

Abstract
The Type III-E RNA-targeting effector complex (gRAMP/Cas7-11) is associated with a caspase-like protein (TPR-CHAT/Csx29) to form Craspase (CRISPR-guided caspase). Here we use cryo-electron microscopy snapshots of Craspase to explain its target RNA cleavage and protease activation mechanisms. Target-guide pairing extending into the 5′ region of the guide RNA displaces a gating loop in gRAMP, which triggers an extensive conformational relay that allosterically aligns the protease catalytic dyad and opens an amino acid sidechain-binding pocket. We further define Csx30 as the endogenous protein substrate that is site-specifically proteolyzed by RNA-activated Craspase. This protease activity is switched off by target RNA cleavage by gRAMP, and is not activated by RNA targets containing a matching protospacer flanking sequence. We thus conclude that Craspase is a target RNA-activated protease with self-regulatory capacity.
One Sentence Summary:
This work generates the high-resolution mechanisms for RNA-guided RNA and protein cleavage by CRISPR-guided Caspase (Craspase).
It has become clear that RNA-guided DNA/RNA degradation is not the sole mechanism for CRISPR-Cas to confer immunity against foreign genetic elements in prokaryotes (1–5). Type III CRISPR-Cas systems in particular present a plethora of alternative mechanisms, including RNA-guided secondary messenger production and signaling (6, 7) to activate a range of immune responses (e.g. collateral RNA damage) (6–8). Type III CRISPR-Cas effectors are typically assembled from multiple protein subunits to enable crRNA binding, target RNA cleavage, DNA cleavage and secondary messenger synthesis (9, 10). Type III-E is a recently identified atypical Type III system. It encodes a large polypeptide (gRAMP) as a fusion of four Cas7-like domains, one Cas11-like domain, and a big insertion domain (BID), but lacks Cas10, the signature component of a canonical Type III system that is required for secondary messenger production (5). Subsequent studies showed that gRAMP ribonucleoprotein (RNP) complex is capable of RNA-guided RNA cleavage at two specific sites (11, 12), six nucleotides (nts) apart (11). Unlike the Type VI CRISPR-Cas effector Cas13, gRAMP does not cause collateral RNA cleavage, and has no cytotoxicity in eukaryotic cells (12).
In Type III-E loci, gRAMP frequently associates with TPR-CHAT, a caspase-like protein with N-terminal TPR repeats (5). Caspases are a family of cysteine proteases controlling programmed cell death (PCD) pathways in eukaryotes (13). Cleavage of gasdermin by caspases, for example, triggers membrane pore formation to cause cell death (14, 15). An equivalent PCD pathway was recently discovered in prokaryotes, where TPR-CHAT was shown to cleave bacterial gasdermin to induce cellular suicide (2). In Type III-E systems, TPR-CHAT and gRAMP form an effector complex named Craspase, for CRISPR-guided caspase (11). This observation raised the possibility that Craspase may function as a CRISPR RNA-guided protease to prevent the spread of phage infection through a suicide mechanism. However, it remains unknown how Craspase is structurally organized, whether TPR-CHAT in Craspase is a protease, and whether its activity is regulated by RNA (16, 17).
gRAMP structures in resting, RNA-bound, and post-cleavage states
To gain insights into the RNA-guided target RNA cleavage mechanisms inside gRAMP, we reconstituted Candidatus “Scalindua brodae” gRAMP (Sb-gRAMP) (11) and determined its cryo-electron microscopy (cryo-EM) structures in different functional states (Figs. 1A, S1). Consistent with previous results (11), Sb-gRAMP bound to the complementary RNA target with better than 25 nM affinity and cleaved it at two distinct locations, after the 3rd (Site 1) and 9th (Site 2) nucleotides (Fig. S1A–C). Single-particle three-dimensional reconstruction produced Sb-gRAMP RNP in four different functional states: a 3.81 Å structure of the resting/apo state, 3.65 Å structure of the non-matching protospacer flanking sequence (PFS) target bound state, 3.76 Å structure of the matching PFS target bound state, and 3.62 Å structure of the post-cleavage state (Fig. 1B–E, S2–S4; Table S1).
Fig. 1. Structural snapshots of Sb-gRAMP RNP in different functional states.

(A) Type III-E operon in Candidatus ‘Scalindua brodae’. Snapshot of (B) Sb-gRAMP at resting state, (C) non-matching PFS RNA bound state, (D) matching PFS RNA bound state, and (E) non-matching PFS RNA post-cleavage state with MgCl2. Top images are cryo-EM densities and bottom images are structural models.
The overall architecture of Sb-gRAMP is similar to that of D. ishimotonii Cas7-11 (Di-Cas7-11), recently reported in the target-bound form (18). The two structures in the same functional state superimpose with an r.m.s.d. of 1.1 Å for Cα atoms, excluding the BID domain, which is less conserved and poorly resolved in the EM density (Fig. S5). Sb-gRAMP also shares some degree of similarity with the canonical Type III-A effector Csm (10, 19–21) in overall architecture, guide RNA display, and target RNA binding mode (Fig. S6). The Sb-gRAMP backbone consists of four non-identical Cas7 domains fused together, instead of three identical Cas7 subunits in Csm (Figs. S6). A Zn-knuckle is present in each of the four Cas7s, which appears to be a shared hallmark among Type III effectors (Fig. S7A). Csm further contains one copy of Csm4 for 5′-handle recognition, two copies of Csm2 as part of the backbone, and one copy of Csm5 for continued guide-target pairing. In contrast, Sb-gRAMP is streamlined: its Cas7.1 has been repurposed for 5′-handle recognition, the single-copy Cas11 domain has been repurposed for target cleavage, and a structurally distinct BID replaces Csm5 (Fig. S6A–H). On the guide RNA side, the ordered 18-nt 5′-handle of the CRISPR RNA (crRNA) in Sb-gRAMP is twice as long as in other Class I CRISPR-Cas systems (Fig. 2A–B). The majority of the handle residues are bound by Cas7.1 and shielded on the top by the linker from Cas11 to Cas7.2 and the Zn-knuckle in Cas7.2 (Fig. S6, S7B–C). Mutagenesis of the Zn-knuckle structure or sequence-specific contacts to the 5′-handle abolished the in vivo RNA silencing activity of Sb-gRAMP, presumably through disruption of RNP assembly (Fig. S8). Surprisingly, Sb-gRAMP differs from Di-Cas7-11 in crRNA biogenesis. In Di-Cas7-11, there is an endoribonuclease center in Cas7.1 for crRNA processing, whereas the equivalent residues in Sb-gRAMP are non-catalytic (Fig. 2C–D) (18). This structural difference rationalizes the observation that the crRNA 5′-handle in Sb-gRAMP is 3-nt longer. We speculate that Sb-gRAMP may rely on certain host nucleases for crRNA biogenesis.
