

Abstract
Ovarian cancer is the most lethal gynecologic malignancy with a stubborn mortality rate of ~65%. The persistent failure of multi-line chemotherapy, and significant tumor heterogeneity, have made it challenging to improve outcomes. A target of increasing interest is the mitochondrion because of its essential role in critical cellular functions, and the significance of metabolic adaptation in chemoresistance. This review describes mitochondrial processes, including metabolic reprogramming, mitochondrial transfer, and the state of mitochondrial networks, in ovarian cancer progression and chemoresistance. The effect of malignant ascites, or excess peritoneal fluid, on mitochondrial function is discussed. The role of photodynamic therapy (PDT) in overcoming mitochondria-mediated resistance, is presented. PDT, a photochemistry-based modality, involves the light-based activation of a photosensitizer leading to the production of short-lived reactive molecular species and spatio-temporally confined photodamage to nearby organelles and biological targets. The consequential effects range from sub-cytotoxic priming of target cells for increased sensitivity to subsequent treatments, such as chemotherapy, to direct cell killing. This review discusses how PDT-based approaches can address key limitations of current treatments. Specifically, an overview of the mechanisms by which PDT alters mitochondrial function, and a summary of preclinical advancements and clinical PDT experience in ovarian cancer are provided.
Graphical abstract
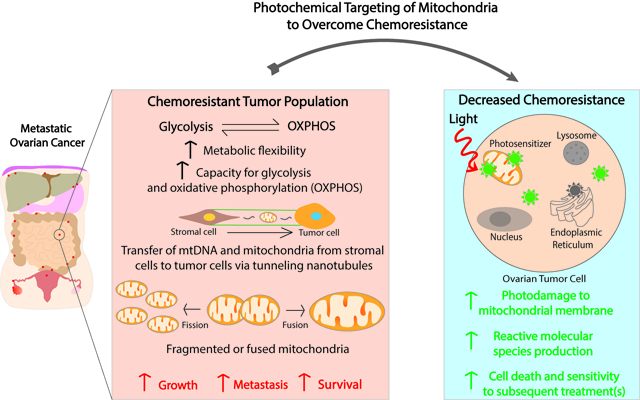
In metastatic ovarian cancer, chemoresistant tumor populations demonstrate increased metabolic flexibility, enhanced capacity for glycolysis and oxidative phosphorylation, increased numbers of mitochondria and of mitochondrial DNA through mitochondrial transfer, and altered mitochondrial dynamics (fission/fusion). Photodynamic therapy reverses chemoresistance in ovarian cancer and synergizes with conventional therapies. The role of mechanism-based combinations using photosensitizers that are, in part, synthesized in mitochondria, or localize to subcellular organelles, including mitochondria, is presented. The effects of photodamage to mitochondria leading to enhanced cell death, as well as priming for increased sensitivity to subsequent treatments, are discussed.
INTRODUCTION
Ovarian cancer is the most lethal gynecologic malignancy with a mortality rate of ~65% (1, 2). In 2022, it is estimated that in the United States there will be 19,880 new cases and 12,810 deaths (2). Worldwide, it is predicted that in 2022, there will be just under 330,000 cases of ovarian cancer, and just under 220,000 patients will succumb to the disease (3). Further, the Global Cancer Observatory projects that by 2040, the number of ovarian cancer cases diagnosed will rise 30% to 428,966 alongside deaths, which are projected to increase by over 40% to 313,617 (1, 3). One of the major contributing factors to such high mortality associated with ovarian cancer is chemoresistance. Currently, the standard of care for ovarian cancer is a combination of platinum and taxane-based chemotherapy (4). While most patients initially respond to platinum-based chemotherapy, nearly 85% will develop resistance and disease recurrence (5–7). Thus, there is a need to understand the mechanisms underlying platinum resistance, which in turn might be exploited as therapeutic targets. In this context, emerging evidence from recent studies suggests that alterations in mitochondrial function contributes to platinum resistance, making these organelles an attractive therapeutic target for ovarian cancer where conventional and targeted therapies have largely failed (8–11).
Photodynamic therapy (PDT), which uses light and light-activatable molecules to generate reactive molecular species (RMS), provides a unique opportunity to target various subcellular compartments, including mitochondria (12, 13). Benzoporphyrin derivative (BPD), Photofrin and protoporphyrin IX (PpIX) are among the clinically approved photosensitizers for which mitochondria have been shown to be sites of preferential localization or synthesis (14–16), and RMS-induced mitochondrial damage is a mechanism of PDT-mediated cell killing. Given the role of mitochondrial dysfunction in chemoresistance, PDT could be leveraged as an enabling modality to sensitize cancer cells to platinum-based chemotherapy through mitochondrial targeting. Moreover, emerging preclinical evidence suggests that PDT can be a suitable strategy for disseminated ovarian cancer treatment. (17–25). Notably, PDT can effectively target platinum-resistant ovarian cancer cells (26), and it has been shown to synergize with platinum-based chemotherapy in a variety of in vitro and in vivo models of ovarian cancer (27–29). Several clinical trials showed technical feasibility of PDT in patients with intraperitoneal carcinomatosis, including in patients with advanced ovarian cancer (30–33). In this review, we will discuss the role of mitochondrial dysfunction in chemoresistance, highlight the progress in implementing PDT for disseminated ovarian cancer and outline several promising photochemical approaches towards overcoming platinum resistance by targeting mitochondria.
Due to the lack of specific screening methods, ovarian cancer often remains undiagnosed until it is disseminated throughout the abdominal cavity. Primary tumors, which most commonly originate from the ovarian surface epithelium and secretory epithelial cells of the fallopian tube, can shed malignant cells directly into the peritoneum (34, 35). This process is often exacerbated by ascites, or excessive fluid buildup, which correlates with disease severity, metastatic spread, and poor treatment outcomes (36). Emerging evidence suggests that fluid shear stress generated by ascitic currents promotes epithelial-mesenchymal transition, chemoresistance and metastatic spread (26, 37). Additionally, our research group recently discovered that sub-cytotoxic doses of the select per- and polyfluoroalkyl substances (PFAS) promote resistance to carboplatin in ovarian cancer cells, suggesting that environmental contaminants contribute to chemoresistance (38). The standard of care for ovarian cancer involves cytoreductive surgery, or removal of the primary tumor and any detectable macroscopic lesions, followed by a combination treatment with platinum- and taxane-based chemotherapy (4, 10, 39, 40). Frequently, this treatment regimen involves the combination of cisplatin or carboplatin (platinum) and paclitaxel (taxane) (4, 5, 41). While most patients receiving this therapeutic regimen respond initially, ~85% of patients present with recurrent disease (5). Platinum resistance is a major barrier to the effective treatment of ovarian cancer (36, 38), and since recurrent ovarian cancer is rarely curable (42), other therapeutic targets should be examined to limit disease progression and increase overall survival in these patients.
One potential therapeutic target worth exploring in the context of ovarian cancer is the mitochondrion. While the primary function of mitochondria is energy metabolism, they play an essential role in cell survival, proliferation, nucleotide, and amino acid metabolism, signaling as well as RMS detoxification. Multiple studies have linked mitochondrial dysfunction to ovarian cancer progression and therapy resistance. Specifically, it has been shown that platinum-resistant ovarian cancer cells display flexible bioenergetic profiles indicative of mitochondria that can effectively adjust to cell function under selection pressures exerted by chemotherapy (43). Moreover, horizontal mitochondrial transfer has been implicated in cell survival and metabolic reprogramming, enhanced metastatic potential and chemoresistance (44). Changes in mitochondrial dynamics such as mitochondrial fission and fusion have also been linked to chemoresistance (45). Interestingly, one platinum-based chemotherapeutic agent currently used in the treatment of ovarian cancer, cisplatin, secondarily targets mitochondria (44, 46–50). Upon administration, the platinum atom of cisplatin forms covalent bonds with purine bases on nuclear DNA (nDNA), forming both intra- and inter-strand crosslinks and preventing DNA replication and transcription (10, 44, 48, 51, 52). While this is one mechanism by which cisplatin leads to tumor cell death, studies have reported that a limited amount, as low as 1% of cisplatin interacts with nDNA (44, 46, 47). The remainder of the administered cisplatin will interact with sulfur donors, proteins, and mitochondria, including mitochondrial DNA (mtDNA) (44, 46–50). Given this emerging evidence of the role of mitochondria in platinum resistance, the use of mitochondria-specific agents that can be administered alone or in combination with platinum-based chemotherapy may be effective at improving ovarian cancer patient outcomes.
One approach that can directly target mitochondria is PDT. The PS molecular structure, the presence of targeting ligands (53), and nanoparticle encapsulation can influence subcellular localization and subsequent photodamage. Notably, several clinical PS have been shown to preferentially localize to mitochondria, which can be leveraged to combat chemoresistance. Above a certain threshold, photodamage to mitochondria results in a rapid loss of ΔΨm and cytochrome c release, which can induce apoptosis (54). However, sub-cytotoxic amounts of RMS can have profound effects on mitochondrial homeostasis, which may be sufficient to overcome the compensatory activation of mitochondrial activity in cancer cells and sensitize them to platinum-based chemotherapy (55, 56). Many preclinical studies have demonstrated the ability of PDT to enhance the efficacy of platinum chemotherapy in a broad range of light doses; however, whether this effect specifically relates to mitochondrial targeting remains to be elucidated. From the clinical standpoint, PDT offers a highly selective and minimally invasive approach to treat disseminated ovarian cancer, strengthening the rationale for exploring photochemical targeting of mitochondria to overcome platinum resistance.
In this review, we will outline several ways in which mitochondrial dysfunction contributes to chemoresistance in ovarian cancer and discuss how photochemical targeting of mitochondria might be utilized to overcome it. Further, we will discuss how PDT fits into the current ovarian cancer treatment paradigms and analyze how recent advancements in photosensitizer design will facilitate its clinical translation. Given the multipronged nature of chemoresistance mechanisms, successful treatment regimens will likely consist of several complementary approaches with non-overlapping toxicities and mechanisms of action. In this context, mitochondria-targeted PDT has the potential to become an essential tool in combating chemoresistance in ovarian cancer.
THE ROLE OF MITOCHONDRIA IN THE DEVELOPMENT OF CHEMORESISTANCE
While mitochondria are best known for their role in bioenergetics, they are also involved in a plethora of cell signaling pathways responsible for cancer development, progression and chemoresistance. It has been shown that most cancer cells carry somatic and/or mtDNA mutations, leading to mitochondrial dysfunction and metabolic rewiring. On the most basic level, cancer cells that have undergone metabolic rewiring are more adept at adapting to changes in their environment (43). Interestingly, this adaptation can involve the activation of both glycolytic and oxidative phosphorylation (OXPHOS) pathways. Moreover, metabolic flexibility in ovarian cancer (i.e., the ability to switch between OXPHOS and glycolysis) to meet high metabolic demands, is associated with chemoresistance (44). The following sections will discuss how increased metabolic activity is implicated in resistance to platinum-based chemotherapeutic agents. We will also examine horizontal mitochondrial transfer, which typically involves the formation of cytoplasmic bridges, through which mitochondria are shuttled between cancer and stromal cells. Recent studies have shown that this process increases the mitochondrial respiratory capacity of cancer cells, facilitating cancer cell survival and promoting chemoresistance. Moreover, changes in mitochondrial dynamics, including fusion and fission, have been observed in chemoresistant cells. Finally, we will discuss how malignant ascites contributes to mitochondrial dysfunction, thereby promoting chemoresistance.
Metabolic state of ovarian cancer
One of the first working hypotheses for the role of mitochondria in cancer cell metabolism was proposed by Otto Warburg. Warburg hypothesized that tumor cells depend more on glycolysis for energy production compared to OXPHOS due to mitochondrial dysfunction (44). This hypothesis, which became known as the “Warburg effect”, formed the basis of the concept of metabolic reprogramming, although it is now accepted that, in cancers, mitochondria are not necessarily dysfunctional despite the metabolic switch to glycolysis (44, 57, 58). Metabolic reprogramming assists tumor cells in supporting their high proliferation rates and survival due to strong channeling towards anabolic processes (59, 60). The ability of tumor cells to accelerate glycolysis to promote growth is primarily due to tumor suppressor dysregulation or oncogene activation that confers glycolytic enzyme hyperactivity (59, 61). For example, GLUT1, which is a glucose transporter, is often overexpressed in high-grade serous ovarian carcinomas, and is associated with increased metastatic potential (59, 62–64). Additionally, hexokinase (HK) is a glycolytic enzyme frequently upregulated in ovarian cancer that is associated with tumorigenesis and cell survival (59, 65–67). Other glycolytic enzymes involved in the Warburg effect and implicated in ovarian cancer are summarized in Table 1.
Table 1.
Glycolytic Proteins/Enzymes Involved in Cancer Progression
| Glycolytic Proteins/Enzyme | Normal Function | Role in Cancer |
|---|---|---|
| Glucose transporter 1 (GLUT1) | Facilitate glucose transfer across a membrane (69) | Facilitates metastasis, indicative of a poor prognosis, marker of hypoxia (63, 64) |
| Hexokinase II (HKII) | Converts glucose to glucose-6-phosphate (60) | Promotes tumorigenesis, survival, chemoresistance, and shift toward glycolytic metabolism (66–68, 70–72) |
| Phosphofructokinase 1 (PFK1) | Converts fructose-6-phosphate to fructose 1,6 bisphosphate (73, 74) | Inhibits apoptosis (73) |
| Pyruvate Kinase | Converts phosphoenolpyruvate to pyruvate (60) | Promotes tumorigenesis, macromolecule synthesis, and metabolic adaptations (75–79) |
| Lactate Dehydrogenase-A (LDH-A) | Converts pyruvate to lactate and NADH to NAD+ (60) | Promotes increased oxidative metabolism and tumor microenvironment acidification (80–83) |
While metabolic reprogramming remains a hallmark of cancer, recent studies have called for re-evaluation of the Warburg effect due to the limited efficacy of anti-glycolytic agents (83). As a result, the “reverse Warburg effect” has been proposed, which states that stromal cells adjacent to tumor cells have elevated levels of aerobic glycolysis and therefore supply energy and metabolites to rapidly proliferating cancer cells (83, 84). One type of stromal cell that may be involved in tumor progression and response to therapy is the cancer stem-like cell (CSC), which plays a key role in both disease initiation and recurrence and is inherently chemoresistant (83, 85). It has also been suggested that cancer cells induce aerobic glycolysis in nearby stromal fibroblasts, enabling their transformation into cancer-associated fibroblasts (CAFs) that can provide cancer cells with energy and metabolites supporting tumor growth and angiogenesis (83, 86). CAFs can also be associated with chemoresistance, and in the context of pancreatic ductal adenocarcinoma, one study found that CAF-induced resistance could be reversed using a combination of oxaliplatin and metformin, a mitochondrial complex I inhibitor (87). The reverse Warburg effect has also been supported clinically, as women with high stroma proportions have significantly worse overall survival rates regardless of ovarian tumor subtype (83, 88).
Also contrary to the Warburg effect, recent studies have found that both glycolysis and OXPHOS pathways are elevated in cancer cells compared to healthy cells (59, 60, 89). This has led researchers to speculate that cancer cells are able to metastasize and evade therapeutic intervention due to their ability to switch between energy substrates and metabolic pathways (59, 90, 91). This bioenergetic flexibility in utilizing glycolysis and OXPHOS for energy production is particularly advantageous because many metabolites and products of glycolysis are required for downstream energy pathways like the tricarboxylic acid (TCA) cycle, pentose phosphate pathway, gluconeogenesis, and fatty acid or amino acid synthesis (59, 60, 92). Flexibility in energy production pathways is also observed in ovarian cancer cells in their enhanced ability to perform glutaminolysis, which converts glutamine into products able to fuel the TCA cycle and energy production. Notably, glutamine metabolism also contributes to antioxidant defenses and nucleotide metabolism, all of which can increase cell proliferation and resistance to oxidative stress. Compared to less invasive ovarian cancer cells, invasive ovarian cancer cells are glutamine-dependent and, in ovarian cancer patients, glutaminolysis is correlated with poor survival (93). Additionally, ovarian tumor cells that overexpress glutaminase, a glutaminolytic enzyme, are platinum-resistant (94). As a result, targeting glutaminolysis has shown some success in mitigating ovarian tumor progression and chemoresistance. For example, Han et al. (95) reported that inhibiting ubiquitin-specific peptidase 13 (USP13), which upregulates enzymes critical for mitochondrial respiration and glutaminolysis, suppressed tumor progression and sensitized ovarian tumor cells to a PI3K/AKT inhibitor. Others have also shown that inhibition of glutaminase in glutaminase-overexpressing cells increased response to Olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor in mice (94). Similar results regarding the sensitization of ovarian cancer cells to treatment after administration of another glutaminase inhibitor have also been reported (96). While inhibiting glutaminolysis may be effective in suppressing tumor progression and mitigating chemoresistance, dual inhibition of glycolysis and glutaminolysis appeared to be a promising route for the treatment of ovarian cancer. In a study by Sun et al. (97), the simultaneous inhibition of glycolysis and glutaminolysis by 2-deoxyglucose and aminooxyacetate, respectively, led to a synergistic decrease in ovarian cancer cell proliferation.
While bioenergetic flexibility assists with maintaining the high proliferation rate of tumor cells, it also helps tumor cells confer resistance to chemotherapeutic agents. One way that this occurs is through the excess production of reactive oxygen species (ROS) and upregulated levels of antioxidants in tumor cells with elevated levels of OXPHOS. During OXPHOS, ROS are produced within the electron transport chain (ETC), and to combat these elevated ROS levels, tumor cells upregulate superoxide dismutase, glutathione, thioredoxin, and peroxiredoxins (44). An important mechanism by which chemotherapeutic drugs work is through the production of ROS (44, 98, 99), meaning that the efficacy of chemotherapeutic drugs may be decreased in tumor cells with elevated OXPHOS levels due to increased levels of ROS and antioxidants. Likewise, cancer cells that activate glycolysis or glutaminolysis can also increase antioxidant defenses by impacting the pentose phosphate pathway and glutathione synthesis, respectively. Under normal conditions, ROS induced by chemotherapy can alter ΔΨm and damage the mitochondrial respiratory chain, leading to apoptosis (44). The same has not been observed in chemoresistant tumor cells, as resistant cell populations with elevated levels of antioxidants are able to counteract drug-induced ROS production and promote their survival (44, 100–104).
