

Abstract
The structural integrity of vaccine antigens is critical to the generation of protective antibody responses, but the impact of protease activity on vaccination in vivo is poorly understood. We characterized protease activity in lymph nodes and found that antigens were rapidly degraded in the subcapsular sinus, paracortex, and interfollicular regions, whereas low protease activity and antigen degradation rates were detected in the vicinity of follicular dendritic cells (FDCs). Correlated with these findings, immunization regimens designed to target antigen to FDCs led to germinal centers dominantly targeting intact antigen, whereas traditional immunizations led to much weaker responses that equally targeted the intact immunogen and antigen breakdown products. Thus, spatially-compartmentalized antigen proteolysis affects humoral immunity and can be exploited to enhance vaccine-induced antibody production against key pathogen structural epitopes.
One-sentence Summary:
Spatially heterogeneous protease activity within lymph nodes modulates humoral responses to vaccination.
After vaccination, humoral immune responses begin by B cells binding with their receptors to cognate antigen, followed by the formation of germinal centers (GCs) where these cells undergo proliferation and affinity maturation, leading to the production of high-affinity antibodies against the target antigen (1–3). Factors determining the makeup of the eventual affinity-matured polyclonal antibody response remain incompletely understood, although the precursor frequency of antigen-specific B cells, the affinity of precursors for the antigen, antigen complexity, follicular helper T cell-derived signals, and antibody feedback all contribute (4–8). In addition, the duration of antigen exposure and the amount of antigen available to B cells plays an important role (9, 10).
We hypothesized that the structural integrity of the antigen in vivo is an additional important factor. To elicit protective responses, antigens need to present neutralizing epitopes that are faithful structural mimics of the target pathogen, which are often complex three-dimensional surfaces (11). Disruption of these epitopes could not only limit the activation of B cells with the capacity to produce neutralizing antibodies but might also create distracting de novo epitopes irrelevant for protective immunity. It has been reported that model protein antigens can be rapidly proteolyzed as they reach the subcapsular sinus (SCS) of lymph nodes (LNs), and this antigen cleavage was linked to protease activity in serum and interstitial fluid (12). Such pathways of antigen breakdown might at least partially explain the substantial proportion of B cells that enter GC reactions but do not detectably bind to the immunizing antigen (7, 9).
By contrast, several lines of evidence suggest that antigen trapped on dendrites of follicular dendritic cells (FDCs) may remain intact over extended time periods. Early studies showed that FDC-bound antigen recovered from LNs after 12 weeks could be recognized by epitope-sensitive monoclonal antibodies (mAbs) and were eluted in size exclusion chromatography in a manner suggesting gross antigen integrity (13). HIV virions deposited on FDCs in mice can be extracted from LNs and functional viral particles recovered over several months, although the quantitative proportion of particles that are infective is not clear (14). FDCs have also been shown to cyclically internalize and recycle trapped antigen, which may protect it from extracellular degradation (15). These data collectively suggest that the follicles and the FDC networks in particular may be sites within LNs where antigens are protected from degradation, whereas regions such as the sinuses may be areas of high proteolytic activity. To our knowledge, however, the nature of protease activity in lymphoid organs has not been studied, and how antigen proteolysis affects the immune response to vaccines is poorly understood.
To shed light on the fate of antigens during the primary immune response, we developed a FRET-based approach to track the integrity of antigens after subunit vaccine immunization, and analyzed the spatial pattern of protease expression and activity in LNs. Unexpectedly, we found a pronounced spatial variation in protease activity, with high levels of antigen breakdown and protease expression in extrafollicular regions of mouse and human lymphoid tissues, but low levels of protease activity and high retention of antigen integrity over time within the FDC network of B cell follicles. Prompted by these findings, we evaluated the impact of antigen localization on the specificity of GC B cell responses, and found evidence that FDC-targeted protein immunizations achieve substantially greater proportions of antigen-specific B cell responses targeting conformationally intact epitopes compared with traditional bolus vaccination.
RESULTS
Monitoring antigen integrity using FRET analysis
To investigate vaccine antigen stability after immunization, we labeled immunogens with paired small-molecule dyes capable of undergoing fluorescence resonance energy transfer (FRET) to detect gross disruptions of antigen structure in situ in tissues. We hypothesized that antigen proteolysis would lead to the separation of FRET donor and acceptor dyes, lowering FRET signals in proportion to the degree of antigen degradation, a process that can be tracked by microscopy or flow cytometry (Fig. 1A). For microscopy-based analysis, we used the acceptor photobleaching method (16), in which the emission of a donor dye (Cy3) is measured before and after the bleaching of an acceptor dye (Cy5) to monitor antigen integrity (fig. S1A). This technique avoids complications of donor and acceptor excitation and emission cross-talk because only the donor emission is analyzed, and it is independent of dye concentrations and ratios. As a test case, we first focused on the clinical vaccine candidate eOD-GT8 60mer (eOD-60mer), an ~30-nm-diameter protein nanoparticle (NP) presenting 60 copies of an HIV Env gp120 engineered outer domain (Fig. 1A). eOD-60mer is used as a priming immunogen to initiate CD4 binding site-directed broadly neutralizing antibodies against HIV (17–21). This NP antigen accommodated labeling with at least 40 dyes per particle (~20 Cy3 and ~20 Cy5 dyes, eOD-60mer40) without disrupting the binding of the broadly neutralizing antibody VRC01 to the CD4-binding site epitope presented by the eOD-GT8 immunogen (fig. S1B). This degree of labeling represents modification of only 3.5% of the total amino acids of the particle immunogen. We previously showed that after a primary immunization, mannose-binding lectin (MBL) present in serum binds to glycans densely displayed on the surface of the eOD-60mer, leading to subsequent complement deposition on the particles and trafficking of the antigen to FDCs in the LN (22). Complement binding to unmodified or dye-labeled eOD-60mer40 incubated in vitro in mouse serum was not statistically different (fig. S1C). eOD-60mer40 coated onto glass coverslips showed a high level of FRET on excitation at 555 nm, but upon photobleaching of the Cy5 dye, emission in the Cy3 donor channel increased by twofold (fig. S1D). Such a change in Cy3 fluorescence was not observed for NPs labeled with only Cy3 or only Cy5, or when Cy3-labeled NPs were mixed with Cy5-labeled NPs and coated on coverslips together (fig. S1, E to H). These changes in acceptor emission before and after photobleaching can be quantified to determine the mean FRET efficiency (E) (see the materials and methods). Of importance for our subsequent application to frozen tissue sections, FRET signals of intact eOD-60mer40 as a fresh protein or after flash freezing were not statistically different (fig. S1, I and J). On the basis of these findings, we focused on NPs labeled with ~40 dyes total for further studies.
Figure 1. FRET-based analysis of antigen integrity reveals spatially compartmentalized antigen degradation in LNs.
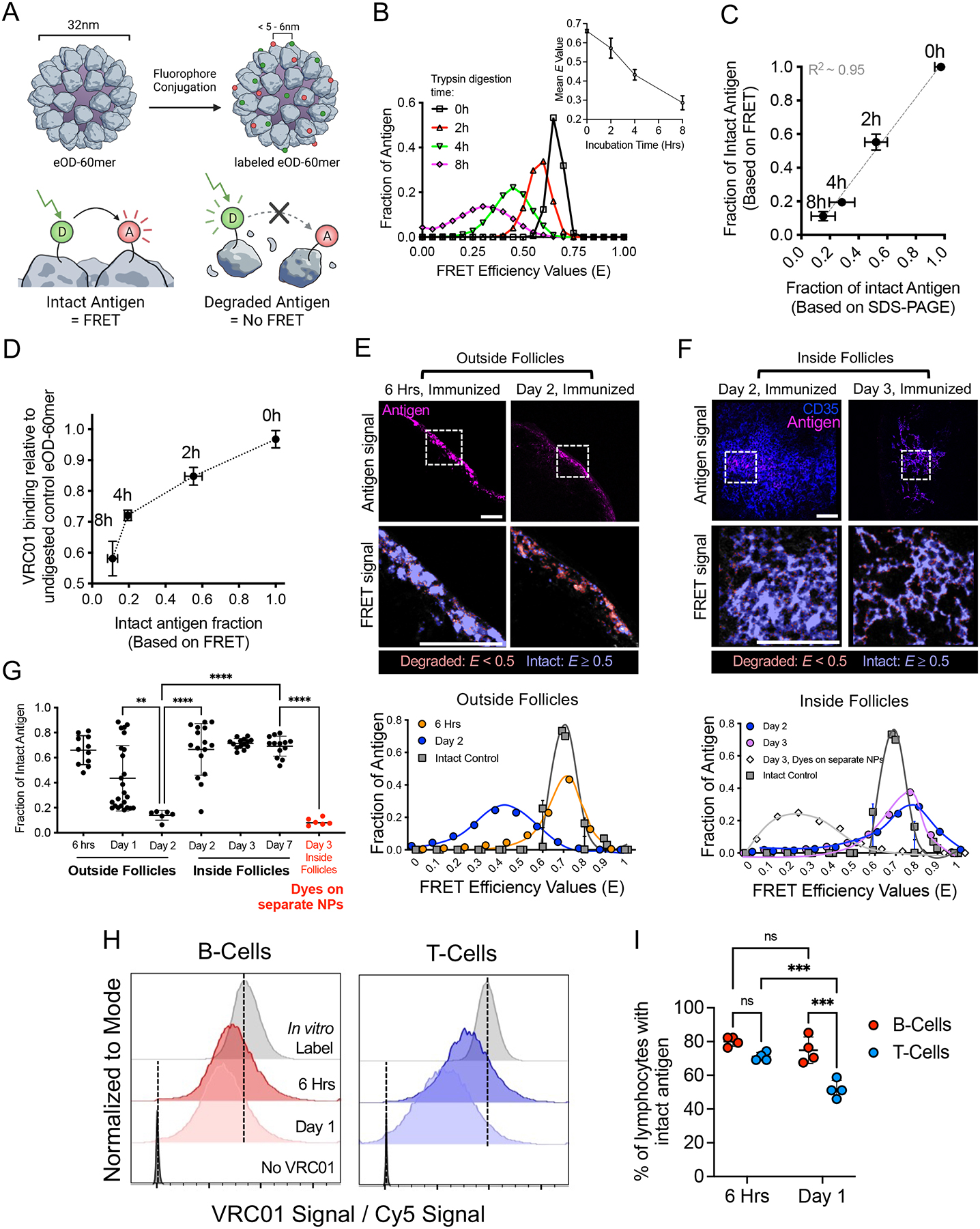
(A) Schematic of eOD-60mer labeled with donor and acceptor dyes to track antigen integrity by FRET. (B to D) FRET analysis of eOD-60mer40 integrity after trypsin digestion (n =3 samples/time point). Shown are representative E histograms and mean E values (B), proportion of intact eOD-60mer40 based on FRET versus SDS-PAGE (C), and VRC01 mAb binding to digested eOD6-mer40 versus FRET-measured proportion of intact eOD60-mer40 (D). (E to G) C57BL/6 mice (n =4 animals/group) were immunized with 10 μg eOD-60mer40 and 5 μg saponin adjuvant. At the indicated time points, LNs were flash frozen and sectioned. (E-F) Representative images of eOD-60mer40 within the SCS [“Outside Follicles”, E] versus B cell follicles [“Inside Follicles”, (F)]. Top images show antigen and CD35 staining; bottom images show intact or degraded antigens in the dashed boxes as false colors at higher magnification. Corresponding E histograms within dashed boxes are shown compared with intact eOD-60mer40 coated on coverslips. “Day 3, Dyes on separate NPs” in (F) indicates mice immunized with saponin adjuvant and 5 μg of two different eOD-60mers each labeled with only Cy3 or Cy5 dyes. Scale bars, 100 μm. (G) Mean fraction of intact eOD-60mer40 at the indicated LN locations at different time points after immunization. Each point represents one region from one tissue section. Data collected from at least eight tissue sections from eight LNs. P values were determined by one-way ANOVA with Tukey’s post test. (H and I) C57BL/6 mice (n =4 animals/group) were immunized with 20 μg of anti-CD45 mAb conjugated to Cy5-labeled eOD monomer and 5 μg of saponin adjuvant. LN cells were isolated at 6 hrs or 1 day after immunization, stained with VRC01 mAb, and analyzed by flow cytometry. (H) Histograms of VRC01 binding normalized to eOD monomer-Cy5 signal showing gating on T cells or B cells. Vertical dashed lines indicate the mean fluorescence intensities of intact eOD-anti-CD45 freshly bound to splenocytes in vitro (positive control) versus cells analyzed in the absence of VRC01 (negative control). (I) Percentage of lymphocytes with intact eOD monomer as determined by VRC01 binding. P values were determined by two-way ANOVA with Tukey’s post test. All plots show mean ± s.d.
To determine how E relates to the structural integrity of the particles, we calculated E distributions of intact or trypsin-digested eOD-60mer40. Proteolyzed eOD-60mer40 showed the development of breakdown products by gel electrophoresis (fig. S1K), which were accompanied by a shift in the distribution of measured E values for the particles to lower values with increasing digestion time (Fig. 1B). The fraction of fully intact antigen as determined by FRET (calculated as the proportion of material with an E value overlapping the undigested control particle E distribution) was highly correlated to the fraction of intact antigen as measured by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 1C). VRC01 binding to eOD-60mer40 captured on ELISA plates also decreased with increasing trypsin digestion time, demonstrating that the loss of FRET signal was not due simply to particle disassembly (Fig. 1D). Altogether, these data suggest that FRET signals from labeled 60mer NPs effectively report on the retention of intact immunogen structure.
Antigens are rapidly proteolyzed in the SCS and extrafollicular regions but protected in follicles
We next used this FRET-based approach to analyze antigen integrity in vivo in mice after immunization. We previously reported that eOD-60mer initially accumulates in the SCS of draining LNs, but over the course of several days NPs are trafficked to the FDC network of follicles in an MBL- and complement-dependent manner, where they are retained for more than a week (22). To assess the structural integrity of the NPs during this process, we immunized mice with eOD-60mer40 and a saponin-based adjuvant, and at various time points after immunization, draining LNs were extracted, flash frozen, and cryosectioned for acceptor photobleaching FRET analysis (fig. S2A). FRET efficiencies were converted to a measure of percent intact antigen by comparing E values measured in the tissue with that of control fresh antigen coated on glass coverslips. At early times (2 to 6 hours after immunization), eOD-60mer40 accumulated in the SCS and could also be detected at very low levels in follicles or extrafollicular regions within the LN parenchyma (at roughly equal levels in these latter two locations, fig. S2, B and C). At later times antigen continued to accumulate in the SCS, but over ~48 hours, antigen levels in the SCS decreased, whereas steadily increasing amounts were detected on FDCs (Fig. 1, E and F, and fig. S2, D and E). At very early times, most eOD-60mer40 was intact regardless of its location (Fig. 1G). However, unexpectedly we observed location-dependent patterns of antigen degradation developing over time. Nanoparticles localized in the sinus or interfollicular regions of the LN exhibited a substantial loss in E between 6 and 48 hours (Fig. 1E, “outside follicles”). By contrast, eOD-60mer40 localized within the FDC network of follicles at the same timepoints exhibited high E values (Fig. 1F, “inside follicles”). Quantification of the proportion of intact NPs from many LNs revealed that most eOD-60mer40 located outside of follicles was degraded within 48 hours, whereas 70 to 80% of the antigen localized to the FDC network remained intact through at least 7 days (Fig. 1G). Imaging of LNs from mice immunized with control mixtures of Cy3-eOD-60mer and Cy5-eOD-60mer showed negligible FRET signals for FDC-localized antigens, suggesting that the sustained FRET signals observed for FDC-localized antigen were not due to inter-particle FRET (Fig. 1G, “dyes on separate NPs”). To determine whether similar spatially-distinct patterns of antigen degradation are observed with a completely different antigen and nanoparticle carrier, we also imaged LNs after injection of FRET-labeled influenza hemagglutinin-ferritin nanoparticles (HA-NPs), representative of a class of NP vaccine in clinical trials (23). These NPs also accumulated on FDCs over time (22, 24) (fig. S2, F and G), and like eOD-60mer, HA-NPs degraded rapidly in extrafollicular regions but were highly protected in follicles (fig. S2H).
