

Abstract
Movement and posture depend on sensory feedback that is regulated by specialized GABAergic neurons (GAD2+) that form axo-axonic contacts onto myelinated proprioceptive sensory axons and are thought to be inhibitory. However, we report here that activating GAD2+ neurons directly with optogenetics or indirectly by cutaneous stimulation actually facilitates sensory feedback to motoneurons in rodents and humans. GABAA receptors located at or near nodes of Ranvier of sensory axons cause this facilitation, by preventing spike propagation failure at the many axon branch points, which is otherwise common without GABA. In contrast, GABAA receptors are generally lacking from axon terminals and so cannot inhibit transmitter release onto motoneurons, unlike GABAB receptors that cause presynaptic inhibition. GABAergic innervation near nodes and branch points allows individual branches to function autonomously, with GAD2+ neurons regulating which branches conduct, adding a computational layer to the neuronal networks generating movement and likely generalizing to other CNS axons.
The ease with which animals move defies the complexity of the underlying neuronal circuits, which include corticospinal tracts (CSTs) that coordinate skilled movement, spinal interneurons that form central patterns generators (CPGs) for walking, and motoneurons that ultimately drive the muscles1. Sensory feedback ensures the final precision of such motor acts, with proprioceptive feedback to motoneurons producing a major part of the muscle activity in routine movement and posture2,3, without which coordination is poor4. Proprioceptive sensory feedback is regulated by specialized GABAergic neurons (GAD2+; abbreviated GABAaxo neurons) that form axo-axonic connections onto the sensory axon terminals5–7. These neurons are thought to produce presynaptic inhibition of sensory feedback to motoneurons8–10 and possibly limit inappropriate sensory feedback3,6. However, during movement the CST, CPG and even sensory neurons all augment GABAaxo neuron activity10–15 right at a time when sensory feedback is known to be increased to ensure precision and postural stability2,3, raising the question of whether GABAaxo neurons have a yet undescribed excitatory action.
The long-standing view that GABAergic neurons and associated axonal GABAA receptors produce presynaptic inhibition of proprioceptive sensory axon terminals in adult mammals actually lacks direct evidence. This is largely because of the difficulty in recording from these small terminals15 and the technical limitations of previously employed reduced spinal cord preparations (immature)6,16 or anesthetized animals, since anesthetics themselves modulate GABAA receptors8,17. Thus, in this paper we used optogenetic approaches to directly target GABAaxo neurons in awake animals and in isolated whole adult spinal cord preparations. Surprisingly, we found that optogenetically activating these GABAergic neurons markedly facilitates sensory axon transmission to motoneurons via axonal GABAA receptors, throwing into doubt the concept of presynaptic inhibition mediated by GABAA receptors.
The mechanism by which GABAA receptors are theorized to produced presynaptic inhibition is rather counterintuitive and based on indirect evidence10,17. That is, sensory axons, like many other axons, have high intracellular chloride concentrations, leading to an outward chloride ion flow through activated GABAA receptors10,18,19. Thus, GABAA receptors cause a depolarization of sensory axons (primary afferent depolarization, PAD)10,15,20–22, which is on face value excitatory, rather than inhibitory, sometimes even evoking axon spikes15. Nevertheless, PAD and associated GABAA receptors have variously been theorized to cause presynaptic inhibition by depolarization-dependent inactivation or shunting of sodium currents at the sensory axon terminals21,23. However, we do not even know if terminals of large myelinated proprioceptive sensory axons express GABAA receptors at all, despite their demonstrated innervation by GABAaxo neurons5. These terminals appear to lack the α5 subunit of extrasynaptic GABAA receptors15 and the more ubiquitous β2/3 subunits of GABAA receptors24, but this leaves open the possibility that they express other GABAA subunits or GABAB receptors. We thus examined this question here and found again that GABAA receptors are generally not at these axon terminals, but are instead near sodium channels (NaV) of the nodes of Ranvier throughout the myelinated regions of the axon, spatially coincident with innervation by GABAaxo neurons, consistent with earlier electron microscopy observations of GABAergic innervation of afferent nodes25 and imaging of α5 subunits15. What then is the function of such GABAA receptors near sodium channels?
An unexplored possibility is that the depolarizing action of GABAA receptors (and GABAaxo neurons) near nodes aids sodium spike propagation between axon nodes. This has not previously been considered, as spikes are thought to securely propagate from node to node, at least in the orthodromic direction15. Myelinated proprioceptive axons branch extensively in the spinal cord15(Fig. 1a) and each branch point poses a theoretical risk for spike propagation failure at downstream nodes26,27. However, branch points are always located at nodes (NaV)15, likely to minimize this failure. Nevertheless, indirect evidence has suggested that propagation failure can occur28–31. Thus, in the present study we sought direct evidence of nodal spike failure near branch points and examined whether GABAA receptors near or at nodes facilitates afferent conduction by preventing this failure. We already know that PAD and GABAA receptors lower the threshold for initiating axon spikes by extracellular stimulation32, and even initiate spikes15, but do not know whether they aid normal spike propagation. We found that spike propagation depends so heavily on GABA that blocking GABA action makes the majority of proprioceptive sensory axons fail to propagate spikes to motoneurons, and thus GABA near sodium channels provides a powerful mechanism to turn on specific nodes and branches to regulate sensory feedback.
Fig. 1 |. Nodal GABAA and terminal GABAB receptors in rats.
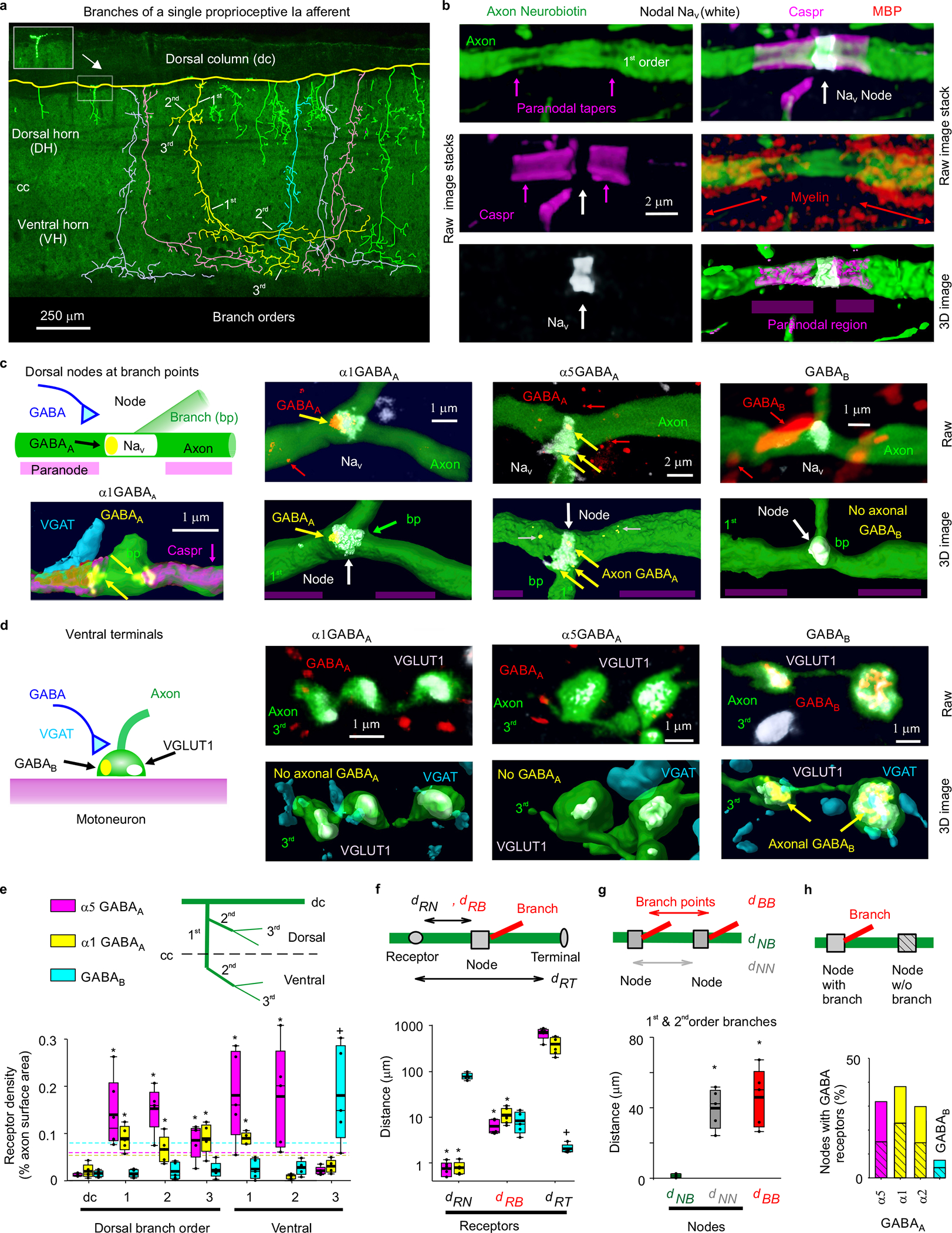
a, Neurobiotin filled proprioceptive Ia axon in the sacrocaudal rat spinal cord (S4 and Ca1), reconstructed from fluorescent images (inset), with branching order indicated and different primary branches distinguished by color. Some ventral branches truncated for clarity (green). Axon diameter not to scale. Central canal: cc. Dorsal columns: dc. Dorsal and ventral horns: DH and VH. b, Node on axon branch in DH immunolabelled for sodium channels (NaV), paranodal Caspr and myelin (MBP), shown with raw images (maximum projection of z-stack), with the paranodal taper indicated, and co-labelling within the axon rendered in 3D (bottom). 1st order branch in DH. c-d, α1 GABAA, α5 GABAA and GABAB receptor immunolabelling in axon branches (raw maximum projection: top row, 3D reconstruction: bottom), with all receptors colocalized with the axon labelled yellow. Receptor clusters specifically in the axon membrane indicated with yellow arrows. Some α5 GABAA receptors are in axon cytoplasm (yellow with gray arrow) or in nearby neurons (red), and not in axon membrane. In (c) nodes identified by NaV (or Caspr) and paranodal taper, and located at branch points (bp, 1st to 2nd bp in DH). In (d) ventral terminal boutons identified by vesicular glutamate transporter 1 (VGLUT1) adjacent to motoneurons. GABAergic contacts identified by vesicular inhibitory amino acid transporter (VGAT). (a-d) representative of 5 rats. e, Receptor densities on axon branches of varying order in dorsal (dorsal and intermediate laminae) and ventral regions. Box plots show the interquartile range (box), median (thin line), mean (thick line), 10 and 90 percentile (whiskers) and extremes (dots). Dashed lines: lower confidence interval, 1 SD below mean maximum density. *significantly more than ventral terminal (3rd order) receptor density, + ventral terminal receptor density significantly more than 1st or 2nd order branch densities, two-sided unpaired t- test, P < 0.05, n = 5 rats each, with 11, 17 and 12 independently filled and reconstructed axons for α1 GABAA, α5 GABAA and GABAB receptors, respectively. f, Distances from GABA receptor clusters in the membrane to nodes (dRN, NaV), branch points (dRB) or ventral terminals at motoneurons (dRT). Distances to 1st and 2nd order dorsal and ventral nodes similar and pooled, as were branch points. *significantly less than dRT. + significantly less than dRN and dRB; one-way ANOVA with Bonferroni correction, P < 0.05, n = 5 rats, with 89, 36 and 70 clusters for α1 GABAA, α5 GABAA and GABAB, respectively. g, Distances between branch points (dBB), nodes (NaV clusters, dNN), and branch points and their nearest node (dNB) for 1st and 2nd order branches. On dorsal columns dNN = 243 ± 117 μm. *significantly larger than dNB; one-way ANOVA with post Bonferroni correction, P < 0.05, n = 5 rats, same axons as (e), with 95 nodes and 57 bp. h, Proportion of nodes with GABA receptors, with and without (hashed) nearby branch points; n = 5 rats each, with 86, 75, 91, and 103 nodes for α5, α1, α2 GABAA and GABAB receptors, respectively.
Results
Nodal GABAA and terminal GABAB receptors.
To confirm and extend previous observations that GABAA receptors are near nodes of proprioceptive sensory axons (group Ia) rather than at ventral terminals15,24, we immunolabelled the most common subunits of synaptic and extrasynaptic GABAA receptors expressed in these axons33, both in rats with neurobiotin filled axons (Fig. 1) and VGLUT1Cre/+ mice with axons labelled by a reporter gene (Extended Data Fig. 1). GABAA receptors containing α5, α1, α2 and γ2 subunits were expressed on these axons, especially near sodium channels (< 6 μm away; Fig. 1c–f, Extended Data Fig. 1). Specifically, GABAA receptors were in the plasma membrane on large myelinated 1st and 2nd order branches in the spinal cord at their nodes (identified by large sodium channel clusters, nearby paranodal Caspr, and/or axonal tapers; Fig. 1a–c,e; Extended Data Fig. 1a,c,f), and on short unmyelinated terminal branches in the dorsal and intermediate laminae (3rd order; Figs. 1a,e). The latter were near the nodes on 1st order branches (< 100 μm away) where they can influence these nodes, consistent with previous observations of axonal GABAergic contacts25,34. In contrast, GABAA receptors were mostly absent from the long unmyelinated ventral terminal branches, where the axon boutons synapse onto motoneurons in the ventral horn (3rd order; Figs. 1a,d,e; Extended Data Fig. 1b,d,g,h), which also generally lacked sodium channels15. This left GABAA receptors on average far from the terminal boutons contacting motoneurons (~500 μm; Fig. 1f) relative to the axon space constant (λS ~90 μm), with the majority of receptors in dorsal and intermediate laminae. Nodes were widely spaced, as were branch points (~50 μm separation, Fig. 1g), but branch points were always near nodes (NaV; Fig. 1c,g), the latter providing additional evidence for nodal GABAA receptors, since these receptors were near branch points (Fig 1f). Nodes sometimes occurred without branching (49%, as in Fig. 1b, rat). Overall, synaptic α1 and α2 and extrasynaptic α5 GABAA receptors were each expressed in about 30% of nodes, roughly equally distributed among branched and unbranched nodes (Fig 1h). Importantly, the majority of nodes were electrotonically close (< 90 μm, λS) to GABAA receptors on a neighboring node (96%) or bouton (80%) on the same axon. In contrast to GABAA receptors, GABAB receptors were found mostly on terminal branches in the ventral horn where boutons had dense receptor expression (Fig. 1d–f, Extended Data Fig. 1e,g,h), and not usually on larger myelinated ventral or dorsal branches, and thus absent from nodes (Figs. 1c,e,h; Extended Data Fig. 1e).
Propagation failure in dorsal horn axon branches.
Considering that GABAA receptors are expressed in large myelinated dorsal branches of proprioceptive axons, we next directly recorded from these branches in the dorsal horn of rat and mouse spinal cords (Figs. 2 and 3) to examine whether spike propagation depends on these receptors. When we stimulated the dorsal root (DR) containing the axon branch, an all-or-nothing spike was recorded in many branches (Figs. 2b, 3d) at the latency of the fastest afferent volley that arrived at the spinal cord (group Ia afferents; EC in Fig. 2b). However, in other axon branches this spike did not occur (~20%), but at the same latency there was a small all-or-nothing residual spike (failure potential, FP; Ia afferents). This FP was indicative of a spike activating a distant node, but failing to propagate further to the recording site, leaving only its passively attenuated potential, with smaller FPs reflecting more distal failure points in the spinal cord (Figs. 2c–g, 3e–f; typically a few nodes away). Failure never occurred in the DR itself (Fig. 2f). The failing branches with FPs were otherwise indistinguishable from non-failing axon branches, exhibiting full spikes (> 60 mV) with current injection pulses (directly evoking spike; or aiding DR spike, Fig. 2cii, g), and low conductances and resting potentials (~ −65 mV, Fig. 2h), ruling out penetration injury. With high repetitive DR stimulation rates all branches (100%) exhibited propagation failure and an associated FP (Fig. 2e–g), again with the FP implying that the spike is reliably initiated in the DR, but incompletely propagates within the spinal cord.
Fig. 2 |. Spike failure.
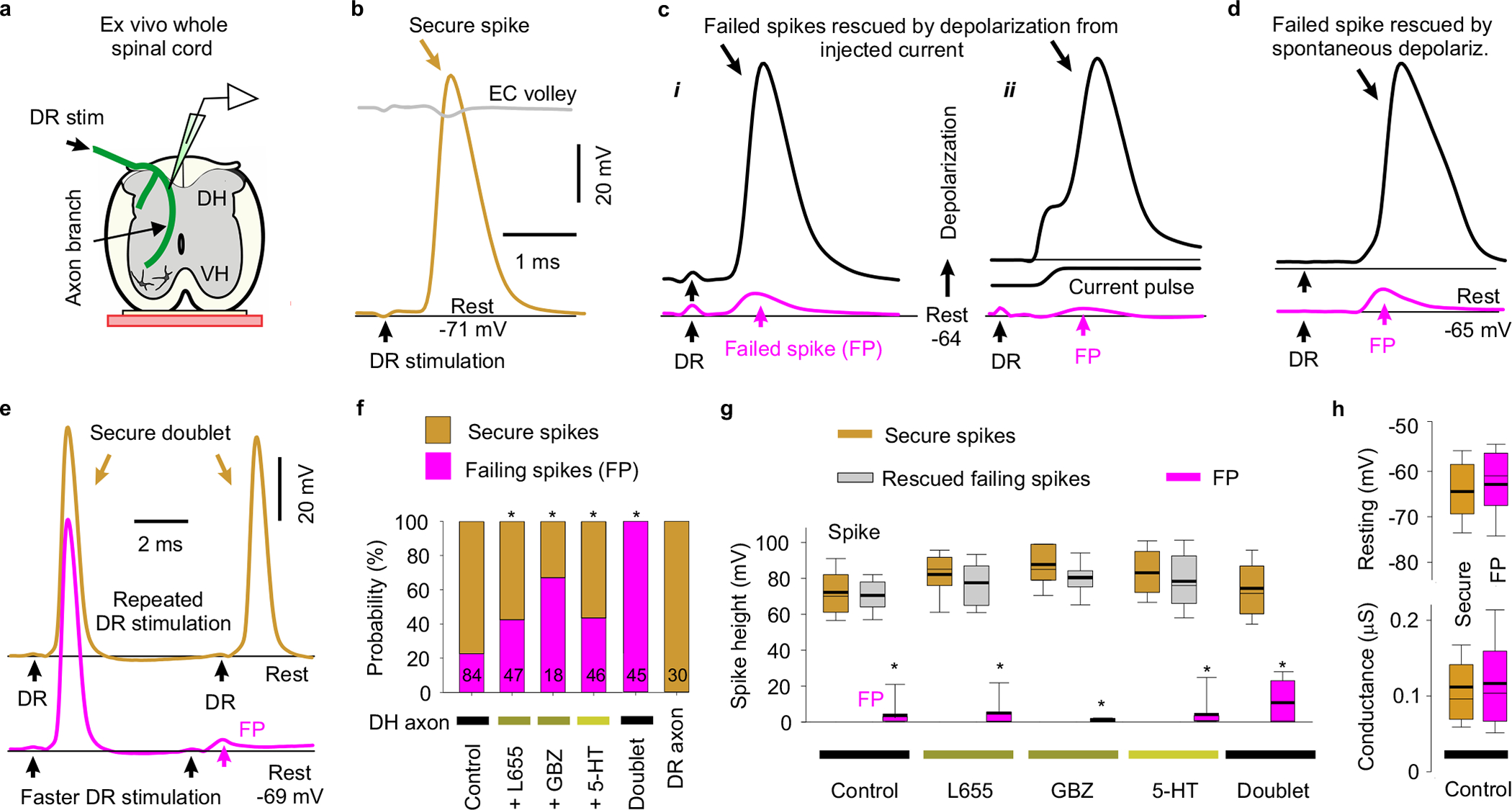
a, Recording from ex vivo whole adult rat spinal cord. b-d, Intracellular recordings from proprioceptive axon branches in the dorsal horn (DH), with dorsal root (DR) stimulation (1.1x T, 0.1 ms; T: afferent volley spike threshold) evoking a spike in some branches (secure, b) and only a failed spike in others (failure potential, FPs; c, d), but depolarization restoring full spikes (black, c and d). Averages of 10 trials at 3 s intervals. Resting potential: thin line. EC: extracellular afferent volley. Axons from S4 sacral DR. e, Fast repeated DR stimulation induced failure in secure spikes (not failing on first stimulation, 1.1xT). Threshold interval (longest) for failure (FP, pink), and just prior to threshold (gold). f, Proportions of DH axon branches (or DR axons) failing to spike with DR stimulation under control resting conditions and with L655708 (0.3 μM), gabazine or bicuculline (50 μM; GBZ), 5-HT (10 μM) or fast repetition (doublet, e). *significantly more than control, χ-squared test, number of axons indicated in bars (n = 84, 47, 18, 46, 45 and 30, from 11 rats each condition). g, Summary of spike and FP heights in secure and failing branches. *significantly smaller than rescued spike, two-sided paired t-test, P < 0.05, n = 11 rats, for axon spikes enumerated in (f). h, Resting membrane potential and conductance for secure and failing branches, not significantly different, two-sided unpaired t-test, P = 0.43 and 0.73, n = 11 rats, for control axons in (f). Box plots show the interquartile range (box), median (thin line), mean (thick line), extremes, 10 and 90 percentile (whiskers).
Fig. 3 |. Nodal facilitation by GABAaxo neurons.
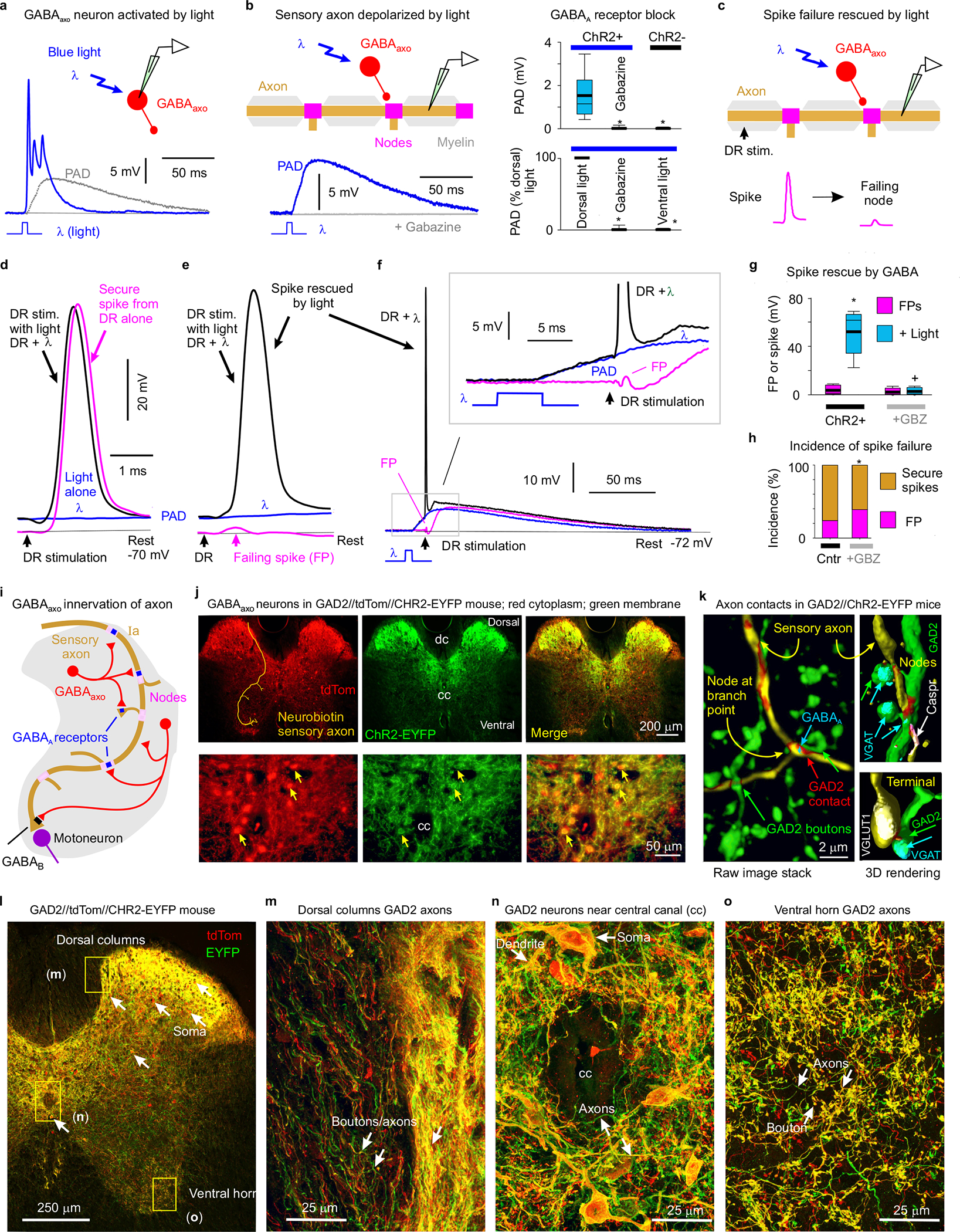
a, Intracellular recording from GABAaxo neuron in ex vivo spinal cord of GAD2//ChR2-EYFP mouse, with ChR2 activated with a light pulse focused on the dorsal horn (5 ms, λ = 447nm laser, 0.7 mW/mm2, 1.5x light threshold to evoke PAD, T) causing a long depolarization and asynchronous spiking (cell isolated in 50 μM gabazine). Average of 10 trials at 0.3 Hz, blue. Cell resting at −61mV. PAD from (b) also shown, grey. b, Intracellular recording from proprioceptive axon branch (in DH, sacral S3, resting at −71 mV; average of 10 trials at 0.3 Hz) with same dorsal light pulse (1.5xT) producing a long depolarization (PAD). Box plots show the interquartile range (box), median (thin line), mean (thick line), extremes, 10 and 90 percentile (whiskers). *significantly less with gabazine or omitting ChR2 (control mice) or focusing light on the ventral rather than dorsal horn (ventral light), two-sided unpaired t-tests with Bonferroni correction, P < 0.05, n = 14 axons each, from 5 mice each. c-g, DR stimulation at rest (1.1xT) evoked a secure spike in some axon branches (d) and not others (e, f, FPs; DH S3 axons). Light evoked PAD (λ, 1.5xT, 10 ms prior) rescued most failed spikes (e, f) and sped up conduction in secure spikes (d). Box plots of FPs and spikes (g); *significant increase with light, two-sided paired t-test, P < 0.05, n = 11 axons from (b); + significant reduction in light effect with 50 μM gabazine or bicuculline, P < 0.05, n =11 axons from (b). h, Incidence of branches with failed DR-evoked spikes. *significant change with gabazine, two-sided χ- squared test, P < 0.05, for n = 45 control axons (Cntr) and n = 27 axons treated with gabazine. i-o, GABAaxo neurons imaged in S3 sacral spinal cord of GAD2//ChR2-EYFP//tdTom mice (j, l-o; green/red, merge yellow; n = 3 mice) or GAD2//ChR2-EYFP mice (k, green, dorsal horn, n = 5 mice). Innervation of neurobiotin filled sensory axons (gold in j and k, as in Fig. 1) by GABAaxo neurons (green; axon contacts labelled red in k) in dorsal horn. Nodes identified by Caspr and paranodal taper, sensory terminals by VGLUT1, GABAaxo terminals by VGAT, and axonal GABAA receptors by the α5GABAA subunit. ChR2-EYFP is mainly expressed on plasma membranes51, whereas tdTom is cytoplasmic (j, l-o). Regions in (l) expanded in (m-o).
Axon spike failure was voltage dependent: in branches with failing spikes (FPs) depolarizations that brought the axon closer to threshold enabled full DR-evoked spikes (via current injection, Fig. 2ci; or spontaneous depolarization, Fig. 2d). Also, in branches without spike failure at rest (secure spikes) a steady hyperpolarizing current induced spike failure (FP), with more branches failing with increasing hyperpolarization (Extended Data Fig. 2). With increasing hyperpolarization, nodes failed progressively more distal to the electrode, causing abrupt drops in the overall spike amplitude with each failure and a characteristic delay in the nodal spike prior to failure, with spike attenuation consistent with λS being about two internodal distances (~90 μm; Extended Data Fig. 2a–d). Simulating spike propagation by applying a brief current pulse to mimic the current arriving from an upstream node (and FP) yielded similar results, with full spikes evoked at rest, but hyperpolarization leading to a spike delay and then failure (Extended Data Fig. 3). Large depolarizations inactivated spikes, though outside of the physiological range (> − 50 mV, Extended Data Fig. 3b–c).
Nodal spike facilitation by GABA.
