

Abstract
KSHV causes primary effusion lymphoma (PEL). Here we review what is known about human gene essentiality in PEL-derived cell lines. We provide an updated list of PEL-specific human gene dependencies, based on the improved definition of core essential genes across human cancer types. Requirements of PEL cell lines for IRF4, BATF, CCND2, CFLAR, MCL1 and MDM2 have been confirmed experimentally. KSHV co-opts IRF4 and BATF to drive super-enhancer (SE)-mediated expression of IRF4 itself, MYC, and CCND2. IRF4-dependency of SE-mediated gene expression are shared with Epstein-Barr Virus (EBV)-transformed lymphoblastoid cell lines (LCLs) and human T cell leukemia virus type 1 (HTLV-1)-transformed adult T-cell leukemia/lymphoma (ATLL) cell lines, as well as several B cell lymphomas of non-viral etiology. LCLs and ATLL cell lines similarly share dependencies on CCND2 and CFLAR with PEL, but also have distinct gene dependencies. Genetic dependencies could be exploited for therapeutic intervention in PEL and other cancers.
Introduction
Primary effusion lymphoma (PEL) is a rare complication of infection by Kaposi’s Sarcoma-associated herpesvirus (KSHV or human herpesvirus 8), accounting for 3–4% of non-Hodgkin lymphomas (NHLs) in human immunodeficiency virus (HIV)-infected patients and <1% of NHLs in HIV-negative patients [1,2]. Since we can only cite the most pertinent references here, we also refer the reader to recent reviews of PEL and KSHV oncogene expression [3,4]. PEL tumor cells are infected by KSHV and lack expression of typical B cell markers or immunoglobulins (Igs), but their expression of plasma cell markers (IRF4, CD138), mRNA expression profiles, rearranged Ig loci and marks of somatic hypermutation indicate a relatively mature post-germinal center B cell origin. The rarity of PEL suggests that KSHV infection and immunodeficiency alone are insufficient for lymphomagenesis. Accordingly, de novo infection of primary human B cells by KSHV does not readily lead to immortalization. Likely cofactors for the development of PEL are co-infection by Epstein-Barr virus (EBV) and cellular driver mutations. Indeed, EBV co-infection is observed in most PEL tumors and evidence for cooperative B cell transformation by EBV and KSHV has been reported in vitro and in vivo [5–8]. A comprehensive understanding of cellular mutations in PEL based on genome or exome sequencing, similarly to other lymphomas, has not yet been achieved, due to the rarity of PEL. However, genomic studies support a role for cellular mutations in PEL lymphomagenesis [9,10]. Moreover, mutations in the tumor suppressors p53, PTEN, and RB, among other genes, have been observed in PEL-derived cell lines and/or individual PEL tumors [11–13]. PEL is currently treated by chemotherapy, similarly to other non-Hodgkin lymphomas. The reported median overall survival of PEL patients remains below 2 years [14].
KSHV gene expression is essential in PEL-derived cell lines
KSHV encodes more than 100 proteins and non-coding RNAs as part of latent (constitutive) or lytic (inducible) gene expression programs. Latency is a viral maintenance state, where gene expression is restricted to several latent genes. The KSHV lytic gene expression cascade is initiated by a single viral transcription factor and culminates in virus production. The majority of PEL tumor cells are latently infected. The latent nuclear antigen (LANA) is expressed in all tumor cells, where it maintains the episomal KSHV genome during cell division and has additional oncogenic roles, including inhibition of p53 [11,15]. Aside from LANA, the major latency locus encodes a dicistronic mRNA for viral homologs of cyclin D2 (vCYC) and FLICE inhibitory protein (vFLIP), pri-miRNA precursors for >20 KSHV miRNAs, and mRNAs for Kaposin proteins. PEL tumors and cell lines also express a viral homolog of interferon regulatory factors (vIRF3) from a separate genetic locus [16]. EBV gene expression in EBV+ PEL follows the latency I program, with expression of the EBV nuclear antigen 1 (EBNA1), the EBER1/2 non-coding RNAs, and multiple miRNAs. EBNA1 functions to maintain the EBV genome, similarly to KSHV LANA. Roles of the KSHV latency genes have been inferred from loss-of-function (LOF) gene dependency studies in PEL cell lines and gain-of-function ectopic expression studies in in vitro and in in vivo models. Specifically, knockdown of LANA, the vCYC/vFLIP dicistron, or vIRF3 impairs survival of PEL cell lines [17,18]. The role of the KSHV miRNAs in PEL is poorly understood, but there is evidence that some miRNAs promote PEL cell viability and/or proliferation [19,20], including miR-K11, a viral mimic of the B cell oncogene miR-155 [21,22]. Ectopic expression of LANA, vFLIP, miR-K11, or the full latency locus induces lymphoproliferative phenotypes in mice [23–25], further supporting their critical role in PEL. Evidence for a dependency on EBV gene expression in EBV+ PEL cell lines comes from a recent study that showed that targeting EBNA1 by CRISPR/Cas9 impaired PEL cell survival [26].
