

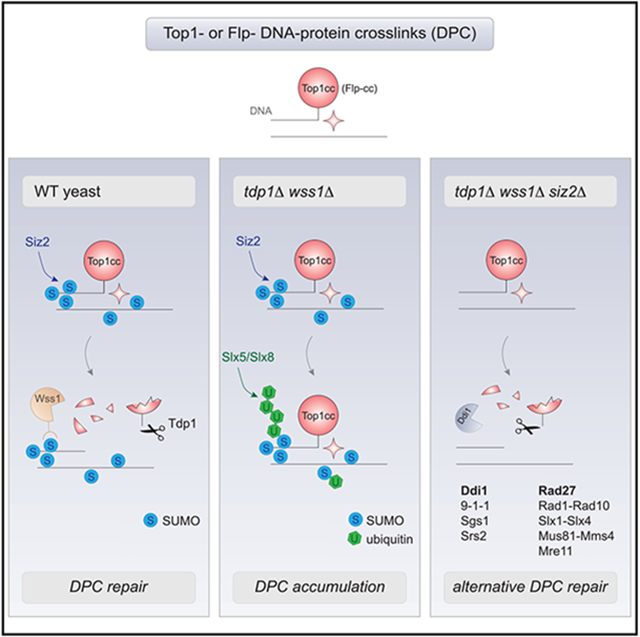
SUMMARY
Endogenous metabolites, environmental agents, and therapeutic drugs promote formation of covalent DNA-protein crosslinks (DPCs). Persistent DPCs compromise genome integrity and are eliminated by multiple repair pathways. Aberrant Top1-DNA crosslinks, or Top1ccs, are processed by Tdp1 and Wss1 functioning in parallel pathways in Saccharomyces cerevisiae. It remains obscure how cells choose between diverse mechanisms of DPC repair. Here, we show that several SUMO biogenesis factors (Ulp1, Siz2, Slx5, and Slx8) control repair of Top1cc or an analogous DPC lesion. Genetic analysis reveals that SUMO promotes Top1cc processing in the absence of Tdp1 but has an inhibitory role if cells additionally lack Wss1. In the tdp1Δ wss1Δ mutant, the E3 SUMO ligase Siz2 stimulates sumoylation in the vicinity of the DPC, but not SUMO conjugation to Top1. This Siz2-dependent sumoylation inhibits alternative DPC repair mechanisms, including Ddi1. Our findings suggest that SUMO tunes available repair pathways to facilitate faithful DPC repair.
Graphical Abstract
In brief
DNA-protein crosslinks (DPCs) are highly toxic DNA lesions that are eliminated by multiple alternative pathways. Serbyn et al. find that SUMO guides the selection of such a pathway required to accomplish DPC repair. DPC-containing loci undergo extensive Siz2-dependent sumoylation that promotes some, but is inhibitory for other, repair mechanisms.
INTRODUCTION
The interaction between two major biopolymers, DNA and protein, is indispensable for the storage and flow of genetic information. DNA-protein complexes are typically dissociated in the order of seconds (Phair et al., 2004), while formation of an aberrant covalent bond abrogates this process. Persistent DNA-protein crosslinks (DPCs) are highly toxic if not resolved in a timely manner, as they cause genome instability and eventually promote cell death (Stingele et al., 2017).
Several classes of enzymes form a covalent bond with DNA as part of their catalytic mechanism. One example is DNA topoisomerases that perform DNA cleavage to rotate DNA and relax the torsional stress arising from replication, transcription, and chromatin remodeling (Pommier et al., 2016). Yeast topoisomerase 1 (Top1) nicks a single DNA strand to form a transient covalent reaction intermediate with the DNA 3′ end. If the subsequent DNA sealing step cannot be completed, Top1 remains trapped on DNA in a covalent cleavage complex (Top1cc). Top1 trapping can occur spontaneously, especially in the presence of a proximal DNA lesion. Top1cc stabilization can also be induced by natural small molecules, such as camptothecin (CPT), or pharmaceutically developed topoisomerase inhibitors (Pommier, 2006). CPT and its derivatives enter the catalytic pocket of Top1, thus inhibiting the DNA religation step and stabilizing a DPC on the nicked 3′ end of DNA (Staker et al., 2002). Several Top1 inhibitors are used in chemotherapies, as the excessive Top1 ccs collide with replication forks, generate DNA damage, and cause cytotoxicity for actively dividing cancer cells (Pommier et al., 2016).
Numerous repair pathways were previously implicated in Top1cc processing. Eukaryotic cells carry a specialized enzyme that directly hydrolyzes the Top1-DNA covalent bond, the phosphodiesterase Tdp1 (human TDP1). Alternatively, Top1ccs can be excised from DNA by several nuclease complexes such as yeast Rad1-Rad10 (hXPF-ERCC1), Slx4-Slx1 (hSLX4-SLX1), Mus81-Mms4 (hMUS81-EME1), Mre11-Rad50-Xrs2 (hMRE11-RAD50-NBS1), and Rad27 (hFEN1) (reviewed in Sun et al., 2020b). Importantly, large proteins such as full-length Top1 are poor in vitro TDP1 substrates and require at least a partial proteolysis (Debéthune et al., 2002). Degradation of the DPC protein moiety can be achieved either by the canonical 26S proteasome machinery (Desai et al., 2001; Larsen et al., 2019; Lin et al., 2008; Zhang et al., 2004) or by the DPC-specific metalloprotease Wss1 in yeast or SPRTN in higher eukaryotes (Lopez-Mosqueda et al., 2016; Maskey et al., 2017; Stingele et al., 2014, 2016; Vaz et al., 2016). Several other proteases were proposed to function in DPC proteolysis, including GCNA, a mammalian SPRTN homolog (Bhargava et al., 2020; Borgermann et al., 2019; Dokshin et al., 2020); the serine protease FAM111A (Kojima et al., 2020); and the yeast aspartic protease Ddi1 (Serbyn et al., 2020). The Cdc48 molecular segregase (p97 in mammals) also assists the Top1cc extraction process (Fielden et al., 2020; Nie et al., 2012; Stingele et al., 2014). Following partial proteolysis, Top1 peptides that remain attached to the single-stranded DNA (ssDNA) break can be further removed by the canonical DNA repair pathways such as nucleotide or base excision repair (NER or BER, respectively) involving the aforementioned nucleases, TDP1, PARP1, PNKP, polymerase β, and DNA ligase. If a ssDNA break persists until S phase, collision of the replication fork with the Top1cc may generate a one-ended double- stranded break (DSB) that can be repaired by homologous recombination (HR) (Pommier et al., 2006).
Mechanisms orchestrating the DPC repair pathway choice remain, however, poorly understood. Emerging evidence indicates that posttranslational modifications (PTMs) such as phosphorylation, ubiquitination, and sumoylation can play a pivotal role in both DPC recognition and processing (Borgermann et al., 2019; Das et al., 2009; Desai et al., 2000; Duxin et al., 2014; Huang et al., 2010; Larsen et al., 2019). Among them, the small ubiquitin-like modifier (SUMO) conjugates to both Top1 and Top1cc repair factors (Chen et al., 2007; Desai et al., 2000; Kanagasabai et al., 2009; Mao et al., 2000b; Sarangi and Zhao, 2015). Top1 sumoylation promotes Top1 exclusion from the highly transcribed rDNA locus (Mao et al., 2000b; Mo et al., 2002), while its role in Top1cc repair is less clear. SUMO conjugation is generally thought to promote the efficient processing of Top1ccs or other DPC types (Borgermann et al., 2019; Chen et al., 2007; Jacquiau et al., 2005; Mao et al., 2000b; Nie et al., 2012; Schellenberg et al., 2017). However, several studies suggest the opposite: SUMO conjugation appears to have a deleterious effect on Top1cc repair (Horie et al., 2002; Sharma et al., 2017). In this work, we show that SUMO conjugation can indeed have a dual role in the Top1cc repair in Saccharomyces cerevisiae. Siz2-mediated sumoylation aids Top1cc repair in the absence of Tdp1 but becomes deleterious if the Wss1 is additionally unavailable. Using a site-specific DPC model, we investigate further how SUMO conjugation modulates detection and processing of Top1ccs or similar DPC types.
RESULTS
A transposon screen identifies multiple components of the SUMO biogenesis pathways as suppressors of tdp1 wss1
Top1-DNA crosslinks accumulate in the tdp1Δ wss1Δ yeast cells lacking the two key Top1cc repair components, the phosphodiesterase Tdp1 and the DPC protease Wss1 (Stingele et al., 2014).To gain insight into the regulatory mechanisms controlling Top1cc processing, we exploited results of a saturated transposon analysis in yeast (SATAY) performed in the tdp1-AID wss1Δ mutant (Serbyn et al., 2020). The tdp1-AID auxin-inducible degron (AID) system (Morawska and Ulrich, 2013) allows for rapid depletion of Tdp1 and permits studying the nearly unviable tdp1Δ wss1Δ double deletion mutant. To identify mutations that suppress synthetic sickness of the double mutant, we compared transposition events in tdp1-AID wss1Δ + auxin and several wild-type (WT)-like libraries (Figure 1A) using the “read_per_gene” value best suited to identify suppressor mutations (Michel et al., 2017). This analysis identified TOP1 among the strongest tdp1-AID wss1Δ suppressors (Figure 1A), supporting previous reports that tdp1Δ wss1Δ synthetic growth defect is mainly due to unrepaired Top1ccs (Balakirev et al., 2015; Stingele et al., 2014). Among other tdp1-AID wss1Δ suppressors were several components of the nuclear pore complex (NPC) (Figure 1A, highlighted in green). We confirmed that deletion of NUP60 indeed rescues the growth phenotype of tdp1Δ wss1Δ (Figure 1B). Remarkably, all identified nucleoporins are present at the nuclear side of the NPC (Figure S1A). In yeast, nuclear pore basket proteins Mlp1, Mlp2, as well as Nup60 and the Nup84 complex, anchor the Ulp1 SUMO protease to the NPC (Palancade et al., 2007; Zhao et al., 2004) (Figure S1A). In the same screen, SUMO biogenesis components were also enriched among tdp1-AID wss1Δ + auxin suppressors (Figure 1A, highlighted in red). We therefore hypothesized that lack of Ulp1 tethering to the NPC promotes tdp1Δ wss1Δ survival. To test this possibility, we took advantage of the fact that the N-terminal domain deletion mutant ulp1-ΔN (Figure 1C) de-localizes the SUMO protease to the nucleoplasm (Texari et al., 2013). Consistent with our hypothesis, Ulp1 de-localization was sufficient to suppress tdp1Δ wss1Δ (Figure 1D). The suppression was dominant (Figure S1B) and required the catalytically active protease domain of Ulp1 (Figures 1C and 1E), indicating that the suppression is caused by Ulp1 desumoylation activity in the nucleoplasm and not by its absence from NPCs.
Figure 1. Mutants of nucleoporins and SUMO biogenesis factors act as suppressors of the tdp1 wss1 double-mutant phenotypes.

(A) Suppressors identified in the tdp1-AID wss1-Δ genetic screen described in Serbyn et al. (2020). tdp1-AID is the shortening of [TDP1-AID*-6HA; pADH-TIR1]. The volcano plot compares sequencing reads in tdp1-AID wss1Δ + auxin and a pool of unrelated SATAY libraries. Fold changes of reads per gene (log2, x axis) and corresponding p values (−log10, y axis) are plotted (Data S1).
(B) Loss of the Nup60 nucleoporin suppresses the growth defect of tdp1Δ wss1Δ. [TDP1/tdp1Δ; WSS1/wss1Δ; NUP60/nup60Δ] diploid was used for the tetrad analysis.
(C) Schematic illustration of the Ulp1 SUMO protease and its mutant variants. NPC, region important for anchoring to the nuclear pore complex; NLS, nuclear localization signal; catalytic, the domain responsible for SUMO maturation and cleavage; C580S, the catalytic active site mutation. In ulp1-ΔN in-frame GFP replaces amino acids 172–340 (Texari et al., 2013).
(D) ulp1-ΔN is a tdp1Δ wss1Δ suppressor. Tetrad analysis of the [TDP1/tdp1Δ; WSS1/wss1Δ; ULP1/ulp1-ΔN] diploid.
(E) The Ulp1 delocalization effect is dominant and depends on its desumoylation activity. Indicated plasmids (as shown in C) expressed from the endogenous ULP1 promoter (Li and Hochstrasser, 2003) were transformed into the [TDP1/tdp1Δ; WSS1/wss1Δ] diploid and analyzed by tetrad analysis.