As an orthogonal measure of antigen integrity, we stained LN sections with VRC01 antibody to detect 60mer with an intact CD4 binding site epitope, and this approach revealed similar results: eOD-60mer in the SCS showed a high level of VRC01 staining at 6 hours after immunization, but this staining was almost completely lost by 48 hours for material outside of the FDC network (fig. S3, A and B). By contrast, NP antigen localized to FDCs retained VRC01 staining even at 3 days after immunization (fig. S3, A and B). Loss of VRC01 staining in extrafollicular sites was not due to an early antibody response blocking the epitope, because the same loss in VRC01 binding was observed in LNs from B cell receptor-transgenic MD4 mice, in which B cells express an antigen receptor specific for an irrelevant antigen (25) (fig. S3C).
It was unclear whether the retention of eOD-60mer integrity reflected a specific protective activity of FDCs or of the follicles in general. To answer this question, we devised an approach to target antigen into both follicles and the paracortex simultaneously. Antibodies to CD45 bind to lymphocytes and remain cell surface localized for multiple days (26). We exploited this biology to target eOD to the surfaces of T cells and B cells in LNs by conjugating dye-labeled monomeric eOD to an anti-CD45 mAb (fig. S4A). The antibody conjugate efficiently labeled CD45+ LN cells in vitro, and a flow cytometry based fluorescence quenching assay (27) showed that >90% of the conjugate bound to the cells was extracellular (fig. S4, B to E). Mice were immunized with Cy5-labeled eOD-anti-CD45 and saponin adjuvant, and LNs were collected 6 hrs or 1 day later for analysis. Approximately half of all B cells and T cells were found to be labeled by the Ab conjugate in this time course (fig. S4, F and G). A portion of these cells were stained with an anti-Cy5 antibody, and the ratio of anti-Cy5 (surface-bound eOD) to Cy5 signals (total eOD) revealed that the most antigen was extracellular on both T and B cells at both time points (fig. S4, H and I). By staining another portion of the cells with VRC01 antibody and quantifying the ratio of VRC01 (intact CD4-binding site) to Cy5 signal (total eOD) compared with cells freshly labeled in vitro showed that ~80% of B cells carried eOD with an intact CD4 binding site at 6 hr, and this remained unchanged at 24 hr (Fig. 1, H and I). By contrast, eOD bound to T cells already trended toward a lower level of intact CD4 binding site at 6 hr, and T cell-bound eOD was 50% degraded by 1 day (Fig. 1, H and I). Thus, both FRET and antibody staining analyses suggest that antigens are rapidly degraded as they enter the SCS, interfollicular regions, and paracortex, but antigen localized to follicles, whether bound to FDCs or not, is protected from rapid proteolysis.
Metalloproteinases are expressed by sinus-lining and stromal cells and contribute to rapid extrafollicular antigen degradation
High-magnification imaging of LNs stained to identify cell membranes 24 hrs after immunization revealed pockets of antigen localized in the interstices between cells, and FRET imaging showed that this antigen was degrading, suggesting a role for extracellular proteolysis (Fig. 2A). To quantify intracellular versus extracellular antigen breakdown in whole LNs, we developed an antigen pull-down assay in which FRET dye labeled antigens in the supernatants of mechanically disrupted LNs or lysates of isolated LN cells were captured on anti-Cy3/Cy5 conjugated beads followed by flow cytometry based FRET measurements (Fig. 2B). For this analysis, cells were excited at the donor dye wavelength and acceptor dye emission was collected, and this FRET signal was normalized by the total fluorescence obtained from exciting the acceptor and collecting its emission (fig. S5A). Total antigen recovered was quantified in parallel by measuring Cy5 fluorescence, calibrated by beads loaded with known quantities of Cy5-labeled eOD-60mer (fig. S5, B and C). These analyses revealed that the total amount of recoverable antigen dropped by 31-fold over 2 days, and at days 1 and 2 nearly equal proportions of antigen were extracellular versus intracellular (Fig. 2C). Focusing on the 6-hourr and 1-day time points, when sufficient antigen was recovered for robust analysis, FRET signals showed that extracellular and intracellular antigen both underwent substantial degradation in this timeframe (Fig. 2D and fig. S5D). Thus, extracellular and intracellular degradation contribute approximately equally to antigen clearance.
Figure 2. After immunization, a significant proportion of antigen remains extracellular and is degraded over time, correlating with the expression of ADAM and MMP proteases by macrophages and stromal cells in the LN.
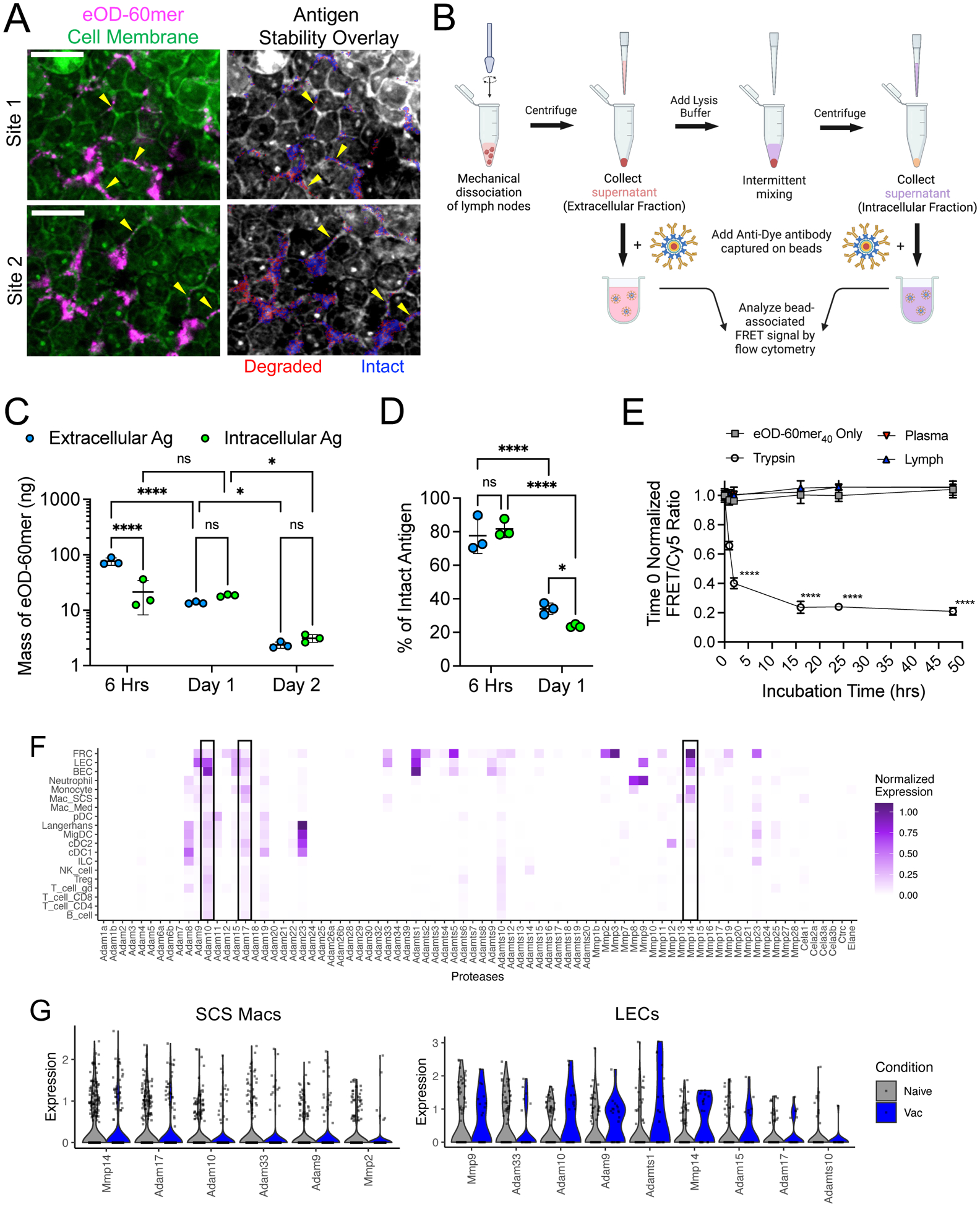
(A) C57BL/6 mice were immunized with 5 μg of saponin adjuvant and 10 μg of eOD-60mer40 and harvested 1 day later for sectioning and confocal imaging. Two representative extrafollicular regions are shown stained with the cell membrane dye, CellMask Green, (left panels) and an overlay of false color binary FRET images indicating intact (blue) and degraded (red) eOD-60mer40 (right panels). Scale bars, 10 μm. (B to D) C57BL/6 mice were immunized as in (A) and draining LNs were isolated (n =3 pools/time point, with each pool containing four LNs from two mice) for measurement of extracellular versus intracellular antigen quantity and integrity by flow cytometry based FRET analysis. Shown are a schematic illustrating the workflow for antigen pull-down assay (B), total mass of antigen recovered from whole LN (C), and % of Intact antigen (D) retrieved at the indicated time points after immunization. ns, not significant; *, q ≤ 0.05; ****, q ≤ 0.0001 by two-way ANOVA for (C) and (D) with two stage step-up method to correct for false discovery rate during multiple comparisons. (E) FRET analysis of eOD-60mer40 integrity after incubation with 10% diluted plasma or lymph from naïve C57BL/6 mice or trypsin at 37°C (n =3 replicates/group). P values were determined by one-way ANOVA followed by Tukey’s post test. (F and G) scRNAseq analysis of protease expression in LNs from naive C57BL/6 mice or mice immunized 6 hours earlier with ovalbumin peptide and CpG. (F) Heatmap of average normalized expression of extracellular protease genes in single cells across LN cell types in immunized mice. (G) Normalized expression levels of the most highly expressed proteases in SCS macrophages and LECs arranged in descending order of average expression; protease genes with an average expression value >0.1 are included. All plotss show mean ± s.d.
We next sought to determine factors governing extracellular antigen degradation. As shown by FRET analysis, eOD-60mer40 incubated with mouse lymph or plasma in vitro remained intact (Fig. 2E). Thus, we examined the extracellular or secreted proteases expressed by LN cells by re-analyzing a single-cell RNA sequencing (scRNA-seq) dataset recently collected from LNs of naïve or immunized mice. We found >30 genes encoding extracellular or secreted proteases expressed in one or more LN cell types, predominantly in stromal cells and myeloid cells (Fig. 2E). Because we observed substantial antigen degradation at the SCS, we focused on sinus-lining SCS macrophages and lymphatic endothelial cells (LECs) (Fig. 2F). The most highly expressed genes in SCS macrophages (in both naïve and vaccinated mice) included Mmp14, Adam17, and Adam10, which encode metalloproteinases. We focused our attention on these three proteases because they were also expressed by LECs and were generally the most broadly and/or highly expressed across multiple cell types in the LN. Mmp9 was also notable as the most highly expressed protease in LECs. Our dataset lacked FDCs, but we analyzed two published LN stroma scRNAseq datasets that included these cells (28, 29). Consistent with our observations of limited antigen degradation in follicles, FDCs expressed very low to minimal levels of extracellular proteases compared with LN fibroblasts or LECs (fig. S5, E and F).
Immunohistochemistry analysis of ADAM17, ADAM10, MMP14, and MMP9 protein expression revealed high levels of these proteases along the subcapsular and medullary sinuses and within isolated regions in the interior of both resting and immunized LNs (Fig. 3A and fig. S6, A and B). By contrast, the FDC network and its immediate vicinity showed low levels of all four proteins. Consistent with the scRNAseq data, all three proteases partially colocalized with CD169+ and LYVE1+ cells, as shown by immunohistochemistry (Fig. 3B and fig. S6, C and D). We also examined the expression patterns of these proteases in human lymphoid tissues. Human tonsil tissue sections showed prominent ridges of ADAM17, MMP9, and MMP14 expression, with more scattered expression of ADAM10 (fig. S6E). However, CD35+ follicles were largely devoid of all four proteases.
Figure 3. Metalloproteinases are expressed by sinus-lining cells and contribute to rapid antigen degradation in LNs.

(A and B) Resting LNs from C57BL/6 mice (n =3 animals/group) were flash frozen and cryosectioned. (A) Anti-CD35 (green) and anti-metalloproteinase or isotype control mAbs (magenta) staining of LN sections and magnified views of the red dashed regions of interest. Scale bar, 200 μm. (B) Magnified images of the SCS stained for CD169+ macrophages (left column) or LYVE1+ LECs (right column) and the indicated proteases. Scale bar, 20 μm. (C) C57BL/6 mice (n =3 animals) were immunized with 5 μg of saponin adjuvant and 10 μg of eOD-60mer40. After 24 hours, LNs were harvested, sectioned, and collectively stained for ADAM10, ADAM17, and MMP14 before FRET imaging. Two representative SCS regions showing protease expression, eOD-60mer localization, and false color overlay of proteases (white), total degraded antigen (green), and degraded antigen colocalized with proteases (red) are shown. Scale bar, 20 μm. Bottom graph quantifies degraded eOD-60mer colocalized with metalloproteinases. Each point represents one region and data were collected from at least six tissue sections from six LNs. LN boundaries are indicated by white (A), green (B), or yellow (C) dashed lines. (D) C57BL/6 mice (n =3 animals/group) were immunized as in (C) and LNs were collected after 2 hours before vibratome slicing. Live LN slices were left untreated or incubated in marimastat (Mari) with or without broad-spectrum protease inhibitors for 6 hours ex vivo before FRET imaging. Each point represents one region and data were collected from at least ten tissue sections from six LNs. P values were determined by one-way ANOVA with Tukey’s post test. (E) C57BL/6 mice were treated with Mari for 5 days and then immunized as in (C)., LNs were analyzed 24 hours after by pull-down assay (n=3 pools/group, each pool containing four LNs from two mice) or FRET imaging (n =3 animals/group). Shown are Mari or vehicle control (VC) dosing and immunization schedule (top panel), intact extracellular antigen recovered by pull-down assay (bottom left panel) and FRET imaging analysis for intact antigen (bottom right panel). Each point represents one region for FRET imaging and data collected from at least six tissue sections from six LNs. P values were determined by Student’s t test. All plots show mean ± s.d.
In vitro, the top three sinus-expressed proteases, ADAM17, ADAM10, and MMP14, all degraded eOD-60mer40, although the FRET profile somewhat differed, likely due to unique patterns of cleavage for each metalloprotease (fig. S7A). We thus examined the relative localization of these proteases and degrading antigen in the sinuses. Six hours after injection, eOD-60mer40 was predominantly colocalized with CD169+ macrophages, followed by LYVE1+ LECs and other cell types not identified by these two markers (fig. S7, B and C). The 60mer also showed substantial colocalization with ADAM17, ADAM10, and MMP14 (fig. S7D). Using high-magnification FRET imaging on immunostained LN sections, we found that ~60% of degraded eOD-60mer40 colocalized with at least one of these three metalloproteinases at 24 hours after immunization (Fig. 3C).