Gene Essentiality Studies Identify Cellular Oncogene Dependencies
Since viral gene dependencies likely reflect oncogenic cellular deregulation, studying host gene dependencies in PEL cells represents an opportunity to identify deregulated genes and pathways. Pooled genetic screening approaches using targeted gene inactivation by CRISPR/Cas9 have dramatically improved the ease, specificity, and sensitivity of genome-wide loss-of-function screening to query gene essentiality [27–31]. In particular, the Cancer Dependency Map (DepMap) Project includes a growing collection of genome-wide loss of function screens and represents the most comprehensive effort to define gene essentiality across cancer types (https://depmap.org/portal/) [32]. Based on data from 1070 CRISPR screens representing 34 different tissue lineages, DepMap currently reports 1982 common essential (“pan-essential”) genes. This designation is made based on their inclusion in the top X depleted genes in 90% or more of the screened cell lines, using an empirical approach to determine the value of X [33]. Other genes are essential in only some cancer cell lines, and thus more likely to reflect the specific biology of the originating cell type or its unique oncogenic drivers. While CRISPR/Cas9 is by far the most established approach to LOF screening, LOF by RNA interference, epigenetic CRISPR interference (CRISPRi), and other CRISPR systems represent alternatives.
Cellular Gene Dependencies in Primary Effusion Lymphoma
To achieve a better understanding of the cellular gene dependencies in PEL, our lab has conducted CRISPR/Cas9 gene essentiality screens in 8 PEL cell lines, including 4 that are co-infected with EBV [34]. This work identified 210 PEL-specific oncogenic dependencies (PSODs), i.e. genes that were preferentially essential in PEL cell lines over non-PEL cell lines. At the time, this designation was made based on 52 screens from 15 non-PEL cancer cell lines. We experimentally validated dependencies on the hematopoietic lineage transcription factors (TFs) interferon regulatory factor 4 (IRF4) and basic leucine zipper ATF-like transcription factor (BATF), the G1/S cyclin D2 (CCND2), the p53 ubiquitin ligase murine double minute 2 (MDM2), as well as anti-apoptotic cellular FLICE-inhibitory protein (cFLIP, encoded by CFLAR) and MCL1 (Fig. 1) [34,35]. Several of the top dependencies are candidates for therapeutic targeting. Specifically, cyclin D-dependent kinase 4/6 (CDK4/6) inhibition by palbociclib is toxic in PEL cells [34,36]. PEL cell lines are also sensitive to specific inhibitors of MCL1, an anti-apoptotic protein of the BCL2 family [34,37]. The implications of IRF4 dependency are discussed below. Consistent with previous reports [38–41], PEL cell lines require MYC and genes involved in mTOR signaling, however, these genes are pan-essential. Importantly, pan-essentiality does not necessarily preclude clinical targeting, since it is difficult to distinguish addiction to overexpression or activation above basic levels from essential housekeeping functions based on CRISPR screens alone.
Fig. 1.
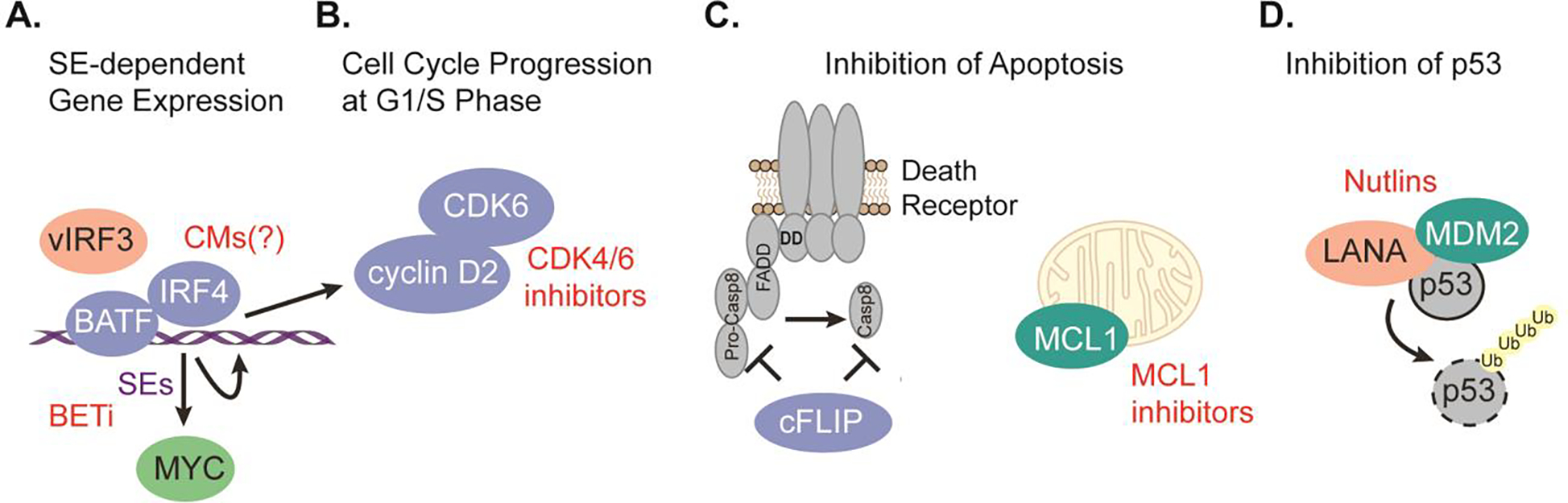
Schematic of the roles of key cellular gene dependencies in PEL cell lines. (A) PEL cell lines depend on the PSODs IRF4 and BATF to drive super-enhancer-mediated expression of common essential MYC, and PSODs IRF4 and CCND2. vIRF3 is involved at least in the regulation of the IRF4-SE. SE-mediated gene expression can be disrupted using BET inhibitors (BETi), while IRF4 can potentially be targeted using CMs (see also Fig 2C). (B) CCND2 dependency most likely involves CDK6 and can be targeted using cyclin-dependent kinase inhibitors. (C) PEL cell lines depend on the PSOD CFLAR, potentially downstream of death receptor signaling, and the skewed pan-essential gene MCL1, which prevents apoptosis via the mitochondrial pathway. MCL1 can be targeted using specific inhibitors. (D) PEL cell lines depend on the p53 inhibitor MDM2. LANA is known to participate in a complex with MDM2 and p53 and contribute to p53 inhibition. The interaction between p53 and MDM2 can be disrupted using Nutlins. Proteins encoded by selectively essential genes are in blue, those encoded by pan-essential genes are in green and essential viral proteins are in orange. Ub: ubiquitin.
