

Abstract
Cellular plasticity lies at the core of cancer progression, metastasis, and resistance to treatment. Stemness and epithelial-mesenchymal plasticity in cancer are concepts that represent a cancer cell’s ability to coopt and adapt normal developmental programs to promote survival and expansion. The cancer stem cell model states that a small subset of cancer cells with stem cell-like properties are responsible for driving tumorigenesis and metastasis while remaining especially resistant to common chemotherapeutic drugs. Epithelial-mesenchymal plasticity describes a cancer cell’s ability to transition between epithelial and mesenchymal phenotypes which drives invasion and metastasis. Recent research supports the existence of stable epithelial/mesenchymal hybrid phenotypes which represent highly plastic states with cancer stem cell characteristics. The cell adhesion molecule CD44 is a widely accepted marker for cancer stem cells, and it lies at a functional intersection between signaling networks regulating both stemness and epithelial-mesenchymal plasticity. CD44 expression is complex, with alternative splicing producing many isoforms. Interestingly, not only does the pattern of isoform expression change during transitions between epithelial and mesenchymal phenotypes in cancer, but these isoforms have distinct effects on cell behavior including the promotion of metastasis and stemness. The role of CD44 both downstream and upstream of signaling pathways regulating epithelial-mesenchymal plasticity and stemness make this protein a valuable target for further research and therapeutic intervention.
Keywords: Epithelial-mesenchymal plasticity, Metastasis, CD44 Isoforms, Cancer Stem Cells
Introduction
Cancer is an extremely complex collection of diseases. Although it has been recognized and characterized since ancient times, the details of its origin and progression are still not completely understood. One major challenge in treating cancer concerns the plasticity underlying cancer progression, metastasis, and resistance to treatment. Cell stemness and epithelial-mesenchymal plasticity (EMP) are phenotypic and transcriptional constructs from normal cell biology that have been adapted by cancer cells to improve proliferation and survival. Both are complex expressions of cellular plasticity driven by multiple intersecting signaling pathways. The cell adhesion protein CD44 plays a unique role in many cancers in that it is both regulated by these signaling pathways and plays an active role in influencing both EMP and stem-like properties.
The cancer stem cell model-one of the most widely accepted models of cancer development— hypothesizes the existence of a small subset of cells within the tumor called tumor initiating cells, or cancer stem cells (CSCs); these are analogous to normal stem cells in that they undergo self-renewal as well as asymmetric division to produce more differentiated progeny [1]. The theoretical framework around the stem cell analogy in cancer was proposed by Reya et al. in 2001, and CSCs were soon thereafter discovered and characterized in a variety of solid tumors including colorectal cancer and pancreatic cancer [1–3]. Because these cells are thought to drive tumor growth and lead to metastasis, they are attractive targets for treatment; however, CSC cells are difficult to target due to their plasticity and heterogeneity, the latter of which has been revealed through single-cell sequencing [4–6]. Adding yet another layer of complication, non-CSCs have been shown to spontaneously convert into CSCs [7]. Understanding the mechanisms underlying this plasticity and the processes that drive the development of cancer cells with stem-like characteristics is vital to the effective treatment of this disease.
Epithelial-mesenchymal transition (EMT) is an important process during both development and tissue remodeling, and it exemplifies a type of normal phenotypic plasticity that is hijacked during oncogenesis [8]. Recently, the transition from an epithelial to mesenchymal phenotype and vice versa (mesenchymal-epithelial transition; MET) is being viewed as less of a binary transformation between two discrete states and more of gradual transition involving a spectrum of hybrid epithelial/mesenchymal (hybrid E/M) states in a process known as epithelial-mesenchymal plasticity (EMP) [9, 10].
Research has begun to show that EMP and cancer stemness share more than just a superficial relationship as manifestations of cellular plasticity. In fact, they share many of the same transcriptional programs and may be intimately related both genetically and phenotypically [11]. Studies have shown that the induction of EMT leads to an increase in stem cell markers and traits [11–13]. EMP and hybrid E/M states may be key to understanding the plasticity important for cancer development, metastasis, and cancer stemness [14, 15].
The expression of the cell adhesion protein CD44 serves as a useful illustration of the intersection between EMP and stemness. It is a canonical marker for CSCs in multiple cancer types, and its expression is specifically affected during transitions between epithelial and mesenchymal phenotypes, leading to changes in cell behavior [16, 17]. CD44 expression is complex, with one gene producing multiple splice variants [17]. It is more accurate and more constructive to think about CD44 as a collection of related proteins with overlapping but distinct physiological impacts. Patterns in CD44 isoform expression change according to shifts along the epithelial-mesenchymal axis, and individual isoforms have different phenotypic consequences. The roles of CD44 in cell adhesion and as an interactor in several signaling pathways important for migration and stemness make it an ideal focal point for studying the relationship between EMP and the CSC phenotype.
Studies on EMP and stemness using controlled in vitro models have often failed to translate to in vivo models or into effective treatments because they do not capture the level of plasticity and heterogeneity in tumors. As we increase our ability to resolve characteristics on a single-cell level, our understanding of EMP and stemness has become more nuanced and complex. Studies of CD44 offer intriguing evidence of isoform shifts related to EMP and stemness, but the techniques used have lacked the resolution capable of capturing the full spectrum of isoform expression. Given this evidence, a deeper and more detailed understanding of CD44 splice variant expression may offer new mechanistic insights and treatment options.
EMT, EMP, and Metastasis
Research has produced competing theories and contradictory evidence regarding the relevance of EMT to cancer progression and metastasis [18]. While many studies have pointed to EMT as a potent driver of metastasis, others have shown that a reciprocal process called mesenchymal-epithelial transition (MET) is necessary for metastasis [19–23]. The role of EMT in metastasis remains ambiguous because the mechanistic details have been difficult to study under physiologically relevant conditions. Much of the research has been conducted on cancer cell lines or manipulated cells, and it is less clear as to whether or when EMT occurs in relation to metastasis under normal conditions in vivo [9, 18]. EMT is a transient process occurring in relatively few cells at any given time making it difficult to detect in vivo or in patient samples. Also, since EMT is a complex process driven by multiple signaling pathways, studies focusing on specific markers or transcription factors may overlook other relevant factors. For example, while one study focusing on EMT transcription factors (EMT-TFs) Snail1 and Twist in the KPC mouse model concluded that EMT was dispensable for metastasis, a follow-up study by another group found that the knockdown of a different EMT-TF Zeb1 strongly inhibited metastasis [24, 25].
Addressing this contradictory evidence has led to a shift in our understanding of EMT and MET from a transformation between two discrete states to a more nuanced and context-dependent transitional process referred to as EMP involving multiple hybrid states and signaling pathways [10, 26]. The concept of EMP captures the cell’s ability to move freely back and forth along a theoretical epithelial-mesenchymal axis, with EMT and MET describing directional shifts along this spectrum (Figure 1) [26, 27]. Hybrid epithelial/mesenchymal cells display aspects of both epithelial and mesenchymal cells including the simultaneous expression of well-established markers for both end states. These hybrid cells have been detected in multiple carcinomas and can be both transitory and stable in nature [14, 22, 28–32]. Recent studies using single cell RNA sequencing have provided evidence for a range of transitory, hybrid E/M phenotypes which are context-dependent, driven by multiple signaling pathways, and have different metastatic and tumorigenic capacities [29, 30, 33]. At this level of detail, EMT or MET are viewed as a multi-step combination of discrete transcriptional events with a large variation in differentially expressed genes and activated transcription factor networks. In one study of 12 distinct EMT time course experiments using different cancer cell lines and inducers of EMT, only 22% of response genes were shared between any two conditions [33]. In other words, there is not a single master transcriptional program responsible for driving EMT even within individual cancers, but several signaling and transcriptional networks that determine the E/M phenotype. Pastushenko et al. used spontaneous mouse models of skin and mammary gland cancers to show that hybrid E/M states exist in vivo; furthermore, it was demonstrated that cells displaying hybrid E/M states are heterogenous, with differences in cellular plasticity, invasiveness, and metastatic potential [34, 35].
Fig. 1. Epithelial-Mesenchymal Plasticity.
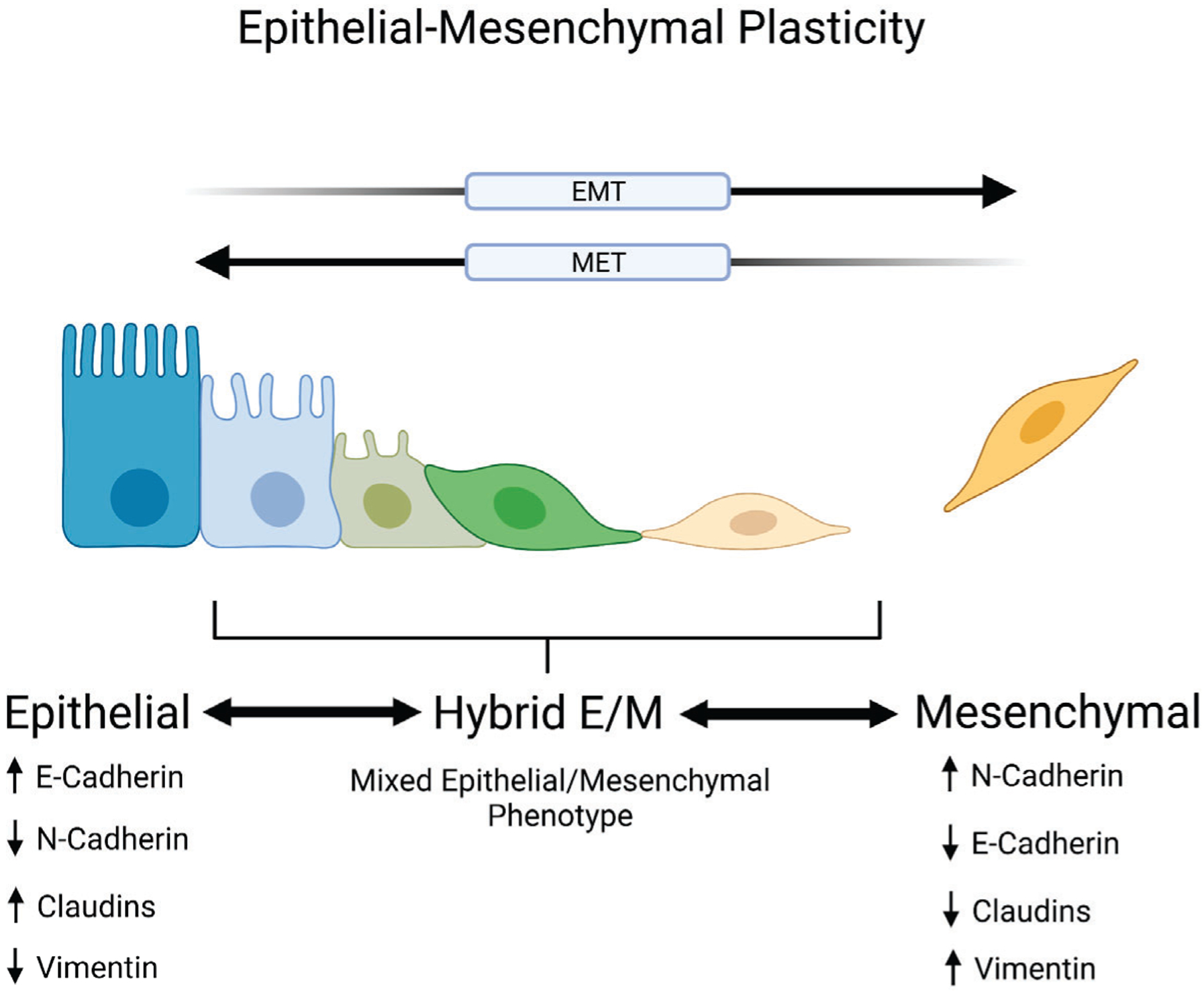
Epithelial-mesenchymal plasticity describes a cell’s ability to transition between epithelial and mesenchymal phenotypes. Epithelial cells are traditionally marked by the expression of junctional proteins such as E-cadherin and claudins while mesenchymal cells are characterized by the expression of N-cadherin and vimentin along with decreased levels of E-cadherin. The transition between these phenotypic end points includes intermediate stages referred to as hybrid epithelial/mesenchymal (E/M) states which display mixed epithelial and mesenchymal markers and characteristics. These hybrid E/M states are associated with increased metastatic potential and cancer stem cell properties. Created with BioRender.com
Importantly, hybrid E/M states have also shown to be more tumorigenic than cells that are either fully epithelial or mesenchymal, with evidence suggesting that the plasticity seen in hybrid E/M states promotes increased metastatic potential and stemness characteristics [28, 34, 36]. In fact, a transcriptional profile developed to detect a high level of epithelial-mesenchymal plasticity was associated with poor prognosis in cancers of the prostate, breast, and lung, demonstrating that this concept is also clinically relevant across different cancer types [37].
