

Abstract
ATP6AP1-CDG is an X-linked disorder typically characterized by hepatopathy, immunodeficiency and an abnormal type II transferrin glycosylation pattern. Here, we present eleven new patients and clinical updates with biochemical characterization on one previously reported patient. We also document intrafamilial phenotypic variability and atypical presentations, expanding the symptomatology of ATP6AP1-CDG to include dystonia, hepatocellular carcinoma, and lysosomal abnormalities on hepatic histology. Three of our subjects received successful liver transplantation. We performed N-glycan profiling of total and fractionated plasma proteins for six patients and show associations with varying phenotypes, demonstrating potential diagnostic and prognostic value of fractionated N-glycan profiles. The aberrant N-linked glycosylation in purified transferrin and remaining plasma glycoprotein fractions normalized in one patient post hepatic transplant, while the increases of Man4GlcNAc2 and Man5GlcNAc2 in purified immunoglobulins persisted. Interestingly, in the single patient with isolated immune deficiency phenotype, elevated high-mannose glycans were detected on purified immunoglobulins without glycosylation abnormalities on transferrin or the remaining plasma glycoprotein fractions. Given the diverse and often tissue specific clinical presentations and the need of clinical management post hepatic transplant in ATP6AP1-CDG patients, these results demonstrate that fractionated plasma N-glycan profiling could be a valuable tool in diagnosis and disease monitoring.
Keywords: ATP6AP1-CDG, liver failure, immunodeficiency, hyperkinetic movement, dystonia, congenital disorder of glycosylation
1. INTRODUCTION
ATPase, H+ Transporting, Lysosomal, Accessory Protein 1 (ATP6AP1) deficiency, also known as ATP6AP1-CDG, CDG IIs, and Immunodeficiency 47 (OMIM 300972), is an X-linked condition where hemizygous males typically present with hepatic dysfunction, immunodeficiency, and an abnormal type II transferrin glycosylation pattern affecting both N- and O-glycosylation.1 The clinical and biochemical phenotype of ATP6AP1-CDG emphasizes the importance of maintaining Golgi homeostasis for glycosylation.2, 3 Glycan assembly is initiated in endoplasmic reticulum (ER), while its further trimming and processing occur in the Golgi apparatus. Of the over 160 CDG types identified to date,4 many also implicate Golgi structure and ER-Golgi trafficking in addition to Golgi homeostasis, which are essential for proper glycosylation.2, 3, 5–7 Interestingly, ATP6AP1 presents in both ER membrane and ER-Golgi intermediate compartment membrane.1
ATP6AP1-CDG was first identified in 2016 in eleven patients from six families.1 Subsequently, eight more ATP6AP1-CDG cases from another six families have been reported in the literature.8–13 Affected patients frequently exhibit coagulopathy, elevated liver enzymes, cholestatic jaundice, low immunoglobulins, recurrent infections, and reduced response to vaccines. Hematologic abnormalities include leukopenia, normocytic anemia and thrombocytopenia.1, 8, 9, 11 Pancreatic insufficiency has been reported with low trypsin and pancreatic elastase, and reduced fat-soluble vitamins.9 Many patients have laboratory abnormalities, including low serum copper and/or ceruloplasmin and hypercholesterolemia.1, 8–13 Neurological manifestations of ATP6AP1-CDG vary among patients and include epilepsy, sensorineural hearing loss (SNHL), intellectual disability and developmental delay.1, 9, 13
Here we report eleven new patients with ATP6AP1-CDG from eight different families and four novel ATP6AP1 variants and provide clinical manifestation updates and glycomic results on a previously reported patient.8 Given the striking hepatic dysfunction and immunodeficiency phenotype of ATP6AP1-CDG and the successful use of liver transplant as a treatment, we profiled N-glycans from fractionated subsets of plasma proteins including purified immunoglobulins, transferrin, and the remaining plasma glycoprotein fractions. Glycosylation abnormalities in different plasma fractions suggest fractionated plasma N-glycan profiling could serve as a valuable tool in diagnosis and disease monitoring.
2. MATERIALS AND METHODS:
2.1. Patients:
Our cohort includes a total of twelve patients from nine different families; one of these patients had been previously reported by Witters et al.8 Written informed consents were obtained from all patients involved in this study. A PubMed search was performed using the terms: ATP6AP1 and ATP6AP1-CDG. Data from all published cases are synthesized in this report (Supplementary table 1).
2.2. Biochemical analysis:
2.2.1. Fractionation of plasma glycoprotein:
Total glycoproteins in 15 μl plasma were fractionated into three groups using affinity columns: 1.) immunoglobulins (IgG), 2.) transferrin and 3.) the remaining glycoproteins depleted of IgG, transferrin and albumin using a protein G (Thermo Fisher Scientific, USA), followed with an IgG plus albumin duo depletion spin column (Thermo Fisher Scientific) and an anti-transferrin affinity spin column as described previously.14 Purified IgG was eluted from the initial protein G column and purified transferrin was eluted from anti-transferrin affinity column. The remaining glycoprotein fraction was collected after passing plasma through a protein G column, IgG plus albumin duo depletion column and an anti-transferrin affinity column sequentially. The purity of intact proteins in each fraction was evaluated by Ultra High-Pressure Liquid Chromatography Electrospray ionization-Quadrupole Time of Flight (UPLC-ESI-QTOF) Mass Spectrometry analysis using a SYNAPT G2-Si ™ system from Waters Corporation (Plymouth Meeting, PA, USA). No cross contamination of glycoproteins in each fraction was detected. Each fraction is lyophilized and resuspended in 30 μl water. N-glycan from 50–60 μg of IgG, 15–20 μg transferrin and 50–60 μg remaining glycoproteins were used for N-glycan analysis respectively.
2.2.2. N-glycan analysis of plasma total and fractionated glycoprotein:
N-glycans were released from each fraction using a rapid PNGaseF digestion, derivatized and purified using Rapifluor ™ N-glycan preparation kit as described previously.15 Briefly, an N-hydroxysuccinimide carbamate tag with a modified quinolone is added to the transient glycosyl amide group at the reducing end of the glycans. The derivatized N-glycans were purified using a 96 well HILIC plate and analyzed using a flow-injection-ESI-QTOF Mass Spectrometry method. A glycopeptide standard with isotope labelled disialoglycan was used as the internal standard. The abundance of each glycan is reported as % total glycans. 40 control plasma collected from 40 identified known non-CDG patients or healthy volunteers with age ranging from 1 week to 72 years old were used to collect the reference ranges. More than one age matched controls were used for each patient in our cohort.
3. RESULTS:
Survival:
The youngest patient in this cohort is 5.5 months old, while the oldest one is 67 years old. Three patients were deceased at the ages of 5.5, 6 and 18 months (P5, P3, and P2, respectively); other patients are alive at the time of this report. The main cause of death was liver failure which was complicated by respiratory syncytial virus (RSV) infection in P2 and SARS-CoV-2 infection in P3.
3.1. Survival
The youngest patient in this cohort is 5.5 months old, while the oldest one is 67 years old. Three patients were deceased at the ages of 5.5, 6, and 18 months (P5, P3, and P2, respectively); other patients are alive at the time of this report. The main cause of death was liver failure, which was complicated by respiratory syncytial virus (RSV) infection in P2 and SARS-CoV-2 infection in P3.
3.2. Molecular variants:
Among the newly reported patients, P1, P6–8, P9 and P10–11 are hemizygous for four novel variants: c.25C>T (p.Arg9*), c.401C>T (p.Pro134Leu), c.294C>A (p.Ser98Arg) and c.230_232delACT (p.Tyr77del), respectively. The other patients are hemizygous for the previously reported recurrent variant: c.1036G>A (p.Glu346Lys). The mode of inheritance was confirmed in 10/11 of the new patients, and de novo variants are reported in six of them.