Since our study in 2018, the DepMap Project has greatly improved the definition of pan-essential genes. Based on these data, 146 of the 210 previously defined PSODs are now designated pan-essential, while 58 still meet our criteria for PSODs (median FDR-adj. p ≤ 0,05 cutoff in the 8 PEL screens, not pan-essential), and 6 genes have unknown status (see Supplementary Table 1). In addition, 18 genes that were not previously included now meet our criteria for PSODs. The 76 “updated PSODs” still include IRF4, BATF, CCND2, and CFLAR. MDM2 and MCL1 are now considered be pan-essential, however, they also belong to a subset of 86 pan-essential genes with highly skewed gene effect distributions in DepMap, i.e. a subset of cell lines display a greater dependency compared to the majority of cancer cell lines. Based on their validated highly significant essentiality in PEL, a more striking dependency of PEL cells on MCL1 and MDM2 compared to most other settings is therefore likely. The known association of LANA with p53 and MDM2 furthermore underscores the critical importance of MDM2 in PEL cell biology. Importantly, MDM2 can in principle be targeted by small molecule inhibitors [11,15,42]. The former PSOD adenylate kinase 2 (AK2) is third example of a gene that is now designated pan-essential but has a skewed essentiality profile. AK2 would be interesting to investigate in PEL, because high levels of adenylate kinase expression in plasma cell malignancies can be exploited for therapeutic intervention [43]. Future studies should investigate the updated PSODs for their roles in PEL, since these genes are particularly likely to include highly specific targets for therapy and to capture the unique biology of PEL. Interestingly, both the previous and updated PSODs include the E3 ubiquitin ligase TRIM43, which has recently been shown to restrict KSHV reactivation [44]. TRIM43 essentiality is detected in only few (9/1070) DepMap screens and could be due to a KSHV reactivation phenotype.
A comparison of non-pan-essential gene dependencies of PEL to those of other transformed cell types could point to commonalities or differences in how oncogenic transformation is achieved that inform future research. It could also help predict whether therapeutic strategies that are in use for other cancers could be effective in PEL. In the following sections, we will provide a limited comparison of gene essentiality in PEL with other cell types that all exhibit dependency on IRF4 as a master transcription factor.
Unique and shared dependencies in virally transformed hematologic cell lines
Besides KSHV, the human tumor viruses EBV and HTLV-1 cause hematologic cancers. EBV causes the EBV-associated subset of Burkitt’s lymphomas and lymphomas in the context of immunodeficiency and/or HIV-infection [45]. In contrast to KSHV, EBV transforms primary human B cells in vitro into lymphoblastoid cell lines (LCLs). LCLs express the full set of EBV latency genes (latency III), similarly to EBV-associated lymphomas in immunodeficient patients. LCLs exhibit dependencies on the expression of several EBV oncoproteins. LCLs share dependencies on IRF4, BATF, CCND2, and CFLAR with PEL, but also have distinct dependencies, for example for genes involved in the activation of PI3K signaling by LMP2A (SYK, BTK, CD19, CD81) [46]. CFLAR dependency of LCLs is overcome by antibody-based inhibition of TNF-α or genetic inactivation of the TNFα receptor (TNFRSF1A) [46]. HTLV-1 causes adult T-cell leukemia/lymphoma (ATL, or ATLL) [47,48]. ATLL cell lines depend on the expression of HTLV-1 HBZ protein, which is the only viral protein that is expressed in all ATLL tumors [49]. Gene essentiality screens in three ATLL cell lines have identified dependencies on IRF4, CCND2, and CFLAR that are shared with PEL and LCLs [49,50]. It is likely that LCLs, PEL, and ATLL also share a dependency on CDK6, although CDK6 narrowly missed our stringent statistical cut-off in PEL (median FDR-adj. p~0.052), and it was shown that LCLs and ATLL cell lines are sensitive to CDK4/6 inhibition [46,50]. ATLL cell lines additionally have single gene dependencies that are not shared with PEL and LCL, for example on BATF3 and JUNB (see below), as well as components of the JAK/STAT signaling pathway (STAT3 and IL10RB).
