

Abstract
Neuroblastoma is the most common and deadliest tumor in infancy. WDR5 (WD Repeat Domain 5), a critical factor supporting an N-myc transcriptional complex via its WBM site and interacting with chromosome via its WIN site, promotes the progression of neuroblastoma, thus making it a potential anti-neuroblastoma drug target. So far, a few WIN site inhibitors have been reported, and the WBM site disruptors are rare to see. In this study we conducted virtual screening to identify candidate hit compounds targeting the WBM site of WDR5. As a result, 60 compounds were selected as candidate WBM site inhibitors. Cell proliferation assay demonstrated 6 structurally distinct WBM site inhibitors, numbering as compounds 4, 7, 11, 13, 19 and 22, which potently suppressed 3 neuroblastoma cell lines (MYCN-amplified IMR32 and LAN5 cell lines, and MYCN-unamplified SK-N-AS cell line). Among them, compound 19 suppressed the proliferation of IMR32 and LAN5 cells with EC50 values of 12.34 and 14.89 μM, respectively, and exerted a moderate inhibition on SK-N-AS cells, without affecting HEK293T cells at 20 μM. Analysis of high-resolution crystal complex structure of compound 19 against WDR5 revealed that it competitively occupied the hydrophobic pocket where V264 was located, which might disrupt the interaction of MYC with WDR5 and further MYC-medicated gene transcription. By performing RNA-seq analysis we demonstrated the differences in molecular action mechanisms of the compound 19 and a WIN site inhibitor OICR-9429. Most interestingly, we established the particularly high synergy rate by combining WBM site inhibitor 19 and the WIN site inhibitor OICR-9429, providing a novel therapeutic avenue for neuroblastoma.
Keywords: neuroblastoma, WDR5, WBM site, protein-protein interaction interrupters, drug combination therapy
Introduction
Neuroblastoma is a type of highly malignant tumor often occurs in children under 5 years old. It originates from neural crest progenitor cells [1]. The estimated incidence is 10.7 cases per million children aged 0–14 years, accounting for approximately 10% of pediatric malignancies [2]. With a remarkable tumor heterogeneity, the prognosis, depending to a great extent on the stage at diagnosis, varies from spontaneous regression to poor outcomes, such as distant metastasis and death [3]. Despite using multimodality treatment regimens, the 5-year overall survival rate for patients with high-risk neuroblastoma is still less than 50% [4], remains a challenging clinical problem. Propelled by the desperate need for novel therapies, numerous efforts have been devoted to the development of molecular targeted drugs, which promise more effective and precise treatment. However, so far, there is no small molecular drugs approved for targeted therapy of neuroblastoma.
WD repeat-containing protein 5 (WDR5) is an extraordinarily highly-conserved protein that regulates diverse cellular processes [5] and is proved to be a critical factor in neuroblastoma tumorigenesis [6, 7] (Fig. 1). Firstly, WDR5 is a co-factor for N-myc [8, 9]. N-myc is encoded by proto-oncogene MYCN, regulating cell proliferation, metabolism, apoptosis, and differentiation [10]. Amplification of MYCN, leading to overexpression of N-myc, is an oncogenic driver of neuroblastoma and strongly related with poor prognosis [11]. As a transcription factor, the function of N-myc is based on its binding to regulatory regions of target genes. The process partially relies on its interaction with WDR5 [8, 9]. Thus, WDR5 is a crucial determinant for the recruitment of N-myc to chromatin. Secondly, WDR5 is a core subunit of the histone methyltransferase complex, RbBP5-WDR5-MLL, catalyzes histone H3 lysine 4 (H3K4) methylation [12]. Since trimethylation of H3K4 is a prerequisite for the transcriptional activation [13], WDR5 also serves as an epigenetic regulator in neuroblastoma. Moreover, WDR5 is demonstrated to be a “reader” for H3Q5ser [14]. H3Q5ser activates transcription of cancer-promoting genes in neuroblastoma cells and is in synergy with H3K4me3 during neuronal cell differentiation. All the above shows that WDR5 is likely to be a therapeutic target for neuroblastoma.
Fig. 1. Schematic illustration of WDR5 function in neuroblastoma tumorigenesis.
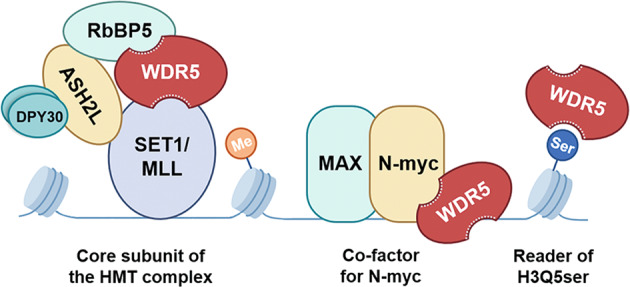
WDR5 serves as a core subunit of the histone methyltransferase (HMT) complex, a co-factor for N-myc and a reader of histone H3Q5 serotonylation (H3Q5ser), thus activating transcription of cancer-promoting genes in neuroblastoma cells.
There are two well-defined pockets that are amenable to small molecular inhibition, the “WDR5-interacting” (WIN) site that mediates the chromatin/MLL1-WDR5 interaction [15] and the “WDR5-binding motif” (WBM) site by which the N-myc/RbBP5-WDR5 connection is mediated [16]. In the past decade, most efforts have been made to discover WIN site inhibitors [17–24], while few studies have centered on the blockade of the WBM site. However, despite the enormous progression made in the development of WIN site inhibitors, the drug is only practical for MLL [25, 26], whereas solid tumors are not sensitive to that.
This study clarifies whether the WBM site is a viable target for treating neuroblastoma by discovering of new WBM site inhibitors through computational methods and experimental technologies. Further experimental studies demonstrated that the WBM site inhibitor could elevate the expression of p53 and down-regulate the level of H3K4me3. Compared with a WIN site molecular binder, the WBM site binder presents more obvious inhibitory activity in treating with neuroblastoma. More interestingly, the present study is the first attempt to investigate the anti-neuroblastoma effects of a combination of a WBM site molecular binder and a WIN site binder. Significantly synergistic tumor-suppressing effects and modulating mechanisms with co-targeting WBM and WIN sites are demonstrated, providing a potential therapeutic avenue for treating neuroblastoma.
