

Abstract
Introduction
We sought insights into neuromyelitis optica spectrum disorder (NMOSD) treatment practices worldwide.
Methods
Neurologists from the USA, Germany, Italy, Brazil, South Korea, and China completed an online survey, contributing clinical records for aquaporin-4 (AQP4) immunoglobulin G (IgG)-seropositive adults with NMOSD, which included patient demographics, diagnosis, maintenance treatment history, relapse occurrence, and severity. Interviewed patients receiving NMOSD maintenance therapy provided information about their diagnosis, treatment, perceptions about relapse severity or disease stability, and treatment switches.
Results
A total of 389 neurologists submitted clinical records for 1185 patients with AQP4-IgG-seropositive NMOSD; 33 patients with NMOSD were interviewed. Approximately 25% (228/910) of patients from the clinical record review (CRR) were initially misdiagnosed; 24% (8/33) of patients interviewed reported formal misdiagnosis. Misdiagnosis was associated with treatment delay and more relapses compared with correct diagnosis (mean 3.3 vs 2.8). Maintenance therapy was not initiated within 2 months for 47% (221/472) of patients from the CRR and 24% (8/33) of interviewed patients. Oral corticosteroids/immunosuppressive therapies were typically the first maintenance treatment initiated, except for the USA, where monoclonal antibodies were equally likely to be prescribed. Relapse severity influenced the decision to initiate/change therapy and use monoclonal antibodies. Of interviewed patients, 76% (25/33) did not recall having a choice of treatment and many did not know the rationale for treatment choice.
Conclusion
Misdiagnosis of NMOSD appears to be common and is associated with a delay in initiation of maintenance therapy, with decisions influenced by relapse severity. Further real-world studies assessing relapse severity in treatment initiation/switch are required to revise NMOSD treatment recommendations.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40120-022-00431-y.
Keywords: Clinical practice, Medical care, Neuromyelitis optica spectrum disorder, Optic neuritis, Real-world evidence, Relapse, Transverse myelitis, Treatment
Key Summary Points
| Why carry out this study? |
| Although current NMOSD treatment guidelines were last updated prior to the approval of several monoclonal antibody drugs, their availability has made decisions around treatment initiation and switch increasingly complex. |
| This study sought to develop a clearer understanding of patient characteristics and other drivers behind treatment initiation, treatment choice, and switch in clinical practice through a global clinical record review and patient interviews. |
| What was learned from the study? |
| Misdiagnosis of NMOSD is common (25% [228/910] of patients from the clinical record review were initially misdiagnosed) and was a factor associated with treatment delay and more frequent relapses (mean 3.3 vs 2.8) compared with correct diagnosis. |
| Oral corticosteroids/immunosuppressive therapies were typically the first-line maintenance therapy of choice for the majority of neurologists, and relapse severity was a key factor in treatment decisions, including use of monoclonal antibodies. |
| This research provides insights into real-world factors driving treatment decisions in this rare disease and highlights the need for further real-world studies assessing relapse severity as a factor in treatment decisions to guide NMOSD treatment recommendations. |
Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is a rare demyelinating disorder of the central nervous system, which is primarily characterized by a relapsing disease course with episodes of optic neuritis and longitudinal extensive transverse myelitis [1, 2]. The discovery of pathogenic aquaporin-4 immunoglobulin G (AQP4-IgG) autoantibodies that target astrocyte water channels and activate the classical complement cascade leading to astrocyte death, demyelination, and subsequent neuronal loss helped to establish NMOSD as distinct from multiple sclerosis (MS) [1, 3–5], leading to revised diagnostic criteria [6].
Relapses in patients with NMOSD can be devastating and lead to bilateral blindness, complete motor paralysis, long-term disability, and, in some cases, death [4, 7, 8]. Relapse prevention is the primary therapeutic goal in NMOSD [9, 10], with the first NMOSD treatment being approved in 2019 [11, 12]. Off-label immunosuppressive therapies (ISTs) and corticosteroids were (and in some regions continue to be) the mainstay of treatment [10–13]. At the time of writing, three monoclonal antibodies have been approved for the treatment of AQP4-IgG-seropositive NMOSD: the anti-complement C5 antibody eculizumab, the anti-CD19 antibody inebilizumab, and the anti-interleukin-6 receptor antibody satralizumab [11, 12, 14–17].
The availability of additional treatment options for NMOSD has meant that treatment choice and decisions around treatment initiation/switch have become increasingly complex. Therefore, a global clinical record review and patient interviews were conducted to develop a clearer understanding of patient characteristics and other drivers behind treatment initiation, treatment choice, and switch in clinical practice.
Methods
Study Design
In this cross-sectional study, neurologists from the USA, Germany, Italy, Brazil, South Korea, and China participated in a 30–60-minute online survey, during which they contributed a minimum of 2–4 patient records relating to adult patients with AQP4-IgG-seropositive NMOSD. Neurologists were recruited via an International Organization for Standardization-certified medical research panel from a range of hospitals (Table 1) and screened on the basis of the following criteria: specializing in neurology for 2–35 years; being responsible for the management and treatment of their patients with NMOSD (not solely maintaining a therapeutic regimen prescribed by another healthcare provider); the number of patients with NMOSD under their care for the last 12 months (≥ 5 in the USA, Brazil, and Italy; ≥ 10 in China; ≥ 3 in Germany and South Korea), at least two of whom were required to be AQP4-IgG-seropositive; some of their patients with AQP4-IgG-seropositive NMOSD receiving maintenance therapy; and their patients with AQP4-IgG-seropositive NMOSD being either newly diagnosed or having undergone a maintenance therapy change within the last 2 years. Data were quality checked daily during fieldwork and removed from the sample if quality control measures were not met (e.g., short interview length, nonsensical responses, repeated selection of the same responses, etc.).
Table 1.
