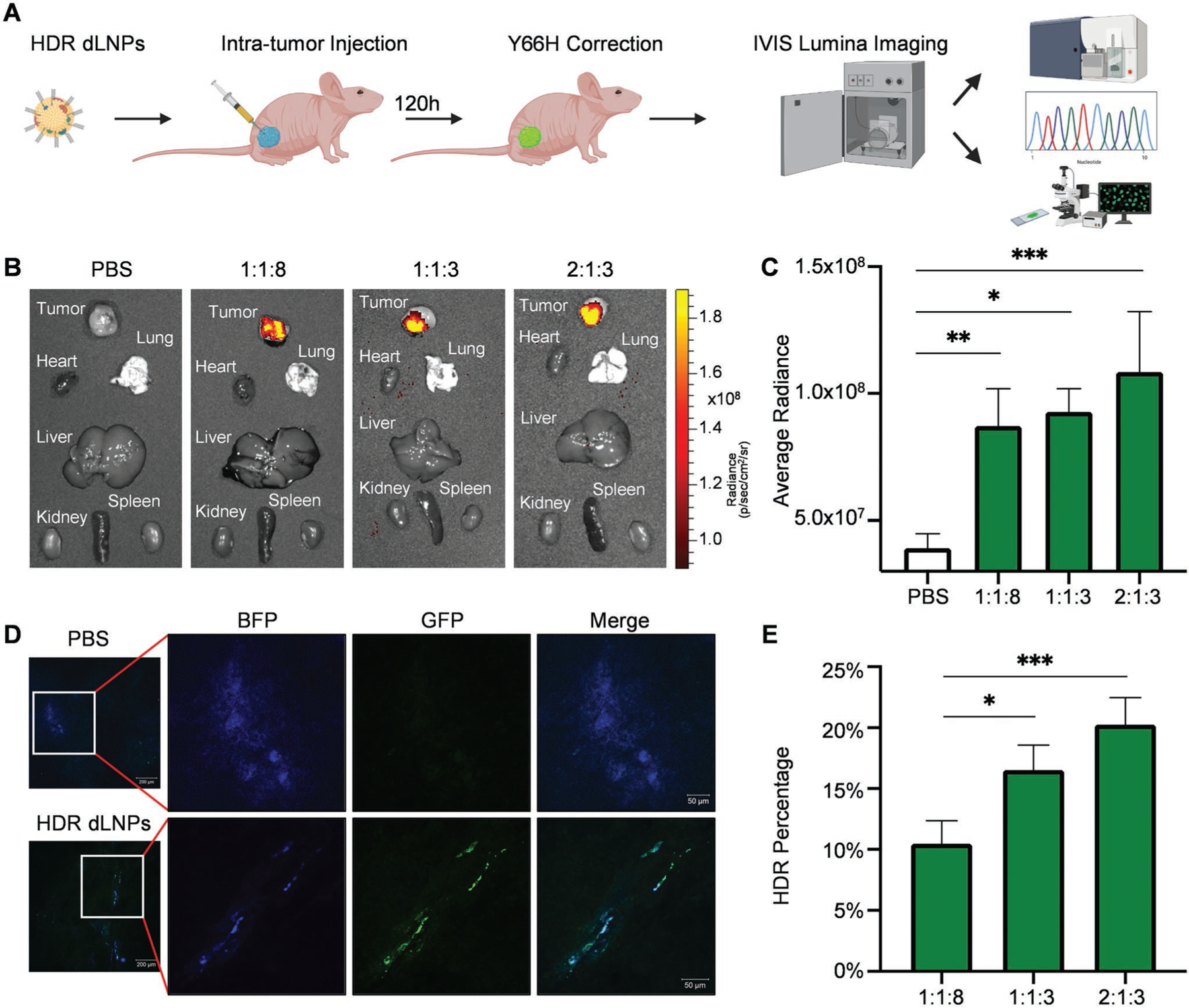
HDR gene correction was achieved in vivo. A) Subcutaneous xenograft HEK293 (Y66H) tumors were resected 5 days after injection of 4A3-SC8 HDR dLNPs for analysis by IVIS Lumina imaging, flow cytometry, confocal microscopy, and DNA sequencing. B) IVIS Lumina imaging of tumors and organs revealed bright GFP signal in the tumors injected with 1:1:8, 1:1:3, and 2:1:3 HDR dLNPs and no detectable GFP signal in the tumors injected with PBS (N = 3). C) Average radiance of tumors in each HDR dLNP group was quantified revealing a significant difference between all three HDR dLNP and PBS injected tumors (for 1:1:8 p = 0.0060, for 1:1:3 p = 0.0106, for 2:1:3 p = 0.0008; statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons test; N = 3). D) Tumors were frozen following resection and sectioned at a thickness of 7 mm for confocal imaging wherein GFP signal was clearly visible in tumors injected with HDR dLNPs. E) gDNA was extracted from whole tumors and then sequenced to obtain HDR correction percentage. Analysis via TIDER revealed average HDR-mediated correction rates of 10.475% for 1:1:8 HDR dLNPs, 16.533% for 1:1:3 HDR dLNPs, and 20.325% for 2:1:3 HDR dLNPs (Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons test, N = 4 with exception of 1:1:3 where N = 3).
