


Keywords: development, maternal fetal health, pregnancy, pulmonary hypoplasia, retinol
Abstract
Congenital diaphragmatic hernia (CDH) is a developmental disorder that results in incomplete diaphragm formation, pulmonary hypoplasia, and pulmonary hypertension. Although a variety of genes have been linked to its etiology, CDH is not a monogenetic disease, and the cause of the condition is still unclear in the vast majority of clinical cases. By comparing human clinical data and experimental rodent data from the literature, we present clear support demonstrating the importance of vitamin A (vitA) during the early window of pregnancy when the diaphragm and lung are forming. Alteration of vitA signaling via dietary and genetic perturbations can create diaphragmatic defects. Unfortunately, vitA deficiency is chronic among people of child-bearing age, and this early window of diaphragm development occurs before many might be aware of pregnancy. Furthermore, there is an increased demand for vitA during this critical period, which exacerbates the likelihood of deficiency. It would be beneficial for the field to further investigate the connections between maternal vitA and CDH incidence, with the goal of determining vitA status as a CDH risk factor. Regular clinical monitoring of vitA levels in child-bearing years is a tractable method by which CDH outcomes could be prevented or improved.
INTRODUCTION
Congenital diaphragmatic hernia (CDH) is a developmental disorder with an unclear etiology, diagnosed via its clinical presentation of a diaphragmatic defect and herniation of the abdominal organs into the pleural cavity. Large cohort studies from various countries report CDH incidence rates of 2–5 per 10,000 births (1–3). Even with treatment, there remains a high incidence of associated mortality and morbidity due to pulmonary hypoplasia and pulmonary hypertension (PH) (4). Some studies report survival at birth as low as 61%; however, co-morbidities often contribute to mortality (5). Irrespective of advances in diagnosis and treatment of CDH (6–10), a recent clinical study showed that for those that survive birth, survival at one year is 93.5% for mild cases of isolated CDH, but only 64% in the severe cases (11). Fetal treatment of CDH is limited and not without risk for the mother and child, thus only applied in the most severe cases (4, 10, 12).
Whereas etiology is still vague, it is suggested that both genetic and environmental factors can drive CDH. Not all cases have genetic abnormalities, and the environmental causative factors are less clear, thus giving genetic factors the main spotlight. In recent years, genetic models have shown that altered signaling in the transient precursor structures to the diaphragm, the pleural peritoneal folds (PPFs), cause improper formation of the diaphragm (13–15). Yet, the genetic mutations examined in these models only represent a small fraction of those associated with human CDH. Overall, mutations account for only a small portion, ∼30%, of CDH cases observed clinically (16), and there is no common monogenetic etiology.
A variety of evidence strongly points to retinoids and the vitamin A (vitA) pathway as highly relevant for the etiology of CDH. The known literature is well summarized in other recent comprehensive and excellent CDH reviews (16–20); however, CDH etiology is not placed explicitly in the context of vitA. Although other mutations and conditions could contribute to a portion of CDH cases, the number of cases and mutations linked to vitA signaling are too many to ignore. This review brings together data from human and mouse studies to highlight the genetic and environmental connections that demonstrate a clear role for vitA processing during diaphragm and lung development and its relation to CDH. We emphasize the importance of sufficient vitA levels at critical times early in pregnancy, coincident with an increased fetal demand for vitA. We propose this hypothesis be more closely examined in clinical and experimental studies and believe CDH risk may be minimized by encouraging sufficient vitA consumption and monitoring during early pregnancy and child-bearing years more broadly.
THE VITAMIN A PATHWAY
VitA is an essential nutrient that is only acquired via dietary intake. VitA can be found as carotenoids in plants, mainly as β-carotene, or in meats, as retinyl esters (REs). REs are absorbed by the gut and transported to the liver within chylomicrons (Fig. 1A). RE is metabolized into retinol in the liver by RE hydrolases (REH) and subsequently released into the circulation. Retinol can also be absorbed by specific cell types such as liver stellate or placental trophoblast cells and reconverted to RE via lecithin retinol acetyltransferase (LRAT) for long-term storage and release. Retinol is hydrophobic and thus must be transported in the bloodstream primarily bound to retinol-binding protein (RBP). RBP complexes with transthyretin (TTR) to further facilitate transport. Homeostatic levels of RBP-retinol in the blood are usually maintained except in extreme cases of malnutrition, including insufficient intake of vitA, disease, or during pregnancy (21–23).
Figure 1.
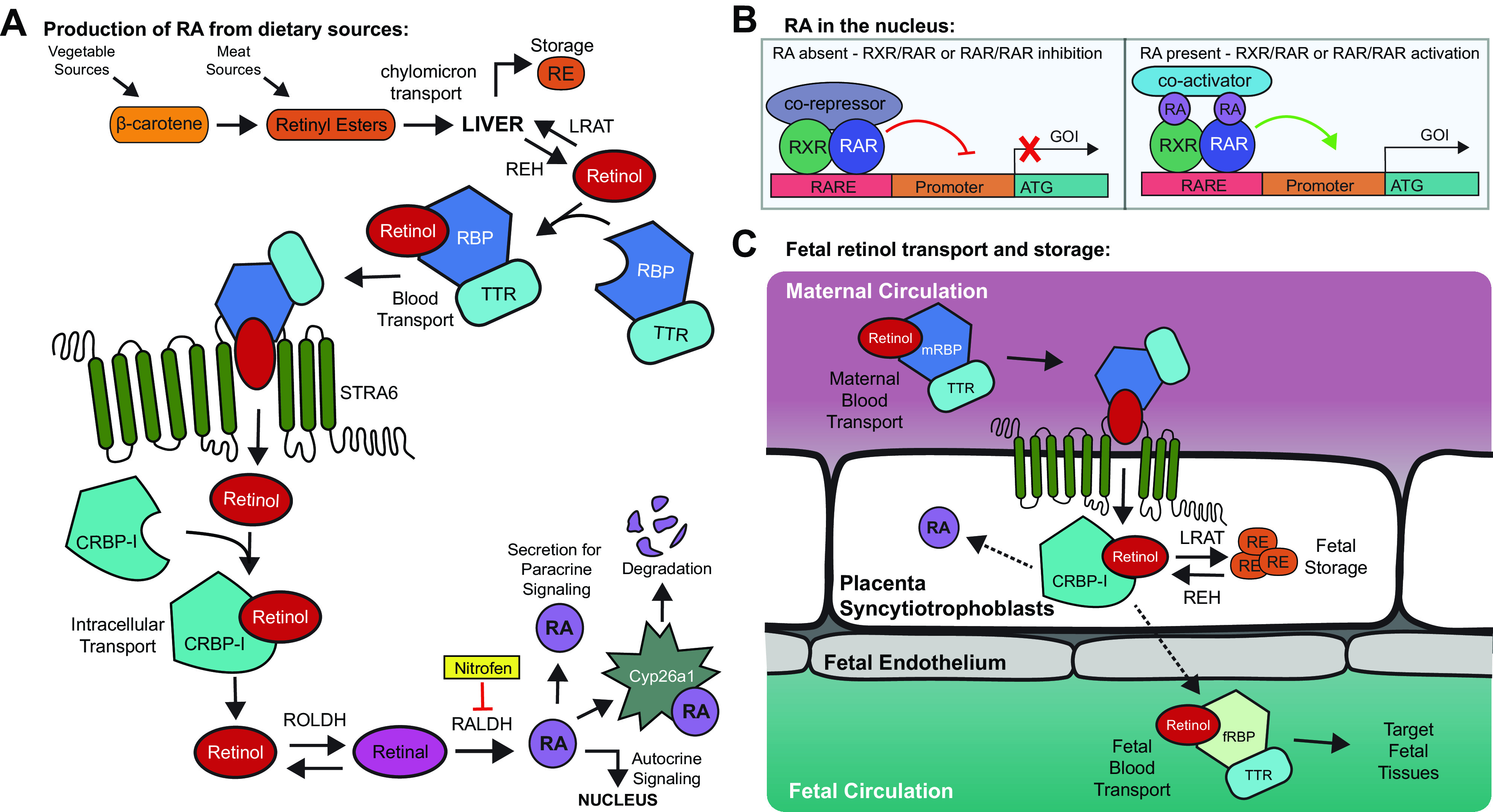
Vitamin A (vitA) is derived from dietary sources and must be transported and converted to retinoic acid (RA). A: vitA is rapidly converted to retinol and is either stored or transported to target cells for conversion to RA. RA can be transported to the nucleus to act on the same cell, be secreted to act in a paracrine fashion, or be otherwise degraded. B: retinoid X receptor (RXR) and retinoic acid receptor (RAR) bind retinoic acid responsive element (RARE) elements in the genome. Without RA, these elements with a corepressor will block transcription. In the presence of RA, these elements with a co-activator will drive transcription. C: retinol binding protein (RBP) cannot cross the placenta. Therefore, maternal-RBP transports retinol to the placenta, where it is used, stored, or secreted to ultimately bind fetal-RBP for transport within the fetal circulation to the target tissues. CRBP-1, cellular retinol binding protein; Cyp26a1, cytochrome p450 family 26a1; fRBP, fetal retinol binding protein; GOI, gene of interest; mRBP, maternal RBP; RALDH, retinal dehydrogenase; RE, retinyl esters; ROLDH, retinol dehydrogenase; STRA6, stimulated by retinoic acid-6; TTR, transthyretin.
Once circulating, RBP-retinol needs to be actively internalized into cells to activate intracellular signaling events (Fig. 1A). STRA6, a protein from the large group of STimulated by Retinoic Acid family, facilitates retinol uptake by cells. Retinol binds to the cellular retinol-binding protein-I (CRBP-I) within the cytoplasm and is oxidized into retinal via retinol dehydrogenase (RDH/ROLDH). Retinal is subsequently and irreversibly dehydrogenated via cytosolic retinal dehydrogenases (RALDH) into the active molecule retinoic acid (RA). RA is a transcriptional regulator and enters the nucleus to bind RA receptors (RARs), which can homodimer, or heterodimer with retinoid X receptors (RXRs) (Fig. 1B). RARs and RXRs are ligand-activated nuclear transcription factors, each with three different isoforms, α, β, and γ. They bind to RA responsive elements (RAREs) present in enhancer and promoter regions of genes. When RA is bound to a RAR/RXR complex, gene transcription is promoted, whereas the RAR/RXR complex can repress gene expression without RA. As the conversion of retinal to RA is irreversible, RA is regulated via catabolism using Cyp26a1, a member of the cytochrome p450 family. Animal models that knockout Cyp26a1 are embryonically lethal, likely due to RA build-up, supporting the requirement of carefully balanced RA signaling (24).