Studies examining the ability of cisplatin to reduce ovarian cancer cell populations found that cisplatin-sensitive cell lines (OVCAR-3, OVCAR-4, and IGROV-1) had higher relative mitochondrial content and basal oxygen consumption rates (OCRs) post-cisplatin exposure compared to resistant cell lines (OVCAR-5, OVCAR-8, and A2780) (41). However, increased OCRs have also been reported in cisplatin-resistant cell lines. Zampieri et al. (10) found that cisplatin-resistant SKOV-3-R cells had higher respiratory spare capacities and increased ETC complex I activity while an additional cisplatin-resistant cell line, COV-362-R, had higher respiratory spare capacities, increased OCR, and increased citrate synthase activity compared to their platinum-sensitive counterparts (SKOV-3 and COV-362, respectively). Additionally, SKOV-3-R cells consumed more glucose and produced more lactate compared to SKOV-3 cells, indicating increased rates of anaerobic glycolysis, and suggesting that cisplatin-resistant cells have an increased capacity for performing OXPHOS and/or glycolysis compared to cisplatin-sensitive cells (10). Kleih et al. (41) also found that platinum-sensitive ovarian cancer cell lines had increased levels of mitochondrial ROS (O−2) following cisplatin exposure compared to platinum-resistant cells. Further exploring the role of ROS in cisplatin-mediated cell death, OVCAR-3 and OVCAR-4 cells incubated with cisplatin in the presence of glutathione, a ROS scavenger, demonstrated significantly increased cell viability. As mitochondrial ROS appear to play an integral role in cisplatin-mediated apoptosis, understanding how to increase mitochondrial ROS levels in platinum-resistant cells may reveal mechanisms to overcome cisplatin resistance. To evaluate one potential mechanism to increase ROS production to overcome cisplatin resistance, Kleih et al. (41) inhibited uncoupling protein 2, since mitochondrial ROS induce uncoupling protein activation, and found that platinum-resistant OVCAR-8 cells had significantly increased cisplatin-induced mitochondrial ROS and apoptosis. This finding illustrates that increasing mitochondrial ROS levels through inhibition of uncoupling proteins may be effective at overcoming cisplatin resistance in ovarian cancer cells.
Bioenergetic adaptions have also been implicated in ovarian cancer cell resistance to chemotherapy by Dar et al. (9). In this study, bioenergetic profiles were measured for a variety of different cell lines, and while most had equal glycolytic properties, as measured by extracellular acidification rate (ECAR), and OXPHOS, measured by OCR, PEO1 and A2780 cells preferred glycolysis while SKOV-3, SKOV-3-IP, and Caov-3 cells favored OXPHOS. Other studies have also reported variable bioenergetic profiles across ovarian cancer cell lines (105). Additionally, in cells with increased glycolysis, proliferation increased, which has been observed previously as well (106). When comparing chemosensitive A2780 and PEO-1 cells with their chemoresistant counterparts, C200 and PEO-4, chemo-sensitive cells had lower overall OXPHOS and glycolysis levels, indicating that chemoresistant cells are more highly metabolically active. Chemoresistant cells also displayed higher mRNA and/or protein levels of OXPHOS and glycolytic genes including cytochrome c oxidase subunit Vb, GLUT1 and LDH. Increased bioenergetic capacities of chemoresistant cells were also demonstrated by increased ΔΨm, ROS levels, and mitochondrial density as well as decreased sensitivity to glucose deprivation, further demonstrating a higher level of “cellular fitness” (9, 105, 107, 108). To determine whether chemotherapy exposure induced a highly metabolically active phenotype in chemosensitive cells, Dar et al. (9) exposed A2780 cells to cisplatin or paclitaxel and found that after cisplatin, but not paclitaxel exposure, cells shifted from a glycolytic phenotype towards a highly metabolically active phenotype.
While the findings reported in the previous two paragraphs resulted from the use of ovarian cancer cell lines, Bindra et al. (109) performed mitochondrial profiling on high-grade ovarian cancer and benign ovarian mass tissues to better understand the role of mitochondrial metabolic function in human ovarian tumors. Results showed that compared to benign tissue, high-grade ovarian cancer tissue had significantly elevated levels of citrate synthase, succinate dehydrogenase, and cytochrome c oxidase activity, as well as increased mitochondrial health indices, computed by dividing the ratio of respiratory chain activity by markers of mitochondrial content. When examining mitochondrial enzyme levels across different stages of ovarian tumors, respiratory chain enzyme activity significantly decreased in stage IV tumors and had approximately half the levels of citrate synthase, succinate dehydrogenase, and cytochrome c oxidase compared to stage I tumors (109).
Overall, these studies support the notion that, to promote survival and evade chemotherapeutic treatment, ovarian cancer cells can adapt their bioenergetic profiles and develop a highly metabolically active phenotype in which cells can preferentially use glycolysis or OXPHOS for energy production. This metabolically active profile has been implicated in resistance to platinum-based chemotherapeutic agents. Thus, preventing bioenergetic shifting in ovarian cancer cells may re-sensitize tumor cells to treatment through limiting their means of energy production.
Mitochondrial transfer in ovarian cancer progression
The shuttling of mitochondria and mtDNA from stromal cells to cancer cells has been implicated in disease progression and the development of chemotherapy resistance. This horizontal transfer permits cancer cells, which typically have limited mtDNA, and therefore limited capacity for performing mitochondrial functions including energy production, to gain mtDNA or complete mitochondria from surrounding cells thereby promoting their survival (44, 110–114). The first study to demonstrate the impact of mitochondrial transfer on cellular function was performed by Spees et al. (112), who found that adult nonhematopoietic stem/progenitor cells from human bone marrow effectively transferred mitochondria to A549 r° recovering their mitochondrial respiratory capacity. Rho 0 (r°) are those depleted of mtDNA that are generally generated by subchronic treatment with low doses of ethidium bromide; as such, they are unable to respire using non-fermentable carbon sources. Other studies have reported similar findings, supporting the notion that mitochondrial transfer rescues the function of damaged mitochondria through the shuttling of necessary molecules or organelles (110, 115). In addition to promoting bioenergetic adaptations in recipient cells, mitochondrial transfer has also been shown to initiate stem cell differentiation and activate inflammatory pathways (44, 110, 116–118). By facilitating these processes, mitochondrial transfer may play a key role in facilitating disease progression (110, 116, 119–122).
Mitochondrial transfer can occur through a variety of mechanisms including through the formation of tunneling nanotubules (TNTs), microvesicles, gap junctions, and cell fusion (110, 123–125). While there are several different mechanisms by which mitochondrial information can be transferred, it is thought that the predominant mechanism is through an active process involving TNTs (44, 123, 124, 126–131). TNTs can be formed through two main mechanisms: 1) two adjacent cells diverging, or 2) fusion of filopodium-like membrane actin protrusions between cells (132–137). Both mechanisms leave a fine tunnel-like structure connecting both cells, known as a TNT (110, 132). After TNTs are formed, mitochondrial transfer between cells is facilitated by Miro1 and Miro2, which are Rho-guanosine triphosphatases (GTPases) that assist mitochondrial movement through the TNT (110, 122, 123, 132, 138–141). These proteins play a critical role in TNT formation, and studies have shown that in mesenchymal stem cells (MSCs), overexpression of Miro1 enabled mitochondrial transfer via TNTs to injure epithelial cells. Conversely, downregulation of Miro1 has been shown to inhibit TNT formation and therefore mitochondrial transfer (123, 142).
While many studies have examined the benefits of mitochondrial transfer on recipient cells, TNT trafficking has been shown to be both unidirectional and bidirectional depending on cell type and cell state (121, 132, 143–155). For example, studies have reported the unidirectional transport of mitochondria between rat pheochromocytoma cells, while other studies have demonstrated bidirectional transport between macrophages connected by a nanotube (132, 133, 143, 156, 157). Understanding the directionality of mitochondrial transfer is critical to understanding how donor and recipient cells are affected by this process.
Cells containing dysfunctional mitochondria can promote the formation of TNTs through various mechanisms, one of which is by sending cell stress signals to adjacent cells. Studies have shown that in injured endothelial cells, phosphatidylserine exposure triggers the formation of TNTs from MSCs (110, 156). The formation of TNTs can also arise from other chemical exposures, including doxorubicin, ethidium bromide, or cigarette smoke as well as by certain medium conditions, such as depleted serum or acidification (110, 124, 145, 146, 158, 159). In certain types of cells, tumor necrosis factor and NF-kB have been shown to induce TNT formation (123, 126, 127), suggesting that a variety of signaling pathways may be involved in this process.
Transfer of mitochondria can be particularly advantageous in conferring cell survival and metabolic reprogramming, thereby enhancing tumor aggression and metastatic potential. It is thought that the transfer of low copy numbers of mtDNA can restore normal mitochondrial function in tumor cells, conferring a major growth and survival advantage (110, 160). Several studies have shown that mitochondrial transfer can be used in vitro and in vivo to promote alterations in bioenergetic profiles in recipient cells (44, 113, 120, 156, 161–163). Other studies have also shown that gap junction channels may play critical roles in mitochondrial transfer. In a study by Islam et al. (120), bone marrow-derived stem cells (mBMSCs) formed connections with alveolar cells through the formation of connexin-43-based gap junction channels. These gap junction channels led to increased calcium communication and the formation of both nanotubules and microvesicles which were not formed in mBMSC cells loaded with a calcium chelator. According to the authors, this suggests a pivotal role of gap junction channel-mediated calcium communication in the formation of nanotubules and microvesicles, which enabled the restoration of ATP and surfactant secretion in injured alveolar cells. Other studies have also implicated connexin-43 gap junctions and related signaling pathways in the formation of TNTs (130, 164–166). Additionally, a recent report by Norris (167) also demonstrated that gap junction internalization is a mechanism by which mitochondrial transfer can occur, thus the multifaceted role of gap junctions and gap junction-mediated signaling in mitochondrial transfer warrants further investigation.
While mitochondrial transfer can facilitate cancer cell survival and increase bioenergetic capacity, it can also facilitate the development of chemoresistance. For example, Pasquier et al. (144) reported that mitochondrial transfer via TNTs from endothelial cells to breast cancer cells significantly enhanced cell resistance to doxorubicin treatment. Similar findings related to chemoresistance have been reported by Moschoi et al. (121), who found that the in vivo transfer of mitochondria from bone marrow-derived stem cells to acute myelogenous leukemia cells conferred chemoresistance. Mitochondrial transfer from MSCs to acute lymphoblastic leukemia cells has also been reported to protect against chemotherapeutic agents like cytarabine and daunorubicin (152). In the context of ovarian cancer, where resistance to platinum-based chemotherapies remains problematic, targeting mitochondrial transfer as a means of overcoming platinum resistance warrants further investigation. Thus, better understanding of how and when mitochondria transfer occurs in ovarian cancer and whether such events would increase or decrease chemoresistance are areas that certainly deserve further exploration.
Mitochondrial dynamics (e.g., fission/fusion) in ovarian cancer
To effectively perform key cellular functions relating to energy generation, ROS production, and regulation of cell signaling and apoptosis (168), mitochondria can adapt their function and organization. These functional or organizational adaptations often lead to differences in mitochondrial morphology, which can vary from small, round, isolated mitochondria < 0.5 mm in length, to elongated, hyperfused mitochondrial networks extending to tens of microns (169, 170). Alterations in the size or shape of mitochondria result from processes known as fission and fusion. Mitochondrial fission occurs when a single mitochondrion divides into two daughter organelles and requires the involvement of specific proteins that localize at the outer mitochondrial membrane including dynamin-related protein 1 (DRP1) and fission protein homolog 1 (FIS1) (44, 171). Conversely, mitochondrial fusion occurs when two mitochondria merge and form one single mitochondrion and is regulated by dynamin family GTPases, outer membrane-anchored dynamin family proteins mitofusins 1 and 2 (MFN1/2), and an inner membrane-anchored protein known as optic atrophy type 1 (OPA-1) (44, 172–176). Fission and fusion also segregate damaged mitochondria for mitophagy as dysfunctional organelles cannot fuse; they also facilitate mitochondrial redistribution during cell division and assist in mitigating cellular stresses (44, 172, 176–180).
As fission and fusion are critically involved in mitochondrial morphological and functional features and can be influenced by microenvironmental cues (59, 181–183), these processes have been implicated in carcinogenesis and therapy resistance (44, 171, 184–190). In the context of therapy resistance, elongated mitochondria are often associated with pro-survival, highly metabolically active cells, and chemoresistant cell populations (179, 191). It is thought that elongated mitochondria can also form during starvation, which can occur during carcinogenesis, to protect the cell against oxidative stress and to maintain ATP production under stress (170, 192–194). Interestingly, increased oxidative stress levels have been associated with membrane depolarization and mitochondrial fission (170, 195). Fragmented mitochondria are often associated with apoptotic cell death and are more commonly found in quiescent cells that depend on glycolysis over OXPHOS (170, 179, 196–198). Studies have found that inhibiting mitochondrial fission led to decreased cytochrome c release and cell death, suggesting that fission plays a critical role in mediating apoptosis (179, 199, 200). Interestingly, mitochondria need to be larger than a certain minimum size for the pro-apoptotic function of the BCL-2 family of proteins to be enabled, providing a potential mechanistic justification for the presence of highly fragmented mitochondria in chemoresistant cells (201).
In the context of ovarian cancer, studies examining the role of mitochondrial dynamics in therapy response have reported somewhat differential findings; however, all studies agree that mitochondrial dynamics are critical in determining ovarian cancer response to chemotherapy. A study by Zampieri et al. (10) showed that compared to cisplatin-sensitive cells (COV-362), cisplatin-resistant cells (COV-362-R) have more individual mitochondria and mitochondrial networks, defined as interconnected mitochondria with at least two branches. Additionally, in COV-362-R cells, cisplatin appeared to increase the numbers of individual mitochondria and mitochondrial networks (10). Other studies have shown that chemotherapy resistant tumor cells of gynecologic or breast origin display increased levels of mitochondrial fusion compared to chemotherapy sensitive cells as well (44, 202, 203).
Conversely, mitochondrial fragmentation in malignant cells has been reported by Grieco et al. (204), who found that mitochondria were fragmented in the spheroid core of malignant MOSE-LTICv cells, and that while the levels of fission and fusion protein were lower in malignant cells compared to benign cells, the ratio of fission to fusion proteins (DRP1:MFN1) increased with malignancy. The authors suggested that the observed mitochondrial fragmentation in malignant spheroids may assist tumor cell aggregates in surviving hypoxic conditions (204). Exploring ovarian cancer cell mitochondrial dynamics under hypoxic conditions, Han et al. (183) reported that mitochondrial fragments, indicative of fission, increased under hypoxic conditions, and that these cells displayed MFN1 downregulation alongside DRP1 activation. Hypoxia-induced fission in ovarian cancer cells was accompanied by increased levels of ROS that decreased upon treatment with antioxidants. Interestingly, when examining the relationship between hypoxia-induced fission and resistance to chemotherapy, Han et al. (183) reported that, treatment with Mdivi-1, a DRP1 GTPase inhibitor, prior to hypoxic exposure prevented hypoxia-induced fission and increased susceptibility of hypoxic ovarian cancer cells to cisplatin. Similar findings were reported in tumor spheroids from malignant ascites, which are known to have enhanced mitochondrial fission (183, 205), post-Mdivi-1 pretreatment, highlighting the value of DRP1 inhibition in preventing mitochondrial fission and increasing tumor cell sensitivity to platinum-based agents (183). It is important to point out that Mdivi-1 has non-fission related effects (206), making it unclear whether the effects of mitochondrial dynamics are driving the phenotypes. While more work is still needed, collectively these findings are in accordance with other studies showing that chemoresistant ovarian cancer cells display fragmented mitochondria compared to chemosensitive lines and that DRP1 inhibition can re-sensitize ovarian cancer cells to treatment (206, 207). Overall, while studies are inconsistent regarding whether fusion or fission is more critical for understanding platinum response, targeting aspects of mitochondrial dynamics and organization may improve response to conventional therapies.
Contribution of malignant ascites to mitochondrial dysfunction in ovarian cancer
Malignant ascites, or the accumulation of excess fluid containing malignant cells, is present in the majority of stage III and stage IV ovarian cancer patients (36). Often, this fluid contains cellular components – tumor cells, fibroblasts, and inflammatory cells, as well as acellular factors – cytokines and metabolites, that create a tumor-promoting microenvironment (36). In fact, the various cellular and acellular factors present in malignant ascites have been implicated in tumor growth, invasion, and chemoresistance (208, 209). Interestingly, recent studies have also suggested a role for malignant ascites, and its components, in mitochondrial dysfunction. For example, a study by Asem et al. (210) reported that the peritoneal compression induced by ascites and the resulting increased intraperitoneal pressure led to enhanced cell adhesion as well as the formation of TNTs between ovarian tumor cells and peritoneal mesothelial cells and increased transport of mitochondria via TNTs from mesothelial cells to tumor cells. Other studies have also reported that malignant ascites can enhance the tumor-promoting nature of peritoneal mesothelial cells by increasing mitochondrial oxidative stress (211). In addition to mesothelial cells, immune cells present in the ascites have also been linked to mitochondrial dysfunction in ovarian cancer. In CD4+ T cells, which represent the predominant leukocyte population in ascites (212–215), exposure to ascites led to IRE1a/XBP1-mediated mitochondrial dysfunction (216). The IRE1a/XBP1 pathway is a conserved branch of the unfolded protein response which is activated by endoplasmic reticulum stress and influences several key regulators of tumorigenesis (217). In addition, ascites exposure decreased glucose uptake, thereby reducing glycolytic capacity, and decreased OCR in a dose-dependent manner (216). Taken together, these findings suggest the ability of cellular components of malignant ascites to contribute to mitochondrial dysfunction in ovarian cancer. Acellular factors, such as cytokines and chemokines, have also been shown to contribute to ovarian cancer cell senescence, which is associated with increased ROS, oxidative DNA damage, and mitochondrial dysfunction (218).
