

Keywords: glycocalyx, heparan sulfate, lung injury, matrix metalloproteinase, surfactant
Abstract
The alveolar epithelial glycocalyx is a dense anionic layer of glycosaminoglycans (GAGs) and proteoglycans that lines the apical surface of the alveolar epithelium. In contrast to the pulmonary endothelial glycocalyx, which has well-established roles in vascular homeostasis and septic organ dysfunction, the alveolar epithelial glycocalyx is less understood. Recent preclinical studies demonstrated that the epithelial glycocalyx is degraded in multiple murine models of acute respiratory distress syndrome (ARDS), particularly those that result from inhaled insults (so-called “direct” lung injury), leading to shedding of GAGs into the alveolar airspaces. Epithelial glycocalyx degradation also occurs in humans with respiratory failure, as quantified by analysis of airspace fluid obtained from ventilator heat moisture exchange (HME) filters. In patients with ARDS, GAG shedding correlates with the severity of hypoxemia and is predictive of the duration of respiratory failure. These effects may be mediated by surfactant dysfunction, as targeted degradation of the epithelial glycocalyx in mice was sufficient to cause increased alveolar surface tension, diffuse microatelectasis, and impaired lung compliance. In this review, we describe the structure of the alveolar epithelial glycocalyx and the mechanisms underlying its degradation during ARDS. We additionally review the current state of knowledge regarding the attributable effect of epithelial glycocalyx degradation in lung injury pathogenesis. Finally, we address glycocalyx degradation as a potential mediator of ARDS heterogeneity, and the subsequent value of point-of-care quantification of GAG shedding to potentially identify patients who are most likely to respond to pharmacological agents aimed at attenuating glycocalyx degradation.
INTRODUCTION
Acute respiratory distress syndrome (ARDS) is a devastating critical illness that affects over 200,000 patients in the United States per year and is associated with substantial morbidity and a mortality rate of 30%–35% (1–3). This complex syndrome is characterized by pulmonary vascular leak, surfactant dysfunction, impaired lung compliance, and hypoxemic respiratory failure (4, 5). Unfortunately, despite decades of preclinical research, clinical trials in ARDS have largely failed to identify pharmacological treatments that improve patient outcomes (6). The inability of promising preclinical data to translate to successful clinical trials is increasingly attributed to the substantial heterogeneity that exists within ARDS. Consequently, randomized studies have generally neglected to match treatment allocation to the mechanisms driving an individual patient’s lung injury (7). The development and personalized implementation of efficacious ARDS therapeutics demands both an understanding of these distinct mechanisms of lung injury and the identification of biomarkers that can determine the predominant driver of injury in each individual with ARDS (8–10).
One potentially targetable mechanism of ARDS pathogenesis is the degradation of the alveolar epithelial glycocalyx, a mesh-like layer composed of glycosaminoglycans (GAGs), proteoglycans, and glycoproteins lining the alveolar surface. In animal models of lung injury (as well as in patients with ARDS), the epithelial glycocalyx is degraded, shedding GAG fragments into the alveolar airspace (11, 12). In humans, the magnitude of this shedding is highly heterogeneous, with some patients with ARDS having little evidence of epithelial glycocalyx degradation (13). However, other patients with ARDS demonstrate substantial airspace GAG shedding, which is predictive of prolonged duration of mechanical ventilation and hospital course (13). GAG shedding can be easily and rapidly identified at the bedside, using a point-of-care assay of noninvasively collected airspace fluid samples. Such assays are capable of rapidly identifying potentially targetable mechanisms of lung injury (so-called “treatable traits”) and may facilitate the implementation of personalized treatment strategies for ARDS (14).
In this review, we will describe the structure of the alveolar epithelial glycocalyx. We then review the existing literature on the mechanisms of epithelial glycocalyx degradation during lung injury and the attributable effect of glycocalyx degradation on ARDS pathogenesis. Finally, we explore the potential role for point-of-care quantification of airspace GAG shedding as a novel, and potentially causal, biomarker in ARDS, which may identify patients most likely to benefit from pharmacological agents aimed at either preventing epithelial glycocalyx degradation or promoting its regeneration.
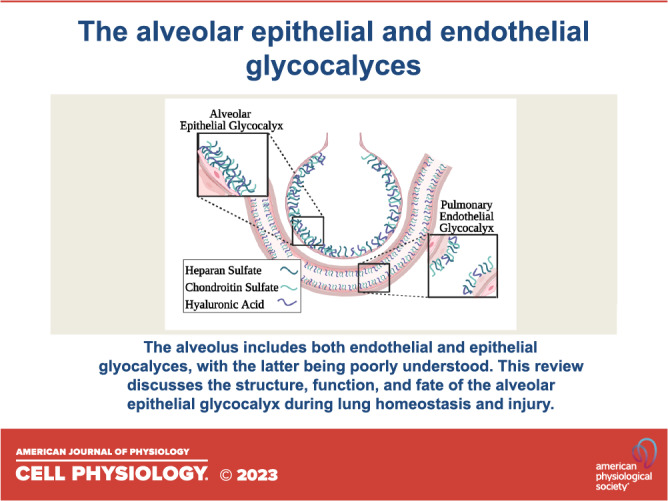
ALVEOLAR EPITHELIAL GLYCOCALYX STRUCTURE
The presence of a continuous layer lining the alveolar epithelium has been appreciated since electron microscopy studies from the 1960s. These foundational studies by Drs. Weibel and Gil noted the presence of a superficial surface film, now known to be comprised of pulmonary surfactant, overlying an alveolar hypophase or “dense basal layer” hypothesized to be composed of mucopolysaccharides (i.e., GAGs) and proteins (15–17). Following these original anatomic descriptions, there have been decades of intense study on the structure and function of surfactant, which is now known to have critical biophysical and immunomodulatory functions (18, 19). In comparison, the alveolar hypophase, now recognized to be the alveolar epithelial glycocalyx, has been understudied (20). Emerging studies now demonstrate that the alveolar epithelial glycocalyx is a critical part of the epithelial surface and that its degradation contributes to lung injury pathogenesis following multiple different pulmonary insults.