In S. cerevisiae, the essential Aos1-Uba2 (E1)and Ubc9 (E2) enzymes conjugate SUMO, while the substrate specificity is defined by several E3 SUMO ligases: Siz1, Siz2, Mms21, and meiotic Zip3. SUMO cleavage is catalyzed by the SUMO proteases Ulp1 and Ulp2; the former is also responsible for C-terminal SUMO cleavage required for its maturation (Johnson, 2004). SUMO-dependent ubiquitin ligases (STUbLs) bind poly-sumoylated substrates through SUMO interaction motifs (SIMs) and ubiquitinate them, thus creating mixed SUMO-ubiquitin (SUMO-Ub) chains (Praefcke et al., 2012). Among suppressors of the tdp1-AID wss1Δ screen, we identified the E3 SUMO ligase Siz2, the SUMO protease Ulp2, and both subunits of the Slx5-Slx8 STUbL (Figure 1A). Several of them were previously isolated through an independent genetic suppressor screen of tdp1Δ wss1Δ, and Slx5 was further characterized to control Top1cc processing (Sharma et al., 2017). In our hands, loss of Slx5, Slx8, or the Ulp2 SUMO protease weakly suppresses tdp1Δ wss1Δ (Figures S1C-S1E). Interestingly, whereas slx5Δ improved the growth of tdp1Δ wss1Δ, the same mutant was deleterious in combination with the single tdp1Δ mutation in the presence of CPT (Figure S1F), suggesting that the Slx5-Slx8 complex may participate in the Wss1-dependent pathway of DPC processing. Together, the above genetic interactions implicate sumoylation and SUMO-dependent ubiquitination in the control of Top1cc processing.
Ulp1 activity in the nucleoplasm promotes tdp1Δ wss1Δ survival and lowers DNA damage levels through degradation of the E3 SUMO ligase Siz2
We next analyzed genetic interactions of the E3 SUMO ligases mutants and tdp1Δ wss1Δ. Loss of Siz2 was not only a potent suppressor of the mutant, consistent with the genetic screen results (Figures 1A and 2A), but also provided CPT resistance to tdp1Δ wss1Δ (Figure S2A). In contrast, the siz1Δ mutation was completely inviable in combination with tdp1Δ wss1Δ, whereas simultaneous deletions of SIZ1 and SIZ2 resulted in partial suppression, and mms21-11 had no or little effect on the double mutant (Figure 2A). Remarkably, Ulp1 de-localization to the nucleoplasm promotedsevere reduction in the protein levels of Siz1 and Siz2, but not Mms21 (Figure 2B). These data suggest an explanation for the puzzling similarity in the genetic interactions of the SUMO conjugation (Siz2) and de-conjugation (Ulp1) factors despite their opposing enzymatic activities (Figures 1D and 2A): Ulp1 activity in the nucleoplasm might rescue tdp1Δ wss1Δ phenotypes by promoting the degradation of the Siz2 SUMO ligase. To test this idea, we set out to restore Siz2 protein levels in ulp1-ΔN tdp1Δ wss1Δ. Siz2 overexpression to WT-like levels negatively impacted growth and CPT sensitivity of the ulp1-ΔN tdp1Δ wss1Δ mutant (Figures 2C and S2B), supporting our hypothesis.
Figure 2. Loss of Ulp1 tethering to the nuclear periphery suppresses tdp1Δ wss1Δ via degradation of the Siz2 SUMO ligase.

(A) The role of E3 SUMO ligases in tdp1Δ wss1Δ suppression. Tetrad analysis of [TDP1/tdp1Δ; WSS1/wss1Δ; SIZ1/siz1Δ; SIZ2/siz2Δ] and [TDP1/tdp1Δ; WSS1/wss1Δ; MMS21/mms21-11] diploids. Representative spores (except mms21-11) were grown on the same plate.
(B) Siz1, Siz2, and Mms21 protein levels in ulp1-ΔN. Cells expressing E3 SUMO ligases genomically tagged with 13Myc at their C terminus were subjected to immunoblotting (IB). For quantification, anti-Myc signal was normalized to Pgk1 and then to WT.
(C) Decreased Siz2 protein levels in ulp1-ΔN are essential to rescue tdp1Δ wss1Δ. Strains were grown on SC-leu to select for plasmids expressing Siz2-13Myc (+) or p415 empty vector (−) and imaged after 72 h. See also Figure S2B for Siz2 protein levels.
(D) DNA damage checkpoint activation measured by Rad53 phosphorylation (gel shift on anti-Rad53 immunoblot).Where indicated, 1 mM auxin was added for 6 h. The anti-Pgk1 control was run in parallel on a different gel.
(E) Cell-cycle progression of the yeast samples as in (D) monitored by fluorescence-activated cell sorting (FACS) analysis.
(F) CPT sensitivity of tdp1Δ, wss1Δ, and siz2Δ in the 12geneΔ0HSR multi-transporter mutant genetic background (Chinen et al., 2011). Yeast strains were grown on CPT-containing or control YEPD and imaged 72 h post-plating.
Since the tdp1Δ wss1Δ strain rapidly accumulates suppressor mutations, we used the auxin-inducible Tdp1 depletion in most of subsequent experiments with the double mutant. As expected, the additional ulp1-ΔN or siz2Δ mutations suppressed tdp1-AID wss1Δ in the presence of auxin (Figure S2D). The slow-growth phenotype of tdp1Δ wss1Δ is associated with hyperactivation of the DNA damage checkpoint and the G2/M cell-cycle arrest. Inactivation of the checkpoint signaling is not sufficient to rescue the growth defect (Stingele et al., 2014), suggesting that the elevated checkpoint does not promote cell death but simply reflects accumulation of unrepaired Top1ccs in tdp1Δ wss1Δ. We therefore reasoned that the rescue of the tdp1Δ wss1Δ growth defect by ulp1-ΔN or siz2Δ must be due to a decrease in Top1cc levels. Consistent with this hypothesis, auxin treatment activated the checkpoint response in tdp1-AID wss1Δ, while the additional ulp1-ΔN or siz2Δ mutations diminished its levels, as measured by the gel shift caused by Rad53 phosphorylation (Figure 2D). Moreover, both siz2Δ and ulp1-ΔN rescued the G2/M-arrest observed in tdp1-AID wss1Δ cells upon auxin treatment (Figure 2E). Based on these observations, we surmise that sumoylation mutants promote more efficient Top1cc repair in tdp1-AID wss1Δ.
Genetic interactions of Siz2 and Top1cc repair factors
The cell wall of canonical laboratory budding yeast strains is poorly permeable to CPT. As a result, CPT fails to impair the growth of individual tdp1Δ, wss1Δ, as well as double siz2Δ wss1Δ and siz2Δ tdp1Δ mutants even at high concentrations (Figure S2C). We previously showed that the 12geneΔ0HSR multi-transporter mutant (Chinen et al., 2011) reveals weak sensitivity of wss1Δ to low dosage of CPT (Serbyn et al., 2020; Figure 2F). In the same genetic background, tdp1Δ and siz2Δ single mutants were also weakly sensitive to CPT (Figures 2F; 1.5 μg/μL CPT). We reasoned that the 12geneΔ0HSR genetic background could reveal synthetic genetic interactions of wss1Δ, tdp1Δ, and siz2Δ in the presence of CPT. Consistent with the near-unviable phenotype of tdp1Δ wss1Δ in a canonical W303 yeast genetic background (Stingele et al., 2014) (Figure S2A), we could not grow tdp1Δ wss1Δ clones in 12geneΔ0HSR in the absence of CPT. We next asked whether siz2Δ similarly suppresses tdp1Δ wss1Δ. The triple mutant has normal fitness in the absence of drugs but, unlike in W303, is sensitive to CPT (Figure 2F), indicating that siz2Δ tdp1Δ wss1Δ is only partially proficient for Top1cc repair. The12geneΔ0HSR background allowed to evaluate the pairwise genetic interactions of siz2Δ. The siz2Δ tdp1Δ mutant showed a pronounced growth defect on CPT, whereas siz2Δ wss1Δ had little additional CPT sensitivity, as compared to the respective single mutants (Figure 2F). These results argue that Siz2 acts independently of Tdp1 but might control the same aspects of Top1cc processing as Wss1. It is worth emphasizing that loss of Wss1, but not Siz2, is nearly synthetic lethal with tdp1Δ, implicating that Siz2 might control Wss1 but is not essential for its function.
Sumoylation changes in ulp1-ΔN and siz2Δ
Since elevated levels of Top1-DNA crosslinks underlie the tdp1Δ wss1Δ severe synthetic sickness (Stingele et al., 2014), and since Top1 is a known substrate for SUMO modification in budding yeast (Balakirev et al., 2015; Chen et al., 2007), we reasoned that SUMO might directly modulate Top1cc processing. It was reported previously that double E3 SUMO ligase mutants siz1Δ mms21-sp and siz1Δ siz2Δ, but not siz2Δmms21-sp, strongly reduce Top1-SUMO conjugates (Chen et al., 2007). To evaluate the sumoylation status of Top1, we tagged the sole essential SUMO-encoding SMT3 gene with 6His-FLAG, allowing the isolation of sumoylated proteins by Ni-NTA beads. One possibility is that Top1-SUMO accumulates in tdp1Δ wss1Δ cells while ulp1-ΔN or siz2Δ mutations reduce Top1-SUMO levels. Contrary to this model, the Top1-13Myc signal in the sumoylated protein fraction is not increased in tdp1-AID wss1Δ + auxin as compared to WT (Figure 3A). Moreover, ulp1-ΔN and siz2Δ do not have the same effect on Top1 sumoylation; the ulp1-ΔN mutation increases Top1-SUMO levels, whereas siz2Δ has no appreciable effect (Figure 3A).
Figure 3. Top1 sumoylation in ulp1-ΔN and siz2Δ.

(A) Relative levels of sumoylated Top1 evaluated by the SUMO assay. SMT3 (gene coding for yeast SUMO) and TOP1 were tagged at genomic loci. Asterisks indicate unmodified Top1-13Myc non-specifically bound to Ni-NTA beads. Tdp1-AID*- HA was depleted by 6 h auxin treatment (note: Tdp1 is 6HA-tagged in tdp1-AID background only).
(B) Sumoylation status of Top1-13Myc or Top1-4KR-13Myc mutants. The SUMO assay was performed as in (A). TOP1-13Myc or top1-4KR-13Myc (K65R, K91R, K92R, K600R) replace the genomic copy of TOP1.
(C) CPT sensitivity of non-sumoylated Top1 mutants. Spot assays were plated on YEPD media and imaged 48 h post-plating.
To further clarify the role for Top1-SUMO species in tdp1-AID wss1Δ suppression, we used the mutant that abrogates Top1 sumoylation. Lysines K65, K91, and K92 were previously implicated in bulk Top1 sumoylation (Chen et al., 2007). A recent study reported that combination of K65R, K91R, and K92R with an additional K600R lysine substitution to arginine partially suppresses the tdp1Δ wss1Δ growth defect (Sharma et al., 2017); we used this mutant, further referred to as top1-4KR, to test the importance of Top1 sumoylation. As expected, top1-4KR markedly decreased Top1 sumoylation in WT and mutant genetic backgrounds (Figure 3B). However, top1-4KR neither improved the growth of tdp1-AID wss1Δ and its resistance to CPT nor affected the suppression effects of ulp1-ΔN and siz2Δ (Figure 3C). Collectively, these results indicate that Top1 is not a Siz2 SUMO ligase target and argue against the model that Top1 sumoylation is responsible for the growth defect of tdp1Δ wss1Δ.
As we did not find any role for Top1-SUMO, we attempted to identify other candidate SUMO targets by quantitative SUMO proteomics (Albuquerque et al., 2013). Global sumoylation changes were examined in two sets of mutants, (1) tdp1-AID wss1Δ (+) auxin versus (−) auxin and (2) ulp1-ΔN tdp1-AID wss1Δ (+) auxin versus tdp1-AID wss1Δ (+) auxin, as summarized in Figure S3A. We were especially interested in the decrease of sumoylation in ulp1-ΔN, as these changes could have resulted from Siz2 degradation. Whereas number of proteins changed their sumoylation status (Figures S3B and S3C; Data S2), we did not identify a strong candidate that might have explained the observed suppression by ulp1-ΔN (see Figure S3 for details). We tested a few known SUMO target proteins, including Cdc14 and PCNA (Figures S4A and S4B), but none of them significantly impacted Top1cc repair or mimicked the suppression by ulp1-ΔN or siz2Δ.