Retinol in maternal circulation reaches the placenta bound to maternal RBP, but maternal RBP cannot cross the placenta (25). Retinol enters placental trophoblast cells and is either converted to RA for use, converted to RE for storage, or is secreted and subsequently transported by fetal RBP to fetal target tissues (Fig. 1C). Maternal-fetal transfer of vitA has been reviewed recently (26). The placenta is thought to be the site of early retinol storage in the embryo, as demonstrated using radiolabeled retinol injections in pregnant rats (27). Interestingly, each embryo seemed to have unequal placental storage, but it is unclear if individual placental storage correlated with individual embryo vitA status. Supplementation with RA or vitA during gestation mildly increases fetal retinol but drastically increases maternal stores, indicating limited storage capacity of the fetus (26). Most pups are born with minimal vitA stores, which increase by 50% after one day of nursing as the liver begins to store reserves (27).
ENVIRONMENTAL PERTURBATION OF VITAMIN A RESULTS IN MODELS OF CDH
A less appreciated proposed etiology of CDH is termed the “retinoid hypothesis” (28). Strong evidence suggests that disruption to the vitA pathway is shared among almost all posterolateral/Bochdalek CDH phenotypes, which represent the most common and most severe form of CDH incidents (29, 30). A significant body of evidence comes from two main animal models used to study CDH. As early as 1941, reports indicated that maternal nutritional deficiency of vitA (VAD) causes a CDH-like phenotype in rats (31–34). Offspring displayed cardiac, lung, and diaphragmatic defects with hernias, as well as edema, paralleling CDH phenotypes in humans. Interestingly, two different rat strains had different initial rates of hernias (2.7% vs. 0%), and incidence was exacerbated with VAD diet (18.9% vs. 0.9%) (31, 34). This indicated that a potential combination of genetic and dietary abnormalities could contribute to incidence, making it difficult to determine a singular etiology of the condition. Notably, only early vitA supplementation, before gestational day (D) 12, rescued lung and diaphragmatic defects (34). Supplementation later in pregnancy either minimally reduced herniation incidence or did not reduce incidence at all (33–36). VAD has since been used as an established experimental model of CDH (37).
The herbicide and teratogen nitrofen (2,4-dichloro-4′-nitrodiphenyl ether) is most commonly used to study CDH since it induces a CDH-like phenotype in rodents (38). As with the VAD model, the timing of nitrofen administration affects the incidence rate of CDH in these animals. In rats, nitrofen administration before D12 causes CDH with high frequency (40%–70%), and after D12, there is minimal effect. In mice, nitrofen is often combined with a second teratogen, bisdiamine, because nitrofen administration alone does not induce CDH (39–42). Coadministration of these chemicals at embryonic day (E)8.5 generates offspring exhibiting CDH (∼50%), and all have pulmonary hypoplasia. Similarly, nitrofen administration later in gestation has minimal or no effect (43). In rats, higher nitrofen doses produce more severe phenotypes (44), and in mice, nitrofen/bisdiamine in combination with VAD increased herniation incidence (39).
A key in vitro study determined that nitrofen directly inhibited the enzyme RALDH2, which is responsible for converting retinal into RA (45). Other teratogens known to induce CDH in rodents (4-biphenyl carboxylic acid, bisdiamine, and SB-210661) were also shown to inhibit RALDH2 activity, demonstrating a shared phenotype and mechanism of action (45). As such, the VAD and teratogen models of CDH involve perturbation of the vitA pathway. Both models support an early critical time window in which vitA is essential for controlling formation of the diaphragm and growth of the lungs. Significantly, when vitA was coadministered with nitrofen in rats, the incidence of CDH dramatically decreased from 54% to 32% (36, 38). Improved results were observed when rodents were directly supplemented with RA (15% incidence), as RA dosing directly bypassed the requirement of RALDH2 processing (38). The RARE-LacZ mouse expresses LacZ under the RA-dependent RARE promoter element and allows for visualization of RA activity. Nitrofen dosing at E9.5 reduced LacZ activity, and LacZ activity increased when co-dosed with RA (41, 46). Collectively, these studies demonstrate the vital role of the vitA pathway in key rodent models of CDH.
GENETIC CONNECTIONS BETWEEN CDH AND RA IN EXPERIMENTAL MODELS
In addition to dietary and environmental perturbation, RA signaling can also be altered genetically by interrupting the myriad of proteins within RA-associated pathways. There are numerous experimental genetic models that result in diaphragmatic defects and phenotypes matching human CDH, which all involve genes directly within RA-associated pathways (Fig. 2). Upstream of RA production, one gene has been widely linked to CDH, Wilm’s Tumor-1 (WT1). WT1 is a zinc-finger transcription factor that can interact with other transcription coregulators to control expression of a myriad of genes. Global and conditional knockout of WT1 produce a CDH-like phenotype with diaphragmatic, cardiac, lung deformities, and hypoplasia (13, 37, 47–52), pleural edema (47, 52, 53), and it is considered a classic genetic model of CDH (37). WT1 was connected upstream of RALDH2/Aldh1a2 in the heart epicardium (53, 54), the diaphragm mesothelium (13), and recently, in the lung mesothelium (47) (Fig. 2i). However, Wt1(−/−) lungs have decreased, but not absent, RALDH2 expression, indicating WT1 is not the sole regulator of RALDH2. This is further supported by the lack of lung agenesis in Wt1(−/−) embryos, a key phenotype of the Aldh1a2(−/−) embryos. In addition, this knockout model produces lung herniation through a diaphragmatic defect into the abdominal cavity (47, 49, 52); inverted from a liver-up herniation into the chest cavity, as seen in other CDH rodent models and humans. Wt1(−/−) embryos present with reduced space available in the pleural cavity, causing the lung to physically interfere with formation of the diaphragm (47). As such, this model may generate a CDH phenotype via a different mechanism than other CDH models.
Figure 2.
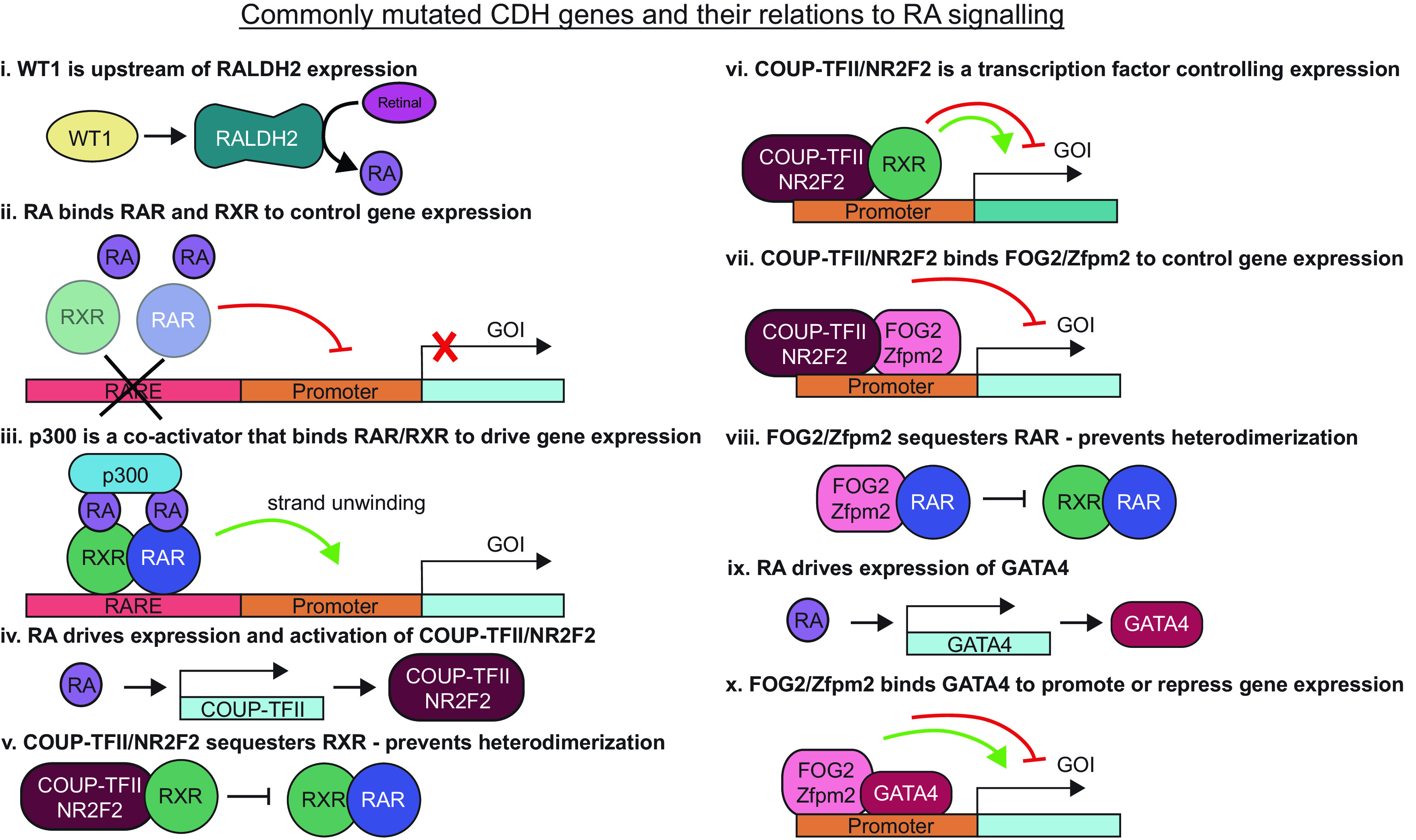
Many genes that produced congenital diaphragmatic hernia (CDH) in experimental models and human cases are related to retinoic acid (RA) signaling. COUP-TFII, chicken ovalbumin upstream promoter transcription factor 2; FOG2, friend of gata-2; GOI, gene of interest; NR2F2, nuclear receptor subfamily 2, group F, member 2; RALDH2, retinal dehydrogenase-2; RAR, retinoic acid receptor; RARE, retinoic acid responsive element; RXR, retinoid X receptor; WT1, Wilm’s tumor-1; Zfpm2, zinc finger protein-2.
