

Abstract
Messenger RNA regulatory elements, such as riboswitches, can display a high degree of flexibility. By characterizing their energy landscapes, and corresponding distributions of 3D configurations, structure-function relationships can be elucidated. Molecular dynamics simulation with enhanced sampling is an important strategy used to computationally access free energy landscapes characterizing the accessible 3D conformations of RNAs. While tertiary contacts are thought to play important roles in RNA dynamics, it is difficult, in explicit solvent, to sample the formation and breakage of tertiary contacts, such as helix-helix interactions, pseudoknot interactions, and junction interactions, while maintaining intact secondary structure elements. To this end, we extend previously developed collective variables and metadynamics efforts, to establish a simple metadynamics protocol, which utilizes only one collective variable, based on multiple tertiary contacts, to characterize the underlying free energy landscape of any RNA molecule. We develop a modified collective variable, the tertiary contacts distance (), which can probe the formation and breakage of all or selectively chosen tertiary contacts of the RNA. The SAM-I riboswitch in the presence of three ionic and substrate conditions was investigated and validated against the structure ensemble previously generated using SAXS experiments. This efficient and easy to implement all-atom MD simulation based approach incorporating metadynamics to study RNA conformational dynamics can also be transferred to any other type of biomolecule.
Keywords: SAM-I Riboswitch, Metadynamics, Free energy, Collective variable, Contact distance
Graphical Abstract
1. Introduction
Understanding the structure and function of RNA has been a key challenge in the life sciences for decades. Improving our understanding towards how RNAs can fold into different shapes and structures to carry out their numerous functions is especially timely now, in light of the numerous vaccines and therapeutic strategies based on messenger RNAs.
Energy landscapes have been highly useful and effective in describing the folding and function of biomolecules [1]. Collective variables have played an important role in revealing order parameters that best describe barriers in the free energy landscape[2]. In particular, collective variables based on the total number of native contacts of a biomolecule have led to a great number of insights into protein folding [2]. Recent efforts using structure-based potentials (native-contact based) have obtained near exhaustive sampling with atomistic resolution [3, 4, 5, 6, 7, 8, 9, 10, 11]. A significant difference between energy landscapes of proteins and of RNA [3, 7, 8, 9, 10, 11] is that RNA landscapes are thought to be more rugged, containing more kinetic traps. Specifically, the landscapes may have several intermediate states separated by energy barriers higher than thermal energy [12, 13, 14]. For systems with such high degrees of freedom (DOFs), standard molecular dynamics (MD) simulations are not necessarily ergodic in the timescale achievable with current computation resources. Enhanced sampling MD simulations are techniques which can help one gain insight into the folding process and also improve understanding of the underlying thermodynamics. In the past, replica exchange based MD (REMD) techniques, such as temperature-REMD [15, 16, 17, 18, 19] and replica exchange with solute tempering (REST2) [20], have extensively been utilized to explore conformation dynamics of biomolecules [19, 21]. Implementing these methods is more straight forward considering a priori knowledge on folding process not required; however, in the case of RNA, helix and base pair melting present important challenges. Enhanced sampling methods based on collective variables, e.g., metadynamics [22, 23, 24, 25, 26, 27, 28, 29, 30], umbrella sampling [31] and adaptive biasing force method [32], facilitate acceleration of sampling along two to three predefined degrees of freedom, and in turn, allow estimates of free energy surfaces as a function of these. Since these systems consist of an extremely large number of degrees of freedom, defining appropriate collective variables for a specific RNA system and applying these enhanced sampling techniques remains a major challenge. In particular, without proper collective variables the convergence of the free energy profiles is more difficult. Moreover, the interplay of key governing factors of RNA dynamics, such as base-pairing, tertiary bridges formation and mono/di-valent ions influence, is still not completely understood at atomistic scale and would be clarified by better understanding of the energy landscape surfaces [33, 34]. In particular, our previous experimental studies have shown that tertiary contacts are important for the operation of the SAM-I riboswitch. Thus, sampling the formation and breakage of tertiary contacts in explicit solvent molecular dynamics simulations is critical for obtaining the corresponding energy landscapes [35]. We show below that, while using the RMSD as a collective variable does not fully sample tertiary contacts, a collective variable based on tertiary contacts is able to sample formation and breakage of tertiary contacts.
Thus, we present a simple simulation strategy which can facilitate investigation of riboswitch free energy landscapes, as well as any other RNA, in general. Riboswitches are regulatory elements embedded in the 5’ untranslated region of mRNA and typically have two domains, the aptamer domain, which binds to a specific metabolite, and an expression platform which regulates the gene expression in response to a conformation change in aptamer [38, 39]. In this study, we have investigated the conformation transitions of the aptamer domain of the SAM-I riboswitch of Thermoanaerobacter tengcongensis (Figure 1A), which regulates expression of genes affiliated with sulphur and methionine metabolism through transcription termination upon binding to S-adenosylmthionine (SAM) metabolite [40, 41, 42, 43]. The SAM bound structure of the SAM-I aptamer domain from Thermoanaerobacter tengcongensis is shown in Figure 1B, and the unbound form adopts a similar secondary structure architecture, but has a 3D ensemble of configurations significantly more extended than the bound state [37, 36]. The SAM-I aptamer domain consists of four helices, P1-P4, connected through a four way junction, with a compact tertiary fold featuring a pseudoknot and kink-turn. This ligand bound structure is also similar to that determined from bacterium Bacillus subtilis yitJ [44]. We refer to this conformation as crystallographic or native closed state hereafter. Using small angle X-ray scattering (SAXS), the SAM-I aptamer domain was demonstrated to undergo Mg2+ ion dependent compaction, adopting conformations closely similar to the native closed state at higher Mg2+ concentrations (Figure 1B) [37]. The ensemble of structures from SAXS experiments in the ligand-free ensemble are shown in Figure 2A, where some conformations in closed configurations have P1 and P3 helices in contact, while, in open configurations in this ensemble, P1 and P3 do not interact. Several other experimental studies also confirmed that Mg2+ ions, especially at physiological concentration, help facillitate spontaneous folding of the SAM-I aptamer domain into well-structured, pre-organized form, ready to bind ligand [44, 45, 46, 47, 9, 48, 11, 49]. These studies also revealed that the addition of SAM swiftly pushes SAM-I into the fully compact form, a thermodynamically more stable closed state. Following these studies, we have employed all-atom explicit solvent metadynamics molecular dynamics simulations to investigate the SAM-I riboswitch in presence of three conditions in this study, i) 150 mM KCl, ii) 150 mM KCl with 7.6 mM MgCl2 and iii) 150 mM KCl, 7.6 mM MgCl2 with a SAM molecule bound to the binding site. SAM-I is progressively expected to achieve the more compact form in the respective conditions. We present a metadynamics based approach to investigate the energetics of conformation dynamics of the SAM-I riboswitch. We have conducted simulations with total time scale of ≈ 55 μs. As our previous experimental studies have shown that tertiary contacts play a key role in the operation of the SAM-I riboswitch, sampling the formation and breakage of tertiary contacts is critical [35]. The primary aim of this study is to design a collective variable to accelerate the dynamics of RNA molecules in order to derive meaningful transition pathways, sampling formation and breakage of tertiary contacts without significantly disrupting the secondary structure (i.e., helical structure of the helices). The commonly used collective variable RMSD captures the fluctations of radius of gyration. The collective variable (fraction of total native contacts) captures folding of the RNA from a completely denatured state and obtains near exhaustive sampling of tertiary contacts for structure-based potentials [3]. For the case of explicit solvent, to specifically sample tertiary contact breakage and formation while maintaining secondary structure, important for functional operation of the riboswitch, we have designed a single collective variable, “tertiary contacts distance ()”, that combines the four major tertiary contact regions and can enhance the formation or breakage of tertiary contacts in RNA. This variable has the advantage over a simple mean average of tertiary contacts in that it produces full sampling of all tertiary contacts while restricting tertiary contact breakage distances (distance between broken tertiary contact regions) to reasonable values, relative to experimentally determined ensembles. This enables us to describe meaningful conformation transitions in three conditions, producing configurations consistent with the SAXS based structure ensemble. Moreover, we have derived the underlying thermodynamics which is in accordance with observations made during previous experiments.