Another way in which malignant ascites can alter mitochondrial function is through the dysregulation of mitochondria-related genes. Sirtuin 3 (SIRT3) is involved in nutrient stress sensing and mitochondrial antioxidant regulation and is suppressed in many tumor types (219–224). Conversely, in detached ovarian tumor cells or tumor cells derived from the malignant ascites of ovarian cancer patients, Kim et al. (224) found that SIRT3 activity was increased. As a result of the increased SIRT3 activity, these cells also displayed increased levels of superoxide dismutase 2 (SOD2) and low levels of mitochondrial superoxide, suppressed glycolytic capabilities, and protection against anoikis. These findings suggest that increased SIRT3 and SOD2 activity are necessary for the survival of anchorage-independent cells, such as those found in malignant ascites, and for metastatic colonization of the peritoneal cavity via the transcoelomic route (224).
Since malignant ascites contains a milieu of cellular and acellular factors known to contribute to ovarian tumor progression, metastatic potential, and response to therapy, it is not surprising that it may also contribute to mitochondrial dysfunction. Although there have been some studies evaluating the effects of ascites fluid and its components on mitochondrial endpoints, further evaluation is warranted as therapeutic targets may be revealed.
PHOTOCHEMICAL TARGETING OF MITOCHONDRIA AS A STRATEGY TO OVERCOME CHEMOTHERAPY RESISTANCE IN OVARIAN CANCER
Localization of a photosensitizer (e.g., subcellular, extracellular, vascular) is among the determinants of the biological mechanisms of PDT (225). There are several mechanisms by which mitochondrial-targeting PDT can change metabolic and signaling states, which may render the cell more susceptible to subsequent treatments (226). Activation of a mitochondria-targeted photosensitizer by light, leads to the production of RMS, damage to the mitochondrial membrane, and photodamage to Bcl-2, an anti-apoptotic regulatory protein (227, 228). Destruction of Bcl-2, combined with the preservation of cytoplasmic pro-apoptotic proteins, tips the balance towards apoptosis (225, 229, 230). Additionally, several studies reported that PDT can alter mitochondrial membrane potential and decrease the activity of mitochondrial enzymes (16, 54). While these PDT effects have not been directly linked to PDT-induced chemosensitization, they could be relevant given the role of mitochondria in chemoresistance. This section provides an overview of various exogenous or endogenous PS that have been shown to localize to, or are partially synthesized in, mitochondria (Fig. 1). We will also discuss how the subcellular localization of a PS can be tuned through its structural modifications, the introduction of targeting ligands and nanoparticle encapsulation. Finally, we will discuss the translational aspects of PDT in the context of disseminated ovarian cancer and highlight the preclinical findings that will improve PDT safety and efficacy profiles.
Figure 1.
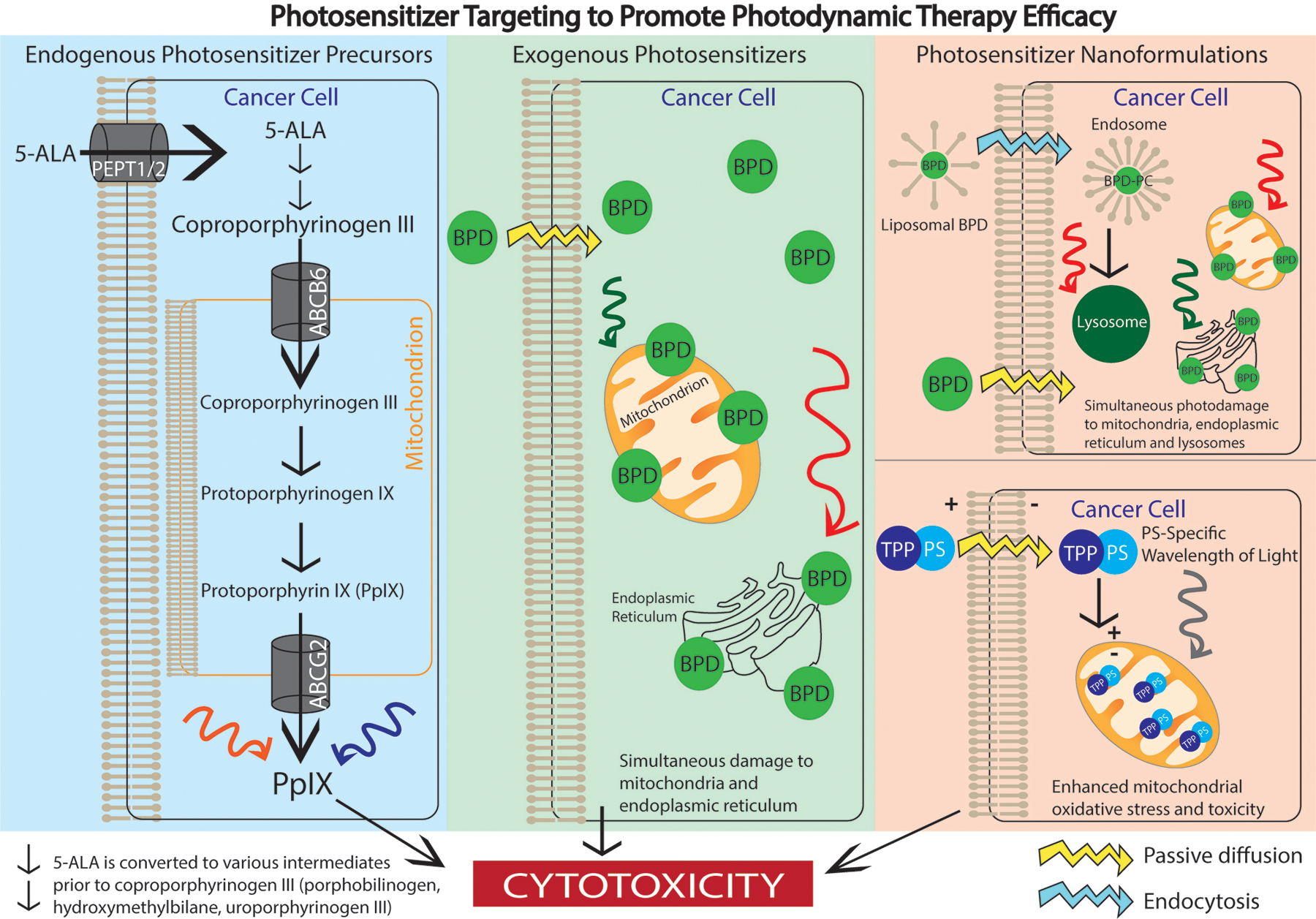
Mitochondria-localized endogenous precursors, exogenous molecule, and nanoformulations can enhance PDT efficacy. Endogenous photosensitizer precursors (blue panel) such as 5-ALA can be administered for ALA-PpIX-PDT. Once 5-ALA is administered, it is transported into malignant cells via the PEPT1/2 transporter and is converted into coproporphyrinogen III. It is then shuttled into the mitochondrion by ABCB6 where it is converted into PpIX. PpIX is an inherently fluorescent molecule, thus irradiation of PpIX can assist with fluorescence-guided resection or PDT-related cytotoxicity. Exogenous photosensitizers (green panel) like BPD can also be used for PDT. Once BPD is administered, it passively diffuses across the cell membrane and preferentially localizes to mitochondria and the endoplasmic reticulum. When irradiated with red or green light, BPD becomes activated and simultaneous photodamage to the mitochondrion and endoplasmic reticulum occurs. In addition to endogenous and exogenous agents, photosensitizer nanoformulations can also be used to enhance PDT efficacy. For example, liposomal BPD, which is endocytosed across the cell membrane, in combination with free BPD, which diffuses across the cell membrane, can be used to cause simultaneous photodamage to the mitochondrion (free BPD), endoplasmic reticulum (free BPD), and lysosomes (liposomal BPD). Other nanoformulations can involve conjugating a mitochondria-localized PS to the lipophilic TPP cation. TPP derivatization enhances mitochondrial oxidative stress and therefore phototoxicity when the PS is activated.
Photochemical targeting of mitochondria with exogenous photosensitizers and nanoformulations
The efficacy of PDT for cancer control depends on the selective initiation of death pathways by photosensitization and light. Mitochondria are high priority targets since a minor amount of photodamage can result in the release of a sufficient level of cytochrome c into the cytoplasm to initiate an apoptotic response. Apoptosis is an ideal route to cell death since the process results in DNA fragmentation and the formation of apoptotic bodies that are then engulfed and digested by macrophages. This prevents the inflammatory effect of necrosis where plasma membrane rupture results in the release of the entire cell contents into the environment of the tumor. It is important to note that the prioritization of one cell death pathway over another to maximize tumor destruction remains an area of discovery. While some tumor types are characterized by apoptosis impairment (231–233), evasion of apoptosis appears to be rare, as indicated by the many clinical successes reported for treatment of different tumor types with PDT. The pathway from the release of cytochrome c into the cytoplasm to apoptosis was first reported by Jiang and Wang (234) and is a well-conserved route to cell death. In addition to inducing mitochondria-mediated apoptosis, PDT can directly impact mitochondria by affecting ΔΨm and cellular respiration enzymes. For example, BPD-PDT induced a rapid loss of the ΔΨm in 1c1c7 murine hepatoma cells (54). Importantly, the loss of ΔΨm was transient, and cells recovered within an hour unless treated with a supralethal PDT dose. Another clinical photosensitizer Photofrin has been shown to impact mitochondrial function by decreasing the activity of succinate dehydrogenase and cytochrome c oxidase, which are the key components of the respiratory electron transport chain (16). We hypothesize that these transient changes in the metabolic state may temporarily sensitize or re-sensitize cells to chemotherapy and contribute to the observed reversal of platinum resistance and synergism with platinum-based agents. Many clinically approved PS have mitochondria among their targets: the list includes Photofrin, benzoporphyrin derivative (BPD), and several others (235). The inherent propensity of some PS to localize to mitochondria stems from their porphyrin structure. Porphyrins are structurally similar to heme and can be efficiently shuttled into mitochondria from cytosol with help of several known transporters (236). While this is true for most PS, there is a group of photosensitizing agents that generally spare mitochondria but do target ER, lysosomes, or other sub-cellular sites; this can also initiate an apoptotic response along with other routes to cell death.
It was reported in 1996 that a combination of two photosensitizing agents could significantly promote PDT efficacy and increase the tissue depth of photokilling in a rat model (237). This was initially attributed to the simultaneous targeting of tumor and tumor vasculature, but it is now known that the enhanced effect was caused by promotion of mitochondrial photodamage by concurrent effects on lysosomes. This resulted in an increase in the efficacy of photons impinging on photosensitized mitochondria, resulting in a greater photokilling efficacy, leading to an increased treatment depth. The mechanism involves a somewhat circuitous route. Photodamage to lysosomes can result in release of lysosomal calcium ions into the cytoplasm where they can activate the protease calpain. Activated calpain can cleave the autophagy-associated protein ATG5 into a truncated form that can bind to mitochondria and promote cytochrome c release after photodamage (238, 239).
Since most PS are fluorescent, their sub-cellular localization can often be visualized directly using widefield or confocal fluorescence microscopy. There is now a series of fluorescent probes for mitochondria that have diverse fluorescence emission spectra that facilitate the selection of a probe that does not fluoresce at the same wavelength as the photosensitizer being examined. There are also probes that can detect changes in ΔΨm. Among the more useful are Mitotracker Red (MTR) and Mitotracker Deep Red (MTDR), which are probes whose fluorescence is dependent on the maintenance of this potential. If the fluorescence of MTR happens to coincide with that of the photosensitizer being examined, there are alternate choices, e.g., Rhod123. Mitotracker green (MTG) fluorescence is independent of ΔΨm and is therefore a good probe for photosensitizer localization since its green fluorescence can readily be distinguished from the red fluorescence of most photosensitizing agents. With fluorescence microscopy, it is therefore possible to explore localization of photosensitizing agents and to detect their effect on ΔΨm. Co-labeling with any of the probes and the photosensitizer in the dark will reveal mitochondrial affinity of the latter. If a probe for ΔΨm is used after irradiation, this will quickly reveal whether there has been a photodynamic effect resulting in a decrease or loss of ΔΨm.
While mitochondrial photodamage is very effective at initiating apoptosis, photodamage to other sites, e.g., lysosomes and ER can confer additional benefits (240–242). Targeting lysosomes has been shown to antagonize the cytoprotective effect of autophagy (239, 240, 243, 244). The subcellular localization of PS offers unparalleled precision in controlling death modes, therefore combining several subcellular photochemical targeting approaches can lead to synergy. One way of tuning photosensitizer subcellular localization is to use nanoformulations. Nanoformulations allow to increase PS delivery payloads, improve PS pharmacokinetic properties, and serve as platforms for molecular targeting and multiagent delivery (245). In some instances, the nanoformulation serves as a carrier for a drug with a natural affinity for mitochondria. When associated with cells, the drug diffuses out of the formulation into the cell and subsequently accumulates in the mitochondria. This is the case for liposomal formulations of the photosensitizer BPD, which in the presence of cells, diffuses out of the liposome and rapidly diffuses into cells, ultimately localizing mitochondria (246–248). Alternatively, when a liposome is used to formulate lysophospholipid conjugates of BPD that stably anchors the photosensitizer to the liposome, the construct enters the cell through phagocytosis and becomes sequestered in the compartments of the endo-lysosomal pathway (246, 248, 249). This intracellular re-routing of PS by lipidation and respective nanoformulation offers unique advantages in selectively inducing differential and synergistic mechanisms of cell death as described above for PS with discrete subcellular fates.
Nanoformulations in and of themselves can be targeted to mitochondria specifically, given that the agent is stably entrapped in the nanoconstruct and amenable to intracellular shuttling by the nanoformulation. The most common approach for targeting nanoformulations to the mitochondria involves their surface functionalization with the lipophilic triphenylphosphonium (TPP) cation and its derivatives (250). TPP has a natural propensity to bind and penetrate mitochondrial membranes due to the electrical potential difference at the mitochondrial membrane. As such, TPP derivatization of drugs, imaging agents, therapeutics and nanoformulations has been shown to allow such entities to target mitochondria. TPP derivatization of photosensitizing nanoformulations has been shown to enhance mitochondrial oxidative stress as a therapeutic strategy (251). One of the earliest reports of using TPP as a mitochondrial targeting strategy for photosensitizing formulations was demonstrated in ovarian cancer cells by Cuchelkar et al. (252). While not strictly a nanoformulation, the authors prepared conjugates of the copolymer N-(2-hydroxypropyl)methacrylamide (HPMA; 48 kDa) with the photosensitizer mesochlorin e6 (Mce6). It was found that TPP derivatization of the polymer-photosensitizer conjugate resulted in its mitochondrial localization in SKOV-3 cells and improved its phototoxicity by up to 3-fold. More recently, nanocomposites comprising silica coated Fe3O4 nanoparticles were loaded with a platinum diamine complex photosensitizer and were functionalized with TPP (253). Derivatization with TPP resulted in 14–17-fold greater phototoxicity in HCT116 and A549 cells, as compared to underivatized nanoparticles. Macrophage membrane camouflaged gold nanodendrites have been functionalized with the photosensitizer indocyanine green and have also been targeted to the mitochondria by TPP functionalization (254). TPP functionalization led to increased co-localization with Mitotracker Green in MDA-MB-231 cells. PDT was induced by 808 nm laser irradiation and photothermal therapy was induced by a 1064 nm laser. The combined effect of photodynamic and photothermal therapy led to optimal cytotoxicity in vitro and in vivo.
In general, targeting mitochondria with agents either directly or by formulation procedures has been shown to be an effective strategy for promoting cancer cell destruction via apoptosis. A valuable facet to this approach is the opportunity to augment cytotoxicity by non-overlapping cell death mechanisms. This can be helpful where one mechanism, e.g., apoptosis, is impaired. With implications in circumventing chemoresistance specifically, targeted damage to mitochondria can open new approaches to design of combination therapies with unique and largely unexplored mechanisms of action.
Targeting mitochondria in ovarian cancer using protoporphyrin IX (PpIX)-mediated PDT in combination with platinum-based chemotherapy
Heme biosynthesis is among the important cellular functions that occur, in part, in mitochondria (255). Heme forms the prosthetic group of many hemoproteins, including hemoglobin and cytochromes (255–257). The heme biosynthesis pathway starts with the generation of aminolevulinic acid (ALA, 5-ALA) from glycine and succinyl CoA by ALA synthase in mitochondria and continues through multiple enzymatic conversions in the cytosol (255). The pathway intersects again with mitochondria as coproporphyrinogen leaves the cytosol and is converted by coproporphyrinogen oxidase into protoporphyrinogen in mitochondria. Protoporphyrinogen is then converted by protoporphyrinogen oxidase into protoporphyrin IX (PpIX), the penultimate molecule of the heme the biosynthesis pathway. The final major step in the pathway involves the insertion of ferrous iron, by ferrochelatase, into PpIX to produce heme (255, 256, 258). Dysregulation, in many cancers, of key heme biosynthetic enzymes, such as ferrochelatase, provides a mechanism for selective accumulation of PpIX in tumor tissue. PpIX is a photoactive molecule, excitation of which, by visible light, leads to the generation of cytotoxic RMS or fluorescence emission for photodynamic therapy (PDT) or fluorescence imaging, respectively (257–259). PpIX levels in target cells can be further enhanced using a variety of methods: administration of exogenous ALA, inhibition of ALA efflux, iron chelation, and differentiation of cancer cells with a concomitant change in metabolism (255). Clinical applications of ALA for PDT (ALA-PpIX-PDT), fluorescence imaging and guided-resection include basal cell carcinomas, actinic keratosis, Bowen’s disease, bladder cancer, and recently glioma (258, 260–262).
Mitochondria also play an important role in cell death mechanisms (263). The release of the mitochondrial protein cytochrome c activates a cascade of caspases that leads to apoptosis. Additionally, the Bcl-2 family of proteins located on the outer membrane of mitochondria play a key role in the regulation of apoptosis (234). Considering the role of mitochondria in both cellular function and cell death, mitochondria have become a therapeutic target for the treatment of various diseases including neurodegenerative disease and cancer. Current research in mitochondrial targeting is focused on drug delivery systems using nanocarriers such as liposomes and polymeric nanoparticles (264–266).
