

Abstract
Phosphoramidite analogues of modified cyclotriphosphates provide a general and step-economical synthesis of nucleoside triphosphates and analogues on scale without the need for protecting groups. These reagents enable rapid access to pure nucleoside oligophosphates and a range of other analogues that were previously difficult to obtain (e.g., NH, CH2, CCl2, and CF2 replacements for O, phosphono- and phosphoimidazolides, -morpholidates, -azidates, and -fluoridates). DFT calculations demonstrate that the selectivity of the cyclotriphosphate opening reactions proceeds via an inline substitution mechanism that displaces the least charged leaving group.
There is a general demand for the fundamental building blocks of our genetic material: the (deoxy)nucleoside triphosphates.1,2 Even more so, to study the myriad of processes nucleoside polyphosphates are additionally required for, general methods to access these molecules and tailored analogues are much sought after.3 Despite a long history of research in the field of chemical nucleoside polyphosphate synthesis, there is still a lack of a general, practical, step-economic, and high-yielding process that at the same time enables analogue generation in a straightforward manner. Such analogues continue to have a significant impact on studies into the biological functions of nucleotides and their derivatives. Detailed discussions of different approaches have been the subject of comprehensive reviews.4–6 A recent study presents in detail the pros and cons of different approaches, which eventually rely on protecting group manipulations and chromatographic steps.7
Herein, a family of reagents is described that enables access to a wide range of modified nucleoside oligophosphates in simple, scalable one-flask operations without the need for protecting groups. These reagents are ease to handle and facilitate modifications in the α-, β-, and γ-positions of the triphosphate chain. They are, in principle, compatible with