

Graphical Abstract
SUMMARY
Background:
Non-alcoholic fatty liver disease (NAFLD) occurs in around one-quarter of the global population and is one of the leading causes of chronic liver disease. The phenotypic manifestation and the severity of NAFLD are influenced by an interplay of environmental and genetic factors. Recently, several inactivating variants in the novel 17-Beta hydroxysteroid dehydrogenase 13 (HSD17B13) gene have been found to be associated with a reduced risk of chronic liver diseases, including NAFLD.
Aims:
To review the existing literature on the epidemiology of HSD17B13 and discuss its role in the natural history, disease pathogenesis and treatment of NAFLD.
Methods:
We extensively searched relevant literature in PubMed, Google Scholar clinicaltrials.gov and the reference list of articles included in the review.
Results:
HSD17B13 is a liver-specific, lipid droplet (LD)-associated protein that has enzymatic pathways involving steroids, proinflammatory lipid mediators and retinol. The estimated prevalence of the most well-characterized HSD17B13 variant (rs72613567) ranges from 5% in Africa to 34% in East Asia. Loss-of-function variants in HSD17B13 are protective against the progression of NAFLD from simple steatosis to non-alcoholic steatohepatitis (NASH), liver fibrosis, cirrhosis, as well as hepatocellular carcinoma (HCC). Emerging data from mechanistic and preclinical studies with RNA interference (RNAi) and small molecule agents indicate that inhibiting HSD17B13 activity may prevent NAFLD progression.
Conclusions:
The loss-of-function polymorphisms of the newly identified HSD17B13 gene mitigate the progression of NAFLD. It is important to understand the exact mechanism by which these variants exert a protective effect and implement the gathered knowledge in the treatment of NAFLD.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) occurs in around one quarter of the global population.1,2 NAFLD is characterized by macrovesicular lipid droplet (LD) accumulation within hepatocytes in the absence of significant alcohol consumption or other secondary causes of steatosis.3,4 NAFLD encompasses an array of histopathological alterations, spanning from non-alcoholic fatty liver (NAFL); the non-progressive form of NAFLD, to non-alcoholic steatohepatitis (NASH), which can progress to cirrhosis, hepatocellular carcinoma (HCC) and hepatic decompensation.2,5 Although NAFLD is closely related to obesity and the metabolic syndrome, NAFLD can develop in non-obese or lean individuals as well.6,7,8
NAFLD is the most common cause of chronic liver disease worldwide and is thought to impact 25% of the global population, with up to 20% of those with the disease progressing to NASH.2,4 Liver fibrosis, which is the major prognostic predictor of mortality in patients with NAFLD, develops in 41% of those with NASH, as determined by meta-analysis of paired biopsy studies.2,9 About 15–20% of patients with NASH is predicted to progress to cirrhosis, while the incidence of HCC in NASH cirrhosis is around 3.8 per 100 person-years.10,11,12,13 In fact, NASH is ranked the most rapidly growing cause of HCC among US patients awaiting liver transplantation.14
The susceptibility to NAFLD is influenced by an interplay of environmental and genetic factors.15 Over the past few years, genome-wide association studies (GWAS) and candidate gene approaches have presented novel insights into the genetic underpinnings of NAFLD pathogenesis. Since the discovery of rs738409 G-allele encoding the I148M variant of patatin-like phospholipase domain-containing protein 3 (PNPLA3) in 2008,16 an increasing number of single nucleotide polymorphisms (SNP) associated with NAFLD have been identified, namely transmembrane 6 superfamily member 2 (TM6SF2),17 membrane bound O-acyltransferase domain-containing 7 (MBOAT7)18,19 and glucokinase regulator (GCKR).20 Robust evidence from genetic studies demonstrates that variants of these genes increase the risk of NAFLD, hepatic fibrosis and HCC in the presence of environmental triggers.21
More recently, HSD17B13 has been identified as a new liver-specific LD-associated protein involved in the pathogenesis of NAFLD. This discovery gathered significant attention, because as opposed to previously described risk variants, genetic polymorphism in the gene coding for HSD17B13 results in loss of enzymatic activity and is linked to protection against NASH.22,23 A genetic variant (rs72613567) in HSD17B13 was the first variant found to be associated with reduced plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, as well as reduced progression from steatosis to steatohepatitis.22 Subsequent genetic studies detected several more HSD17B13 loss-of-function variants that were protective against the full spectrum of NAFLD in diverse populations.17,18,19,20,26 According to 1000 Genomes Project, the global frequency of the HSD17B13 minor allele varies from 23% in intergenic variant rs6834314 to 18% in variants rs72613567, rs9992651 and rs13118664, 6% in 143404524 and 2% in rs62305723.22,23,24,25,31,32 Although the precise mechanism by which HSD17B13 polymorphism protects against NAFLD awaits clarification, anti-HSD17B13 therapies are under ongoing investigation in clinical trials.33,34 In this review, we summarize the global epidemiology of HSD17B13 and highlight its role in pathogenesis and treatment of NAFLD.
