


Keywords: branched-chain amino acids, cellular redox, fatty acid oxidation, lipogenesis, mitochondria
Abstract
Metabolic and molecular interactions between branched-chain amino acid (BCAA) and lipid metabolism are evident in insulin-resistant tissues. However, it remains unclear whether insulin resistance is a prerequisite for these relationships and whether BCAAs or their metabolic intermediates can modulate hepatic lipid oxidation and synthesis. We hypothesized that BCAAs can alter hepatic oxidative function and de novo lipogenesis, independent of them being anaplerotic substrates for the mitochondria. Mice (C57BL/6NJ) were reared on a low-fat (LF), LF diet plus 1.5X BCAAs (LB), high-fat (HF) or HF diet plus 1.5X BCAAs (HB) for 12 wk. Hepatic metabolism was profiled utilizing stable isotopes coupled to mass spectrometry and nuclear magnetic resonance, together with fed-to-fasted changes in gene and protein expression. A greater induction of lipid oxidation and ketogenesis on fasting was evident in the BCAA-supplemented, insulin-sensitive livers from LB mice, whereas their rates of hepatic de novo lipogenesis remained lower than their LF counterparts. Onset of insulin resistance in HF and HB mice livers blunted these responses. Whole body turnover of BCAAs and their ketoacids, their serum concentrations, and the ketogenic flux from BCAA catabolism, all remained similar between fasted LF and LB mice. This suggested that the impact of BCAAs on lipid metabolism can occur independent of them or their degradation products fueling anaplerosis through the liver mitochondria. Furthermore, the greater induction of lipid oxidation in the LB livers accompanied higher mitochondrial NADH/NAD+ ratio and higher fed-to-fasting phosphorylation of AMPKα and ACC. Taken together, our results provide evidence that BCAA supplementation, under conditions of insulin sensitivity, improved the feeding-to-fasting induction of hepatic lipid oxidation through changes in cellular redox, thus providing a favorable biochemical environment for flux through β-oxidation and lower de novo lipogenesis.
NEW & NOTEWORTHY Branched-chain amino acids (BCAAs) have been shown to modulate lipid metabolic networks in various tissues, especially during insulin resistance. In this study we show that the dietary supplementation of BCAAs to normal, insulin-sensitive mice resulted in higher mitochondrial NADH:NAD+ ratios and AMPK activation in the liver. This change in the cellular redox status provided an optimal metabolic milieu to increase fatty acid oxidation while keeping the rates of de novo lipogenesis lower in the BCAA-supplemented mice livers.
INTRODUCTION
Elevated circulating levels of BCAAs and defects in their catabolic networks are central features of insulin-resistant tissues during obesity, diabetes mellitus, kidney, and liver dysfunction (1–3). More importantly, these alterations in BCAA metabolism coexist with those in the biochemical networks of lipid oxidation and synthesis in insulin-resistant tissues (4–8). Several hypotheses have been postulated to explain the crosstalk between BCAA and lipid metabolism in tissues. Larger pools of circulating BCAAs during insulin resistance and the parallel selective induction of their catabolic networks have the potential to increase the pool sizes of the BCAA catabolic intermediates (branched-chain ketoacids, succinyl CoA, propionyl CoA, acetyl-CoA, ketones, amino acids), which in turn can fuel anaplerotic flux through the mitochondrial tricarboxylic acid (TCA) cycle (9–12). Higher levels of some of the BCAA catabolic intermediates including propionyl CoA and succinyl CoA also have the potential to decrease the efficiency of lipid oxidation and promote lipid accumulation and lipotoxicity. Furthermore, in differentiating adipocytes (4) and during mitochondrial dysfunction of the muscle (7), BCAA degradation networks can be a source of acetyl-CoA for de novo lipogenesis (DNL), promoting lipid accumulation in these tissues. Furthermore, supplementation of BCAAs to insulin-resistant mice and humans has also been shown to improve metabolic health (13–16). Overall, these are all evidence of the significant biochemical and molecular interactions between BCAA metabolism and different networks of lipid metabolism.
The liver is a hub of intermediary metabolism, which integrates substrate oxidation through mitochondrial energetics with the cytoplasmic anabolic networks of DNL and gluconeogenesis. Indeed, dysfunctional lipid oxidation and sustained rates of DNL are central features of nonalcoholic fatty liver disease (NAFLD) (17–22). Mechanisms regulating these critical hepatic networks are of major interest in the management and treatment of NAFLD. In this context, it is of relevance to identify how BCAA metabolism interacts with the hepatic networks of free fatty acid oxidation and DNL. Liver tissue has limited expression of branched-chain aminotransferases (BCAT), the first enzyme in the BCAA degradation pathway (23–25). Nonetheless, the expression of the branched-chain α-keto-acid dehydrogenase (BCKDH) in the liver is high allowing the liver to utilize systemic BCAA keto acids and nonessential amino acids as anaplerotic substrates for the mitochondrial TCA cycle (23, 25). Furthermore, ketogenic amino acids in the diets (leucine, isoleucine, and lysine) have been shown to lower hepatic lipogenesis, possibly by diverting mitochondrial acetyl-CoA toward oxidation through the TCA cycle (26, 27). The interactions between shared molecular mediators of BCAA and lipid metabolism [e.g., mammalian target of rapamycin complex 1 (mTORC1), AMP-activated protein kinase, peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1α (PGC-1α), and PPARα] can also be a contributing factor to the relationship between BCAA and lipid metabolism in the liver (28–32). Although several of these biochemical and molecular nodes of interactions are postulated, it remains unclear whether changes in lipid oxidation and DNL are central features of BCAA-supplemented livers. Furthermore, it remains unclear whether insulin resistance is a prerequisite for the manifestation of the biochemical interactions between BCAA and lipid metabolism. We hypothesized that the supplementation of BCAAs can alter hepatic lipid oxidation and DNL, independent of their biochemical role toward providing anaplerotic substrates for the liver mitochondria, and under the normal physiology of insulin sensitivity.
MATERIALS AND METHODS
Animals and Diets
All the animal experiments were conducted in accordance with the protocols approved by the Institutional Animal Care and Use Committee at the University of Maryland, College Park. Male C57BL/6NJ mice, aged 4–6 wk (Jackson Laboratory, Bar Harbor, ME), were reared on either a low-fat diet (LF; 10% fat calories), a low-fat diet supplemented with BCAAs (LB; LF + 150% BCAAs), high-fat diet (HF; 60% fat calories) or a high-fat diet supplemented with BCAAs (HB; HF + 150% BCAA) for 12 wk. These custom-made diets were purchased from Research Diets Inc. New Brunswick, NJ, and their dietary compositions are provided in Supplemental Table S1 (https://doi.org/10.6084/m9.figshare.21574797). Mice were fed ad libitum and maintained on a 12-h light/dark cycle. Metabolic profiling as detailed in the following sections was conducted under fed or overnight fasted (14–16 h) conditions.
Determination of Whole Body Nutrient Turnover Rates
Following 12 wk of dietary treatment, mice were surgically implanted with jugular vein catheters and were allowed to recover for 4–6 days before infusion of a cocktail of stable isotope tracers. The stable isotope infusions were conducted in half of the mice from each group under fed conditions and in the other half following an overnight fast. The mixture of stable isotope tracers (Cambridge Isotope Laboratories Inc., Tewksbury, MA) contained stable isotopes, [13C6]leucine (1.25 mg/mL), [13C6]sodium 2-keto-3-methyl-pentanoate (0.5 mg/mL), [13C5] α-ketoisovaleric acid, sodium salt (0.8 mg/mL), [13C6]glucose (15 mg/mL), and [13C4]β-hydroxybutyrate (3 mg/mL), toward the determination of their respective turnover rates. This stable isotope mixture was infused as a bolus (0.3 mL/h) for the first 10 min, followed by continuous infusion (0.12 mL/h) for another 80 min, to attain an isotopic steady state. Following 90 min of these infusions, the blood and tissues were collected under isoflurane anesthesia. The blood was spun at 9,000 rpm for 10 min at 4°C to separate the plasma, and the tissues were flash-frozen in liquid nitrogen before storage at −80°C for further analysis.
Metabolism of Freshly Isolated Liver Mitochondria
Toward profiling, the metabolism of isolated mitochondria, mice from the four dietary treatments were anesthetized under isoflurane and fresh livers were collected. The liver was immediately rinsed with 1X ice-cold phosphate-buffered saline (PBS) and ∼800 mg of the liver was excised and used to isolate mitochondria. The liver tissue was minced in 3 mL of MSHE buffer [70 mM sucrose, 210 mM mannitol, 5 mM HEPES, 1 mM EGTA, and 0.5% bovine serum albumin (BSA); pH 7.2] and was homogenized using a Dounce homogenizer (2–4 strokes, 5–10 turns each stroke). The homogenized tissue was transferred to a 15 mL falcon tube and the homogenizer was rinsed with an additional 4 mL of MSHE buffer, which was then added to the tissue homogenate. The tissue homogenate was spun at 800 g for 10 min at 4°C. The supernatant was transferred through a double-layered cheesecloth and was centrifuged at 8,000 g for 10 min at 4°C to obtain the crude mitochondrial pellet. The pellet was then washed and resuspended in 3 mL of MSHE buffer and spun at 8,000 g for 10 min at 4°C. This step was repeated two times before the supernatant was completely removed and the mitochondrial pellet resuspended in 100 µL of MSHE buffer without BSA. The mitochondrial protein content was estimated following 40 times dilution of mitochondrial pellet using Pierce protein assay kit (Thermo Fisher Scientific, Waltham, MA). For the in vitro incubation studies detailed below, 500 µg of mitochondrial protein was used as an experimental unit.