Fig. 2. crRNA accommodation and target RNA recognition mechanisms by Sb-gRAMP.
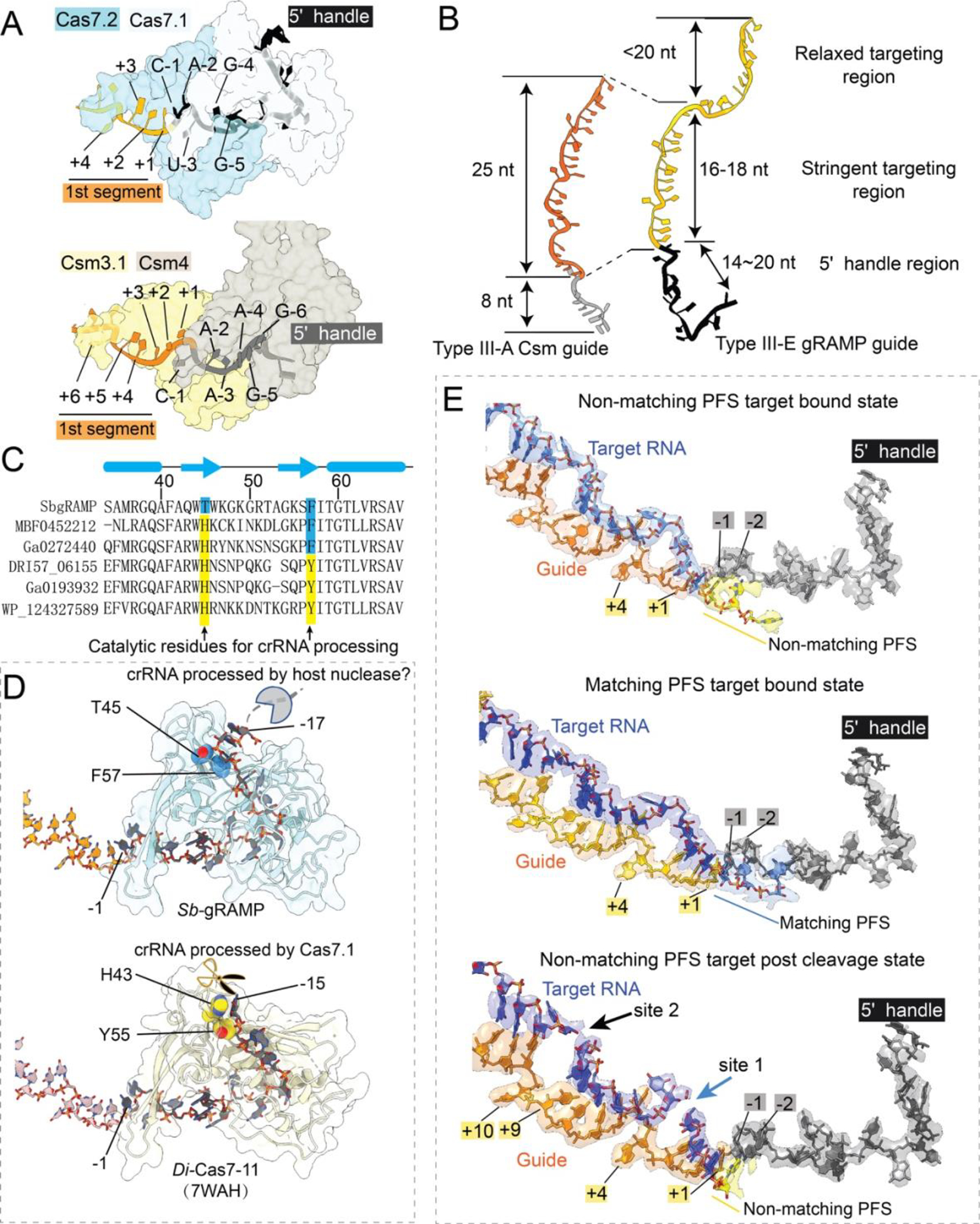
(A) Accommodation of crRNA 5’-handle and (B) spacer region in Type III-E Sb-gRAMP and Type III-A Csm. (C) Primary sequence and (D) 3D structural alignment at the pre-crRNA cleavage center. Catalytic residues in Di-Cas7-11 are colored in yellow; equivalent residues in Sb-gRAMP are in blue. (E) Extracted Cryo-EM density from non-matching PFS RNA (left), matching PFS RNA (middle) and non-matching PFS RNA post-cleavage state (right).
Notably, the last two handle nucleotides (5′-A−2C−1-3′) are base-pairing competent because they are displayed like a guide (Fig. 2A, S6H–G). Type I, III, and IV effectors display the crRNA spacer (guide region) in 6-nt segments, with the 6th nucleotide pinned down by the thumb loop of Cas7; the target is hence recognized in 5-nt segments with the 6th nucleotide unspecified. Sb-gRAMP contains major exceptions. The first 5-nt segment contains the last two nucleotides of the 5′-handle and the first three nucleotides of the spacer, a scenario only observed in Type III-E (18) (Fig. 2A, 2E, S6H–G). The third segment deviates from the normal again, as an unconventional knotted protein loop from Cas7.4 divides the displayed bases to a 3-nt block and a 6-nt block. The two blocks are divided by a single peptide crossover rather than a β-hairpin thumb, therefore no nucleotide is pinned underneath and the base-pairing in the 3rd segment is not interrupted. The following crRNA nucleotides are displayed by the dynamic BID domain (aa 1031–1385) which is only resolved to low-resolution and therefore docked with an AlphaFold predicted model (22) (Fig. 1B).