EMP in Metastasis
Studies have shown that while EMT allows for invasion, intravasation, and the dissemination of cancer cells throughout the body, fully mesenchymal cells are less able to establish distant metastases [38–40]. In these studies, a reversion to an epithelial phenotype through MET was necessary for colonization and proliferation. In other words, cells must acquire mesenchymal characteristics to detach and migrate, but must then reacquire epithelial traits in order to establish macrometastatic lesions. Multiple experimental models have provided evidence that this dynamic occurs during metastasis [19–22, 38, 39, 41–43]. For example, in prostate cancer, cells exhibiting mesenchymal properties were more invasive and capable of escaping the primary tumor, while cells with more epithelial characteristics displayed increased tumor-initiating capacity and established more distant metastases [42]. Beerling et al. demonstrated evidence of cells undergoing EMT followed by MET during metastasis in vivo using a spontaneous ductal mammary carcinoma mouse model [41]. Importantly, these cells were not artificially induced to initiate EMT. This study showed that a small population of epithelial cancer cells within a tumor undergo spontaneous EMT to become more invasive and migratory, but that once established at a distant metastatic site, they revert to an epithelial state within one or two cell divisions. Another study by Li et al. used a novel variation on Cre-loxP lineage tracing to permanently label cells from a specific lineage (in this case, c-Kit+ mammary luminal epithelial cells) in a spontaneous mammary carcinoma mouse model which have undergone EMT [43]. In this study, a majority of lung metastases had been labeled by the activation of N-cadherin. Interestingly, N-cadherin itself was no longer expressed in the lung metastases, providing further evidence that EMT is a transient process, and that MET is necessary for colonization. In addition, knocking out N-cadherin in this model significantly reduced metastasis to the lungs, providing evidence that the transitory activation of N-cadherin in these cells during EMT is necessary for metastasis. These studies are significant because they provide direct evidence for epithelial-mesenchymal plasticity in metastasis while at the same time demonstrating that artificial EMT induction may fail to recapitulate the transient and dynamic nature of the process since the overexpression of EMT-TFs may inhibit plasticity.
The key to understanding EMP and metastatic potential lies within the plasticity inherent to hybrid E/M states as well as extracellular factors present in the primary tumor or metastatic niche [9]. Studies have shown that circulating tumor cells as well as cells from metastasizing tumors exhibit mixed epithelial and mesenchymal traits and that these are dynamic, reversible states [34, 44]. Evidence from in silico, in vitro, and in vivo studies support the idea that hybrid E/M phenotypes have much higher metastatic potential than cells at either end of the EMT spectrum [45].
EMP and Stemness
Similar to metastatic potential, the role of EMT in stemness is still debated [9]. Multiple studies have shown a connection between EMT and the acquisition of stem-like characteristics [11, 12, 46–48]. In these studies, the induction of EMT using EMT-TFs such as Snail, Twist, or Zeb1 increase the expression of CSC markers, sphere-forming ability, and tumorigenicity. However, other studies have challenged this notion by showing that a fully mesenchymal phenotype is associated with a decrease in stemness and that knocking down EMT-TFs can increase stemness in these cases [38, 42].
Models built around epithelial-mesenchymal plasticity attempt to unify these seemingly conflicting findings [9, 10]. Rather than linking either EMT or MET with the acquisition of stem cell characteristics, studies have shown that hybrid E/M states represent the highest level of plasticity and promote stemness characteristics [32, 34, 36, 49]. In these studies, cells displaying characteristics of both epithelial and mesenchymal cells exhibit higher levels of stem cell markers and tumor-forming abilities. In basal breast cancer, Kroger et al. showed that tumorigenicity and stemness were associated with a stable subpopulation of cells displaying a mixture of both epithelial and mesenchymal markers [36]. This phenotype was driven by both Snail and canonical Wnt signaling. Interestingly, they also showed that driving these cells towards a fully mesenchymal phenotype through the overexpression of Zeb-1 was associated with the loss of stemness. This supports the idea that stemness is linked to the flexibility inherent to hybrid E/M states and that stemness is lost during full transdifferentiation. Strauss et al. found that a subset of ovarian cancer cells with this hybrid phenotype had a capacity for self-renewal as well as the ability to differentiate into either epithelial or mesenchymal-type cells depending on the external factors such as stress or co-culture with epithelial or mesenchymal cells [32]. Again, this transdifferentiation into either fully epithelial or mesenchymal states led to a loss of stemness. Pastuskenko et al. showed that several subpopulations of distinct hybrid E/M phenotypes with varying levels of tumorigenicity and stemness exist within skin and mammary tumors [34]. In clusters of circulating tumor cells, Quan et al. showed that 60% of CSCs leading collective invasion displayed a hybrid E/M phenotype [49].
Another theory proposes that heterogenous CSC populations are generated through epithelial-mesenchymal plasticity, producing populations of cycling and non-cycling CSCs [50, 51]. In this model, cycling CSCs display a more epithelial phenotype, undergo self-renew, are more proliferative, and differentiate into diverse cancer cells to form the bulk tumor. Non-cycling CSCs display a more mesenchymal phenotype, are more invasive and metastatic, have higher levels of macroautophagy, and are more resistant to therapeutic intervention [50]. In this way, these two cell types fulfill different roles in tumor development, and epithelial-mesenchymal plasticity allows for flexibility in responding to different environmental challenges such as nutrient stress or treatment-induced toxicity.
Targeting EMP
Given the early research connecting EMT to metastasis and resistance to therapy, there has been extensive translational research into targeting EMT [52]. However, given the plasticity of cancer cells outlined by the EMP model, therapies aimed at reversing EMT can have unwanted consequences [18]. For example, the epithelization of the tumor at a metastatic site can promote proliferation and colonization. On the other hand, targeting metastatic colonization by inhibiting MET may lead to a more invasive, mesenchymal phenotype. In addition, many common treatment regimens can induce EMP, often leading to an enrichment in EMT markers. For example, a combination of docetaxel and hormonal therapy can induce EMT in prostate cancer and lead to relapse [53]. This phenomenon was also seen in breast cancer after chemotherapy [54, 55] In these cases, chemotherapies which target highly-proliferative epithelial-like cells induce a shift towards a more resistant, mesenchymal phenotype. In effect, targeting either the epithelial or mesenchymal end state can induce EMP, leading to a more plastic, stem-like state.
Directly targeting cells which exist in hybrid E/M states would arrest cells that are the most resistant to conventional treatments and have the highest metastatic potential; however, there is no consistent marker for this hybrid state that could serve as a therapeutic target. Several studies have begun to characterize the unique and dynamic nature of CD44 expression during transitions between epithelial and mesenchymal states. The complex regulation of CD44 isoforms is driven by the same transcriptional and signaling networks that control EMP. Identifying isoforms which are specific or enriched in this hybrid state could serve as an attractive target for therapy.
CD44 in EMP and Stemness
CD44 is a cell surface adhesion molecule that was first discovered by Gallatin et al. as a lymphocyte homing receptor in 1983 [56]. It is a non-kinase, class-I transmembrane glycoprotein that is expressed in various isoforms across most vertebrate cell types. It is highly expressed in embryonic stem cells and is involved in normal developmental processes such as organogenesis. In adult tissues, it is involved in hematopoiesis, various immune functions such as lymphocytic homing and activation, and mediates interactions between cells and their microenvironment [57, 58]. The physiological role of CD44 is carried out through multiple mechanisms: interaction with components of the extracellular matrix, functioning as a binding platform for growth factors and matrix metalloproteinases, acting as a coreceptor for receptor tyrosine kinases (RTKs), and through interaction with elements of the cytoskeleton [57]. In other words, CD44 acts as a connection between the extracellular matrix and the cytoskeleton as well as an organizing element for signaling platforms on the plasma membrane. Through these mechanisms, it is involved in cell growth, survival, differentiation, cell-cell interactions, migration, and adhesion (Table 1) [57, 59]. In the context of cancer, CD44 promotes EMT, motility, resistance to apoptosis, and drug resistance. These characteristics make it not only a marker for stemness, but a functional contributor to the stem cell phenotype [59]. The aberrant expression of CD44 has been implicated in the progression of several cancers including of the breast, colon, and pancreas, and it is broadly recognized as a marker for the CSC phenotype [2, 3, 59–61].
Table 1.
Known Physiological Roles of CD44 Isoforms
| CD44 Isoform | Physiological Role | Role in Cancer | References |
|---|---|---|---|
| CD44s | Regulates cell growth, survival, differentiation, cell-cell interactions, migration, and adhesion | Overexpression promotes metastatic potential and stemness. | [17, 57, 59] |
| CD44v3 | Binds heparin-binding growth factors which can impact receptor tyrosine kinase signaling | Promotes tumor growth, migration, and metastasis in head and neck cancer. | [67, 78, 84] |
| CD44v6 | Binds and sequesters hepatocyte growth factor to activate the receptor tyrosine kinase c-MET. | Promotes metastasis in pancreatic adenocarcinoma, colorectal cancer, and breast cancer. | [80–83] |
| CD44v8-10 | Interacts with xCT glutamate-cysteine transporter to regulate glutathione levels and redox status | Enhances metastatic colonization in lung cancer | [40] |
Several recent studies as detailed in the following section and summarized in Table 2 have demonstrated the role of CD44 in EMP, metastasis, and cell stemness across multiple cancer types. There are consistent and predictable patterns of change in the expression of CD44 isoforms during shifts between epithelial and mesenchymal states which themselves affect cellular phenotype (including stemness). It is important to note that while most studies focused solely on EMT and the early stages of metastasis, those that looked at metastatic colonization identified a reversion to CD44 variant isoforms in line with a transition to a more epithelial phenotype [40, 62]. Together, these results suggest CD44s is associated with EMT and early events in metastasis (i.e., invasion and migration); however, cells must undergo a reciprocal MET involving an isoform switch to CD44v in order to establish new metastatic sites and proliferate. This mirrors patterns seen in studies of EMP in metastasis.
Table 2.
CD44 Isoform Switching in Cancer
| Cancer Type | Isoform Switching | Role of CD44 | Reference |
|---|---|---|---|
| Ovarian cancer | CD44v to CD44s in EMT |
|
[89, 95] |
| Lung cancer | CD44v to CD44s in EMT |
|
[96] |
| Prostate cancer | CD44v to CD44s in EMT |
|
[94, 97] |
| Hepatocellular carcinoma | CD44v to CD44s in EMT |
|
[99, 100, 139, 140] |
| Gallbladder cancer | CD44v9 to CD44s in EMT |
|
[102] |
| Colon cancer | CD44v9 to CD44s in EMT |
|
[104] |
| Pancreatic cancer | CD44v to CD44s in EMT CD44s to CD44v in MET |
|
[62, 71] |
| Breast cancer | CD44v to CD44s in EMT CD44s to CD44v in MET |
|
[40, 71] |
CD44 Gene Structure and Isoform Expression
The expression of CD44 is complex, with one highly conserved gene capable of being diversely expressed as multiple splice variants (Figure 2a) [63]. This complexity has resulted in different nomenclature and exon numbering systems for CD44 across the literature and genomic databases [64]. The exons are commonly numbered based on the homologous CD44 gene in mice which encodes 20 exons, including ten standard exons (exons 1–5 and exons 16–20) and 10 variable exons (exons v1–v10). In the human CD44 gene, the v1 exon (exon 6) is present, but contains a mutation resulting in a stop codon after the 17th amino acid and is not found in translated protein [65]. As a result, only 9 variant exons are encoded in humans, and the largest CD44 isoform contains variant exons v2–10. In addition, although it is often not addressed in the literature, alternative splicing also occurs in exons 19 and 20 due to a premature stop codon occurring in exon 19, resulting in two versions of the intracellular c-terminal region. Most isoforms include exon 20 with exon 19 spliced out, while a truncated version of the intracellular domain containing only exon 19 is found in an isoform called CD44st [63, 64]. As a result, there is no isoform that contains both exons 19 and 20. Exons 7–15 (v2–v10) may be included into an extracellular juxtamembrane stem region of the protein through alternative splicing to create different translated variants. The “standard” version of CD44 (CD44s) contains no variant exons in the stem region and is expressed across most cell types, while larger variant isoforms (collectively referred to as CD44v) are expressed primarily in limited epithelial cells such as keratinocytes as well as in many cancer cells [66].