Incorporation of 13C into Mitochondrial Intermediates from [13C16] Palmitate
Freshly isolated mitochondria (500 µg), from all the four dietary groups, were incubated in 1 mL of MAS-3 respiration buffer (115 mM KCl, 10 mM, KH2PO4, 2 mM MgCl2, 3 mM HEPES, 1 mM EGTA, and 0.2% fat-free BSA; pH 7.2), with 5 mM glutamate, 2.5 mM malate, and 1 mM carnitine along with 20 µM of, either [13C16] palmitate or unlabeled palmitate. The mitochondria were incubated for 5 and 10 min at 37°C. Immediately following the incubations, the tubes were centrifuged at 8,000 rpm for 5 min at 4°C, to pellet the mitochondria. The respiration buffer supernatant was pipetted out and frozen at −80°C for analysis of ketones, produced by the mitochondria. The mitochondrial pellet was then washed with 1× cold PBS and centrifuged again at 8,000 rpm for 2 min, the supernatant was discarded, and the pellet was frozen at −80°C for future analysis of mitochondrial intermediates by gas-chromatography mass spectrometry (GC-MS) as explained below.
Incorporation of 13C into Mitochondrial Intermediates from [13C6] Ketoisocaproic Acid
In a separate set of experiments, liver mitochondria were isolated, as described in the previous section, from mice reared on LF and LB diets for 12 wk. The freshly isolated liver mitochondria were incubated with 1 mL MAS-3 buffer with 5 mM glutamate, 2.5 mM malate, 1 mM carnitine, 25 µM palmitate, and 25 µM [13C6] ketoisocaproic acid or unlabeled ketoisocaproic acid (the keto acid of leucine) for 10 min. The media and the mitochondrial pellet were collected and frozen for GC-MS analysis of ketones and the mitochondrial intermediates.
Analysis of Amino Acids, TCA Cycle Intermediates, and β-Hydroxybutyrate by GC-MS
In general, samples intended for the determination of metabolite levels/pool sizes were spiked in with a known amount of stable isotope-labeled internal standard mixture, before processing for GC-MS analysis. Samples intended for the determination of metabolite enrichment derived from a 13C-labeled precursor ([13C16] palmitate and [13C6] ketoisocaproic acid), or for the determination of stable isotope dilution following tracer infusion, were processed without the addition of an internal standard. Briefly, following the addition of the stable isotope-labeled internal standard, serum (25 µL) samples were deproteinized with 750 µL of cold acetonitrile. The mitochondrial incubation media (250 µL) was also processed by deproteinization with 750 µL of cold acetonitrile, for the determination of β-hydroxybutyrate concentration or enrichment. The supernatant was separated by centrifugation at 13,000 rpm for 15 min and was dried down under a stream of nitrogen gas.
For the metabolite analysis from the isolated mitochondrial pellet, 250 µg of mitochondrial protein was used to extract the TCA cycle intermediates and the amino acids. Following deproteinization with 750 µL of cold acetonitrile, the supernatant was collected and dried under nitrogen.
The dry samples were derivatized, first by the addition of 20 µL of 2% methoxamine hydrochloride in pyridine and microwaving for 90 s, followed by the addition of 30 µl of N-methyl-N-tert butyldimethylsilyl tri fluoroacetamide (MTBSTFA) and heating at 90°C for 1 h. The derivatized samples were run on a HP-5MS UI GC column (30 m × 0.25 mm × 0.25 μm; Agilent, Santa Clara, CA). Metabolite fragments of interest were detected following electrical ionization and single ion monitoring (SIM) on a 5973 N-Mass Selective Detector with 6890-Series GC (Agilent, Santa Clara, CA) (33). Metabolite levels/pool sizes were calculated relative to their respective stable isotope labeled internal standards. Enrichment arising from a labeled precursor in a metabolite fragment ion of interest was calculated as the ratio of the enriched fragment/(enriched fragment + unlabeled fragment), after subtracting off the natural abundance enrichment in the fragment.
A separate set of plasma samples for the 12-wk LF and LB group was processed for the determination of the β-hydroxybutyrate and acetoacetate concentration, toward the calculation of the NADH/NAD+ ratio. For this, serum (25 µL) was spiked with 1 M Sodium borodeuteride (NaBD4) to reduce the unstable acetoacetate to its stable β-hydroxybutyrate, with a deuterium tag. Following this conversion, [13C4] β-hydroxybutyrate was spiked as the internal standard before processing. The sample mix was incubated for 5 min at room temperature followed by the addition of 200 µL of cold 12% sulfosalicylic acid. The samples were spun for 30 s at 13,000 rpm and were followed by the addition of 800 µL more of cold 12% sulfosalicylic acid. The samples were then centrifuged at 13,000 rpm for 15 min at 4°C and the supernatant was transferred to 10 mL glass tubes. The samples were then mixed with 1 mL of 4 M hydrochloric acid and then 1 mL of saturated NaCl. The β-hydroxybutyrate and acetoacetate were extracted with 3 mL ethyl acetate for 1 h with continuous shaking on a shaker plate. The sample mixture was centrifuged for 10 min at room temperature and the supernatant was dried under nitrogen. The dry samples were derivatized and run on the GC-MS similar to the analysis of amino acids and TCA cycle intermediates. Mitochondrial NADH/NAD+ was determined from the plasma β-hydroxybutyrate/acetoacetate ratio, based on β-hydroxybutyrate dehydrogenase equilibrium (34, 35). The cytosolic NADH/NAD+ was measured from the liver pyruvate/lactate ratio based on lactate dehydrogenase equilibrium (35, 36).
Determination of Hepatic De Novo Lipogenesis by 2H NMR
Mice maintained on LF, LB, HF, and HB diets for 12 wk were intraperitoneally injected with saline deuterium oxide (D2O; 27 µL/g body wt). These mice were then provided with 4% D2O in drinking water for the next 4 days. Mice were then sacrificed under isoflurane anesthesia and livers and plasma were collected and stored at −80°C for further analysis. Approximately 250 mg of liver tissue was used for the Folch extraction of total lipids. Extraction was done with 10 mL of 2:1 chloroform: methanol together with 5 mL of 50 mM NaCl, for 1 h with frequent vortexing. Following extraction, the samples were spun at 3,000 rpm for 10 min and the bottom chloroform layer was transferred into glass tubes and dried completely. The dried lipids were reconstituted in 600 µL of chloroform containing a pyrazine internal standard (2 mg/mL, 2.5% d4-pyrazine, and 97.5% unlabeled pyrazine) (37). The samples were then transferred to 5 mm NMR tubes for analysis on a Bruker Ultrashield Advance III system equipped with a CPTXI 600S3 H-C/N-D-05 Z cryoprobe. Proton and deuterium lock channels were used to acquire 1H and 2H NMR spectra, respectively. The 1H spectra of lipids were acquired at 600 MHz, with 2-s delay (d1), 64 number of scans (NS), and the 2H spectra were acquired at a frequency of 92.14 MHz with an acquisition time of 2 s and 4,096 scans. All the spectra were analyzed using TopSpin3.2 software. Body water enrichment was determined using 2H NMR, as previously described (38). De novo lipogenesis (%) was calculated as (2H methyl enrichment/2H body water enrichment) × 100.
Reactive Oxygen Species Generation by Isolated Mitochondria
Mitochondria (64 µg) were incubated with horseradish peroxidase (0.2 U/mL; HRP) and Amplex Red reagent (100 µM; 10-acetyl-3,7-dihydroxyphenoxazine), prepared in MAS-3 buffer containing 5 mM glutamate and 2.5 mM malate. The rate of hydrogen peroxide (H2O2) released by the isolated mitochondria was measured by recording the real-time oxidation of Amplex Red to the fluorescent resorufin, detected with 530 nm excitation and 590 nm emission wavelengths, using a Cytation 5 spectrophotometer (BioTek Instruments, Inc., Winooski, VT).
Oxygen Consumption by the Isolated Mitochondria
Mitochondrial oxygen consumption was measured using an oxygraph oxygen electrode (Hansatech Instruments, Norfolk, UK). Isolated mitochondria (500 μg) were suspended in 1 mL of MAS-3 buffer containing 5 mM glutamate and 2.5 mM malate. Basal and ADP stimulated (100 μM ADP) respiration rates were determined.
Hepatic Gene Expression Profiles
Frozen liver (∼10–15 mg) was lysed with 600 µL TRIZOL reagent (Invitrogen, Carlsbad, CA) to isolate mRNA using a miniprep kit (Bio-Rad Laboratories Inc., Hercules, CA). cDNA was prepared from 1 µg of mRNA using a cDNA synthesis kit (Bio-Rad, Hercules, CA). Quantitative real-time PCR was performed using 25 ng of cDNA, 150 nM of each primer, and 5 μL of SYBR Green PCR master mix (Invitrogen, Carlsbad, CA) with cyclophilin as the housekeeping gene. The samples were run in triplicates on a Bio-Rad CFX Real-Time system (C1000 Touch Thermal Cycler). The list of primers and their sequences is provided in Supplemental Table S2 (https://doi.org/10.6084/m9.figshare.21574821).
Western Blot Analysis
Frozen liver (∼10–15 mg) and mitochondrial pellet were lysed with 1× RIPA lysis buffer containing protease and phosphatase inhibitors followed by centrifugation at 15,000 rpm for 15 min at 4°C. The total protein was measured using the Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA). The proteins were separated using Bolt 8% Bis-tris Plus gels (Invitrogen, Carlsbad, CA) and transferred onto a nitrocellulose membrane. The membrane was incubated with the primary antibody overnight followed by incubation with the secondary antibody. Primary antibodies against CPT1A, PPARA, COXIV, ACC, pACCSer79, AMPK, pAMPKThr172, GAPDH (Cell Signaling Technology, Danvers MA), and LCAD/ACADL (Abcam, Cambridge, UK) were used for this study.
Biochemical Assays
Serum insulin and c-peptide were measured by enzyme-linked immunoassay (Crystal Chem USA, IL). Serum nonesterified fatty acid (NEFA) concentrations were determined using the HR Series NEFA-HR2 kit (WAKO diagnostics, CA). Liver triglycerides were measured using serum triglyceride determination kit (Sigma Aldrich, St. Louis, MO). Liver pAMPKThr172 levels were measured using an enzyme-linked immunosorbent assay (Invitrogen, Waltham, MA), and NADH and NAD levels in the liver tissue were measured using NAD/NADH calorimetric assay kit (Millipore Sigma, Burlington, MA). All assays were conducted following the manufacturer’s protocols.