SUMO levels at the DPC site modulate the speed of DPC repair
Siz2 sumoylates multiple proteins involved in DNA replication and repair in response to certain genotoxin treatments (Cremona et al., 2012; Psakhye and Jentsch, 2012). If Top1cc also induces multiple sumoylation events at the damage site, the synthetic rescue effects of ulp1-ΔN and siz2Δ may not be attributable to a unique substrate. To test this idea, we used the Flp-nick system that generates a site-specific DPC bound to DNA through a chemical bond identical to Top1cc (Nielsen et al., 2009). In this system, galactose induces expression of pGAL10-flp-H305L-3HA, and the produced protein is targeted to the Flp recognition target (FRT) sequence. The H305L mutation prevents the resolution of a recombination intermediate, leaving a covalent crosslink between Flp and DNA, or Flp-cc (Figure 4A). The top1Δ mutation allows to study Flp-nick defects independently of the Top1cc-related phenotypes. We have previously shown that tdp1Δ wss1Δ top1Δ cells are sensitive to Flp-cc induction, as reflected by the growth defect on galactose (Serbyn et al., 2020) (Figure 4B). Both ulp1-ΔN and siz2Δ mutations rescued this growth defect (Figure 4B), confirming that suppression by these SUMO pathway mutants is recapitulated in the Flp-nick system. Consistent with our hypothesis, chromatin immunoprecipitation targeting SUMO showed that Flp-cc induction was accompanied by a local increase in sumoylation as a function of proximity to the DPC-containing FRT locus (Figure 4C, compare raffinose and galactose). The tdp1Δ wss1Δ top1Δ mutant accumulated even higher SUMO levels in galactose, and the increase was only observed in the presence of the FRT-binding site (Figure 4C). Remarkably, additional ulp1-ΔN and siz2Δ mutations significantly decreased the SUMO signal at the FRT locus (Figure 4C).
Figure 4. Excessive sumoylation by Siz2 in the vicinity of the DPC delays Flp-cc repair.

(A) Schematic representation of the Flp-nick system described in Nielsen et al. (2009). Expression of the pGAL10-flp-H305L construct is induced by galactose; the mutant Flp recombinase is targeted to the FRT locus artificially introduced 2.7 kb upstream of the ARS607 replication origin into chromosome VI; the system generates a DPC (Flp-cc) on the nicked DNA. Yeast cells are devoid of 2μ plasmid, a natural substrate of the Flp recombinase.
(B) ulp1-ΔN and siz2Δ mutations rescue growth defects caused by Flp-nick induction in tdp1Δ wss1Δ top1Δ. Cells were pre-grown in YEP-2% raffinose medium prior to plating on SC-2% glucose or SC-2% galactose plates. Images were taken 60 h post-plating.
(C) Sumoylation of the FRT locus. Non-synchronous yeast cultures were either grown in YEP + 2% raffinose or additionally induced with 3% galactose for 2 h. Total SUMO (6His-Flag-Smt3) was immunoprecipitated with an anti-FLAG antibody; ChIP-qPCR signals at the FRT locus (relative to intergenic) are plotted as the mean ± SEM of three to six replicates. ***p < 0.001; ****p < 0.0001; ns, non-significant (compared to WT, two-way ANOVA, Tukey’s multiple comparison test).
(D) Ubiquitination of the FRT locus. Ubiquitin antibody was used for chromatin immunoprecipitation experiment, performed and analyzed as in (C).
(E) Experimental design of Flp-nick induction and repression in different cell-cycle stages. raf, raffinose; gal, galactose; glu, glucose; α-f, alpha factor; ns, non-synchronous. Bottom: cell-cycle analysis of WT-like (Flp-nick bar1Δ) strain monitored by FACS. See Figure S5C for FACS analyses of other mutants.
(F) Kinetics of Flp-cc repair in different mutants. Yeast strains were grown as shown in (E). In addition to indicated mutations, all strains are in bar1Δ Flp-nick genetic background. Flp-H305L-3HA levels at FRT were monitored by ChIP without crosslink using an anti-HA antibody. qPCR primers map 0.2 kb downstream of FRT. Data are presented as the mean ± SEM of five independent replicates. The percentage of input for each genotype was normalized to maximum galactose induction. See also Figure S5A for relative Flp-H305L-3HA levels after maximum induction with galactose.
We detected significant accumulation of ubiquitin at the vicinity of FRT in tdp1Δ wss1Δ top1Δ upon Flp-nick induction (Figure 4D). Similar to SUMO, ulp1-ΔN or siz2Δ mutations reverted ubiquitin increase (Figure 4D), indicating that ubiquitination of the FRT locus is SUMO dependent. Therefore, we next tested the role of SUMO-dependent ubiquitin ligases Slx5 or Slx8. The slx5Δ mutation had no impact on SUMO levels in tdp1Δ wss1Δ top1Δ (Figure 4C), whereas a striking decrease in ubiquitin signal was seen in STUbL mutants (Figure 4D). STUbL mutants slx5Δ and slx8Δ are very weak genetic suppressors of tdp1Δ wss1Δ phenotypes (Figures S1C, S1D, and S1F) (Sharma et al., 2017), suggesting that excessive SUMO rather than ubiquitin is a crucial toxic mark in the proximity of the DPC that inhibits repair in the absence of Tdp1 and Wss1.
If the excessive sumoylation at the FRT locus is inhibitory for repair, then ulp1-ΔN or siz2Δ should accelerate the kinetics of Flp-cc turnover. To test this possibility, cells were synchronized by α-factor in G1, and then Flp-H305L-3HA expression was induced by galactose, followed by the simultaneous glucose repression and release into the cell cycle (Figure 4E). The levels of Flp-H305L-3HAcrosslinked to FRT were assessed in galactose (maximum induction in G1), after 30 min glucose release (corresponding to G1/early S), or 2 h glucose release (G2) (Figure 4F). Because starting Flp-cc levels in galactose considerably differed between the mutants (Figure S5A), the results were presented as the percentage of remaining signal relative to maximum galactose induction. As reported before (Serbyn et al., 2020), tdp1Δ wss1Δ top1Δ slows down the speed of Flp-cc repair. Shortly after Flp-cc transcriptional repression by glucose, the additional siz2Δ or ulp1-ΔN mutations show comparable or even higher levels of Flp-H305L-3HA that remained crosslinked to DNA (Figure 4F, left). Strikingly, however, after 2 h of glucose release, both siz2Δ and ulp1-ΔN visibly accelerated Flp-cc repair in tdp1Δ wss1Δ top1Δ (Figure 4F, right). Note that, while siz2Δ improved Flp-cc processing, the repair remained slower than in WT-like strain, consistent with the genetic data (Figure 2F). From this, we concluded that SUMO pathway mutants promote more efficient DPC repair of a site-specific DPC in cells lacking Tdp1 and Wss1.
We noted that the 30-min time point corresponds to G1 cell-cycle stage (Figures 4E and S5C), suggesting that SUMO may inhibit repair only upon S-phase entry. We therefore asked how G1 arrest would impact the kinetics of Flp-cc repair in different mutants. As before, Flp-cc was induced in α-factor-synchronized cells, but this time, cells were retained in G1 after shift to glucose (Figures S5B and S5C). Both WT and tdp1Δ wss1Δ top1Δ cells were proficient for Flp-cc repair even in the absence of replication, albeit with slower kinetics as compared to non-arrested cells (Figure S5D). In G1-arrested cells, siz2Δ tdp1Δ wss1Δ top1Δ (but not ulp1-ΔN tdp1Δ wss1Δ top1Δ) maintained the ability to accelerate Flp-cc repair (Figure S5D), arguing against the hypothesis that excessive sumoylation is only deleterious for cells progressing through S and/or G2 phases. The lack of ulp1-ΔN-dependent suppression could reflect the additional phenotypes of the ulp1 mutant other than Siz2 degradation.
We addressed next how the cell-cycle stage impacts SUMO dynamics at FRT. Remarkably, in G1 phase, SUMO species were turned over more efficiently in WT-like cells, but not in the tdp1Δ wss1Δ top1Δ mutant (Figure S5E). Together, the above results suggest that sumoylation promotes DPC processing in G1, and this at least partially depends on Wss1 or Tdp1.
Alternative Top1cc repair pathways function in tdp1Δ wss1Δ siz2Δ
Ulp1- and Siz2-dependent suppression of tdp1Δ wss1Δ implies that the loss of sumoylation activates alternative repair pathways that cope with Top1ccs. To identify those pathways, a SATAY transposon screen was performed on tdp1Δ wss1Δ siz2Δ, as shown in Figure S6A. The transposon coverage in the mutant tdp1Δ wss1Δ siz2Δ SATAY library was compared to a pool of several libraries WT for these genes (Figure 5A) and (Data S3). Since TDP1, WSS1, and SIZ2 were deleted and therefore could not be targeted by transposons, these genes appeared as top hits in the screen. Potential synthetic lethal genetic interactors of tdp1Δ wss1Δ siz2Δ are highlighted on the volcano plot (Figure 5A). Among them, several known Top1ccs- and DNA double-strand break repair factors were found, which are analyzed next.
Figure 5. A genetic screen uncovers factors essential for tdp1Δ wss1Δ siz2Δ survival.

(A) A volcano plot summarizing results of the SATAY transposon screen of tdp1Δ wss1Δ siz2Δ. The fold change in the number of transposons per yeast gene body in tdp1Δ wss1Δ siz2Δ library was obtained by comparison to a pool of six unrelated libraries (log2, x axis). Respective p values (−log10, y axis) are plotted. Selected DNA damage repair factors are highlighted in colors. For validations and additional genetic analyses, see Figure S6 and Tables S1 and S2.
(B) Genetic interaction and CPT sensitivity of ddi1Δ and tdp1Δ, wss1Δ, siz2Δ. Yeast were spotted on control or CPT-containing YEPD plates and imaged after 48 h of growth.
(C) Loss of DDI1 delays Flp-cc repair kinetics. Flp-H305L-3HA levels at FRT monitored by ChIP-qPCR as in Figure 4F. The graph shows means ± SEM of three to five replicates; *p < 0.05; ***p<0.001 (two-way ANOVA, Tukey’s multiple comparison test).
(D) Ddi1 levels at the FRT locus after Flp-cc induction. Ddi1-TAP recruitment was monitored by ChIP-qPCR. Experiment was performed as in Figure 4E, except that cells were not synchronized with alpha factor prior to and during Flp induction. Data are analyzed and plotted as in (C).
Several structure-specific endonucleases are thought to eliminate Top1ccs from DNA in the absence of the direct Top1-DNA bond cleavage by Tdp1 (Sun et al., 2020b). Consistently, the genes encoding the Rad27 and Rad1 nucleases were among the least targeted in the tdp1Δ wss1Δ siz2Δ SATAY screen (Figure 5A). To further test the importance of structure-specific nucleases, we deleted RAD27 or MRE11 or disrupted several other nuclease complexes found in the screen or previously implicated in Top1cc processing (Rad1-Rad10, Slx4-Slx1, and Mus81-Mms4). Most of these mutations modestly affected growth and CPT resistance of the tdp1Δ wss1Δsiz2Δ strain (summarized in Tables S1 and S2). In contrast, RAD27, a gene that encodes a flap nuclease with a dual role in DNA replication and DNA damage repair, was essential for tdp1Δ wss1Δ siz2Δ viability (Figure S6B). Combination of individual or double tdp1Δ, wss1Δ, or siz2Δ mutations with rad27Δ was not detrimental for cell growth. These data suggest that Rad27 is indispensable for Top1cc repair when Tdp1-, Wss1-, and Siz2-dependent pathways are not accessible.
DNA helicases are known to play important roles in diverse DNA repair pathways, including the removal of DPCs. A number of them were identified in the SATAY screen (Figure 5A). The ReqQ helicase Sgs1 (BLM and WRN in mammals) works with Top3 to resolve Holliday junctions and promote the restart of replication forks collapsed at Top1ccs (Pichierri et al., 2000; Vance and Wilson, 2002). The sgs1Δ mutation was previously reported to be deleterious in wss1Δ (Mullen et al., 2010); loss of this helicase in tdp1Δ wss1Δ siz2Δ did not cause additional growth defects as compared to sgs1Δ wss1Δ (Figure S6C). Another helicase identified in the screen, the Srs2 anti-recombinase, is recruited to the site of damage via interactions with sumoylated PCNA (Pfander et al., 2005) and disrupts Rad51 nucleoprotein filaments formation (Krejci et al., 2003). Interestingly, the combination of srs2Δ and siz2Δ alone was deleterious in the presence of the Top1cc-inducing drug CPT, a phenotype reflected also in the context of tdp1Δ wss1Δ siz2Δ (Figure S6D). These findings suggest an involvement of these helicases in processing Top1ccs in tdp1Δ wss1Δ siz2Δ, confirming the findings from the SATAY screen.
The DNA damage checkpoint was shown to play a role in the response to CPT-induced DNA damage (Pommier et al., 2006; Simon et al., 2000). Consistent with this idea, the 9-1-1 complex (Ddc1, Rad17, and Mec3) and its loader, Rad24, were all identified in the SATAY screen (Figure 5A). Further genetic analysis showed that the rad17Δ and rad9Δ checkpoint response mutants both sensitize tdp1Δ wss1Δ siz2Δ to CPT, although they did not appreciably affect the growth property of this triple mutant (Figures S6E and S6F).