Since sensory axons are tonically depolarized by spontaneous GABA activity15, we wondered whether this GABAA current aids propagation. Blocking extrasynaptic α5 GABAA receptors (with L655708) or all GABAA receptors (with gabazine) increased the incidence of spike failure (to ~45% and 65%, respectively; Fig. 2f) and sensitivity to hyperpolarization (Extended Data Fig. 2e–h), without altering overall spike properties (Fig. 2g). Application of 5-HT to mimic natural brainstem-derived 5-HT also increased failure (Fig. 2f), likely via its indirect inhibition of GABAA receptor activity35.
Nodal spike facilitation by GABAaxo neuron activation.
To examine whether GABAaxo neurons facilitate spike propagation, we expressed light-sensitive channelrhodopsin-2 (ChR2) in GAD2+ neurons in adult GAD2CreER/+;R26LSL-ChR2-EYFP mice (termed GAD2//ChR2-EYFP mice, Fig. 3). A brief light pulse (5 – 10 ms) produced a long-lasting depolarization and spiking in these GABAaxo neurons (Fig. 3a), followed by a longer lasting GABAA-mediated depolarization (PAD) of proprioceptive axons at a monosynaptic latency that was blocked by gabazine (Fig. 3a–b). In these mice, spikes in proprioceptive axons failed with a similar incidence as observed in rats (Figs. 3c–h), but the light-evoked PAD prevented this failure (Fig. 3e–g), similar to direct depolarization. Occasionally, spikes were only partially rescued by PAD (< 60 mV spikes; Fig. 3g), suggestive of PAD restoring conduction in some, but not all, nodes. In branches with secure non-failing spikes, light had minor effects (Fig. 3d), but blocking GABAA receptors again increased the incidence of spike failure (Fig. 3h).
In GAD2//ChR2-EYFP or GAD2//ChR2-EYFP//tdTom mice the EYFP and tdTom reporters labelled GABAergic neurons (Fig. 3k; VGAT+, GAD2+ and VGLUT1−) residing near the central canal and throughout much of the dorsal horn (Fig. 3i–o, Extended Data Fig. 4). These neurons densely innervated the dorsal horn (Fig. 3j,l,n; Extended Data Fig. 4a), and less densely innervated both the ventral horn and dorsal columns with terminal boutons (Fig. 3l,m,o), allowing GABAergic innervation of sensory axons along their entire length. They made both synaptic and perisynaptic contacts along proprioceptive Ia sensory axons labelled either intracellularly with neurobiotin or peripherally with a viral vector, both at nodes and sensory axon terminals (Figs. 3k, and 1e, Extended Data Fig. 4), confirming their identity as GABAaxo neurons. About 25% of nodes were directly innervated by GABAergic GAD2+ neurons (GABAaxo neurons; Extended Data Fig. 4). Furthermore, the majority of nodes were electrotonically close (< λS) to such GABAergic contacts on a neighbouring node or an unmyelinated branch on the same axon (98% - 77%; Extended Data Fig. 4). Also, most nodes (95%) had nearby GABAaxo terminal boutons not contacting the axon (within 5 μm, GAD2+ or VGAT+; Extended Data Fig. 4), potentially providing extrasynaptic GABA.
Consistent with the predominantly dorsal GAD2+ innervation of nodes (Fig 3) and lack of terminal GABAA receptors (Fig 1), PAD was evoked by light focused on the dorsal horn, but not on the ventral horn (Fig 3b, bottom). Furthermore, light-evoked PAD improved axon conductance even after silencing neuronal circuits with CNQX (50 μM, n = 11/11 axons in two mice, as in Fig 3e–g).
Computer simulation of branch point failure and GABA action.
To establish that spike failure arises at the branch points where GABA can influence them, we generated a computer simulation of a proprioceptive sensory axon arbour in the spinal cord (Extended Data Fig. 5)25. With simulated DR stimulation, spike failure occurred distal to complex branch points (at nodes N2 and N3 in Extended Data Fig. 5a–b) that had associated increases in net conductance, which shunted the nodal currents. Simulated nodal GABAA receptor activation rescued these failed spikes, with increasing GABAA receptor activation (gGABA) preventing more branch point failures (Extended Data Fig. 5c). Importantly, a single well placed GABA contact (at either N2 or N3, or on a nearby bouton) rescued conduction in an entire branch. In contrast, when we moved all these GABAA receptors to the ventral terminals, then their activation did not rescue failed spikes (Extended Data Fig. 5d). This is because GABAA-induced depolarizations (PAD) were attenuated sharply with distance (λS ~90 μm); so only PAD generated near nodes, and not far away at ventral terminals, was visible at the dorsal columns (Extended Data Fig. 5a,g–h), in agreement with previous terminal recordings15.
Spike facilitation by sensory evoked GABAaxo activity
We next examined whether natural activation of GABAaxo neurons affects proprioceptive axon conduction (Fig 4). GABAaxo neurons are indirectly activated by sensory activity via two variants of a trisynaptic circuit, where sensory axons drive excitatory neurons that activate GABAaxo neurons and cause PAD: one driven by cutaneous afferents and the other by proprioceptive afferents (Extended Data Figs. 6 and 7)15. As expected, following DR stimulation these circuits caused fast synaptic and slower extrasynaptic GABAA receptor mediated depolarizations of proprioceptive axons (termed sensory-evoked phasic PAD and tonic PAD, respectively15) that were blocked by GABAA receptor antagonists, and mimicked by optogenetic activation of GABAaxo neurons (Fig. 4a–d).
Fig. 4 |. Sensory driven nodal facilitation.
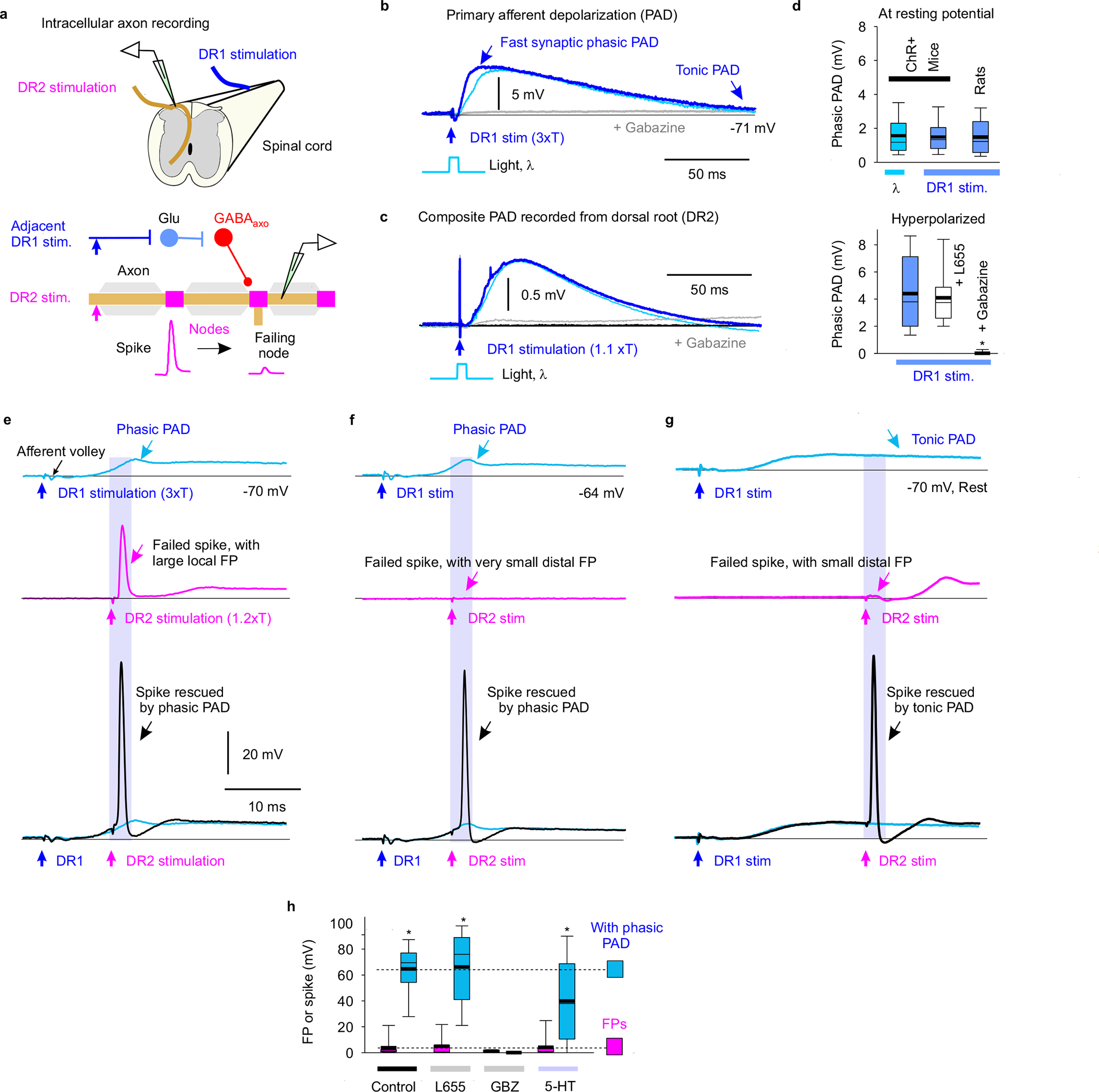
a, Experimental setup to indirectly activate GABAaxo neurons by DR stimulation (DR1) by trisynaptic PAD circuits (detailed in Extended Data Figs. 6–7) in ex vivo spinal cords of rats or GAD2//ChR2-EYFP mice. b-c, Depolarization (PAD) of a proprioceptive axon branch (b, intracellular in sacral S3 DH) or multiple axons in a DR (c, grease gap recording; sacral S3 DR; DR2) from stimulating the adjacent S4 DR (1.1 – 3xT, 0.1 ms pulse; DR1; T: afferent volley spike threshold) or applying a light pulse to activate GABAaxo neurons (5 ms, 447nm, 0.7 mW/mm2, as in Fig. 3b), both blocked by gabazine (50 μM), in GAD2//ChR2 mouse. Thin line resting potential. d, Summary box plots of peak phasic PAD evoked in axons by adjacent DR stimulation (DR1) or light, at rest (top, n = 16 axons from 6 rats or n = 14 axons from 5 mice, the latter from Fig. 3b) and with hyperpolarization (−10 mV, bottom, same rats and axons), and effects of applied gabazine (50 μM; n = 14 axons from 5 rats) or L655708 (0.3 μM; n = 14 axons from 5 rats). Box plots show the interquartile range (box), median (thin line), mean (thick line), extremes, 10 and 90 percentile (whiskers). *significant difference from pre-drug (blue, lower plot), two-sided unpaired t-test, P < 0.05. e-g, DR axon branches (sacral S3 DH) exhibiting spike failure (FPs, magenta) following stimulating their DR (S3 DR, 1.2xT, 0.1 ms; DR2) in rats at rest. Spikes rescued by PAD evoked by prior conditioning of adjacent DR (S4 or contralateral S3 DR, at 3xT; DR1). Rescue occurred with fast synaptic depolarizations (phasic PAD; e-f) and tonic depolarizations (tonic PAD, g), both for local FPs (large, e) or distal FPs (small, f-g). h, FP or spike heights before and during DR evoked phasic PAD (n = 17 axons from 11 rats) as in e-f, and actions of L655708 (n = 12 axons from 5 rats, 0.3 μM), gabazine (n = 12 axons from 5 rats, 50 μM) and 5-HT (n = 12 axons from 5 rats, 10 μM). *significant increase in spike with PAD, two-sided paired t-test, P < 0.05.
Like with direct GABAaxo activation, spike propagation failure was prevented by sensory-evoked phasic PAD, regardless of whether the failure was spontaneous (Figs. 4e–f,h), 5-HT-induced (Fig. 4h), or repetition-induced (Extended Data Fig. 7b–f). The latter is particularly important because sensory axons naturally fire at high rates, where they are vulnerable to spike failure (Fig. 2e–f). This action of phasic PAD was abolished by gabazine but not L655708, supporting a synaptic origin (Fig. 4h). Slow extrasynaptic GABAergic depolarization (tonic PAD; L655708-sensitive15) further facilitated spike propagation (Fig. 4g), especially as it built up with repeated DR stimulation (at 1 Hz; Extended Data Fig. 5b). Cutaneous (Extended Data Fig. 6), proprioceptive (Extended Data Fig. 7) or mixed afferent (Fig. 4e–h) -evoked PAD all helped prevent spike failure.
In secure non-failing axon branches sensory-evoked PAD (or optogenetic GABAaxo activation) sped up the spikes and lowered their threshold (rheobase current; Fig. 3d and Extended Data Fig. 8a–d), as predicted from computer simulations (Extended Data Fig. 5e). Importantly, spike height was only slightly reduced during PAD (~1% or 1 mV) indicating that GABAA receptor conductances have minimal shunting action on nearby spikes (Fig. 3 and Extend Data Fig. 8a–d).
Failure of axon conduction to motoneurons and rescue by PAD.
To quantify the overall failure of spikes to conduct from the DR to the sensory axon terminals we measured whether axon branches not conducting during failure were not refractory to subsequent stimulation with a microelectrode in the ventral horn (Extended Data Fig. 9). This method indicated that about 50 – 80% of sensory axons failed to conduct to their ventral terminals under resting conditions, especially in long axons, whereas sensory-evoked PAD decreased failure to < 30%. Similar conclusions were reached by directly recording the extracellular afferent volley in the ventral horn produced by the spikes propagating from a DR stimulation to the motoneurons, which was consistently increased by PAD (Extended Data Fig. 10).
Facilitation of sensory feedback by GABAA receptors.
To examine the functional role of GABA in regulating sensory feedback to motoneurons, we recorded monosynaptic excitatory postsynaptic potentials (EPSPs) from motoneurons in response to proprioceptive sensory axon stimulation (Fig. 5). This EPSP was inhibited by optogenetically silencing GABAaxo neurons with light in mice expressing archaerhodopsin-3 (Arch3, induced in GAD2CreER/+;R26LSL-Arch3-GFP mice; abbreviated GAD2//Arch3, Fig. 5a–b,d), consistent with a tonic GABAA receptor tone facilitating spike propagation in axons. Likewise, the EPSP was reduced when sensory axon conduction was reduced by blocking endogenous GABAA receptor tone with antagonists, despite increasing motoneuron and polysynaptic reflex excitability (the latter minimized with APV, Fig. 5c,d). GABAB antagonists slightly increased the EPSP, suggesting a tonic GABAB-mediated presynaptic inhibition (Fig. 5d), though much smaller than the tonic GABAA-mediated nodal facilitation that dominates when all GABA was reduced (in GAD2//Arch3 mice).
Fig. 5 |. Facilitation of monosynaptic sensory transmission by GABA.
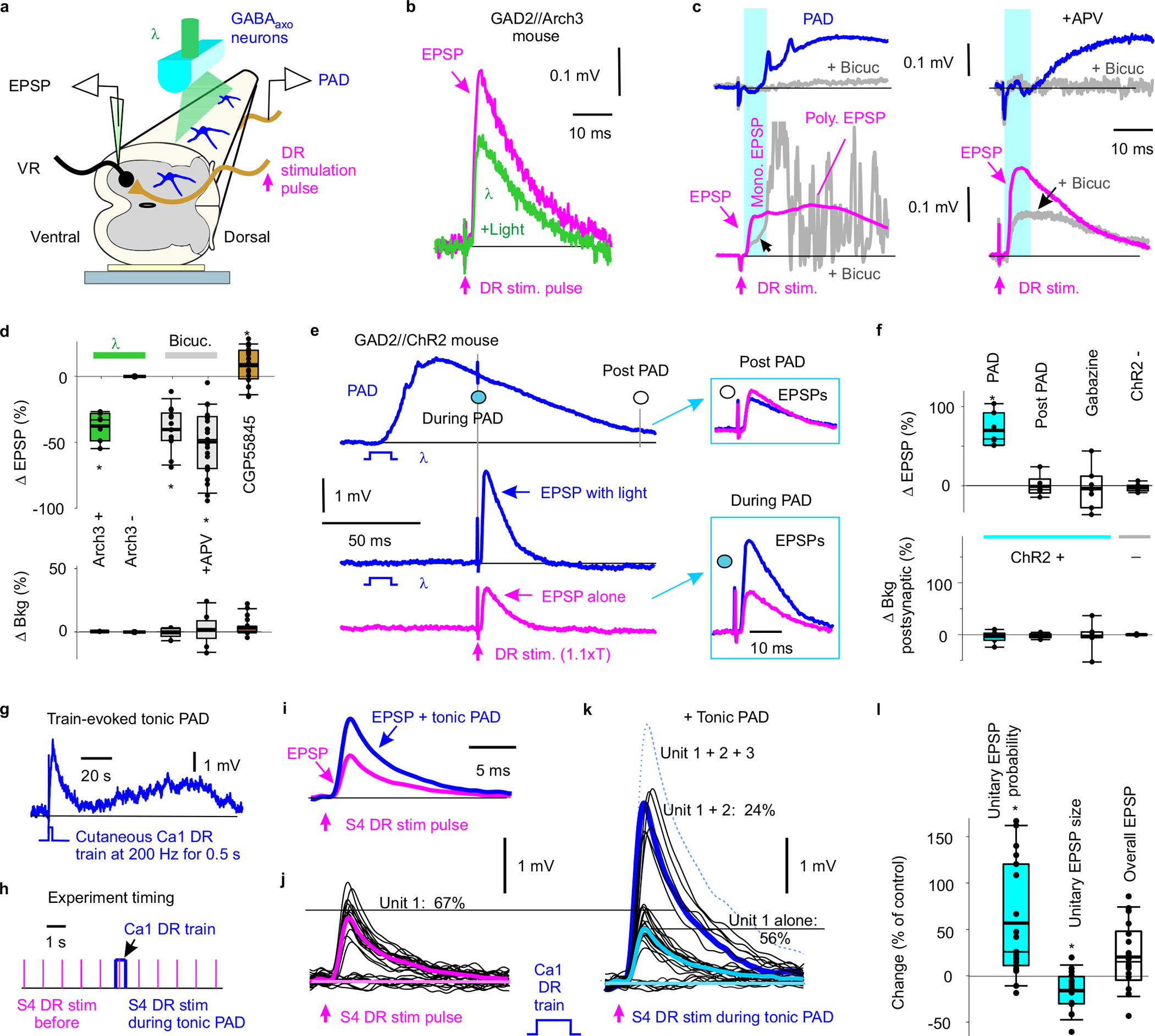
a, Ex vivo recording from motoneurons while illuminating GABAaxo neurons with light λ. b-d, Composite monosynaptic EPSP from motoneuron pool (recorded in sacral S4 VR) evoked by a DR stimulation pulse alone (S4 DR, 0.1 ms, 1.1xT, magenta; T: EPSP threshold). Actions of optogenetic silencing GABAaxo neurons with light (a-b, 532nm, 5 mW/mm2, 80 ms, in n = 7 composite EPSPs from 4 GAD2//Arch3 mice), blocking GABAA receptors (c, with bicuculline, 50 μM, with and without NMDA antagonist APV, 50 μM; n = 23 from 10 mice and 13 EPSPs from 5 mice, respectively; rats similar, n = 5), or blocking GABAB receptors (d, CGP55845, 0.3 μM, n = 20 composite EPSPs in 20 mice). PAD shown for reference, recorded on S3 DR (c, top). Summary box plots of changes in EPSP and background postsynaptic activity (Bkg, over 10 ms prior to EPSP) with light or drugs, and with Arch3+ and Arch3− mice (d). * significant change, Δ, two-sided paired t-test, P < 0.05. GAD2//Arch3 mice are VGAT+. e-f, Composite EPSP (evoked in S4 or S3 motoneurons, as in a-b) before, during and post PAD (recorded simultaneously on S3 DR) evoked by light activation of GABAaxo neurons (10 ms, 1.1xT, 447nm, 0.5 mW/mm2, 60 ms and 140 ms pre EPSP, ISI) in GAD2//ChR2 mice. Box plots of changes in EPSP and Bkg (10 ms prior) with light in ChR2+ mice without and with gabazine (50 μM, during PAD), and in ChR2− mice (60 ms ISI). * significant change, two-sided paired t-test with Bonferroni correction, P < 0.05, for each n = 7 composite EPSPs from 7 mice each; specifically, no significant change in Bkg with PAD, P = 0.55. g, Tonic PAD (L655708 sensitive) recorded in sacral S4 proprioceptive Ia axon in response to 0.5 s, 200 Hz DR stimulation train applied to the largely cutaneous Ca1 DR of caudal cord (3xT, DR2) in rat. h-i, Average EPSP in S4 motoneuron (intracellular recording, EPSP evoked by S4 DR stimulation at 3 s intervals used for average; DR1) before and during tonic PAD (i) evoked by the brief DR train of (h), at matched postsynaptic potentials. j-k, Individual trials used to make EPSP averages in (i) (at 1 s intervals, h), with large all or nothing unitary EPSPs (thick lines unitary averages; dotted single occurrence of Unit 3). Lowpass filtered at 3 kHz. l, Changes in unitary EPSP probability and size, and overall EPSP with tonic PAD. * significant change, two-sided paired t-test, P < 0.05, n = 18 motoneurons from 5 rats. Box plots show the interquartile range (box), median (thin line), mean (thick line), 10 and 90 percentile (whiskers) and extremes (dots).
Consistent with GABAA receptors and PAD facilitating axon conduction, the monosynaptic EPSP was facilitated during, but not after, depolarizing proprioceptive axons (evoking PAD) with an optogenetic activation of GABAaxo neurons in GAD2//ChR2 mice (10 ms light conditioning stimulation; Fig. 5e–f). The EPSP was also facilitated by naturally activating GABAaxo neurons by a sensory conditioning stimulation (Supplementary Fig. 1), including with a conditioning stimulation of cutaneous and/or proprioceptive afferents (Supplementary Fig. 1a,b,e). The latter indicates that proprioceptive activity primes subsequent proprioceptive reflex transmission (self-facilitation). GABAA receptor antagonists (gabazine), but not GABAB antagonists (CGP55845), blocked the EPSP facilitation with sensory (Supplementary Fig. 1e) or light (Fig. 5f) conditioning.
The facilitation of the EPSP by conditioning-evoked PAD arose from axonal GABAA receptors, rather than from postsynaptic actions on the motoneurons, since it occurred with weak conditioning stimuli that produced only a transient background postsynaptic depolarization that terminated before the EPSP testing (at 60 ms; Figs. 5e, Supplementary Fig. 1b,g), followed by a slight hyperpolarization that if anything would reduce the EPSP (shunting the synaptic current, Supplementary Fig. 1h). Increasing the DR conditioning intensity produced large background depolarizing conductances in the motoneurons during the EPSP testing, which led to postsynaptic inhibition of the EPSP (shunting inhibition; Supplementary Fig. 1d,g) and post activation depression, masking the effect of nodal facilitation. Importantly, sometimes PAD itself induced afferent spikes (Extended Data Fig. 8e; termed DRR spikes), and following these spikes, the EPSP was always smaller than when these spikes were not present (n = 8/8 mice). This is because these DRR spikes themselves triggered EPSPs, leading to a post activation depression, as noted by Eccles8, and thus we minimized DRR activity by keeping the conditioning-evoked PAD small.
Sensory conditioning was particularly effective when it was repeated to mimic natural firing, which increased tonic PAD for minutes (Fig. 5g). This facilitated the EPSP for ~3 min after a brief fast DR repetition (200 Hz, 0.5 s conditioning, Fig. 5i, Supplementary Fig. 1e, Tonic), and ~1 min after slower repetition (0.1 Hz, 2 min conditioning, Supplementary Fig. 1e, After effect), both long outlasting postsynaptic effects from each conditioning pulse (< 1 s). This was blocked by L655708 or gabazine (Supplementary Fig. 1e). Interestingly, optogenetic activation of GABAaxo neurons did not produce a similar after effect, consistent with this tonic PAD and associated nodal facilitation being mediated by extrasynaptic GABA spillover from other sources of GABA15.
Increases in the probability of unitary EPSPs.
We often noticed large all-or-nothing EPSPs (unitary EPSPs) spontaneously fluctuating on and off during repeated EPSP testing, leading to discrete large changes in the total EPSP size and time course (Fig. 5j–k). We thought this might be due to spontaneous branch point failures, rather than quantal changes in transmitter release that produce much smaller fluctuations36, as previously suggested29. Indeed, when we increased the axon conduction by activating the GABAaxo neurons and PAD (via a cutaneous conditioning train) the probability of unitary EPSPs occurring increased (Fig. 5k–l), and this sometimes recruited further large unitary EPSPs (Fig. 5k). In contrast, the size of the underlying unitary EPSP was not increased by this conditioning (Fig. 5j–l), ruling out decreases in terminal presynaptic inhibition or postsynaptic inhibition contributing to the increased overall EPSP (Fig. 5i,l). The unitary EPSP actually decreased slightly, likely from GABAB receptors causing presynaptic inhibition. In contrast, the increased unitary EPSP probability arose from GABAA receptors causing nodal facilitation.
Facilitation of sensory axon transmission in awake mice.
To determine whether GABAaxo neurons increase sensory feedback to motoneurons in awake mice we activated these neurons with light applied through a window chronically implanted over the spinal cord of GAD2//ChR2 mice (Fig. 6), and assessed the monosynaptic reflex (MSR) recorded in tail muscles in response to nerve stimulation (counterpart of EPSPs; Fig 6a–c). As expected, the MSR was facilitated by a conditioning light pulse, but only during, and not after, the expected time of phasic PAD induced on sensory axons (Fig. 6b–d,j). This light-induced facilitation occurred both at rest and when there was a background voluntary contraction, with the latter matched with and without light, again ruling out postsynaptic depolarization related differences in MSR (Fig. 6d). Light alone caused a brief pause in ongoing EMG (~30 ms post-light; Fig. 6b), indicative of postsynaptic inhibition, which masked nodal facilitation at short intervals.
Fig. 6 |. Facilitation of reflexes in awake mice.

a, Recording tail muscle EMG and evoking monosynaptic reflexes (MSR) with tail nerve stimulation (1.1xT, 0.2 Hz; T: reflex threshold) in GAD2//ChR2 mice, while activating GABAaxo neurons (PAD) with light (λ = 447 nm, 10 ms pulse, 1.5xT, 5 mW/mm2) applied through a chronically implanted window over the spinal cord. A backpack (pink plastic) was clipped to the window structure at the time of recording to hold the fibre optic cable. b-c, Effect of light pulse λ on active background EMG (Active Bkg condition in b) and the MSR evoked 60 ms later, the latter expanded in (c). MSR tested with (c, top; Active Bkg, 30% max) and without (c, bottom; Rest) background EMG. Thin black lines in (b) are individual trial examples at 10 s intervals (0.1 Hz); thick lines: averages. d, Changes in MSR with light activation of GABAaxo neurons at matched postsynaptic background (Bkg measured over 20 ms prior to MSR; lack of change in Bkg). Measured in active and resting (no Bkg) states, in ChR2+ and ChR2− mice (rest only), and during (60 ms ISI) and post PAD (200ms ISI at rest only). ISI: interstimulus interval. * significant change, two- sided paired t-tests with Bonferroni correction, P < 0.05, n = 5 mice each, with specifically no significant change in Bkg with PAD, P = 0.62 and 0.68 for active and rest, respectively. Box plots show the interquartile range (box), median (thin line), mean (thick line), 10 and 90 percentile (whiskers) and extremes (dots).
Facilitation of MSR by PAD in awake rats and humans.
When we evoked PAD by cutaneous sensory stimulation in awake rats (Supplementary Fig. 2) or humans (Supplementary Fig. 3) the MSR reflex recorded in the tail or lower leg (soleus) was again increased and L655708 sensitive, consistent with the increased EPSPs seen in rats in vitro (Fig 5). This generalizes our main finding to lumbar spinal circuits that control the leg and to humans. Importantly, the probability of a single motor unit (MU) contributing to the human MSR was increased by cutaneous conditioning (Supplementary Fig. 3fi–ii). This occurred without an increase in the estimated EPSP amplitude or rise time (PSF; see Methods; Supplementary Fig. 3Fiii) or motoneuron depolarization prior to the MSR testing (Supplementary Fig. 3Fiv), consistent with an increased probability of unitary EPSPs and decreased branch point failure, as in rats (Fig. 5).
Discussion
Our results demonstrate that GABAA receptors do not cause presynaptic inhibition of the monosynaptic connection from sensory axons to motoneurons, with receptors expressed too far from the axon terminals to influence transmitter release15,24, contrary to long-standing assumptions8,21,23, and throwing into doubt the general concept of GABAA-mediated presynaptic inhibition. Instead, we find that axonal GABAA receptors and associated GABAergic contacts are near nodes of Ranvier at the many branch points of these myelinated axons within the spinal cord, and help prevent conduction failure by depolarizing failing nodes closer to spike threshold (nodal facilitation; summarized in Fig 7; see also31). This differs from the excitatory role of GABAA (and glycine) receptors on transmitter release observed in terminals of other axons in the mammalian brain20,21,37. We show that branch point failure in spike transmission to motoneurons is unexpectedly common, depending on the branching structure and firing frequency, especially when GABAA receptors are not active.