IRF4 is a transcriptional master regulator in PEL and other IRF4-dependent cancers
The invariant requirement for IRF4 places PEL into a group of IRF4-dependent hematologic cancers that includes multiple myeloma (MM) [51], ATLL [49], the activated B cell-like subtype of diffuse large B cell lymphoma (ABC-DLBCL) [52,53], and anaplastic large cell lymphoma [54]. While IRF4 overexpression can result from genetic mutations or translocations, recent studies suggest that IRF4 overexpression in KSHV or HTLV-1-associated hematological cancers and LCLs results from viral oncogene expression [35,46,49]. IRF4 is important at several stages of B and T cell differentiation, including plasma cell differentiation following IRF4-induced upregulation of PRDM1 [55,56]. In IRF4 dependent cancer cell lines, IRF4 is involved in super-enhancer (SE)-mediated gene expression. SEs are extended regions of enhancer elements that drive high expression of genes that specify cell identity in normal cells and oncogenic programs in cancer [57]. In PEL cell lines IRF4 occupies most SEs and IRF4-dependency has been validated for the SEs that promote the expression of IRF4 itself, CCND2, and MYC [35,58,59]. In PEL cells, IRF4 cooperates with BATF and the viral vIRF3 on several sites, including the IRF4-SE in the DUSP22 locus (Fig. 1A) [35]. Viral oncoproteins also deregulate IRF4 expression and function in LCLs and ATLL. In LCLs, IRF4 and BATF co-occupy SEs together with the EBV EBNA2/3A/3C/LP transcription factors, to drive overexpression of IRF4, MYC and other genes [46,60–63]. A subset of the EBV-SEs overlaps IRF4-occupied SEs in PEL. In LCLs, IRF4 and BATF additionally repress the BCL2L11 locus [46], which encodes pro-apoptotic Bim. The transcriptional program in LCLs differs from that in PEL in that LCL require NF-κB transcription factors, RBPJ and IRF2, which have only weak and variable (NF-κB) or undetectable (RBPJ, IRF2) requirements in PELs. In LCLs, one essential role of IRF2 is to repress the IRF4 effector PRDM1, since inactivation of PRDM1 partially rescued LCLs from IRF2 dependency [46]. Inhibition of PRDM1 may at least in part be required to prevent EBV reactivation during plasma cell differentiation. Interestingly, In ATLL, HBZ cooperates with BATF3/JUNB complexes to promote IRF4 expression and BATF3 and IRF4 promote the SE-dependent expression of MYC and other genes [49,50]. In MM, IRF4 expression depends on the cellular transcription factors IKZF1/3 and IRF4 cooperates with FLI1 on SEs [64–66]. Interestingly, MM cell lines exhibit a dependency on PRDM1, likely reflecting their advanced B cell differentiation stage. In sum, studies of IRF4-dependent lymphoid cancer cells have identified key similarities in the role of IRF4 in SE-mediated gene expression, but also important differences in how IRF4 overexpression is achieved and how downstream effects are modulated.
Targeting IRF4 dependency for therapeutic intervention
SE-dependent gene expression can be disrupted by inhibitors of BET family proteins [67], which recognize acetylated histones present in SEs. SEs drive important essential genes (e.g. IRF4, CCND2, MYC) in PEL cells and accordingly BET-inhibitors kill PEL cells in vitro and in vivo [35,41,59]. Similar findings have been reported for LCLs, ATLL, and other IRF4-dependent cancers [49,63]. In MM and DLBCL, IRF4 dependency can furthermore be targeted using cereblon modulators (CMs), including lenalidomide and pomalidomide. CMs bind the substrate recognition pocket of cereblon (CRBN), a substrate adaptor of cullin 4 ring E3 ubiquitin ligase complexes (CRL4), and redirect CRCL4CRBN to neosubstrates, resulting in their polyubiquitination and proteasomal degradation (Fig. 2A). CM toxicity in MM and DLBCL can be explained due to degradation of the neosubstrates IKZF1/3, which are in turn required for expression of IRF4 (Fig. 2B) [64,65,68]. In 5q-myelodysplatic syndrome, where the haploid expression of the pomalidomide-CRCL4CRBN neosubstrate casein kinase 1α (CK1α) creates a therapeutic window [69], the mechanism of CM action is independent of IRF4. It has been shown that CMs are toxic in PEL cell lines [40]. While an IKZF1-IRF4 axis was initially proposed to operate in PEL [40], our subsequent study failed to confirm dependencies of PEL cell lines on IKZF1 or IKZF3 (Fig. 2C) [70]. Although IRF4 expression is reduced after CM treatment of PEL cell lines, the underlying mechanisms are unknown. CK1α is pan-essential and was validated as an essential gene in PEL cell lines [70]. However, although CM toxicity depends on CRL4CRBN [71], even the combined re-expression of CK1α and IRF4 provided only partial rescue from toxicity [70]. Thus, the complete set of relevant neosubstrates and the mechanism of reduced IRF4 expression in CM treated PEL cells remain unknown. In addition to its toxicity, pomalidomide also has immunomodulatory properties in PEL [72]. Future studies and an ongoing clinical trial that includes lenalidomide in combination with cytotoxic chemotherapy (ClinicalTrials.gov Identifier: NCT02911142) should establish if CMs represent a viable treatment strategy in PEL.
Fig. 2. Mechanisms of CM toxicity.