Off-targeting prevention and RNA cleavage mechanisms in Sb-gRAMP
By capturing three additional functional states, we have the temporal resolution to interpret the target recognition and cleavage mechanisms by Sb-gRAMP. We found that the long linker from Cas11 to Cas7.2 (G375-E412, here named the gating loop) has acquired important functions for RNase regulation (Fig. 3A–B, S9). Its N-terminal portion (G375-G397) senses RNA substrate binding and controls RNase activities. In resting state, the gating loop blocks the first segment of the guide RNA and the nearby Site 1 cleavage center. This conformation is incompatible with target-guide pairing at the first segment and the gating loop has to be displaced to enable cleavage at Site 1 (Fig. 3A). We therefore envision that the target-guide pairing initiates from the third and second segments and propagates into the first segment (Fig. S9), as observed for other Type III systems (23). In follow-up experiments, we found Sb-gRAMP’s RNase activity was optimal against a target with 18-nt complementarity from the 5′-end of the spacer portion; 12-nt or shorter complementarity abolished cleavage and 24-nt or longer complementarity attenuated cleavage (Fig. S10). This suggests that at least some base-pairing along all three segments of the guide RNA, displayed by Cas7.2-Cas7.4, is required for efficient RNA cleavage. In contrast, additional base-paring with crRNA at the BID is not required or may even be counterproductive (Fig. S10). This is consistent with the previous observation that the 3′-end of the crRNA in the endogenous Sb-gRAMP is often as short as 20 nt (11), and that the BID is dispensable for Cas7-11 activity in human cells (18).
Fig. 3. Target validation and cleavage mechanisms by Sb-gRAMP RNP.

(A) Models depicting the gate closed structure in resting state (left) and gate open structure in target RNA bound state (middle). Superposition is shown as the right panel. (B) Sequence alignment at the gating loop region. Conserved residues are highlighted in burgundy red. (C) Structural comparison of the resting and non-matching PFS RNA bound states. Vector length is proportional to residue movement distance. Hinge motion in Cas11 is pronounced. (D) EMSA (top) and urea-PAGE (bottom) to evaluate the impact of gating loop disruption on the binding and cleavage of partially matching RNA targets. (E) Mechanistic model depicting the essential role of the gating loop in target validation. (F) Structural basis for Site 1 cleavage. (G) Structural basis for Site 2 cleavage. (H) Impact of Site 1 (in blue) and Site 2 (in black) mutations on target RNA cleavage efficiency.
Sb-gRAMP was further incubated with two kinds of RNA targets whose PFS was either matching (complementary) or non-matching with the 5′-handle in the crRNA, as complementarity in this region may be indicative of a self-target (i.e. anti-sense transcript from the CRISPR locus) and thus perhaps leads to alternative structural configurations in Sb-gRAMP. However, our structures reveal that regardless of the PFS status, RNA binding induces the same set of conformational changes in Sb-gRAMP. Where the guide nucleotides are pinned down by the Cas7 thumbs, the corresponding target nucleotides (4th and 10th) flip outwards. Rotation of the backbone orients their 2′-OH towards the previous phosphate, forming the so-called “in-line” conformation, which is necessary for RNA cleavage. For target RNA with a matching PFS, the first segment consists of five base-pairs, starting from the last two nucleotides of the 5′-handle and ending with the 3rd nucleotide in the spacer portion (Fig. 2E). The rest of the PFS is not traceable in the EM map. For target RNA with a non-matching PFS, only three base-pairs are found between the target RNA and the spacer portion of the guide. While the first two nucleotides of the PFS do not form hydrogen bonds with the two 5′-handle residues on the opposite side, they remain stacked to complete the first target-guide segment (Fig. 2E). In both PFS matched and non-matched conditions, the impinging gating loop in Sb-gRAMP is pushed away from the first segment and becomes entirely disordered (Fig. 3A). Concurrently, the cleavage center at Site 1 is exposed and further enhanced by a hinge motion in Cas11 (Fig. 3C, S11A), which aligns catalytic residues among Cas11 and Cas7.2. It should be noted that stacking from the additional 2-nt PFS is not a prerequisite to activate Sb-gRAMP, as RNA substrates lacking nucleotides in the PFS region were found to be cleaved efficiently (11, 18). To validate these structural findings, we replaced the tip of the gating loop with a flexible linker to evaluate its importance in target RNA recognition (Fig. S9D–E). Wild-type Sb-gRAMP did not bind or cleave RNA that only base-pairs with the first 9-nt of the crRNA guide. In contrast, the gating loop mutant bound this target RNA efficiently and subsequently cleaved it (Fig. 3D). These experiments suggest that the gating loop plays a pivotal role in preventing off-targeting. Overall, our RNA-bound Sb-gRAMP structures support a mechanistic model in which the resting Sb-gRAMP exists in an autoinhibited state to avoid sequence-nonspecific RNA binding and cleavage. Target RNA is validated via crRNA-pairing in a directional fashion from the 3′ to 5′ region of the guide. Upon completion of target binding, movement of the gating loop initiates a chain of allosteric events to switch on the RNase centers in gRAMP (Fig. 3E; Movie S1).
We further attempted to interpret the cleavage mechanism by comparing the pre- and post-cleavage states (Fig. 1, 3F–G). EM densities suggest the RNA substrate was cleaved after the 3rd and 9th nucleotides (Site 1 and Site 2, respectively) (Fig. 2E), which is consistent with previous reports (11, 18). Since cleavage is metal-dependent, we identified multiple candidate residues around the cleavage sites that may contribute to metal coordination (generally acidic residues), proton shuttling (generally polar residues), and transition state stabilization (generally positively charged residues) (Fig. 3F–G). In subsequent mutagenesis testing (Fig. S11B–D), RNA cleavage at Site 1 was abolished by alanine substitutions to D547 in Cas7.2 and R294, D298, Y367, and K371 in Cas11 (Fig. 3H). Since Site 1 is assembled from residues in both Cas11 and Cas7.2, it may only become active after target binding-induced hinge motion in Cas11. Cleavage at Site 2 was abolished by Cas7.3 mutations D698A (11) and D806A, but not by Cas11 mutations R323A and H328A (Fig. 3H). An allosteric effect was noticed: Site 1 disruptive mutations D547A and D298A impaired Site 2 cleavage as well, and Site 2 mutation H328A impaired Site 1 cleavage instead. These mutants appeared to weaken or alter the RNA-binding mode of Sb-gRAMP, as revealed by electrophoretic mobility shift assay (EMSA) (Fig. 3H, S11C). Sb-gRAMP containing the double mutations R294A/D698A or D547/D806A was efficient in RNA binding but completely inactive in RNA cleavage (Fig. S11C). Such dead-gRAMP variants could be useful in RNA editing, tagging, or tracing applications.
