

Abstract
Metabolic reprogramming is recognized as a hallmark of cancer. Lipids are the essential biomolecules required for membrane biosynthesis, energy storage, and cell signaling. Altered lipid metabolism allows tumor cells to survive in the nutrient-deprived environment. However, lipid metabolism remodeling in renal cell carcinoma (RCC) has not received the same attention as in other cancers. RCC, the most common type of kidney cancer, is associated with almost 15,000 death in the USA annually. Being refractory to conventional chemotherapy agents and limited available targeted therapy options has made the treatment of metastatic RCC very challenging. In this article, we review recent findings that support the importance of synthesis and metabolism of cholesterol, free fatty acids (FFAs), and polyunsaturated fatty acids (PUFAs) in the carcinogenesis and biology of RCC. Delineating the detailed mechanisms underlying lipid reprogramming can help to better understand the pathophysiology of RCC and to design novel therapeutic strategies targeting this malignancy.
Keywords: Metabolic reprogramming, Cholesterol, Free fatty acids, PUFA, Renal cell carcinoma
1. Introduction
Renal cell carcinoma (RCC) is the most common type of kidney cancer. In 2020, the USA had approximately 74,000 new cases and 15,000 deaths from RCC [1]. This cancer originates from renal tubular epithelial cells and the most common subtypes are clear cell RCC (ccRCC), papillary RCC, and chromophobe RCC. Multiple risk factors such as smoking, obesity, and chronic kidney disease (CKD) have been associated with this malignancy [2].
Surgery is the usual treatment in the majority of patients with localized RCC. However, in 30% of cases, cancer will recur after successful treatment for localized tumors [3]. Five-year survival for localized RCC exceeds 90%, whereas this number for patients with distant metastases is only 13% [4]. Metastatic RCC (mRCC) is refractory to conventional chemotherapy drugs, and effective therapies are limited [2]. Metastatic RCC requires systemic therapy with immunotherapy and/or molecularly targeted therapy [5].
First-line treatment for ccRCC includes immunotherapy with programmed cell death 1 protein (PD-1) checkpoint inhibitors such as nivolumab and pembrolizumab in combination with molecular targeted therapy. The most commonly used molecularly targeted medications in RCC are tyrosine kinase inhibitors which block vascular endothelial growth factor receptors (VEGFR) like axitinib, cabozantinib, and lenvatinib [6–9]. Combination of nivolumab and ipilimumab as anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) antibodies can also be considered in the first-line setting [10]. Agents targeting mammalian (mechanistic) target of rapamycin (mTOR) like everolimus can be used mainly in patients with mRCC who have progressed after one previous VEGF-targeted therapy [11]. While targeted therapy options are very limited in this type of malignancy, improved knowledge of disease biology is critically needed for the identification of novel targets.
The prospective analysis of more than 300,000 participants in the National Institutes of Health and American Association for Retired Persons (NIH-AARP) Diet and Health Study showed that excess body weight is a risk factor for development of RCC in both men and women [12]. Another large cohort study with 348,550 participants from 8 countries of the European Prospective Investigation into Cancer and Nutrition (EPIC) revealed the same association [13]. Weight gain between young adulthood (18–35 years of age) and midlife (35–50 years of age) was strongly related to risk of RCC. Moreover, a positive association between waist-to-hip ratio and RCC was observed in women [12]. Mechanistically, RCC is characterized by a high degree of metabolic reprogramming including changes in lipid metabolism (Table 1) [14]. It has been shown that increased endogenous lipid synthesis or exogenous uptake is necessary for neoplastic cell survival and proliferation [14]. Specifically, dysregulation of lipid metabolism is among the most prominent changes in RCC. Furthermore, imbalance in lipid metabolism is likely associated with RCC aggressiveness.
Table 1.
Lipid reprogramming in RCC. This table depicts lipid dysregulation in RCC and the proteins that are involved in different pathways
| Lipid reprogramming in RCC tumors | Mechanism | References |
|---|---|---|
| Decreased cholesterol biosynthesis |
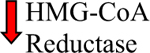
|
59,65 |
| Increased cholesterol uptake |
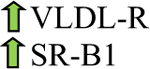
|
78,81,82 |
| Increased fatty acid biosynthesis |

|
82,88,89,92,94,95 |
| Increased fatty acid uptake |
|
98,99 |
| Reduced fatty acid catabolism |
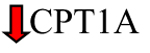
|
101 |
| Reduced Lipolysis |
|
125 |
| LDs Accumulation | HIF-α stabilization | 101,129,130 |
In this review, we focus on lipid metabolism dysregulation associated with RCC and ccRCC as the most common subtype. We highlight the recent evidence implicating deregulated lipid metabolism in RCC development, including alterations in the metabolism of cholesterol, free fatty acids (FFAs), and polyunsaturated fatty acids (PUFAs), as well as lipid storage in RCC, and we will describe how delineating these pathways can offer novel targets for prevention and treatment.
2. Lipid biosynthesis, metabolism, and homeostasis in cancers
The abnormal growth of tumors usually leads to a limited supply of nutrients. To adapt to these challenging conditions, cancer cells often reprogram the metabolism of glucose, proteins, nucleic acids, and lipids. Lipids including sterols, acylglycerols, phospholipids, and triacylglycerol represent a complex group of hydrophobic biomolecules. These biomolecules serve a plethora of biological functions. They are not only responsible for the structural integrity of biological membranes but also are important for energy metabolism and storage, as well as playing a significant role in signal transduction [15].
2.1. Cholesterol
Cholesterol, a member of the sterol category of lipids, is a crucial component of cell membranes. It plays an important role in controlling membrane fluidity and assembly and function of lipid rafts which contain multiple signaling cascades such as RAS, AKT, or SRC that are involved in cancer development [16].
Besides its direct uptake from the diet, cholesterol is synthesized through the mevalonate pathway. This pathway starts with the condensation of acetyl-Coenzyme A (CoA) with acetoacetyl-CoA to generate 3-hydroxy-3-methylglutaryl (HMG)-CoA (Fig. 1). The conversion of HMG-CoA to mevalonate by HMG-CoA reductase is the rate-limiting step in cholesterol biosynthesis. Mevalonate undergoes subsequent reactions to form the isoprenoid farnesyl-pyrophosphate (FPP). FPP is the precursor for squalene that is further converted to cholesterol. FPP is also used to produce geranylgeranyl-pyrophosphate (GGPP). Both FPP and GGPP are essential for post-translational modifications (PTMs) of various proteins, a process called prenylation [17]. Rho GTPases are well-studied prenylated signaling proteins that are part of Ras superfamily. Once Rho GTPase is prenylated, it is delivered and bound to plasma membrane which protect it from degradation and protein misfolding [18]. Increased activity of these proteins is associated with cancer progression and PTM prenylation has been identified as a contributor to tumorigenesis [19, 20].
Fig. 1.

Lipid metabolism. Acetyl-CoA is the starting material in cholesterol and FFA biosynthesis. Cholesterol and FFA can also be provided from exogenous resources. The red boxes are the proteins involved in lipid regulation
HMG-CoA reductase inhibitor medications, known as statins, can inhibit the production of mevalonate that is the precursor of cholesterol [21–23]. Many studies have investigated antitumor activity of statins, specifically, their ability to inhibit the active form of oncoproteins such as Rho and Ras and their downstream pathways controlling proliferation, migration, invasion, and survival of cancer cells including glioma, prostate, breast, and pancreatic cancer cell lines [24–28].
Intracellular cholesterol can also be acquired through receptor-mediated uptake of plasma lipoproteins. Low-density lipoprotein receptor (LDL-R) is the major receptor involved in exogenous cholesterol uptake. However, very-low-density lipoprotein (VLDL) and high-density lipoprotein (HDL) are other sources of extracellular cholesterol [29, 30]. Once lipoprotein binds to its receptor, it forms an endosome within the membrane which translocates into cells. Following internalization and transport into the lysosome, the cholesterol ester is hydrolyzed by lysosomal acid lipase (LAL) to release the free cholesterol (Fig. 1) [31].
Hypercholesterolemia and a high cholesterol diet have been associated with an increased risk of malignancy in both clinical and experimental studies [32, 33]. Targeting cholesterol metabolism has received increasing attention as a new preventive or therapeutic approach for cancers such as colorectal and prostate malignancies [34], but has not been extensively evaluated in RCC.
2.2. Free fatty acids and polyunsaturated fatty acids
Fatty acids (FAs) are the main building blocks of various lipid species such as acylglycerols, phospholipids, and triacylglycerol. Generally, de novo FA biosynthesis occurs in the liver, adipose tissue, and lactating breasts. Other tissues acquire FAs from the bloodstream either as free fatty acids (FFAs) or within lipoprotein forms [35]. However, tumors require a constant supply of FAs for cell proliferation and survival which leads to upregulated de novo FA synthesis or lipogenesis and uptake in cancer cells [36, 37].