The alveolar epithelial glycocalyx, like the glycocalyces of other endothelial and epithelial cells throughout the body, is predominantly composed of long unbranched GAGs that are anchored to the apical cell membrane by transmembrane or glycosylphosphatidylinositol-anchored proteoglycans (21). The main GAG species that make up this structure are heparan sulfate (HS, a linear polysaccharide composed of repeating units of N-acetyl glucosamine and a hexuronic acid—either glucuronic acid or its epimer, iduronic acid), chondroitin sulfate (CS, a linear polysaccharide composed of repeating units of N-acetyl galactosamine and a hexuronic acid), and hyaluronic acid (HA, an unsulfated linear polysaccharide composed of N-acetyl galactosamine and glucuronic acid) (22, 23) (Fig. 1). HS and CS are synthesized on a protein backbone in the Golgi apparatus by a complex series of steps including chain polymerization (mediated by exostosins-1 and -2), sulfation (mediated by sulfotransferases such as N-deacetylase/N-sulfotransferase-1 and -2), and epimerases (22). These sulfated GAG chains and their attached protein backbones, which include HS proteoglycans (HSPGs) or CS proteoglycans (CSPGs), such as syndecans, are then transported to the cellular surface, where they form the glycocalyx (24). Once extracellular, glycocalyx HS may undergoing additional modification by extracellular sulfatases such as sulf-1 and -2; this postsynthetic control of HS sulfation suggests homeostatic importance of glycocalyx sulfation. Indeed, sulfation of HS and CS at specific sites imparts geographic domains of negative charge across these molecules, allowing electrostatic binding to cognate positively charged residues of proteins. GAG binding modifies protein function, allowing GAGs to regulate downstream cellular signaling (25). HA, in contrast to HS and CS, is unsulfated and does not bind to proteoglycans (26). HA is synthesized by hyaluronan synthases, which are enzymes with multiple transmembrane domains that are located on the inner surface of the plasma membrane. During synthesis, the growing HA polymer is extruded through the membrane to the extracellular space, where it then anchors to the cell surface by binding to HA synthase enzymes or cell surface receptors such as CD44 (27). The extracellular HA also forms complexes with other GAGs, which enhances the structural stability of the glycocalyx (28). Together this mesh-like anionic layer supports many homeostatic functions of both endothelial and epithelial cells throughout the body. A detailed schematic of the structure of the epithelial glycocalyx is provided in an excellent review by Ochs et al. (20).
Figure 1.

Structure of the alveolar epithelial glycocalyx. The epithelial glycocalyx is a layer of glycosaminoglycans (GAGs) that line the epithelial surface. Heparan sulfate (HS) is a linear polysaccharide composed of repeating units of glucosamine and a hexuronic acid (either glucosamine or its epimer, iduronic acid). Chondroitin sulfate (CS) is a linear polysaccharide composed of repeating units of galactosamine and a hexuronic acid. Both HS and CS are attached to a proteoglycan backbone and can be sulfated at specific sites as indicated. Hyaluronic acid (HA) is a linear polysaccharide that is composed of galactosamine and glucuronic acid. It binds to cluster differentiation 44 (CD44) and is not covalently linked to proteoglycans of the epithelial surface and is not sulfated. Gal, galactosamine; GalNAc, N-acetyl galactosamine; GlcA, glucuronic acid; GlcNAc, N-acetyl glucosamine; IdoA, iduronic acid; Xyl, xylulose. The schematic figure was created using BioRender.
MECHANISMS OF ALVEOLAR EPITHELIAL GLYCOCALYX DEGRADATION
Recent studies have demonstrated that the alveolar epithelial glycocalyx is degraded in multiple murine models of lung injury, suggesting that it may play an important role in the pathogenesis of ARDS (11–13). Haeger et al. (12) found that GAGs, including HS and CS, are shed into the airspaces, but not the circulation, of mice following the administration of intratracheal lipopolysaccharide (LPS). These results are supported by in vitro experiments in which LPS induced HS shedding in cultured A549 epithelial cells (29). Interestingly, these findings are not unique to LPS, as HS is also shed into the bronchoalveolar lavage (BAL) fluid in murine models of bleomycin-induced lung injury, where full-length and highly sulfated HS was observed to persist in the airspaces for 3 wk after the initial injury (11). Furthermore, similar epithelial glycocalyx degradation occurs after influenza infection, which induces a profound and prolonged GAG shedding into the airspaces (30). In contrast to the substantial airspace GAG shedding observed in these models characterized by initial epithelial injury (so-called “direct” lung injury), we observed little evidence of alveolar epithelial glycocalyx degradation following cecal ligation and puncture (CLP), a model characterized primarily by pulmonary endothelial dysfunction (so-called “indirect” lung injury) (Fig. 2). Taken together, these preclinical findings suggest that direct forms of lung injury induce epithelial glycocalyx degradation, with minimal effect on the pulmonary endothelial glycocalyx. Conversely, indirect forms of lung injury induce prominent endothelial glycocalyx degradation with minimal damage to the epithelial glycocalyx. This compartmentalization of glycocalyx degradation in direct versus indirect lung injury is supported by observational studies of critically ill humans (31, 32).
Figure 2.

The epithelial and endothelial glycocalyces of the lung during health and lung injury. A: during homeostasis the lung has both a pulmonary endothelial glycocalyx and an alveolar epithelial glycocalyx. Lung injury from inhaled insults (“direct lung injury”) is associated with degradation of the alveolar epithelial glycocalyx. Conversely, lung injury from systemic insults (“indirect lung injury”) is largely characterized by degradation of the pulmonary endothelial glycocalyx. Notably, patients with acute respiratory distress syndrome (ARDS) often have multiple simultaneous insults, which can lead to degradation of both the endothelial and epithelial glycocalyces. B: direct pulmonary insults in mice (lipopolysaccharide, influenza, and bleomycin) induces shedding of heparan sulfate (HS) into the alveolar lining fluid (bronchoalveolar lavage, corrected for urea dilution) (11, 12, 30). C: indirect lung injury in mice (cecal ligation and puncture, CLP) does not induce shedding of HS into the alveolar lining fluid. CLP airspace samples pooled (n = 3/group). D: CLP does induce shedding of HS from the endothelial glycocalyx into the plasma (34). For B and D, n > 5 mice/group. Bars = standard error of the mean. Individual comparisons by t test; multiple comparisons by ANOVA. *P < 0.05. FFU, focus forming unit; IN, intranasal; IT, intratracheal; LPS, lipopolysaccharide. The schematic figure (A) was created using BioRender.