With visual confirmation that HDR had been accomplished in vivo, tumors were further analyzed via DNA sequencing to more accurately quantify HDR correction throughout the tumor at the single nucleotide scale. Since visual fluorescence imaging may only analyze areas within close proximity to the injection site, this may unintentionally overestimate gene editing efficacy. Although such image quantification is commonly done in the literature and useful, we reasoned that DNA extraction from the entire tumor tissue would more accurately reflect total editing in a more unbiased fashion. Moreover, since the HEK293 (Y66H) cells were created via transduction with a lentivirus, the copy number, and insertion site of the mutated GFP reporter sequence may vary between cells. Thus, DNA sequencing may eliminate these variables by quantifying all copies of the mutant GFP sequence in cells. DNA sequencing of genomic DNA extracted from tumors revealed HDR editing of up to 23% in the group treated with 2:1:3 HDR dLNPS after analysis using TIDER (Tracking of Insertions, DEletions, and Recombination events) for assessing gene editing (Figure 4E). To the best of our knowledge, in vivo HDR corrections rates have to date been limited to 1–5%, which in some cases required combination of viral and non-viral delivery or repeated local injections.[2d,18] Here we show that a single injection of HDR dLNPs into the tumor at a dose of 0.5 mg kg−1 yielded >20% HDR-mediated gene correction. Moreover, dLNPs are able to overcome the avascularity, large size, and stiffness of the tumors to mediate HDR in vivo.
In summary, we have developed a one-pot, non-viral delivery platform capable of inducing HDR-mediated correction of a single amino acid mutation in vivo. These results improve the fundamental understanding of non-viral gene editing with respect to the components necessary for achieving HDR, and lay the foundation for accomplishing gene editing and correction in numerous genetic diseases that are present in cells and tissues with a high turnover rate. With further advancement, non-viral all-in-one nanoparticles may advance treatment options for those with many crippling genetic diseases and cancer.
