

Abstract
Objective:
Inflammatory bowel diseases (IBD) cause chronic intestinal damage and extracellular matrix (ECM) remodeling. The ECM may play an active role in inflammation by modulating immune cell functions, including cell adhesion, but this hypothesis has not been tested in IBD.
Design:
Primary human intestinal myofibroblast (HIMF)-derived ECM from IBD and controls, 3D decellularized colon or ECM molecule-coated scaffolds were tested for their adhesiveness for T cells. Matrisome was analysed via proteomics. Functional integrin blockade was used to investigate the underlying mechanism. Analysis of the pediatric Crohn’s disease (CD) RISK inception cohort was used to explore an altered ECM gene expression as a potential predictor for a future complicated disease course.
Results:
HIMF-derived ECM and 3D decellularized colonic ECM from IBD bound more T cells compared to control. Control HIMFs exposed to the pro-inflammatory cytokines Iinterleukin-1β (IL-1β) and tumor necrosis factor (TNF) increased, and to transforming growth factor-β1 (TGF-β1) decreased ECM adhesiveness to T cells. Matrisome analysis of the HIMF-derived ECM revealed collagen VI as a major culprit for differences in T cell adhesion. Collagen VI knockdown in HIMF reduced adhesion T cell as did the blockage of integrin αvβ1. Elevated gene expression of collagen VI in biopsies of pediatric CD patients was linked to risk for future stricturing disease.
Conclusion:
HIMF-derived ECM in IBD binds a remarkably enhanced number of T cells, which is dependent on Collagen VI and integrin αvβ1. Collagen VI expression is a risk factor for a future complicated CD course. Blocking immune cells retention may represent a novel approach to treatment in IBD.
Keywords: Inflammatory bowel disease, Extracellular matrix, Intestinal T cells, Cell adhesion
Introduction
Crohn’s disease (CD) and ulcerative colitis (UC), the two major forms of inflammatory bowel disease (IBD), have an unknown etiology and a chronic variable clinical evolution, but both lead to structural tissue damage [1]. In addition to the classical notion of immune-mediated tissue damage, there is now considerable evidence that non-immune cells are also intimately involved in IBD pathogenesis and the ensuing structural abnormalities [2]. Epithelial, endothelial, and mesenchymal cells all produce and secrete extracellular matrix (ECM), a complex mixture of glycoproteins and glycosaminoglycans, deposited throughout the bowel wall in CD and predominantly in the submucosa in UC [3]. Once deposited, ECM is not static, as it undergoes constant remodeling that alters its composition both qualitatively and quantitatively, resulting in a dynamic modulation of its biological impact, as noted during injury and healing of IBD tissue [3,4]. Importantly, in addition to its mechanical properties, ECM exerts a series of critical biological activities that modulate multiple immune cell functions, including leukocyte binding, adhesion and recruitment [5]. Distinct collagen types, the major constituent of the ECM, have diverse roles and are predominantly located in the interstitial and basement membrane matrix [6]. Among the 28 different types of collagens, collagen VI, a interconnecting collagen, has gained recent attention as a driver and disease biomarker in inflammation and fibrosis [7].
Regardless of the type of IBD, inflammatory infiltrates develop due to trafficking of immune cells from the microcirculation into the interstitial space [8] where they end up residing in direct contact with the ECM. Activated human intestinal myofibroblasts (HIMF) are considered the major source the intestinal ECM in both non-IBD and IBD tissue [9], and the key cell type responsible for the structural and functional changes occurring locally in the ECM [2]. The interaction of immune cells and ECM is mediated by various kinds of cell surface molecules, among which integrins are of most investigated [10]. However, limited information is available about the interaction between immune cells and the ECM in IBD. The observation that current IBD treatments, which fundamentally only target the immune component of both CD and UC, have reached a therapeutic ceiling makes targeting the ECM and its immune cell interactions a reasonable and innovative proposition to improve patient outcomes [11].
We hypothesized that the interaction between ECM and T cells via integrins is altered in IBD, leading to an excessive binding and retention of T cells in the inflamed mucosa. We further hypothesized that this adaptation results from a distinctively altered composition of the ECM in IBD and is mediated by specific integrins. This would help explain the chronic nature of inflammation in IBD and its subsequent tissue damaging effects.
Materials and methods
Procurement of intestinal tissues
The laboratory standardized process for tissue procurement and isolation and culture of primary human intestinal myofibroblasts as well as lamina propria T cells (LPT) has been described previously [12–15]. A detailed description can be found in Supplementary Materials and Methods.
Generation of primary human intestinal myofibroblast derived 2-dimensional extracellular matrix-coated plates
Forty thousand HIMFs/well were plated in a 24-well cluster plate (Corning, New York, NY, USA) and, after reaching confluence, medium was replaced and the culture continued for additional 10 days. At the end of this period, HIMF were washed with Hank’s balanced salt solution (HBSS) and sequentially exposed to 0.5% Triton-X (Sigma, St. Louis, MO, USA) and 0.025 N ammonium hydroxide (Sigma, St. Louis, MO, USA) for 2 min at room temperature. The residual layer of ECM adherent to the plastic surface was then washed 4 times with HBSS and stored at 4 °C until used (Fig. 1A). In some experiments, ECM was generated by HIMF stimulated by exposure to various cytokines, including interleukin-1β (IL-1β) (10 U/mL), tumor necrosis factor (TNF) (10 U/mL), interferon-γ (IFN-γ) (100 U/mL) or transforming growth factor-β1 (TGF-β1) (1 ng/mL) (all purchased from R&D, Minneapolis, MN, USA), which were added during the 10-day period of ECM deposition.
Fig. 1.

Inflammatory bowel disease human intestinal myofibroblast-derived extracellular matrix displays enhanced adhesiveness for T cells.
(A) Diagram of the T cell with extracellular matrix (ECM) adhesion assay. (B) An increased number of T cells (MOLT4 on the left, peripheral blood T cells in the middle and lamina propria T cells from a CD patient on the right panel) adhered to inflammatory bowel disease (IBD) human intestinal myofibroblast (HIMF) derived ECM compared to that from non-IBD control (n = 5, t test). (C) No differences in cell number (left) or produced ECM protein amount (right) between HIMF from normal control (NL), ulcerative colitis (UC) and Crohn’s disease (CD) groups were noted during cell culture (n = 5, t test). (D) The adhesiveness of T cells to ECM was increased upon stimulation of NL HIMF with interleukin (IL)-1β and tumor necrosis factor (TNF) but decreased with transforming growth factor (TGF)-β1 in matrix derived from NL HIMF. T cell to ECM adhesion remained unchanged in IBD HIMF exposed to IL-1β and TNF, but was reduced upon HIMF stimulation with interferon (IFN)-γ and TGF-β1 (n = 5, t test). Representative images of T cells adhering to HIMF derived ECM are shown below. *, p < 0.05, **, p < 0.01, ***, p < 0.001, ****, p < 0.0001.
Decellularization of colonic tissue sections
Using a slightly modified previously described decellularization protocol [16], fresh tissues harvested from surgical specimen were rinsed in HBSS and mucus and blood clots removed. They were then immersed in double-distilled water (ddH2O) with penicillin, streptomycin and amphotericin B (PSF, Loza, Basel, Switzerland) at room temperature overnight and subsequently incubated in ddH2O with PSF for 8 hours. They were then immersed in 4% sodium deoxycholate (SDC, Abcam, Cambridge, MA, USA) at 4°C overnight with an 7h wash in ddH2O the following day. Tissues were then incubated with 0.2mg/ml RNase and 0.1mg/ml DNase (both Worthington-Biochem, Lakewood, NJ, USA) in 50mmol/L MgCl2 (Sigma, St. Louis, MO, USA) at 37°C overnight, followed by sterile phosphate-buffered saline (PBS) washes twice a day for 10 days. Decellularized tissues were embedded in optical coherence tomography compound (OCT) and frozen at −80 °C. OCT embedded blocks were sectioned at 10μm and attached into 3-well chamber slides (IBIDI GMBH, Martinsried, Germany) for future adhesion experiments. Decellularized IBD tissue was obtained from the inflamed bowel segments.
Adhesion assay
HIMF derived 2D ECM-coated plates, ECM-coated plates and decellularized tissue were overlaid with 1 × 106/ml T cells (MOLT4 or peripheral blood T cells [PBT]) in Roswell Park Memorial Institute 1640 medium (RPMI 1640). In specific experiments, 5 ng/ml phorbol 12-myristate 13-acetate (PMA, Sigma, St. Louis, MO, USA) was added for 1 h. After 3 h at 37 °C, non-adherent T cells were removed by gentle aspiration and wells rinsed 3 times with Ca++- and Mg++-containing HBSS. T cells were either pre-labeled by calcein (Thermo Fisher Scientific, Waltham, MA, USA) or were stained after adhesion and fixation (Diff Quick Stain Set, Dade Diagnostics, Aguada, PR). All experiments were performed in duplicates or triplicates. At least 3 random high-power fields (Olympus IX71 microscope, Olympus Scientific Solutions Technologies Inc, Waltham, MA) in each well were obtained. Number of cells were counted using ImageJ (version 1.8.0, National Institutes of Health & LOCI, Madison, WI).
Matrisome analysis of human intestinal myofibroblast derived extracellular matrix
The ECM derived from HIMF was generated as described above. Deposited ECM from untreated NL, UC and CD HIMF as well as NL HIMF that were untreated or stimulated with 10 U/mL TNF and 1 ng/mL TGF-β1 was digested in 8M urea Tris-HCl buffer (Sigma, St. Louis, MO, USA) and subjected to liquid chromatography mass spectrometry (LC-MS) as previously described [12]. The protein false discovery rate (FDR) rate was set to 1%. Label-free quantitation (LFQ) intensities were determined using PD2.2. In order to focus on the function of ECM components in T cell adhesion, the whole dataset identified by proteomics was then matched with the matrisome database (MatrisomeDB), for further analysis[17]. A detailed description of the proteomics workflow can be found in Supplementary Materials and Methods.
Isolation and purification of peripheral blood T cells, calcein labelling, RNA interference, ECM coating, blocking experiments, immunohistochemistry, immunofluorescence, quantitative reverse transcriptase polymerase chain reaction, the cohort description of the Rapid Disease Progression in Children with Crohn’s Disease (RISK) study and statistical analysis can be found in Supplementary Materials and Methods.
Results
Enhanced adhesiveness of T cells to extracellular matrix derived from IBD intestinal myofibroblasts
To investigate whether ECM produced by HIMF from the IBD mucosa displayed a different capacity to bind T cells compared to ECM produced by non-IBD control (NL) HIMF, we generated 2-dimensional (2D) ECM scaffolds from NL, UC and CD HIMF (Fig. 1A). Given that the focus of this investigation was on the HIMF derived ECM properties, we used the well characterized MOLT4 T lymphoblast cell line for most of the experiments. MOLT4 was originally derived from human acute lymphoblastic leukemia [18]. ECM deposited by UC and CD HIMF bound a greater number of MOLT4 cells compared to ECM generated by NL HIMF (Fig. 1B). Comparable results were also found for PBT and CD derived LPT adhesion to the HIMF derived 2D ECM (Fig. 1B). To rule out the possibility that the enhanced T cell binding of IBD ECM was simply due to a greater proliferation of HIMF while producing ECM, the numbers of NL, UC and CD HIMF were measured at various time points. Similar numbers of HIMF were present in all groups at 3, 7 and 10 days of culture (Fig. 1C, left). We additionally measured the amount of ECM protein produced by HIMF, and no differences could be detected for ECM produced by NL, UC and CD HIMF (Fig. 1C, right). Together, these results suggest that ECM produced by IBD HIMF has a higher adhesiveness to T cells compared to NL HIMF derived ECM. Due to the comparable results in the adhesion assay using MOLT4, PBT or CD LPT, we used MOLT4 as the representative cell line for further experiments (unless otherwise stated). While not the focus of this investigation we performed preliminary experiments testing adhesion of LPT derived from NL, UC and CD patients on HIMF derived 2D ECM scaffolds. Those results can be found in Supplementary Fig. 1B and Supplementary Results.