Fig. 7 |. Graphical summary of nodal facilitation.
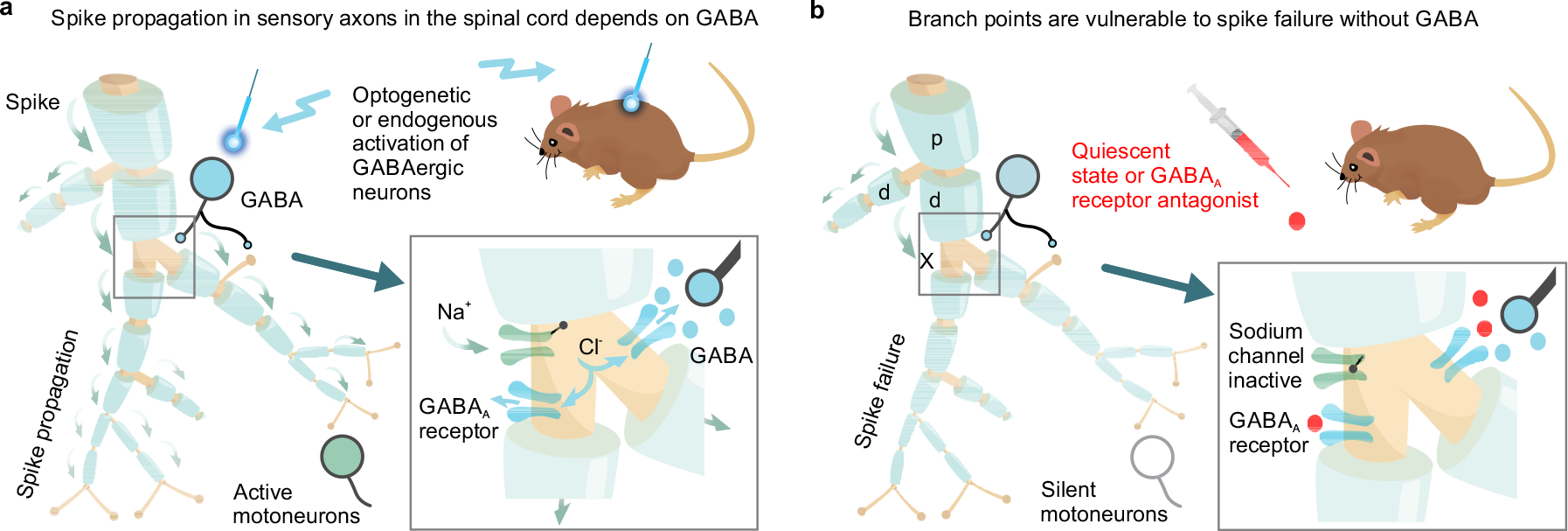
a, Spike propagation (green arrows) in myelinated proprioceptive afferents is facilitated by GABAA receptors at or near nodes, depolarizing axons closer to spike threshold via outward chloride currents. GABAergic neurons (GABAaxo) provide synaptic or perisynaptic innervation of nodes and nearby afferent boutons, and optogenetic or endogenous activation of these neurons increases sensory transmission to motoneurons. b, Without this GABAA receptor activity, spikes fail to propagate into some of the branches of proprioceptive sensory axons in the spinal cord, due to branch point failure. Failure is initiated when a parent branch, p, cannot provide enough current to drive the daughter branches, d, with at least one of the daughters failing, especially if that branch has further branch points, producing a large conductive load. Overall, nodal facilitation by GABAaxo neurons allows selective regulation of conduction in individual branches, increasing the computational complexity of sensory circuits.
Our computer simulations demonstrate that spike conduction failure is only initiated at particularly vulnerable branch points, as previously suggested31, and thus only nodes at or downstream of these failure points require GABAergic facilitation, consistent with the observation that GABAA receptors and GABAergic contacts are only at or near a fraction of nodes25. For this, GABAA receptors need only be within ~90 μm of the node (λS), at another node or even on one of the short unmyelinated terminal branches in dorsal and intermediate regions connected to the node. Ultrastructural imaging of the spinal cord has demonstrated that GABAA receptors often lack presynaptic contacts24, consistent with many GABA receptors being activated extrasynaptically from spillover of nearby GABA, and accounting for the fewer number of nodes with GABAergic contacts than nodes with GABA receptors. We cannot rule out the possibility that the oligodendrocytes at the paranode also influence GABAergic control of the axon, since they express GABA receptors and GABA38, and GABAaxo contacts sometimes straddle the paranodal myelin and the node, maybe forming a tripartite neuron-glia-axon arrangement. The concept of nodal facilitation that we describe here may generalize to other large central axons, such as pyramidal cells, that are innervated by GABAergic neurons, branch extensively (and so may fail), and have depolarizing actions of GABA19,21,39,40, allowing selective recruitment of specific axon branches and functional pathways, especially for high frequency firing (as in Extended Data Fig. 7).
The characteristically long-lasting sensory driven PAD is important since it allows us to indirectly study the action of GABAaxo neurons in humans. Specifically, the expected time-course of PAD evoked by cutaneous conditioning in humans is associated with a potent reflex facilitation over the same time-course, as in awake rodents. This suggests a substantial ongoing spike failure prior to facilitation that can be alleviated by GABAaxo neuron activity (PAD). Indeed, we found that during PAD the probability of EPSPs occurring (and MU firing) is increased without changing the EPSP size, as estimated by PSFs in humans. The latter rules out changes in presynaptic inhibition with PAD that grades the EPSP size, including ruling out previous arguments that MSR facilitation by cutaneous conditioning is due to a removal of presynaptic inhibition41,42.
A pressing question that remains is how can nearly a century of research on sensory transmission and presynaptic inhibition be reconciled with GABA-mediated nodal facilitation and reflex facilitation (summarized in Supplementary Table 1)? Sensory axon conduction failure has repeatedly been noted from indirect observations29–31,43, but GABAA receptors and PAD were previously thought to cause, rather than prevent, conduction failure30, even though computer simulations showed physiological GABA levels unlikely to block spike propagation25, as we confirmed. Furthermore, the fundamental assumption that GABAA receptors cause presynaptic inhibition that reduces transmitter release from sensory axons was from the outset circumspect, based mainly on the observation that a conditioning stimulation on a flexor nerve caused an inhibition of the MSR evoked in extensor muscles that was somewhat correlated to the time-course of PAD caused by this conditioning in flexor afferents8. However, in retrospect this PAD is too brief to account for the much longer (up to 1 s) inhibition caused by this conditioning8,10,44, and GABAB receptor antagonists block much of this MSR inhibition16,44 (see also Fig 5d), suggesting that GABAB receptors may be responsible for presynaptic inhibition of the MSR. Indeed, the well known dense innervation of the these sensory axon terminals by GABAaxo neurons5 combined with our observation that GABAB receptors are strongly expressed on their axon terminals indicate that GABAB receptors are primarily responsible for presynaptic inhibition in proprioceptive axons, as in other neurons, where typically GABAB inhibits and GABAA facilitates synaptic transmission20,21. The predominant GABAB receptor action in proprioceptive axon terminals does not rule out GABAA-mediated action in other sensory axons that have terminal GABAA receptor expression, such as cutaneous Aβ afferents15.
Anatomical studies suggest that GABAaxo neuron activation is likely accompanied by some postsynaptic inhibition, since most GABAaxo contacts on afferent terminals also contact motoneurons, in a triad7,45. Indeed, we find that GABAaxo neuron activation produces an inhibition of motoneurons (Fig. 6b) and associated MU firing that masks, and at times overwhelms, the facilitation of the MSR by GABAA receptors (as with muscle vibration; Supplementary Fig. 3), and thus is readily mistaken for presynaptic inhibition. The argument that presynaptic inhibition with conditioning should be evident from reductions in the EPSP without changing its time course46 now seems untenable, especially as conditioned unitary EPSPs differ markedly in shape and conditioning increases the number of unitary EPSPs contributing to the EPSP, as different axon branches are recruited (Fig. 5k)29.
Early on Barron and Matthews22 and later others6,15 established that sensory-evoked PAD (or light-evoked) excites axons by directly inducing spiking, including spikes in the sensory axons mediating the MSR itself, raising a further contradiction with presynaptic inhibition. While these PAD-triggered spikes only sometimes fully propagate antidromically out the DR47, they are more likely to conduct orthodromically15 where they activate the motoneurons6,8, making these axons and their motoneuron synapse refractory to subsequent testing8. This contributes to a long-lasting post activation depression of the MSR pathway that is GABAA-mediated (sensitive to GABAA antagonists, like PAD), likely explaining much of the inhibition related to PAD that was mistaken for GABAA-mediated presynaptic inhibition6,16,17.
Functionally, nodal facilitation and regulation of branch point failure by GABAaxo-driven GABAA receptors acts like a global switching system that recruits entire silent sensory or motor circuits. This system works in concert with terminal presynaptic inhibition (including GABAB receptor action) that locally fine tunes reflex gains to optimize the stability and compliance of the limbs3,6. The direct activation of GABAaxo neurons and associated PAD by cortical (CST) and spinal (CPG) circuits11,12,48, and inhibition by the brainstem (e.g. 5-HT)35,49, suggests that nodal facilitation is under explicit central control during reaching and locomotion. The widespread action of PAD, occurring simultaneously over many spinal segments,10,15 implies that nodal facilitation acts over large regions of the spinal cord to ready sensory axons for action during cortical, spinal or sensory evoked activity, reminiscent of the Jendrassik maneuver50, ensuring that adequate sensory feedback aids postural stability and walking. More generally, our results imply that each axonal branch point has the capacity to function separately, depending on its GABAergic innervation, increasing the complexity of sensory processing in the spinal cord.
Methods
Adult mice, rats and humans used.
Recordings were made from large proprioceptive group Ia sensory afferents, GABAergic neurons, motoneurons and muscles in adult mice (2.5 – 6 months old, both female and male equally; strains detailed below) and rats (3 – 8 months old, female only, Sprague-Dawley). Animals were caged in groups of 2 – 4 and maintained in a 12 hr light/dark cycle, in stable conditions of temperature and humidity, with food and water ad libitum. All experimental procedures were approved by the University of Alberta Animal Care and Use Committee, Health Sciences division (ACUC Protocol numbers AUP00000224 and AUP00002891) in accordance with the Canadian CCAC guidelines. Recordings were also made from the soleus muscle of neurologically intact adult humans (female and male equally), aged 21 to 58, with written informed consent prior to participation, without compensation. Experiments were approved by the Health Research Ethics Board of the University of Alberta (Protocols 00023530 and 00076790) and conformed to the Declaration of Helsinki. No effects of sex were noted and data from both sexes were combined for analysis. See Supplementary Methods for human subject details.
Mice used for optogenetics and imaging.
We evaluated GABAergic neurons in a strain of mice with Cre expressed under the endogenous Gad2 promotor region. Gad2 encodes the Glutamate decarboxylase 2 enzyme GAD2 (also called GAD65), which is unique to axoaxonic contacting GABAergic neurons that project to the ventral horn, whereas all GABAergic neurons express GAD15. These GAD2+ neurons were activated or inhibit optogenetically using channelrhodopsin-2 (ChR2)52,53 or archaerhodopsin-3 (Ach3)54,55, respectively. The following mouse strains were employed (Extended Data Table 2):
Gad2tm1(cre/ERT2)Zjh mice (abbreviated Gad2CreER mice; The Jackson Laboratory, Stock # 010702; CreERT2 fusion protein expressed under control of the endogenous Gad2 promotor)56,
B6;129S-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze mice (abbreviated R26LSL-ChR2-EYFP mice; The Jackson Laboratory, Stock # 012569; ChR2-EYFP fusion protein expressed under the R26::CAG promotor in cells that co-express Cre because a loxP-flanked STOP cassette, LSL, prevents transcription of the downstream ChR2-EYFP gene)57,
B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze and B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze mice (abbreviated R26LSL-tdTom mice; The Jackson Laboratory, Stock # 007914 and #007909; tdTomato fluorescent protein expressed under the R26::CAG promotor in cells that co-express Cre)58,
B6;129S-Gt(ROSA)26Sortm35.1(CAG-aop3/GFP)Hze mice (abbreviated R26LSL-Arch3-GFP mice; The Jackson Laboratory Stock # 012735; Arch3-GFP fusion protein expressed under the R26::CAG promotor in cells that co-express Cre)57,
B6;129S-Slc17a7tm1.1(cre)Hze mice (abbreviated VGLUT1Cre mice; The Jackson Laboratory, Stock # 023527; Cre protein expressed under control of the endogenous Vglut1 promotor; spinal cords kindly donated by Dr. Francisco J. Alvarez)59 and
EIIa-cre × Gabra5-floxed in a C57BL/6 mouse background, with cre bred out to yield α5 GABAA receptor knockout mice (termed Gabra5 KO mice; produced by RA Pearce laboratory). Gabra5-floxed mice are specifically C57BL/6-Gabra5tm2.1Uru/J mice and possess loxP sites flanking exons 4–5 of the Gabra5 receptor subunit gene. EIIa-cre mice are specifically 6.FVB-Tg(EIIa-cre)C5379Lmgd/J (The Jackson Laboratory, Stock # 003724).
Heterozygous GAD2CreER mice (i.e., GAD2CreER/+ mice) were crossed with homozygous reporter strains to generate GAD2CreER/+; R26LSL-ChR2-EYFP, GAD2CreER/+; R26LSL-tdTom and GAD2CreER/+; R26LSL-Arch3-GFP mice that we abbreviate: GAD2//ChR2, GAD2//tdTom and GAD2//Arch3 mice. Offspring without the GAD2CreER mutation, but with the effectors ChR2, Arch3 or tdTom were used as controls. We also used mice bred by crossing homozygous VGLUT1Cre mice with R26lsl-tdTom reporter mice to obtain mice with VGLUT1 labelled sensory axons60.
CreER is an inducible form of Cre that requires tamoxifen to activate 61, which we applied in adult mice to prevent developmental issues of earlier induction of Cre. Specifically, mice were injected at 4 – 6 weeks old with two doses of tamoxifen separated by two days, and studied > 1 month later, long after washout of tamoxifen. Each injection was 0.2 mg/g wt (i.p.) of tamoxifen dissolved in a corn oil delivery vehicle (Sigma C8267). These tamoxifen-treated mice were denoted GAD2//ChR2+ and GAD2//Arch3+, and non treated mice were used as controls and denoted GAD2//ChR2− and GAD2//Arch2−. Mice with Gad2-cre not reacting to light were excluded from analysis only if it was confirmed that the tamoxifen did not induce ChR2 by post hoc imaging of the reporter. For all mice, genotyping was performed according to the Jackson Laboratories protocols by PCR of ear biopsies using primers specific for the appropriate mutant and wild type alleles for each of the mouse lines (see Supplementary Table 2 for primer details).
Ex vivo recording from axons and motoneurons in whole adult spinal cords.
Mice or rats were anaesthetized with urethane (for mice 0.11 g/100 g, with a maximum dose of 0.065 g; and for rats 0.18 g/100 g, with a maximum dose of 0.45 g), a laminectomy was performed, and then the entire sacrocaudal spinal cord was rapidly removed and immersed in oxygenated modified artificial cerebrospinal fluid (mACSF)62–64. This preparation is particularly useful as the small sacrocaudal spinal cord is the only portion of the adult spinal cord that survives whole ex vivo, allowing axon conduction to be assessed along large distances. Furthermore, this segment of cord innervates the axial muscles of the tail that are readily assessable for reflex recording in awake animals, and has proven to be a useful model of motor function in normal and injured spinal cords, with very similar spinal circuitry, reflex and motoneuron properties to those seen in the hindlimb of other preparations, including having reciprocal inhibition, Ia afferent innervation of muscle spindles and monosynaptic reflexes63,65–67. Interestingly, the rat motoneuron firing rates in the sacral region are more similar to those in human hindlimb motoneurons than the much higher firing rats seen in rat hindlimb motoneurons, and thus the sacral cord has proven to be a useful model of lumbar motoneuron function in humans68. Spinal roots were removed, except the sacral S3, S4 and caudal Ca1 ventral and dorsal roots on both sides of the cord. After 1.5 hours in the dissection chamber (at 20° C), the cord was transferred to a recording chamber containing normal ACSF (nACSF) maintained at 23 – 32°C, with a flow rate > 3 ml/min. A one-hour period in nACSF was given to wash out the residual anaesthetic prior to recording, at which time the nACSF was recycled in a closed system. The cord was secured onto tissue paper at the bottom of a rubber (Silguard) chamber by insect pins in connective tissue and cut root fragments. The dorsal surface of the cord was usually oriented upwards when making intracellular recording from afferents in the dorsal horn, whereas the cord was oriented with its left side upwards when making recordings from motoneurons or afferent terminals in the ventral horn. The laser beam used for optogenetics was focused vertically downward on the GAD2 neurons, as detailed below.
Optogenetic regulation of GABAaxo neurons.
The GAD2//ChR2 or GAD2//Arch3 mice were used to optogenetically excite or inhibit GAD2+ neurons (with 447 nm D442001FX and 532 nM LRS-0532-GFM-00200–01 lasers from Laserglow Technologies, Toronto), respectively69. Light was derived from the laser passed through a fibre optic cable (MFP_200/220/900–0.22_2m_FC-ZF1.25 and MFP_200/240/3000–0.22_2m_FC-FC, Doric Lenses, Quebec City) and then a half cylindrical prism the length of about two spinal segments (8 mm; 3.9 mm focal length, Thor Labs, Newton, USA,), which collimated the light into a narrow long beam (200 μm wide and 8 mm long). This narrow beam was usually focused longitudinally on the left side of the spinal cord roughly at the level of the dorsal horn, to target the epicentre of GABAaxo neurons, which are dorsally located (Fig. 3). ChR2 rapidly depolarizes neurons 53, and thus we used 5 – 10 ms light pulses to activate GABAaxo neurons, as confirmed by direct recordings from these neuron (see below). Light was always kept at a minimal intensity, 1.1x T, where T is the threshold to evoke a light response in sensory axons, which made local heating from light unlikely. Arch3 is a proton pump that is activated by green light, leading to a hyperpolarization and slowly increased pH (over seconds), both of which inhibit the neurons 53,70. Thus, we used longer light pulses (~200 ms) to inhibit GABAaxo neurons.
To directly confirm the presence of functional ChR2 expression in GABAaxo neurons of GAD2//ChR2 mice we recorded from them with similar methods and intracellular electrodes used to record from motoneurons (see below). Electrodes were advanced into these cells through the dorsal horn (with the dorsal surface oriented upwards), and their identity established by a direct response to light activation of the ChR2 construct (5 – 10 ms light pulse, 447 nm), without a synaptic delay (<1 ms) and continued light response after blocking synaptic transmission.
Dorsal and ventral root stimulation.
Dorsal and ventral roots (DR and VR) were mounted on silver-silver chloride wires above the nASCF of the recording chamber and covered with grease (a 3:1 mixture of petroleum jelly and mineral oil) for monopolar stimulation 15,65,71. This grease was surrounded by a more viscous synthetic high vacuum grease to prevent oil leaking into the bath flow. Bipolar stimulation was also used at times to reduce the stimulus artifact during recording from ventral roots (detailed below). Roots were stimulated with a constant current stimulator (Isoflex, Israel) with short pulses (0.1 ms). Note that proprioceptive afferents are relatively selectively activated by low intensity DR stimulation (1.1 – 1.5 × afferent volley threshold, T) and cutaneous afferents are additionally activated by higher intensity DR stimulation (2 – 3xT). DRs were dissected to be as long as possible, and the distal end of this root was stimulated, so it was ~20 mm way from the spinal cord. In this way the DR stimulation site itself (at wire, and threshold for stimulation) could not be affected by axonal depolarizations in the spinal cord, since dorsal root potentials from spinal events (PAD) are only observed very close to the cord (within a few mm, see below), and drop exponentially in size with distance15.
Intracellular recording from sensory axon branches in the dorsal horn.
Electrode preparation and amplifier.
Recording from fine afferent collaterals in the spinal cord without damaging them or disturbing their intracellular milieu required specialized sharp intracellular electrodes15,62. Glass capillary tubes (603000 A-M Systems; Sequim, USA) were pulled with a Sutter P-87 puller (Flaming-Brown; Sutter Instrument, Novato, USA), filled through their tips with 2 M K-acetate mixed with varying proportions of 2 M KCl (to make KCl concentrations ranging of 0, 100, 500, and 1000 mM) or 500 mM KCl in 0.1 Trizma buffer with 5 – 10% neurobiotin (Vector Labs, Birmingame, USA). Electrodes were then beveled from an initial resistance of 40 – 150 MΩ to 30 – 40 MΩ using a rotary beveller (Sutter BV-10). GABAergic chloride-mediated potentials (PAD) and their reversal potentials were the same with different concentrations of KCl, without passing large amounts of negative current15, indicating that the ultra-sharp tips impeded passive fluid exchange between the electrode and intracellular milieu, with in particular electrode Cl- not affecting the axon; thus, recordings were mostly made with electrodes with 1 M K-acetate and 1 M KCl, when not filling cells with neurobiotin.
Intracellular recording and current injection were performed with an Axoclamp2B amplifier (Axon Inst. and Molecular Devices, San Jose, USA). Recordings were low pass filtered at 10 kHz and sampled at 30 kHz (Clampex and Clampfit; Molecular Devices, San Jose, USA). Sometimes recordings were made in discontinuous-single-electrode voltage-clamp (gain 0.8 –2.5nA/mV; for Ca PICs) or discontinuous-current-clamp modes (switching rate 7 kHz), as indicated (the latter only when injecting current, for example during recording of input resistance or the voltage dependence of spikes). Electrodes were advanced into myelinated afferents of the sacrocaudal spinal cord with a stepper motor (Model 2662, Kopf, USA, 10 μm steps at maximal speed, 4 mm/s), usually at the boundary between the dorsal columns and dorsal horn gray matter, where axons bundle together densely, as they branch and descend to the ventral horn (Extended Data Fig. 4A). Prior to penetrating afferents, we recorded the extracellular (EC) afferent volley following dorsal root (DR) stimulation (0.1 ms pulses, 3xT, T: afferent volley threshold, where T = ~3 uA, repeated at 1 Hz), to determine the minimum latency and threshold of afferents entering the spinal cord. The group Ia afferent volley occurs first with a latency of 0.5 – 1.0 ms, depending on the root length (which were kept as long as possible, 10 – 20 mm), corresponding to a conduction velocity of about 16 – 24 m/s, as previously described for in vitro conduction at 23 C 15,72. Axon penetrations were usually in the myelinated portion of the axon between nodes, rather than at nodes, because the chance of penetrating a node is low since they only make up a small fraction of the total axon length (Fig. 1). The spikes from the two nodes adjacent to the electrode were readily detected separately when testing for the spike threshold with current injection pulses (20 ms; rheobase test), because just at threshold the current sometimes evoked a spike from just one node and not the other, which usually halved the total spike height, consistent with the penetration being about halfway between the two nodes separated by about a space constant distance.
Proprioceptive afferent identification.
Upon penetration, afferents were identified with direct orthodromic spikes evoked from DR stimulation. We focused on the lowest threshold proprioceptive group Ia afferents (or possibly Ib), identified by their direct response to DR stimulation, very low threshold (< 1.5 × T, T: afferent volley threshold), short latency (group Ia latency, coincident with onset of afferent volley), and antidromic response to ventral horn afferent terminal microstimulation (~ 10 μA stimulation via tungsten microelectrode to activate Ia afferent terminals; tested in some afferents, detailed below)15. Post hoc these were confirmed to be large proprioceptive Ia afferents by their unique extensive terminal branching around motoneurons73, unlike large cutaneous Aβ afferents that do not project to the ventral horn. Clean axon penetrations without injury occurred abruptly with a sharp pop detected on speakers attached to the recorded signal, the membrane potential settling rapidly to near – 70 mV, and > 70 mV spikes usually readily evoked by DR stimulation or brief current injection pulses (1 – 3 nA, 20 ms, 1 Hz). Sensory axons also had a characteristic >100 ms long depolarization following stimulation of a dorsal root (primary afferent depolarization, PAD, at 4 – 5 ms latency, detailed below and in Supplementary Methods) and short spike afterhyperpolarization (AHP ~ 10 ms), which further distinguished them from other axons or neurons. Injured axons had higher resting potentials (> − 60 mV), poor spikes (< 60 mV) and low resistance (to current pulse; Rm < 10 MΩ), and were discarded.
Quantification of spike conduction failure in the dorsal horn.
Sometimes healthy intracellular penetrations were made into a sensory axon branch (e.g. < −60 mV rest, large PAD), but dorsal root stimulation did not evoke a full spike, even though a full > 60 mV spike could be readily evoked by intracellular current injection. Instead, DR stimulation evoked a partial spike at the latency and threshold of group Ia afferents, indicating that this was a branch of a Ia afferent that failed to fully conduct spikes to the electrode, with only the passively attenuated spike from the last node to spike prior to conduction failure recorded at the electrode (failure potential, FP; also referred to as electronic residue by Luscher74). The size of the FP reflected how far away the spike failure occurred, with spatial attenuation corresponding to a space constant of about 90 μm (see Results), and so FPs became exponentially smaller with distance from failure. Occasionally axons were penetrated with undetectable DR evoked spikes or FPs, but otherwise they had characteristics of a Ia afferent (PAD, Rm similar). These were likely afferents with FPs too distal to detect, but were usually excluded from the main analysis to avoid ambiguity, though this underestimates the incidence of failure. However, some of these axons exhibited short latency, low threshold DR spikes when depolarized by a prior DR stimulation (PAD) of an adjacent DR, in which case they were unequivocally Ia afferents and included in the analysis (Fig. 4f).
When we found that PAD evoked in sensory axons can prevent failure of spikes to propagate in the cord after DR stimulation (Fig 2), we worried that PAD somehow influenced the initiation of the spike by the DR stimulation at the silver wire it was mounted on. However, we ruled this out by stimulating dorsal roots as far away from the spinal cord as possible (20 mm), where PAD has no effect on the wire, due to the exponential attenuation of its dorsal root potential with distance (see below), and found that PAD still facilitated sensory axon spike transmission to motoneurons.
Both during extracellular and intracellular recording the group Ia afferent volley (small negative field) was observed as the first event after DR stimulation (the latter subthreshold to a spike), though this was usually small in relation to intracellular events and ignored. However, this was sometimes removed from the intracellular record by subtracting the extracellular potential recorded just outside the same axon to determine the actual transmembrane potential 15. This was necessary to see the very smallest FPs following DR stimulation in some afferents, as the negative volley from other nearby afferents obscured the FPs.
Neurobiotin filling of axons.
Some of the proprioceptive afferents that we recorded intracellularly were subsequently filled with neurobiotin by passing a very large positive 2 – 4 nA current with 90% duty cycle (900 ms on, 100 ms off) for 10 – 20 min. Prior to penetrating and filling axons with neurobiotin filled electrodes, a small negative holding current was maintained on the electrodes to avoid spilling neurobiotin outside axons.
Quantification of spike conduction failure in the ventral horn
Wall’s method.
To measure whether spikes fail during propagation to their fine terminals in the ventral horn we examined whether failed axon segments were relatively less refractory to activation after spike conduction failure, using a double pulse method adapted from Wall30,75. The essence of the method is that after DR activation all nodes that generate spikes become relatively refractory for a few ms, whereas nodes that fail to spike are not refractory to activation. Thus, a microelectrode placed near these failing nodes more readily activates them if they fail rather than generate spikes with DR stimulation and orthodromic conduction. For this we placed a tungston microelectrode (12 MΩ, #575400, A-M Systems, Sequim, USA) in the ventral horn near the axons terminals on motoneurons, to activate the branches/nodes of the axon projecting to the motoneuron that may have failed (VH stimulation).
Spikes from VH or DR stimulation were recorded intracellularly in a proprioceptive Ia axon penetrated in the dorsal columns directly above the VH stimulation site or in an adjacent segment, with two combinations of double axon stimulations. First, we applied two rapidly repeated VH stimuli (VH doublet; two 0.1 ms pulses) at a ~4 ms interval to make the axon relatively refractory to stimulation and determine both the threshold current to activate the first spike (TVH1, with VH1 stimulation) and the higher threshold current to overcome this the inactivation and generate a second spike (TVH2, with VH2 stimulation). Second, we repeated this double spike activation, but with the first activation from a supra-threshold DR stimulation (at 1.5x DR threshold) and the second from a VH stimulation at the TVH2 intensity from B (DR-VH pair). In this case the VH stimulation readily activates the axon spike if the orthodromic DR evoked spike does not propagate to the ventral horn, leaving the silent portion of the axon non refractory. Accordingly, we also determined the threshold current to activate the VH after the DH in this arrangement (termed TDR,VH), which was lower than TVH2. For comparison to the spike inactivation with VH doublets, we adjusted the DR-VH pair timing slightly so that the pairs of spikes (or expected spikes, at vertical lines) are separated by the same interval (~ 4 ms) when they reach the recording site, to compensate for DR conduction delays. The putative spike failure with DR stimulation happens at a node somewhere between the recording site and the VH, because we only studied axons that securely conducted single DR pulses to the recording site, and thus failure was not directly visible.