Consistent with this compartmentalization of glycocalyx shedding, the mechanisms driving endothelial and epithelial glycocalyx degradation are distinct. In sepsis-associated endothelial glycocalyx degradation, GAG shedding is mediated by activation of heparanase (an HS-specific glucuronidase), which directly degrades HS and releases small octasaccharide fragments into the plasma (33, 34). This contrasts with direct lung injury-associated alveolar epithelial glycocalyx degradation, during which the shed GAGs are largely intact (>20 saccharides in length) and highly sulfated (11, 12). The presence of full-length GAGs within the alveolar space suggests that epithelial glycocalyx degradation is mediated by enzymes that cleave the proteoglycans anchoring GAGs to the epithelial surface, rather than glucuronidases (such as heparanase) or other glycan-targeted enzymes that directly cleaves GAGs into small fragments (22). Indeed, inducible deletion of heparanase in mice after intratracheal bleomycin had no effect on persistent airspace GAG shedding after bleomycin (11). Similarly, constitutive deletion of heparanase did not prevent the magnitude of lung injury after intratracheal LPS (35). Notably, these results are in contradiction to the findings of Li et al. (29), who reported that pharmacological inhibition of heparanase attenuated LPS-induced damage to tight junctions in both A549 cells and murine LPS-induced lung injury. In addition, although much of the published literature has focused on mechanisms of HS shedding, it is important to note that CS and HA are also shed into the airspaces during lung injury (13). CS shedding could also be a consequence of syndecan shedding, which can have CS side chains (36). Alternatively, shedding of CS and HA may also be a consequence of shedding of versican, a large CS-containing proteoglycan that interacts with HA via its core protein (27, 37).
Recent work highlights that epithelial glycocalyx degradation during direct lung injury is likely mediated by multiple redundant proteases. For example, LPS and bleomycin both induce expression of matrix metalloproteinases (MMPs), which are proteases capable of cleaving proteoglycans (such as syndecan-1) anchoring GAGs to the epithelial surface (11, 12, 38–40). LPS is associated with an induction of both MMP 9 and (to a lesser extent) MMP 2; intriguingly, genetic knockout of MMP 9 does not attenuate epithelial glycocalyx shedding, perhaps due to compensatory upregulation of MMP 2 (12). Similarly, bleomycin induced an early activation of alveolar MMP 2, followed by later induction of MMP 9 (11). In addition, work by Gill et al. (40) demonstrated that MMP7 is an important protease that promotes shedding of syndecan-1 from the epithelial glycocalyx. Together, these findings suggest that broad MMP inhibition may be necessary to attenuate glycocalyx shedding in direct lung injury. Indeed, doxycycline, a broad inhibitor of MMPs and related proteases, attenuates the severity of epithelial glycocalyx shedding and/or lung injury in LPS, bleomycin, or influenza infection (11, 12, 41). Other proteases, such as a disintegrin-like metalloproteinases (ADAMs), likely also contribute to the extent of epithelial glycocalyx degradation during lung injury. Pruessmeyer et al. (42) demonstrated that ADAM17 mediates shedding of syndecan-1 and -4 from the epithelial cell surface in response to tumor necrosis factor (TNF)α and interferon (IFN)γ. Together, this body of work suggests that a complex system in which a redundant induction of proteases, including MMPs and ADAMs, mediate epithelial glycocalyx degradation following direct lung injury.
CONSEQUENCES OF ALVEOLAR EPITHELIAL GLYCOCALYX DEGRADATION DURING ARDS
Epithelial Barrier Dysfunction
To determine the attributable effect of epithelial glycocalyx degradation in lung injury, our group developed a murine model of targeted, enzymatic (bacterial heparinase-induced) degradation of the epithelial glycocalyx. Using this model, we observed that targeted epithelial glycocalyx degradation was sufficient to induce an increase in BAL protein content. This increased alveolar permeability to protein surprisingly occurred in the absence of alveolar inflammation or the development of pulmonary edema, suggestive that epithelial heparan sulfate degradation was not simply causing nonspecific injury to the alveolar septum (12). Interestingly, these findings were specific to HS degradation, as similar targeted degradation of epithelial CS with chondroitinases did not impair epithelial barrier function in mice (12). Although the precise mechanisms by which epithelial glycocalyx shedding causes epithelial barrier dysfunction are unknown, work by Brauer et al. (43) demonstrated that cell membrane-associated syndecan-1 regulates the response to influenza by facilitating cytoprotective signaling that limits bronchial epithelial apoptosis and attenuates lung injury. These findings suggest that epithelial glycocalyx degradation may impair barrier function by increasing epithelial apoptosis, which could explain the findings of increased barrier permeability in the absence of a significant inflammatory response that was reported during targeted epithelial glycocalyx degradation (12). In addition, it is critical to note that the importance of the epithelial glycocalyx to barrier integrity is not limited to the lung, as work by multiple groups has demonstrated the importance of the glycocalyx to barrier regulation in other epithelial interfaces (including the gut and the eye) (44, 45).
Surfactant Dysfunction
Surfactant is a complex mixture of lipids (90%) and proteins (10%) secreted by alveolar type II epithelial cells into the alveolar space, where it serves to reduce surface tension at the air-liquid interface to prevent alveolar collapse during exhalation (46). The alveolar epithelial glycocalyx is interposed between epithelial cells and surfactant; however, technical challenges in visualization of the epithelial glycocalyx structures have limited investigation into the potential interactions between these two structures (20). We recently reported that targeted degradation of HS from the alveolar epithelial glycocalyx impairs surfactant function, leading to microatelectasis and impaired lung compliance (12, 13). Interestingly, no loss of compliance was induced by the addition of exogenous GAG fragments (HS or CS) to uninjured mice, suggesting that surfactant dysfunction arises from injury to the native glycocalyx, rather than the presence of GAG fragments in the airspaces.