Effect of cytokines on T cell adhesiveness to ECM derived from intestinal myofibroblasts
Knowledge of the modulatory effect of cytokines on ECM composition and ECM biological activity is extremely limited. We hence investigated the effect of various cytokines on the adhesive function of ECM produced by NL and IBD HIMFs. While it is not feasible to test all cytokines known to be differentially regulated in IBD we deliberately selected four of them, with established major roles in inflammation and ECM deposition: TNF [19,20], IFN-γ [21,22] and IL-1β [23,24] are major pro-inflammatory cytokines in IBD, while TGF-β1 [2,25] has well established matrix remodeling properties. NL HIMFs exposed to the pro-inflammatory activity of IL-1β and TNF, produced an ECM with marked increase in adhesiveness, but IBD HIMF activated by the same two cytokines, did not further increase the ECM adhesive capacity (Fig. 1D). IFN-γ and TGF-β1 are two cytokines abundantly produced in the inflammatory milieu of the IBD mucosa, but with distinct immunomodulatory activities and effects on ECM production [26], with IFN-γ downregulating and TGF-β1 upregulating ECM production. Surprisingly, treatment of NL HIMF with IFN-γ had no effect on the ability of the secreted ECM to bind T cells. In contrast, ECM produced by UC and CD HIMF exposed to IFN-γ exhibited reduced adhesive capacity compared to that of untreated IBD HIMF (Fig. 1D). Finally, HIMF exposed to TGF-β1, the main pro-fibrotic growth factor, induced a drop of T cell binding capacity by the ECM from all groups, and this was particularly evident for ECM generated by IBD HIMF (Fig. 1D). This indicates that the increased adhesiveness of ECM derived from IBD HIMF could be reproduced by exposure of NL HIMF to TNF and IL-1β. Surprisingly, ECM from HIMF exposed to TGF-β1, despite being the major profibrotic growth factor, showed reduced adhesiveness to T cells, suggesting that not the amount of ECM, but potentially its composition may be relevant for T cell binding.
Collagen VI is associated with the T cell to extracellular matrix adhesion pattern in myofibroblast derived matrix scaffolds
HIMF derived ECM is complex mixture of multiple proteins. We next determined which components in HIMF-derived ECM could contribute to its enhanced adhesiveness in IBD to derive candidates that may portend the adhesiveness to T cells. For this purpose, we used proteomics analysis focusing on the core matrisome [17] (Fig. 2A), the main constituent of the interstitial ECM. It comprises a collection of ECM proteins [17], such as glycoproteins, collagens and proteoglycans. Principal component analysis (PCA) of 2D HIMF derived ECM scaffolds, revealed limited global differences among NL, UC and CD ECM (Fig. 2B). Most of the top 25 abundant matrisome protein components were shared between groups, but unique expression was found for LAMC1, NID2 in NL, LAMA4, THBS1, COL18A1, PXDN in UC and COL3A1, LTBP2 in CD (Fig. 2C). Fibronectin and tenascin were the top two ECM proteins identified in all conditions (Fig. 2F). Given the observed differences in T cell adhesion pattern observed between NL and IBD ECM and to identify ECM candidates that may contribute to this adhesion we first focused on the matrisome components showing an increase in UC and CD compared to NL. This was done as IBD HIMF derived ECM had an increased adhesives to T cells compared to NL HIMF. In total, 13 matrisome factors were identified to be elevated in IBD ECM compared to control, namely COL6A1, COL6A2, COL6A3, COL4A1, COL5A1, COL8A1, COL12A1, COL18A1, EDIL3, CTGF, FNDC1, NTN1 and SRPX2.
Fig. 2.
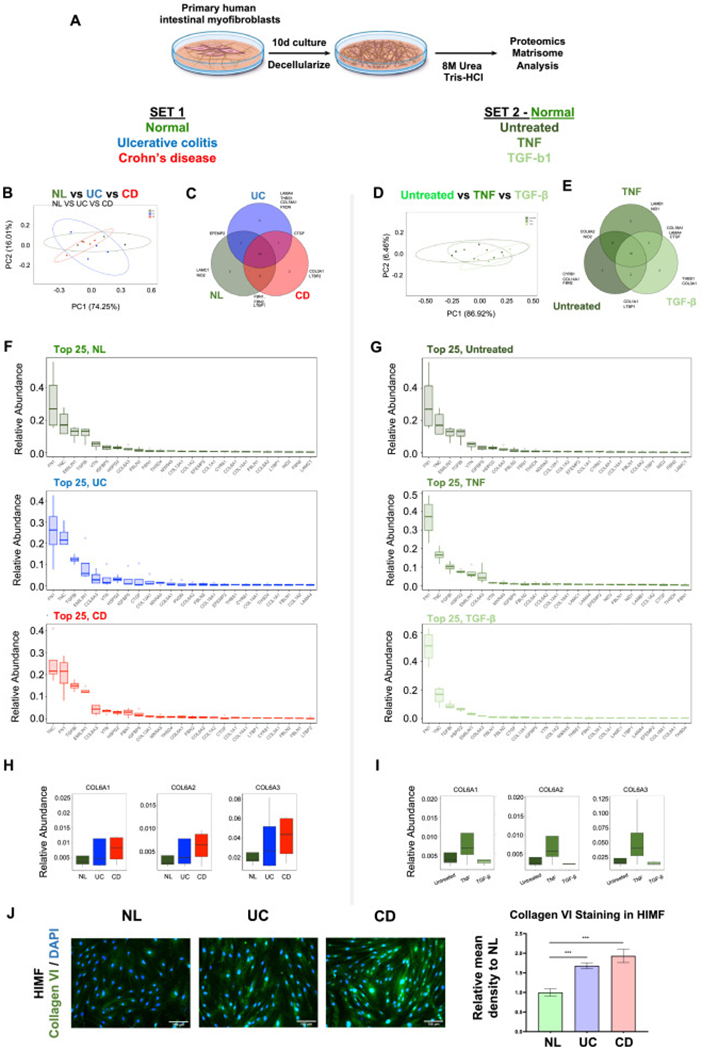
Matrisome analysis in human intestinal myofibroblast derived matrix reveals distinct extracellular matrix production from control to inflammatory bowel disease, and from untreated to cytokine stimulation.
(A) Schematic overview of the process of matrisome analysis. (B) Principal component analysis (PCA) analysis of the human intestinal myofibroblast (HIMF) matrisome showing modest differences among normal control (NL), ulcerative colitis (UC) and Crohn’s disease (CD) groups. (C) Unique extracellular matrix (ECM) components were identified among NL, UC and CD groups. (D) PCA analysis showing minimal differences among untreated, tumor necrosis factor (TNF) stimulated or transforming growth factor (TGF)-β1 exposed NL HIMF in respect to matrisome expression. (E) Unique ECM components were identified among untreated, TNF stimulated, TGF-β1 stimulated NL HIMF. (F) Top 25 abundant matrisome ECM molecules in NL, UC and CD groups. (G) Top 25 abundant matrisome ECM molecules in untreated, TNF stimulated and TGF-β1 stimulated groups. (H) Collagen VI protein expression in UC and CD compared to NL matrisome. (I) Collagen VI protein expression in untreated NL HIMF, or NL HIMF exposed to TNF or TGF-β matrisome. (n = 4 in each group). (J) Immunofluorescence staining of cultured HIMFs showed increased expression of collagen VI α1 in UC and CD compared to NL (n = 6, t test). ***, p < 0.001.
To further elucidate the matrisome candidates responsible for an increase in T cell adhesion we cultured NL HIMF with TNF (increased T cell adhesion) or TGF-β1 (decreased T cell adhesion) and again performed a matrisome analysis. PCA revealed limited differences, with most core matrisome components being shared by untreated or stimulated HIMF (Fig. 2D and E). Untreated HIMF uniquely produced CYR61, COL14A1 and FBN2, only TNF-treated HIMF produced LAMB1 and NID1, and only TGF-β-treated HIMF produced THBS1 and COL3A1 (Fig. 2E). Again, fibronectin and tenascin were the top two ECM proteins that were identified in all conditions (Fig. 2G). We next aimed to recapitulate the originally observed T cell adhesion patterns (Fig. 1B and D). We hence selected matrisome factors that were elevated in the UC and CD ECM (Fig. S2A). Among those, we identified specific factors produced by NL HIMF that increased with TNF treatment and those that decreased with TGF-β1 treatment (Fig. S2B). COL6A1, COL6A2 and COL6A3 followed this pattern (Fig. 2H and I), and their relative abundance matched the adhesion profiles observed in Fig. 1B and D. Of note, COL6A4, COL6A5 and COL6A6 were not detectable in our matrisome analysis. The entire set of ECM molecules that were identified in different groups are summarized in Fig. S2A and B. Hence the proteomic matrisome analysis rendered collagen VI as one possible candidate leading to the increased T cell adhesion to IBD HIIMF derived ECM. This provided our rationale for further exploring the expression and functional relevance of collagen VI in T cell adhesion in IBD.
To confirm our findings of collagen VI upregulation in HIMF derived ECM we performed immunofluorescence staining for collagen VI a1 in HIMF monolayers using selective antibodies. Collagen VI α1 was elevated in both freshly isolated UC and CD HIMF compared to NL HIMF (Fig. 2J) supporting our proteomics results. This expression analysis also confirms HIMF as a source of collagen VI in IBD.
To evaluate, whether the spontaneous cytokine expression by HIMF is different between NL, UC and CD we performed a flow cytometry cytokine assay of HIMF supernatants that were conditioned for 48h. We found expression of IL-6, IL-8 and MCP-1 with no significant difference between NL, UC or CD. A trend was noted for increased MCP-1 expression in UC and CD compared to NL. IL-1β, IL-10 and TNF were not detectable (Fig. S3A).
Collagen VI increases adhesion of T cells
To corroborate our findings of the relevance of collagen VI for T cell adhesion we investigated its direct interaction with MOLT4 in vitro. In fact, T cells adhered to collagen VI-coated plates in a concentration-dependent manner compared to uncoated plates, and adhesion dramatically increased by 30-fold in T cells pre-activated with phorbol myristate acetate (PMA) (Fig. 3A and B). As a control we used collagen I and IV, which are both known to be increased in IBD and which also increased adherence of T cells to a comparable degree in our adhesion assay (Figs. 3B and S3B and C). However, neither collagen I nor IV showed differences in the proteomics results that matched the originally observed T cell adhesion patterns (Fig. 1D). We next used LPT isolated from a CD patient and tested their adhesion to collagens I, IV and VI. We found increased adhesion of LPT to all three collagens, with the strongest adhesive capacity exerted by collagen VI (Fig. S3D).