Craspase architecture and component interfaces
To gain mechanistic insights into how the putative RNA-guided protease system may work, we reconstituted Craspase in its apo (resting) state, the matching PFS-containing RNA target bound state, and the non-matching PFS-containing target bound state, and generated their corresponding cryo-EM structures at 3.7 Å, 2.6 Å, and 2.7 Å resolutions, respectively (Fig. S12–S14). The TPR-CHAT binding surface is on top of the buried crRNA 5′-handle in Sb-gRAMP, architecturally similar to where the cOA synthetase (Csm1/Cas10) binds in canonical Type III-A effector complexes (Fig. 4A–B, S15A, Movie S2). TPR-CHAT consists of an N-terminal TPR domain (aa 1–323), a dynamic mid-region (aa 324–399), and a C-terminal cysteine protease from the caspase family (aa 400–717). The domain arrangement of TPR-CHAT resembles that of separase (24, 25), an essential eukaryotic protein that cleaves the cohesin ring to allow chromosome segregation (Fig. S15B–D). Like separase, the CHAT domain contains a N-terminal pseudo-caspase domain, a long dimeric coiled coil mid-insertion, and a C-terminal active-protease domain (24, 25). Although structurally distinct, the two caspase domains pack in a similar fashion as the eukaryotic caspase dimers do (26). In TPR-CHAT, the β-sheet structure in the pseudo-caspase domain interacts with the TPR domain and the mid-region serves as the sole anchoring point of CHAT onto Sb-gRAMP. The TPR repeats belong to the so-called solenoid domains, which are assembled from repeating structural units and often mediate protein-protein or protein-ligand interactions (27). The seven TPR repeats in TPR-CHAT pack side-by-side to form a C-shaped architecture, with the 7th TPR repeat packing against the β-sheet of the globular CHAT domain. Together, TPR-CHAT adopts the rough shape of a padlock, with TPR being the shackle and CHAT the body (Fig. S15B). In the Craspase structure without target RNA (apo-Craspase), the shackle of the padlock captures a long “switch helix” (aa338–362) in the middle. The switch helix is captured by the molecular contacts from the inward facing loops in the TPR repeats. When wedged in the shackle, the switch helix pins down a loop-helix-loop structure underneath (aa 324–337). Together, they mediate an extensive set of molecular contacts to multiple regions inside the padlock (Fig. S15B), including contacts to the tips of two long β-hairpins (sensor hairpins) that further extend all the way to the protease center in CHAT (Fig. S15E).
Fig. 4. Structural basis for Craspase assembly.
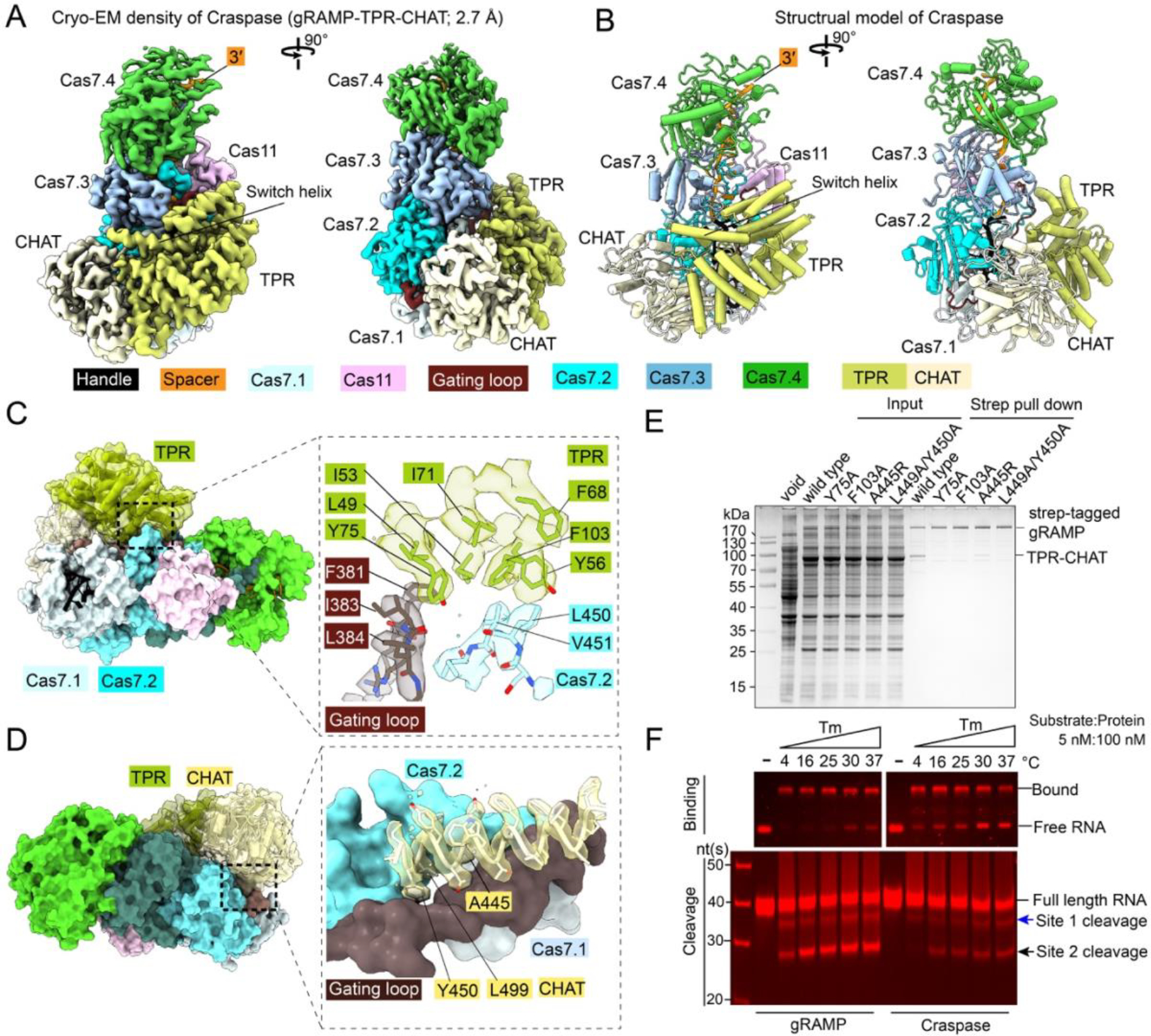
(A) 2.7 Å cryo-EM density and (B) structural model of Craspase (gRAMP-TPR-CHAT). (C) Location and zoom-in view of the molecular contacts between gRAMP and TPR. Interface residues and corresponding cryo-EM densities are shown. (D) Location and zoom-in view of the molecular contacts between gRAMP and CHAT. (E) Strep-tag affinity purifications quantifying the impact of interface mutations on Craspase complex formation. (F) EMSA (top) and urea-PAGE (bottom) to quantify activity differences in RNA binding and cleavage by gRAMP and Craspase.