The clinical relevance of these findings is supported by an observational human study in which airspace GAG fragments were predictive of duration of respiratory failure (13). In addition, patients with lysosomal storage diseases, such as mucopolysaccharidosis type IIIA, exhibit impaired surfactant metabolism and decreased lung function (47). Although the precise mechanisms by which the epithelial glycocalyx supports surfactant function are uncertain at this time, we observed that heparin, a highly sulfated form of HS, can directly bind to surfactant proteins A, B, and D (13). Surfactant proteins A and D both belong to the collectin family of proteins, which contain a carbohydrate recognition domain capable of binding to glycans (48). We speculate that an intact epithelial glycocalyx could serve to tether surfactant to the epithelium, ensuring optimal surfactant distribution necessary for the maintenance of alveolar recruitment.
Impaired Epithelial Repair
In addition to these acute effects on pulmonary physiology, alveolar epithelial glycocalyx degradation impedes recovery from injury. During bleomycin-induced lung injury, a model in which lung injury is followed by a prolonged period of recovery, HS fragments persist in the air spaces for 3 wk after the initial insult. Treatment with doxycycline, a broad MMP inhibitor, starting 7 days after bleomycin administration attenuated persistent GAG shedding and improved lung function, suggestive that ongoing glycocalyx degradation may impede the process of epithelial repair (11). Although the exact mechanisms underlying these findings are unknown, work by Altemeier et al. (49) demonstrates that syndecan-1 is important for lung epithelial migration and adhesion, which suggests that shedding of syndecan-1 may contribute to impaired epithelial repair. In addition, work by Parimon et al. (50, 51) demonstrates that syndecan-1 shedding influences the progression or resolution of lung injury in both lung fibrosis, by regulating epithelial reprogramming through extracellular vesicles, and lung tumorigenesis, by regulating exosomal miRNAs. Furthermore, given that the epithelial glycocalyx is necessary for barrier function, this prolonged GAG shedding may delay recovery of normal alveolar-capillary integrity (12). Finally, shed GAG fragments are also biologically active and capable of binding to and sequestering lung-reparative growth factors, such as fibroblast growth factor (FGF) and hepatocyte growth factor (HGF), thus providing another possible mechanism by which persistent glycocalyx degradation impairs lung recovery (11).
Increased Severity of Secondary Bacterial Pneumonia
In addition to the direct effects of alveolar epithelial glycocalyx degradation on lung injury and repair, epithelial glycocalyx degradation may influence host-pathogen interactions. Although it is well known that viral pneumonias place the lung at increased susceptibility for severe secondary bacterial pneumonias, the mechanisms by which this occurs are incompletely understood (52). Our group recently reported that the influenza-injured lung microenvironment induces methicillin-resistant staph aureus (MRSA) to upregulate the pore forming cytotoxin leucocidin A/B (LukAB), which contributes to the increased severity of secondary MRSA pneumonia. Interestingly, alveolar epithelial HS that is shed during influenza infection binds and activates LukAB, contributing to the severity of postinfluenza MRSA pneumonia (30). Studies of syndecan-1 shedding in corneal infections suggest that epithelial glycocalyx shedding may promote secondary bacterial infection via additional mechanisms, such as promoting Streptococcal adhesion to cryptic fibronectin sites on the epithelial surface (53) or sequestration of host-protective cationic antimicrobial peptides (54).
THE ROLE OF HYALURONIC ACID IN THE EPITHELIAL GLYCOCALYX
As described earlier, HA differs from HS and CS in that it is both unsulfated and not covalently bound to the epithelial surface. Given these structural differences, it is not surprising that HA also has distinct functions within the epithelial glycocalyx (27, 55). Like other GAGs, epithelial HA is shed in multiple forms of lung injury, including inflammatory lung disease and pulmonary fibrosis, via the action of hyaluronidases and inflammatory stimuli, such as reactive oxygen species (13, 55, 56). Furthermore, in the aftermath of injury, the lung generates a provisional HA glycocalyx which can have both protective and deleterious effects in a context-dependent manner (27, 55, 57–59). For example, increased HA production and/or impaired HA clearance causes an ongoing innate inflammatory response that is mediated by HA-binding proteins including tumor necrosis factor (TNF)-stimulated gene 6 (57, 60). Here, increased HA leads to the generation of heavy-chain-HA complexes, which can be degraded into smaller LMW fragments that can engage cell receptors, including CD44, TLR4, and TLR2, and impact downstream biological effects including inflammation and fibrosis (58). In addition, recent work has also demonstrated that imbalances in HA impact the resolution of influenza in mice, during which HA complexes with the HA-binding protein inter-α-inhibitor, ultimately leading to ongoing inflammation and decreased lung function, an effect that can be abrogated with administration of exogenous hyaluronidases (59). The clinical importance of epithelial HA in inflammatory lung disease is also highlighted by recent work by Kratochvil et al. (61), which demonstrated that HA is a major component of the respiratory secretions of patients with COVID-19. Finally, in addition to these inflammatory consequences of HA shedding during lung injury, HA fragments and associated proteins, such as inter-alpha-inhibitor, have been shown to influence epithelial sodium channel activity, which could impact alveolar fluid clearance and ARDS pathogenesis (62, 63).
ALVEOLAR EPITHELIAL GLYCOCALYX DEGRADATION AS A MEDIATOR OF ARDS HETEROGENEITY
The failure of strong preclinical data to translate into efficacious ARDS therapies is increasingly attributed to the mechanistic heterogeneity of this complex illness (3, 7, 64). Calfee et al. (65) identified two distinct phenotypes of ARDS (8), each of which may experience different (and possibly opposing) treatment responses to therapeutics. This work inspired interest in precision medicine approaches to ARDS care, in which rapid, biomarker-based identification of the specific mechanisms driving lung injury would allow for personalization of targeted therapies (10, 14). Epithelial glycocalyx degradation may be one such heterogeneous and potentially targetable mechanism that contributes to lung injury in patients with ARDS. Interestingly, the heterogeneity that has been noted in GAG shedding in ARDS depends on both injury-specific factors (direct vs. indirect) as well as unexpected patient-level factors (such as sex) (13).