Since PpIX is synthesized, in part, in mitochondria, ALA-PpIX-PDT can be used as a mitochondria-targeted treatment method (267). Several studies have examined the effectiveness of ALA-PpIX-PDT in the context of ovarian cancer. For example, Spörri et al. compared the effect of ALA incubation, as well as the efficacy of ALA-PpIX-PDT, on an endothelial cell line, HUVEC, and on tumor cells derived from human ovarian cystadenocarcinoma (268). One of the major findings of the study was that tumor cells accumulate at least 500 times more PpIX compared to HUVEC cells, which was in accordance with previous findings revealing that PpIX selectively accumulates in malignant cells. Evaluating the effectiveness of ALA-PpIX-PDT in ovarian cancer further, an in vivo study by Ascencio et al. showed that ALA-PpIX-PDT using both green (532 nm) and red (630 nm) light promoted tumor necrosis on peritoneal metastatic ovarian cancer models (269).
Cells can also acquire resistance to PDT, including ALA-PpIX-PDT (270). Yokoyama et al. conducted an in vivo study to investigate the efficacy of ALA-PpIX-PDT, as well as the mechanism of resistance to ALA-PpIX-PDT in the context of ovarian cancer (271). The results of the study revealed that subcutaneous ovarian cancer tumors developed using MCAS and TOV21G cell lines were resistant to ALA-PpIX-PDT, while the tumors developed using other cell lines could be successfully treated with ALA-PpIX-PDT. Further analysis showed that the ALA-PpIX-PDT-resistant cell lines expressed significantly lower levels of glutathione transferase Omega-1 (GSTO1) compared to the ALA-PpIX-PDT sensitive cell lines. Previous research has shown that GSTO1 is associated with the inhibition of the conversion of PpIX to heme. Considering that cellular PpIX levels are a key determinant for the efficacy of ALA-PpIX-PDT, these results suggest that decreased GSTO1 expression can be associated with resistance to ALA-PpIX-PDT in ovarian cancer. In another study investigating the efficacy of ALA-PpIX-PDT on ovarian clear cell carcinoma cell lines, Teshigawara et al. showed that resistance to ALA-PpIX-PDT is associated with the expression ATP-binding cassette super-family G member 2 (ABCG2) protein, a transmembrane transporter protein that exports PpIX from mitochondria to the cytoplasm (272). Based on the findings of Yokoyama et al. and Teshigawara et al., the efficacy of ALA-PpIX-PDT may be increased by transferring the GSTO1 gene to tumor cells or by using ABCG2 inhibitors, respectively, to promote PpIX accumulation in ovarian tumor cells.
Although resistance to chemotherapy and PDT pose challenges to cancer treatment, minimal cross-resistance (resistance to platinum-based chemotherapy concurrent with resistance to PDT) has been reported (270). Combining mitochondria-targeted PDT with chemotherapy can, therefore, be an effective strategy to complement the mechanisms of conventional agents with non-overlapping toxicities (29, 273–275). While this approach has been explored in the context of various photosensitizers, ALA-PpIX-based priming in the context of ovarian cancer remains understudied.
Translational considerations and clinical scenarios
Robust preclinical evidence of PDT synergy with platinum-based chemotherapy creates a strong rationale for the integration of PDT with current ovarian cancer treatment protocols (27, 226). Oncology-related applications of PDT were initially limited to largely superficial cancers and premalignant conditions that were easily accessible by light. Recent advancements in fiber optics and integration of these technologies with laparoscopic surgery workflows have enabled light delivery to deeper, or less readily accessible, malignancies (276, 277). In the case of disseminated ovarian cancer, light can be delivered intraperitoneally using a flat-cut fiber or a diffusion wand. Notably, the tissue penetration depth of visible light ranges from 3–10 mm, depending on the wavelength (278), which may be advantageous when treating intraperitoneal carcinomatosis to help spare the deeper layers of the abdominal wall (279). Given that most disseminated ovarian cancer nodules are confined within the peritoneum, PDT provides an opportunity for locoregional priming of metastatic tumor nodules to enhance the efficacy of platinum-based therapy with non-overlapping toxicities. This section will summarize the progress towards PDT clinical translation for disseminated ovarian cancer treatment and discuss how recent advances in photosensitizer design may address existing clinical challenges.
The first Phase I trial was conducted at the National Cancer Institute (NCI) to test PDT feasibility and safety in patients with disseminated intraperitoneal malignancies, including ovarian carcinomatosis (31). Patients were intravenously injected with Porfimer sodium 48 hours before the treatment, after which the disseminated lesions were resected or debulked. The peritoneal cavity was then irradiated with 630 nm light using a flat-cut fiber. Light dosimetry was performed using several photodiodes placed into various areas within the peritoneum, including the right and left upper quadrants, right and left peritoneal gutters, and pelvis. This photodiode placement enabled real-time light dosimetry, ensuring that each relevant area within the peritoneum received adequate irradiation. The peritoneum was filled with 0.2% Intralipid™, which served as a light-scattering medium. While this study showed that intraperitoneal PDT is feasible in a clinical setting, it also revealed significant adverse effects. Four out of forty-six patients who received light irradiation developed intestinal fistulae and bowel perforation, which were the main dose-limiting toxicities. Another significant adverse effect was the capillary leak syndrome, which necessitated fluid resuscitation for the first 4–5 days postoperatively, and, in some cases, mechanical ventilation (280). Despite these toxicities, PDT slightly prolonged a median survival time, and three out of 25 ovarian cancer patients remained disease-free 36 months post-treatment. These results warranted a Phase II trial at the University of Pennsylvania, which included 33 ovarian cancer patients, 37 patients with gastrointestinal malignancies, and 30 sarcoma patients (32, 33, 280–287). This study followed a similar treatment approach, wherein patients underwent cytoreductive surgery followed by Porfimer sodium PDT at the maximum tolerated dose determined in Phase I. Similar to the NCI trial, patients suffered from capillary leak syndrome, bowel fistulae/anastomotic leaks and other adverse effects. Consistent with the Phase I trial, PDT in ovarian cancer patients resulted in a prolonged median failure-free survival from 2.1 to 3 months and overall survival from 20.1 to 22 months, suggesting some benefit from this treatment. Despite the overall treatment feasibility and a minor increase in overall survival, the lack of treatment selectivity and adverse toxicities halted any subsequent trials.
The suboptimal PDT therapeutic window observed in these studies stems from the insufficient photosensitizer tumor-to-tissue ratio (281, 282). Clinical studies showed that the ~2.31 ratio of mean Porfimer sodium concentration in tumor versus bowel, a toxicity-limiting organ for intraperitoneal PDT, was insufficient to achieve PDT selectivity (281). These results suggest that preclinical cancer models overestimated the photosensitizer tumor-to-tissue ratio, and more sophisticated targeting methods are needed to enhance PDT selectivity in humans (21). Over the past few decades, considerable progress has been achieved in targeted photosensitizer design, including the development of photosensitizer-antibody conjugates, as well as molecular and nanophotosensitizers that preferentially accumulate in cancer cells and/or localize to the desired subcellular compartments.
One of the targeted PDT approaches that have been gaining momentum in the clinic is photoimmunotherapy, or the use of photosensitizer-antibody conjugates. Photochemical targeting of the epidermal growth factor receptor (EGFR) with photosensitizers conjugated to anti-EGFR antibodies and antibody fragments has been particularly successful (19–21, 27). For example, a chlorin derivative conjugated to OC 125 antibody showed an increased tumor photosensitizer concentration by 2 times compared to its free version, resulting in a more favorable tumor-to-intestine ratio of 3.5 (21). This enabled highly effective photoimmunotherapy, leaving only 5% of cancer cells viable in an OVCAR-5 model of malignant ascites. EGFR-targeting antibody-photosensitizer conjugates not only enhance cancer-specific photosensitizer accumulation and improve tumor-to-tissue ratio but also exert additional cytotoxicity by inhibiting ligand binding and preventing receptor dimerization (288). Moreover, next-generation photosensitizer-antibody conjugates enable tumor-selective photosensitizer activation. In a seminal study, Savellano and Hasan produced BPD-C225 anti-EGFR antibody conjugates with improved chemical purity and aqueous solubility by first conjugating a small number of lysine residues to polyethylene glycol chains (289). This method reduced BPD-antibody aggregation, improved solubility and facilitated BPD conjugation. Importantly, photosensitizer loading ratios could be precisely controlled, and it was found that higher BPD-antibody ratios corresponded to a higher degree of fluorescence self-quenching. In a more recent study, Spring and co-authors utilized this elegant approach to activatable photoimmunoconjugate design using BPD and an anti-EGFR antibody Cetuximab (25). By tuning the photosensitizer-to-antibody ratio, the authors yielded agents with a varying degree of self-quenching. Specifically, conjugating one BPD molecule to one Cetuximab did not affect BPD’s fluorescence and PDT activity. However, conjugating seven BPD molecules to a single antibody resulted in fluorescence and PDT quenching, which could be restored upon cell internalization and lysosomal proteolysis. This unique approach resulted in low PS background fluorescence and PDT activity outside the EGFR-expressing micrometastasis, enabling highly selective tumor detection and PDT. Importantly, this study showed a favorable tumor-to-bowel ratio of 9.2 based on fluorescence, resulting in safer and more effective treatment.
The advancements in targeted photosensitizer design have already been partially implemented in the clinic. EGFR-targeted photoimmunotherapy using an IRDye 700DX-Cetuximab immunoconjugate has recently been evaluated in a global Phase III trial in patients with unresectable recurrent head and neck cancer, including patients that failed on platinum-based therapies (ClinicalTrials.gov Identifier: NCT03769506). Another rapidly developing direction is the use of targeted fluorescent agents for more complete and accurate fluorescence-guided tumor resection. For example, intraoperative visualization of ovarian cancer nodules was conducted with a folate receptor-targeting fluorescein isothiocyanate conjugate (290). Fluorescence guidance with a targeted agent enabled more sensitive cancerous lesions detection compared to bright-field illumination. Since most PS are fluorescent, combining targeted PDT with the standard of care workflows would not only potentiate chemotherapy but also enable surgical guidance. Overall, recent clinical success of targeted PDT combined with the preclinical evidence of PDT synergy with platinum-based chemotherapy creates a strong foundation for its clinical translation in the context of disseminated ovarian cancer.
SUMMARY AND CONCLUSIONS
Mitochondria play a critical role in cancer cell survival, energy production and chemoresistance, therefore, they represent an important therapeutic target. Recent evidence suggests that mitochondrial dysfunction is one of the critical drivers of platinum resistance in ovarian cancer, a key factor in the high mortality associated with this deadly disease. In this review, we discussed the role of mitochondria in ovarian cancer progression and therapy response and outlined several promising therapeutic strategies that could counteract it. Specifically, the upregulation of mitochondrial transfer in platinum-resistant ovarian cancer cells suggests the development of inhibitory therapies targeted at TNT formation may be useful in overcoming therapy resistance. We discussed reported methods of mitochondrial transfer and its effect on resistance, however, there are also other potential means of mitochondrial transfer such as through extracellular vesicles that remains to be further explored in the context of resistance. Additionally, mitochondrial transfer promotes bioenergetic flexibility in cancer cells, which has been linked to invasive potential and onset of platinum resistance. Although targeting glycolysis alone has not proven effective in ovarian cancer, inhibiting the ability of ovarian cancer cells to switch between OXPHOS and glycolysis for energy production may render the cells susceptible to platinum-based chemotherapy. Recent studies identified several potential pharmacological approaches to target mitochondrial function. For example, genetic silencing of the mitochondrial BNIP3 protein or pharmacological inhibition of autophagosome formation was sufficient to re-sensitize ovarian cancer cells to cisplatin (291). Alternatively, mitochondrial dynamics may also be exploited for the treatment of ovarian cancer, since studies have shown that inhibitors of regulatory proteins involved in mitochondrial fission enhance cisplatin-mediated death (292). However, these approaches have not been tested in vivo, and their cancer selectivity and clinical utility remains to be investigated.
PDT represents a mechanistically distinct alternative to pharmacological targeting approaches that can trigger mitochondrial damage with exquisite spatial and temporal precision. PS that preferentially localize to mitochondria can be used to generate RMS, induce mitochondrial membrane damage and photodamage to Bcl-2, and trigger cytochrome C release into the cytoplasm, thereby promoting an apoptotic response. For tumor eradication, mitochondria are a particularly good target because the small amount of damage inflicted by PDT yields a major result in apoptosis, as this is an irreversible death pathway. While some clinical PS have the inherent ability to localize to mitochondria, many mitochondrial targeting strategies have been explored preclinically, including the design of targeted small molecules, peptides and nanoformulations (293). A plethora of studies have demonstrated that PDT potentiates platinum chemotherapy in ovarian cancer in vitro and in vivo, resulting in more effective destruction of disseminated intraperitoneal nodules. However, the role of mitochondrial targeting in these outcomes remains to be elucidated. From a clinical perspective, intraperitoneal PDT is feasible in patients with advanced ovarian cancer, and we believe that recent advances in targeted photosensitizer design have the potential to address key dose-limiting toxicities noted in previous studies. Moreover, since most PS are fluorescent, PDT can be readily combined with fluorescence-guided surgery, resulting in more complete and accurate tumor resection. Overall, there is a strong mechanistic and clinical rationale for exploring photochemical targeting of mitochondria as a means of overcoming chemoresistance and improving the management of ovarian cancer.
ACKNOWLEDGEMENTS:
The authors would like to thank Walfre Franco for reviewing this manuscript and providing feedback. This research was supported by the National Institutes of Health (NIH), National Institute of Environmental Health Sciences (NIEHS) (Z01-ES102785 to SEF), a pre-doctoral traineeship from (National Research Service Award T32 ES007126 to BPR) from NIEHS, a NIH T32 award to the Certificate in Translational Medicine Program at UNC-Chapel Hill: grant number GM122741 (to BPR), as well as funding from the NC Translational and Clinical Sciences Institute (NC TraCS) at UNC-Chapel Hill supported by the National Center for Advancing Translational Sciences (NCATS), NIH through Grant Award Number UL1TR002489 (to IR), the Center for Environmental Health and Susceptibility (CEHS) at UNC-Chapel Hill supported by the NIEHS through Grant Award Number P30ES010126 (to IR), and UNC-NC State Joint Department of Biomedical Engineering Startup Funds (to IR). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Biographies

Brittany Rickard received her B.S. in Pharmacology and Toxicology from the Philadelphia College of Pharmacy at the University of the Sciences. She is currently a doctoral candidate in the Curriculum in Toxicology and Environmental Medicine at the University of North Carolina at Chapel Hill. Her doctoral research, performed under the guidance of her co-advisors Dr. Imran Rizvi and Dr. Suzanne Fenton, is focused on understanding the effects of perfluoroalkyl substances (PFAS) on ovarian cancer cell response to chemotherapy and how photodynamic therapy (PDT), a light-based treatment modality, can be used to overcome chemoresistance resulting from environmental exposures.

Dr. Marta Overchuk is a postdoctoral fellow in the Joint Department of Biomedical Engineering at the University of North Carolina at Chapel Hill and North Carolina State University working under Dr. Imran Rizvi and Dr. Frances Ligler’s supervision. Here she explores PDT as a means of combating increased chemoresistance in ovarian cancer cells subjected to flow-induced shear stress. Marta received her PhD from the University of Toronto in Biomedical Engineering under Dr. Gang Zheng’s supervision. In her PhD, Marta utilized targeted photosensitizers to improve the safety and efficacy of PDT as well as enhance tumor chemotherapy delivery.

Dr. Girgis Obaid is an NIH-NCI K99/R00 funded Assistant Professor of Bioengineering at the University of Texas at Dallas. He obtained his Ph.D. in Nanochemistry at the University of East Anglia, Norwich (U.K.), studying molecular targeted nanomedicines for photodynamic cancer therapy. His Postdoctoral Fellowship and Instructorship at the Wellman Center for Photomedicine (Massachusetts General Hospital, Harvard Medical School) with Dr. Tayyaba Hasan, focused on molecular targeted photoactivable combination therapies for cancers of the pancreas and head and neck. He currently focuses on optical molecular imaging, photodynamic therapy and nanotechnology for personalized treatment and image guided surgical removal of solid tumors.

Mustafa Kemal Ruhi is an assistant professor in the Institute of Biomedical Engineering at Boğaziçi University, Istanbul, where he also received his PhD. Before starting his lab at Boğaziçi, Dr. Ruhi worked as a postdoctoral research associate under Dr. Imran Rizvi in the Joint Department of Biomedical Engineering at the University of North Carolina at Chapel Hill and North Carolina State University. Dr. Ruhi’s research interests include photodynamic therapy (PDT)-based combination regimens for cancer as well as targeted PDT approaches to enhance the efficacy of chemotherapy.

Dr. Utkan Demirci, a professor with tenure at Stanford University, serves as the interim director and division chief at Canary Center for Cancer Early Detection, Department of Radiology. Dr. Demirci is a fellow-elect of the American Institute of Medical and Biological Engineering. Received his Ph.D. from Stanford University in Electrical Engineering, M.S. degree in Management Science & Engineering. Has published +300 peer-reviewed articles, abstracts, and proceedings, 24 book chapters and editorials, 7 edited books, and over 25 patents. Serves as an editorial board member for various peer-reviewed journals. Co-founded several startups and serves as an advisor/board member to other companies.

Dr. Suzanne “Sue” Fenton is a scientist leading the Reproductive Endocrinology group in the Mechanistic Toxicology Branch at the National Institute of Environmental Health Sciences. Her laboratory has expertise in discovery of chemicals or environmental factors contributing to mammary gland developmental defects and cancer susceptibility, pregnancy-related disease, and persistent adverse health effects in developmentally exposed rodent offspring. She has received several NIH and EPA-based awards for her research on perfluorinated chemicals and endocrine disruptors.