Ddi1 protease promotes earlier Flp-cc repair
The Ddi1 protease was previously implicated in yeast DPC processing (Serbyn et al., 2020), and this gene was another top candidate in our screen (Figure 5A). We further found that Ddi1 is not essential for tdp1Δ wss1Δ siz2Δ survival but supports CPT resistance of the mutant (Figure 5B). Consistently, repair of the Flp-cc after 2 h of glucose release is delayed, but not fully abrogated, in the absence of the DDI1 gene (Figure 5C). In the absence of Tdp1 and Wss1, Ddi1 could be detected in the proximity of the FRT locus (Serbyn et al., 2020), so we tested how sumoylation loss might impact the kinetics of Ddi1 recruitment. We measured Ddi1-TAP levels at the FRT locus in non-synchronous cells prior to Flp-cc induction (raf), at maximum induction of Flp (gal), or after transcriptional repression of Flp-H305L-3HA (glu) (Figure 5D). In the absence of Siz2, Ddi1 was recruited to the FRT locus earlier than in wss1Δ tdp1Δ top1Δ (Figure 5D, 30 min release). After 2 h release, Ddi1-TAP levels further increase in tdp1Δ wss1Δ top1Δ, whereas the signal was no more detected in tdp1Δ wss1Δ top1Δ siz2Δ (Figures 5D, 2 h release). These results recapitulate faster processing kinetics of Flp-cc in tdp1Δ wss1Δ top1Δ siz2Δ (Figure 4F), further supporting that Ddi1 may function as a backup Top1cc proteolysis pathway. Since Ddi1 was recently described as a ubiquitin-dependent protease (Yip et al., 2020), we checked ubiquitin levels in the same conditions but could not correlate Ddi1 and ubiquitin increase in tdp1Δ wss1Δ top1Δ siz2Δ (Figure S6G). These data argue that Ddi1 recruitment does not depend on Slx5/8-dependent ubiquitination of the Flp-cc locus.
Taken together, multiple DNA repair and checkpoint pathways play a role in improving the growth of tdp1Δ wss1Δ siz2Δ and its response to CPT-induced DPCs. They include endonucleases, helicases, the Ddi1 protease, and the DNA damage checkpoint genes. Thus, reducing sumoylation at DPCs in the tdp1Δ wss1Δ mutant appears to trigger the activation of an extensive network of DNA repair and DNA damage signaling pathways that collectively counter the toxic effect of DPCs.
DISCUSSION
DPC adducts, including Top1ccs, are formed during normal cell metabolism but do not threaten cell survival, owing to multiple DPC repair mechanisms involved in their elimination. The redundancy in such DNA repair pathways becomes especially important when cells are exposed to diverse industrial, household, and environmental agents or chemotherapeutic drugs that induce DPCs. In this study, we show that SUMO helps to create a balance between the redundant Top1cc repair pathways in budding yeast. We reveal that the E3 ligase Siz2 conjugates SUMO in the proximity of a damaged site. Our genetic findings suggest that SUMO stimulates repair by promoting the recruitment of the DPC protease Wss1. At the same time, we show that excessive Siz2-dependent SUMO conjugation inhibits alternative repair mechanisms and additionally identified at least some of them in a genetic screen. How SUMO could cooperate with some or compete with other DPC repair mechanisms to coordinate them in time and space is discussed below.
Favorable effects of SUMO conjugation around the DPC locus
The cellular response to DPC-induced DNA damage can be divided into several steps: (1) DPC detection, (2) proteolysis, and (3) elimination of the remaining DNA damage (Figure 6). The SUMO signal increases in the vicinity of the DPC-containing locus upon damage formation (Figure 4C) and likely facilitates DPC detection (Figure 6, step 1). Conjugation of SUMO was previously documented in several organisms and model DPC adducts such as TOP1, TOP2, M.HpaII, and DNMT1 (Borgermann et al., 2019; Larsen et al., 2019; Liu et al., 2021; Mao et al., 2000a, 2000b; Schellenberg et al., 2017), suggesting a general response mechanism. SUMO could mark the covalently trapped protein directly and/or be conjugated to the other factors residing in close proximity to the DPC. This was observed for the Top1cc and Flp-cc models used in this study (Figures 3A and 4C) (Chen et al., 2007; Xiong et al., 2009) and seen previously for the trapped mammalian DNMT1 (Borgermann et al., 2019). Our data allow us to distinguish between the two events and suggest that SUMO conjugation by Siz2 around the DPC, but not to Top1, is detrimental for Top1cc repair in tdp1Δ wss1Δ. One interpretation of these data is that multiple SUMO conjugation events around a DPC create a sumoylated domain required to recruit relevant factors and establish a platform for efficient DNA repair. Accordingly, Siz2 sumoylates numerous DNA repair proteins in response to other genotoxin treatments such as methyl methanesulfonate (MMS) or zeocin (Chung and Zhao, 2015; Psakhye and Jentsch, 2012). Consistently, our SILAC-MS analysis and the subsequent candidate approach does not point to a single Siz2 target relevant for DPC repair (Figure S3).
Figure 6. A hypothetical model summarizing the interplay between PTMs and different DPC repair factors.

Topoisomerase 1 (Top1) is covalently trapped on DNA as a result of an aberrant enzymatic reaction, forming a DPC, or Top1cc. Top1cc and proximally located proteins are modified by SUMO (S) and ubiquitin (U). (1) The E3 SUMO ligases Siz1 and Mms21 facilitate Top1 sumoylation; Siz2 sumoylates proteins at the DPC lesion site. DPCs are additionally ubiquitinated in a SUMO-dependent manner (by the Slx5-Slx8 complex) or independently of SUMO (by unknown E3 ligases in yeast). (2) SUMO stimulates Wss1 recruitment; ubiquitin and mixed SUMO-Ub chains are recognized by Cdc48 (Wss1 partner); ubiquitin is also recognized by the alternative DPC proteases Ddi1 and 26S proteasome. Either of these three proteases allows the initial degradation of a protein trapped in DPC. (3) Partial or full Top1cc proteolysis opens access for Tdp1 cleavage (preferred pathway) or allows DNA cleavage by various structural endonucleases, such as Rad27, Rad1-Rad10, Slx1-Slx4, Mus81-Mms4, and Mre11. SUMO and ubiquitin may additionally influence the downstream DPC repair. Excessive sumoylation that persists in the absence of Wss1 and Tdp1, or when these factors become rate-limiting, will inhibit efficient Top1cc processing (not shown).
In line with this model, loss of SUMO acceptor lysines in Top1 has no or only a minor effect on Top1cc repair in tdp1Δ wss1Δ (Figures 3B and 3C). Of note, we did not observe the partial suppression of tdp1Δ wss1Δ by top1-4KR found in another recent study (Sharma et al., 2017). Here, we edited the genomic TOP1 locus, whereas Sharma et al. (2017) complemented top1Δ with Top1 expressed from a plasmid, which might explain the discrepancy with our results. Moreover, we cannot completely exclude the role of residual high-molecular-weight Top1-SUMO species detectable in all to date available KR mutants (Chen et al., 2007), raising the possibility that poly-sumoylation of Top1 plays some role in Top1cc removal.
What factor could SUMO recruit to the DPC locus? The Wss1 DPC protease is an obvious candidate, as it has strong SUMO-binding affinity indispensable for its in vivo proteolytic function in yeast (Balakirev et al., 2015; Mullen et al., 2010; Stingele et al., 2014). Emerging evidence suggests that SUMO controls the SprT family of proteases in higher eukaryotes. Although the Wss1 ortholog SPRTN lacks an obvious SIM domain, a recent preprint nevertheless reported that SUMO stimulates its proteolytic activity (Vaz et al., 2020). Another Wss1 ortholog, GCNA, has a SUMO-binding domain and is targeted to DPC sites in a SUMO-dependent manner (Borgermann et al., 2019; Dokshin et al., 2020). Consistently, our genetic data (Figure 2F) indicate that Wss1 and the E3 SUMO ligase Siz2 might control the same aspects of DPC processing. Despite our attempts, we could not detect Wss1 at the DPC-containing FRT locus, and whether in vivo SUMO helps to recruit yeast Wss1 requires additional clarification. Cdc48, a Wss1 partner essential for Top1cc repair, recognizes mixed SUMO-Ub signals through an adaptor protein (Nie et al., 2012); it could therefore also stimulate Wss1 recruitment to a sumoylated locus. All these observations collectively suggest that SUMO at the DPC locus is likely to promote Wss1 recruitment directly or via Cdc48 (Figure 6).
The Slx5-Slx8 STUbL is another complex that is directed by SUMO to DPC sites. STUbLs are described to facilitate Top1cc processing in budding and fission yeast, as well as in mammalian cells (Nie et al., 2017; Sharma et al., 2017; Sun et al., 2020a). We report here that DPC-containing sites undergo STUbL-dependent ubiquitination (Figure 4D). The Slx5-Slx8 STUbL promotes repair in a pathway parallel to Tdp1 (Figure S1F), as also observed in fission yeast (Nie et al., 2017). Mixed SUMO-Ub chains generated by the Slx5-Slx8 complex could, on the one hand, recruit Cdc48-Wss1 (Figure 6), as suggested by the epistatic relationship of wss1Δ with the tdp1Δ slx5Δ mutant (Figure S1F). On the other hand, ubiquitin could stimulate Top1cc degradation by 26S proteasome (Figure 6), as proposed for the mammalian Slx5-Slx8 functional ortholog RNF4 (Liu et al., 2021; Sun et al., 2020a).
Deleterious effects of Siz2-dependent SUMO conjugation on Top1cc processing
Wss1 is a low-abundance protein in yeast (Stingele et al., 2015); its availability could therefore be rate-limiting for DPC repair. When cells are challenged with an excess of DPCs, they may require an alternative DPC protease. Ddi1 and 26S proteasome could at least partially substitute for Wss1 function (Serbyn et al., 2020) (Figure 5C), and both of them target ubiquitinated proteins for degradation (Finley et al., 2012; Yip et al., 2020). Ubiquitin and SUMO are known to compete for the acceptor lysines (Hendriks et al., 2014). Thus, excessive sumoylation by Siz2, especially in tdp1Δ wss1Δ (Figure 4C), could block the access of alternative proteases to the lesion. In agreement with this model, Ddi1 helps to resolve Top1ccs and is detected earlier at the Flp-cc locus in tdp1Δ wss1Δ siz2Δ (Figures 5B and 5C). We only detected SUMO-dependent ubiquitination at Flp-cc, and this ubiquitination is not relevant for Ddi1 recruitment (Figures 5D and S6G). One explanation might be that ultra-long polyubiquitin chains required to stimulate Ddi1 proteolytic activity (Yip et al., 2020), unlike SUMO, are only conjugated to crosslinked proteins and therefore present in very low quantities that are not readily detectable by chromatin immunoprecipitation.
At least some repair pathways identified in the tdp1Δ wss1Δ siz2Δ transposon screen could assist Ddi1 or 26S proteasome in downstream damage processing (Figure 6). Structure-specific nucleases may be required to clear the peptide remnants, especially when Tdp1 is absent or not accessible and fails to perform direct digestion of the Top1-DNA bond. The identified nucleases and helicases are known to recognize and resolve aberrant replication and recombination intermediates that appear during or after DNA replication, consistent with our earlier observations that Ddi1 signal appears at the DPC locus in a replication-dependent manner (Serbyn et al., 2020). Moreover, almost every factor described above is a SUMO target itself (Sarangi and Zhao, 2015). Molecular effects of sumoylation greatly vary depending on the substrate and sometimes could be inhibitory for repair; we cannot exclude that excessive SUMO additionally hampers one or several downstream DPC repair steps.
Spatial and temporal control of DPC sumoylation
If several mechanisms compete for DPC repair, cells may have separated them in time and/or space. One possibility is that DPCs could be differently processed depending on the cell-cycle stage. For example, in a Xenopus cell-free system, non-replicating extracts accumulate SUMO and mixed SUMO-Ub chains that promote DPC resolution (Liu et al., 2021), whereas SUMO-independent ubiquitination is one of the earliest events accompanying replication-coupled DPC resolution (Larsen et al., 2019). Consistently, we observe SUMO enrichment at the DPC-containing locus that is turned over more rapidly in G1 than in the presence of replication (Figure S5E). One model is that SUMO helps to cope with low or moderate DPC levels in G1 by clearing them via STUbLs and proteasome (Borgermann et al., 2019; Sun et al., 2020a). Sumoylated non-processed DPCs that are carried to S phase will direct Wss1 for replication-coupled proteolysis typical for Wss1/SPRTN family (Reinking et al., 2020; Stingele et al., 2014). In the absence of Wss1 (as in tdp1Δ wss1Δ), SUMO and SUMO-dependent ubiquitination by STUbLs will inhibit repair by the alternative pathways. In WT cells, a similar scenario might occur when low-abundant Wss1 becomes rate-limiting for repair of excessive DPCs.
Intranuclear positioning is known to determine the fate of diverse damaged loci, including heterochromatic DSBs, collapsed replication forks, eroded telomeres, and repeated DNA sequences. SUMO stimulates the motion of the respective loci to the nuclear periphery to define the succeeding DNA repair pathway choice (Amaral et al., 2017; Géli and Lisby, 2015). We and others showed that proper NPC anchoring of the Ulp1 SUMO protease impacts Top1cc processing (Figures 1 and 2C) (Chen et al., 2007), suggesting that DPC positioning inside the nucleus may also be detrimental for damage processing. It remains to be determined whether hyper-sumoylation of DPCs could be directly reverted at the NPC by Ulp1 and how such an event may affect Top1cc processing.