Supplementary Material
Acknowledgements
D.J.S. acknowledges financial support from the National Institutes of Health (NIH) National Institute of Biomedical Imaging and Bioengineering (NIBIB) (R01 EB025192-01A1), the Cystic Fibrosis Foundation (CFF) (SIEGWA18XX0), the Cancer Prevention and Research Institute of Texas (CPRIT) (RP190251, RP160157), the American Cancer Society (ACS) (RSG-17-012-01), and the Welch Foundation (I-1855). T.W. acknowledges financial support from a CPRIT Training Grant (RP160157). The authors acknowledge the UTSW Small Animal Imaging Shared Resource and the UTSW Tissue Resource, which are supported in part by the National Institutes of Health, National Cancer Institute Support Grant (P30CA142543), and the Moody Foundation Flow Cytometry Facility. All experiments were approved by the Institutional Animal Care & Use Committee (IACUC) of The University of Texas Southwestern Medical Center and were consistent with local, state and federal regulations as applicable. Some graphical elements in the table of contents figure, Figure 1, Figure 3A, and Figure 4A were created with Biorender.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].a) Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E, Science 2012, 337, 816; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cong L, Ran FA, Cox D, Lin SL, Barretto R, Habib N, Hsu PD, Wu XB, Jiang WY, Marraffini LA, Zhang F, Science 2013, 339, 819; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mali P, Yang LH, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM, Science 2013, 339, 823; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Doudna JA, Charpentier E, Science 2014, 346, 1258096; [DOI] [PubMed] [Google Scholar]; e) Sander JD, Joung JK, Nat. Biotechnol 2014, 32, 347; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Wang HX, Li M, Lee CM, Chakraborty S, Kim HW, Bao G, Leong KW, Chem. Rev 2017, 117, 9874. [DOI] [PubMed] [Google Scholar]
- [2].a) Long CZ, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, Olson EN, Science 2016, 351, 400; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Min Y-L, Bassel-Duby R, Olson EN, Annu. Rev. Med 2019, 70, 239; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Min YL, Li H, Rodriguez-Caycedo C, Mireault AA, Huang J, Shelton JM, McAnally JR, Amoasii L, Mammen PPA, Bassel-Duby R, Olson EN, Sci. Adv 2019, 5, eaav4324; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jo DH, Song DW, Cho CS, Kim UG, Lee KJ, Lee K, Park SW, Kim D, Kim JH, Kim JS, Kim S, Kim JH, Lee JM, Sci. Adv 2019, 5, eaax1210; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wei T, Cheng Q, Farbiak L, Anderson DG, Langer R, Siegwart DJ, ACS Nano 2020, 14, 9243; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Li L, Hu S, Chen X, Biomaterials 2018, 171, 207; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Xu CF, Chen GJ, Luo YL, Zhang Y, Zhao G, Lu ZD, Czarna A, Gu Z, Wang J, Adv. Drug Delivery Rev 2019, 168, 3; [DOI] [PubMed] [Google Scholar]; h) Wang L, Zheng W, Liu S, Li B, Jiang X, ChemBio-Chem 2019, 20, 634. [DOI] [PubMed] [Google Scholar]
- [3].a) Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F, Nat. Protoc 2013, 8, 2281; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Platt RJ, Chen SD, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, Graham DB, Jhunjhunwala S, Heidenreich M, Xavier RJ, Langer R, Anderson DG, Hacohen N, Regev A, Feng GP, Sharp PA, Zhang F, Cell 2014, 159, 440; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sanchez-Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A, Joshi NS, Subbaraj L, Bronson RT, Xue W, Jacks T, Nature 2014, 516, 428; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Knott GJ, Doudna JA, Science 2018, 361, 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Lin S, Staahl B, Alla RK, Doudna JA, eLife 2014, 3, e04766; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Glass Z, Lee M, Li Y, Xu Q, Trends Biotechnol. 