Geographical distribution of neurologists participating in the online survey and patients participating in telephone interviews
| USA | Germany | Italy | Brazil | South Korea | China | |
|---|---|---|---|---|---|---|
| Neurologists (N = 389) | ||||||
| Neurologists, n | 100 | 60 | 51 | 51 | 50 | 77 |
| Type of hospital/practice neurologists were recruited from, n (%) | ||||||
| Academic hospital, n (%) | 38 (38) | 32 (53) | 40 (78) | – | 19 (38) | – |
| Non-academic hospital, n (%) | 7 (7) | 6 (10) | 10 (20) | – | 7 (14) | – |
| Group practice/office, n (%) | 44 (44) | 14 (23) | 0 (0) | – | 12 (24) | – |
| Solo practice/office, n (%) | 11 (11) | 8 (13) | 1 (2) | – | 12 (24) | – |
| Private practice (Brazil only), n (%) | – | – | – | 11 (22) | – | – |
| Public practice (Brazil only), n (%) | – | – | – | 3 (6) | – | – |
| Private and public practice (Brazil only), n (%) | – | – | – | 37 (73) | – | – |
| Tier 3A hospitala (China only), n (%) | – | – | – | – | – | 77 (100) |
| Patient clinical records, n | 293 | 170 | 181 | 165 | 153 | 223 |
| Patient proportion of total sample, % | 25% | 14% | 15% | 14% | 13% | 19% |
| Proportion of patients misdiagnosed prior to NMOSD diagnosis, n (%) | 45 (21) | 39 (30) | 40 (26) | 34 (26) | 6 (5) | 64 (39) |
| Patients participating in telephone interviews (N = 33) | ||||||
| Patients, n | 8 | 5 | 5 | 5 | 5 | 5 |
| Proportion of total sample, % | 24% | 15% | 15% | 15% | 15% | 15% |
NMOSD, neuromyelitis optica spectrum disorder
aTier 3 hospitals in China are medical centers with comprehensive medical, teaching, and scientific research capabilities, a bed capacity exceeding 500, and provide high-level specialist care across regions, cities, and provinces. Based on the level of service provision, size, medical technology, medical equipment, and management and medical quality, these three grades are further subdivided into three subsidiary levels: A, B, and C
A separate sample of patients with NMOSD was also recruited to participate in 30-minute qualitative telephone interviews to understand their perceptions of their NMOSD and its management. None of the patients included in the qualitative interviews were the same patients as those included in the clinical record review. Patients were recruited by phone and email from the same six countries as the participating neurologists via physician referrals, patient panels, and patient groups/associations. Data were quality checked by patient engagement and whether their responses during the interview matched those during screening. Both neurologists and patients received an honorarium for their participation.
At the time of the clinical record review, satralizumab had not been approved in any of the six assessed countries, whereas eculizumab had been approved in the USA and the EU [18, 19], and inebilizumab had been approved in the USA [15]. Satralizumab was approved by the US Food and Drug Administration on 14 August 2020, 2–3 months prior to the patient interviews [16].
Study Population
Participating neurologists submitted clinical records for patients at least 18 years old with confirmed NMOSD who tested seropositive for AQP4-IgG and were (1) newly diagnosed with NMOSD within the last 2 years, but not the last 3 months; (2) diagnosed with NMOSD for at least 2 years and had their maintenance therapy changed within the past 2 years (treatment switch or add-on); or (3) the most recent patients with NMOSD seen by the neurologist. Newly diagnosed patients were included to understand decisions around treatment initiation, drivers for choice of first maintenance therapy, and triggers to initiate therapy. Patients diagnosed with NMOSD at least 2 years ago were included to understand triggers for switching maintenance therapy. Patients most recently seen in clinical practice were included to represent a “random” patient and ensure a representative sample for “sizing” of behaviors (i.e., how often switching occurs) or disease/patient characteristics.
Patients participating in telephone interviews were at least 18 years old, had a confirmed NMOSD diagnosis, and were receiving NMOSD maintenance therapy at the time of the interview. Patients were recruited via patient panels (USA only), patient groups or organizations, and/or physician referrals. Eligible patients were at least 18 years old and either (1) diagnosed with NMOSD within the last 2 years (newly diagnosed) or (2) diagnosed with NMOSD for at least 2 years and had a switch in maintenance therapy within the last 2 years. All patients must have had at least one relapse in the past 2 years, be receiving maintenance therapy for NMOSD, and have no work history in a health-related, pharmaceutical industry, or market-research-related field. As a result of the qualitative nature of the patient interviews, self-reported patient AQP4-IgG status was not always established.
Data Collection and Assessments
The clinical record review was designed to collect information on patient demographics, diagnosis, maintenance treatment history, and relapse severity. Time between first symptoms and diagnosis, date of diagnosis, diagnostic criteria, previous misdiagnoses, AQP4-IgG status, and timing of AQP4-IgG testing were also recorded, along with details of previous and current therapies and time of prescription (including any periods without therapy), rationale for choice of first maintenance therapy, and rationale behind each subsequent treatment selection.
Relapse history and characteristics were assessed by recording the number of relapses, dates of most recent relapse and implications (i.e., change in maintenance treatment and reason for change) and additional details, including clinical signs/symptoms, relapse duration (i.e., time between the symptom onset and maximum recovery), acute treatment at relapse, duration of hospitalization (if applicable), extent of relapse recovery, change in function/disability due to relapse, and perceived relapse severity and rationale. As no well-established definitions of relapse severity exist, the severity classifications applied by physicians completing the survey were based on clinician judgement.
Patient interviews were designed to collect information about patient journey from diagnosis to treatment, perceptions about relapse severity or disease stability, perceived barriers or drivers to treatment switches, and other factors that might influence treatment choice. The online survey used in the clinical record review and the patient interview guide are available in the Supplementary Material.
Statistical Analysis
Sample sizes were based on the maximum feasible sample of neurologists that could be achieved in each country within the 4-week fieldwork period. Statistical analysis involved cross-tabulations with significance testing. P values were calculated using SPSS software, version 26 (IBM SPSS Statistics, IBM Corporation, Armonk, NY, USA), with a two-sided significance level of p < 0.05.
The chi-squared (χ2) test was used to detect an association between two categorical variables. Pairwise comparisons of proportions were performed using a z-test when the test variable was categorical, and pairwise comparisons of means were performed using a t-test when the variable was scale based. Bonferroni corrections were conducted for the z- and t-tests.
For the qualitative patient interviews, verbatim English transcripts were first developed from the patient interview audios and analyzed via thematic analysis to identify common patterns.