Discovery of the link between HSD17B13 polymorphism and progression of NAFLD
In 2011, a GWAS in 61,089 individuals of Caucasian and Asian Indian ethnicity revealed that intergenic SNP (rs6834314) near HSD17B13 was strongly associated with plasma ALT concentrations, a marker of hepatocyte injury.35 In a subsequent comparative LD proteomic study of 21 human liver biopsies, Su et al. determined that HSD17B13 was a pathogenic protein involved in NAFLD.36
An important breakthrough towards identifying the role of HSD17B13 in NAFLD pathogenesis occurred in 2018, when an exome-wide association study revealed that a splice variant (rs72613567) in HSD17B13 conferred a protective effect against the development of NASH and advanced fibrosis.22 This variant (minor allele) is the product of an insertion of adenine adjacent to the donor splice site of exon 6 (T > TA), which disturbs mRNA splicing and generates truncated unstable proteins with decreased enzymatic activity. Among 46,544 obese individuals (median BMI 30 kg/m2), the presence of the splice variant (minor allele) was found to be associated with reduced levels of ALT (P=4.2×10−12) and AST (P=6.2×10−10). Subsequent studies detected five more independent variants in or near HSD17B13 gene that mitigate NASH progression and chronic liver injury: rs62305723 (encodes proline to serine mutation at AA position 260), rs6834314 (an intergenic variant in strong linkage disequilibrium with rs72613567),23 rs9992651 and rs13118664 (both non-coding variants in strong linkage disequilibrium with rs72613567),25 and rs143404524 (deletion and frameshift at codon 192).24 In all instances, improper splicing, insertion, deletion, or nonsynonymous mutations led to loss-of-function mutations in HSD17B13 gene and decrease in progression to advanced disease. However, while the above-mentioned polymorphisms in HSD17B13 are protective against disease progression to NASH and advanced fibrosis, they do not prevent the development of simple steatosis.22,23,24,25 These variants are discussed in more detail in Section 4.
The role of HSD17B13 in the progression of NAFLD
Characterization of HSD17B13
The 17β-hydroxysteroid dehydrogenase (HSD17B) family encompasses enzymes that catalyze the conversion between 17-ketosteroids and 17β-hydroxysteroids. At present, 15 HSD17Bs have been identified in mammals,37 all belonging to short-chain dehydrogenase/reductase (SDR) superfamily, with the exception of HSD17B5, which is a member of aldo-ketoreductase family.38 These enzymes display divergent expression patterns, variable tissue targeting, and subcellular localizations. Along with playing key roles in sex steroid metabolism, the HSD17B enzymes are involved in the metabolism of fatty acids, cholesterol, bile acids and retinoids. One of the latest additions to the HSD17B family occurred in 2007, when Liu et al. isolated HSD17B13 from an adult human liver cDNA library, originally named SCDR9.39 In 2008, Horiguchi et al. searched the data base for the paralogue of LD-associated protein HSD17B11 and identified HSD17B13 as a candidate protein.40 The authors further demonstrated that HSD17B13 targeted LDs and was expressed exclusively in the liver, in comparison to HSD17B11, which was induced in mouse liver and intestine.