Materials and methods
Virtual screening
The compounds from three commercial databases, Specs (https://www.specs.net/), TargetMol (https://www.targetmol.com/) and MCE (https://www.medchemexpress.cn/), were prepared using LigPrep [27] tool by Schrödinger. The three-dimensional molecular structures were generated for establishment of a compound library. The compound adopted as template in the ligand-based screening was a known WBM site inhibitor, named as 1-Cyclopentyl-2-(methylsulfonyl)-4-nitro-1H-imidazole (PDB: 6UOZ) [28]. The structure of the template compound and its binding mode to WDR5 were shown in Supplementary Fig. S1. The three-dimensional structure of WDR5 used in the receptor-based screening was obtained from the Protein Data Bank (PDB) code 6U5Y [29]. To search for compounds similar to the template, the 3D similarity method SHAFTS [30] was applied. The top 1000 matched structures were then docked into the crystal structure of WDR5 in the Glide (Grid-based ligand docking with energetics) [31]. For the receptor-based screening, the prepared library was imported to the Glide to conduct high-throughput virtual screening. The top 10% compounds were then screened and re-ranked using Standard Precision (SP) docking to get more accurate prediction. All compounds selected by virtual screening as above then underwent manual screening.
Cell culture
Human HEK293T and IMR32 cells were obtained from Cell Bank, Chinese Academy of Sciences (Shanghai, China). Human LAN5 cell line was purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany). Human SK-N-AS cell line was purchased from American Tissue Culture Collection (ATCC, Manassas, VA, USA). HEK293T, IMR32 and SK-N-AS cells were cultured in DMEM (Gibco, Grand Island, NY, USA). LAN5 cells were cultured in IMDM (Gibco). The base medium was supplemented with 10% (v/v) FBS (Gibco), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco). Cells were maintained at 37 °C in a 5% CO2 incubator.
Cell proliferation assay
Cell proliferation was measured using the Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan) according to the manufacturer’s protocol. Cells were seeded into 96-well plates at a density of 5 × 103 (HEK293T), 1.2 × 104 (SK-N-AS) or 2 × 104 (IMR32 and LAN5) cells per well. After 24 h incubation, medium was replaced with fresh medium (0.1% DMSO) or the compounds of various concentrations (0.625, 1.25, 2.5, 5, 10, and 20 μM) with final DMSO concentration of 0.1%. Cell viability was assessed after a 72 h incubation. CCK-8 reagent (10 μL) was added to each well, followed by incubation at 37 °C for 2 h. A microplate reader (PerkinElmer, Waltham, MA, USA) was used to measure the absorbance values at 450 nm. GraphPad Prism software was used to generate EC50 values. Error bars on proliferation curves represent standard errors of the mean.
Protein expression, purification, and crystallization
The pET28a expression vectors encoding WDR5 (residues 22–334) with N-terminal 6× His tag was transformed into Rosetta™ 2(DE3) Competent Cells (TransGen Biotech, Beijing, China). Cells were grown in LB medium at 37 °C until OD600 = 1.0 and protein was expressed at 16 °C for 20 h after 0.2 mM isopropyl-β-D-thiogalactoside (IPTG) was added. After collecting by centrifugation, cells were suspended with lysis buffer (50 mM NaH2PO4, pH 7.5, 1 M NaCl, 10 mM imidazole, 0.5 mM TCEP, 1 × EDTA-free protease inhibitor cocktails (AbMole, Houston, TX, USA)). After high pressure crushing, the lysate was cleared by centrifugation at 4 °C, and the supernatant was collected and incubated with Ni2+-NTA beads (Smart-Lifesciences, Changzhou, China) for 30 min. The mixture was loaded into column and washed with lysis buffer, and eluted with buffer containing 300 mM imidazole. Fractions were further purified using size exclusion chromatography (Superdex 200 Increase 10/300, GE, Fairfield, CO, USA) with buffer of 25 mM HEPES, pH 7.0, 250 mM NaCl, 0.5 mM TCEP. Protein was concentrated to 10 mg/mL for crystallization.
Crystallization of apo WDR5 was performed at 18 °C using the hanging drop vapor-diffusion method, by mixing equal volumes (1.5:1.5 μL) of the protein and crystallization solution (30% (w/v) pEG3350, 0.1 M Bis-Tris, pH 5.8, 0.2 M ammonium acetate). Crystals were formed within 3 days and then soaked into crystallization buffer with 5 mM compounds for 6 h. Using crystallization buffer containing 20% glycerol as cryoprotectant, the crystals were flash-frozen into liquid nitrogen.
Structure determination and refinement
X-ray diffraction data were collected at beamline BL18U1 at the Shanghai Synchrotron Radiation Facility (SSRF). Data were processed with HKL3000 [32]. The structure was solved by molecular replacement using Phaser module of CCP4 [33] program suit with a search model of 6UJL [28]. The structure was built using COOT [34] and refined with TLS and simulated-annealing protocol using PHENIX [35].
Surface plasmon resonance (SPR) analysis
Prior to determining the binding affinity of each compound, recombinant WDR5 protein was diluted to 20 μg/mL in sodium acetate at pH 4.5 and immobilized on series S sensor CM5 chip (Cytiva, Marlborough, MA, USA) using standard amine coupling to a level of 10,000 response units (RU). Compound stock solutions (10 mM) were diluted with 1× HBS-EP buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3.0 mM EDTA, and 0.05% (v/v) Tween-20) in a nine-concentration series ranging from 0.195 μM to 100 μM with final DMSO concentration of 5%. Three startup cycles with running buffer were performed first, and analyte was injected and run through the chip with association time of 100 s and disassociation time of 100 s and washed with 50% DMSO in each cycle. Solvent correction with different DMSO concentration varying from 4.5% to 5.8% was performed every 48th cycle. Raw data were reduced, double-referenced, and solvent-corrected using Biacore 8 K Evaluation Software and KD values of each compound were calculated using steady-state affinity model with a constant Rmax.
RNA sequencing (RNA-seq)
LAN5 cells were seeded into 6-well plates at a density of 1.2 × 106 cells per well. After a treatment of 20 μM compound 19, 20 μM OICR-9429 or 0.1% DMSO only for 72 h, cells were washed in PBS then lysed in 500 μL Trizol. RNA isolation, library preparation and RNA-seq was performed by Majorbio (Shanghai, China) using NovaSeq 6000 platform. Prior to sequencing, RNA integrity was assessed by 2100 Bioanalyzer and agarose gel electrophoresis. The concentration was determined by NanoDrop.