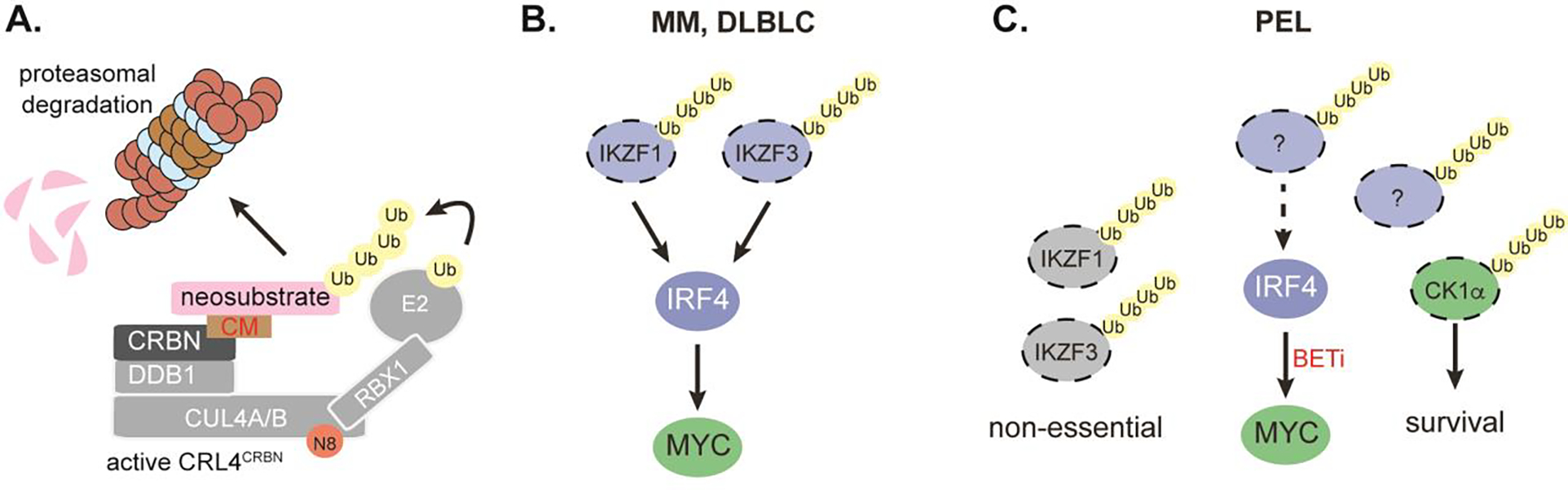
(A) CMs bind to the substrate binding pocket of the cereblon (CRBN) substrate adaptor of the CRL4 E3 ubiquitin ligase complex and lead to recruitment of neosubstrates, which undergo CRL4CRBN-mediated polyubiquitination and subsequent proteasomal degradation. Cullin 4A/B, DDB1, and RBX1 are other components of CRL4CRBN, which also requires neddylation (N8) for activity. RBX1 recruits an E2 ubiquitin ligase to the complex. (B) In multiple myeloma (MM) and DLBCL, the CM-induced degradation of IKZF1/3 leads to downregulation of IRF4 and MYC and fully accounts for CM toxicity. (C) In PEL, CM-induced degradation of IKZF1/3 is observed, but inconsequential. CM-induced degradation of CK1α is toxic. IRF4 down-downregulation is observed and depends on CRL4CRBN, but the underlying mechanisms are indirect and unknown. Combined restoration of IRF4 and CK1α expression is insufficient to rescue CM toxicity, pointing to additional mechanisms of CM toxicity. Proteins encoded by selectively essential genes are in blue, those encoded by pan-essential genes are in green. Neosubstrates bear ubiquitin (Ub) chains.
Summary and Key Questions for future studies
Recent studies have provided important insights into human gene essentiality in various cancers, including those driven by viral infection. In PEL, these studies have identified key dependencies on IRF4, BATF, CCND2, CFLAR, MDM2, and MCL1. Future work should seek to further link cellular and viral gene dependencies and to elucidate underlying molecular functions. It will also be interesting to compare mechanisms of KSHV-mediated transformation in PEL with that in other models of KSHV-mediated transformation, such as KSHV-transformed rat mesenchymal cells and endothelial cell models of KS [73,74]. Finally, genetic vulnerabilities should continue to be considered for development of therapeutic strategies.
Supplementary Material
(A) List of the original 210 PSODs including their updated designations as pan-essential genes, updated PSODs, pan-essential genes with skewed gene effect distributions, or unknowns. (B) List of the 76 updated PSODs, including a column to indicate whether these genes were originally designated as PSODs. (C) List of genes that are pan-essential but have highly skewed gene effect distributions in DepMap and score as essential in the 8 PEL screens (median FDR-adj. p ≤ 0.05). These genes are candidates for preferentially essential pan-essential genes in PEL. Based on Manzano et al. (2018) [34] and DepMap release 2022, quarter 1.
Acknowledgements
We apologize to the authors of many important studies that could not be cited due to space limitations. We would like to thank Dr. Masao Nakagawa for helpful discussions. This work was supported by NCI R01 CA247619 and R01 CA247619-01A1S1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM: Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 1995, 332:1186–1191. [DOI] [PubMed] [Google Scholar]
- 2.Nador RG, Cesarman E, Chadburn A, Dawson DB, Ansari MQ, Sald J, Knowles DM: Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus. Blood 1996, 88:645–656. [PubMed] [Google Scholar]
- 3. Cesarman E, Chadburn A, Rubinstein PG: KSHV/HHV8-mediated hematologic diseases. Blood 2022, 139:1013–1025. **This review includes an up to date discussion of the features and treatment of PEL.