Gene expression, tissue distribution and structure of HSD17B13
The HSD17B13 gene on chromosome 4 encodes a 300-amino acid protein that shares high sequence similarity (78%) with HSD17B11.39 The active enzyme has NAD/NADH binding motif TGxxxGxR (x indicates any amino acid residue) and the active center motif YxxxK that are characteristic for the members of SDR superfamily. HSD17B13 is highly observed in liver, as demonstrated in both human and mice tissue distribution studies.36,39,40,41 Expression in ovary, kidney, brain, lung, skeletal muscle, testis is also detectable at a much lower level. The protein is targeted from endoplasmic reticulum (ER) to LDs by its N-terminal region. Ma et al. showed N-terminal 28-amino acid sequence of HSD17B13 to be sufficient for its LD targeting by using in vitro mutagenesis.42 Furthermore, they identified three crucial fragments within 106-amino acid N-terminal to be critical for LD targeting, which are transmembrane amino acid 4–16 hydrophobic domain, amino acid 22–28 PAT-like domain and amino acid 69–106 alpha-helix/beta-sheet/alpha-helix structure. Interestingly, distinct cellular distribution patterns were observed when particular N-terminal domain underwent deletion. Whilst hydrophobic domain deletion reallocated HSD17B13 enzyme to mitochondria, deletion of PAT-like domain lowered protein stability. Lastly, HSD17B13 was retained and degraded in the ER when the deletion of alpha-helix/beta-sheet/alpha-helix fragment occurred, suggesting the structure’s possible role in proper folding and transportation of the protein from ER to LDs.
Regulation of HSD17B13 expression
The mechanisms regulating the transcription of HSD17B13 are still unclear. In 2010, Rotinen et al. reported that promoter region of HSD17B13 gene contains CCAAT boxes and binding sites for CCAAT enhancer binding factors (C/EBPs), indicating the possible involvement of C/EBPs in HSD17B13 transcriptional activity.43 Su et al.’s findings in murine hepatocyte cell lines demonstrated that liver x receptor-α (LXR-α) , a nuclear receptor which regulates the expression of genes involved in lipid metabolism, induces HSD17B13 expression via sterol regulatory binding protein-1c (SREBP-1c) (Figure 1).44 In turn, HSD17B13 promotes SREBP-1c maturation, generating a vicious positive feedback loop,36 which potentially contributes to the hepatic lipogenesis. SREBP-1c is a transcription factor that controls lipogenic gene expression and aberrant expression of SREBP-1c and LXR-a is associated with de novo lipogenesis, obesity and NAFLD.45,46,47 In contrast, HSD17B13 is dominantly expressed in peroxisome proliferator-activated receptor-alpha knockout (PPARα) mice models, hinting at the suppression of HSD17B13 expression by PPARα.40 This notion is consistent with the data from a gene expression study, in which mice exposed to PPARα agonist fenofibrate displayed reduction in HSD17B13 gene expression.48 PPARα is a nuclear receptor that regulates genes involved in fatty acid beta-oxidation and has a beneficial effect in preventing liver from steatosis, inflammation and fibrosis.49 To summarize, overexpression of HSD17B13 is associated with the higher levels of regulators of hepatic lipogenic gene expression, whereas the presence of modulators of fatty acid oxidation suppresses HSD17B13 expression.
Figure 1. Proposed role of HSD17B13 in modulating disease progression in NAFLD.