Figure 1:

(A) Secondary structure of the aptamer domain of SAM-I riboswitch from Thermoanaerobacter tengcongensis bacterium, highlighting the tertiary architecture [36]. Different structural domains are colored as well as labelled as follows: P, helices; J, joining loops; PK, pseudoknot; KT, kink-turn. (B) Three dimensional structure of SAM-I aptamer domain in cartoon representation is depicted with the SAM ligand in van der Waals representation (PDB ID: 3IQR) [37].
Figure 2:

(A) Superimposition of seven conformations of SAM-I based on the SAXS experiments [37]. (B) Four tertiary contacts forming regions along the SAM-I structure are illustrated as surface and labelled according to the secondary structure elements involved. Additional labels d1 to d4 correspond to collective variables used during WTmetaD simulations. (C) Depiction of the normalization of collective variables d1 to d4 to new ones to using Eq. 6. (D) Time evolution of collective variable for eight walkers during a WTmetaD simulation performed for the system having a SAM bound SAM-I in the presence of 150 mM KCl and 7.6 mM MgCl2.
2. Theory
Here, we have employed the well-tempered metadynamics technique and the multiple-walker metadynamics technique. In the well-tempered metadynamics approach [50, 51, 52], a history-dependent bias potential () which is a sum of Gaussian hills, is added during simulation along the predefined number of collective variables . This potential is given as
| (1) |
where are the widths of Gaussian hills, and are previously visited points along the collective variable space at time interval of and is the initial height of the Gaussian. To assure a smooth convergence of the bias potential in well-tempered scheme, height is progressively decayed using a scaling factor . The denotes the Boltzmann constant and the tuning temperature limits the sampling of collective variable space up to energy barriers of height , where is the system temperature. Together with well-tempered approach, we also used multiple-walker metadyanmics technique which runs multiple interacting replicas in parallel to speedup the bias potential construction [53], collectively, denoted as WTmetaD simulations hereafter. All simulations in this study were conducted using PLUMED 2.7.2 [54] patched with GROMACS 2021.4 [55], following the protocol established previously [56, 57, 58].
Upon completion of simulations, one dimensional (1D) free energy surfaces are estimated according to typical metadynamics protocols, using the Tiwary-Parrinello reweighting scheme [59]
| (2) |
where , is the Dirac delta function, and the time-dependent offset, which can be estimated by [59, 56]
| (3) |
Subsequently, free energy profiles for number of independent runs are determined and the average profile is calculated using following expression [60]
| (4) |
The error is calculated using standard error propagation scheme.
3. Results and Discussion
Following the system setup of the SAM-I riboswitch in three different ionic and substrate conditions (see section Materials and Methods), we choose collective variables, an important step in WTmetaD simulations. Collective variables are functions of atomic coordinates which describe the process under investigation in low dimensional space and should have the ability to distinguish initial, final and intermediate metastable states as well as, in the case of metadynamics, promote transitions between states. In the case of RNA, it is not clear which collective variables perform the best to elevate the sampling of the molecule with the size of SAM-I [21]. Moreover, how realistic conformation dynamics can be achieved for such molecules using these collective variables has not been benchmarked. Therefore, we compare routinely used collective variables with collective variables based on tertiary contacts defined in this study. Importantly, we aim to find a single collective variable which is easy to implement and capable of producing realistic dynamics for any RNA in the contect of metadynamics.
The SAM-I aptamer has four regions where tertiary contacts are formed(Figure 2B). The structure ensemble of SAM-I from SAXS experiments shown in Figure 2A hints that these tertiary contacts need to be broken to achieve the conformation transitions. Moreover, it was also illustrated in our previous wetlab experiments that stability in the tertiary contacts mainly controls the collapse and the subsequent switching from aptamer to expression platform conformation in SAM-I [35]. In principle, this criteria may be sufficient to instigate the conformation changes in any RNA. Thus, we seek a collective variable that can bias tertiary contact formation and breakage in RNAs. In this direction, we have employed four collective variables in this study. First, we start with two of the most commonly used collective variables utilized for the conformation sampling of RNA molecules previously: the root mean squared deviation (RMSD) and total number of native contacts () [2]. RMSD was calculated using all atoms of the SAM-I molecule and a WTmetaD simulation was performed using eight walkers for a duration of 250 ns each, resulting in total timescale of 2 μs. Note that for all four collective variables tested in this investigation, the simulation time remains constant and the system setup of SAM-I containing SAM in binding pocket placed in ionic solution of 150 mM KCl and 7.6 mM MgCl2 was utilized. The second collective variable which is also known as fraction of native contacts [2] is calculated using all native tertiary contact pairs of RNA in this study. When two atoms and from residues and form a contact, provided the distance between these atoms is below 0.45 nm, for number of pairs of native contacts is defined as [2, 61]
| (5) |
where is a smoothing factor set to 50 nm−1 and accounts for the fluctuations during the contact formation, which is taken to be 1.8. is the distance between atoms and in the native state, while is the distance between atoms and for instantaneous state . The third and fourth collective variables are based upon the principle of biasing the distance between tertiary contacts. At first, we define four preliminary collective variables, d1 to d4, respectively for four contact forming regions of SAM-I (see Figure 2B). These collective variables correspond to inter-domain distances as follow: d1, P1 and P3; d2, J1/2, J3/4 and J4/1; d3, J1/2 and P3; d4, J3/4 and P2. For each collective variable, first the center of mass for each domain is calculated (e.g., P1 and P3 in the case of d1) choosing only atoms involved in tertiary contact formation, followed by the distance between them. Tackling all four collective variables at once during WTmetaD simulations is difficult, because the bias potential construction becomes computationally very expensive as the cost grows exponentially with addition of each collective variable. Thus, we define the third collective variable dmean, which calculates the mean of collective variables d1 to d4. This collective variable was seen to perform well (discussed below), but controlling the sampling of underlying four collective variables was not possible. In other words, if the simulation oversamples one of the collective variable and the other three are not even being sampled, there is no way other than adding half-harmonic upper walls on individual collective variables to assure controlled sampling. However, this makes the free energy estimation process extremely tedious at later stages as the metadynamics and harmonic biases are added on different collective variables. To tackle this, by taking the inspiration from the collective variables and dmean, we design the fourth collective variable, the tertiary contacts distance, . First, collective variables d1 to d4 are transformed to to . Here, original collective variables are normalized such that the renormalized variables range from 0 to 1 (Figure 2C). The mean of these new coordinates is used to obtain :
| (6) |
where the sum is over , the number of normalized distances , which are calculated as follows. For collective variable number , is the cut-off distance. That is, is the value of at which its sampling needs to be restricted, where is the instantaneous value of collective variable for configuration . The switching distance, , is the value of from where the normalized collective variable will be smoothly switched to zero while reaching the cut-off distance. How smoothly the switching occurs depends on the smoothing parameter , taken to be 5 nm−1 for all four collective variables. To prevent sampling of the collective variable d1 at 4 nm, c1 is set to 4 nm and to 3.5 nm, while for remaining three collective variables is set to 2 nm and to 1.5 nm to prevent sampling at 2 nm, where . The is a constant with a value of 1 nm−1 always, which makes the numerator and denominator dimensionless. Similar to , is normalized such that . The graphical depiction of the normalization of all four collective variables can be seen in Figure 2C. Notably, the values of collective variables to go slightly below zero in the switching region, which is possible to tackle by taking the absolute of numerator in Eq. 6, i.e., . Nevertheless, the normalization will not be perfect in both cases.