Cell cycle analysis
Cell cycle was monitored using BD Cycletest™ Plus DNA Reagent Kit (BD Biosciences, Franklin Lakes, NJ, USA) in accordance with the manufacturer’s protocol and flow cytometry. LAN5 cells were seeded into 6-well plates at a density of 1.2 × 106 cells per well. Cells were trypsinized and harvested after treated with 10 μM and 20 μM compound 19 for 72 h. Followed by washing with PBS, solution A, B, C of the kit was added to stain the cells with PI. The PI signal was measured by flow cytometry. The results were analyzed by FlowJo software.
Apoptosis assay
Cell apoptosis was quantified using FITC Annexin V Apoptosis Detection Kit (BD Biosciences) following the manufacturer’s protocol and flow cytometry. Cells were collected after a 72-h treatment of 10 μM and 20 μM compound 19 and washed twice with cold PBS. Subsequently, cells were resuspended in 1× Binding Buffer and mixed with FITC Annexin V and PI. Cells were subjected to a flow cytometer (Beckman Coulter, Brea, CA, USA) after an incubation for 15 min in the dark. The results were analyzed by ModFit software.
Western blot
LAN5 cells were seeded into 6-well plate at a density of 1.2 × 106 cells per well. The adherent cells were treated with compound 19 or OICR-9429. After 24 h, cells were lysed in RIPA buffer (Thermo Scientific, Waltham, MA, USA) containing protease and phosphatase inhibitors. Total protein fractions were boiled in loading buffer for 5 min and then equal quantities (20 μg) were separated by 4%–12% NuPage Bis-Tris gels (Invitrogen, Frederick, MD, USA). The proteins were transferred to PVDF membranes (Millipore, Boston, MA, USA) for 30 min in transfer buffer (Tanon, Shanghai, China). The membranes were blocked in blocking buffer (Takara Biotechnology, Dalian, China) for 1 h at the room temperature, incubated with primary antibodies against p53 (1:1000; #HA201-01, TransGen Biotech), H3K4me3 (1:1000; #9751, Cell Signaling Technology, Danvers, MA, USA) and GAPDH (1:1000; #5174, Cell Signaling Technology) overnight at 4 °C. The anti-rabbit IgG (1:10,000; W4011, Promega, Madison, WI, USA), conjugated with HRP were used as secondary antibodies. Proteins were visualized with Enhanced Chemiluminescence Kit (EpiZyme, Shanghai, China) and detected by Amersham Imager 680 (GE).
Drug combination testing
IMR32 and LAN5 cells were plated in 96-well plates, followed by treatment of drug combination for 72 h. A total of seven concentration gradients (0, 0.625, 1.25, 2.5, 5, 10, and 20 μM) were set for compound 19 and three (0, 10, and 20 μM) for OICR-9429. Cell viability was monitored by CCK-8 assay, of which protocol was described above. The result was analyzed and visualized by the Bliss independent model using SynergyFinder 2.0 (https://synergyfinder.fimm.fi/) [36].
Statistical analysis
Statistical analysis of RNA-seq was performed by Majorbio. RNA-Seq reads were aligned to the human reference genome hg19 using STAR (Version 2.7.1a) [37] and quantified by RSEM (Version 1.3.3) [38]. Differential analysis was performed by DESeq2 (Version 1.24.0) [39]. The significantly changed genes were assessed with an adjusted P-value < 0.05. Other statistical analysis was conducted by GraphPad Prism (version 8.01). The results were presented as mean ± standard deviation (SD). T-test was applied to compare outcomes between two groups. P value < 0.05 was considered statistically significant.
Results
Discovery of WBM site inhibitors
According to our limited knowledge, WBM site inhibitors are rare to see. In this study, a virtual screening [40] was performed to identify candidate hit compounds by targeting the WBM site of WDR5. To enhance the success of drug discovery, both of ligand-based and target-based techniques were used to search through three commercial databases, Specs (https://www.specs.net/), TargetMol (https://www.targetmol.com/) and MCE (https://www.medchemexpress.cn/). Ligand-based virtual screening relies on computing the similarities of the chemical scaffolds’ structural information and physicochemical properties between a known active molecule and candidates. A known WBM site inhibitor, named as 1-Cyclopentyl-2-(methylsulfonyl)-4-nitro-1H-imidazole (its dissociation rate with WDR5: KD = 0.10 ± 0.01 mM) [28] was adopted as our template compound. Ligand-based methods often lack of full consideration of protein structure information. For the compensation, a receptor-based screening was also used in this study. Molecular docking is the most used receptor-based screening method [41], and it predicts the binding model of a compound against the binding site of a target, helping to estimate the binding affinity between them. The WBM site was determined by referring to the binding interface of WDR5-MYC complex structure (PDB: 6U5Y) [29]. Those compounds that show potential in competitively occupying the WBM site of WDR5 were selected. The detailed process of the screening is shown in Fig. 2a. As a result, sixty compounds (Supplementary Table S1) were selected as candidate WBM site inhibitors for further experimental validations.
Fig. 2. The discovery of WBM site inhibitors.

a Flowchart of the screening process. b Chemical structures of the four active compounds.
Cell growth inhibitory activities of these sixty compounds against neuroblastoma were tested by CCK-8 assay with the same concentration of 20 μM for 72 h. The cell proliferation assay used one MYCN-unamplified neuroblastoma cell line (SK-N-AS) and two MYCN-amplified cell lines (LAN5 and IMR32). In addition, the selectivity for each compound was determined by comparing the cytotoxicity for cancer-cell lines to a non-cancerous cell line HEK293T. There were six compounds, compound 4, 7, 11, 13, 19, and 22, possessed anti-tumor activities against MYCN-amplified neuroblastoma cells (Supplementary Table S2). Finally, four druggable compounds, compound 11, 13, 19, and 22 were further studied in the following part, of which the chemical structures were shown in Fig. 2b and the estimated ADMET properties were shown in Supplementary Table S3.