Fig. 2. Structures of the CD44 gene and protein.
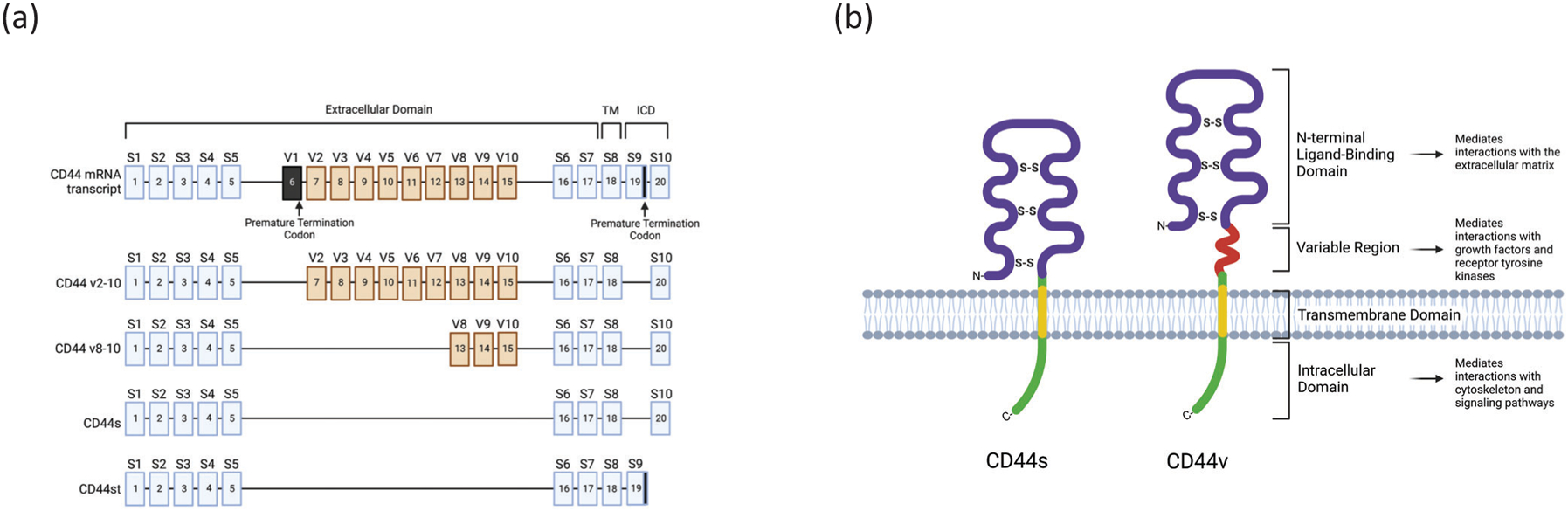
(a) The full structure of the human CD44 gene. Exons 1–5 and exons 16–18 are constitutively expressed while exons v2–v10 and exons 19–20 undergo alternative splicing. Exons 1–17 encode the extracellular domain, exon 18 encodes the transmembrane domain (TM), and exons 19–20 are alternatively spliced to form the intracellular domain (ICD). Exon v1 is present in the human gene, but a mutation resulting in a premature termination codon prevents this exon from being expressed. Exon 19 contains a premature termination codon which blocks the downstream translation of exon 20 and is spliced out in most isoforms but is retained in CD44st. (b) A schematic of the CD44s isoform and a generalized CD44v isoform which includes additional amino acid sequences in the stem region. Created with BioRender.com
Alternative splicing results in hundreds of different possible exon combinations and isoforms being transcribed from a single gene, and each isoform may have specific physiological functions [67]. The control of alternative splicing is multifaceted, and it involves a collection of interacting elements within the spliceosome complex, each of which is subject to regulation [68]. In CD44, there are many factors that control the alternative splicing of specific variant exons [69]. For example, epithelial splicing regulatory proteins (ESRPs) play a role in the regulation of variable exons v6–v10 and can be regulated by EMT-related transcription factors such as Zeb1 and Snail [70–72]. ESRP1/2 bind to cis-regulatory elements in mRNA and coordinate with other RNA-binding proteins and splicing elements in a combinatorial manner to induce changes in exon inclusion [70]. The position of the ESRP binding sites relative to an exon determine whether the exon is included or skipped, and CD44 pre-mRNA contains several ESRP1 binding sites which mediate exon inclusion [72, 73]. The serine/arginine repetitive matrix protein SRm160 can regulate the inclusion of exons v2 to v9 and can be regulated in a Ras-dependent manner [74]. Transformer-2 protein homolog beta, Tra2B1, specifically regulates the inclusion of v4 and v5 [75]. The serine-arginine RNA-binding protein SC35 promotes v6 inclusion by recognizing SC35 response elements in the v6 exon and flanking introns [76]. In addition to these RNA-binding proteins, epigenetic factors such as histone modifications can also influence exon inclusion [69]. In summary, CD44 variant expression is driven by the sum of multiple influences converging on the spliceosome complex. As detailed elsewhere in this review, alternative splicing plays a major role in epithelial-mesenchymal plasticity in cancer.
CD44 Protein Structure
The CD44 protein is composed of four domains: a globular amino-terminal domain (exons 1–5), an extracellular stem region (exons 16 and 17 plus any variant exons), a transmembrane region (exon 18), and a c-terminal cytoplasmic tail (exon 19 or 20) (Figure 2b) [57]. In the standard CD44s isoform, the amino-terminal domain is linked to the transmembrane segment by a short 46-amino acid stem structure [57]. In alternatively-spliced variants, the stem structure may contain up to an additional 381 amino acids [77].
The amino-terminal domain is responsible for interactions with elements of the extracellular matrix. While hyaluronic acid (HA) is the most specific ligand for CD44, it can also bind and interact with other elements of the extracellular matrix such as collagen, chondroitin, fibronectin, and osteopontin [17]. Conformational changes induced by ligand binding lead to changes in interactions with the cytoskeleton and other signaling molecules.
The variable region of the stem domain confers a wide range of functional variation on CD44 as summarized in Table 1. For example, the v3 exon can be modified with the glycosaminoglycan heparin sulfate which allows it to bind heparin-binding growth factors such as heparin-binding epidermal growth factor, vascular endothelial growth factor, hepatocyte growth factor (HGF), basic fibroblast growth factor, keratinocyte growth factor, and amphiregulin which can in turn impact RTK signaling [67, 78]. The v8–10 region regulates the redox status of cancer cells by interacting and stabilizing the xCT glutamate-cysteine transporter resulting in increased levels of glutathione [79]. The v6 exon contains a peptide motif which binds and sequesters HGF. This complex then activates the RTK c-MET in a two-step process. First, the v6/HGF complex is required for c-MET activation and autophosphorylation. Then, the CD44 cytoplasmic domain along with the ERM (ezrin, radixin and moesin) protein ezrin is necessary for signal transduction to mitogen-activated protein kinase kinase (MEK) and extracellular signal-regulated kinase (ERK) [80]. The expression of specific variant exons of CD44 is associated with cancer progression and metastasis in multiple types of cancer [17]. CD44v6 is known to promote metastasis in pancreatic adenocarcinoma, colorectal cancer, and breast cancer [81–83]. Variable exon 3 is associated with tumor cell growth, migration, and metastasis in head and neck cancer [84]. In lung cancer, CD44v8-10 enhances metastatic colonization [40].
The cytoplasmic tail of CD44 is important for interactions with elements of the cytoskeleton and various signaling pathways. Both ankyrin and ERM proteins—well-characterized cytoskeletal adaptor proteins—have binding motifs within the 70 amino acid cytoplasmic domain of CD44 [85, 86]. ERM proteins bind to positively-charged amino acid residues in the membrane-proximal region of the cytoplasmic domain just below the ankyrin-binding regions and link CD44 to actin filaments in the cytoskeleton [87]. Ligand binding within the CD44 extracellular domain leads to conformational changes in CD44 that promote ERM binding. ERM activation then promotes changes in the cytoskeletal structure leading to alterations in cell morphology and motility. In addition, these proteins enable crosstalk with multiple signaling pathways such as PI3K/Akt and Ras-activated MAPK which are important proliferation and survival pathways relevant to cancer [17].
CD44 also undergoes extensive post-translational modification. It has many potential glycosylation sites including several within the variable exons which can affect functions such as its ability to bind HA or growth factors in the extracellular environment [57, 88]. In fact, due to heavy glycosylation, the apparent molecular weight of CD44s is around 80 kDa in gel electrophoresis despite the fact that its calculated weight is only 39 kDa [64, 77]. These extensive post-transcriptional and post-translational modifications allow this widely expressed protein to perform a variety of functions in response to diverse physiological and pathological situations.
CD44 Isoform Switching in EMP and Stemness
CD44 plays an integral role in the EMT process and operates both upstream and downstream of EMT-related transcriptional programs [17, 62]. Increased expression of CD44 can induce the expression of EMT-related transcription factors such as Zeb1 and Snail [89]. In addition, Snail itself can induce the expression of CD44 [46]. Interestingly, it has been demonstrated in several forms of cancer that during EMT, CD44 undergoes a switch from the expression of the variant forms of the protein (CD44v) which are associated with a more epithelial phenotype to the smaller standard form (CD44s), as illustrated in Figure 3 and summarized in Table 2 [90]. In the context of EMT, multiple studies have implicated ESRP1 as a major regulator CD44 isoform expression, and its activity leads to the inclusion of variant exons into the stem region of the protein [70, 90]. Both ESPR1 and ESPR2 regulate the post-transcriptional processing of a large network of proteins important for maintaining an epithelial phenotype, and their global loss results in EMT [73, 91]. Reinke et al. have shown that the transcription factor Snail drives EMT by repressing the transcription of ESRP1 and altering the splicing of several mRNA species including CD44 [72]. Importantly, ESRP1 overexpression inhibited Snail-induced EMT, and this was rescued by the ectopic expression of CD44s, suggesting that EMT is in fact mediated in some cases through the alternative splicing of CD44. Transforming growth factor β (TGF-β), a key driver of EMT, has also been shown to induce isoform switching to CD44s through the inhibition of ESRPs [92]. In addition, TGF-β can also regulate CD44 isoform switching through an interaction between SMAD3 and the RNA-binding protein poly-(rC) binding protein 1 (PCBP1) [93, 94]. Changes in CD44 isoform expression during transitions between epithelial and mesenchymal phenotypes have been documented across multiple types of cancer.
Fig. 3. Schematic representation of CD44 expression in the context of epithelial-mesenchymal plasticity.