A ~75X35 Å2 area of the Cas7.1 surface in Sb-gRAMP is buried by TPR-CHAT (Fig. 4C–D). However, the actual physical contacts between TPR-CHAT and Sb-gRAMP are limited to two surface patches 50 Å apart. On the TPR side, a hydrophobic patch in the first and second TPR repeats makes hydrophobic and mainchain hydrogen bond contacts to a portion of the gating loop (F381, I383, and L384), and a nearby Cas7.2 loop (L450, V451) (Fig. 4C). A more extensive and mostly hydrophobic interface is found between one of the coiled coil helix in the CHAT domain (aa 434–450) and two regions of Sb-gRAMP, namely the C-terminal portion of the gating loop (aa 396–403) and the Zn-knuckle of Cas7.2 (Fig. 4D). In particular, Y450 and L499 of CHAT insert into a hydrophobic pocket on the Sb-gRAMP surface, promoting shape complementarity at the interface. The interaction between gRAMP and TPR-CHAT was completely disrupted by Y75A and F103A mutations in the TPR interface, and severely impaired by A445R and L449A/Y450A mutations in the CHAT interface (Fig. 4E). An important observation is that the gating loop of Sb-gRAMP, which plays a pivotal role in regulating the RNase activity of Sb-gRAMP through conformational changes, is sandwiched between Sb-gRAMP and TPR-CHAT (Fig. S16A). Whereas the entire gating loop becomes unstructured in the RNA-bound Sb-gRAMP structure, only the tip of it is rearranged in the RNA-bound Craspase (Fig. 3A, S17). Given this conformational restriction, we speculated that the energetic barrier for RNase activation may be higher in Craspase compared to Sb-gRAMP. Indeed, RNA binding was consistently weaker at different temperatures and the cleavage was slower in Craspase compared to Sb-gRAMP (Fig. 4F, S16B–C).
RNA-guided protease activation mechanism in Craspase
When Craspase is in the resting state, the catalytic dyad in the TPR-CHAT protease center, Cys627 and His585, are 6.6 Å apart (Fig. S18). As this exceeds hydrogen bonding distance by a large margin, C627 could not be deprotonated by H585, hence could not initiate the nucleophilic attack on the peptide substrate. Our structure therefore suggests TPR-CHAT in the apo Craspase is an inactive protease. When Craspase is bound to a target RNA with a matching PFS (Fig. 5A), a perfectly base-paired first segment is formed between guide and target. Constrained by the base-pairing from the first two PFS residues to the guide, the remaining PFS nucleotides point towards the bottom of TPR. Although their densities are difficult to model, possible phosphate densities suggest that the PFS travels underneath TPR (Fig. 5C, Movies S3–S5). This path may have perturbed the conformation dynamics of the sensing β-hairpin in CHAT, as its tip that may contact PFS becomes disordered. This coincides with a backbone twitch at the protease center, on the opposite end of the sensing hairpin (aa 626–631) (Fig. 5D). Notably, C627 and H585 reside on the two strands of the sensing hairpin. The allosteric change shortens their distance from 6.6 to 5.2 Å (Fig. S18B). This distance, however, is still too far to allow H585-mediated C627 deprotonation. Moreover, the nearby sidechain-binding pocket found in the apo structure is closed after the structural rearrangement (Fig. 5D). Therefore, the matching PFS RNA bound Craspase is not expected to be proteolytically active, either.
Fig. 5. Structural basis for Craspase protease activation.