WBM site inhibitors suppress cell proliferation of neuroblastoma in vitro
To further investigate the anti-tumor activities of our hit compounds, we tested the antiproliferation effects of the above four active compounds at six concentrations (0.625, 1.25, 2.5, 5, 10, and 20 μM) for 72 h on HEK293T, SK-N-AS, LAN5, and IMR32 cell lines. The CCK-8 assay showed those compounds inhibited the proliferation of neuroblastoma cells in a dose-dependent manner. Specifically, MYCN-amplified cells, as IMR32 and LAN5 cell lines, were more sensitive to the treatment of these compounds. Compound 11 could effectively inhibit the proliferation of IMR32 and LAN5 cells with EC50 values of 8.59 μM and 7.01 μM, respectively (Fig. 3, Supplementary Table S4). While the antiproliferation activities of compound 11 to HEK293T and MYCN-unamplified cell line as SK-N-AS were at same level with EC50 values of 12.10 μM and 4.40 μM, respectively. Same as compound 11, compounds 13 and 22 non-selectively inhibited the proliferation of all cell lines (Fig. 3b–d). Compound 19 had no cytotoxicity to HEK293T cells even at a concentration of 20 μM and a moderate antiproliferation effect on SK-N-AS cells (Fig. 3c), while the EC50 values against IMR32 and LAN5 cell lines were 12.34 μM and 14.89 μM, respectively (Supplementary Table S4).
Fig. 3. The validation of the four active compounds at cellular and molecular level.

Dose-response curves of cell viability in HEK293T, SK-N-AS, IMR32, and LAN5 cell lines after 72-h treatment and SPR sensorgrams of the compounds 11 (a), 13 (b), 19 (c) and 22 (d) tested by CCK-8 assay and Biacore 8 K, respectively.
Assessment of compound-WDR5 binding capability
In order to further explore the antiproliferation mechanism of the compounds, we measured the binding affinities of the four active WBM site inhibitors by SPR. All four compounds could bind to WDR5 with KD values of 27.6 μM, 16.8 μM, 36.8 μM, and 18.2 μM, respectively (Fig. 3). Although the binding of compound 19 to WDR5 was not the strongest among these four, it possessed the most obviously specific cytotoxic activity against neuroblastoma cells, thus compound 19 was chosen for further study to avoid bias caused by off-target effects. Besides, we tested the inhibitory effect of a WIN site binder, OICR-9429 (KD = 93 ± 28 nM) [21], on the same four cell lines to compare the anti-neuroblastoma activities between the WBM and WIN site inhibitor (Supplementary Fig. S2). Surprisingly, such a stronger molecular binder of WDR5 only present a weak ability in inhibiting proliferation over the LAN5 cell line, having no effects on the other cell lines.
Crystal structure of WDR5 in complex with compound 19
To gain insight of the binding mode of compound 19 with WDR5, we solved the crystal structure of WDR5 complexed with compound 19 at a quite high resolution of 1.41 Å (PDB: 7WVK, Supplementary Table S5). Like reported co-crystal structures of MYC-MbIIIb, KANSL2, RbBP5 peptides with WDR5, compound 19 was embedded into the hydrophobic groove at the WBM site (Fig. 4a). The 1,4-dichlorobenzene group sticked into a hydrophobic pocket and stabilized by van der Waals interactions formed with the side chains of F266, V268, K272 and I274. The sulfone group engaged in polar interactions as hydrogen bonds with N225 (Fig. 4b), further strengthening the binding affinity between the compound and WDR5. Previous studies [8, 9] indicated that the binding conformations of MYC-MbIIIb, KANSL2, RbBP5 peptides with WDR5 were virtually identical. The invariant core motif of MYC-MbIIIb called as “IDVV motif” was highly similar to the corresponding regions of KANSL2 and RbBP5 (Fig. 4c). And residue V264 was the vital component for the binding of MYC with WDR5. Analyzing the superimposed structures of compound 19 and MYC-MbIIIb with WDR5 at WBM site showed compound 19 competitively occupied the hydrophobic pocket where V264 was located, which may disrupt the interaction of MYC with WDR5 and further MYC-medicated gene transcription (Fig. 4d).
Fig. 4. X-ray crystal structure of WDR5 in complexed with compound 19.

a A surface representation of compound 19 and MYC-MbIIIb, KANSL2, RbBP5 peptides interactions with WDR5 at WBM site. b Detailed interactions of compound 19 with pivotal residues of WDR5 is shown as sticks and hydrogen bonds are shown as black dash lines. c Superimposition of the conserved MbIIIb, KANSL2 and RbBP5 in complex with WDR5, and the sequence alignment of the peptides. d Superimposed binding modes of compound 19 and MYC-MbIIIb with WDR5 at WBM site.
Compound 19 increases the expression of ribosome genes
To investigate the underlying mechanism, RNA-seq was conducted to identify transcriptional profiles of the control and treatment groups. In order to compare the molecular mechanism of action between the WBM site and WIN site inhibitor, transcriptional profiles of cells treated with OICR-9429 were also analyzed. The overlap of genes affected by the WBM site or the WIN site inhibitor accounted for less than 30% of all differentially expressed genes in the compound 19 group (Fig. 5a).
Fig. 5. Transcriptional profiles of LAN5 cells treated with the WBM and WIN site inhibitors.

LAN5 cells were treated with DMSO, compound 19 and OICR-9429 for 24 h. a Venn diagram, showing the overlap of differential genes after compound 19 or OICR-9429 treatment. b The top 10 KEGG enrichment pathways for genes induced and suppressed by compound 19 or OICR-9429. c Heatmap displaying ribosome genes affected by OICR-9429 and compound 19 and their log2-fold change in two groups.
KEGG [42] enrichment analysis was performed to explore the targeted pathway of the WBM and WIN site inhibitor. The top ten KEGG pathways up- or down-regulated by the compounds were shown in Fig. 5b. Compound 19 affected a series of biological activities which were closely related to survival of tumor cells such as ribosome pathway, oxidative phosphorylation, p53 signaling pathway, cell cycle, RNA processing DNA replication and repair. Furthermore, we can see that the enriched pathways in compound 19 and OICR-9429 groups were relatively different, despite there is some overlap, i.e., both show the abilities of inducing the ribosome pathway, but suppressing cell cycle, RNA transport and DNA replication, etc. It indicates that the compound 19 and OICR-9429 may have complementary biological activities.
Ribosome topped the list of enriched KEGG pathways in both compound 19 and OICR-9429 groups. However, we found that forty-three ribosome genes were significantly induced in the compound 19 group, while 60% (48/80) of the differential ribosome genes were down-regulated and 40% (32/80) up-regulated in the OICR-9429 group. Furthermore, the heatmap (Fig. 5c) showed that almost of those 80 genes were increased in the compound 19 treatment group, presenting an obvious difference with OICR-9429 treated group, which further demonstrates the different molecular action mechanisms of them.