Ethics Statement
This was a market research study and, as such, it was conducted in accordance with the European Pharmaceutical Market Research Association’s (EphMRA) industry code of conduct. Market research as defined in this code of conduct does not require clinical research ethics committee or independent review board approval. All subjects provided informed consent to participate in the study (in accordance with the EphMRA code of conduct), and research was compliant with all international and national data protection laws. Quote responses provided are vignettes of real answers that have been amended to maintain anonymity of respondents without altering the theme of their statements.
Results
Patient Characteristics and Geographical Distribution
The online survey was conducted between 14 July and 15 August 2020. Initially, 1097 neurologists participated in the online survey, of whom 668 completed the screening questions but did not qualify to participate on the basis of their responses and the patient types they managed (Supplementary Material). Therefore, 429 neurologists completed the survey, 40 of whom (9%) were removed during the quality check, resulting in a final sample of 389 neurologists (Table 1). These 389 neurologists provided records for 1185 AQP4-IgG-seropositive patients with NMOSD, comprising 472 patients diagnosed with NMOSD within the past 2 years (but not the last 3 months) who were either receiving their first maintenance therapy or had not yet been initiated on a maintenance therapy; 655 patients with a maintenance therapy change (switch or add-on) within the past 2 years; and 58 patients diagnosed over 2 years ago who had not changed their maintenance therapy in the past 2 years.
In the patient interviews, 53 patients were screened, and 34 patients interviewed between 20 October and 27 November 2020 (Table 1), with each interview lasting approximately 30 minutes. One patient was removed during the quality check process, as she was not receiving NMOSD maintenance therapy, leaving a final sample of 33 patients. Patient demographics are shown in the Supplementary Material. Twenty-one patients (64%) were newly diagnosed with NMOSD, and the remaining 12 patients were not newly diagnosed.
Diagnosis
Neurologist Clinical Record Review
Of patients for whom misdiagnosis information was available (n = 910), approximately 25% (228/910) of patients with NMOSD reported by neurologists were initially misdiagnosed, most commonly in China and least commonly in South Korea (Table 1). Of the 222/228 patients where the condition patients were previously misdiagnosed with was known, 44% (n = 97), 10% (n = 23), 8% (n = 17), and 7% (n = 16) were diagnosed with MS, idiopathic myelitis, optic neuritis, and stroke/transient ischemic attack. Other misdiagnoses occurred in less than 5% of patients. Of 228 patients initially misdiagnosed, 72 patients (32%) were correctly diagnosed with NMOSD within 2 months of misdiagnosis.
Misdiagnosis rates remained relatively consistent over time. Approximately one-third of patients with NMOSD diagnosed more than 36 months ago (32%; n = 59/187) were initially misdiagnosed versus 20% (n = 45/226), 26% (n = 75/289), and 24% (n = 49/208) of patients who were diagnosed 25–36 months ago, 12–24 months ago, and less than 12 months ago, respectively.
In those initially misdiagnosed, time between first observation of symptoms to initiation of first maintenance therapy was longer than for patients not misdiagnosed (21.2 vs 9.9 months; p < 0.05). Patients who were misdiagnosed experienced more relapses in total (assessed over the course of patients’ lives up to survey initiation) versus those not misdiagnosed (mean number of total relapses 3.3 vs 2.8, respectively; p < 0.05). A higher proportion of patients who were misdiagnosed had a severe attack (at their most recent relapse) versus those who were not misdiagnosed (23% vs 10%).
Patient Interviews
Prior to their NMOSD diagnosis, 60% (20/33) of patients were informally told that their symptoms were due to conditions other than NMOSD, e.g., tiredness, stress, gastric or ophthalmological issues. Of those, eight patients reported being formally misdiagnosed before their NMOSD diagnosis, typically with MS, but also optic neuritis or gastritis. Misdiagnosis led to an average 5-year delay (range 1–20 years) in receiving a correct NMOSD diagnosis, with a delay of 20 years being reported in one case. During this time, patients continued to experience relapses and disability. On receiving a diagnosis of NMOSD (following a misdiagnosis), three patients directly reported feeling angry and scared:
I was scared and angry because I had only just accepted my [wrong] diagnosis and was then diagnosed with something else. I was aware of what NMO was and was terrified of going blind.
Drivers for Treatment Initiation
Neurologist Clinical Record Review
Neurologists did not initiate maintenance therapy within 2 months of diagnosis for 47% (221/472) of newly diagnosed patients (Fig. 1i), with the main reasons being “stable” disease, patient refusal, and cost/access restrictions (Fig. 1ii). The proportion citing cost/access restrictions differed by country (Fig. 1iii).
Fig. 1.
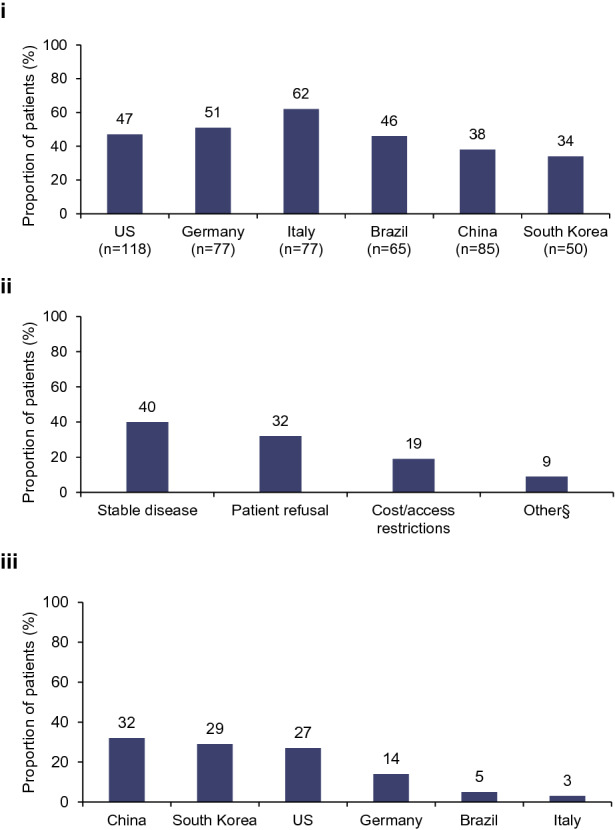
Proportion of (i) newly diagnosed patients by country not initiating therapy within 2 months of diagnosis, (ii) reasons for not initiating maintenance therapy within 2 months of diagnosis, and (iii) cost/access restrictions as a reason for newly diagnosed patients not initiating therapy within 2 months of diagnosis by country. §Other reasons included a need to wait until after acute treatment and logistics, such as scheduling infusions. Data presented in (i) are for all newly diagnosed patients who had not been initiated on therapy within 2 months of diagnosis by country as a proportion of all newly diagnosed patients (N = 472). Data presented in (ii) are reasons given for not initiating therapy within 2 months of diagnosis as a proportion of all patients that were receiving therapy at the time of the survey (N = 151); stable disease (n = 60), patient refusal (n = 49), cost/access restrictions (n = 28), other (n = 14). Data presented in (iii) are for all newly diagnosed patients receiving treatment at the time of the survey that mentioned cost/access restrictions as a reason for not being initiated on therapy within 2 months of diagnosis (N = 28); China (n = 7), South Korea (n = 4), the USA (n = 12), Germany (n = 3), Brazil (n = 1), Italy (n = 1)
There was an association between time to treatment initiation and initial attack severity (p < 0.05 for interaction). Experiencing an initial attack (i.e., the attack leading to diagnosis) that was classed as severe was more common in patients who were immediately initiated on therapy than in those not initiated on maintenance therapy within 2 months of diagnosis (13% vs 5%; Fig. 2). In contrast, experiencing an initial attack that was classed as mild was more common in patients not initiated on maintenance therapy within 2 months of diagnosis than in those immediately initiated on therapy (53% vs 42%; Fig. 2). Evidence suggests that a delay between diagnosis and maintenance therapy initiation is becoming less common. For patients diagnosed within 12 months preceding the survey, 39% (109/276) were not initiated on maintenance therapy within 2 months of diagnosis, whereas 63% (155/248) who were diagnosed over 36 months ago were not initiated on maintenance therapy within 2 months of diagnosis (p < 0.05).