Janine Santos received her PhD in Genetics and Molecular Biology at the Federal University of Rio Grande do Sul in Porto Alegre, Brazil,which was followed by a post-doctoral period at the National Institute of Environmental Health Sciences (NIEHS) where her work focused on the outcomes of mitochondrial DNA damage to cell biology. She is currently at the Mechanistic Toxicology Branch at the Division of the National Toxicology Program at NIEHS where her group is interested in understanding the broad impact of mitochondrial function to cellular physiology and health outcomes associated with environmental exposures, including from a genomics scale.

David Kessel. Professor of Pharmacology at Wayne State University School of Medicine. Involved in assorted aspects of photobiology since about 1978. Past President of the International Photodynamic Association and occasional member of the ASP Council. Recent photograph taken at the Houses of Parliament (London) with the Jubilee Cap (Royal Purple, commemorating the long reign of Queen Elizabeth II).

Imran Rizvi is a tenure-track Assistant Professor in the Joint Department of Biomedical Engineering at the University of North Carolina at Chapel Hill and North Carolina State University. The Rizvi Lab focuses on developing photodynamic therapy-based combinations to target resistance in ovarian cancer using 3D and in vivo models. Previously, he was an Assistant Professor (tenure-track) at Harvard Medical School and an Assistant Biomedical Engineer at the Wellman Center for Photomedicine, Massachusetts General Hospital. He holds a Ph.D. in Engineering Sciences from Dartmouth College, a master’s degree in Tumor Biology from Georgetown University, and a B.A. from Johns Hopkins University.
Footnotes
This article is part of a Special Issue celebrating the 50th Anniversary of the American Society for Photobiology.
REFERENCES
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A and Bray F (2021) Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians 71, 209–249. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Fuchs HE and Jemal A (2022) Cancer statistics, 2022. CA: A Cancer Journal for Clinicians 72, 7–33. [DOI] [PubMed] [Google Scholar]
- 3.Globocan (2020) Ovarian Cancer: Key Stats.
- 4.Della Pepa C, Tonini G, Pisano C, Di Napoli M, Cecere SC, Tambaro R, Facchini G and Pignata S (2015) Ovarian cancer standard of care: are there real alternatives? Chinese journal of cancer 34, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foley OW, Rauh-Hain JA and del Carmen MG (2013) Recurrent epithelial ovarian cancer: An update on treatment. ONCOLOGY (United States) 27. [PubMed] [Google Scholar]
- 6.Liu J, Jiao X and Gao Q (2020) Neoadjuvant chemotherapy-related platinum resistance in ovarian cancer. Drug Discovery Today 25, 1232–1238. [DOI] [PubMed] [Google Scholar]
- 7.Cowan R, Chi D, Kehoe S, Nankivell M and Leary A (2016) Primary Surgery or Neoadjuvant Chemotherapy in Advanced Ovarian Cancer: The Debate Continues…. American Society of Clinical Oncology Educational Book, 153–162. [DOI] [PubMed] [Google Scholar]
- 8.Blagden SP (2015) Harnessing pandemonium: The clinical implications of tumor heterogeneity in ovarian cancer. Frontiers in Oncology 5, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dar S, Chhina J, Mert I, Chitale D, Buekers T, Kaur H, Giri S, Munkarah A and Rattan R (2017) Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Scientific Reports 7, 8760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zampieri LX, Grasso D, Bouzin C, Brusa D, Rossignol R and Sonveaux P (2020) Mitochondria Participate in Chemoresistance to Cisplatin in Human Ovarian Cancer Cells. Mol Cancer Res 18, 1379–1391. [DOI] [PubMed] [Google Scholar]
- 11.Alharbi M, Lai A, Sharma S, Kalita-de Croft P, Godbole N, Campos A, Guanzon D, Salas-Burgos A, Carrion F, Zuñiga FA, Perrin L, He Y, Pejovic T, Winters C, Morgan T, Hooper JD, Rice GE and Salomon C (2021) Extracellular Vesicle Transmission of Chemoresistance to Ovarian Cancer Cells Is Associated with Hypoxia-Induced Expression of Glycolytic Pathway Proteins, and Prediction of Epithelial Ovarian Cancer Disease Recurrence. Cancers (Basel) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson BC and Patterson MS (2008) The physics, biophysics and technology of photodynamic therapy. Phys Med Biol 53, R61–109. [DOI] [PubMed] [Google Scholar]
- 13.Kessel D and Luo Y (1998) Mitochondrial photodamage and PDT-induced apoptosis. Journal of Photochemistry and Photobiology B: Biology 42, 89–95. [DOI] [PubMed] [Google Scholar]
- 14.Peng TI, Chang CJ, Guo MJ, Wang YH, Yu JS, Wu HY and Jou MJ (2005) Mitochondrion-targeted photosensitizer enhances the photodynamic effect-induced mitochondrial dysfunction and apoptosis. Ann N Y Acad Sci 1042, 419–428. [DOI] [PubMed] [Google Scholar]
- 15.Kessel D (2017) Subcellular Targeting as a Determinant of the Efficacy of Photodynamic Therapy. Photochem Photobiol 93, 609–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh G, Jeeves WP, Wilson BC and Jang D (1987) Mitochondrial photosensitization by Photofrin II. Photochem Photobiol 46, 645–649. [DOI] [PubMed] [Google Scholar]
- 17.Cramer G, Lewis R, Gymarty A, Hagan S, Mickler M, Evans S, Punekar SR, Shuman L, Simone CB 2nd, Hahn SM, Busch TM, Fraker D and Cengel KA (2020) Preclinical Evaluation of Cetuximab and Benzoporphyrin Derivative-Mediated Intraperitoneal Photodynamic Therapy in a Canine Model. Photochem Photobiol 96, 684–691. [DOI] [PubMed] [Google Scholar]
- 18.del Carmen MG, Rizvi I, Chang Y, Moor ACE, Oliva E, Sherwood M, Pogue B and Hasan T (2005) Synergism of epidermal growth factor receptor-targeted immunotherapy with photodynamic treatment of ovarian cancer in vivo. Journal of the National Cancer Institute 97, 1516–1524. [DOI] [PubMed] [Google Scholar]
- 19.Goff BA, Bamberg M and Hasan T (1991) Photoimmunotherapy of Human Ovarian Carcinoma Cells ex Vivo1. Cancer Research 51, 4762–4767. [PubMed] [Google Scholar]
- 20.Goff BA, Blake J, Bamberg MP and Hasan T (1996) Treatment of ovarian cancer with photodynamic therapy and immunoconjugates in a murine ovarian cancer model. Br J Cancer 74, 1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goff BA, Hermanto U, Rumbaugh J, Blake J, Bamberg M and Hasan T (1994) Photoimmunotherapy and biodistribution with an OC125-chlorin immunoconjugate in an in vivo murine ovarian cancer model. Br J Cancer 70, 474–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molpus KL, Hamblin MR, Rizvi I and Hasan T (2000) Intraperitoneal photoimmunotherapy of ovarian carcinoma xenografts in nude mice using charged photoimmunoconjugates. Gynecol Oncol 76, 397–404. [DOI] [PubMed] [Google Scholar]
- 23.Sato K, Hanaoka H, Watanabe R, Nakajima T, Choyke PL and Kobayashi H (2015) Near infrared photoimmunotherapy in the treatment of disseminated peritoneal ovarian cancer. Mol Cancer Ther 14, 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song K, Kong B, Li L, Yang Q, Wei Y and Qu X (2007) Intraperitoneal photodynamic therapy for an ovarian cancer ascite model in Fischer 344 rat using hematoporphyrin monomethyl ether. Cancer Sci 98, 1959–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spring BQ, Abu-Yousif AO, Palanisami A, Rizvi I, Zheng X, Mai Z, Anbil S, Sears RB, Mensah LB, Goldschmidt R, Erdem SS, Oliva E and Hasan T (2014) Selective treatment and monitoring of disseminated cancer micrometastases in vivo using dual-function, activatable immunoconjugates. Proceedings of the National Academy of Sciences of the United States of America 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nath S, Pigula M, Khan AP, Hanna W, Ruhi MK, Dehkordy FM, Pushpavanam K, Rege K, Moore K, Tsujita Y, Conrad C, Inci F, del Carmen MG, Franco W, Celli I, Nath S, Pigula M, Khan AP, Hanna W, Ruhi MK, Dehkordy FM, Pushpavanam K, Rege K, Moore K, Tsujita Y, Conrad C, Inci F, del Carmen MG, Franco W, Celli JP, Demirci U, Hasan T, Huang H-C and Rizvi I (2020) Flow-induced Shear Stress Confers Resistance to Carboplatin in an Adherent Three-Dimensional Model for Ovarian Cancer: A Role for EGFR-Targeted Photoimmunotherapy Informed by Physical Stress. Journal of clinical medicine 9, 924–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duska LR, Hamblin MR, Miller JL and Hasan T (1999) Combination Photoimmunotherapy and Cisplatin: Effects on Human Ovarian Cancer Ex Vivo. JNCI: Journal of the National Cancer Institute 91, 1557–1563. [DOI] [PubMed] [Google Scholar]
- 28.Molpus KL, Kato D, Hamblin MR, Lilge L, Bamberg M and Hasan T (1996) Intraperitoneal photodynamic therapy of human epithelial ovarian carcinomatosis in a xenograft murine model. Cancer Res 56, 1075–1082. [PubMed] [Google Scholar]
- 29.Rizvi I, Celli JP, Evans CL, Abu-Yousif AO, Muzikansky A, Pogue BW, Finkelstein D and Hasan T (2010) Synergistic enhancement of carboplatin efficacy with photodynamic therapy in a three-dimensional model for micrometastatic ovarian cancer. Cancer Research 70, 9319–9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Azaïs H, Mordon S and Collinet P (2017) [Intraperitoneal photodynamic therapy for peritoneal metastasis of epithelial ovarian cancer. Limits and future prospects]. Gynecol Obstet Fertil Senol 45, 249–256. [DOI] [PubMed] [Google Scholar]
- 31.DeLaney TF, Sindelar WF, Tochner Z, Smith PD, Friauf WS, Thomas G, Dachowski L, Cole JW, Steinberg SM and Glatstein E (1993) Phase I study of debulking surgery and photodynamic therapy for disseminated intraperitoneal tumors. Int J Radiat Oncol Biol Phys 25, 445–457. [DOI] [PubMed] [Google Scholar]
- 32.Hahn SM, Fraker DL, Mick R, Metz J, Busch TM, Smith D, Zhu T, Rodriguez C, Dimofte A, Spitz F, Putt M, Rubin SC, Menon C, Wang HW, Shin D, Yodh A and Glatstein E (2006) A phase II trial of intraperitoneal photodynamic therapy for patients with peritoneal carcinomatosis and sarcomatosis. Clin Cancer Res 12, 2517–2525. [DOI] [PubMed] [Google Scholar]
- 33.Hendren SK, Hahn SM, Spitz FR, Bauer TW, Rubin SC, Zhu T, Glatstein E and Fraker DL (2001) Phase II trial of debulking surgery and photodynamic therapy for disseminated intraperitoneal tumors. Ann Surg Oncol 8, 65–71. [DOI] [PubMed] [Google Scholar]
- 34.Lim D and Oliva E (2013) Precursors and pathogenesis of ovarian carcinoma. Pathology 45, 229–242. [DOI] [PubMed] [Google Scholar]
- 35.Seidman JD and Khedmati F (2008) Exploring the Histogenesis of Ovarian Mucinous and Transitional Cell (Brenner) Neoplasms and Their Relationship With Walthard Cell Nests: A Study of 120 Tumors. Archives of Pathology & Laboratory Medicine 132, 1753–1760. [DOI] [PubMed] [Google Scholar]
- 36.Rickard BP, Conrad C, Sorrin AJ, Ruhi MK, Reader JC, Huang SA, Franco W, Scarcelli G, Polacheck WJ, Roque DM, Del Carmen MG, Huang HC, Demirci U and Rizvi I (2021) Malignant Ascites in Ovarian Cancer: Cellular, Acellular, and Biophysical Determinants of Molecular Characteristics and Therapy Response. Cancers (Basel) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rizvi I, Gurkan UA, Tasoglu S, Alagic N, Celli JP, Mensah LB, Mai Z, Demirci U and Hasan T (2013) Flow induces epithelial-mesenchymal transition, cellular heterogeneity and biomarker modulation in 3D ovarian cancer nodules. PNAS 110, 1974–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rickard BP, Tan X, Fenton SE and Rizvi I (2022) Select Per- and Polyfluoroalkyl Substances (PFAS) Induce Resistance to Carboplatin in Ovarian Cancer Cell Lines. Int J Mol Sci 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ledermann JA, Raja FA, Fotopoulou C, Gonzalez-Martin A, Colombo N and Sessa C (2013) Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 24 Suppl 6, vi24–32. [DOI] [PubMed] [Google Scholar]
- 40.Stewart C, Ralyea C and Lockwood S (2019) Ovarian Cancer: An Integrated Review. Seminars in Oncology Nursing 35, 151–156. [DOI] [PubMed] [Google Scholar]
- 41.Kleih M, Böpple K, Dong M, Gaißler A, Heine S, Olayioye MA, Aulitzky WE and Essmann F (2019) Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis 10, 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ushijima K (2010) Treatment for recurrent ovarian cancer-at first relapse. Journal of oncology 2010, 497429–497429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jia D, Park JH, Jung KH, Levine H and Kaipparettu BA (2018) Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cocetta V, Ragazzi E and Montopoli M (2019) Mitochondrial Involvement in Cisplatin Resistance. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maycotte P, Marín-Hernández A, Goyri-Aguirre M, Anaya-Ruiz M, Reyes-Leyva J and Cortés-Hernández P (2017) Mitochondrial dynamics and cancer. Tumour Biol 39, 1010428317698391. [DOI] [PubMed] [Google Scholar]
- 46.Siddik ZH (2003) Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 22, 7265–7279. [DOI] [PubMed] [Google Scholar]
- 47.Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bellan A, Castedo M and Kroemer G (2014) Systems biology of cisplatin resistance: past, present and future. Cell Death & Disease 5, e1257–e1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makovec T (2019) Cisplatin and beyond: molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol Oncol 53, 148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS and Doetsch PW (2013) Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS One 8, e81162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olivero OA, Chang PK, Lopez-Larraza DM, Semino-Mora MC and Poirier MC (1997) Preferential formation and decreased removal of cisplatin-DNA adducts in Chinese hamster ovary cell mitochondrial DNA as compared to nuclear DNA. Mutat Res 391, 79–86. [DOI] [PubMed] [Google Scholar]
- 51.Fichtinger-Schepman AMJ, Van der Veer JL, Den Hartog JHJ, Lohman PHM and Reedijk J (1985) Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation. Biochemistry 24, 707–713. [DOI] [PubMed] [Google Scholar]
- 52.Pascoe JM and Roberts JJ (1974) Interactions between mammalian cell DNA and inorganic platinum compounds—I: DNA interstrand cross-linking and cytotoxic properties of platinum(II) compounds. Biochemical Pharmacology 23, 1345–1357. [DOI] [PubMed] [Google Scholar]
- 53.Mahalingam SM, Ordaz JD and Low PS (2018) Targeting of a Photosensitizer to the Mitochondrion Enhances the Potency of Photodynamic Therapy. ACS Omega 3, 6066–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kessel D (2014) Reversible effects of photodamage directed toward mitochondria. Photochem Photobiol 90, 1211–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hilf R (2007) Mitochondria are targets of photodynamic therapy. Journal of Bioenergetics and Biomembranes 39, 85–89. [DOI] [PubMed] [Google Scholar]
- 56.Agostinis P, Buytaert E, Breyssens H and Hendrickx N (2004) Regulatory pathways in photodynamic therapy induced apoptosis. Photochemical & Photobiological Sciences 3, 721–729. [DOI] [PubMed] [Google Scholar]
- 57.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 58.Pavlova NN and Thompson CB (2016) The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han Y, Cho U, Kim S, Park IS, Cho JH, Dhanasekaran DN and Song YS (2018) Tumour microenvironment on mitochondrial dynamics and chemoresistance in cancer. Free Radic Res 52, 1271–1287. [DOI] [PubMed] [Google Scholar]
- 60.Hay N (2016) Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer 16, 635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Annibaldi A and Widmann C (2010) Glucose metabolism in cancer cells. Curr Opin Clin Nutr Metab Care 13, 466–470. [DOI] [PubMed] [Google Scholar]
- 62.Younes M, Brown RW, Stephenson M, Gondo M and Cagle PT (1997) Overexpression of Glut1 and Glut3 in stage I nonsmall cell lung carcinoma is associated with poor survival. Cancer 80, 1046–1051. [DOI] [PubMed] [Google Scholar]
- 63.Lidgren A, Bergh A, Grankvist K, Rasmuson T and Ljungberg B (2008) Glucose transporter-1 expression in renal cell carcinoma and its correlation with hypoxia inducible factor-1 alpha. BJU Int 101, 480–484. [DOI] [PubMed] [Google Scholar]
- 64.Pizzuti L, Sergi D, Mandoj C, Antoniani B, Sperati F, Chirico A, Di Lauro L, Valle M, Garofalo A, Vizza E, Corrado G, Tomao F, Rinaldi M, Carpano S, Maugeri-Saccà M, Conti L, Digiesi G, Marchetti P, De Maria R, Giordano A, Barba M, Carosi MA and Vici P (2018) GLUT 1 receptor expression and circulating levels of fasting glucose in high grade serous ovarian cancer. J Cell Physiol 233, 1396–1401. [DOI] [PubMed] [Google Scholar]
- 65.Mathupala SP, Ko YH and Pedersen PL (2006) Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25, 4777–4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB and Hay N (2004) Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell 16, 819–830. [DOI] [PubMed] [Google Scholar]
- 67.Ha JH, Radhakrishnan R, Jayaraman M, Yan M, Ward JD, Fung KM, Moxley K, Sood AK, Isidoro C, Mukherjee P, Song YS and Dhanasekaran DN (2018) LPA Induces Metabolic Reprogramming in Ovarian Cancer via a Pseudohypoxic Response. Cancer Res 78, 1923–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thorens B and Mueckler M (2010) Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab 298, E141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, Jha AK, Smolen GA, Clasquin MF, Robey B and Hay N (2013) Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 24, 213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, Hawkins C and Guha A (2011) Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med 208, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang XY, Zhang M, Cong Q, Zhang MX, Zhang MY, Lu YY and Xu CJ (2018) Hexokinase 2 confers resistance to cisplatin in ovarian cancer cells by enhancing cisplatin-induced autophagy. Int J Biochem Cell Biol 95, 9–16. [DOI] [PubMed] [Google Scholar]