Compound 19 inhibits the cell cycle progression and DNA replication
According to KEGG enrichment analysis, cell cycle was significantly inhibited by compound 19. In addition, DNA replication, which is highly correlated with cell cycle, was also disrupted (Fig. 5b). To clarify the influence of the WBM site inhibitor on cell cycle, LAN5 cells were treated with 10 μM and 20 μM compound 19 for 72 h, and then flow cytometry was performed to monitor the influence on the cell cycle. Although 10 μM compound 19 had little impact, it is interested to be seen that the proportions of cells in the G0/G1 phase significantly increased at the 20 μM group, indicating that this compound could induce a G0/G1 phase arrest (Fig. 6a).
Fig. 6. Anti-tumor mechanism of the WBM site inhibitor.

a Compound 19 caused G0/G1 arrest in LAN5 cells detected by flow cytometry. b Compound 19 induced apoptosis in LAN5 cells determined by flow cytometry. c Compound 19 activated the expression of p53 measured by WB, while OICR-9429 had little effect on p53 expression. d The level of H3K4me3 in cells treated with compound 19 or OICR-9429 for 24 h was measured by WB. The result indicated that compound 19 was a more potent inhibitor for H3K4mes.
Compound 19 elevates the expression of p53 and induces cell apoptosis
The KEGG enrichment analysis also revealed that compound 19 activated the p53 signaling pathway. As we know, the activation of p53 leads to cell apoptosis [43], so we measured the apoptosis rate of cells treated with the compound for 72 h by Annexin V/PI double staining. The apoptotic cells respectively increased from 12.18% to 26.78% and 42.13% in the 10 μM and 20 μM groups (Fig. 6b). To further verify the activation of the p53 signaling pathway, we applied WB to detect the expression of p53 in LAN5 cells treated with 10, 20 and 40 μM compound 19 for 24 h. The protein level of p53 was evidently elevated even in the 10 μM group (Fig. 6c), demonstrating that compound 19 was able to stimulate the expression of p53. On the contrary, OICR-9429 was found to had little impact on p53 expression in LAN5 cells even though the concentration was up to 40 μM.
Compound 19 reduces the level of H3K4me3 in neuroblastoma cells
Since WDR5 is an essential component of the RbBP5-WDR5-SET1/MLL methyltransferase complex [12], the blockade of the WBM or WIN site is supposed to hinder the methylation of H3K4. We detected the levels of H3K4me3 in LAN5 cells separately treated with DMSO, compound 19, and OICR-9429 for 24 h. Surprisingly, compound 19 presented a markedly stronger inhibition on H3K4me3 over OICR-9429 (Fig. 6d). We can see that 20 μM of compound 19 was enough to cause an obvious reduction of H3K4me3 while 40 μM of OICR-9429 had little effect.
Compound 19 and OICR-9429 synergistically inhibit neuroblastoma
Theoretically, the WBM and WIN site inhibitors bind to different binding interfaces of WDR5. We thus wondered if the combination of them would boost the anti-tumor activity. The concentration gradient for compound 19 was 0 to 20 μM (0, 0.625, 1.25, 2.5, 5, 10, and 20 μM), and we set the concentration of OICR-9429 at 0, 10 and 20 μM. After applying combinations of the two compounds for 72 h, we assessed the cell viability using CCK-8 reagent. A dose–response matrix was obtained (Fig. 7) and analyzed by the Bliss independence model [44]. The Bliss synergy scores were visualized in the form of 2D plot. A positive Bliss score represents a synergistic effect between two drugs. The overall synergy scores were 9.677 for LAN5 cells and 11.281 for IMR32 cells, meaning that compound 19 and OICR-9429 exhibited a synergistic effect in most combination groups (Fig. 7).
Fig. 7. Synergistic effect between the WBM and WIN site inhibitors.

IMR32 and LAN5 cells were treated with combinations of compound 19 and OICR-9429. A dose–response matrix was obtained through CCK-8 assay and analyzed by the Bliss independence model. The result demonstrated that compound 19 and OICR-9429 acted synergistically in treating neuroblastoma.
Discussion
Among sixty potential WBM site inhibitors discovered by ligand- and receptor-based virtual screening, four compounds, compound 11, 13, 19 and 22, were confirmed to be able to suppress proliferation of MYCN-amplified neuroblastoma cells and bind to WDR5. Binding affinities of the four active compounds were identified by SPR, ranging from 16.8 μM to 36.8 μM (Fig. 3). The WBM site is a typical protein-protein interaction (PPI) binding site which is shallow and challengeable to design a strong molecular binder [45]. Such hits have already presented potential for further development.
Based on the results from CCK-8 assay, we found that MYCN-amplified cells were extremely more susceptible to our discovered WDR5 binders than MYCN-unamplified cells (Fig. 3a). We thus speculated that interfering with the interaction between WDR5 and N-myc was involved in the anti-tumor mechanism of WBM site inhibitors. This is consistent with the previous finding that mutations disrupting WDR5 binding can perturb MYC-driven cancer [8]. Additionally, we observed an apparent off-target effect of compounds 11, 13, and 22 on 293 T cells, implying the existence of uncertain mechanisms. Thus, we chose compound 19 for further studies of mechanism of action in case of confounding factors.
The binding mode of compound 19 to WDR5 was determined by X-ray diffraction, providing the structural basis for understanding the mechanism of molecular action for this compound. Superposing the complex structures of compound 19 with the PPI complexes of MYC-MbIIIb, KANSL2, and RbBP5 with WDR5 confirmed that the compound 19 competitively occupied their binding region on WDR5. Taking the PPI system of MYC-MbIIIb and WDR5 as an example, a tight hydrophobic contact network formed between the residue V264 of MYC-MbIIIb and F266 and V268 of WDR5 was destroyed by a replacement of a new hydrophobic core formed between the 1,4-dichlorobenzene group of compound 19 and those two residues. This disruption of the binding of MYC or other cofactors at the WBM site may lead to a modulation of downstream gene transcription.