We performed four metadynamics simulation studies, each with eight walker simulations. In study 1, we biased the metadynamics using RMSD. In study 2, we biased the metadynamics using . In study 3, we biased the metadynamics using dmean. In study 4, we biased the metadynamics using (see section Materials and Methods for more details on simulation parameters). In every simulation, we tracked the evolution of all four collective variables. In study 1, when the RMSD was biased, sampling with maximum RMSD values of ≈ 1.7 nm was achieved (see Figure S1A in SI), much lower than the upper limit set to 3 nm. In study 1 (RMSD), analyzing the evolution of the other three collective variables shows that tertiary contacts are not broken. Visualization of trajectories indicates the deformation of secondary structure of SAM-I, e.g., melting of helices, suggesting RMSD is not an optimal collective variable. This behavior did not change when only SAM-I backbone atoms were used to calculate RMSD (data not shown). In study 2, with being biased, we witnessed complete sampling in the range of 0 to 1, indicating all tertiary contacts in four regions are diminished (Figure S1B in SI). However, evolution of other collective variables suggests that in most of the trajectories, tertiary contacts forming domains do not move further after detaching from each other. In other words, the distance between them did not increase soon after the detachment, producing structures less extended in comparison to the SAXS results. This inspired us to find a collective variable which can elevate the distance between the tertiary contacts forming elements to recapitulate the SAXS configurational ensemble. In study 3, we show that biasing the collective variable dmean clearly satisfies this requirement. In study 3 ( dmean), as can be seen in Figure S1C in SI, the sampling of all four collective variable was improved compared to two previous studies, where RMSD (study 1) and (study 2) were biased. The only remaining issue with dmean was that the underlying collective variables d1 to d4 were not sampled evenly. In study 3, we restricted the sampling of dmean to 2.5 nm, with anticipation of limiting sampling of d1 to 4 nm, limiting the sampling of d2 to 2 nm, limiting the sampling of d3 to 2 nm, and limiting the sampling of d4 to 2 nm. However, we observed exploration up to values of d1 ≈ 6, d2 ≈ 1, d3 ≈ 3 and d4 ≈ 1 nm for respective collective variables during a simulation (Figure S2A in SI). In study 4, biasing with , a complete sampling in the range of 0 to 1 was achieved, as shown in Figure 2D. Moreover, biaising produced excellent sampling of the remaining three collective variables (RMSD, , dmean) (Figure S1D in SI). Most importantly, we could restrict the sampling of underlying collective variables (d1, d2, d3, d4) more precisely using the collective variable , as can be seen in Figure S2B in SI.
Next, we have performed the WTmetaD simulations biasing the collective variable for three experimental condition systems: (i) 150 mM KCl, 0 mM MgCl2, without SAM; (ii) 150 mM KCl, 7.6 mM MgCl2, without SAM; and (iii) 150 mM KCl, 7.6 mM MgCl2, with SAM bound at binding site. For each system, 2 μs long simulations were performed eight times. Upon completion of simulations, eight one dimensional (1D) free energy surfaces as a function of collective variable were estimated using Eq.2, and the average free energy profiles with Eq.4. Final free energy surfaces are shown in Figure 3A. In all three scenarios, the lowest free energy minimum is observed at , which corresponds to the crystallographic state shown on Figure 1B having all tertiary contacts intact. This state is undoubtedly the most probable state in all three conditions. In the presence of only 150 mM KCl (system (i)), the RNA is more extended, and several metastable states are observed. All four tertiary contacts are broken at the end of the simulations, i.e., . From the native state, the free energy barrier to achieve the extended state is approximately 18 kcal/mol, assuming there are no substates along the transition pathway. In the presence of 150 mM KCl and 7.6 mM MgCl2 (system (ii)), the free energy profile looks similar until , where a subsequently a steep rise in the energy occurs. The energy barrier to attain the extended state is ≈ 30 kcal/mol, which is 12 kcal/mol higher compared to system (i). This is consistent with previous studies showing that Mg2+ ions facillitate compaction of the SAM-I aptamer, producing conformations closer to the crystallographic form. In the presence of both salts and SAM in the binding cavity, the unfolding barrier was approximately 35 kcal/mol higher, which is 5 kcal/mol more with respect to the same ionic conditions but without SAM at binding site. More interestingly, energies were much higher throughout the landscape when compared with two previous scenarios, suggesting escape from the native state is much less likely. Again in agreement with previous outcomes, binding of SAM further drives the SAM-I aptamer closer to the native state.