- 4.Wen KW, Wang L, Menke JR, Damania B: Cancers associated with human gammaherpesviruses. FEBS J 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kliche S, Kremmer E, Hammerschmidt W, Koszinowski U, Haas J: Persistent infection of Epstein-Barr virus-positive B lymphocytes by human herpesvirus 8. J Virol 1998, 72:8143–8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trivedi P, Takazawa K, Zompetta C, Cuomo L, Anastasiadou E, Carbone A, Uccini S, Belardelli F, Takada K, Frati L, et al. : Infection of HHV-8+ primary effusion lymphoma cells with a recombinant Epstein-Barr virus leads to restricted EBV latency, altered phenotype, and increased tumorigenicity without affecting TCL1 expression. Blood 2004, 103:313–316. [DOI] [PubMed] [Google Scholar]
- 7.McHugh D, Caduff N, Barros MHM, Ramer PC, Raykova A, Murer A, Landtwing V, Quast I, Styles CT, Spohn M, et al. : Persistent KSHV Infection Increases EBV-Associated Tumor Formation In Vivo via Enhanced EBV Lytic Gene Expression. Cell Host Microbe 2017, 22:61–73 e67. [DOI] [PubMed] [Google Scholar]
- 8.Faure A, Hayes M, Sugden B: How Kaposi’s sarcoma-associated herpesvirus stably transforms peripheral B cells towards lymphomagenesis. Proc Natl Acad Sci U S A 2019, 116:16519–16528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roy D, Sin SH, Damania B, Dittmer DP: Tumor suppressor genes FHIT and WWOX are deleted in primary effusion lymphoma (PEL) cell lines. Blood 2011, 118:e32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luan SL, Boulanger E, Ye H, Chanudet E, Johnson N, Hamoudi RA, Bacon CM, Liu H, Huang Y, Said J, et al. : Primary effusion lymphoma: genomic profiling revealed amplification of SELPLG and CORO1C encoding for proteins important for cell migration. J Pathol 2010, 222:166–179. [DOI] [PubMed] [Google Scholar]
- 11.Petre CE, Sin SH, Dittmer DP: Functional p53 signaling in Kaposi’s sarcoma-associated herpesvirus lymphomas: implications for therapy. J Virol 2007, 81:1912–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boulanger E, Marchio A, Hong SS, Pineau P: Mutational analysis of TP53, PTEN, PIK3CA and CTNNB1/beta-catenin genes in human herpesvirus 8-associated primary effusion lymphoma. Haematologica 2009, 94:1170–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Platt G, Carbone A, Mittnacht S: p16INK4a loss and sensitivity in KSHV associated primary effusion lymphoma. Oncogene 2002, 21:1823–1831. [DOI] [PubMed] [Google Scholar]
- 14.Lurain K, Polizzotto MN, Aleman K, Bhutani M, Wyvill KM, Goncalves PH, Ramaswami R, Marshall VA, Miley W, Steinberg SM, et al. : Viral, immunologic, and clinical features of primary effusion lymphoma. Blood 2019, 133:1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarek G, Kurki S, Enback J, Iotzova G, Haas J, Laakkonen P, Laiho M, Ojala PM: Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. J Clin Invest 2007, 117:1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rivas C, Thlick AE, Parravicini C, Moore PS, Chang Y: Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J Virol 2001, 75:429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Godfrey A, Anderson J, Papanastasiou A, Takeuchi Y, Boshoff C: Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood 2005, 105:2510–2518. [DOI] [PubMed] [Google Scholar]
- 18.Wies E, Mori Y, Hahn A, Kremmer E, Sturzl M, Fleckenstein B, Neipel F: The viral interferon-regulatory factor-3 is required for the survival of KSHV-infected primary effusion lymphoma cells. Blood 2008, 111:320–327. [DOI] [PubMed] [Google Scholar]
- 19.Ju E, Li T, Liu Z, da Silva SR, Wei S, Zhang X, Wang X, Gao SJ: Specific Inhibition of Viral MicroRNAs by Carbon Dots-Mediated Delivery of Locked Nucleic Acids for Therapy of Virus-Induced Cancer. ACS Nano 2020, 14:476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Happel C, Ziegelbauer JM: Kaposi’s Sarcoma-Associated Herpesvirus MicroRNAs Target GADD45B To Protect Infected Cells from Cell Cycle Arrest and Apoptosis. J Virol 2017, 91:e02045–02016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi JT, Braich R, Manoharan M, Soutschek J, Ohler U, et al. : A viral microRNA functions as an orthologue of cellular miR-155. Nature 2007, 450:1096–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skalsky RL, Samols MA, Plaisance KB, Boss IW, Riva A, Lopez MC, Baker HV, Renne R: Kaposi’s sarcoma-associated herpesvirus encodes an ortholog of miR-155. J Virol 2007, 81:12836–12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boss IW, Nadeau PE, Abbott JR, Yang Y, Mergia A, Renne R: A Kaposi’s sarcoma-associated herpesvirus-encoded ortholog of microRNA miR-155 induces human splenic B-cell expansion in NOD/LtSz-scid IL2Rgammanull mice. J Virol 2011, 85:9877–9886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dahlke C, Maul K, Christalla T, Walz N, Schult P, Stocking C, Grundhoff A: A microRNA Encoded by Kaposi Sarcoma-Associated Herpesvirus Promotes B-Cell Expansion In Vivo. PLoS One 2012, 7:e49435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bravo Cruz AG, Damania B: In Vivo Models of Oncoproteins Encoded by Kaposi’s Sarcoma-Associated Herpesvirus. J Virol 2019, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bigi R, Landis JT, An H, Caro-Vegas C, Raab-Traub N, Dittmer DP: Epstein-Barr virus enhances genome maintenance of Kaposi sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A 2018, 115:E11379–E11387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanjana NE, Shalem O, Zhang F: Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014, 11:783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. : Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM: Identification and characterization of essential genes in the human genome. Science 2015, 350:1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME, Vaimberg EW, Goodale A, Root DE, Piccioni F, et al. : Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat Commun 2018, 9:5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. : High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163:1515–1526. [DOI] [PubMed] [Google Scholar]
- 32. Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. : Defining a Cancer Dependency Map. Cell 2017, 170:564–576 e516. ** The DepMap project represents the most comprehensive study of gene essentiality. DepMap datasets were used in preparation of this review and can be found on here: https://depmap.org/portal/
- 33.Dempster JM, Pacini C, Pantel S, Behan FM, Green T, Krill-Burger J, Beaver CM, Younger ST, Zhivich V, Najgebauer H, et al. : Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat Commun 2019, 10:5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Manzano M, Patil A, Waldrop A, Dave SS, Behdad A, Gottwein E: Gene essentiality landscape and druggable oncogenic dependencies in herpesviral primary effusion lymphoma. Nat Commun 2018, 9:3263. ** Original study of gene essentiality in PEL. The updated list of PSODs is based on the screens in this study.