2018, 36, 173; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pickar-Oliver A, Gersbach CA, Nat. Rev. Mol. Cell Biol 2019, 20, 490; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yeh CD, Richardson CD, Corn JE, Nat. Cell Biol 2019, 21, 1468; [DOI] [PubMed] [Google Scholar]; e) Anzalone AV, Koblan LW, Liu DR, Nat. Biotechnol 2020, 38, 824; [DOI] [PubMed] [Google Scholar]; f) Li B, Niu Y, Ji W, Dong Y, Trends Pharmacol. Sci 2020, 41, 55; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R, Nat. Rev. Drug Discovery 2021, 20, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Pinder J, Salsman J, Dellaire G, Nucleic Acids Res. 2015, 43, 9379; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Song J, Yang DS, Xu J, Zhu TQ, Chen YE, Zhang JF, Nat. Commun 2016, 7, 10548; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gutschner T, Haemmerle M, Genovese G, Draetta GF, Chin L, Cell Rep. 2016, 14, 1555; [DOI] [PubMed] [Google Scholar]; d) Jasin M, Haber JE, DNA Repair 2016, 44, 6; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Gallagher DN, Haber JE, ACS Chem. Biol 2018, 13, 397; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Aird EJ, Lovendahl KN, St Martin A, Harris RS, Gordon WR, Commun. Biol 2018, 1, 54; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Nambiar TS, Billon P, Diedenhofen G, Hayward SB, Taglialatela A, Cai KH, Huang JW, Leuzzi G, Cuella-Martin R, Palacios A, Gupta A, Egli D, Ciccia A, Nat. Commun 2019, 10, 3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Chandler RJ, LaFave MC, Varshney GK, Trivedi NS, Carrillo-Carrasco N, Senac JS, Wu W, Hoffmann V, Elkahloun AG, Burgess SM, Venditti CP, J. Clin. Invest 2015, 125, 870; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chandler RJ, Sands MS, Venditti CP, Hum. Gene Ther 2017, 28, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Lattanzi A, Meneghini V, Pavani G, Amor F, Ramadier S, Felix T, Antoniani C, Masson C, Alibeu O, Lee C, Porteus MH, Bao G, Amendola M, Mavilio F, Miccio A, Mol. Ther 2019, 27, 137; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mout R, Ray M, Lee YW, Scaletti F, Rotello VM, Bioconjugate Chem. 2017, 28, 880; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu C, Zhang L, Liu H, Cheng K, J. Controlled Release 2017, 266, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Zhou K, Nguyen LH, Miller JB, Yan Y, Kos P, Xiong H, Li L, Hao J, Minnig JT, Zhu H, Siegwart DJ, Proc. Natl. Acad. Sci. USA 2016, 113, 520; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang S, Zhou K, Luo X, Li L, Tu HC, Sehgal A, Nguyen LH, Zhang Y, Gopal P, Tarlow BD, Siegwart DJ, Zhu H, Dev. Cell 2018, 44, 447; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang S, Nguyen LH, Zhou K, Tu HC, Sehgal A, Nassour I, Li L, Gopal P, Goodman J, Singal AG, Yopp A, Zhang Y, Siegwart DJ, Zhu H, Gastroenterology 2018, 154, 1421; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cheng Q, Wei T, Jia Y, Farbiak L, Zhou K, Zhang S, Wei Y, Zhu H, Siegwart DJ, Adv. Mater 2018, 30, 1805308; [DOI] [PubMed] [Google Scholar]; e) Zhou K, Johnson LT, Xiong H, Barrios S, Minnig JT, Yan Y, Abram B, Yu X, Siegwart DJ, Mol. Pharmaceutics 2020, 17, 1575; [DOI] [PubMed] [Google Scholar]; f) Cheng Q, Wei T, Farbiak L, Johnson LT, Dilliard SA, Siegwart DJ, Nat. Nanotechnol 2020, 15, 313; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Wei T, Cheng Q, Min YL, Olson EN, Siegwart DJ, Nat. Commun 2020, 11, 3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Zelphati O, SzokaJr FC., Proc. Natl. Acad. Sci. USA 1996, 93, 11493; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Harvie P, Wong FM, Bally MB, Biophys. J 1998, 75, 1040; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hafez IM, Maurer N, Cullis PR, Gene Ther. 2001, 8, 