automated solid phase synthesis. The approach proceeds through modified (deoxy)cyclotriphosphates as central intermediates (Scheme 1), which are then linearized with nucleophiles.8–16 Such intermediates have previously been difficult to obtain in pure form. Moreover, despite their role as key intermediates, the mechanism and selectivity of this reaction, i.e., linearization vs branching, still remain poorly characterized (Scheme 2) and are therefore analyzed by DFT calculations in this publication.
Scheme 1.
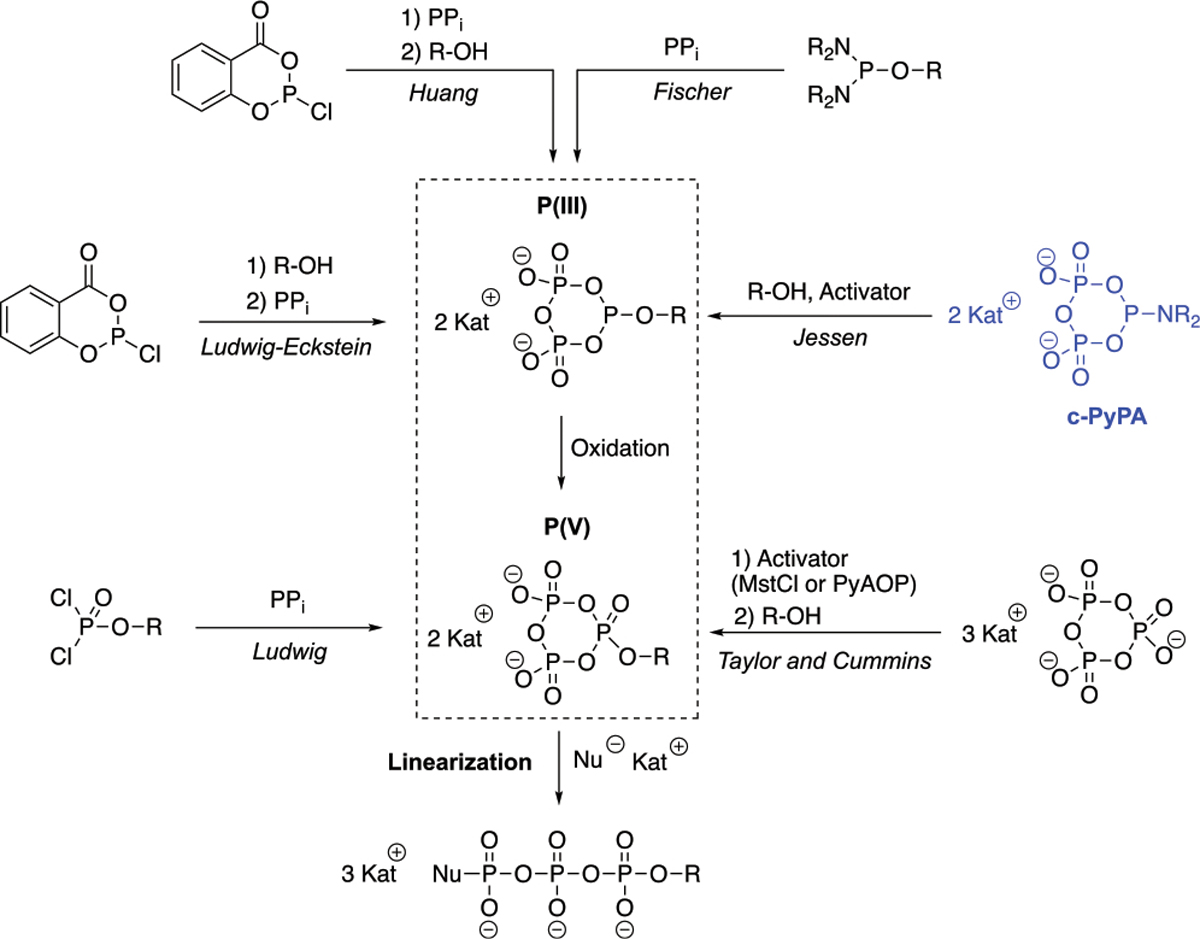
Overview of Synthetic Approaches to Modified Oligophosphates via (Deoxy)cyclotriphosphates
Scheme 2.
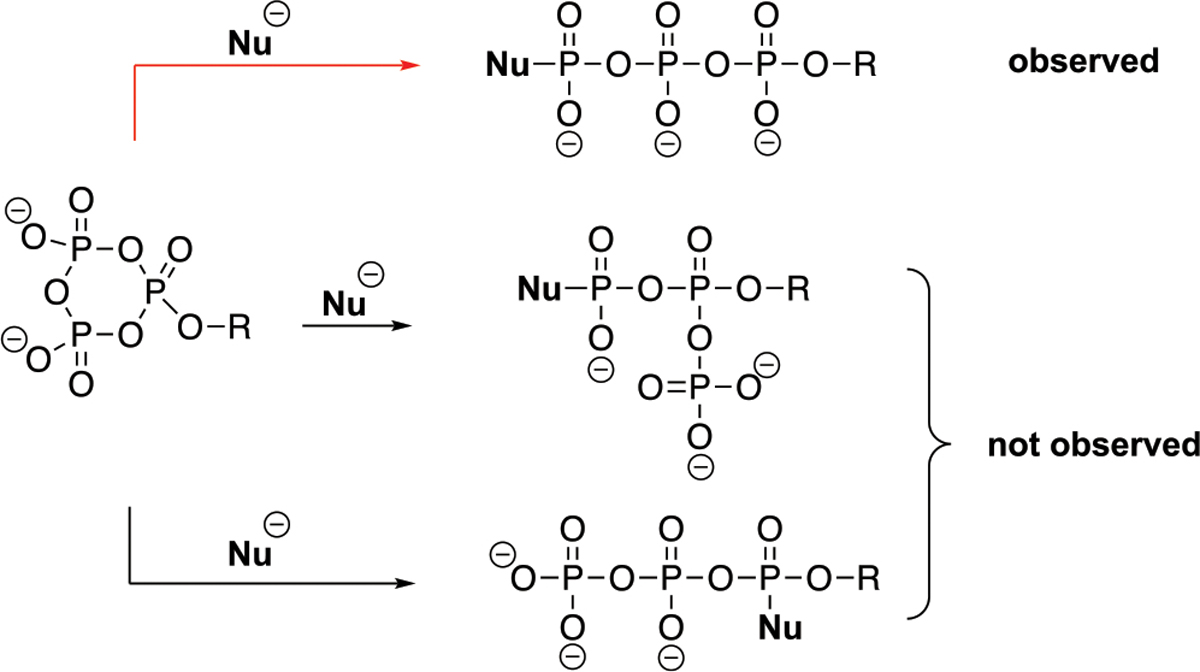
Ring-Opening of the Cyclotriphosphate Can Lead to Linearization with the Nucleophile Residing in the Terminal Position (Top, Observed) or the α-Position (Bottom, Not Observed), or Branching (Middle, Not Observed)
We recently disclosed the synthesis of a stable prototype reagent that represents a phosphoramidite analogue of cyclotriphosphate (a cyclic pyrophosphoryl P-amidite: cPyPA, Scheme 1, reagent highlighted in blue). This reagent was applied to the synthesis of polyphosphate analogues to study polyphosphate metabolism.17 The application of cPyPA to generate modified nucleoside triphosphates is now discussed. The ease of its preparation starting from pyrophosphate also suggested that pyrophosphate analogues could be used to generate a whole family of new reagents (Scheme 3, box) that can be used to position nonhydrolyzable units in between the β and γ phosphate. These reagents can furthermore be applied to access activated and modified nucleoside triphosphates, facilitating the one-flask synthesis of, e.g., phosphoimidazolides, -morpholidates, and -azidates. These can subsequently serve again as building blocks for longer oligophosphate chains, as, e.g., found in dye-linked nucleoside hexaphosphates (Scheme 4).18
Scheme 3. Overview of the Reaction Scope of cPyPA and Analoguesa.