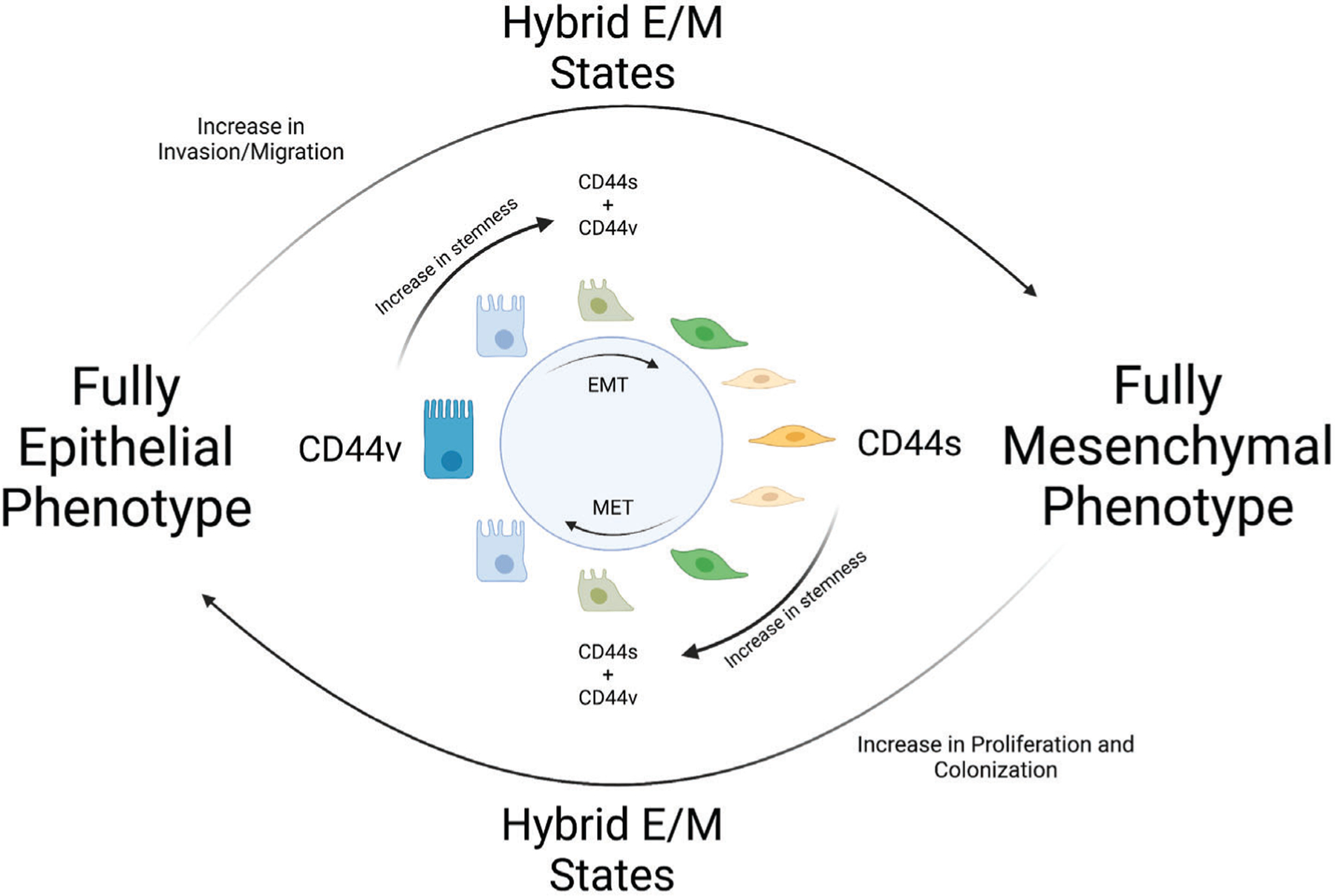
In cancer, CD44v isoforms are associated with a more epithelial phenotype while CD44s is associated with mesenchymal characteristics. Changes in CD44 splicing lead to shifts in isoform expression during EMT and MET. These shifts in isoform expression in turn promote changes in metastatic potential and stemness characteristics. Created with BioRender.com
In ovarian cancer, CD44 is associated with both EMT and stemness, and it regulates EMT transcription factors including Snail and Zeb1 [89]. A follow-up study specifically identified the standard isoform of CD44 as a driver of EMT and stemness [95]. TGF-β treatment led to a downregulation of ESRP1 with a subsequent shift from CD44v to CD44s, and the expression of CD44s was associated with properties of stemness including stem cell marker expression, increased aldehyde dehydrogenase activity, a quiescent metabolic state, and increased efflux ability.
In lung cancer as well, Zeb1 was shown to promote EMT by directly binding to E-box elements of the ESRP1 promotor and repressing its expression [96]. The loss of ESRP1 resulted in a shift from CD44v isoforms to CD44s which led to an increase in cell-surface expression of CD44, increased tumorigenicity, and increased invasive potential. Interestingly, Zeb1 was also able to promote EMT through this mechanism in immortalized human bronchial epithelial cells (HBEC) in addition to transformed cell lines [96].
In a proteomic analysis of prostate CSCs isolated after TGF-β treatment, PCBP-1 was highly downregulated when compared to normal cells [97]. It was subsequently shown that its downregulation was important for the maintenance of the prostate CSC populations. The downregulation of PCBP1 led to a shift from CD44 variants to CD44s, and this was essential for both EMT and the increased expression of stem cell markers as a result of TGF-β treatment [94]. CD44 knockdown not only inhibited EMT after TGF-β treatment, the phenotype was rescued specifically by CD44s expression but not CD44v expression. In addition, CD44s, but not CD44v, promoted proliferation, migration, and invasion both in vitro and in vivo.
In hepatocellular carcinoma (HCC), the standard isoform is also associated with a mesenchymal phenotype. In one study, CD44s correlated with EMT marker expression and increased invasiveness, and EMT induced by TGF-β stimulation increased CD44s [98]. Interestingly, CD44s knockdown inhibited TGF-β-induced EMT, suggesting that CD44s is an essential downstream element in this process. Clinically, high CD44s expression in HCC tumors was correlated with a more mesenchymal phenotype and poor prognosis. Further investigation revealed a connection between CD44s, EMT, and the CSC phenotype [99]. In this study, CD44 expression was high in circulating tumor cells compared to the primary tumor and was associated with EMT. Analysis showed that CD44s is the primary isoform in HCC, and the expression of this isoform contributes to EMT as well as CSC characteristics such as sphere-forming ability and resistance to anoikis. In addition, a study of HCC patient samples revealed that high CD44s expression was significantly associated with an EMT expression profile and was associated with local recurrence as well as intrahepatic dissemination after local ablation therapy [100]. Another study has shown that in c-Met+ HCC, tumor initiation and stemness is regulated through a c-MET/PIK3/Akt/CD44s pathway [101].
In the NOZ gallbladder cancer cell line, cells could be separated into two populations: a CD44-high group and CD44-low group [102]. The CD44-high group was largely CD44s positive and CD44v9 negative, while the CD44-low group displayed opposite expression. The CD44s-positive cells displayed a more mesenchymal phenotype and were more invasive, while CD44v9 cells were more epithelial and more tumorigenic. In patient samples, both CD44s-high and CD44v9-high tumors were associated with poor prognosis, though high expression of CD44s was associated with less-differentiated tumors and distant metastasis. Overall, these data point to different functions of CD44 variants with different clinical implications.
In colon cancer as well, CD44 expression is associated with poor prognosis and EMT. A recent meta-analysis of 48 CRC patient studies showed that total CD44 expression is associated with poor differentiation, metastasis, and poor overall survival [103]. In SW480 cells, increased expression of CD44 resulted in the increased expression of mesenchymal markers, changes in cell morphology, and invasive properties [104]. There have been several studies on CD44 subtypes in CRC, with sometimes conflicting conclusions as to correlations with metastasis and prognosis [105–109]. CD44v6 in particular has been shown to promote metastasis and the CSC phenotype [82]. With regard to EMT, in a study of 14 CRC cell lines and 150 CRC patient tumor samples, CD44 isoform switch from CD44v9 to CD44s was significantly associated with a mesenchymal phenotype [110]. In patient samples from this study, both mesenchymal phenotype and high CD44s to CD44v9 ratio were significantly correlated with poor survival.
In a study of pancreatic and breast cancer, it was shown that CD44s and Zeb1 regulate each other to promote a mesenchymal phenotype and stemness characteristics [71]. Zeb1 represses ESRP1 which results in a shift from the variant forms of CD44 to the standard form. CD44s then promotes the expression of Zeb1, resulting in a sustained positive feedback loop which induces EMT. It should be noted that while Zeb1 expression impacted the relative levels of specific CD44 isoforms, it did not affect the overall expression of CD44. Importantly, this increase in Zeb1 and CD44s expression resulted in increased sphere-forming ability, increased resistance to cytotoxic drugs, and was correlated with an increase in tumor recurrence in clinical samples; these effects were specific to the standard isoform. In another study using breast cancer cells, however, increased expression of CD44 variants was associated with enhanced colonization and metastasis formation in the lungs [40]. Combined, the results of these two breast cancer studies align well with the current model of EMP and metastasis—with CD44s being associated with EMT and invasion while CD44v is associated with an epithelial phenotype and colonization.
In pancreatic adenocarcinoma, both the level and pattern of CD44 isoform expression determine phenotypic and tumorigenic properties [62]. Cells expressing the CD44s isoform had a more mesenchymal phenotype and were more invasive, resistant to gemcitabine, and enriched for stem cell markers and stem cell characteristics such as sphere-forming ability. Interestingly, the in vivo model revealed that 6 weeks after engraftment, tumors reverted to a more epithelial phenotype, with higher expression of E-cadherin and CD44v. This is evidence of a CD44 isoform shift during the MET that is required for rapid proliferation and colonization.
Regulation of CD44 Beyond Splicing in EMP
Many of the same signaling networks that regulate EMP and stemness also regulate CD44 expression itself. For example, NF-kB is known to promote the expression of EMT-TFs such as Twist1, Slug, and Zeb2/Sip1 [111]. CD44 also contains NF-kB binding elements in a regulatory region upstream of its promotor which drives its expression [112, 113]. In addition, the Wnt/B-catenin pathway is a known activator of both EMT and the CSC phenotype, and the Wnt effector TCF4 binds to the promotor of CD44 and activates its expression [114–116].
Reciprocally, CD44 itself interacts with signaling pathways that impact EMP and stemness. Through its interactions with RTKs and ERM proteins, CD44 can affect MAPK, PI3K/Akt, and Wnt signaling [17]. CD44 positively regulates Wnt signaling by physically associating with LRP6 and modulating its membrane localization [117]. A recent study has shown that Fat 1 loss-of-function in squamous cell carcinoma promotes a hybrid E/M state and increased stemness through a CAMK2–CD44–SRC–YAP–ZEB1 axis [35]. In addition, as detailed earlier, specific CD44 variants can impact downstream signaling in different ways. CD44s has been shown to activate the PI3K/Akt pathway which in turn promotes EMT and stemness programs [90, 118, 119]. CD44 isoforms containing the variant exons 3 or 6 can promote EMT and stemness characteristics through its interaction with different growth factors or growth factor receptors [57, 67, 78, 120].
CD44 is unique in that it is regulated on both transcriptional and post-transcriptional levels during transitions between epithelial and mesenchymal phenotypes, and the resulting isoforms have differing effects on cell behavior which can enhance both metastasis and stem cell-like features. While new technologies have begun to discriminate differences in the molecular mechanisms underlying EMP and tumor-initiating ability, they share many important regulatory networks [34, 35, 41]. Jolly et al. have created a mathematical model describing interconnected feedback loops with CD44, Zeb1, ESRP1, and hyaluronic acid synthase 2 which help to drive EMP and maintain hybrid E/M states [121]. This model ascribes different activities to CD44s and CD44v isoforms based on an earlier study by Preca et al. (CD44s promotes Zeb1 expression while CD44v inhibits it), and the fine-tuning of their expression is integral to promoting different metastatic and stemness properties [71, 121]. The overall pattern of CD44 isoform expression fits well into the framework of EMP, and understanding the specific expression patterns of CD44 isoforms in hybrid E/M phenotypes may help to clarify the enhanced metastatic potential and stemness characteristics of these cells. To date, many studies have characterized general changes in total CD44 expression or shifts in overall splicing patterns during EMT or MET, but few have detailed the specific isoforms beyond investigations into particular variant exons such as v3 or v6. More sophisticated techniques must be used to understand the spatiotemporal regulation of CD44 isoform expression in hybrid states.