The de novo lipogenesis (DNL) pathway starts from citrate that is generated from glucose and glutamine metabolism in the Krebs cycle. Citrate is cleaved by ATP-citrate lyase (ACLY) to generate acetyl-CoA which is an important building block for FA synthesis (Fig. 1). Acetyl-CoA is later converted to Malonyl-CoA by acetyl-CoA carboxylase (ACC). Both of these molecules are substrates for FA synthase (FASN) needed to generate palmitate (C16:0), the most abundant saturated FA in cells [37]. This saturated 16-carbon FA can be then elongated and desaturated to produce other FAs. Stearoyl-CoA desaturase (SCD) adds a double bond to the Δ9 position of FAs. Oleate (C18:1) is the most prevalent monounsaturated fatty acid (MUFA) generated by SCD (Fig. 1) [38]. While unsaturated and mono-saturated FAs are synthesized in cells, two 18-carbon polyunsaturated fatty acids (PUFAs), linoleic acid (LA, omega-6) and α-linolenic acid (ALA, omega-3), are considered essential FAs that must be supplemented via the diet. These essential dietary PUFAs are desaturated by FA desaturase 1 and 2 (FADS1, FADS2) and elongated by ELOVL FA elongase 5(ELOVL5) to form the long-chain PUFAs arachidonic acid (AA, omega-6) and eicosapentaenoic acid (EPA, omega-3) (Fig. 2). Additional elongation and desaturation of EPA finally result in the production of docosahexaenoic acid (DHA, omega3) [39]. PUFAs are important molecules involved in numerous cell functions. These are substantial components of cell membranes, affecting membrane fluidity. They also serve as active molecules in cell signaling, inflammation, and cell death. The metabolism of AA, the most studied omega-6 PUFA, generates eicosanoids such as prostaglandin E2 (PGE2) (Fig. 2) which is considered important contributor to cancer progression [40, 41]. On the other hand, eicosanoids produced from omega-3 PUFAs like PGE3 have been shown to inhibit tumor growth [42–44].
Fig. 2.

PUFA metabolism pathway. Linoleic acid and α-Linolenic acid are the essential PUFA provided from food. Other PUFA can either be obtained from diet or synthesized in the body. Blue boxes represent the enzymes involved in PUFA metabolism
Cancer cells require increased exogenous FA uptake to sustain their lipid demands. This can be achieved by increased expression of CD36 known as fatty acid translocase. CD36 is a multifunctional transmembrane glycoprotein which has been proposed as a prognostic marker in many cancers [45]. FA binding proteins (FABPs) also promote FA uptake. FABPs are intracellular lipid chaperones that facilitate movement of FFAs intracellularly and serve different roles in tumorigenesis (Fig. 1) [46]. For example, increased FABP3 expression has been shown to have an inhibitory effect on cell proliferation and promotes apoptosis in embryonic cancer cells [47]. On the other hand, elevated FABP5 enhances tumor progression in pancreatic and colon cancers [48, 49]. Moreover, in ovarian cancer, high FABP4 expression was associated with metastasis [50].
FAs are a valuable source of cellular energy. In cytoplasm, FFAs are conjugated to CoA, followed by a modification to acyl-carnitine by carnitine palmitoyltransferase 1 (CPT1) which is rate-limiting to FA entry into mitochondria (Fig. 1). Once FAs are transported into mitochondria, they are oxidized by β-oxidation generating ATP (Fig. 1). One palmitate molecule (C16:0) can yield 130 molecules of ATP. Several studies have shown that β-oxidation is required, especially under metabolic stress, to render tumors more adaptable to nutrient deprivation [37].
2.3. Lipid reservoir
Excess FAs are incorporated into triacylglycerol or triglyceride (TG) for energy storage in form of lipid droplets (LDs) that can be mobilized by fatty acid oxidation to generate ATP (Fig. 1). Therefore, TGs act as a reservoir for excessive FAs in the cell. Diacylglycerol acyltransferases (DGAT1 and DGAT2) involved in TG synthesis have been studied in tumor carcinogenesis, cancer aggressiveness, chemotherapy resistance, and cancer stem cell invasiveness [51]. Adipose triglyceride lipase (ATGL) is a rate-limiting enzyme in the hydrolysis of TG. Several types of malignancies have been associated with decreased levels of ATGL or deregulated expression of its protein partners [52].
Another component of LD is cholesteryl ester (CE). CE is the product of fatty acid esterification to cholesterol by acetyl-CoA acetyltransferase (ACAT) (Fig. 1). Increased CE is correlated with tumor aggressiveness [53, 54]. In fact, CE accumulation is a hallmark of ccRCC [55].
2.4. Lipid metabolism regulation
Sterol regulatory element-binding proteins (SREBPs) are transcription factors that regulate enzymes involved in cholesterol and fatty acid biosynthesis. When there are sufficient intracellular lipid levels, these proteins are inactive and they are retained in the endoplasmic reticulum (ER) bound to SREBP cleavage-activating protein (SCAP). Insufficient lipid levels in ER activate SREBPs. Their activation requires proteolytic cleavage in Golgi, and translocation into the nucleus. Once inside the nucleus, they bind to the promoter regions of SREBP target genes and initiate gene expression. This process is highly regulated by cellular levels of sterols, insulin, growth factors, and mammalian target of rapamycin (mTOR) signaling [56]. SREBP-1 mainly regulates FA and TG synthesis while SREBP-2 selectively induces the expression of enzymes involved in cholesterol biosynthesis including HMG-CoA reductase (Fig. 1) [57].
Liver X receptor (LXR), a nuclear transcription factor receptor, is another regulator of lipogenesis in cancer. LXR regulates multiple genes related to cholesterol homeostasis and fatty acid biosynthesis and works as a cholesterol sensor in the cell. Oxysterols, derivatives of cholesterol, activate the formation of the LXR complex with retinoid X receptor α (RXRα). Upon activation, this complex induces the expression of genes involved in cholesterol efflux from cells such as the ATP-binding cassette transporter ABCA1 (Fig. 1). In addition, LXR can activate the expression of several lipogenic genes such as FASN and SCD [58].
Peroxisome proliferator-activated receptors (PPARs) are also important regulators for lipid metabolism and storage; e.g., PPAR-α plays a key role in lipid β-oxidation in the liver, while PPAR-γ regulates lipid uptake and storage in the adipose tissue. However, their roles in RCCs are relatively unclear.
3. Cholesterol and RCC
ccRCC, the major RCC subtype, is characterized by lipid and glycogen accumulation, implicating altered fatty acid and glucose metabolism in its etiology. ccRCC cells contain high levels of triglyceride and also cholesterol mainly in the form of CE [59–62]. It can be speculated that cholesterol accumulation is one of the reasons why ccRCC is refractory to treatment. An in vitro study showed that the addition of LDL cholesterol to ccRCC cells in culture compromised the efficacy of sunitinib [63]. One of the major mechanisms for increased intracellular cholesterol involves cellular uptake in form of lipoprotein or free cholesterol. Cellular cholesterol can be also de novo synthesized from acetyl-CoA via the mevalonate pathway (Fig. 1). In this pathway, HMG-CoA reductase is the rate-limiting step responsible for production of mevalonate which ultimately is converted into cholesterol [64]. Histologic examination of RCC primary specimens showed decreased levels of HMG-CoA reductase [59]. The TCGA-KIRC project dataset also revealed that HMG-CoA reductase gene expression is significantly lower in primary ccRCCs compared to normal tissue [65]. Therefore, cholesterol accumulation in ccRCCs is likely the result of increased uptake rather than excessive biosynthesis from acetate.
3.1. Circulating cholesterol
In clinical studies, preoperative lower serum total cholesterol levels are significantly associated with aggressive tumor characteristics, poor prognosis, and worse overall survival outcomes after surgical removal of tumor [66, 67]. A meta-analysis of nine cohort studies consisting of 15,609 patients concluded that preoperative serum total cholesterol levels are an independent prognostic predictor for patients with surgically treated RCC [68]. Blood cholesterol increases during treatment with the mTOR inhibitor temsirolimus are shown to be a potential predictor of drug efficacy and is associated with longer overall survival [69].
3.2. Cholesterol biosynthesis and statins
The mevalonate pathway has a pivotal regulatory role in cell growth and proliferation. As described before, statins inhibit HMG-CoA reductase and prevent the formation of FPP to reduce cholesterol biosynthesis (Fig. 1) [70]. A few studies demonstrated that statin treatment has an inhibitory effect on RCC. Thompson et al. showed that statins have a profound cytotoxic effect on von Hippel-Lindau (VHL) gene deficient RCC cell lines (Table 2) [71]. The majority of RCC tumors carry a defective copy of the VHL gene that is responsible for ubiquitination and degradation of hypoxia-induced factor (HIF) (Fig. 3) [72]. Therefore, the HIF signaling pathway is overactivated in RCC regardless of oxygenation status. Overactivation of this pathway contributes to the sensitivity to statins. It appears that statins inhibit tumor initiation and growth of xenografts via inhibition of GTPase isoprenylation [71].
Table 2.
Inhibitors targeting lipid metabolism in RCC as cited in the main text.
| Compound | Structure | Mechanism of Action | Observed effect | References | |
|---|---|---|---|---|---|
| In vitro | In vivo | ||||
| Statins a |
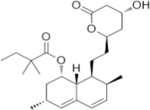
|
HMG-CoA reductase inhibitor | Growth inhibition of 786-O, RCC4 and RCC10 cell lines | Tumor growth inhibition of 786-OT1 xenograft | 71,74 |
| LXR-623 |
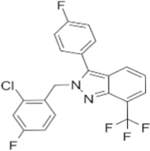
|
LXR agonist | Growth inhibition of 786-O and ACHN cell line | 78 | |
| SR9243 |
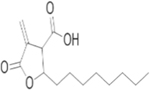
|
LXR inverse agonist | Growth inhibition of 786-O and ACHN cell lines |
Tumor growth inhibition of 786-O xenograft |
78 |
| C75 |
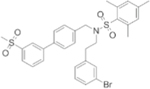
|
FASN inhibitor | Growth inhibition of 769P, Caki-1 and KU20–01 cell lines | Tumor volume reduction of Caki-1 xenografts | 97 |
Molecular structure of Simvastatin is shown here as statin representative
Fig. 3.