We quantified the spike failure based on the following considerations: If the DR-evoked spike entirely fails to propagate to the VH, then the threshold for subsequently activating the ventral horn (TDR,VH) should be the same as the threshold without any prior activation (TVH1 = TDR,VH), whereas if it does not fail, then the threshold for activating the ventral horn should be the same as with a VH doublet (TVH2 = TDR,VH). In between these two extreme scenarios, the DR evoked spike may only partially fail to propagate spikes to the ventral horn (by only some of its branches failing or conducting only partially to the VH); in this case TDR,VH should be between TVH1 and TVH2, with the difference TVH2 - TVH1 representing the range of possible thresholds between full failure and full conduction. Thus, overall the failure was quantified as: Conduction failure = (TVH2 - TDR,VH) / (TVH2 - TVH1) × 100%, which is 100% at full failure and 0% with no failure. This estimate is predicated on the assumption that the failed spikes are only relatively refractory to conduction and increased stimulation can overcome this failure, which is reasonable for the interspike intervals we used, and means that the computed % failure reflects the number of nodes that failed to spike, with more dorsal branch point failures giving more failed nodes. On the other hand, we used interspike intervals that were short enough for the DR stimulation not to evoke PAD that affected the subsequent spike threshold (~ 4 ms), in contrast to the longer intervals where PAD can help DR doublet firing (DR-DR in Extended Data Fig. 7, ~ 5 – 10 ms).
Extracellular recording from sensory axon terminals.
To directly record spike conduction in proprioceptive afferent terminal branches in the VH we used our intracellular glass pipette electrode (~30 MΩ) positioned just outside these axons (extracellular, EC), to avoid penetration injury in these fine axon branches. The DR was stimulated near threshold for spikes (1.1xT, T: afferent volley threshold) to evoke the EC response in a few single axons near the electrode, and many trials were averaged to remove noise from these small signals (20 – 50 trials at 3 s intervals). The EC field was multiphasic as previously described for other axons76–78, with a small initial positive field resulting from passively conducted axial current from sodium spikes at distant nodes (closer to the DR; outward current at electrode), some of which fail to propagate spikes to the VH recording site, making this field a measure of conduction failure76,78. Following this, a larger negative field arises, resulting from spikes arising at nodes near the electrode (inward current), making this negative field a measure of secure conduction. A relatively large stimulus artifact is present prior to these fields, due to the small size of the EC fields themselves, and we truncated this.
We conducted three control experiments to confirm the relation of these EC fields to spike conduction. First, in the dorsal horn where we can readily intracellularly record from large proprioceptive axon branches, we compared intracellular (IC) recordings from axons to EC recordings just outside the same axon, to confirm that the DR evoked spike (IC) arrives at about the time of the negative EC field. Second, we locally applied TTX to the DR near the recording site (10 μl bolus of 100 μM TTX over DR) which eliminated the negative field and left only the initial positive field, confirming that the positive field is from distal nodes upstream of the TTX block, and generated by passive axial current conduction. This is important, since some investigators have argued on theoretical grounds that the positive field can instead result from the closed-end electrical properties of axons at their terminals79, rather than spike failure, though others have refuted this78. Finally, we improved nodal spike conduction by reducing the divalent cations Mg++ and Ca++ in the bath medium, since divalent cations normally cause a gating or guarding action on the sodium channel, the latter by one charge binding to the membrane and the other raising the local extracellular positive charge, and overall raising the local voltage drop across the channel and its spike threshold80. This decreased the failure-related initial positive field and increased the main EC negative field, indicating improved conduction, and again confirming the use of these fields as measures of conduction, similar to previous conclusions for the motor endplate76 and mathematical consideration of axon cable properties 81.
To quantify the EC fields we estimated the overall conduction to the recording site as: Conduction Index = nf / (nf + pf) × 100%, where pf and nf are the positive and negative EC field amplitudes. This conduction index approaches 100% for full conduction (pf ~=0) and 0% for no conduction (nf = 0). The absolute EC field potential amplitudes are highly variable between different recordings sites, and thus are difficult to quantify across animals and sites, whereas this ratio of field amplitudes (nf / (nf + pf) ) eliminates the variability, and can effectively be viewed as a normalization of the negative field (nf) by the total field peak-to-peak size (nf + pf).
Intracellular recording from motoneurons.
The same intracellular glass electrode, stepper motor and amplifier used for recording sensory axons were used for intracellular recording from motoneurons, except that the electrodes were bevelled to a lower resistance (30 MΩ). The electrode was advanced into motoneurons with fast 2 μm steps and brief high frequency currents (capacitance overcompensation) guided by audio feedback from a speaker. After penetration, motoneuron identification was made with antidromic ventral root stimulation, and noting ventral horn location, input resistance and time constant (> 6 ms for motoneurons)63. The monosynaptic excitatory postsynaptic potentials (EPSPs) and associated currents (EPSCs) were measured in motoneurons following stimulation of dorsal roots (at 1.1– 1.5 xT, 0.1 ms, 3 – 10 s trial intervals). These were identified as monosynaptic by their rapid onset (first component), lack of variability in latency (< 1 ms jitter), persistence at high rates (10 Hz) and appearance in isolation at the threshold for DR stimulation (< 1.1xT; T, Threshold for EPSP, which also equals afferent volley threshold), unlike polysynaptic EPSPs which varying in latency, disappear at high rates, and mostly need stronger DR stimulation to activate.
Dorsal and ventral root grease gap recording.
In addition to recording directly from single proprioceptive axons and motoneurons, we employed a grease gap method to record the composite intracellular response of many sensory axons or motoneurons by recording from dorsal and ventral roots, respectively, as previously detailed for similar sucrose and grease gap methods, where a high impedance seal on the axon reduces extracellular currents, allowing the recording to reflect intracellular potentials15,81–83. We mounted the freshly cut roots onto silver-silver chloride wires just above the bath, and covered them in grease over about a 2 mm length, as detailed above for monopolar recordings. Return and ground wires were in the bath and likewise made of silver-silver chloride. Specifically for sensory axons, we recorded from the central ends of dorsal roots cut within about 2 – 4 mm of their entry into the spinal cord, to give the compound potential from all afferents in the root (dorsal roots potential, DRP, corresponding to outward sink current from inward PAD source current in source-sink analysis), which has previously been shown to correspond to PAD, though it is attenuated compared to the intracellular recordings of PAD15. The signal attenuation has two reasons. First the voltage PAD is attenuated along the length of nerve in the bath, as detailed in the next paragraph. Second, the grease does not completely remove the extracellular fluid around the nerve, even though we deliberately allowed the nerve to dry for a few seconds before greasing, and this causes a conductance that shunts or short circuits the recorded signal, reducing it by about half76,83. For optogenetic experiments we additionally added silicon carbide powder (9 % wt, Tech-Met, Markham) to the grease to make it opaque to light and minimize light induced artifactual current in the silver-silver chloride recording wire during optogenetic activation of ChR2 (detailed below). Likewise, we covered our bath ground and recording return wires with a plastic shield to prevent stray light artifacts. The dorsal root recordings were amplified (2,000 times), high-pass filtered at 0.1 Hz to remove drift, low-pass filtered at 10 kHz, and sampled at 30 kHz (Axoscope 8; Axon Instruments/Molecular Devices, Burlingame, CA).
These grease gap recordings of PAD on sensory afferents reflect only the response of largest diameter axons in the dorsal root, mainly group I proprioceptive afferents, because of the following considerations. First, the largest axons in peripheral nerves have a nodal spacing of about 1 mm84,85, and length constants λS are estimated to be similar, at about 1 – 2 times the nodal spacing86, Further, in our recordings we were only able to get the grease to within about 2 mm of the spinal cord. Thus, the centrally generated signal (PAD) is attenuated exponentially with distance × along the axon length in the bath (x = 2 mm). This is proportional to exp(− x / λS) (see 81), which is 1 / e2 = 0.11 for x = 2 λS, as is approximately the case here. This makes a central PAD of about 4 mV appear as a ~0.4 mV potential on the root recording (DRP, 10 times smaller)15. Furthermore, the nodal spacing and λS decrease linearly with smaller axon diameters81,84, making the voltages recorded on the smaller afferents contribute to much less of the compound root potential (halving the diameter attenuates PAD instead by 1/e4 or 0.012, which is 99% attenuation). Finally, unmyelinated sensory axons attenuate voltages over a much shorter distance than myelinated axons, since that membrane resistance drops markedly without myelin and λS is proportional to (where is axial resistance; Stein 1980). Thus, any centrally generated change in potential in these small axons is unlikely to contribute to the recorded signal 2 mm away.
The composite EPSPs in many motoneurons were likewise recorded from the central cut end of ventral roots mounted in grease (grease gap), which has also previously been shown to yield reliable estimates of the EPSPs, though again attenuated by the distance from the motoneurons87. The monosynaptic EPSPs were again identified as monosynaptic by their rapid onset (first component, ~1 ms after afferent volley arrives in the ventral horn; see below), lack of variability in latency (< 1 ms jitter), persistence at high rates (10 Hz) and appearance in isolation at the threshold (T) for evoking EPSPs with DR stimulation (< 1.1xT, T ~ afferent volley threshold), unlike polysynaptic reflexes which varying in latency, disappear at high rates, and mostly need stronger DR stimulation to activate.
Analysis of synaptic responses in sensory axons and motoneurons.
When we recorded from sensory axons of an associated dorsal root (directly or via the dorsal roots) stimulation of an adjacent dorsal root (not containing the recorded axon; 0.1 ms, 1 – 3xT; T: threshold to evoke PAD or EPSPs, same as afferent volley threshold) evoked a characteristic large and long depolarization of the afferents, previously demonstrated to be mediated by GABAergic input onto the sensory axons15. This depolarization is termed primary afferent depolarization (PAD). PAD occurs at a minimal latency of 4 – 5 ms following the afferent volley, consistent with its minimally trisynaptic origin13,15, making it readily distinguishable from earlier events on the axon (Supplementary Methods). PAD has a fast synaptic component evoked by a single DR stimulation (rising within 30 ms and decaying exponentially over < 100 ms; termed phasic PAD) and a slower longer lasting extrasynaptic component (starting at about 30 ms and lasting many seconds) that is enhanced by repeated DR stimulation (tonic PAD, especially with cutaneous stimulation)15. We used this sensory activation of PAD or direct optogenetic activation of PAD to examine the action of GABA on sensory axon spike transmission to motoneurons, usually evoking phasic PAD about 10 – 60 ms prior to spikes or associated EPSPs on motoneurons (during phasic PAD), though we also examined longer lasting effects of tonic PAD evoked by repeated DR stimulation. Sometimes PAD is so large that it directly evokes spikes on the afferents, and these travel out the dorsal root, and thus they have been termed dorsal root reflexes (DRRs)15,22. We usually minimized these DRRs by keeping the DR stimulus that evokes PAD low (1.1 – 3.0 xT), though there were inevitably some DRRs, as they even occur in vivo in cats and humans8,47,88.
When we recorded from motoneurons (directly or via ventral roots) stimulation of proprioceptive afferents in a dorsal root (0.1 ms, 1.1–1.5xT, T: EPSP threshold, which is similar to afferent volley threshold) evoked a monosynaptic EPSP, and associated monosynaptic reflex (MSR, spikes from EPSP). This EPSP is depressed by fast repetition (rate depended depression, RDD) 89, and thus to study the EPSP we evoked it at long intervals (10 s, 0.1 Hz rate) where RDD was less. However, even with this slow repetition rate (0.1 Hz), at the start of testing the first EPSP was often not similar to the steady state EPSP after repeated testing. Thus, to avoid RDD we usually ran the 0.1 Hz EPSP testing continuously throughout the experiment, at least until a steady state response was reached (after 10 minutes). We then examined the action of activating (or inhibiting) GABAaxo neurons on this steady state EPSP, by introducing light or sensory conditioning that activated these neurons at varying intervals (inter-stimulus intervals, ISIs) prior to each EPSP stimulation (control, GAD2//ChR2 mice and GAD2//Arch3 mice). We averaged the EPSP from ~10 trials (over 100 s) just before conditioning and then 10 trials during conditioning, and then computed the change in the peak size of the monosynaptic EPSP with conditioning from these averages. After conditioning was completed EPSP testing continued and any residual changes in the EPSP was computed from the 10 trials following conditioning (after-effect). Finally, EPSP testing continued over many minutes after which the original steady state EPSP was established. The background motoneuron potential, membrane resistance (Rm) and time constant just prior to the EPSP was also assessed before and after conditioning to examine whether there were any postsynaptic changes that might contribute to changes in the EPSP with conditioning. Along with the VR recordings, we simultaneously recorded PAD from DRs by similar averaging methods (10 trials of conditioning), to establish the relation of changes in EPSPs with associated sensory axon depolarization PAD.
Drugs and solutions
Two kinds of artificial cerebrospinal fluid (ACSF) were used in these experiments: a modified ACSF (mACSF) in the dissection chamber prior to recording and a normal ACSF (nACSF) in the recording chamber. The mACSF was composed of (in mM) 118 NaCl, 24 NaHCO3, 1.5 CaCl2, 3 KCl, 5 MgCl2, 1.4 NaH2PO4, 1.3 MgSO4, 25 D-glucose, and 1 kynurenic acid. Normal ACSF was composed of (in mM) 122 NaCl, 24 NaHCO3, 2.5 CaCl2, 3 KCl, 1 MgCl2, and 12 D-glucose. Both types of ACSF were saturated with 95% O2–5% CO2 and maintained at pH 7.4. The drugs sometimes added to the ACSF were APV (NMDA receptor antagonist), CNQX (AMPA antagonist), gabazine (GABAA antagonist), bicuculline (GABAA, antagonist), L655708 (α5 GABAA, antagonist), CGP55845 (GABAB antagonist; all from Tocris, USA), 5-HT, kynurenic acid (all from Sigma-Aldrich, USA), and TTX (TTX-citrate; Toronto Research Chemicals, Toronto). Drugs were first dissolved as a 10 – 50 mM stock in water or DMSO before final dilution in ACSF. DMSO was necessary for dissolving gabazine, L655708, bicuculline and CGP55845, but was kept at a minimum (final DMSO concentration in ACSF < 0.04%), which by itself had no effect on reflexes or sensory axons in vehicle controls. L655708 was particularly difficult to dissolve and precipitated easily, especially after it had been exposed a few times to air; so immediately after purchase we dissolved the entire bottle and froze it at −40°C in single use 5 – 20 μl aliquots, and upon use it was first diluted in 100 μl distilled water before dispersing it into ACSF.
Recording monosynaptic reflexes in awake mice and rats, and PAD activation.
Window implant over spinal cord.
In GAD2//ChR2+ mice and control GAD2//ChR− mice a glass window was implanted over the exposed spinal cord to gain optical access to the sacrocaudal spinal cord69. Briefly, mice were given Meloxicam (1 mg/kg, s.c.) and then anesthetized using ketamine hydrochloride (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.). Using aseptic technique, a dorsal midline incision was made over the L2 to L5 vertebrae. Approximately 0.1 ml of Xylocaine (1%) was applied to the surgical area and then rinsed. The animals were suspended from a spinal-fork stereotaxic apparatus (Harvard Apparatus) and the muscles between the spinous and transverse processes were resected to expose the L2 to L5 vertebrae. The tips of modified staples were inserted along the lateral edge of the pedicles and below the lateral processes of L2 and L5, and glued in place using cyanoacrylate. A layer of cyanoacrylate was applied to all of the exposed tissue surrounding the exposed vertebrae followed by a layer of dental cement to cover the cyanoacrylate and to form a rigid ring around the exposed vertebrae. A modified paperclip was implanted in the layer of dental cement to serve as a holding point for surgery. A laminectomy was performed at L3 and L4 to expose the spinal cord caudal to the transection site. Approximately 0.1 ml of Xylocaine (1%) was applied directly to the spinal cord for 2 – 3 s, and then rinsed. A line of Kwik-Sil (World-Precision Instruments) was applied to the dura mater surface along the midline of the spinal cord and a glass window was immediately placed over the exposed spinal cord. The window was glued in place along the outer edges using cyanoacrylate followed by a ring of dental cement. Small nuts were mounted onto this ring to later bolt on a backpack to apply the laser light (on the day of experimentation). Saline (1 ml, s.c.) and buprenorphine (0.03 mg/kg, s.c.) was administered post-operatively, and analgesia was maintained with buprenorphine (0.03 mg/kg, s.c.) every 12 hours for two days. Experimentation started 1 week after the window implant when the mouse was fully recovered.
Percutaneous EMG wire implant and fibre optic cable attachment.
On the day of experimentation, the mouse was briefly sedated with isoflurane (1.5 %) and fine stainless steel wires (AS 613, Cooner Wire, Chatsworth, USA) were percutaneously implanted in the tail for recording EMG and stimulating the caudal tail trunk nerve (wires de-insulated by 2 mm at their tip and inserted in the core of 27 gauge needle that was removed after insertion)63. A pair of wires separated by 8 mm were inserted at base of the tail for recording EMG the tail muscles, and second pair of wires was inserted nearby for bipolar activation of the caudal trunk nerve to evoke reflexes. A fifth ground wire was implanted between the EMG and stimulation wires. Following this a backpack was bolted into the nuts imbedded in the dental cement ring around the window. This backpack held and aligned a light fibre optic cable that was focused on the centre of the S3 – S4 sacral spinal cord. The Cooner wires were secured to the skin with drops of cyanoacrylate and taped onto the backpack so that the mouse could not chew them. The isoflurane was removed, and the mouse quickly recovered from the anesthesia and was allowed to roam freely around an empty cage during recording, or was sometimes lightly restrained by hand or by a sling. The fibre optic cable was attached to a laser (447 nM, same above) and the Cooner wires attached to the same models of amplifiers and stimulators used for ex vivo monosynaptic testing detailed above.
MSR testing.
The monosynaptic reflex (MSR) was recorded in the tail EMG at ~6 ms latency after stimulating the caudal tail trunk nerve at a low intensity that just activated proprioceptive afferents (0.2 ms current pulses, 1.1 xT, T: Threshold to evoke MSR, which is near afferent threshold), usually near the threshold to activate motor axons and an associated M-wave (that arrived earlier). See latency detail in Supplementary Methods. We studied the tail MSR reflex because our ex vivo recordings were made in the corresponding sacral spinal cord of adult mice and rats, which is the only portion of the spinal cord that survives whole ex vivo, due to its small diameter 65. This reflex was verified to be of monosynaptic latency because it was the first reflex to arrive, had little onset jitter, and had the same latency as the F wave (the F wave is evoked by a strong stimulation of all motor axons, at 5xT, which causes a direct motoneuron response on the same axons, while the monosynaptic EPSP is blocked by collision at this intensity) 90. The MSR also underwent rate dependent depression (RDD) with fast repeated stimulation and so was synaptic and not a direct muscle response (M-wave), which occurred earlier at sufficient intensity to recruit the motor axons.
Conditioning of the MSR by optogenetic activation of GABAaxo neurons.
As with in vitro EPSP testing, the MSR was tested repeatedly at long 5 – 10 s intervals until a steady state MSR was achieved. Then testing continued but with a conditioning light pulse applied just prior to the MSR stimulation (40 – 120 ms), to examine the effect of PAD evoked during this time frame on sensory transmission to motoneurons. Background EMG just prior to MSR testing was assessed to estimate the postsynaptic activity on the motoneurons. The changes in MSR and background EMG with light were quantified by comparing the average response before and during the light application, computed from the mean rectified EMG at 6 – 11 ms after the nerve stimulation (MSR) and over 20 ms prior to the nerve stimulation (background just prior to the MSR, Bkg). Because awake mice spontaneously varied their EMG, we plotted the relation between the MSR and the background EMG, with as expected a positive linear relation between these two variables91, computed by fitting a regression line. In trials with conditioning light applied the same plot of EMG vs background EMG was made and a second regression line computed. The change in the MSR with conditioning at a fixed matched background EMG level was then computed for each mouse by measuring the difference between the regression line responses at the fixed background EMG. This ruled out changes in MSRs being due to postsynaptic changes. Two background levels were assessed: rest (0%) and 30% of maximum EMG, expressed as a percentage of the control pre-conditioning MSR. The change in background EMG with light was computed by comparing the EMG just prior to the light application (over 20 ms prior) to the EMG just prior to the MSR (over 20 ms prior, Bkg), and expressed as a percentage of the maximum EMG.
Viral labelling of sensory afferents.
Large diameter peripheral afferents were labelled by viral injections43,92, providing an additional method of examining central afferent projections in the spinal cord. Adeno-associated viral vectors (AAVs) with the transgene encoding the cytoplasmic fluorophore tdTom under the CAG promoter were injected IP into anesthetized P1–2 mice (AAV9-CAG-tdTom, 5.9 × 1012 vg/ml; 2–4 μl per injection; UNC Vector Core). To improve transduction efficiencies, viral vectors were incubated with LAH4 peptide (200 μM) at 37 °C for ~45 min immediately prior to injection. Mice were perfused for immunolabelling > 60 days post injection (adult mice). This injection method yields a sparce Golgi-like labelling of about 5% of afferents in each spinal segment, without central labelling of other neurons (with the exception of one or two motoneurons labelled per spinal segment), allowing afferents to be traced to the motoneurons for morphological identification as Ia afferents.
Immunohistochemisty.
Tissue fixation and sectioning.
After sensory axons were injected with neurobiotin ex vivo in mouse and rat sacrocaudal spinal cords, the cords were left in the recording chamber in oxygenated nACSF for an additional 4 – 6 hr to allow time for diffusion of the neurobiotin throughout the axon. Then the spinal cord was immersed in 4% paraformaldehyde (PFA; in phosphate buffer) for 20–22 hours at 4°C, cryoprotected in 30% sucrose in phosphate buffer for 24–48 hours. Alternatively, afferents were labelled genetically in VGLUT1Cre/+; R26lsl-tdTom mice or by a AAV9-CAG-tdTom viral injection, which were euthanized with Euthanyl (BimedaMTC; 700 mg/kg) and perfused intracardially with 10 ml of saline for 3 – 4 min, followed by 40 ml of 4% paraformaldehyde (PFA; in 0.1 M phosphate buffer at room temperature), over 15 min (Gabra5-KO mice also fixed similarly). Then spinal cords of these mice were post-fixed in PFA for 1 hr at 4°C, and then cryoprotected in 30% sucrose in phosphate buffer (~48 hrs). Following cryoprotection all cords were embedded in OCT (Sakura Finetek, Torrance, CA, USA), frozen at −60C with 2-methylbutane, cut on a cryostat NX70 (Fisher Scientific) in sagittal or transverse 25 μm sections, and mounted on slides. Slides were frozen until further use.
Immunolabelling.
The tissue sections on slides were first rinsed with phosphate buffered saline (PBS, 100 mM, 10 min) and then again with PBS containing 0.3% Triton X-100 (PBS-TX, 10 min rinses used for all PBS-TX rinses). For the sodium channel antibody, we additionally performed antigen retrieval by incubating slides three times for 10 min each with a solution of 0.2% sodium borohydride (NaBH4, Fisher, S678–10) in PB, followed by a PBS rinse (4× 5 min), because this antibody is sensitive to over-fixation. We verified that this sodium channel antibody labels axon nodes just as well in our tissue treated with the antigen retrieval, compared to in control tissue that was only lightly fixed (PFA perfusion, followed by no postfixation). Next, for all tissue, nonspecific binding was blocked with a 1 h incubation in PBS-TX with 10% normal goat serum (NGS; S-1000, Vector Laboratories, Burlingame, USA) or normal donkey serum (NDS; ab7475, Abcam, Cambridge, UK). Sections were then incubated for at least 20 hours at room temperature with a combination of the following primary antibodies in PBS-TX with 2% NGS or NDS: rabbit anti-α5 GABAA receptor subunit (1:200; TA338505, OriGene Tech., Rockville, USA; same antibody as SAB2100878, Sigma-Aldrich, St. Louis, USA; verified with Western blot and IHC93,94, and knockout detailed below), rabbit anti-α1 GABAA receptor subunit (1:300; 06–868, Sigma-Aldrich, St. Louis, USA; verified by Western blot, IHC, and α1 GABAA knockout95,96), guinea pig anti-α2 GABAA receptor subunit (1:500; 224 104, Synaptic Systems, Goettingen, Germany; verified with Western blot, IHC, lack of labelling with loss of GABRA2 quantified with RT-qPCR97, and labelling in HEK239 cells transfected with GABRA2 cDNA98), chicken anti-γ2 GABAA receptor subunit (1:500; 224 006, Synaptic Systems, Goettingen, Germany; verified with Western blot, IHC and receptor colocalization with gephrine99), rabbit anti-GABAB1 receptor subunit (1:500; 322 102, Synaptic Systems, Goettingen, Germany), mouse anti-Neurofilament 200 (NF200) (1:2000; N0142, Sigma-Aldrich, St. Louis, USA), guinea pig anti-Neurofilament M (NFM, 1:500; 171 204, Synaptic Systems), guinea pig anti-VGLUT1 (1:1000; AB5905, Sigma-Aldrich, St. Louis, USA), rabbit anti-Caspr (1:500; ab34151, Abcam, Cambridge, UK), mouse anti-Caspr (1:500; K65/35, NeuroMab, Davis, USA), chicken anti-Myelin Basic Protein (MBP) (1:200; ab106583, Abcam, Cambridge, UK), guinea pig anti-GAD2/GAD65 (1:500; 198 104; Synaptic Systems); chicken anti-VGAT (1:500; 131 006, Synaptic Systems, Goettingen, Germany), rabbit anti-VGAT (1:500; AB5062P, Sigma-Aldrich, St. Louis, USA), rabbit anti-EYFP (1:500; orb256069, Biorbyt, Riverside, UK), goat anti-RFP (1:500; orb334992, Biorbyt, Riverside, UK), rabbit anti-RFP (1:500; PM005, MBL International, Woburn, USA), rabbit anti-GFP (1:500, A11122, ThermoFisher Scientific, Waltham, USA), and mouse anti-Pan Sodium Channel (1:500; S8809, Sigma-Aldrich, St. Louis, USA). The latter is a pan-sodium antibody, labelling an intracellular peptide sequence common to all known vertebrate sodium channels. Genetically expressed EYFP, tdTom (RFP) and GFP were amplified with the above antibodies, rather than rely on the endogenous fluorescence. When anti-mouse antibodies were applied in mice tissue, the M.O.M (Mouse on Mouse) immunodetection kit was used (M.O.M; BMK-2201, Vector Laboratories, Burlingame, USA) prior to applying antibodies. This process included 1h incubation with a mouse Ig blocking reagent. Primary and secondary antibody solutions were diluted in a specific M.O.M diluent. See Supplementary Table 2.
The following day, tissue was rinsed with PBS-TX (3× 10 min) and incubated with fluorescent secondary antibodies. The secondary antibodies used included: goat anti-rabbit Alexa Fluor 555 (1:200; A32732, ThermoFisher Scientific, Waltham, USA), goat anti-rabbit Alexa Fluor 647 (1:500, ab150079, Abcam, Cambridge, UK), goat ant-rabbit Pacific orange (1:500; P31584, ThermoFisher Scientific, Waltham, USA), goat anti-mouse Alexa Fluor 647 (1:500; A21235, ThermoFisher Scientific, Waltham, USA), goat anti-mouse Alexa Fluor 488 (1:500; A11001, ThermoFisher Scientific, Waltham, USA), goat anti-mouse Alexa Fluor 555 (1:500; A28180, ThermoFisher Scientific, Waltham, USA), goat anti-guinea pig Alexa Fluor 647 (1:500; A21450, ThermoFisher Scientific, Waltham, USA), goat anti-chicken Alexa Fluor 405 (1:200; ab175674, Abcam, Cambridge, UK), goat anti-chicken Alexa Fluor 647 (1:500; A21449, ThermoFisher Scientific, Waltham, USA), donkey anti-goat Alexa Fluor 555 (1:500; ab150130, Abcam, Cambridge, UK), donkey anti-rabbit Alexa Fluor 488 (1:500; A21206, ThermoFisher Scientific, Waltham, USA), Streptavidin-conjugated Alexa Fluor 488 (1:200; 016–540-084, Jackson immunoResearch, West Grove, USA) or Streptavidin-conjugated Cyanine Cy5 (1:200; 016–170-084, Jackson immunoResearch, West Grove, USA ) in PBS-TX with 2% NGS or NDS, applied on slides for 2 h at room temperature. The latter streptavidin antibodies were used to label neurobiotin filled afferents. After rinsing with PBS-TX (2 times × 10 min/each) and PBS (2 times × 10 min/each), the slides were covered with Fluoromount-G (00–4958-02, ThermoFisher Scientific, Waltham, USA) and coverslips (#1.5, 0.175 mm, 12–544-E; Fisher Scientific, Pittsburg, USA).