Fig. 3.

Collagen VI increases matrix adhesiveness
T cells adhesion to extracellular matrix (ECM) coated plates was tested. (A) The adhesiveness of T cells was enhanced with increasing concentrations of collagen VI. Phorbol myristate acetate (PMA) pre-treated T cells showed a higher adherence compared to untreated T cells. (B) The increased adhesiveness of T cells was not only observed in in collagen VI coated plates but also in collagen I and collagen IV coated plates and adhesion increased in all groups after exposure of T cells to PMA (n = 4~6, t test). (C) Human intestinal myofibroblasts (HIMFs) were transfected with small interfering (si)RNA targeting COL6A1 prior to generation of 2D ECM scaffolds. T cell adhesion was robustly decreased in the extracellular matrix (ECM) derived from HIMFs which were transfected by COL6A1 siRNA compared to scrambled siRNA (n = 6, t test). *, p < 0.05, **, p < 0.01, ***, p < 0.001.
We then silenced the central COL6A1 chain in HIMF via siRNA knockdown prior to submitting the HIMF produced ECM to the T cell adhesion assay. Knockdown efficiency can be found in Fig. S3E. COL6A1 depleted HIMF derived 2D ECM scaffolds reduced T cell adhesion by ~50% compared to scrambled siRNA control (Fig. 3C).
T cells bind to the intestinal myofibroblast derived extracellular matrix via integrins
To assess mechanisms of T cell binding, we next blocked integrin function on the surface of T cells, first by using the tripeptide Arg-Gly-Asp (RGD), a conserved amino acid sequence occupying a binding site for specific integrins [27]. RGD dose-dependently inhibited MOLT4 adhesion to 2D HIMF derived 2D ECM scaffolds. The non-integrin binding Arg-Gly-Glu (RGE) peptide served as negative control (Fig. 4A). We next blocked the central RGD binding integrin [27], integrin β1, which dose-dependently and robustly reduced T cell with ECM adhesion (Fig. 4B). Testing all potential integrins would not be feasible within the scope of this investigation. Hence, among the integrin β1 binding partners we selected integrins αv (due to its ability to form heterodimers with multiple β-integrins), integrin α5 (integrin β1 and RGD binding) and as a control integrin α3 (integrin β1 but not RGD binding) (Fig. 4C to E). Integrins αv, α3 and α5 were present on MOLT4 as shown by flow cytometry (Fig. S4A). Intergin αv, but not α3 or α5 dose dependently reduced T cell binding to HIMF ECM. As an additional control and given their involvement in IBD pathogenesis we blocked integrins α4 and β7 on MOLT4 prior to their adhesion to HIMF derived 2D ECM. No difference in adhesion was noted whether integrins α4 or β7 were inhibited (Fig. S4B) compared to isotype control antibody. This set of experiments shows that integrin binding sites of the HIMF deposited ECM are responsible for T cell adhesion, further corroborating our finding that the type of deposited ECM is responsible for the adhesiveness to T cells.
Fig. 4.
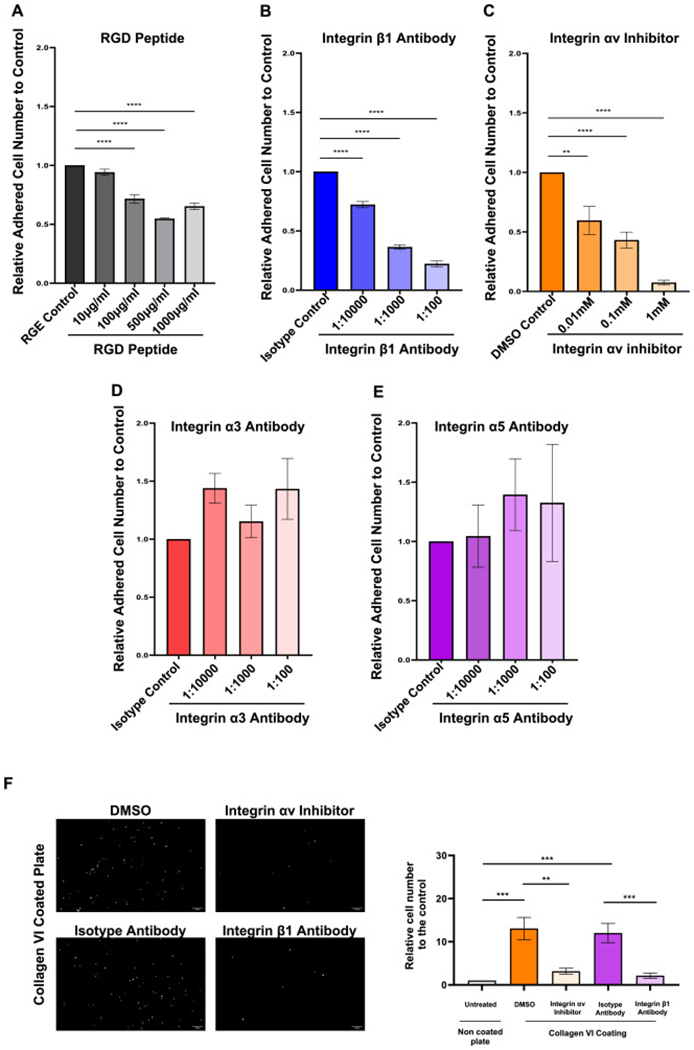
The adhesiveness for T cells is influenced by different cytokines and mediated by integrin αvβ1.
T cells adhesion to normal human intestinal myofibroblast (HIMF) derived extracellular matrix (ECM) was tested in the presence or absence of integrin function modulating factors. (A) Arg-Gly-Asp (RGD) inhibited the matrix adhesiveness for T cells in a concentration dependent manner (n = 5, t test). (B)&(C) Blockage of integrin αv and β1 inhibited the adhesiveness in a concentration dependent manner (n = 5, t test). (D)&(E) Blockage of integrin α3 and α5 did not alter the adhesiveness of HIMF derived ECM for T cells (n = 3, t test). (F) The T cell adhesion to collagen VI coated plates was dramatically inhibited by the blockage of integrin αv or integrin β1 (n = 8, t test). **, p < 0.01, ***, p < 0.001, ****, p < 0.0001.
After identifying Integrins αv and β1 as a candidate for T cell to ECM binding we next wondered whether those integrins are also relevant for T cell binding to collagen VI. This is plausible since collagen VI contains an RGD motif [28]. Consistent with our earlier findings, blocking integrins αv and β1 on MOLT4 T cells dramatically suppressed their adhesion to collagen VI (Fig. 4F). We again confirmed these findings with LPT derived from NL and CD patients, in which blocking integrin β1 reduced their adhesion to collagen VI (Fig. S4C). This data supports that T cells bind to HIMF derived collagen VI via an integrin mediated mechanism and that collagen VI is one ECM molecule mediating T cell to ECM adhesion in IBD.
Increased expression of collagen VI in inflammatory bowel disease tissues
We then explored the expression of collagen VI as well as its cellular source in intestinal tissues. Immunohistochemistry (IHC) staining of IBD and control tissues for collagen VI α1 indicated consistently stronger expression in all tissue layers of UC and CD compared to NL by automatic quantification as well as blinded scoring using a prespecified semiquantitative expression scale (Figs. 5A,B and S5A). We additionally detected an upregulation of collagens VI α2 and α3 in UC and CD (Fig. S5B). The major source of collagen VI were mesenchymal cells located in the muscularis mucosa, submucosa and around vessels and their number as well as individual staining intensity were increased in UC and CD compared to NL (Figs. 5B and S5B). In UC and CD, abundant CD3 positive immune cells (brown color) encased by collagen VI α1-positive ECM (red color) were identified in the submucosa, suggesting a close T cell with ECM interaction (Fig. 5B, black arrows). Analysis of a publicly available single-cell RNA sequencing dataset derived from 18 patients with UC and 12 healthy controls [29] validated our results, by showing exclusive expression of COL6A1, COL6A2 and COL6A3 in stromal cells but not epithelial or immune cells (Fig. 5C). Inflammatory fibroblasts and myofibroblasts were a major source of COL6A1, COL6A2 and COL6A3 (Fig. 5C, red boxes). A second single-cell RNA sequencing dataset confirmed that stromal cells had a high expression of collagen VI [30] (Fig. S6).
Fig. 5.
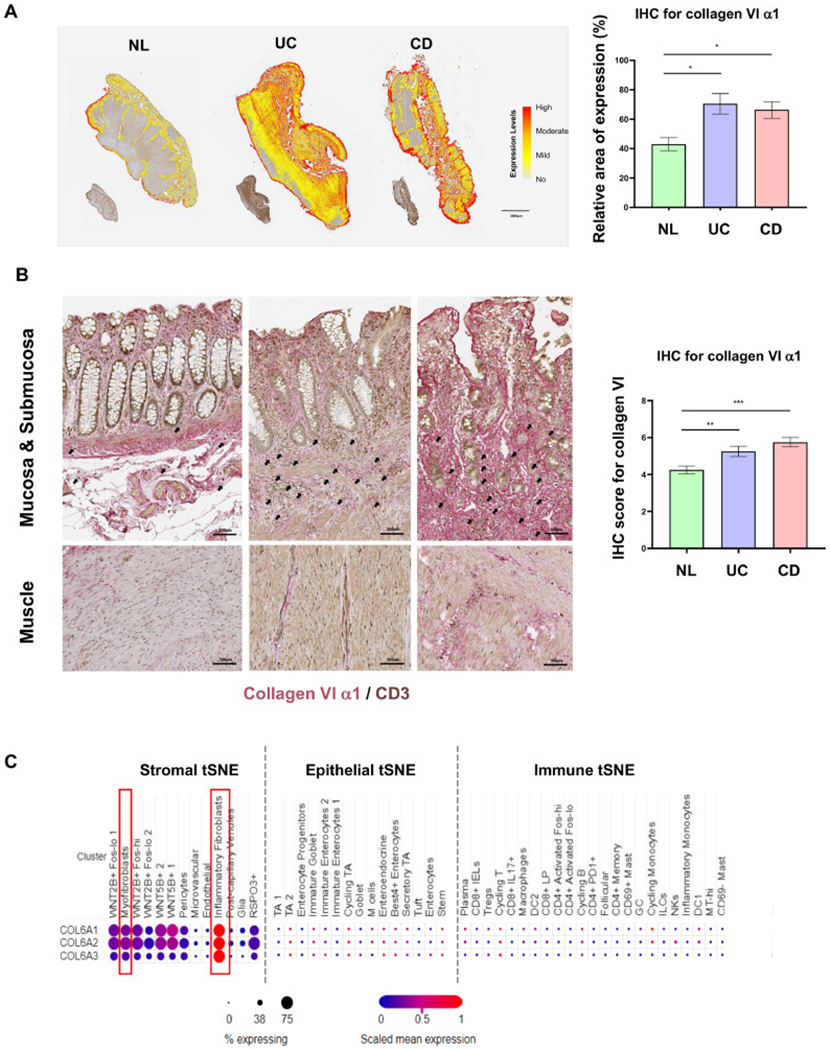
The expression of collagen VI is elevated in inflamed colon tissue in inflammatory bowel disease.