- 72.Yalcin A, Clem BF, Imbert-Fernandez Y, Ozcan SC, Peker S, O’Neal J, Klarer AC, Clem AL, Telang S and Chesney J (2014) 6-Phosphofructo-2-kinase (PFKFB3) promotes cell cycle progression and suppresses apoptosis via Cdk1-mediated phosphorylation of p27. Cell Death Dis 5, e1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Costa Leite T, Da Silva D, Guimarães Coelho R, Zancan P and Sola-Penna M (2007) Lactate favours the dissociation of skeletal muscle 6-phosphofructo-1-kinase tetramers down-regulating the enzyme and muscle glycolysis. Biochem J 408, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gao X, Wang H, Yang JJ, Liu X and Liu ZR (2012) Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell 45, 598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and Cantley LC (2008) The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452, 230–233. [DOI] [PubMed] [Google Scholar]
- 76.Chao TK, Huang TS, Liao YP, Huang RL, Su PH, Shen HY, Lai HC and Wang YC (2017) Pyruvate kinase M2 is a poor prognostic marker of and a therapeutic target in ovarian cancer. PLoS One 12, e0182166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang YH, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB, Cantley LC, Vander Heiden MG and Scadden DT (2014) Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 158, 1309–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kobierzycki C, Piotrowska A, Latkowski K, Zabel M, Nowak-Markwitz E, Spaczynski M, Kedzia W, Pula B, Podhorska-Okolow M and Dziegiel P (2018) Correlation of Pyruvate Kinase M2 Expression with Clinicopathological Data in Ovarian Cancer. Anticancer Res 38, 295–300. [DOI] [PubMed] [Google Scholar]
- 79.Feron O (2009) Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol 92, 329–333. [DOI] [PubMed] [Google Scholar]
- 80.Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, Feron O and Dewhirst MW (2008) Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 118, 3930–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B and Gillies RJ (2006) Acid-mediated tumor invasion: a multidisciplinary study. Cancer Res 66, 5216–5223. [DOI] [PubMed] [Google Scholar]
- 82.Gatenby RA and Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4, 891–899. [DOI] [PubMed] [Google Scholar]
- 83.Suh DH, Kim HS, Kim B and Song YS (2014) Metabolic orchestration between cancer cells and tumor microenvironment as a co-evolutionary source of chemoresistance in ovarian cancer: a therapeutic implication. Biochem Pharmacol 92, 43–54. [DOI] [PubMed] [Google Scholar]
- 84.Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, Witkiewicz A, Lin Z, Balliet R, Howell A and Sotgia F (2010) Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor micro-environment. Cancer Biol Ther 10, 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim TH, Suh DH, Kim MK and Song YS (2014) Metformin against cancer stem cells through the modulation of energy metabolism: special considerations on ovarian cancer. Biomed Res Int 2014, 132702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F and Lisanti MP (2009) The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 8, 3984–4001. [DOI] [PubMed] [Google Scholar]
- 87.Broekgaarden M, Anbil S, Bulin A-L, Obaid G, Mai Z, Baglo Y, Rizvi I and Hasan T (2019) Modulation of redox metabolism negates cancer-associated fibroblasts-induced treatment resistance in a heterotypic 3D culture platform of pancreatic cancer. Biomaterials 222, 119421–119421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Labiche A, Heutte N, Herlin P, Chasle J, Gauduchon P and Elie N (2010) Stromal compartment as a survival prognostic factor in advanced ovarian carcinoma. Int J Gynecol Cancer 20, 28–33. [DOI] [PubMed] [Google Scholar]
- 89.Pfeiffer T and Morley A (2014) An evolutionary perspective on the Crabtree effect. Front Mol Biosci 1, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R and Spiegelman BM (2006) Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127, 397–408. [DOI] [PubMed] [Google Scholar]
- 91.Andrzejewski S, Klimcakova E, Johnson RM, Tabariès S, Annis MG, McGuirk S, Northey JJ, Chénard V, Sriram U, Papadopoli DJ, Siegel PM and St-Pierre J (2017) PGC-1α Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab 26, 778–787.e775. [DOI] [PubMed] [Google Scholar]
- 92.Pedersen PL (2007) Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr 39, 211–222. [DOI] [PubMed] [Google Scholar]
- 93.Yang L, Moss T, Mangala LS, Marini J, Zhao H, Wahlig S, Armaiz-Pena G, Jiang D, Achreja A, Win J, Roopaimoole R, Rodriguez-Aguayo C, Mercado-Uribe I, Lopez-Berestein G, Liu J, Tsukamoto T, Sood AK, Ram PT and Nagrath D (2014) Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol Syst Biol 10, 728–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shen Y-A, Hong J, Asaka R, Asaka S, Hsu F-C, Suryo Rahmanto Y, Jung J-G, Chen Y-W, Yen T-T, Tomaszewski A, Zhang C, Attarwala N, DeMarzo AM, Davidson B, Chuang C-M, Chen X, Gaillard S, Le A, Shih I-M and Wang T-L (2020) Inhibition of the MYC-Regulated Glutaminase Metabolic Axis Is an Effective Synthetic Lethal Approach for Treating Chemoresistant Ovarian Cancers. Cancer research 80, 4514–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Han C, Yang L, Choi HH, Baddour J, Achreja A, Liu Y, Li Y, Li J, Wan G, Huang C, Ji G, Zhang X, Nagrath D and Lu X (2016) Amplification of USP13 drives ovarian cancer metabolism. Nature communications 7, 13525–13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Masamha CP and LaFontaine P (2018) Molecular targeting of glutaminase sensitizes ovarian cancer cells to chemotherapy. J Cell Biochem 119, 6136–6145. [DOI] [PubMed] [Google Scholar]
- 97.Sun L, Yin Y, Clark LH, Sun W, Sullivan SA, Tran A-Q, Han J, Zhang L, Guo H, Madugu E, Pan T, Jackson AL, Kilgore J, Jones HM, Gilliam TP, Zhou C and Bae-Jump VL (2017) Dual inhibition of glycolysis and glutaminolysis as a therapeutic strategy in the treatment of ovarian cancer. Oncotarget 8, 63551–63561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.de Sá Junior PL, Câmara DAD, Porcacchia AS, Fonseca PMM, Jorge SD, Araldi RP and Ferreira AK (2017) The Roles of ROS in Cancer Heterogeneity and Therapy. Oxid Med Cell Longev 2017, 2467940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van Gisbergen MW, Voets AM, Starmans MH, de Coo IF, Yadak R, Hoffmann RF, Boutros PC, Smeets HJ, Dubois L and Lambin P (2015) How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutat Res Rev Mutat Res 764, 16–30. [DOI] [PubMed] [Google Scholar]
- 100.Shadel GS and Horvath TL (2015) Mitochondrial ROS signaling in organismal homeostasis. Cell 163, 560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Che M, Wang R, Li X, Wang H-Y and Zheng XFS (2016) Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discovery Today 21, 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Okon IS and Zou M-H (2015) Mitochondrial ROS and cancer drug resistance: Implications for therapy. Pharmacological Research 100, 170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bansal A and Simon MC (2018) Glutathione metabolism in cancer progression and treatment resistance. Journal of Cell Biology 217, 2291–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gandin V and Fernandes AP (2015) Metal- and Semimetal-Containing Inhibitors of Thioredoxin Reductase as Anticancer Agents. Molecules 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dier U, Shin D-H, Hemachandra LPMP, Uusitalo LM and Hempel N (2014) Bioenergetic Analysis of Ovarian Cancer Cell Lines: Profiling of Histological Subtypes and Identification of a Mitochondria-Defective Cell Line. PLOS ONE 9, e98479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jose C, Bellance N and Rossignol R (2011) Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochimica et Biophysica Acta (BBA) - Bioenergetics 1807, 552–561. [DOI] [PubMed] [Google Scholar]
- 107.Caneba CA, Bellance N, Yang L, Pabst L and Nagrath D (2012) Pyruvate uptake is increased in highly invasive ovarian cancer cells under anoikis conditions for anaplerosis, mitochondrial function, and migration. American Journal of Physiology-Endocrinology and Metabolism 303, E1036–E1052. [DOI] [PubMed] [Google Scholar]
- 108.Fabian C, Koetz L, Favaro E, Indraccolo S, Mueller-Klieser W and Sattler UGA (2012) Protein profiles in human ovarian cancer cell lines correspond to their metabolic activity and to metabolic profiles of respective tumor xenografts. The FEBS Journal 279, 882–891. [DOI] [PubMed] [Google Scholar]
- 109.Bindra S, McGill MA, Triplett MK, Tyagi A, Thaker PH, Dahmoush L, Goodheart MJ, Ogden RT, Owusu-Ansah E, R Karan K, Cole S, Sood AK, Lutgendorf SK and Picard M (2021) Mitochondria in epithelial ovarian carcinoma exhibit abnormal phenotypes and blunted associations with biobehavioral factors. Scientific Reports 11, 11595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Torralba D, Baixauli F and Sánchez-Madrid F (2016) Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front Cell Dev Biol 4, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rogers RS and Bhattacharya J (2013) When cells become organelle donors. Physiology (Bethesda) 28, 414–422. [DOI] [PubMed] [Google Scholar]
- 112.Spees JL, Olson SD, Whitney MJ and Prockop DJ (2006) Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci U S A 103, 1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Berridge MV, Crasso C and Neuzil J (2018) Mitochondrial Genome Transfer to Tumor Cells Breaks The Rules and Establishes a New Precedent in Cancer Biology. Mol Cell Oncol 5, e1023929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, Stepanova A, Iommarini L, Mastroleo C, Daly L, Galkin A, Thakur BK, Soplop N, Uryu K, Hoshino A, Norton L, Bonafé M, Cricca M, Gasparre G, Lyden D and Bromberg J (2017) Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc Natl Acad Sci U S A 114, E9066–e9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Patananan AN, Wu TH, Chiou PY and Teitell MA (2016) Modifying the Mitochondrial Genome. Cell Metab 23, 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B, Pesdar EA, Sobol M, Filimonenko A, Stuart S, Vondrusova M, Kluckova K, Sachaphibulkij K, Rohlena J, Hozak P, Truksa J, Eccles D, Haupt LM, Griffiths LR, Neuzil J and Berridge MV (2015) Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 21, 81–94. [DOI] [PubMed] [Google Scholar]
- 117.Acquistapace A, Bru T, Lesault PF, Figeac F, Coudert AE, le Coz O, Christov C, Baudin X, Auber F, Yiou R, Dubois-Randé JL and Rodriguez AM (2011) Human mesenchymal stem cells reprogram adult cardiomyocytes toward a progenitor-like state through partial cell fusion and mitochondria transfer. Stem Cells 29, 812–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K and Hauser CJ (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X and Lo EH (2016) Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S and Bhattacharya J (2012) Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 18, 759–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Moschoi R, Imbert V, Nebout M, Chiche J, Mary D, Prebet T, Saland E, Castellano R, Pouyet L, Collette Y, Vey N, Chabannon C, Recher C, Sarry J-E, Alcor D, Peyron J-F and Griessinger E (2016) Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 128, 253–264. [DOI] [PubMed] [Google Scholar]
- 122.Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Kumar M, Rehman R, Tiwari BK, Jha KA, Barhanpurkar AP, Wani MR, Roy SS, Mabalirajan U, Ghosh B and Agrawal A (2014) Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. Embo j 33, 994–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li C, Cheung MKH, Han S, Zhang Z, Chen L, Chen J, Zeng H and Qiu J (2019) Mesenchymal stem cells and their mitochondrial transfer: a double-edged sword. Biosci Rep 39, BSR20182417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li X, Zhang Y, Yeung SC, Liang Y, Liang X, Ding Y, Ip MS, Tse HF, Mak JC and Lian Q (2014) Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am J Respir Cell Mol Biol 51, 455–465. [DOI] [PubMed] [Google Scholar]
- 125.Saha T, Dash C, Jayabalan R, Khiste S, Kulkarni A, Kurmi K, Mondal J, Majumder PK, Bardia A, Jang HL and Sengupta S (2022) Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nature Nanotechnology 17, 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang Y, Yu Z, Jiang D, Liang X, Liao S, Zhang Z, Yue W, Li X, Chiu SM, Chai YH, Liang Y, Chow Y, Han S, Xu A, Tse HF and Lian Q (2016) iPSC-MSCs with High Intrinsic MIRO1 and Sensitivity to TNF-α Yield Efficacious Mitochondrial Transfer to Rescue Anthracycline-Induced Cardiomyopathy. Stem Cell Reports 7, 749–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jiang D, Gao F, Zhang Y, Wong DSH, Li Q, Tse H.-f., Xu G, Yu Z and Lian Q (2016) Mitochondrial transfer of mesenchymal stem cells effectively protects corneal epithelial cells from mitochondrial damage. Cell Death & Disease 7, e2467–e2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mahrouf-Yorgov M, Augeul L, Da Silva CC, Jourdan M, Rigolet M, Manin S, Ferrera R, Ovize M, Henry A, Guguin A, Meningaud J-P, Dubois-Randé J-L, Motterlini R, Foresti R and Rodriguez A-M (2017) Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death & Differentiation 24, 1224–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jackson MV, Morrison TJ, Doherty DF, McAuley DF, Matthay MA, Kissenpfennig A, O’Kane CM and Krasnodembskaya AD (2016) Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells 34, 2210–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yao Y, Fan X-L, Jiang D, Zhang Y, Li X, Xu Z-B, Fang S-B, Chiu S, Tse H-F, Lian Q and Fu Q-L (2018) Connexin 43-Mediated Mitochondrial Transfer of iPSC-MSCs Alleviates Asthma Inflammation. Stem Cell Reports 11, 1120–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li X, Michaeloudes C, Zhang Y, Wiegman CH, Adcock IM, Lian Q, Mak JCW, Bhavsar PK and Chung KF (2018) Mesenchymal stem cells alleviate oxidative stress-induced mitochondrial dysfunction in the airways. Journal of Allergy and Clinical Immunology 141, 1634–1645.e1635. [DOI] [PubMed] [Google Scholar]
- 132.Zampieri LX, Silva-Almeida C, Rondeau JD and Sonveaux P (2021) Mitochondrial Transfer in Cancer: A Comprehensive Review. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rustom A, Saffrich R, Markovic I, Walther P and Gerdes H-H (2004) Nanotubular Highways for Intercellular Organelle Transport. Science 303, 1007–1010. [DOI] [PubMed] [Google Scholar]
- 134.Önfelt B, Nedvetzki S, Yanagi K and Davis DM (2004) Cutting Edge: Membrane Nanotubes Connect Immune Cells. The Journal of Immunology 173, 1511. [DOI] [PubMed] [Google Scholar]
- 135.Sowinski S, Jolly C, Berninghausen O, Purbhoo MA, Chauveau A, Köhler K, Oddos S, Eissmann P, Brodsky FM, Hopkins C, Onfelt B, Sattentau Q and Davis DM (2008) Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat Cell Biol 10, 211–219. [DOI] [PubMed] [Google Scholar]
- 136.Sherer NM, Lehmann MJ, Jimenez-Soto LF, Horensavitz C, Pypaert M and Mothes W (2007) Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nature Cell Biology 9, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hase K, Kimura S, Takatsu H, Ohmae M, Kawano S, Kitamura H, Ito M, Watarai H, Hazelett CC, Yeaman C and Ohno H (2009) M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nature Cell Biology 11, 1427–1432. [DOI] [PubMed] [Google Scholar]
- 138.Paliwal S, Chaudhuri R, Agrawal A and Mohanty S (2018) Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. Journal of Biomedical Science 25, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chang KT, Niescier RF and Min KT (2011) Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proc Natl Acad Sci U S A 108, 15456–15461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Brickley K and Stephenson FA (2011) Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neurons. J Biol Chem 286, 18079–18092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Quintero OA, DiVito MM, Adikes RC, Kortan MB, Case LB, Lier AJ, Panaretos NS, Slater SQ, Rengarajan M, Feliu M and Cheney RE (2009) Human Myo19 Is a Novel Myosin that Associates with Mitochondria. Current Biology 19, 2008–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ahmad T, Mukherjee S, Pattnaik BR, Kumar MN, Singh S, Rehman R, Jha A, Wani MR, Mabalirajan U, Ghosh B, Roy SS and Agrawal A (2013) Miro 1 Knockdown in Stem Cells Inhibits Mitochondrial Donation Mediated Rescue of Bronchial Epithelial Injury. Biophysical Journal 104. [Google Scholar]
- 143.Wang X and Gerdes HH (2015) Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death & Differentiation 22, 1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii S, Le Foll F and Rafii A (2013) Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. Journal of Translational Medicine 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cho YM, Kim JH, Kim M, Park SJ, Koh SH, Ahn HS, Kang GH, Lee JB, Park KS and Lee HK (2012) Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PLoS One 7, e32778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lou E, Fujisawa S, Morozov A, Barlas A, Romin Y, Dogan Y, Gholami S, Moreira AL, Manova-Todorova K and Moore MA (2012) Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS One 7, e33093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Caicedo A, Fritz V, Brondello J-M, Ayala M, Dennemont I, Abdellaoui N, de Fraipont F, Moisan A, Prouteau CA, Boukhaddaoui H, Jorgensen C and Vignais M-L (2015) MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Scientific Reports 5, 9073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lin H-Y, Liou C-W, Chen S-D, Hsu T-Y, Chuang J-H, Wang P-W, Huang S-T, Tiao M-M, Chen J-B, Lin T-K and Chuang Y-C (2015) Mitochondrial transfer from Wharton’s jelly-derived mesenchymal stem cells to mitochondria-defective cells recaptures impaired mitochondrial function. Mitochondrion 22, 31–44. [DOI] [PubMed] [Google Scholar]