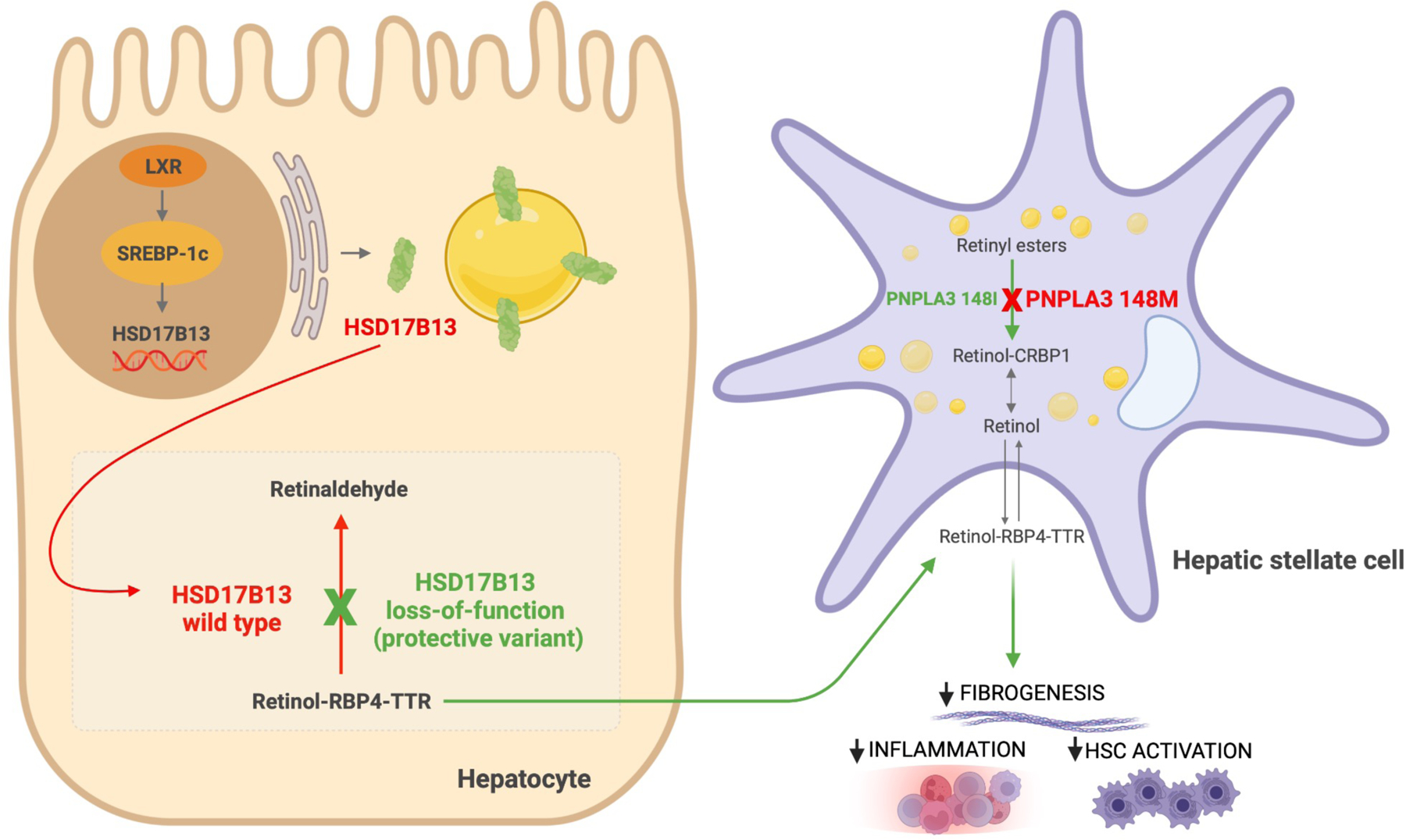
HSD17B13 expression is induced by liver x receptor-α (LXR-α) through sterol regulatory binding protein-1c (SREBP-1c) in the hepatocyte nucleus. HSD17B13 is then targeted from the endoplasmic reticulum to lipid droplets. Wild type HSD17B13 catalyzes the conversion of retinol to retinaldehyde. Genetic polymorphism in HSD17B13 results in a loss of this enzymatic activity, which increases retinol-retinol binding protein (RBP4)-transthyretin (TTR) transport from hepatocytes. PNPLA3 is located on lipid droplets, and it has hydrolase activity towards retinyl esters in hepatic stellate cells (HSCs). The I148M mutation results in a loss of retinyl esterase activity with retinol retention in HSCs. Retinoids play an important role in hepatic immunomodulation and suppression of HSC-mediated fibrogenesis. Abbreviations: CRBP1, cellular retinol-binding protein 1.