Statistical Analysis
All the data are reported as means ± standard error (SE). Comparisons between the 12-wk dietary groups (LF, LB, HF, and HB) and the fed versus fasted (LF and LB) groups were conducted using a two-way ANOVA to test the main effects and interaction of two factors (either diet and BCAA supplementation or diet and feeding/fasting) on a dependent variable, followed by Tukey’s pairwise mean comparison test. The fed-to-fasted fold-changes between dietary treatments were analyzed using an unpaired Student’s t test. Means are considered significantly different at P ≤ 0.05. The differences between means are considered a trend at P ≤ 0.1. All statistical analyses were conducted, and the graphs were plotted utilizing Prism 7 (GraphPad Software Inc., San Diego, CA).
RESULTS
Hepatic Lipid Oxidation, Induced by an Overnight Fast, Was Higher in the BCAA-Supplemented Mice
Fed-to-fasted induction of hepatic genes and proteins involved in lipid oxidation was profiled after 12 wk of BCAA supplementation, on low-fat and high-fat diets. Following 12 wk on the diets, fed-to-fasted induction of genes involved in lipid oxidation was higher in the low-fat BCAA-supplemented mice (LB), compared with their LF counterparts (Fig. 1, A–E). Although the fasting-induced gene expression following BCAA did not reach significance for Cd36, a cellular importer of fatty acids (Fig. 1A), carnitine palmitoyltransferase 1a (Cpt1a; P ≤ 0.05, Fig. 1B), medium- and long-chain acyl dehydrogenase enzymes (Mcad and Lcad; P ≤ 0.05, Fig. 1, C and D) and Ppara, a transcriptional regulator of lipid oxidation (P ≤ 0.05, Fig. 1E) were all higher in the LB livers. A similar induction (P ≤ 0.05) in liver proteins (LCAD, CPT1A, PPARA) involved in the regulation of lipid oxidation was also evident after fasting, in the BCAA-supplemented mice reared on a low-fat diet (Fig. 1, F and G). Supplementation of BCAAs on a high-fat diet for 12 wk blunted the changes in gene expression observed on a low-fat plus BCAA diet (Fig. 1, A–E). Statistically significant upregulation of protein expression following fasting was only evident for CPT1A, after 12 wk of BCAA supplementation on the high-fat diet (Fig. 1, H and I). Furthermore, changes in ketone metabolism (Fig. 1, J–L) supported the aforementioned results in that the BCAA-supplemented mice in a low-fat diet had higher fasting-induced levels of serum β-hydroxybutyrate (Fig. 1K) and higher fed-to-fasted induction of Hmgcs2 (Fig. 1L; P ≤ 0.05), a key regulator of ketogenesis. Taken together, these results demonstrate the ability of BCAAs to upregulate hepatic lipid oxidation under a low-fat, insulin-sensitive dietary environment, whereas insulin resistance seems to blunt this effect.
Figure 1.
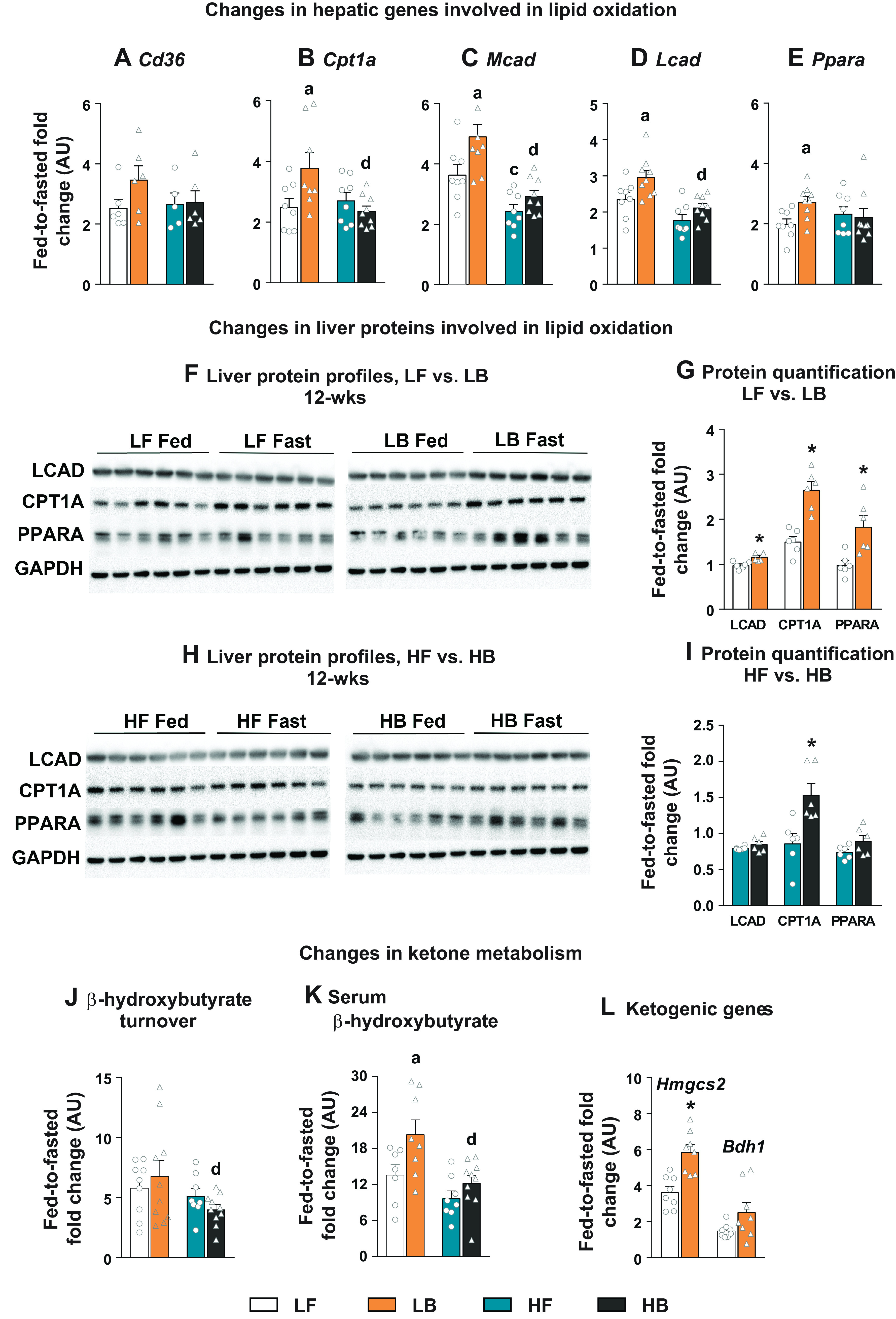
Hepatic lipid oxidation in BCAA-supplemented mice. Fed-to-fasted fold-change in the expression of hepatic genes involved in lipid oxidation including (A) Cd36, (B) Cpt1a, (C) Mcad, (D) Lcad, and (E) Ppara. F: the changes in protein expression of LCAD, CPT1A and PPARA, after 12 wk on LF and LB diets with their densitometry analysis (G). H: the changes in protein expression of LCAD, CPT1A, and PPARA, after 12 wk on HF and HB diets with their densitometry analysis (I). Changes in ketone metabolism: fed-to-fasted fold-changes in the turnover rate of β-hydroxybutyrate (J); fed-to-fasted fold-changes in the circulating levels of β-hydroxybutyrate (K); and fed-to-fasted fold-changes in expression of genes involved in ketogenesis (Hmgcs2 and Bdh1; L). For all the fed-to-fasted comparisons, the normalization is relative to their respective “fed” LF or “fed” HF counterparts. All the values are represented as means ± SE, with n = 5–10/group. Results are considered statistically significant at P ≤ 0.05 following two-way ANOVA followed by pairwise mean comparisons represented as follows: “a”—LF vs. LB; “b”—HF vs. HB; “c”—LF vs. HF; “d”—LB vs. HB. Densitometry analysis between 12-wk (LF vs. LB and HF vs. HB), were determined following a Student’s t test and is represented by “*.” Cd36, cluster of differentiation 36; Cpt1a, carnitine palmitoyl transferase 1; Mcad, medium-chain acyl dehydrogenase; Lcad, long-chain acyl dehydrogenase; Ppara, peroxisome proliferator-activated receptor α; Hmgcs2, 3-hydroxy-3-methylglutaryl-CoA synthase 2 (mitochondrial) and Bdh1; 3-hydroxybutyrate dehydrogenase 1; AU, arbitrary units; BCAA, branched-chain amino acid.
Freshly Isolated Liver Mitochondria from Fasted BCAA-Supplemented Mice Were Primed to Oxidize Lipids
To test whether the mitochondrial machinery was truly primed to oxidize lipids, freshly isolated liver mitochondria were allowed to respire in a media containing [13C16]palmitate, and the incorporation of the 13C was tracked into β-hydroxybutyrate isotopomers and TCA cycle intermediates (Fig. 2A). There was a steady increase (P ≤ 0.05) in the M + 2 and M + 4 isotopomers of β-hydroxybutyrate from [13C16]palmitate in all four groups, both with the time of incubation (5 vs. 10 min) and between the mitochondria isolated from fed versus fasted livers (Supplemental Table S3: https://doi.org/10.6084/m9.figshare.21574851). More interestingly, the fold-change in the M + 2 and M + 4 isotopomer enrichment of β-hydroxybutyrate from fed-to-fasted states were significantly higher (P ≤ 0.05) in the 12-wk BCAA-supplemented groups (both LB and HB), relative to their respective LF and HF counterparts (Fig. 2B). The aforementioned results from the 12-wk dietary groups point to the greater ability of the liver mitochondria isolated from the BCAA-supplemented mice to oxidize free fatty acids into ketones. Complete oxidation of acetyl-CoA occurs in the TCA cycle and thus can result in the 13C enrichment of TCA cycle intermediates from [13C16]palmitate (Fig. 2A). However, when we determined the 13C incorporation into citrate and other TCA cycle intermediates, we did not find any significant differences between the BCAA-supplemented and nonsupplemented groups (Fig. 2C and Supplemental Tables S4 and S5: https://doi.org/10.6084/m9.figshare.21574860 and https://doi.org/10.6084/m9.figshare.21574866). In agreement with the higher fed-to-fasted increase in β-hydroxybutyrate 13C enrichment, the fed-to-fasted fold-change in the expression of LCAD and CPT1A was also higher in the BCAA-supplemented mice (LF vs. LB; P ≤ 0.05, Fig. 2D). Comparison between the HF versus HB groups following 12 wk of dietary challenges, only revealed significant differences in the fed-to-fasted induction for LCAD (P ≤ 0.05, Fig. 2D). Taken together, these results demonstrate that the BCAA supplementation primed the mitochondrial machinery to oxidize lipids.