Fig. 2.
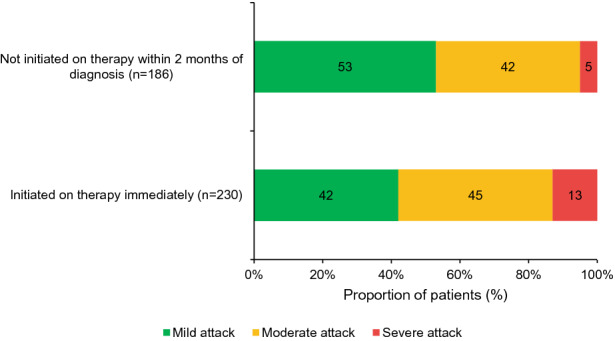
Proportion of newly diagnosed patients who were either initiated on therapy immediately or had initiation of maintenance therapy delayed by over 2 months, by initial attack severity. Data are presented for all newly diagnosed patients for whom severity of the initial attack was recorded (N = 416). The χ2 test was used to test for an association between time to initiation on treatment and severity of initial attack, and there was a strong association between these variables (p < 0.05)
For newly diagnosed patients with mild symptoms, 38% (146/389) of neurologists strongly agreed that IST alone (e.g., azathioprine, mycophenolate mofetil, cyclophosphamide, mitoxantrone) was an appropriate treatment option, whereas for patients with moderate-to-severe symptoms, 32% (123/389) of neurologists said the same. Oral corticosteroids/ISTs were typically the first maintenance treatment initiated in most countries, with higher use in Brazil, China, and South Korea than in other countries (Fig. 3i). Only in the USA were neurologists equally as likely to prescribe monoclonal antibodies as oral corticosteroids/ISTs.
Fig. 3.
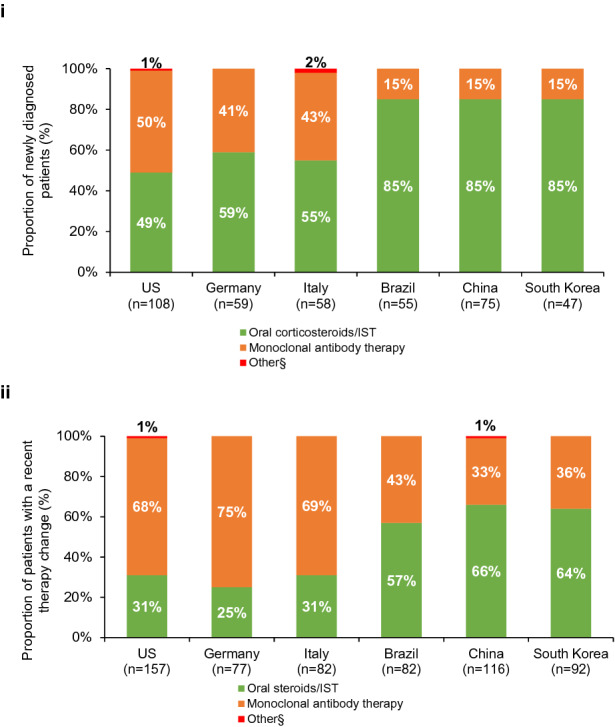
Preferred treatment option when (i) initiating treatment and (ii) following a treatment change. IST, immunosuppressive therapy. §Other treatments were chronic intravenous immunoglobulin (n = 1) in the USA and mitoxantrone (n = 1) in Italy for newly diagnosed patients and chronic intravenous immunoglobulin in the USA (n = 2) and China (n = 1) in patients with a recent therapy change. Data presented in (i) are for all newly diagnosed patients who had been initiated on any treatment at any point (N = 402). Data presented in (ii) are for all patients who had a recent therapy change and were still receiving therapy at the time of the study (N = 606)
In newly diagnosed patients with recorded initial attack severity, who were initiated on and were still receiving therapy at the time of the study (n = 354), there was an association between treatment choice (oral corticosteroids/ISTs versus monoclonal antibodies) and initial attack severity (p < 0.05 for interaction). A higher proportion of patients initiated on oral corticosteroids/ISTs had an initial attack classified as mild than patients initiated on a monoclonal antibody (52% [122/236] vs 25% [30/118]). In contrast, a higher proportion of patients initiated on monoclonal antibodies had a moderate initial attack than those who were initiated on oral corticosteroids/ISTs (65% [77/118] vs 37% [87/236]). Of all the core clinical characteristics analyzed at disease onset in newly diagnosed patients that received therapy with oral corticosteroids/ISTs or monoclonal antibodies at any point (n = 400), only acute transverse myelitis was associated with the decision to start patients on monoclonal antibodies over oral corticosteroids/ISTs (47% [61/131] vs 29% [78/269]; p < 0.05; Fig. 4).