After conversion by RALDH2, RA can control gene expression by complexing with RAR and RXR transcription factors (Fig. 2ii). When RAR or RXR is mutated, RA-dependent gene expression cannot proceed normally. Rxra(−/−) mutant mice die in utero (55, 56). This points to a specific role for RXRα during development that the other RXR isoforms cannot compensate. Rxrg(−/−) animals present with pericardial edema and irregular visceral lining by E12.5, along with lung hypoplasia. Rara(−/−);b2(−/−) and Rxra(−/−);Rara(−/−) compound mutants displayed pulmonary hypoplasia, and some showed left lung agenesis (56, 57). Deviations from lung stereotypy and CDH occurred in Rara(−/−);b2(−/−) and in Rara(−/−);b2(+/−) mutants (57). The phenotypes in these RXR or RAR mutants are similar to those seen in VAD rodents (55, 56) and the Wt1(−/−) model (47, 52, 53).
Control of gene expression via RAR/RXR involves binding supporting proteins, coactivators, or corepressors, which can directly interact with transcriptional machinery or alter chromatin accessibility. p300 is a coactivator recruited to genetic locations via RARs to promote gene transcription (Fig. 2iii). p300 is a histone acetyltransferase (HAT) that can bind other HATs and assemble protein complexes to drive DNA relaxation and transcription by hyperacetylating histones. In rats dosed with nitrofen at D9.5, p300 mRNA and immunoreactivity were significantly reduced at D18.5 (58). Mutant p300 mice have a nonfunctional acetyltransferase domain, and >33% of p300(+/mutant) embryos die between E12.5 and E15.5, or otherwise at birth from respiratory failure. To date, p300 has not been directly linked to CDH, yet mice with mutant p300 have many similarities to other CDH models (47, 52, 53, 55, 56), including pulmonary hypoplasia, pleural edema, and cardiac deformities (59). Diaphragmatic defects were not investigated, but it was noted that the mesothelium in these embryos appeared to delaminate from the underlying tissue. Laminin is a key component of the basement membrane of the mesothelium and laminin was also altered in the GATA4 mutants. It would be valuable to examine this model for diaphragmatic defects and signs of altered RA-based signaling.
Although p300 is a coactivator, supporting transcription factors can also repress RAR/RXR signaling, such as COUP-TFII (chicken ovalbumin upstream promoter transcription factor 2), also called NR2F2 (nuclear receptor subfamily 2, group F, member 2). COUP-TFII/NR2F2 is an orphan nuclear receptor in the steroid thyroid hormone receptor superfamily that acts as a transcriptional protein. It is particularly interesting as its activation and expression can be regulated by RA, it regulates other RA signaling by sequestering RXR, thus inhibiting RAR/RXR heterodimerization, and it can directly impact transcription of other proteins related to vitA signaling (60–62) (Fig. 2, iv–vi). This gene is expressed in the primitive foregut mesenchyme, developing posthepatic mesenchymal place (PHMP) in humans (PPFs in rodents), developing lung, and septum transversum (63). COUP-TFII/NR2F2 knockout mutant mice die by E10.5 and have heart and vascular deformities (64). There was a ∼50% CDH incidence rate with homozygous COUP-TFII/NR2F2 knockout using an Nkx3.2-Cre, but no CDH was present in heterozygous mutants (63). Lung defects were not examined but are possible given the expression pattern of Nkx3.2. Of note, the chromosomal region where COUP-TFII/NR2F2 is located is recurrently deleted in human cases of CDH (65–68).
COUP-TFII/NR2F2 physically interacts with another gene linked to CDH downstream of RA signaling, Friend of GATA2 (FOG2) (63, 69) (Fig. 2, vii). FOG2, also called ZFPM2 (zinc finger protein member-2), is a multi-zinc finger transcriptional co-repressor. FOG2 can bind to RARs and the GATA family of transcription factors, particularly GATA4 (70). GATA4 is induced by RA and binds promoter regions of genes to control expression (Fig. 2, vii–x). FOG2/ZFPM2 expression is localized to the pulmonary mesenchyme, pulmonary mesothelium, and PPFs (71), whereas GATA4 is expressed in the pulmonary mesenchyme and mesothelium, pleural cavity mesothelium, heart, septum transversum, gastric epithelium, and PPFs (72). FOG2/ZFPM2 and GATA4 have overlapping expression in the lung mesothelium and PPFs, where many other RA signaling genes are also expressed (73). These genes have long been associated with CDH in clinical cases and animal models (66). One clinical study demonstrated 14 different sequence changes in FOG2/ZFPM2 in ∼4.7% (13/275) human cases of CDH (74).
Initially, the role of FOG2/ZFPM2 during lung and diaphragm development was investigated using the “lil” mouse (71). This mouse resulted from random mutations in the FOG2/ZFPM2 gene that produced a truncated protein and was termed the “lil” mouse due to the little lung phenotype. Mice presented with bilateral pulmonary hypoplasia, abnormal diaphragms, and cardiac deformities. All mutant lungs were missing their accessory lobes and had developmentally delayed medial lobes. Lungs were hypoplastic before the diaphragmatic defect, consistent with the CDH “dual hit” hypothesis. However, when cultured ex vivo, “lil” lungs had normal branching patterns similar to Wt1(−/−) mutant lungs (47), indicating a recovery of the hypoplastic branching phenotype ex vivo. The “lil” mutation displayed a variable penetrance of the phenotype, which the authors note could be due to low levels of normal transcript (71). However, this could alternatively be explained due to variable RA signaling or vitA storage of the individual embryos and dams.
GATA4 and FOG2/ZFPM2 are strongly dependent on each other, making it difficult to study the specific roles independently. Gata4(−/−) embryos die by E10.5, indicating the overall importance of this gene in development (72). Mice that were heterozygous for GATA4 containing a deletion of exon 2 could survive until birth yet presented with congenital defects, including CDH. In these mice, the liver herniated into the thoracic cavity as hernia “sacs”, indicating an incomplete diaphragm defect. In clinical cases, this sac herniation is considered a better prognosis (75). These mice also presented with ectopic smooth muscle actin expression in mesothelial and submesothelial cells (72), which was also present in p300 mutants (59). To explore the specific role of GATA4-FOG2 interactions, one group used a mouse model with a mutated GATA4 to disrupt the GATA-FOG binding, but preserves FOG-independent GATA function (76). The lung phenotypes were similar to the “lil” embryos (71), indicating that GATA4-FOG2 binding is essential for lung formation but diaphragmatic development has not been investigated in this model. Clinical CDH cases have also been linked to pathogenic alterations in GATA4 using whole exome signaling (77).
Importantly, modification to RA signaling either directly (dietary or teratogenic) or indirectly (genes upstream or downstream) produce CDH-like phenotypes, making altered vitA processing and signaling a compelling cause of CDH. One paper examined commonly deleted and mutated gene regions in clinical cases of CDH and identified many regulators of the RA pathway in these regions (66). These data suggest that a combination of genetic and environmental factors at a critical point in development can create a perfect storm to drive CDH presentation.
VITAMIN A CONNECTIONS TO THE DUAL HIT HYPOTHESIS
Most studies looking for CDH etiology have focused on understanding the diaphragmatic defect, with pulmonary complications often marked as a consequence rather than a contributing factor. Pulmonary hypoplasia was initially discussed as a secondary defect due to the herniated organs imposing space constraints (78–80). However, the “dual hit” hypothesis was suggested after noting primary lung defects before diaphragmatic defects and herniation in rodents (81). This hypothesis is supported by clinical cases where minimal organ displacement is observed, yet lung hypoplasia and abnormal lung branching still occurs (82).
One fundamental similarity between the lung and diaphragm is the mesothelium, an epithelial-like tissue that surrounds many visceral organs. RALDH2 is expressed in the lung only by mesothelial cells (83, 84) and is expressed by the mesothelium of the PPFs and heart (epicardium) (41). These tissue layers all originate from the coelomic epithelium (85, 86). In addition, RARs and RXRs are expressed in these tissue layers, indicating that the mesothelium is likely a central site for RA processing and signaling, and thus could be a significant driver of CDH (41, 87).
The evidence is convincing that defective vitA processing in the lung mesothelium could be the “first hit” causing pulmonary hypoplasia in CDH presentation. Yet, the role of retinoids in controlling lung growth is often unappreciated. Despite various studies examining the role of RA in lung development (83, 88–94), and reviewed in detail (88), RA signaling is usually not considered as extensively as other molecules as a significant regulator of lung growth. FGF10 is commonly considered the central molecule required for growth because lung agenesis occurs without FGF10 (95–98). Importantly, RA has been linked upstream of FGF10 (89–91, 94).
In the VAD rat model, 4% displayed lung agenesis and 31% had CDH. Yet, a single dose of supplemental vitA before D12 completely prevented lung agenesis and pulmonary abnormalities, independent of the diaphragm (33). Furthermore, lung agenesis also occurs with RALDH2 knockout (90, 99). There is conflicting data regarding RA’s effects on lung development; some studies show RA inhibits branching (83, 92), and others have shown RA can stimulate branching (88, 93, 94), and even rescue nitrofen-induced hypoplasia (100). This evidence suggests an important role for RA in lung development with more work needed to elaborate its function.
INHERITED MUTATIONS DO NOT ALWAYS PRODUCE CLINICAL CASES OF CDH
A variety of syndromes are associated with diaphragmatic defects and hernias, and other comorbidities. Still, the highest CDH frequency occurs in syndromes associated with mutations in RA-related genes (16). External to these syndromic mutations, specific mutations related to RA signaling have been found clinically in patients with CDH. Whereas some of these mutations appear to be de novo (20), curiously, some of them are not, and are instead inherited from one or both parents. In some cases, these inherited mutations can be traced through many generations. Yet, none of the relatives carrying the mutation present with a clinical diagnosis of CDH. In one case, a parent and grandparent carrying the mutation were identified to have minor diaphragmatic defects and small, nonpathogenic hernias only after a screening following the child’s CDH diagnosis (77). In another study, four relatives had isolated CDH traced back to a Fog2 mutation, yet this mutation was also present in unaffected family members (74). Another case showed three children with CDH shared a Fog2 mutation with their mother, yet the mother did not present with CDH. These cases could be explained by a second, unidentified mutation inherited from the opposite lineage tree to cause CDH. This second mutation would be absent in the other relatives, indicating that the single mutation is insufficient to induce disease. However, another intriguing possibility is that these mutations in RA-related genes predispose the child to incidence of CDH. A subsequent environmental insult, such as insufficient maternal vitA intake or processing, may induce disease, especially if the mother also carried the mutation. This would suggest that adequate vitA could prevent disease. Rodent studies have shown that even in cases where key vitA storage and transport genes are deleted, proper embryogenesis can occur as long as sufficient maternal dietary vitA is sustained (26, 39, 101). Therefore, mutation of genes in combination with maternal vitA deficiency, particularly as the lung and diaphragm form, could have the potential to result in an increased likelihood of CDH.