3.3. Clinical findings:
Clinical manifestations of the patients in this study are summarized in table 1, which also contains updates of the previously reported patient by Witters et al. (P12).8 Hepatic dysfunction and immunodeficiency are the most common manifestations in newly reported patients of this study (9/11 and 9/10, respectively). The hepatic manifestations were evident during the first year of life, ranging in severity from isolated elevated liver enzymes to cirrhosis and hepatic failure. Three patients (P6, P10 and P11) received orthotopic liver transplant at the ages of 9 months, 10 months, and 7 months, respectively. The post-transplant course for P6 was complicated by hepatic artery thrombosis (HAT) which required emergent exploration on two occasions, repeat thrombectomies and anastomotic revision. He also had Klebsiella bacteremia with acute cholangitis and developed long-term complications of biliary strictures requiring a temporary biliary drain. These three patients continue to show evidence of exocrine pancreatic insufficiency post-transplant which is treated with pancrelipase. P6 also continues to have untreated intermittent hypogammaglobulinemia.
Table 1:
Summary of clinical manifestations and laboratory findings of the study subjects, including the updates (Bolded) for a previously reported patients.8
| P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | P11 | P12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |||
| DNA variant | c.25C>T | c.1036G>A | c.1036G>A | c.1036G>A | c.1036G>A | c.401C>T | c.401C>T | c.401C>T | c.294C>A | c.230_232delACT | c.230_232delACT | c.649T>A |
| Protein variant | p.Arg9* (R9*) | p.Glu346Lys (E346K) | p.Glu346Lys (E346K) | p.Glu346Lys (E346K) | p.Glu346Lys (E346K) | p.Pro134Leu (P134L) | p.Pro134Leu (P134L) | p.Pro134Leu (P134L) | p.Ser98Arg (S98R) | p.Tyr77del (Y77del) | p.Tyr77del (Y77del) | p.Tyr217Asn (Y217N) |
| Novel variant | Yes | No | No | No | No | Yes | Yes | Yes | Yes | Yes | Yes | - |
| Inheritance | De Novo | De novo | De novo | De novo | Maternally inherited | Maternally inherited | Maternally inherited | NR | Maternally inherited | De novo | De novo | Maternally inherited |
| Survival | Alive, 4 years | Died, 18 months | Died, 6 months | Alive, 19 years | Died, 5.5 months | Alive, 28 months | Alive, 5 years | Alive, 67 years | Alive, 13 years | Alive, 5 years and 9 months | Alive, 5 years and 9 months | Alive, 5 years |
| Age/Mode of presentation | 11 months/DD and dystonia | 10 months/hypoglycemia, metabolic acidosis, seizure like activity |
2 months/feeding difficulties, bilious vomiting, jaundice | 1 year/idiopathic cirrhosis | 28 days/jaundice that was treated with phototherapy and phenobarbital | 6 months/liver failure | Diagnosed via familial targeted variant testing | Diagnosed via familial targeted variant testing | Recurrent infections | 2 months/jaundice | 2 months/jaundice | 2 weeks/UTI E. coli sepsis |
| Perinatal history | Normal | Dilated kidneys (resolved), neonatal jaundice | Normal | Neonatal jaundice | High maternal serum AFP | Normal | Normal | NR | Normal | Cystic hygroma, cardiomegaly, dilated ascending aorta | Cystic hygroma, cardiomegaly, dilated ascending aorta | Normal |
| Hepatic abnormalities | No | Yes, elevated liver enzymes, liver failure, hepatosplenomegaly | Yes, elevated liver enzymes, acute hepatic failure, hepatosplenomegaly, cholestasis | Yes, elevated liver enzymes, cirrhosis, portal hypertension, hepatosplenomegaly | Yes, elevated liver enzymes, acute hepatic failure, cholestasis, hepatosplenomegaly | Yes, hepatic failure, elevated liver enzymes, cholestasis, hepatosplenomegaly S/P liver transplant at 9 months of age | Yes, mildly elevated liver enzymes | Yes, elevated alkaline phosphatase | No | Yes, elevated liver enzymes, chronic hepatic failure, hepatomegaly, cirrhosis S/P liver transplant at 10 months of age | Yes, elevated liver enzymes, chronic hepatic failure, hepatomegaly S/P liver transplant at 7 months of age | Yes, Elevated liver enzymes, hepatomegaly (resolved), splenomegaly, cholestasis (resolved) |
| Other GI findings | Feeding difficulties (resolved) | FTT | FTT | Gallstones | - | Exocrine pancreatic insufficiency | - | Chronic idiopathic diarrhea | - | Exocrine pancreatic insufficiency | Exocrine pancreatic insufficiency | Slow weight gain, exocrine pancreatic insufficiency |
| Coagulopathy | No | Yes, high INR, low antithrombin and protein C activity | Yes | No | Yes | Yes, mildly low factors XI, X, IX, V and II | No | No | No | Yes, resolved post-transplant | Yes, resolved post-transplant | Slight decrease of factor XI |
| Immunodeficiency | No | NR | Yes, recurrent infections | Yes, recurrent infections and low immunoglobulin | Yes, recurrent infections | Yes, recurrent infections pre-liver transplant (streptococcal pneumonia bacteremia, E. coli bacteremia and E. coli peritonitis) | Yes, low immunoglobulin | Yes, low immunoglobulin | Yes, recurrent infections and low immunoglobulin | Yes, frequent URIs and ear infections normal immunoglobulin | Yes, frequent URIs, bronchiolitis, pneumonia and ear infections normal immunoglobulin | Yes, recurrent infections, low immunoglobulin |
| Neurological findings | Hyperkinetic movement of hands and feet, complex febrile seizure at 34 months | Seizure in the setting of hypoglycemia only | No | Complex partial epilepsy | No | No | No | SNHL | No | Hypotonia (resolved) | Hypotonia (resolved) | No |
| Developmental Delay | Yes, mainly motor and speech | Yes | No | Yes | No | No | No | No | No | Yes, resolved | Yes, resolved but continues to require speech therapy | No |
| Hematological findings | No | Pancytopenia | Thrombocytopenia, anemia (required multiple transfusions), spherocytosis | Pancytopenia, required regular PRBC transfusions, Beta thalassemia carrier | Thrombocytopenia, hemolytic anemia, required multiple transfusions vacuolated lymphocytes on peripheral blood smear | Anemia | Anemia | Lymphopenia and anemia | No | Hemolytic anemia requiring frequent transfusion, thrombocytopenia (resolved post liver transplant) | Hemolytic anemia requiring frequent transfusion, thrombocytopenia (resolved post liver transplant) | Slight dysplasia of myeloid and erythroid lineages and a typical cells with vacuolization and inclusions on bone marrow biopsy |
| Cutis laxa | No | No | No | No | No | Yes, on anterior chest and abdomen | No | No | No | Yes | Yes | Yes (improved with age) |
| Dysmorphism | No | Microcephaly | No | Thin upper lip, prominent nose | No | Full cheeks | Mild dolichocephaly | No | No | Mild trigonocephaly, low set and posteriorly rotated ears with prominent lobes, inverse epicanthal folds, high palate, webbed neck, pectus carinatum, wide-spaced nipples, clinodactyly of the fifth toe bilaterally | Midline occipital plagiocephaly, long face, low set and posteriorly rotated ears with prominent lobes, inverse epicanthal folds, high palate, webbed neck, pectus carinatum, wide-spaced nipples, clinodactyly of the fifth toe bilaterally | Yes, deep-set eyes |