Fig. 4.
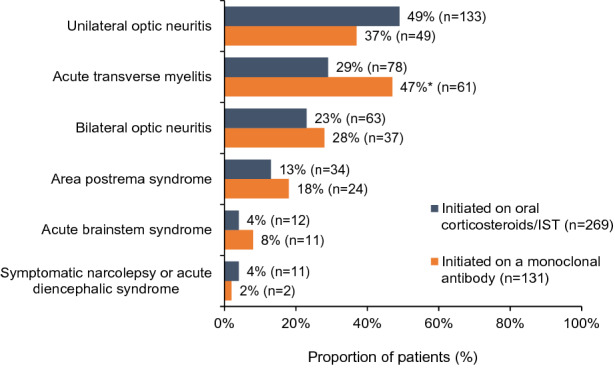
Association between core clinical characteristics at disease onset and treatment initiation with an oral corticosteroid/IST or a monoclonal antibody in newly diagnosed patients who received therapy at any point (N = 400). IST, immunosuppressive therapy. *p < 0.05 (corrected p value via SPSS). The z-test was used for pairwise comparisons of proportions, and the Bonferroni correction was applied to this test
Patient Interviews
Of interviewed patients, 24% (8/33) were not initiated on NMOSD maintenance therapy within 2 months of diagnosis. However, in five of these patients, this was due to logistics (e.g., specialist appointments, insurance, vaccinations, etc.) rather than a physician decision to use a “watch and wait” approach. Of the 33 patients interviewed, only one patient (3%) in the USA appeared to describe a true “watch and wait” approach, whereby her neurologist intended to see how her disease progressed before administering treatment.
Twenty-five patients (76%), most commonly in China, did not recall being given a choice of treatment and often did not know why a specific treatment had been chosen for them. Patients considered the decision to initiate treatment to be driven by their neurologist’s guidance, as they often felt overwhelmed at diagnosis and struggled to participate in treatment decisions:
I completely relied upon my doctor’s prescription when I was diagnosed. I didn’t know anything about the different treatments.
I did not actively contribute to choosing a medication because I didn’t know the different options. It was entirely my doctor’s decision.
Comments made by patients during their interviews demonstrated a clear belief among their neurologists that oral corticosteroids/ISTs are sufficient for newly diagnosed patients. Eight of the 33 interviewed patients (24%) claim to have been initiated on a monoclonal antibody: 4/5 patients from Germany, 3/8 patients from the USA, and 1/5 patients from South Korea. Twenty-four patients (73%), including the US patient on the “watch and wait” approach, were initiated on oral corticosteroid/ISTs. The remaining patient in the USA was initiated on long-term intravenous immunoglobulin. The eight patients treated with monoclonal antibodies typically had a severe initial attack or symptoms, up to and including paralysis (three patients were paralyzed by the initial attack; one is still using a wheelchair and two are now ambulatory). Two of the eight patients initiated on a monoclonal antibody were already receiving a monoclonal antibody (rituximab) prior to their formal NMOSD diagnosis because of misdiagnosis with MS. Cost/access restrictions were seen as a key barrier to first-line monoclonal antibody use by patients (n = 6), most commonly in Brazil (n = 4) and China (n = 2).
Drivers for Treatment Switch
Neurologist Clinical Record Review
Lack of efficacy, which was primarily determined by the occurrence of a relapse (with magnetic resonance imaging [MRI] confirmation), MRI findings (with/without symptoms), relapse severity, and insufficient recovery from a relapse drove 54% of all treatment changes, whereas lack of tolerability and adverse events drove 19% and 15% of treatment changes, respectively (Fig. 5). However, 48% of “random” patients (i.e., the most recent patient seen for a routine clinical visit) who had a relapse since diagnosis (n = 136) did not change their therapy despite experiencing a relapse.
Fig. 5.
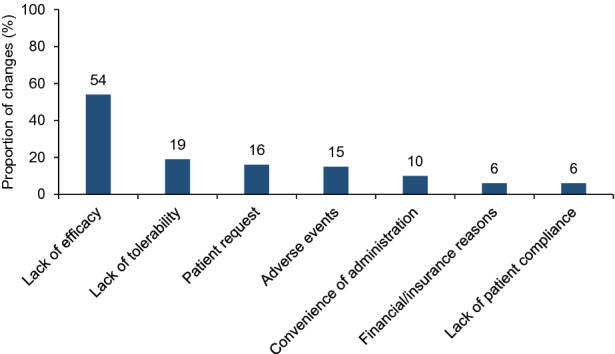
Key triggers (named by more than 5% of respondents) driving a change in maintenance therapy (N = 831). N = 831 corresponds to the number of treatment switches. A total of 606 patients had a therapy change and were still receiving therapy at the time of the study, but, of these, some patients had more than one therapy switch
The most common reasons for not switching treatments (in more than 10% of neurologists) included a mild relapse (20%, n = 13), “stable” patient with good quality of life (QoL) (17%, n = 11), the patient already receiving a good therapy (16%, n = 10), and patient choice (11%, n = 7).
Of the 65 patients from the clinical record review who did not have their treatment changed after a relapse, 38 (58%) were noted as having a mild relapse as their most recent relapse, with 49% (n = 32) making a full recovery post-relapse. Patients who did not have a treatment change after relapse (n = 65) were also less likely (p < 0.05) to have experienced the following symptoms, as well as residual disability resulting from their most recent relapse versus patients who did have a treatment change after relapse (n = 71): vision impairment, orbital pain, fatigue and neuropathic pain, or a decline in daily living activities.
Among patients who had a recent therapy change (N = 606), those in Brazil, China, and South Korea were more likely to remain on oral corticosteroids/ISTs compared with the USA, Germany, and Italy (Fig. 3ii). Escalation to monoclonal antibody treatment was more likely to occur versus escalation to another oral corticosteroid/IST if treatment change was driven by a lack of efficacy (63% [274/437 switches] vs 45% [148/332 switches]; p < 0.05) than if driven by adverse events or financial issues, etc.