AN EARLY WINDOW OF SUSCEPTIBILITY TO VITAMIN A DEFICIENCY IN PREGNANCY
The time of susceptibility to VAD or nitrofen in rodent models is equivalent to approximately weeks 4–8 of human gestation (102–104). This early critical window of susceptibility explains the myriad of data in rodent models where later administration of nitrofen cannot cause CDH, nor can supplemental vitA or RA prevent CDH. Of significant importance, in rodents, this critical period also corresponds with a substantial decrease in maternal retinol levels, perhaps due to the increased use of retinol by the fetus during this time (105–107) (Fig. 3).
Figure 3.

Maternal retinol levels drop during the critical window of lung and diaphragm formation in the mouse. After the first week of gestation, maternal retinol levels drop drastically, particularly around the initiation of lung branching off of the foregut. Maternal retinol levels are lowest at the point when the diaphragm typically closes. [Adapted from Satre et al. (105), with permission.]
Therefore, we and others hypothesize a critical window, approximately weeks 4–8, during which vitA is essential for proper diaphragm and lung development (28, 33, 66, 108). Clinical studies have attempted to investigate maternal vitA status and connections to CDH. As humans cannot synthesize vitA, we rely on diet for appropriate levels or hepatic stores. Therefore, fetuses gather all their vitA from maternal sources (109), yet retinol levels are not routinely measured during pregnancy. In an attempt to correlate vitA status and CDH, newborn blood was sampled and compared with maternal blood collected earlier at 38 wk. CDH newborns had plasma retinol and RBP levels 50% lower than normal (110, 111). Surprisingly, one study found retinol and RBP were lower in CDH newborns but higher in CDH mothers, perhaps indicating a maternal-fetal transport issue and a maternal attempt at compensation. This data is supported by a similar study that showed CDH newborns had significantly lower measured retinol and RBP than controls without any difference in maternal retinol levels (112). They also showed an increased CDH incidence in the lower 15% of measured values, indicating that lower serum RBP and retinol levels in newborns corresponded to increased CDH risk (112). However, the blood sampled was well past the early critical window, thus making it challenging to assess maternal vitA status when the lungs and diaphragm were forming.
VITAMIN A DEFICIENCY IS AN ENDEMIC PROBLEM
Globally, 20 million women are considered vitA “insufficient,” and 7 million are considered “deficient” (113); however, countries recommend different daily intake levels, meaning “deficiency” is also designated by political boundaries. Even more startling, vitA “deficiency,” as per WHO standards, is chronic in females during their reproductive years, especially in low-to-middle-income countries (LMICs) (114–117). According to WHO, serum retinol levels are clinically “deficient” in pregnant people for the same levels that are only “insufficient” in nonpregnant people due to the increased vitA required during pregnancy (Fig. 4) (114, 118).
Figure 4.

Vitamin A deficiency is a common problem during early pregnancy. A: as per WHO standards, retinol levels for nonpregnant people are greater than those for pregnant people due to the increased demand for retinol during pregnancy (118). B: using the National Health and Nutrition Examination Survey (NHANES), ∼7% of people of child-bearing age have retinol levels that would be considered insufficient or deficient during pregnancy (117). C: during early pregnancy, ∼38% of people had retinol levels that would be considered insufficient or deficient (107).
This also begs the question of whether the incidence of CDH is increased in demographics where nutritional vitA status is decreased, such as in malnourished populations. There are minimal reports of increased incidence of CDH in low-to-middle-income countries (119); however, in these areas, there is also a lack of detailed studies documenting these specific birth defects, and a lack of national registries (120). In addition, if a child with CDH survives past birth, CDH is often associated with various comorbidities, which could lead to a lack of proper CDH diagnosis. The symptoms could be confounded as general cardio-pulmonary malformations (121).
A recent global CDH incidence report specifically notes the increased incidence of CDH in Iran (5.7/10,000 births) and Malta (5.4/10,000 births), with a global average CDH incidence of 2.8/10,000 births (119). When this is compared with the WHO data on global vitA deficiency in pregnant women during a similar reporting period, the proportion of pregnant women with deficient serum retinol levels is in the “moderate” category in Iran (10%–20%), and “severe” in Malta (≥20% of the population) (114). Other LMICs represented in this study had 2%–10% or <2% of the populations’ pregnant people presenting with retinol deficiency. These data place a strong correlation between vitA status and CDH incidence. Further studies should focus on determining if increased incidence is also found in other pregnant populations categorized by WHO as “moderate” or “severe,” in these regions.
Although the teratogenic effects of excess vitA during pregnancy are quite severe (26, 122), many prenatal supplements in combination with daily diet do not approach the conservative limits of vitA for pregnant people (>2.42–3 mg or >8,000–10,000 IU). For context, this pregnancy upper-limit is more than three times the recommended daily consumption for nonpregnant people. Prenatal supplements recommend a daily dose of 0.7–1 mg or 2,300–3,300 IU of retinol, with many supplements containing less than this daily dose (122). In normal, healthy adults, large excess intake of retinol, single doses >500 mg or elevated chronic intake, can cause hypervitaminosis A (122). Given the large disparities between teratogenic limits and adult limits, there can be a fetal toxic effect without overt maternal toxicity. Extremely high doses of vitA can cause infertility or spontaneous abortion in rodent models, further emphasizing the need to monitor optimal levels carefully (26).
REGULAR CLINICAL MONITORING OF VITAMIN A LEVELS IN CHILD-BEARING YEARS COULD IMPROVE CDH OUTCOMES
The diaphragm is formed by approximately week 8 of gestation, yet CDH is diagnosed after the first anatomical scan in the second trimester (approximately week 24) with a fraction diagnosed postpartum (10, 12, 108, 123). This is due to limited early imaging resolution of the thorax via routine ultrasound, and typically MRI is only performed after CDH diagnosis (6, 124). Plasma retinol status is a typical output of a routine nutritional blood panel. However, the first maternal blood draw during pregnancy usually occurs between ∼11 and 14 wk, after diaphragm closure, and does not usually test nutritional status. Studies have attempted to identify the vitA status of mothers early in pregnancy via dietary questionnaires and approximation of past retinoid intake (104, 120, 125). However, this method is unreliable as it requires mothers to list the exact intake of food and supplements over the preceding months while trying to control for a variety of factors that could confound the data (obesity status, socioeconomic status, presence of severe morning sickness, etc.). One study did determine that in normal-weight mothers, lower dietary vitA was associated with CDH risk (104). The myriad of phenotypes observed in clinical CDH cases is likely multifactorial, resulting from variable vitA deficiency at an early critical gestational time window or a combination of genetic and dietary defects.
Supplemental vitA or RA has been shown in rodent models to improve CDH outcomes when also given during this early critical period (33–36). In the absence of other confounding fetal defects, dietary vitA is adequate to sustain proper embryonic development even when maternal hepatic stores are depleted or defective (25, 126, 127). This is further supported by clinical cases where a genetic mutation is inherited, yet no relatives present with pathological CDH, indicating a contributing factor either prevented or caused the condition in the child (74, 77). Rather than excess vitA, which can be teratogenic, maintaining sufficient levels of vitA in pregnancy is essential and could be enough to reduce CDH incidence.
Prepregnancy vitA measurements could inform status during the early weeks of pregnancy, and subsequent nutritional testing could be added to monitor levels throughout pregnancy. For planned pregnancies, a prepregnancy indication of maternal serum retinol would be helpful to ensure sufficient vitA status during this critical window. For unplanned pregnancies, a blood draw at routine annual gynecological examinations would allow for monitoring and supplementation of inadequate vitA levels (Fig. 4). As many do not know they are pregnant until ∼5.5 wk gestation (128), regular monitoring could give opportunity to provide critical correction of vitA inadequacy before the critical CDH window closes. However, we acknowledge the practical and fiscal implications of such monitoring. As an initial step, investigators could mine data where serum retinol was measured preconception or during early pregnancy, potentially from patients undergoing preconception or infertility counseling, and correlate these measured values with CDH incidence.
Beyond improving CDH outcomes, vitA deficiency is linked to various other developmental defects, making careful maintenance of levels during child-bearing years generally valuable. Prenatal vitamins contain retinol and likely provide sufficient levels for proper fetal development, but are not always consumed. Rodent studies indicate that extra supplementation of retinol later in pregnancy cannot remedy CDH. Thus, early monitoring and prevention of vitA deficiency in pregnancy has the potential to be the most beneficial approach.
CONCLUDING REMARKS
CDH is a devastating disease with significant mortality and morbidity and unclear etiology. Although only a fraction (<30%) of human CDH cases are linked to genetic causes (16), the majority of attention is placed on these mutations. Furthermore, there is no common monogenetic cause linking these cases. With a lack of clear genetic etiology, an environmental cause, such as vitA deficiency, is a tractable causative factor of CDH. VitA is critical for the production of RA, and RA is responsible for coordinating a variety of developmental processes. When the VAD model was first identified to experimentally create diaphragmatic hernias in 1949, it was proposed that vitA could be a risk factor for the condition in humans (34), and others have postulated this since (103, 129–131). Yet, this possibility has not been thoroughly examined in a quantitative clinical study. Classic experimental models of CDH result from direct perturbation of RA signaling, by either depriving vitA in the VAD model, or preventing RA synthesis using nitrofen/bisdiamine. Moreover, cases with genetic abnormalities often involve genes connected to vitA processing or signaling. VitA is only derived from dietary sources, and insufficient vitA status is endemic in people of child-bearing age. In addition, the increased vitA demand during early pregnancy can quickly result in a deficiency. Future studies should look to measure serum retinol early in pregnancy to determine a causative link between vitA status and human CDH incidence and severity. The critical window of diaphragm development and full formation occurs very early in gestation, concurrent or before awareness of the pregnancy. Therefore, for CDH cases not linked to genetic causes, early routine blood panel testing and dietary prevention strategies may reduce CDH incidence.