Standard negative controls in which the primary antibody was either 1) omitted or 2) blocked with its antigen (quenching) were used to confirm the selectivity of the antibody staining, and no specific staining was observed in these controls. Previous tests detailed by the manufactures further demonstrate the antibody specificity, including quenching, immunoblots (Western blots), co-immunoprecipitation, and/or receptor knockout. Most antibodies had been previously verified for selectivity, as detailed in the manufacture’s literature and other publications (see above)15, but we verified this for the GABA receptor antibodies with quenching (Supplementary Fig. 4). For antibody quenching, the peptides used to generate the antibodies, including anti-α5 GABAA receptor subunit (AAP34984, Aviva Systems Biology, San Diego, USA), anti-α1 GABAA receptor subunit (224–2P, Synaptic Systems, Goettingen, Germany) and anti-γ2 GABAA receptor subunit (224–1P, Synaptic Systems, Goettingen, Germany), were mixed with the antibodies at a 10:1 ratio and incubated for 20 h and 4°C. This mixture was then used instead of the antibody in the above staining procedure. Control receptor knockout experiments were also performed on for the anti-α5 GABAA antibody, with this antibody producing no receptor labelling in brain tissue from α5 GABAA knockout mice (Gabra5 KO mice; Supplementary Fig. 4).
Confocal and epifluorescence microscopy
Image acquisition was performed by confocal (Leica TCS SP8 Confocal System) and epifluorescence (Leica DM 6000 B) microscopy for high magnification 3D reconstruction and low magnification imaging, respectively. All the confocal images were taken with a 63x (1.4 NA) oil immersion objective lens and 0.1 μm optical sections that were collected into a z-stack over 10–20 μm. Excitation and recording wavelengths were set to optimize the selectivity of imaging the fluorescent secondary antibodies. The same parameters of laser intensity, gain and pinhole size was used to take pictures for each animal, including the negative controls. Complete sagittal sections were imaged with an epifluorescence 10x objective lens using the Tilescan option in Leica Application Suite X software (Leica Microsystems CMS GmbH, Germany). Sequential low power images were used to reconstruct the afferent extent over the whole spinal cord, using CorelDraw (Ottawa, Canada), and to identify locations where confocal images were taken.
3D reconstruction of afferents and localization of GABA receptors.
The fluorescently labelled afferents (neurobiotin, tdTom), GABA receptors, VGLUT1, VGAT, NF200, Caspr, MBP and sodium channels were analyzed by 3D confocal reconstruction software in the Leica Application Suite X (Leica Microsystems CMS GmbH) 15. To be very conservative in avoiding nonspecific antibody staining, a threshold was set for each fluorescence signal at a minimal level where no background staining was observed in control tissue with the primary antibody omitted, less 10%. Signals above this threshold were rendered in 3D for each antibody. Any GABA receptor, Caspr or NaV expression within the volume of the neurobiotin filled axon (binary mask set by threshold) was labelled in 3D reconstructions (yellow, pink and white respectively in Fig. 1). Receptor density within the axon membrane surface area was quantified using the same Leica software. Receptors are usually cycled in and out of the membrane63 and so receptors within the axon cytoplasm provide additional evidence of the presence of axonal GABA receptors, distinct from receptors that may be in the postsynaptic contacts of the afferents. Thus, we also computed the receptor densities in the axon volume and found qualitatively the same distribution (at nodes and terminals) as with surface density calculations and thus only reported the surface membrane density. Receptor densities were measured for all orders of branch sizes (1st, 2nd, 3rd etc.; see below), for both branches dorsal to the central canal (dorsal) and ventral to the central canal (ventral). Nodes were identified with dense bands of Caspr or Na channel labelling (and lack of MBP). Branch points were also identified. Many receptors and channels lay outside the afferent, and near the afferent these were difficult to manually identify without the 3D reconstruction software, making the 3D reconstruction the only practical method to fully quantify the receptors over the entire afferent. We also optimized the reconstruction of the neurobiotin filled afferents following the methods of Fenrich100, including brightening and widening the image edges slightly (1 voxel, 0.001 μm3) when necessary to join broken segments of the afferent in the final afferent reconstruction, to account for the a priori knowledge that afferents are continuous and neurobiotin signals tend to be weaker at the membrane (image edges) and in fine processes. Finally, we also counted the proportion of nodes and ventral boutons innervating motoneurons that contain GABA receptors clusters using the 3D viewer, as an additional quantification of the receptor distribution. Limitations in the sensitivity of receptor antibody labeling leave open the possibility that we missed small quantities of receptors. Thus, while we find that most proprioceptive afferent terminal boutons lack GABAA receptors15 (see Results) there may well still be small quantities of receptors. However, these receptors are unlikely to have much functional impact, since previous direct recording from ventral terminals boutons show little PAD at the time when PAD is observed in more dorsal portions of the same axons15.
GABA receptors usually occurred in the axons in distinct clusters. The distances from these receptor clusters to nodes or branch points was measured and average distances computed (from centers of clusters to centre of nodes), from high power confocal images evenly sampled across the axon arbour. Some nodes did not branch, so receptors at these nodes were fairly far from the nearest branch (~20 μm), making the average receptor to branch point distance larger than the receptor to node distance, the latter which were small because GABAA receptors were mainly only at nodes (see Results). The average distance between the receptor clusters and the nearest axon terminals on the motoneurons was also computed, but this was complicated by the very large distances often involved, forcing us to compute the distances from low power images and relate these to the high power images of receptors sampled relatively evenly along the axon arbour. For this distance calculation, to avoid sampling bias in the high power images, we only admitted images from axon branch segments (1st, 2nd and 3rd order, detailed below) that had a receptor density within one standard deviation (SD) of the mean density in branch types with the highest density (1st or 2nd order ventral branches for GABAA receptors and 3rd order ventral terminal branches for GABAB receptors; i.e. images from axons branches with density above the dashed confidence interval lines in Fig. 1e were included; this SD computed from densities of pooled axons from all rats, rather than single rat averages, to better reflect the axon density variability). This eliminated very large distances being included from branch segments with relatively insignificant receptor densities. We also confirmed these calculations by computing the weighted sum of all the receptor distances weighted by the sum of the receptor density for each branch type (and divided by the sum of all receptor densities), which further eliminated sampling bias. This gave similar average distance results to the above simpler analysis.
Sensory axon branch order terminology.
The branches of proprioceptive axons were denoted as follows: dorsal column branches, 1st order branches that arose of the dorsal column and project toward the motoneurons, 2nd order branches that arose from the 1st order branches, and 3rd order branches that arose from the 2nd order branches. Higher order branches occasionally arose from the 3rd order branches, but these were collectively denoted 3rd order branches. First and second order branches were myelinated with large dense clusters of sodium channels at the nodes in the myelin gaps, which were characteristically widely spaced. As the second order branches thinned near the transition to 3rd order branches, they became unmyelinated, and at this point sodium channel clusters were smaller and more closely spaced (~6 μm apart). These thinned branches gave off 3rd order (and higher) unmyelinated terminal branches with chains of characteristic terminal boutons that terminated on motoneurons. The 1st order branches gave off 2nd order branches along most of their length as they traversed the cord from the dorsal columns to the motoneurons, but we separately quantified 1st, 2nd and 3rd order branches in more dorsal (including dorsal and intermediate laminae) and ventral regions of the cord.
Node identification.
Nodes in myelinated axon segments nodes were identified either directly via direct Na channel clusters and paranodal Caspr, or indirectly by their characteristic paranodal taper. That is, in the paranodal region the neurobiotin filled portion of the axon tapered to a smaller diameter, which also made the intracellular neurobiotin label less dense (Fig. 1b, black regions in taper). This taper forces the axial current densities to increase at the nodes, presumably assisting spike initiation, and consistent with previous reconstructions of myelinated proprioceptive afferents101.
GAD2 neuron labelling.
GABAaxo neurons that express GAD2 were visualized by genetically tagging them with Cre-ER driven fluorescent reporters. Usually we used the ChR2-EYFP reporter to both insert ChR2 and label with EYFP. This ChR2 construct is membrane bound and so does not fill soma or large processes making cells sometimes hard to visualize. Thus, in some animals we additionally included the Cre driven tdTom reporter, which is a cytoplasmic reporter that fills the entire cell to help visualize the complete anatomy of the entire GABAaxo neuron (Fig 3). In this case, GAD2 neurons should have both EYFP (green in Fig 3) and tdTom (red) reporter labelling. However, the balance of green and red expression intensity was variable, with some processes with more EYFP and others with more tdTom, leading to some axons more one color than the other. This was likely due to a number of factors. First, membrane bound fluorophores are easier to see in small diameter axons or dendrites, because of a higher membrane-to-cytoplasm ratio, making red more visible in small axons. Second, variability in tissue penetration of the antibodies we used to amplify the reporter signals and more intense ChR2-EYFP labelling (green) in axons may have led to variable red and green intensity. Finally, genetic variability in the Cre-ER driven reporter expression, which only occurs transiently after the tamoxifen administration, may explain why a small proportion of neurons are either just green or just red, with one reporter not expressed by this transient Cre expression. Expression of only one reporter happened in only a small proportion of neurons, but when it did our double reporter method is an advantage in visualizing these neurons.
Computer simulations
All computer models and simulations were implemented in NEURON ver7.5 102. The geometry and myelination pattern of the model were extracted from a previous study that used serial-section electron microscopy to generate about 15,000 photomicrographs to reconstruct a large myelinated proprioceptive Ia afferent collateral in the cat (Nicol and Walmsley, 1991)101. This structure was used in a prior modeling study 25. Four classes of segment were defined in the model: myelinated internodes, nodes, unmyelinated bridges, and terminal boutons. Data from 18 of the 83 segments were missing from the original study. The missing data were estimated using mean values of the same segment class. The cable properties of the model were determined from diameter-dependent equations previously used for models of myelinated axons103 and included explicit representation of myelinated segments using the double cable approach103–105. Hodgkin-Huxley style models of voltage gated sodium (transient and persistent) and potassium channels were adopted from a previous study, at 37°C103. All three voltage-gated conductances were colocalized to unmyelinated nodes and segments throughout the modelled axon collateral. The density of sodium and potassium conductances was adjusted to match the size and shape of experimentally recorded action potentials. To be conservative, sodium channels were placed at each node and bouton (gNa = 1 S/cm2), even though bouton immunolabelling for these channels was not common in our terminal bouton imaging (Fig. 1), since disperse weak sodium channel labelling may have been missed. Removing these bouton sodium channels did not qualitatively change our computer simulation results. Current clamp stimulation was applied to the middle of the first myelinated segment (pulse width 0.1 ms, amplitude 2 nA; near dorsal root) to initiate propagating action potentials in the model. Voltage at multiple sites of interest along the collateral was measured to assess propagation of action and graded potentials through branch points. Transient chloride conductance (i.e. GABAA receptors) was modeled using a double-exponential point process (Eq. 1); parameters were manually fit to experimental data. GABAA receptors were localized to nodes at branch points to match experimental data. The amplitude and time course of the modeled PAD (also termed PAD) was measured from the first myelinated internode segment, similar to the location of our intra-axonal recordings.
| (Eq. 1) |
The parameters at all synapses were the same: time constant of rise (τrise) = 6ms, time constant of decay (τdecay) = 50ms, default maximum conductance (gmax) = 1.5nS (varied depending on simulation, see figure legends), and chloride reversal potential (ECl−) = −25mV (i.e. 55 mV positive to the resting potential to match our experimental data)15. Space constants (λS) were computed for each segment of the afferent, from subthreshold current injections (100 ms) on the distal end of each branch segment and fitting an exponential decay (with space constant λS) to the passive depolarization along its length, and then repeating this with current injected in the proximal end to get a second λS, and finally averaging these two space constants.
Statistics and reproducibility
Data were analyzed in Clampfit 10 (Axon Instruments, USA), Excel version 2206 (Microsoft, USA) and Sigmaplot 14.5 (Systat Software, USA). A Student’s t-test or one-way ANOVA (as appropriate) was used to test for statistical differences between two or more independent comparisons, respectively, with a significance level of P < 0.05, all two-sided. Tests were paired for pre and post treatment data, but otherwise were unpaired. Multiple comparisons with t-tests or ANOVA were followed by a post-hoc Bonferroni correction or Tukey test to determine which pairs of measures likely differed, while compensating for the multiple comparisons indicated. A priori knowledge was used to determine the measures compared, since there is no need to compare experiments on separate issues, even though they we often put many experiments on one graph to save space. For example, we plotted GABAA and GABAB receptor antagonist data together, but we know a priori that GABAA and GABAB receptors are different, and thus reflex responses with and without GABAA receptor antagonists or with and without GABAB receptor antagonists were only compared with a t-test, rather than an ANOVA, since we have no interest in whether the responses to GABAA and GABAB receptors are different. Also, for an ANOVA to be used, the repeated measures should be independent, otherwise it weakens the statistical test. Thus, when we have a priori knowledge that measures are dependent, then comparison to a control factor only requires a t-test, rather than an ANOVA. Power analysis was used a priori to design experiments and determine sample sizes n (with α = 0.05, β = 0.2, r = 0.8–1.6, t-test or ANOVA, where r = effect size, based on previous studies15). Animals were randomly allocated to different groups for the in vitro and in vivo experiments using a block design. Data collection and analysis were not performed blind to the conditions of the experiments, but the collection and analysis was automated so that the experimenter had no influence on the outcome. A Kolmogorov-Smirnov test for normality was applied to the data set, with a P < 0.05 level set for significance. Data sets were found to be normally distributed, as is required for a t-tests and ANOVA, but significance was often confirmed with Wilcoxon Signed Rank Tests with P < 0.05. Categorical data was instead analyzed using two-sided Chi-squared tests, with Yate’s continuity correction used for 2 × 2 contingency tables and again significant difference set at P < 0.05. Effects in male and female animals were similar and grouped together in analysis. For in vivo experiments, a single data point is taken from the average response in each subject/animal and n values indicate subject number. Axons and motoneurons were recorded ex vivo from widely separated locations (one segment apart or contralateral) within the whole spinal cord, and are considered independent; so statistics were performed across all neurons (n) from all animals, though all main effects were confirmed to occur in each animal, and comparing across animal averages also showed significant changes (n animal numbers). Data are indicated as box plots representing the interquartile range and median (thin line), whiskers (error bars) representing the 90th and 10th percentile interpolated between nearest points (usually maxima, Cleveland method), and dots representing maxima. Mean also shown as thick line in boxes.
Extended Data
Extended Data Fig. 1 |. Nodal and not terminal GABAA receptors in mice and rats.
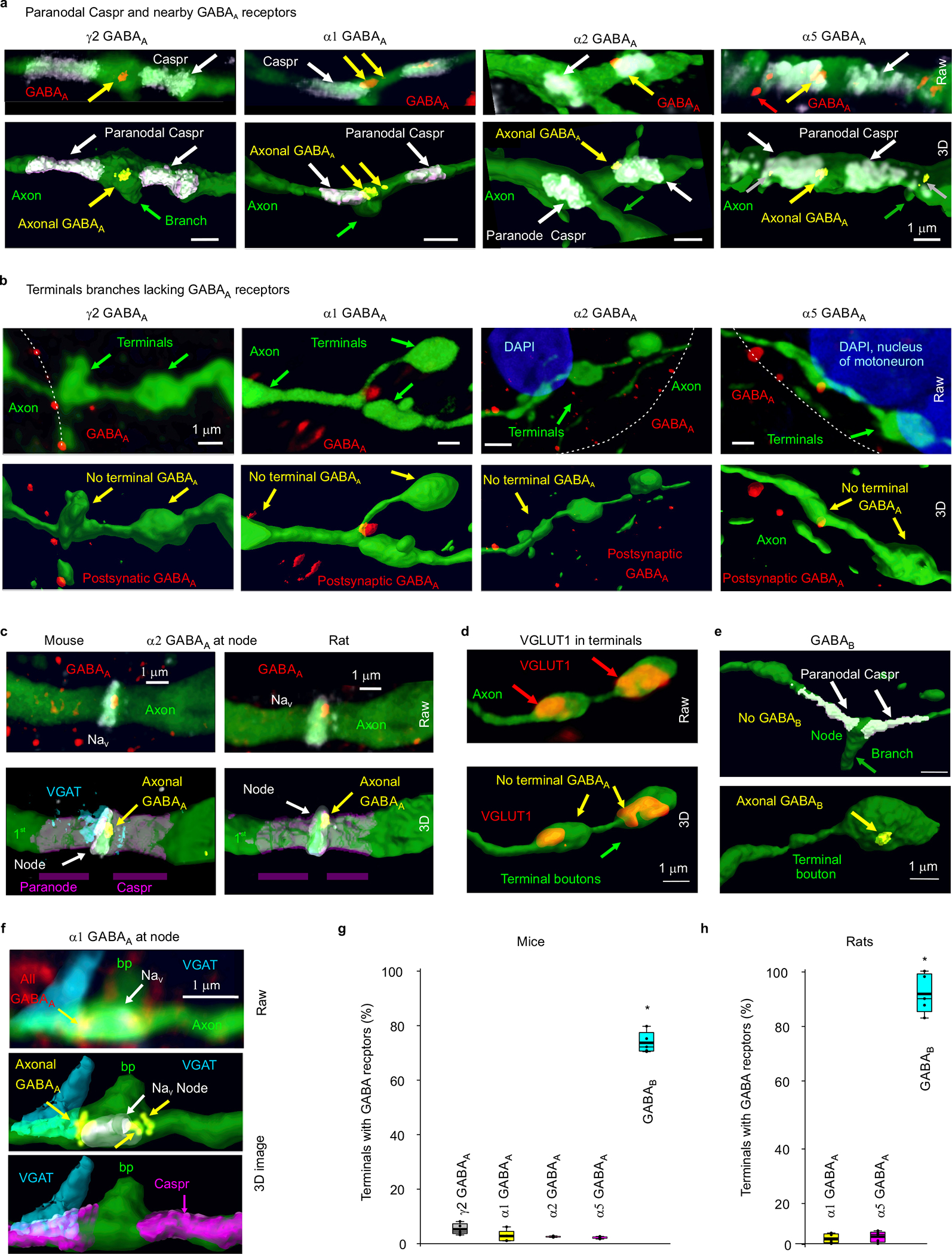
a-b, In the sacrocaudal spinal cord of mice we examined the distribution of GABAA receptor subunits on nodes and terminals of sensory axons, including extrasynaptic α5 subunits, synaptic α1 and α2 subunits, and ubiquitous γ2 subunits (e.g. forming the common α1βγ2 or α5βγ2 receptors, though less common extrasynaptic α1βδ have been reported)106–109. We genetically labelled primary sensory axons by their expression of the vesicular glutamate transporter VGLUT1 with a reporter in VGLUT1Cre/+; R26LSL-tdTom mice (tdTom reporter displayed as green, for consistency with Fig. 1). VGLUT1 is mainly only expressed in sensory axons60, especially ventral proprioceptive afferents, as other afferents do not reach the ventral horn15. Axons were reconstructed in 3D as detailed in Fig. 1. 3D reconstructed nodes of Ranvier on myelinated 2nd order ventral branches are shown identified by paranodal Capsr immunolabelling (a, bottom), along with raw confocal image stacks (maximum projection from z-stack) prior to 3D reconstruction (a, top; receptors red). As in rats, nodes were often near branch points (green arrow). Terminal boutons from the ventral horn are likewise shown with raw and 3D rendered images (b). GABA receptors colocalized with the axon are labelled yellow in the 3D reconstructions. Receptor clusters specifically in the plasma membrane are indicated by yellow arrows in (a). Similar to rats, the α5, α1, and γ2 GABAA receptor subunits were found on large axon branches (1st and 2nd order) in the dorsal, intermediate, and ventral cord, near nodes (a), but not on ventral horn terminal boutons (b, 3rd order). As also seen in rats (Fig 1e), synaptic GABAA receptors were usually in single large membrane bound clusters at nodes, whereas extrasynaptic α5 receptors were often broken up into multiple clusters, with the largest clusters in the membrane (yellow arrows), and smaller cytoplasmic clusters several μm from the edge of the node under the paranodal Caspr (grey arrows; Fig 1f). Cytoplasmic α5 receptors have been reported previously95. Many receptors were in neighboring neurons (red arrows), and in our previous publication15 these and cytoplasmic axon receptors may have been mistaken for nodal receptors in the axon membrane, though this is corrected here with evaluation of higher resolution confocal images. The presence of these α5, α1, and γ2 GABAA subunits is consistent with their mRNA previously reported in the dorsal root ganglion110. Also, the finding of α1 subunits on these axons is consistent with the recent observation that α1 is only on myelinated sensory axons, rather than unmyelinated C fibres111. c, Synaptic α2 GABAA receptor labelling in mouse and rat axons (rat axon labelled as in Fig 1) with nodes labelled with both antibodies to sodium channels NaV and Caspr, to confirm the relation of receptors to the node and paranodal region. Receptors are often at the transition between the node and paranodal Caspr, as here. Putative GABAergic synaptic contact labelled with VGAT. d, Ventral terminal boutons in VGLUT1Cre/+; R26LSL-tdTom mouse (with reporter labelling the complete axon, green) immunolabelled with VGLUT1 to verify that these are afferent terminals, which have vesicular VGLUT1+ protein expression. Immunolabelling for γ2 GABAA receptors again showed that terminals lacked this ubiquitous subunit that makes up most GABAA receptors. e, Immunolabelling for GABAB receptors on 3D reconstructed sensory axons, with same format and mice of (a-b). GABAB receptors were generally absent from nodes identified by paranodal Caspr, but present on ventral terminal boutons, as in rats (Fig. 1). Similar results (a- e) were obtained from n = 5 mice. f, Synaptic α1 GABAA receptor labelling at a rat Ia afferent node labelled with both antibodies to sodium channels NaV and Caspr, to confirm the relation of receptors to the node and paranodal region in rat, like in mouse (c, same node as in left of Fig 1c, but NaV and raw images included). Putative GABAergic synaptic contact labelled with VGAT, where left red arrow shows GABAA receptor contacting VGAT at edge of node. VGAT is near, but does not contact Caspr (in 3D view), but may well contact the paranodal myelin loops, since oligodendrocytes express GABA receptors38. Node is at dorsal 2nd order axon branch point. (a-f) representative of 5 mice. g-h, Box plots of the proportion of ventral terminal boutons with GABA receptors in both mice (g, VGLUT1+) and rats (h), again showing that few boutons contain GABAA receptors, whereas many contain GABAB receptors. * significantly more GABAB than GABAA receptors, ANOVA with Bonferroni correction, P < 0.05, n = 5 animals per condition, with 70 – 120 terminals examined per animal and receptor. Box plots show the interquartile range (box), median (thin line), mean (thick line), 10 and 90 percentiles (whiskers), and extremes (dots).
Extended Data Fig. 2 |. Voltage dependence of nodal spike propagation failure following dorsal root stimulation.
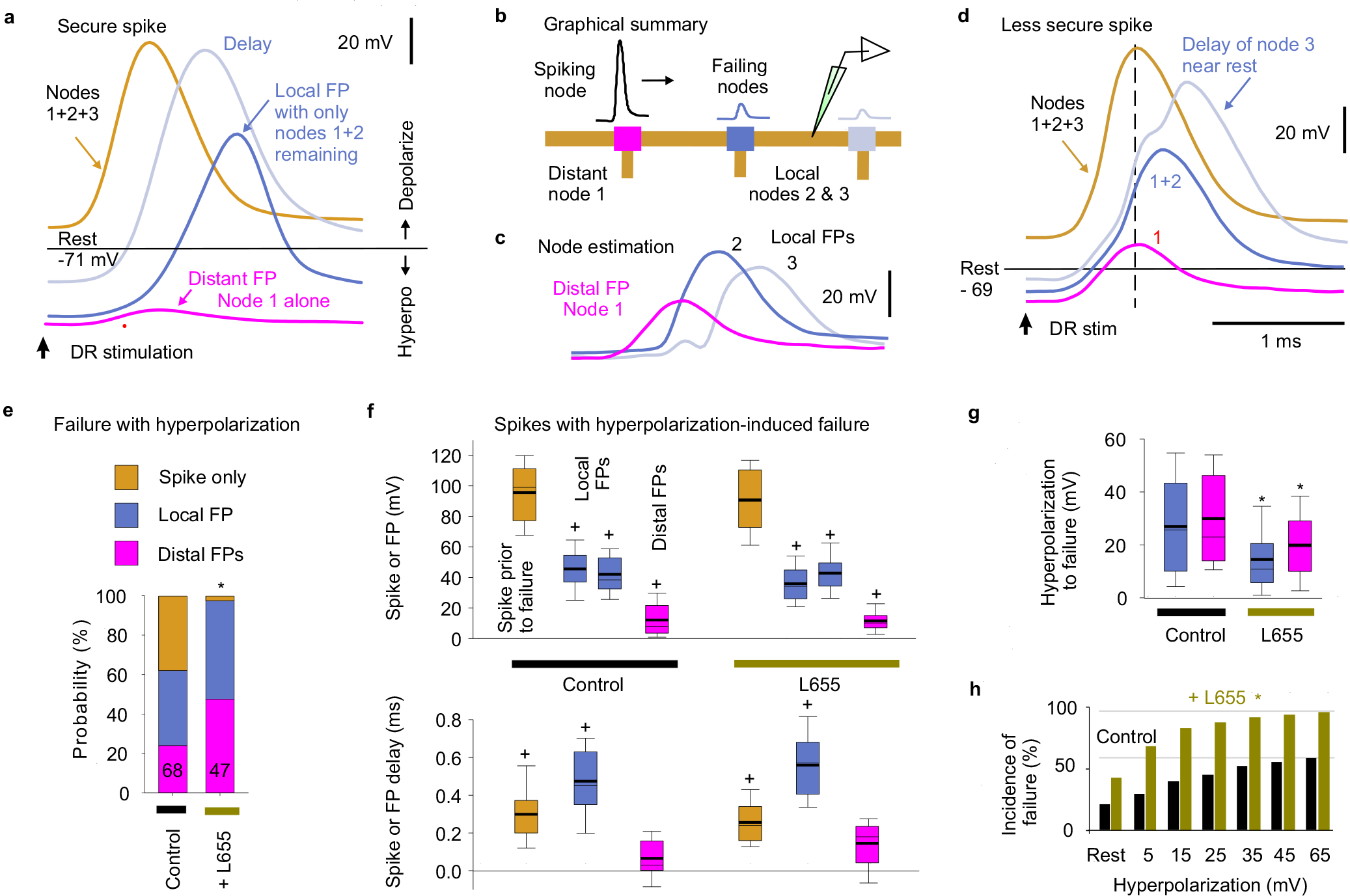
a-d, Intracellular recording from proprioceptive Ia afferent branches in the rat dorsal horn with secure spikes at rest, evoked by DR stimulation (1.1xT, 0.1 ms; T: afferent volley spike threshold; sacral S4 DR; a, d). Spike failure was induced by increasing hyperpolarization (failure near rest in d, but not a), with a delay and then abrupt loss of height, reflecting failure of successively further away nodes (a, d). Of the two local nodes adjacent to the electrode, one failed first with hyperpolarization, leaving the attenuated spike from the other node (local FP, about 1 λS away), which eventually failed as well with further hyperpolarization, leaving a much smaller FP from more distal nodes (distal FP). Spike failure in a node was always proceeded by a delay in the nodal spike. Estimated contributions from local nodes (b-c) were computed by subtraction from traces in (d). Spike attenuation was down to about 1/e by the second failure, consistent with the space constant λS being two internodal distances (c), or about 90 μm. Note that due to attenuation of injected current with distance, larger hyperpolarizations were needed to stop spikes with more distal vulnerable nodes (secure spikes), as observed by smaller distal FPs (a verses d), and some axon spikes could not be stopped (Spike only, quantified in e). Data collected in discontinuous current clamp (DCC) mode so electrode rectification during the injected hyperpolarizing current did not affect potential (DCC switching rate 7 kHz, low pass filtered at 3 kHz to remove switching artifact, which also removed stimulus artifact). e, Distribution of spiking in branches with full spikes only, one local nodal spike (local FP), or a distal nodal spike (distant FP) remaining after maximal hyperpolarization, and * significant change when blocking α5 GABAA receptors with L655708 (0.1 – 0.3 μM) using two-sided χ-squared test, P < 0.05; n = 68 control and n = 47 L655708 treated axon branches from 7 rats. f, Box plots of spike or FP heights and delays in branches and rats indicated in (e), measured just prior to spike failure (or at maximal hyperpolarization for secure spikes) and after failure (for local and distal FPs, as induced in a-d). Delay measured relative to peak of spike at rest. + FP significantly different than spike at rest, n= 7 rats, two-sided paired t-test, P < 0.05. g, Box plots of the hyperpolarization needed to induce failure, in axon branches and rats from (e). * significant change with L655708, two-sided unpaired t-test, P < 0.05, n from (e). Box plots show the interquartile range (box), median (thin line), mean (thick line), extremes, 10 and 90 percentiles (whiskers). h, Incidence of failure at varying potentials (% of total spikes from e). * significantly more failure with L655708, two-sided χ-squared test, n from (e), P < 0.05. Note that secure spikes in rats and mice are not overall different when measure by spike height at rest (Figs. 2g and 3g). However, the spike height shown here in (f) is the height while the cell is hyperpolarized far from rest, and so is much larger than at rest, since spikes generally overshoot to near the reversal potential for sodium, and so spike heights while hyperpolarizing are not comparable to the spikes at rest in mice or rats (measured from holding potential to peak). Thus, during hyperpolarization the nodes produce larger overall spikes, including FPs from local nodes (a-f). Also, during hyperpolarization from current injection, the two adjacent nodes to the electrode fail, unlike during natural failures, and thus when one nodes fails the spike only halves in height, leaving the spike from the second adjacent node (a-b). Thus, these local FPs are large in relation to the FPs from distal nodes with natural failure (Fig 2).