To functionally test the role of extracellular protease activity in antigen breakdown, we prepared live vibratome sections of LNs isolated from mice 2 hours after immunization with eOD-60mer40 (a timepoint when most antigen in the LN is still intact) and incubated tissue slices in the presence or absence of protease inhibitors. In LN slices cultured without inhibitors, eOD-60mer degradation proceeded, with ~60% of antigen intact relative to the starting material after 6 hrs, whereas LNs cultured with the metalloproteinase-specific inhibitor marimastat reduced this degradation by ~50% (Fig. 3D). Moreover, media containing marimastat supplemented with a broad-spectrum protease inhibitor cocktail did not yield significantly more intact antigen (Fig. 3D). We next treated mice with marimastat before and during immunization with eOD-60mer40. As shown in Fig. 3E, marimastat treatment led to a 40% increase in the total amount of intact extracellular antigen recovered by our antigen pull-down assay 24 hours after immunization compared with vehicle control. In parallel, microscopy-based FRET analysis of LN sections similarly showed that significantly more eOD-60mer40 was intact within the SCS after marimastat treatment (Fig. 3E). Collectively, these data suggest that proteases, particularly metalloproteinases, contribute to rapid extrafollicular antigen breakdown in LNs.
Extracellular protease activity is enriched in sinus-lining macrophages and stromal cells but low within follicles
We next sought to understand why antigen localized to FDCs was protected from proteolysis. We observed low staining of ADAM17, ADAM10, and MMP14 in follicles, but this data could not rule out expression of other proteases in the vicinity of FDCs. We thus used an imaging zymography approach to visualize protease activity more comprehensively on live tissue sections (Fig. 4A) (30). Live LN vibratome sections were incubated with two fluorescent peptide probes. The first, an activatable zymography probe (AZP), is composed of a 5-carboxyfluorescein dye linked to a cationic polyarginine (polyR) peptide, followed by a broad-spectrum protease-cleavable substrate sequence and an anionic polyglutamic acid (polyE) peptide that complexes with the polyR sequence. When proteases cleave the substrate site of the AZP, the polyR sequence is freed and electrostatically binds to nearby cells, labeling the site of protease activity (Fig. 4A). The AZP selected was cleaved by metalloproteinases including ADAM17, ADAM10, and MMP14, as well as aspartic and cysteine protease cathepsins reported to be extracellularly active and expressed by immune cells (fig. S7E) (30–32). A second control probe, a Cy5-conjugated polyR peptide, provides a map of overall electrostatic-binding sites in the tissue. Tissue areas labeled with both probes identify proteolytically active sites, whereas locations labeled by the control peptide but not the AZP indicate areas devoid of protease activity.
Figure 4. Protease activity is spatially heterogeneous in LNs, with high levels in the SCS and low activity in B cell follicles.
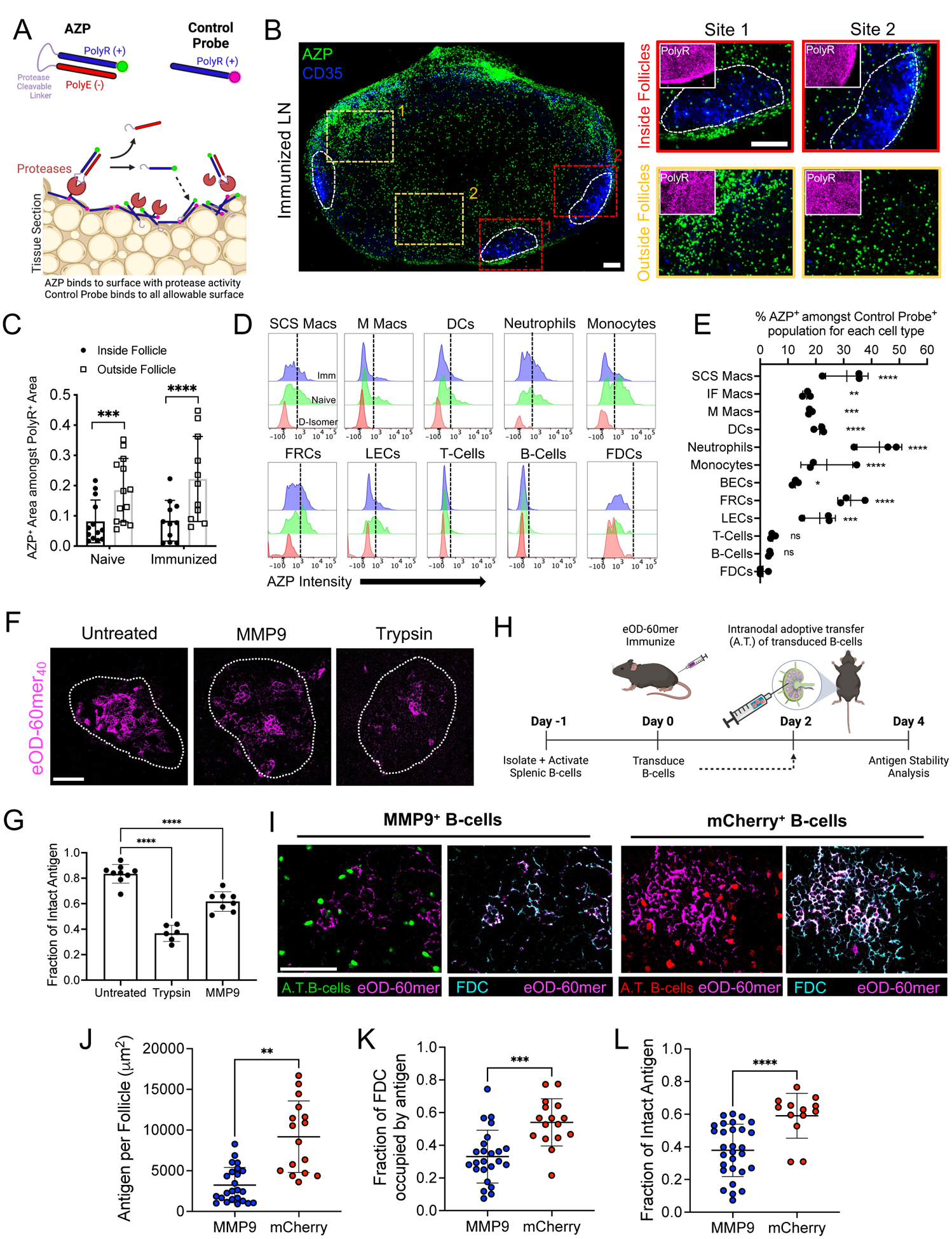
(A) Schematic illustrating AZPs and protease activity. (B to E) Live LN slices from C57BL/6 mice (n =3 animals/group), either naïve or immunized with eOD-60mer and 5 μg of saponin adjuvant, were incubated with AZP and control probes. (B) Confocal microscopy of LN slices (left) showing AZP+ cells (green), CD35+ FDCs (blue), and magnified views (right) of the boxed regions “Inside Follicle” or “Outside Follicle.” Insets show control polyR probe binding (magenta). White contours denote follicles. (C) Quantification of AZP binding inside follicles versus outside follicles. Each point represents one LN section, collected from a total of 11 to 13 sections from six LNs. P values determined by Student’s t test. (D) Representative flow histograms of AZP and D-isomer probe staining for cells from immunized (Imm) or resting (Naïve) LNs. Dashed lines denote AZP+ and AZP− populations. (E) Quantification of AZP+ cells. P values were determined by one-way ANOVA with Dunnett’s post test against FDCs. (F and G) C57BL/6 mice (n =3 animals/group) were immunized with 5 μg of saponin adjuvant and 10 μg of eOD-60mer40. Live LN slices were generated 3 days later and incubated with trypsin, MMP9, or left untreated. Representative images of eOD-60mer40 remaining on FDCs (F) and fraction of intact antigen on FDCs based on FRET imaging (G). White lines show follicle boundaries. Scale bar, 100 μm. Each point represents one region from one tissue section. Data were collected from at least six tissue sections from six LNs. P values based on one-way ANOVA with Dunnett’s post test against untreated sections. (H to L) C57BL/6 mice (n =3/group) were immunized as in (F) and 2 days later, MMP9- or mCherry-expressing polyclonal B-cells were adoptively transferred. (H) Timeline for retroviral transduction, adoptive transfer, and analysis. (I) Images of transferred MMP9+ (left panel) and mCherry+ control B cells (right panel) near FDCs bearing eOD-60mer40. Scale bar, 100 μm. (J to L) Image analysis for total eOD-60mer40 in the follicle (J), fraction of FDC network with eOD-60mer40 (K), and integrity of FDC-localized eOD-60mer40 (L). P values were determined by Student’s t test. All plots show mean ± s.d.
Confocal imaging of LN tissue sections from naïve or immunized animals showed AZP labeling of discrete cells across the entire tissue, with particular concentrations of protease activity at the SCS, except within the immediate vicinity of the FDC networks (Fig. 4B and fig. S7F). Incubation of LN sections with a control uncleavable D-isomer form of the same AZP peptide sequence showed substantially lower fluorescence signal, suggesting that AZPs were not internalized and required extracellular proteolysis to bind to cells (fig. S7, G to I). Quantification of AZP signal within FDC regions versus adjacent LN tissue revealed substantially lower protease activity in the follicles of both resting and immunized LNs (Fig. 4C).
Flow cytometry analysis of single cells extracted from polyR- and AZP-treated LN sections revealed that 80% or more of all lymphocyte and stromal cell populations examined were positive for control polyR binding (fig. S8, A to C) (33). Gating on cells positive for polyR binding (indicating sufficient binding sites for AZP-based protease detection), we found that neutrophils, SCS macrophages, and fibroblastic reticular cells had the highest levels of AZP binding above background labeling by D-isomer control AZP in both immunized (Fig. 4, D and E, and fig. S8D) and resting LNs (fig. S8E). All macrophage subsets examined (CD169+CD11bhiF4/80low subcapsular; CD169+CD11blowF4/80low interfollicular; and CD169+F4/80hi medullary macrophages) had some proportion of AZP+ cells, but SCS macrophages showed the most prominent protease activity (Fig. 4, D and E, and fig. S8D). Lymphatic endothelial cells (LECs), DCs, and monocytes also exhibited lower but clearly detectable protease activity, whereas FDCs, B cells, and T cells showed very low levels of AZP labeling (Fig. 4, D and E).
We next assessed other potential mechanisms of antigen protection within follicles. Because eOD-60mer is recognized by MBL and becomes complement-decorated in the presence of serum, it seemed possible that MBL and/or complement binding might have a shielding effect by blocking proteolytic attack on FDC-localized antigen. However, in an in vitro trypsin digestion assay, eOD-60mer was proteolyzed with destruction of the VRC01 epitope regardless of serum-derived MBL or C3 binding on the NP surface (fig. S9A). A second possibility was that antibodies produced very early after immunization form immune complexes to sterically inhibit protease attack, or that FDCs capturing immune complexes rapidly internalize and recycle it on their dendrites, providing a degree of protection from protease exposure in the follicles. We immunized MD4 mice in which B cells transgenically express an antigen receptor specific for an irrelevant antigen with eOD-60mer40, and found that antigen localized to FDCs after 2 days was largely non degraded, which was similar to what we found in wild-type mice, suggesting that antigen protection is not mediated by antibodies at least during these early time points (fig. S9B).
To assess whether FDCs could protect antigen in the face of artificially enforced protease activity in follicles, we immunized mice with eOD-60mer40 and adjuvant, extracted LNs 3 days later, when antigen was concentrated on the FDC networks, and incubated live LN slices with trypsin or recombinant matrix metalloproteinase 9 (MMP9) to expose FDC-localized antigen to protease attack. Both trypsin and MMP9 treatment led to a significant loss of intact eOD-60mer within 4 hours, suggesting that antigen localized to FDCs is not intrinsically protease resistant (Fig. 4, F and G). To determine whether low protease activity in follicles was important for the retention of intact antigen in vivo, we devised a strategy to introduce active extracellular proteolysis adjacent to FDCs. For this polyclonal B cells were transduced to express a constitutively active form of MMP9 (34) together with mNeonGreen as a reporter or mCherry reporter only as a control (fig. S10, A to D), and injected intranodally into mice that had been immunized 2 days earlier with eOD-60mer40 (Fig. 4H). Two days after B cell transfer, LNs were recovered for FRET imaging. As shown in Fig. 4I, engineered B cells dispersed into follicles in and around the FDC networks. Image analysis revealed that transfer of MMP9-expressing B cells led to 65% less antigen retained on FDCs after 2 days (Fig. 4, J and K), and that 37% less of the retained antigen was intact (Fig. 4L). Therefore, although additional factors may play a role, the lack of active protease activity in the vicinity of FDCs appears to be an important contributor to the long lifetime of immunogens captured in the FDC networks.
Antigen localization to FDCs selectively enhances B cell responses to intact antigen but not breakdown products
We hypothesized that rapid antigen degradation in extrafollicular regions of the LN could limit responses to the intact antigen, whereas rapid delivery of antigen to FDCs might enhance responses to the native immunogen. To test this idea, we performed experiments using a stabilized HIV Env gp140 SOSIP trimer called N332-GT2 (35) in soluble trimer and protein nanoparticle forms. Labeling conditions were identified (six dyes per trimer on average, trimer6) that allowed for FRET tracking of N332-GT2 integrity with minimal perturbation of the antigenicity profile of the trimer (fig. S11A). Similar to our findings with eOD-60mer, the E of protease-treated trimer6 was highly correlated with the proportion of intact antigen, as determined by PAGE analysis in vitro (fig. S11, B to D). Loss of FRET signal also correlated with reduced binding of structure-sensitive antibodies (fig. S9E). Concomitantly, antibodies that recognize epitopes of the gp120 inner domain buried in the interior of properly folded trimers bound to trypsin-digested trimer6 (fig. S9E). The trimer was stable in serum or lymph for at least 48 hours, although it was degraded by trypsin or recombinant MMP9 (fig. S9F). This trimer was also produced as a NP by fusion of the trimer sequence with ferritin subunits (abbreviated trimer-NPs) (35). For N332-GT2 nanoparticles, 40 dyes could be conjugated to each particle (trimer-NP40) without affecting the binding of trimer structure-specific mAbs (fig. S9G).
We first examined the localization and in vivo stability of trimers and trimer-NPs after immunization (Fig. 5A). We previously showed that ferritin-based trimer-NPs are rapidly transported to FDCs through the same MBL/complement pathway as the eOD-60mer immunogen, whereas soluble trimer distributes more diffusely in LNs before clearance (22, 36). Here, bolus-injected soluble trimer6 or trimer-NP40 were detected in the SCS for up to 2 days, but trimer-NP40 also accumulated on FDCs starting 2 days after immunization (Fig. 5B). We also tested an “escalating-dose” (ED) immunization (10, 37), giving the same total trimer6/adjuvant dose but administered as seven injections of increasing dose over 2 weeks (Fig. 5A). ED immunization is another approach that promotes antigen deposition on FDCs through immune complex formation as the dosing progresses (10, 37). Two days after the last ED injection (day 14) of trimer6, substantial antigen was localized on the FDC network (Fig. 5B). Flow cytometry analysis of LNs pooled from multiple mice revealed that FDCs were by far the major cell type where antigen accumulated after immunization (Fig. 5, C and D, and fig. S12A), and trimer-NP or ED immunizations increased the amount of FDC-trapped antigen by 24-fold and 120-fold over bolus immunization, respectively (Fig. 5E). FRET analysis indicated that substantial portions of extrafollicular antigen (60% of trimer and 80% trimer-NP) were degraded within 2 days, whereas trimer-NP40 or ED-administered trimer6 localized to FDCs remained predominately intact (Fig. 5F). Thus, immunization with HIV Env trimers using ED or nanoparticle regimens promotes follicular targeting and increases the level of intact antigen retained within the LN.