GRANTS
This work was supported by National Institutes of Health Grants R01HL133163, R01HL145147, R21ES027962, and F31HL140781 (to R. M. Gilbert).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.M.G. and J.P.G. conceived and designed research; R.M.G. and J.P.G. prepared figures; R.M.G. drafted manuscript; R.M.G. and J.P.G. edited and revised manuscript; R.M.G. and J.P.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We extend our gratitude to Dr. Matthew Hoffman, MPH, FACOG for helpful discussions of the manuscript.
REFERENCES
- 1. Balayla J, Abenhaim HA. Incidence, predictors and outcomes of congenital diaphragmatic hernia: a population-based study of 32 million births in the United States. J Matern Fetal Neonatal Med 27: 1438–1444, 2014. doi: 10.3109/14767058.2013.858691. [DOI] [PubMed] [Google Scholar]
- 2. Mesas Burgos C, Ehrén H, Conner P, Frenckner B. Maternal risk factors and perinatal characteristics in congenital diaphragmatic hernia: a nationwide population-based study. Fetal Diagn Ther 46: 385–391, 2019. doi: 10.1159/000497619. [DOI] [PubMed] [Google Scholar]
- 3. McAteer JP, Hecht A, De Roos AJ, Goldin AB. Maternal medical and behavioral risk factors for congenital diaphragmatic hernia. J Pediatr Surg 49: 34–38, 2014. Discussion 38. doi: 10.1016/j.jpedsurg.2013.09.025. [DOI] [PubMed] [Google Scholar]
- 4. Olutoye Ii OO, Short WD, Gilley J, Hammond Ii JD, Belfort MA, Lee TC, King A, Espinoza J, Joyeux L, Lingappan K, Gleghorn JP, Keswani SG. The cellular and molecular effects of fetoscopic endoluminal tracheal occlusion in congenital diaphragmatic hernia. Front Pediatr 10: 925106, 2022. doi: 10.3389/fped.2022.925106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Colvin J, Bower C, Dickinson JE, Sokol J. Outcomes of congenital diaphragmatic hernia: a population-based study in Western Australia. Pediatrics 116: e356–e363, 2005. [Erratum in Pediatrics 117: 1870, 2006]. doi: 10.1542/peds.2004-2845. [DOI] [PubMed] [Google Scholar]
- 6. Prayer F, Metzelder M, Krois W, Brugger PC, Gruber GM, Weber M, Scharrer A, Rokitansky A, Langs G, Prayer D, Unger E, Kasprian G. Three-dimensional reconstruction of defects in congenital diaphragmatic hernia: a fetal MRI study. Ultrasound Obstet Gynecol 53: 816–826, 2019. doi: 10.1002/uog.20296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grivell RM, Andersen C, Dodd JM. Prenatal interventions for congenital diaphragmatic hernia for improving outcomes. Cochrane Database Syst Rev 2015: CD008925, 2015. doi: 10.1002/14651858.CD008925.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Russo FM, De Coppi P, Allegaert K, Toelen J, van der Veeken L, Attilakos G, Eastwood MP, David AL, Deprest J. Current and future antenatal management of isolated congenital diaphragmatic hernia. Semin Fetal Neonatal Med 22: 383–390, 2017. doi: 10.1016/j.siny.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 9. Russo FM, Cordier A-G, Basurto D, Salazar L, Litwinska E, Gomez O, Debeer A, Nevoux J, Patel S, Lewi L, Pertierra A, Aertsen M, Gratacos E, Nicolaides K, Benachi A, Deprest J. Fetoscopic endoluminal tracheal occlusion reverses the natural history of right-sided congenital diaphragmatic hernia: a European multicenter experience. eLife 5: e16009, 2016. doi: 10.7554/eLife.16009. [DOI] [PubMed] [Google Scholar]
- 10. Verla MA, Style CC, Olutoye OO. Prenatal intervention for the management of congenital diaphragmatic hernia. Pediatr Surg Int 34: 579–587, 2018. doi: 10.1007/s00383-018-4270-0. [DOI] [PubMed] [Google Scholar]
- 11. Style CC, Olutoye OO, Belfort MA, Ayres NA, Cruz SM, Lau PE, Shamshirsaz AA, Lee TC, Olutoye OA, Fernandes CJ, Cortes MS, Keswani SG, Espinoza J. Fetal endoscopic tracheal occlusion reduces pulmonary hypertension in severe congenital diaphragmatic hernia. Ultrasound Obstet Gynecol 54: 752–758, 2019. doi: 10.1002/uog.20216. [DOI] [PubMed] [Google Scholar]
- 12. Basurto D, Russo FM, Van der Veeken L, Van der Merwe J, Hooper S, Benachi A, De Bie F, Gomez O, Deprest J. Prenatal diagnosis and management of congenital diaphragmatic hernia. Best Pract Res Clin Obstet Gynaecol 58: 93–106, 2019. doi: 10.1016/j.bpobgyn.2018.12.010. [DOI] [PubMed] [Google Scholar]
- 13. Carmona R, Cañete A, Cano E, Ariza L, Rojas A, Muñoz-Chápuli R. Conditional deletion of WT1 in the septum transversum mesenchyme causes congenital diaphragmatic hernia in mice. eLife 5: e16009, 2016. doi: 10.7554/eLife.16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Merrell AJ, Ellis BJ, Fox ZD, Lawson JA, Weiss JA, Kardon G. Muscle connective tissue controls development of the diaphragm and is a source of congenital diaphragmatic hernias. Nat Genet 47: 496–504, 2015. doi: 10.1038/ng.3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sefton EM, Gallardo M, Kardon G. Developmental origin and morphogenesis of the diaphragm, an essential mammalian muscle. Dev Biol 440: 64–73, 2018. doi: 10.1016/j.ydbio.2018.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kardon G, Ackerman KG, McCulley DJ, Shen Y, Wynn J, Shang L, Bogenschutz E, Sun X, Chung WK. Congenital diaphragmatic hernias: from genes to mechanisms to therapies. Dis Model Mech 10: 955–970, 2017. doi: 10.1242/dmm.028365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Edel GG, Schaaf G, Wijnen RMH, Tibboel D, Kardon G, Rottier RJ. Cellular origin(s) of congenital diaphragmatic hernia. Front Pediatr 9: 1385, 2021. doi: 10.3389/fped.2021.804496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cannata G, Caporilli C, Grassi F, Perrone S, Esposito S. Management of congenital diaphragmatic hernia (CDH): role of molecular genetics. Int J Mol Sci 22: 6353, 2021. doi: 10.3390/ijms22126353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Donahoe PK, Longoni M, High FA. Polygenic causes of congenital diaphragmatic hernia produce common lung pathologies. Am J Pathol 186: 2532–2543, 2016. doi: 10.1016/j.ajpath.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brosens E, Peters NCJ, van Weelden KS, Bendixen C, Brouwer RWW, Sleutels F, Bruggenwirth HT, van Ijcken WFJ, Veenma DCM, Otter S-D, Wijnen RMH, Eggink AJ, van Dooren MF, Reutter HM, Rottier RJ, Schnater JM, Tibboel D, de Klein A. Unraveling the genetics of congenital diaphragmatic hernia: an ongoing challenge. Front Pediatr 9: 800915, 2021. doi: 10.3389/fped.2021.800915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clagett-Dame M, Knutson D. Vitamin A in reproduction and development. Nutrients 3: 385–428, 2011. doi: 10.3390/nu3040385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Quadro L, Blaner WS, Salchow DJ, Vogel S, Piantedosi R, Gouras P, Freeman S, Cosma MP, Colantuoni V, Gottesman ME. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J 18: 4633–4644, 1999. doi: 10.1093/emboj/18.17.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bastos Maia S, Rolland Souza AS, Costa Caminha M, de F, Lins da Silva S, Callou Cruz R, de SBL, Carvalho dos Santos C, Batista Filho M. Vitamin A and pregnancy: a narrative review. Nutrients 11: 681, 2019. doi: 10.3390/nu11030681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Niederreither K, Abu-Abed S, Schuhbaur B, Petkovich M, Chambon P, Dollé P. Genetic evidence that oxidative derivatives of retinoic acid are not involved in retinoid signaling during mouse development. Nat Genet 31: 84–88, 2002. doi: 10.1038/ng876. [DOI] [PubMed] [Google Scholar]
- 25. Quadro L, Hamberger L, Gottesman ME, Colantuoni V, Ramakrishnan R, Blaner WS. Transplacental delivery of retinoid: the role of retinol-binding protein and lipoprotein retinyl ester. Am J Physiol Endocrinol Physiol 286: E844–E851, 2004. doi: 10.1152/ajpendo.00556.2003. [DOI] [PubMed] [Google Scholar]
- 26. Quadro L, Spiegler EK. Maternal-fetal transfer of vitamin A and its impact on mammalian embryonic development. Subcell Biochem 95: 27–55, 2020. doi: 10.1007/978-3-030-42282-0_2. [DOI] [PubMed] [Google Scholar]
- 27. Ismadi SD, Olson JA. Dynamics of the fetal distribution and transfer of vitamin A between rat fetuses and their mother. Int J Vitam Nutr Res 52: 112–119, 1982. [PubMed] [Google Scholar]
- 28. Greer JJ, Babiuk RP, Thebaud B. Etiology of congenital diaphragmatic hernia: the retinoid hypothesis. Pediatr Res 53: 726–730, 2003. doi: 10.1203/01.PDR.0000062660.12769.E6. [DOI] [PubMed] [Google Scholar]
- 29. Longoni M, Pober BR, High FA, Congenital diaphragmatic hernia overview. In: GeneReviews®, edited by Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, Amemiya A.. Seattle: University of Washington, 2006, p. 1993–2023. [PubMed] [Google Scholar]
- 30. Zani A, Chung WK, Deprest J, Harting MT, Jancelewicz T, Kunisaki SM, Patel N, Antounians L, Puligandla PS, Keijzer R. Congenital diaphragmatic hernia. Nat Rev Dis Primers 8: 37, 2022. doi: 10.1038/s41572-022-00362-w. [DOI] [PubMed] [Google Scholar]
- 31. Andersen DH. Incidence of congenital diaphragmatic hernia in the young of rats bred on a diet deficient in vitamin A. Am J Dis Child 62: 888–889, 1941. [Google Scholar]