To investigate the mechanism of WBM site inhibitors, including the downstream modulation mechanisms, RNA-seq was conducted to compare the transcriptional profiles between the compound 19 group and the control group. In KEGG pathway enrichment analysis, forty-three up-regulated genes were enriched in the ribosome pathway (Fig. 5b). In the previous studies, it has been confirmed that WDR5 is highly correlated with the expression of ribosomal proteins through binding to the genes and activating the transcription [18, 46]. The treatment of a high-affinity WIN site inhibitor caused a decline in a wide range of ribosome genes in MV4:11 leukemic cells [18], however, in this study large amounts of ribosome genes in LAN5 cells treated with the WBM site inhibitor were up-regulated. It indicates that WBM site and WIN site play a different roles in mediating the biological function of WDR5 via interacting with other binding proteins, etc. Interfering protein-protein interactions of WDR5 from its different sites, i.e., WBM site and WIN site, leads to a different regulation of signaling pathway. When we block the WBM site and keep the WIN site active, up-regulation of the expression of ribosomal proteins via WIN site is enhanced. The elevation of ribosome gene transcription might be a secondary consequence of WBM site inhibition owing to the augmentation of WIN site function. Additionally, the cells applied in previous studies were MV4:11 leukemic cells, while our study focused on neuroblastoma cells. The contrary tendency may result from not only the functional difference between WBM site and WIN site but also the distinction between cell types. Since ribosomal proteins have diverse biological functions, their impacts on tumor progression are hence distinct. For example, RPL35 has been proved to accelerate proliferation of neuroblastoma cells through enhancing aerobic glycolysis [47], whereas RPL11 is a typical anti-tumor ribosomal protein which leads to MDM2 inactivation and subsequent p53 activation [48]. Therefore, the up-regulation of extensive ribosomal proteins caused by compound 19 may be a favorable factor of tumor growths, but at the same time a contributing factor for anti-tumor activities.
There were thirty-eight down-regulated genes enriched in the cell cycle pathway and nineteen in the DNA replication pathway. The WBM site inhibitor, compound 19, hindered cell cycle progression by repressing the cell cycle- and DNA replication-related genes. The flow cytometry assay demonstrated that compound 19 arrested LAN5 cells at the G1 phase (Fig. 6a). Since a previous study showed that the H3K4 methylation was required for DNA replication [49], the reduction of MLL-WDR5-RbBP5 methyltransferase complexes resulting from compound 19 might be involved in the regulation of DNA replication and cell cycle as well.
The KEGG pathway enrichment analysis also revealed that the p53-mediated apoptosis played a role in the anti-tumor effect of a WBM site inhibitor. The expression of p53 was then verified to be elevated after compound 19 treatment through WB (Fig. 6c). Furthermore, the flow cytometry analysis showed that the apoptotic rate of the treated group was significantly higher (Fig. 6b). As to the effect of WDR5 on the p53 signaling pathway, a previous study has proved that WDR5 activates the MDM2 gene transcription by inducing histone H3K4 trimethylation at its promoter region [7]. Given that MDM2 is a p53-specific E3 ubiquitin ligase, the upregulation of MDM2 leads to the increased degradation of p53 [50]. For that reason, an impairment of WDR5 function caused by WBM site inhibitors is supposed to alleviate the p53 inhibition by MDM2 and induce apoptosis in the treated cells.
Our study also focused on whether the WBM site or WIN site was a preferable therapeutic target for neuroblastoma. We compared the effect of compound 19 to a verified WIN site inhibitor, OICR-9429. Despite having a high binding affinity with WDR5, OICR-9429 did not show any stronger inhibitory activity against neuroblastoma cells which was in line with the previous reported studies [8]. Through RNA-seq, we found that the effects of the WBM site and WIN site inhibitor on cell cycle and DNA replication were similar, indicating that both sites were essential in this respect. However, the effects of the two inhibitors on ribosome genes and the p53 signaling pathway were quite different. Compared to the compound 19 treatment group, where nearly all differentially expressed ribosome genes were up-regulated, more than half of the genes were down-regulated in the OICR-9429 treatment group. Although OICR-9429 inhibited the expression of 48 ribosome genes, there were still thirty-three genes that were induced in LAN5 cells. It indicates that LAN5 cells, compared to MV4:11 leukemic cells, can resist the down-regulation of ribosome genes caused by the WIN site inhibitor. Regarding the effect on the p53 signaling pathway, both RNA-seq and WB showed that 20 μM OICR-9429 did not significantly stimulate the expression of p53. These results indicated that the WBM site inhibitor was more powerful in activating the p53 signaling pathway than the WIN site inhibitor.
Intuitively speaking, WIN site was thought to be a more essential site related with methyltransferase activity of WDR5 than WBM site because of its direct interaction with chromosome. However, in this study, it was observed that H3K4me3 was decreased with the treatment of compound 19, but not for OICR-9429, at the same concentration of 20 μM. It was inevitable that a WIN site binder could break the WDR5-MLL1 interaction and the RbBP5-WDR5-SET1/MLL complex assembly [21], but H3K4me3 in neuroblastoma cells was slightly affected. Although OICR-9429 is a potent WIN site inhibitor, it is applied solely to the treatment of MLL fusion gene-driven acute leukemia so far. In view of the crucial role of MLL in the development of MLL fusion gene-driven acute leukemia, the dysfunction of MLL is unlikely to be remedied. However, other malignant tumors may be able to compensate the effect of WIN site inhibitors by unknown mechanism, which explains why WIN site inhibitors have a poorer therapeutic effect against solid tumors than against leukemia. On the contrary, the disruption of the binding between WDR5 and RbBP5 by the compound 19 showed catalysis suppression of H3K4me3. Given that, the WBM site was proved to be an essential component of the methyltransferase activity, and the decline of H3K4me3 caused by WBM site inhibition was less likely to be overcome by compensatory effect than WIN site inhibition in neuroblastoma.
Now that compound 19 and OICR-9429 targeted WDR5 at two different sites, we tested the therapeutic effects when two types of inhibitors were simultaneously applied. The result revealed that the combination of compound 19 and OICR-9429 had an obvious synergistic effect in treating neuroblastoma. The reason might lie in the thorough disturbance of WDR5 function, which deprived neuroblastoma cells of the ability to resist the compounds. However, when neuroblastoma cells were treated with either the WBM or WIN site inhibitor, the function of the other site was preserved and was likely to participate in the compensatory response. Thus, the combination therapy affected the compensatory function of neuroblastoma cells and enhanced their sensitivity to the two compounds.