In conclusion, the SUMO posttranslational modification tunes available alternative DPC repair pathways. Substantial additional work is required to understand all mechanistic details and identify the exact sumoylation event(s) capable of altering DPC processing. Given the evolutionary conservation of mechanisms involving the SUMO modification in DNA damage response, these new aspects of DPC biology are anticipated to be relevant for different domains of life. Last, the suppressive phenotypes of SUMO pathway mutants could help to understand the resistance to Top1 inhibitors often observed in chemotherapy.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Françoise Stutz (francoise.stutz@unige.ch)
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and code availability
Sequencing have been deposited at European Nucleotide Archive (ENA) repository and are publicly available as of the date of publication. Proteomics data have been deposited at the MassIVE ProteomeXchange Consortium Member and are publicly available as of the date of publication. This paper analyzes existing, publicly available sequencing data. These accession numbers for the datasets are listed in the key resources table. Original western blot and spot assay images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Secondary Goat Anti-Mouse Immunoglobulins/HRP, 1:5000 | Dako | P0447; RRID:AB_2617137 |
| Goat Anti-Rabbit Immunoglobulins/HRP, WB:1:5000 | Dako | P0448;: RRID:AB_2617138 |
| Anti-HA (anti-HA.11 Epitope Tag Antibody clone 16B12), WB:1:2000‘ | BioLegend | 901502, lot B276381 or B242905; RRID:AB_2565006 |
| Anti-Pgk1 antibody [22C5D8], WB:1:3000 | Abcam | ab113687, GR3195775-3; RRID:AB_10861977 |
| Anti-Rad53 (monoclonal, K-medium), WB:1:100 | Gift from Maksym Shyian (D. Shore laboratory) | Mab clone EL7.E1 |
| Anti-Flag M2, WB:1:5000 | Sigma | F3165, lot SLBQ7119V or SLCC4005: RRID:AB259529 |
| Anti-Myc, WB: 1:5000 | Abcam | Ab32; lot GR310953-2 or GR3232358-2; RRID:AB_303599 |
| Anti-Ubiquitin FK2 | Calbiochem | ST1200-100UG, lots 31671118, 3291266, 3443858; RRID:AB_2043482 |
| Anti-SUMO (polyclonal), WB: 1:10000 | Helle Ulrich laboratory: (Papouli et al., 2005) | N/A |
| Anti-PCNA (polyclonal), WB: 1:2500 | Helle Ulrich laboratory: (Papouli et al., 2005) | N/A |
| Bacterial and virus strains | ||
| E. coli DH5α | N/A | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| D(+)-Glucose | Carl Roth | Art.-Nr. HN063 |
| D(+)-Raffinose pentahydrate | Carl Roth | Art.-Nr. 5241.3 |
| D(+)-Galactose | USBiological | G1030 |
| Yeast Nitrogen Base | Qbiogene | 4027-532 |
| Bacto Peptone | BD Biosciences | Ref. 211677 |
| Yeast Extract | BD Biosciences | Ref. 212750 |
| Ammonium Sulfate | Carl Roth | Art.-Nr. 3746.1 |
| 5-Fluoroorotic Acid | Toronto Research Chemicals | F595000 |
| RNase A, PureLink, (20 mg/mL) | Invitrogen | 12091-021 |
| DpnII | NEB | R0543L |
| NlaIII | NEB | R0125L |
| T4 Ligase | Thermo Scientific | EL0011 |
| Linear Acrylamide | Ambion | AM9520 |
| Taq polymerase | NEB | M0267L |
| Salmon sperm ssDNA | Sigma-Aldrich | D1626 |
| PEG4000 | Carl Roth | Art.-Nr. 0156.1 |
| Zymolyase - 20T | Amsbio | 120491-1 |
| Phusion High-Fidelity DNA Polymerase | Thermo Scientific | F530L |
| Auxin (3-indoleacetic acid) | Sigma-Aldrich | I2886 |
| Camptothecin (CPT) | Lucerna-Chem | Cat. 0215973225 |
| WesternBright ECL HRP substrate | Advansta | K-12045 |
| WesternBright Sirius HRP substrate | Advansta | K-12043 |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 4693159001 |
| NEM (N-Ethylmaleimide) | Sigma-Aldrich | E1271 |
| Dynabeads Protein G for Immunoprecipitation | Invitrogen | 10009D |
| Dynabeads Pan Mouse IgG | Invitrogen | 11041 |
| Alpha Factor | PRIMM | 201307-00007 |
| Trypsin | Promega | V5113 |
| Propidium Iodide | Sigma | P4170 |
| Proteinase K | Carl Roth | 7528.3 |
| Ni-NTA agarose | QIAGEN | 30210 |
| Critical commercial assays | ||
| The Bio-Rad protein assay | Bio-Rad | 500-0006 |
| Phire Green Hot Start II PCR Master Mix | Thermo Scientific | F126L |
| NEBuilder® HiFi DNA Assembly Cloning Kit | NEB | E5520 |
| NucleoSpin® Gel and PCR Clean-up | Macherey-Nagel | Ref. 740609.250 |
| MiSeq Reagent Kit v3 | Illumina | MS-102 |
| SYBR Select Master Mix for CFX | Applied biosystems | 4472942 |
| MinElute PCR Purification Kit | QIAGEN | 28006 |
| Deposited data | ||
| Sequencing data of the tdp1Δ wss1Δ siz2Δ transposon screen | This Manuscript | ENA: PRJEB47156 |
| Sequencing data of “wss1D Tdp1-AID*-6HA TIR1 + auxin” transposon screen | Serbyn et al. (2020), Mol.Cell | ENA: PRJEB31382 |
| The SUMO mass spectrometry proteomics data | This Manuscript | MassIVE: MSV000088129 |
| Unprocessed imaging (immunoblots, spot assays, tetrad analyses) and FACS data | This Manuscript | Mendeley Data: https://data.mendeley.com/datasets/h7bxnynd7s/1 |
| Experimental models: Organisms/strains | ||
| S. cerevisiae: Strain background: W303 | EUROSCARF | BMA64 |
| S. cerevisiae: Strain background: BY4741 | EUROSCARF | Y00000 |
| Oligonucleotides | ||
| See Table S3 for the list of oligonucleotides | This Manuscript | N/A |
| Recombinant DNA | ||
| See Table S3 for full list of plasmids | This Manuscript | N/A |
| Software and algorithms | ||
| Prism 9.1.2 | GraphPad Software, Inc | RRID:SCR_002798 |
| RStudio Version 1.1.456 | RStudio, Inc | RRID:SCR_000432 |
| Fiji | ImageJ | RRID:SCR_002285 |
| SnapGene 3.2.1 | GSL Biotech LLC | RRID:SCR_015052 |
| Kaluza | Beckman Coulter | RRID:SCR_016182 |
| Other | ||
| 0.5 mm Glass Beads | BioSpec Products | Cat. No 11079105 |
| MagNA Lyser Instrument | Roche | 03358976001 |
| Bioruptor Twin | Diagenode | UCD-400 |
| Gallios 8 colors 2 Lasers flow cytometer | Beckman Coulter | B43619 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Saccharomyces cerevisiae
Yeast strains
Saccharomyces cerevisiae yeast strains were derived from W303 [leu2-3,112; trp1-1; can1-100; ura3-1; ade2-1; his3-11,15] or S288C (BY4741) [his3Δ1; leu2Δ;0 lys2Δ0; met15Δ0; ura3Δ0; MATa] genetic backgrounds. An exhaustive list of strains with respective genotypes is available in Table S3. Additional details of strain construction are available on request.
Yeast growth media
Yeast cultures were grown at 30°C in YEP- or SC- based liquid media or plates supplemented with 20 g/l agar. 2% glucose (default), 2% raffinose, or 2%–3% galactose was used as a source of sugar. Selection against the URA3 marker was performed in the presence of 1 mg/ml of 5-Fluoroorotic acid (5-FOA). Selection for dominant markers was performed on YEPD-based medium supplemented with 200 μg/ml G418, 200 μg/ml clonNAT or 50 μg/ml Hygromycin B. Prototrophs were selected on SC media lacking the respective amino acids or nucleobases.
YEP medium: 1% yeast extract, 2% peptone.
YEPD medium: YEP, 2% glucose.
SC medium: 1.7 g/l yeast nitrogen base, 5 g/l ammonium sulfate, 2% agar, 2% glucose, 0.87 g/l dropout mix. Dropout mix composition: 0.8 g adenine, 0.8 g uracil, 0.8 g tryptophan, 0.8 g histidine, 0.8 g arginine, 0.8 g methionine, 1.2 g tyrosine, 2.4 g leucine, 1.2 g lysine, 2 g phenylalanine, 8 g threonine.
SD+2% galactose-adenine agar plates used for the transposon screen: 1.7 g/l yeast nitrogen base, 5 g/l ammonium sulfate, 2% agar, 2% galactose, 0.03 g/l isoleucine, 0.15 g/l valine, 0.02 g/l arginine, 0.02 g/l histidine, 0.1 g/l leucine, 0.03 g/l lysine, 0.02 g/l methionine, 0.05 g/l phenylalanine, 0.2 g/l threonine, 0.04 g/l tryptophan, 0.03 g/l tyrosine, 0.02 g/l uracil, 0.1 g/l glutamate, 0.1 g/l aspartate.
Escherichia coli
DH5α E. coli bacterial strains were grown at 37°C in LB medium or on LB-2% agar plates supplemented with 50 μg/ml of ampicillin for plasmid selection.
METHOD DETAILS
Yeast techniques
Construction of yeast strains
De novo mutations were introduced into yeast genomes by transformation. Oligonucleotides, template plasmid or genomic DNA used for PCR and transformation are listed in Table S3. Genome editing was verified by colony PCR and/or sequencing. An epitope tag insertion was additionally checked by immunoblotting. Mutants with multiple genomic modifications were obtained by genetic crosses. A complete list of all yeast strains (arranged by Figures) is available in Table S3.
Yeast transformation
To edit yeast genomic DNA or to transform plasmid DNA (pDNA), log-phase growing yeast cultures were resuspended in LiTE buffer (100 mM LiAc, 10 mM Tris pH 7.5, 1 mM EDTA) and mixed with 100 μg/ml salmon sperm ssDNA, 37.28% [w/v] PEG4000, and purified PCR fragment or pDNA. Cells were incubated for 1-2 h at 30°C, then supplemented with 6% DMSO and a heat shock was performed for 10 min at 42°C. Single colonies were isolated by growth on selective medium.
Genetic crosses and tetrad analysis
To perform a yeast genetic cross, two strains of opposite mating type were mixed and grown overnight on rich YEPD medium, selected for diploid markers on corresponding media, and sporulated for 4-5 days on KAC plates (20 g/l potassium acetate, 2.2 g/l yeast extract, 0.5 g/l glucose, 0.87 g/l dropout mix, 20 g/l agar, pH 7.0) until visible tetrads were formed. Asci were digested with 0.5 μg/ml zymolyase 20T, spores were separated by dissection and grown for 2-4 days on rich YEPD media. Genotyping was performed by replica-plating on selective auxotrophic or dominant markers (when available) or made by colony PCR. In some cases, diploids were transformed with pDNA prior to sporulation. The presence of pDNA in colonies formed from spores was tested by growth on selective markers. Tetrads presented in the same figure panels were grown on the same plate unless otherwise indicated.
Colony PCR
Genotyping of yeast and bacterial strains was performed using the Phire Green Hot Start II PCR Master Mix supplemented with the relevant oligonucleotides according to manufacturer’s recommendations.
Spot assay
10x serial dilutions of log-phase growing yeast cultures were spotted on agar plates supplemented with 1 mM auxin or indicated amounts of CPT. To induce pGAL10-Flp-H305L expression, cells were pre-grown in YEP + 2% raffinose and spotted on 2% glucose- or 2% galactose-containing media.
Flp-nick induction for ChIP
Induction of the pGAL10-flp-H305L or pGAL10-flp-H305L-3HA expression in the Flp-nick or Flp-nick-HA genetic backgrounds (see Table S3 for full genotypes) was performed essentially as described in Nielsen et al. (2009). Cells were pre-cultured overnight in YEP + 2% raffinose, then diluted to OD600 = 0.15-0.4 in fresh YEP + 2% raffinose media and grown for 3-4 h before galactose induction to enter log phase. Flp-H305L constructs were induced for 2 hours with 3% galactose that was directly added to YEP + 2% raffinose medium. For Flp-nick induction in G1, cells were synchronized for 1-2 h with 200 ng/ml α-factor prior to galactose induction; additional 100 ng/ml α-factor was re-added together with galactose. All Flp-nick strains used for G1 arrest contained the bar1Δ mutation. To remove galactose, two washes using 1/5 yeast culture volume of cold YEP medium were performed. Next, cells were re-suspended in YEP-2% glucose to repress pGAL1-flp-H305L (glucose release). For the glucose release in G1, 200 ng/ml α-factor was added to the YEP medium used for washes, as well as to YEP-2% glucose.