(A) Collagen VI α1 immunohistochemistry of full thickness intestinal tissue sections in ulcerative colitis (UC) and Crohn’s disease (CD), compared to normal control (NL). Automatic quantification by HALO software showed significantly higher expression in UC and CD compared to NL (n = 5, t test). (B) Collagen VI α1 expression on immunohistochemistry was elevated in the mucosa, submucosa and muscle layers in UC and CD, compared to NL. Under high magnification CD3 positive immune cells (brown) were found encased by and in close proximity to collagen VI α1 (red) in the submucosa of inflammatory bowel disease (IBD) tissues (black arrows). The increase in collagen VI α1 in intestinal tissues was confirmed by blinded scoring of full thickness colonic sections using a prespecified scoring system (n = 12, t test). (C) Open access single cell dataset [29] revealed that COL6A1, COL6A2 and COL6A3 were mainly expressed in stromal cells in UC, especially in inflammatory fibroblasts, but not in epithelial and immune cells in UC. *, p < 0.05, **, p < 0.01, ***, p < 0.001.
Increased adhesion of T cells to decellularized IBD tissue is increased in inflammatory bowel disease and is inhibited by αv and β1 integrin blockade
We next assessed whether the identified integrins responsible for the adhesion of T cells to HIMF ECM and collagen VI are also relevant in the setting of native intestinal wall ECM. To mimic in vivo adhesion events we generated decellularized 3-dimensional (3D) IBD colonic tissue sections. Decellularization of intestinal resection tissues was confirmed by a sharp loss of GAPDH and β-actin mRNA in the decellularized compared to native tissues and loss of immunofluorescence staining for DAPI (cell nuclei) while retaining structure and amount of collagen VI (Figs. 6A and S7). In concordance with and reproducing our initial findings (Fig. 1), CD and UC tissue ECM sections displayed an increased capacity to bind T cells compared to non-IBD controls (Fig. 6B). Strikingly, again blockade of both αv and β1 integrins decreased T cell adhesion by ~70% compared to controls (Fig. 6C). This lends relevance to our finding of selective integrins being important for T cell with ECM adhesion.
Fig. 6.
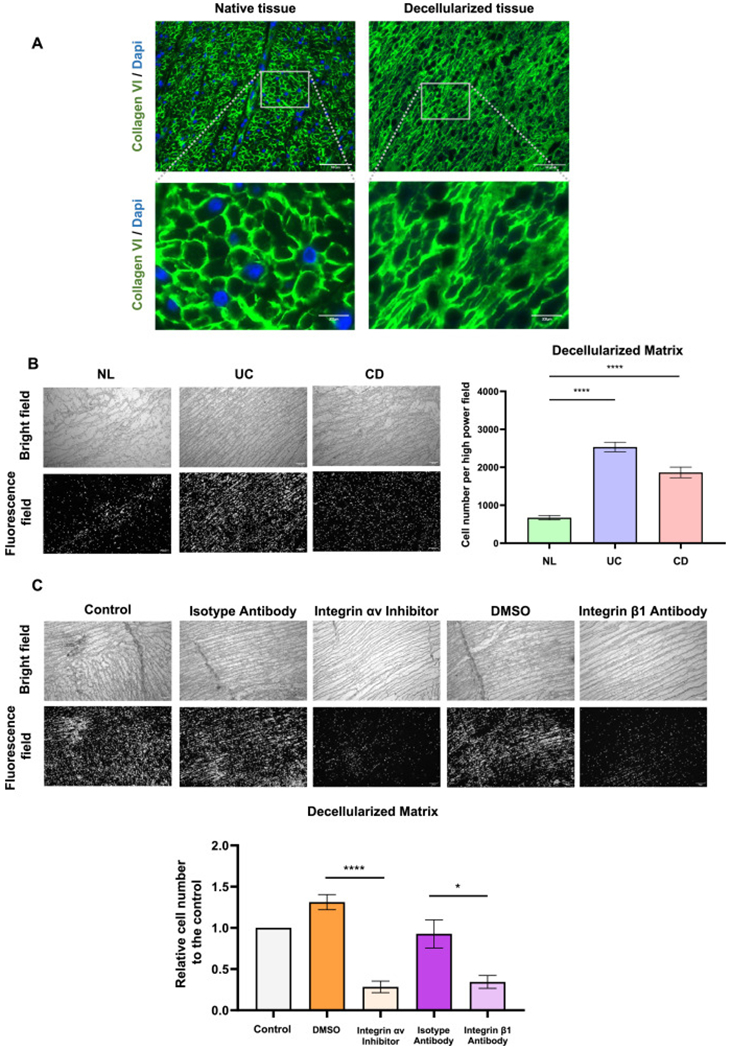
T cell adhesiveness of inflammatory bowel disease decellularized tissue is inhibited by the blockage of integrin αv or integrin β1 in decellularized intestinal matrix
(A) Immunofluorescence staining showing successful removal of cellular components of the decellularized intestinal tissue as indicated by nuclear DAPI stain with extracellular matrix (ECM) structure and collagen VI amount remaining intact. (B) A higher amount of T cells adhered to ulcerative colitis (UC) and Crohn’s disease (CD) decellularized intestinal tissue sections compared to normal control (NL) tissues (n = 6, t test). (C) The adhesiveness of T cells to decellularized tissue dramatically decreased by the blockage of integrin αv or integrin β1 (n = 6, t test). *, p < 0.05, ****, p < 0.0001.
Integrin αv is elevated in CD4±PD1± and CD8±IL17± T cells in ulcerative colitis
Given that the expression of integrin subsets on T cells in the inflamed intestinal mucosa has been described prior, we performed an analysis of a single-cell RNA sequencing dataset derived from patients with uninflamed, inflamed UC and healthy controls [29]. The integrin αv gene, one integrin we identified to be mediating T cell with ECM adhesion (HIMF derived ECM as well as 3D decellularized intestinal ECM), was expressed at low levels in T cell subsets in the healthy intestinal mucosa. Levels of integrin αv remained low in T regulatory cells, irrespective of uninflamed or inflamed ulcerative colitis (UC). They however increased in frequency and average expression in CD4+ PD1+ T cells in the inflamed UC mucosa compared to uninflamed UC and healthy and in addition an increase in frequency and average expression was noted in CD8+ IL17+ and CD4+ Activated Fos-lo T cells in UC compared to healthy (Fig. S8). Integrins α3 and α5 had a lower expression in this group compared to integrin αv (Fig. S8). This re-analysis of available datasets indicated that integrin αv is present on intestinal T cells and is upregulated in intestinal inflammation in T cell subsets with pro-inflammatory properties, but not in T regulatory cells.
Collagen VI gene expression is associated with fibrostenotic Crohn’s disease
Finally, to investigate a possible pathogenic implication of collagen VI-mediated increased adhesiveness for T cells we examined its expression in ileal biopsy tissue of CD patients by probing the transcriptomes of the RISK inception cohort [31,32]. Patients with an purely inflammatory CD phenotype were followed until development of complications (internal penetrating disease: Montreal classification B3 or B2+B3) or stricturing disease (Montreal classification B2; Table S1). Given the focus of our paper, our analysis was restricted to core matrisome genes corresponding to our proteomics analysis (Fig. 2). PCA showed modest discrimination between the control and the future stricturing groups (Fig. 7A). However, COL6A1, COL6A2 and COL6A3 were among the top 20 contributing variables associated with complications, and explaining 80.8% of the variations by contribution plot (Fig. 7B). Both COL6A2 and COL6A3 gene expression were increased in those that went on to develop stricturing CD, while COL6A1 showed a non-significant nominal increase (p = 0.068; Fig. 7C and Table S2). Cox regression analysis indicated that both COL6A1 and COL6A2 were risk factors for future stricturing complications (Fig. 7D and Table 1). This human observational study suggests that collagen VI gene expression is linked with future complicated disease courses.
Fig. 7.
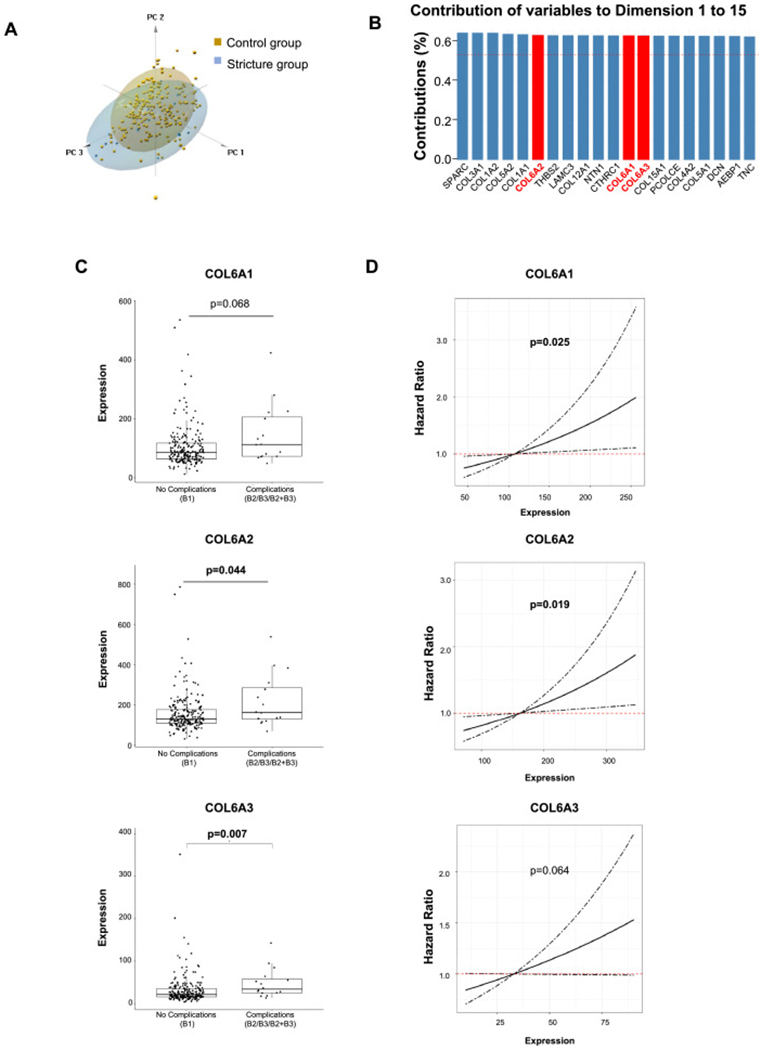
Collagen VI gene expression levels are elevated in pediatric Crohn’s disease patients who develop strictures and predicts future stricturing disease.
(A) Principal component analysis (PCA) showed modest differences between stricture and control groups. Dimension 1 to 3 were plotted. Scree plot showing the first dimension (component) explained 28.1% variations and the second dimension explained 10.6% variations. (B) COL6A1, COL6A2 and COL6A3 were among the top 20 variables that contribute to the differences between fibrostenotic and control groups from dimension 1 to 15, which explained 80.8% the variations. (C) Both levels of COL6A2 and COL6A3 were higher in the future fibrostenotic group than those in the control group. The level of COL6A1 was nominally higher in fibrostenotic group but did not reach statistical significance. (D) COL6A1 and COL6A2 were risk factors for the future development of strictures. The risk for developing strictures nominally increased with the increase of COL6A3 level but did not reach statistical significance. n=218 in control group and n=16 in stricture group.