HIF pathway in ccRCC. VHL mutation, mTOR activation, and hypoxia cause an increase of HIFs in the cell. This event ultimately leads to LD accumulation
mTOR inhibitors have been approved for the treatment of metastatic RCC because the mTOR pathway is highly activated in this type of malignancy, but unfortunately, the efficacy of these drugs is not as expected due to resistance. Inhibiting mTOR induces feedback activation of the phosphatidylinositol 3 kinase (PI3K)/AKT pathway that results in resistance. Expression levels of phospho-S6 and phospho-AKT can be used as predictive biomarkers of responses to mTOR inhibitors [73]. Hagiwara et al. demonstrated that statins synergistically inhibit the growth of RCC cell lines when combined with mTOR inhibitors (Table 2). Statins sensitize cells to mTOR inhibitors by suppressing KRAS and Rac1 prenylation. This results in hypophosphorylation and activation of retinoblastoma protein, resulting in induction of G1 arrest upon combination therapy [74]. Importantly, statins were shown to improve survival outcomes of patients with metastatic RCC who are treated with targeted therapy such as mTOR inhibitors [75]. Moreover, in a population-based cohort study, use of statins showed to reduce the risk of RCC with hazard ratio of 0.64(95% CI, 0.38 to 0.87) [76]. Hence, statin can be considered for further clinical study in patients with risk factors for RCC or in treatment of RCC.
3.3. Cholesterol uptake
Cholesterol uptake is mediated through different receptors including LDL-R, VLDL-R, and scavenger receptor B1 (SR-B1) (Fig. 1) [29, 30]. A genome-wide association study (GWAS) on RCC demonstrated that a single nucleotide polymorphism (SNP) which maps to the SR-B1 gene is associated with a higher risk of RCC [77]. Indeed, cancer tissues containing excessive cholesterol showed an increased level of SR-B1, a receptor for uptake of HDL-cholesterol [78]. In a mouse xenograft model, lowering HDL-cholesterol intake significantly reduced cholesterol levels in the tumor which suggests that cholesterol accumulation is partly provided through HDL uptake [79]. Conversely, LDL-R, the main receptor involved in cholesterol uptake, is low in malignant renal tissues [80]. However, VLDL-R, an alternative receptor for the uptake of lipoproteins, is upregulated in ccRCC tissues [81, 82]. Following internalization, lipoprotein is hydrolyzed by lysosomal acid lipase (LAL) in lysosomes to release free cholesterol. LAL is upregulated in ccRCC cells and it is associated with lower patient survival [83]. When Wang et al. attempted to block LAL and the utilization of cholesterol esters, this resulted in impaired proliferation and survival of ccRCC cells [83]. These studies demonstrate that lipid accumulation in ccRCC is more dependent on increased lipid uptake than on cholesterol biosynthesis.
3.4. Cholesterol storage
To protect cells from the toxic effects of high free cholesterol levels resulting from LAL hydrolysis, ACAT re-esterifies free cholesterol with FAs for storage inside the cell (Fig. 1). High levels of CE in RCC tumors result from increased activity of this enzyme [59]. In breast cancer, cholesterol biosynthesis is reduced when ACAT mRNA levels and cholesterol esters are elevated [84]. It can be speculated that higher uptake of cholesterol regulates the retention of SREBPs in ER and degradation of HMG-CoA reductase [85].
3.5. Cholesterol metabolism regulation
LXR regulates multiple genes involved in cholesterol transport. LXR facilitates regulatory feedback in the presence of high cholesterol. In one study, Wu et al. evaluated the effects of a LXR agonist in ccRCC cells [78]. Targeting LXR downregulates LDL-R and upregulates expression of ABCA1 also known as cholesterol efflux regulatory protein. This regulatory effect results in reduced intracellular cholesterol and apoptosis of neoplastic cells without an effect on normal renal tubular epithelial HK2 cells. Interestingly, this LXR-agonist showed a large difference in the killing of two RCC cell lines including 786-O, representing a primary ccRCC tumor, and ACHN, with characteristics of metastatic papillary RCC. Expression of HMG-CoA reductase is higher in ACHN compared to 786-O cells, suggesting that ACHN is more dependent on the intracellular biosynthesis of cholesterol. Therefore, a higher dose of the LXR agonist was required to inhibit cell growth of ACHN than 786-O cells. This suggests that ccRCC tumor cells might be more dependent on exogenous cholesterol than other RCC tumors and normal cells, such that LXR may offer a new therapeutic target for ccRCC (Table 2) [78].
In summary, ccRCC, especially VHL-deficient tumors, are more dependent on exogenous cholesterol compared to other RCC subtypes. Blocking the uptake of cholesterol, by increasing the cholesterol efflux or decreasing influx into tumors, resulting in higher plasma cholesterol may be a useful strategy for treating RCC patients.
4. FFA metabolism and RCC
4.1. FA biosynthesis
FAs are essential for tumor cell growth to maintain membrane sustainability and provide energy sources during rapid proliferation. Hence, FA biosynthesis is upregulated in tumor cells irrespective of the levels of circulating lipids [86]. Lipidomic studies have shown an increase in utilization of FAs in ccRCC [87]. Moreover, aerobic glycolysis and glutamine utilization are well-known metabolic reprogramming pathways in ccRCC [14]. Teng et al. reported that ACLY is highly expressed in RCC tumors compared to adjacent normal tissue. They also used siRNAs to downregulate ACLY in vitro and demonstrated that ACLY knockdown can inhibit RCC cell proliferation and induce apoptosis [88]. In addition, increased levels of ACC protein are associated with worse clinical outcomes of RCC patients [89]. These findings suggest that ACLY and ACC expression levels are partially responsible for increased lipogenesis in RCC. While pharmacological inhibition of these two enzymes attenuated cell growth in other cancers such as lung and prostate cancers [90, 91], chemical agents targeting ACLY and ACC have not been studied in RCC.
Similar to several types of cancer such as breast, prostate, and lung cancer, FASN is overexpressed in ccRCC. FASN overexpression commonly occurs in cancers with higher risk of both disease recurrence and death including ccRCC. FASN hyperactivity is associated with aggressiveness and poor prognosis in ccRCC [92]. The product of FASN (palmitate) is desaturated by SCD to generate MUFA (Fig. 1). Studies revealed that SCD is overexpressed in many malignant cells [93]. In patients with ccRCC, SCD expression is higher in malignant renal cells than in adjacent normal tissues, and patients with higher SCD have worse overall survival [94, 95]. Inhibiting lipogenic enzymes such as FASN and SCD leads to reduced neoplastic cell proliferation and induction of apoptosis [96]. For example, administration of C75, a pharmacological inhibitor of FASN, significantly inhibited cell growth and triggered programmed cell death in vitro and in a xenograft model of RCC (Table 2) [97].
4.2. FA metabolism regulation
Similar to cholesterol metabolism, stimulating LXR promotes fatty acid production. Treatment with an inverse agonist of LXR (Table 2) can downregulate LXR-mediated genes responsible for fatty acid synthesis including FASN and SCD. This leads to impaired cell growth by depleting the FFA content of cells, and also induces cell death in cancer cells but not in normal epithelial kidney cells [78].
4.3. FA transport
Newly synthesized FAs require FABPs for transport into cells (Fig. 1). FABP-5 is upregulated in ccRCC and is associated with poor overall survival in patients [98]. Knockdown of FABP-5 significantly decreased cell proliferation although this did not affect cell migration and invasion of ccRCC cell lines [98]. In addition, FABP-5 levels positively correlate with lipoprotein lipase (LPL) that hydrolyzes FAs from lipoprotein, resulting in increased intracellular FAs which promote tumor progression [99].
4.4. FA oxidation
The excess FAs stored in lipid droplets (LDs) can undergo oxidation and provide a valuable source of ATP. CPT1A, the rate-limiting step in FA oxidation, has shown to be upregulated in several cancers such as prostate, lung, gastric, and breast [100]. On the contrary, Du et al. found that CPT1A is downregulated in ccRCCs compared to normal kidney tissue. As this is observed in VHL defective ccRCC cells, this suggests that CPT1A is suppressed in a VHL-dependent manner. Indeed, restoring CPT1A levels in ccRCC cell lines reduces the number of lipid droplets and inhibits tumor growth in vivo [101].
Increased de novo FFA synthesis or FFA uptake promotes RCC tumorigenesis. In ccRCC tumors, fatty acid catabolism (FA oxidation) is reduced. Treatments that influence the FA accumulation in cells by reducing FA biosynthesis or uptake, or by increasing FA oxidation, could be promising combination therapies in RCC patients.
4.5. PUFA and RCC
In modern western diets, the ratio of the consumed dietary essential PUFAs LA (omega-6) to ALA (omega-3) has surged from 5:1 to more than 10:1 [102, 103]. LA is the predominant PUFA in western diets and this imbalance of omega-6 and omega-3 PUFAs can impact human health and disease [104]. This also suggests the importance of studying the role of dietary intervention in cancer risk and therapy. EPA and DHA are the major omega-3 PUFAs and their high dietary intake has been proposed to reduce the risk of cancer [105–107]. Serum profiling of 112 patients with RCC prior to surgical resection demonstrated that patients with metastatic disease had lower DHA levels compared to patients without metastasis. Furthermore, patients with DHA levels below the median value showed shorter cancer-specific survival compared to patients with higher levels of DHA in the serum [108].