Function of HSD17B13 in healthy liver and NAFLD
The liver is the primary organ responsible for lipid homeostasis. Hepatocytes, the parenchymal cells of the liver, instigate mobilization of lipids for energy and store excess lipids in the form of LDs. Imbalances in the process of this physiological equilibrium can lead to hepatic steatosis. Plasma free fatty acid from high adipose tissue lipolysis, increased de novo lipogenesis, dietary fatty acids, reduced fatty acid oxidation as well as decreased secretion of very-low-density lipoproteins (VLDL) drive excess triglyceride (TG) accumulation in LDs.50 LDs are highly dynamic organelles comprised of neutral lipid core (triacylglycerols and cholesterol esters), which is surrounded by monolayer of phospholipids and sphingomyelin. The outermost surface of LDs is covered by distinct proteins whose principal roles encompass maintaining bioactivity of LDs and control of lipid trafficking and flux.51 HSD17B13 is newly identified liver-specific protein that localizes to the surface of LDs (Figure 1). Overexpression of HSD17B13 in cultured hepatocyte cell lines leads to the increase in number and size of LDs,36 potentially by stabilizing intracellular TG content.52 By contrast, enzymatically inactive HSD17B13 variants confer a protective effect against NAFLD progression to NASH. However, the physiological function of HSD17B13 and the mechanism by which HSD17B13 variants mediate protection from chronic liver damage remain to be fully elucidated. 3D structure of HSD17B13 homodimers, modeled by HSD17B11 template, predicted the catalytic tetrad Asn-144/Ser-172/Tyr-185/Lys-189, substrate-binding sites Lys-153, Leu-156, Leu-199, Glu-202, and Lys-208 and putative homodimer interaction sites Arg-97/Tyr-101 to be essential domains for the enzymatic activity of HSD17B13.42 Based on in vitro recombinant protein and cell-based assays, steroids, proinflammatory lipid mediators such as leukotriene B3 and B4 and retinol have been found to be the potential enzymatic substrates of HSD17B13.22,23,53 In agreement with these findings, morbidly obese (BMI 42 kg/m2) NAFLD patients undergoing bariatric surgery carrying HSD17B13 rs72613567 variant had significant downregulation of proinflammatory genes and plasma cytokine IL-6.22
Rare variants in HSD17B13 were found to perturb the plasma TG and high-density lipoprotein (HDL) levels in response to fenofibrate treatment.48 Recent plasma lipidomics analysis in children with NAFLD demonstrated that HSD17B13 rs72613567 variant was positively associated with very long-chain polyunsaturated TG and negatively associated with medium-chain monosaturated TG,54 providing further support for the role of HSD17B13 in lipid metabolism. Moreover, rs72613567 was found to increase the concentration of phospholipids in the liver, including phosphatidylcholines and phosphatidylethanolamines.55 Phospholipids are essential component of LD membrane and decreased levels of phospholipids have been proven to be linked to NAFL and NASH.56,57,58
The discovery of hepatic retinol dehydrogenase activity of HSD17B13 in in vitro cell-based assays23 was a step towards unraveling the biological role of HSD17B13 in NAFLD pathogenesis. Granted proper LD targeting and cofactor binding, HSD17B13 catalyzes the conversion of retinol to retinaldehyde in hepatocytes and genetic polymorphism in HSD17B13 results in a loss of this enzymatic activity (Figure 1).23,25 The liver is the central hub for vitamin A metabolism and NAFLD has a strong inverse correlation with hepatic retinoid levels.59 Retinyl esters from diet are taken up by hepatocytes and are hydrolyzed to form retinol. Retinol, bound to retinol binding protein 4 (RBP4), is then transported to hepatic stellate cells (HSCs) via an unknown mechanism and stored as retinyl esters in LDs. A growing body of evidence indicates that vitamin A metabolites play an important role in hepatic mitochondrial fatty acid β-oxidation, immunomodulation, and suppression of fibrogenesis60,61,62,63,64. Recent transcriptome analyses revealed the hyperdynamic state of hepatic retinol metabolism in NAFLD patients65 and retinyl ester accumulation in HSCs of NAFLD mouse models,66 further emphasizing the role of disturbed vitamin A metabolism in the development of NAFLD.