Conclusion
In conclusion, this study has discovered a potent sulfonamide compound (compound 19) that can effectively bind to the WBM site of WDR5 and specifically kill neuroblastoma cells with high copies of MYCN. It could elevate the expression of p53, down-regulate the level of H3K4me3, inhibit the progression of cell cycle and DNA replication, and induce apoptosis. In comparison with a WIN site molecular binder, the WBM site binder presents more obvious inhibitory activity in treating neuroblastoma. Additionally, synergistic tumor-suppressing effects between the WBM and WIN sites inhibitors are demonstrated, providing a potential therapeutic avenue for treating neuroblastoma (Fig. 8).
Fig. 8. Model of anti-tumor mechanism of compound 19.

Compound 19 binds to the WBM site of WDR5 and hinders the interaction of WDR5 with its ligand, thus regulating the transcription of genes which are related to ribosomes, cell cycle, DNA replication and apoptosis. Through combining with the WIN site inhibitor, OICR-9492, the anti-tumor effect of WDR5 inhibitors is boosted, providing a potential therapeutic avenue for treating neuroblastoma.
Supplementary information
Acknowledgements
This study was sponsored by Shanghai Municipal Science and Technology Commission (21Y11912200 to Kai Li), the Cyrus Tang Foundation, Clinical Research Plan of SHDC (SHDC2020CR2009A), Shanghai Municipal Key Clinical Specialty (shslczdzk05703), Hengjie Special Support Plan (2022 to Kai Li), Lingang Laboratory (LG202102-01-03), National Natural Science Foundation of China (82003654), Shanghai Science and Technology Development Funds (20QA1406400), and the Science and Technology Commission of Shanghai Municipality Grants (20431900100, 2043190012 and 20430780300); as well as the startup package from ShanghaiTech University. We also thank the HPC Platform of ShanghaiTech University and Shanghai Supercomputer Center for computing time.
Author contributions
FB, KL, and LHM conceptualized and designed the study. PXR and LW performed virtual screening. QLH, XLZ, YC, and WHL performed molecular biology experiments and analyzed the data. QLH and XLZ drafted the manuscript, which was revised and embellished by FB. FB, and KL supervised the study. All authors read and approved the article.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Qi-lei Han, Xiang-lei Zhang
Contributor Information
Kai Li, Email: likai2727@163.com.
Fang Bai, Email: baifang@shanghaitech.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-022-00999-z.
References
- 1.Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Prim. 2016;2:16078. doi: 10.1038/nrdp.2016.78. [DOI] [PubMed] [Google Scholar]
- 2.Twist CJ, Schmidt ML, Naranjo A, London WB, Tenney SC, Marachelian A, et al. Maintaining outstanding outcomes using response- and biology-based therapy for intermediate-risk neuroblastoma: a report from the children’s oncology group study anbl0531. J Clin Oncol. 2019;37:3243–55. doi: 10.1200/JCO.19.00919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kholodenko IV, Kalinovsky DV, Doronin II, Deyev SM, Kholodenko RV. Neuroblastoma origin and therapeutic targets for immunotherapy. J Immunol Res. 2018;2018:7394268. doi: 10.1155/2018/7394268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castel V, Cañete A, Navarro S, García-Miguel P, Melero C, Acha T, et al. Outcome of high-risk neuroblastoma using a dose intensity approach: improvement in initial but not in long-term results. Med Pediatr Oncol. 2001;37:537–42. doi: 10.1002/mpo.1248. [DOI] [PubMed] [Google Scholar]
- 5.Migliori V, Mapelli M, Guccione E. On WD40 proteins: propelling our knowledge of transcriptional control? Epigenetics. 2012;7:815–22. doi: 10.4161/epi.21140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang F, Zhang J, Ke X, Peng W, Zhao G, Peng S, et al. WDR5-Myc axis promotes the progression of glioblastoma and neuroblastoma by transcriptional activating CARM1. Biochem Biophys Res Commun. 2020;523:699–706. doi: 10.1016/j.bbrc.2019.12.101. [DOI] [PubMed] [Google Scholar]
- 7.Sun Y, Bell JL, Carter D, Gherardi S, Poulos RC, Milazzo G, et al. WDR5 supports an N-myc transcriptional complex that drives a protumorigenic gene expression signature in neuroblastoma. Cancer Res. 2015;75:5143–54. doi: 10.1158/0008-5472.CAN-15-0423. [DOI] [PubMed] [Google Scholar]
- 8.Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, et al. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol Cell. 2015;58:440–52. doi: 10.1016/j.molcel.2015.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas LR, Adams CM, Wang J, Weissmiller AM, Creighton J, Lorey SL, et al. Interaction of the oncoprotein transcription factor MYC with its chromatin cofactor WDR5 is essential for tumor maintenance. Proc Natl Acad Sci USA. 2019;116:25260–8. doi: 10.1073/pnas.1910391116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rickman DS, Schulte JH, Eilers M. The expanding world of N-myc-driven tumors. Cancer Discov. 2018;8:150–63. doi: 10.1158/2159-8290.CD-17-0273. [DOI] [PubMed] [Google Scholar]
- 11.Otte J, Dyberg C, Pepich A, Johnsen JI. MYCN function in neuroblastoma development. Front Oncol. 2021;10:624079. doi: 10.3389/fonc.2020.624079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ernst P, Vakoc CR. WRAD: enabler of the SET1-family of H3K4 methyltransferases. Brief Funct Genomics. 2012;11:217–26. doi: 10.1093/bfgp/els017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guccione E, Martinato F, Finocchiaro G, Luzi L, Tizzoni L, Dall’ Olio V, et al. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol. 2006;8:764–70. doi: 10.1038/ncb1434. [DOI] [PubMed] [Google Scholar]