CD44 as a Therapeutic Target
The role of CD44 and its variants during the EMT process make it a potential candidate for identifying the hybrid E/M state. Moreover, its expression on the membrane and its variant regions in the extracellular domain make it an easily accessible therapeutic target [122, 123]. The potential of CD44 as a target in cancer has long been recognized, and several strategies have been developed including monoclonal antibodies (mAb), pharmacological inhibitors, peptides and aptamers, decoys, and HA oligomers as summarized in Table 3 [124]. Anti-CD44 mAbs in various stages of testing and development include Bivatuzumab and U36 (both specific for CD44v6) as well as pan-CD44 mAbs such as RG7356 and RO5429083 [17, 125–127]. In addition, in vitro studies have shown the effectiveness of specifically blocking CD44-HA interactions in cancer cells using the mAbs IM7 and KM201 [128, 129]. The compound silibinin has been shown to decrease CD44 promotor activity and can act as a competitive antagonist for CD44-HA binding [130, 131]. Peptides and aptamers have also been developed to inhibit the expression of CD44 or its interaction with other proteins and have been used to target specific regions such as the v10 exon in breast cancer [132–134]. Decoys such as soluble CD44 HA-binding domain or HA oligomers have also been used to interfere with CD44 interactions with the extracellular matrix or other proteins in cancer cells [135, 136].
Table 3.
CD44 as a Therapeutic Target
| Therapies | Targeted isoform | Cancers studied | Mode of action | References |
|---|---|---|---|---|
| Monoclonal antibodies | ||||
| Chimeric mAb U36 (cmAb U36) | CD44v6 | Head and neck squamous cell carcinoma | Radiolabeled cmAbU36 showed anti-tumor effect by binding and internalization in tumor cells. | [125, 126] |
| Humanized mAb BIWA 4 (bivatuzumab) | CD44v6 | Head and neck squamous cell carcinoma | Radiolabeled bivatuzumab showed anti-tumor effect by binding and internalization in tumor cells. | [126] |
| Bivatuzumab mertansine | CD44v6 | Head and neck squamous cell carcinoma |
|
[141] |
| RG7356 | Pan-CD44 | Phase I trial in several solid malignancies |
|
[127] |
| RO5429083 | Pan-CD44 | Phase I trial in several solid malignancies |
|
[142] |
| mAbs IM7 | Pan-CD44 | Breast cancer cells | Blocks CD44-HA interaction. Induces CD44 shedding from the cell surface. | [128] |
| LA-chitosan-IM7 | Pan-CD44 | Ovarian cancer | Blocks CD44-HA interaction and CD44-mediated proliferation. Effective in treating cancers in vitro and in vivo while reducing the toxicity of mAbs IM7. | [143] |
| KM201 | Pan-CD44 | Human vascular endothelial cells | Blocks CD44-HA interaction | [129] |
| Natural compound | ||||
| Silibinin | CD44v7-10 | Prostate cancer | Blocks CD44-HA interaction | [130] |
| Silibinin | Cd44v6 | Colon cancer | Attenuates stemness of colon cancer stem cells | [144] |
| Aptamers | ||||
| DNA aptamer | CD44v10 | Breast cancer | Inhibits breast cancer cell migration | [132] |
| RNA aptamers | Pan-CD44 | Breast Cancer | Targets CD44-positive cells, including cancer stem cells, for detection, sorting, enrichment, and drug delivery purposes. | [133] |
Although each of these strategies have shown promise, CD44 isoforms are widely expressed in tissues throughout the body, and a lack of specificity has led to off-target effects. Even treatments designed to target a specific variant exon known to be highly expressed in cancer cells such as v6 can have undesired effects. Clinical trials of Bivatuzumab conjugated to the cytotoxic agent mertansine were halted after off-target binding to keratinocytes led to serious reactions and one fatal outcome [137]. Rather than targeting either pan-CD44 or specific variant exons which can exist in multiple isoforms including those normally transcribed in the body, a more promising strategy would be to target more specific epitopes on isoforms that are found only in cancer cells. Moreover, attacking isoforms which are present on cells exhibiting a hybrid E/M phenotype could specifically target cells which are least sensitive to traditional treatment and most likely to lead to metastasis and relapse.
Limitations in the Study of CD44
Understanding the role of CD44 expression in cancer is complicated by limitations in characterizing the heterogeneous nature of CD44 variant expression. Alternative splicing can produce hundreds of different possible exon combinations in the variable stem region [67]. Commercial antibodies and primers often target regions of the protein that are common to all isoforms, and caution must therefore be exercised when drawing conclusions about the overall expression of CD44 without consideration of the specific variants involved. Immunoblotting with pan-CD44 antibodies allows for a gross distinction between low molecular weight isoforms such as CD44s and higher molecular weight forms that include multiple variant exons; however, changes in molecular weight are much smaller when fewer variant exons are included, and post-translational modifications such as glycosylation can confound conclusions based on small changes in molecular weight alone. In addition, antibodies that are targeted towards specific variant exons do not provide information about additional variant exons that are expressed along with the target. Given that these variable regions have different functional impacts in the cell, it is important to dissect and understand the details of CD44 expression. Studies using RT-PCR and sequencing have begun to give a more nuanced view of CD44 heterogeneity [67, 138]. For example, in a detailed study of CD44 isoform expression in colorectal cancer cell lines, 26 different splice variants were found [67]. Interestingly, this study also found that splicing patterns change during metastasis, specifically in the expression of variant exons 3 and 6, without changes in overall CD44 expression. Studies that are limited to the investigation of overall CD44 expression will fail to detect these subtle shifts.
Conclusion
The connection between CD44 expression and EMP, and its resulting impacts on metastasis and stemness, make CD44 an ideal candidate for study as well as for targeted treatment. As detailed in this review, there is evidence for changes in the expression of CD44 isoforms during cancer progression and metastasis in diverse cancers. Many studies have demonstrated that a shift in CD44 expression from variant forms to the standard isoform is associated with invasion, a more mesenchymal phenotype, and CSC-like characteristics. On the other hand, other studies have shown that variant forms of CD44 are associated with metastatic colonization and stemness characteristics. Although these findings seem contradictory at first, they align well with the more dynamic concept of EMP and hybrid E/M states. Shifts towards CD44s may be associated with EMT and the early stages of metastasis, while a reversion towards variant isoforms may be important for MET, colonization, and proliferation. Many cancer cells display a mixed expression of CD44 isoforms, and more detailed study is needed to elucidate the dynamic of CD44 isoform expression as it relates specifically to plasticity, hybrid E/M states, and stemness.
Implications and Future Directions.
A detailed investigation of the patterns of CD44 isoform expression during cancer development and metastasis is needed to understand the relationship between the expression of specific CD44 variants and important events during carcinogenesis. Much like how single-cell studies have revolutionized our understanding of EMP, a higher-resolution survey of CD44 expression across different cancers and cellular phenotypes may reveal new dimensions to its role in promoting aggressive and resistant disease. Technical challenges remain in fully characterizing CD44 expression at a single-cell level in larger cell populations, but studies using traditional reverse-transcription PCR along with sequencing hint at unique patterns of expression involving dozens of splice variants in cells with different behavioral characteristics. The expression of individual variant exons has been associated with aggressive or metastatic disease in some cancers, but the study of individual exons does not capture the full heterogeneity of expression. CD44 isoform expression is known to change as cancer cells undergo shifts between epithelial and mesenchymal phenotypes during metastasis; however, the details of expression in intermediate hybrid E/M states are unknown. Technologies such as full-length single cell transcriptomics may help to correlate specific isoform expression patterns with particular cellular phenotypes including hybrid E/M cells. Ultimately, this knowledge could lead to new opportunities for treatment.
Importantly, the variable region of CD44 is exposed to the extracellular environment and could easily be targeted with antibody-based therapies. Some isoforms of CD44 are widely expressed in normal cells, but if unique combinations of variant exon splicing exist in hybrid E/M phenotypes, this could lead to targeted therapies aimed specifically at cells exhibiting increased levels of stemness and/or metastatic characteristics associated with highly plastic hybrid epithelial/mesenchymal states while avoiding unwanted off-target effects.
Acknowledgement:
This study was supported by BX002086 (VA merit), CA250383 (NIH/NCI), CA216746 (NIH/NCI) and Nebraska Research Initiative (NRI) to P.D.
Footnotes
Conflicts of interest/Competing interests - There are no potential conflicts of interest to disclose.
Consent for publication: All authors have agreed to publish this manuscript.