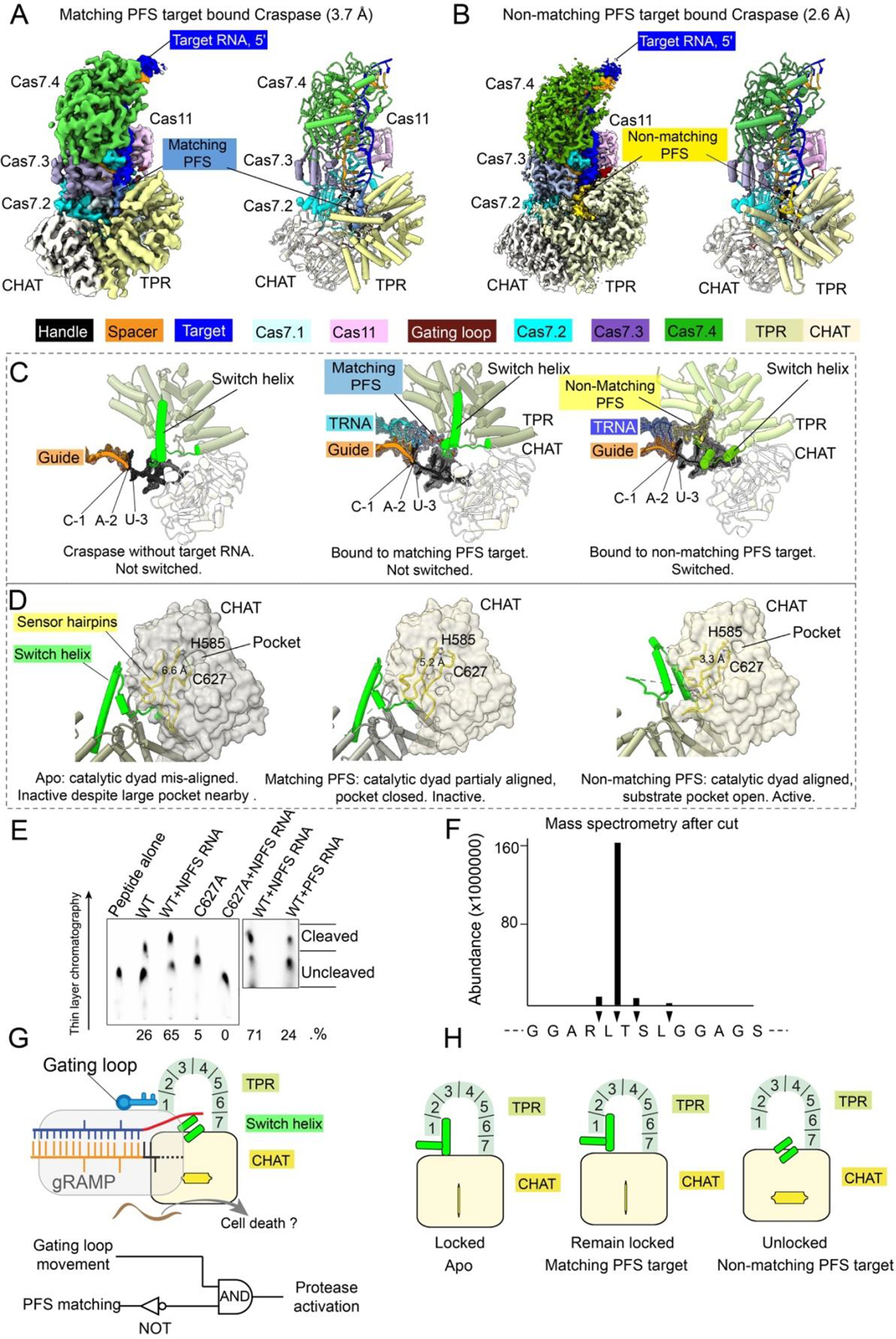
(A) 3.7 Å cryo-EM density (left) and structural model (right) of matching PFS RNA bound Craspase. (B) 2.6 Å cryo-EM density (left) and structural model (right) of non-matching PFS RNA bound Craspase. (C) Close-up views of switch helix in resting state (left), matching PFS RNA bound state (middle), and non-matching PFS RNA bound state (right). Switch helix highlighted in green and the density of crRNA and target RNA (TRNA) are shown in mesh. (D) Conformation of the switch helix and sensor hairpin in three states. Changing status in the catalytic dyad and the nearby side-chain binding pocket in CHAT (grey surface) highlighted. (E) TLC-based peptide cleavage assay by Craspase. (F) Cleavage site mapping by mass-spectrometry. (G) Top: model depicting non-matching PFS RNA induced Craspase activation. Bottom: Logic gate diagram illustrating the protease activation mechanism. (H) Model depicting TPR-CHAT status in the apo/resting, matching PFS RNA bound state, and non-matching PFS RNA bound states.
A greater set of conformational changes take place when RNA target containing a non-matching PFS is bound by Craspase (Fig. 5B). Lacking sequence complementarity to the first 2-nt of PFS, the base-pairing in the first guide-target segment is incomplete and the gating loop is only partially dislodged (Fig. S17). While the first nucleotide of PFS forms a partially frayed A•C pair, the rest of PFS pivots toward the surface of TPR (Fig. 5C). The switch helix is dislodged from the shackle of the padlock, possibly due to clashes with the non-matching PFS. This helix and the preceding loop-helix-loop connection rotates 90 degree and packs against CHAT as a coiled coil structure (Fig. 5D, S19). The sensor hairpin undergoes a larger set of long-range allosteric alterations. Consequently, C627 and H585 become oriented within hydrogen bonding distance (3.3 Å) (Fig. 5D), and a hydrophobic pocket opens nearby (Fig. S18C). The entire CHAT domain further undergoes a rigid-body movement. As the result, the cleft between Sb-gRAMP and TPR-CHAT widens, which may enable the peptide substrate to access binding surfaces (Fig. 5D).
Based on the observed structural features in the protease center, we designed candidate peptides to probe for potential RNA-guided peptidase activity in Craspase. We noticed that one designed peptide showed Craspase-dependent cleavage in thin-layer chromatography assays (Fig. S20A–B, 5E). Consistent with our mechanistic predictions, the activity was stronger in the presence of a non-matching PFS RNA substrate than a matching PFS substrate (Fig. 5E). This peptide could also be cleaved by Craspase in the context of an inter-domain protein linker, and the cleavage was stimulated by non-PFS target RNA (Fig. S20C–F). Mass spectrometry (MS) revealed that the cleavage took place after a leucine residue (Fig. S21). Judging by the fact that only one of the two leucine residues in the peptide was selectively cleaved (Fig. 5F), and that the cleavage activity was low and only partially RNA-dependent, Craspase clearly specifies additional sequences nearby.
The above mechanistic analysis defines how RNA-guided RNA recognition regulates the protease activity of Craspase (Fig. 5G). Sequence complementarity in the target RNA is a prerequisite, which is indirectly read out from the gating loop movement. A NOT logic gate is also in place to avoid activation by a self-RNA. Craspase is only activated when both conditions are true. The structural feature performing the logic calculation is the switch helix: its movement triggers a stepwise conformational relay that allosterically unlocks the TPR-CHAT padlock and switches on the protease activity (Fig. 5H).