The ability to rapidly identify a subgroup of patients in whom lung injury is driven by epithelial glycocalyx degradation would be highly useful, as it would enable predictive and/or prognostic enrichment of clinical trials aimed at determining the efficacy of therapies preventing glycocalyx degradation. GAGs are a promising biomarker in part due to their durability: they are unaffected by proteases and are durable in freeze-thaw cycles (66). Although mass spectrometry with multiple reaction monitoring is the gold standard for GAG quantification, this approach is expensive and relies on a high level of technical expertise. Our group has recently optimized a simple and inexpensive ($2 per sample) colorimetric assay (dimethylmethylene blue) of GAGs that is accurate in both urine (where it reflects excreted circulating GAGs and thus endothelial glycocalyx degradation) and HME fluid (where it reflects alveolar epithelial glycocalyx degradation) (13, 67). Assays such as these could be useful in identifying a subgroup of patients most likely to benefit from therapeutic strategies aimed at preventing GAG shedding, such as MMP inhibitors or MMP-blocking monoclonal antibodies (68, 69).
CONCLUSIONS
The alveolar epithelial glycocalyx is a complex and understudied structure with importance to both lung homeostasis and lung injury pathogenesis. During injury, multiple inflammatory stimuli activate MMPs and other (likely redundant) proteases, cleaving HSPGs and CSPGs that anchor the glycocalyx to the epithelial surface. The subsequent shedding of the epithelial glycocalyx has important consequences that contribute to the onset and progression of lung injury, including alveolar hyperpermeability, disruption of surfactant function, increased bacterial virulence, and impaired epithelial cell repair. Additional observational studies in humans are needed to better understand the contribution of epithelial glycocalyx degradation to lung injury heterogeneity, potentially allowing the epithelial glycocalyx to serve as a therapeutic target in precision medicine approaches for ARDS care.
GRANTS
This work was supported by the National Heart, Lung, and Blood Institute Grants R01 HL125371 (to E. P. Schmidt) and F32 HL162230 (to A. N. Rizzo) and the Congressionally Directed Medical Research Program Grant W81XWH-18-1-0682 (to E. P. Schmidt).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.N.R. and E.P.S. conceived and designed research; A.N.R. performed experiments; A.N.R. and E.P.S. analyzed data; A.N.R. and E.P.S. interpreted results of experiments; A.N.R. prepared figures; A.N.R. and E.P.S. drafted manuscript; A.N.R. and E.P.S. edited and revised manuscript; A.N.R. and E.P.S. approved final version of manuscript.
ACKNOWLEDGMENTS
Figures and graphical abstract created with BioRender and published with permission.
REFERENCES
- 1. Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, Gattinoni L, van Haren F, Larsson A, McAuley DF, Ranieri M, Rubenfeld G, Thompson BT, Wrigge H, Slutsky AS, Pesenti A; LUNG SAFE Investigators, ESICM Trials Group. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 315: 788–800, 2016. doi: 10.1001/jama.2016.0291. [DOI] [PubMed] [Google Scholar]
- 2.ARDS Definition Task Force, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, Slutsky AS. Acute respiratory distress syndrome: the Berlin Definition. JAMA 307: 2526–2533, 2012. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 3. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, Herridge M, Randolph AG, Calfee CS. Acute respiratory distress syndrome. Nat Rev Dis Primers 5: 18, 2019. doi: 10.1038/s41572-019-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet 2: 319–323, 1967. doi: 10.1016/s0140-6736(67)90168-7. [DOI] [PubMed] [Google Scholar]
- 5. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349, 2000. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 6. Matthay MA, McAuley DF, Ware LB. Clinical trials in acute respiratory distress syndrome: challenges and opportunities. Lancet Respir Med 5: 524–534, 2017. doi: 10.1016/S2213-2600(17)30188-1. [DOI] [PubMed] [Google Scholar]
- 7. Beitler JR, Thompson BT, Baron RM, Bastarache JA, Denlinger LC, Esserman L, Gong MN, LaVange LM, Lewis RJ, Marshall JC, Martin TR, McAuley DF, Meyer NJ, Moss M, Reineck LA, Rubin E, Schmidt EP, Standiford TJ, Ware LB, Wong HR, Aggarwal NR, Calfee CS. Advancing precision medicine for acute respiratory distress syndrome. Lancet Respir Med 10: 107–120, 2022. doi: 10.1016/S2213-2600(21)00157-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA; NHLBI ARDS Network. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med 2: 611–620, 2014. doi: 10.1016/S2213-2600(14)70097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Prescott HC, Calfee CS, Thompson BT, Angus DC, Liu VX. Toward smarter lumping and smarter splitting: rethinking strategies for sepsis and acute respiratory distress syndrome clinical trial design. Am J Respir Crit Care Med 194: 147–155, 2016. doi: 10.1164/rccm.201512-2544CP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reilly JP, Calfee CS, Christie JD. Acute respiratory distress syndrome phenotypes. Semin Respir Crit Care Med 40: 19–30, 2019. doi: 10.1055/s-0039-1684049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. LaRivière WB, Liao S, McMurtry SA, Oshima K, Han X, Zhang F, Yan S, Haeger SM, Ransom M, Bastarache JA, Linhardt RJ, Schmidt EP, Yang Y. Alveolar heparan sulfate shedding impedes recovery from bleomycin-induced lung injury. Am J Physiol Lung Cell Mol Physiol 318: L1198–L1210, 2020. doi: 10.1152/ajplung.00063.