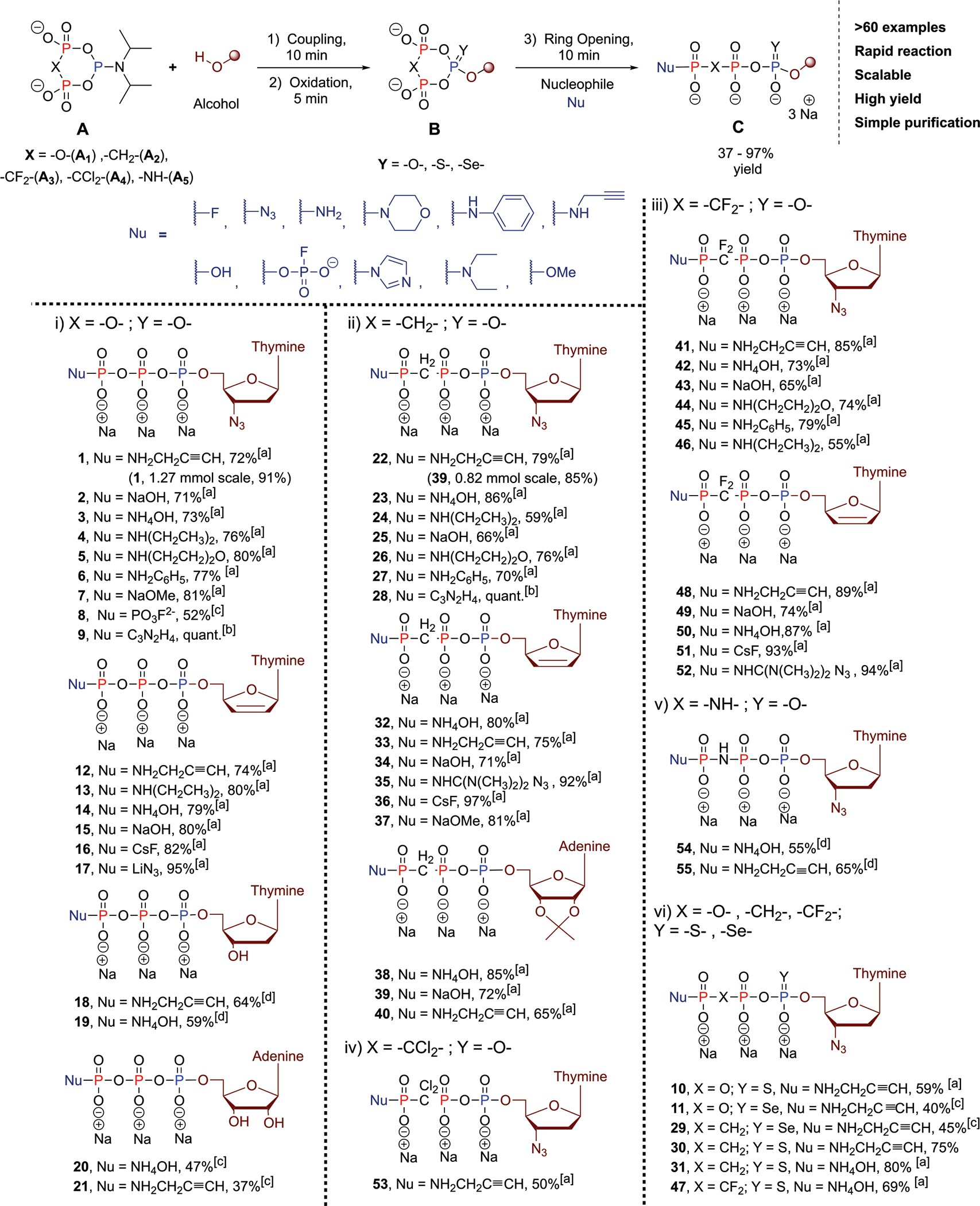
a[a] The triphosphates were directly precipitated from the reaction mixture by using NaClO4 in acetone without further purification. [b] The reaction mixture was characterized by NMR, and the product was directly used for the next transformations. [c] The product was precipitated from the reaction mixture and purified by anion exchange chromatography (SAX). [d] The product was precipitated from the reaction mixture and subsequently purified by Reverse Phase MPLC.