- 32. Warkany J, Roth CB. Congenital malformations induced in rats by maternal vitamin A deficiency. II. Effect of varying the preparatory diet upon the yield of abnormal young: four figures. J Nutr 35: 1–11, 1948. doi: 10.1093/jn/35.1.1. [DOI] [Google Scholar]
- 33. Wilson JG, Roth CB, Warkany J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am J Anat 92: 189–217, 1953. doi: 10.1002/aja.1000920202. [DOI] [PubMed] [Google Scholar]
- 34. Andersen DH. Effect of diet during pregnancy upon the incidence of congenital hereditary diaphragmatic hernia in the rat. Am J Pathol 25: 163–185, 1949. [PMC free article] [PubMed] [Google Scholar]
- 35. Baptista MJ, Melo-Rocha G, Pedrosa C, Gonzaga S, Teles A, Estevão-Costa J, Areias JC, Flake AW, Leite-Moreira AF, Correia-Pinto J. Antenatal vitamin A administration attenuates lung hypoplasia by interfering with early instead of late determinants of lung underdevelopment in congenital diaphragmatic hernia. J Pediatr Surg 40: 658–665, 2005. doi: 10.1016/j.jpedsurg.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 36. Thébaud B, Tibboel D, Rambaud C, Mercier JC, Bourbon JR, Dinh-Xuan AT, Archer SL. Vitamin A decreases the incidence and severity of nitrofen-induced congenital diaphragmatic hernia in rats. Am J Physiol Lung Cell Mol Physiol 277: L423–L429, 1999. doi: 10.1152/ajplung.1999.277.2.L423. [DOI] [PubMed] [Google Scholar]
- 37. Clugston RD, Klattig J, Englert C, Clagett-Dame M, Martinovic J, Benachi A, Greer JJ. Teratogen-induced, dietary and genetic models of congenital diaphragmatic hernia share a common mechanism of pathogenesis. Am J Pathol 169: 1541–1549, 2006. doi: 10.2353/ajpath.2006.060445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Babiuk RP, Thébaud B, Greer JJ. Reductions in the incidence of nitrofen-induced diaphragmatic hernia by vitamin A and retinoic acid. Am J Physiol Lung Cell Mol Physiol 286: L970–L973, 2004. doi: 10.1152/ajplung.00403.2003. [DOI] [PubMed] [Google Scholar]
- 39. Rocke AW, Clarke TG, Dalmer TRA, McCluskey SA, Rivas JFG, Clugston RD. Low maternal vitamin A intake increases the incidence of teratogen induced congenital diaphragmatic hernia in mice. Pediatr Res 91: 83–91, 2022. doi: 10.1038/s41390-021-01409-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nguyen TM, Jimenez J, Rendin LE, Müller C, Westergren-Thorsson G, Deprest J, Toelen J. The proportion of alveolar type 1 cells decreases in murine hypoplastic congenital diaphragmatic hernia lungs. PLoS One 14: e0214793, 2019. [Erratum in PLoS One 14: e0217322, 2019]. doi: 10.1371/journal.pone.0214793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Clugston RD, Zhang W, Alvarez S, de Lera AR, Greer JJ. Understanding abnormal retinoid signaling as a causative mechanism in congenital diaphragmatic hernia. Am J Respir Cell Mol Biol 42: 276–285, 2010. doi: 10.1165/rcmb.2009-0076OC. [DOI] [PubMed] [Google Scholar]
- 42. Babiuk RP, Greer JJ. Diaphragm defects occur in a CDH hernia model independently of myogenesis and lung formation. Am J Physiol Lung Cell Mol Physiol 283: L1310–L1314, 2002. doi: 10.1152/ajplung.00257.2002. [DOI] [PubMed] [Google Scholar]
- 43. Cilley RE, Zgleszewski SE, Krummel TM, Chinoy MR. Nitrofen dose-dependent gestational day-specific murine lung hypoplasia and left-sided diaphragmatic hernia. Am J Physiol Lung Cell Mol Physiol 272: L362–L371, 1997. doi: 10.1152/ajplung.1997.272.2.L362. [DOI] [PubMed] [Google Scholar]
- 44. Kluth D, Kangah R, Reich P, Tenbrinck R, Tibboel D, Lambrecht W. Nitrofen-induced diaphragmatic hernias in rats: an animal model. J Pediatr Surg 25: 850–854, 1990. doi: 10.1016/0022-3468(90)90190-K. [DOI] [PubMed] [Google Scholar]
- 45. Mey J, Babiuk RP, Clugston R, Zhang W, Greer JJ. Retinal dehydrogenase-2 is inhibited by compounds that induce congenital diaphragmatic hernias in rodents. Am J Pathol 162: 673–679, 2003. doi: 10.1016/S0002-9440(10)63861-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen M, MacGowan A, Ward S, Bavik C, Greer JJ. The activation of the retinoic acid response element is inhibited in an animal model of congenital diaphragmatic hernia. Biol Neonate 83: 157–161, 2003. doi: 10.1159/000068932. [DOI] [PubMed] [Google Scholar]
- 47. Gilbert RM, Schappell LE, Gleghorn JP. Defective mesothelium and limited physical space are drivers of dysregulated lung development in a genetic model of congenital diaphragmatic hernia. Development 148: dev199460, 2021. doi: 10.1242/dev.199460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cano E, Carmona R, Muñoz-Chápuli R. Wt1-expressing progenitors contribute to multiple tissues in the developing lung. Am J Physiol Lung Cell Mol Physiol 305: L322–L332, 2013. doi: 10.1152/ajplung.00424.2012. [DOI] [PubMed] [Google Scholar]
- 49. Paris ND, Coles GL, Ackerman KG. Wt1 and β-catenin cooperatively regulate diaphragm development in the mouse. Dev Biol 407: 40–56, 2015. doi: 10.1016/j.ydbio.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kling DE, Schnitzer JJ. Vitamin A deficiency (VAD), teratogenic, and surgical models of congenital diaphragmatic hernia (CDH). Am J Med Genet C Semin Med Genet 145: 139–157, 2007. doi: 10.1002/ajmg.c.30129. [DOI] [PubMed] [Google Scholar]
- 51. Norden J, Grieskamp T, Christoffels VM, Moorman AFM, Kispert A. Partial absence of pleuropericardial membranes in Tbx18- and Wt1-deficient mice. PLoS One 7: e45100, 2012. doi: 10.1371/journal.pone.0045100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R. WT-1 is required for early kidney development. Cell 74: 679–691, 1993. doi: 10.1016/0092-8674(93)90515-R. [DOI] [PubMed] [Google Scholar]
- 53. von Gise A, Zhou B, Honor LB, Ma Q, Petryk A, Pu WT. WT1 regulates epicardial epithelial to mesenchymal transition through β-catenin and retinoic acid signaling pathways. Dev Biol 356: 421–431, 2011. doi: 10.1016/j.ydbio.2011.05.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guadix JA, Ruiz-Villalba A, Lettice L, Velecela V, Muñoz-Chápuli R, Hastie ND, Pérez-Pomares JM, Martínez-Estrada OM. Wt1 controls retinoic acid signalling in embryonic epicardium through transcriptional activation of Raldh2. Development 138: 1093–1097, 2011. doi: 10.1242/dev.044594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sucov HM, Dyson E, Gumeringer CL, Price J, Chien KR, Evans RM. RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev 8: 1007–1018, 1994. doi: 10.1101/gad.8.9.1007. [DOI] [PubMed] [Google Scholar]
- 56. Kastner P, Mark M, Ghyselinck N, Krezel W, Dupé V, Grondona JM, Chambon P. Genetic evidence that the retinoid signal is transduced by heterodimeric RXR/RAR functional units during mouse development. Development 124: 313–326, 1997. doi: 10.1242/dev.124.2.313. [DOI] [PubMed] [Google Scholar]
- 57. Mendelsohn C, Lohnes D, Décimo D, Lufkin T, LeMeur M, Chambon P, Mark M. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 120: 2749–2771, 1994. doi: 10.1242/dev.120.10.2749. [DOI] [PubMed] [Google Scholar]
- 58. Takahashi H, Friedmacher F, Fujiwara N, Hofmann A, Takahashi T, Puri P. Downregulation of p300 gene expression in airway mesenchyme of nitrofen-induced hypoplastic lungs. Pediatr Surg Int 30: 431–435, 2014. doi: 10.1007/s00383-014-3466-1. [DOI] [PubMed] [Google Scholar]
- 59. Shikama N, Lutz W, Kretzschmar R, Sauter N, Roth J-F, Marino S, Wittwer J, Scheidweiler A, Eckner R. Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation. EMBO J 22: 5175–5185, 2003. doi: 10.1093/emboj/cdg502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Qiu Y, Krishnan V, Pereira FA, Tsai SY, Tsai MJ. Chicken ovalbumin upstream promoter-transcription factors and their regulation. J Steroid Biochem Mol Biol 56: 81–85, 1996. doi: 10.1016/0960-0760(95)00225-1. [DOI] [PubMed] [Google Scholar]
- 61. Tsai SY, Tsai MJ. Chick ovalbumin upstream promoter-transcription factors (COUP-TFs): coming of age. Endocr Rev 18: 229–240, 1997. doi: 10.1210/edrv.18.2.0294. [DOI] [PubMed] [Google Scholar]
- 62. Litchfield LM, Klinge CM. Multiple roles of COUP-TFII in cancer initiation and progression. J Mol Endocrinol 49: R135–R148, 2012. doi: 10.1530/JME-12-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. You L-R, Takamoto N, Yu C-T, Tanaka T, Kodama T, Demayo FJ, Tsai SY, Tsai M-J. Mouse lacking COUP-TFII as an animal model of Bochdalek-type congenital diaphragmatic hernia. Proc Natl Acad Sci USA 102: 16351–16356, 2005. doi: 10.1073/pnas.0507832102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pereira FA, Qiu Y, Zhou G, Tsai MJ, Tsai SY. The orphan nuclear receptor COUP-TFII is required for angiogenesis and heart development. Genes Dev 13: 1037–1049, 1999. doi: 10.1101/gad.13.8.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Klaassens M, van Dooren M, Eussen HJ, Douben H, den Dekker AT, Lee C, Donahoe PK, Galjaard RJ, Goemaere N, de Krijger RR, Wouters C, Wauters J, Oostra BA, Tibboel D, de Klein A. Congenital diaphragmatic hernia and chromosome 15q26: determination of a candidate region by use of fluorescent in situ hybridization and array-based comparative genomic hybridization. Am J Hum Genet 76: 877–882, 2005. doi: 10.1086/429842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Goumy C, Gouas L, Marceau G, Coste K, Veronese L, Gallot D, Sapin V, Vago P, Tchirkov A. Retinoid pathway and congenital diaphragmatic hernia: hypothesis from the analysis of chromosomal abnormalities. Fetal Diagn Ther 28: 129–139, 2010. doi: 10.1159/000313331. [DOI] [PubMed] [Google Scholar]