There was an association between treatment change (oral corticosteroids/ISTs vs monoclonal antibodies) and whether an MRI was conducted to confirm patients’ most recent relapse (p < 0.05 for interaction). Of patients with available MRI data regarding their most recent relapse who had a treatment change, a higher proportion of patients who were escalated to a monoclonal antibody had an MRI confirming their most recent relapse versus patients who had a treatment change to another oral corticosteroid/IST (83% [213/257] vs 67% [128/190]). Choice of treatment change (oral corticosteroids/ISTs vs monoclonal antibodies) was associated with relapse severity (p < 0.05 for interaction). A higher proportion of patients who had their treatment escalated to a monoclonal antibody experienced a severe relapse than those switched to a different oral corticosteroid/IST (19% vs 8%; Fig. 6), whereas a higher proportion of patients who had their treatment changed to a different oral corticosteroid/IST had a mild relapse than those escalated to a monoclonal antibody (51% vs 29%; Fig. 6).
Fig. 6.
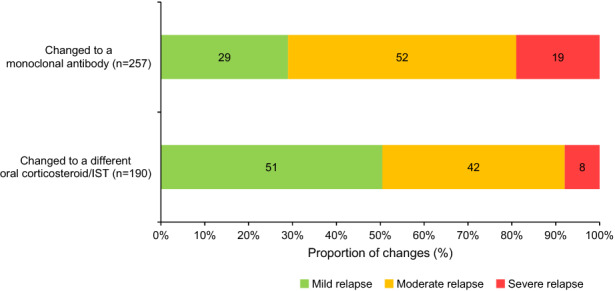
Severity of the most recent relapse driving a change in treatment. IST, immunosuppressive therapy. Data shown are for patients for whom information was collected regarding their most recent relapse who had a treatment change to either a monoclonal antibody or an oral corticosteroid/IST. The χ2 test was used to test for an association between treatment change and severity of relapse, and there was a considerable association between these variables (p < 0.05)
Patient interviews
Six of the 21 newly diagnosed patients and eight of the remaining 12 “ongoing” patients (i.e., not newly diagnosed) reported recent/upcoming therapy changes. Seventeen therapy changes (either a treatment switch or an add-on) in 14 patients were discussed in total.
Twelve of the 33 patients involved in the interviews had not had their maintenance therapy changed after experiencing a relapse following their diagnosis. In this setting, the decision not to switch treatment was perceived by patients (10/12) as being driven by their neurologists:
My doctor believed that my symptoms were mild enough that I should stick with the same medication, but if I had had a severe relapse, like before, I would have changed medication.
I would ask for advice, but I would not ask outright for a change in medication. It’s not really my place to decide when I need a change in medication; that is for my doctor.
The lack of treatment change was a source of frustration for some patients (n = 2), but patients who experienced a mild relapse and were otherwise tolerating therapy generally agreed with the decision of their neurologists (n = 5).
Patients also reported relapse to be a key driver of treatment switching, affecting approximately 60% (10/17) of therapy changes. At the time of the relapse, nine of these patients were receiving oral corticosteroids/ISTs and one patient was treated with a monoclonal antibody (who then switched to another monoclonal antibody). Although patients were typically willing to tolerate some side effects if they felt that their therapy was otherwise efficacious, severe or persistent side effects (i.e., constant nausea, vomiting, or migraines) were likely to prompt a discussion between patients and their neurologists about a treatment switch, and was the reason behind 24% (4/17) of therapy changes. Awareness of alternate therapies was also a driver behind switching treatments in 6% (1/17) of cases, but most patients said that, although this would prompt a discussion with their neurologists (18/33), a treatment change may be advised against if they were perceived to be doing well on treatment (8/18).
Nineteen patients indicated that a hypothetical therapy with a more convenient administration and dosing regimen would prompt them to ask their neurologists for a change in therapy, although method of administration and dosing regimen were not drivers of treatment changes in these interviews.
Discussion
This international analysis was conducted across six countries worldwide in patients with AQP4-IgG-seropositive NMOSD. Collection of data from neurologists in this manner and scale allows for better understanding of current clinical management in this rare disease; concurrent patient interviews also provide insight into how the patients’ views compare with those of the neurologists.
NMOSD diagnosis has been challenging because of the rarity of the disease and the overlapping clinical phenotype with MS [1, 20]. We observed high misdiagnosis rates in this analysis, with 25% of patients with NMOSD initially receiving a misdiagnosis of MS or other conditions. Of those initially misdiagnosed, 32% of patients received a correct diagnosis within 2 months, which may be due to some of these patients testing positive for AQP4-IgG during this timeframe. However, 68% of patients remained misdiagnosed for at least 2 months, highlighting that misdiagnosis remains a challenge, even with AQP4-IgG testing available to aid differential diagnosis [6].
Misdiagnosis led to a delay in treatment initiation and misdiagnosed patients experienced more relapses in total versus those who were not misdiagnosed. Relapses often occur early in the disease, and in clusters after the initial attack, potentially causing substantial neurological disability [20–22]. Therefore, these findings reinforce the need for early and accurate diagnosis of NMOSD.
Encouragingly, the number of patients not initiating maintenance therapy within 2 months of diagnosis seems to be decreasing with time, although this approach was still adopted in a high proportion (47%) of patients recorded during the clinical record review. Neurologists cited patient refusal as a reason for not initiating therapy in more than 30% of newly diagnosed patients: China (50%), Italy (38%), Germany (38%), the USA (27%), Brazil (25%), and South Korea (14%).
Long-term disability with relapses in NMOSD is well documented [4, 7, 9, 21], further underlining the importance of the patient–neurologist relationship, the need for appropriate physician/patient education, and active management.
A treatment initiation delay was more likely to occur for patients whose initial attack was classified as mild rather than severe. Where treatment was prescribed for newly diagnosed patients, oral corticosteroids/ISTs continued to be the first-line therapy of choice for most neurologists, in line with existing treatment guidelines [10, 23–25]. However, current treatment guidelines are not based on class I evidence and were last updated before the approval of the monoclonal antibodies, eculizumab, inebilizumab, and satralizumab [11, 12, 14–17].
Initial attack severity and presence of acute transverse myelitis at disease manifestation were the biggest drivers for initiation of oral corticosteroids/ISTs or monoclonal antibodies as first-line maintenance therapy. This is consistent with MRI imaging playing an important role in NMOSD decision-making and disease assessment [26].