Scheme 4.
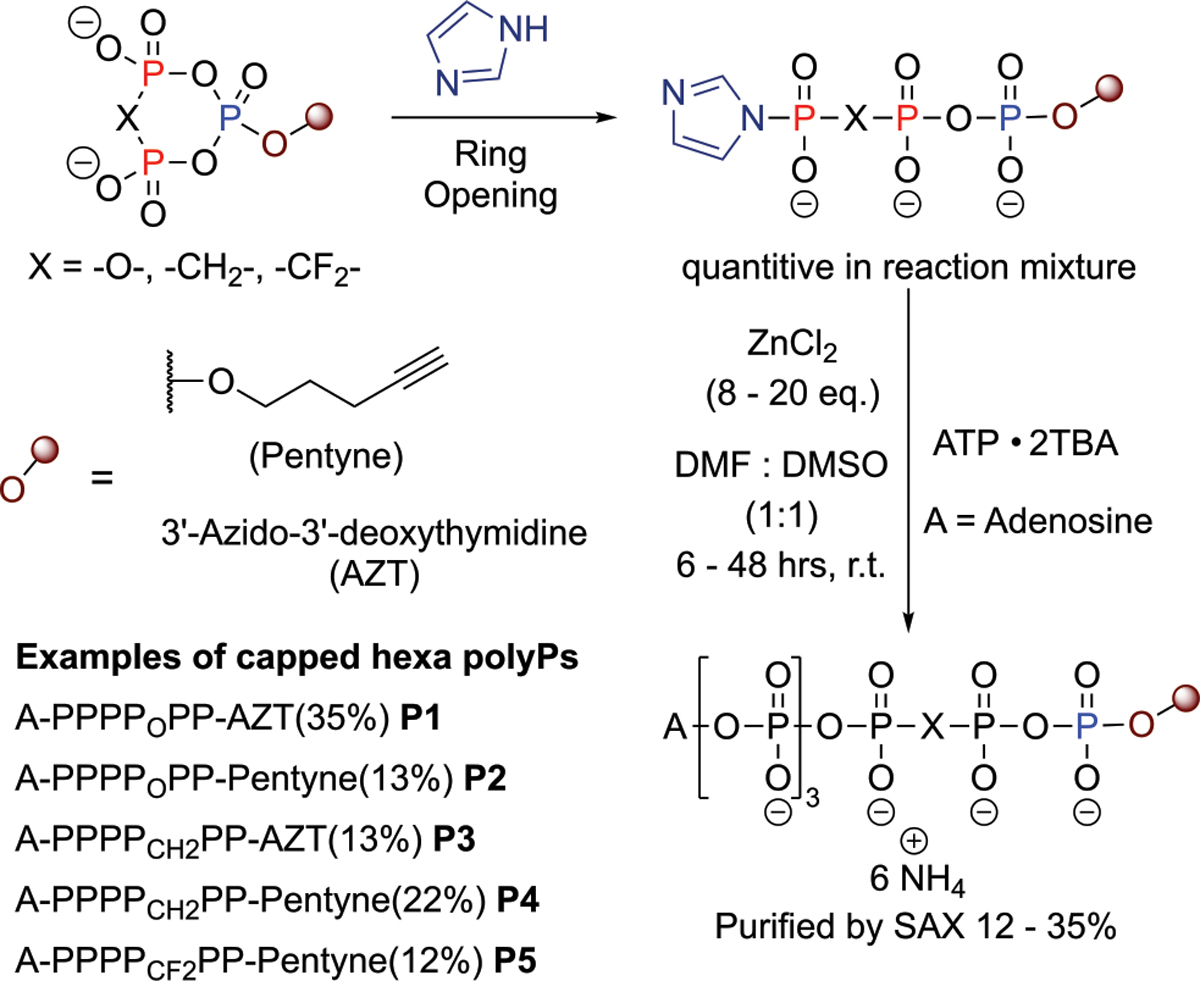
Synthesis of Capped Hexaphosphates from γ-Imidazolide Triphosphates
The reactions of pyrophosphate and its nonhydrolyzable analogues, i.e., imidodiphosphate,19 methylene diphosphonate, dichloromethylene diphosphonate, and difluoromethylene diphosphonate with (diisopropyl-amido)dichlorophosphite delivered cPyPA and its previously unknown imidodiphosphate and phosphonate analogues cPyNHPA, cPyCH2PA, cPyCCl2PA, and cPyCF2PA. All reagents except cPyNHPA were stable under argon atmosphere in dry solvents for several weeks and were obtained in pure form in solution. Reactions of these reagents with different alcohols (nucleosides, aliphatic alcohols) provided access to mixed P(III)–P(V) deoxycyclotriphosphate esters within a few minutes that were then oxidized, again in a few minutes, to the modified cyclotriphosphates (or phosphonates in the case of cPyCH2PA, cPyCCl2PA, and cPyCF2PA). Such rapid conversions remain the domain of P(III) reagents and cannot be achieved with the less reactive P(V) reagents.6
Oxidation was achieved with mCPBA, Beaucagés reagent, or KSeCN, thereby additionally enabling selective modifications of P-α. Ring opening with different nucleophiles (e.g., aliphatic, cyclic, and aromatic amines, ammonia, imidazole, azide, morpholine, hydroxide, alkoxides, fluoride, and fluorophosphate) gave linearized products with the nucleophiles exclusively found in the terminal position. Of note, linearization occurred not only with the cyclotriphosphates, but also with the phosphonates and imidophosphates in comparable efficiency. Scheme 3 gives an overview of the structures that were obtained highlighting the generality of the approach. No Staudinger reduction with azides is induced by the P(III) reagents as demonstrated with azidothymidine (AZT) as starting material, where the azido group remained untouched.
The ease of cyclotriphosphate generation provide significant advantages over prior methods20–22 and enables selective access to a wide range of modified nucleoside analogues including nonhydrolyzable bridges in between P-β and P-γ. Triphosphorylations of 2′,3′-protected nucleosides and of nucleosides without additional reactive alcohols (e.g., AZT, d4T) are straightforward and result in quantitative conversions to the modified cyclotriphosphates, which, after ring opening with, e.g., hydroxide, can be collected by simple precipitation. Unprotected nucleosides with a reactive 3′ OH