Several studies showed that co-administration of omega-3 PUFA with chemotherapy drugs is an effective adjuvant in cancer therapy [109]. Regorafenib is a multi-kinase inhibitor used in advanced RCC. In addition to blocking vascular endothelial growth factor receptors to prevent angiogenesis, this drug has an inhibitory effect on soluble epoxide hydrolase (sEH). The DHA metabolite, epoxydocosapentaenoic acid (EDP), is one of the substrates for this enzyme and is converted to a stable diol form. EDP has anti-angiogenesis properties. The combination of DHA and regorafenib was shown to have a synergistic effect in tumor inhibition likely because EDP is increased in cells due to blockage of epoxide hydrolase by regorafenib. As the result, the combination of DHA and regorafenib led to a reduction in angiogenesis and tumor invasiveness in vitro and in an animal model [110].
FADS1, a key rate-limiting enzyme in PUFA metabolism, is responsible for biosynthesis of EPA and AA from essential fatty acids (Fig. 2) and its activity is highly dependent on the diet [111]. Studies have shown that FADS1 is aberrantly expressed in many cancers, including colon, pancreas, breast, and laryngeal cancers. Furthermore, the knockdown of FADS1 can inhibit cancer growth [112–116]. Moreover, a lipidomic study revealed that FADS1 is overexpressed in ccRCC specimens. Surprisingly, in the same study, the levels of free PUFAs including AA, EPA, and DHA were lower in malignant tissues [82]. Free PUFAs can be incorporated into phospholipids and TGs, and additional analysis of the lipidomic data revealed that levels of PUFA-phosphatidylethanolamine (PE)/ether-PEs and PUFA-phosphatidylcholine (PC)/ether-PCs were higher in ccRCC tumors than in normal renal tissues. Moreover, PUFA-phospholipids were increased in high-grade compared to low-grade tumor samples [117]. Whether increased PUFA-phospholipids have an impact on tumorigenesis needs further investigation. It is important to note that the nutrient source and the omega-6/omega-3 ratio in the diet can have tremendous potential in cancer prevention and therapy response.
5. Hypoxia, lipid metabolism, and RCC
VHL tumor suppressor gene is located on chromosome 3p25–26 [118]. VHL protein is responsible for ubiquitinylating and degradation of HIFs under normoxic condition [119]. The loss of VHL is the most common feature of ccRCC [120] and can occur through genetic mutation, epigenetic mechanisms, and post translational changes [2, 121]. Oxygen deprivation or absence of VHL leads to increased level of HIFs (Fig. 3). HIFs regulate metabolic pathways related to cellular adaptation to hypoxia such as angiogenesis, glycolysis, and the Krebs cycle to maintain oxygen homeostasis [122]. In addition, HIFs are key regulators of lipid metabolism [123–126]. HIF-1α and HIF-2α are detected in various malignancies and are widely associated with poor prognosis [127]. The 2p21 gene locus to which Endothelial PAS Domain Protein 1 (EPAS1) gene maps and encodes HIF-2α is associated with increased RCC susceptibility that is identified in genome-wide association studies (GWAS) of RCC [77].
5.1. Hypoxia and lipid droplets
Increase in levels of HIFs contributes to the accumulation of LDs within ccRCC cells (Fig. 3). Surplus of FFA specially PUFA can react with reactive oxygen species (ROS) and cause lipid peroxidation which is harmful to the cells [128]. Channeling FAs to LDs is a mechanism for the cells to prevent lipid peroxidation [129]. These cytoplasmic vesicles are needed for energy homeostasis and release of lipid species during membrane synthesis and cell proliferation [129]. HIF-1α and HIF-2α knockdown lead to a reduction in lipid droplets in ccRCC cell lines [101, 129]. Perilipin2, LD coat protein, is abundantly expressed in non-adipose tissue. This protein is overexpressed in ccRCC tumor samples and is a marker of cellular lipid accumulation. Qiu et al. demonstrated that upregulation of Perilipin2 is due to HIF-2α activation (Fig. 3). The HIF-2α/Perilipin2 lipid storage axis suggests a model to protect tumor cells against endoplasmic reticulum stress [130]. A transgenic mouse model of ccRCC with expression of constitutively activated HIF-1 α also showed elevated level of perilipin2 that indicates lipid storage in LDs [131].Taken together, LD accumulation supports a potential drug target for cancer.
5.2. Hypoxia and TG
Hypoxia also reduces lipolysis in cancer cells by inhibiting ATGL, an important lipase on LDs controlling TG homeostasis (Fig. 3). Hence, it promotes LD formation, attenuates reactive oxygen species (ROS) production, and sustains cancer cell survival [125]. Blocking TG synthesis by inhibiting DGAT enhances apoptosis and represses the proliferation of ccRCC tumor cells in vivo. A low level of oxygen increases the incorporation of saturated fatty acid in TGs to protect the cells from the toxic buildup of free saturated FAs in vitro. These data suggest that TG incorporation into LDs plays an important role in reducing the availability of specific FAs for metabolism, and promotes cell viability [132].
5.3. Hypoxia and PUFAs
HIF-2α also plays an important role in regulating cellular PUFA levels. HIF-2α depletion demonstrates a remarkable reduction in free PUFAs and PUFA-TGs compared with TGs containing saturated/mono-unsaturated fatty acyl chains (SFA/MUFA-TAGs) in vitro. In addition, HIF-2α depletion results in a lower amount of PE-containing PUFA chains (AA and DHA). While HIF-2α activity strongly affects the level of PUFAs, free SFA/MUFAs are less impacted by HIF-2α activity [117].
5.4. Other effects of hypoxia
In the previous sections, we reviewed how cholesterol uptake is increased in RCC cells due to overexpression of VLDL receptors which result in accumulation of lipid droplets. This exogenous uptake of cholesterol is reduced when HIF-1α is downregulated with siRNA [81]. Moreover, we discussed that lipogenesis genes like FASN and SCD are upregulated in RCC. In other cancers such as breast cancer, FASN is upregulated by hypoxia, followed by activation of SREBP-1, the major transcriptional regulator of the FASN gene [126]. Nonetheless, the association of hypoxia and FASN, and SCD upregulation in RCC, requires further investigation.
6. Targeting lipid metabolism in RCC
Lipid metabolism reprogramming in RCC (Table 1) offers unique vulnerabilities and opportunities for therapeutic targeting. Whereas clinical trials have targeted lipid metabolism in cancers such as breast, colon, and lung [46, 133], research on RCC is limited to preclinical studies. FASN has received widespread attention as a therapeutic target since it is overexpressed in most cancer types [37]. The oral FASN inhibitor, TVB-2640 was tested in a phase I clinical trial in patients with advanced-stage solid tumors. The combination therapy of TVB-2640 with taxane increased the time to progression specifically KRAS mutated in non-small cell lung, breast, and ovarian cancers. FASN inhibition was associated with predictable and manageable safety profile with non-serious, reversible adverse events [134]. This therapeutic is used in combination therapy in two ongoing phase II clinical trials on astrocytoma and HER2-positive breast cancer [135, 136]. Since FA biosynthesis is upregulated in RCC, FASN inhibitor can be considered for clinical trial in this cancer type.
Another well-known example involves HMG-CoA reductase inhibitors. Statins are being repurposed for cancer patient care and are being tested extensively in clinical trials for assorted cancers [133]. Although a cohort study showed that use of statins is associated with a reduced risk of RCC [76], there are no clinical studies demonstrating the effects of statins in treating RCC.
As mentioned in previous section, HIFs play crucial role in pathophysiology of ccRCC and they are directly influencing lipid metabolism. An ongoing phase II clinical trial is evaluating the efficacy of HIF-2α, PT2385, on VHL disease-associated ccRCC tumors [137]. The result of this clinical trial may provide useful information on design of study blocking HIF-2α and the effect of that on lipid metabolism in progression of ccRCC.
In this review, we summarized studies that showed targeting lipid metabolism in RCC can be beneficial in treating this disease (Table 2). Collectively, these offer compelling evidence in support of the need for more clinical studies that target lipid metabolism in this cancer type.
7. Summary and future directions
Studies discussed here highlight altered lipid metabolism as an important metabolic phenotype of RCC. During kidney damage, renal epithelial cells reprogram their metabolic pathways to increase lipid accumulation. Understanding the mechanisms behind this condition can provide us with new insights into the basis for increased cancer risk associated with kidney disease. We provided a graphic summary of lipid reprogramming in RCC (Table 1). There is a close relationship between lipid metabolism and oncogenic signaling to promote cell proliferation and survival. Novel therapeutic approaches to target fatty acid or cholesterol homeostasis of cancer cells, either through blocking biosynthesis or uptake, have shown promising results in vitro and in vivo. This review was designed to facilitate a greater understanding of lipid biology in cancer to identify potential treatment strategies to overcome the limitations of chemotherapy in metastatic RCC, particularly ccRCC. Although 70% of all RCC malignancies are ccRCC, more research clearly needs to be done in papillary and chromophobe RCC, as well.
Funding
This study is supported in part by the NIH/NIDDK grants (R01DK106540 and R01DK124612) (W.L.), the start-up fund of the Office of Vice President for Research of Wayne State University (W.L.), the Eunice and Milton Ring Endowed Chair for Cancer Research (L.H.M), and a NCI T32-CA009531 (L.H.M.) Pre-doctoral Training fellowship to G.H.