Construction of recombinant pDNA
Plasmid DNA was constructed either by standard digestion-based cloning or using NEB Builder HiFi DNA Assembly Master Mix. A list oligonucleotides and PCR templates used to generate products for cloning and genome editing is provided in Table S3. Additional details of pDNA construction and genomic DNA modification are available on request.
Flow cytometry analysis (FACS)
0.5-1 mL aliquots of cell cultures at OD600 = 0.3-1 were collected to monitor cell cycle progression by flow cytometry cell sorting analysis (FACS). Cultures were pelleted, re-suspended in 70% EtOH, and stored at 4°C for up to 1-2 weeks. To prepare samples for FACS, fixed cells were centrifuged at 3800 g for 2 min, washed once with 300 μL of NaCi buffer (50 mM NaCi, pH 7.2) and centrifuged for 10 min at 3800 g. Pellets were resuspended in 250 μL of NaCi buffer supplemented with 0.4 mg/ml RNaseA and incubated for 1-3 h at 37°C. DNA was stained with 25 μg/ml propidium iodide for 1 h at 37°C and sonicated 5 times for 5 s with a Bioruptor Twin. Flow cytometric analysis was performed in NaCi buffer using a Gallios flow cytometer. FACS profiles were analyzed by Kaluza software.
Chromatin immunoprecipitation (ChIP)
ChIP analysis was performed as detailed in (Serbyn et al., 2020). Flp-nick cultures were grown as described above. 100 mL of yeast cell culture at OD600 = 0.7-1.2 was fixed with 1% formaldehyde for 15 min, neutralized by 250 mM glycine for 5 min at room temperature, kept for 10 min or longer (up to 2 h) on ice, then pelleted, washed twice with cold 1x PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) and frozen in liquid nitrogen. Pellets were resuspended in FA lysis buffer (50 mM HEPES-KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate (w/v)) supplemented with freshly added inhibitor (1 tablet of mini EDTA-free protease inhibitor cocktail per 10 mL buffer) and 25 mM N-ethylmaleimide (NEM). Cells were mixed with 500 μL glass beads and disrupted in the MagNA Lyser Instrument (5 times for 30 s at 6000 rpm with 1 min pause) at 4°C. The soluble fraction was separated by centrifugation (30 min at 18000 g at 4°C) and further discarded. Chromatin pellets were resuspended in 1 mL of the fresh FA+inhibitor+NEM buffer. DNA was fragmented by sonication in the Bioruptor Twin, 20 cycles of 30 s – 30 s. Lysates were centrifuged for 15 min, 18000 g at 4°C to remove insoluble fraction. Protein concentration in the soluble fraction was defined by the Bio-Rad protein assay. Equivalent quantities of total protein (0.5-1.5 mg) were used for immunoprecipitation with 1 μL of anti-Flag antibody (SUMO ChIPs, to bind 6His-Flag-Smt3), 1 μL of anti-Ubiquitin antibody (Ubiquitin ChIPs), 1 μL of anti-HA (Flp-H305L-3HA ChIPs); 1/10 of each sample (input) was kept separately before mixing with antibody. Immunoprecipitations were performed overnight at 4°C followed by 3 h coupling to 25 μL of washed protein G Dynabeads. Ddi1-TAP was directly precipitated using 25 ul of washed Dynabeads Pan Mouse IgG (to directly bind Ddi1-TAP via Protein A). IPs were washed once with 500 μL of FA buffer, twice with FA-500 (50 mM HEPES-KOH, pH 7.5, 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate (w/v)), twice with Buffer III (10 mM Tris-HCl pH 8, 1 mM EDTA, 250 mM LiCl, 1% IGEPAL, 1% sodium deoxycholate (w/v)) and once with TE (50 mM Tris-HCl pH 7.5, 10 mM EDTA). Complexes were then separated from beads at 65°C by two 8 min sequential incubations with 100 μL of elution buffer B (50 mM Tris-HCl pH 7.5, 1% SDS, 10 mM EDTA). The volume of inputs was adjusted to 200 μL with TE buffer. Proteins in ChIP and input samples were digested with 0.75 mg/ml Proteinase K for 2 h at 42°C. Decrosslinking was performed for at least 12 h at 65°C. DNA was purified using either the MinElute PCR purification kit or the Nucleospin Gel and PCR clean up kit (using NTB) according to the manufacturer’s recommendations. DNA was eluted from the column twice with 30 μL sterile H2O or the elution buffer from a kit. 2 μL eluates were used for 10 μL qPCR reactions with SYBR Select Master Mix. Oligonucleotide pairs and respective concentrations are listed in Table S3 (FRT+0.2 kb, FRT+0.5 kb FRT+1 kb, intergenic). For all experiments, percent of inputs were calculated. Where indicated, percent of inputs were normalized to the intergenic signal.
ChIP without crosslink
Since the flp-H305L mutation covalently crosslinks Flp protein to DNA, the formaldehyde crosslinking step was omitted in Flp-H305L-3HA ChIP experiments. Instead, yeast cell cultures were spun, washed once with 1/5 volume of 1xPBS buffer and pellets were directly frozen in liquid nitrogen and stored at −80°C until further processing using the ChIP protocol described above. For Flp-H305L-3HA ChIPs, no NEM was added to the lysis buffer.
SATAY library generation
The SATAY transposon screen was performed as in (Michel et al., 2017). Generation of the tdp1-AID wss1Δ + auxin library (Figure 1) is described in (Serbyn et al., 2020). The tdp1Δ wss1Δ siz2Δ SATAY library was generated using the strain FSY7292 [tdp1Δ::HPH; wss1Δ::HIS3; siz2Δ::NATMX6; ade2D::KANMX6; MATa] as detailed in (Michel et al., 2017). Briefly, the pBK257-transformed yeast strain was grown in SC-ura+2% raffinose/0.2% glucose to saturation and plated on SD-adenine+2% galactose agar plates. The library was grown for 3 weeks, then yeast colonies were collected, pooled, diluted to an approximate concentration of 2.5x106 cells/ml in 2 l of SC+2%glucose-adenine medium, grown until saturation, pelleted and frozen. By our estimation, the original library contained approximately 2.2 x 106 clones. 500mg of yeast pellet was used to extract genomic DNA, digest with DpnII and NlaIII, circularize, and perform PCR with P5_MiniDs and MiniDs_P7 oligonucleotides. The DNA sequencing library contained equal amounts of DpnII- and NlaIII-digested DNA and was sequenced using the MiSeq v3 chemistry adding 3.4 μL of 100 μM 688_minidsSEQ1210 primer. The SATAY sequencing dataset of tdp1Δ wss1Δ siz2Δ generated during this study is available at European Nucleotide Archive (ENA) under ID code PRJEB47156 and could be accessed by the following URL: https://www.ebi.ac.uk/ena/browser/view/PRJEB47156
The sequencing data were analyzed as described in Michel et al. (2017). The coordinates and number of associated sequencing reads were saved as .bed and .wig files for uploading into a genome browser. The “read_per_gene” value (Figure 1A) takes into consideration the fact that abundant transposons mutants can be detected by multiple sequencing reads. The “tn” value (Figure 5B) counts number of positions per yeast gene body where transposons were inserted and is best suited to identify synthetic sickness genetic interactions (Michel et al., 2017).
Protein extraction
To extract proteins, 5-10 ODs of exponentially growing yeast cultures were fixed with 6.25% trichloroacetic acid, kept on ice for 10 min or more, pelleted, washed twice with 100% acetone and dried under vacuum. Dry pellets were resuspended in urea buffer (50 mM Tris-Cl pH 7.5, 5 mM EDTA, 6 M Urea, 1% SDS), mixed with 200 μL of 0.5 mm glass beads and homogenized in the MagNA Lyser 5 times for 45 s at 4°C. Lysates were then incubated for 10 min at 65°C and centrifuged for 10 min at 18000 g to remove foam. Lysates were mixed with 1.5x volume of sample buffer (3% SDS, 15% glycerol, 0.1 M Tris pH 6.8, 0.0133% bromophenol blue, 0.95 M 2-mercaptoethanol) and boiled for 10 min.
Immunoblotting
Protein extracts were resolved by SDS-PAGE using 6%–14% gels, transferred to nitrocellulose or PVDF membranes, blocked with 5% dry milk dissolved in TBS-T (150 mM NaCl, 20 mM Tris-HCl, 0.05% Tween, pH 7.4). Primary and secondary antibodies were diluted in TBS-T containing 5% dry milk as indicated in the Key Resource Table. Immunoblot signals were revealed with WesternBright ECL or Sirius HRP substrate; images were taken with the Li-COR Odyssey Imaging System and quantified using Gel Analyze tool of the ImageJ v. 2.1.0.
SUMO assay
To isolate SUMO proteins, the genomic copy of yeast SMT3 was N-terminally tagged with 6His-Flag. Exponentially growing yeast cultures were fixed with 5% trichloroacetic acid, cooled on ice for approximately 1 h, pelleted, washed twice with 100% acetone, dried under vacuum and stored at −80°C or processed immediately. Dry yeast pellets were re-suspended in 1 mL of freshly prepared G-buffer (100 mM sodium phosphate pH = 8, 10 mM Tris-HCl pH = 8, 6M guanidinium, 10 mM 2-mercaptoethanol, 0.1% Triton X-100, 5 mM MG132, 25 mM NEM, EDTA-free protease inhibitor cocktail), mixed with 500 μL of 0.5 mm glass beads and homogenized in the MagNA Lyser 5 times for 45 s at 4°C. Recovered lysates were centrifuged for 20 min, 18000 g at room temperature. The concentration of proteins in the soluble fraction was measured by the Bio-Rad protein assay. Total proteins (3-5 mg in 1 mL of buffer) were mixed with 80 μL of Ni-NTA agarose pre-washed twice with water and once with G-buffer and rotated on a wheel for 2 h at room temperature. After binding, Ni-NTA resins were washed once with 500 μL of G-buffer, three times with fresh U-buffer (8 M urea, 100 mM sodium phosphate pH = 6.4, 10 mM Tris-HCl pH = 6.4, 10 mM 2-mercaptoethanol, 0.1% Triton X-100, EDTA-free protease inhibitor cocktail). Bound proteins were next eluted from agarose beads by 5 min boiling with 40 μL 3x sample buffer (125 mM Tris-HCl pH = 6.8, 4% SDS, 286 mM 2-mercaptoethanol, 0.02 mg/ml bromophenol blue, 20% glycerol). To prepare input fractions, 40 μL of total cell lysates were diluted 10 times in water, supplemented with 10% trichloroacetic acid, incubated 30 min, centrifuged at room temperature for 30 min, 18000 g and re-suspended in sample buffer. Ni-NTA bound and input fractions were further analyzed by immunoblotting.
The sumoylation status of 6His-PCNA was evaluated as described previously (Davies and Ulrich, 2012). SUMO antibody was a gift from the Helle Ulrich lab (Papouli et al., 2005).
SILAC-MS of sumoylated proteins
The enrichment of sumoylated proteins in preparation for quantitative mass spectrometry (SILAC-MS) analysis was performed as previously described (Albuquerque et al., 2013) but with the following exceptions: each mutant strain was grown in 1-l of synthetic media containing either light or heavy stable isotope-labeled lysine and arginine until an OD600 of 0.3, at which point auxin (1 mM final concentration) was added to deplete Tdp1-AID*-6HA. Cells were harvested following 6 hours of auxin treatment and lysed under denaturing condition as previously described (Albuquerque et al., 2013). Following lysis, protein concentrations were determined by Bradford Reagent (Bio-Rad) and were normalized prior to mixing and subsequent purification of sumoylated proteins. Nanoflow LC-MS/MS analysis was performed on a Thermo Scientific Ultimate 3000 Nano-LC System and a Thermo Scientific Orbitrap Fusion Lumos mass spectrometer; acquired via NIH S10 OD023498. The mass spectrometry proteomics raw data to analyze global sumoylation changes in this study have been deposited to the ProteomeXchange Consortium Member MassIVE (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) with the dataset identifier MSV000088129. Data analysis for SILAC-labeled samples was performed as previously described, with the exception that all proteins were required to have a minimum of 3 unique peptides (Suhandynata et al., 2019). Data S2 contains the list of identified sumoylated proteins. A list of “DNA repair” genes (GO:0006281) was extracted from the R package “org.Sc.sgd.db,” v.3.7.0.
QUANTIFICATION AND STATISTICAL ANALYSIS
Prism 9 was used to generate graphs and quantify p values using statistical methods as indicated in the respective figure legends where “n” represents the number of biological replicates. Volcano plots from Figures 1A and 5A were generated with R Studio plotting Data S1 and Data S3 respectively.
Supplementary Material
Highlights.