1188; [DOI] [PubMed] [Google Scholar]; d) Sahay G, Querbes W, Alabi C, Eltoukhy A, Sarkar S, Zurenko C, Karagiannis E, Love K, Chen D, Zoncu R, Buganim Y, Schroeder A, Langer R, Anderson DG, Nat. Biotechnol 2013, 31, 653; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U, Manygoats K, Seifert S, Andree C, Stoter M, Epstein-Barash H, Zhang L, Koteliansky V, Fitzgerald K, Fava E, Bickle M, Kalaidzidis Y, Akinc A, Maier M, Zerial M, Nat. Biotechnol 2013, 31, 638; [DOI] [PubMed] [Google Scholar]; f) Wittrup A, Ai A, Liu X, Hamar P, Trifonova R, Charisse K, Manoharan M, Kirchhausen T, Lieberman J, Nat. Biotechnol 2015, 33, 870; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Li WJ, Szoka FC, Pharm. Res 2007, 24, 438. [DOI] [PubMed] [Google Scholar]
- [10].a) Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao YJ, Pirzada ZA, Eckert MR, Vogel J, Charpentier E, Nature 2011, 471, 602; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gasiunas G, Barrangou R, Horvath P, Siksnys V, Proc. Natl. Acad. Sci. USA 2012, 109, E2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nguyen DN, Roth TL, Li PJ, Chen PA, Apathy R, Mamedov MR, Vo LT, Tobin VR, Goodman D, Shifrut E, Bluestone JA, Puck JM, Szoka FC, Marson A, Nat. Biotechnol 2020, 38, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mojica FJM, Diez-Villasenor C, Garcia-Martinez J, Almendros C, Microbiology 2009, 155, 733. [DOI] [PubMed] [Google Scholar]
- [13].Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan AH, Moineau S, Nature 2010, 468, 67. [DOI] [PubMed] [Google Scholar]
- [14].a) Miller JB, Zhang S, Kos P, Xiong H, Zhou K, Perelman SS, Zhu H, Siegwart DJ, Angew. Chem., Int. Ed 2017, 56, 1059; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miller JB, Kos P, Tieu V, Zhou K, Siegwart DJ, ACS Appl. Mater. Interfaces 2018, 10, 2302. [DOI] [PubMed] [Google Scholar]
- [15].a) Li B, Luo X, Deng B, Wang J, McComb DW, Shi Y, Gaensler KM, Tan X, Dunn AL, Kerlin BA, Dong Y, Nano Lett. 2015, 15, 8099; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kauffman KJ, Dorkin JR, Yang JH, Heartlein MW, DeRosa F, Mir FF, Fenton OS, Anderson DG, Nano Lett. 2015, 15, 7300; [DOI] [PubMed] [Google Scholar]; c) Miller JB, Siegwart DJ, Nano Res. 2018, 11, 5310; [Google Scholar]; d) Ball RL, Hajj KA, Vizelman J, Bajaj P, Whitehead KA, Nano Lett. 2018, 18, 3814; [DOI] [PubMed] [Google Scholar]; e) Hajj KA, Ball RL, Deluty SB, Singh SR, Strelkova D, Knapp CM, Whitehead KA, Small 2019, 15, 1805097; [DOI] [PubMed] [Google Scholar]; f) Patel S, Ashwanikumar N, Robinson E, DuRoss A, Sun C, Murphy-Benenato KE, Mihai C, Almarsson O, Sahay G, Nano Lett. 2017, 17, 5711; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Hao J, Kos P, Zhou K, Miller JB, Xue L, Yan Y, Xiong H, Elkassih S, Siegwart DJ, J. Am. Chem. Soc 2015, 137, 9206. [DOI] [PubMed] [Google Scholar]
- [16].Yan Y, Liu L, Xiong H, Miller JB, Zhou K, Kos P, Huffman KE, Elkassih S, Norman JW, Carstens R, Kim J, Minna JD, Siegwart DJ, Proc. Natl. Acad. Sci. USA 2016, 113, E5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE, Nat. Biotechnol 2016, 34, 339. [DOI] [PubMed] [Google Scholar]
- [18].a) Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, Koteliansky V, Sharp PA, Jacks T, Anderson DG, Nat. Biotechnol 2014, 32, 551; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yin H, Song C-Q, Dorkin JR, Zhu LJ, Li Y, Wu Q, Park A, Yang J, Suresh S, Bizhanova A, Gupta A, Bolukbasi MF, Walsh S, Bogorad RL, Gao G, Weng Z, Dong Y, Koteliansky V, Wolfe SA, Langer R, Xue W, Anderson DG, Nat. Biotechnol 2016, 34, 328; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA, Mackley VA, Chang K, Rao A, Skinner C, Shobha T, Mehdipour M, Liu H, Huang WC, Lan F, Bray NL, Li S, Corn JE, Kataoka K, Doudna JA, Conboy I, Murthy N, Nat. Biomed. Eng 2017, 1, 889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.