group gave partial phosphorylation also in that position (10–20% depending on the substrate). Nonetheless, cPyPA and analogues could be used to modify T and A, directly without protecting groups providing access to modified nucleoside triphosphates in one flask operations in overall yields of 37–64%, which is usually not possible with other approaches that have to rely on protecting groups.6,7 While step economy was significantly improved without protecting groups, an additional ion chromatography purification step was required. The separated 3′-NTPs were also isolated and characterized (see the Supporting Information) and are thus accessible.
Some selected examples will be briefly discussed to emphasize the scope of the approach: ring-opening with imidazole as a nucleophile enables the generation of terminally modified P-imidazolides7,23 (e.g., 9, 28; for more examples, see the Supporting Information). 9 and 28 can be activated with Zn2+, and reaction with ATP resulted in the formation of hexaphosphates with modified termini (Scheme 4). The use of tetramethylguanidinium azide as a nucleophile provides access to phosphoazidates 35 and 52 which are potential photoaffinity probes with minimal structural perturbation.24 Ring opening with fluorophosphates generates nucleoside tetraphosphates, such as 8, with terminal fluoro substitution as potential electrophilic traps and mechanistic probes.25 All these reactions were compatible with the whole family of cPyPA reagents. These advantages render cPyPA and its nonhydrolyzable derivatives novel benchmark reagents in the field of nucleotide analogue synthesis.
Selective nucleophilic attack and opening of the cyclotriphosphate to a linear product are highlights of the method. The present empirical results, over 60+ derivatives, indicate that attack occurs at the sterically least hindered site and elimination follows with preferential cleavage of the anhydride bond that results in the least charge accumulation on the leaving group. Discussions of the mechanism of related phosphotransfer reactions26–31 present a full spectrum of associative, concerted, or dissociative possibilities. B97-D/Def2-TZVPPD(water) calculations support a concerted SN2 type mechanism in cyclotriphosphate ring opening with amine nucleophiles (Figure 1). The transition state structures found leading to the branched and linear products with ammonia as a reference nucleophile predict barriers of 32.0 and 23.5 kcal/mol, respectively.