SUMO controls elimination of Top1cc and Flp-cc DNA-protein crosslinks (DPCs)
Sumoylation around a DPC site, but not the crosslinked protein, modulates DPC repair
Siz2 sumoylates DPC sites and hampers repair in cells lacking Tdp1 and Wss1
Sumoylation triggers DPC ubiquitination and inhibits alternative repair pathways
ACKNOWLEDGMENTS
We thank Helle Ulrich for sharing the parental auxin degron strain, PCNA plasmid constructs, and anti-PCNA and anti-SUMO antibodies; Lotte Bjergbaek for the Flp-nick system; Mark Hochstrasser for Ulp1 constructs; Benoît Palancade for siz1Δ, siz2Δ, and mms21-11 strains; and Takeo Usui for the 12geneΔ0HSR strain. We are grateful for Geraldine Silvano’s technical assistance. We thank Maksym Shyian, Simon Boulton, Johannes Walter, Vincent Dion, Daria Gudkova, and all members of the Stutz laboratory for their comments, discussions, suggestions, and critical reading of the manuscript. This work was supported by funds from the Swiss National Science Foundation (grants 31003A 153331 and 31003A_182344 to F.S.) and the Canton of Geneva. The Kornmann lab is supported by a grant from the Swiss National Science Foundation (31003A_179549) and the Wellcome Trust (214291/Z/18/Z). R.T.S. was supported by a postdoctoral fellowship from the National Cancer Institute (T32 CA009523). H.Z. was supported by National Institutes of Health grants R01 GM116897 and S10 OD023498.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110034.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following references appear in the supplemental information: Costanzo et al. (2016) and Doe et al. (2002).
REFERENCES
- Albuquerque CP, Wang G, Lee NS, Kolodner RD, Putnam CD, and Zhou H (2013). Distinct SUMO ligases cooperate with Esc2 and Slx5 to suppress duplication-mediated genome rearrangements. PLoS Genet. 9, e1003670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral N, Ryu T, Li X, and Chiolo I (2017). Nuclear Dynamics of Heterochromatin Repair. Trends Genet. 33, 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakirev MY, Mullally JE, Favier A, Assard N, Sulpice E, Lindsey DF, Rulina AV, Gidrol X, and Wilkinson KD (2015). Wss1 metalloprotease partners with Cdc48/Doa1 in processing genotoxic SUMO conjugates. eLife 4, e06763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava V, Goldstein CD, Russell L, Xu L, Ahmed M, Li W, Casey A, Servage K, Kollipara R, Picciarelli Z, et al. (2020). GCNA Preserves Genome Integrity and Fertility Across Species. Dev. Cell 52, 38–52.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgermann N, Ackermann L, Schwertman P, Hendriks IA, Thijssen K, Liu JC, Lans H, Nielsen ML, and Mailand N (2019). SUMOylation promotes protective responses to DNA-protein crosslinks. EMBO J. 38, e101496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XL, Silver HR, Xiong L, Belichenko I, Adegite C, and Johnson ES (2007). Topoisomerase I-dependent viability loss in saccharomyces cerevisiae mutants defective in both SUMO conjugation and DNA repair. Genetics 177, 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinen T, Ota Y, Nagumo Y, Masumoto H, and Usui T (2011). Construction of multidrug-sensitive yeast with high sporulation efficiency. Biosci. Biotechnol. Biochem 75, 1588–1593. [DOI] [PubMed] [Google Scholar]
- Chung I, and Zhao X (2015). DNA break-induced sumoylation is enabled by collaboration between a SUMO ligase and the ssDNA-binding complex RPA. Genes Dev. 29, 1593–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, et al. (2016). A global genetic interaction network maps a wiring diagram of cellular function. Science 353, aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona CA, Sarangi P, Yang Y, Hang LE, Rahman S, and Zhao X (2012). Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the mec1 checkpoint. Mol. Cell 45, 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das BB, Antony S, Gupta S, Dexheimer TS, Redon CE, Garfield S, Shiloh Y, and Pommier Y (2009). Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA-PK. EMBO J. 28, 3667–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AA, and Ulrich HD (2012). Detection of PCNA Modifications in Saccharomyces cerevisiae. In DNA Repair Protocols, Bjergbæk L, ed. (Totowa, NJ: Humana Press; ), pp. 543–567. [DOI] [PubMed] [Google Scholar]
- Debéthune L, Kohlhagen G, Grandas A, and Pommier Y (2002). Processing of nucleopeptides mimicking the topoisomerase I-DNA covalent complex by tyrosyl-DNA phosphodiesterase. Nucleic Acids Res. 30, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai SD, Mao Y, Sun M, Li TK, Wu J, and Liu LF (2000). Ubiquitin, SUMO-1, and UCRP in camptothecin sensitivity and resistance. Ann. N Y Acad. Sci 922, 306–308. [DOI] [PubMed] [Google Scholar]
- Desai SD, Li TK, Rodriguez-Bauman A, Rubin EH, and Liu LF (2001). Ubiquitin/26S proteasome-mediated degradation of topoisomerase I as a resistance mechanism to camptothecin in tumor cells. Cancer Res. 61, 5926–5932. [PubMed] [Google Scholar]
- Doe CL, Ahn JS, Dixon J, and Whitby MC (2002). Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J. Biol. Chem 277, 32753–32759. [DOI] [PubMed] [Google Scholar]
- Dokshin GA, Davis GM, Sawle AD, Eldridge MD, Nicholls PK, Gourley TE, Romer KA, Molesworth LW, Tatnell HR, Ozturk AR, et al. (2020). GCNA Interacts with Spartan and Topoisomerase II to Regulate Genome Stability. Dev. Cell 52, 53–68.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxin JP, Dewar JM, Yardimci H, and Walter JC (2014). Repair of a DNA-protein crosslink by replication-coupled proteolysis. Cell 159, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielden J, Wiseman K, Torrecilla I, Li S, Hume S, Chiang SC, Ruggiano A, Narayan Singh A, Freire R, Hassanieh S, et al. (2020). TEX264 coordinates p97- and SPRTN-mediated resolution of topoisomerase 1-DNA adducts. Nat. Commun 11, 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D, Ulrich HD, Sommer T, and Kaiser P (2012). The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics 192, 319–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géli V, and Lisby M (2015). Recombinational DNA repair is regulated by compartmentalization of DNA lesions at the nuclear pore complex. BioEssays 37, 1287–1292. [DOI] [PubMed] [Google Scholar]
- Hendriks IA, D’Souza RC, Yang B, Verlaan-de Vries M, Mann M, and Vertegaal AC (2014). Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol 21, 927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie K, Tomida A, Sugimoto Y, Yasugi T, Yoshikawa H, Taketani Y, and Tsuruo T (2002). SUMO-1 conjugation to intact DNA topoisomerase I amplifies cleavable complex formation induced by camptothecin. Oncogene 21, 7913–7922. [DOI] [PubMed] [Google Scholar]
- Huang TH, Chen HC, Chou SM, Yang YC, Fan JR, and Li TK (2010). Cellular processing determinants for the activation of damage signals in response to topoisomerase I-linked DNA breakage. Cell Res. 20, 1060–1075. [DOI] [PubMed] [Google Scholar]
- Jacquiau HR, van Waardenburg RC, Reid RJ, Woo MH, Guo H, Johnson ES, and Bjornsti MA (2005). Defects in SUMO (small ubiquitin-related modifier) conjugation and deconjugation alter cell sensitivity to DNA topoisomerase I-induced DNA damage. J. Biol. Chem 280, 23566–23575. [DOI] [PubMed] [Google Scholar]
- Johnson ES (2004). Protein modification by SUMO. Annu. Rev. Biochem 73, 355–382. [DOI] [PubMed] [Google Scholar]
- Kanagasabai R, Liu S, Salama S, Yamasaki EF, Zhang L, Greenchurch KB, and Snapka RM (2009). Ubiquitin-family modifications of topoisomerase I in camptothecin-treated human breast cancer cells. Biochemistry 48, 3176–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Y, Machida Y, Palani S, Caulfield TR, Radisky ES, Kaufmann SH, and Machida YJ (2020). FAM111A protects replication forks from protein obstacles via its trypsin-like domain. Nat. Commun 11, 1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci L, Van Komen S, Li Y, Villemain J, Reddy MS, Klein H, Ellenberger T, and Sung P (2003). DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423, 305–309. [DOI] [PubMed] [Google Scholar]
- Larsen NB, Gao AO, Sparks JL, Gallina I, Wu RA, Mann M, Räschle M, Walter JC, and Duxin JP (2019). Replication-Coupled DNA-Protein Crosslink Repair by SPRTN and the Proteasome in Xenopus Egg Extracts. Mol. Cell 73, 574–588.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SJ, and Hochstrasser M (2003). The Ulp1 SUMO isopeptidase: distinct domains required for viability, nuclear envelope localization, and substrate specificity. J. Cell Biol 160, 1069–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CP, Ban Y, Lyu YL, Desai SD, and Liu LF (2008). A ubiquitin-proteasome pathway for the repair of topoisomerase I-DNA covalent complexes. J. Biol. Chem 283, 21074–21083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JCY, Kühbacher U, Larsen NB, Borgermann N, Garvanska DH, Hendriks IA, Ackermann L, Haahr P, Gallina I, Guérillon C, et al. (2021). Mechanism and function of DNA replication-independent DNA-protein crosslink repair via the SUMO-RNF4 pathway. EMBO J. 40, e107413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Mosqueda J, Maddi K, Prgomet S, Kalayil S, Marinovic-Terzic I, Terzic J, and Dikic I (2016). SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. eLife 5, e21491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Desai SD, and Liu LF (2000a). SUMO-1 conjugation to human DNA topoisomerase II isozymes. J. Biol. Chem 275, 26066–26073. [DOI] [PubMed] [Google Scholar]
- Mao Y, Sun M, Desai SD, and Liu LF (2000b). SUMO-1 conjugation to topoisomerase I: A possible repair response to topoisomerase-mediated DNA damage. Proc. Natl. Acad. Sci. USA 97, 4046–4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey RS, Flatten KS, Sieben CJ, Peterson KL, Baker DJ, Nam HJ, Kim MS, Smyrk TC, Kojima Y, Machida Y, et al. (2017). Spartan deficiency causes accumulation of Topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic Acids Res. 45, 4564–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AH, Hatakeyama R, Kimmig P, Arter M, Peter M, Matos J, De Virgilio C, and Kornmann B (2017). Functional mapping of yeast genomes by saturated transposition. eLife 6, e23570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo YY, Yu Y, Shen Z, and Beck WT (2002). Nucleolar delocalization of human topoisomerase I in response to topotecan correlates with sumoylation of the protein. J. Biol. Chem 277, 2958–2964. [DOI] [PubMed] [Google Scholar]
- Morawska M, and Ulrich HD (2013). An expanded tool kit for the auxin-inducible degron system in budding yeast. Yeast 30, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen JR, Chen CF, and Brill SJ (2010). Wss1 is a SUMO-dependent isopeptidase that interacts genetically with the Slx5-Slx8 SUMO-targeted ubiquitin ligase. Mol. Cell. Biol 30, 3737–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie M, Aslanian A, Prudden J, Heideker J, Vashisht AA, Wohlschlegel JA, Yates JR 3rd, and Boddy MN (2012). Dual recruitment of Cdc48 (p97)-Ufd1-Npl4 ubiquitin-selective segregase by small ubiquitin-like modifier protein (SUMO) and ubiquitin in SUMO-targeted ubiquitin ligase-mediated genome stability functions. J. Biol. Chem 287, 29610–29619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie M, Moser BA, Nakamura TM, and Boddy MN (2017). SUMO-targeted ubiquitin ligase activity can either suppress or promote genome instability, depending on the nature of the DNA lesion. PLoS Genet. 13, e1006776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen I, Bentsen IB, Lisby M, Hansen S, Mundbjerg K, Andersen AH, and Bjergbaek L (2009). A Flp-nick system to study repair of a single protein-bound nick in vivo. Nat. Methods 6, 753–757. [DOI] [PubMed] [Google Scholar]