Figure 5: Immunizations targeting antigen to FDCs preserve antigen integrity in vivo.
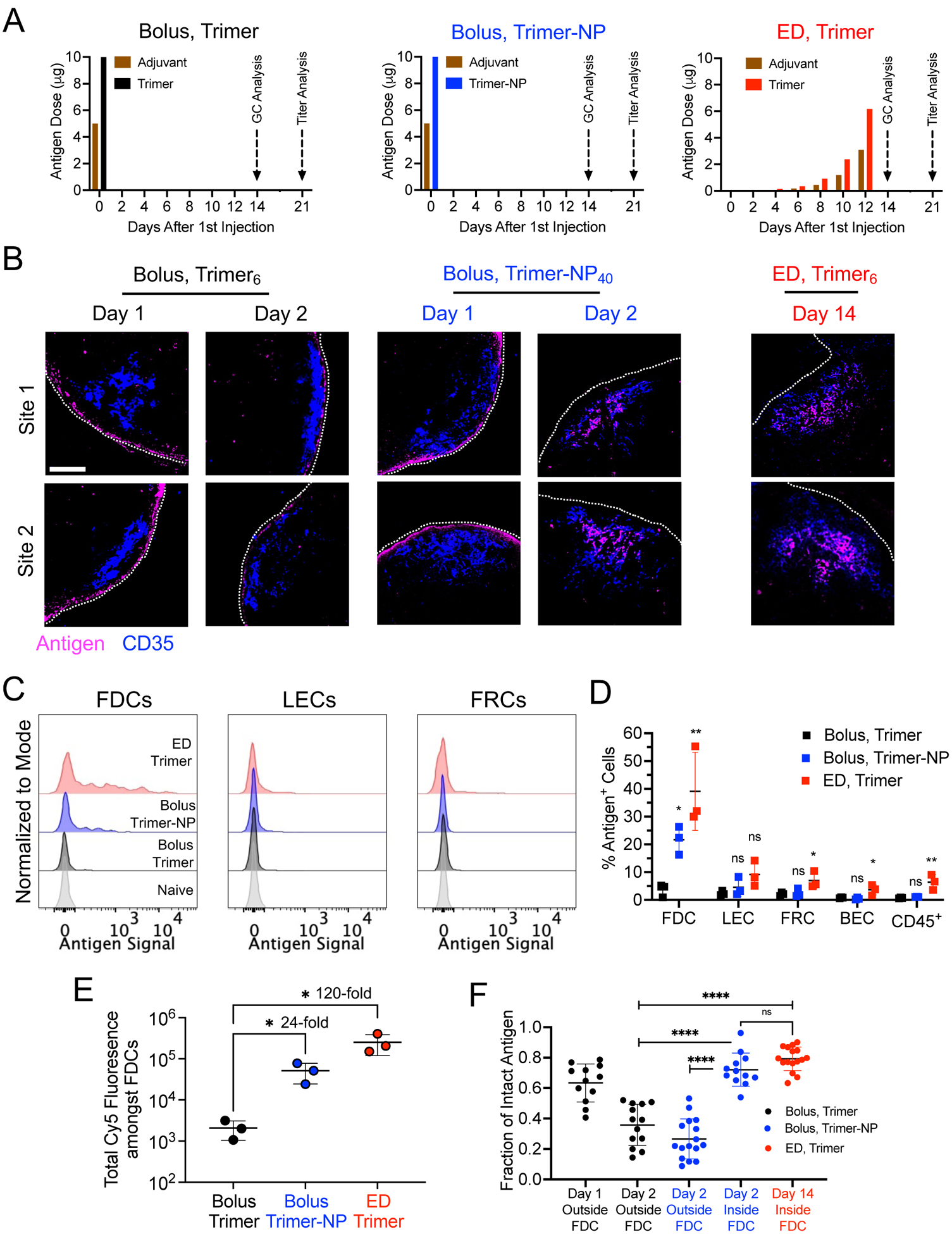
(A) Administration schedules for bolus and escalating dose (ED) immunization regimens using soluble trimer and Trimer-NP immunogens. Saponin adjuvant was used for all conditions. (B) C57BL/6 mice (n =3 animals/group) were immunized with indicated dye-labeled antigens and dosing scheme. At the indicated time points, LNs were flash frozen, cryosectioned, and imaged for antigen localization. Shown are representative images of two distinct follicles illustrating antigen (magenta) and CD35 (blue). Scale bar, 100 μm. (C to E) Flow cytometry analysis of LN cells (n =3 pools/group, each pool containing four LNs from two mice) isolated after immunization with fluorescently-labeled trimer (day 2 for bolus trimer and trimer-NP injection, or day 14 after the initial injection of ED trimer). Shown are representative histograms of antigen intensities amongst LN cells (C), the quantification of antigen+ population (D), and the sum of all antigen intensities amongst all antigen+ FDCs (E) for the indicated immunization conditions. P values determined by one-way ANOVA with Dunnett’s post test for pair-wise comparisons against Bolus Trimer. (F) C57BL/6 mice (n =4 animals/group) were immunized as in (B) and LNs were prepared for FRET imaging at the specified time points. The fractions of intact antigens in the LN at the indicated locations and times. Each point represents one region from one tissue section. Data were collected from at least eight tissue sections from eight LNs. P values were determined by one-way ANOVA with Tukey’s post test. All plots show mean ± s.d.
Bolus immunization with trimer-NP or ED immunization with trimer led to increased total numbers of GC B cells compared with bolus trimer immunization, by 8- and 27-fold, respectively (Fig. 6A, B). Staining with N332-GT2 trimer probes to identify cells capable of binding to the intact antigen at day 14 revealed ~200-fold and ~40-fold increases in the number of GC B cells specific for the intact trimer after ED immunization and trimer-NP immunization, respectively, compared with bolus trimer injection (Fig. 6B). Trimer probe staining of GC B cells from mice immunized with an irrelevant antigen, ovalbumin, showed low background staining (Fig. 6B). The proportion of trimer-specific GC B cells after bolus trimer vaccination remained relatively constant through 21 days after immunization and never reached the levels primed by trimer-NP or ED vaccination (fig. S12B).
Figure 6: Targeting of immunogens to FDCs amplifies GC B cell responses against intact antigens without increasing the response to antigen breakdown products.
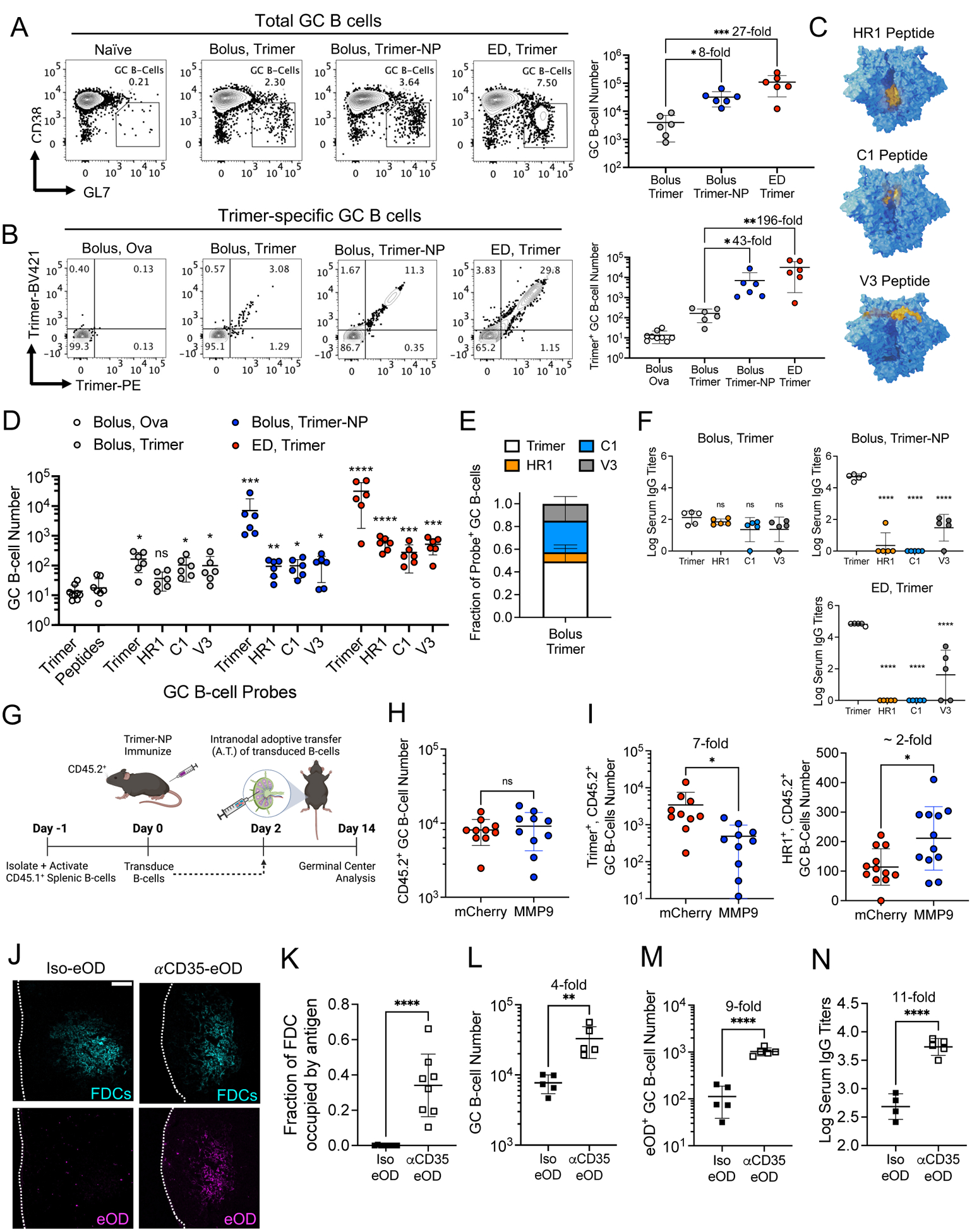
C57BL/6 mice (n =5–10 animals/group) were immunized with the indicated antigens and saponin adjuvant according to the dosing schemes. (A to E) LNs were harvested at day 14 for flow cytometry analysis. (A) Representative flow plots showing GC B cells (left) and their quantification (right). (B) Representative flow plots of intact trimer-specific GC B cells (left) and their quantification (right). *q ≤ 0.05; **q ≤ 0.01; ***q ≤ 0.001 by Kruskal-Wallis one-way ANOVA followed by two stage step-up method for comparison against bolus trimer. (C) Structural models of the HIV Env trimer highlighting (in yellow) the location of the HR1, C1, and V3 peptides used as probes to identify breakdown product specific GC B cells. (D) Enumeration of intact trimer and off-target peptide specific GC B cells after bolus, trimer-NP, ED trimer immunization, or bolus ovalbumin-immunized mice (control). ns, not significant; *, q ≤ 0.05; **q ≤ 0.01; ***q ≤ 0.001; ****, q ≤ 0.0001 by Kruskal-Wallis one-way ANOVA with two stage step-up method for comparison against bolus ovalbumin. (E) Distribution of GC B cells recognizing indicated probes after bolus trimer immunization. (F) Serum IgG titers against intact trimer or breakdown product on day 21. P values determined by one-way ANOVA and Dunnett’s post test against intact trimer. (G to I) MMP+ or control mCherry+ polyclonal B cells were adoptively transferred intranodally into mice (n =10 animals/group) immunized with 10 μg of trimer-NP and 5 μg of saponin adjuvant, followed by flow analysis. Shown are the experimental timeline (G) and enumeration of total host CD45.2+ GC B cells (H), intact trimer [(I), left], and HR1 peptide specific GC B cells [(I), right]. P values were determined by Student’s t test. (J to M) C57BL/6 mice (n =4–5 animals/group) were immunized with 2.5 μg of Iso-eOD or αCD35-eOD and 5 μg of saponin adjuvant, followed by histology and flow cytometry. Shown are images of labeled antigen in the FDC networks (J) and its quantification (K) 2 days after immunization. Scale bar, 100 μm. (L to N) Flow cytometry analysis enumerating GC B cells (L) and eOD-specific GC B cells at day 14 (M), and eOD-specific serum IgG titers at day 21 (N). P values were determined by Student’s t test. All plots show mean ± s.d.
We also investigated the frequency of GC B cells reacting to trimer breakdown products, by staining GC B cells with HR1, C1, or V3 loop peptide probes, representing epitopes buried in the intact trimer (Fig. 6, C and D, and fig. S12C). Fourteen days after immunization, low but statistically significant levels of GC B cells recognizing the C1 and V3 epitopes were detectable after bolus trimer immunization (Fig. 6D and fig. S12C), and collectively more B cells targeted these three breakdown product epitopes than intact trimer (Fig. 6E). Strikingly, trimer-NP and ED immunizations greatly amplified the on-target GC B cell response against intact trimer, but had minimal effect on the magnitude of GC responses targeting breakdown product epitopes (Fig. 6D, fig. S12C). Further, compared with bolus trimer immunizations, NP or ED yielded 1000-fold higher serum immunoglobulin (IgG) titers recognizing the intact trimer 3 weeks after immunization (Fig. 6F and fig. S12D). Low titers against intact trimer observed after bolus trimer immunization were of the same order of magnitude as responses to the HR1/C1/V3 breakdown product epitopes (Fig. 6F and fig. S12D).
To determine whether having a low level of protease activity in follicles is important for the efficacy of FDC-targeting immunizations, we again adoptively transferred MMP9-expressing polyclonal B cells to introduce protease activity into the follicles. MMP9-expressing or control mCherry-expressing B cells were injected intranodally 2 days after immunization with trimer-NPs, and 12 days later GC responses were analyzed (Fig. 6G). As shown in Fig. 6, H and I, introduction of MMP9+ B cells did not affect the total GC response, but GC B cells binding to the intact trimer were reduced by sevenfold, and HR1 peptide-binding cells were increased by about twofold. Thus, low protease activity in follicles is important for optimal on-target B cell responses to FDC-targeted antigens.
Enhancements in GC responses targeting the intact immunogen elicited by NP or ED immunization likely reflect, in part, the fact that GC B cells are encountering antigen in a multivalent form (either the NP or as immune complexes in the case of ED immunization) and/or costimulatory effects of complement decorating the antigen with these immunizations. To test the impact of FDC localization in the absence of these additional factors, we performed immunizations targeting monomeric eOD antigen to FDCs by creating a fusion of eOD and an anti-CD35 ScFv (αCD35-eOD) or an isotype control ScFv (Iso-eOD) (fig. S12E). αCD35-eOD accumulated on FDCs within 2 days after immunization, whereas isotype-eOD remained localized primarily in the sinus and extrafollicular regions (Fig. 6, J and K). Immunization with αCD35-eOD elicited about fourfold more total GC B cells and about ninefold more eOD-specific GC B cells than Iso-eOD (Fig. 6, L and M, and fig. S12F and G). Moreover, serum IgG titers at 21 days after injection were 11-fold higher for the FDC-targeted antigen (Fig. 6K and fig. S12G). Therefore, FDC localization can have a substantial impact on the priming of B cells against intact antigen structures even for monomeric antigens.