Craspase proteolytically cleaves Csx30 in an RNA-dependent manner
Type III-E loci encode three other well-conserved proteins: the putative sigma-factor RpoE and two proteins of unknown function, denoted Csx30 and Csx31 (5, 11, 12). As a protease and its target are often co-localized in the genome (2, 8), we tested Craspase protease activity against these proteins in co-expression experiments (Fig. 6A). Full-length Csx30 was strongly reduced in the presence of target bound Craspase, whereas full-length RpoE and Csx31 levels were unaffected (Fig. 6B). This effect was alleviated when Craspase carried inactivated cysteine-histidine residues (H585A and C627A) (Fig. 6B), suggesting that Craspase possesses proteolytic activity against Csx30. This observation was confirmed in vitro, where purified Craspase processes Csx30 into two distinct fragments (Fig. 6C; Table S2), demonstrating that Csx30 is a natural protein target of Craspase. Mutational analysis of the amino acids encompassing the cleavage site showed that L407 in Csx30 is important for Craspase activity (Table S3; Table S4; Fig. S22A–B). Cleavage by Craspase after a leucine residue is consistent with MS (Fig. S22A) and the peptide cleavage experiments (Fig 5E–F). Corroborating the structural insights, proteolytic digestion could only be observed in the presence of target RNA with non-matching PFS, whereas no cleavage fragments accumulated with non-target RNA or target RNA with matching PFS (Fig. 6C; Fig. 6D). As Craspase cleaves bound RNA only under bivalent cation conditions (11), we reasoned that the peptidase in target bound Craspase would stay active in the absence of magnesium ions. We indeed observed a marked increase in Csx30 processing under magnesium poor conditions compared to magnesium rich conditions (Fig. 6E), suggesting that target RNA cleavage switches off the peptidase. This is further supported by the finding that the peptidase activity of a nuclease-dead variant of Craspase is not impaired in the presence of magnesium ions (Fig. 6E), rendering Craspase R294A D698A a ‘stay-on’ variant. Binding of a complementary ssDNA, which is not cleaved by Craspase (11, 12), does not activate the peptidase (Fig. 6E). These findings combined support a model (Fig. 6F) in which the peptidase activity of Craspase is switched-on upon target RNA binding to cleave Csx30 after L407, separating a large N-terminal fragment of ~47 kDa from a small C-terminal fragment of ~19 kDa small fragment. Due to the low sequence and structural similarity to known proteins, a prediction of the function of the two protein fragments cannot be made with confidence (Fig. S22C). However, based on analogous defense systems, processed Csx30 fragments likely enable an immune response, possibly by eliciting toxicity to the native host cell. Craspase then self-regulates through target RNA cleavage to switch the peptidase off, thereby timing the duration of the immune response and possibly recycling the Craspase complex to bind new target RNAs.
Fig. 6. Craspase proteolytically cleaves Csx30 in an RNA-dependent manner.

(A) Genetic context for RpoE, Csx31 and Csx30 co-expression with Craspase wild-type (WT) or mutant (MT; H585A C627A) and a target RNA in E. coli BL21-AI. (B) Protein gel showing the eluted protein content from Streptavidin purifications of Tag-RpoE, Tag-Csx31 and Tag-Csx30 after co-expression with either Craspase WT or Craspase MT (H585A C627A). Colored arrows indicate the expected size for full length protein. (C) Protein gels after Craspase WT or Craspase MT (H585A C627A) incubation with Csx30 in the presence of target RNA or non-target RNA. Protein cleavage products are indicated with a red asterisk. (D) Protein gel after Craspase WT incubation with target RNA containing either a non-matching PFS (NPFS) or matching PFS (PFS). (E) Left: protein gel after incubation of Tag-Csx30 with target RNA and Craspase WT or Craspase D698A R294A, with or without prior incubation with MgCl2. Right: protein gel after incubation of Tag-Csx30 with target ssDNA and Craspase D698A R294A. (F) Model for Craspase functionality. Once unbound Craspase has bound a target RNA, the peptidase activity is activated. This results in proteolytic cleavage of Csx30 between L407 and D408. Upon target RNA cleavage by Craspase, the peptidase activity is shut off.
Discussion
A new frontier in CRISPR-Cas biology has emerged, in which the RNA-guided effectors control physiological responses using mechanisms other than nucleic acid degradation. Here we define how the Craspase protease is allosterically activated by target RNA recognition and inactivated by target RNA cleavage to cleave the native substrate Csx30 in a binary fashion. We tuned its dynamic response range using mechanism-inspired mutants, which will pave the way for biotechnological and therapeutic applications. Our observations point to the possibility that the cleavage sequence in the native protein substrate is read out in the context of the 3D structure, which is also the case for the molecular recognition of gasdermins by eukaryotic caspases (26, 28). We wait for follow-up studies to reveal the missing recognition codes in substrate recognition and cleavage.
Despite the large structural distinctions, our studies revealed that Type III-E systems share fundamental mechanistic similarities with canonical CRISPR-Cas Type III systems. Analogous to Cas10 activation in other Type III effectors, Craspase only turns on the protease activity in response to non-self RNA targets, whereas it does not differentiate self and non-self RNA targets at the RNA cleavage level. This, combined with the observation that Craspase switches off protease activity upon target RNA cleavage, suggests that the protease activity may only be desired temporarily in the cell, which points to a possible ominous consequence of turning on the Craspase pathway. Does Csx30 proteolysis lead to cell dormancy or possibly programmed cell death? Due to the lack of homology to known proteins, it is difficult to infer the physiological function of Csx30 with confidence. Based on the AlphaFold (22) predicted structure, we speculate that proteolysis may relieve a physical sequestration or trigger a conformational change in Csx30, converting it to the active form (Fig. S22C–D). An analogous scenario was described for bacterial gasdermin, which only induced its anti-viral effect after site-specific cleavage by TPR-CHAT (2). Potential involvement of other Craspase-associated proteins, RpoE and Csx31, needs to be assessed in future experiments. However, unraveling the biological details is complicated by the difficulty of working with the native host Candidatus “Scalindua brodae” (29). Alternative model organisms may be needed for future functional dissections. On the application side, the fact that the Craspase peptidase is only active in the presence of a specific RNA species renders it useful for both in vivo (e.g. gene expression profiling) and in vitro (e.g. RNA diagnostics) biotechnological applications. This represents a major expansion of the range of biomolecular engineering possibilities of CRISPR-Cas effectors.