Figure 1.
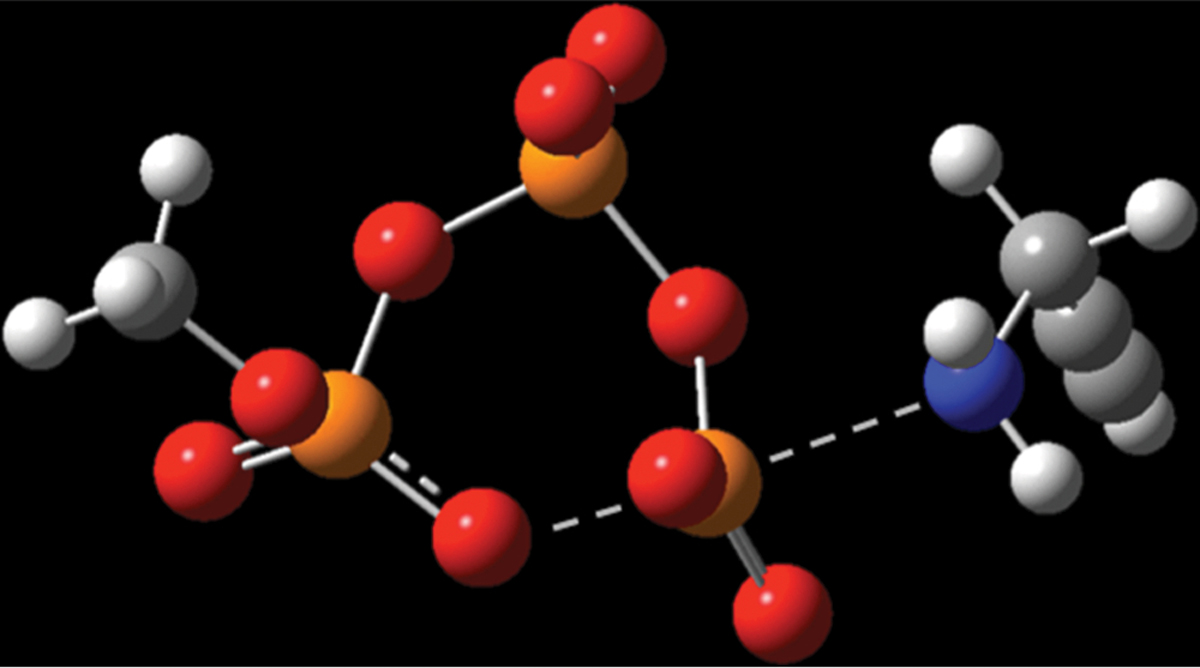
B97-D/Def2-TZVPPD (water) structure for the proposed transition state of methylcyclotriphosphate reacting with propargylamine. Illustration of colors: red, oxygen; orange, phosphorus; blue, nitrogen; gray, carbon; white, hydrogen.
The transition state leading to linear product with propargyl amine as nucleophile predicts a barrier of 23.2 kcal/mol, essentially the same as that with ammonia and consistent with the empirical reaction energetics. Thus, computations predict a selective reaction in which the nucleophile attacks at the least hindered phosphate opposite to the leaving group on which the lowest charge would be accumulated (Figure 1).
In summary, a new family of reagents has been developed and applied in the synthesis of modified oligophosphate analogues with a focus on nucleotides. These reagents are highly versatile, thereby enabling researchers to assemble diverse modified nucleotides on demand for studies into the function of these central building blocks of biology. The modularity of the approach in combination with high yields, short reactions times, step economy, scalability, and avoidance of protecting groups offers significant new opportunities in the field of nucleotide research.
Supplementary Material
ACKNOWLEDGMENTS
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – EXC-2193/1-390951807. H.J.J. and P.A.W. thank the HFSPO (Human Frontier Science Program RGP0025/2016) for financial support.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b08273.