Figure 2.
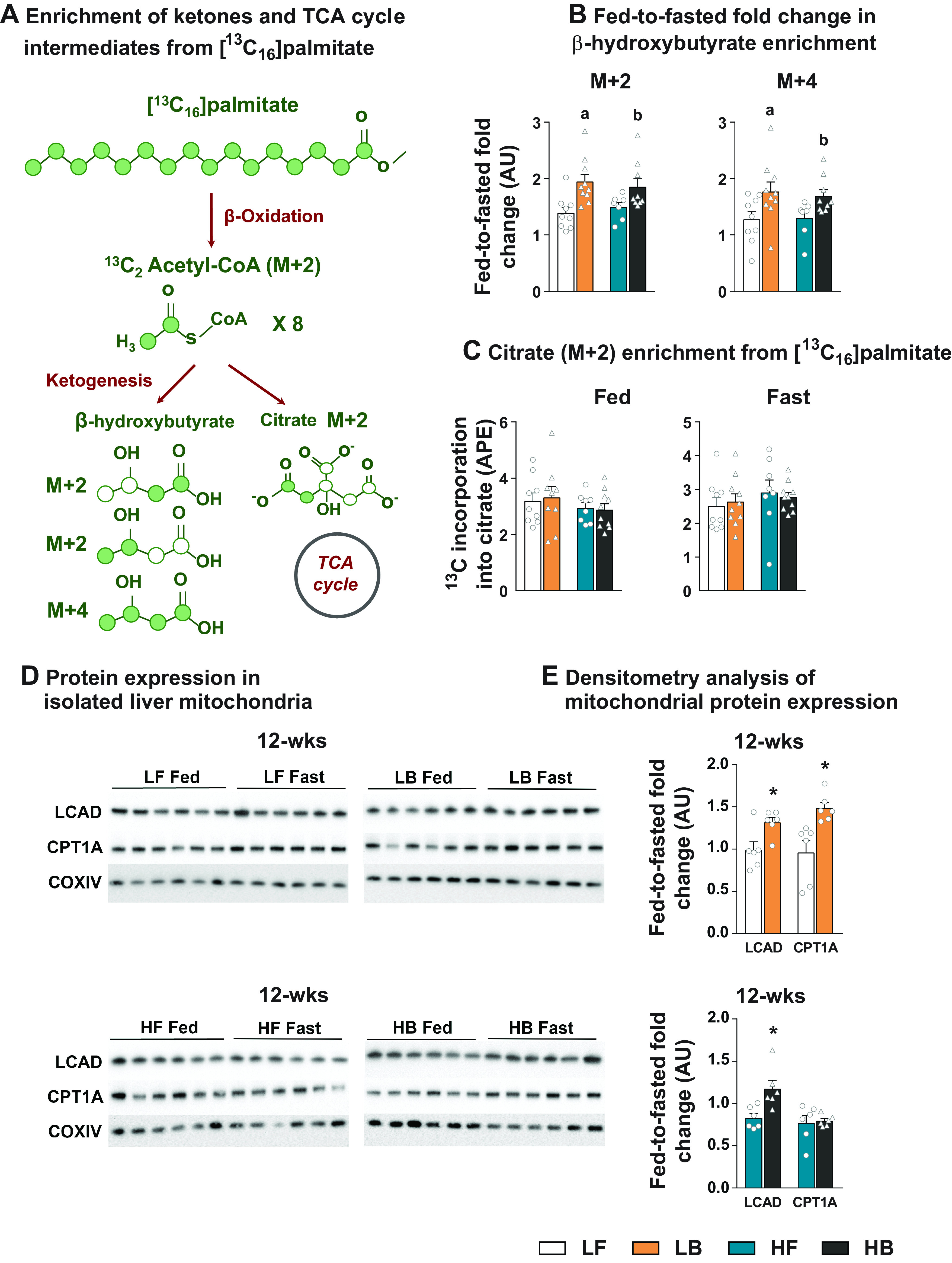
Profiling the metabolism of freshly isolated liver mitochondria. Schematic representing the shuttling of 13C enrichment from [13C16]palmitate into β-hydroxybutyrate isotopomers and TCA cycle intermediates, in isolated mitochondria incubated in a defined respiration buffer (A). Fed-to-fasted fold-change in β-hydroxybutyrate isotopomers (M + 2 and M + 4) enriched by 13C from [13C16] palmitate (B). 13C incorporation from [13C16]palmitate into the TCA cycle intermediate, Citrate isotopomer (M + 2) under fed and fasting conditions (C). Expression of proteins (LCAD and CPT1A) in the mitochondria isolated from the livers of LF, LB, HF, HB dietary groups following 12 wk on diets, under fed and fasting conditions (D) and their densitometry analysis (E). All the values are represented as means ± SE, with n = 6–10/group. For all the fed-to-fasted comparisons, the normalization is relative to their respective “fed” LF or “fed” HF counterparts. Results are considered statistically significant at P ≤ 0.05 following two-way ANOVA, followed by pairwise mean comparisons represented as follows: “a”—LF vs. LB; “b”—HF vs. HB. Densitometry analysis between 12-wk LF vs. LB and HF vs. HB was determined following a t test and is represented by “*”. AU, arbitrary units; BCAA, branched-chain amino acid; HB, high-fat diet with BCAA; HF, high-fat diet; LB, low-fat diet with BCAA; LF, low-fat diet.
Supplementation of BCAAs Resulted in the Suppression of De Novo Lipogenesis in an Insulin-Sensitive Liver
The significant impact of BCAA supplementation on hepatic lipid oxidation, especially in the low-fat diets, prompted us to explore the impact of BCAA supplementation on hepatic DNL. Representative 2H-NMR spectra of the hepatic lipids obtained following keeping the mice on 4% deuterium-labeled water for 4 days are presented in Fig. 3A. Enrichment of 2H in the body water, which is the precursor pool for the incorporation of 2H into lipids, remained similar between all the four groups (Fig. 3B). However, 2H enrichment in the methyl groups of the hepatic lipids (Fig. 3C) and the rate of DNL calculated from these 2H enrichments (Fig. 3D) were significantly lower in the BCAA-supplemented mice livers from a low-fat diet (P ≤ 0.05). The lower expression of lipogenic genes in the liver of the LB mice (Fig. 3E) was in line with the lower rates of DNL in their livers. The high-fat diet resulted in a dramatic suppression of DNL when compared with their LF counterparts (P ≤ 0.05). Due to the significant high-fat diet-mediated suppression of DNL, no further suppression of DNL was evident in the livers of mice supplemented with BCAAs on a high-fat diet (HB; Fig. 3, D and E).
Figure 3.

Hepatic de novo lipogenesis in BCAA-supplemented mice. Representative 2H-NMR spectra of hepatic lipids obtained following 12 wk of dietary supplementation with LF, LB, HF, and HB diet and 4 days of administering 4% D2O in drinking water (A). Enrichment of 2H in the body water (B), enrichment of 2H in the methyl groups of lipids (C), and rate of hepatic de novo lipogenesis (D) following 12 wk of LF, LB, HF, and HB dietary regime. Expression of lipogenic genes (Acly, Acc1, Fasn, Elovl6, and Scd1) in “fed” livers, after 12 wk on LF, LB, HF, and HB diets (E). Liver triglyceride levels after 12 wk on LF, LB, HF, and HB diets (F). All the values are represented as means ± SE, with n = 6–9/group. Results are considered statistically significant at P ≤ 0.05 following two-way ANOVA followed by pairwise mean comparisons represented as follows: “a”—LF vs. LB; “b”—HF vs. HB; “c”—LF vs. HF; “d”—LB vs. HB. Acc1, acetyl-CoA carboxylase α; Acly, ATP citrate lyase; AU, arbitrary units; BCAA, branched-chain amino acid; Elovl6, elongation of very long-chain fatty acid elongase 6; Fasn, fatty acid synthase; Scd1, stearoyl-CoA desaturase 1.
Determining the Anaplerotic Impact of the BCAAs and Their Keto Acids on the Hepatic Mitochondria
We investigated whether the anaplerotic impact of the BCAA keto acids is a significant modulator of mitochondrial lipid metabolism in the LB and HB livers. Supplementation of BCAAs under a low-fat diet did not result in elevated levels of circulating BCAAs or their whole body turnover (measured from the plasma dilution of [13C6]leucine), under fed or fasting conditions (Fig. 4, A and B; Supplemental Fig. S1, A and B: https://doi.org/10.6084/m9.figshare.21574731). The circulating levels of all the three BCAAs were lower with fasting, compared with their fed counterparts (P ≤ 0.05; Fig. 4, A and B; Supplemental Fig. S1, A and B). Despite these differences in the serum BCAA concentrations, the whole body turnover rate of [13C6]leucine remained similar between LF and LB groups, irrespective of feeding or fasting conditions (Fig. 4B). On a high-fat diet, BCAA supplementation resulted in higher circulating levels of only leucine, under fed conditions (Fig. 4A). However, the fed levels of all the serum BCAAs in both the HF and HB mice dropped significantly with fasting (P ≤ 0.05) but remained similar between the two fasted groups. The whole body turnover rate measured with [13C6]leucine, although not statistically significant, was higher in the fed BCAA-supplemented mice (HB). The whole body leucine turnover rates in the HB mice dropped significantly with fasting (P ≤ 0.05) but remained similar to those in the HF-fasted mice.