Abbreviations
- HMG
3-Hydroxy-3-methylglutaryl
- CoA
Acetyl-Co enzyme
- ACAT
Acetyl-CoA acetyltransferase
- ACC
Acetyl-CoA carboxylase
- ATGL
Adipose triglyceride lipase
- ALA
α-Linolenic acid
- AA
Arachidonic acid
- ABCA1
ATP-binding cassette transporter
- ACLY
ATP-citrate lyase
- CPT1
Carnitine palmitoyltransferase 1
- CE
Cholesteryl ester
- CKD
Chronic kidney disease
- COX
Cyclooxygenase
- ccRCC
Clear cell RCC
- DNL
De novo Lipogenesis
- DG
Diacylglycerol
- DGAT
Diacylglycerol acyltransferase
- DHA
Docosahexaenoic acid
- EPA
Eicosapentaenoic acid
- ELOVL5
ELOVL fatty acid elongase 5
- ER
Endoplasmic reticulum
- EPAS1
Endothelial PAS domain protein 1
- sEH
Epoxide hydrolase
- EDP
Epoxydocosapentaenoic acid
- FPP
Farnesyl-pyrophosphate
- FA
Fatty acid
- FABPs
Fatty acid binding proteins
- FADS1
Fatty acid desaturase 1
- FADS2
Fatty acid desaturase 2
- FASN
Fatty acid synthase
- FFAs
Free fatty acids
- GWAS
Genome-wide association study
- GGPP
Geranylgeranyl-pyrophosphate
- HDL
High-density lipoprotein
- HIF
Hypoxia-induced factors
- LA
Linoleic acid
- LDs
Lipid droplets
- LPL
Lipoprotein lipase
- LXR
Liver X receptor
- LDL-R
Low-density lipoprotein recepto
- LAL
Lysosomal acid lipase
- mTOR
Mammalian target of rapamycin
- mRCC
Metastatic RCC
- MUFA
Monounsaturated fatty acid
- PPARs
Peroxisome proliferator-activated receptors
- PC
Phosphatidylcholine
- PE
Phosphatidylethanolamine
- PI3K
Phosphatidylinositol 3 kinase
- PUFAs
Polyunsaturated fatty acids
- PTM
Post-translational modification
- PGE2
Prostaglandin E2
- ROS
Reactive oxygen species
- RCC
Renal cell carcinoma
- RXRα
Retinoid X receptor α
- SR-B1
Scavenger receptor B1
- SNP
Single nucleotide polymorphism
- SEH
Soluble epoxide hydrolase
- SCAP
SREBP cleavage-activating protein
- SCD
Stearoyl-CoA desaturase
- SREBPs
Sterol regulatory element binding proteins
- TGF-β
Transforming growth factor-β
- TG
Triglyceride
- VEGF
Vascular endothelial growth factor
- VLDL
Very-low-density lipoprotein
- VHL
Von Hippel-Lindau
Footnotes
Conflict of interest The authors declare no competing interests.
Declarations
Ethics approval No ethical approvals are required for this review.
Consent to participate Informed consent is not required for this review.
References
- 1.Siegel RL, Miller KD, & Jemal A (2020). Cancer statistics, 2020. CA: A Cancer Journal for Clinicians, 70(1), 7–30. 10.3322/caac.21590 [DOI] [PubMed] [Google Scholar]
- 2.Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, Heng DY, Larkin J, & Ficarra V (2017). Renal cell carcinoma. Nature Reviews. Disease Primers, 3, 17009. 10.1038/nrdp.2017.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chin AI, Lam JS, Figlin RA, & Belldegrun AS (2006). Surveillance strategies for renal cell carcinoma patients following nephrectomy. Reviews in Urology, 8(1), 1–7. [PMC free article] [PubMed] [Google Scholar]
- 4.Howlader N, Krapcho M, Miller D, Bishop K, Kosary CL, & Yu M (2017). SEER cancer statistics review. In Cronin KA(Ed.), SEER cancer statistics review (pp. 1975–2014). National Cancer Institute. [Google Scholar]
- 5.Hofmann F, Hwang EC, Lam TB, Bex A, Yuan Y, Marconi LS, & Ljungberg B (2020). Targeted therapy for metastatic renal cell carcinoma. Cochrane Database of Systematic Reviews, (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, Pouliot F, Alekseev B, Soulières D, & Melichar B (2019). Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. New England Journal of Medicine, 380(12), 1116–1127. [DOI] [PubMed] [Google Scholar]
- 7.Powles T, Plimack ER, Soulières D, Waddell T, Stus V, Gafanov R, Nosov D, Pouliot F, Melichar B, & Vynnychenko I (2020). Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. The Lancet Oncology, 21(12), 1563–1573. [DOI] [PubMed] [Google Scholar]
- 8.Choueiri TK, Powles T, Burotto M, Escudier B, Bourlon MT, Zurawski B, Oyervides Juárez VM, Hsieh JJ, Basso U, & Shah AY (2021). Nivolumab plus cabozantinib versus sunitinib for advanced renal-cell carcinoma. New England Journal of Medicine, 384(9), 829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Motzer R, Alekseev B, Rha S, Porta C, Eto M, Powles T, Grünwald V, Hutson TE, Kopyltsov E, & Méndez-Vidal MJ (2021). Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. New England Journal of Medicine, 384(14), 1289–1300. [DOI] [PubMed] [Google Scholar]
- 10.Albiges L, Tannir NM, Burotto M, McDermott D, Plimack ER, Barthélémy P, Porta C, Powles T, Donskov F, & George S (2020). Nivolumab plus ipilimumab versus sunitinib for first-line treatment of advanced renal cell carcinoma: Extended 4-year follow-up of the phase III CheckMate 214 trial. ESMO Open, 5(6), e001079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Motzer RJ, Hutson TE, Glen H, Michaelson MD, Molina A, Eisen T, Jassem J, Zolnierek J, Maroto JP, & Mellado B (2015). Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. The Lancet Oncology, 16(15), 1473–1482. [DOI] [PubMed] [Google Scholar]
- 12.Adams KF, Leitzmann MF, Albanes D, Kipnis V, Moore SC, Schatzkin A, & Chow W (2008). Body size and renal cell cancer incidence in a large US cohort study. American Journal of Epidemiology, 168(3), 268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pischon T, Lahmann PH, Boeing H, Tjønneland A, Halkjær J, Overvad K, Klipstein-Grobusch K, Linseisen J, Becker N, & Trichopoulou A (2006). Body size and risk of renal cell carcinoma in the european prospective investigation into cancer and nutrition (EPIC). International Journal of Cancer, 118(3), 728–738. [DOI] [PubMed] [Google Scholar]
- 14.Weiss RH (2018). Metabolomics and metabolic reprogramming in kidney cancer. Seminars in Nephrology, 38(2), 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snaebjornsson MT, Janaki-Raman S, & Schulze A (2020). Greasing the wheels of the cancer machine: The role of lipid metabolism in cancer. Cell Metabolism, 31(1), 62–76. [DOI] [PubMed] [Google Scholar]
- 16.Ding X, Zhang W, Li S, & Yang H (2019). The role of cholesterol metabolism in cancer. American Journal of Cancer Research, 9(2), 219. [PMC free article] [PubMed] [Google Scholar]
- 17.Göbel A, Rauner M, Hofbauer LC, & Rachner TD (2020). Cholesterol and beyond-the role of the mevalonate pathway in cancer biology. Biochimica Et Biophysica Acta (BBA)-Reviews on Cancer, 1873(2), 1883514. [DOI] [PubMed] [Google Scholar]
- 18.Hodge RG, & Ridley AJ (2016). Regulating rho GTPases and their regulators. Nature Reviews Molecular Cell Biology, 17(8), 496–510. [DOI] [PubMed] [Google Scholar]
- 19.Castellano E, & Santos E (2011). Functional specificity of ras isoforms: So similar but so different. Genes & Cancer, 2(3), 216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haga RB, & Ridley AJ (2016). Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases, 7(4), 207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldstein JL, & Brown MS (1990). Regulation of the mevalonate pathway. Nature, 343(6257), 425–430. [DOI] [PubMed] [Google Scholar]
- 22.Sinensky M (2000). Recent advances in the study of prenylated proteins. Biochimica Et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids, 1484(2–3), 93–106. [DOI] [PubMed] [Google Scholar]
- 23.Russell DW (1992). Cholesterol biosynthesis and metabolism. Cardiovascular Drugs and Therapy, 6(2), 103–110. [DOI] [PubMed] [Google Scholar]
- 24.Iannelli F, Lombardi R, Milone MR, Pucci B, De Rienzo S, Budillon A, & Bruzzese F (2018). Targeting mevalonate pathway in cancer treatment: Repurposing of statins. Recent Patents on Anti-Cancer Drug Discovery, 13(2), 184–200. [DOI] [PubMed] [Google Scholar]
- 25.Misirkic M, Janjetovic K, Vucicevic L, Tovilovic G, Ristic B, Vilimanovich U, Harhaji-Trajkovic L, SumaracDumanovic M, Micic D, & Bumbasirevic V (2012). Inhibition of AMPK-dependent autophagy enhances in vitro antiglioma effect of simvastatin. Pharmacological Research, 65(1), 111–119. [DOI] [PubMed] [Google Scholar]
- 26.Parikh A, Childress C, Deitrick K, Lin Q, Rukstalis D, & Yang W (2010). Statin-induced autophagy by inhibition of geranylgeranyl biosynthesis in prostate cancer PC3 cells. The Prostate, 70(9), 971–981. [DOI] [PubMed] [Google Scholar]
- 27.Kang S, Kim E, & Moon A (2009). Simvastatin and lovastatin inhibit breast cell invasion induced by H-ras. Oncology Reports, 21(5), 1317–1322. [DOI] [PubMed] [Google Scholar]
- 28.Ding N, Cui X, Gao Z, Huang H, Wei X, Du Z, Lin Y, Shih WJ, Rabson AB, & Conney AH (2014). A triple combination of atorvastatin, celecoxib and tipifarnib strongly inhibits pancreatic cancer cells and xenograft pancreatic tumors. International Journal of Oncology, 44(6), 2139–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Go G, & Mani A (2012). Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. The Yale Journal of Biology and Medicine, 85(1), 19. [PMC free article] [PubMed] [Google Scholar]