Table 1.
Multivariate COX analysis for stricturing complications adjusted to early treatment.
| Clinical Outcomes (Sample Size, n) | Factors | Hazard Risk (95% CI) | p value |
|---|---|---|---|
| Stricturing complications (B2) (n = 229) | COL6A1 | 0.94 (1.00–1.01) | 0.025 |
| Early treatment | 0.99 (0.30–2.94) | 0.915 | |
| COL6A2 | 1.00 (1.00–1.01) | 0.019 | |
| Early treatment | 0.93 (0.30–2.91) | 0.898 | |
| COL6A3 | 1.01 (1.00–1.02) | 0.064 | |
| Early treatment | 0.96 (0.31–3.01) | 0.947 |
Discussion
Trafficking controls multiple functions of immune cells, including homing, adhesion, retention and recirculation [8]. The process of trafficking is tightly regulated by multiple molecules like integrins, chemokines and endothelial cell adhesion molecules which maintain homeostasis and prevent excessive immune responses in the gut [33]. This balance is perturbed in states of chronic inflammation as occurs in IBD. The pathogenesis of IBD is believed to include adhesion of T cells to endothelial cells, their migration from the circulation into tissues, and their retention in situ [34]. Mechanisms underlying enhanced T cell retention in IBD remain unclear and understanding its modulation may lead to a novel therapeutic approach to control inflammation in IBD.
The ECM is a group of proteins universally expressed in different tissues and organs, providing physical support and structural integrity [35]. Despite increasing evidence from other organs indicating the ECM serving not only a structural role but also actively participating in disease initiation and progression [36–38], the biological functions of the ECM in IBD have been largely overlooked [5]. This is surprising, as in IBD, an altered amount and composition of the ECM are present, mainly due to activation of HIMF exposed to the chronic inflammatory mucosal milieu [9]. As a major component of the interstitial and basement membrane ECM, collagens exert both structural and bioregulatory roles in the tissue, including cell adhesion, proliferation, migration and differentiation [6,7]. Alteration of the collagen amount, type of collagen present or spatial relationship with immune cells could fuel pathological conditions, especially fibrosis, which is characterized by the remodeling and excessive deposition of ECM within tissues [6,7]. Among the multiple functions of collagens, cell adhesion mediated via collagen adhesion receptors has been found of importance in fibrogenesis [6,39], lending conceptual support to cell with ECM interactions being critical in IBD pathogenesis.
Our results show an increased adhesiveness of T cells to IBD HIMF-derived ECM, which is neither due to different proliferative capacities of HIMF, nor the produced total amount of ECM, but rather due to distinct ECM composition. Importantly this increased adhesion was observed when not only the T cell line MOLT4, but also when PBT and LPT were used. HIMF exposed to TNF, IFN-γ, IL-1β and TGF-α1 change both the quantity and quality of the ECM they secrete [9], but its functional relevance is unknown. NL HIMF exposed to pro-inflammatory TNF and IL-1β produced an ECM more adhesive for T cells, and to a degree comparable to spontaneously increased baseline adhesion of ECM secreted by IBD HIMF. This suggests that inflammation is a driver altering ECM functional properties. Surprisingly, IFN-γ and TGF-β1 did not alter or decreased ECM adhesiveness, respectively. Since TGF-β1 is a major activator of HIMF to produce ECM [40] this indicates that not only the amount, but also the composition of the ECM leads to functional abnormalities. While the implications of this finding for intestinal fibrosis need to be determined one may speculate that the increased amount of ECM per se may not drive further T cell adhesion, but that TGF-β1 may rather mediate the opposite.
Our systematic investigation into HIMF derived matrisome identified collagen VI as one matrisome component associated with the pattern of T cell to ECM adhesion observed in the 2D ECM scaffolds. While this is likely not the only ECM molecule responsible for an increased adhesion of T cells to the gut ECM, it piqued our interest given its expression pattern in relation to the adhesive properties of the ECM. In health this molecule is found mainly in the gut submucosa [41,42], but it is also present in the muscularis propria, secreted by smooth muscle cells [12]. We confirmed these observations and additionally found that collagen VI is increased in the IBD mucosa/submucosa and muscularis propria compared to non-IBD controls, which is consistent with previous studies [43,44]. While not providing a direct proof, CD3 positive immune cells (T cells) were found in close proximity to collagen VI in the gut tissue, which is suggestive of a direct physical and biological interaction. HIMF are the major source of collagen VI in IBD as shown by IHC and interrogating two publicly available single cell RNA sequencing datasets [29,30]. The highest gene expression was detected in myofibroblasts and inflammatory fibroblasts, both α-SMA positive, which is compatible with our in vitro HIMF cultures. HIMF retained their higher expression of collagen VI in vitro, validating our proteomics results.
Collagen VI is an important component of the interstitial ECM in virtually all tissues [45]. Mutations in the essential collagen VI coding genes lead to serious clinical disorders, including Bethlem myopathy, Ullrich congenital muscular dystrophy and metabolic diseases [45]. Fibroblasts are the best characterized source of collagen VI [46], which is consistent with our study findings. Collagen VI production is regulated by multiple factors, including cytokines, such as IL-1β, IL-4, IL-10, IL-36, granulocyte-macrophage colony-stimulating factor (GM-CSF) and TGF- β1 with TGF-β1 being considered the main regulator [43,46–48]. Interestingly our proteomics results did not show an upregulation of collagen VI by TGF-β1, which supports the prior observation that collagen VI regulation may be independent of other ECM components, such as collagen I, III or fibronectin [46]. Future work will determine, if and how combinations of cytokines and growth factors drive collagen VI expression in HIMF. Biological functions of collagen VI have been identified including cell with ECM linkage [49], interaction with and binding to other ECM molecules [41] and activating signaling pathways, such as phosphoinositide 3-kinase [49].
The functional role of collagen VI in IBD, especially its interaction with T cells, has not been explored. A key finding in our study is that collagen VI is one ECM molecule responsible for a dose dependent adhesiveness of T cells to IBD ECM. Upon its knockdown in HIMF, the produced ECM reduced T cell adhesion by ~50%. While approximately half of the adhesive capacity of the ECM was retained, we still feel this is relevant given that fibronectin and tenascin-C were the most abundantly expressed ECM molecules. The reduced adhesion upon collagen VI knockdown might be explained by the lower amount of collagen VI or its changed ability to bind to other ECM components and modulate their properties [49], e.g. the meshwork of fibronectin created by cultured fibroblasts is altered by COL6A1 knock down [50]. It has to be noted that collagen VI is not the only ECM molecule with the capacity to bind ECM as for instance shown in our study for collagens I and IV. Rather, our study identified collagen VI as one important contributor to this phenomenon. Collagen VI has been initially described as a trimer composed of α1-3 chains, but recently three additional chains (α4-6) that may replace α3 have been reported [51]. Collagen chains α5&6 may have important biological functions in Duchenne muscular dystrophy [52]. To our knowledge no study on the adhesive function of the α4-6 chains to immune cells is available and those chains were absent in our HIMF proteomics dataset. This could mean that their expression levels were too low to be detectable or those chains were absent.
Of special interest is the more recently described C-terminus of the released C5 domain of type VI collagen α3 chain, termed PRO-C6 or endotrophin [7,53–55]. Endotrophin has been shown to drive fibrosis, inflammation and insulin resistance [7]. These functions may be highly relevant for IBD pathogenesis. One may speculate that endotrophin is elevated in IBD and could play a role beyond increase in immune cell binding. Currently ongoing studies attempt to unravel this phenomenon [7,53,56]. Aside T-cells, other immune cells adhere to the ECM with important functional implications. This includes roles of the ECM in neutrophil chemotaxis, degranulation and phagocytosis [57] or the inflammatory response of macrophages [58]. Ongoing work by our group is assessing the effect of collagen VI on those cell types.
Our results indicate that the T cell with ECM adhesion is mediated by RGD binding integrins. This is relevant as triple-helical collagen VI, among other collagens, such as collagens I, IV and V [59–62], also contains this motif [28]. Of note, collagen VI contains 11 RGD motifs, with three in the α1 chain, three in the α2 chain and five in the α3 chain [63]. Based on our data we cannot conclude which α-chains are responsible for the adhesive effect of T-cells to the collagen VI. Specific integrins, such as αv and β1, contain RGD binding pockets [64]. Several different anti-integrin therapies, mainly targeting integrin α4β7, have been approved for IBD [65–67]. We identified integrin αvβ1, but not integrin integrin α4β7, as a key integrin in T cell with ECM adhesion in IBD, including adhesion to collagen VI. This could be observed not only on HIMF derived 2D matrix but also on collagen VI coated surfaces and, importantly, directly on the exact same ECM environment T cells encounter in the intestinal wall, as modeled through decellularized intestinal 3D ECM. Further investigations need to show, which integrins and if integrin αvβ1 mediates T cell binding to collagens I and IV. In addition, it would be relevant to determine which collagen VI chain lends its RGD motif for integrin binding. This supports the notion that, unlike anti-α4 or anti-α4α7 biologics which prevent T cell homing in the gut [68], inhibition of integrin αvβ1 could prevent the adhesion of T cells to the intestinal interstitial ECM and promote the recirculation of T cells out of the gut. This concept, however, would need to be confirmed in further studies. A combination of both anti-α4β7 and anti-αvβ1 therapies could be considered in patients who exhibit poor response to single anti-integrin therapy by further reducing T cell number and alleviating inflammation locally in the gut.
To this end it is important to note that T cells are crucial in mediating the inflammatory response, but their functions vary depending on their subtype [69,70]. Interestingly, low expression of integrin αv was noted in healthy intestinal T regulatory cells, a T cell type important for immunological self-tolerance and negative regulation of the inflammatory response48, and integrin αv remained low in T regulatory cells in uninflamed and inflamed UC. To the contrary, integrin αv was expressed in the highest proportion and levels in CD4+ PD1+ and CD8+ IL17+ T cells in UC, but not in the healthy intestinal mucosa, and both of these T cell subtypes are considered pro-inflammatory [71,72]. One may speculate that this high expression of integrin αv leads to increased adherence to HIMF produced ECM scaffolds and ultimately the 3D intestinal ECM environment and hence promotes inflammation. While the scope of this project is focused on the ECM and its composition mediating T cell adhesion, ongoing studies are exploring, (1) if integrin modulation on T cells or deletion of collagen VI in vitro and in vivo alter intestinal inflammation, (2) if the adhesiveness of distinct T cell subtypes to collagen VI as well as their integrin expression profile is different when tested on 2D or 3D ECM scaffolds and (3) if the increased adhesiveness of the IBD ECM for T cells also applies to other cell types, such as B cells or myeloid cell types.