Relapses did not always result in treatment changes for the patient, with the decision typically being neurologist driven, whereas patients had little input. Neurologists indicated that a treatment change may not be initiated if the patient has a relapse that was considered to be mild or stable with good QoL. Furthermore, use of oral corticosteroids/ISTs as first-line maintenance therapy occurred in many countries, particularly Brazil, China, and South Korea, where cost of and/or access to monoclonal antibodies may be a bigger issue than in other countries. Escalation to a monoclonal antibody was more likely when a treatment change was driven by a lack of efficacy and if the patient had experienced a severe relapse. The availability of international, multicenter, randomized, placebo-controlled trial data in NMOSD means that it is perhaps time to revisit the current standards of care and provide recommendations on which therapies should be initiated/switched and when, based on the more complete picture that we now have of treatment of the disease [14–16, 27].
Hypothetical investigations into patient preferences indicated that a more convenient route of administration or dosing regimen might prompt them to discuss the possibility of switching treatments with their neurologists. As more therapies are approved and the number of treatment options increases, administration, dosing, and efficacy may play a bigger role in treatment decisions and may prompt patients to be more active in the treatment decision-making process.
This study was limited by data being collected retrospectively, and the inability to consistently obtain complete data for all patients when performing a clinical record review. The review was also restricted to three pre-specified categories. However, requiring patients with NMOSD to be AQP4-IgG-seropositive for the clinical record review allowed for a homogenous cohort in which patients with less well understood mechanisms of disease were not included (e.g., patients with double-seronegative NMOSD or myelin oligodendrocyte glycoprotein antibody-associated disease). Future studies should explore these patient groups, as they may have distinct disease characteristics.
With regards to the patient interviews, AQP4-IgG status was not recorded; as such, the patient perspectives that we describe may represent a broader population than in the clinical record review.
There is a risk of bias associated with physician self-reporting of clinical records (e.g., selection bias, recall bias, and desirability bias), and this may have skewed the population towards including patients with more “favorable” results. Similarly, as physician referral was one method of recruiting patients for the telephone interviews, those referred were more likely to be “satisfied” patients, rather than those who viewed their management less positively. These biases should be taken into consideration when interpreting the findings of this study.
No well-established definitions of relapse severity exist, so definitions applied by physicians completing the survey were subjective and based on clinician judgement. However, data from this study provide us with an opportunity to understand real-world classification of relapse severity [26]. Furthermore, this study provides a snapshot of current clinical practice, although patients were not followed up after therapy initiation or switch, so the effect of treatment changes could not be ascertained from this analysis.
Overall, this global clinical record review, in conjunction with patient interviews, has helped to further understand how AQP4-IgG-seropositive NMOSD is managed in real-world clinical practice worldwide. Misdiagnosis of NMOSD remains a concern, indicating that greater education is required to ensure that this rare disease is recognized, particularly as a delay in diagnosis may be associated with increased disability caused by relapses and negatively affect patients’ emotional well-being. In this analysis, relapse severity influenced decisions about whether to initiate treatment, what treatment to prescribe, and whether a change in treatment was required, and this should be further investigated with clear definitions of relapse severity to provide revised recommendations for treatment decisions in NMOSD. Furthermore, oral corticosteroids/ISTs are still typically the first-line maintenance treatment for many neurologists, and only after these therapies are deemed to be ineffective are monoclonal antibodies generally prescribed, despite their efficacy and safety being demonstrated in randomized, placebo-controlled, phase III trials.
Conclusions
Investigating patient characteristics and other drivers behind treatment initiation, treatment choice, and switch in clinical practice is important for improving knowledge of current clinical management, as well as how the patients’ views compare with those of neurologists. Our findings suggest that NMOSD misdiagnosis is high and associated with a delay in treatment initiation. In NMOSD, relapses often occur early in the disease, and therefore our findings reinforce the need for early and accurate diagnosis of NMOSD and initiation of the correct therapy. Use of oral corticosteroids/ISTs continues to be the mainstay of first-line maintenance treatment in countries where the cost of and/or access to monoclonal antibodies may pose a bigger issue than in other countries. Relapse severity was a factor that guided the neurologist’s decisions on treatment initiation/switch, and future studies using real-world evidence to assess relapse severity in treatment initiation/switch are required to improve understanding of this disease and revise NMOSD treatment recommendations.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We thank all neurologists and patients that participated in this study. We also thank the following team members at Roche for their critical review: Cynthia Tso and Nicole Antonio.
Funding
The study and the journal’s Rapid Service Fee were funded by F. Hoffmann-La Roche.
Medical Writing Assistance
Medical writing assistance was provided by Patricia Lobo, BSc, of ApotheCom, London, UK. Support for this assistance was funded by F. Hoffmann-La Roche.
Authorship
All named authors meet the International Committee of Medical Journal Editors criteria for authorship for this article, take responsibility for the integrity of the work as a whole and have given their approval for this version to be published.
Author Contributions
Carly Welsh designed and conceptualized the study and collected and analyzed the data. The first draft of the manuscript was written by Patricia Lobo. All authors were involved in data interpretation, critical revision of the manuscript for important intellectual content, and approved the final manuscript.
Disclosures
This study was sponsored by F. Hoffmann-La Roche. Ju-Hong Min received speaker honorarium from Bayer Schering, Merck, Biogen Idec, and Sanofi Genzyme, and personal compensation for consulting from Samsung Bioepis and Roche. Marco Capobianco received personal compensation for consulting from Biogen, Roche, Novartis, Sanofi, and Merck. Patricia Lobo is an employee of ApotheCom, who are paid to provide medical writing assistance for F. Hoffmann-La Roche. Carly Welsh, Gabrielle deFiebre, Marco Lana-Peixoto and Jiawei Wang report no disclosures. Dean M. Wingerchuk received personal compensation for consulting from Roche, Genentech, UCB Pharma, Horizon, VielaBio, Biogen, Mitsubishi Tanabe, and research support paid to Mayo Clinic from Alexion. Marius Ringelstein received speaker honoraria from Novartis, Bayer Vital GmbH, Roche, Alexion, and Ipsen, and travel reimbursement from Bayer Schering, Biogen Idec, Merz, Genzyme, Teva, Roche, and Merck, none related to this study.