- 30.Shen W, Azhar S, & Kraemer FB (2018). SR-B1: A unique multifunctional receptor for cholesterol influx and efflux. Annual Review of Physiology, 80, 95–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li F, & Zhang H (2019). Lysosomal acid lipase in lipid metabolism and beyond. Arteriosclerosis, Thrombosis, and Vascular Biology, 39(5), 850–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du Q, Wang Q, Fan H, Wang J, Liu X, Wang H, Wang Y, & Hu R (2016). Dietary cholesterol promotes AOM-induced colorectal cancer through activating the NLRP3 inflammasome. Biochemical Pharmacology, 105, 42–54. [DOI] [PubMed] [Google Scholar]
- 33.Kitahara CM, De González AB, Freedman ND, Huxley R, Mok Y, Jee SH, & Samet JM (2011). Total cholesterol and cancer risk in a large prospective study in korea. Journal of Clinical Oncology, 29(12), 1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silvente-Poirot S, & Poirot M (2012). Cholesterol metabolism and cancer: The good, the bad and the ugly. Current Opinion in Pharmacology, 6(12), 673–676. [DOI] [PubMed] [Google Scholar]
- 35.Santos CR, & Schulze A (2012). Lipid metabolism in cancer. The FEBS Journal, 279(15), 2610–2623. 10.1111/j.1742-4658.2012.08644.x [DOI] [PubMed] [Google Scholar]
- 36.Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, & Li J (2014). Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Reports, 9(1), 349–365. [DOI] [PubMed] [Google Scholar]
- 37.Röhrig F, & Schulze A (2016). The multifaceted roles of fatty acid synthesis in cancer. Nature Reviews Cancer, 16(11), 732. [DOI] [PubMed] [Google Scholar]
- 38.Paton CM, & Ntambi JM (2009). Biochemical and physiological function of stearoyl-CoA desaturase. American Journal of Physiology-Endocrinology and Metabolism, 297(1), E28–E37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chilton FH, Dutta R, Reynolds LM, Sergeant S, Mathias RA, & Seeds MC (2017). Precision nutrition and omega-3 polyunsaturated fatty acids: A case for personalized supplementation approaches for the prevention and management of human diseases. Nutrients, 9(11), 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cen B, Lang JD, Du Y, Wei J, Xiong Y, Bradley N, Wang D, & DuBois RN (2020). Prostaglandin E2 induces miR675–5p to promote colorectal tumor metastasis via modulation of p53 expression. Gastroenterology, 158(4), 971–984.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang D, Fu L, Sun H, Guo L, & DuBois RN (2015). Prostaglandin E2 promotes colorectal cancer stem cell expansion and metastasis in mice. Gastroenterology, 149(7), 1884–1895.e4. 10.1053/j.gastro.2015.07.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan Y, Davidson LA, Callaway ES, Goldsby JS, & Chapkin RS (2014). Differential effects of 2-and 3-series E-prostaglandins on in vitro expansion of Lgr5 colonic stem cells. Carcinogenesis, 35(3), 606–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang P, Chan D, Felix E, Cartwright C, Menter DG, Madden T, Klein RD, Fischer SM, & Newman RA (2004). Formation and antiproliferative effect of prostaglandin E3 from eicosapentaenoic acid in human lung cancer cells. Journal of Lipid Research, 45(6), 1030–1039. [DOI] [PubMed] [Google Scholar]
- 44.Szymczak M, Murray M, & Petrovic N (2008). Modulation of angiogenesis by ω−3 polyunsaturated fatty acids is mediated by cyclooxygenases. Blood, the Journal of the American Society of Hematology, 111(7), 3514–3521. [DOI] [PubMed] [Google Scholar]
- 45.Enciu A, Radu E, Popescu ID, Hinescu ME, & Ceafalan LC (2018). Targeting CD36 as biomarker for metastasis prognostic: How far from translation into clinical practice? BioMed Research International, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koundouros N, & Poulogiannis G (2019). Reprogramming of fatty acid metabolism in cancer. British Journal of Cancer, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song GX, Shen YH, Liu YQ, Sun W, Miao LP, Zhou LJ, Liu HL, Yang R, Kong XQ, & Cao KJ (2012). Overexpression of FABP3 promotes apoptosis through inducing mitochondrial impairment in embryonic cancer cells. Journal of Cellular Biochemistry, 113(12), 3701–3708. [DOI] [PubMed] [Google Scholar]
- 48.Kawaguchi K, Senga S, Kubota C, Kawamura Y, Ke Y, & Fujii H (2016). High expression of fatty acid-binding protein 5 promotes cell growth and metastatic potential of colorectal cancer cells. FEBS Open Bio, 6(3), 190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Senga S, Kawaguchi K, Kobayashi N, Ando A, & Fujii H (2018). A novel fatty acid-binding protein 5-estrogen-related receptor α signaling pathway promotes cell growth and energy metabolism in prostate cancer cells. Oncotarget, 9(60), 31753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gharpure KM, Pradeep S, Sans M, Rupaimoole R, Ivan C, Wu SY, Bayraktar E, Nagaraja AS, Mangala LS, & Zhang X (2018). FABP4 as a key determinant of metastatic potential of ovarian cancer. Nature Communications, 9(1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hernández-Corbacho MJ, & Obeid LM (2019). A novel role for DGATs in cancer. Advances in Biological Regulation, 72, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vegliante R, Di Leo L, Ciccarone F, & Ciriolo MR (2018). Hints on ATGL implications in cancer: Beyond bioenergetic clues. Cell Death & Disease, 9(3), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yue S, Li J, Lee S, Lee HJ, Shao T, Song B, Cheng L, Masterson TA, Liu X, & Ratliff TL (2014). Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metabolism, 19(3), 393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Gonzalo-Calvo D, López-Vilaró L, Nasarre L, Perez-Olabarria M, Vázquez T, Escuin D, Badimon L, Barnadas A, Lerma E, & Llorente-Cortés V (2015). Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: A molecular and clinicopathological study. BMC Cancer, 15(1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Drabkin HA, & Gemmill RM (2012). Cholesterol and the development of clear-cell renal carcinoma. Current Opinion in Pharmacology, 12(6), 742–750. 10.1016/j.coph.2012.08.002 [DOI] [PubMed] [Google Scholar]
- 56.Shimano H, & Sato R (2017). SREBP-regulated lipid metabolism: Convergent physiology—divergent pathophysiology. Nature Reviews Endocrinology, 13(12), 710. [DOI] [PubMed] [Google Scholar]
- 57.Eberlé D, Hegarty B, Bossard P, Ferré P, & Foufelle F (2004). SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie, 86(11), 839–848. [DOI] [PubMed] [Google Scholar]
- 58.Wang B, & Tontonoz P (2018). Liver X receptors in lipid signalling and membrane homeostasis. Nature Reviews Endocrinology, 14(8), 452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gebhard RL, Clayman RV, Prigge WF, Figenshau R, Staley NA, Reesey C, & Bear A (1987). Abnormal cholesterol metabolism in renal clear cell carcinoma. Journal of Lipid Research, 28(10), 1177–1184. [PubMed] [Google Scholar]
- 60.Tugnoli V, Trinchero A, & Tosi MR (2004). Evaluation of the lipid composition of human healthy and neoplastic renal tissues. The Italian Journal of Biochemistry, 53(4), 169–182. [PubMed] [Google Scholar]
- 61.Hoffmann K, Blaudszun J, Brunken C, Hopker WW, Tauber R, & Steinhart H (2005). Lipid class distribution of fatty acids including conjugated linoleic acids in healthy and cancerous parts of human kidneys. Lipids, 40(10), 1057–1062. 10.1007/s11745-005-1469-y [DOI] [PubMed] [Google Scholar]
- 62.Yoshimura K, Chen LC, Mandal MK, Nakazawa T, Yu Z, Uchiyama T, Hori H, Tanabe K, Kubota T, Fujii H, Katoh R, Hiraoka K, & Takeda S (2012). Analysis of renal cell carcinoma as a first step for developing mass spectrometry-based diagnostics. Journal of the American Society for Mass Spectrometry, 23(10), 1741–1749. 10.1007/s13361-012-0447-2 [DOI] [PubMed] [Google Scholar]
- 63.Naito S, Makhov P, Astsaturov I, Golovine K, Tulin A, Kutikov A, Uzzo RG, & Kolenko VM (2017). LDL cholesterol counteracts the antitumour effect of tyrosine kinase inhibitors against renal cell carcinoma. British Journal of Cancer, 116(9), 1203–1207. 10.1038/bjc.2017.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buhaescu I, & Izzedine H (2007). Mevalonate pathway: A review of clinical and therapeutical implications. Clinical Biochemistry, 40(9–10), 575–584. [DOI] [PubMed] [Google Scholar]
- 65.Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, & Brooks AN (2020). Visualizing and interpreting cancer genomics data via the xena platform. Nature Biotechnology, 38(6), 675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ohno Y, Nakashima J, Nakagami Y, Gondo T, Ohori M, Hatano T, & Tachibana M (2014). Clinical implications of preoperative serum total cholesterol in patients with clear cell renal cell carcinoma. Urology, 83(1), 154–158. 10.1016/j.urology.2013.08.052 [DOI] [PubMed] [Google Scholar]