Figure 3:

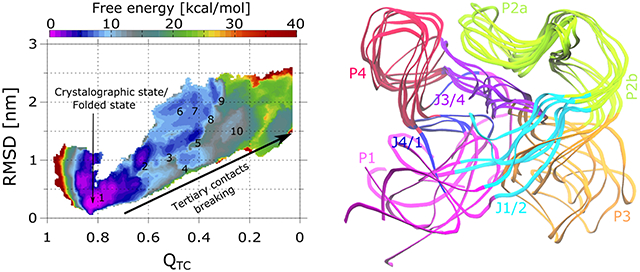
(A) 1D free energy surfaces as a function of collective variable from WTmetaD simulations performed for SAM-I in three different ionic/ligand conditions. Error bars indicate the statistical error determined after averaging eight independent runs in each case. (B) Three panels depict 2D free energy landscapes with respect to collective variables and RMSD determined from same three sets of simulations. Metastable states found along the landscapes are numbered. (C) Superimposition of the representative conformations of SAM-I from six metastable states (1-5 and 10) identified along the 2D free energy surface of the system having only 150 mM KCl. (D) 1D free energy profiles with respect to collective variables d1 to d4, which represent the four tertiary bonds forming areas, derived for all three SAM-I setups and depicted in three panels. The numbers indicate the energy difference between the native state (d1−4 ≈ 0.5 nm) and the upper bound of each collective variable (d1 ≈ 3.9 nm; d2−4 ≈ 1.9 nm). Note that in all panels, free energy estimates for few upper/lower bound values of collective variables are not depicted, because they do not seem to be converged due to the boundary effect.
To gain better understanding of metastable states, we have estimated 2D free energy surfaces with respect to and RMSD using Eq. 2, where is the 2D Dirac delta function. First, eight 2D free energy surfaces were estimated from eight independent runs, and subsequently average free energy landscapes were determined using Eq. 4. Resulting free energy profiles for all three systems are shown in Figure 3B. In each case, the lowest energy native state can be seen at and RMSD = 0.25 nm. For system (i) (150 mM KCl, 0 mM MgCl2, without SAM), 10 prominent metastable states are visible as SAM-I approaches the extended form, highlighted along the 2D free energy surface in Figure 3. Metastable states with RSMD less than 1.7 nm, i.e., substates 1, 2, 3, 4, 5 and 10, have similar conformations as seen with the SAXS derived structure ensemble (see Figures 3C and 2A for the comparison). One can see the large scale changes in P1, P3, J1/2, and J4/1, while remaining regions remain stable. For substates 7 to 9, we see further deviations in the mobile domains of the SAM-I aptamer (Figure S3A in SI), which is much larger than observed in SAXS data. For system (ii) (150 mM KCl, 7.6 mM MgCl2, without SAM), less collective variable space with respect to RMSD is being sampled. The free energy surface is more compact compared to the previous scenario and only two prominent minima are visible. Interestingly, only the area along the free energy profile where minima 1 to 5 were seen in the previous case, i.e., and RMSD ∈ [0,1.7] nm, is energetically favorable. Six representative conformations found from this region (Figure S3B in SI) are similar to those obtained in the previous system (i) and SAXS based structure ensemble. This is interesting, considering the SAXS experiments were conducted in the same ionic conditions. Collectively, all these outcomes also support the previous claim that the SAM-I aptamer domain in the MgCl2 solution remains as an ensemble of structures having a fold closer to the native state. This study reveals that same is not necessarily true in the absence of MgCl2, as the SAM-I aptamer can quite easily access the conformations much different than the native state, as shown in Figure S3A in SI. In system (iii) (150 mM KCl, 7.6 mM MgCl2, with SAM bound at binding site), we observed only one minimum affiliated to the native state on the free energy surface, while other all regions are energetically much less favourable. This again shows that SAM binding is the key step for attaining the native fold of the SAM-I aptamer. Interestingly, the sampling of collective variable space with respect to RMSD is relatively higher compared to the previous case (ii) having no SAM, but the same ionic environment. We find that in this case, fewer chelated (inner shell) Mg2+ ions along the SAM-I surfaces are observed (Figure S4 in SI), especially on P1 and J1/2. These same regions display more fluctuations as shown in Figure S3B and C in SI. On the other hand, there are no dramatic changes in outer and inner shell K+ ions as well as the outer shell Mg2+ ions, indicating inner sphere Mg2+ ions have a prominent role in the RNA dynamics. From the theoretical perspective, these outcomes indicate that Mg2+ binding with the RNA also needs to be biased to further improve the free energy estimates.
At the next stage, we have estimated the free energy landscapes using the above mentioned reweighting procedure with respect to collective variables d1 to d4 to understand how the interactions between the tertiary bridges forming domains vary, shown in Figure 3D. The collective variables correspond to inter-domain distances as follow: d1, P1 and P3; d2, J1/2, J3/4 and J4/1; d3, J1/2 and P3; d4, J3/4 and P2. In the presence of 150 mM KCl (system i), the free energy barrier observed for breaking the tertiary bridges in these four regions are, 7, 7, 17 and 23 kcal/mol, respectively. That confirms that P1 and P3, and the three-way junction (J1/2-J3/4-J4/1) dissociation is much easier, as observed in the SAXS ensemble. Whereas, tertiary bridges between J3/4 and P2 (PK) as well as J1/2 and P3 are more difficult to interrupt. With the addition of 7.6 mM Mg2+ ions (system ii), the energy barrier for P1-P3 deformation increased slightly, i.e., 9 kcal/mol, while for the remaining regions increased dramatically to 33, 29 and 34 kcal/mol, suggesting that the three-way junction, which was much weaker during the previous condition (i), is strengthened with addition of Mg2+ ions. In the third setup (system iii) having SAM at the binding site in addition of potassium and magnesium, we observed a dramatic rise in the energy barrier to 21 kcal/mol for P1-P3 disengagement, confirming that SAM strongly enhances the binding affinity between P1 and P3 and helps to attain the closed state. The free energy barriers have also slightly gone higher for J1/2-P3 and J3/4-P2 dissociation to 35 kcal/mol in both cases, indicating that the stability of the P1-P3 bridge increases the rigidity of the whole SAM-I aptamer structure. One notable exception is the three-way junction J1/2-J3/4-J4/1, where the energy barrier for the disruption is reduced to 18 kcal/mol. As mentioned above, there was a loss in the number of chelated Mg2+ ions near the loop J1/2 and the stem P1 (Figure S4B in SI), causing instability in this region. In the presence of chelated Mg2+ ions, these regions are more likely to remain rigid.
Taken together, the outcomes from systems (i)-(iii) suggest that the SAM-I aptamer can adopt the SAM-bound native state like conformations in the presence of only KCl solution (system i), but the probability is very low to remain in that form. The RNA will have diverse conformations ranging from native state like forms to extended conformations with no tertiary contacts. The pseudoknot will most likely to form, while remaining domains are expected to remain loose. The insertion of Mg2+ ions (system ii) plays an important role in driving the SAM-I aptamer towards the native form. Again, SAM-I adopts a variety of states, but not as diverse as seen with no Mg2+ ions. We observed that the chelated Mg2+ ions impose a significant rigidity throughout the SAM-I structure. We also confirm that SAM has a significant influence on enforcing the SAM-I aptamer toward the native state by bridging the P1 and P3 with more affinity. This also has an indirect effect on improving the rigidity of other regions of SAM-I.