One of the enthralling findings related to HSD17B13’s function is the interplay between HSD17B13 polymorphism and PNPLA3 I148M. Intriguingly, HSD17B13 rs72613567 was shown to mitigate the risk of liver injury associated with PNPLA3 I148M mutation and decrease PNPLA3 mRNA expression among NAFLD patients.22,67 Another HSD17B13 intergenic variant rs6834314 attenuated the effect of PNPLA3 I148M on advanced hepatic fibrosis in 290 Japanese patients with biopsy-proven NAFLD.67 Furthermore, in a cohort of 328 non-morbidly obese Type 2 diabetes mellitus patients with NAFLD, PNPLA3 I148M genotype no longer predicted liver stiffness in carriers of HSD17B13 rs72613567.68 The positive trend between PNPLA3 I148M and HCC was completely blunted in hepatitis C patients who were carriers of HSD17B13 rs72613567,69 but data in NAFLD patients are lacking. Wild type PNPLA3 harbors retinyl ester hydrolase activity and I148M mutation of PNPLA3 results in enhanced retinyl ester accumulation in HSCs, alongside increased risk of HSC-mediated hepatic fibrogenesis.70,71,72 Impaired conversion of retinyl ester to retinol leads to decreased serum retinol and RBP4 levels in individuals carrying PNPLA3 I148M. Interestingly, in two murine models NASH was associated with reduced levels of hepatic retinol and RBP4, with the significant increase in hepatic retinyl ester storage and HSD17B13 expression.66 In a co-culture system, high HSD17B13 expression in hepatocytes indirectly induces HSC activation and downregulation of HSD17B13 in HFD-fed mice is associated with decreased HSC activation.52 Although speculative, these findings suggest that the protective effect of HSD17B13 loss-of-function variants on NAFLD-related liver damage might be partly via reduced HSC activity as a result of HSD17B13’s depleted retinol dehydrogenase enzymatic function, which leads to increase in hepatic retinol availability across the liver (Figure 1). This hypothesis needs to be validated and the potential mechanisms of HSD17B13 investigated. Taken together, current data indicate a strong link between HSD17B13, PNPLA3, HSCs and retinol metabolism in NAFLD pathogenesis.
Global prevalence of HSD17B13 variants
The demographic and ethnic characteristics of NAFLD vary across the globe, with the highest estimated prevalence in South and North America (35%), followed by Europe and Asia (30%), and the lowest in Africa (29%).2 A recent meta-analysis determined that the prevalence of NAFLD increased from 21.9% in 1991 to 37.3% in 2019.73 In the USA, the prevalence of NAFLD, NASH and NASH cirrhosis is highest in Hispanics, followed by non-Hispanic white individuals, whereas the lowest prevalence is observed in African Americans.74,75,76
The frequency of HSD17B13 protective variants differs across populations, which might potentially contribute to the interethnic variation in the prevalence and severity of NAFLD. The global prevalence of the most studied HSD17B13 variant (rs72613567) is 18%.28 It is most frequent among East Asians (34%) and Europeans (24%), followed by South Asians and Americans (16%) and less common in Africans (5%) according to the 1000 Genomes project (Figure 2). Data regarding the minor allele frequency of rs72613567 in NAFLD patients are limited. The prevalence of this risk-reducing variant among NAFLD patients is highest in China (34%),77 followed by the US (27%),78 and the least common in Argentina (16%) (Figure 3).79 It is less frequent in Hispanic patients (9%) compared to non-Hispanic patients (23%) with NAFLD in the US.80
Figure 2. Worldwide prevalence of HSD17B13 rs72613567 minor allele in general population.