- 149.Chang J-C, Chang H-S, Wu Y-C, Cheng W-L, Lin T-T, Chang H-J, Kuo S-J, Chen S-T and Liu C-S (2019) Mitochondrial transplantation regulates antitumour activity, chemoresistance and mitochondrial dynamics in breast cancer. Journal of Experimental & Clinical Cancer Research 38, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Elliott RL, Jiang XP and Head JF (2012) Mitochondria organelle transplantation: introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res Treat 136, 347–354. [DOI] [PubMed] [Google Scholar]
- 151.Marlein CR, Piddock RE, Mistry JJ, Zaitseva L, Hellmich C, Horton RH, Zhou Z, Auger MJ, Bowles KM and Rushworth SA (2019) CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res 79, 2285–2297. [DOI] [PubMed] [Google Scholar]
- 152.Burt R, Dey A, Aref S, Aguiar M, Akarca A, Bailey K, Day W, Hooper S, Kirkwood A, Kirschner K, Lee SW, Lo Celso C, Manji J, Mansour MR, Marafioti T, Mitchell RJ, Muirhead RC, Cheuk Yan Ng K, Pospori C, Puccio I, Zuborne-Alapi K, Sahai E and Fielding AK (2019) Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 134, 1415–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Salaud C, Alvarez-Arenas A, Geraldo F, Belmonte-Beitia J, Calvo GF, Gratas C, Pecqueur C, Garnier D, Pérez-Garcià V, Vallette FM and Oliver L (2020) Mitochondria transfer from tumor-activated stromal cells (TASC) to primary Glioblastoma cells. Biochemical and Biophysical Research Communications 533, 139–147. [DOI] [PubMed] [Google Scholar]
- 154.Ippolito L, Morandi A, Taddei ML, Parri M, Comito G, Iscaro A, Raspollini MR, Magherini F, Rapizzi E, Masquelier J, Muccioli GG, Sonveaux P, Chiarugi P and Giannoni E (2019) Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 38, 5339–5355. [DOI] [PubMed] [Google Scholar]
- 155.Wang J, Liu X, Qiu Y, Shi Y, Cai J, Wang B, Wei X, Ke Q, Sui X, Wang Y, Huang Y, Li H, Wang T, Lin R, Liu Q and Xiang AP (2018) Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J Hematol Oncol 11, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liu K, Ji K, Guo L, Wu W, Lu H, Shan P and Yan C (2014) Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res 92, 10–18. [DOI] [PubMed] [Google Scholar]
- 157.Önfelt B, Nedvetzki S, Benninger RKP, Purbhoo MA, Sowinski S, Hume AN, Seabra MC, Neil MAA, French PMW and Davis DM (2006) Structurally Distinct Membrane Nanotubes between Human Macrophages Support Long-Distance Vesicular Traffic or Surfing of Bacteria. The Journal of Immunology 177, 8476. [DOI] [PubMed] [Google Scholar]
- 158.Yasuda K, Park HC, Ratliff B, Addabbo F, Hatzopoulos AK, Chander P and Goligorsky MS (2010) Adriamycin nephropathy: a failure of endothelial progenitor cell-induced repair. Am J Pathol 176, 1685–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang Y, Cui J, Sun X and Zhang Y (2011) Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ 18, 732–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dickinson A, Yeung KY, Donoghue J, Baker MJ, Kelly RD, McKenzie M, Johns TG and St John JC (2013) The regulation of mitochondrial DNA copy number in glioblastoma cells. Cell Death Differ 20, 1644–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Plotnikov EY, Babenko VA, Silachev DN, Zorova LD, Khryapenkova TG, Savchenko ES, Pevzner IB and Zorov DB (2015) Intercellular Transfer of Mitochondria. Biochemistry (Mosc) 80, 542–548. [DOI] [PubMed] [Google Scholar]
- 162.Dong LF, Kovarova J, Bajzikova M, Bezawork-Geleta A, Svec D, Endaya B, Sachaphibulkij K, Coelho AR, Sebkova N, Ruzickova A, Tan AS, Kluckova K, Judasova K, Zamecnikova K, Rychtarcikova Z, Gopalan V, Andera L, Sobol M, Yan B, Pattnaik B, Bhatraju N, Truksa J, Stopka P, Hozak P, Lam AK, Sedlacek R, Oliveira PJ, Kubista M, Agrawal A, Dvorakova-Hortova K, Rohlena J, Berridge MV and Neuzil J (2017) Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Vallabhaneni KC, Haller H and Dumler I (2012) Vascular smooth muscle cells initiate proliferation of mesenchymal stem cells by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev 21, 3104–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tishchenko A, Azorín DD, Vidal-Brime L, Muñoz MJ, Arenas PJ, Pearce C, Girao H, Ramón YCS and Aasen T (2020) Cx43 and Associated Cell Signaling Pathways Regulate Tunneling Nanotubes in Breast Cancer Cells. Cancers (Basel) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li X (2019) Gap junction protein connexin43 and tunneling nanotubes in human trabecular meshwork cells. Int J Physiol Pathophysiol Pharmacol 11, 212–219. [PMC free article] [PubMed] [Google Scholar]
- 166.Qin Y, Jiang X, Yang Q, Zhao J, Zhou Q and Zhou Y (2021) The Functions, Methods, and Mobility of Mitochondrial Transfer Between Cells. Frontiers in oncology 11, 672781–672781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Norris RP (2021) Transfer of mitochondria and endosomes between cells by gap junction internalization. Traffic 22, 174–179. [DOI] [PubMed] [Google Scholar]
- 168.Vyas S, Zaganjor E and Haigis MC (2016) Mitochondria and Cancer. Cell 166, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kuznetsov AV and Margreiter R (2009) Heterogeneity of mitochondria and mitochondrial function within cells as another level of mitochondrial complexity. International journal of molecular sciences 10, 1911–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Willems PH, Rossignol R, Dieteren CE, Murphy MP and Koopman WJ (2015) Redox Homeostasis and Mitochondrial Dynamics. Cell Metab 22, 207–218. [DOI] [PubMed] [Google Scholar]
- 171.Wieder SY, Serasinghe MN, Sung JC, Choi DC, Birge MB, Yao JL, Bernstein E, Celebi JT and Chipuk JE (2015) Activation of the Mitochondrial Fragmentation Protein DRP1 Correlates with BRAF(V600E) Melanoma. J Invest Dermatol 135, 2544–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Quintana-Cabrera R, Quirin C, Glytsou C, Corrado M, Urbani A, Pellattiero A, Calvo E, Vázquez J, Enríquez JA, Gerle C, Soriano ME, Bernardi P and Scorrano L (2018) The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nature Communications 9, 3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hoppins S and Nunnari J (2009) The molecular mechanism of mitochondrial fusion. Biochim Biophys Acta 1793, 20–26. [DOI] [PubMed] [Google Scholar]
- 174.Eisner V, Lenaers G and Hajnóczky G (2014) Mitochondrial fusion is frequent in skeletal muscle and supports excitation-contraction coupling. J Cell Biol 205, 179–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE and Shirihai OS (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. The EMBO Journal 27, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Boldogh IR, Nowakowski DW, Yang H-C, Chung H, Karmon S, Royes P and Pon LA (2003) A Protein Complex Containing Mdm10p, Mdm12p, and Mmm1p Links Mitochondrial Membranes and DNA to the Cytoskeleton-based Segregation Machinery. Molecular Biology of the Cell 14, 4618–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.van der Bliek AM, Shen Q and Kawajiri S (2013) Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol 5, a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Grandemange S, Herzig S and Martinou J-C (2009) Mitochondrial dynamics and cancer. Seminars in Cancer Biology 19, 50–56. [DOI] [PubMed] [Google Scholar]
- 179.Kong B, Tsuyoshi H, Orisaka M, Shieh DB, Yoshida Y and Tsang BK (2015) Mitochondrial dynamics regulating chemoresistance in gynecological cancers. Ann N Y Acad Sci 1350, 1–16. [DOI] [PubMed] [Google Scholar]
- 180.Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke I, Merkwirth C, Ehses S, Krause F, Chan DC, Alexander C, Bauer C, Youle R, Langer T and Martinou JC (2009) SLP-2 is required for stress-induced mitochondrial hyperfusion. Embo j 28, 1589–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Altieri DC (2017) Mitochondria on the move: emerging paradigms of organelle trafficking in tumour plasticity and metastasis. British journal of cancer 117, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ferree A and Shirihai O (2012) Mitochondrial dynamics: the intersection of form and function. Adv Exp Med Biol 748, 13–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Han Y, Kim B, Cho U, Park IS, Kim SI, Dhanasekaran DN, Tsang BK and Song YS (2019) Mitochondrial fission causes cisplatin resistance under hypoxic conditions via ROS in ovarian cancer cells. Oncogene 38, 7089–7105. [DOI] [PubMed] [Google Scholar]
- 184.Vafai SB and Mootha VK (2012) Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374–383. [DOI] [PubMed] [Google Scholar]
- 185.Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C and Archer SL (2012) Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. The FASEB Journal 26, 2175–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW and Tu Y (2013) Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32, 4814–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Anderson GR, Wardell SE, Cakir M, Yip C, Ahn YR, Ali M, Yllanes AP, Chao CA, McDonnell DP and Wood KC (2018) Dysregulation of mitochondrial dynamics proteins are a targetable feature of human tumors. Nat Commun 9, 1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Anderson RG, Ghiraldeli LP and Pardee TS (2018) Mitochondria in cancer metabolism, an organelle whose time has come? Biochim Biophys Acta Rev Cancer 1870, 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.von Eyss B, Jaenicke LA, Kortlever RM, Royla N, Wiese KE, Letschert S, McDuffus LA, Sauer M, Rosenwald A, Evan GI, Kempa S and Eilers M (2015) A MYC-Driven Change in Mitochondrial Dynamics Limits YAP/TAZ Function in Mammary Epithelial Cells and Breast Cancer. Cancer Cell 28, 743–757. [DOI] [PubMed] [Google Scholar]
- 190.Porporato PE, Filigheddu N, Pedro JMB, Kroemer G and Galluzzi L (2018) Mitochondrial metabolism and cancer. Cell Res 28, 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Skulachev VP (2001) Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem Sci 26, 23–29. [DOI] [PubMed] [Google Scholar]
- 192.Mishra P and Chan DC (2016) Metabolic regulation of mitochondrial dynamics. J Cell Biol 212, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Giacomello M, Pyakurel A, Glytsou C and Scorrano L (2020) The cell biology of mitochondrial membrane dynamics. Nature Reviews Molecular Cell Biology 21, 204–224. [DOI] [PubMed] [Google Scholar]
- 194.Gomes LC, Di Benedetto G and Scorrano L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nature cell biology 13, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wu S, Zhou F, Zhang Z and Xing D (2011) Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. Febs j 278, 941–954. [DOI] [PubMed] [Google Scholar]
- 196.Collins TJ, Berridge MJ, Lipp P and Bootman MD (2002) Mitochondria are morphologically and functionally heterogeneous within cells. Embo j 21, 1616–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hackenbrock CR (1966) Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol 30, 269–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hackenbrock CR (1972) Energy-linked ultrastructural transformations in isolated liver mitochondria and mitoplasts. Preservation of configurations by freeze-cleaving compared to chemical fixation. J Cell Biol 53, 450–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL and Youle RJ (2001) The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1, 515–525. [DOI] [PubMed] [Google Scholar]
- 200.Dhingra R and Kirshenbaum LA (2014) Regulation of Mitochondrial Dynamics and Cell Fate. Circulation Journal 78, 803–810. [DOI] [PubMed] [Google Scholar]
- 201.Renault TT, Floros KV, Elkholi R, Corrigan KA, Kushnareva Y, Wieder SY, Lindtner C, Serasinghe MN, Asciolla JJ, Buettner C, Newmeyer DD and Chipuk JE (2015) Mitochondrial shape governs BAX-induced membrane permeabilization and apoptosis. Mol Cell 57, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kong B, Wang Q, Fung E, Xue K and Tsang BK (2014) p53 is required for cisplatin-induced processing of the mitochondrial fusion protein L-Opa1 that is mediated by the mitochondrial metallopeptidase Oma1 in gynecologic cancers. J Biol Chem 289, 27134–27145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Farrand L, Kim JY, Im-Aram A, Suh JY, Lee HJ and Tsang BK (2013) An improved quantitative approach for the assessment of mitochondrial fragmentation in chemoresistant ovarian cancer cells. PLoS One 8, e74008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Grieco JP, Allen ME, Perry JB, Wang Y, Song Y, Rohani A, Compton SLE, Smyth JW, Swami NS, Brown DA and Schmelz EM (2020) Progression-Mediated Changes in Mitochondrial Morphology Promotes Adaptation to Hypoxic Peritoneal Conditions in Serous Ovarian Cancer. Front Oncol 10, 600113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kim B, Jung JW, Jung J, Han Y, Suh DH, Kim HS, Dhanasekaran DN and Song YS (2017) PGC1α induced by reactive oxygen species contributes to chemoresistance of ovarian cancer cells. Oncotarget 8, 60299–60311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Qian W, Wang J, Roginskaya V, McDermott LA, Edwards RP, Stolz DB, Llambi F, Green DR and Van Houten B (2014) Novel combination of mitochondrial division inhibitor 1 (mdivi-1) and platinum agents produces synergistic pro-apoptotic effect in drug resistant tumor cells. Oncotarget 5, 4180–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Catanzaro D, Gaude E, Orso G, Giordano C, Guzzo G, Rasola A, Ragazzi E, Caparrotta L, Frezza C and Montopoli M (2015) Inhibition of glucose-6-phosphate dehydrogenase sensitizes cisplatin-resistant cells to death. Oncotarget 6, 30102–30114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ahmed N and Stenvers KL (2013) Getting to know ovarian cancer ascites: opportunities for targeted therapy-based translational research. Frontiers in oncology 3, 256–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kim S, Kim B and Song YS (2016) Ascites modulates cancer cell behavior, contributing to tumor heterogeneity in ovarian cancer. Cancer science 107, 1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Asem M, Young A, Oyama C, ClaureDeLaZerda A, Liu Y, Ravosa MJ, Gupta V, Jewell A, Khabele D and Stack MS (2020) Ascites-induced compression alters the peritoneal microenvironment and promotes metastatic success in ovarian cancer. Scientific Reports 10, 11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Pakuła M, Mikuła-Pietrasik J, Stryczyński Ł, Uruski P, Szubert S, Moszyński R, Szpurek D, Sajdak S, Tykarski A and Książek K (2018) Mitochondria-related oxidative stress contributes to ovarian cancer-promoting activity of mesothelial cells subjected to malignant ascites. Int J Biochem Cell Biol 98, 82–88. [DOI] [PubMed] [Google Scholar]
- 212.Bamias A, Tsiatas ML, Kafantari E, Liakou C, Rodolakis A, Voulgaris Z, Vlahos G, Papageorgiou T, Tsitsilonis O, Bamia C, Papatheodoridis G, Politi E, Archimandritis A, Antsaklis A and Dimopoulos MA (2007) Significant differences of lymphocytes isolated from ascites of patients with ovarian cancer compared to blood and tumor lymphocytes. Association of CD3+CD56+ cells with platinum resistance. Gynecol Oncol 106, 75–81. [DOI] [PubMed] [Google Scholar]
- 213.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L and Zou W (2004) Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10, 942–949. [DOI] [PubMed] [Google Scholar]
- 214.Lukesova S, Vroblova V, Tosner J, Kopecky J, Sedlakova I, Čermáková E, Vokurkova D and Kopecky O (2015) Comparative study of various subpopulations of cytotoxic cells in blood and ascites from patients with ovarian carcinoma. Contemp Oncol (Pozn) 19, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Knutson KL, Maurer MJ, Preston CC, Moysich KB, Goergen K, Hawthorne KM, Cunningham JM, Odunsi K, Hartmann LC, Kalli KR, Oberg AL and Goode EL (2015) Regulatory T cells, inherited variation, and clinical outcome in epithelial ovarian cancer. Cancer Immunol Immunother 64, 1495–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Song M, Sandoval TA, Chae C-S, Chopra S, Tan C, Rutkowski MR, Raundhal M, Chaurio RA, Payne KK, Konrad C, Bettigole SE, Shin HR, Crowley MJP, Cerliani JP, Kossenkov AV, Motorykin I, Zhang S, Manfredi G, Zamarin D, Holcomb K, Rodriguez PC, Rabinovich GA, Conejo-Garcia JR, Glimcher LH and Cubillos-Ruiz JR (2018) IRE1α–XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 562, 423–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Cubillos-Ruiz JR, Bettigole SE and Glimcher LH (2017) Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 168, 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Pakuła M, Mały E, Uruski P, Witucka A, Bogucka M, Jaroszewska N, Makowska N, Niklas A, Moszyński R, Sajdak S, Tykarski A, Mikuła-Pietrasik J and Książek K (2020) Deciphering the Molecular Mechanism of Spontaneous Senescence in Primary Epithelial Ovarian Cancer Cells. Cancers 12, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.McGlynn LM, McCluney S, Jamieson NB, Thomson J, MacDonald AI, Oien K, Dickson EJ, Carter CR, McKay CJ and Shiels PG (2015) SIRT3 & SIRT7: Potential Novel Biomarkers for Determining Outcome in Pancreatic Cancer Patients. PloS one 10, e0131344–e0131344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Desouki MM, Doubinskaia I, Gius D and Abdulkadir SA (2014) Decreased mitochondrial SIRT3 expression is a potential molecular biomarker associated with poor outcome in breast cancer. Human pathology 45, 1071–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Yang B, Fu X, Shao L, Ding Y and Zeng D (2014) Aberrant expression of SIRT3 is conversely correlated with the progression and prognosis of human gastric cancer. Biochem Biophys Res Commun 443, 156–160. [DOI] [PubMed] [Google Scholar]