Figure 4.

Alterations in the levels and turnover rates of BCAAs and their keto acids, and changes in genes involved in BCAA degradation. Serum leucine concentration (A), whole body turnover rates of leucine (B), serum α-ketoisocaproate concentration (C), and whole body turnover rates of α-keto-β-methylvalerate (D), after 12 wk on LF, LB, HF, and HB diets. E: the fed-to-fasted fold-change in the expression of genes associated with BCAA degradation (Bckdha, Bckdhd, Bckdk, and Pp2cm) after 12 wk on LF, LB, HF, and HB diets. All the values are represented as means ± SE, with n = 7–16/group. Results are considered statistically significant at P ≤ 0.05 following two-way ANOVA and pairwise mean comparisons. For the groups on the low-fat diet, the mean comparisons are represented as follows: “d”—LB Fed vs. LB Fast. For the groups on the high-fat diet, the mean comparisons are represented as follows: “a”—HF Fed vs. HB Fed; “b”—HF-Fed vs. HF-Fast; “d”—HB Fed vs. HB Fast. For the hepatic gene expression profiles, comparisons are represented as follows: “a”—LF vs. LB; “b”—HF vs. HB; “c”—LF vs. HF; “d”—LB vs. HB. AU, arbitrary units; BCAA, branched-chain amino acid; Bckdk, branched-chain keto-acid dehydrogenase kinase; BCAA, branched-chain amino acid; Bckdha/b, branched-chain keto-acid dehydrogenase E1 subunit α and β; HB, high-fat diet with BCAA; HF, high-fat diet; LB, low-fat diet with BCAA; LF, low-fat die; Pp2cm, protein phosphatase 2 C mitochondrial.
The associated alterations in the serum concentrations and turnover rates of the BCAA keto acids under the low-fat, and high-fat diets, and plus/minus BCAA supplementation, presented a complicated picture. However, the major take-home result from these data (Fig. 4, C and D; Supplemental Fig. S1, C–E) is the observation that the fasted serum concentrations and turnover rates of all the BCAA keto acids remained similar between the BCAA-supplemented versus. nonsupplemented groups, under both low-fat and the high-fat diets. This observation highlighted the possibility that the observed fed-to-fasted induction of lipid oxidation and ketogenesis in the BCAA-supplemented mice (Figs. 1 and 2) are occurring, independent of the anaplerotic impact of these BCAA-derived keto acids on the hepatic mitochondria. Furthermore, among the genes responsible for upregulating BCAA degradation, only the fed-to-fasted expression of Bckdhb was higher in the LB livers (Fig. 4E), demonstrating no clear differences between LF and LB livers.
Ketogenic Flux from α-Ketoisocaproic Acid Remained Similar between the Hepatic Mitochondria, Isolated from Fasted LF and LB Livers
The aforementioned results in Fig. 4 still presented the possibility that BCAA degradation rates are higher in the fasted LB mice and raised the possibility that it could affect mitochondrial metabolism. Leucine being a ketogenic BCAA, and further considering the ability of the liver mitochondria to extensively oxidize the keto acid of leucine, α-ketoisocaproic acid, we tested the contribution of this keto-acid carbons toward the synthesis of β-hydroxybutyrate. This was done to test whether the ketogenic amino acids are in any way responsible for the higher rate of ketone synthesis, observed in the fasted LB mice livers and isolated mitochondria (Figs. 1 and 2). The general scheme of 13C incorporation from [13C6]ketoisocaproic acid into ketones and TCA cycle intermediates is illustrated in Fig. 5A. The 13C enrichment in the β-hydroxybutyrate isotopomers was significantly higher in the LB liver mitochondria during fed conditions (P ≤ 0.05; Fig. 5B), as would be expected under BCAA-rich diets. However, the 13C enrichment in β-hydroxybutyrate isotopomers remained similar between the LF and LB groups under fasted conditions (Fig. 5B). These patterns of 13C enrichment in β-hydroxybutyrate occurred without any significant changes in the levels of β-hydroxybutyrate produced by the liver mitochondria of LF and LB groups (Fig. 5C). Furthermore, the citrate M + 2 enrichment was the lowest in the fasted LB mitochondria (Fig. 5D), with no significant 13C-enrichment observed in any other TCA cycle intermediates. These results suggest that the ketogenic role of α-ketoisocaproic acid is not a major determinant of the observed changes in lipid oxidation and ketogenesis (Figs. 1 and 2) following BCAA supplementation. Taken together, the results from Figs. 4 and 5 indicate that the anaplerotic impact of the BCAA keto acids on the liver mitochondria is not entirely responsible for the observed changes in hepatic lipid oxidation in the BCAA-supplemented livers.
Figure 5.

Ketogenic flux generated by α-ketoisocaproic acid in isolated mitochondria from the BCAA-supplemented mice. Schematic of 13C enrichment into β-hydroxybutyrate isotopomers and citrate from [13C6] α-ketoisocaproic acid (A).13C incorporation into β-hydroxybutyrate isotopomers (M + 1, M + 2, M + 3, M + 4) from [13C6] α-ketoisocaproic acid in 12-wk LF and LB diet fed mice (B). Concentration of β-hydroxybutyrate in the incubation media, produced by liver mitochondria, isolated from 12-wk LF and LB diet fed mice (C). 13C incorporation into mitochondrial citrate (M + 2) from [13C6] α-ketoisocaproic acid, in 12-wk LF and LB diet fed mice (D). All the values are represented as means ± SE, with n = 8–10/group. Results are considered significant at P ≤ 0.05 following two-way ANOVA between LF-fed, LB-fed, LF-fast and LB-fast dietary groups, followed by pairwise mean comparisons represented by the following alphabets: “a”—LF-Fed vs. LB Fed; “d”—LB Fed vs. LB Fast. BCAA, branched-chain amino acid; HB, high-fat diet with BCAA; HF, high-fat diet; LB, low-fat diet with BCAA; LF, low-fat diet.
The Cellular Redox Status of the BCAA-Supplemented Livers Were Favorable toward Sustaining Mitochondrial Lipid Oxidation
To test whether BCAA supplementation altered cellular redox status, we determined multiple indices of hepatocellular redox. Lactate to pyruvate ratio, an indicator of the cytosolic redox state (35, 36) remained similar between all the groups (Fig. 6A). Interestingly, the NADH/NAD+ ratio, determined from direct measurements of NADH and NAD in the liver tissue, was higher in the fasted BCAA-supplemented livers (P ≤ 0.05; Fig. 6B). The interconversion of acetoacetate and β-hydroxybutyrate in the hepatocyte mitochondria is an NAD+/NADH-dependent reaction and thus is a good indicator of the redox status of the hepatocyte mitochondria (34, 35) (Fig. 6C). The ratio of β-hydroxybutyrate to acetoacetate was significantly higher in the fasted LB mice (P ≤ 0.05; Fig. 6D). Furthermore, the ratio of Bdh1 and Bdh2 gene expression, the two genes which regulate the interconversion of acetoacetate and β-hydroxybutyrate (Fig. 6C), were also higher in the fasted LB livers (P ≤ 0.05; Fig. 6E), substantiating the higher NADH to NAD+ ratio in these livers. We also tested the impact of BCAA supplementation on the glutathione redox cycle (Fig. 6F). Although the ratio of reduced to oxidized glutathione increased with fasting, those ratios remained similar between LF and LB livers (Fig. 6G). However, there was a significant trend (P ≤ 0.1), in terms of the higher gene expression of glutathione peroxidase (Gpx) and glutathione reductase (Gsr) in the livers of the LB mice (Fig. 6H). Taken together, these results demonstrate that BCAA supplementation altered the redox status of the hepatocyte mitochondria.
Figure 6.

Impact of BCAA supplementation on hepatocellular redox. Cytosolic NADH/NAD+ ratio in the fed and fasted livers, as determined from the lactate to pyruvate ratio in 12-wk LF and LB diet fed mice (A). B: the NADH/NAD+ ratio determined from direct measurements of NAD+ and NADH in the liver tissue, under fed and fasted conditions in the12-wk dietary LF and LB groups. Schematic depicting the interconversion of ketones (acetoacetate and β-hydroxybutyrate), driven by Bdh1 and Bdh2 expression (C). Hepatic mitochondrial NADH/NAD+ under fed and fasted conditions, determined from the ratio of β-hydroxybutyrate to acetoacetate after 12 wk on LF and LB diets (D). Relative Bdh1 to Bdh2 mRNA expression in the liver of LF and LB mice under fed and fasted conditions (E). Schematic depicting the glutathione redox cycle (F). Ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG) in 12 wk LF and LB diet fed mice (G). Fed-to-fasted fold-change in the gene expression of Gsr and Gpx1 after 12-wk dietary supplementation with LF and LB diet (H). All the values are represented as means ± SE, with n = 5–9/group. Results are considered significant at P ≤ 0.05 following two-way ANOVA, followed by pairwise mean comparisons represented by the following alphabet: “d”—LB Fed vs. LB Fast. Comparisons with a symbol “*” represents a Student’s t test with a P ≤ 0.05, and those with a symbol “#” represents a Student t test with a P ≤ 0.1. BCAA, branched-chain amino acid; Bdh1/2; 3-hydroxybutyrate dehydrogenase 1/2; Gpx1, glutathione peroxidase 1; Gsr, glutathione reductase; HB, high-fat diet with BCAA; HF, high-fat diet; LB, low-fat diet with BCAA; LF, low-fat diet.