Supplementary Material
Acknowledgements
Funding
National Institutes of Health grant GM118174 (AK)
Department of Defense through the National Defense Science & Engineering Graduate Fellowship Program (GS)
NSF MRSEC program grant DMR-1719875 (Cornell Center for Materials Research Shared Facilities)
DOE Office of Biological and Environmental Research grant KP1607011 (Laboratory for BioMolecular Structure (LBMS))
Netherlands Organisation for Scientific Research (NWO VICI; VI.C.182.027 to S.J.J.B.) European Research Council (ERC) CoG under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 101003229 to S.J.J.B.)
Footnotes
Competing Interests
S.P.B.v.B. and S.J.J.B. are inventors on patent application N2028346 and PCT/NL2022/050296 submitted by Delft University of Technology that covers uses of gRAMP and Craspase. A provisional patent application related to this research has been filed by Cornell University.
Data and materials availability:
The resting-gRAMP coordinates and cryo-EM density map have been deposited in the Protein Data Bank (PDB:8D97) and the Electron Microscopy Data Bank (EMD-27257); gRAMP/non matching PFS RNA bound (PDB:8D8N, EMD-27252); gRAMP/matching PFS RNA bound (PDB:8D9E, EMD-27259); gRAMP/non matching PFS RNA post cleavage state (PDB:8D8I, EMD-27263); Craspase complex (PDB:8D8F, EMD-27260); Craspase/matching PFS RNA complex (PDB:8D8H, EMD-27262); Craspase/non-matching PFS RNA complex (PDB:8D8G, EMD-27261) Plasmids used in this study are available upon request.
References and Notes
- 1.Doron S et al. , Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson AG et al. , Bacterial gasdermins reveal an ancient mechanism of cell death. Science 375, 221–225 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah SA et al. , Comprehensive search for accessory proteins encoded with archaeal and bacterial type III CRISPR-cas gene cassettes reveals 39 new cas gene families. RNA Biol 16, 530–542 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makarova KS et al. , Evolutionary and functional classification of the CARF domain superfamily, key sensors in prokaryotic antivirus defense. Nucleic Acids Res 48, 8828–8847 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makarova KS et al. , Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nature reviews. Microbiology 18, 67–83 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kazlauskiene M, Kostiuk G, Venclovas C, Tamulaitis G, Siksnys V, A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science 357, 605–609 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Niewoehner O et al. , Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature 548, 543–548 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Rouillon C et al. , SAVED by a toxin: Structure and function of the CRISPR Lon protease. bioRxiv, 2021.2012.2006.471393 (2021). [Google Scholar]
- 9.Molina R, Sofos N, Montoya G, Structural basis of CRISPR-Cas Type III prokaryotic defence systems. Curr Opin Struct Biol 65, 119–129 (2020). [DOI] [PubMed] [Google Scholar]
- 10.You L et al. , Structure Studies of the CRISPR-Csm Complex Reveal Mechanism of Co-transcriptional Interference. Cell 176, 239–253 e216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Beljouw SPB et al. , The gRAMP CRISPR-Cas effector is an RNA endonuclease complexed with a caspase-like peptidase. Science 373, 1349–1353 (2021). [DOI] [PubMed] [Google Scholar]
- 12.Ozcan A et al. , Programmable RNA targeting with the single-protein CRISPR effector Cas7-11. Nature 597, 720–725 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Shi Y, Caspase activation: revisiting the induced proximity model. Cell 117, 855–858 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Ding J et al. , Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Shi J et al. , Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Hochstrasser ML, Nunez JK, CRISPR meets caspase. Nat Microbiol 6, 1481–1482 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Catchpole RJ, Terns MP, New Type III CRISPR variant and programmable RNA targeting tool: Oh, thank heaven for Cas7-11. Mol Cell 81, 4354–4356 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato K et al. , Structure and engineering of the type III-E CRISPR-Cas7-11 effector complex. Cell, (2022). [DOI] [PubMed] [Google Scholar]
- 19.Jia N et al. , Type III-A CRISPR-Cas Csm Complexes: Assembly, Periodic RNA Cleavage, DNase Activity Regulation, and Autoimmunity. Mol Cell 73, 264–277 e265 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo M et al. , Coupling of ssRNA cleavage with DNase activity in type III-A CRISPR-Csm revealed by cryo-EM and biochemistry. Cell Res 29, 305–312 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sridhara S et al. , Structural and biochemical characterization of in vivo assembled Lactococcus lactis CRISPR-Csm complex. Commun Biol 5, 279 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jumper J et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steens JA et al. , SCOPE enables type III CRISPR-Cas diagnostics using flexible targeting and stringent CARF ribonuclease activation. Nat Commun 12, 5033 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin Z, Luo X, Yu H, Structural basis of cohesin cleavage by separase. Nature 532, 131–134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boland A et al. , Cryo-EM structure of a metazoan separase-securin complex at near-atomic resolution. Nat Struct Mol Biol 24, 414–418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Z et al. , Caspase-1 Engages Full-Length Gasdermin D through Two Distinct Interfaces That Mediate Caspase Recruitment and Substrate Cleavage. Immunity 53, 106–114 e105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blatch GL, Lassle M, The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays 21, 932–939 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Wang K et al. , Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell 180, 941–955 e920 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Awata T et al. , Physiological characterization of an anaerobic ammonium-oxidizing bacterium belonging to the “Candidatus scalindua” group. Appl Environ Microbiol 79, 4145–4148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA, cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Scheres SH, RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pettersen EF et al. , UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liebschner D et al. , Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams CJ et al. , MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci 27, 293–315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ, Basic local alignment search tool. J Mol Biol 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The resting-gRAMP coordinates and cryo-EM density map have been deposited in the Protein Data Bank (PDB:8D97) and the Electron Microscopy Data Bank (EMD-27257); gRAMP/non matching PFS RNA bound (PDB:8D8N, EMD-27252); gRAMP/matching PFS RNA bound (PDB:8D9E, EMD-27259); gRAMP/non matching PFS RNA post cleavage state (PDB:8D8I, EMD-27263); Craspase complex (PDB:8D8F, EMD-27260); Craspase/matching PFS RNA complex (PDB:8D8H, EMD-27262); Craspase/non-matching PFS RNA complex (PDB:8D8G, EMD-27261) Plasmids used in this study are available upon request.