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haeger SM, Liu X, Han X, McNeil JB, Oshima K, McMurtry SA, Yang Y, Ouyang Y, Zhang F, Nozik-Grayck E, Zemans RL, Tuder RM, Bastarache JA, Linhardt RJ, Schmidt EP. Epithelial heparan sulfate contributes to alveolar barrier function and is shed during lung injury. Am J Respir Cell Mol Biol 59: 363–374, 2018. doi: 10.1165/rcmb.2017-0428OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rizzo AN, Haeger SM, Oshima K, Yang Y, Wallbank AM, Jin Y, Lettau M, McCaig LA, Wickersham NE, McNeil JB, Zakharevich I, McMurtry SA, Langouët-Astrié CJ, Kopf KW, Voelker DR, Hansen KC, Shaver CM, Kerchberger VE, Peterson RA, Kuebler WM, Ochs M, Veldhuizen RA, Smith BJ, Ware LB, Bastarache JA, Schmidt EP. Alveolar epithelial glycocalyx degradation mediates surfactant dysfunction and contributes to acute respiratory distress syndrome. JCI Insight 7: e154573, 2022. doi: 10.1172/jci.insight.154573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meyer NJ, Calfee CS. Novel translational approaches to the search for precision therapies for acute respiratory distress syndrome. Lancet Respir Med 5: 512–523, 2017. doi: 10.1016/S2213-2600(17)30187-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Low FN. Electron microscopy of the rat lung. Anat Rec 113: 437–449, 1952. doi: 10.1002/ar.1091130406. [DOI] [PubMed] [Google Scholar]
- 16. Weibel ER, Gil J. Electron microscopic demonstration of an extracellular duplex lining layer of alveoli. Respir Physiol 4: 42–57, 1968. doi: 10.1016/0034-5687(68)90006-6. [DOI] [PubMed] [Google Scholar]
- 17. Gil J, Weibel ER. Improvements in demonstration of lining layer of lung alveoli by electron microscopy. Respir Physiol 8: 13–36, 1969. doi: 10.1016/0034-5687(69)90042-5. [DOI] [PubMed] [Google Scholar]
- 18. Knudsen L, Ochs M. The micromechanics of lung alveoli: structure and function of surfactant and tissue components. Histochem Cell Biol 150: 661–676, 2018. doi: 10.1007/s00418-018-1747-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han S, Mallampalli RK. The role of surfactant in lung disease and host defense against pulmonary infections. Ann Am Thorac Soc 12: 765–774, 2015. doi: 10.1513/AnnalsATS.201411-507FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ochs M, Hegermann J, Lopez-Rodriguez E, Timm S, Nouailles G, Matuszak J, Simmons S, Witzenrath M, Kuebler WM. On top of the alveolar epithelium: surfactant and the glycocalyx. Int J Mol Sci 21: 3075, 2020. doi: 10.3390/ijms21093075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tarbell JM, Cancel LM. The glycocalyx and its significance in human medicine. J Intern Med 280: 97–113, 2016. doi: 10.1111/joim.12465. [DOI] [PubMed] [Google Scholar]
- 22. Haeger SM, Yang Y, Schmidt EP. Heparan sulfate in the developing, healthy, and injured lung. Am J Respir Cell Mol Biol 55: 5–11, 2016. doi: 10.1165/rcmb.2016-0043TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mikami T, Kitagawa H. Biosynthesis and function of chondroitin sulfate. Biochim Biophys Acta 1830: 4719–4733, 2013. doi: 10.1016/j.bbagen.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 24. Xu D, Esko JD. Demystifying heparan sulfate-protein interactions. Annu Rev Biochem 83: 129–157, 2014. doi: 10.1146/annurev-biochem-060713-035314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bernfield M, Götte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem 68: 729–777, 1999. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 26. Fraser JR, Laurent TC, Laurent UB. Hyaluronan: its nature, distribution, functions and turnover. J Intern Med 242: 27–33, 1997. doi: 10.1046/j.1365-2796.1997.00170.x. [DOI] [PubMed] [Google Scholar]
- 27. Wight TN. Provisional matrix: a role for versican and hyaluronan. Matrix Biol 60–61: 38–56, 2017. doi: 10.1016/j.matbio.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sullivan RC, Rockstrom MD, Schmidt EP, Hippensteel JA. Endothelial glycocalyx degradation during sepsis: causes and consequences. Matrix Biol Plus 12: 100094, 2021. doi: 10.1016/j.mbplus.2021.100094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li J, Qi Z, Li D, Huang X, Qi B, Feng J, Qu J, Wang X. Alveolar epithelial glycocalyx shedding aggravates the epithelial barrier and disrupts epithelial tight junctions in acute respiratory distress syndrome. Biomed Pharmacother 133: 111026, 2021. doi: 10.1016/j.biopha.2020.111026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Langouët-Astrié C, Oshima K, McMurtry SA, Yang Y, Kwiecinski JM, LaRivière WB, Kavanaugh JS, Zakharevich I, Hansen KC, Shi D, Zhang F, Boguslawski KM, Perelman SS, Su G, Torres VJ, Liu J, Horswill AR, Schmidt EP. The influenza-injured lung microenvironment promotes MRSA virulence, contributing to severe secondary bacterial pneumonia. Cell Rep 41: 111721, 2022. doi: 10.1016/j.celrep.2022.111721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murphy LS, Wickersham N, McNeil JB, Shaver CM, May AK, Bastarache JA, Ware LB. Endothelial glycocalyx degradation is more severe in patients with non-pulmonary sepsis compared to pulmonary sepsis and associates with risk of ARDS and other organ dysfunction. Ann Intensive Care 7: 102, 2017. doi: 10.1186/s13613-017-0325-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schmidt EP, Li G, Li L, Fu L, Yang Y, Overdier KH, Douglas IS, Linhardt RJ. The circulating glycosaminoglycan signature of respiratory failure in critically ill adults. J Biol Chem 289: 8194–8202, 2014. doi: 10.1074/jbc.M113.539452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, Zemans RL, Bowman JC, Koyanagi DE, Yunt ZX, Smith LP, Cheng SS, Overdier KH, Thompson KR, Geraci MW, Douglas IS, Pearse DB, Tuder RM. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med 18: 1217–1223, 2012. doi: 10.1038/nm.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang Y, Haeger SM, Suflita MA, Zhang F, Dailey KL, Colbert JF, Ford JA, Picon MA, Stearman RS, Lin L, Liu X, Han X, Linhardt RJ, Schmidt EP. Fibroblast growth factor signaling mediates pulmonary endothelial glycocalyx reconstitution. Am J Respir Cell Mol Biol 56: 727–737, 2017. doi: 10.1165/rcmb.2016-0338OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morris A, Wang B, Waern I, Venkatasamy R, Page C, Schmidt EP, Wernersson S, Li J-P, Spina D. The role of heparanase in pulmonary cell recruitment in response to an allergic but not non-allergic stimulus. PLoS One 10: e0127032, 2015. doi: 10.1371/journal.pone.0127032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alexopoulou AN, Multhaupt HAB, Couchman JR. Syndecans in wound healing, inflammation and vascular biology. Int J Biochem Cell Biol 39: 505–528, 2007. doi: 10.1016/j.biocel.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 37. Day AJ, de la Motte CA. Hyaluronan cross-linking: a protective mechanism in inflammation? Trends Immunol 26: 637–643, 2005. doi: 10.1016/j.it.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 38. Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell 111: 635–646, 2002. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- 39. Zhang D, Zhang J-T, Pan Y, Liu X-F, Xu J-W, Cui W-J, Qiao X-R, Dong L. Syndecan-1 shedding by matrix metalloproteinase-9 signaling regulates alveolar epithelial tight junction in lipopolysaccharide-induced early acute lung injury. J Inflamm Res 14: 5801–5816, 2021. doi: 10.2147/JIR.S331020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gill SE, Nadler ST, Li Q, Frevert CW, Park PW, Chen P, Parks WC. Shedding of syndecan-1/CXCL1 complexes by matrix metalloproteinase 7 functions as an epithelial checkpoint of neutrophil activation. Am J Respir Cell Mol Biol 55: 243–251, 2016. doi: 10.1165/rcmb.2015-0193OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ng HH, Narasaraju T, Phoon MC, Sim MK, Seet JE, Chow VT. Doxycycline treatment attenuates acute lung injury in mice infected with virulent influenza H3N2 virus: involvement of matrix metalloproteinases. Exp Mol Pathol 92: 287–295, 2012. doi: 10.1016/j.yexmp.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 42. Pruessmeyer J, Martin C, Hess FM, Schwarz N, Schmidt S, Kogel T, Hoettecke N, Schmidt B, Sechi A, Uhlig S, Ludwig A. A disintegrin and metalloproteinase 17 (ADAM17) mediates inflammation-induced shedding of syndecan-1 and -4 by lung epithelial cells. J Biol Chem 285: 555–564, 2010. doi: 10.1074/jbc.M109.059394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brauer R, Ge L, Schlesinger SY, Birkland TP, Huang Y, Parimon T, Lee V, McKinney BL, McGuire JK, Parks WC, Chen P. Syndecan-1 attenuates lung injury during influenza infection by potentiating c-Met signaling to suppress epithelial apoptosis. Am J Respir Crit Care Med 194: 333–344, 2016. doi: 10.1164/rccm.201509-1878OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Coulson-Thomas VJ, Chang S-H, Yeh L-K, Coulson-Thomas YM, Yamaguchi Y, Esko J, Liu C-Y, Kao W. Loss of corneal epithelial heparan sulfate leads to corneal degeneration and impaired wound healing. Invest Ophthalmol Vis Sci 56: 3004–3014, 2015. doi: 10.1167/iovs.14-15341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen S, He Y, Hu Z, Lu S, Yin X, Ma X, Lv C, Jin G. Heparanase mediates intestinal inflammation and injury in a mouse model of sepsis. J Histochem Cytochem 65: 241–249, 2017. doi: 10.1369/0022155417692536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bersten AD, Davidson K, Nicholas TE, Doyle IR. Respiratory mechanics and surfactant in the acute respiratory distress syndrome. Clin Exp Pharmacol Physiol 25: 955–963, 1998. doi: 10.1111/j.1440-1681.1998.tb02352.x. [DOI] [PubMed] [Google Scholar]
- 47. Paget TL, Parkinson-Lawrence EJ, Trim PJ, Autilio C, Panchal MH, Koster G, Echaide M, Snel MF, Postle AD, Morrison JL, Pérez-Gil J, Orgeig S. Increased alveolar heparan sulphate and reduced pulmonary surfactant amount and function in the mucopolysaccharidosis IIIA mouse. Cells 10: 849, 2021. doi: 10.3390/cells10040849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Crouch EC. Collectins and pulmonary host defense. Am J Respir Cell Mol Biol 19: 177–201, 1998. doi: 10.1165/ajrcmb.19.2.140. [DOI] [PubMed] [Google Scholar]
- 49. Altemeier WA, Schlesinger SY, Buell CA, Brauer R, Rapraeger AC, Parks WC, Chen P. Transmembrane and extracellular domains of syndecan-1 have distinct functions in regulating lung epithelial migration and adhesion. J Biol Chem 287: 34927–34935, 2012. doi: 10.1074/jbc.M112.376814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Parimon T, Brauer R, Schlesinger SY, Xie T, Jiang D, Ge L, Huang Y, Birkland TP, Parks WC, Habiel DM, Hogaboam CM, Gharib SA, Deng N, Liu Z, Chen P. Syndecan-1 controls lung tumorigenesis by regulating miRNAs packaged in exosomes. Am J Pathol 188: 1094–1103, 2018. doi: 10.1016/j.ajpath.2017.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Parimon T, Yao C, Habiel DM, Ge L, Bora SA, Brauer R. Syndecan-1 promotes lung fibrosis by regulating epithelial reprogramming through extracellular vesicles. JCI Insight 5: e129359, 2019. doi: 10.1172/jci.insight.129359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chertow DS, Memoli MJ. Bacterial coinfection in influenza: a grand rounds review. JAMA 309: 275–282, 2013. doi: 10.1001/jama.2012.194139. [DOI] [PubMed] [Google Scholar]