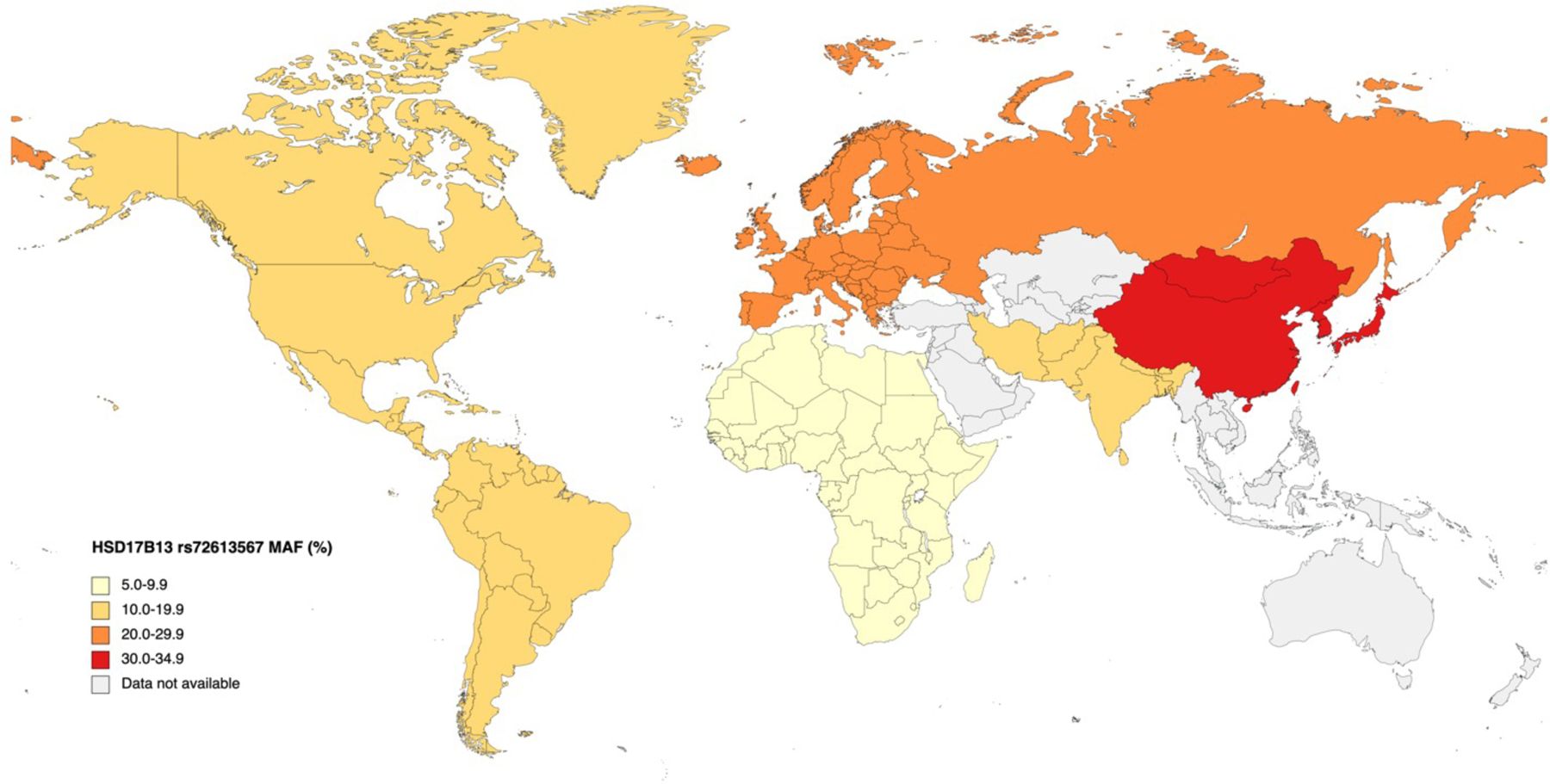
Data based on MAF in the general population are as follows: Africa, America, East Asia, Europe, South Asia: 1000 Genomes project.28 Abbreviations: MAF, minor allele frequency.
Figure 3. Worldwide prevalence of HSD17B13 rs72613567 minor allele in patients with NAFLD.