Of direct clinical relevance, elevated gene expression of collagen VI in ileal mucosal biopsies of uncomplicated pediatric CD patients close to diagnosis [32] was associated with future development of complications. While the predictive ability of collagen VI gene expression was not strong enough to be used as a clinically relevant test, this data supports the biologic relevance of collagen VI in IBD. A comparable dataset in adult CD patients is not available at this time, but one may speculate that, given the often aggressive nature of pediatric CD compared to adult CD, a different matrisome composition may be found. This may also have direct impact on disease progression. A multicenter study evaluating this question is in progress. Data on a functional link between collagen VI and IBD is still limited and restricted to measuring collagen VI or its split products in the circulation. A previous study reported elevated expression of the collagen VI α3 chain in the serum of IBD patients compared to controls [73]. The serum levels of the type VI collagen α3 chain fragment C6Ma3 and endotrophin were associated with clinical and endoscopic disease activity in IBD [53]. Collagen VI or its fragments have also been used as a biomarker for fibrosis of various organs [7]. For example, TGF-β1 induced the production of collagen VI in primary human lung fibroblasts [54]. Endotrophin levels in the plasma were associated with poor prognosis in acute-on-chronic liver failure, with fibrosis progression [55].
Our study, however, not only thoroughly evaluated the levels of collagen VI in the intestinal wall and its cellular sources, but also its functional relevance. This, again, does not mean that collagen VI is the only matrisome component important for IBD inflammation and prognosis, but given that three collagen VI genes appeared on our unbiased analysis as relevant, suggests that it may be one important part of the ECM.
Several questions for future research programs remain. First, collagen VI or its split products may provide outside-in signaling cues to immune cells influencing their intracellular programs in relation to inflammation. Second, T cells excessively adhered to Collagen VI could also stimulate the production and deposition of Collagen VI by HIMFs, forming a positive forward feedback loop promoting the progression of IBD. Third, although the role of collagen VI in intestinal fibrosis has not been explored, the enhanced adhesiveness of T cells to collagen VI might be one of the potential mechanisms for fibrogenesis in the gut. Blockage of T cells adhesion to collagen VI would provide a new target in the future therapy for IBD-related intestinal fibrosis. Finally, since the clinical cohort is of pediatric patients and the event rate is relatively small, the finding of collagen VI association with complicated future CD courses should be extended to adult patients, and also evaluated in UC.
Supplementary Material
Summary Box.
What is already known about this subject?
The extracellular matrix (ECM) in inflammatory bowel diseases (IBD) is altered and may influence response to anti-inflammatory therapy. Immune cell adhesion to the ECM has been reported in other chronic inflammatory diseases.
What are the new findings?
Human intestinal myofibroblast (HIMF) derived ECM from IBD patients has a higher adhesiveness for T cells compared to ECM from non-IBD HIMF. This enhanced adhesiveness is modulated by different cytokines including TNF and TGF-β1 and integrin αvβ1. Collagen VI is a key ECM component mediating enhanced adhesiveness and increased Collagen VI gene expression in intestinal biopsies is linked with future stricturing in pediatric Crohn’s disease.
How might it impact on clinical practice in the foreseeable future?
Targeting T cell adhesion to Collagen VI or integrin αvβ1 could be a potential anti-inflammatory therapy in IBD.
Acknowledgements
We would like to thank Dr. Judith Drazba, Dr. John Peterson, Andrelie Branicky, and Apryl Helmick from the LRI Microscopy and Image Core, for help in microscopy and image analysis. We acknowledge the support of the Departments of Colorectal Surgery and Pathology of the Cleveland Clinic. Tissue samples were provided by the Human Tissue Procurement Facility of the Cleveland Clinic through the services of the Biorepository Core funded by 2 P30 DK097948-06.
-Furthermore, we would like to thank the RISK investigator group for contribution of the biosamples:
-Thomas D. Walters: Hospital for Sick Children, University of Toronto, Toronto, ON, Canada;
-Robert N. Baldassano: The Children’s Hospital of Philadelphia, Philadelphia, PA, USA;
-Joshua D Noe: Medical College of Wisconsin, Milwaukee, WI. USA;
-Joel Rosh: Goryeb Children’s Hospital/Atlantic Health, Morristown, NJ, USA;
-James Markowitz: Cohen Children’s Medical Center of New York, New Hyde Park, NY, USA;
-Jennifer L. Dotson: Nationwide Children’s Hospital, Columbus, OH, USA;
-David R. Mack: 11Children’s Hospital of Eastern Ontario, University of Ottawa, Ottawa, ON, Canada;
-Richard Kellermayer: Texas Children’s Hospital, Baylor College School of Medicine, Houston, TX, USA;
-Anne M. Griffiths: Hospital for Sick Children, University of Toronto, Toronto, ON, Canada;
-Melvin B. Heyman: University of California San Francisco, San Francisco, CA, USA;
-Susan S. Baker: University at Buffalo, Buffalo, NY, USA;
-Dedrick Moulton: Monroe Carell Jr Children’s Hospital, Nashville, TN;
-Ashish S. Patel: UT Southwestern Medical Center at Dallas, Dallas, TX, USA;
-Ajay S. Gulati: University of North Carolina, Chapel Hill, NC, USA;
-Steven J Steiner: Riley Children’s Hospital, Indianapolis, IN, USA;
-Neal LeLeiko: Hasbro Children’s Hospital, Providence, RI, USA;
-Anthony Otley: IWK Health Centre, Halifax, NS, Canada;
-Maria Oliva-Hemker: John Hopkins University, Baltimore, MD, USA;
-David Ziring: Cedars-Sinai Medical Center, Los Angeles, CA, USA;
-Ranjana Gokhale: University of Chicago Comer Children’s Hospital, Chicago, IL, USA;
-Sandra Kim: Children’s Hospital of Pittsburgh of UPMC, Pittsburgh, PA, USA;
-Stephen L. Guthery: University of Utah and Intermountain Primary Children’s Hospital, Salt Lake City, UT, USA;
-Stanley A Cohen: Children’s Center for Digestive Health Medicine, Atlanta, GA, USA;
-Scott Snapper: Children’s Hospital - Boston, Boston, MA, USA;
-Michael Stephens: Mayo clinic, Rochester, MN, USA;
-Marla Dubinsky: Mount Sinai Hospital New York, NY, USA;
-Jeffrey S. Hyams: Connecticut Children’s Medical Center, Hartford, CT, USA.
Funding
This work was supported by the Helmsley Charitable Trust through the Stenosis Therapy and Anti-Fibrotic Research (STAR) Consortium (No. 3081 to F.R.), the Crohn’s and Colitis Foundation (No. 569125 to F.R.), the National Institute of Health (NIDDK R01DK123233 to F.R.), the Cleveland Clinic through the LabCo program to F.R. and the National Institute of Health (NIDDK 2 P30 DK097948) to C.F. the National Science Foundation of China (82200573) to S.L., the National Science Foundation of China (81970483, 82170537 and 82222010) to R.M.
Declaration of Competing Interest
F.R. is consultant to Agomab, Allergan, AbbVie, Boehringer-Ingelheim, Celgene, Cowen, Genentech, Gilead, Gossamer, Guidepoint, Helmsley, Index Pharma, Jannsen, Koutif, Metacrine, Morphic, Pfizer, Pliant, Prometheus Biosciences, Receptos, RedX, Roche, Samsung, Takeda, Techlab, Thetis, UCB, 89Bio. C.F. received speaker fees from UCB, Genentech, Sandoz, Janssen and he is consultant for Athos Therapeutics, Inc.
Abbreviations:
- 2D ECM
2 dimensional extracellular matrix
- 3D ECM
3 dimensional extracellular matrix
- CD
Crohn’s disease
- ddH2O
Double-distilled water
- DMEM
Dulbecco’s modified Eagle medium
- DMSO
Dimethyl sulfoxide
- ECM
Extracellular matrix
- EDTA
Ethylenediaminetetraacetic acid
- FBS
Fetal bovine serum
- FCS
Fetal calf serum
- FDR
False discovery rate
- FFPE
Formalin-fixed, paraffin-embedded
- HBSS
Hank’s balanced salt solution
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HIMF
Human intestinal myofibroblast
- HPLC
High performance liquid chromatography
- IBD
Inflammatory bowel diseases
- IFN-γ
Interferon-γ
- IHC
Immunohistochemistry
- IL-1β
Interleukin-1β
- IL-13
Interleukin-13
- LC-MS
Liquid chromatrography mass spectrometry
- LFQ
Label-free quantitation
- NL
Normal control
- OCT
Optical coherence tomography compound
- PBMC
Peripheral blood mononuclear cell
- PBS
Phosphate-buffered saline
- PBT
Peripheral blood T cells
- PCA
Principle component analysis
- PMA
Phorbol 12-myristate 13-acetate
- RPMI 1640
Roswell Park Memorial Institute 1640 medium
- PSF
2500U potassium penicillin, 2500μg streptomycin sulfate, 625μg Amphotericin B
- qPCR
Quantitative polymerase chain reaction
- RGD
Arg-Gly-Asp
- RGE
Arg-Gly-Glu
- SDC
Sodium deoxycholate
- SEM
Standard error of mean
- siRNA
Small interfering RNA
- TFA
Trifluoroacetic acid
- TGF-β1
Transforming growth factor-β1
- Th
T helper
- TNF
Tumor necrosis factor
- Treg
T regulator
- UC
Ulcerative colitis
Footnotes
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.matbio.2022.09.001.
CRediT authorship contribution statement
Si-Nan Lin: Visualization, Data curation, Data curation, Formal analysis, Data curation, Writing – review & editing. Alessandro Musso: Visualization, Data curation, Data curation, Formal analysis, Data curation. Jie Wang: Visualization, Data curation, Formal analysis. Pranab K. Mukherjee: Data curation, Formal analysis. Ren Mao: Investigation. Jiannan Li: Investigation. Shuai Zhao: Investigation. Michael Elias: Investigation. Yael Haberman: Data curation. Lee A. Denson: Data curation. Subra Kugathasan: Investigation. Min-Hu Chen: Investigation. Doug Czarnecki: Investigation. Dina Dejanovic: Investigation. Hongnga T. Le: Investigation. Jyotsna Chandra: Investigation. Jeremy Lipman: Investigation. Scott R. Steele: Investigation. Quang Tam Nguyen: Investigation. Claudio Fiocchi: Visualization, Investigation, Funding acquisition, Writing – review & editing. Florian Rieder: Visualization, Writing – review & editing, Funding acquisition, Writing – review & editing.
Data Availability
Data will be made available on request.