| Cardiac findings | No | Borderline prolonged QT interval | Small ASD (ostium secundum type) | No | Bradycardia, mild left ventricular hypertrophy | No | No | No | No | Large secundum ASD (surgically repaired), dilated ascending aorta | Large secundum ASD (surgically repaired), dilated ascending aorta | Dilation of sinus aorta (stable on follow up) |
| Low serum copper/ceruloplasmin level | No | NR | NR | No | NR | Yes, normal copper, low ceruloplasmin | Yes, low copper and ceruloplasmin | NR | No | Yes | Yes | Yes (transiently resolved) |
| Hypercholesterolemia | No | NR | Yes | No | Yes | No | NR | NR | No | No | No | Yes (transiently resolved) |
| Elevated CK | No | Yes | Yes, mild increase | No | NR | No | NR | NR | NR | NR | NR | No |
| Liver imaging | Normal | Cirrhosis with multiple regenerating hepatic nodules and focal steatosis within the left hepatic lobe | NR | NR | US: hepatic parenchymal echogenicity | NR | NR | Normal US | Normal CT | US: echogenic liver with hypoechoic lesions CT: multiple heterogeneously enhancing regions and dominant hypoenhancing lesion | US: homogeneous liver with multiple varices | MRI: Focal steatosis |
| Liver biopsy | NR | Macro-regenerative nodule, abundant glycogen in the cytoplasm, pushing other organelles to the cell margins | NR | Cirrhosis | Fibrosis, lipid-laden (foamy) macrophages | Cirrhosis | NR | NR | NR | Steatosis, evolving cirrhosis, multiple nodules of varying sizes with well-differentiated fibrolamellar hepatocellular neoplasms of two nodules | Cirrhosis, cholestasis, swollen hepatocytes with intra-cellular eosinophilic globules associated with scattered large droplet fat Electron microscopy: prominent and enlarged lysosomes contain electron-dense clumped and whorled material | Micronodular cirrhosis with ductular reaction, steatosis, PAS-positive globules |
| Others | Normal brain and spinal MRI | Osteopenia on X-ray | Left ureter dilation | Congenital kyphosis with abnormal vertebrae, Moya Moya angiopathy, tethered cord, inguinal hernia, left cryptorchidism, right renal cyst | - | - | - | OSA | - | Umbilical and bilateral inguinal hernia, hypermobility of the elbow and distal digit joints, chordee hypospadia, right diaphragmatic hernia (post-liver transplant) | Umbilical and left inguinal hernia, hypermobility of the elbow and distal digit joints, chordee with orthotopic meatus, undescended left testicle | Unilateral cryptorchidism, small umbilical hernia, inguinal hernia, anterior diaphragmatic hernia (Morgagni) |
| Cause of death | NA | RSV infection | SARS-CoV-2 infection | NA | Hepatic encephalopathy and cerebral edema | NA | NA | NA | NA | NA | NA | NA |
Abbreviations: GI, gastrointestinal; CK, creatine kinase; DD, developmental delay; NR, not reported; MRI, magnetic resonance imaging; NA, not applicable; FTT, failure to thrive; INR, international normalized ratio; RSV, respiratory syncytial virus; ASD, atrial septal defect; PRBC, Packed red blood cells; AFP, alpha-fetoprotein; US, ultrasound; S/P, status post; SNHL, sensorineural hearing loss; OSA, obstructive sleep apnea; CT, computerized tomography; URI, upper respiratory infection; UTI, urinary tract infection; PAS, periodic acid-Schiff.
Most patients showed evidence of immunodeficiency in the forms of hypogammaglobulinemia, inadequate vaccine response and/or recurrent infections such as otitis media, streptococcal pneumonia and E. coli infections with the exception of P1. Two patients (P4 and P9) are on immunoglobulin therapy, while other patients with hypogammaglobulinemia (P6–8, P10 and P11) are on regular monitoring only with no required therapy. P12 initially presented with recurrent infections in the setting of normal immunoglobulin levels;8 however, the patient developed chronic cough and hypogammaglobulinemia on longer term follow-up, and he was managed with subcutaneous immunoglobulin injections and prophylactic antibiotic.
Neurological involvement has been reported in 6/11 patients. The degree of developmental delay is variable and improved with age. The twin siblings (P10 and P11) had hypotonia at younger ages that later resolved. P1 presented with hyperkinetic movement with no hepatic dysfunction or immunodeficiency. A comprehensive neurologic, genetic, and metabolic evaluation excluded other causes of dystonia. P4, who is Caucasian, was found to have tethered cord and cerebral Moyamoya angiopathy; these findings were not previously reported in ATP6AP1-CDG and their presence in this patient could be coincidental and not secondary to the underlying condition. However, the incidence rate of Moyamoya angiopathy is highest among East Asian population but considered of rare occurrence in the patient population.16 Late onset sensorineural hearing loss was only identified in the oldest patient (P8) of this study and he is the only old adult in our cohort.
Dysmorphism (7/12) and cardiac involvement (6/12) were less frequently reported findings. The identified dysmorphic features were not consistent among the patients with ATP6AP1-CDG. Maternal inheritance of ATP6AP1 variant was confirmed in 4/10 patients of three families. Intrafamilial phenotypic variability was noted among the patients. The proband (P6) of family 6 (Figure 1) presented in early infancy with hepatic dysfunction that required liver transplantation. The brother (P7) was diagnosed via targeted variant testing, and evaluation revealed mildly elevated liver enzymes and subclinical hypogammaglobulinemia. The variant was subsequently detected in their maternal grandfather (P8) who experienced previously idiopathic chronic diarrhea (5–10 stools a day), hypogammaglobulinemia, and elevated alkaline phosphatase for over 30 years; and P8’s brother died in infancy with unknown cause. P10 and P11 are monochorionic diamniotic twins; they presented with early hepatic involvement and received liver transplant at the 10 and 7 months of age, respectively. However, P11 demonstrated higher degree of dysmorphism and developmental delay and more frequent infections than his twin brother, and he continues to require speech therapy. Liver biopsy findings of these twins were also variable. Six separate nodules of varying sizes were identified on gross examination of the liver explant of P10, two of these nodules showed parallel fibrous lamellae, eosinophilic hepatocytes with enlarged nuclei, and a paucireticulin pattern, with immunohistochemical features consistent with well-differentiated fibrolamellar hepatocellular carcinoma. P11’s liver biopsy revealed swollen hepatocytes with intracellular eosinophilic globules and scattered large fat droplets. Electron microscopy was remarkable for prominent enlarged lysosomes with abnormal storage of whorled materials. These findings are reminiscent of the changes in sphingomyelin lipidosis associated with Niemann-Pick disease.
Figure 1:
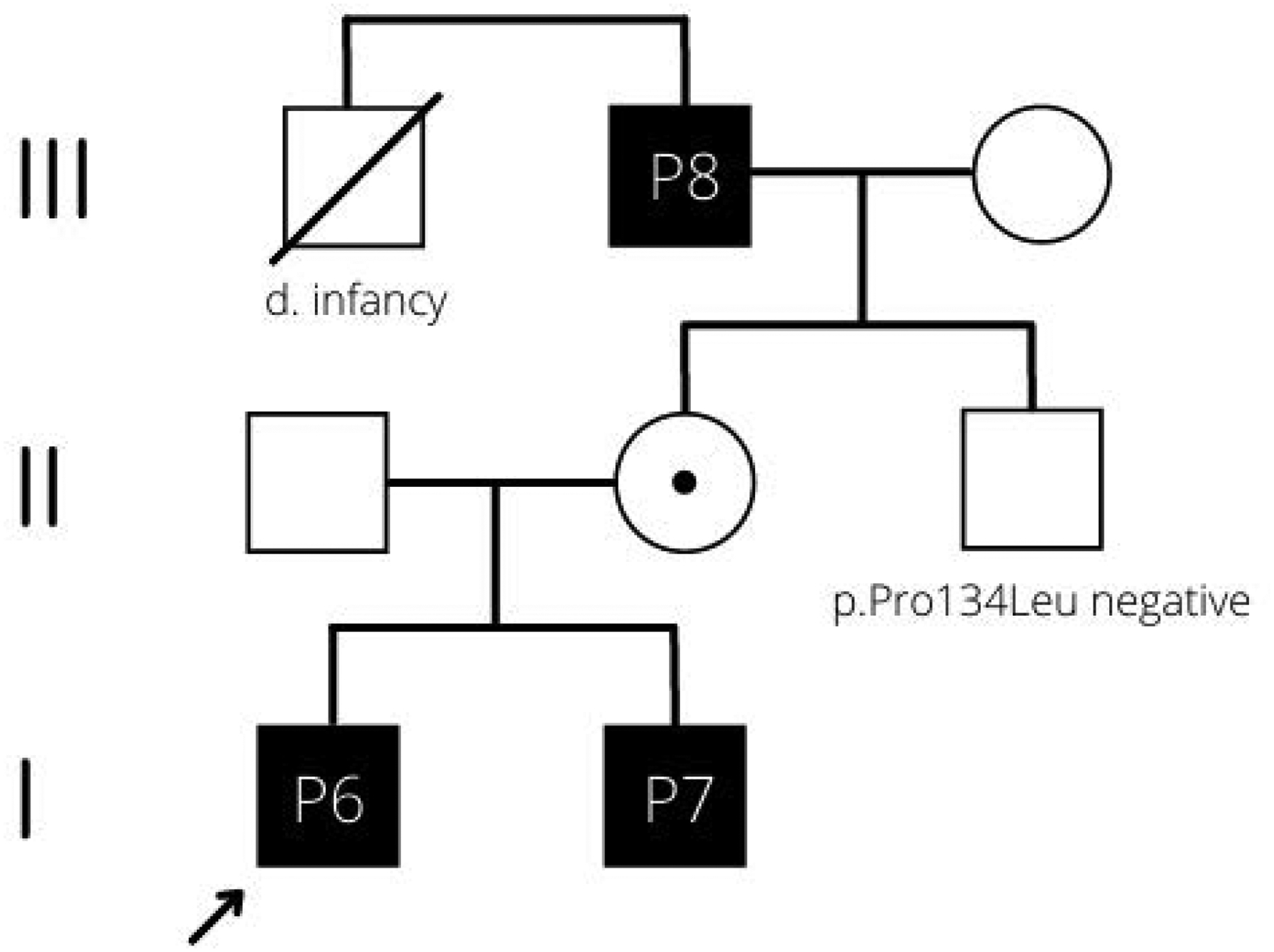
Pedigree structure of family 6. The arrow indicates the index patient (P6).