These changes in cellular redox happened without any significant changes in mitochondrial reactive oxygen species generation (Supplemental Fig. S2A: https://doi.org/10.6084/m9.figshare.21574755) or alterations in insulin sensitivity in the LF versus. LB groups, as demonstrated by similar rates of endogenous glucose production (Supplemental Fig. S2B), similar hepatic insulin resistance indices (Supplemental Fig. S2C) and similar fed-to-fasted suppression of AKT phosphorylation (Supplemental Fig. S2D). The metabolic characteristics between LF and the LB animals, following 12 wk on the diets, remained similar, except for the higher body weight (P ≤ 0.05) and fed adipose tissue weight (P ≤ 0.05) in the LB mice (Supplemental Table S6: https://doi.org/10.6084/m9.figshare.21574869). Interestingly, the fold induction from basal to ADP stimulated oxygen consumption was significantly higher in the LB liver mitochondria during fed and fasted states (P ≤ 0.05; Supplemental Fig. S3C: https://doi.org/10.6084/m9.figshare.21574773). This, however, occurred without any significant changes in mitochondrial OXPHOS protein expression (Supplemental Fig. S3, A and B). These results suggest that BCAA supplementation under a low-fat diet did not adversely impact insulin sensitivity.
Fed-to-Fasting Phosphorylation of AMPK and ACC Were Higher in BCAA-Supplemented Livers
Considering the known cross talk between hepatocellular redox and their master regulators namely AMPK and ACC, we determined their phosphorylation rates in the LF and LB livers. As expected, fasting induced the phosphorylation of AMPK at Thr172 (P ≤ 0.05; Fig. 7, A and B). More significantly, the induction of AMPK phosphorylation with fasting was higher in the LB livers (P ≤ 0.05; Fig. 7B). A similar trend was evident when we quantified the hepatic p-AMPK levels using an ELISA (Fig. 7D). Levels of phosphorylated ACCSer79 were significantly higher in the fasted LB livers, compared with their LF counterparts (P ≤ 0.05; Fig. 7C).
Figure 7.

Phosphorylation of AMPK and ACC in livers of BCAA-supplemented mice. A: Western blot images of pAMPKThr172, AMPK, pACCSer79 and ACC in LF and LB livers. B: quantification of p-AMPKThr172: AMPK and fed-to-fasted fold-changes in phosphorylation. C: quantification of p-ACCSer79: ACC and fed-to-fasted fold-changes in phosphorylation. D: hepatic p-AMPK levels and fed-to-fasted fold-changes in p-AMPK content determined by ELISA on liver tissue from LF and LB mice. All the values are represented as means ± SE, with n = 6/group. Results are considered significant at P ≤ 0.05 following two-way ANOVA, followed by pairwise mean comparisons represented by the following alphabets: “b”—LF Fed vs. LF Fast; “c”—LF Fast vs. LB Fast; “d”—LB Fed vs. LB Fast. Comparisons with a symbol “*” represents a Student’s t test with a P ≤ 0.05. AU, arbitrary units; BCAA, branched-chain amino acid; HB, high-fat diet with BCAA; HF, high-fat diet; LB, low-fat diet with BCAA; LF, low-fat diet.
DISCUSSION
Our results provide evidence that BCAAs and/or their metabolic intermediates can modulate lipid oxidation and de novo lipogenesis in the liver while maintaining insulin sensitivity. The modulation of hepatic lipid oxidation following dietary BCAA supplementation appears to be mediated through changes in hepatocellular redox, modulated by signaling through AMPK and ACC. The supplementation of BCAAs provided the hepatocyte a more reduced biochemical milieu inside the mitochondria which favored oxidation of free fatty acids while maintaining lower rates of DNL. These results are relevant toward understanding the relationships between BCAAs and hepatic lipid metabolism under the following contexts. First, elevated levels of BCAAs are robust indices of insulin resistance and strong predictors of future type-2 diabetes onset (2, 39). Second, as dysfunctional lipid metabolism is a central feature of hepatic insulin resistance and NAFLD (18–20), there is a strong interest in identifying mechanisms modulating lipid metabolism in the liver (40). Third, identifying the mechanisms responsible for the biochemical and molecular relationships between the metabolic intermediates of BCAA metabolism and various networks of lipid metabolism, especially during insulin resistance (3, 4, 6, 8, 41, 42) is of major interest.
Indeed, hepatic mitochondrial oxidative metabolism encompasses multiple central metabolic networks including β-oxidation of free fatty acids, generation of ketones from acetyl-CoA, complete oxidation of acetyl-CoA in the TCA cycle, utilization of the reducing equivalents to fuel OXPHOS and ATP synthesis. With 12 wk of chronic BCAA supplementation in our studies, the fed-to-fasted fold induction of genes and proteins involved in free fatty acid oxidation (via. β-oxidation) and ketogenesis were higher in the BCAA-supplemented livers. Furthermore, fresh liver mitochondria isolated from BCAA-supplemented mice livers, retained the ability to oxidize free fatty acids into ketones at a higher rate, compared with their unsupplemented counterparts. Interestingly, although the incorporation of 13C from [13C16]palmitate into β-hydroxybutytrate was higher in the mitochondria isolated from the BCAA-supplemented livers, with fasting, there were no differences in the 13C enrichment of any TCA cycle intermediates, between the BCAA-supplemented and unsupplemented groups. The ability of freshly isolated mitochondria from BCAA-supplemented mice to synthesize more β-hydroxybutytrate suggest that their mitochondrial machinery was primed to oxidize lipids. Taken together, these results point out the impact of BCAA supplementation on mitochondrial oxidative function and that this impact is primarily at the level of β-oxidation and ketogenesis.
The impact of BCAA supplementation on lipid oxidation was the most obvious in mice reared on a low-fat diet, which maintained insulin sensitivity with BCAA supplementation. Most of the metabolic responses observed on a low-fat BCAA-supplemented diets were not apparent under a high-fat diet. This was especially true for hepatic DNL. Although there was a significant reduction in the rate of DNL with BCAA supplementation on a low-fat diet, this effect was not evident on a high-fat diet. Here, it is worthwhile to note that the high-fat diet had already diminished the rates of DNL to such a significant extent, that any further impact of BCAA supplementation could not be ascertained. Here it is worthwhile to point out the fact that diets rich in fat calories, commonly utilized to induce insulin resistance can result in suppression of DNL, an integral component of human NAFLD (17, 33). Thus, although the ability to suppress DNL by ketogenic amino acids (leucine, isoleucine, lysine) has been previously documented (26, 27), better diet-induced insulin-resistant animal models, which sustain higher rates of hepatic DNL, are needed to probe the impact of BCAA supplementation on DNL and mitochondrial oxidative dysfunction during NAFLD. Branched-chain amino acid-supplemented mice on the low-fat diet also seem to have higher adipose tissue mass. Branched-chain amino acids have been shown to favor adipocyte differentiation by being carbon substrates for lipid synthesis(4). In fact, it could also be plausible that this increase in white adipose tissue, in the BCAA-supplemented mice, could provide more lipolysis-derived free fatty acids to the liver for oxidation. However, the serum fatty acid levels remained similar between the LF and LB mice. This led us to believe that the overall impact of BCAA supplementation we observed is due to mechanisms other than just higher flow of free fatty acids into the liver for oxidation.
The biochemical impact of the intermediates of BCAA catabolism on the mitochondria is well established (9–11). Catabolism of BCAAs occurs in multiple tissues, the first set of transamination reactions generating their corresponding keto acids and multiple nonessential amino acids (12, 43) (BCAT activity) and the subsequent steps (mediated by BCKDH) breaking down these keto acids into metabolic intermediates which can fuel anaplerotic reactions through the mitochondrial TCA cycle (23–25). The first-pass catabolism of the BCAAs is considered insignificant in the liver due to the low BCAT expression (23). However, the liver can utilize circulating BCAA keto acids and the nonessential amino acids, arising from BCAT activity in other tissues (especially muscle), to fuel hepatic TCA cycle anaplerosis (23, 25, 43). Furthermore, acute infusion (4 h) of BCAAs has been shown to modulate TCA cycle function and ketogenesis in the liver (3). Although the biochemical impact of BCAA catabolic networks on the mitochondrial TCA cycle metabolism is clear, we hypothesized that these biochemical mechanisms are more active during physiological conditions, where the availability of the BCAAs and their catabolic intermediates are abundant (e.g., fed conditions, acute infusion of BCAAs, etc.). Indeed, serum levels and whole body turnover rates of the BCAAs were lower with fasting and remained similar between the BCAA-supplemented and nonsupplemented mice. Taken together with the observed fed-to-fasting induction of lipid oxidation and ketogenesis in our BCAA-supplemented mice, this pointed to a mechanism independent of the anaplerotic impact of BCCA intermediates. This interpretation is further strengthened by the fact that there was minimal incorporation of the 13C from either [13C16]palmitate or [13C5]α-ketoisocaproate (keto acid of leucine) into the metabolic intermediates of the TCA cycle, especially in the fasted liver mitochondria. Thus, the fed-to-fasted induction of lipid oxidation and ketogenesis in the BCAA-supplemented livers occurred concurrent to the lower availability of BCAAs and keto-acid carbon substrates, during fasting. This in turn suggests that the modulation of lipid oxidation and DNL observed in our studies occurred independently of the anaplerotic impact of the BCAAs on the hepatic mitochondria.
The interplay between the activation of 5'-AMP-activated protein kinase (AMPK), a master regulator of cellular redox, lipid oxidation, and DNL, is well known. In multiple tissues, including the liver, AMPK activation has been shown to increase fatty acid oxidation via PPARA- and PGC1A-dependent mechanisms, while reducing lipid accumulation through the inhibitory phosphorylation of ACC (44, 45). Reactivation of AMPK activity during NAFLD has been proposed as a strategy to lower hepatic lipid content through activating lipid oxidation and inhibiting lipogenesis (46, 47). The ability of the BCAA-supplemented livers to induce fed-to-fasted lipid oxidation and ketogenesis, while simultaneously maintaining elevated ratios of mitochondrial NADH/NAD+, pointed us to the AMPK-ACC-dependent mechanism of action. Indeed, the phosphorylation of both AMPK and ACC was higher in the BCAA-supplemented livers as was the fed-to-fasted induction of Ppara gene expression. Based on these results, we postulate a model (Fig. 8) where chronic dietary supplementation of BCAAs, under the setting of insulin sensitivity, alters the hepatocellular redox by increasing the NADH:NAD+ ratio. The higher NADH:NAD+ ratio in the BCAA-supplemented livers then acts as a stimulus for the higher rates of phosphorylation of AMPKα, which in turn, through the well-known PGC1α/PPARα dependent mechanisms, primed the liver to sustain higher fed-to-fasted induction of the mitochondrial oxidative networks (β-oxidation and ketogenesis). Under such a metabolic milieu, and together with higher phosphorylation of ACC, the acetyl-CoA generated from the oxidation of fatty acids are diverted toward ketogenesis and away from de novo lipid synthesis and complete oxidation through the TCA cycle.