Discussion
Elicitation of protective antibody responses by vaccination is predicated on the activation of B cells wwith antigen receptors that recognize protective epitopes present on a targeted pathogen. Elegant advances in protein engineering have led to the generation of vaccine immunogens that faithfully mimic native viral envelope proteins and exhibit greatly enhanced structural stability relative to the native antigens (11, 38). However, despite the critical importance of immunogen structure to the specificity of the resulting immune response, the lifetime of structurally intact antigen in vivo after immunization is poorly understood. Here, we applied a FRET imaging based approach to track the fate of protein immunogens in mice, and made several important findings. First, we found that LN protease activity is spatially heterogeneous, with high protease levels and activity found in stromal cells and macrophages, especially in the sinuses, but low levels of protease activity in B cell follicles. Second, consistent with this observation, antigens arriving in the SCS and extrafollicular regions of LNs rapidly begin degrading, with significant losses in integrity within 24 to 48 hrs. By contrast, immunogens localized to FDCs within this early time window remain largely intact and are preserved for more than a week. As a result of this spatial compartmentalization of protease activity, traditional bolus immunization using soluble HIV envelope antigens, which does not efficiently deliver antigen to B cell follicles, led to weak GC B cell responses to the structurally intact immunogen, which were matched in magnitude by responses against irrelevant breakdown products. However, using immunizations that promoted antigen accumulation on FDCs, responses to intact antigen could be greatly amplified without concomitant increases in responses to off-target antigen breakdown products.
We hypothesize that spatially heterogeneous protease activity within LNs is part of an evolutionary strategy to halt pathogen dissemination at draining LNs before systemic spread, while preserving the ability to produce effective structure-specific antibodies if needed. In this concept, LNs serve a first line of defense function as immediate pathogen filtration and elimination units, with adaptive immunity developing over time in parallel only if immediate microbe clearance cannot be achieved. This idea was first proposed in the setting of local viral or bacterial infections, in which draining LN innate immune responses were important for blocking systemic dissemination of pathogens (39). Extracellular LN protease activity may similarly contribute to attenuating infection, because it is known that metalloproteases can inactivate some viruses (40, 41). However, the adaptive immune response also needs to be capable of “seeing” functional pathogen epitopes to generate useful antibody responses–motivating the necessity for a site where B cells can encounter intact antigen.
Although protease activity in LNs is understudied, antigen degradation within the SCS has been reported. Jenkins and colleagues observed that hen egg lysozyme conjugated to polymer microspheres could be cleaved and acquired by hen egg lysozyme specific B cells within 4 hours of injection, which the authors ascribed to proteolytic activity in lymph (12). However, we found that incubation of eOD-60mer or stabilized HIV Env trimers in plasma or lymph resulted in minimal degradation of the antigens. Healthy peripheral lymph has been shown by proteomic analysis to contain a relatively narrow set of proteases, including collagenase type IV, carboxypeptidase B2 and carboxypeptidase N (42, 43), representing a much narrower range of proteases than we detected in the LN itself.
Antigen retention and protection within FDCs was first suggested more than three decades ago in seminal work by Tew et al. Immunogen stability was shown indirectly through chromatography after retrieval of radiolabeled antigen from whole LNs (13). However, with the emerging evidence that lymphatic endothelial cells can also store antigen in a complement receptor 2 independent manner, antigen retrieved from whole LN extracts may not be specific to FDCs (44). mAbs have been used to detect antigens within FDCs (45), but this approach can only report on individual epitopes and is limited by the potential for vaccine-elicited antibody responses to confound interpretation. Experiments such as these have suggested that the FDC network provides a sanctuary site that not only retains antigen but also protects it from degradation (46); however, direct evidence in support of this idea has remained sparse. Here, AZP-based spatial zymography assays applied to live tissue sections suggested that extracellular protease activity is low in follicles relative to the SCS and interfollicular regions of LNs. Prior work has shown that FDCs cyclically present and internalize captured antigens from their surfaces, which would be expected to aid in protecting antigen locally (15). However, when we challenged LN sections with recombinant proteases or transferred protease-expressing B cells into follicles in vivo, FDC-localized antigen underwent degradation. Nevertheless, it seems likely that antigen recycling and low protease activity in follicles work together to preserve antigen on FDCs over prolonged periods.
In summary, the studies presented here provide evidence for rapid proteolysis and clearance of antigens from extrafollicular regions of LNs after immunization, but also show long-lived retention of structurally intact antigen by follicular dendritic cells. In the absence of efficient follicle targeting, a significant proportion of B cells specific for irrelevant breakdown products are primed by vaccination. Such a substantial response against irrelevant epitopes may be problematic for difficult pathogens such as HIV, in which B cells capable of maturing to produce broadly neutralizing antibodies are very rare (19), because competition in GCs can overwhelm these rare precursors (4, 6). These data provide clear motivation for immunization strategies that promote antigen delivery to the FDC network. Although we have demonstrated two approaches to achieve this, nanoparticle delivery and escalating dosing immunization, a variety of approaches may be capable of achieving this goal. These include immunization with immune complexes (47), use of antigen-complement fusions (48), and extended antigen delivery through nucleic acid vaccines (49). Such approaches may be particularly important for the generation of protective antibody responses against difficult pathogens such as HIV and for the generation of broadly neutralizing antibody responses to other variable pathogens.
Materials and Methods
Mice
Six-nine week old female C57BL/6 mice were purchased from Jackson Laboratory (Strain No: 000664). Tg(IghelMD4)4Ccg/J breeding pairs were purchased from Jackson Laboratory (Stock No: 002595) and mated in-house. Their progeny was genotyped (Transnetyx) and female mice between ages of 8–11 weeks old were used for immunization studies.
Saponin Adjuvant preparation
The saponin adjuvant used in this study is an ISCOMs-like self-assembled nanoparticle comprised of cholesterol, phospholipid, and Quillaja saponin as previously described (22). Briefly, under sterile conditions, the following solutions were made: 0.5 mL of 20 mg/mL of cholesterol (700000P, Avanti) in 20% MEGA-10 detergent (D6277, Sigma Aldrich), 0.5 mL of DPPC (850355C, Avanti) in 20% MEGA-10 detergent, and 0.5 mL of 100 mg/mL Quil-A adjuvant (vac-quil, Invivogen) in deionized H20 were prepared. DPPC solution was mixed with the cholesterol followed by the addition of Quil-A saponin in rapid succession. This mixture was diluted with PBS to a concentration of 1 mg/mL cholesterol and 2% MEGA-10, prior to overnight equilibration at 25°C. The lipids/saponin/surfactant solution was then dialyzed against PBS using a 10 kDa MWCO membrane for 5 days at 25°C and filter sterilized using a 0.2 Supor syringe filter. For further purification, the adjuvant solution was concentrated using 50 kDa MWCO Amicon Ultra-filters (UFC905008, Millipore Sigma) and purified by size exclusion chromatography using a Sephacryl S-500 HR size exclusion column. For quality control, the final saponin adjuvant was characterized by Limus Amebocyte Lystae assay (QCL-1000, Lonza) for low endotoxin levels. The adjuvant concentration was determined using a cholesterol quantification kit (MAK043, Sigma).
Recombinant immunogen production
eOD-60mer was produced recombinantly as previously described (18). The eOD-60mer gene comprised of eOD-GT8 monomer fused to Lumazine Synthase was synthesized by Integrated DNA Technologies, cloned into phLsec plasmid, and transfected into Expi293F (A14527, Thermo Fisher Scientific) cells. After 6 days cell culture supernatant was harvested by centrifugation and filtration through a 0.2 μm filter. The eOD-60mer was affinity purified by incubating with Galanthus Nivalis Lectin-conjugated agarose beads (AL-1243-5, Vector Laboratories) overnight under gentle agitation at 4°C and eluted with lectin elution buffer containing 1 M Methyl a-D-mannopyranoside (M6882, Millipore Sigma). The resulting solution was dialyzed in PBS and further purified by size exclusion chromatography using Sephacryl S-500 HR resin.
N332-GT2 trimers were expressed in FreeStyle 293F cells (Invitrogen, Cat no. R79007) and purified in two steps by affinity chromatography using a GE HisTrap column and size-exclusion chromatography using a GE S200 Increase column as described previously (35, 50). N332-GT2 nanoparticles were expressed and purified as previously described (35).
Antigen labeling and characterization
Protein antigens (eOD-60mer, HIV Env trimer, trimer-NPs, and αCD35-eOD) at 1 mg/mL in PBS were diluted at a 1:1 volume ratio in 0.2 M Sodium Bicarbonate buffer (S8875, Sigma-Aldrich) pH 8.4 and kept on ice. Fresh 1 mg/mL stock solutions of Sulfo-Cyanine 3 (21320, Lumiprobe) and -Cyanine 5 (23320, Lumiprobe) NHS esters were made in 0.2 M Sodium Bicarbonate (pH 8.4) and added to the antigen solutions. This reaction was allowed to proceed for 16 hours at 4°C then samples were desalted by passing through a Zeba Spin Desalting column (89882 and 87766, Thermofisher Scientific) equilibrated in PBS twice. Labeled antigens were sterilized by filtering through 0.22 μm pore size Spin-X centrifuge tube filters (CLS8160, Millipore Sigma) and stored at 4°C until use. Antigen degree of labeling was determined by measuring the absorbance at 280, 568, and 646 nm wavelengths to measure total protein, Cy3 dye, and Cy5 dye, respectively. To calculate the concentration of the different constituents, extinction coefficient values of 41200, 113215, 141390, 85550, 162000, and 271000 M−1cm−1 were used for one subunit of eOD-60mer, N332-GT2 Trimer, one subunit N332-GT2 Trimer-ferritin (24mer), αCD35-eOD, sulfo-cy3 NHS ester, and sulfo-cy5 NHS ester, respectively. The degree of labeling for either the nanoparticle or soluble antigens was calculated based on the ratio of antigen concentration to Cy3 concentration or to Cy5 concentration.
Complement binding assay for labeled eOD-60mer
High binding ELISA plates (07-200-37, Fisher Scientific) were coated with 1 μg/mL of labeled eOD-60mer40 and blocked with 2% BSA in PBS. Serum was collected from naïve mice using collection tube with Serum Gel (41.1500.005, Sarstedt), diluted in PBS (3% v/v), and incubated in ELISA plates for 1 hour at 37°C. Complement binding was detected by biotinylated anti-C3 antibody (NB200-540B, Novus Biologicals) followed by streptavidin-HRP (3310-9-1000, Mabtech AB). The plates were developed with 1-Step Ultra TMB-ELISA Substrate Solution (34028, Thermo Fisher Scientific) and the reaction was stopped by adding with 2 N sulfuric acid (BDH7500-1).
Antigenicity profiling of labeled immunogens
Antigenicity profiles of immunogens following dye conjugation were assessed through binding of structure-sensitive monoclonal antibodies (mAbs) to plate-bound immunogens by ELISA. eOD-60mer with varying amounts of dyes conjugated were directly coated onto Corning High Binding plates (07-200-37, Fisher Scientific) at 2 μg/mL and blocked with 2% bovine serum albumin (A8022, Sigma-Aldrich) dissolved in PBS. For trimers or trimer-NPs, plates were coated with 2 μg/mL Galanthus Nivalis Lectin (L8275, Sigma-Aldrich) prior to blocking and capture of 2 μg/mL antigen. Antigenicity profiles were evaluated by adding indicated human monoclonal antibodies at titrated concentrations to plate-bound antigen, followed by detection with mouse anti-human secondary antibody conjugated to HRP (1721050, Bio-Rad Laboratories). HRP binding was determined based on reaction with TMB substrate (34028, Thermo Fisher Scientific) and was stopped by addition of 2 M sulfuric acid (BCH7500-1, VWR) at 1:1 volume ratio. The optical density of the mixture was read out at 450 nm minus the absorbance at 540 nm according to the manufacturer’s instructions.
In vitro eOD-60mer40 digestion assays
Agarose bead-immobilized TPCK-treated trypsin, 150 μL, (20230, Thermo Fisher Scientific) was prepared and equilibrated in 0.1 M ammonium bicarbonate buffer (S8875, Sigma-Aldrich) pH 8 according to the manufacturer’s instructions. These beads were mixed with 100 μL of 20 μg/mL FRET dye-labeled Ags diluted in ammonium bicarbonate buffer and incubated in 37°C. The digested antigens were isolated at specified time points by centrifugation to remove the trypsin beads and characterized using three different assays to identify changes in molecular weight, FRET efficiency, and antigenicity. Molecular weight changes of the antigen were evaluated by reducing SDS-PAGE and protein bands were visualized with high sensitivity Flamingo Fluorescent Protein Gel Stain (1610491, Bio-Rad Laboratories). Intact antigen fractions were determined by digital imaging of gels followed by ImageJ analysis. FRET efficiency and antigenicity changes were obtained by directly coating the recovered antigens onto glass coverslips for imaging or plates for ELISA, respectively.
In addition to trypsin, eOD-60mer40 was digested with 150 μg/mL of recombinant MMP9 (909-MM-010, R&D Systems), 150 μg/mL of MMP14 (918-MP-010, R&D Systems), or 100 μg/mL of ADAM17 (2978-AD-010, R&D Systems) according to the manufacturers’ instructions with minor modifications. For MMP9, 2.5 μM ZnCl2 was added to the assay buffer and 1.5 mM p-aminophenylmercuric (APMA) was used to activate 150 μg/mL of MMP9. For MMP14, 150 μg/mL of the protease was activated by 3.5 μg/mL of Furin (1503-FE, R&D Systems) for 2 hours at 37 °C in activation buffer containing 50 mM Tris, 2.5 mM CalCl2, and 0.25% w/v Brij-35 adjusted to pH 9. For 60mer digestion, 1 μM ZnCl2 was added to the MMP14 activation buffer. For ADAM17, freshly reconstituted protease at 100 μg/mL concentration and eOD-60mer40 were both desalted in Zeba columns equilibrated with DI water before mixing together to initiate cleavage. For all cases, 10 μg/mL of eOD-60mer40 was incubated with individual metalloproteinases or left in control conditions (APMA containing assay buffer for MMP9, Furin containing assay buffer for MMP14, or alone in DI water for ADAM17) for 48 hours at 37 °C. The digested 60mer was diluted to 1.5 μg/mL and analyzed on a TECAN Infinite 200Pro plate reader for solution-based FRET measurements (Ex/Em: 555/665 nm) and Cy5 (Ex/Em: 630/665 nm) fluorescence measurement or coated onto high binding plates for VRC01 mAb (1 μg/mL) binding ELISA.
Antigen stability within lymph and plasma
Lymph was collected from the mesenteric lymphatic duct as previously described (51) with a slightly modified terminal procedure. In brief, C57BL/6 mice received 200 μl of commercial olive oil via oral gavage 60 minutes prior to lymph collection to allow for easier visualization of the lymphatic ducts. At the time of collection, mice were anesthetized with isofluorane (4% for induction, 2.5% for maintenance) and their abdomen was shaven and prepared with ethanol and Betadine scrubs. A 3 cm incision was made in the skin and peritoneum along the midline. Intestine and colon were retracted to the side using sterile cotton swabs to expose the superior mesenteric artery and the adjacent mesenteric lymph ducts. The needle of a precision syringe (30 G, Hamilton) was inserted in one of the lymph ducts with the bevel pointing towards the distal portion of duct and 5–10 μl of lymph were collected by gently retracting the plunger. For plasma collection, naïve C57BL/6 mice were bled retro-orbitally and processed in MiniCollect Tubes with EDTA (450480, MiniCollect).
To examine antigen stability, 10 μg/mL eOD-60mer40 or Trimer6 was incubated in diluted plasma or lymph (10% v/v in PBS) or 150 μg/mL of porcine trypsin (T4549, Millipore Sigma) at 37°C. The FRET (Ex/Em: 555/665 nm) and Cy5 (Ex/Em: 630/665 nm) signals were recorded at specified time points using a TECAN plate reader.