Compliance with Ethics Guidelines
This was a market research study and, as such, it was conducted in accordance with the European Pharmaceutical Market Research Association’s (EphMRA) industry code of conduct. Market research as defined in this code of conduct does not require clinical research ethics committee or independent review board approval. All subjects provided informed consent to participate in the study (in accordance with the EphMRA code of conduct), and research was compliant with all international and national data protection laws. Quote responses provided are vignettes of real answers that have been amended to maintain anonymity of respondents without altering the theme of their statements.
Data Availability
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1.Papadopoulos MC, Bennett JL, Verkman AS. Treatment of neuromyelitis optica: state-of-the-art and emerging therapies. Nat Rev Neurol. 2014;10(9):493–506. doi: 10.1038/nrneurol.2014.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Juryńczyk M, Tackley G, Kong Y, et al. Brain lesion distribution criteria distinguish MS from AQP4-antibody NMOSD and MOG-antibody disease. J Neurol Neurosurg Psychiatry. 2017;88(2):132–136. doi: 10.1136/jnnp-2016-314005. [DOI] [PubMed] [Google Scholar]
- 3.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364(9451):2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- 4.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6(9):805–815. doi: 10.1016/S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- 5.Jarius S, Wildemann B. Aquaporin-4 antibodies (NMO-IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain Pathol. 2013;23(6):661–683. doi: 10.1111/bpa.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66(10):1485–1489. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]
- 7.Oh J, Levy M. Neuromyelitis optica: an antibody-mediated disorder of the central nervous system. Neurol Res Int. 2012;2012:460825. doi: 10.1155/2012/460825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitley J, Leite MI, Nakashima I, et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain. 2012;135(Pt 6):1834–1849. doi: 10.1093/brain/aws109. [DOI] [PubMed] [Google Scholar]
- 9.Kessler RA, Mealy MA, Levy M. Treatment of neuromyelitis optica spectrum disorder: acute, preventive, and symptomatic. Curr Treat Options Neurol. 2016;18(1):2. doi: 10.1007/s11940-015-0387-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimbrough DJ, Fujihara K, Jacob A, et al. Treatment of neuromyelitis optica: review and recommendations. Mult Scler Relat Disord. 2012;1(4):180–187. doi: 10.1016/j.msard.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cree BAC, Bennett JL, Kim HJ, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. 2019;394(10206):1352–1363. doi: 10.1016/S0140-6736(19)31817-3. [DOI] [PubMed] [Google Scholar]
- 12.Pittock SJ, Berthele A, Fujihara K, et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381(7):614–625. doi: 10.1056/NEJMoa1900866. [DOI] [PubMed] [Google Scholar]
- 13.Weinshenker BG, Wingerchuk DM. Neuromyelitis spectrum disorders. Mayo Clin Proc. 2017;92(4):663–679. doi: 10.1016/j.mayocp.2016.12.014. [DOI] [PubMed] [Google Scholar]
- 14.Traboulsee A, Greenberg BM, Bennett JL, et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020;19(5):402–412. doi: 10.1016/S1474-4422(20)30078-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.US Food and Drug Administration. FDA approves new therapy for rare disease affecting optic nerve, spinal cord. June 11, 2020. https://www.fda.gov/news-events/press-announcements/fda-approves-new-therapy-rare-disease-affecting-optic-nerve-spinal-cord. Accessed 21 July 2022.
- 16.US Food and Drug Administration. FDA approves treatment for rare disease affecting optic nerves, spinal cord. August 17, 2020. https://www.fda.gov/news-events/press-announcements/fda-approves-treatment-rare-disease-affecting-optic-nerves-spinal-cord. Accessed 21 July 2022.
- 17.Yamamura T, Kleiter I, Fujihara K, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381(22):2114–2124. doi: 10.1056/NEJMoa1901747. [DOI] [PubMed] [Google Scholar]
- 18.European Medicines Agency. Public summary of opinion on orphan designation: Eculizumab for the treatment of neuromyelitis optica spectrum disorders. https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/13/1185-eculizumab-treatment-neuromyelitis-optica-spectrum-disorders_en.pdf. Accessed July 21, 2022.
- 19.FDA approves first treatment for neuromyelitis optica spectrum disorder, a rare autoimmune disease of the central nervous system 2019. https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-neuromyelitis-optica-spectrum-disorder-rare-autoimmune-disease-central. Accessed 21 July 2022.
- 20.Etemadifar M, Salari M, Mirmosayyeb O, et al. Efficacy and safety of rituximab in neuromyelitis optica: review of evidence. J Res Med Sci. 2017;22:18. doi: 10.4103/1735-1995.200275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome) Neurology. 1999;53(5):1107–1114. doi: 10.1212/WNL.53.5.1107. [DOI] [PubMed] [Google Scholar]
- 22.ZhangBao J, Zhou L, Li X, et al. The clinical characteristics of AQP4 antibody positive NMO/SD in a large cohort of Chinese Han patients. J Neuroimmunol. 2017;302:49–55. doi: 10.1016/j.jneuroim.2016.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Sellner J, Boggild M, Clanet M, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17(8):1019–1032. doi: 10.1111/j.1468-1331.2010.03066.x. [DOI] [PubMed] [Google Scholar]
- 24.Scott TF, Frohman EM, De Seze J, Gronseth GS, Weinshenker BG. Evidence-based guideline: clinical evaluation and treatment of transverse myelitis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2011;77(24):2128–2134. doi: 10.1212/WNL.0b013e31823dc535. [DOI] [PubMed] [Google Scholar]
- 25.Trebst C, Jarius S, Berthele A, et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS) J Neurol. 2014;261(1):1–16. doi: 10.1007/s00415-013-7169-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Capobianco M, Ringelstein M, Welsh C, et al. Characterization of disease severity and stability in NMOSD: a global clinical record review with patient interviews. Neurol Ther. 2023 doi: 10.1007/s40120-022-00432-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang C, Zhang M, Qiu W, et al. Safety and efficacy of tocilizumab versus azathioprine in highly relapsing neuromyelitis optica spectrum disorder (TANGO): an open-label, multicentre, randomised, phase 2 trial. Lancet Neurol. 2020;19(5):391–401. doi: 10.1016/S1474-4422(20)30070-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.