3.4. Biochemical findings:
Given the prominent clinical phenotypes of immunodeficiency and hepatic disease being common among ATP6AP1-CDG patients, we evaluated N-glycans on fractionated plasma glycoproteins: Immunoglobulin G, transferrin and the remaining glycoprotein fraction. Fractionated glycosylation studies were performed on six affected individuals (P1, P4, P6, P7, P9 and P12). The results are summarized in table 2. We identified elevated N-linked Man4 and Man5, along with under-galactosylation and/or under-sialyation of complex-type glycans as consistent abnormalities in the tested samples. Mildly or atypically affected patients (P1 and P9) demonstrated mild biochemical abnormalities in a subset of protein fractions, mainly isolated under-sialylation in the transferrin fraction for P1 who presented with dystonia with no immune deficiency or other cardinal features, and high mannose glycan changes in IgG fraction for P9 who in contrast to P1, only manifested with immunodeficiency. P9 has been on IVIG therapy before he was referred to the genetic service, and plasma glycosylation studies for P9 were only done on samples obtained 2 weeks and 2 months post-intravenous immunoglobulin therapy. Severely affected patients with hepatic involvement (P6 and P12) showed significant abnormalities in all three fractions. In particular, the changes of both high mannose and complex-type glycans are present in all fractions from P12 before starting the immunoglobulin therapy. P4, another severely affected patient, received his intravenous immunoglobulin (IVIG) infusion 1 day before the sample was drawn, which likely masked some IgG abnormalities. P4 also receives packed red blood cell (pRBC) transfusion routinely. Since the plasma was collected before his next pRBC transfusion, we expect effects on the last transfusion to be small. Nonetheless, both the transferrin and the remaining glycoprotein fraction of this patient showed under-galactosylation and significant under-sialylation. Carbohydrate deficient transferrin (CDT) and apolipoprotein assay performed by mass spectrometry (LC-ESI- MS) of the intact glycoproteins14 in both P1 and P4 showed increased under-sialylated transferrin and essentially normal Apolipoprotein CIII O-glycosylation (Table 3). The siblings, P6 and P7, demonstrated similar severe glycosylation defects which is consistent with the shared genotype. Interestingly, during P6 initial presentation, Tri-sialo/Di-oligosaccharide transferrin ratio (a marker for undersialylation) is lower than the ratio in P7 or P8 with normal Apolipoprotein CIII-1/CIII-2 ratio, while CIII-0/CIII-2 ratio is increased14. Post-liver transplantation, these ratios were trending down and totally normalized within 1.5 months (Table 3). In consistent with this intact glycoprotein analysis, the PNGaseF released N-glycan abnormalities of his total plasma glycoprotein, transferrin and remaining glycoprotein fraction resolved, but despite the use of immunosuppressant, the IgG glycosylation remains abnormal comparing to either non-CDG patients with status post-liver transplant or normal population (Table 2). The other tested sibling (P7), who is clinically milder, however showed similar marked glycosylation changes, although to a lesser degree in the IgG fraction, including elevated Man4 and Man5. Under-galactosylation of complex-type glycans were detected in all plasma fractions with under-sialylation being more obvious than P6. The sample from P8 is not available for fractionation studies. However, CDT and Apolipoprotein analysis results for P7 and P8 are similar or somewhat more “severe” than pre-transplant values of P8 despite variable clinical phenotype (Table 3).
Table 2:
N-glycan profile of total and fractionated plasma protein for six patients; the values are demonstrated as percentage of total glycan.
| N-glycans (%total) | P1 | P4 1 day Post-IVIG |
P6 Pre-Liver Transplant |
P7 | P9 2 Weeks Post-IVIG |
P9 2 Months Post-IVIG |
P12 Pre-Immunoglobul in Therapy |
Reference Range | P6 Post-Liver Transplant |
P6 >6 months Post-Liver Transplant |
Reference Range Non-CDG Post-Liver Transplant |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Plasma Total Protein | |||||||||||
Hex4HexNAc2 (Man4)
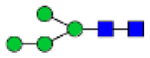
|
0.88 | 0.72 | 1.52 | 1.37 | 0.60 | 0.90 | 4.24 | 0.22 −1.20 | 0.73 | 0.97 | 1.07 – 1.32 |
Hex5HexNAc2 (Man5)
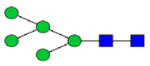
|
2.32 | 2.12 | 4.10 | 3.58 | 1.52 | 2.23 | 10.84 | 0.81 −3.13 | 1.93 | 2.60 | 2.98 – 3.54 |
|
M-Gal
|
1.85 | 4.44 | 5.31 | 6.20 | 1.34 | 1.14 | 6.68 | 0.67 −1.88 | 1.00 | 1.12 | 0.98 – 1.15 |
|
M-sialo
|
22.20 | 14.05 | 13.72 | 19.28 | 15.93 | 12.90 | 11.61 | 7.75 −16.55 | 8.17 | 11.24 | 6.14 – 10.85 |
| Selected Ratios – Plasma Total Protein | |||||||||||
| Man5/Man6 | 1.31 | 1.43 | 2.20 | 1.94 | 1.03 | 1.21 | 3.82 | 0.76 – 1.22 | 0.88 | 0.87 | 1.05 – 1.30 |
| Man5/Man9 | 2.73 | 2.59 | 7.74 | 5.42 | 2.11 | 3.91 | 13.22 | 1.51 – 3.61 | 2.17 | 2.26 | 5.73 – 6.33 |
| Transferrin Fraction | |||||||||||
Hex4HexNAc2 (Man4)
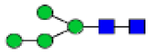
|
0.34 | 0.11 | 0.62 | 0.15 | 0.03 | 0.22 | 1.21 | 0.15 – 0.30 | 0.17 | 0.24 | 0.20 – 0.48 |
Hex5HexNAc2 (Man5)
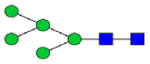
|
0.57 | 0.26 | 1.42 | 0.35 | 0.36 | 0.49 | 3.12 | 0.06 – 0.56 | 0.44 | 0.64 | 0.52 – 1.18 |