- 35. Manzano M, Gunther T, Ju H, Nicholas J, Bartom ET, Grundhoff A, Gottwein E: Kaposi’s Sarcoma-Associated Herpesvirus Drives a Super-Enhancer-Mediated Survival Gene Expression Program in Primary Effusion Lymphoma. mBio 2020, 11. *This study implicates vIRF3, IRF4, and BATF in SE-mediated gene expression in PEL.
- 36.Wu Y, Shrestha P, Heape NM, Yarchoan R: CDK4/6 inhibitors sensitize gammaherpesvirus-infected tumor cells to T-cell killing by enhancing expression of immune surface molecules. J Transl Med 2022, 20:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quentmeier H, Geffers R, Hauer V, Nagel S, Pommerenke C, Uphoff CC, Zaborski M, Drexler HG: Inhibition of MCL1 induces apoptosis in anaplastic large cell lymphoma and in primary effusion lymphoma. Sci Rep 2022, 12:1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sin SH, Roy D, Wang L, Staudt MR, Fakhari FD, Patel DD, Henry D, Harrington WJ Jr., Damania BA, Dittmer DP: Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling. Blood 2007, 109:2165–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uddin S, Hussain AR, Al-Hussein KA, Manogaran PS, Wickrema A, Gutierrez MI, Bhatia KG: Inhibition of phosphatidylinositol 3’-kinase/AKT signaling promotes apoptosis of primary effusion lymphoma cells. Clin Cancer Res 2005, 11:3102–3108. [DOI] [PubMed] [Google Scholar]
- 40.Gopalakrishnan R, Matta H, Tolani B, Triche T Jr., Chaudhary PM: Immunomodulatory drugs target IKZF1-IRF4-MYC axis in primary effusion lymphoma in a cereblon-dependent manner and display synergistic cytotoxicity with BRD4 inhibitors. Oncogene 2016, 35:1797–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tolani B, Gopalakrishnan R, Punj V, Matta H, Chaudhary PM: Targeting Myc in KSHV-associated primary effusion lymphoma with BET bromodomain inhibitors. Oncogene 2014, 33:2928–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mullard A: p53 programmes plough on. Nat Rev Drug Discov 2020, 19:497–500. [DOI] [PubMed] [Google Scholar]
- 43.Nayar U, Sadek J, Reichel J, Hernandez-Hopkins D, Akar G, Barelli PJ, Sahai MA, Zhou H, Totonchy J, Jayabalan D, et al. : Identification of a nucleoside analog active against adenosine kinase-expressing plasma cell malignancies. J Clin Invest 2017, 127:2066–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Full F, van Gent M, Sparrer KMJ, Chiang C, Zurenski MA, Scherer M, Brockmeyer NH, Heinzerling L, Sturzl M, Korn K, et al. : Centrosomal protein TRIM43 restricts herpesvirus infection by regulating nuclear lamina integrity. Nat Microbiol 2019, 4:164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shannon-Lowe C, Rickinson A: The Global Landscape of EBV-Associated Tumors. Front Oncol 2019, 9:713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ma Y, Walsh MJ, Bernhardt K, Ashbaugh CW, Trudeau SJ, Ashbaugh IY, Jiang S, Jiang C, Zhao B, Root DE, et al. : CRISPR/Cas9 Screens Reveal Epstein-Barr Virus-Transformed B Cell Host Dependency Factors. Cell Host Microbe 2017, 21:580–591 e587. ** Original study of gene essentiality in LCLs.
- 47.Miura M, Naito T, Saito M: Current Perspectives in Human T-Cell Leukemia Virus Type 1 Infection and Its Associated Diseases. Front Med (Lausanne) 2022, 9:867478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watanabe T: Adult T-cell leukemia: molecular basis for clonal expansion and transformation of HTLV-1-infected T cells. Blood 2017, 129:1071–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nakagawa M, Shaffer AL 3rd, Ceribelli M, Zhang M, Wright G, Huang DW, Xiao W, Powell J, Petrus MN, Yang Y, et al. : Targeting the HTLV-I-Regulated BATF3/IRF4 Transcriptional Network in Adult T Cell Leukemia/Lymphoma. Cancer Cell 2018, 34:286–297 e210. ** This study identifies roles for HBZ, IRF4, and BATF in SE-mediated gene regulation in ATLL.