- 14.Zhao J, Chen W, Pan Y, Zhang Y, Sun H, Wang H, et al. Structural insights into the recognition of histone H3Q5 serotonylation by WDR5. Sci Adv. 2021;7:eabf4291. doi: 10.1126/sciadv.abf4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song JJ, Kingston RE. WDR5 interacts with mixed lineage leukemia (MLL) protein via the histone H3-binding pocket. J Biol Chem. 2008;283:35258–64. doi: 10.1074/jbc.M806900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Odho Z, Southall SM, Wilson JR. Characterization of a novel WDR5-binding site that recruits RbBP5 through a conserved motif to enhance methylation of histone H3 lysine 4 by mixed lineage leukemia protein-1. J Biol Chem. 2010;285:32967–76. doi: 10.1074/jbc.M110.159921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tian J, Teuscher KB, Aho ER, Alvarado JR, Mills JJ, Meyers KM, et al. Discovery and structure-based optimization of potent and selective WD repeat domain 5 (WDR5) inhibitors containing a dihydroisoquinolinone bicyclic core. J Med Chem. 2020;63:656–75. doi: 10.1021/acs.jmedchem.9b01608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aho ER, Wang J, Gogliotti RD, Howard GC, Phan J, Acharya P, et al. Displacement of WDR5 from chromatin by a WIN site inhibitor with picomolar affinity. Cell Rep. 2019;26:2916–28. doi: 10.1016/j.celrep.2019.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang F, Jeon KO, Salovich JM, Macdonald JD, Alvarado J, Gogliotti RD, et al. Discovery of potent 2-Aryl-6,7-dihydro-5 H-pyrrolo[1,2- a]imidazoles as WDR5-WIN-site inhibitors using fragment-based methods and structure-based design. J Med Chem. 2018;61:5623–42. doi: 10.1021/acs.jmedchem.8b00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karatas H, Li Y, Liu L, Ji J, Lee S, Chen Y, et al. Discovery of a highly potent, cell-permeable macrocyclic peptidomimetic (MM-589) targeting the WD repeat domain 5 protein (WDR5)-mixed lineage leukemia (MLL) protein-protein interaction. J Med Chem. 2017;60:4818–39. doi: 10.1021/acs.jmedchem.6b01796. [DOI] [PubMed] [Google Scholar]
- 21.Getlik M, Smil D, Zepeda-Velázquez C, Bolshan Y, Poda G, Wu H, et al. Structure-based optimization of a small molecule antagonist of the interaction between WD repeat-containing protein 5 (WDR5) and mixed-lineage leukemia 1 (MLL1) J Med Chem. 2016;59:2478–96. doi: 10.1021/acs.jmedchem.5b01630. [DOI] [PubMed] [Google Scholar]
- 22.Senisterra G, Wu H, Allali-Hassani A, Wasney GA, Barsyte-Lovejoy D, Dombrovski L, et al. Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5. Biochem J. 2013;449:151–9. doi: 10.1042/BJ20121280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolshan Y, Getlik M, Kuznetsova E, Wasney GA, Hajian T, Poda G, et al. Synthesis, optimization, and evaluation of novel small molecules as antagonists of WDR5-MLL interaction. CS Med Chem Lett. 2013;4:353–7. doi: 10.1021/ml300467n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karatas H, Townsend EC, Cao F, Chen Y, Bernard D, Liu L, et al. High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein-protein interaction. J Am Chem Soc. 2013;135:669–82. doi: 10.1021/ja306028q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marschalek R. Mechanisms of leukemogenesis by MLL fusion proteins. Br J Haematol. 2011;152:141–54. doi: 10.1111/j.1365-2141.2010.08459.x. [DOI] [PubMed] [Google Scholar]
- 26.Marschalek R. MLL leukemia and future treatment strategies. Arch Pharm (Weinheim) 2015;348:221–8. doi: 10.1002/ardp.201400449. [DOI] [PubMed] [Google Scholar]
- 27.Chen IJ, Foloppe N. Drug-like bioactive structures and conformational coverage with the LigPrep/ConfGen suite: comparison to programs MOE and catalyst. J Chem Inf Model. 2010;50:822–39. doi: 10.1021/ci100026x. [DOI] [PubMed] [Google Scholar]
- 28.Chacón Simon S, Wang F, Thomas LR, Phan J, Zhao B, Olejniczak ET, et al. Discovery of WD repeat-containing protein 5 (WDR5)-MYC inhibitors using fragment-based methods and structure-based design. J Med Chem. 2020;63:4315–33. doi: 10.1021/acs.jmedchem.0c00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macdonald JD, Chacón Simon S, Han C, Wang F, Shaw JG, Howes JE, et al. Discovery and optimization of salicylic acid-derived sulfonamide inhibitors of the WD repeat-containing protein 5-MYC protein-protein interaction. J Med Chem. 2019;62:11232–59. doi: 10.1021/acs.jmedchem.9b01411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu X, Jiang H, Li H. SHAFTS: A hybrid approach for 3D molecular similarity calculation. 1. Method and assessment of virtual screening. J Chem Inf Model. 2011;51:2372–85. doi: 10.1021/ci200060s. [DOI] [PubMed] [Google Scholar]
- 31.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–49. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 32.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–66. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 33.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–42. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–54. doi: 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- 36.Ianevski A, Giri AK, Aittokallio T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020;48:W488–W93. doi: 10.1093/nar/gkaa216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Costa-Silva J, Domingues D, Lopes FM. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS One. 2017;12:e0190152. doi: 10.1371/journal.pone.0190152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.da Silva Rocha SFL, Olanda CG, Fokoue HH, Sant’Anna CMR. Virtual screening techniques in drug discovery: review and recent applications. Curr Top Med Chem. 2019;19:1751–67. doi: 10.2174/1568026619666190816101948. [DOI] [PubMed] [Google Scholar]
- 41.Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules. 2015;20:13384–421. doi: 10.3390/molecules200713384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X, Simpson ER, Brown KA. p53: Protection against tumor growth beyond effects on cell cycle and apoptosis. Cancer Res. 2015;75:5001–7. doi: 10.1158/0008-5472.CAN-15-0563. [DOI] [PubMed] [Google Scholar]
- 44.Bliss CI. The calculation of microbial assays. Bacteriol Rev. 1956;20:243–58. doi: 10.1128/br.20.4.243-258.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guarnaccia AD, Tansey WP. Moonlighting with WDR5: A cellular multitasker. J Clin Med. 2018;7:21. doi: 10.3390/jcm7020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a cancer dependency map. Cell. 2017;170:564–76. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu W, Yu N, Li F, Gao P, Lin S, Zhu Y. RPL35 promotes neuroblastoma progression via the enhanced aerobic glycolysis. Am J Cancer Res. 2021;11:5701–14. [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng J, Lang Y, Zhang Q, Cui D, Sun H, Jiang L, et al. Structure of human MDM2 complexed with RPL11 reveals the molecular basis of p53 activation. Genes Dev. 2015;29:1524–34. doi: 10.1101/gad.261792.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu F, Wu X, Yin F, Chia-Fang Lee C, Yu M, Mihaylov IS, et al. Regulation of DNA replication and chromosomal polyploidy by the MLL-WDR5-RBBP5 methyltransferases. Biol Open. 2016;5:1449–60. doi: 10.1242/bio.019729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res. 2003;1:1001–8. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.