Antigen degradation after incubation with serum
To determine if complement deposition onto antigen prevents enzymatic degradation, 20 μg/mL eOD-60mer was incubated for 15 minutes at 37°C with 10% fresh serum from naïve mice or left alone in PBS. This solution was mixed with trypsin conjugated agarose beads and digested overnight at 37°C. Complement binding and structural integrity of the nanoparticles after digestion was assessed by ELISA using 1 μg/mL anti-C3 and human VRC01 monoclonal antibody, respectively.
Mouse immunizations
All animal studies were carried out under an IACUC-approved animal protocol following local, state, and NIH guidelines for care and use of animals. C57BL/6 mice were anesthetized and immunized with 10 μg of indicated antigens in the presence or absence of 5 μg saponin adjuvant subcutaneously at the left and right sides of the tail base.
Tissue processing
Inguinal lymph nodes (LNs) extracted from euthanized mice were submerged into cryomolds containing O.C.T. (23-730-571, Fisher Scientific) compound and dipped into 2-methylbutane (M32631, Millipore Sigma) pre-chilled in liquid nitrogen for 5–10 minutes prior. All frozen tissues were cryosectioned on a Leica CM1950 at 10 μm thickness, adhered to Superfrost Plus microscope slides (12-550-15, Fisher Scientific), and stored in −80°C until use.
Confocal Microscopy
For all experiments, imaging was performed on a Leica SP8 confocal microscope equipped with a white light laser and spectral emission filter to detect emission wavelengths between 470 and 670 nm with a minimum bandwidth of 10 nm. All images were recorded with a 25X water immersion lens and for assessing antigen drainage in the LNs, laser settings were kept constant across different time points for each immunogen.
FRET imaging and analysis
To assess antigen integrity, an acceptor photobleaching method was used to compared Cy3 dye emission intensity before and after photobleaching the Cy5 dye. FRET imaging was carried out on a Leica SP8 Confocal Microscope with a 25x objective, imaging selected square regions with dimensions ranging from 175 to 250 μm. Cy3 signal was recorded by exciting the dye at 555 nm and collecting its emission between 565 and 615 nm wavelengths, while Cy5 signal was recorded after exciting the dye at 640 nm and collecting emission between 660 and 720 nm wavelengths. To photobleach the Cy5 dye, regions of interest in LN sections were excited with 640 nm wavelengths lasers at maximum power until the Cy5 intensity was less than 10% of its pre-bleached values; the duration of the bleaching process varied between 90 and 180 seconds. Cy3 and Cy5 fluorescence signals were then collected again post-acceptor bleaching. FRET efficiencies (E) of antigens were calculated using a custom MATLAB code (52) based on established methods (16). FRET efficiency E at each pixel (Eij) is given by:
where Cy3Bef and Cy3Aft are Cy3 emissions before and after Cy5 photobleaching. Correction factors α and γ account for incomplete bleaching of the Cy5 (0 ≤ α ≤ 1) and unintended photobleaching of Cy3 (γ ≥ 1), respectively. α values were experimentally determined from the ratio of Cy5 emission signals before and after photobleaching, while γ was determined to be ~1.05 based on coverslip-coated antigens conjugated only with Cy3. Moreover, correction factors, δ, crosstalk from acceptor emission into donor emission channel, and ε, crosstalk of photodegraded acceptor product into donor emission channel, that are listed in the full equation for E are found to be 0.004 ± 0.00094 and 0.0062 ± 0.00077, respectively, from monolayer experiments with antigens labeled only with Cy5. Thus, both δ and ε were approximated to be zero. To quantify E, antigen locations were first identified by using a binary mask generated from thresholding intensity values in the Cy5 image prior to photobleaching. To this end, non-immunized LNs were imaged under same conditions to approximate the background fluorescence value of Cy5 needed for thresholding. This mask was applied to the remaining images to quantify Cy3Bef from the Cy3 image before photobleaching, Cy3Aft from the Cy3 image after photobleaching, and α from the Cy5 images before and after photobleaching. These parameters were used to generate a histogram of E values for antigens found within each imaging area. To determine the fraction of degraded antigen, this procedure was repeated for an intact control sample comprised of FRET dye-conjugated antigen coated onto a glass coverslip. To quantify the amount of fully intact antigen at each time point/condition was determined by quantify the fraction of antigen+ pixels in each imaged region with Eij values larger than the minimal E value detected in the intact control sample.
For examining fractions of intact antigens co-localized with specified protein markers, the acceptor photobleaching procedure was carried out on LN sections that were fixed, immunostained with antibodies, and mounted in PBS. In addition to the 4 images required for calculating E, fluorescence images of cell markers were recorded and overlaid onto regions of intact and degraded antigens.
Anti-CD45 antibody conjugated eOD-monomer for extracellular antigen stability analysis
Anti-CD45 mAb (103102, BioLegend) was conjugated to Cy5-labeled eOD-GT8 monomer with an added free N-terminal (53) cysteine using Sulfo-SMCC linker (A39268, Thermofisher Scientific). Sulfo-SMCC linker was first added to 1 mg/mL mAb solubilized in PBS at 20-fold molar excess. This reaction was carried out for 45 minutes at room temperature and was terminated by removing unreacted linkers with PBS equilibrated Zeba columns. This solution was temporarily maintained at 4°C.
The dye-labeled eOD monomer was next conjugated to Sulfo-SMCC tethered anti-CD45 mAb. To this end, 1 mg/mL of the Cy5-labeled eOD monomer, maintained in 1 x TBS (BP24711, Fisher Scientific), was reduced by adding two-fold molar excess of TCEP (20490, Thermofisher Scientific). This reaction was stopped after 15 minutes at room temperature by using PBS equilibrated Zeba desalting columns. The reduced eOD monomer was immediately mixed with Sulfo-SMCC linker tethered antibody at 1:1 mass ratio and was allowed to react overnight under mild agitation at 4°C. This mixture was repeatedly washed through Amicon ultra-centrifugal units with 100 kDa MW cutoff (UFC510024, Millipore Sigma) to remove unreacted eOD monomer. The resulting antibody-eOD monomer construct was sterilized by passing through centrifugal filters with 0.2 μm pores (CLS8160-96EA, Millipore Sigma) and kept at 4°C.
For immunization of C57BL/6 mice, indicated doses of saponin adjuvant and antibody-eOD conjugate was administered subcutaneously at the tail base and inguinal LNs were harvested and mechanically processed at specified time points. The isolated cells were first stained with Live/Dead Aqua (L34957, Thermofisher Scientific), anti-B220 BV421 antibody (103239, BioLegend), and anti-CD3ε antibody (100321, BioLegend) before splitting into two distinct pools.
To detect surface bound or intact eOD monomer, each pool of cells was stained with either biotinylated anti-Cy3/Cy5 mAb (C3117, Millipore Sigma) or biotinylated VRC01 mAb, respectively, followed by exposure to streptavidin-BUV737 (612775, BD Biosciences), and analysis by flow cytometry.
Bulk antigen stability analysis using pull-down assay
Pairs of Inguinal LNs from two C57BL/6 mice immunized with eOD-60mer40 were harvested and pooled into prechilled tubes each containing 120 μL of 0.5% wt/v Bovine serum albumin (A8022, Sigma Aldrich), 2x Halt protease inhibitor cocktail (78430, Thermofisher Scientific) and 5 mM EDTA (15575020, Thermofisher Scientific) solubilized in PBS. The LNs were mechanically dissociated using a Biomasher II microtissue homogenizers (K7496250010, Fisher Scientific) and kept at 4°C for 30 minutes under gentle agitation and away from light. To obtain the extracellular protein fraction, the homogenized LN solution was centrifuged for 5 minutes at 500 × g before transferring 100 μL of the supernatant into a V-bottom 96 well plate (12-565-215, Fisher Scientific). In addition, 100 μL of intact antigen standards containing 300, 30, 3, or 0.3 ng of eOD-60mer40 or eOD-60mer conjugated with only 20 Cy5 dyes (eOD-60mer20 Cy5) were added to the 96 well plate. To obtain the intracellular protein fraction from the LNs, 120 μL cell lysis solution comprised of T-PER (78510, Thermofisher Scientific), 2x Halt protease inhibitor cocktail, and 5 mM EDTA was added to the each of the centrifuged tubes with pelleted cells and tissue debris. These tubes were vortexed vigorously to dissociate the pellets and maintained under moderate agitation for 1 hour at room temperature and away from light. The tubes were then centrifuged at 1500 × g for 10 minutes and 100 μL of the supernatant containing intracellular proteins were transferred to the V-bottom 96 well plate. For all wells containing LN samples and standards, 7.5 μL of 1 mg/mL of biotinylated anti-Cy3/Cy5 antibody was added and the plate was kept on a shaker overnight at 4°C.
The resulting antibody-antigen complexes were next captured onto microparticles. Specifically, 32.5 μL of streptavidin-conjugated magnetic particles (88816, Thermofisher Scientific) were first transferred to individual wells in a 96 well V-bottom plate and mixed with 170 μL of PBS. Using a 96 well side-skirted magnet (12027, Thermofisher Scientific), the beads were pulled to the bottom of the wells and the supernatant was removed. The pelleted beads were resuspended in 105 μL solutions containing LN extracts or eOD-60mer standards bound to biotinylated anti-dye antibodies and kept on a shaker overnight at 4°C.
The beads bound with eOD-60mer40 were next analyzed using the BD FACSymphony A3 flow cytometer where the FRET (561 nm Excitation, 670/30 nm Emission) and acceptor signals (640 nm Excitation, 670/30 nm Emission) of the captured antigens were recorded. To quantify the mass of antigen within LNs, a standard curve was first generated based on the Cy5 mean fluorescence intensities (MFIs) from eOD-60mer40 standards with known masses. Next, the raw mass values determined based on the standard curve was scaled by a correction factor to account for the inefficiency of eOD-60mer40 retrieval from LN samples. Specifically, this factor (approx. 1.8) was calculated by dividing the Cy5 signal from beads incubated with 3 ng eOD-60mer40 standard kept in BSA/protease inhibitor solution with the Cy5 signal from the same standard mass mixed into mechanically dissociated naïve LN solution or lysate. The reported 60mer masses detected in LNs are the product of the correction factor and values obtained from the standard curve.
To evaluate the fraction of intact 60mer bound to the beads, normalized FRET (nFRET), defined as mean value of FRET divided by Cy5 intensity of all the particles, was first quantified for eOD-60mer40 and eOD-60mer20,Cy5 standards (made in PBS with BSA and inhibitors) that are used to represent 100% and 0% intact antigen, respectively. The nFRET value for eOD-60mer40 is expected to be upper bound since all of the donor and acceptor dyes are present on the NP. In contrast, nFRET value for eOD-60mer20,Cy5 serves as the lower bound since FRET does not occur without donor (Cy3) dyes. Moreover, nFRET was observed to vary with the amount of 60mer bound on the beads thus this value was quantified for beads incubated with serially diluted eOD-60mer40 and eOD-60mer20,Cy5 standards. The resulting data points were fitted to sigmoidal curves that provided the upper (100% intact) and lower (0% intact) limits of nFRET values for beads bound to any amount of 60mer. By assuming that the change in nFRET value between the upper and lower limits was linearly related to the % of intact antigen, the percent of intact 60mer retrieved from LNs, whose antigen amount was determined as previously described, was quantified.
Vibratome sectioning
Low melting point agarose (A4018, Sigma Aldrich) solution was prepared by adding 0.4 g of agarose into 20 mL of PBS to achieve a concentration of 2% wt/v. This mixture was microwaved at 30 seconds intervals until the agarose was fully dissolved and the solution was equilibrated at 37 °C for at least 45 minutes. Inguinal LNs were harvested from immunized or naïve mice and placed onto a 35 mm diameter Petri dish. The agarose solution was carefully added to this dish and incubated at 4 °C for 10 minutes. A rectangular block encompassing the LNs were excised and mounted onto the sample holder of a Leica Vibratome VT1200s using Vetbond tissue adhesive (3M). The sample was held in PBS pre-chilled to 4°C and sectioned to obtain 250 μm thick live tissue sections that were immediately transferred to a petri dish containing ice cold RPMI 1640. All slices were collected and maintained in this solution until further use.
In vitro AZP assay
The sequences for the custom peptide (CPC Scientific) probes are shown below (30).
| Quenched AZP | (5FAM)-GPVPLSLVMG-K(CPQ2)-(Peg2)-GC |
| D-Isomer | UeeeeeeeeXGpvplslvmGrrrrrrrrrX-k(5-FAM)-amide |
| AZP | UeeeeeeeeXGPVPLSLVMGrrrrrrrrrX-k(5-FAM)-amide |
| PolyR | rrrrrrrrrX-k(Cy5)-amide |
| Upper case: L-Isomer | Lower case: D-Isomer |
| U: Succinoyl | X: 6-aminohexanoyl |
| 5-FAM: 5-Carboxyfluorescein | Cy5: Cyanine 5 |
| Peg2: polyethylene glycol | CPQ2: Quencher |
To analyze the cleavage of peptide sequence in the AZP probe by different recombinant proteases, a quenched version of the probe that consisted of a peptide sequence flanked by quencher CPQ2 and fluorophore FAM was used. This reaction was carried out by incubating 1 μM of the quenched AZP probe with 1 μg/mL of recombinant MMP14 (918-MP-010, R&D Systems), ADAM17 (2978-AD-10, R&D Systems), ADAM10 (946-AD-020, R&D Systems), Cathepsin D (1029-AS-010, R&D Systems), Cathepsin S (50769-M08H, Sino Biologicals), or Cathepsin L (1515-CY-010, R&D Systems). The buffer pH for digestion was adjusted based on manufacturer’s protocol and were 8.5, 9.0, 9.0, 3.5, 4.5, and 6.0 for MMP14, ADAM17, ADAM10, Cathepsin D, Cathepsin S, and Cathepsin L, respectively. The FAM signal (Ex/Em: 485/535 nm) was recorded on a plate reader initially and after 60 minutes of incubation at 37°C.
For visualizing protease activity within tissues, LNs were harvested from naïve mice or mice immunized with eOD-60mer and saponin at indicated time points. These tissues were embedded within 2% agarose gel containing 5 μM AZP and PolyR control probes, vibratome sliced to obtain 250 μm thick live sections, and maintained within RMPI 1640 supplemented with 2 μM ZnCl2. Parallel tissue sections were also incubated in 5 μM uncleavable D-Isomer probe and PolyR as a control. After 2 hours incubation at 37°C, the tissue-gel constructs were washed twice with PBS before fixation in 4% paraformaldehyde (PFA) overnight at 4°C. Excess PFA was removed by washing in PBS and these sections were imaged directly by confocal microscopy. From the images, areas of positive AZP probe signal normalized to areas with positive PolyR probe signal was calculated for inside versus outside of the follicles.
To immunophenotype proteolytically active cells, live LN vibratome sections were placed into RMPI medium containing the following combination of probes at 2 μM concentration: (i) AZP and PolyR probes, (ii) D-Isomer and PolyR probe, and (iii) AZP probe only. These slices were maintained in the probe media for 2 hours at 37°C prior to washing away the excess probe by performing two cycles of transferring the slices to and maintaining in fresh PBS for 15 minutes at 37°C. The LN sections were subsequently processed for flow cytometry analysis. To assess protease activity, different cellular phenotypes were first identified and AZP+ cells were determined only amongst the PolyR+ cells within each cell type. Gating for PolyR and AZP probe binding was determined based on cells extracted from slices incubated in probe media conditions (iii) and (ii), respectively (Figure S5A).