Figure 8.

Modulation of hepatocellular redox, lipid oxidation and de novo lipogenesis by BCAAs. Chronic supplementation of BCAAs maintains hepatic insulin sensitivity while altering mitochondrial NADH:NAD+ ratio, activating fed-to-fasted phosphorylation of AMPKα and ACC. The higher NADH:NAD+ ratio and the activation of AMPKα primes the liver to oxidize free fatty acids, whereas the inhibitory phosphorylation of ACC helps downregulate DNL in the liver. BCAA, branched-chain amino acid.
The question of whether the higher NADH:NAD+ ratio in the BCAA-supplemented animals is a cause or consequence of AMPK activation is debatable and needs further investigation. Furthermore, our results also do not provide an answer to how BCAAs alter the NADH:NAD+ ratio and AMPK activity. To add to this conundrum, leucine deprivation has also been shown to induce AMPK activity and improve insulin sensitivity (48). Here, the possibility that the drop in BCAA levels observed during feeding-to-fasting is a possible impetus for AMPK activation is worth exploring. Lastly, the impact of hepatic insulin resistance on the interactions between BCAAs and lipid metabolism observed under an insulin-sensitive environment needs to be further explored. For this, we need to reconsider the specific dietary conditions provided to the rodent models of NAFLD to induce hepatic insulin resistance. This is due to the fact that the metabolic networks, which are most responsive to BCCA action on a low-fat diet, are the same ones which are significantly downregulated by a high-fat-rich macronutrient environment. Despite all these, our results provide evidence that BCAA supplementation can bring together a concurrence of events where higher mitochondrial NADH:NAD+ ratios together with AMPK activation can provide an optimal metabolic milieu to increase fatty acid oxidation and lower de novo lipogenesis. This in turn suggests that nutritional or therapeutic manipulation of BCAA-mediated mechanisms, which can modulate lipid metabolic networks, could help alleviate metabolic dysfunction in an insulin-resistant liver.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.21574797.
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.21574821.
Supplemental Table S3: https://doi.org/10.6084/m9.figshare.21574851.
Supplemental Table S4: https://doi.org/10.6084/m9.figshare.21574860.
Supplemental Table S5: https://doi.org/10.6084/m9.figshare.21574866.
Supplemental Table S6: https://doi.org/10.6084/m9.figshare.21574869.
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.21574731.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.21574755.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.21574773.
GRANTS
The work was supported by National Institutes of Health (NIH) grant RO1-DK112865 (to N.E.S.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.E.S. conceived and designed research; C.S., V.M., C.E.R., K.P., D.Z., and N.E.S. performed experiments; C.S., V.M., C.E.R., K.P., D.Z., and N.E.S. analyzed data; C.S. and N.E.S. interpreted results of experiments; C.S., V.M., C.E.R., and N.E.S. prepared figures; C.S. and N.E.S. drafted manuscript; C.S. and N.E.S. edited and revised manuscript; C.S., V.M., C.E.R., K.P., D.Z., and N.E.S. approved final version of manuscript.
REFERENCES
- 1. Biswas D, Dao KT, Mercer A, Cowie AM, Duffley L, El Hiani Y, Kienesberger PC, Pulinilkunnil T. Branched-chain ketoacid overload inhibits insulin action in the muscle. J Biol Chem 295: 15597–15621, 2020. doi: 10.1074/jbc.RA120.013121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, Rochon J, Gallup D, Ilkayeva O, Wenner BR, Yancy WS, Eisenson H, Musante G, Surwit RS, Millington DS, Butler MD, Svetkey LP. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 9: 311–326, 2009. [Erratum in Cell Metab 9: 565–566, 2009]. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sunny NE, Kalavalapalli S, Bril F, Garrett TJ, Nautiyal M, Mathew JT, Williams CM, Cusi K. Cross-talk between branched-chain amino acids and hepatic mitochondria is compromised in nonalcoholic fatty liver disease. Am J Physiol Endocrinol Physiol 309: E311–E319, 2015. doi: 10.1152/ajpendo.00161.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Green CR, Wallace M, Divakaruni AS, Phillips SA, Murphy AN, Ciaraldi TP, Metallo CM. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat Chem Biol 12: 15–21, 2016. doi: 10.1038/nchembio.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Halama A, Horsch M, Kastenmüller G, Möller G, Kumar P, Prehn C, Laumen H, Hauner H, Hrabĕ de Angelis M, Beckers J, Suhre K, Adamski J. Metabolic switch during adipogenesis: from branched chain amino acid catabolism to lipid synthesis. Arch Biochem Biophys 589: 93–107, 2016. doi: 10.1016/j.abb.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 6. Lerin C, Goldfine AB, Boes T, Liu M, Kasif S, Dreyfuss JM, De Sousa-Coelho AL, Daher G, Manoli I, Sysol JR, Isganaitis E, Jessen N, Goodyear LJ, Beebe K, Gall W, Venditti CP, Patti M-E. Defects in muscle branched-chain amino acid oxidation contribute to impaired lipid metabolism. Mol Metab 5: 926–936, 2016. doi: 10.1016/j.molmet.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sánchez-González C, Nuevo-Tapioles C, Herrero Martín JC, Pereira MP, Serrano Sanz S, Ramírez de Molina A, Cuezva JM, Formentini L. Dysfunctional oxidative phosphorylation shunts branched-chain amino acid catabolism onto lipogenesis in skeletal muscle. EMBO J 39: e103812, 2020. doi: 10.15252/embj.2019103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. White PJ, McGarrah RW, Grimsrud PA, Tso SC, Yang WH, Haldeman JM, Grenier-Larouche T, An J, Lapworth AL, Astapova I, Hannou SA, George T, Arlotto M, Olson LB, Lai M, Zhang GF, Ilkayeva O, Herman MA, Wynn RM, Chuang DT, Newgard CB. The BCKDH kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP-citrate lyase. Cell Metab 27: 1281–1293.e7, 2018. doi: 10.1016/j.cmet.2018.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adams SH, Hoppel CL, Lok KH, Zhao L, Wong SW, Minkler PE, Hwang DH, Newman JW, Garvey WT. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J Nutr 139: 1073–1081, 2009. doi: 10.3945/jn.108.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. She P, Van Horn C, Reid T, Hutson SM, Cooney RN, Lynch CJ. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am J Physiol Endocrinol Physiol 293: E1552–E1563, 2007. doi: 10.1152/ajpendo.00134.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. She P, Zhou Y, Zhang Z, Griffin K, Gowda K, Lynch CJ. Disruption of BCAA metabolism in mice impairs exercise metabolism and endurance. J Appl Physiol (1985) 108: 941–949, 2010. doi: 10.1152/japplphysiol.01248.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. White PJ, Lapworth AL, McGarrah RW, Kwee LC, Crown SB, Ilkayeva O, An J, Carson MW, Christopher BA, Ball JR, Davies MN, Kjalarsdottir L, George T, Muehlbauer MJ, Bain JR, Stevens RD, Koves TR, Muoio DM, Brozinick JT, Gimeno RE, Brosnan MJ, Rolph TP, Kraus WE, Shah SH, Newgard CB. Muscle-liver trafficking of BCAA-derived nitrogen underlies obesity-related glycine depletion. Cell Rep 33: 108375, 2020. doi: 10.1016/j.celrep.2020.108375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cummings NE, Williams EM, Kasza I, Konon EN, Schaid MD, Schmidt BA, Poudel C, Sherman DS, Yu D, Arriola Apelo SI, Cottrell SE, Geiger G, Barnes ME, Wisinski JA, Fenske RJ, Matkowskyj KA, Kimple ME, Alexander CM, Merrins MJ, Lamming DW. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J Physiol 596: 623–645, 2018. doi: 10.1113/JP275075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karusheva Y, Koessler T, Strassburger K, Markgraf D, Mastrototaro L, Jelenik T, Simon MC, Pesta D, Zaharia OP, Bódis K, Bärenz F, Schmoll D, Wolkersdorfer M, Tura A, Pacini G, Burkart V, Müssig K, Szendroedi J, Roden M. Short-term dietary reduction of branched-chain amino acids reduces meal-induced insulin secretion and modifies microbiome composition in type 2 diabetes: a randomized controlled crossover trial. Am J Clin Nutr 110: 1098–1107, 2019. doi: 10.1093/ajcn/nqz191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maida A, Chan JSK, Sjøberg KA, Zota A, Schmoll D, Kiens B, Herzig S, Rose AJ. Repletion of branched chain amino acids reverses mTORC1 signaling but not improved metabolism during dietary protein dilution. Mol Metab 6: 873–881, 2017. doi: 10.1016/j.molmet.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yap YW, Rusu PM, Chan AY, Fam BC, Jungmann A, Solon-Biet SM, Barlow CK, Creek DJ, Huang C, Schittenhelm RB, Morgan B, Schmoll D, Kiens B, Piper MDW, Heikenwälder M, Simpson SJ, Bröer S, Andrikopoulos S, Müller OJ, Rose AJ. Restriction of essential amino acids dictates the systemic metabolic response to dietary protein dilution. Nat Commun 11: 2894, 2020. doi: 10.1038/s41467-020-16568-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kattapuram N, Zhang C, Muyyarikkandy MS, Surugihalli C, Muralidaran V, Gregory T, Sunny NE. Dietary macronutrient composition differentially modulates the remodeling of mitochondrial oxidative metabolism during NAFLD. Metabolites 11: 272, 2021. doi: 10.3390/metabo11050272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel WT, Schlensak M, Roden M. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab 21: 739–746, 2015. doi: 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 19. Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 146: 726–735, 2014. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Patterson RE, Kalavalapalli S, Williams CM, Nautiyal M, Mathew JT, Martinez J, Reinhard MK, McDougall DJ, Rocca JR, Yost RA, Cusi K, Garrett TJ, Sunny NE. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am J Physiol Endocrinol Metab 310: E484–E494, 2016. doi: 10.1152/ajpendo.00492.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Satapati S, Sunny NE, Kucejova B, Fu X, He TT, Méndez-Lucas A, Shelton JM, Perales JC, Browning JD, Burgess SC. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J Lipid Res 53: 1080–1092, 2012. doi: 10.1194/jlr.M023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab 14: 804–810, 2011. doi: 10.1016/j.cmet.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hutson SM, Sweatt AJ, LaNoue KF. Branched-chain amino acid metabolism: implications for establishing safe intakes. J Nutr 135: 1557S–1564S, 2005. [Erratum in J Nutr 135(8):2009, 2005]. doi: 10.1093/jn/135.6.1557S. [DOI] [PubMed] [Google Scholar]