- 67.Lee H, Kim YJ, Hwang EC, Kang SH, Hong S, Chung J, Kwon TG, Kwak C, Kim HH, Oh JJ, Lee SC, Hong SK, Lee SE, Byun S, & KOrean Renal Cell Carcinoma (KORCC) Group. (2017). Preoperative cholesterol level as a new independent predictive factor of survival in patients with metastatic renal cell carcinoma treated with cyto-reductive nephrectomy. BMC Cancer, 17(1), 364. 10.1186/s12885-017-3322-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li B, Huang D, Zheng H, Cai Q, Guo Z, & Wang S (2020). Preoperative serum total cholesterol is a predictor of prognosis in patients with renal cell carcinoma: A meta- analysis of observational studies. International Braz J Urol : Official Journal of the Brazilian Society of Urology, 46(2), 158–168. 10.1590/S1677-5538.IBJU.2019.0560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee CK, Marschner IC, Simes RJ, Voysey M, Egleston B, Hudes G, & De Souza P (2012). Increase in cholesterol predicts survival advantage in renal cell carcinoma patients treated with temsirolimus. Clinical Cancer Research, 18(11), 3188–3196. [DOI] [PubMed] [Google Scholar]
- 70.Mullen PJ, Yu R, Longo J, Archer MC, & Penn LZ (2016). The interplay between cell signalling and the mevalonate pathway in cancer. Nature Reviews Cancer, 16(11), 718–731. [DOI] [PubMed] [Google Scholar]
- 71.Thompson JM, Alvarez A, Singha MK, Pavesic MW, Nguyen QH, Nelson LJ, Fruman DA, & Razorenova OV (2018). Targeting the mevalonate pathway suppresses VHL-deficient CC-RCC through an HIF-dependent mechanism. Molecular Cancer Therapeutics, 17(8), 1781–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li L, & Kaelin WG (2011). New insights into the biology of renal cell carcinoma. Hematology/Oncology Clinics, 25(4), 667–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Battelli C, & Cho DC (2011). mTOR inhibitors in renal cell carcinoma. Therapy, 8(4), 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hagiwara N, Watanabe M, Iizuka-Ohashi M, Yokota I, Toriyama S, Sukeno M, Tomosugi M, Sowa Y, Hongo F, & Mikami K (2018). Mevalonate pathway blockage enhances the efficacy of mTOR inhibitors with the activation of retinoblastoma protein in renal cell carcinoma. Cancer Letters, 431, 182–189. [DOI] [PubMed] [Google Scholar]
- 75.McKay RR, Lin X, Albiges L, Fay AP, Kaymakcalan MD, Mickey SS, Ghoroghchian PP, Bhatt RS, Kaffenberger SD, & Simantov R (2016). Statins and survival outcomes in patients with metastatic renal cell carcinoma. European Journal of Cancer, 52, 155–162. [DOI] [PubMed] [Google Scholar]
- 76.Chou Y, Lin C, Wong C, Chou W, Chang J, & Sun C (2020). Statin use and the risk of renal cell carcinoma: National cohort study. Journal of Investigative Medicine, 68(3), 776–781. [DOI] [PubMed] [Google Scholar]
- 77.Purdue MP, Johansson M, Zelenika D, Toro JR, Scelo G, Moore LE, Prokhortchouk E, Wu X, Kiemeney LA, & Gaborieau V (2011). Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13. 3. Nature Genetics, 43(1), 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu G, Wang Q, Xu Y, Li J, Zhang H, Qi G, & Xia Q (2019). Targeting the transcription factor receptor LXR to treat clear cell renal cell carcinoma: Agonist or inverse agonist? Cell Death & Disease, 10(6), 416. 10.1038/s41419-019-1654-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim J, Thompson B, Han S, Lotan Y, McDonald JG, & Ye J (2019). Uptake of HDL-cholesterol contributes to lipid accumulation in clear cell renal cell carcinoma. Biochimica Et Biophysica Acta. Molecular and Cell Biology of Lipids, 1864(12), 158525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Clayman RV, Bilhartz LE, Spady DK, Buja LM, & Dietschy JM (1986). Low density lipoprotein-receptor activity is lost in vivo in malignantly transformed renal tissue. FEBS Letters, 196(1), 87–90. [DOI] [PubMed] [Google Scholar]
- 81.Sundelin JP, Ståhlman M, Lundqvist A, Levin M, Parini P, Johansson ME, & Borén J (2012). Increased expression of the very low-density lipoprotein receptor mediates lipid accumulation in clear-cell renal cell carcinoma. PLoS ONE, 7(11), e48694. 10.1371/journal.pone.0048694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saito K, Arai E, Maekawa K, Ishikawa M, Fujimoto H, Taguchi R, Matsumoto K, Kanai Y, & Saito Y (2016). Lipidomic signatures and associated transcriptomic profiles of clear cell renal cell carcinoma. Scientific Reports, 6, 28932. 10.1038/srep28932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang J, Tan M, Ge J, Zhang P, Zhong J, Tao L, Wang Q, Tong X, & Qiu J (2018). Lysosomal acid lipase promotes cholesterol ester metabolism and drives clear cell renal cell carcinoma progression. Cell Proliferation, 51(4), e12452. 10.1111/cpr.12452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Antalis CJ, Arnold T, Rasool T, Lee B, Buhman KK, & Siddiqui RA (2010). High ACAT1 expression in estrogen receptor negative basal-like breast cancer cells is associated with LDL-induced proliferation. Breast Cancer Research and Treatment, 122(3), 661–670. [DOI] [PubMed] [Google Scholar]
- 85.Lee JN, Song B, DeBose-Boyd RA, & Ye J (2006). Sterol-regulated degradation of insig-1 mediated by the membrane-bound ubiquitin ligase gp78. Journal of Biological Chemistry, 281(51), 39308–39315. [DOI] [PubMed] [Google Scholar]
- 86.Menendez JA, & Lupu R (2007). Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nature Reviews Cancer, 7(10), 763–777. [DOI] [PubMed] [Google Scholar]
- 87.Ganti S, Taylor SL, Aboud OA, Yang J, Evans C, Osier MV, Alexander DC, Kim K, & Weiss RH (2012). Kidney tumor biomarkers revealed by simultaneous multiple matrix metabolomics analysis. Cancer Research, 72(14), 3471–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Teng L, Chen Y, Cao Y, Wang W, Xu Y, Wang Y, Lv J, Li C, & Su Y (2018). Overexpression of ATP citrate lyase in renal cell carcinoma tissues and its effect on the human renal carcinoma cells in vitro. Oncology Letters, 15(5), 6967–6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cancer Genome Atlas Research Network. (2013). Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature, 499(7456), 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, Verhoeven G, & Swinnen JV (2007). Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Research, 67(17), 8180–8187. [DOI] [PubMed] [Google Scholar]
- 91.Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, & Thompson CB (2005). ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell, 8(4), 311–321. [DOI] [PubMed] [Google Scholar]
- 92.Horiguchi A, Asano T, Asano T, Ito K, Sumitomo M, & Hayakawa M (2008). Fatty acid synthase over expression is an indicator of tumor aggressiveness and poor prognosis in renal cell carcinoma. The Journal of Urology, 180(3), 1137–1140. [DOI] [PubMed] [Google Scholar]
- 93.Mounier C, Bouraoui L, & Rassart E (2014). Lipogenesis in cancer progression (review). International Journal of Oncology, 45(2), 485–492. 10.3892/ijo.2014.2441 [DOI] [PubMed] [Google Scholar]
- 94.Wang J, Xu Y, Zhu L, Zou Y, Kong W, Dong B, Huang J, Chen Y, Xue W, & Huang Y (2016). High expression of stearoyl-CoA desaturase 1 predicts poor prognosis in patients with clear-cell renal cell carcinoma. PloS One, 11(11), e0166231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Von Roemeling CA, Marlow LA, Wei JJ, Cooper SJ, Caulfield TR, Wu K, Tan WW, Tun HW, & Copland JA (2013). Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clinical Cancer Research, 19(9), 2368–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mason P, Liang B, Li L, Fremgen T, Murphy E, Quinn A, Madden SL, Biemann H, Wang B, & Cohen A (2012). SCD1 inhibition causes cancer cell death by depleting monounsaturated fatty acids. PloS One, 7(3), e33823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Horiguchi A, Asano T, Asano T, Ito K, Sumitomo M, & Hayakawa M (2008). Pharmacological inhibitor of fatty acid synthase suppresses growth and invasiveness of renal cancer cells. The Journal of Urology, 180(2), 729–736. [DOI] [PubMed] [Google Scholar]
- 98.Lv Q, Wang G, Zhang Y, Han X, Li H, Le W, Zhang M, Ma C, Wang P, & Ding Q (2019). FABP5 regulates the proliferation of clear cell renal cell carcinoma cells via the PI3K/AKT signaling pathway. International Journal of Oncology, 54(4), 1221–1232. 10.3892/ijo.2019.4721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wu G, Zhang Z, Tang Q, Liu L, Liu W, Li Q, & Wang Q (2020). Study of FABP’s interactome and detecting new molecular targets in clear cell renal cell carcinoma. Journal of Cellular Physiology, 235(4), 3776–3789. 10.1002/jcp.29272 [DOI] [PubMed] [Google Scholar]
- 100.Melone MAB, Valentino A, Margarucci S, Galderisi U, Giordano A, & Peluso G (2018). The carnitine system and cancer metabolic plasticity. Cell Death & Disease, 9(2), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Du W, Zhang L, Brett-Morris A, Aguila B, Kerner J, Hoppel CL, Puchowicz M, Serra D, Herrero L, Rini BI, Campbell S, & Welford SM (2017). HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nature Communications, 8(1), 1769–1778. 10.1038/s41467-017-01965-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Simopoulos AP (2011). Importance of the omega-6/omega-3 balance in health and disease: Evolutionary aspects of diet. Healthy agriculture, healthy nutrition, healthy people (pp. 10–21). Karger Publishers. [DOI] [PubMed] [Google Scholar]