4. Conclusions
In this study, we put forward a metadynamics simulations based strategy to study the energy landscapes of RNAs using the SAM-I riboswitch as an example. The collective variable, tertiary contacts distance , developed in this study, is demonstrated to work efficiently to bias the tertiary bridging in RNA, and is shown to perform better then traditionally used collective variables like RMSD and total fraction of native contacts for the case of explicit solvent metadynamics. The collective variable dmean is also found to work nicely, and in future can be used for cases where one wants to induce large scale conformation transitions, especially for systems with no a priori knowledge. In the case of SAM-I, we only biased the sampling of tertiary bridges using to understand the local conformation changes in a better fashion, but this collective variable is also capable of elevating the sampling of all other contacts in RNAs, e.g., canonical base pairing, to study global conformation dynamics. However, this will be more difficult to achieve and the quality of the outcomes will be needed to validate in a more systematic manner. This collective variable also allows one to choose selective tertiary bridges for biasing, if all are not required, as well as making it feasible to restrict sampling space for a particular bridge. For the SAM-I aptamer, we demonstrate that inducing the sampling of tertiary bridge breaking was sufficient to obtain a picture on its conformation transitions, which we validated using SAXS experiments based structure ensemble. Most importantly, we could derive the underlying free energy landscapes and the energy barriers for overall structural transitions of SAM-I as well as for local regions. The overall mechanisms for conformation changes observed here are also in excellent agreement with the previous studies. Overall, the strategy is easy to implement, computationally cost effective and capable of handling most RNA molecules. We also believe this strategy can easily be transferred to other biomolecules.
5. Materials and Methods
5.1. System Setup and MD Simulations
We started with the crystal structure of the SAM-I aptamer domain bound with SAM from Thermonaerobacter tengcongensis organism (PDB ID: 3IQR) [37]. The mutated residue 94 was changed back from adenine to guanine. Subsequently, we have built systems in three different conditions, i) in presence of 150 mM KCl and absence of SAM, ii) in presence of 150 mM KCl, 7.6 mM MgCl2 and absence of SAM, iii) in presence of 150 mM KCl, 7.6 mM MgCl2 and SAM present in the binding pocket. For the first and second systems, SAM was removed from the binding site, while for second and third systems five Ba2+ ions bound to SAM-I were replaced with Mg2+. Then systems were solvated with the TIP3P water molecules and ions were added to match the above mentioned ionic conditions. During the equilibration stage, ions are equilibrated. Here, the bulk is defined as exclusion of RNA as well the surrounding water within the distance of 1.2 nm. To achieve the bulk ionic concentration of 150 mM KCl and 7.6 mM MgCl2, we have iteratively changed the number of ions after following the below described equilibration protocol. We repeated the process until the bulk concentration of ions reaches closed to the desired ones. All the resulting systems were contained approximately 104,000 atoms.
Following the energy minimization step, all systems were equilibrated using the following strategy [56, 57, 58]. First, equilibration was conducted in the NVT ensemble for 1 ns with a time step of 1 fs using velocity rescaling thermostat [62] to keep the temperature at 300 K. At this step, all atoms of RNA and ligand were fixed applying the position restraints with a force constant of k = 1000 kJ mol−1 nm−2. Then, 2 ns equilibration in NPT ensemble was conducted with 1 fs time step applying the position restraints same as previous step, where Parrinello-Rahman barostat [63] to maintain the pressure at 1 bar, and Nosé-Hoover thermostat [64] for keeping the temperature at 300 K were used. In following all steps, equilibration was conducted in NPT ensemble. At next step, 10 ns equilibration was carried out using 2 fs time step by keeping the remaining settings same as the previous step. Subsequently, the system was equilibrated during 20 ns where the position restraints were only applied to backbone atoms of RNA and all atoms of the SAM molecule. Then, another 100 ns equilibration was conducted with the position restraints kept only on the phosphorus atoms of RNA and all atoms of the SAM molecule. At last, 100 ns simulation was performed without any position restraints.
All MD simulations were performed using the GROMACS package 2021.4 [55]. We have used amber ff99bsc0χOL3 force field parameters for the RNA molecule [65, 66, 67, 68]. For the SAM molecule, the force field parameters were obtained from the previous study [69]. Force field parameters for the ions K+, Cl− and Mg2+ (MicroMg) optimized with TIP3P water were taken from the recent studies [70, 71]. The cutoff for the short-range electrostatics and the van der Waals interactions was set to 1.2 nm, and the particle-mesh Ewald method [72] was utilized for the long-range electrostatics with a grid size of 0.12 nm. Hydrogen containing bonds were constrained using the LINCS algorithm [73].
5.2. WTmetaD Simulations
The metadynamics simulations were performed using the PLUMED 2.7.2 plugin [54] patched to GROMACS package 2021.4 [55]. WTmetaD simulations based strategy developed in the previous studies to investigate the antibiotics/substrates transport across the bacterial channels is implemented here [56, 57, 58]. A total of eight walkers were used in WTmetaD simulations, and the simulation time was set to 250 ns for each walker, resulting in total time scale of 2 μs. For the SAM-I system with SAM bound in binding pocket and having 150 mM KCl and 7.6 mM MgCl2, four independent WTmetaD simulations for timescale of 2 μs were performed having four collective variables (RMSD, , dmean and ) being biased respectively. For all three SAM-I systems, simulations were conducted biasing the collective variable , and repeated eight times for the same time duration. All the metadynamics simulations related parameters are indicated in Table 1.
Table 1:
Parameters used during the WTmetaD simulations performed using four different collective variables. denote the Gaussian width, the Gaussian height, the tuning temperature, the bias deposition time interval, Uwall is the value of the collective variable at which the half-harmonic restraint will be activated, and is the force constant for this restraint.
| Collective variable | Uwall | |||||
|---|---|---|---|---|---|---|
| RMSD | 0.01 [nm] | 1 | 8700 | 2 | 3 [nm] | 800 |
| 0.005 | 1 | 8700 | 2 | - | - | |
| dmean | 0.01 [nm] | 1 | 8700 | 2 | 2.5 [nm] | 800 |
| 0.0025 | 1 | 8700 | 2 | - | - |
Supplementary Material
Movie_MG.
Movie_SAM-I.
Highlights:
Metadynamics simulations-based approach developed to explore conformation dynamics of riboswitches.
New collective variable “tertiary contacts distance, ” is designed to break and form tertiary contacts in RNAs.
Free energy landscapes for SAM-I conformation transitions are determined in various conditions.
Conformation transition mechanism is deciphered for SAM-I and validated with previous SAXS experiments-based structure ensemble.