Extended Data Fig. 3 |. Voltage dependence of spike failure with simulated nodal currents.
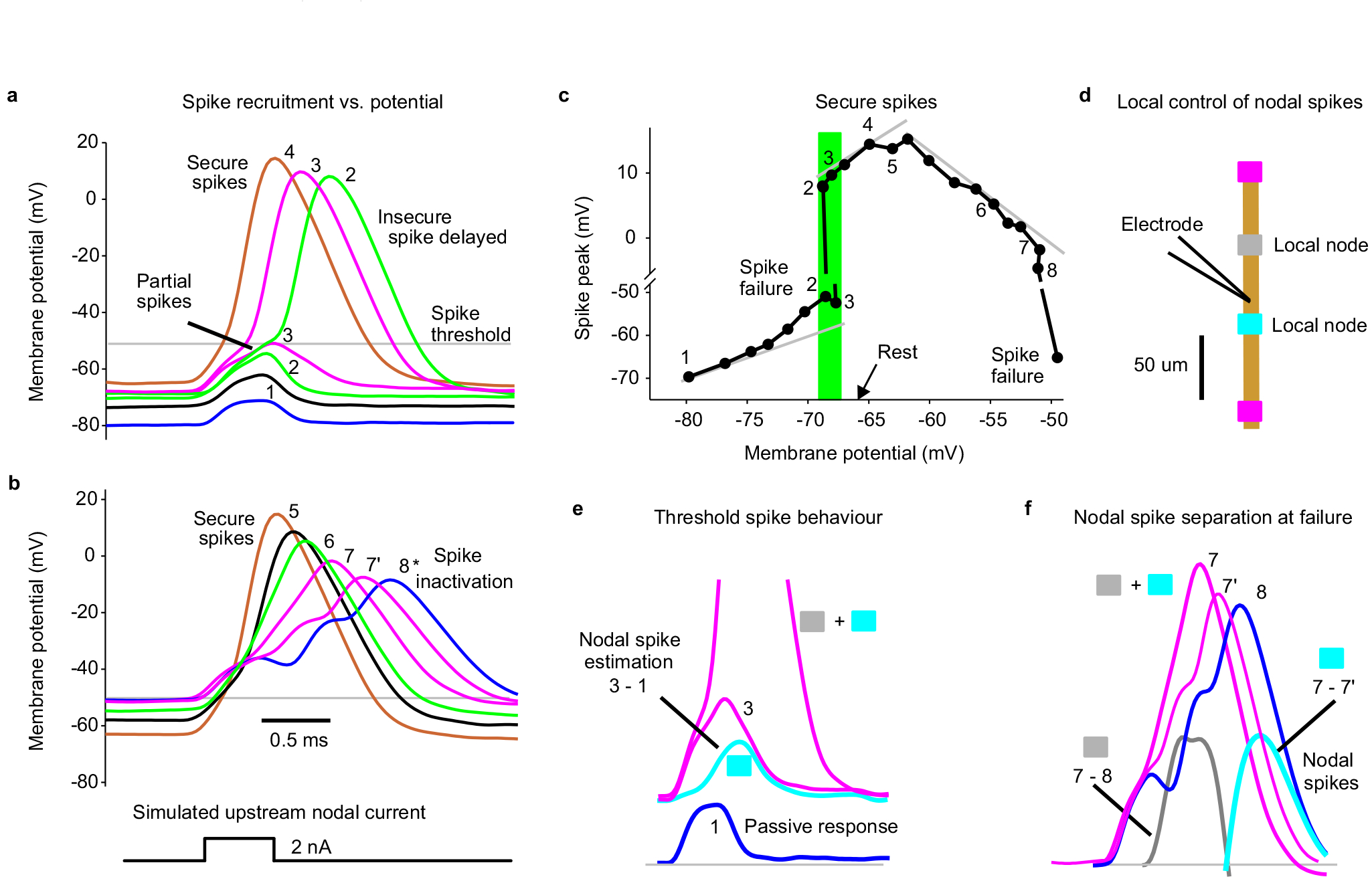
Simulating spike propagation failure in a proprioceptive axon by applying a brief intracellular current injection to mimic the current arriving from an upstream node (and FP), yielded full spikes evoked at rest, but nearby nodal spikes delayed and then failing as the membrane was held progressively more hyperpolarized with a steady bias current. Also, large steady depolarizations inactivated these spikes, though well outside of the physiological range (> −50 mV). a, Intracellular recording from proprioceptive Ia afferent branch in the rat dorsal horn (sacral S4 axon). A brief current injection pulse (0.5 ms) was applied to simulate the current arriving from distal nodes during normal axon spike conduction, and repeated at 1s intervals. During these pulses the membrane was held at varying potentials for 1 – 2 s with steady current injection, with numbers and colours denoting a given holding potential (in DCC mode). At the most hyperpolarized levels spikes failed to be evoked and only the passive response is seen, like a FP (blue, 1). As the potential was depolarized to near the axon’s resting potential (−67 mV) partial spikes occurred (green, 2 and 3), likely from a single adjacent node activating, and then delayed broad spikes occurred, as both adjacent nodes were activated. At more depolarized levels the spikes arose more rapidly and increased in height to full secure spikes (4). b, In the same axon as (a), at holding potentials well above those seen physiologically (near −50 mV, lower plots) spikes started to exhibit sodium channel inactivation and failure, with a decrease in spike height and delay (7 – 8) and eventually full failure (shown in c). Adjacent nodes started failing at slightly different times with different delays, broadening the spike and eventually separating into two distinct nodal spikes (8*). c, Spike heights plotted as a function of holding potential, including those spikes illustrated in (a and b), with spike number-labels indicated. Left grey line indicates passive leak current response, and shows deviation from passive response near rest. Shaded green region shows all or nothing failure or spikes near the resting potential. Middle grey line shows a region of secure spikes with relatively invariant spikes. Right grey line shows spike inactivation with large depolarizations and outright failure near - 50 mV. Note the split vertical axis. Similar voltage dependence of spike failure occurred for n = 5/5 axons showed similar results, from 4 rats. This demonstrates two modes of spike failure: 1) spikes that fail at rest or at hyperpolarized potentials and 2) spikes that fail with large depolarizations above rest. The latter is likely not physiological, since even the largest PAD that we have observed (5 – 10 mV; Fig. 4d) does not depolarize axons to −50 mV, since axons rest near −70 mV, and PAD is only large at hyperpolarized levels (Fig. 4d) and decreases steeply as the potential approaches the reversal potential for chloride (−15 mV)15. d, Schematic of recording arrangement and relation to adjacent nodes for data in (e) and (f). e, Expansion of responses 1 (blue) and 3 (pink) from (a), and difference (cyan) to show the first local nodal spike height at threshold, recorded at electrode. Active nodes from schematic in (d) shown with shaded boxes. f, Spikes near sodium inactivation from (b) (7 and 8), with differences indicating local nodal spikes (grey: 7 – 8, and cyan: 7 – 7’, both truncated to only show estimated nodal spike). Nodes likely arranged as in (d).
Extended Data Fig. 4 |. GABAergic innervation of nodes and nearby boutons in viral vector labelled proprioceptive afferents.
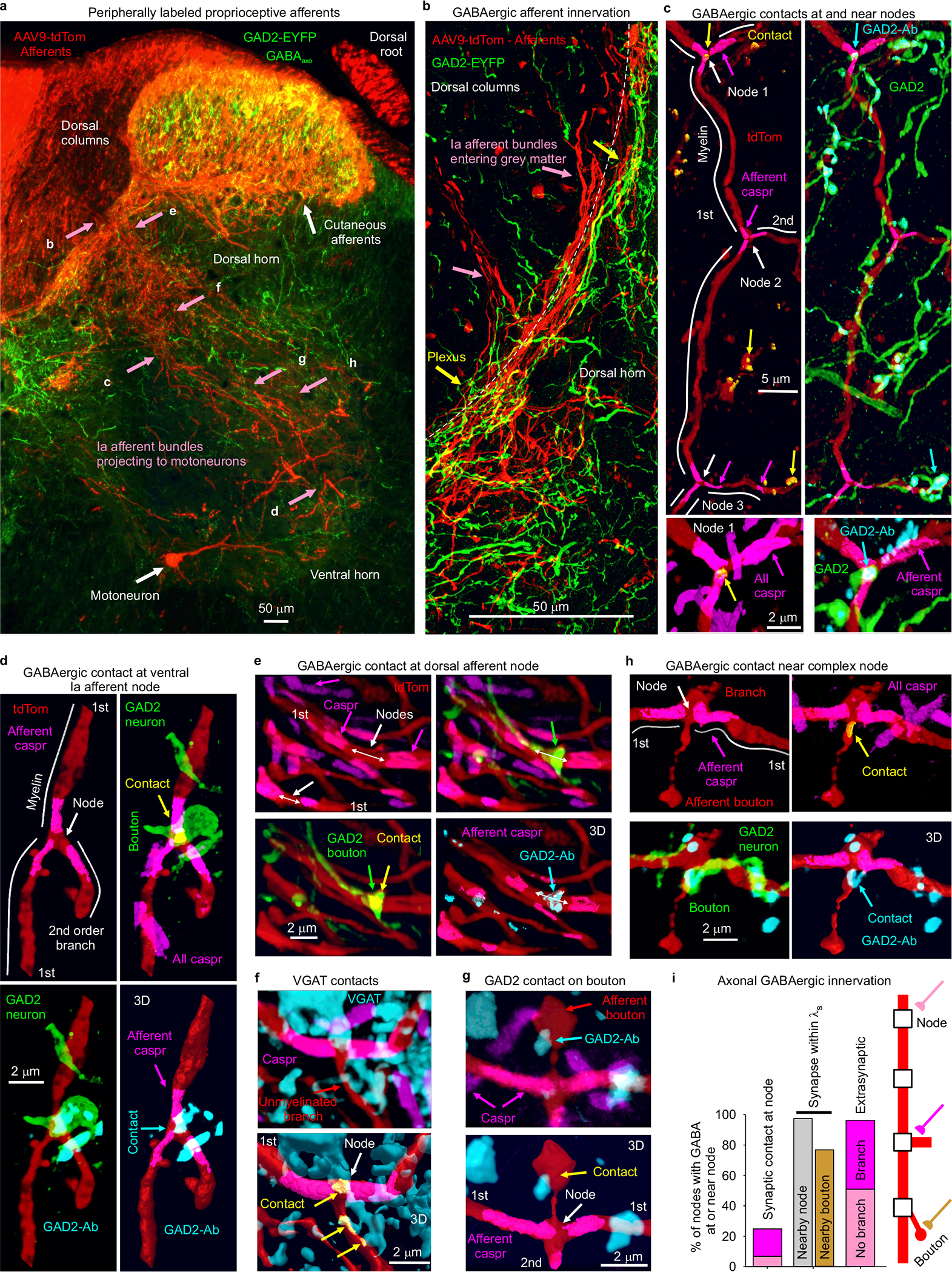
a, Sensory afferents labelled in the spinal cord and DR of adult GAD2//ChR2-EYFP mouse (GAD2-EYFP) by a peripheral adeno- associated virus (AAV9-tdTom); confocal image of a transverse spinal cord section. Large myelinated proprioceptive Ia afferents were identified by characteristic extensive ventral horn branching and innervation of motoneurons (as in Fig 1a). These Ia afferents left the dorsal columns in bundles that were readily traced to the ventral horn in serial sections, with an S-shaped projection path from the dorsal columns to the motoneurons (as in Fig 3j)112. The relatively sparce afferent labelling helped facilitate this tracing of afferent innervation. The peripheral AAV9-tdTom injection occasionally also labelled one motoneuron in a transverse (lower white arrow), though this only occurred a couple times per spinal cord segment, making them possible to distinguished from afferents elsewhere. No other central neurons were labelled, indicated that the viral vector only affected peripheral sensory and motor axons, as we previously found43. EYFP+ cells (GABAaxo neurons) were only in the dorsal and intermediate laminae (GAD2-EYFP+), with projections into the ventral horn and dorsal columns along the entire path of the Ia afferent bundles, as detailed in Fig 3. The approximate regions where images in (b-h) were taken are indicated in (a), but in different transverse sections, oriented similarly. b, GABAaxo neurons formed a dense plexus that wound around Ia afferents as they branched out of the dorsal columns into the grey matter (at dashed line). c, Main 1st order myelinated branch of a Ia afferent in intermediate laminae with three nodes at branch points labelled with axonal caspr, approximate myelin regions marked with white lines, and GABAaxo neurons seen wrapping the axon (top images). Node 1 had a GAD2-EYFP+ contact that had presynaptic GAD2 labelled with an antibody (GAD2-Ab, cyan; yellow marks contact of afferent, GAD2-EYFP and GAD2-Ab computed in 3D), whereas Nodes 2 and 3 lacked contacts, though the short branch arising off of Node 3 had a nearby unmyelinated terminal branch with a GAD2+ contact. GAD2 (GAD65) is known to be closely associated with GABAergic terminals and vesicular GABA and highly enriched in terminals compared to elsewhere in neurons5,7,113,114, and thus the GAD2-Ab provides a presynaptic contact label, consistent with the intense GAD2-Ab labelling we observed in GAD2-EYFP boutons (also bassoon+), and similar to the GAD65-intense boutons on afferents previously detailed7. Expanded view of Node 1 shown at bottom left, where all caspr labelling is displayed, including in overlapping nearby axons, whereas in other images only the axonal caspr in the 3D volume of the afferent is shown for clarity, together with raw images from other antibodies. Expanded view of Node 1 contact shown on bottom right, where all GAD2-ab labelling is shown, whereas in other images only GAD2-Ab in the GAD2-EYFP neuron volume is shown. Not all GAD2-Ab was in GAD2-EYFP+ neurons, likely due to variability in tamoxifen induced cre or transport of EYFP in GAD2//Ch2R-EYFP mice. d, Nodal contact in the ventral horn, shown with similar format to (c), with again a GABAaxo neuron wrapping around the axon and specifically making a contact at the node (yellow; EYFP+, GAD2-Ab+). White lines: estimated myelin regions. 1st order afferent. e, Two nodes on two 1st order Ia afferent branches in the dorsal horn plexus of (b), again delineated by paranodal caspr, one of which had a GABAaxo neuron contact (GAD2- EYFP, marked yellow again) with presynaptic GAD2-Ab (cyan, shown in 3D volume of GABAaxo neuron) and the other did not. f-g, Nodes from 1st order Ia afferent branches, identified by paranodal Caspr, in control wild-type mice, and so lacking EYFP, but inhibitory innervation examined with VGAT or GAD2 antibodies. In (f) the node had a direct VGAT+ contact and nearby contacts on the small unmyelinated branch arising from the node (contacts computed in 3D labelled yellow). In (g) the node lacked a direct GABAergic contact, but had a nearby GAD2-Ab+ contact on a bouton of a short unmyelinated branch arising from the node. h, Complex node in intermediate laminae with enlarged inter-myelin region and two unmyelinated branches, one with a GAD2-Ab+ contact on the neck of the branch, and the other with contacts on nearby boutons (above top of image). Same format as (d-e). Similar to enlarged nodal boutons of Walmsley25. (a-h) representative of n = 3 mice. i, Quantification of the fraction of total nodes with direct synaptic GABAergic contacts (nodal contacts, GAD2-Ab+, ~25%), nearby contacts at a node or unmyelinated terminal branch/bouton on the same axon (within λS = 90 μm; 98 – 77%; as in c) and putative extrasynaptic innervation (within 5 μm, ~95%) from GABAaxo neurons, with presynaptic GABA inferred from GAD2-Ab immunolabelling, in n = 3 mice from n = 43 – 53 nodes, each on 1st and 2nd order branches of Ia afferents. VGAT+ contacts on or near nodes occurred with similar incidence (28% of nodes had synaptic contacts, from n = 50 nodes in n = 3 mice). Similar incidence of contacts occurred in rat afferents labelled with neurobiotin (n = 5 rats). Synaptic contacts occurred at unbranched or branched nodes, as did extrasynaptic innervation, with about half the nodes branched overall. Synaptic contacts that occurred on short unmyelinated afferent terminal branches arising from the node usually occurred at a bouton and are labelled as: Nearby bouton. Synaptic GAD2+ contacts occurred most frequently at or near complex branch points where a parent branch split into large daughter branches (d) or the daughter branches had large boutons (g-h), which theoretically increased the local conductance and probability of spike failure downstream to the branch point (as in Extended Data Fig. 5).
Extended Data Fig. 5 |. Computer simulation of branch point failure and rescue by GABA.
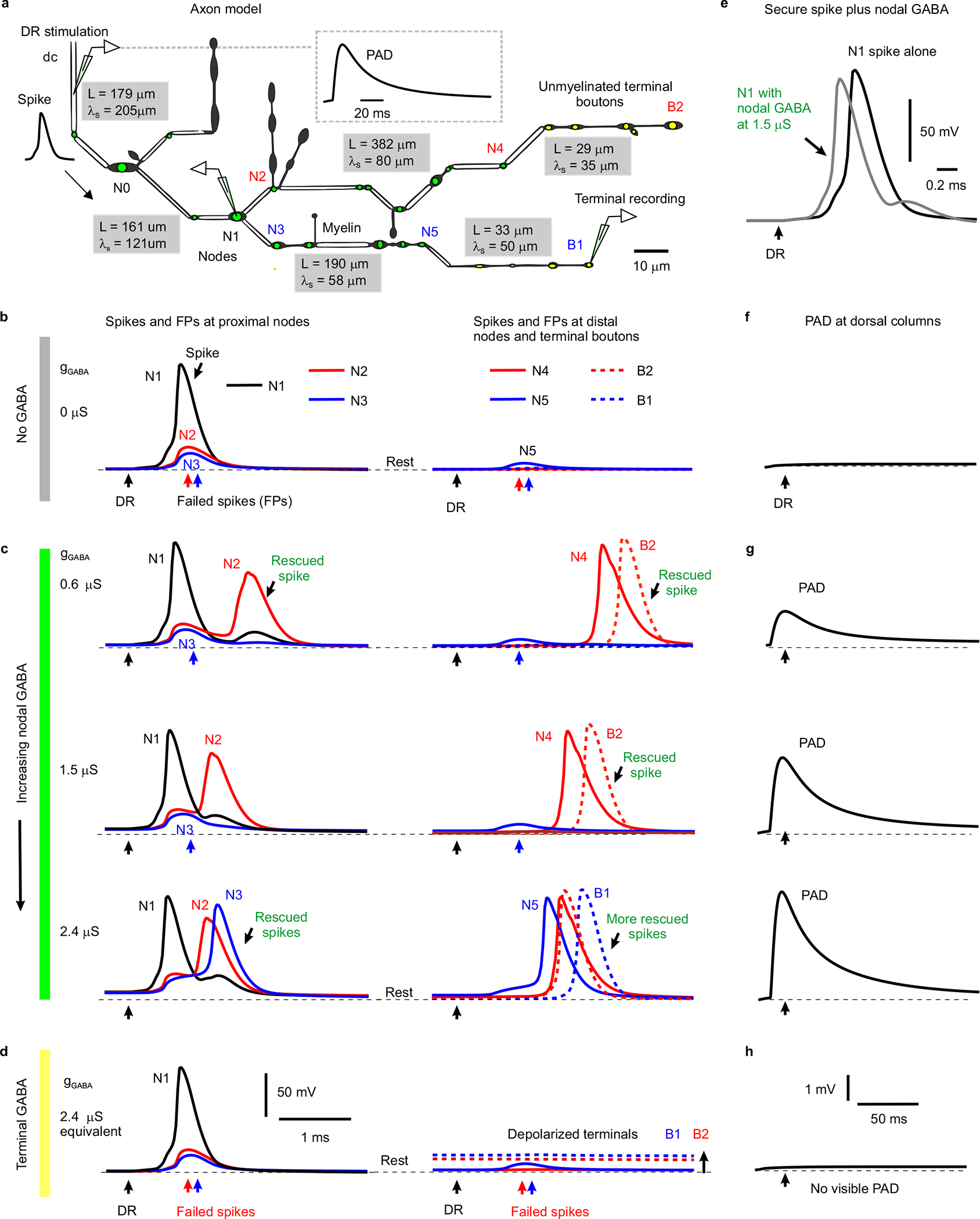
a, Model of a 3D reconstructed proprioceptive afferent, drawn to scale, except myelinated branch lengths all shortened an order of magnitude. Double line segments are myelinated (white) and the rest unmyelinated. Adapted from anatomical studies of Walmsley25. Nodes are indicated with a green dot, and ventrally projecting terminal boutons indicated with a yellow dot. As in our axons of Fig. 1, branch points were always at nodes. GABAA receptors of equal conductance (nS) were placed at each node and associated branch points, and total dorsal columns (dc) depolarization from phasically activating these receptors is shown in inset. The branch lengths (L) and computed space constants (λS) are indicated in gray boxes for each segment of the afferent, the latter computed from subthreshold current injections into each segment. From left to right the gray boxes are for segments spanning from the dorsal columns (dc) to N0, N0 to N1, N1 to N5, N1 to N4, N4 to B2 and N5 to B1. Average space constant was λS = 91 μm, similar to in other axons39. b, Responses to simulated dorsal root (DR) stimulation (0.1 ms pulse, 2 nA, at black arrows) computed at various downstream branch points (nodes) and terminal boutons in the spinal cord, with resting potential indicated by thin dashed line (−82 mV). A sodium spike propagated to the branch point at node N1, but failed to invade into the downstream branches, leaving nodes N2 and N3 with only a passive depolarization from the N1 spike (failure potential, FP). Further downstream nodes and terminals experienced vary little depolarization during this failure (N5, B1 and B2). In this case the GABA conductance was set to zero (gGABA = 0, control), simulating a lack of GABA tone. Note that only the nodes beyond the parent branch at node N1 failed due to the conductance increases in large daughter branches to nodes N2 and N3 that drew more current than node N1 could provide (shunting conductances). Node 3 is particularly interesting as it does not itself branch, though a neighbouring node has a small branch, both contributing to the overall conductance and related failure, and Node 2 has two adjacent branches contributing to its conductance. Other branch points with relatively smaller conductance increases (N0, simpler branching) did not fail to conduct spikes. Generally speaking, if the upstream node of a parent branch cannot provide enough current to activate the nodes of its daughter branches then spikes fail, and this is especially likely with multiple sequential branch points, like in N1 – N2. However, failure even occurs at daughter nodes than themselves lack branches (N3), so GABA receptors are useful in aiding spikes at unbranched nodes. c, As seen experimentally, nodal GABAA receptor activation to produce PAD prior to the DR stimulation (~ 10 ms prior; as in Fig. 4f) rescued spikes from failing to propagate. That is, with GABAA receptors placed just at nodes and associated branch points a weak phasic activation of these receptors (conductance gGABA = 0.6 – 1.5 μS per node shown) rescued conduction down the branch to node N2, with full nodal spikes seen at the distal node N4 and the terminal bouton B2 (DR stimulation at peak of PAD, detailed in (g) with black arrows indicating DR stimulation timing). A larger GABA receptor activation (2.4 nS) additionally rescued spike conduction down the branch to node N3, with full spike conduction to the distal node N5 and the terminal bouton B1. Note that increasing GABA conductance sped up the arrival of distal spikes (e.g. at N4 and B2), by up to 1 ms, suggesting substantial variation in sensory transmission times induced by GABA, as we see experimentally. Also note that this nodal GABA depolarized the nodes (N1 – N3) relative to rest (thin dashed line), thus assisting spike initiation. In contrast, nodal GABA did not depolarize the terminal boutons (B1 and B2), consistent with our recent direct recordings from terminals15. Sensitivity analysis revealed similar results with a wide range of sodium channel and GABA receptor conductances, though increasing sodium conductance sufficiently prevented failure all together (like in Extended Data Fig. 10f). Interestingly, when we put GABA receptors only at node N2 (or at the unmyelinated bouton immediately above N2) then the spike propagating though N2 to the terminal bouton B2 was rescued with the same GABA conductance (0.6 μS) as in the main simulation (c). Likewise with GABA receptors only at node N3 and nowhere else, then the spike was rescued at that node N3 (at 2.4 μS, as in c). d, When instead we removed all nodal GABA receptors and instead place them on terminal boutons (near B1 and B2, yellow, with equivalent total conductance, 2.4 μS condition), then activating them did not rescue the spike propagation failure, since the associated depolarization of nodes is too attenuated at the failure point (N1-N3; no change from resting potential). The GABA receptors did depolarize the terminal boutons (B1 and B2, thick dashed lines) substantially relative to the resting potential (thin dashed lines), but this depolarization was sharply attenuated in more proximal nodes (N1–3). e, Reduction of spike height (shunt) and speeding of spike onset with increasing GABA conductance at a non-failing node (N1; model with nodal and not terminal bouton GABA conductances, c), consistent with actual recordings from axons in Fig. 3d and Extended Data Fig. 8a. f-h, PAD recorded at the dorsal columns (dc) during conditions in (b-d), respectively, as experimentally recorded dorsal PAD. A phasic GABA induced depolarization (PAD) was generated by changing GABA conductances, gGABA, as detailed in Methods, and GABA receptor location varied as in (c-d). DRs were stimulated at the peak of this PAD in (c-d). Note that nodal (g) but not terminal (h) GABAA receptors caused a visible depolarization (PAD) at the dc, due to less electrotonic attenuation over a shorter distance to the dc.
Extended Data Fig. 6 |. Cutaneous driven trisynaptic circuits mediating PAD and assisting repetitive firing.
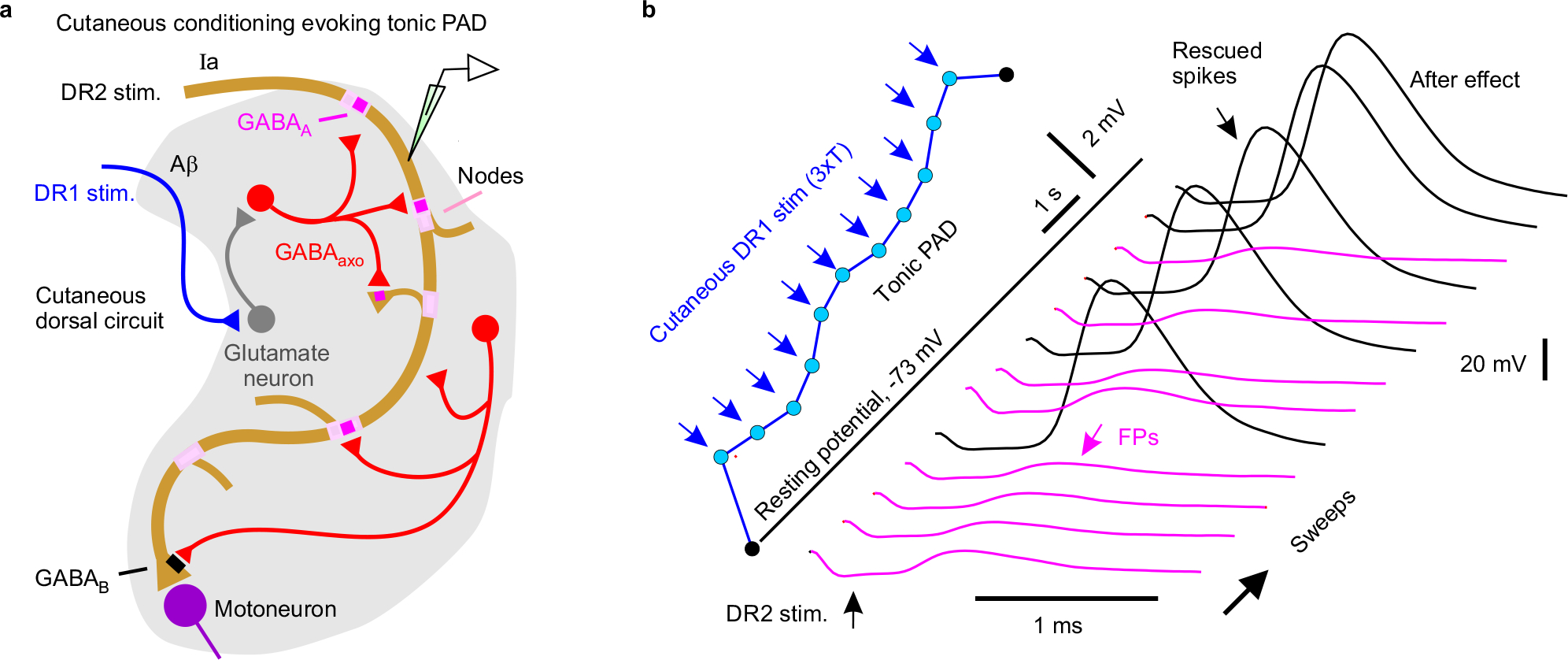
a, Cutaneous driven dorsal trisynaptic circuit mediating PAD. A minimally trisynaptic circuit is classically known to depolarize afferents via GABAaxo neurons. This circuit involves sensory afferents activating excitatory intermediary neurons (glutamatergic) that in turn activate GABAaxo neurons that return to innervate sensory axons13,115. Even though GABAaxo neurons are small5 this circuit influences afferents over widespread regions of the spinal cord15. Specifically, the activation of a small group of sensory axons in just one DR or nerve causes this trisynaptic circuit to produce a widespread activation of many axons across the spinal cord, even many segments away and across the midline15. This allowed us to activate PAD from adjacent roots without directly activating an axon in a particular root, as detailed in Fig. 4 and the rest of this figure. One variant of this classic trisynaptic circuit specifically involves cutaneous stimulation activating dorsal intermediary neurons13,14 that activates GABAaxo neurons (likely dI4 neurons115) that in turn innervate cutaneous afferents, which we term the cutaneous dorsal circuit. While this cutaneous dorsal circuit also synaptically innervates some proprioceptive afferents116 (a), its main action on proprioceptive afferents is to produce a pronounced extrasynaptic spillover of GABA that depolarizes these afferents tonically via α5 GABAA receptors (termed: tonic PAD, L655708 sensitive), especially with repetitive cutaneous nerve stimulation (1 – 200 Hz) that leads to minutes of depolarization15, and we see similar tonic PAD here (detailed next). b, Intracellular recording from a proprioceptive axon branch in rat dorsal horn (sacral S4 axon, DR2). The axon branch spontaneously exhibited spike propagation failure when its was stimulated alone (denoted DR2 stimulation, repeated at 1 Hz, 1.1xT, 0.1 ms; T: afferent volley spike threshold), with only a small failure potential (FP) visible (lower pink traces). Activation of a largely cutaneous DR (caudal Ca1 DR, innervating the tip of the tail, stimulation at intensity for cutaneous afferents, 3xT, 0.1 ms; denoted DR1) evoked a slowly rising tonic PAD when repeated at 1 Hz (blue). When the axon stimulation (DR2 stimulation) was combined with the repeated cutaneous stimulation (DR1, 60 ms prior to each DR2 stimulation) the slowly building PAD prevented spike failure (black spikes), and this outlasted the cutaneous stimulation (after effect). Similar results obtained in n = 20/20 axons tested from 10 rats.