|
M-Gal
|
0.87 | 2.75 | 5.60 | 6.16 | 0.56 | 0.96 | 9.25 | 0.47 – 1.26 | 1.19 | 0.64 | 0.69 – 0.88 |
|
M-sialo
|
14.70 | 15.62 | 14.45 | 24.60 | 5.51 | 7.65 | 20.05 | 3.75 – 10.12 | 5.73 | 6.09 | 5.21 – 7.11 |
|
D-sialo
|
62.76 | 61.54 | 47.15 | 50.24 | 73.36 | 68.33 | 36.46 | 60.60 – 75.6 | 68.36 | 73.77 | 66.9 – 70.9 |
| IgG Fraction | |||||||||||
Hex4HexNAc2 (Man4)
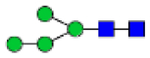
|
0.10 | 0.07 | 0.30 | 0.16 | 0.10 | 0.14 | 0.34 | 0.04 – 0.14 | 0.16 | 0.18 | 0.06 – 0.11 |
Hex5HexNAc2 (Man5)
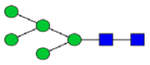
|
0.27 | 0.19 | 0.79 | 0.48 | 0.27 | 0.32 | 0.87 | 0.08 – 0.33 | 0.32 | 0.46 | 0.17 – 0.30 |
|
M-Gal
|
0.40 | 0.42 | 0.42 | 0.40 | 0.74 | 0.35 | 0.90 | 0.24 – 0.60 | 0.55 | 0.51 | 0.27 – 0.65 |
|
M-sialo
|
0.93 | 0.86 | 0.62 | 0.71 | 1.67 | 0.88 | 1.83 | 0.48 – 1.75 | 0.97 | 0.59 | 0.53 – 0.85 |
| Selected Ratios – IgG Fraction | |||||||||||
| Man5/Man6 | 0.56 | 1.27 | 3.76 | 2.29 | 1.23 | 1.68 | 2.56 | 0.18 – 1.14 | 1.68 | 2.30 | 0.50 – 1.29 |
| Man5/Man9 | 1.29 | 0.90 | 4.16 | 2.40 | 1.00 | 0.91 | 3.78 | 0.36 – 2.17 | 1.14 | 1.48 | 0.72 – 1.07 |
| Remaining Fraction | |||||||||||
Hex4HexNAc2 (Man4)
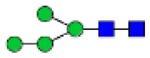
|
0.99 | 1.19 | 2.06 | 1.65 | 0.68 | 0.96 | 3.73 | 0.53 – 1.56 | 0.65 | 0.78 | 0.79 – 1.01 |
Hex5HexNAc2 (Man5)
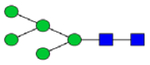
|
2.68 | 3.09 | 5.37 | 4.65 | 1.88 | 2.50 | 10.51 | 1.43 – 4.00 | 1.80 | 2.09 | 2.11 – 2.73 |
|
M-Gal
|
1.79 | 4.57 | 5.72 | 5.91 | 1.06 | 0.93 | 8.42 | 0.55 – 2.41 | 0.83 | 1.46 | 1.05 – 1.90 |
|
M-sialo
|
21.85 | 16.98 | 15.48 | 19.85 | 17.18 | 14.98 | 14.42 | 11.2 – 25.7 | 7.58 | 12.48 | 10.1 – 14.9 |
|
D-sialo
|
47.68 | 45.96 | 38.79 | 34.77 | 58.39 | 50.25 | 26.39 | 36.1 – 61.1 | 62.12 | 56.28 | 52.3 – 56.7 |
| Selected Ratios – Remaining Fraction | |||||||||||
| Man5/Man6 | 1.84 | 1.75 | 2.95 | 1.96 | 1.42 | 0.98 | 5.42 | 0.88 – 1.95 | 0.97 | 1.17 | 1.07 – 1.26 |
| Man5/Man9 | 5.47 | 3.91 | 12.20 | 9.69 | 4.09 | 3.57 | 13.83 | 1.74 – 5.88 | 3.67 | 2.95 | 2.78 – 3.90 |
Note: Significant out-of-range results are presented in bold for increased values and in italic for decreased values. The reference range consists of values of 40 normal controls. Post-transplant sample results for P6 were also compared to a reference range consists of values of three non-CDG post-liver transplant patients. Results of P9 include samples at 2 weeks and 2 months post immunoglobulin therapy.
Abbreviations: IVIG, intravenous immunoglobulin.
Table 3:
The results of carbohydrate deficient transferrin and Apolipoprotein CIII for P1, P4 and P6–9. The table shows the serial measurements for P6 pre and post liver transplant. The out-of-range results are colored in red.
| Patient | Mono-oligo/Di-Oligosaccharide | A-oligo/Di-Oligosaccharide | Tri-Sialo/Di-Oligosaccharide | Apo-CIII-1/CIII-2 | Apo-CIII-0/CIII-2 | |
|---|---|---|---|---|---|---|
| Reference Range | <0.06 | <0.011 | <0.05 | <2.91 | <0.48 | |
| P1 | 0.016 | 0.033 | 0.21 | 2.92 | 0.3 | |
| P4 | 0.04 | 0.003 | 0.18 | 2.15 | 0.44 | |
| P6 (Pre-Liver Transplant) | Day 26 | 0.11 | 0.01 | 0.27 | 1.6 | 0.65 |
| Day 1 | 0.1 | 0.007 | 0.34 | 1.62 | 0.76 | |
| P6 (Post-Liver Transplant) | Day 2 | 0.07 | 0.006 | 0.1 | 0.88 | 0.18 |
| Day 3 | 0.05 | 0.003 | 0.06 | 0.58 | 0.11 | |
| Day 3 | 0.06 | 0.002 | 0.08 | 0.74 | 0.15 | |
| Day 5 | 0.09 | 0.007 | 0.12 | 0.65 | 0.25 | |
| Day 6 | 0.05 | 0.004 | 0.08 | 1.11 | 0.35 | |
| Day 44 | 0.04 | 0.003 | 0 | 1.33 | 0.25 | |
| P7 | 0.07 | 0.007 | 0.49 | 4.42 | 0.62 | |
| P8 | 0.07 | 0.011 | 0.53 | 4.42 | 1.32 | |
| P9 | 0.06 | 0.005 | 0.03 | 0.89 | 0.08 | |
4. DISCUSSION:
The V-ATPase is an ATP-dependent proton pump that regulates the pH of many intracellular compartments. It has important functions in endocytosis, intracellular transport, cell growth and transformation and entry of certain viruses and toxins into the cell. It is also essential in targeting newly synthesized lysosomal enzymes from the Golgi to the lysosomes.17 The V-ATPase complex is composed of two multi-protein domains, peripheral V1 and integral V0,18 and two accessory proteins ATP6AP1 (also known as Ac45) and ATP6AP2 (also known as (pro)renin receptor).19 ATP6AP1 is ubiquitously expressed with the highest expression found in the brain, neuroendocrine cells and osteoclasts.1, 20, 21
Several CDG have been associated with pathogenic variants in genes encoding various subunits of the vacuolar (H+)-ATPase (V-ATPase) complex. Mutations in ATP6V0A2, ATP6V1A and ATP6V1E1 genes result in autosomal recessive defects in protein glycosylation and share cutis laxa as a common clinical feature22, 23. Disorders due to genetic defects of accessory subunits follow an X-linked mode of inheritance. ATP6AP2-CDG presents with cutis laxa, liver disease, immunodeficiency, cognitive impairment and ataxia.24 ATP6AP1-CDG was first described in 2016 by Jansen et al. with liver dysfunction and immunodeficiency as the prominent features.1