- 222.Zhang CZ, Liu L, Cai M, Pan Y, Fu J, Cao Y and Yun J (2012) Low SIRT3 expression correlates with poor differentiation and unfavorable prognosis in primary hepatocellular carcinoma. PLoS One 7, e51703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Zhou Y, Cheng S, Chen S and Zhao Y (2018) Prognostic and clinicopathological value of SIRT3 expression in various cancers: a systematic review and meta-analysis. Onco Targets Ther 11, 2157–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kim YS, Gupta Vallur P, Jones VM, Worley BL, Shimko S, Shin D-H, Crawford LC, Chen C-W, Aird KM, Abraham T, Shepherd TG, Warrick JI, Lee NY, Phaeton R, Mythreye K and Hempel N (2020) Context-dependent activation of SIRT3 is necessary for anchorage-independent survival and metastasis of ovarian cancer cells. Oncogene 39, 1619–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kessel D and Luo Y (1999) Photodynamic therapy: a mitochondrial inducer of apoptosis. Cell Death Differ 6, 28–35. [DOI] [PubMed] [Google Scholar]
- 226.Spring BQ, Rizvi I, Xu N and Hasan T (2015) The role of photodynamic therapy in overcoming cancer drug resistance. Photochemical and Photobiological Sciences 14, 1476–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Xue LY, Chiu SM and Oleinick NL (2001) Photochemical destruction of the Bcl-2 oncoprotein during photodynamic therapy with the phthalocyanine photosensitizer Pc 4. Oncogene 20, 3420–3427. [DOI] [PubMed] [Google Scholar]
- 228.Kessel D and Arroyo AS (2007) Apoptotic and autophagic responses to Bcl-2 inhibition and photodamage. Photochem Photobiol Sci 6, 1290–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Lindsay J, Esposti MD and Gilmore AP (2011) Bcl-2 proteins and mitochondria--specificity in membrane targeting for death. Biochim Biophys Acta 1813, 532–539. [DOI] [PubMed] [Google Scholar]
- 230.Schellenberg B, Wang P, Keeble JA, Rodriguez-Enriquez R, Walker S, Owens TW, Foster F, Tanianis-Hughes J, Brennan K, Streuli CH and Gilmore AP (2013) Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol Cell 49, 959–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Sjöström J and Bergh J (2001) How apoptosis is regulated, and what goes wrong in cancer. BMJ (Clinical research ed.) 322, 1538–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Wong RS (2011) Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res 30, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Fulda S (2010) Evasion of apoptosis as a cellular stress response in cancer. Int J Cell Biol 2010, 370835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Jiang X and Wang X (2004) Cytochrome C-mediated apoptosis. Annu Rev Biochem 73, 87–106. [DOI] [PubMed] [Google Scholar]
- 235.Kessel D (2019) Apoptosis, Paraptosis and Autophagy: Death and Survival Pathways Associated with Photodynamic Therapy. Photochem Photobiol 95, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Krishnamurthy PC, Du G, Fukuda Y, Sun D, Sampath J, Mercer KE, Wang J, Sosa-Pineda B, Murti KG and Schuetz JD (2006) Identification of a mammalian mitochondrial porphyrin transporter. Nature 443, 586–589. [DOI] [PubMed] [Google Scholar]
- 237.Cincotta L, Szeto D, Lampros E, Hasan T and Cincotta AH (1996) Benzophenothiazine and benzoporphyrin derivative combination phototherapy effectively eradicates large murine sarcomas. Photochem Photobiol 63, 229–237. [DOI] [PubMed] [Google Scholar]
- 238.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T and Simon HU (2006) Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 8, 1124–1132. [DOI] [PubMed] [Google Scholar]
- 239.Kessel D and Evans CL (2016) Promotion of Proapoptotic Signals by Lysosomal Photodamage: Mechanistic Aspects and Influence of Autophagy. Photochem Photobiol 92, 620–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kessel D (2021) Death Pathways Associated with Photodynamic Therapy. Photochemistry and Photobiology 97, 1101–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Rodriguez ME, Zhang P, Azizuddin K, Delos Santos GB, Chiu S.-m., Xue L.-y., Berlin JC, Peng X, Wu H, Lam M, Nieminen A-L, Kenney ME and Oleinick NL (2009) Structural factors and mechanisms underlying the improved photodynamic cell killing with silicon phthalocyanine photosensitizers directed to lysosomes versus mitochondria. Photochemistry and photobiology 85, 1189–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Chiarante N, García Vior MC, Rey O, Marino J and Roguin LP (2018) Lysosomal permeabilization and endoplasmic reticulum stress mediate the apoptotic response induced after photoactivation of a lipophilic zinc(II) phthalocyanine. Int J Biochem Cell Biol 103, 89–98. [DOI] [PubMed] [Google Scholar]
- 243.Wei MF, Chen MW, Chen KC, Lou PJ, Lin SY, Hung SC, Hsiao M, Yao CJ and Shieh MJ (2014) Autophagy promotes resistance to photodynamic therapy-induced apoptosis selectively in colorectal cancer stem-like cells. Autophagy 10, 1179–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Duan X, Chen B, Cui Y, Zhou L, Wu C, Yang Z, Wen Y, Miao X, Li Q, Xiong L and He J (2018) Ready player one? Autophagy shapes resistance to photodynamic therapy in cancers. Apoptosis 23, 587–606. [DOI] [PubMed] [Google Scholar]
- 245.Bhandari C, Guirguis M, Savan NA, Shrivastava N, Oliveira S, Hasan T and Obaid G (2021) What NIR photodynamic activation offers molecular targeted nanomedicines: Perspectives into the conundrum of tumor specificity and selectivity. Nano Today 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Obaid G, Bano S, Mallidi S, Broekgaarden M, Kuriakose J, Silber Z, Bulin AL, Wang Y, Mai Z, Jin W, Simeone D and Hasan T (2019) Impacting Pancreatic Cancer Therapy in Heterotypic in Vitro Organoids and in Vivo Tumors with Specificity-Tuned, NIR-Activable Photoimmunonanoconjugates: Towards Conquering Desmoplasia? Nano Lett 19, 7573–7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Rizvi I, Nath S, Obaid G, Ruhi MK, Moore K, Bano S, Kessel D and Hasan T (2019) A Combination of Visudyne and a Lipid-anchored Liposomal Formulation of Benzoporphyrin Derivative Enhances Photodynamic Therapy Efficacy in a 3D Model for Ovarian Cancer. [DOI] [PMC free article] [PubMed]
- 248.Rizvi I, Obaid G, Bano S, Hasan T and Kessel D (2018) Photodynamic therapy: Promoting in vitro efficacy of photodynamic therapy by liposomal formulations of a photosensitizing agent. Lasers in Surgery and Medicine 50, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Obaid G, Jin W, Bano S, Kessel D and Hasan T (2019) Nanolipid Formulations of Benzoporphyrin Derivative: Exploring the Dependence of Nanoconstruct Photophysics and Photochemistry on Their Therapeutic Index in Ovarian Cancer Cells. Photochem Photobiol 95, 364–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Zielonka J, Joseph J, Sikora A, Hardy M, Ouari O, Vasquez-Vivar J, Cheng G, Lopez M and Kalyanaraman B (2017) Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem Rev 117, 10043–10120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Jou MJ (2008) Pathophysiological and pharmacological implications of mitochondria-targeted reactive oxygen species generation in astrocytes. Adv Drug Deliv Rev 60, 1512–1526. [DOI] [PubMed] [Google Scholar]
- 252.Cuchelkar V, Kopecková P and Kopecek J (2008) Novel HPMA copolymer-bound constructs for combined tumor and mitochondrial targeting. Mol Pharm 5, 776–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Bai X, Zhu Y, Wang H, Li J and Zhang Z (2021) Triphenylphosphonium-functionalized nanocomposites as carriers of a platinum diimine complex for photodynamic therapy. Photodiagnosis and Photodynamic Therapy 34, 102223. [DOI] [PubMed] [Google Scholar]
- 254.Sun J, Wang J, Hu W, Wang Y, Chou T, Zhang Q, Zhang B, Yu Z, Yang Y, Ren L and Wang H (2021) Camouflaged Gold Nanodendrites Enable Synergistic Photodynamic Therapy and NIR Biowindow II Photothermal Therapy and Multimodal Imaging. ACS Applied Materials & Interfaces 13, 10778–10795. [DOI] [PubMed] [Google Scholar]
- 255.Maytin EV and Hasan T (2020) Vitamin D and Other Differentiation-promoting Agents as Neoadjuvants for Photodynamic Therapy of Cancer. Photochem Photobiol 96, 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Ajioka RS, Phillips JD and Kushner JP (2006) Biosynthesis of heme in mammals. Biochim Biophys Acta 1763, 723–736. [DOI] [PubMed] [Google Scholar]
- 257.Pogue BW, Elliott JT, Kanick SC, Davis SC, Samkoe KS, Maytin EV, Pereira SP and Hasan T (2016) Revisiting photodynamic therapy dosimetry: reductionist & surrogate approaches to facilitate clinical success. Phys Med Biol 61, R57–89. [DOI] [PubMed] [Google Scholar]
- 258.Yang X, Palasuberniam P, Kraus D and Chen B (2015) Aminolevulinic Acid-Based Tumor Detection and Therapy: Molecular Mechanisms and Strategies for Enhancement. Int J Mol Sci 16, 25865–25880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Kennedy JC and Pottier RH (1992) Endogenous protoporphyrin IX, a clinically useful photosensitizer for photodynamic therapy. J Photochem Photobiol B 14, 275–292. [DOI] [PubMed] [Google Scholar]
- 260.Krammer B and Plaetzer K (2008) ALA and its clinical impact, from bench to bedside. Photochem Photobiol Sci 7, 283–289. [DOI] [PubMed] [Google Scholar]
- 261.Hadjipanayis CG and Stummer W (2019) 5-ALA and FDA approval for glioma surgery. J Neurooncol 141, 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson BC and Golab J (2011) Photodynamic therapy of cancer: an update. CA Cancer J Clin 61, 250–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Bock FJ and Tait SWG (2020) Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol 21, 85–100. [DOI] [PubMed] [Google Scholar]
- 264.D’Souza GG, Wagle MA, Saxena V and Shah A (2011) Approaches for targeting mitochondria in cancer therapy. Biochim Biophys Acta 1807, 689–696. [DOI] [PubMed] [Google Scholar]
- 265.Wang Z, Guo W, Kuang X, Hou S and Liu H (2017) Nanopreparations for mitochondria targeting drug delivery system: Current strategies and future prospective. Asian J Pharm Sci 12, 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Yousif LF, Stewart KM and Kelley SO (2009) Targeting mitochondria with organelle-specific compounds: strategies and applications. Chembiochem 10, 1939–1950. [DOI] [PubMed] [Google Scholar]
- 267.Sorrin AJ, Kemal Ruhi M, Ferlic NA, Karimnia V, Polacheck WJ, Celli JP, Huang HC and Rizvi I (2020) Photodynamic Therapy and the Biophysics of the Tumor Microenvironment. Photochem Photobiol 96, 232–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Sporri S, Chopra V, Egger N, Hawkins HK, Motamedi M, Dreher E and Schneider H (2001) Effects of 5-aminolaevulinic acid on human ovarian cancer cells and human vascular endothelial cells in vitro. J Photochem Photobiol B 64, 8–20. [DOI] [PubMed] [Google Scholar]
- 269.Ascencio M, Delemer M, Farine MO, Jouve E, Collinet P and Mordon S (2007) Evaluation of ALA-PDT of ovarian cancer in the Fisher 344 rat tumor model. Photodiagnosis Photodyn Ther 4, 254–260. [DOI] [PubMed] [Google Scholar]
- 270.Casas A, Di Venosa G, Hasan T and Al B (2011) Mechanisms of resistance to photodynamic therapy. Curr Med Chem 18, 2486–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Yokoyama Y, Shigeto T, Miura R, Kobayashi A, Mizunuma M, Yamauchi A, Futagami M and Mizunuma H (2017) Differences in the sensitivity of ovarian cancer to photodynamic therapy and the mechanisms for those differences. Oncol Lett 13, 4933–4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Teshigawara T, Mizuno M, Ishii T, Kitajima Y, Utsumi F, Sakata J, Kajiyama H, Shibata K, Ishizuka M and Kikkawa F (2018) Novel potential photodynamic therapy strategy using 5-Aminolevulinic acid for ovarian clear-cell carcinoma. Photodiagnosis Photodyn Ther 21, 121–127. [DOI] [PubMed] [Google Scholar]
- 273.Anbil S, Pigula M, Huang HC, Mallidi S, Broekgaarden M, Baglo Y, De Silva P, Simeone DM, Mino-Kenudson M, Maytin EV, Rizvi I and Hasan T (2020) Vitamin D Receptor Activation and Photodynamic Priming Enables Durable Low-dose Chemotherapy. Mol Cancer Ther 19, 1308–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Huang HC, Rizvi I, Liu J, Anbil S, Kalra A, Lee H, Baglo Y, Paz N, Hayden D, Pereira S, Pogue BW, Fitzgerald J and Hasan T (2018) Photodynamic Priming Mitigates Chemotherapeutic Selection Pressures and Improves Drug Delivery. Cancer Res 78, 558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Zuluaga MF and Lange N (2008) Combination of photodynamic therapy with anti-cancer agents. Curr Med Chem 15, 1655–1673. [DOI] [PubMed] [Google Scholar]
- 276.Wilson BC, Patterson MS and Burns DM (1986) Effect of photosensitizer concentration in tissue on the penetration depth of photoactivating light. Lasers in Medical Science 1, 235–244. [Google Scholar]
- 277.Wilson BC and Weersink RA (2020) The Yin and Yang of PDT and PTT. Photochemistry and Photobiology 96, 219–231. [DOI] [PubMed] [Google Scholar]
- 278.Wilson BC, Jeeves WP and Lowe DM (1985) In vivo and post mortem measurements of the attenuation spectra of light in mammalian tissues. Photochem Photobiol 42, 153–162. [DOI] [PubMed] [Google Scholar]
- 279.Sindelar WF, DeLaney TF, Tochner Z, Thomas GF, Dachoswki LJ, Smith PD, Friauf WS, Cole JW and Glatstein E (1991) Technique of photodynamic therapy for disseminated intraperitoneal malignant neoplasms. Phase I study. Arch Surg 126, 318–324. [DOI] [PubMed] [Google Scholar]
- 280.Canter RJ, Mick R, Kesmodel SB, Raz DJ, Spitz FR, Metz JM, Glatstein EJ, Hahn SM and Fraker DL (2003) Intraperitoneal Photodynamic Therapy Causes a Capillary-Leak Syndrome. Annals of Surgical Oncology 10, 514–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Hahn SM, Putt ME, Metz J, Shin DB, Rickter E, Menon C, Smith D, Glatstein E, Fraker DL and Busch TM (2006) Photofrin uptake in the tumor and normal tissues of patients receiving intraperitoneal photodynamic therapy. Clin Cancer Res 12, 5464–5470. [DOI] [PubMed] [Google Scholar]
- 282.Busch TM, Hahn SM, Wileyto EP, Koch CJ, Fraker DL, Zhang P, Putt M, Gleason K, Shin DB, Emanuele MJ, Jenkins K, Glatstein E and Evans SM (2004) Hypoxia and Photofrin uptake in the intraperitoneal carcinomatosis and sarcomatosis of photodynamic therapy patients. Clin Cancer Res 10, 4630–4638. [DOI] [PubMed] [Google Scholar]
- 283.Wilson JJ, Jones H, Burock M, Smith D, Fraker DL, Metz J, Glatstein E and Hahn SM (2004) Patterns of recurrence in patients treated with photodynamic therapy for intraperitoneal carcinomatosis and sarcomatosis. Int J Oncol 24, 711–717. [PubMed] [Google Scholar]
- 284.Menon C, Kutney SN, Lehr SC, Hendren SK, Busch TM, Hahn SM and Fraker DL (2001) Vascularity and uptake of photosensitizer in small human tumor nodules: implications for intraperitoneal photodynamic therapy. Clin Cancer Res 7, 3904–3911. [PubMed] [Google Scholar]
- 285.Bauer TW, Hahn SM, Spitz FR, Kachur A, Glatstein E and Fraker DL (2001) Preliminary report of photodynamic therapy for intraperitoneal sarcomatosis. Ann Surg Oncol 8, 254–259. [DOI] [PubMed] [Google Scholar]
- 286.Dimofte A, Zhu TC, Hahn SM and Lustig RA (2002) In vivo light dosimetry for motexafin lutetium-mediated PDT of recurrent breast cancer. Lasers Surg Med 31, 305–312. [DOI] [PubMed] [Google Scholar]
- 287.Wang HW, Zhu TC, Putt ME, Solonenko M, Metz J, Dimofte A, Miles J, Fraker DL, Glatstein E, Hahn SM and Yodh AG (2005) Broadband reflectance measurements of light penetration, blood oxygenation, hemoglobin concentration, and drug concentration in human intraperitoneal tissues before and after photodynamic therapy. J Biomed Opt 10, 14004. [DOI] [PubMed] [Google Scholar]
- 288.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P and Ferguson KM (2005) Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 7, 301–311. [DOI] [PubMed] [Google Scholar]
- 289.Savellano MD and Hasan T (2003) Targeting cells that overexpress the epidermal growth factor receptor with polyethylene glycolated BPD verteporfin photosensitizer immunoconjugates. Photochem Photobiol 77, 431–439. [DOI] [PubMed] [Google Scholar]
- 290.van Dam GM, Themelis G, Crane LMA, Harlaar NJ, Pleijhuis RG, Kelder W, Sarantopoulos A, de Jong JS, Arts HJG, van der Zee AGJ, Bart J, Low PS and Ntziachristos V (2011) Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-α targeting: first in-human results. Nature Medicine 17, 1315–1319. [DOI] [PubMed] [Google Scholar]
- 291.Vianello C, Cocetta V, Catanzaro D, Dorn GW, De Milito A, Rizzolio F, Canzonieri V, Cecchin E, Roncato R, Toffoli G, Quagliariello V, Di Mauro A, Losito S, Maurea N, Cono S, Sales G, Scorrano L, Giacomello M and Montopoli M (2022) Cisplatin resistance can be curtailed by blunting Bnip3-mediated mitochondrial autophagy. Cell Death & Disease 13, 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Woo SM, Min K.-j. and Kwon TK (2020) Inhibition of Drp1 Sensitizes Cancer Cells to Cisplatin-Induced Apoptosis through Transcriptional Inhibition of c-FLIP Expression. Molecules 25, 5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Li X, Zhao Y, Zhang T and Xing D (2021) Mitochondria-Specific Agents for Photodynamic Cancer Therapy: A Key Determinant to Boost the Efficacy. Advanced Healthcare Materials 10, 2001240. [DOI] [PubMed] [Google Scholar]