- 53. Jinno A, Hayashida A, Jenkinson HF, Park PW. Syndecan-1 promotes streptococcus pneumoniae corneal infection by facilitating the assembly of adhesive fibronectin fibrils. mBio 11: e01907-20, 2020. doi: 10.1128/mBio.01907-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hayashida A, Amano S, Gallo RL, Linhardt RJ, Liu J, Park PW. 2-O-Sulfated domains in syndecan-1 heparan sulfate inhibit neutrophil cathelicidin and promote Staphylococcus aureus corneal infection. J Biol Chem 290: 16157–16167, 2015. doi: 10.1074/jbc.M115.660852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tighe RM, Garantziotis S. Hyaluronan interactions with innate immunity in lung biology. Matrix Biol 78-79: 84–99, 2019. doi: 10.1016/j.matbio.2018.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Garantziotis S, Savani RC. Hyaluronan biology: a complex balancing act of structure, function, location and context. Matrix Biol 78–79: 1–10, 2019. doi: 10.1016/j.matbio.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 11: 1173–1179, 2005. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 58. Lauer ME, Dweik RA, Garantziotis S, Aronica MA. The rise and fall of hyaluronan in respiratory diseases. Int J Cell Biol 2015: 712507, 2015. doi: 10.1155/2015/712507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bell TJ, Brand OJ, Morgan DJ, Salek-Ardakani S, Jagger C, Fujimori T, Cholewa L, Tilakaratna V, Östling J, Thomas M, Day AJ, Snelgrove RJ, Hussell T. Defective lung function following influenza virus is due to prolonged, reversible hyaluronan synthesis. Matrix Biol 80: 14–28, 2019. doi: 10.1016/j.matbio.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Day AJ, Milner CM. TSG-6: a multifunctional protein with anti-inflammatory and tissue-protective properties. Matrix Biol 78–79: 60–83, 2019. doi: 10.1016/j.matbio.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 61. Kratochvil MJ, Kaber G, Demirdjian S, Cai PC, Burgener EB, Nagy N, Barlow GL, Popescu M, Nicolls MR, Ozawa MG, Regula DP, Pacheco-Navarro AE, Yang S, de Jesus Perez VA, Karmouty-Quintana H, Peters AM, Zhao B, Buja ML, Johnson PY, Vernon RB, Wight TN, Milla CE, Rogers AJ, Spakowitz AJ, Heilshorn SC, Bollyky PL. Biochemical, biophysical, and immunological characterization of respiratory secretions in severe SARS-CoV-2 infections. JCI Insight 7: e152629, 2022. doi: 10.1172/jci.insight.152629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lazrak A, Jurkuvenaite A, Ness EC, Zhang S, Woodworth BA, Muhlebach MS, Stober VP, Lim Y-P, Garantziotis S, Matalon S. Inter-α-inhibitor blocks epithelial sodium channel activation and decreases nasal potential differences in ΔF508 mice. Am J Respir Cell Mol Biol 50: 953–962, 2014. doi: 10.1165/rcmb.2013-0215OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gavina M, Luciani A, Villella VR, Esposito S, Ferrari E, Bressani I, Casale A, Bruscia EM, Maiuri L, Raia V. Nebulized hyaluronan ameliorates lung inflammation in cystic fibrosis mice. Pediatr Pulmonol 48: 761–771, 2013. doi: 10.1002/ppul.22637. [DOI] [PubMed] [Google Scholar]
- 64. Martin TR, Zemans RL, Ware LB, Schmidt EP, Riches DWH, Bastarache L, Calfee CS, Desai TJ, Herold S, Hough CL, Looney MR, Matthay MA, Meyer N, Parikh SM, Stevens T, Thompson BT. New insights into clinical and mechanistic heterogeneity of the acute respiratory distress syndrome: summary of the Aspen Lung Conference 2021. Am J Respir Cell Mol Biol 67: 284–308, 2022. doi: 10.1165/rcmb.2022-0089WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Calfee CS, Delucchi KL, Sinha P, Matthay MA, Hackett J, Shankar-Hari M, McDowell C, Laffey JG, O'Kane CM, McAuley DF, Johnston AJ, Paikray A, Yates C, Polgarova P, Price E, McInerney A, Zamoscik K, Dempsey G, Seasman C, Gilfeather L, Hemmings N, O'Kane S, Johnston P, Pokorny L, Nutt C, O'Neill O, Prashast P, Smalley C, Jacob R, O'Rourke J. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med 6: 691–698, 2018. doi: 10.1016/S2213-2600(18)30177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jandik KA, Kruep D, Cartier M, Linhardt RJ. Accelerated stability studies of heparin. J Pharm Sci 85: 45–51, 1996. doi: 10.1021/js9502736. [DOI] [PubMed] [Google Scholar]
- 67. Schmidt EP, Overdier KH, Sun X, Lin L, Liu X, Yang Y, Ammons LA, Hiller TD, Suflita MA, Yu Y, Chen Y, Zhang F, Cothren Burlew C, Edelstein CL, Douglas IS, Linhardt RJ. Urinary glycosaminoglycans predict outcomes in septic shock and acute respiratory distress syndrome. Am J Respir Crit Care Med 194: 439–449, 2016. doi: 10.1164/rccm.201511-2281OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Marshall DC, Lyman SK, McCauley S, Kovalenko M, Spangler R, Liu C, Lee M, O’Sullivan C, Barry-Hamilton V, Ghermazien H, Mikels-Vigdal A, Garcia CA, Jorgensen B, Velayo AC, Wang R, Adamkewicz JI, Smith V. Selective allosteric inhibition of MMP9 is efficacious in preclinical models of ulcerative colitis and colorectal cancer. PLoS One 10: e0127063, 2015. doi: 10.1371/journal.pone.0127063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sela-Passwell N, Kikkeri R, Dym O, Rozenberg H, Margalit R, Arad-Yellin R, Eisenstein M, Brenner O, Shoham T, Danon T, Shanzer A, Sagi I. Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat Med 18: 143–147, 2012. doi: 10.1038/nm.2582. [DOI] [PubMed] [Google Scholar]