References
- [1].Guan Q, A comprehensive review and update on the pathogenesis of inflammatory bowel disease, J. Immunol. Res 2019(2019) 7247238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rieder F, Fiocchi C, Rogler G, Mechanisms, management, and treatment of fibrosis in patients with inflammatory bowel diseases, Gastroenterology 152 (2) (2017) 340–350 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mortensen JH, Lindholm M, Langholm LL, Kjeldsen J, Bay-Jensen AC, Karsdal MA, Manon-Jensen T, The intestinal tissue homeostasis - the role of extracellular matrix remodeling in inflammatory bowel disease, Expert Rev. Gastroenterol. Hepatol 13 (10) (2019) 977–993. [DOI] [PubMed] [Google Scholar]
- [4].Shimshoni E, Yablecovitch D, Baram L, Dotan I, Sagi I, ECM remodelling in IBD: innocent bystander or partner in crime? The emerging role of extracellular molecular events in sustaining intestinal inflammation, Gut 64 (3) (2015) 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Petrey AC, de la Motte CA, The extracellular matrix in IBD: a dynamic mediator of inflammation, Curr. Opin. Gastroenterol 33 (4) (2017) 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Karsdal MA, Nielsen SH, Leeming DJ, Langholm LL, Nielsen MJ, Manon-Jensen T, Siebuhr A, Gudmann NS, Ronnow S, Sand JM, Daniels SJ, Mortensen JH, Schuppan D, The good and the bad collagens of fibrosis - their role in signaling and organ function, Adv. Drug. Deliv. Rev 121 (2017) 43–56. [DOI] [PubMed] [Google Scholar]
- [7].Williams L, Layton T, Yang N, Feldmann M, Nanchahal J, Collagen VI as a driver and disease biomarker in human fibrosis, FEBS J. (2021). [DOI] [PubMed] [Google Scholar]
- [8].Neurath MF, Targeting immune cell circuits and trafficking in inflammatory bowel disease, Nat. Immunol 20 (8) (2019) 970–979. [DOI] [PubMed] [Google Scholar]
- [9].Li J, Mao R, Kurada S, Wang J, Lin S, Chandra J, Rieder F, Pathogenesis of fibrostenosing Crohn’s disease, Transl. Res 209 (2019) 39–54. [DOI] [PubMed] [Google Scholar]
- [10].Fernandes NRJ, Reilly NS, Schrock DC, Hocking DC, Oakes PW, Fowell DJ, CD4(+) T cell interstitial migration controlled by fibronectin in the inflamed skin, Front. Immunol 11 (2020) 1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Privitera G, Pugliese D, Lopetuso LR, Scaldaferri F, Neri M, Guidi L, Gasbarrini A, Armuzzi A, Novel trends with biologics in inflammatory bowel disease: sequential and combined approaches, Therap. Adv. Gastroenterol 14 (2021) 17562848211006669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mao R, Doyon G, Gordon IO, Li J, Lin S, Wang J, Le THN, Elias M, Kurada S, Southern B, Olman M, Chen M, Zhao S, Dejanovic D, Chandra J, Mukherjee PK, West G, Van Wagoner DR, Fiocchi C, Rieder F, Activated intestinal muscle cells promote preadipocyte migration: a novel mechanism for creeping fat formation in Crohn’s disease, Gut (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhao S, Dejanovic D, Yao P, Bhilocha S, Sadler T, Schirbel A, West G, Doyon G, Lopez R, Mao R, Kurada S, El Ouali S, Grassl G, Fox PL, Cruise M, Worthley DL, de la Motte C, Fiocchi C, Rieder F, Selective deletion of MyD88 signaling in alpha-SMA positive cells ameliorates experimental intestinal fibrosis via post-transcriptional regulation, Mucosal. Immunol (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rieder F, Georgieva M, Schirbel A, Artinger M, Zugner A, Blank M, Brenmoehl J, Scholmerich J, Rogler G, Prostaglandin E2 inhibits migration of colonic lamina propria fibroblasts, Inflamm. Bowel Dis 16 (9) (2010) 1505–1513. [DOI] [PubMed] [Google Scholar]
- [15].Musso A, Condon TP, West GA, De La Motte C, Strong SA, Levine AD, Bennett CF, Fiocchi C, Regulation of ICAM-1-mediated fibroblast-T cell reciprocal interaction: implications for modulation of gut inflammation, Gastroenterology 117 (3) (1999) 546–556. [DOI] [PubMed] [Google Scholar]
- [16].Giuffrida P, Curti M, Al-Akkad W, Biel C, Crowley C, Frenguelli L, Telese A, Hall A, Tamburrino D, Spoletini G, Fusai G, Tinozzi FP, Pietrabissa A, Corazza GR, De Coppi P, Pinzani M, Di Sabatino A, Rombouts K, Mazza G, Decellularized human gut as a natural 3D platform for research in intestinal fibrosis, Inflamm. Bowel Dis 25 (11) (2019) 1740–1750. [DOI] [PubMed] [Google Scholar]
- [17].Shao X, Taha IN, Clauser KR, Gao YT, Naba A, MatrisomeDB: the ECM-protein knowledge database, Nucleic Acids Res. 48 (D1) (2020) D1136–D1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Han T, Minowada J, A unique ‘leukaemic’ T lymphoid cell line: absence of stimulating effect in mixed lymphocyte reaction. Lack of MLR-S in leukaemic T lymphoid cells, Clin. Exp. Immunol 15 (4) (1973) 535–541. [PMC free article] [PubMed] [Google Scholar]
- [19].Schmitt H, Billmeier U, Dieterich W, Rath T, Sonnewald S, Reid S, Hirschmann S, Hildner K, Waldner MJ, Mudter J, Hartmann A, Grutzmann R, Neufert C, Munster T, Neurath MF, Atreya R, Expansion of IL-23 receptor bearing TNFR2+ T cells is associated with molecular resistance to anti-TNF therapy in Crohn’s disease, Gut 68 (5) (2019) 814–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G, Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies, Immunity 10 (3) (1999) 387–398. [DOI] [PubMed] [Google Scholar]
- [21].Langer V, Vivi E, Regensburger D, Winkler TH, Waldner MJ, Rath T, Schmid B, Skottke L, Lee S, Jeon NL, Wohlfahrt T, Kramer V, Tripal P, Schumann M, Kersting S, Handtrack C, Geppert CI, Suchowski K, Adams RH, Becker C, Ramming A, Naschberger E, Britzen-Laurent N, Sturzl M, IFN-gamma drives inflammatory bowel disease pathogenesis through VE-cadherin-directed vascular barrier disruption, J. Clin. Invest 129 (11) (2019) 4691–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Peterson LW, Artis D, Intestinal epithelial cells: regulators of barrier function and immune homeostasis, Nat. Rev. Immunol 14 (3) (2014) 141–153. [DOI] [PubMed] [Google Scholar]
- [23].Mao L, Kitani A, Strober W, Fuss IJ, The role of NLRP3 and IL-1beta in the pathogenesis of inflammatory bowel disease, Front. Immunol 9 (2018) 2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ligumsky M, Simon PL, Karmeli F, Rachmilewitz D, Role of interleukin 1 in inflammatory bowel disease–enhanced-production during active disease, Gut 31 (6) (1990) 686–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yun SM, Kim SH, Kim EH, The molecular mechanism of transforming growth factor-beta signaling for intestinal fibrosis: a mini-review, Front. Pharmacol 10 (2019) 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Friedrich M, Pohin M, Powrie F, Cytokine networks in the pathophysiology of inflammatory bowel disease, Immunity 50(4) (2019) 992–1006. [DOI] [PubMed] [Google Scholar]
- [27].Barczyk M, Carracedo S, Gullberg D, Integrins, Cell Tissue Res. 339 (1) (2010) 269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pfaff M, Aumailley M, Specks U, Knolle J, Zerwes HG, Timpl R, Integrin and Arg-Gly-asp dependence of cell adhesion to the native and unfolded triple helix of collagen type VI, Exp. Cell. Res 206 (1) (1993) 167–176. [DOI] [PubMed] [Google Scholar]
- [29].Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, Herbst RH, Rogel N, Slyper M, Waldman J, Sud M, Andrews E, Velonias G, Haber AL, Jagadeesh K, Vickovic S, Yao J, Stevens C, Dionne D, Nguyen LT, Villani AC, Hofree M, Creasey EA, Huang H, Rozenblatt-Rosen O, Garber JJ, Khalili H, Desch AN, Daly MJ, Ananthakrishnan AN, Shalek AK, Xavier RJ, Regev A, Intra- and inter-cellular rewiring of the human colon during ulcerative colitis, Cell 178 (3) (2019) 714–730 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kinchen J, Chen HH, Parikh K, Antanaviciute A, Jagielowicz M, Fawkner-Corbett D, Ashley N, Cubitt L, Mellado-Gomez E, Attar M, Sharma E, Wills Q, Bowden R, Richter FC, Ahern D, Puri KD, Henault J, Gervais F, Koohy H, Simmons A, Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease, Cell 175 (2) (2018) 372–386 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kugathasan S, Denson LA, Walters TD, Kim MO, Marigorta UM, Schirmer M, Mondal K, Liu C, Griffiths A, Noe JD, Crandall WV, Snapper S, Rabizadeh S, Rosh JR, Shapiro JM, Guthery S, Mack DR, Kellermayer R, Kappelman MD, Steiner S, Moulton DE, Keljo D, Cohen S, Oliva-Hemker M, Heyman MB, Otley AR, Baker SS, Evans JS, Kirschner BS, Patel AS, Ziring D, Trapnell BC, Sylvester FA, Stephens MC, Baldassano RN, Markowitz JF, Cho J, Xavier RJ, Huttenhower C, Aronow BJ, Gibson G, Hyams JS, Dubinsky MC, Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: a multicentre inception cohort study, Lancet 389 (10080) (2017) 1710–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Haberman Y, Minar P, Karns R, Dexheimer PJ, Ghandikota S, Tegge S, Shapiro D, Shuler B, Venkateswaran S, Braun T, Ta A, Walters TD, Baldassano RN, Noe JD, Rosh J, Markowitz J, Dotson JL, Mack DR, Kellermayer R, Griffiths AM, Heyman MB, Baker SS, Moulton D, Patel AS, Gulati AS, Steiner SJ, LeLeiko N, Otley A, Oliva-Hemker M, Ziring D, Gokhale R, Kim S, Guthery SL, Cohen SA, Snapper S, Aronow BJ, Stephens M, Gibson G, Dillman JR, Dubinsky M, Hyams JS, Kugathasan S, Jegga AG, Denson LA, Mucosal inflammatory and wound healing gene programs reveal targets for stricturing behavior in pediatric Crohn’s disease, J. Crohns Colitis (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zundler S, Becker E, Schulze LL, Neurath MF, Immune cell trafficking and retention in inflammatory bowel disease: mechanistic insights and therapeutic advances, Gut 68 (9) (2019) 1688–1700. [DOI] [PubMed] [Google Scholar]
- [34].Kanai T, Nemoto Y, Tomita T, Totsuka T, Watanabe M, Hibi T, Persistent retention of colitogenic CD4+ memory T cells causes inflammatory bowel diseases to become intractable, Inflamm. Bowel Dis 15 (6) (2009) 926–934. [DOI] [PubMed] [Google Scholar]
- [35].Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK, Extracellular matrix structure, Adv. Drug. Deliv. Rev 97 (2016) 4–27. [DOI] [PubMed] [Google Scholar]