- 24. Hutson SM, Wallin R, Hall TR. Identification of mitochondrial branched chain aminotransferase and its isoforms in rat tissues. J Biol Chem 267: 15681–15686, 1992. doi: 10.1016/S0021-9258(19)49589-6. [DOI] [PubMed] [Google Scholar]
- 25. Neinast MD, Jang C, Hui S, Murashige DS, Chu Q, Morscher RJ, Li X, Zhan L, White E, Anthony TG, Rabinowitz JD, Arany Z. Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cell Metab 29: 417–429.e4, 2019. doi: 10.1016/j.cmet.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nishikata N, Shikata N, Kimura Y, Noguchi Y. Dietary lipid-dependent regulation of de novo lipogenesis and lipid partitioning by ketogenic essential amino acids in mice. Nutr Diabetes 1: e5, 2011. doi: 10.1038/nutd.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Noguchi Y, Nishikata N, Shikata N, Kimura Y, Aleman JO, Young JD, Koyama N, Kelleher JK, Takahashi M, Stephanopoulos G. Ketogenic essential amino acids modulate lipid synthetic pathways and prevent hepatic steatosis in mice. PLoS One 5: e12057, 2010. doi: 10.1371/journal.pone.0012057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chotechuang N, Azzout-Marniche D, Bos C, Chaumontet C, Gausserès N, Steiler T, Gaudichon C, Tome D. mTOR, AMPK, and GCN2 coordinate the adaptation of hepatic energy metabolic pathways in response to protein intake in the rat. Am J Physiol Endocrinol Physiol 297: E1313–E1323, 2009. doi: 10.1152/ajpendo.91000.2008. [DOI] [PubMed] [Google Scholar]
- 29. Hatazawa Y, Tadaishi M, Nagaike Y, Morita A, Ogawa Y, Ezaki O, Takai-Igarashi T, Kitaura Y, Shimomura Y, Kamei Y, Miura S. PGC-1α-mediated branched-chain amino acid metabolism in the skeletal muscle. PLoS One 9: e91006, 2014. doi: 10.1371/journal.pone.0091006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Y, Xiong Z, Yan W, Gao E, Cheng H, Wu G, Liu Y, Zhang L, Li C, Wang S, Fan M, Zhao H, Zhang F, Tao L. Branched chain amino acids exacerbate myocardial ischemia/reperfusion vulnerability via enhancing GCN2/ATF6/PPAR-α pathway-dependent fatty acid oxidation. Theranostics 10: 5623–5640, 2020. doi: 10.7150/thno.44836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martin SB, Reiche WS, Fifelski NA, Schultz AJ, Stanford SJ, Martin AA, Nack DL, Radlwimmer B, Boyer MP, Ananieva EA. Leucine and branched-chain amino acid metabolism contribute to the growth of bone sarcomas by regulating AMPK and mTORC1 signaling. Biochem J 477: 1579–1599, 2020. doi: 10.1042/BCJ20190754. [DOI] [PubMed] [Google Scholar]
- 32. Zaganjor E, Yoon H, Spinelli JB, Nunn ER, Laurent G, Keskinidis P, Sivaloganathan S, Joshi S, Notarangelo G, Mulei S, Chvasta MT, Tucker SA, Kalafut K, van de Ven RAH, Clish CB, Haigis MC. SIRT4 is an early regulator of branched-chain amino acid catabolism that promotes adipogenesis. Cell Rep 36: 109345, 2021. doi: 10.1016/j.celrep.2021.109345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Muyyarikkandy MS, McLeod M, Maguire M, Mahar R, Kattapuram N, Zhang C, Surugihalli C, Muralidaran V, Vavilikolanu K, Mathews CE, Merritt ME, Sunny NE. Branched chain amino acids and carbohydrate restriction exacerbate ketogenesis and hepatic mitochondrial oxidative dysfunction during NAFLD. FASEB J 34: 14832–14849, 2020. doi: 10.1096/fj.202001495R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krebs HA, Veech RL. Equilibrium relations between pyridine nucleotides and adenine nucleotides and their roles in the regulation of metabolic processes. Adv Enzyme Regul 7: 397–413, 1969. doi: 10.1016/0065-2571(69)90030-2. [DOI] [PubMed] [Google Scholar]
- 35. Satapati S, Kucejova B, Duarte JAG, Fletcher JA, Reynolds L, Sunny NE, He T, Nair LA, Livingston KA, Fu X, Merritt ME, Sherry AD, Malloy CR, Shelton JM, Lambert J, Parks EJ, Corbin I, Magnuson MA, Browning JD, Burgess SC. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J Clin Invest 125: 4447–4462, 2015. [Erratum in J Clin Invest 126: 1605, 2016]. doi: 10.1172/JCI82204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Go S, Kramer TT, Verhoeven AJ, Oude Elferink RPJ, Chang JC. The extracellular lactate-to-pyruvate ratio modulates the sensitivity to oxidative stress-induced apoptosis via the cytosolic NADH/NAD+ redox state. Apoptosis 26: 38–51, 2021. doi: 10.1007/s10495-020-01648-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Delgado TC, Pinheiro D, Caldeira M, Castro MM, Geraldes CF, López-Larrubia P, Cerdán S, Jones JG. Sources of hepatic triglyceride accumulation during high-fat feeding in the healthy rat. NMR Biomed 22: 310–317, 2009. doi: 10.1002/nbm.1327. [DOI] [PubMed] [Google Scholar]
- 38. Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin C-T, Price JW, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119: 573–581, 2009. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, O'Donnell CJ, Carr SA, Mootha VK, Florez JC, Souza A, Melander O, Clish CB, Gerszten RE. Metabolite profiles and the risk of developing diabetes. Nat Med 17: 448–453, 2011. doi: 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sunny NE, Bril F, Cusi K. Mitochondrial adaptation in nonalcoholic fatty liver disease: novel mechanisms and treatment strategies. Trends Endocrinol Metab 28: 250–260, 2017. doi: 10.1016/j.tem.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 41. Biswas D, Pulinilkunnil T. Disrupted branched-chain amino acid catabolism impair cardiac insulin signaling and is associated with adverse cardiometabolic outcomes. J Mol Cell Cardiol 153: 93–94, 2021. doi: 10.1016/j.yjmcc.2020.12.011. [DOI] [PubMed] [Google Scholar]
- 42. Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab 15: 606–614, 2012. doi: 10.1016/j.cmet.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Okun JG, Rusu PM, Chan AY, Wu Y, Yap YW, Sharkie T, Schumacher J, Schmidt KV, Roberts-Thomson KM, Russell RD, Zota A, Hille S, Jungmann A, Maggi L, Lee Y, Blüher M, Herzig S, Keske MA, Heikenwalder M, Müller OJ, Rose AJ. Liver alanine catabolism promotes skeletal muscle atrophy and hyperglycaemia in type 2 diabetes. Nat Metab 3: 394–409, 2021. doi: 10.1038/s42255-021-00369-9. [DOI] [PubMed] [Google Scholar]
- 44. Foretz M, Even PC, Viollet B. AMPK activation reduces hepatic lipid content by increasing fat oxidation in vivo. Int J Mol Sci 19: 2826, 2018. doi: 10.3390/ijms19092826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee WJ, Kim M, Park HS, Kim HS, Jeon MJ, Oh KS, Koh EH, Won JC, Kim MS, Oh GT, Yoon M, Lee KU, Park JY. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARα and PGC-1. Biochem Biophys Res Commun 340: 291–295, 2006. doi: 10.1016/j.bbrc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 46. Boudaba N, Marion A, Huet C, Pierre R, Viollet B, Foretz M. AMPK re-activation suppresses hepatic steatosis but its downregulation does not promote fatty liver development. EBioMedicine 28: 194–209, 2018. doi: 10.1016/j.ebiom.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, Gao B, Wierzbicki M, Verbeuren TJ, Shaw RJ, Cohen RA, Zang M. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 13: 376–388, 2011. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xiao F, Huang Z, Li H, Yu J, Wang C, Chen S, Meng Q, Cheng Y, Gao X, Li J, Liu Y, Guo F. Leucine deprivation increases hepatic insulin sensitivity via GCN2/mTOR/S6K1 and AMPK pathways. Diabetes 60: 746–756, 2011. doi: 10.2337/db10-1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.21574797.
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.21574821.
Supplemental Table S3: https://doi.org/10.6084/m9.figshare.21574851.
Supplemental Table S4: https://doi.org/10.6084/m9.figshare.21574860.
Supplemental Table S5: https://doi.org/10.6084/m9.figshare.21574866.
Supplemental Table S6: https://doi.org/10.6084/m9.figshare.21574869.
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.21574731.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.21574755.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.21574773.
Data Availability Statement
Data will be made available upon reasonable request.