Acknowledgements
We acknowledge generous support from the National Institutes of Health (NIH) [RO1-GM110310 to J.N.O., RO1-GM110310 to K.Y.S.];
References
- [1].Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG, Funnels, pathways, and the energy landscape of protein folding: a synthesis, Proteins 21 (3) (1995) 167–95. doi: 10.1002/prot.340210302. URL http://www.ncbi.nlm.nih.gov/pubmed/7784423 [DOI] [PubMed] [Google Scholar]
- [2].Clementi C, Nymeyer H, Onuchic JN, Topological and energetic factors: What determines the structural details of the transition state ensemble and “en-route” intermediates for protein folding? an investigation for small globular proteins, Journal of molecular biology 298 (5) (2000) 937–953. doi: 10.1006/jmbi.2000.3693. [DOI] [PubMed] [Google Scholar]
- [3].Whitford PC, Schug A, Saunders J, Hennelly SP, Onuchic JN, Sanbonmatsu KY, Nonlocal helix formation is key to understanding s-adenosylmethionine-1 riboswitch function, Biophys J 96 (2) (2009) L7–9. doi: 10.1016/j.bpj.2008.10.033. URL https://www.ncbi.nlm.nih.gov/pubmed/19167285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Whitford PC, Noel JK, Gosavi S, Schug A, Sanbonmatsu KY, Onuchic JN, An all-atom structure-based potential for proteins: bridging minimal models with all-atom empirical forcefields, Proteins 75 (2) (2009) 430–41. doi: 10.1002/prot.22253. URL https://www.ncbi.nlm.nih.gov/pubmed/18837035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ratje AH, Loerke J, Mikolajka A, Brunner M, Hildebrand PW, Starosta AL, Donhofer A, Connell SR, Fucini P, Mielke T, Whitford PC, Onuchic JN, Yu Y, Sanbonmatsu KY, Hartmann RK, Penczek PA, Wilson DN, Spahn CM, Head swivel on the ribosome facilitates translocation by means of intra-subunit trna hybrid sites, Nature 468 (7324) (2010) 713–6. doi: 10.1038/nature09547. URL https://www.ncbi.nlm.nih.gov/pubmed/21124459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Whitford PC, Geggier P, Altman RB, Blanchard SC, Onuchic JN, Sanbonmatsu KY, Accommodation of aminoacyl-trna into the ribosome involves reversible excursions along multiple pathways, RNA 16 (6) (2010) 1196–204. doi: 10.1261/rna.2035410. URL https://www.ncbi.nlm.nih.gov/pubmed/20427512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hayes RL, Noel JK, Mandic A, Whitford PC, Sanbonmatsu KY, Mohanty U, Onuchic JN, Generalized manning condensation model captures the rna ion atmosphere, Phys Rev Lett 114 (25) (2015) 258105. doi: 10.1103/PhysRevLett.114.258105. URL https://www.ncbi.nlm.nih.gov/pubmed/26197147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Roy S, Lammert H, Hayes RL, Chen B, LeBlanc R, Dayie TK, Onuchic JN, Sanbonmatsu KY, A magnesium-induced triplex pre-organizes the sam-ii riboswitch, PLoS Comput Biol 13 (3) (2017) e1005406. doi: 10.1371/journal.pcbi.1005406. URL https://www.ncbi.nlm.nih.gov/pubmed/28248966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hayes RL, Noel JK, Whitford PC, Mohanty U, Sanbonmatsu KY, Onuchic JN, Reduced model captures mg2+-rna interaction free energy of riboswitches, Biophysical journal 106 (7) (2014) 1508–1519. doi: 10.1016/j.bpj.2014.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Roy S, Onuchic JN, Sanbonmatsu KY, Cooperation between magnesium and metabolite controls collapse of the sam-i riboswitch, Biophys J 113 (2) (2017) 348–359. doi: 10.1016/j.bpj.2017.06.044. URL https://www.ncbi.nlm.nih.gov/pubmed/28746845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Roy S, Hennelly SP, Lammert H, Onuchic JN, Sanbonmatsu KY, Magnesium controls aptamer-expression platform switching in the sam-i riboswitch, Nucleic acids research 47 (6) (2019) 3158–3170. doi: 10.1093/nar/gky1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ma H, Proctor DJ, Kierzek E, Kierzek R, Bevilacqua PC, Gruebele M, Exploring the energy landscape of a small rna hairpin, Journal of the American Chemical Society 128 (5) (2006) 1523–1530. doi: 10.1021/ja0553856. [DOI] [PubMed] [Google Scholar]
- [13].Chen S-J, Rna folding: Conformational statistics, folding kinetics, and ion electrostatics, Annu. Rev. Biophys 37 (2008) 197–214. doi: 10.1146/annurev.biophys.37.032807.125957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sun T.-t., Zhao C, Chen S-J, Predicting cotranscriptional folding kinetics for riboswitch, The Journal of Physical Chemistry B 122 (30) (2018) 7484–7496. doi: 10.1021/acs.jpcb.8b04249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sugita Y, Okamoto Y, Replica-exchange molecular dynamics method for protein folding, Chem. Phys. Lett 314 (1) (1999) 141–151. doi: 10.1016/S0009-2614(99)01123-9. [DOI] [Google Scholar]
- [16].Garcia AE, Sanbonmatsu KY, Exploring the energy landscape of a beta hairpin in explicit solvent, Proteins 42 (3) (2001) 345–54. doi:. URL https://www.ncbi.nlm.nih.gov/pubmed/11151006 [DOI] [PubMed] [Google Scholar]
- [17].Garcia AE, Sanbonmatsu KY, Alpha-helical stabilization by side chain shielding of backbone hydrogen bonds, Proc Natl Acad Sci U S A 99 (5) (2002) 2782–7. doi: 10.1073/pnas.042496899. URL https://www.ncbi.nlm.nih.gov/pubmed/11867710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sanbonmatsu KY, Garcia AE, Structure of met-enkephalin in explicit aqueous solution using replica exchange molecular dynamics, Proteins 46 (2) (2002) 225–34. doi: 10.1002/prot.1167. URL https://www.ncbi.nlm.nih.gov/pubmed/11807951 [DOI] [PubMed] [Google Scholar]
- [19].Yang S, Onuchic JN, García AE, Levine H, Folding time predictions from all-atom replica exchange simulations, Journal of molecular biology 372 (3) (2007) 756–763. doi: 10.1016/j.jmb.2007.07.010. [DOI] [PubMed] [Google Scholar]
- [20].Wang L, Friesner RA, Berne BJ, Replica exchange with solute scaling: A more efficient version of replica exchange with solute tempering (rest2), J. Phys. Chem. B 115 (30) (2011) 9431–9438. doi: 10.1021/jp204407d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mlỳnskỳ V, Bussi G, Exploring rna structure and dynamics through enhanced sampling simulations, Current opinion in structural biology 49 (2018) 63–71. doi: 10.1016/j.sbi.2018.01.004. [DOI] [PubMed] [Google Scholar]