- 67. Matsunami N, Shanmugam H, Baird L, Stevens J, Byrne JL, Barnhart DC, Rau C, Feldkamp ML, Yoder BA, Leppert MF, Yost HJ, Brunelli L. Germline but not somatic de novo mutations are common in human congenital diaphragmatic hernia. Birth Defects Res 110: 610–617, 2018. doi: 10.1002/bdr2.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. High FA, Bhayani P, Wilson JM, Bult CJ, Donahoe PK, Longoni M. De novo frameshift mutation in COUP-TFII (NR2F2) in human congenital diaphragmatic hernia. Am J Med Genet A 170: 2457–2461, 2016. doi: 10.1002/ajmg.a.37830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Doi T, Sugimoto K, Puri P. Prenatal retinoic acid up-regulates pulmonary gene expression of COUP-TFII, FOG2, and GATA4 in pulmonary hypoplasia. J Pediatr Surg 44: 1933–1937, 2009. doi: 10.1016/j.jpedsurg.2009.04.027. [DOI] [PubMed] [Google Scholar]
- 70. Crispino JD, Lodish MB, Thurberg BL, Litovsky SH, Collins T, Molkentin JD, Orkin SH. Proper coronary vascular development and heart morphogenesis depend on interaction of GATA-4 with FOG cofactors. Genes Dev 15: 839–844, 2001. doi: 10.1101/gad.875201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ackerman KG, Herron BJ, Vargas SO, Huang H, Tevosian SG, Kochilas L, Rao C, Pober BR, Babiuk RP, Epstein JA, Greer JJ, Beier DR. Fog2 is required for normal diaphragm and lung development in mice and humans. PLoS Genet 1: 58–65, 2005. doi: 10.1371/journal.pgen.0010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jay PY, Bielinska M, Erlich JM, Mannisto S, Pu WT, Heikinheimo M, Wilson DB. Impaired mesenchymal cell function in Gata4 mutant mice leads to diaphragmatic hernias and primary lung defects. Dev Biol 301: 602–614, 2007. doi: 10.1016/j.ydbio.2006.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Clugston RD, Zhang W, Greer JJ. Gene expression in the developing diaphragm: significance for congenital diaphragmatic hernia. Am J Physiol Lung Cell Mol Physiol 294: L665–L675, 2008. doi: 10.1152/ajplung.00027.2008. [DOI] [PubMed] [Google Scholar]
- 74. Longoni M, Russell MK, High FA, Darvishi K, Maalouf FI, Kashani A, Tracy AA, Coletti CM, Loscertales M, Lage K, Ackerman KG, Woods SA, Ward-Melver C, Andrews D, Lee C, Pober BR, Donahoe PK. Prevalence and penetrance of ZFPM2 mutations and deletions causing congenital diaphragmatic hernia. Clin Genet 87: 362–367, 2015. doi: 10.1111/cge.12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bouchghoul H, Marty O, Fouquet V, Cordier A-G, Senat M-V, Saada J, Mokhtari M, Le Sache N, Martinovic J, Benachi A. Congenital diaphragmatic hernia has a better prognosis when associated with a hernia sac. Prenat Diagn 38: 638–644, 2018. doi: 10.1002/pd.5326. [DOI] [PubMed] [Google Scholar]
- 76. Ackerman KG, Wang J, Luo L, Fujiwara Y, Orkin SH, Beier DR. Gata4 is necessary for normal pulmonary lobar development. Am J Respir Cell Mol Biol 36: 391–397, 2007. doi: 10.1165/rcmb.2006-0211RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yu L, Wynn J, Cheung YH, Shen Y, Mychaliska GB, Crombleholme TM, Azarow KS, Lim FY, Chung DH, Potoka D, Warner BW, Bucher B, Stolar C, Aspelund G, Arkovitz MS, Chung WK. Variants in GATA4 are a rare cause of familial and sporadic congenital diaphragmatic hernia. Hum Genet 132: 285–292, 2013. doi: 10.1007/s00439-012-1249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nelson CM, Gleghorn JP, Pang M-F, Jaslove JM, Goodwin K, Varner VD, Miller E, Radisky DC, Stone HA. Microfluidic chest cavities reveal that transmural pressure controls the rate of lung development. Development 144: 4328–4335, 2017. doi: 10.1242/dev.154823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nelson CM, Gleghorn JP. Sculpting organs: mechanical regulation of tissue development. Annu Rev Biomed Eng 14: 129–154, 2012. doi: 10.1146/annurev-bioeng-071811-150043. [DOI] [PubMed] [Google Scholar]
- 80. Gilbert RM, Morgan JT, Marcin ES, Gleghorn JP. Fluid mechanics as a driver of tissue-scale mechanical signaling in organogenesis. Curr Pathoiol Rep 44: 199–208, 2016. doi: 10.1007/s40139-016-0117-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Keijzer R, Liu J, Deimling J, Tibboel D, Post M. Dual-hit hypothesis explains pulmonary hypoplasia in the nitrofen model of congenital diaphragmatic hernia. Am J Pathol 156: 1299–1306, 2000. doi: 10.1016/S0002-9440(10)65000-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ackerman KG, Vargas SO, Wilson JA, Jennings RW, Kozakewich HPW, Pober BR. Congenital diaphragmatic defects: proposal for a new classification based on observations in 234 patients. Pediatr Dev Pathol 15: 265–274, 2012. doi: 10.2350/11-05-1041-OA.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Malpel S, Mendelsohn C, Cardoso WV. Regulation of retinoic acid signaling during lung morphogenesis. Development 127: 3057–3067, 2000. doi: 10.1242/dev.127.14.3057. [DOI] [PubMed] [Google Scholar]
- 84. Fernandes-Silva H, Araújo-Silva H, Correia-Pinto J, Moura RS. Retinoic acid: a key regulator of lung development. Biomolecules 10: 152, 2020. doi: 10.3390/biom10010152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Koopmans T, Rinkevich Y. Mesothelial to mesenchyme transition as a major developmental and pathological player in trunk organs and their cavities. Commun Biol 1: 170, 2018. doi: 10.1038/s42003-018-0180-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ariza L, Carmona R, Cañete A, Cano E, Muñoz-Chápuli R. Coelomic epithelium-derived cells in visceral morphogenesis. Dev Dyn 245: 307–322, 2016. doi: 10.1002/dvdy.24373. [DOI] [PubMed] [Google Scholar]
- 87. Båvik C, Ward SJ, Ong DE. Identification of a mechanism to localize generation of retinoic acid in rat embryos. Mech Dev 69: 155–167, 1997. doi: 10.1016/s0925-4773(97)00167-6. [DOI] [PubMed] [Google Scholar]
- 88. Fernandes-Silva H, Vaz-Cunha P, Barbosa VB, Silva-Gonçalves C, Correia-Pinto J, Moura RS. Retinoic acid regulates avian lung branching through a molecular network. Cell Mol Life Sci 74: 4599–4619, 2017. doi: 10.1007/s00018-017-2600-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chen F, Cao Y, Qian J, Shao F, Niederreither K, Cardoso WV. A retinoic acid-dependent network in the foregut controls formation of the mouse lung primordium. J Clin Invest 120: 2040–2048, 2010. doi: 10.1172/JCI40253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Desai TJ, Chen F, Lü J, Qian J, Niederreither K, Dollé P, Chambon P, Cardoso WV. Distinct roles for retinoic acid receptors alpha and beta in early lung morphogenesis. Dev Biol 291: 12–24, 2006. doi: 10.1016/j.ydbio.2005.10.045. [DOI] [PubMed] [Google Scholar]
- 91. Desai TJ, Malpel S, Flentke GR, Smith SM, Cardoso WV. Retinoic acid selectively regulates Fgf10 expression and maintains cell identity in the prospective lung field of the developing foregut. Dev Biol 273: 402–415, 2004. doi: 10.1016/j.ydbio.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 92. Cardoso WV, Williams MC, Mitsialis SA, Joyce-Brady M, Rishi AK, Brody JS. Retinoic acid induces changes in the pattern of airway branching and alters epithelial cell differentiation in the developing lung in vitro. Am J Respir Cell Mol Biol 12: 464–476, 1995. doi: 10.1165/ajrcmb.12.5.7742011. [DOI] [PubMed] [Google Scholar]
- 93. Schuger L, Varani J, Mitra R, Gilbride K. Retinoic acid stimulates mouse lung development by a mechanism involving epithelial-mesenchymal interaction and regulation of epidermal growth factor receptors. Dev Biol 159: 462–473, 1993. doi: 10.1006/dbio.1993.1256. [DOI] [PubMed] [Google Scholar]
- 94. Chen F, Desai TJ, Qian J, Niederreither K, Lü J, Cardoso WV. Inhibition of Tgf beta signaling by endogenous retinoic acid is essential for primary lung bud induction. Development 134: 2969–2979, 2007. doi: 10.1242/dev.006221. [DOI] [PubMed] [Google Scholar]
- 95. Volckaert T, Campbell A, Dill E, Li C, Minoo P, De Langhe S. Localized Fgf10 expression is not required for lung branching morphogenesis but prevents differentiation of epithelial progenitors. Development 140: 3731–3742, 2013. doi: 10.1242/dev.096560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Park WY, Miranda B, Lebeche D, Hashimoto G, Cardoso WV. FGF-10 is a chemotactic factor for distal epithelial buds during lung development. Dev Biol 201: 125–134, 1998. doi: 10.1006/dbio.1998.8994. [DOI] [PubMed] [Google Scholar]
- 97. Weaver M, Dunn NR, Hogan BL. Bmp4 and Fgf10 play opposing roles during lung bud morphogenesis. Development 127: 2695–2704, 2000. doi: 10.1242/dev.127.12.2695. [DOI] [PubMed] [Google Scholar]