Including this study, a total of 30 patients with ATP6AP1-CDG are reported. A summary of the clinical features of all the patients is depicted in figure 2. Hepatopathy and immunodeficiency are the most commonly documented systemic involvement, followed by hematological and neurological abnormalities. Interestingly, P1 presented with developmental delay and dystonic movement in the form of chorea and athetosis of hands and feet with no cardinal features of hepatic involvement and immunodeficiency. This is the first report of a hyperkinetic movement disorder occurring in ATP6AP1-CDG. However, several other CDG types have been reported to involve hyperkinetic movement disorders.25–28 They are typically non-progressive and of early onset, and they are also thought to be an underappreciated manifestation in CDG patients.25 Complex hyperkinesia also characterizes NGLY1-CDDG, which is a disorder impairing the cytosolic deglycosylation of glycoproteins28, 29 a process that activates essential transcriptional factors.30 Similarly, V-type ATPass complex and assembly factors are also known to control transcriptional factors through regulation of cellular ion levels.31
Figure 2:
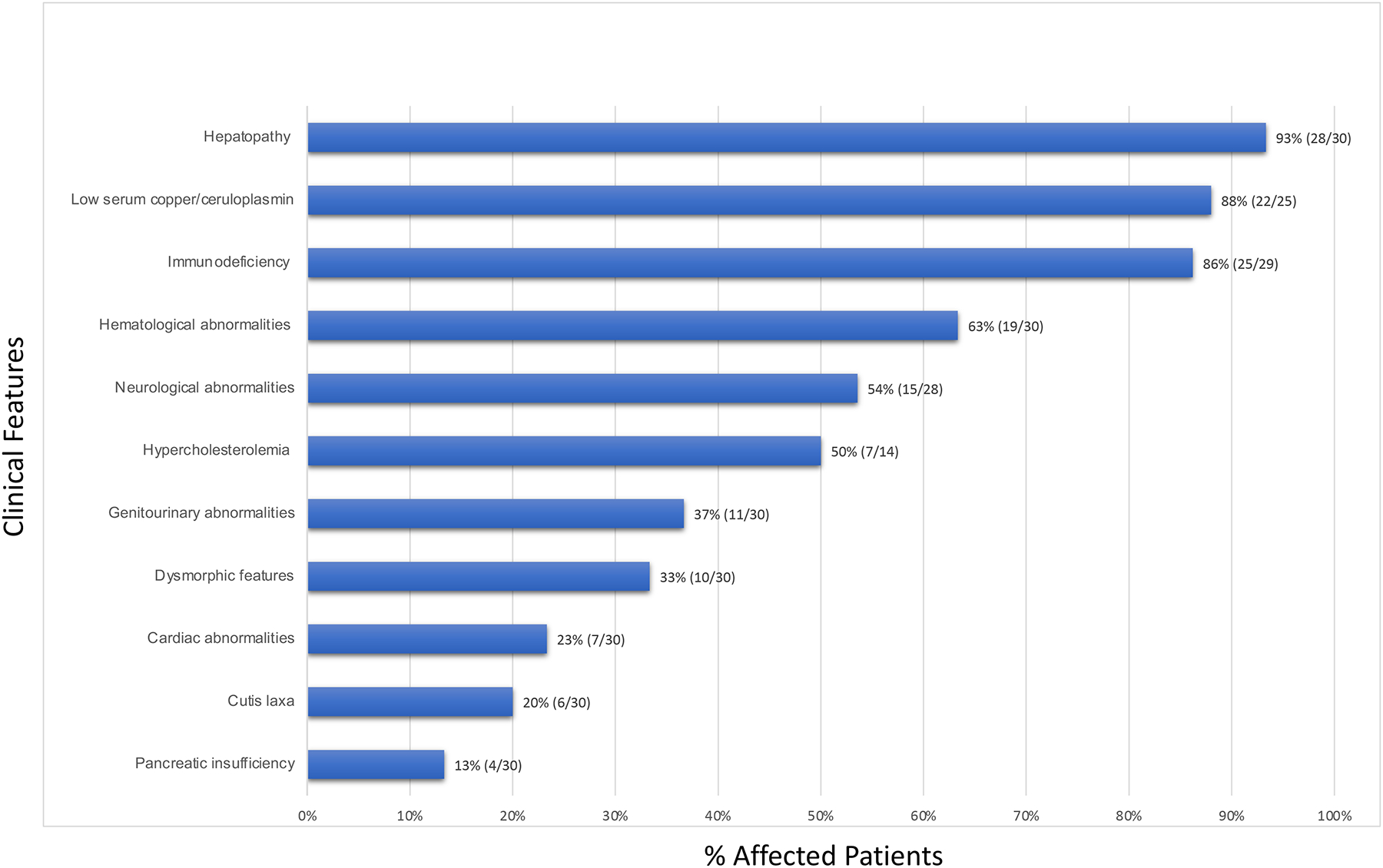
Clinical summary of all reported ATP6AP1-CDG patients (including this study) demonstrated as percentage of affected patients (number of affected patients/total number of reported or tested patients).
Connective tissue involvement such as cutis laxa, joint hypermobility, and inguinal hernia are less frequently observed. These features were thought to be partially related to reduction of plasma copper and/or ceruloplasmin level9, 10 which was identified in 22/25 of ATP6AP1-CDG patients. Ceruloplasmin is a liver secreted glycoprotein and its reduction has been reported in other CDGs with hepatic involvement.32 Given that the ceruloplasmin is the main plasma copper binding protein, the serum copper level will be low if the ceruloplasmin production is impaired. Lysyl oxidase, a copper-dependent enzyme, is essential for extracellular elastin and collagen crossing,33 and its activity could be impaired by low copper level. However, no abnormalities of elastin fibers were detected on skin biopsy of an ATP6AP1-CDG patient manifested with cutis laxa,8 and the exact mechanism needs to be established. CDG screening via CDT and N-glycans analysis could be considered for patients with unexplained low copper/ceruloplasmin levels. Progressive hair loss is another rarely reported finding identified in three patients34 but was not seen in the older patients of our cohort. The reported dysmorphic features (10/30) are variable among the patients and have no consistent pattern (9, 11, 12 and P2, P4, P6–7 and P10–12).
Some features improve with age and include hepatic involvement and cutis laxa8, 9, 34 as well as developmental delay (P1, P4, P10 and 11). Improvement of visceral and cutaneous manifestations has been previously reported in other CDG types.23, 35, 36 However, hair loss and SNHL are apparently progressive over time.34 P8, who is 67 years old at the time of evaluation, has a history of SNHL with onset at age 65, which could be age-related. Proteinuria also appeared in older patients with no associated renal dysfunction34, but it was not documented in this study. Laboratory abnormalities including low plasma copper level and hypercholesterolemia of P12 normalized when the cholestasis resolved8 but reappeared on longer term follow up.
All reported patients to date are males. Heterozygous females are generally asymptomatic and lack the main characteristic manifestations of the disease. The only potential manifestations from heterozygous females described in a single extended pedigree were proteinuria and SNHL (possibly age-related) in 2/3 and 1/3 carrier mothers, respectively.1, 34 Their glycosylation studies were reported as normal,34 although fractionated N-glycan analysis was not performed. In our study, the de novo variant rate is higher than previously reported (60% vs 11%), and glycosylation study by routine CDT analysis from the heterozygous mother of P6 and P7 was normal and her sample was not available for plasma N-glycan analysis; none of the female carriers had reported significant past medical history.
Given the paucity of the reported cases with this disorder, an evident genotype-phenotype correlation is hard to establish. The p.Glu346Lys (E346K) is the most commonly reported variant (de novo or inherited) from 8 families of different ethnicity and associated with early death in 5 out of 11 patients (1 and P2, P3 and P5). Detection of this variant in our severely affected patients (P2-P5) is also supportive for its severity. The p.Leu74Pro variant was found in two patients with severe phenotype who died in infancy due to liver failure; but they were from the same family.10 The p.Leu311Gln variant has been associated with early mortality in one infant and severe hepatic involvement of his affected brother who underwent liver transplantation.11, 37 The p.Arg9* variant creates a premature stop codon in exon 1 of the ATP6AP1 gene. While this transcript appears to be the mostly highly expressed in all tissues, there is an alternative transcript that excludes a small region of exon 1 for which this variant would be noncoding. Additionally, there are potential alternative in-frame start sites using methionine at codons 12 and 23 in exon 1, which could bypass this variant and produce a nearly full length protein with truncated signal peptide, potentially explaining the milder phenotype and the normal glycosylation in IgG and the remaining glycoprotein fractions of P1.