- [22].Laio A, Parrinello M, Escaping free-energy minima, Proc. Natl. Acad. Sci. USA 99 (20) (2002) 12562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bussi G, Laio A, Parrinello M, Equilibrium free energies from nonequilibrium metadynamics, Phys Rev Lett 96 (9) (2006) 090601. doi: 10.1103/PhysRevLett.96.090601. URL https://www.ncbi.nlm.nih.gov/pubmed/16606249 [DOI] [PubMed] [Google Scholar]
- [24].Bussi G, Gervasio FL, Laio A, Parrinello M, Free-energy landscape for beta hairpin folding from combined parallel tempering and metadynamics, J Am Chem Soc 128 (41) (2006) 13435–41. doi: 10.1021/ja062463w. URL https://www.ncbi.nlm.nih.gov/pubmed/17031956 [DOI] [PubMed] [Google Scholar]
- [25].Barducci A, Bussi G, Parrinello M, Well-tempered metadynamics: a smoothly converging and tunable free-energy method, Phys Rev Lett 100 (2) (2008) 020603. doi: 10.1103/PhysRevLett.100.020603. URL https://www.ncbi.nlm.nih.gov/pubmed/18232845 [DOI] [PubMed] [Google Scholar]
- [26].Branduardi D, Bussi G, Parrinello M, Metadynamics with adaptive gaussians, J Chem Theory Comput 8 (7) (2012) 2247–54. doi: 10.1021/ct3002464. URL https://www.ncbi.nlm.nih.gov/pubmed/26588957 [DOI] [PubMed] [Google Scholar]
- [27].Gil-Ley A, Bottaro S, Bussi G, Empirical corrections to the amber rna force field with target metadynamics, J Chem Theory Comput 12 (6) (2016) 2790–8. doi: 10.1021/acs.jctc.6b00299. URL https://www.ncbi.nlm.nih.gov/pubmed/27153317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kuhrova P, Best RB, Bottaro S, Bussi G, Sponer J, Otyepka M, Banas P, Computer folding of rna tetraloops: Identification of key force field deficiencies, J Chem Theory Comput 12 (9) (2016) 4534–48. doi: 10.1021/acs.jctc.6b00300. URL https://www.ncbi.nlm.nih.gov/pubmed/27438572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cunha RA, Bussi G, Unraveling mg(2+)-rna binding with atomistic molecular dynamics, RNA 23 (5) (2017) 628–638. doi: 10.1261/rna.060079.116. URL https://www.ncbi.nlm.nih.gov/pubmed/28148825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mlynsky V, Bussi G, Understanding in-line probing experiments by modeling cleavage of nonreactive rna nucleotides, RNA 23 (5) (2017) 712–720. doi: 10.1261/rna.060442.116. URL https://www.ncbi.nlm.nih.gov/pubmed/28202709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kumar S, Rosenberg J, Bouzida D, Swendsen R, Kollman P, The Weighted Histogram Analysis Method for Free-energy Calculations on Biomolecules. I. The Method, J. Comput. Chem 13 (8) (1992) 1011–1021. [Google Scholar]
- [32].Darve E, Rodriguez Gomez D, Pohorille A, Adaptive biasing force method for scalar and vector free energy calulations, J. Chem. Phys 128 (14) (2008) 144120. doi: 10.1063/1.2829861. [DOI] [PubMed] [Google Scholar]
- [33].Sponer J, Bussi G, Krepl M, Banáš P, Bottaro S, Cunha RA, Gil-Ley A, Pinamonti G, Poblete S, Jurečka P, et al. , Rna structural dynamics as captured by molecular simulations: A comprehensive overview, Chemical reviews 118 (8) (2018) 4177–4338. doi: 10.1021/acs.chemrev.7b00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yu T, Chen S-J, Hexahydrated mg2+ binding and outer-shell dehydration on rna surface, Biophysical journal 114 (6) (2018) 1274–1284. doi: 10.1016/j.bpj.2018.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hennelly SP, Sanbonmatsu KY, Tertiary contacts control switching of the sam-i riboswitch, Nucleic acids research 39 (6) (2011) 2416–2431. doi: 10.1093/nar/gkq1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Montange RK, Batey RT, Structure of the s-adenosylmethionine riboswitch regulatory mrna element, Nature 441 (7097) (2006) 1172–1175. doi: 10.1038/nature04819. [DOI] [PubMed] [Google Scholar]
- [37].Stoddard CD, Montange RK, Hennelly SP, Rambo RP, Sanbonmatsu KY, Batey RT, Free state conformational sampling of the sam-i riboswitch aptamer domain, Structure 18 (7) (2010) 787–797. doi: 10.1016/j.str.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Winkler WC, Breaker RR, Genetic control by metabolite-binding riboswitches, Chembiochem 4 (10) (2003) 1024–1032. [DOI] [PubMed] [Google Scholar]
- [39].Nudler E, Mironov AS, The riboswitch control of bacterial metabolism, Trends in biochemical sciences 29 (1) (2004) 11–17. doi: 10.1016/j.tibs.2003.11.004. [DOI] [PubMed] [Google Scholar]
- [40].Grundy FJ, Henkin TM, The s box regulon: A new global transcription termination control system for methionine and cysteine biosynthesis genes in gram-positive bacteria, Molecular microbiology 30 (4) (1998) 737–749. doi: 10.1046/j.1365-2958.1998.01105.x. [DOI] [PubMed] [Google Scholar]
- [41].Epshtein V, Mironov AS, Nudler E, The riboswitch-mediated control of sulfur metabolism in bacteria, Proceedings of the National Academy of Sciences 100 (9) (2003) 5052–5056. doi: 10.1073/pnas.0531307100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].McDaniel BAM, Grundy FJ, Artsimovitch I, Henkin TM, Transcription termination control of the s box system: Direct measurement of s-adenosylmethionine by the leader rna, Proceedings of the National Academy of Sciences 100 (6) (2003) 3083–3088. doi: 10.1073/pnas.0630422100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Winkler WC, Nahvi A, Sudarsan N, Barrick JE, Breaker RR, An mrna structure that controls gene expression by binding s-adenosylmethionine, Nature Structural & Molecular Biology 10 (9) (2003) 701–707. doi: 10.1038/nsb967. [DOI] [PubMed] [Google Scholar]
- [44].Lu C, Ding F, Chowdhury A, Pradhan V, Tomsic J, Holmes WM, Henkin TM, Ke A, Sam recognition and conformational switching mechanism in the bacillus subtilis yitj s box/sam-i riboswitch, Journal of molecular biology 404 (5) (2010) 803–818. doi: 10.1016/j.jmb.2010.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Heppell B, Blouin S, Dussault A-M, Mulhbacher J, Ennifar E, Penedo JC, Lafontaine DA, Molecular insights into the ligand-controlled organization of the sam-i riboswitch, Nature chemical biology 7 (6) (2011) 384–392. doi: 10.1038/nchembio.563. [DOI] [PubMed] [Google Scholar]