- [36].Bonnans C, Chou J, Werb Z, Remodelling the extracellular matrix in development and disease, Nat. Rev. Mol. Cell Biol 15 (12) (2014) 786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Koelink PJ, Overbeek SA, Braber S, Morgan ME, Henricks PA, Abdul Roda M, Verspaget HW, Wolfkamp SC, te Velde AA, Jones CW, Jackson PL, Blalock JE, Sparidans RW, Kruijtzer JA, Garssen J, Folkerts G, Kraneveld AD, Collagen degradation and neutrophilic infiltration: a vicious circle in inflammatory bowel disease, Gut 63 (4) (2014) 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hatoum OA, Heidemann J, Binion DG, The intestinal microvasculature as a therapeutic target in inflammatory bowel disease, Ann. N Y Acad. Sci 1072 (2006) 78–97. [DOI] [PubMed] [Google Scholar]
- [39].Coelho NM, McCulloch CA, Contribution of collagen adhesion receptors to tissue fibrosis, Cell Tissue Res. 365 (3) (2016) 521–538. [DOI] [PubMed] [Google Scholar]
- [40].Meng XM, Nikolic-Paterson DJ, Lan HY, TGF-beta: the master regulator of fibrosis, Nat. Rev. Nephrol 12 (6) (2016) 325–338. [DOI] [PubMed] [Google Scholar]
- [41].Gatseva A, Sin YY, Brezzo G, Van Agtmael T, Basement membrane collagens and disease mechanisms, Essays Biochem. 63 (3) (2019) 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ji Y, Zhou J, Sun T, Tang K, Xiong Z, Ren Z, Yao S, Chen K, Yang F, Zhu F, Guo X, Diverse preparation methods for small intestinal submucosa (SIS): Decellularization, components, and structure, J. Biomed. Mater. Res. A 107 (3) (2019) 689–697. [DOI] [PubMed] [Google Scholar]
- [43].Scheibe K, Kersten C, Schmied A, Vieth M, Primbs T, Carle B, Knieling F, Claussen J, Klimowicz AC, Zheng J, Baum P, Meyer S, Schurmann S, Friedrich O, Waldner MJ, Rath T, Wirtz S, Kollias G, Ekici AB, Atreya R, Raymond EL, Mbow ML, Neurath MF, Neufert C, Inhibiting interleukin 36 receptor signaling reduces fibrosis in mice with chronic intestinal inflammation, Gastroenterology 156 (4) (2019) 1082–1097 e11. [DOI] [PubMed] [Google Scholar]
- [44].Moriggi M, Pastorelli L, Torretta E, Tontini GE, Capitanio D, Bogetto SF, Vecchi M, Gelfi C, Contribution of extracellular matrix and signal mechanotransduction to epithelial cell damage in inflammatory bowel disease patients: a proteomic study, Proteomics 17 (2017) 23–24. [DOI] [PubMed] [Google Scholar]
- [45].Lamande SR, Bateman JF, Collagen VI disorders: Insights on form and function in the extracellular matrix and beyond, Matrix Biol. 71-72 (2018) 348–367. [DOI] [PubMed] [Google Scholar]
- [46].Williams L, Layton T, Yang N, Feldmann M, Nanchahal J, Collagen VI as a driver and disease biomarker in human fibrosis, FEBS J. 289 (13) (2022) 3603–3629. [DOI] [PubMed] [Google Scholar]
- [47].Fernandez M, Minguell JJ, G-CSF regulates the expression of mRNA for collagen type VI and collagen VI production in human bone marrow stromal cells, Hematology 2 (3) (1997)219–227. [DOI] [PubMed] [Google Scholar]
- [48].Jarisch A, Krieg T, Hunzelmann N, Regulation of collagen expression by interleukin-1 beta is dependent on donor age, Acta Derm. Venereol 76 (4) (1996) 287–290. [DOI] [PubMed] [Google Scholar]
- [49].Cescon M, Gattazzo F, Chen P, Bonaldo P, Collagen VI at a glance, J. Cell Sci 128 (19) (2015) 3525–3531. [DOI] [PubMed] [Google Scholar]
- [50].Sabatelli P, Bonaldo P, Lattanzi G, Braghetta P, Bergamin N, Capanni C, Mattioli E, Columbaro M, Ognibene A, Pepe G, Bertini E, Merlini L, Maraldi NM, Squarzoni S, Collagen VI deficiency affects the organization of fibronectin in the extracellular matrix of cultured fibroblasts, Matrix Biol. 20 (7) (2001) 475–486. [DOI] [PubMed] [Google Scholar]
- [51].Fitzgerald J, Rich C, Zhou FH, Hansen U, Three novel collagen VI chains, alpha4(VI), alpha5(VI), and alpha6(VI), J. Biol. Chem. 283 (29) (2008) 20170–20180. [DOI] [PubMed] [Google Scholar]
- [52].Sabatelli P, Gualandi F, Gara SK, Grumati P, Zamparelli A, Martoni E, Pellegrini C, Merlini L, Ferlini A, Bonaldo P, Maraldi NM, Paulsson M, Squarzoni S, Wagener R, Expression of collagen VI alpha5 and alpha6 chains in human muscle and in Duchenne muscular dystrophy-related muscle fibrosis, Matrix Biol. 31 (3) (2012) 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lindholm M, Godskesen LE, Manon-Jensen T, Kjeldsen J, Krag A, Karsdal MA, Mortensen JH, Endotrophin and C6Ma3, serological biomarkers of type VI collagen remodelling, reflect endoscopic and clinical disease activity in IBD, Sci. Rep 11 (1) (2021) 14713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ronnow SR, Dabbagh RQ, Genovese F, Nanthakumar CB, Barrett VJ, Good RB, Brockbank S, Cruwys S, Jessen H, Sorensen GL, Karsdal MA, Leeming DJ, Sand JMB, Prolonged Scar-in-a-Jar: an in vitro screening tool for anti-fibrotic therapies using biomarkers of extracellular matrix synthesis, Respir. Res 21 (1) (2020)108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kerbert AJC, Gupta S, Alabsawy E, Dobler I, Lonsmann I, Hall A, Nielsen SH, Nielsen MJ, Gronbaek H, Amoros A, Yeung D, Macnaughtan J, Mookerjee RP, Macdonald S, Andreola F, Moreau R, Arroyo V, Angeli P, Leeming DJ, Treem W, Karsdal MA, Jalan R, Biomarkers of extracellular matrix formation are associated with acute-on-chronic liver failure, JHEP Rep. 3 (6) (2021) 100355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Karsdal MA, Henriksen K, Genovese F, Leeming DJ, Nielsen MJ, Riis BJ, Christiansen C, Byrjalsen I, Schuppan D, Serum endotrophin identifies optimal responders to PPARgamma agonists in type 2 diabetes, Diabetologia 60 (1) (2017) 50–59. [DOI] [PubMed] [Google Scholar]
- [57].Borgquist JD, Quinn MT, Swain SD, Adhesion to extracellular matrix proteins modulates bovine neutrophil responses to inflammatory mediators, J. Leukoc. Biol 71 (5) (2002) 764–774. [PubMed] [Google Scholar]
- [58].Meli VS, Atcha H, Veerasubramanian PK, Nagalla RR, Luu TU, Chen EY, Guerrero-Juarez CF, Yamaga K, Pandori W, Hsieh JY, Downing TL, Fruman DA, Lodoen MB, Plikus MV, Wang W, Liu WF, YAP-mediated mechanotransduction tunes the macrophage inflammatory response, Sci. Adv 6 (49) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Taubenberger AV, Woodruff MA, Bai H, Muller DJ, Hutmacher DW, The effect of unlocking RGD-motifs in collagen I on pre-osteoblast adhesion and differentiation, Biomaterials 31 (10) (2010) 2827–2835. [DOI] [PubMed] [Google Scholar]
- [60].Pedchenko V, Zent R, Hudson BG, Alpha(v)beta3 and alpha(v)beta5 integrins bind both the proximal RGD site and non-RGD motifs within noncollagenous (NC1) domain of the alpha3 chain of type IV collagen: implication for the mechanism of endothelia cell adhesion, J. Biol. Chem 279 (4) (2004) 2772–2780. [DOI] [PubMed] [Google Scholar]
- [61].Ruggiero F, Champliaud MF, Garrone R, Aumailley M, Interactions between cells and collagen V molecules or single chains involve distinct mechanisms, Exp. Cell. Res 210 (2) (1994)215–223. [DOI] [PubMed] [Google Scholar]
- [62].Marcelino J, McDevitt CA, Attachment of articular cartilage chondrocytes to the tissue form of type VI collagen, Biochim. Biophys. Acta 1249 (2) (1995) 180–188. [DOI] [PubMed] [Google Scholar]
- [63].Chu ML, Conway D, Pan TC, Baldwin C, Mann K, Deutzmann R, Timpl R, Amino acid sequence of the triple-helical domain of human collagen type VI, J. Biol. Chem 263 (35) (1988) 18601–18606. [PubMed] [Google Scholar]
- [64].Moreno-Layseca P, Icha J, Hamidi H, Ivaska J, Integrin trafficking in cells and tissues, Nat. Cell Biol 21 (2) (2019) 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Targan SR, Feagan BG, Fedorak RN, Lashner BA, Panaccione R, Present DH, Spehlmann ME, Rutgeerts PJ, Tulassay Z, Volfova M, Wolf DC, Hernandez C, Bornstein J, Sandborn WJ, R. International Efficacy of Natalizumab in Crohn’s Disease, G. Remission Trial, Natalizumab for the treatment of active Crohn’s disease: results of the ENCORE trial, Gastroenterology 132 (5) (2007) 1672–1683. [DOI] [PubMed] [Google Scholar]
- [66].Feagan BG, Rutgeerts P, Sands BE, Hanauer S, Colombel JF, Sandborn WJ, Van Assche G, Axler J, Kim HJ, Danese S, Fox I, Milch C, Sankoh S, Wyant T, Xu J, Parikh A, Vedolizumab as induction and maintenance therapy for ulcerative colitis, N. Engl. J. Med 369 (8) (2013) 699–710. [DOI] [PubMed] [Google Scholar]
- [67].Sandborn WJ, Feagan BG, Rutgeerts P, Hanauer S, Colombel JF, Sands BE, Lukas M, Fedorak RN, Lee S, Bressler B, Fox I, Rosario M, Sankoh S, Xu J, Stephens K, Milch C, Parikh A, Vedolizumab as induction and maintenance therapy for Crohn’s disease, N. Engl. J. Med 369 (8) (2013) 711–721. [DOI] [PubMed] [Google Scholar]
- [68].Li H, Huang SY, Shi FH, Gu ZC, Zhang SG, Wei JF, alpha4beta7 integrin inhibitors: a patent review, Expert Opin. Ther. Pat 28 (12) (2018) 903–917. [DOI] [PubMed] [Google Scholar]
- [69].Hirahara K, Nakayama T, CD4+ T-cell subsets in inflammatory diseases: beyond the Th1/Th2 paradigm, Int. Immunol 28 (4) (2016) 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Cronkite DA, Strutt TM, The Regulation of Inflammation by Innate and Adaptive Lymphocytes, J. Immunol. Res 2018 (2018) 1467538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Goods BA, Hernandez AL, Lowther DE, Lucca LE, Lerner BA, Gunel M, Raddassi K, Coric V, Hafler DA, Love JC, Functional differences between PD-1+ and PD-1-CD4+ effector T cells in healthy donors and patients with glioblastoma multiforme, PLoS One 12 (9) (2017) e0181538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Gunderson AJ, Mohammed J, Horvath FJ, Podolsky MA, Anderson CR, Glick AB, CD8(+) T cells mediate RAS-induced psoriasis-like skin inflammation through IFN-gamma, J. Invest. Dermatol. 133 (4) (2013) 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Holm Nielsen S, Mortensen JH, Willumsen N, Rasmussen DGK, Mogensen DJ, Di Sabatino A, Mazza G, Jorgensen LN, Giuffrida P, Pinzani M, Klinge L, Kjeldsen J, Leeming DJ, Karsdal MA, Genovese F, A fragment of collagen type VI alpha-3 chain is elevated in serum from patients with gastrointestinal disorders, Sci. Rep 10 (1) (2020) 5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.