References
- [1].Reya T, Morrison SJ, Clarke MF, and Weissman IL, “Stem cells, cancer, and cancer stem cells,” (in eng), Nature, vol. 414, no. 6859, pp. 105–11, Nov 1 2001, doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- [2].Dalerba P et al. , “Phenotypic characterization of human colorectal cancer stem cells,” Proceedings of the National Academy of Sciences, vol. 104, no. 24, p. 10158, 2007, doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Li C et al. , “Identification of pancreatic cancer stem cells,” (in eng), Cancer Res, vol. 67, no. 3, pp. 1030–7, Feb 1 2007, doi: 10.1158/0008-5472.can-06-2030. [DOI] [PubMed] [Google Scholar]
- [4].Zheng H et al. , “Single-cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma,” (in eng), Hepatology, vol. 68, no. 1, pp. 127–140, Jul 2018, doi: 10.1002/hep.29778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Boesch M et al. , “Heterogeneity of Cancer Stem Cells: Rationale for Targeting the Stem Cell Niche,” (in eng), Biochim Biophys Acta, vol. 1866, no. 2, pp. 276–289, Dec 2016, doi: 10.1016/j.bbcan.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hirata A, Hatano Y, Niwa M, Hara A, and Tomita H, “Heterogeneity in Colorectal Cancer Stem Cells,” (in eng), Cancer Prev Res (Phila), vol. 12, no. 7, pp. 413–420, Jul 2019, doi: 10.1158/1940-6207.capr-18-0482. [DOI] [PubMed] [Google Scholar]
- [7].Chaffer CL et al. , “Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state,” (in eng), Proc Natl Acad Sci U S A, vol. 108, no. 19, pp. 7950–5, May 10 2011, doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Singh M, Yelle N, Venugopal C, and Singh SK, “EMT: Mechanisms and therapeutic implications,” (in eng), Pharmacol Ther, vol. 182, pp. 80–94, Feb 2018, doi: 10.1016/j.pharmthera.2017.08.009. [DOI] [PubMed] [Google Scholar]
- [9].Lu W and Kang Y, “Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis,” (in eng), Dev Cell, vol. 49, no. 3, pp. 361–374, May 6 2019, doi: 10.1016/j.devcel.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nieto MA, Huang RY, Jackson RA, and Thiery JP, “EMT: 2016,” (in eng), Cell, vol. 166, no. 1, pp. 21–45, Jun 30 2016, doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- [11].Mani SA et al. , “The epithelial-mesenchymal transition generates cells with properties of stem cells,” (in eng), Cell, vol. 133, no. 4, pp. 704–15, May 16 2008, doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li J and Zhou BP, “Activation of β-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters,” (in eng), BMC Cancer, vol. 11, p. 49, Feb 1 2011, doi: 10.1186/1471-2407-11-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sato R, Semba T, Saya H, and Arima Y, “Concise Review: Stem Cells and Epithelial-Mesenchymal Transition in Cancer: Biological Implications and Therapeutic Targets,” (in eng), Stem Cells, vol. 34, no. 8, pp. 1997–2007, Aug 2016, doi: 10.1002/stem.2406. [DOI] [PubMed] [Google Scholar]
- [14].Jolly MK et al. , “Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis,” (in eng), Front Oncol, vol. 5, p. 155, 2015, doi: 10.3389/fonc.2015.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zheng X, Dai F, Feng L, Zou H, and Xu M, “Communication Between Epithelial-Mesenchymal Plasticity and Cancer Stem Cells: New Insights Into Cancer Progression,” (in eng), Front Oncol, vol. 11, p. 617597, 2021, doi: 10.3389/fonc.2021.617597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xu H et al. , “The role of CD44 in epithelial-mesenchymal transition and cancer development,” (in eng), Onco Targets Ther, vol. 8, pp. 3783–92, 2015, doi: 10.2147/ott.s95470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen C, Zhao S, Karnad A, and Freeman JW, “The biology and role of CD44 in cancer progression: therapeutic implications,” Journal of Hematology & Oncology, vol. 11, no. 1, p. 64, 2018/May/10 2018, doi: 10.1186/s13045-018-0605-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Williams ED, Gao D, Redfern A, and Thompson EW, “Controversies around epithelial-mesenchymal plasticity in cancer metastasis,” (in eng), Nat Rev Cancer, vol. 19, no. 12, pp. 716–732, Dec 2019, doi: 10.1038/s41568-019-0213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, and Williams ED, “Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2,” (in eng), Cancer Res, vol. 66, no. 23, pp. 11271–8, Dec 1 2006, doi: 10.1158/0008-5472.can-06-2044. [DOI] [PubMed] [Google Scholar]
- [20].Saha B et al. , “Overexpression of E-cadherin protein in metastatic breast cancer cells in bone,” (in eng), Anticancer Res, vol. 27, no. 6b, pp. 3903–8, Nov-Dec 2007. [PubMed] [Google Scholar]
- [21].Kowalski PJ, Rubin MA, and Kleer CG, “E-cadherin expression in primary carcinomas of the breast and its distant metastases,” (in eng), Breast Cancer Res, vol. 5, no. 6, pp. R217–22, 2003, doi: 10.1186/bcr651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chao Y, Wu Q, Acquafondata M, Dhir R, and Wells A, “Partial mesenchymal to epithelial reverting transition in breast and prostate cancer metastases,” (in eng), Cancer Microenviron, vol. 5, no. 1, pp. 19–28, Apr 2012, doi: 10.1007/s12307-011-0085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Thiery JP, Acloque H, Huang RY, and Nieto MA, “Epithelial-mesenchymal transitions in development and disease,” (in eng), Cell, vol. 139, no. 5, pp. 871–90, Nov 25 2009, doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- [24].Zheng X et al. , “Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer,” (in eng), Nature, vol. 527, no. 7579, pp. 525–530, Nov 26 2015, doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Krebs AM et al. , “The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer,” (in eng), Nat Cell Biol, vol. 19, no. 5, pp. 518–529, May 2017, doi: 10.1038/ncb3513. [DOI] [PubMed] [Google Scholar]
- [26].Bornes L, Belthier G, and van Rheenen J, “Epithelial-to-Mesenchymal Transition in the Light of Plasticity and Hybrid E/M States,” (in eng), Journal of clinical medicine, vol. 10, no. 11, p. 2403, 2021, doi: 10.3390/jcm10112403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bhatia S, Monkman J, Toh AKL, Nagaraj SH, and Thompson EW, “Targeting epithelial-mesenchymal plasticity in cancer: clinical and preclinical advances in therapy and monitoring,” (in eng), Biochem J, vol. 474, no. 19, pp. 3269–3306, Sep 20 2017, doi: 10.1042/bcj20160782. [DOI] [PubMed] [Google Scholar]
- [28].Jolly MK et al. , “Stability of the hybrid epithelial/mesenchymal phenotype,” (in eng), Oncotarget, vol. 7, no. 19, pp. 27067–84, May 10 2016, doi: 10.18632/oncotarget.8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Puram SV et al. , “Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer,” (in eng), Cell, vol. 171, no. 7, pp. 1611–1624.e24, Dec 14 2017, doi: 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Karacosta LG et al. , “Mapping lung cancer epithelial-mesenchymal transition states and trajectories with single-cell resolution,” (in eng), Nat Commun, vol. 10, no. 1, p. 5587, Dec 6 2019, doi: 10.1038/s41467-019-13441-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Grosse-Wilde A et al. , “Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival,” PLoS One, vol. 10, no. 5, p. e0126522, 2015, doi: 10.1371/journal.pone.0126522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Strauss R et al. , “Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity,” (in eng), PLoS One, vol. 6, no. 1, p. e16186, Jan 14 2011, doi: 10.1371/journal.pone.0016186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cook DP and Vanderhyden BC, “Context specificity of the EMT transcriptional response,” (in eng), Nat Commun, vol. 11, no. 1, p. 2142, May 1 2020, doi: 10.1038/s41467-020-16066-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pastushenko I et al. , “Identification of the tumour transition states occurring during EMT,” Nature, vol. 556, no. 7702, pp. 463–468, 2018/April/01 2018, doi: 10.1038/s41586-018-0040-3. [DOI] [PubMed] [Google Scholar]
- [35].Pastushenko I et al. , “Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis,” (in eng), Nature, vol. 589, no. 7842, pp. 448–455, Jan 2021, doi: 10.1038/s41586-020-03046-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kröger C et al. , “Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells,” (in eng), Proc Natl Acad Sci U S A, vol. 116, no. 15, pp. 7353–7362, Apr 9 2019, doi: 10.1073/pnas.1812876116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Stylianou N et al. , “A molecular portrait of epithelial-mesenchymal plasticity in prostate cancer associated with clinical outcome,” (in eng), Oncogene, vol. 38, no. 7, pp. 913–934, Feb 2019, doi: 10.1038/s41388-018-0488-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ocaña OH et al. , “Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1,” (in eng), Cancer Cell, vol. 22, no. 6, pp. 709–24, Dec 11 2012, doi: 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- [39].Tsai JH, Donaher JL, Murphy DA, Chau S, and Yang J, “Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis,” (in eng), Cancer Cell, vol. 22, no. 6, pp. 725–36, Dec 11 2012, doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yae T et al. , “Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell,” Nat Commun, vol. 3, p. 883, Jun 6 2012, doi: 10.1038/ncomms1892. [DOI] [PubMed] [Google Scholar]
- [41].Beerling E et al. , “Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity,” (in eng), Cell Rep, vol. 14, no. 10, pp. 2281–8, Mar 15 2016, doi: 10.1016/j.celrep.2016.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Celià-Terrassa T et al. , “Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells,” (in eng), J Clin Invest, vol. 122, no. 5, pp. 1849–68, May 2012, doi: 10.1172/jci59218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li Y et al. , “Genetic Fate Mapping of Transient Cell Fate Reveals N-Cadherin Activity and Function in Tumor Metastasis,” (in eng), Dev Cell, vol. 54, no. 5, pp. 593–607.e5, Sep 14 2020, doi: 10.1016/j.devcel.2020.06.021. [DOI] [PubMed] [Google Scholar]
- [44].Yu M et al. , “Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition,” (in eng), Science, vol. 339, no. 6119, pp. 580–4, Feb 1 2013, doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jolly MK et al. , “Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas,” Pharmacology & Therapeutics, vol. 194, pp. 161–184, 2019/February/01/ 2019, doi: 10.1016/j.pharmthera.2018.09.007. [DOI] [PubMed] [Google Scholar]
- [46].Dang H, Ding W, Emerson D, and Rountree CB, “Snail1 induces epithelial-to-mesenchymal transition and tumor initiating stem cell characteristics,” (in eng), BMC Cancer, vol. 11, p. 396, Sep 19 2011, doi: 10.1186/1471-2407-11-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wellner U et al. , “The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs,” (in eng), Nat Cell Biol, vol. 11, no. 12, pp. 1487–95, Dec 2009, doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- [48].Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, and Puisieux A, “Generation of breast cancer stem cells through epithelial-mesenchymal transition,” (in eng), PLoS One, vol. 3, no. 8, p. e2888, Aug 6 2008, doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Quan Q et al. , “Cancer stem-like cells with hybrid epithelial/mesenchymal phenotype leading the collective invasion,” (in eng), Cancer Sci, vol. 111, no. 2, pp. 467–476, Feb 2020, doi: 10.1111/cas.14285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Marcucci F, Ghezzi P, and Rumio C, “The role of autophagy in the cross-talk between epithelial-mesenchymal transitioned tumor cells and cancer stem-like cells,” (in eng), Mol Cancer, vol. 16, no. 1, p. 3, Jan 30 2017, doi: 10.1186/s12943-016-0573-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Garg M, “Epithelial Plasticity, Autophagy and Metastasis: Potential Modifiers of the Crosstalk to Overcome Therapeutic Resistance,” (in eng), Stem Cell Rev Rep, vol. 16, no. 3, pp. 503–510, Jun 2020, doi: 10.1007/s12015-019-09945-9. [DOI] [PubMed] [Google Scholar]