- [46].Hayes RL, Noel JK, Mohanty U, Whitford PC, Hennelly SP, Onuchic JN, Sanbonmatsu KY, Magnesium fluctuations modulate rna dynamics in the sam-i riboswitch, Journal of the American Chemical Society 134 (29) (2012) 12043–12053. doi: 10.1021/ja301454u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hennelly SP, Novikova IV, Sanbonmatsu KY, The expression platform and the aptamer: Cooperativity between mg2+ and ligand in the sam-i riboswitch, Nucleic acids research 41 (3) (2013) 1922–1935. doi: 10.1093/nar/gks978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Manz C, Kobitski AY, Samanta A, Keller BG, Jäschke A, Nienhaus GU, Single-molecule fret reveals the energy landscape of the full-length sam-i riboswitch, Nature chemical biology 13 (11) (2017) 1172–1178. [DOI] [PubMed] [Google Scholar]
- [49].Manz C, Kobitski AY, Samanta A, Nienhaus K, Jäschke A, Nienhaus GU, Exploring the energy landscape of a sam-i riboswitch, Journal of Biological Physics 47 (4) (2021) 371–386. doi: 10.1007/s10867-021-09584-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Barducci A, Bussi G, Parrinello M, Well-tempered metadynamics: A smoothly converging and tunable free-energy method, Phys. Rev. Lett 100 (2) (2008) 020603. doi: 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- [51].Dama JF, Parrinello M, Voth GA, Well-tempered metadynamics converges asymptotically, Phys. Rev. Lett 112 (24) (2014) 240602. doi: 10.1103/physrevlett.112.240602. [DOI] [PubMed] [Google Scholar]
- [52].Tiwary P, Dama JF, Parrinello M, A perturbative solution to metadynamics ordinary differential equation, J. Chem. Phys 143 (23) (2015) 234112. doi: 10.1063/1.4937945. [DOI] [PubMed] [Google Scholar]
- [53].Raiteri P, Laio A, Gervasio GL, Micheletti C, Parrinello M, Efficient reconstruction of complex free energy landscapes by multiple walkers metadynamics, J. Chem. Phys. B 110 (8) (2006) 3533–3539. doi: 10.1021/jp054359r. [DOI] [PubMed] [Google Scholar]
- [54].Tribello GA, Bonomi M, Branduardi D, Camilloni C, Bussi G, PLUMED 2: New feathers for an old bird, Comput. Phys. Commun 185 (2) (2014) 604–613. doi: 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- [55].Hess B, Kutzner C, Spoel DVD, Lindahl E, GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation, J. Chem. Theory Comput 4 (3) (2008) 435–447. [DOI] [PubMed] [Google Scholar]
- [56].Prajapati JD, Solano CJF, Winterhalter M, Kleinekathöfer U, Characterization of ciprofloxacin permeation pathways across the porin ompc using metadynamics and a string method, J. Chem. Theory Comput 13 (2017) 4553–4566. doi: 10.1021/acs.jctc.7b00467. [DOI] [PubMed] [Google Scholar]
- [57].Prajapati JD, Solano CJF, Winterhalter M, Kleinekathöfer U, Enrofloxacin permeation pathways across the porin ompc, J. Phys. Chem. B 122 (4) (2018) 1417–1426. doi: 10.1021/acs.jpcb.7b12568. [DOI] [PubMed] [Google Scholar]
- [58].Golla VK, Prajapati JD, Joshi M, Kleinekathöfer U, Exploration of free energy surfaces across a membrane channel using metadynamics and umbrella sampling, J. Chem. Theory Comput 16 (4) (2020) 2751–2765. doi: 10.1021/acs.jctc.9b00992. [DOI] [PubMed] [Google Scholar]
- [59].Tiwary P, Parrinello M, A time-independent free energy estimator for metadynamics, J. Phys. Chem. B 119 (3) (2014) 736–742. doi: 10.1021/jp504920s. [DOI] [PubMed] [Google Scholar]
- [60].Atkovska K, Hub JS, Energetics and mechanism of anion permeation across formate-nitrite transporters, Sci. Rep 7 (1) (2017) 12027. doi: 10.1038/s41598-017-11437-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Best RB, Hummer G, Eaton WA, Native contacts determine protein folding mechanisms in atomistic simulations, Proceedings of the National Academy of Sciences 110 (44) (2013) 17874–17879. doi: 10.1073/pnas.1311599110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Bussi G, Donadio D, Parrinello M, Canonical sampling through velocity rescaling, J. Chem. Phys 126 (1) (2007) 014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- [63].Parrinello M, Rahman A, Polymorphic transitions in single crystals: A new molecular dynamics method, J. Appl. Phys 52 (12) (1981) 7182–7190. [Google Scholar]
- [64].Cheng A, Merz KM, Application of the Nosé-hoover chain algorithm to the study of protein dynamics, J. Phys. Chem 100 (5) (1996) 1927–1937. [Google Scholar]
- [65].Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA, A second generation force field for the simulation of proteins, nucleic acids, and organic molecules, J. Am. Chem. Soc 117 (19) (1995) 5179–5197. arXiv: 10.1021/ja00124a002, doi: 10.1021/ja00124a002. [DOI] [Google Scholar]
- [66].Wang J, Cieplak P, Kollman PA, How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules?, J. Comput. Chem 21 (12) (2000) 1049–1074. [Google Scholar]
- [67].Pérez A, Marchán I, Svozil D, Sponer J, Cheatham TE III, Laughton CA, Orozco M, Refinement of the amber force field for nucleic acids: Improving the description of conformers, Biophys. J 92 (11) (2007) 3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Zgarbová M, Otyepka M, Šponer J, Mládek A, Banáš P, Cheatham TE III, Jurecka P, Refinement of the cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles, Journal of chemical theory and computation 7 (9) (2011) 2886–2902. doi: 10.1021/ct200162x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Saez DA, Vöhringer-Martinez E, A consistent s-adenosylmethionine force field improved by dynamic hirshfeld-i atomic charges for biomolecular simulation, Journal of computer-aided molecular design 29 (10) (2015) 951–961. doi: 10.1007/s10822-015-9864-1. [DOI] [PubMed] [Google Scholar]
- [70].Mamatkulov S, Schwierz N, Force fields for monovalent and divalent metal cations in tip3p water based on thermodynamic and kinetic properties, The Journal of Chemical Physics 148 (7) (2018) 074504. doi: 10.1063/1.5017694. [DOI] [PubMed] [Google Scholar]
- [71].Grotz KK, Cruz-León S, Schwierz N, Optimized magnesium force field parameters for biomolecular simulations with accurate solvation, ion-binding, and water-exchange properties, Journal of chemical theory and computation 17 (4) (2021) 2530–2540. doi: 10.1021/acs.jctc.0c01281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG, A smooth particle mesh ewald method, J. Chem. Phys 103 (19) (1995) 8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- [73].Hess B, Bekker H, Berendsen HJC, Fraaije JGEM, LINCS: A linear constraint solver for molecular simulations, J. Comput. Chem 18 (12) (1997) 1463–1472. doi:. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.