Intrafamilial variability was pronounced in family 6 of this study who carry the newly reported variant (p.Pro134Leu). The proband (P6) presented with severe hepatic involvement that required liver transplantation. However, his older brother (P7) and maternal grandfather (P8) were clinically mild, and the variant was detected via targeted molecular testing. Intrafamilial phenotypic variability has been reported in other types of CDG.38, 39 Although ATP6AP1 is an X-linked gene, it has a pseudogene ATP6AP1L on chromosome 5, which may contribute to the heterogeneity within families due to several possibilities including possible mitotic intragenic recombination.40 Overall, the degree of glycosylation changes between siblings in this family are similar. We suspect that the different clinical severity among siblings may not be entirely due to the pathogenic variant, but rather it could be partly triggered by postnatal stresses, such as viral infection. P10 and P11, who are monochorionic diamniotic twins with de novo p.Tyr77del variant, expressed some degree of clinical and histologic variability as well. P11 demonstrated a more clinically significant course and his hepatic biopsy findings revealed lysosomal abnormalities concerning for storage disorders. In contrast, P10 had more prominent steatosis, and hepatocellular carcinoma was identified on liver explant. These findings were not previously reported in ATP6AP1-CDG and may indicate an increased risk of hepatic neoplasm development in this disorder.
Including this study, four cases of ATP6AP1-CDG with hepatic involvement who underwent successful liver transplantation are reported.37 The patients in this study tolerated the procedure well with complications of HAT and biliary stricture for P6, and right diaphragmatic hernia for P10. They also demonstrated long term post-transplant survival of 19 months for P6, more than 4 years for P10, and 5 years for P11.
A defective Golgi structure has been identified in a previous report.10 Our findings of under-galactosylation and -sialyation and the increases of small high mannose-type glycans species in this study support the association of Golgi dysfunction in ATP6AP1-CDG. However, increases of small high mannose glycans are more commonly seen in patients with defect in ER associated glycan assembling. In addition, not all the affected patients present with both hepatic disease and immune deficiency. In our cohort, P1 does not have any immune deficiency. Consistent with this, the glycosylation of IgG fraction in P1 is normal (Table 2). Conversely, P9 presents with isolated immune deficiency and does not have any hepatic dysfunction. In this particular patient, the glycosylation in transferrin and the remaining glycoprotein fraction, most of which are made in liver, is normal, while increased Man4 and Man5/6 ratio were detected in the IgG fraction (Table 2). It is interesting that mildly increased Man4 and Man5 were also detected in the transferrin fraction of P1. These findings suggest an association between the tissue specific phenotype and glycan abnormalities, which would potentially help in prognosis and directing individualized care. Additionally, persistent N-glycan abnormalities in the IgG fraction when the CDT and transferrin N-glycan fraction normalized after liver transplant in P6 illustrates the diagnostic utility of fractionated N-glycan analysis in individuals post liver transplant. The use of immunosuppressant post-transplant often depresses the glycosylation and IgG protein level in plasma and make it difficult to detect abnormalities even when reference ranges of non-CDG patients post liver transplant are used. We found that N-glycan analysis of IgG purified from patient plasma can overcome this issue by normalize the amount of IgG protein used and allows us to monitor glycosylation changes in our patients. An essentially normal IgG N-glycan fraction one day post IVIG therapy in P4 may indicate a source of false negatives for this assay. P4 was also on regular pRBC transfusion (pRBC), and we cannot rule out the possibility that his milder glycosylation changes in transferrin being related to ongoing therapy. However under-galactosylation is readily detected in the plasma and the remaining fraction from P4 consistent with the known severe mutation he carries.
In conclusion, we present eleven additional cases of ATP6AP1-CDG, six of them caused by four novel genetic variants. This study emphasizes the utility of both plasma/serum carbohydrate deficient transferrin and N-glycan biochemical analysis as well as molecular genetic analysis. The biochemical studies in this report demonstrated new findings of under-galactosylation and/or under- sialyation of complex-type glycans and increases of Man4 and Man5 in different fractions of plasma glycoproteins. This new approach provides a sensitive method of detecting the underlying glycosylation abnormalities and may be useful disease biomarkers.
Supplementary Material
Supplementary Table 1: Overview of the previously reported ATP6AP1-CDG patients.
* Updates on these patients were reported by Lipinski et al.28
Abbreviations: GI, gastrointestinal; SNHL, sensorineural hearing loss; NR, not reported; B/L, bilateral; N/A, not applicable; FH, family history; FTT, failure to thrive; S/P, status post; IVIG, Intravenous immune globulin; MRI, magnetic resonance imaging; ASD, atrial septal defect; US, ultrasound.
Synopsis:
This study expands the phenotype of ATP6AP1-CDG, including identifying intrafamilial phenotypic variability in a multi-generational pedigree, and reports novel gene variants. We also identify glycosylation abnormalities in purified immunoglobulins, transferrin and remaining glycoprotein fractions correlating with disease phenotypes.
Funding details:
This work was supported by The Rocket Fund and NIH grants R01DK99551 (HHF) and U54 NS115198 (CL, HHF, KMR, ACE, and MH) from the National Institute of Neurological Diseases and Stroke (NINDS) and the National Center for Advancing Translational Sciences (NCATS), and the Rare Disorders Clinical Research Network (RDCRN), at the National Institute of Health. P.W. was funded by the Fonds Wetenschappelijk Onderzoek-Vlaanderen (Fundamenteel Klinisch Mandaat 18B4322N).
Footnotes
Conflict of Interest: Hana Alharbi, Earnest James Paul Daniel Jenny Thies, Irene Chang, Dana L. Goldner, Bobby G. Ng, Peter Witters, Amal Aqul, Frances Velez-Bartolomei, Evelyn Hsu, Elizabeth Kichula, Esther Lee, Charles Lourenco, Sheri A Poskanzer, Sara Rasmussen, Katelyn Saarela, YunZu M. Wang, Kimiyo M. Raymond, Matthew J Schultz, Christina Lam, Andrew C. Edmondson, and Miao He declare that they have no conflicts of interest. Gregory M. Enns has served as a consultant for Glycomine, Inc. Hudson H. Freeze is a consultant for Avalo Therapeutics.
Ethics approval and Informed Consent: All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. This study was approved by the institutional review board of the respective institutions. Written informed consent was obtained from all patients’ guardians before inclusion in the study.
Animal rights: This article does not contain any studies with animal subjects performed by any of the authors.
Data availability statement:
Additional supporting information may be found online in the Supporting Information section at the end of this article.