Ex vivo modulation of protease activity within LN slices
To examine the impact of proteases on FDC-localized antigens, mice were first immunized with saponin and eOD-60mer40. After 3 days, LNs were harvested and vibratome sliced to 250 μm thickness and were left untreated in RPMI1640 medium or exposed to either 200 μg/mL porcine trypsin (T4549, Millipore Sigma) or 10 μg/mL recombinant full length murine MMP9 without additional chemical activation by p-Aminophenylmercuric acetate (ab39309, Abcam). After 4 hours at 37°C, tissue slices were washed in PBS and fixed overnight in 10% Neutral Buffered Formalin (HT5011-1CS, Sigma-Aldrich) at 4°C prior to confocal imaging.
Similarly, the effect of protease inhibitors on antigen degradation was assessed by immunizing mice with saponin and eOD-60mer40, followed by harvesting of LNs 2 hours later for vibratome sectioning. These live slices were left untreated in RPMI1640 medium or incubated with 100 μg/mL of Marimastat (M2699, Millipore Sigma) or Halt Broad-spectrum protease inhibitor without EDTA (78430, Thermo Fisher Scientific) at a five-fold dilution of the stock solution. After 6 hours in 37°C, the tissue slices were washed in PBS and fixed overnight in 10% Neutral Buffered Formalin (NBF) at 4°C.
In both ex vivo experiments, the fixed sections were incubated in fresh PBS for 1 hour at 25°C followed by flash freezing and cryosectioning. The sections adhered to microscope slides were mounted in PBS and imaged immediately.
Effect of in vivo metalloproteinase inhibition on antigen stability within LNs
Marimastat (Selleck Chemicals) was dissolved in DMSO to achieve a concentration of 52 mg/mL. This stock solution was diluted to 9 mg/mL in PBS, sonicated for 5 minutes, and injected intraperitoneally (IP) at a volume of 50 μL per mouse resulting in a ~25 mg/kg dose. Control mice were administered matching concentrations of DMSO diluted in PBS. These injections were carried out twice daily for 4 days. On the 5th day, Marimastat was administered 2 hours prior to and 6 hours following immunization with 5 μg saponin adjuvant and 10 μg eOD-60mer40. Draining LNs were extracted 24 hours after immunization for antigen pull-down assay and FRET imaging.
MMP9 transfer plasmid design
Gene fragments encoding the full length human MMP9 with a G100L mutation (to achieve functional proteolysis without a requirement for pro-peptide cleavage) (34) and C-terminal HA affinity tag was purchased from Integrated DNA Technologies. This gene block was inserted into a MIGR1 retroviral transfer plasmid followed by an IRES sequence and the gene encoding for mNeongreen reporter protein.
Retrovirus production
Phoenix Eco cells (CRL-3214, ATCC) were plated at a density of 75,000 cells/cm2 in a 10 cm diameter tissue culture dish and maintained in 10 mL of DMEM containing 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 1% penicillin/streptomycin (complete media) for 24 hours. To produce retroviruses, these cells were transfected with retro-viral vectors using lipofectamine 3000 (L3000015, Thermofisher Scientific) according to the manufacturer’s instructions. In brief, 1.5 mL of Opti-MEM (31985062, Thermofisher Scientific) containing 41 μL Lipofectamine 3000 reagent was mixed with 1.5 mL of Opti-MEM containing 35 μL of P3000 reagent, 4.5 μg of pCL-Eco packaging plasmid (12371, Addgene), and 13.5 μg of transfer plasmid containing constitutively active MMP9/mNeongreen or mCherry inserts. This mixture was maintained at room temperature for 20 minutes. Prior to transfection, the culture media for Phoenix Eco cells was replaced with 6 mL of Opti-MEM and Lipofectamine-plasmid mixture was added dropwise over the surface of the culture dish. The Phoenix Eco cells were transfected overnight before replacing the Opti-MEM with 10 mL of complete media. The generated viruses were harvested after 48 hours by collecting the media, centrifuging at 2000 × g for 5 minutes, and filtering through 0.45 μm PVDF syringe filters (24–242, Genesee Scientific). The retrovirus was used immediately after or flash frozen in liquid nitrogen for extended storage.
Transduction of splenic B-cells and adoptive transfer
Spleens were harvested from naïve C57BL/6 mice or CD45.1+ C57BL/6 mice (Strain: 002014, Jackson Laboratory) and B-cells were isolated using a commercially available negative selection kit (19854, STEMCELL Technologies) according to manufacturer’s instructions. The isolated B-cells were resuspended at a density of 1 million cells/mL in base media, distributed at 2 mL per well in 6-well plates, and activated for 24 hours with 10 μg/mL LPS (L2630-10MG, Millipore Sigma) and 25 ng/mL IL-4 (404-ML-025, R&D Systems). The base media was comprised of RPMI 1640, 10% v/v FBS, 55 μM β-Mercaptoethanol (21-985-023, Fisher Scientific), and 1% v/v penicillin/streptomycin.
To transduce the B-cells using retrovirus, 2 mL of 20 μg/mL RetroNectin (T100B, Takara Bio) was first coated onto each well of non-tissue culture treated 6-well plates (351146, Corning) on the same day that splenic B-cells were isolated. The plates were kept overnight at 4°C before removing the RetroNectin solution and blocking with sterile 2% BSA solubilized in PBS for 30 minutes at room temperature. The plates were next washed once with PBS before adding 1.3 mL of supernatant with MMP9/mNeongreen- or mCherry-encoding retrovirus per well (see Retrovirus production) and centrifuging the plate at 2000 × g for 2 hours at 32°C to attach the virus onto RetroNectin coated plates. Afterwards, the supernatant was removed, wells were washed three times with PBS, and activated B-cells were transferred to the wells to undergo transduction for 48 hours. The expression of MMP9 within the culture media was quantified by using a commercially available ELISA kit (ab100610, Abcam). The activity of secreted MMP9 was validated by concentrating the culture media by 8-fold, transferring the supernatant onto anti-HA tag Ab (901533, BioLegend) coated ELISA plates pre-blocked with 2% BSA in PBS, and exposing the captured MMP9 in the wells to 1 μM quenched AZP probe. The change in FAM intensity was recorded using a plate reader as previously described.
For adoptive transfer, transduced B-cells were collected from the wells, centrifuged, supernatant aspirated, and pellet resuspended in 15 mL of base media. The washing process was repeated three more times to completely remove LPS and after the final wash, the B-cells were resuspended in PBS at a density of 2.5 million cells per 10 μL. This cell mixture was kept on ice. To perform intranodal injections into the inguinal LNs, mice were anesthetized and the area between the hind thigh and abdomen was shaved and depilated with a mild hair removal cream. Left and right superficial inguinal LNs were visualized with the help of a gooseneck fiber optic illuminator as defined opaque spots that are 5–7 mm superior and lateral to the corresponding inguinal (4th) nipple. The LN was gently pushed up against the skin, which was pulled taut to allow for controlled insertion of the needle. The needle of a precision syringe (30 G, Hamilton) was inserted under the skin and pushed 1–2 mm into the LN along its long axis. For each LN, 10 μl of B-cell suspension was slowly injected and the entry of this solution into the tissue was confirmed by the visible swelling of the LN.
Single cell RNA-sequencing analysis
The scRNA-seq data were collected and analyzed in a separate study. The data were used here to identify cell type-specific expression of proteases. Briefly, scRNA-seq (10X Genomics) was performed according to manufacturer’s instructions on dissociated cells from LNs of mice that were either untreated or 6 hours following vaccination with 200 μg ovalbumin peptide and 20 μg CpG 1826 adjuvant. Cell Ranger was used to process sequence data. Gene expression is log-normalized for each cell such that the total unit for each cell sums to 10,000. Clusters of the major cell types were derived using the Louvain clustering algorithm and annotated based on known markers. Genes encoding extracellular or secreted proteases, including ADAMs, ADAMTSs, MMPs and elastases, were included in the analysis. For FDCs, scRNA-seq analysis was carried out in a similar manner based on published datasets (28, 29).
Immunofluorescence staining
Frozen sections were retrieved from −80°C, quickly thawed, and incubated in 10% neutral buffered formalin solution for 8.5 minutes at 25°C. The sections were washed 3 times in PBS with 10-minute incubation time between each wash. Excess PBS was removed after the final wash before incubating the slides in blocking buffer, comprised of 2% BSA and 2% Triton X-100 in PBS. After 30 minutes, the blocking buffer was aspirated and the slides were stained in the following primary antibody solutions also made in blocking buffer for ~16 hours at 4°C: 1:75 anti-ADAM17 (NBP2-67179, Novus Biologicals), 1:75 anti-ADAM10 (NBP176973, Novus Biologicals), 1:75 anti-MMP14 (ab51074, Abcam), 1:75 anti-CD35 (740029, BD Biosciences), 1:75 anti-LYVE1 (53-0443-82, Thermo Fisher Scientific), 1:75 anti-CD169 (142421, BioLegend). These slides were washed in PBS 3 times for 10 min, stained with 1:200 diluted secondary antibodies solution in blocking buffer (ab150063 Abcam, ab150061, Abcam) for 4 hours at room temperature, and washed again in PBS. To mount the slides, 5 μL of PBS was added directly onto the stained tissues prior to gently placing a 20×20 mm coverslip on top of the PBS droplet to sandwich the section. The coverslip was sealed using CoverGrip coverslip sealant (23005, Biotium) and imaged immediately. Slides were stored at 4°C for maximum of 1 week.
To visualize 5 distinct markers within the same LN, modifications were made to the above staining protocol. Cryosectioned LNs were first fixed and blocked with 10% v/v normal Donkey serum and 0.1% wt/v Triton X-100 constituted in PBS. The slides were then sequentially stained with the following antibodies and reagents at listed dilutions for indicated times.
| Staining Order | Antibodies and Reagents | Durations |
|---|---|---|
| 1 | anti-ADAM17 mAb (NBP2-67179, Novus Biologicals), 1:75 | 1 hr |
| 2 | donkey anti-rabbit polyclonal Ab conjugated to BV421 (406410, BioLegend), 1:250 | 1 hr |
| 3 | anti-ADAM10 mAb conjugated to AF647 (NBP2-12015AF647, Novus Biologicals), 1:75 anti-MMP14 mAb conjugated to AF555 (ab302548, Abcam), 1:75 anti-CD169 mAb conjugated to AF488 (142419, BioLegend), 1:75 biotinylated anti-LYVE1 mAb (13-0443-82, Thermofisher Scientific), 1:75 |
1 hr |
| 4 | streptavidin conjugated to BV510 (405233, BioLegend), 1:150 | 0.5 hr |
The slides were washed thrice in PBS after each staining and were mounted as previously described after streptavidin binding. The LNs were imaged on a Leica SP8 scanning confocal where 3 distinct scan sequences were created to eliminate fluorophore cross talk. For the first sequence, the LN section was excited with 405 nm laser and emission between 410–450 nm and 620–710 nm recorded to acquire ADAM17 and LYVE1+ LECs signal. For the second sequence, the tissue was excited with 488 and 650 nm laser and emission between 498–530 nm and 660–770 nm were recorded to detect CD169+ subcapsular macrophages (SSMs) and ADAM10, respectively. For the final sequence, the slide was excited with 555 nm laser and emission between 565–620 nm were obtained to visualize MMP14. The colocalization of the proteases with SSMs or LECs were quantified using coloc2 plugin on ImageJ. Regions of interests were created on either SSM or LEC images and colocalization with each protease was represented using Mander’s coefficient which required images to have p > 0.95 after Costes analysis.
Follicle targeting anti-CD35 ScFv-eOD Monomer construct
Follicle targeting of antigens was achieved by using an anti-CD35 monoclonal antibody (clone: 8C12) that specifically recognized complement receptor 1 (CR1) that is highly expressed by FDCs. To express this antibody efficiently, the framework sequences within the variable region of both light and heavy chains of the original clone, 8C12, (54) were mutated towards rat germline amino acid sequences using the SabPred toolbox (55). This construct was expressed as a single chain variable fragment (ScFv) fused to eOD monomer (αCD35-eOD). eOD monomer fused to anti-GFP ScFv was used as a control construct (Iso-eOD). Gene fragments encoding these fusion proteins and flanked with overhangs designed for insertion into NotI restriction enzyme linearized GWIZ vector were purchased from Integrated DNA Technologies. Plasmid assembly and protein expression was carried out as previously described.
αCD35-eOD:
HHHHHHGGDTITLPCRPAPPPHCSSNITGLILTRQGGYSNDNTVIFRPSGGDWRDIARCQIAGTVVSTQLFLNGSLAEEEVVIRSEDWRDNAKSICVQLNTSVEINCTGAGHCNISRAKWNNTLKQIASKLREQYGNKTIIFKPSSGGDPEFVNHSFNCGGEFFYCDSTQLFNSTWFNSTGGGGSGGGGSGGGGSDIVMTQTPSSLAVSAGEKVTMSCKSSQSLLYSKNKKNYLAWYQQKPGQSPKLLISWASSRESGVPDRFTGSGSGTDFTLTISSVQAEDLAVYYCEQYYNIPYTFGGGTKLELKRGGGGSGGGGSGGGGSQVKLQESGGGLVQPGRSLKLSCAASGFTFSNYDMAWVRQAPTKGLEWVASINYDGSSTYYRDSVKGRFTISRDNAKSTLYLQMDSLRSEDTATYYCTTLYNWYVMDAWGQGTTV TVSS
Iso-eOD:
HHHHHHGGDTITLPCRPAPPPHCSSNITGLILTRQGGYSNDNTVIFRPSGGDWRDIARCQIAGTVVSTQLFLNGSLAEEEVVIRSEDWRDNAKSICVQLNTSVEINCTGAGHCNISRAKWNNTLKQIASKLREQYGNKTIIFKPSSGGDPEFVNHSFNCGGEFFYCDSTQLFNSTWFNSTGGGGSGGGGSGGGGSDIQLTQSPAIMSPSLGERVTMTCTASSSVGSSYLHWFQQKPGSSPKLWIYSTSNLASGVPARFSGSGSGTSYSLTISRMEAEDAATYYCHQYHRTPYTFGGGTKLEIKRAGGGGSGGGGSGGGGSEVQLQQSGPELVKPGSSMKISCKASGYSFTGYTMNWVKQSHGQNLEWIGLINPYNGGTNYNQKFKGKATLTVDKSSSTAYMELLGLTSEDSAVYYCTRGNSDFSAWFAYWGQGTSVTVSS
Underline: eOD-monomer
Red: ScFv construct
Human tonsil tissue acquisition and processing
Adult human tonsil samples sourced from standard surgical procedures performed for clinical care was provided by the Massachusetts Eye and Ear Infirmary at the Massachusetts General Hospital. Usage for scientific research was approved by the Massachusetts General Hospital Institutional Review Board under Protocol P2020-003521. Tonsil samples were deidentified and processed within 2–3 h after removal. Acquired tonsil tissues were flash frozen, cryosectioned, and immunostained in the same manner as murine LNs.