- 50. Ishio T, Kumar S, Shimono J, Daenthanasanmak A, Dubois S, Lin Y, Bryant B, Petrus MN, Bachy E, Huang DW, et al. : Genome-wide CRISPR screen identifies CDK6 as a therapeutic target in adult T-cell leukemia/lymphoma. Blood 2022, 139:1541–1556. ** Original study of gene essentiality in ATLL
- 51.Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H, et al. : IRF4 addiction in multiple myeloma. Nature 2008, 454:226–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang Y, Shaffer AL 3rd, Emre NC, Ceribelli M, Zhang M, Wright G, Xiao W, Powell J, Platig J, Kohlhammer H, et al. : Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell 2012, 21:723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Care MA, Cocco M, Laye JP, Barnes N, Huang Y, Wang M, Barrans S, Du M, Jack A, Westhead DR, et al. : SPIB and BATF provide alternate determinants of IRF4 occupancy in diffuse large B-cell lymphoma linked to disease heterogeneity. Nucleic Acids Res 2014, 42:7591–7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schleussner N, Merkel O, Costanza M, Liang HC, Hummel F, Romagnani C, Durek P, Anagnostopoulos I, Hummel M, Johrens K, et al. : The AP-1-BATF and -BATF3 module is essential for growth, survival and TH17/ILC3 skewing of anaplastic large cell lymphoma. Leukemia 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, Ludwig T, Rajewsky K, Dalla-Favera R: Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol 2006, 7:773–782. [DOI] [PubMed] [Google Scholar]
- 56.Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H: Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity 2006, 25:225–236. [DOI] [PubMed] [Google Scholar]
- 57.Sur I, Taipale J: The role of enhancers in cancer. Nat Rev Cancer 2016, 16:483–493. [DOI] [PubMed] [Google Scholar]
- 58. Park A, Oh S, Jung KL, Choi UY, Lee HR, Rosenfeld MG, Jung JU: Global epigenomic analysis of KSHV-infected primary effusion lymphoma identifies functional MYC superenhancers and enhancer RNAs. Proc Natl Acad Sci U S A 2020, 117:21618–21627. *This study implicates defines the MYC SE in PEL
- 59. Wang C, Zhang L, Ke L, Ding W, Jiang S, Li D, Narita Y, Hou I, Liang J, Li S, et al. : Primary effusion lymphoma enhancer connectome links super-enhancers to dependency factors. Nat Commun 2020, 11:6318. *This study implicates IRF4 in SE-mediated gene expression in PEL and estbalishes super-enhancer connectivities.
- 60.Jiang S, Zhou H, Liang J, Gerdt C, Wang C, Ke L, Schmidt SCS, Narita Y, Ma Y, Wang S, et al. : The Epstein-Barr Virus Regulome in Lymphoblastoid Cells. Cell Host Microbe 2017, 22:561–573 e564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmidt SC, Jiang S, Zhou H, Willox B, Holthaus AM, Kharchenko PV, Johannsen EC, Kieff E, Zhao B: Epstein-Barr virus nuclear antigen 3A partially coincides with EBNA3C genome-wide and is tethered to DNA through BATF complexes. Proc Natl Acad Sci U S A 2015, 112:554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang S, Willox B, Zhou H, Holthaus AM, Wang A, Shi TT, Maruo S, Kharchenko PV, Johannsen EC, Kieff E, et al. : Epstein-Barr virus nuclear antigen 3C binds to BATF/IRF4 or SPI1/IRF4 composite sites and recruits Sin3A to repress CDKN2A. Proc Natl Acad Sci U S A 2014, 111:421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou H, Schmidt SC, Jiang S, Willox B, Bernhardt K, Liang J, Johannsen EC, Kharchenko P, Gewurz BE, Kieff E, et al. : Epstein-Barr virus oncoprotein super-enhancers control B cell growth. Cell Host Microbe 2015, 17:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, Kaelin WG Jr.: The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343:305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al. : Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343:301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jin Y, Chen K, De Paepe A, Hellqvist E, Krstic AD, Metang L, Gustafsson C, Davis RE, Levy YM, Surapaneni R, et al. : Active enhancer and chromatin accessibility landscapes chart the regulatory network of primary multiple myeloma. Blood 2018, 131:2138–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA: Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153:320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mo Z, Wood S, Namiranian S, Mizukoshi R, Weng S, Jang IS, Fontanillo C, Baughman JM, Silva-Torres A, Slade M, et al. : Deciphering the mechanisms of CC-122 resistance in DLBCL via a genome-wide CRISPR screen. Blood Adv 2021, 5:2027–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kronke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, Chamberlain PP, Mani DR, Man HW, Gandhi AK, et al. : Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 2015, 523:183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patil A, Manzano M, Gottwein E: CK1alpha and IRF4 are essential and independent effectors of immunomodulatory drugs in primary effusion lymphoma. Blood 2018, 132:577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patil A, Manzano M, Gottwein E: Genome-wide CRISPR screens reveal genetic mediators of cereblon modulator toxicity in primary effusion lymphoma. Blood Adv 2019, 3:2105–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shrestha P, Davis DA, Jaeger HK, Stream A, Aisabor AI, Yarchoan R: Pomalidomide restores immune recognition of primary effusion lymphoma through upregulation of ICAM-1 and B7–2. PLoS Pathog 2021, 17:e1009091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gruffaz M, Yuan H, Meng W, Liu H, Bae S, Kim JS, Lu C, Huang Y, Gao SJ: CRISPR-Cas9 Screening of Kaposi’s Sarcoma-Associated Herpesvirus-Transformed Cells Identifies XPO1 as a Vulnerable Target of Cancer Cells. mBio 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Holmes DL, Vogt DT, Lagunoff M: A CRISPR-Cas9 screen identifies mitochondrial translation as an essential process in latent KSHV infection of human endothelial cells. Proc Natl Acad Sci U S A 2020, 117:28384–28392. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) List of the original 210 PSODs including their updated designations as pan-essential genes, updated PSODs, pan-essential genes with skewed gene effect distributions, or unknowns. (B) List of the 76 updated PSODs, including a column to indicate whether these genes were originally designated as PSODs. (C) List of genes that are pan-essential but have highly skewed gene effect distributions in DepMap and score as essential in the 8 PEL screens (median FDR-adj. p ≤ 0.05). These genes are candidates for preferentially essential pan-essential genes in PEL. Based on Manzano et al. (2018) [34] and DepMap release 2022, quarter 1.