References
- 1.Jansen EJR, Timal S, Ryan M, et al. ATP6AP1 deficiency causes an immunodeficiency with hepatopathy, cognitive impairment and abnormal protein glycosylation. Nature Communications. 2016; 7:11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang X, Wang Y. Glycosylation quality control by the golgi structure. J Mol Biol. 2016; 428:3183–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freeze HH, Ng BG. Golgi glycosylation and human inherited diseases. Cold Spring Harb Perspect Biol. 2011; 3:a005371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freeze HH, Jaeken J, Matthijs G. CDG or not CDG. J Inherit Metab Dis. 2022;. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaeken J, Péanne R. What is new in CDG? J Inherit Metab Dis. 2017; 40:569–586. [DOI] [PubMed] [Google Scholar]
- 6.Ferreira CR, van Karnebeek CDM, Vockley J, Blau N. A proposed nosology of inborn errors of metabolism. Genet Med. 2019; 21:102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verheijen J, Tahata S, Kozicz T, Witters P, Morava E. Therapeutic approaches in congenital disorders of glycosylation (CDG) involving N-linked glycosylation: An update. Genet Med. 2020; 22:268–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Witters P, Breckpot J, Foulquier F, Preston G, Jaeken J, Morava E. Expanding the phenotype of metabolic cutis laxa with an additional disorder of N-linked protein glycosylation. Eur J Hum Genet. 2018; 26:618–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dimitrov B, Himmelreich N, Hipgrave Ederveen AL, et al. Cutis laxa, exocrine pancreatic insufficiency and altered cellular metabolomics as additional symptoms in a new patient with ATP6AP1-CDG. Mol Genet Metab. 2018; 123:364–374. [DOI] [PubMed] [Google Scholar]
- 10.Ondruskova N, Honzik T, Vondrackova A, et al. Severe phenotype of ATP6AP1-CDG in two siblings with a novel mutation leading to a differential tissue-specific ATP6AP1 protein pattern, cellular oxidative stress and hepatic copper accumulation. J Inherit Metab Dis. 2020; 43:694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tvina A, Thomsen A, Palatnik A. Prenatal and postnatal phenotype of a pathologic variant in the ATP6AP1 gene. Eur J Med Genet. 2020; 63:103881. [DOI] [PubMed] [Google Scholar]
- 12.Quelhas D, Martins E, Azevedo L, et al. Congenital disorders of glycosylation in portugal-two decades of experience. J Pediatr. 2021; 231:148–156. [DOI] [PubMed] [Google Scholar]
- 13.Yang X, Lv Z, Tang Q, et al. Congenital disorder of glycosylation caused by mutation of ATP6AP1 gene (c.1036G>A) in a chinese infant: A case report. World J Clin Cases. 2021; 9:7876–7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lacey JM, Bergen HR, Magera MJ, Naylor S, O’Brien JF. Rapid determination of transferrin isoforms by immunoaffinity liquid chromatography and electrospray mass spectrometry. Clin Chem. 2001; 47:513–518. [PubMed] [Google Scholar]
- 15.Chen J, Li X, Edmondson A, et al. Increased clinical sensitivity and specificity of plasma protein N-glycan profiling for diagnosing congenital disorders of glycosylation by use of flow injection-electrospray ionization-quadrupole time-of-flight mass spectrometry. Clin Chem. 2019; 65:653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujimura M, Bang OY, Kim JS. Moyamoya disease. Front Neurol Neurosci. 2016; 40:204–220. [DOI] [PubMed] [Google Scholar]
- 17.Nishi T, Forgac M. The vacuolar (H+)-ATPases--nature’s most versatile proton pumps. Nat Rev Mol Cell Biol. 2002; 3:94–103. [DOI] [PubMed] [Google Scholar]
- 18.Forgac M Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007; 8:917–929. [DOI] [PubMed] [Google Scholar]
- 19.Jansen EJR, Martens GJM. Novel insights into V-ATPase functioning: Distinct roles for its accessory subunits ATP6AP1/Ac45 and ATP6AP2/(pro) renin receptor. Curr Protein Pept Sci. 2012; 13:124–133. [DOI] [PubMed] [Google Scholar]
- 20.Holthuis JC, Jansen EJ, Schoonderwoert VT, Burbach JP, Martens GJ. Biosynthesis of the vacuolar H+-ATPase accessory subunit Ac45 in xenopus pituitary. Eur J Biochem. 1999; 262:484–491. [DOI] [PubMed] [Google Scholar]
- 21.Feng H, Cheng T, Pavlos NJ, et al. Cytoplasmic terminus of vacuolar type proton pump accessory subunit Ac45 is required for proper interaction with V0 domain subunits and efficient osteoclastic bone resorption. J Biol Chem. 2008; 283:13194–13204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kornak U, Reynders E, Dimopoulou A, et al. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat Genet. 2008; 40:32–34. [DOI] [PubMed] [Google Scholar]
- 23.Van Damme T, Gardeitchik T, Mohamed M, et al. Mutations in ATP6V1E1 or ATP6V1A cause autosomal-recessive cutis laxa. Am J Hum Genet. 2017; 100:216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rujano MA, Cannata Serio M, Panasyuk G, et al. Mutations in the X-linked ATP6AP2 cause a glycosylation disorder with autophagic defects. J Exp Med. 2017; 214:3707–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mostile G, Barone R, Nicoletti A, et al. Hyperkinetic movement disorders in congenital disorders of glycosylation. Eur J Neurol. 2019; 26:1226–1234. [DOI] [PubMed] [Google Scholar]
- 26.Bögershausen N, Shahrzad N, Chong J, et al. Recessive TRAPPC11 mutations cause a disease spectrum of limb girdle muscular dystrophy and myopathy with movement disorder and intellectual disability. Am J Hum Genet. 2013; 93:181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altassan R, Péanne R, Jaeken J, et al. International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: Diagnosis, treatment and follow up. J Inherit Metab Dis. 2019; 42:5–28. [DOI] [PubMed] [Google Scholar]
- 28.Lam C, Wolfe L, Need A, Shashi V, Enns G. NGLY1-Related Congenital Disorder of Deglycosylation. In: Adam MP, Ardinger HH, Pagon RA, et al. (eds). GeneReviews®. Seattle (WA): University of Washington, Seattle, 1993:. [PubMed] [Google Scholar]
- 29.Need AC, Shashi V, Hitomi Y, et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet. 2012; 49:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomlin FM, Gerling-Driessen UIM, Liu Y, et al. Inhibition of NGLY1 inactivates the transcription factor Nrf1 and potentiates proteasome inhibitor cytotoxicity. ACS Cent Sci. 2017; 3:1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miles AL, Burr SP, Grice GL, Nathan JA. The vacuolar-ATPase complex and assembly factors, TMEM199 and CCDC115, control HIF1α prolyl hydroxylation by regulating cellular iron levels. Elife. 2017; 6: 10.7554/eLife.22693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heywood WE, Bliss E, Mills P, et al. Global serum glycoform profiling for the investigation of dystroglycanopathies & congenital disorders of glycosylation. Mol Genet Metab Rep. 2016; 7:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith-Mungo LI, Kagan HM. Lysyl oxidase: Properties, regulation and multiple functions in biology. Matrix Biol. 1998; 16:387–398. [DOI] [PubMed] [Google Scholar]
- 34.Lipiński P, Rokicki D, Bogdańska A, Lesiak J, Lefeber DJ, Tylki-Szymańska A. ATP6AP1-CDG: Follow-up and female phenotype. JIMD Rep. 2020; 53:80–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lam C, Krasnewich DM. PMM2-CDG. In: Adam MP, Ardinger HH, Pagon RA, et al. (eds). GeneReviews®. Seattle (WA): University of Washington, Seattle, 1993:. [PubMed] [Google Scholar]
- 36.Mohamed M, Kouwenberg D, Gardeitchik T, Kornak U, Wevers RA, Morava E. Metabolic cutis laxa syndromes. J Inherit Metab Dis. 2011; 34:907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gumm AJ, Basel DG, Thakrar P, Suchi M, Telega G. Liver failure and x-linked immunodeficiency type 47. Pediatric transplantation. 2020; 24:e13808–n/a. [DOI] [PubMed] [Google Scholar]
- 38.Riess S, Reddihough DS, Howell KB, et al. ALG3-CDG (CDG-id): Clinical, biochemical and molecular findings in two siblings. Mol Genet Metab. 2013; 110:170–175. [DOI] [PubMed] [Google Scholar]
- 39.Rymen D, Winter J, Van Hasselt PM, et al. Key features and clinical variability of COG6-CDG. Mol Genet Metab. 2015; 116:163–170. [DOI] [PubMed] [Google Scholar]
- 40.Kane MS, Davids M, Adams C, et al. Mitotic intragenic recombination: A mechanism of survival for several congenital disorders of glycosylation. Am J Hum Genet. 2016; 98:339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Overview of the previously reported ATP6AP1-CDG patients.
* Updates on these patients were reported by Lipinski et al.28
Abbreviations: GI, gastrointestinal; SNHL, sensorineural hearing loss; NR, not reported; B/L, bilateral; N/A, not applicable; FH, family history; FTT, failure to thrive; S/P, status post; IVIG, Intravenous immune globulin; MRI, magnetic resonance imaging; ASD, atrial septal defect; US, ultrasound.
Data Availability Statement
Additional supporting information may be found online in the Supporting Information section at the end of this article.