- [52].Shibue T and Weinberg RA, “EMT, CSCs, and drug resistance: the mechanistic link and clinical implications,” (in eng), Nat Rev Clin Oncol, vol. 14, no. 10, pp. 611–629, Oct 2017, doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Marín-Aguilera M et al. , “Epithelial-to-mesenchymal transition mediates docetaxel resistance and high risk of relapse in prostate cancer,” (in eng), Mol Cancer Ther, vol. 13, no. 5, pp. 1270–84, May 2014, doi: 10.1158/1535-7163.mct-13-0775. [DOI] [PubMed] [Google Scholar]
- [54].Creighton CJ et al. , “Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features,” (in eng), Proc Natl Acad Sci U S A, vol. 106, no. 33, pp. 13820–5, Aug 18 2009, doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Redfern AD, Spalding LJ, and Thompson EW, “The Kraken Wakes: induced EMT as a driver of tumour aggression and poor outcome,” (in eng), Clin Exp Metastasis, vol. 35, no. 4, pp. 285–308, Apr 2018, doi: 10.1007/s10585-018-9906-x. [DOI] [PubMed] [Google Scholar]
- [56].Gallatin WM, Weissman IL, and Butcher EC, “A cell-surface molecule involved in organ-specific homing of lymphocytes,” Nature, vol. 304, no. 5921, pp. 30–34, 1983/July/01 1983, doi: 10.1038/304030a0. [DOI] [PubMed] [Google Scholar]
- [57].Ponta H, Sherman L, and Herrlich PA, “CD44: from adhesion molecules to signalling regulators,” (in eng), Nat Rev Mol Cell Biol, vol. 4, no. 1, pp. 33–45, Jan 2003, doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- [58].Oppenheimer-Marks N, Davis LS, and Lipsky PE, “Human T lymphocyte adhesion to endothelial cells and transendothelial migration. Alteration of receptor use relates to the activation status of both the T cell and the endothelial cell,” (in eng), J Immunol, vol. 145, no. 1, pp. 140–8, Jul 1 1990. [PubMed] [Google Scholar]
- [59].Zöller M, “CD44: can a cancer-initiating cell profit from an abundantly expressed molecule?,” Nature Reviews Cancer, vol. 11, no. 4, pp. 254–267, 2011/April/01 2011, doi: 10.1038/nrc3023. [DOI] [PubMed] [Google Scholar]
- [60].Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, and Clarke MF, “Prospective identification of tumorigenic breast cancer cells,” (in eng), Proc Natl Acad Sci U S A, vol. 100, no. 7, pp. 3983–8, Apr 1 2003, doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Patrawala L et al. , “Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells,” (in eng), Oncogene, vol. 25, no. 12, pp. 1696–708, Mar 16 2006, doi: 10.1038/sj.onc.1209327. [DOI] [PubMed] [Google Scholar]
- [62].Zhao S et al. , “CD44 Expression Level and Isoform Contributes to Pancreatic Cancer Cell Plasticity, Invasiveness, and Response to Therapy,” (in eng), Clinical cancer research : an official journal of the American Association for Cancer Research, vol. 22, no. 22, pp. 5592–5604, 2016, doi: 10.1158/1078-0432.CCR-15-3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Screaton GR, Bell MV, Jackson DG, Cornelis FB, Gerth U, and Bell JI, “Genomic structure of DNA encoding the lymphocyte homing receptor CD44 reveals at least 12 alternatively spliced exons,” (in eng), Proc Natl Acad Sci U S A, vol. 89, no. 24, pp. 12160–4, Dec 15 1992, doi: 10.1073/pnas.89.24.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Azevedo R et al. , “CD44 glycoprotein in cancer: a molecular conundrum hampering clinical applications,” (in eng), Clinical proteomics, vol. 15, pp. 22–22, 2018, doi: 10.1186/s12014-018-9198-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Screaton GR, Bell MV, Bell JI, and Jackson DG, “The identification of a new alternative exon with highly restricted tissue expression in transcripts encoding the mouse Pgp-1 (CD44) homing receptor. Comparison of all 10 variable exons between mouse, human, and rat,” (in eng), J Biol Chem, vol. 268, no. 17, pp. 12235–8, Jun 15 1993. [PubMed] [Google Scholar]
- [66].Roy Burman D, Das S, Das C, and Bhattacharya R, “Alternative splicing modulates cancer aggressiveness: role in EMT/metastasis and chemoresistance,” (in eng), Mol Biol Rep, vol. 48, no. 1, pp. 897–914, Jan 2021, doi: 10.1007/s11033-020-06094-y. [DOI] [PubMed] [Google Scholar]
- [67].Bánky B, Rásó-Barnett L, Barbai T, Tímár J, Becságh P, and Rásó E, “Characteristics of CD44 alternative splice pattern in the course of human colorectal adenocarcinoma progression,” Molecular Cancer, vol. 11, no. 1, p. 83, 2012/November/14 2012, doi: 10.1186/1476-4598-11-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bonnal SC, López-Oreja I, and Valcárcel J, “Roles and mechanisms of alternative splicing in cancer — implications for care,” Nature Reviews Clinical Oncology, vol. 17, no. 8, pp. 457–474, 2020/August/01 2020, doi: 10.1038/s41571-020-0350-x. [DOI] [PubMed] [Google Scholar]
- [69].Prochazka L, Tesarik R, and Turanek J, “Regulation of alternative splicing of CD44 in cancer,” (in eng), Cell Signal, vol. 26, no. 10, pp. 2234–9, Oct 2014, doi: 10.1016/j.cellsig.2014.07.011. [DOI] [PubMed] [Google Scholar]
- [70].Warzecha CC, Sato TK, Nabet B, Hogenesch JB, and Carstens RP, “ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing,” (in eng), Mol Cell, vol. 33, no. 5, pp. 591–601, Mar 13 2009, doi: 10.1016/j.molcel.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Preca BT et al. , “A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells,” Int J Cancer, vol. 137, no. 11, pp. 2566–77, Dec 1 2015, doi: 10.1002/ijc.29642. [DOI] [PubMed] [Google Scholar]
- [72].Reinke LM, Xu Y, and Cheng C, “Snail represses the splicing regulator epithelial splicing regulatory protein 1 to promote epithelial-mesenchymal transition,” (in eng), J Biol Chem, vol. 287, no. 43, pp. 36435–42, Oct 19 2012, doi: 10.1074/jbc.M112.397125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Warzecha CC et al. , “An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition,” (in eng), Embo j, vol. 29, no. 19, pp. 3286–300, Oct 6 2010, doi: 10.1038/emboj.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Cheng C and Sharp PA, “Regulation of CD44 alternative splicing by SRm160 and its potential role in tumor cell invasion,” (in eng), Molecular and cellular biology, vol. 26, no. 1, pp. 362–370, 2006, doi: 10.1128/MCB.26.1.362-370.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Watermann DO, Tang Y, Zur Hausen A, Jäger M, Stamm S, and Stickeler E, “Splicing factor Tra2-beta1 is specifically induced in breast cancer and regulates alternative splicing of the CD44 gene,” (in eng), Cancer Res, vol. 66, no. 9, pp. 4774–80, May 1 2006, doi: 10.1158/0008-5472.can-04-3294. [DOI] [PubMed] [Google Scholar]
- [76].Loh TJ et al. , “SC35 promotes splicing of the C5-V6-C6 isoform of CD44 pre-mRNA,” (in eng), Oncol Rep, vol. 31, no. 1, pp. 273–9, Jan 2014, doi: 10.3892/or.2013.2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Goodison S, Urquidi V, and Tarin D, “CD44 cell adhesion molecules,” (in eng), Mol Pathol, vol. 52, no. 4, pp. 189–96, Aug 1999, doi: 10.1136/mp.52.4.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Grimme HU et al. , “Colocalization of basic fibroblast growth factor and CD44 isoforms containing the variably spliced exon v3 (CD44v3) in normal skin and in epidermal skin cancers,” (in eng), Br J Dermatol, vol. 141, no. 5, pp. 824–32, Nov 1999, doi: 10.1046/j.1365-2133.1999.03154.x. [DOI] [PubMed] [Google Scholar]
- [79].Ishimoto T et al. , “CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(−) and thereby promotes tumor growth,” (in eng), Cancer Cell, vol. 19, no. 3, pp. 387–400, Mar 8 2011, doi: 10.1016/j.ccr.2011.01.038. [DOI] [PubMed] [Google Scholar]
- [80].Orian-Rousseau V, Chen L, Sleeman JP, Herrlich P, and Ponta H, “CD44 is required for two consecutive steps in HGF/c-Met signaling,” (in eng), Genes Dev, vol. 16, no. 23, pp. 3074–86, Dec 1 2002, doi: 10.1101/gad.242602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Matzke-Ogi A et al. , “Inhibition of Tumor Growth and Metastasis in Pancreatic Cancer Models by Interference With CD44v6 Signaling,” (in eng), Gastroenterology, vol. 150, no. 2, pp. 513–25.e10, Feb 2016, doi: 10.1053/j.gastro.2015.10.020. [DOI] [PubMed] [Google Scholar]
- [82].Ma L, Dong L, and Chang P, “CD44v6 engages in colorectal cancer progression,” (in eng), Cell Death Dis, vol. 10, no. 1, p. 30, Jan 10 2019, doi: 10.1038/s41419-018-1265-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Bellerby R et al. , “Overexpression of Specific CD44 Isoforms Is Associated with Aggressive Cell Features in Acquired Endocrine Resistance,” (in eng), Front Oncol, vol. 6, p. 145, 2016, doi: 10.3389/fonc.2016.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wang SJ, Wreesmann VB, and Bourguignon LY, “Association of CD44 V3-containing isoforms with tumor cell growth, migration, matrix metalloproteinase expression, and lymph node metastasis in head and neck cancer,” (in eng), Head Neck, vol. 29, no. 6, pp. 550–8, Jun 2007, doi: 10.1002/hed.20544. [DOI] [PubMed] [Google Scholar]
- [85].Lokeshwar VB, Fregien N, and Bourguignon LY, “Ankyrin-binding domain of CD44(GP85) is required for the expression of hyaluronic acid-mediated adhesion function,” (in eng), J Cell Biol, vol. 126, no. 4, pp. 1099–109, Aug 1994, doi: 10.1083/jcb.126.4.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Yonemura S, Hirao M, Doi Y, Takahashi N, Kondo T, and Tsukita S, “Ezrin/radixin/moesin (ERM) proteins bind to a positively charged amino acid cluster in the juxta-membrane cytoplasmic domain of CD44, CD43, and ICAM-2,” (in eng), J Cell Biol, vol. 140, no. 4, pp. 885–95, Feb 23 1998, doi: 10.1083/jcb.140.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Legg JW and Isacke CM, “Identification and functional analysis of the ezrin-binding site in the hyaluronan receptor, CD44,” (in eng), Curr Biol, vol. 8, no. 12, pp. 705–8, Jun 4 1998, doi: 10.1016/s0960-9822(98)70277-5. [DOI] [PubMed] [Google Scholar]
- [88].Vuorio J et al. , “N-Glycosylation can selectively block or foster different receptor–ligand binding modes,” Scientific Reports, vol. 11, no. 1, p. 5239, 2021/March/04 2021, doi: 10.1038/s41598-021-84569-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Zhou J et al. , “CD44 Expression Predicts Prognosis of Ovarian Cancer Patients Through Promoting Epithelial-Mesenchymal Transition (EMT) by Regulating Snail, ZEB1, and Caveolin-1,” Front Oncol, vol. 9, p. 802, 2019, doi: 10.3389/fonc.2019.00802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Brown RL et al. , “CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression,” (in eng), J Clin Invest, vol. 121, no. 3, pp. 1064–74, Mar 2011, doi: 10.1172/jci44540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Göttgens EL, Span PN, and Zegers MM, “Roles and Regulation of Epithelial Splicing Regulatory Proteins 1 and 2 in Epithelial-Mesenchymal Transition,” (in eng), Int Rev Cell Mol Biol, vol. 327, pp. 163–194, 2016, doi: 10.1016/bs.ircmb.2016.06.003. [DOI] [PubMed] [Google Scholar]
- [92].Horiguchi K et al. , “TGF-β drives epithelial-mesenchymal transition through δEF1-mediated downregulation of ESRP,” (in eng), Oncogene, vol. 31, no. 26, pp. 3190–201, Jun 28 2012, doi: 10.1038/onc.2011.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Tripathi V et al. , “Direct Regulation of Alternative Splicing by SMAD3 through PCBP1 Is Essential to the Tumor-Promoting Role of TGF-β,” (in eng), Mol Cell, vol. 64, no. 3, pp. 549–564, Nov 3 2016, doi: 10.1016/j.molcel.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Chen Q et al. , “TGF-β1 promotes epithelial-to-mesenchymal transition and stemness of prostate cancer cells by inducing PCBP1 degradation and alternative splicing of CD44,” (in eng), Cell Mol Life Sci, vol. 78, no. 3, pp. 949–962, Feb 2021, doi: 10.1007/s00018-020-03544-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Bhattacharya R, Mitra T, Ray Chaudhuri S, and Roy SS, “Mesenchymal splice isoform of CD44 (CD44s) promotes EMT/invasion and imparts stem-like properties to ovarian cancer cells,” J Cell Biochem, vol. 119, no. 4, pp. 3373–3383, Apr 2018, doi: 10.1002/jcb.26504. [DOI] [PubMed] [Google Scholar]
- [96].Larsen JE et al. , “ZEB1 drives epithelial-to-mesenchymal transition in lung cancer,” J Clin Invest, vol. 126, no. 9, pp. 3219–35, Sep 1 2016, doi: 10.1172/JCI76725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Chen Q et al. , “Poly r(C) binding protein-1 is central to maintenance of cancer stem cells in prostate cancer cells,” Cell Physiol Biochem, vol. 35, no. 3, pp. 1052–61, 2015, doi: 10.1159/000373931. [DOI] [PubMed] [Google Scholar]