Experimental methods, chromatograms, computational details, and NMR spectra (PDF)
REFERENCES
- (1).Kamerlin SCL; Sharma PK; Prasad RB; Warshel A Why nature really chose phosphate. Q. Rev. Biophys. 2013, 46 (1), 1–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Westheimer FH Why Nature Chose Phosphates. Science 1987, 235 (4793), 1173–1178. [DOI] [PubMed] [Google Scholar]
- (3).Jessen HJ; Ahmed N; Hofer A Phosphate esters and anhydrides - recent strategies targeting nature’s favoured modifications. Org. Biomol. Chem. 2014, 12 (22), 3526–3530. [DOI] [PubMed] [Google Scholar]
- (4).Burgess K; Cook D Syntheses of nucleoside triphosphates. Chem. Rev. 2000, 100 (6), 2047–60. [DOI] [PubMed] [Google Scholar]
- (5).Dutta AK; Captain I; Jessen HJ New Synthetic Methods for Phosphate Labeling. Top. Curr. Chem. 2017, 375 (3), 51. [DOI] [PubMed] [Google Scholar]
- (6).Roy B; Depaix A; Perigaud C; Peyrottes S Recent Trends in Nucleotide Synthesis. Chem. Rev. 2016, 116 (14), 7854–97. [DOI] [PubMed] [Google Scholar]
- (7).Bala S; Liao JY; Ngor AK; Yik E; Chaput JCP (V) Reagents for the Scalable Synthesis of Natural and Modified Nucleoside Triphosphates. J. Am. Chem. Soc. 2019, 141 (34), 13286–13289. [DOI] [PubMed] [Google Scholar]
- (8).Nahum V; Zundorf G; Levesque SA; Beaudoin AR; Reiser G; Fischer B Adenosine 5 ‘-O-(1-boranotriphosphate) derivatives as novel P2Y(1) receptor agonists. J. Med. Chem. 2002, 45 (24), 5384–5396. [DOI] [PubMed] [Google Scholar]
- (9).Mohamady S; Taylor SD Synthesis of Nucleoside Triphosphates from 2’-3′-Protected Nucleosides Using Trimetaphosphate. Org. Lett. 2016, 18 (3), 580–3. [DOI] [PubMed] [Google Scholar]
- (10).Azevedo C; Singh J; Steck N; Hofer A; Ruiz FA; Singh T; Jessen HJ; Saiardi A Screening a Protein Array with Synthetic Biotinylated Inorganic Polyphosphate To Define the Human PolyPome. ACS Chem. Biol. 2018, 13 (8), 1958–1963. [DOI] [PubMed] [Google Scholar]
- (11).Ludwig J A new route to nucleoside 5′-triphosphates. Acta Biochim. Biophys. Acad. Sci. Hung. 1981, 16 (3–4), 131–3. [PubMed] [Google Scholar]
- (12).Ludwig J; Eckstein F Rapid and Efficient Synthesis of Nucleoside 5′-O-(1-Thiotriphosphates), 5′-Triphosphates and 2’,3′-Cyclophosphorothioates Using 2-Chloro-4h-1,3,2-Benzodioxaphosphorin-4-One. J. Org. Chem. 1989, 54 (3), 631–635. [Google Scholar]
- (13).Shepard SM; Cummins CC Functionalization of Intact Trimetaphosphate: A Triphosphorylating Reagent for C, N, and O Nucleophiles. J. Am. Chem. Soc. 2019, 141 (5), 1852–1856. [DOI] [PubMed] [Google Scholar]
- (14).Caton-Williams J; Lin L; Smith M; Huang Z Convenient synthesis of nucleoside 5′-triphosphates for RNA transcription. Chem. Commun. 2011, 47 (28), 8142–4. [DOI] [PubMed] [Google Scholar]
- (15).Li P; Dobrikov M; Liu H; Shaw BR Synthesis of acyclothymidine triphosphate and alpha-P-boranotriphosphate and their substrate properties with retroviral reverse transcriptase. Org. Lett. 2003, 5 (14), 2401–3. [DOI] [PubMed] [Google Scholar]
- (16).Mordhorst S; Singh J; Mohr MKF; Hinkelmann R; Keppler M; Jessen HJ; Andexer JN Several Polyphosphate Kinase 2 Enzymes Catalyse the Production of Adenosine 5′-Polyphosphates. ChemBioChem 2019, 20 (8), 1019–1022. [DOI] [PubMed] [Google Scholar]
- (17).Singh J; Steck N; De D; Hofer A; Ripp A; Captain I; Keller M; Wender PA; Bhandari R; Jessen HJ A Phosphoramidite Analogue of Cyclotriphosphate Enables Iterative Polyphosphorylations. Angew. Chem., Int. Ed. 2019, 58 (12), 3928–3933. [DOI] [PubMed] [Google Scholar]