- 103.Blasbalg TL, Hibbeln JR, Ramsden CE, Majchrzak SF, & Rawlings RR (2011). Changes in consumption of omega-3 and omega-6 fatty acids in the united states during the 20th century. The American Journal of Clinical Nutrition, 93(5), 950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chilton FH, Murphy RC, Wilson BA, Sergeant S, Ainsworth H, Seeds MC, & Mathias RA (2014). Dietgene interactions and PUFA metabolism: A potential contributor to health disparities and human diseases. Nutrients, 6(5), 1993–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bostick RM, Potter JD, Kushi LH, Sellers TA, Steinmetz KA, McKenzie DR, Gapstur SM, & Folsom AR (1994). Sugar, meat, and fat intake, and non-dietary risk factors for colon cancer incidence in iowa women (united states). Cancer Causes & Control, 5(1), 38–52. [DOI] [PubMed] [Google Scholar]
- 106.Gago-Dominguez M, Yuan JM, Sun CL, Lee HP, & Yu MC (2003). Opposing effects of dietary n-3 and n-6 fatty acids on mammary carcinogenesis: The singapore chinese health study. British Journal of Cancer, 89(9), 1686–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Takezaki T, Inoue M, Kataoka H, Ikeda S, Yoshida M, Ohashi Y, Tajima K, & Tominaga S (2003). Diet and lung cancer risk from a 14-year population-based prospective study in japan: With special reference to fish consumption. Nutrition and Cancer, 45(2), 160–167. [DOI] [PubMed] [Google Scholar]
- 108.Tasaki S, Horiguchi A, Asano T, Kuroda K, Sato A, Asakuma J, Ito K, Asano T, & Asakura H (2016). Preoperative serum docosahexaenoic acid level predicts prognosis of renal cell carcinoma. Molecular and Clinical Oncology, 5(1), 69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vaughan VC, Hassing MR, & Lewandowski PA (2013). Marine polyunsaturated fatty acids and cancer therapy. British Journal of Cancer, 108(3), 486–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim J, Ulu A, Wan D, Yang J, Hammock BD, & Weiss RH (2016). Addition of DHA synergistically enhances the efficacy of regorafenib for kidney cancer therapy. Molecular Cancer Therapeutics, 15(5), 890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sprecher H, Luthria DL, Mohammed BS, & Baykousheva SP (1995). Reevaluation of the pathways for the biosynthesis of polyunsaturated fatty acids. Journal of Lipid Research, 36(12), 2471–2477. [PubMed] [Google Scholar]
- 112.Xu Y, Yang X, Wang T, Yang L, He Y, Miskimins K, & Qian SY (2018). Knockdown delta-5-desaturase in breast cancer cells that overexpress COX-2 results in inhibition of growth, migration and invasion via a dihomo-γ-linolenic acid peroxidation dependent mechanism. BMC Cancer, 18(1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xu Y, Yang X, Zhao P, Yang Z, Yan C, Guo B, & Qian SY (2016). Knockdown of delta-5-desaturase promotes the anti-cancer activity of dihomo-γ-linolenic acid and enhances the efficacy of chemotherapy in colon cancer cells expressing COX-2. Free Radical Biology and Medicine, 96, 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang X, Xu Y, Brooks A, Guo B, Miskimins KW, & Qian SY (2016). Knockdown delta-5-desaturase promotes the formation of a novel free radical byproduct from COX-catalyzed ω−6 peroxidation to induce apoptosis and sensitize pancreatic cancer cells to chemotherapy drugs. Free Radical Biology and Medicine, 97, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhao R, Tian L, Zhao B, Sun Y, Cao J, Chen K, Li F, Li M, Shang D, & Liu M (2020). FADS1 promotes the progression of laryngeal squamous cell carcinoma through activating AKT/mTOR signaling. Cell Death & Disease, 11(4), 272–275. 10.1038/s41419-020-2457-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yang X, Xu Y, Wang T, Shu D, Guo P, Miskimins K, & Qian SY (2017). Inhibition of cancer migration and invasion by knocking down delta-5-desaturase in COX-2 overexpressed cancer cells. Redox Biology, 11, 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, Tseng YY, Deasy R, Kost-Alimova M, Dančík V, Leshchiner ES, Viswanathan VS, Signoretti S, Choueiri TK, Boehm JS, Wagner BK, Doench JG, Clish CB, Clemons PA, & Schreiber SL (2019). A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nature Communications, 10(1), 1617–1619. 10.1038/s41467-019-09277-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, & Geil L (1993). Identification of the von hippel-lindau disease tumor suppressor gene. Science (New York, NY), 260(5112), 1317–1320. 10.1126/science.8493574 [DOI] [PubMed] [Google Scholar]
- 119.Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, & Oldfield EH (2003). Von hippel-lindau disease. The Lancet, 361(9374), 2059–2067. 10.1016/S0140-6736(03)13643-4 [DOI] [PubMed] [Google Scholar]
- 120.Hu SL, Chang A, Perazella MA, Okusa MD, Jaimes EA, Weiss RH, & American Society of Nephrology Onco-Nephrology Forum. (2016). The nephrologist’s tumor: Basic biology and management of renal cell carcinoma. Journal of the American Society of Nephrology : JASN, 27(8), 2227–2237. 10.1681/ASN.2015121335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, & Bencko V (2008). Improved identification of von hippel-lindau gene alterations in clear cell renal tumors. Clinical Cancer Research, 14(15), 4726–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Masson N, & Ratcliffe PJ (2014). Hypoxia signaling pathways in cancer metabolism: The importance of co-selecting interconnected physiological pathways. Cancer & Metabolism, 2(1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu R, Feng Y, Deng Y, Zou Z, Ye J, Cai Z, Zhu X, Liang Y, Lu J, Zhang H, Luo Y, Han Z, Zhuo Y, Xie Q, Hon CT, Liang Y, Wu CL, & Zhong W (2021). A HIF1α-GPD1 feedforward loop inhibits the progression of renal clear cell carcinoma via mitochondrial function and lipid metabolism. Journal of Experimental & Clinical Cancer Research : CR, 40(1), 188–196. 10.1186/s13046-021-01996-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, Zhang Q, Wakelam MJO, Karpe F, Schulze A, & Harris AL (2014). Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Reports, 9(1), 349–365. [DOI] [PubMed] [Google Scholar]
- 125.Zhang X, Saarinen AM, Hitosugi T, Wang Z, Wang L, Ho TH, & Liu J (2017). Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. eLife, 6, e31132. 10.7554/eLife.31132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Furuta E, Pai SK, Zhan R, Bandyopadhyay S, Watabe M, Mo Y, Hirota S, Hosobe S, Tsukada T, & Miura K (2008). Fatty acid synthase gene is up-regulated by hypoxia via activation of akt and sterol regulatory element binding protein-1. Cancer Research, 68(4), 1003–1011. [DOI] [PubMed] [Google Scholar]
- 127.Keith B, Johnson RS, & Simon MC (2012). HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nature Reviews Cancer, 12(1), 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Olzmann JA, & Carvalho P (2019). Dynamics and functions of lipid droplets. Nature Reviews Molecular Cell Biology, 20(3), 137–155. 10.1038/s41580-018-0085-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Koizume S, & Miyagi Y (2016). Lipid droplets: A key cellular organelle associated with cancer cell survival under normoxia and hypoxia. International Journal of Molecular Sciences, 17(9), 1430. 10.3390/ijms17091430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Qiu B, Ackerman D, Sanchez DJ, Li B, Ochocki JD, Grazioli A, Bobrovnikova-Marjon E, Diehl JA, Keith B, & Simon MC (2015). HIF2α-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discovery, 5(6), 652–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.van der Mijn JC, Fu L, Khani F, Zhang T, Molina AM, Barbieri CE, Chen Q, Gross SS, Gudas LJ, & Nanus DM (2020). Combined metabolomics and genome-wide transcriptomics analyses show multiple HIF1α-induced changes in lipid metabolism in early stage clear cell renal cell carcinoma. Translational Oncology, 13(2), 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ackerman D, Tumanov S, Qiu B, Michalopoulou E, Spata M, Azzam A, Xie H, Simon MC, & Kamphorst JJ (2018). Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Reports, 24(10), 2596–2605. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Longo J, van Leeuwen JE, Elbaz M, Branchard E, & Penn LZ (2020). Statins as anticancer agents in the era of precision medicine. Clinical Cancer Research, 26(22), 5791–5800. [DOI] [PubMed] [Google Scholar]
- 134.Falchook G, Infante J, Arkenau H, Patel MR, Dean E, Borazanci E, Brenner A, Cook N, Lopez J, & Pant S (2021). First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine, 34, 100797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.US national library of medicine. (2017). TVB- 2640 in combination with bevacizumab in patients with first relapse of high grade astrocytoma. Unpublished manuscript. https://ClinicalTrials.gov/show/NCT03032484
- 136.US national library of medicine. (2017). FASN inhibitor TVB-2640, paclitaxel, and trastuzumab in treating patients with HER2 positive advanced breast cancer. Unpublished manuscript. https://ClinicalTrials.gov/show/NCT03179904
- 137.US national library of medicine. (2017). PT2385 for the treatment of von hippel-lindau disease-associated clear cell renal cell carcinoma. Unpublished manuscript. https://clinicaltrials.gov/ct2/show/NCT03108066