Extended Data Fig. 7 |. Proprioceptive driven trisynaptic circuit for PAD enabling high frequency spike transmission.
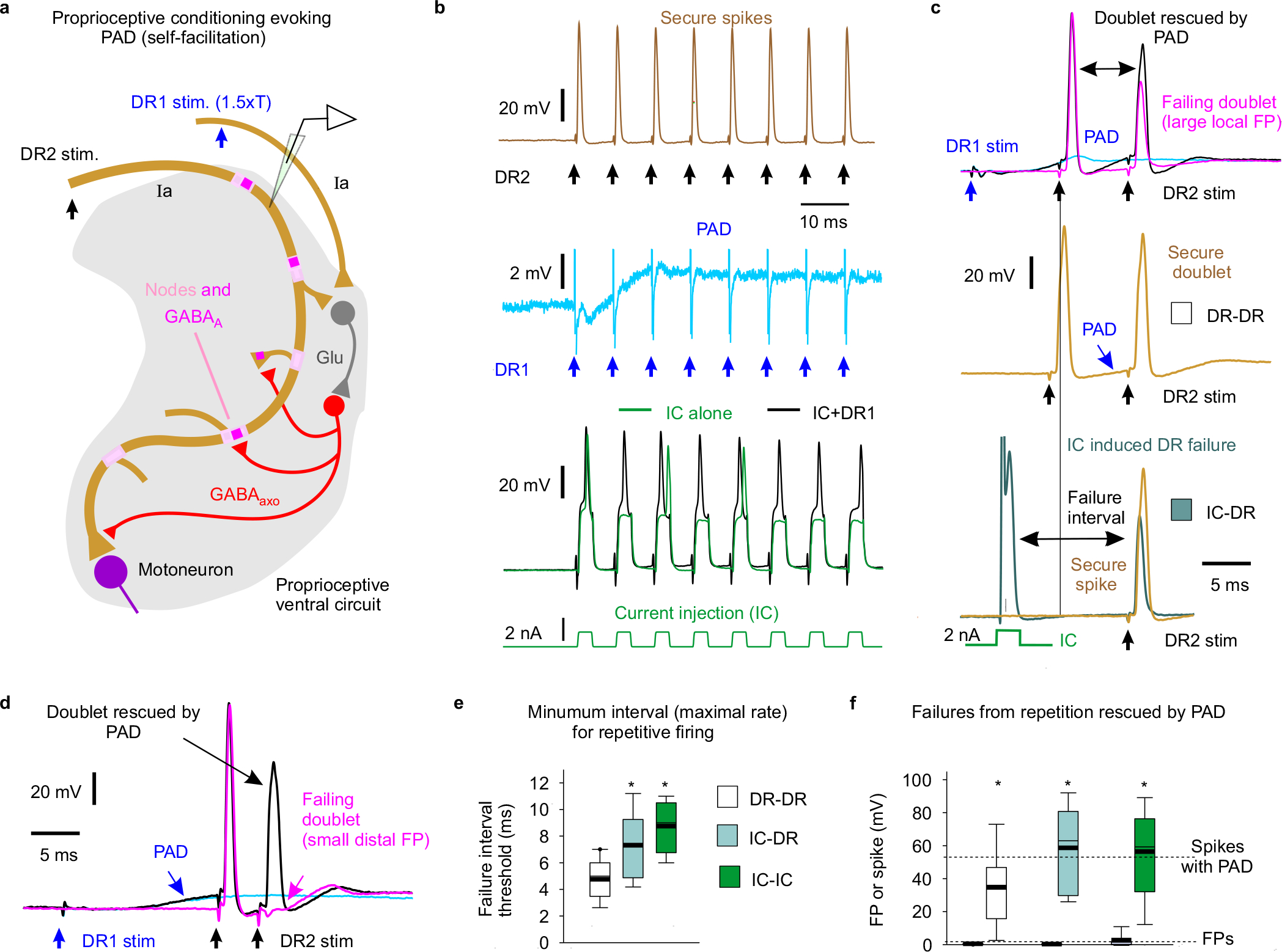
a, Proprioceptive driven ventral trisynaptic circuit mediating PAD. Another variant of the classic trisynaptic circuit involves proprioceptive afferents activating excitatory intermediary neurons (glutamatergic) that then activate GABAaxo neurons that innervate these same afferents, including ventral terminal regions of the afferents13,15,115. This circuit is more ventrally located compared to the cutaneous dorsal PAD circuit13, and thus, we term it the proprioceptive ventral PAD circuit. It likely involves GABAaxo neurons that are from the dILA population115. Importantly, the circuit is recurrent, with proprioceptive afferents causing self-facilitation of themselves (via homonymous PAD). It produces a fast phasic axon depolarization (phasic PAD, fast synaptic; Fig. 4c), as well as a slower tonic depolarization (tonic PAD, Fig. 4c, likely from extrasynaptic GABA spillover), as detailed previously15. Since proprioceptive sensory axons naturally fire at high rates117 where they are vulnerable to spike failure (Fig. 2e–f), we examined the action of self- facilitation by GABA on this failure. b-f, During rapid repetitive stimulation of a DR to evoke spikes in a proprioceptive axon there was an inevitable activation of PAD from low threshold proprioceptive axons (homonymous PAD, b). This PAD helped spikes fire at high physiological rates of up to 200–300 Hz (5 – 3 ms intervals) before spike inactivation and failure occurred because, in absence of PAD, isolated repetitive activation of the axon with intracellular current pulses (IC) led to failure at much lower firing rates (~100 Hz; longer spike intervals; b, e), even after just two stimuli (doublets; c, e). Additional PAD evoked by simultaneous stimulation of an adjacent DR (2xT; T, spike threshold observed from afferent volley) reduced failure from fast repeated IC stimuli (b, f), repeated DR stimuli (doublet, c, d, f) or hybrid IC-DR stimulation pairs (f). Legend details are as follows: b, Intracellular recording from proprioceptive axon in rat dorsal horn (sacral S4 axon) with spikes securely evoked by fast repeated DR stimulation (top, 1.1xT, 0.1 ms, sacral S4 DR, denoted DR2; resting potential −68 mV), but spikes failing intermittently with repeated intracellular current injection (IC) at the same rate (bottom green), due to sodium channel inactivation. The reason that spike failure does not occur with the fast DR stimulation is that it is accompanied by a build up of tonic PAD (from self-activation) that helps prevent failure, because adding to the IC stimulation a simultaneous conditioning stimulation of other proprioceptive afferents in an adjacent DR (DR1 stim at 1.5xT, 0.1 ms, S3 DR) prevents spike failure (black trace, bottom), via the proprioceptive ventral circuit (a). This DR1 conditioning stimulation does not directly activate spikes in the axon, but it causes a fast depolarization (phasic PAD) that rapidly helps spikes (as early as 6 – 10 ms later), and a building tonic depolarization (tonic PAD) with repetition that further helps later spikes in the stimulation train (DR1 stimulation alone blue, middle trace). Similar results obtained in n = 7/7 axons from 4 rats. Likely similar tonic PAD and associated increased spike conduction helps explain post-tetanic potentiation of the monosynaptic EPSP, as previously suggested118. c, Repeated DR stimulation at higher rates eventually causes spike failure in proprioceptive axons (sodium spike relatively refractory), as shown in the top panel where a double stimulation (doublet, S4 DR, denoted DR2-DR2, 1.1xT, 0.1 ms, resting at −75 mV) exhibits failure on the second spike (with large FP indicated, magenta). However, additional PAD provided by stimulating an adjacent DR (DR1; 1.5xT, group I intensity, 0.1 ms) about 10 ms earlier helps prevent this spike failure (black trace; blue trace: PAD alone). When the same axon was stimulated slightly slower (with a longer doublet interval, second plot, DR-DR) failure did not occur, which we designate the failure interval threshold, which is quantified in (e). The self-activated PAD caused by the first DR stimulation in this doublet helped prevent failure in second DR stimulation because replacing the first DR stimulation with an intracellular stimulation (IC, 2 nA) to activate the spike leads to failure of the second spike evoked by the DR stimulation at much longer intervals (lower trace, IC-DR). d, Another example of a failed doublet spike (DR2-DR2 stim, 1.1xT, 0.1 ms) that was rescued by PAD evoked by adjacent DR stimulation (DR1 1.5xT, 0.1 ms, resting at −78 mV), as in the top plots of (c), except that in this case the failure is at a more distal node, since the FP is small. e, Failure interval threshold (minimum firing interval prior to failure, or maximal firing rate) with DR doublets (DR- DR), IC doublets (IC-IC) or IC-DR pair stimulation. Note the shorter intervals possible with the PAD evoked by the first stimulation (DR- DR). * significantly longer than minimum DR doublet interval (DR- DR), one-way ANOVA with post hoc Tukey test for multiple comparisons, n = 18 axons each condition from 5 rats, P < 0.05. f, Quantification of the FP heights that were induced by a fast doublet (DR-DR or IC-IC; n = 14 axons each from 5 rats, at failure threshold interval) or IC and DR stimulation (IC-DR; n = 11 axons from 5 rats), and the rescue of spikes by PAD evoked by adjacent DR stimulation for each condition. * significant increase in height with PAD, two- sided paired t-test, P < 0.05. Box plots show the interquartile range (box), median (thin line), mean (thick line), extremes, 10 and 90 percentiles (whiskers).
Extended Data Fig. 8 |. Other excitatory actions of GABAA receptors on proprioceptive axons.
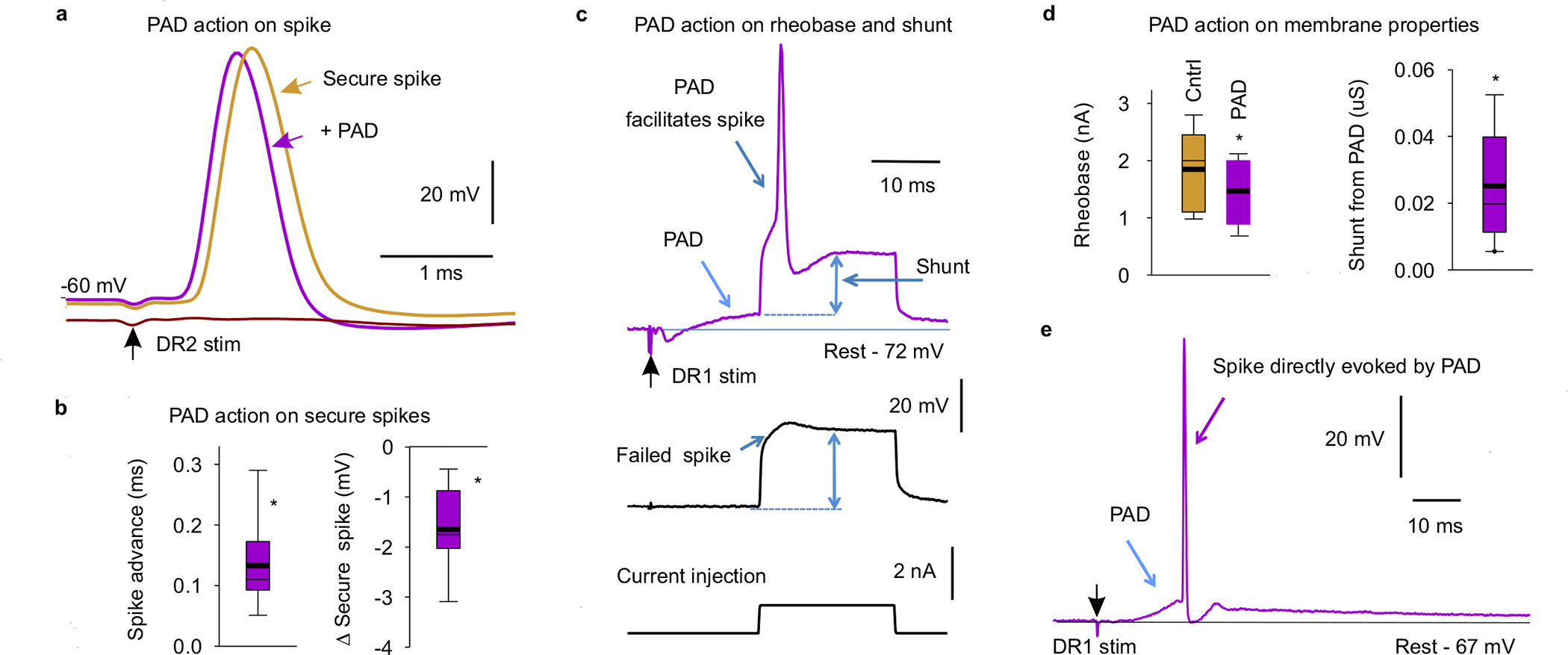
a, Intracellular recording from a rat sacral S3 proprioceptive Ia afferent branch in the dorsal horn with a secure spike evoked by S3 DR stimulation at rest (DR2, 1.1xT, 0.1 ms, - 60 mV rest, rat; T: spike threshold in afferent volley). Sensory-evoked PAD initiated by stimulating an adjacent DR (DR1; S4 DR; 2xT, 0.1 ms pulse, as in Fig. 4) 10 ms prior to the DR2 stimulation only moderately influenced the spike (DR1 stimulation time). It sped up the spike latency and rise time, reduced the fall time and slightly reduced the spike height. Hyperpolarization induced spike failure (lower trace), as in Extend Data Fig. 2a. b, Summary box plots of change in spike peak latency (advance) and height with prior sensory PAD activation as in (a). *significant change, two-sided paired t-test, P < 0.05, n = 26 axons, from 9 rats. c, Intracellular recording from an S3 proprioceptive Ia afferent branch in the dorsal horn with a brief current injection pulse just subthreshold to initiating a spike (near rheobase, bottom trace) at rest, only initiating a passive response with a small failed spike (middle trace). However, prior activation of PAD by stimulating an adjacent DR (DR1 as in A; S4 DR, 2xT, 0.1ms top trace) allowed the same current pulse to evoke a spike (above rheobase). The passive response to the current injection (double blue arrow; resistance R = V/I) was decreased during the PAD, corresponding to an increase in conductance, that contributed to a shunt (reduction) of the currents generating the spike, though this only caused about a 1% drop in spike height (1 mV; b). DCC recording mode, as in Extend Data Fig. 2a–d. d, Summary box plots of rheobase (current threshold from c) before and during PAD, and change in shunt (conductance = 1/R) with PAD, as in (c). Box plots show the interquartile range (box), median (thin line), mean (thick line), extremes, 10 and 90 percentiles (whiskers). * significant change with PAD, two-sided paired t-test, P < 0.05, n = 37 axons from 11 rats. e, By itself sensory evoked PAD sometimes initiated a spike on its rising phase, when the DR stimulation was large enough, demonstrating a direct excitatory action of GABAA receptors, as previously reported15. These spikes propagate antidromically toward the DR; and are thus termed dorsal root reflexes (DRR). Example shown of intracellular recording from rat sacral S3 proprioceptive afferent branch in the dorsal horn, with PAD produced by a DR1 stimulation (S4 DR stimulation 3.5xT, 0.1 ms) evoking a spike that propagates out the DR (DRR). These DRR occurred in n = 11/120 axons from 15 rats (9% incidence). These PAD evoked spikes occur with a variable latency of 10 – 30 ms15 and thus make axons refractory for about 30 ms after the DR stimulation119. PAD evokes spikes with a high incidence (79%)15, but these spikes fail to propagate antidomically, yielding the 9% incidence we see. However, these spikes are more likely to travel othodromically (up to 79% incidence) and evoke EPSPs in motoneurons via the monosynaptic pathway8, and thus also produce a post activation depression of the EPSPs for many seconds. We thus kept the PAD low when examining the effects of PAD on EPSPs, to avoid these spikes and their subsequent inhibitory action (in Figs. 5 – 6).
Extended Data Fig. 9 |. Estimating the overall spike conduction failure from the dorsal root to the motoneurons.
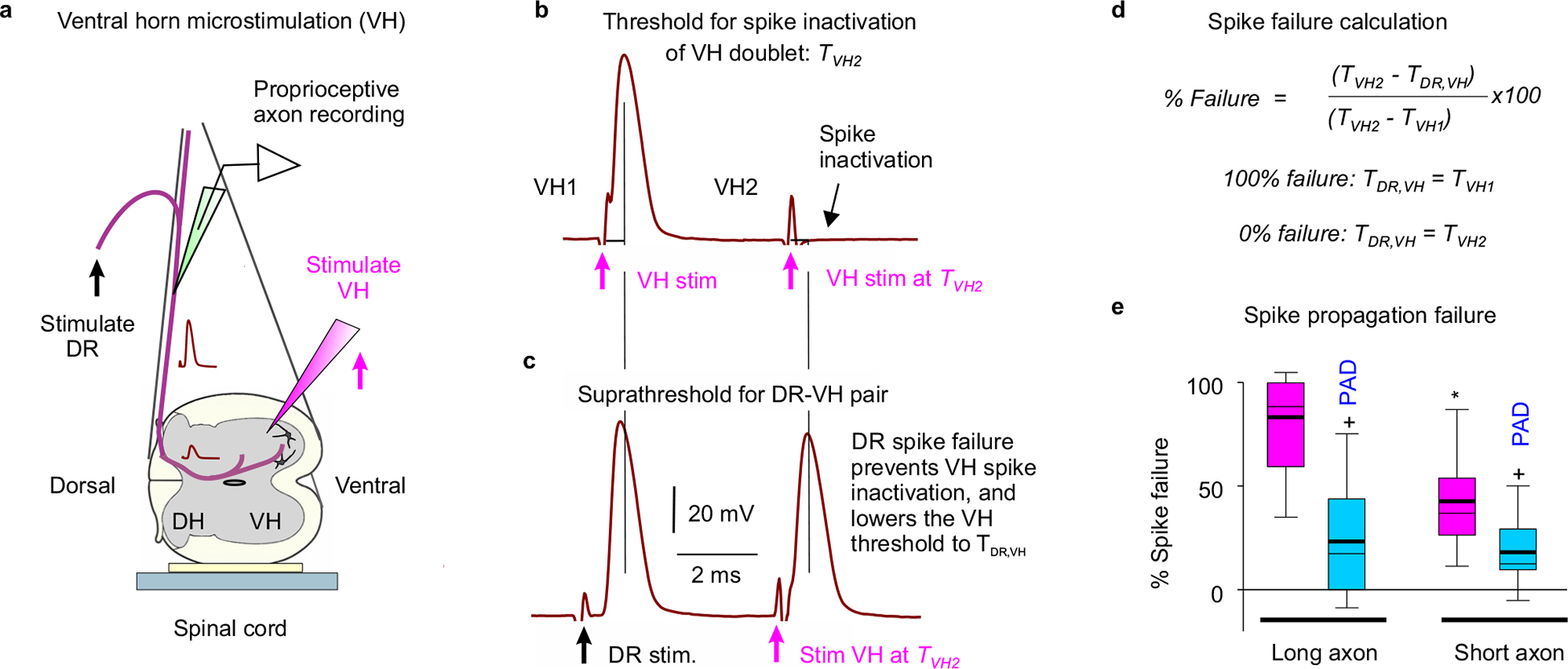
a, Experimental setup to indirectly measure sensory axon conduction failure following DR stimulation, by examining whether failed axon segments are relatively less refractory to activation after failure, using a double pulse method adapted from Wall30. A tungsten microelectrode (12 MΩ) was placed in the ventral horn (VH) near the sensory axon terminals on motoneurons (S3 or S4 VH), to activate the branches/nodes of the axon projecting to the motoneuron that may have failed (VH stimulation). Spikes from VH or DR stimulation were recorded intracellularly in a proprioceptive axon penetrated in the dorsal columns. b, VH threshold in refractory period. Rapidly repeated VH stimulation (VH doublet; two 0.1 ms pulses) at an interval short enough to produce spike inactivation on the second stimulation (4 ms), with stimulus current adjusted to threshold for inactivation, TVH2. This TVH2 (~15 uA) was always higher than the threshold VH stimulation for evoking a spike with the first stimulation, TVH1 (~10 uA). Recorded in sacral S4 afferent resting at - 72 mV, with doublets repeated at 3 s intervals to determine current thresholds. c, VH threshold after DR stimulation. Similar repeated activation of the axon in (b), but with the first activation from a DR stimulation (at 1.5x DR threshold) and the second from VH stimulation at the TVH2 intensity from (b). In this case the VH stimulation readily activated the axon spike, likely because the DR- evoked spike did not propagate to the VH, leaving the silent portion of the axon non refractory. Thus, this VH stimulation evoked spikes with a lower current than TVH2, with this lower threshold denoted TDR,VH (~ 12 μA). This DR – VH stimulation interval was deliberately set too short for the involvement of PAD (which rises in > 4 ms; Fig. 4). d, Computation of spike failure based on changes in VH stimulation thresholds. If the DR-evoked spike entirely fails to propagate to the VH, then the threshold for subsequently activating the VH (TDR,VH) should be the same as the threshold without any prior activation (TVH1 = TDR,VH), whereas if it does not fail then the threshold for activating the VH should be the same as with a VH doublet (TVH2 = TDR,VH). In between these two extreme scenarios, the DR evoked spike may only partially fail to propagate spikes to the VH; in this case TDR,VH should be between TVH1 and TVH2, with the difference TVH2 - TVH1 representing the range of possible thresholds between full failure and conduction. Overall the % conduction failure can be thus quantified as: (TVH2 - TDR,VH)/(TVH2 - TVH1) * 100%, which is 100% at full failure and 0% with no failure. e, Average spike conduction failure to the VH in proprioceptive axons, and decrease following a DR conditioning stimulation that depolarized the axon (PAD). Box plots of failure estimated as in (b-d). Prior DR conditioning to produce PAD (via adjacent S4 or Ca1 DR stimulation at 3xT) reduced the failure estimated 20 ms later by the paired-pulse conduction testing (repeating DR – VH stimulations of b-d). DR conditioning itself lowered the thresholds for VH activation (n = 5 rats), as previously reported32. We studied two lengths of axons: long axons (intersegmental, n = 11 axons, from 5 rats) with the VH stimulation one segment away from the recording site, and short axons (segmental, n = 12, from 5 rats) with the VH stimulation near the recording site, in the same segment. Box plots show the interquartile range (box), median (thin line), mean (thick line), extremes, 10 and 90 percentiles (whiskers). + significantly less failure with PAD and * significantly less failure with short compared to long axons, two-sided paired and unpaired t-tests, respectively, P < 0.05.
Extended Data Fig. 10 |. Axonal spike conduction estimated from extracellular recordings.