- 98. Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, Yagishita N, Matsui D, Koga Y, Itoh N, Kato S. Fgf10 is essential for limb and lung formation. Nat Genet 21: 138–141, 1999. [Erratum in Nat Genet 51: 921, 2019]. doi: 10.1038/5096. [DOI] [PubMed] [Google Scholar]
- 99. Niederreither K, Subbarayan V, Dollé P, Chambon P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet 21: 444–448, 1999. doi: 10.1038/7788. [DOI] [PubMed] [Google Scholar]
- 100. Montedonico S, Nakazawa N, Puri P. Retinoic acid rescues lung hypoplasia in nitrofen-induced hypoplastic foetal rat lung explants. Pediatr Surg Int 22: 2–8, 2006. doi: 10.1007/s00383-005-1571-x. [DOI] [PubMed] [Google Scholar]
- 101. Chen F, Marquez H, Kim Y-K, Qian J, Shao F, Fine A, Cruikshank WW, Quadro L, Cardoso WV. Prenatal retinoid deficiency leads to airway hyperresponsiveness in adult mice. J Clin Invest 124: 801–811, 2014. doi: 10.1172/JCI70291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Clugston RD, Zhang W, Greer JJ. Early development of the primordial mammalian diaphragm and cellular mechanisms of nitrofen-induced congenital diaphragmatic hernia. Birth Defects Res A Clin Mol Teratol 88: 15–24, 2010. doi: 10.1002/bdra.20613. [DOI] [PubMed] [Google Scholar]
- 103. Montedonico S, Nakazawa N, Puri P. Congenital diaphragmatic hernia and retinoids: searching for an etiology. Pediatr Surg Int 24: 755–761, 2008. doi: 10.1007/s00383-008-2140-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Michikawa T, Yamazaki S, Sekiyama M, Kuroda T, Nakayama SF, Isobe T, Kobayashi Y, Iwai-Shimada M, Suda E, Kawamoto T, Nitta H; Japan Environment and Children’s Study Group. Maternal dietary intake of vitamin A during pregnancy was inversely associated with congenital diaphragmatic hernia: the Japan Environment and Children’s Study. Br J Nutr 122: 1295–1302, 2019. doi: 10.1017/S0007114519002204. [DOI] [PubMed] [Google Scholar]
- 105. Satre MA, Ugen KE, Kochhar DM. Developmental changes in endogenous retinoids during pregnancy and embryogenesis in the mouse. Biol Reprod 46: 802–810, 1992. doi: 10.1095/biolreprod46.5.802. [DOI] [PubMed] [Google Scholar]
- 106. Berggren Söderlund M, Fex GA, Nilsson-Ehle P. Concentrations of retinoids in early pregnancy and in newborns and their mothers. Am J Clin Nutr 81: 633–636, 2005. doi: 10.1093/ajcn/81.3.633. [DOI] [PubMed] [Google Scholar]
- 107. Chen H, Qian N, Yan L, Jiang H. Role of serum vitamin A and E in pregnancy. Exp Ther Med 16: 5185–5189, 2018. doi: 10.3892/etm.2018.6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kirby E, Keijzer R. Congenital diaphragmatic hernia: current management strategies from antenatal diagnosis to long-term follow-up. Pediatr Surg Int 36: 415–429, 2020. doi: 10.1007/s00383-020-04625-z. [DOI] [PubMed] [Google Scholar]
- 109. Quadro L. A gold standard to accurately assess vitamin a status: are we there yet? J Nutr 146: 1929–1930, 2016. doi: 10.3945/jn.116.238311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Spiegler E, Kim Y-K, Wassef L, Shete V, Quadro L. Maternal-fetal transfer and metabolism of vitamin A and its precursor β-carotene in the developing tissues. Biochim Biophys Acta 1821: 88–98, 2012. doi: 10.1016/j.bbalip.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Major D, Cadenas M, Fournier L, Leclerc S, Lefebvre M, Cloutier R. Retinol status of newborn infants with congenital diaphragmatic hernia. Pediatr Surg Int 13: 547–549, 1998. doi: 10.1007/s003830050399. [DOI] [PubMed] [Google Scholar]
- 112. Beurskens LW, Tibboel D, Lindemans J, Duvekot JJ, Cohen-Overbeek TE, Veenma DCM, Klein A. D, Greer JJ, Steegers-Theunissen RPM. Retinol status of newborn infants is associated with congenital diaphragmatic hernia. Pediatrics 126: 712–720, 2010. doi: 10.1542/peds.2010-0521. [DOI] [PubMed] [Google Scholar]
- 113. West KP. Vitamin A deficiency disorders in children and women. Food Nutr Bull 24: S78–90, 2003. doi: 10.1177/15648265030244S204. [DOI] [PubMed] [Google Scholar]
- 114.WHO. Global Prevalence of Vitamin A Deficiency in Populations at Risk 1995–2005: WHO Global Database on Vitamin A Deficiency. Geneva: World Health Organization, 2009. [Google Scholar]
- 115.National Institutes of Health. Vitamin A and Carotenoids (Online). https://ods.od.nih.gov/factsheets/VitaminA-HealthProfessional/ [2021 Apr 16].
- 116. Timoneda J, Rodríguez-Fernández L, Zaragozá R, Marín MP, Cabezuelo MT, Torres L, Viña JR, Barber T. Vitamin A deficiency and the lung. Nutrients 10: 1132, 2018. doi: 10.3390/nu10091132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hanson C, Lyden E, Abresch C, Anderson-Berry A. Serum retinol concentrations, race, and socioeconomic status in of women of childbearing age in the United States. Nutrients 8: 508, 2016. doi: 10.3390/nu8080508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.WHO. Guideline: Vitamin A Supplementation in Pregnant Women. Geneva: World Health Organization, 2011. [PubMed] [Google Scholar]
- 119. Politis MD, Bermejo-Sánchez E, Canfield MA, Contiero P, Cragan JD, Dastgiri S, de Walle HEK, Feldkamp ML, Nance A, Groisman B, Gatt M, Benavides-Lara A, Hurtado-Villa P, Kallén K, Landau D, Lelong N, Lopez-Camelo J, Martinez L, Morgan M, Mutchinick OM, Pierini A, Rissmann A, Šípek A, Szabova E, Wertelecki W, Zarante I, Bakker MK, Kancherla V, Mastroiacovo P, Nembhard WN; International Clearinghouse for Birth Defects Surveillance and Research. Prevalence and mortality in children with congenital diaphragmatic hernia: a multicountry study. Ann Epidemiol 56: 61–69.e3, 2021. doi: 10.1016/j.annepidem.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Beurskens LWJE, Schrijver LH, Tibboel D, Wildhagen MF, Knapen MFCM, Lindemans J, de Vries J, Steegers-Theunissen RPM. Dietary vitamin A intake below the recommended daily intake during pregnancy and the risk of congenital diaphragmatic hernia in the offspring. Birth Defects Res A Clin Mol Teratol 97: 60–66, 2013. doi: 10.1002/bdra.23093. [DOI] [PubMed] [Google Scholar]
- 121. Comptour A, Rouzaire M, Belville C, Bouvier D, Gallot D, Blanchon L, Sapin V. Nuclear retinoid receptors and pregnancy: placental transfer, functions, and pharmacological aspects. Cell Mol Life Sci 73: 3823–3837, 2016. doi: 10.1007/s00018-016-2332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Tzimas G, Nau H. The role of metabolism and toxicokinetics in retinoid teratogenesis. Curr Pharm Des 7: 803–831, 2001. doi: 10.2174/1381612013397708. [DOI] [PubMed] [Google Scholar]
- 123. Deprest J, Brady P, Nicolaides K, Benachi A, Berg C, Vermeesch J, Gardener G, Gratacos E. Prenatal management of the fetus with isolated congenital diaphragmatic hernia in the era of the TOTAL trial. Semin Fetal Neonatal Med 19: 338–348, 2014. doi: 10.1016/j.siny.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 124. Oliver ER, DeBari SE, Adams SE, Didier RA, Horii SC, Victoria T, Hedrick HL, Adzick NS, Howell LJ, Moldenhauer JS, Coleman BG. Congenital diaphragmatic hernia sacs: prenatal imaging and associated postnatal outcomes. Pediatr Radiol 49: 593–599, 2019. doi: 10.1007/s00247-018-04334-9. [DOI] [PubMed] [Google Scholar]
- 125. Yang W, Shaw GM, Carmichael SL, Rasmussen SA, Waller DK, Pober BR, Anderka M, National Birth Defects Prevention Study. Nutrient intakes in women and congenital diaphragmatic hernia in their offspring. Birth Defects Res A Clin Mol Teratol 82: 131–138, 2008. doi: 10.1002/bdra.20436. [DOI] [PubMed] [Google Scholar]
- 126. Quadro L, Blaner WS, Hamberger L, Novikoff PM, Vogel S, Piantedosi R, Gottesman ME, Colantuoni V. The role of extrahepatic retinol binding protein in the mobilization of retinoid stores. J Lipid Res 45: 1975–1982, 2004. doi: 10.1194/jlr.M400137-JLR200. [DOI] [PubMed] [Google Scholar]
- 127. Quadro L, Hamberger L, Gottesman ME, Wang F, Colantuoni V, Blaner WS, Mendelsohn CL. Pathways of vitamin A delivery to the embryo: insights from a new tunable model of embryonic vitamin A deficiency. Endocrinology 146: 4479–4490, 2005. doi: 10.1210/en.2005-0158. [DOI] [PubMed] [Google Scholar]
- 128. Branum AM, Ahrens KA. Trends in timing of pregnancy awareness among US women. Matern Child Health J 21: 715–726, 2017. doi: 10.1007/s10995-016-2155-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Beurskens LWJE, Tibboel D, Steegers-Theunissen RPM. Role of nutrition, lifestyle factors, and genes in the pathogenesis of congenital diaphragmatic hernia: human and animal studies. Nutr Rev 67: 719–730, 2009. doi: 10.1111/j.1753-4887.2009.00247.x. [DOI] [PubMed] [Google Scholar]
- 130. Coste K, Beurskens LWJE, Blanc P, Gallot D, Delabaere A, Blanchon L, Tibboel D, Labbé A, Rottier RJ, Sapin V. Metabolic disturbances of the vitamin A pathway in human diaphragmatic hernia. Am J Physiol Lung Cell Mol Physiol 308: L147–L157, 2015. doi: 10.1152/ajplung.00108.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wynn J, Yu L, Chung WK. Genetic causes of congenital diaphragmatic hernia. Semin Fetal Neonatal Med 19: 324–330, 2014. doi: 10.1016/j.siny.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]