- [98].Mima K et al. , “CD44s regulates the TGF-β-mediated mesenchymal phenotype and is associated with poor prognosis in patients with hepatocellular carcinoma,” (in eng), Cancer Res, vol. 72, no. 13, pp. 3414–23, Jul 1 2012, doi: 10.1158/0008-5472.can-12-0299. [DOI] [PubMed] [Google Scholar]
- [99].Okabe H et al. , “CD44s signals the acquisition of the mesenchymal phenotype required for anchorage-independent cell survival in hepatocellular carcinoma,” Br J Cancer, vol. 110, no. 4, pp. 958–66, Feb 18 2014, doi: 10.1038/bjc.2013.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Mima K et al. , “High CD44s expression is associated with the EMT expression profile and intrahepatic dissemination of hepatocellular carcinoma after local ablation therapy,” J Hepatobiliary Pancreat Sci, vol. 20, no. 4, pp. 429–34, Apr 2013, doi: 10.1007/s00534-012-0580-0. [DOI] [PubMed] [Google Scholar]
- [101].Dang H, Steinway SN, Ding W, and Rountree CB, “Induction of tumor initiation is dependent on CD44s in c-Met+ hepatocellular carcinoma,” (in eng), BMC Cancer, vol. 15, p. 161, Mar 21 2015, doi: 10.1186/s12885-015-1166-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Miwa T, Nagata T, Kojima H, Sekine S, and Okumura T, “Isoform switch of CD44 induces different chemotactic and tumorigenic ability in gallbladder cancer,” Int J Oncol, vol. 51, no. 3, pp. 771–780, Sep 2017, doi: 10.3892/ijo.2017.4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Wang Z et al. , “The Prognostic and Clinical Value of CD44 in Colorectal Cancer: A Meta-Analysis,” (in eng), Front Oncol, vol. 9, p. 309, 2019, doi: 10.3389/fonc.2019.00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Cho SH et al. , “CD44 enhances the epithelial-mesenchymal transition in association with colon cancer invasion,” Int J Oncol, vol. 41, no. 1, pp. 211–8, Jul 2012, doi: 10.3892/ijo.2012.1453. [DOI] [PubMed] [Google Scholar]
- [105].Fernández JC et al. , “CD44s expression in resectable colorectal carcinomas and surrounding mucosa,” (in eng), Cancer Invest, vol. 22, no. 6, pp. 878–85, 2004, doi: 10.1081/cnv-200039658. [DOI] [PubMed] [Google Scholar]
- [106].Bendardaf R et al. , “Comparison of CD44 expression in primary tumours and metastases of colorectal cancer,” (in eng), Oncol Rep, vol. 16, no. 4, pp. 741–6, Oct 2006. [PubMed] [Google Scholar]
- [107].Kunimura T, Yoshida T, Sugiyama T, and Morohoshi T, “The Relationships Between Loss of Standard CD44 Expression and Lymph Node, Liver Metastasis in T3 Colorectal Carcinoma,” (in eng), J Gastrointest Cancer, vol. 40, no. 3–4, pp. 115–8, 2009, doi: 10.1007/s12029-009-9100-0. [DOI] [PubMed] [Google Scholar]
- [108].Huh JW et al. , “Expression of standard CD44 in human colorectal carcinoma: association with prognosis,” (in eng), Pathol Int, vol. 59, no. 4, pp. 241–6, Apr 2009, doi: 10.1111/j.1440-1827.2009.02357.x. [DOI] [PubMed] [Google Scholar]
- [109].Sadeghi A, Roudi R, Mirzaei A, Zare Mirzaei A, Madjd Z, and Abolhasani M, “CD44 epithelial isoform inversely associates with invasive characteristics of colorectal cancer,” (in eng), Biomark Med, vol. 13, no. 6, pp. 419–426, Apr 2019, doi: 10.2217/bmm-2018-0337. [DOI] [PubMed] [Google Scholar]
- [110].Mashita N et al. , “Epithelial to mesenchymal transition might be induced via CD44 isoform switching in colorectal cancer,” (in eng), J Surg Oncol, vol. 110, no. 6, pp. 745–51, Nov 2014, doi: 10.1002/jso.23705. [DOI] [PubMed] [Google Scholar]
- [111].Pires BR et al. , “NF-kappaB Is Involved in the Regulation of EMT Genes in Breast Cancer Cells,” (in eng), PLoS One, vol. 12, no. 1, p. e0169622, 2017, doi: 10.1371/journal.pone.0169622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Smith SM and Cai L, “Cell specific CD44 expression in breast cancer requires the interaction of AP-1 and NFκB with a novel cis-element,” (in eng), PLoS One, vol. 7, no. 11, p. e50867, 2012, doi: 10.1371/journal.pone.0050867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Smith SM, Lyu YL, and Cai L, “NF-κB affects proliferation and invasiveness of breast cancer cells by regulating CD44 expression,” (in eng), PLoS One, vol. 9, no. 9, p. e106966, 2014, doi: 10.1371/journal.pone.0106966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Dongre A and Weinberg RA, “New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer,” (in eng), Nat Rev Mol Cell Biol, vol. 20, no. 2, pp. 69–84, Feb 2019, doi: 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- [115].Zhan T, Rindtorff N, and Boutros M, “Wnt signaling in cancer,” (in eng), Oncogene, vol. 36, no. 11, pp. 1461–1473, Mar 2017, doi: 10.1038/onc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Wang L et al. , “ATDC induces an invasive switch in KRAS-induced pancreatic tumorigenesis,” (in eng), Genes Dev, vol. 29, no. 2, pp. 171–83, Jan 15 2015, doi: 10.1101/gad.253591.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Schmitt M, Metzger M, Gradl D, Davidson G, and Orian-Rousseau V, “CD44 functions in Wnt signaling by regulating LRP6 localization and activation,” Cell Death Differ, vol. 22, no. 4, pp. 677–89, Apr 2015, doi: 10.1038/cdd.2014.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Xia P and Xu X-Y, “PI3K/Akt/mTOR signaling pathway in cancer stem cells: from basic research to clinical application,” (in eng), American journal of cancer research, vol. 5, no. 5, pp. 1602–1609, 2015. [PMC free article] [PubMed] [Google Scholar]
- [119].Grille SJ et al. , “The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines,” (in eng), Cancer Res, vol. 63, no. 9, pp. 2172–8, May 1 2003. [PubMed] [Google Scholar]
- [120].Todaro M et al. , “CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis,” (in eng), Cell Stem Cell, vol. 14, no. 3, pp. 342–56, Mar 6 2014, doi: 10.1016/j.stem.2014.01.009. [DOI] [PubMed] [Google Scholar]
- [121].Jolly MK et al. , “Interconnected feedback loops among ESRP1, HAS2, and CD44 regulate epithelial-mesenchymal plasticity in cancer,” (in eng), APL Bioeng, vol. 2, no. 3, p. 031908, Sep 2018, doi: 10.1063/1.5024874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Senbanjo LT and Chellaiah MA, “CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells,” Front Cell Dev Biol, vol. 5, p. 18, 2017, doi: 10.3389/fcell.2017.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Chen C, Zhao S, Karnad A, and Freeman JW, “The biology and role of CD44 in cancer progression: therapeutic implications,” J Hematol Oncol, vol. 11, no. 1, p. 64, May 10 2018, doi: 10.1186/s13045-018-0605-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Orian-Rousseau V and Ponta H, “Perspectives of CD44 targeting therapies,” Arch Toxicol, vol. 89, no. 1, pp. 3–14, Jan 2015, doi: 10.1007/s00204-014-1424-2. [DOI] [PubMed] [Google Scholar]
- [125].Sauter A et al. , “Pharmacokinetics, immunogenicity and safety of bivatuzumab mertansine, a novel CD44v6-targeting immunoconjugate, in patients with squamous cell carcinoma of the head and neck,” Int J Oncol, vol. 30, no. 4, pp. 927–35, Apr 2007. [PubMed] [Google Scholar]
- [126].Rupp U et al. , “Safety and pharmacokinetics of bivatuzumab mertansine in patients with CD44v6-positive metastatic breast cancer: final results of a phase I study,” Anticancer Drugs, vol. 18, no. 4, pp. 477–85, Apr 2007, doi: 10.1097/CAD.0b013e32801403f4. [DOI] [PubMed] [Google Scholar]
- [127].Menke-van der Houven van Oordt CW et al. , “First-in-human phase I clinical trial of RG7356, an anti-CD44 humanized antibody, in patients with advanced, CD44-expressing solid tumors,” Oncotarget, vol. 7, no. 48, pp. 80046–80058, Nov 29 2016, doi: 10.18632/oncotarget.11098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Uchino M et al. , “Nuclear beta-catenin and CD44 upregulation characterize invasive cell populations in non-aggressive MCF-7 breast cancer cells,” BMC Cancer, vol. 10, p. 414, Aug 10 2010, doi: 10.1186/1471-2407-10-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Murphy JF, Lennon F, Steele C, Kelleher D, Fitzgerald D, and Long AC, “Engagement of CD44 modulates cyclooxygenase induction, VEGF generation, and proliferation in human vascular endothelial cells,” FASEB J, vol. 19, no. 3, pp. 446–8, Mar 2005, doi: 10.1096/fj.03-1376fje. [DOI] [PubMed] [Google Scholar]
- [130].Handorean AM, Yang K, Robbins EW, Flaig TW, and Iczkowski KA, “Silibinin suppresses CD44 expression in prostate cancer cells,” Am J Transl Res, vol. 1, no. 1, pp. 80–6, Jan 1 2009. [PMC free article] [PubMed] [Google Scholar]
- [131].Patel S et al. , “Silibinin, A Natural Blend In Polytherapy Formulation For Targeting Cd44v6 Expressing Colon Cancer Stem Cells,” Scientific Reports, vol. 8, no. 1, p. 16985, 2018/November/19 2018, doi: 10.1038/s41598-018-35069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Iida J et al. , “DNA aptamers against exon v10 of CD44 inhibit breast cancer cell migration,” PLoS One, vol. 9, no. 2, p. e88712, 2014, doi: 10.1371/journal.pone.0088712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Ababneh N et al. , “In vitro selection of modified RNA aptamers against CD44 cancer stem cell marker,” Nucleic Acid Ther, vol. 23, no. 6, pp. 401–7, Dec 2013, doi: 10.1089/nat.2013.0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Pecak A et al. , “Anti-CD44 DNA Aptamers Selectively Target Cancer Cells,” Nucleic Acid Ther, vol. 30, no. 5, pp. 289–298, Oct 2020, doi: 10.1089/nat.2019.0833. [DOI] [PubMed] [Google Scholar]
- [135].Ahrens T et al. , “Soluble CD44 inhibits melanoma tumor growth by blocking cell surface CD44 binding to hyaluronic acid,” Oncogene, vol. 20, no. 26, pp. 3399–408, Jun 7 2001, doi: 10.1038/sj.onc.1204435. [DOI] [PubMed] [Google Scholar]
- [136].Pall T, Gad A, Kasak L, Drews M, Stromblad S, and Kogerman P, “Recombinant CD44-HABD is a novel and potent direct angiogenesis inhibitor enforcing endothelial cell-specific growth inhibition independently of hyaluronic acid binding,” Oncogene, vol. 23, no. 47, pp. 7874–81, Oct 14 2004, doi: 10.1038/sj.onc.1208083. [DOI] [PubMed] [Google Scholar]
- [137].Tijink BM et al. , “A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus,” Clin Cancer Res, vol. 12, no. 20 Pt 1, pp. 6064–72, Oct 15 2006, doi: 10.1158/1078-0432.CCR-06-0910. [DOI] [PubMed] [Google Scholar]
- [138].Raso-Barnett L, Banky B, Barbai T, Becsagh P, Timar J, and Raso E, “Demonstration of a melanoma-specific CD44 alternative splicing pattern that remains qualitatively stable, but shows quantitative changes during tumour progression,” (in eng), PloS one, vol. 8, no. 1, pp. e53883–e53883, 2013, doi: 10.1371/journal.pone.0053883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Mima K et al. , “CD44s regulates the TGF-beta-mediated mesenchymal phenotype and is associated with poor prognosis in patients with hepatocellular carcinoma,” Cancer Res, vol. 72, no. 13, pp. 3414–23, Jul 1 2012, doi: 10.1158/0008-5472.CAN-12-0299. [DOI] [PubMed] [Google Scholar]
- [140].Dang H, Steinway SN, Ding W, and Rountree CB, “Induction of tumor initiation is dependent on CD44s in c-Met(+) hepatocellular carcinoma,” BMC Cancer, vol. 15, p. 161, Mar 21 2015, doi: 10.1186/s12885-015-1166-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Riechelmann H et al. , “Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma,” Oral Oncol, vol. 44, no. 9, pp. 823–9, Sep 2008, doi: 10.1016/j.oraloncology.2007.10.009. [DOI] [PubMed] [Google Scholar]
- [142].Roche H-L, “A Study of RO5429083 in Patients With Metastatic and/or Locally Advanced, CD44-Expressing, Malignant Solid Tumors,” Clinicaltrails.gov, Clinical trial 2016. [Google Scholar]
- [143].Yang Y, Zhao X, Li X, Yan Z, Liu Z, and Li Y, “Effects of anti-CD44 monoclonal antibody IM7 carried with chitosan polylactic acid-coated nano-particles on the treatment of ovarian cancer,” Oncol Lett, vol. 13, no. 1, pp. 99–104, Jan 2017, doi: 10.3892/ol.2016.5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Patel S et al. , “Silibinin, A Natural Blend In Polytherapy Formulation For Targeting Cd44v6 Expressing Colon Cancer Stem Cells,” Sci Rep, vol. 8, no. 1, p. 16985, Nov 19 2018, doi: 10.1038/s41598-018-35069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]