- (18).Eid J; Fehr A; Gray J; Luong K; Lyle J; Otto G; Peluso P; Rank D; Baybayan P; Bettman B; Bibillo A; Bjornson K; Chaudhuri B; Christians F; Cicero R; Clark S; Dalal R; Dewinter A; Dixon J; Foquet M; Gaertner A; Hardenbol P; Heiner C; Hester K; Holden D; Kearns G; Kong X; Kuse R; Lacroix Y; Lin S; Lundquist P; Ma C; Marks P; Maxham M; Murphy D; Park I; Pham T; Phillips M; Roy J; Sebra R; Shen G; Sorenson J; Tomaney A; Travers K; Trulson M; Vieceli J; Wegener J; Wu D; Yang A; Zaccarin D; Zhao P; Zhong F; Korlach J; Turner S Real-time DNA sequencing from single polymerase molecules. Science 2009, 323 (5910), 133–8. [DOI] [PubMed] [Google Scholar]
- (19).Larsen M; Willett R; Yount RG Imidodiphosphate and pyrophosphate: possible biological significance of similar structures. Science 1969, 166 (3912), 1510–1. [DOI] [PubMed] [Google Scholar]
- (20).Michelson AM; Todd AR Nucleotides 0.4. A Novel Synthesis of Adenosine Triphosphate. J. Chem. Soc. 1949, No. Oct, 2487–2490. [Google Scholar]
- (21).Grachev MA; Mustaev AA Cyclic Adenosine-5′-Trimetaphosphate Phosphorylates a Histidine Residue Nearby the Initiating Substrate Binding-Site of Escherichia-Coli DNA-Dependent Rna-Polymerase. FEBS Lett. 1982, 137 (1), 89–94. [DOI] [PubMed] [Google Scholar]
- (22).Knorre DG; Kurbatov VA; Samukov VV General method for the synthesis of ATP gamma-derivatives. FEBS Lett. 1976, 70 (1), 105–8. [DOI] [PubMed] [Google Scholar]
- (23).Wanat P; Walczak S; Wojtczak BA; Nowakowska M; Jemielity J; Kowalska J Ethynyl, 2-Propynyl, and 3-Butynyl C-Phosphonate Analogues of Nucleoside Di- and Triphosphates: Synthesis and Reactivity in CuAAC. Org. Lett. 2015, 17 (12), 3062–5. [DOI] [PubMed] [Google Scholar]
- (24).Breslow R; Feiring A; Herman F Intermolecular Insertion Reactions of Phosphoryl Nitrenes. J. Am. Chem. Soc. 1974, 96 (18), 5937–5939. [Google Scholar]
- (25).Baranowski MR; Nowicka A; Rydzik AM; Warminski M; Kasprzyk R; Wojtczak BA; Wojcik J; Claridge TD; Kowalska J; Jemielity J Synthesis of fluorophosphate nucleotide analogues and their characterization as tools for (1)(9)F NMR studies. J. Org. Chem. 2015, 80 (8), 3982–97. [DOI] [PubMed] [Google Scholar]
- (26).Allen KN; Dunaway-Mariano D Phosphoryl group transfer: evolution of a catalytic scaffold. Trends Biochem. Sci. 2004, 29 (9), 495–503. [DOI] [PubMed] [Google Scholar]
- (27).Blackburn GM; Williams NH; Gamblin SJ; Smerdon SJ Comment on ″The pentacovalent phosphorus intermediate of a phosphoryl transfer reaction″. Science 2003, 301 (5637), 1184 author reply 1184. [DOI] [PubMed] [Google Scholar]
- (28).Lahiri SD; Zhang G; Dunaway-Mariano D; Allen KN The pentacovalent phosphorus intermediate of a phosphoryl transfer reaction. Science 2003, 299 (5615), 2067–71. [DOI] [PubMed] [Google Scholar]
- (29).Ma BY; Meredith C; Schaefer HF The Quest for a Metaphosphate Intermediate - the Mechanisms for Hydrolysis of Pyrophosphates with and without Catalysis. J. Phys. Chem. 1995, 99 (11), 3815–3822. [Google Scholar]
- (30).Herschlag D; Jencks WP Evidence that metaphosphate monoanion is not an intermediate in solvolysis reactions in aqueous solution. J. Am. Chem. Soc. 1989, 111 (19), 7579–7586. [Google Scholar]
- (31).Lassila JK; Zalatan JG; Herschlag D Biological phosphoryl-transfer reactions: understanding mechanism and catalysis. Annu. Rev. Biochem. 2011, 80, 669–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.