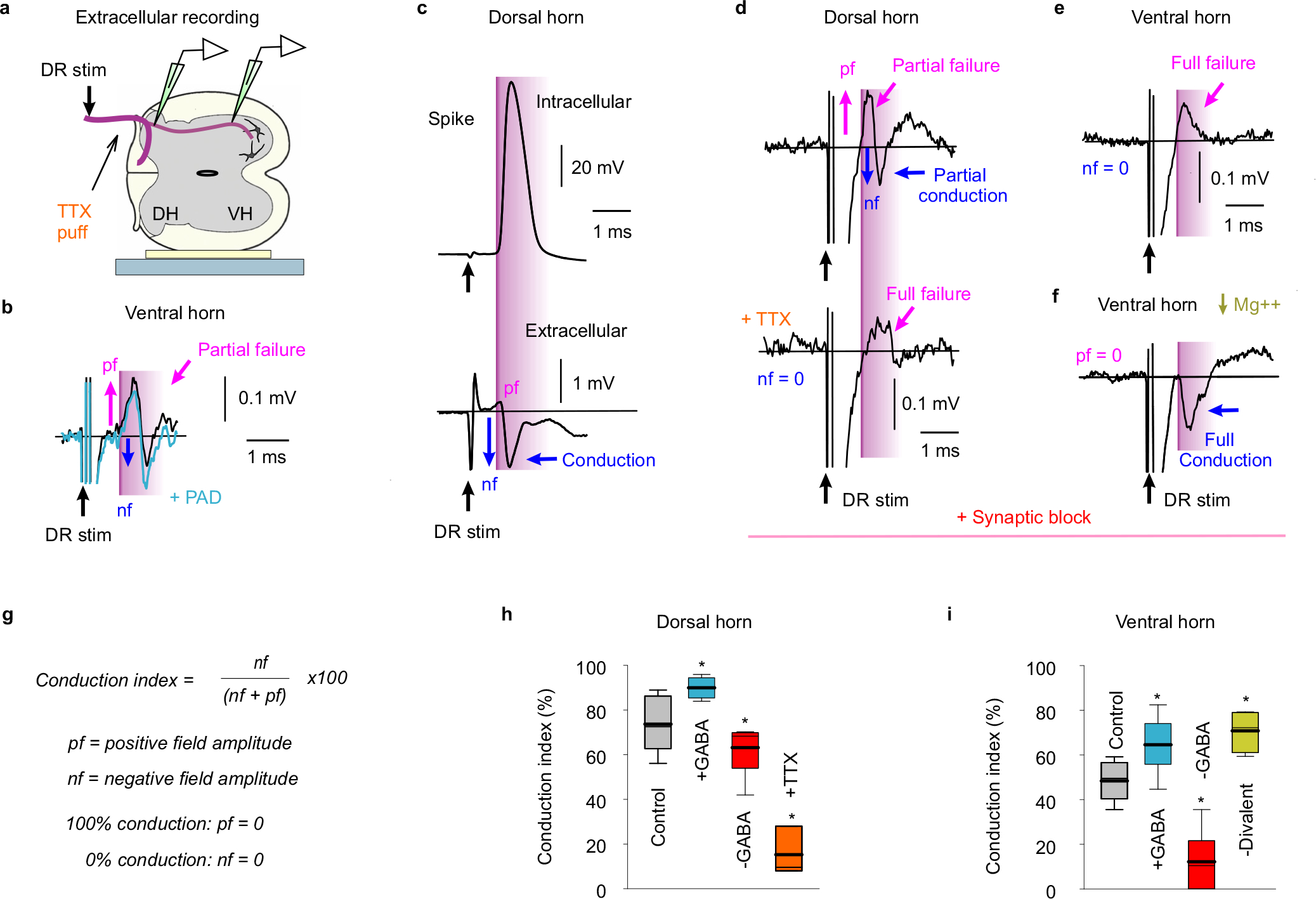
a, Experimental setup to directly record spike conduction failure in proprioceptive axon terminals in the ventral horn (VH) following DR stimulation. Extracellular (EC) recordings from axon terminals in VH, with glass electrode positioned just outside these axons, and for comparison EC recording in the dorsal horn (DH). b, EC field recorded in VH after DR stimulation (S4 DR, 1.1xT; T: afferent volley threshold), with a relatively large initial positive field (magenta arrows, pf) resulting from passively conducted axial current from sodium spikes at distant nodes (closer to the DR; outward current at electrode), some of which fail to propagate spikes to the VH recording site; thus, this field is a measure of conduction failure, as demonstrated in (c-f) below. Following this, a negative field arises (blue arrow, nf), resulting from spikes arising at nodes near the electrode (inward current); thus, this field is a measure of secure conduction. Reducing conduction failure by depolarizing the axon (+PAD) with a prior conditioning stimulation of an adjacent DR (Ca1, 2xT, 30 ms prior), decreased the positive field (pf) and increased the negative field (nf), consistent with increased conduction to the terminals, and in retrospect the same as Sypert et al. (1980)120 saw in cat (their Fig. 4). Large stimulus artifacts prior to these fields are truncated. c, Control recordings from proprioceptive axons in dorsal horn (DH) to confirm the relation of the EC negative field (nf) to spike conduction. Intracellular (IC) recording from axon (sacral S4, resting at −64 mV) and EC recording just outside the same axon, showing the DR evoked spike (IC) arriving at about the time of the negative EC field (nf). There is likely little spike failure in this axon or nearby axons, due to the very small initial positive field (pf). EC fields are larger in DH compared to VH (G, 10x), and thus the artifact is relatively smaller. d, Locally blocking nodes with TTX to confirm the relation of the positive EC field to spike failure. EC recording from proprioceptive axon in the dorsal horn (S4), with an initial positive field (pf) followed by a negative field (nf), indicative of mixed failure and conduction. A local puff of TTX (10 μl of 100 μM) on the DR just adjacent to the recording site to transiently block DR conduction eliminated the negative field (nf) and broadened the positive field (pf), consistent with distal nodes upstream of the TTX block generating the positive field via passive axial current conduction, and closer nodes not spiking. Recordings were in the presence of synaptic blockade (with glutamate receptor blockers, kynurenic acid, CNQX and APV, at doses of 1000, 100 and 50 μM respectively), to prevent TTX spillover having an indirect action by blocking neuronal circuit activity, including GABAaxo neuron activity. This synaptic blockade itself contributed to some spike failure, consistent with a block of GABAaxo neuron activity, as there was a more prominent positive field (pf) compared to without blockade in (c). e, EC field recorded from terminals of proprioceptive axons in the ventral horn near motoneurons (S4), in the presence of an excitatory synaptic block that largely eliminates most neuronal circuit behaviour (with kynurenic acid, CNQX and APV, as in d). In this synaptic block negative fields were generally absent (nf = 0), and only prominent positive fields (pf) occurred (as with TTX block), suggesting that conduction to the VH often completely failed when circuit behavior is blocked, which likely indirectly reduces GABAaxo neuronal circuit activity and its associated facilitation of nodal conduction. f, Rescue of spike conduction to the ventral horn by increasing sodium channel excitability by reducing the divalent cations Mg++ and Ca ++ in the bath medium80. Same EC field recording as in e, but with divalent cations reduced (Mg++, 0 mM; Ca++, 0.1 mM). The positive field was largely eliminated (pf = 0) and replaced by a negative field (nf), consistent with elimination of conduction failure, and proving that the positive field is not a trivial property of axon terminals78,79,120. g, Conduction index computed from positive (pf) and negative (nf) field amplitudes as: nf / (nf + pf) × 100%, which approaches 100% for full conduction (pf ~0; as in c) and 0% for no conduction (nf = 0; as in e). h-i, Summary of conduction index estimated from EC field potentials, shown with box plots. Without drugs present in the recording chamber, the axon conduction from the DR to the dorsal horn was about 70% (h, n = 17 axon fields, from 10 rats), consistent with Fig. 2, whereas conduction from the DR to the VH was only about 50% (i, n = 11, from 5 rats), suggesting substantial failure at the many branch points in the axon projections from the dorsal horn to the motoneurons. Increasing GABAaxo neuron activity with DR conditioning (PAD, 30 – 60 ms prior) increased conduction (+GABA, in both the DH and VH, n = 5 and 9 from 5 rats, as in b), whereas decreasing GABA and all circuit activity in a synaptic blockade decreased conduction (-GABA, in both DH and VH, n = 5 and 6 from 5 rats, as in d-e). TTX (n = 5 from 2 rats, h) or removal of divalent cations (Mg++ and Ca++, -Divalent, n = 5 from 2 rats in synaptic block, i) reduced or increased conduction, respectively (as in d and f). Box plots show the interquartile range (box), median (thin line), mean (thick line), extremes, 10 and 90 percentiles (whiskers).* significant difference from control pre-drug or pre-conditioning, two-sided paired t-test, P < 0.05.
Supplementary Material
Acknowledgements
We thank Leo Sanelli, Jennifer Duchcherer, Babak Afsharipour and Christopher K. Thompson for technical assistance, and Shawn Hochman, CJ Heckman, FJ Alvarez and Tia Bennett for discussions and editing the manuscript. VGLUT1Cre mice cords were kindly donated by Dr. FJ Alvarez. We thank Prof Uwe Rudolph (McLean Hospital, currently University of Illinois Urbana-Champaign) for providing Gabra5-floxed mice. This research was supported by the Canadian Institutes of Health Research (MOP 14697 and PJT 165823 D.J.B.) and the US National Institutes of Health (NIH, R01NS47567, D.J.B. and K.F.; R01GM118801, R.A.P.). These funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Competing Interests Statement
All authors declare no competing interests.
Code availability
The computer code used to perform the axon simulations (Extended Data Fig. 5) are publicly available on the github repository: https://github.com/kelvinejones/noah-axon.git
Data availability
All data are available in the manuscript or the supplementary materials. Raw data are available upon request to the corresponding authors. This study did not generate large data sets or new unique reagents.
References (Main text-only references)
- 1.Goulding M Circuits controlling vertebrate locomotion: moving in a new direction. Nature reviews. Neuroscience 10, 507–518 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Capaday C & Stein RB Amplitude modulation of the soleus H-reflex in the human during walking and standing. The Journal of neuroscience : the official journal of the Society for Neuroscience 6, 1308–1313 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett DJ, De Serres SJ & Stein RB Gain of the triceps surae stretch reflex in decerebrate and spinal cats during postural and locomotor activities. The Journal of physiology 496 (Pt 3), 837–850 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andrechek ER, et al. ErbB2 is required for muscle spindle and myoblast cell survival. Mol Cell Biol 22, 4714–4722 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Betley JN, et al. Stringent specificity in the construction of a GABAergic presynaptic inhibitory circuit. Cell 139, 161–174 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fink AJ, et al. Presynaptic inhibition of spinal sensory feedback ensures smooth movement. Nature 509, 43–48 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hughes DI, et al. P boutons in lamina IX of the rodent spinal cord express high levels of glutamic acid decarboxylase-65 and originate from cells in deep medial dorsal horn. Proceedings of the National Academy of Sciences of the United States of America 102, 9038–9043 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eccles JC, Eccles RM & Magni F Central inhibitory action attributable to presynaptic depolarization produced by muscle afferent volleys. The Journal of physiology 159, 147–166 (1961). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelman HS & MacDermott AB Presynaptic ionotropic receptors and control of transmitter release. Nature reviews. Neuroscience 5, 135–145 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Rudomin P & Schmidt RF Presynaptic inhibition in the vertebrate spinal cord revisited. Experimental brain research 129, 1–37 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Rossignol S, Beloozerova I, Gossard JP & Dubuc R Presynaptc mechanisms during locomotion. in Presynaptic Inhibition and Neuron Control (eds. Rudmon P, Romo R & Mendell LM) 385–397 (Oxford University Press, New York, 1998). [Google Scholar]
- 12.Ueno M, et al. Corticospinal Circuits from the Sensory and Motor Cortices Differentially Regulate Skilled Movements through Distinct Spinal Interneurons. Cell Rep 23, 1286–1300 e1287 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jankowska E, McCrea D, Rudomin P & Sykova E Observations on neuronal pathways subserving primary afferent depolarization. Journal of neurophysiology 46, 506–516 (1981). [DOI] [PubMed] [Google Scholar]
- 14.Zimmerman AL, et al. Distinct Modes of Presynaptic Inhibition of Cutaneous Afferents and Their Functions in Behavior. Neuron 102, 420–434 e428 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lucas-Osma AM, et al. Extrasynaptic alpha5GABAA receptors on proprioceptive afferents produce a tonic depolarization that modulates sodium channel function in the rat spinal cord. J Neurophysiol 120, 2953–2974 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fink AJ Exploring a behavioral role for presynaptic inhibition at spinal sensory-motor synapses. PhD Thesis, Columbia University, 1–293 (2013). [Google Scholar]
- 17.Stuart GJ & Redman SJ The role of GABAA and GABAB receptors in presynaptic inhibition of Ia EPSPs in cat spinal motoneurones. The Journal of physiology 447, 675–692 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bardoni R, et al. Pre- and postsynaptic inhibitory control in the spinal cord dorsal horn. Annals of the New York Academy of Sciences 1279, 90–96 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szabadics J, et al. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science 311, 233–235 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Howell RD & Pugh JR Biphasic modulation of parallel fibre synaptic transmission by co-activation of presynaptic GABAA and GABAB receptors in mice. The Journal of physiology 594, 3651–3666 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trigo FF, Marty A & Stell BM Axonal GABAA receptors. The European journal of neuroscience 28, 841–848 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Barron DH & Matthews BH The interpretation of potential changes in the spinal cord. The Journal of physiology 92, 276–321 (1938). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cattaert D & El Manira A Shunting versus inactivation: analysis of presynaptic inhibitory mechanisms in primary afferents of the crayfish. The Journal of neuroscience : the official journal of the Society for Neuroscience 19, 6079–6089 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alvarez FJ, Taylor-Blake B, Fyffe RE, De Blas AL & Light AR Distribution of immunoreactivity for the beta 2 and beta 3 subunits of the GABAA receptor in the mammalian spinal cord. J Comp Neurol 365, 392–412 (1996). [DOI] [PubMed] [Google Scholar]
- 25.Walmsley B, Graham B & Nicol MJ Serial E-M and simulation study of presynaptic inhibition along a group Ia collateral in the spinal cord. Journal of neurophysiology 74, 616–623 (1995). [DOI] [PubMed] [Google Scholar]
- 26.Debanne D, Campanac E, Bialowas A, Carlier E & Alcaraz G Axon physiology. Physiol Rev 91, 555–602 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Goldstein SS & Rall W Changes of action potential shape and velocity for changing core conductor geometry. Biophysical journal 14, 731–757 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burke RE & Glenn LL Horseradish peroxidase study of the spatial and electrotonic distribution of group Ia synapses on type-identified ankle extensor motoneurons in the cat. The Journal of comparative neurology 372, 465–485 (1996). [DOI] [PubMed] [Google Scholar]
- 29.Henneman E, Luscher HR & Mathis J Simultaneously active and inactive synapses of single Ia fibres on cat spinal motoneurones. The Journal of physiology 352, 147–161 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wall PD & McMahon SB Long range afferents in rat spinal cord. III. Failure of impulse transmission in axons and relief of the failure after rhizotomy of dorsal roots. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 343, 211–223 (1994). [DOI] [PubMed] [Google Scholar]
- 31.Swadlow HA, Kocsis JD & Waxman SG Modulation of impulse conduction along the axonal tree. Annu Rev Biophys Bioeng 9, 143–179 (1980). [DOI] [PubMed] [Google Scholar]
- 32.Wall PD Excitability changes in afferent fibre terminations and their relation to slow potentials. The Journal of physiology 142, i3–21 (1958). [PMC free article] [PubMed] [Google Scholar]
- 33.Wu H, et al. Distinct subtypes of proprioceptive dorsal root ganglion neurons regulate adaptive proprioception in mice. Nat Commun 12, 1026 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watson AH & Bazzaz AA GABA and glycine-like immunoreactivity at axoaxonic synapses on 1a muscle afferent terminals in the spinal cord of the rat. The Journal of comparative neurology 433, 335–348 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Lucas-Osma AM, et al. 5-HT1D receptors inhibit the monosynaptic stretch reflex by modulating C-fiber activity. Journal of neurophysiology 121, 1591–1608 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Redman S Quantal analysis of synaptic potentials in neurons of the central nervous system. Physiol Rev 70, 165–198 (1990). [DOI] [PubMed] [Google Scholar]
- 37.Zbili M & Debanne D Past and Future of Analog-Digital Modulation of Synaptic Transmission. Frontiers in cellular neuroscience 13, 160 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Serrano-Regal MP, et al. Oligodendrocyte Differentiation and Myelination Is Potentiated via GABAB Receptor Activation. Neuroscience 439, 163–180 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Zorrilla de San Martin J, Trigo FF & Kawaguchi SY Axonal GABAA receptors depolarize presynaptic terminals and facilitate transmitter release in cerebellar Purkinje cells. The Journal of physiology 595, 7477–7493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burke KJ Jr. & Bender KJ Modulation of Ion Channels in the Axon: Mechanisms and Function. Frontiers in cellular neuroscience 13, 221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rudomin P, Nunez R, Madrid J & Burke RE Primary afferent hyperpolarization and presynaptic facilitation of Ia afferent terminals induced by large cutaneous fibers. Journal of neurophysiology 37, 413–429 (1974). [DOI] [PubMed] [Google Scholar]
- 42.Aimonetti JM, Vedel JP, Schmied A & Pagni S Mechanical cutaneous stimulation alters Ia presynaptic inhibition in human wrist extensor muscles: a single motor unit study. The Journal of physiology 522 Pt 1, 137–145 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, et al. Branching points of primary afferent fibers are vital for the modulation of fiber excitability by epidural DC polarization and by GABA in the rat spinal cord. J Neurophysiol 124, 49–62 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Curtis DR & Lacey G GABA-B receptor-mediated spinal inhibition. Neuroreport 5, 540–542 (1994). [DOI] [PubMed] [Google Scholar]
- 45.Pierce JP & Mendell LM Quantitative ultrastructure of Ia boutons in the ventral horn: scaling and positional relationships. The Journal of neuroscience : the official journal of the Society for Neuroscience 13, 4748–4763 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCrea DA, Shefchyk SJ & Carlen PL Large reductions in composite monosynaptic EPSP amplitude following conditioning stimulation are not accounted for by increased postsynaptic conductances in motoneurons. Neuroscience letters 109, 117–122 (1990). [DOI] [PubMed] [Google Scholar]
- 47.Beloozerova I & Rossignol S Antidromic discharges in dorsal roots of decerebrate cats. I. Studies at rest and during fictive locomotion. Brain research 846, 87–105 (1999). [DOI] [PubMed] [Google Scholar]
- 48.Russ JB, Verina T, Comer JD, Comi AM & Kaltschmidt JA Corticospinal tract insult alters GABAergic circuitry in the mammalian spinal cord. Front Neural Circuits 7, 150 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nelson SG, Collatos TC, Niechaj A & Mendell LM Immediate increase in Ia-motoneuron synaptic transmission caudal to spinal cord transection. Journal of neurophysiology 42, 655–664 (1979). [DOI] [PubMed] [Google Scholar]
- 50.Zehr EP & Stein RB Interaction of the Jendrassik maneuver with segmental presynaptic inhibition. Experimental brain research 124, 474–480 (1999). [DOI] [PubMed] [Google Scholar]
- 51.Boyden ES, Zhang F, Bamberg E, Nagel G & Deisseroth K Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci 8, 1263–1268 (2005). [DOI] [PubMed] [Google Scholar]
References (Methods-only references)
- 52.Pinol RA, Bateman R & Mendelowitz D Optogenetic approaches to characterize the long-range synaptic pathways from the hypothalamus to brain stem autonomic nuclei. Journal of neuroscience methods 210, 238–246 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang F, et al. The microbial opsin family of optogenetic tools. Cell 147, 1446–1457 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chow BY, et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 463, 98–102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kralj JM, Douglass AD, Hochbaum DR, Maclaurin D & Cohen AE Optical recording of action potentials in mammalian neurons using a microbial rhodopsin. Nature methods 9, 90–95 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taniguchi H, et al. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71, 995–1013 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Madisen L, et al. A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat Neurosci 15, 793–802 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harris JA, et al. Anatomical characterization of Cre driver mice for neural circuit mapping and manipulation. Front Neural Circuits 8, 76 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Todd AJ, et al. The expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in neurochemically defined axonal populations in the rat spinal cord with emphasis on the dorsal horn. The European journal of neuroscience 17, 13–27 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Feil R, Wagner J, Metzger D & Chambon P Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun 237, 752–757 (1997). [DOI] [PubMed] [Google Scholar]
- 62.Harvey PJ, Li Y, Li X & Bennett DJ Persistent sodium currents and repetitive firing in motoneurons of the sacrocaudal spinal cord of adult rats. Journal of neurophysiology 96, 1141–1157 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murray KC, et al. Recovery of motoneuron and locomotor function after spinal cord injury depends on constitutive activity in 5-HT2C receptors. Nature medicine 16, 694–700 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murray KC, et al. Polysynaptic excitatory postsynaptic potentials that trigger spasms after spinal cord injury in rats are inhibited by 5-HT1B and 5-HT1F receptors. Journal of neurophysiology 106, 925–943 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Street LJ, et al. Synthesis and biological evaluation of 3-heterocyclyl-7,8,9,10-tetrahydro-(7,10-ethano)-1,2,4-triazolo[3,4-a]phthalazine s and analogues as subtype-selective inverse agonists for the GABA(A)alpha5 benzodiazepine binding site. Journal of medicinal chemistry 47, 3642–3657 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Bennett DJ, et al. Spasticity in rats with sacral spinal cord injury. J Neurotrauma 16, 69–84 (1999). [DOI] [PubMed] [Google Scholar]
- 67.Bennett DJ, Li Y, Harvey PJ & Gorassini M Evidence for plateau potentials in tail motoneurons of awake chronic spinal rats with spasticity. Journal of neurophysiology 86, 1972–1982 (2001). [DOI] [PubMed] [Google Scholar]
- 68.Heckmann CJ, Gorassini MA & Bennett DJ Persistent inward currents in motoneuron dendrites: implications for motor output. Muscle Nerve 31, 135–156 (2005). [DOI] [PubMed] [Google Scholar]
- 69.Lin S, et al. Locomotor-related V3 interneurons initiate and coordinate muscles spasms after spinal cord injury. Journal of neurophysiology 121, 1352–1367 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.El-Gaby M, et al. Archaerhodopsin Selectively and Reversibly Silences Synaptic Transmission through Altered pH. Cell Rep 16, 2259–2268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li Y, et al. Pericytes impair capillary blood flow and motor function after chronic spinal cord injury. Nature medicine 23, 733–741 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Y, Harvey PJ, Li X & Bennett DJ Spastic long-lasting reflexes of the chronic spinal rat studied in vitro. Journal of neurophysiology 91, 2236–2246 (2004). [DOI] [PubMed] [Google Scholar]
- 73.Vincent JA, et al. Muscle proprioceptors in adult rat: mechanosensory signaling and synapse distribution in spinal cord. Journal of neurophysiology 118, 2687–2701 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Luscher C, Streit J, Lipp P & Luscher HR Action potential propagation through embryonic dorsal root ganglion cells in culture. II. Decrease of conduction reliability during repetitive stimulation. Journal of neurophysiology 72, 634–643 (1994). [DOI] [PubMed] [Google Scholar]
- 75.Wall PD Some unanswered questions about the mechanims and function of presynaptic inhibition. in Presynaptic Inhibition and Neuron Control (eds. Rudmon P, Romo R & Mendell LM) 228–241 (Oxford University Press, New York, 1998). [Google Scholar]
- 76.Hubbard JI, Llinas R & Quastel DMJ Electrophysiological Analysis of Synaptic Transmission, (Edward Arnold Ltd, London, 1969). [Google Scholar]
- 77.Munson JB & Sypert GW Properties of single central Ia afferent fibres projecting to motoneurones. The Journal of physiology 296, 315–327 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dudel J The Mechanism of Presynaptic Inhibition at the Crayfish Neuromuscular Junction. Pflugers Arch Gesamte Physiol Menschen Tiere 284, 66–80 (1965). [DOI] [PubMed] [Google Scholar]
- 79.Katz B & Miledi R Propagation of Electric Activity in Motor Nerve Terminals. Proc R Soc Lond B Biol Sci 161, 453–482 (1965). [DOI] [PubMed] [Google Scholar]
- 80.Armstrong CM & Cota G Calcium ion as a cofactor in Na channel gating. Proceedings of the National Academy of Sciences of the United States of America 88, 6528–6531 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stein RB Nerve and Muscle : Membranes, Cells, and Systems, (Springer US, Boston, MA, 1980). [Google Scholar]
- 82.Luscher HR, Ruenzel P, Fetz E & Henneman E Postsynatpic population potentials recorded from ventral roots perfused with isotonic sucrose: connections of groups Ia and II spindle afferent fibers with large populations of motoneurons. Journal of neurophysiology 42, 1146–1164 (1979). [DOI] [PubMed] [Google Scholar]
- 83.Leppanen L & Stys PK Ion transport and membrane potential in CNS myelinated axons I. Normoxic conditions. Journal of neurophysiology 78, 2086–2094 (1997). [DOI] [PubMed] [Google Scholar]
- 84.Rushton WA A theory of the effects of fibre size in medullated nerve. The Journal of physiology 115, 101–122 (1951). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Arbuthnott ER, Boyd IA & Kalu KU Ultrastructural dimensions of myelinated peripheral nerve fibres in the cat and their relation to conduction velocity. The Journal of physiology 308, 125–157 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blight AR Computer simulation of action potentials and afterpotentials in mammalian myelinated axons: the case for a lower resistance myelin sheath. Neuroscience 15, 13–31 (1985). [DOI] [PubMed] [Google Scholar]
- 87.Fedirchuk B, et al. Spontaneous network activity transiently depresses synaptic transmission in the embryonic chick spinal cord. The Journal of neuroscience : the official journal of the Society for Neuroscience 19, 2102–2112 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shefner JM, Buchthal F & Krarup C Recurrent potentials in human peripheral sensory nerve: possible evidence of primary afferent depolarization of the spinal cord. Muscle Nerve 15, 1354–1363 (1992). [DOI] [PubMed] [Google Scholar]
- 89.Boulenguez P, et al. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nature medicine 16, 302–307 (2010). [DOI] [PubMed] [Google Scholar]
- 90.Stalberg E, et al. Standards for quantification of EMG and neurography. Clin Neurophysiol 130, 1688–1729 (2019). [DOI] [PubMed] [Google Scholar]
- 91.Matthews PB Observations on the automatic compensation of reflex gain on varying the pre-existing level of motor discharge in man. The Journal of physiology 374, 73–90 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Foust KD, Poirier A, Pacak CA, Mandel RJ & Flotte TR Neonatal intraperitoneal or intravenous injections of recombinant adeno-associated virus type 8 transduce dorsal root ganglia and lower motor neurons. Hum Gene Ther 19, 61–70 (2008). [DOI] [PubMed] [Google Scholar]
- 93.Loeza-Alcocer E, et al. alpha(5)GABA(A) receptors mediate primary afferent fiber tonic excitability in the turtle spinal cord. Journal of neurophysiology 110, 2175–2184 (2013). [DOI] [PubMed] [Google Scholar]
- 94.Bravo-Hernandez M, et al. The alpha5 subunit containing GABAA receptors contribute to chronic pain. Pain 157, 613–626 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu K, Castellano D, Tian Q & Lu W Distinct regulation of tonic GABAergic inhibition by NMDA receptor subtypes. Cell Rep 37, 109960 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhou C, et al. Altered cortical GABAA receptor composition, physiology, and endocytosis in a mouse model of a human genetic absence epilepsy syndrome. J Biol Chem 288, 21458–21472 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhou W, et al. Loss of function of NCOR1 and NCOR2 impairs memory through a novel GABAergic hypothalamus-CA3 projection. Nat Neurosci 22, 205–217 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brown LE, et al. gamma-Aminobutyric Acid Type A (GABAA) Receptor Subunits Play a Direct Structural Role in Synaptic Contact Formation via Their N-terminal Extracellular Domains. J Biol Chem 291, 13926–13942 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dzyubenko E, et al. Inhibitory control in neuronal networks relies on the extracellular matrix integrity. Cell Mol Life Sci 78, 5647–5663 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fenrich KK, Zhao EY, Wei Y, Garg A & Rose PK Isolating specific cell and tissue compartments from 3D images for quantitative regional distribution analysis using novel computer algorithms. Journal of neuroscience methods 226, 42–56 (2014). [DOI] [PubMed] [Google Scholar]
- 101.Nicol MJ & Walmsley B A serial section electron microscope study of an identified Ia afferent collateral in the cat spinal cord. The Journal of comparative neurology 314, 257–277 (1991). [DOI] [PubMed] [Google Scholar]
- 102.Hines ML & Carnevale NT NEURON: a tool for neuroscientists. Neuroscientist 7, 123–135 (2001). [DOI] [PubMed] [Google Scholar]
- 103.McIntyre CC, Richardson AG & Grill WM Modeling the excitability of mammalian nerve fibers: influence of afterpotentials on the recovery cycle. Journal of neurophysiology 87, 995–1006 (2002). [DOI] [PubMed] [Google Scholar]
- 104.Cohen CCH, et al. Saltatory Conduction along Myelinated Axons Involves a Periaxonal Nanocircuit. Cell 180, 311–322 e315 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stephanova DI & Bostock H A distributed-parameter model of the myelinated human motor nerve fibre: temporal and spatial distributions of action potentials and ionic currents. Biol Cybern 73, 275–280 (1995). [DOI] [PubMed] [Google Scholar]
- 106.Olsen RW & Sieghart W GABA A receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology 56, 141–148 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chua HC & Chebib M GABAA Receptors and the Diversity in their Structure and Pharmacology. Adv Pharmacol 79, 1–34 (2017). [DOI] [PubMed] [Google Scholar]
- 108.Chuang SH & Reddy DS Genetic and Molecular Regulation of Extrasynaptic GABA-A Receptors in the Brain: Therapeutic Insights for Epilepsy. J Pharmacol Exp Ther 364, 180–197 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lagrange AH, Hu N & Macdonald RL GABA beyond the synapse: defining the subtype-specific pharmacodynamics of non-synaptic GABAA receptors. The Journal of physiology 596, 4475–4495 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Persohn E, Malherbe P & Richards JG In situ hybridization histochemistry reveals a diversity of GABAA receptor subunit mRNAs in neurons of the rat spinal cord and dorsal root ganglia. Neuroscience 42, 497–507 (1991). [DOI] [PubMed] [Google Scholar]
- 111.Paul J, Zeilhofer HU & Fritschy JM Selective distribution of GABA(A) receptor subtypes in mouse spinal dorsal horn neurons and primary afferents. The Journal of comparative neurology 520, 3895–3911 (2012). [DOI] [PubMed] [Google Scholar]
- 112.Ishizuka N, Mannen H, Hongo T & Sasaki S Trajectory of group Ia afferent fibers stained with horseradish peroxidase in the lumbosacral spinal cord of the cat: three dimensional reconstructions from serial sections. The Journal of comparative neurology 186, 189–211 (1979). [DOI] [PubMed] [Google Scholar]
- 113.Pan ZZ Transcriptional control of Gad2. Transcription 3, 68–72 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Esclapez M, Tillakaratne NJ, Kaufman DL, Tobin AJ & Houser CR Comparative localization of two forms of glutamic acid decarboxylase and their mRNAs in rat brain supports the concept of functional differences between the forms. The Journal of neuroscience : the official journal of the Society for Neuroscience 14, 1834–1855 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lalonde RL & Bui T Do spinal circuits still require gating of sensory information by presynaptic inhibition after spinal cord injury? Current Opinion in Physiol. 21, 113–118 (2020). [Google Scholar]
- 116.Rudomin P Presynaptic selection of afferent inflow in the spinal cord. Journal of physiology, Paris 93, 329–347 (1999). [DOI] [PubMed] [Google Scholar]
- 117.Prochazka A & Gorassini M Ensemble firing of muscle afferents recorded during normal locomotion in cats. The Journal of physiology 507 (Pt 1), 293–304 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Luscher HR, Ruenzel P & Henneman E Composite EPSPs in motoneurons of different sizes before and during PTP: implications for transmission failure and its relief in Ia projections. Journal of neurophysiology 49, 269–289 (1983). [DOI] [PubMed] [Google Scholar]
- 119.Eccles JC, Magni F & Willis WD Depolarization of central terminals of Group I afferent fibres from muscle. The Journal of physiology 160, 62–93 (1962). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sypert GW, Munson JB & Fleshman JW Effect of presynaptic inhibition on axonal potentials, terminal potentials, focal synaptic potentials, and EPSPs in cat spinal cord. Journal of neurophysiology 44, 792–803 (1980). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the manuscript or the supplementary materials. Raw data are available upon request to the corresponding authors. This study did not generate large data sets or new unique reagents.