Flow Cytometry
For analysis of AZP binding and protease expression, inguinal LNs from immunized or naïve mice or live LN sections were incubated in 1 mL of digestion solution comprised of RPMI1640 medium containing 0.8 U/mL Dispase (07913, Stemcell Technologies), 0.1 mg/mL Collagenase D (11088858001, Sigma-Aldrich), and 0.1 mg/mL of DNAse (10104159001, Sigma-Aldrich). After 20 minutes incubation at 37 °C, the LNs were repeatedly passed through a 1 mL pipette tip to mechanically rupture the tissue. Care was taken to collect ~ 1 mL of the solution containing only the liberated cells prior to transferring to ice cold PBS containing 5 mM EDTA and 2% fetal bovine serum (stop buffer). The remaining tissue debris was further processed by a second incubation with 1 mL of fresh digest buffer for 20 minutes at 37 °C and transfer of the supernatant to stop buffer. This solution was passed through a 40 μm pore size filter, centrifuged, and transferred into a V-bottom 96 well plate for flow staining. For germinal center analysis, cells were isolated by mechanically crushing the LNs against a filter cap with 70 μm pore size and generously pipetting cold PBS containing 1% BSA and 5 mM EDTA (FACS buffer) onto the porous mesh. The cells were centrifuged, resuspended in flow cytometry buffer, passed through a 40 μm pore size filter, and transferred to a V-bottom 96 well plate.
The isolated cells were stained with Live/Dead fixable aqua stain (L34957, Thermo Fisher Scientific) for 10 minutes at 25°C prior to washing twice in flow cytometry buffer. Cells were then incubated with Fc Block for 10 minutes at 4°C before staining with antibodies listed in the Key Resource table for 20 additional minutes at 4°C. For on-target trimer and off-target probe specific GC B cell analysis, cells stained with antibodies were distributed evenly and exposed to biotinylated trimer or peptide probes preincubated with streptavidin (30 minutes at molar ratio of 1:4 at 25°C) conjugated to PE (405203, BioLegend), BV421 (405226, BioLegend), or APC (40520L7, BioLegend). For AZP analysis, the stained cells were immediately analyzed (28). For protease expression analysis, cells stained with surface markers were fixed and permeabilized using transcription factor staining buffer kit (00-5523-00, Invitrogen) followed by incubation with anti-protease antibody for 30 minutes at 25°C. After washing twice with wash buffer, the cells were incubated with donkey anti-rabbit antibody conjugated to Alexa Fluor 488 dye to detect the proteases. For compensation, UltraComp eBeads™ Plus compensation beads (01-3333-42, Thermo Fisher Scientific) were used to bind to dye-labeled antibodies. Stained cells were analyzed using a BD FACSymphony A3.
Probe synthesis for characterizing Trimer Breakdown product specific GC B cells
Peptides corresponding to and buried within BG505 MD39 were synthesized by GenScript, Inc. Sequences of the peptides are as follows:
HR1 peptide-biotin, GIKQLQARVLAVEHYLGAGK-biotin;
C1 peptide-biotin, DQSLKPAVKLTPLGAGK-biotin;
V3 peptide-biotin, TRPNNNTVKSIRIGPGQAFYYTGGAGK-biotin.
ELISA analysis of serum antibody responses
Blood collected from immunized mice through retro-orbital bleeding was processed in collection tube with separator gel (41.1500.005, Sarstedt) to obtain serum samples. To analyze on-target antibody response, high binding ELISA plates (07-200-37, Fisher Scientific) were coated with 1 μg/mL trimer and blocked with 2% BSA in PBS. For off-target responses, plates were first coated with 2 μg/mL streptavidin (434302, Thermo Fisher Scientific), blocked with 2% BSA in PBS, and incubated with 2 μg/mL of HR1, C1, or V3 peptides diluted in blocking buffer. To detect antigen-specific IgG responses, dilutions of serum were added to the wells and incubated for 1.5 hours at 25°C. Plates were washed 3 times in PBS containing 0.2% Tween-20, then anti-mouse IgG secondary antibody conjugated to HRP (172–1011, Bio-Rad Laboratories), diluted 1:5000 in blocking buffer as per manufacturer instructions, was added to the wells. After 1 hour incubation, plates were again washed, developed with TMB, and stopped with sulfuric acid. Endpoint titers are reported as inverse dilutions where absorbance at 450 nm minus the reference absorbance at 540 nm equals 0.1.
Quantification and statistical analysis
Information pertaining to statistical analysis such as n (number of mice or tissue slices or image regions) and p or q values are indicated in the figure legends. All plots within the figures show mean with standard deviation. No methods were used to determine if data met assumptions of statistical approach. GraphPad Prism 9 was used to calculate statistical significance from tests specified within each figure legend. All results are from at least two independent experiments. Unless specified otherwise, significance from statistical tests within the figures are denoted by *, **, ***, and **** to indicate p values less than or equal to 0.05, 0.01, 0.001, and 0.0001, respectively. Non-significant results are represented by n.s.
Supplementary Material
Acknowledgements
We thank the Koch Institute Swanson Biotechnology Center’s Flow Cytometry and Microscopy core facilities for their technical support. We thank Dr. Daniel Lingwood for the generously providing the Hemagglutinin-Ferritin nanoparticle antigen. We also thank Dr. Wanessa du Fresne von Hohenesche of Geneva Antibody Facility for providing insights on improving expression efficiency of follicle targeting antibody construct.
Funding:
D.J.I. received financial support from Scripps Consortium for HIV/AIDS Vaccine Development Grant UM1 AI144462, National Institute of Health Grant P01AI048240, Ragon Institute of MGH, MIT, and Harvard, Marble Center for Cancer Nanomedicine, and Howard Hughes Medical Institute. S.N.B. was supported by Koch Institute Support Grant P30-CA14051, National Institute of Environmental Health Sciences Grant P30-ES002109, Marble Center for Cancer Nanomedicine, and Howard Hughes Medical Institute. W.R.S was supported by Scripps Consortium for HIV/AIDS Vaccine Development Grant UM1 AI144462, Scripps Center for HIV/AIDS Vaccine Immunology and Immunogen Discovery, UM1 Al100663, Bill and Melinda Gates Foundation Collaboration for AIDS Vaccine Discovery (NAC INV-007522 and INV-008813), and IAVI Neutralizing Antibody Center at TSRI. A.P.S. was supported by NIH Molecular Biophysics Training Grant and National Science Foundation Graduate Research Fellowship. J.D.K. was supported by Ludwig Center at MIT’s Koch Institute Fellowship. D.S.K. received financial support from Ragon Institute of MGH, MIT, and Harvard. A.A. was supported by National Institute of Health Training Grant 5T32AI007386.
Competing Interests:
S.N.B. reports compensation for cofounding, consulting for, and/or board membership in Glympse Bio, Satellite Bio, CEND Therapeutics, Catalio Capital, Intergalactic Therapeutics, Port Therapeutics, Vertex Pharmaceuticals, and Moderna, and receives sponsored research funding from Johnson & Johnson, Revitope, and Owlstone.W.R.S. is an inventor on patents filed by Scripps and IAVI on the eOD-GT8 60mer, eOD-GT8 monomer, and BG505 MD39 trimer immunogens. N.H. holds equity in BioNtech and consults for Related Sciences/Danger Bio. D.J.I and A.A. are inventors on a patent application related to CD35-targeted immunogens.
Footnotes
Data and materials availability:
All data is available in the main text, the supplementary materials or deposited at Zenodo (52) and Dryad Repository (56).
References and Notes
- 1.Pollard AJ, Bijker EM, Publisher Correction: A guide to vaccinology: from basic principles to new developments. Nat Rev Immunol 21, 129 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shlomchik MJ, Luo W, Weisel F, Linking signaling and selection in the germinal center. Immunol Rev 288, 49–63 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Victora GD, Nussenzweig MC, Germinal centers. Annu Rev Immunol 30, 429–457 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Abbott RK et al. , Precursor Frequency and Affinity Determine B Cell Competitive Fitness in Germinal Centers, Tested with Germline-Targeting HIV Vaccine Immunogens. Immunity 48, 133–146 e136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bannard O, Cyster JG, Germinal centers: programmed for affinity maturation and antibody diversification. Curr Opin Immunol 45, 21–30 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Dosenovic P et al. , Anti-HIV-1 B cell responses are dependent on B cell precursor frequency and antigen-binding affinity. Proc Natl Acad Sci U S A 115, 4743–4748 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuraoka M et al. , Complex Antigens Drive Permissive Clonal Selection in Germinal Centers. Immunity 44, 542–552 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McNamara HA et al. , Antibody Feedback Limits the Expansion of B Cell Responses to Malaria Vaccination but Drives Diversification of the Humoral Response. Cell Host Microbe 28, 572–585 e577 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Cirelli KM, Crotty S, Germinal center enhancement by extended antigen availability. Curr Opin Immunol 47, 64–69 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tam HH et al. , Sustained antigen availability during germinal center initiation enhances antibody responses to vaccination. Proc Natl Acad Sci U S A 113, E6639–E6648 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham BS, Gilman MSA, McLellan JS, Structure-Based Vaccine Antigen Design. Annu Rev Med 70, 91–104 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Catron DM, Pape KA, Fife BT, van Rooijen N, Jenkins MK, A protease-dependent mechanism for initiating T-dependent B cell responses to large particulate antigens. J Immunol 184, 3609–3617 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tew JG, Mandel TE, Burgess AW, Retention of intact HSA for prolonged periods in the popliteal lymph nodes of specifically immunized mice. Cell Immunol 45, 207–212 (1979). [DOI] [PubMed] [Google Scholar]
- 14.Smith BA et al. , Persistence of infectious HIV on follicular dendritic cells. J Immunol 166, 690–696 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Heesters BA et al. , Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity 38, 1164–1175 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roszik J, Szollosi J, Vereb G, AccPbFRET: an ImageJ plugin for semi-automatic, fully corrected analysis of acceptor photobleaching FRET images. BMC Bioinformatics 9, 346 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dosenovic P et al. , Immunization for HIV-1 Broadly Neutralizing Antibodies in Human Ig Knockin Mice. Cell 161, 1505–1515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jardine J et al. , Rational HIV immunogen design to target specific germline B cell receptors. Science 340, 711–716 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jardine JG et al. , HIV-1 broadly neutralizing antibody precursor B cells revealed by germline-targeting immunogen. Science 351, 1458–1463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jardine JG et al. , HIV-1 VACCINES. Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science 349, 156–161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sok D et al. , Priming HIV-1 broadly neutralizing antibody precursors in human Ig loci transgenic mice. Science 353, 1557–1560 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tokatlian T et al. , Innate immune recognition of glycans targets HIV nanoparticle immunogens to germinal centers. Science 363, 649–654 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Houser KV et al. , Safety and immunogenicity of a ferritin nanoparticle H2 influenza vaccine in healthy adults: a phase 1 trial. Nat Med 28, 383–391 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Read BJ et al. , Mannose-binding lectin and complement mediate follicular localization and enhanced immunogenicity of diverse protein nanoparticle immunogens. Cell Rep 38, 110217 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mason DY, Jones M, Goodnow CC, Development and follicular localization of tolerant B lymphocytes in lysozyme/anti-lysozyme IgM/IgD transgenic mice. Int Immunol 4, 163–175 (1992). [DOI] [PubMed] [Google Scholar]
- 26.Tang L et al. , Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol 36, 707–716 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt MM, Thurber GM, Wittrup KD, Kinetics of anti-carcinoembryonic antigen antibody internalization: effects of affinity, bivalency, and stability. Cancer Immunol Immunother 57, 1879–1890 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kapoor VN et al. , Gremlin 1(+) fibroblastic niche maintains dendritic cell homeostasis in lymphoid tissues. Nat Immunol 22, 571–585 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Rodda LB et al. , Single-Cell RNA Sequencing of Lymph Node Stromal Cells Reveals Niche-Associated Heterogeneity. Immunity 48, 1014–1028 e1016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soleimany AP et al. , Activatable Zymography Probes Enable In Situ Localization of Protease Dysregulation in Cancer. Cancer Res 81, 213–224 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jakos T, Pislar A, Jewett A, Kos J, Cysteine Cathepsins in Tumor-Associated Immune Cells. Front Immunol 10, 2037 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yadati T, Houben T, Bitorina A, Shiri-Sverdlov R, The Ins and Outs of Cathepsins: Physiological Function and Role in Disease Management. Cells 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soleimany AP et al. , Multiscale profiling of enzyme activity in cancer. Nat Commun 13, 5745 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fisher KE et al. , Engineering autoactivating forms of matrix metalloproteinase-9 and expression of the active enzyme in cultured cells and transgenic mouse brain. Biochemistry 41, 8289–8297 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Steichen JM et al. , A generalized HIV vaccine design strategy for priming of broadly neutralizing antibody responses. Science 366, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin JT et al. , Combined PET and whole-tissue imaging of lymphatic-targeting vaccines in non-human primates. Biomaterials 275, 120868 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cirelli KM et al. , Slow Delivery Immunization Enhances HIV Neutralizing Antibody and Germinal Center Responses via Modulation of Immunodominance. Cell 180, 206 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ng’uni T, Chasara C, Ndhlovu ZM, Major Scientific Hurdles in HIV Vaccine Development: Historical Perspective and Future Directions. Front Immunol 11, 590780 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kastenmuller W, Torabi-Parizi P, Subramanian N, Lammermann T, Germain RN, A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell 150, 1235–1248 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dabo AJ, Cummins N, Eden E, Geraghty P, Matrix Metalloproteinase 9 Exerts Antiviral Activity against Respiratory Syncytial Virus. PLoS One 10, e0135970 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernandes JP et al. , Breast Tumor-Associated Metalloproteases Restrict Reovirus Oncolysis by Cleaving the sigma1 Cell Attachment Protein and Can Be Overcome by Mutation of sigma1. J Virol 93, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hansen KC, D’Alessandro A, Clement CC, Santambrogio L, Lymph formation, composition and circulation: a proteomics perspective. Int Immunol 27, 219–227 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Clement CC et al. , Protein expression profiles of human lymph and plasma mapped by 2D-DIGE and 1D SDS-PAGE coupled with nanoLC-ESI-MS/MS bottom-up proteomics. J Proteomics 78, 172–187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tamburini BA, Burchill MA, Kedl RM, Antigen capture and archiving by lymphatic endothelial cells following vaccination or viral infection. Nat Commun 5, 3989 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bachmann MF, Odermatt B, Hengartner H, Zinkernagel RM, Induction of long-lived germinal centers associated with persisting antigen after viral infection. J Exp Med 183, 2259–2269 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cyster JG, B cell follicles and antigen encounters of the third kind. Nat Immunol 11, 989–996 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Wang XY, Wang B, Wen YM, From therapeutic antibodies to immune complex vaccines. NPJ Vaccines 4, 2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT, C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science 271, 348–350 (1996). [DOI] [PubMed] [Google Scholar]
- 49.Pardi N et al. , Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release 217, 345–351 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steichen JM et al. , HIV Vaccine Design to Target Germline Precursors of Glycan-Dependent Broadly Neutralizing Antibodies. Immunity 45, 483–496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banan B et al. , Intestinal Lymph Collection via Cannulation of the Mesenteric Lymphatic Duct in Mice. J Surg Res 260, 399–408 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aung Aereas. FRET Efficiency Calculation for acceptor photobleaching approach. Zenodo: 10.5281/zenodo.7327790. [DOI] [Google Scholar]
- 53.Moyer TJ et al. , Author Correction: A Engineered immunogen binding to alum adjuvant enhances humoral immunity. Nat Med 26, 804 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lima WC et al. , The ABCD database: a repository for chemically defined antibodies. Nucleic Acids Res 48, D261–D264 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dunbar J et al. , SAbPred: a structure-based antibody prediction server. Nucleic Acids Res 44, W474–478 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aung Aereas et al. (2023), Data for: Low protease activity in B cell follicles promotes retention of intact antigens after immunization. Dryad, Dataset: 10.5061/dryad.51c59zwcx. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available in the main text, the supplementary materials or deposited at Zenodo (52) and Dryad Repository (56).