- Palancade B, Liu X, Garcia-Rubio M, Aguilera A, Zhao X, and Doye V (2007). Nucleoporins prevent DNA damage accumulation by modulating Ulp1-dependent sumoylation processes. Mol. Biol. Cell 18, 2912–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papouli E, Chen S, Davies AA, Huttner D, Krejci L, Sung P, and Ulrich HD (2005). Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell 19, 123–133. [DOI] [PubMed] [Google Scholar]
- Pfander B, Moldovan GL, Sacher M, Hoege C, and Jentsch S (2005). SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 436, 428–433. [DOI] [PubMed] [Google Scholar]
- Phair RD, Scaffidi P, Elbi C, Vecerová J, Dey A, Ozato K, Brown DT, Hager G, Bustin M, and Misteli T (2004). Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell. Biol 24, 6393–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichierri P, Franchitto A, Mosesso P, and Palitti F (2000). Werner’s syndrome cell lines are hypersensitive to camptothecin-induced chromosomal damage. Mutat. Res 456, 45–57. [DOI] [PubMed] [Google Scholar]
- Pommier Y (2006). Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer 6, 789–802. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Barcelo JM, Rao VA, Sordet O, Jobson AG, Thibaut L, Miao ZH, Seiler JA, Zhang H, Marchand C, et al. (2006). Repair of topoisomerase I-mediated DNA damage. Prog. Nucleic Acid Res. Mol. Biol 81, 179–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Sun Y, Huang SN, and Nitiss JL (2016). Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol 17, 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praefcke GJ, Hofmann K, and Dohmen RJ (2012). SUMO playing tag with ubiquitin. Trends Biochem. Sci 37, 23–31. [DOI] [PubMed] [Google Scholar]
- Psakhye I, and Jentsch S (2012). Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 151, 807–820. [DOI] [PubMed] [Google Scholar]
- Reinking HK, Kang HS, Götz MJ, Li HY, Kieser A, Zhao S, Acampora AC, Weickert P, Fessler E, Jae LT, et al. (2020). DNA Structure-Specific Cleavage of DNA-Protein Crosslinks by the SPRTN Protease. Mol. Cell 80, 102–113.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarangi P, and Zhao X (2015). SUMO-mediated regulation of DNA damage repair and responses. Trends Biochem. Sci 40, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberg MJ, Lieberman JA, Herrero-Ruiz A, Butler LR, Williams JG, Muñoz-Cabello AM, Mueller GA, London RE, Cortés-Ledesma F, and Williams RS (2017). ZATT (ZNF451)-mediated resolution of topoisomerase 2 DNA-protein cross-links. Science 357, 1412–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbyn N, Noireterre A, Bagdiul I, Plank M, Michel AH, Loewith R, Kornmann B, and Stutz F (2020). The Aspartic Protease Ddi1 Contributes to DNA-Protein Crosslink Repair in Yeast. Mol. Cell 77, 1066–1079.e9. [DOI] [PubMed] [Google Scholar]
- Sharma P, Mullen JR, Li M, Zaratiegui M, Bunting SF, and Brill SJ (2017). A Lysine Desert Protects a Novel Domain in the Slx5-Slx8 SUMO Targeted Ub Ligase To Maintain Sumoylation Levels in Saccharomyces cerevisiae. Genetics 206, 1807–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, Szankasi P, Nguyen DK, Ludlow C, Dunstan HM, Roberts CJ, Jensen EL, Hartwell LH, and Friend SH (2000). Differential toxicities of anticancer agents among DNA repair and checkpoint mutants of Saccharomyces cerevisiae. Cancer Res. 60, 328–333. [PubMed] [Google Scholar]
- Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB Jr., and Stewart L (2002). The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl. Acad. Sci. USA 99, 15387–15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, Schwarz MS, Bloemeke N, Wolf PG, and Jentsch S (2014). A DNA-dependent protease involved in DNA-protein crosslink repair. Cell 158, 327–338. [DOI] [PubMed] [Google Scholar]
- Stingele J, Habermann B, and Jentsch S (2015). DNA-protein crosslink repair: proteases as DNA repair enzymes. Trends Biochem. Sci 40, 67–71. [DOI] [PubMed] [Google Scholar]
- Stingele J, Bellelli R, Alte F, Hewitt G, Sarek G, Maslen SL, Tsutakawa SE, Borg A, Kjær S, Tainer JA, et al. (2016). Mechanism and Regulation of DNA-Protein Crosslink Repair by the DNA-Dependent Metalloprotease SPRTN. Mol. Cell 64, 688–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, Bellelli R, and Boulton SJ (2017). Mechanisms of DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol 18, 563–573. [DOI] [PubMed] [Google Scholar]
- Suhandynata RT, Quan Y, Yang Y, Yuan WT, Albuquerque CP, and Zhou H (2019). Recruitment of the Ulp2 protease to the inner kinetochore prevents its hyper-sumoylation to ensure accurate chromosome segregation. PLoS Genet. 15, e1008477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Miller Jenkins LM, Su YP, Nitiss KC, Nitiss JL, and Pommier Y (2020a). A conserved SUMO pathway repairs topoisomerase DNA-protein cross-links by engaging ubiquitin-mediated proteasomal degradation. Sci. Adv 6, eaba6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Saha S, Wang W, Saha LK, Huang SN, and Pommier Y (2020b). Excision repair of topoisomerase DNA-protein crosslinks (TOP-DPC). DNA Repair (Amst.) 89, 102837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Texari L, Dieppois G, Vinciguerra P, Contreras MP, Groner A, Letourneau A, and Stutz F (2013).The nuclear pore regulates GAL1 gene transcription by controlling the localization of the SUMO protease Ulp1. Mol. Cell 51, 807–818. [DOI] [PubMed] [Google Scholar]
- Vance JR, and Wilson TE (2002). Yeast Tdp1 and Rad1-Rad10 function as redundant pathways for repairing Top1 replicative damage. Proc. Natl. Acad. Sci. USA 99, 13669–13674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz B, Popovic M, Newman JA, Fielden J, Aitkenhead H, Halder S, Singh AN, Vendrell I, Fischer R, Torrecilla I, et al. (2016). Metalloprotease SPRTN/DVC1 Orchestrates Replication-Coupled DNA-Protein Crosslink Repair. Mol. Cell 64, 704–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz B, Ruggiano A, Popovic M, Rodriguez-Berriguete G, Kilgas S, Singh AN, Higgins GS, Kiltie AE, and Ramadan K (2020). SPRTN protease and SUMOylation coordinate DNA-protein crosslink repair to prevent genome instability. bioRxiv, 2020.2002.2014.949289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L, Chen XL, Silver HR, Ahmed NT, and Johnson ES (2009). Deficient SUMO attachment to Flp recombinase leads to homologous recombination-dependent hyperamplification of the yeast 2 microm circle plasmid. Mol. Biol. Cell 20, 1241–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip MCJ, Bodnar NO, and Rapoport TA (2020). Ddi1 is a ubiquitin-dependent protease. Proc. Natl. Acad. Sci. USA 117, 7776–7781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HF, Tomida A, Koshimizu R, Ogiso Y, Lei S, and Tsuruo T (2004). Cullin 3 promotes proteasomal degradation of the topoisomerase I-DNA covalent complex. Cancer Res. 64, 1114–1121. [DOI] [PubMed] [Google Scholar]
- Zhao X, Wu CY, and Blobel G (2004). Mlp-dependent anchorage and stabilization of a desumoylating enzyme is required to prevent clonal lethality. J. Cell Biol 167, 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing have been deposited at European Nucleotide Archive (ENA) repository and are publicly available as of the date of publication. Proteomics data have been deposited at the MassIVE ProteomeXchange Consortium Member and are publicly available as of the date of publication. This paper analyzes existing, publicly available sequencing data. These accession numbers for the datasets are listed in the key resources table. Original western blot and spot assay images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Secondary Goat Anti-Mouse Immunoglobulins/HRP, 1:5000 | Dako | P0447; RRID:AB_2617137 |
| Goat Anti-Rabbit Immunoglobulins/HRP, WB:1:5000 | Dako | P0448;: RRID:AB_2617138 |
| Anti-HA (anti-HA.11 Epitope Tag Antibody clone 16B12), WB:1:2000‘ | BioLegend | 901502, lot B276381 or B242905; RRID:AB_2565006 |
| Anti-Pgk1 antibody [22C5D8], WB:1:3000 | Abcam | ab113687, GR3195775-3; RRID:AB_10861977 |
| Anti-Rad53 (monoclonal, K-medium), WB:1:100 | Gift from Maksym Shyian (D. Shore laboratory) | Mab clone EL7.E1 |
| Anti-Flag M2, WB:1:5000 | Sigma | F3165, lot SLBQ7119V or SLCC4005: RRID:AB259529 |
| Anti-Myc, WB: 1:5000 | Abcam | Ab32; lot GR310953-2 or GR3232358-2; RRID:AB_303599 |
| Anti-Ubiquitin FK2 | Calbiochem | ST1200-100UG, lots 31671118, 3291266, 3443858; RRID:AB_2043482 |
| Anti-SUMO (polyclonal), WB: 1:10000 | Helle Ulrich laboratory: (Papouli et al., 2005) | N/A |
| Anti-PCNA (polyclonal), WB: 1:2500 | Helle Ulrich laboratory: (Papouli et al., 2005) | N/A |
| Bacterial and virus strains | ||
| E. coli DH5α | N/A | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| D(+)-Glucose | Carl Roth | Art.-Nr. HN063 |
| D(+)-Raffinose pentahydrate | Carl Roth | Art.-Nr. 5241.3 |
| D(+)-Galactose | USBiological | G1030 |
| Yeast Nitrogen Base | Qbiogene | 4027-532 |
| Bacto Peptone | BD Biosciences | Ref. 211677 |
| Yeast Extract | BD Biosciences | Ref. 212750 |
| Ammonium Sulfate | Carl Roth | Art.-Nr. 3746.1 |
| 5-Fluoroorotic Acid | Toronto Research Chemicals | F595000 |
| RNase A, PureLink, (20 mg/mL) | Invitrogen | 12091-021 |
| DpnII | NEB | R0543L |
| NlaIII | NEB | R0125L |
| T4 Ligase | Thermo Scientific | EL0011 |
| Linear Acrylamide | Ambion | AM9520 |
| Taq polymerase | NEB | M0267L |
| Salmon sperm ssDNA | Sigma-Aldrich | D1626 |
| PEG4000 | Carl Roth | Art.-Nr. 0156.1 |
| Zymolyase - 20T | Amsbio | 120491-1 |
| Phusion High-Fidelity DNA Polymerase | Thermo Scientific | F530L |
| Auxin (3-indoleacetic acid) | Sigma-Aldrich | I2886 |
| Camptothecin (CPT) | Lucerna-Chem | Cat. 0215973225 |
| WesternBright ECL HRP substrate | Advansta | K-12045 |
| WesternBright Sirius HRP substrate | Advansta | K-12043 |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 4693159001 |
| NEM (N-Ethylmaleimide) | Sigma-Aldrich | E1271 |
| Dynabeads Protein G for Immunoprecipitation | Invitrogen | 10009D |
| Dynabeads Pan Mouse IgG | Invitrogen | 11041 |
| Alpha Factor | PRIMM | 201307-00007 |
| Trypsin | Promega | V5113 |
| Propidium Iodide | Sigma | P4170 |
| Proteinase K | Carl Roth | 7528.3 |
| Ni-NTA agarose | QIAGEN | 30210 |
| Critical commercial assays | ||
| The Bio-Rad protein assay | Bio-Rad | 500-0006 |
| Phire Green Hot Start II PCR Master Mix | Thermo Scientific | F126L |
| NEBuilder® HiFi DNA Assembly Cloning Kit | NEB | E5520 |
| NucleoSpin® Gel and PCR Clean-up | Macherey-Nagel | Ref. 740609.250 |
| MiSeq Reagent Kit v3 | Illumina | MS-102 |
| SYBR Select Master Mix for CFX | Applied biosystems | 4472942 |
| MinElute PCR Purification Kit | QIAGEN | 28006 |
| Deposited data | ||
| Sequencing data of the tdp1Δ wss1Δ siz2Δ transposon screen | This Manuscript | ENA: PRJEB47156 |
| Sequencing data of “wss1D Tdp1-AID*-6HA TIR1 + auxin” transposon screen | Serbyn et al. (2020), Mol.Cell | ENA: PRJEB31382 |
| The SUMO mass spectrometry proteomics data | This Manuscript | MassIVE: MSV000088129 |
| Unprocessed imaging (immunoblots, spot assays, tetrad analyses) and FACS data | This Manuscript | Mendeley Data: https://data.mendeley.com/datasets/h7bxnynd7s/1 |
| Experimental models: Organisms/strains | ||
| S. cerevisiae: Strain background: W303 | EUROSCARF | BMA64 |
| S. cerevisiae: Strain background: BY4741 | EUROSCARF | Y00000 |
| Oligonucleotides | ||
| See Table S3 for the list of oligonucleotides | This Manuscript | N/A |
| Recombinant DNA | ||
| See Table S3 for full list of plasmids | This Manuscript | N/A |
| Software and algorithms | ||
| Prism 9.1.2 | GraphPad Software, Inc | RRID:SCR_002798 |
| RStudio Version 1.1.456 | RStudio, Inc | RRID:SCR_000432 |
| Fiji | ImageJ | RRID:SCR_002285 |
| SnapGene 3.2.1 | GSL Biotech LLC | RRID:SCR_015052 |
| Kaluza | Beckman Coulter | RRID:SCR_016182 |
| Other | ||
| 0.5 mm Glass Beads | BioSpec Products | Cat. No 11079105 |
| MagNA Lyser Instrument | Roche | 03358976001 |
| Bioruptor Twin | Diagenode | UCD-400 |
| Gallios 8 colors 2 Lasers flow cytometer | Beckman Coulter | B43619 |