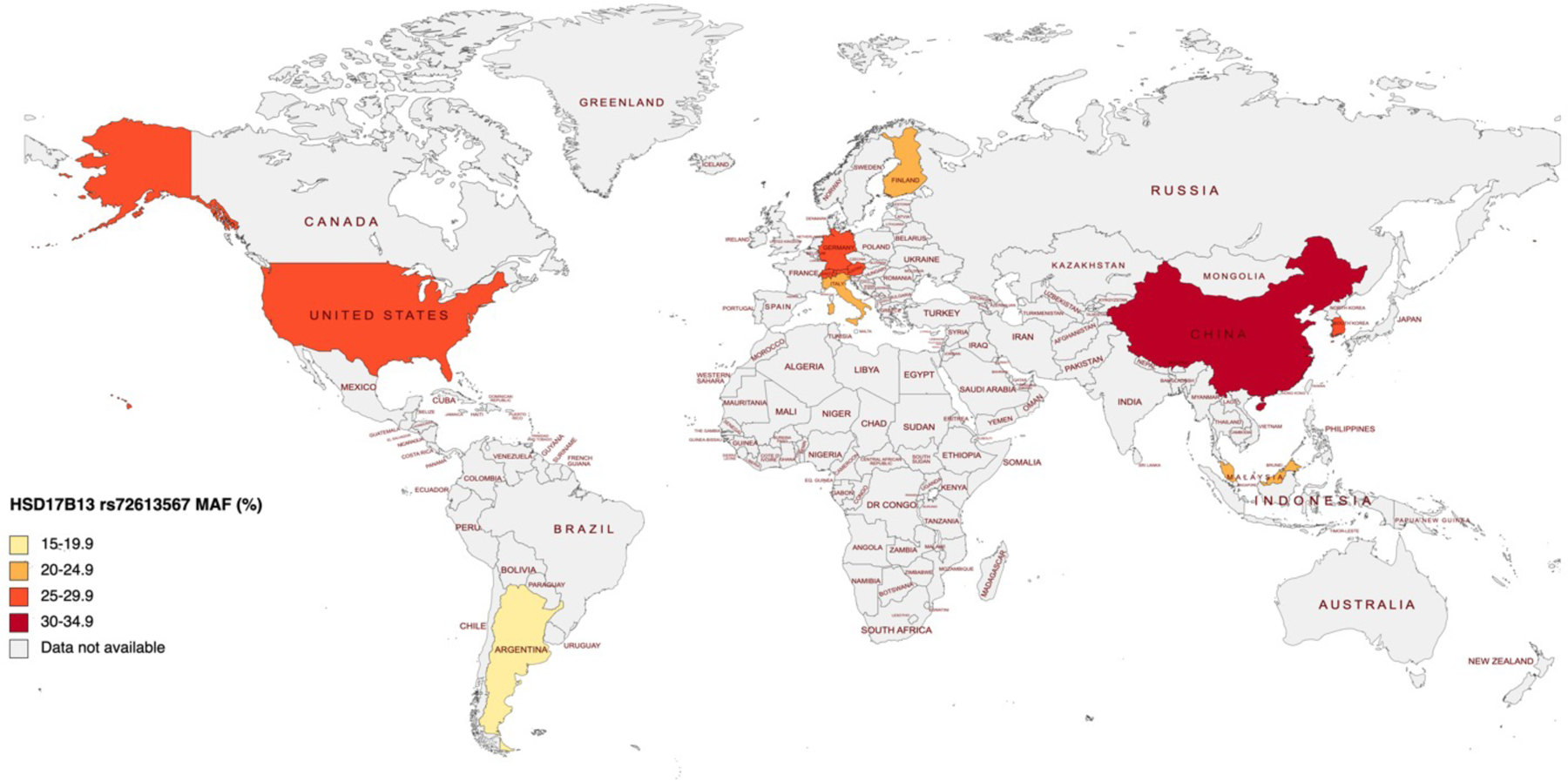
Data based on MAF in patients with NAFLD are as follows: Argentina, data from Pirola79; Austria, calculated from genotype frequency112; China, data from Sun77; Finland, calculated from genotype frequency55; Germany, calculated from genotype frequency112; Italy, data from Anstee25; Malaysia, data from Ting86; South Korea, data from Koo113; Switzerland, calculated from genotype frequency112; US, data from Serper78. Abbreviations: MAF, minor allele frequency.
Another hepatoprotective variant rs143404524 has low prevalence overall (6%), but is common among Africans (22%) in 1000 Genomes Project.31 Study conducted in the US general population showed that rs143404524 variant was more prevalent among African Americans (18.7%), in contrast to Hispanics (2.4%) and whites (0.2%).24 More recently, a study investigating the prevalence of HSD17B13 loss-of-function variants in an ancestrally diverse cohort in the US provided insight into the complex variability of HSD17B13 polymorphism across multiple ethnicities.81 This study was conducted in 29,585 individuals and demonstrated that the variants rs72613567 and rs143404524 were most prevalent among East/Southeast Asians (32%) and African Americans (19%), respectively.
The minor allele frequency of intergenic variant rs6834314 is 23%, rising up to 34% in East Asians and 25% in Europeans, but it is less common in Hispanic Americans (17%) and South Asians(17%).27 Additionally, the variants rs9992651 and rs13118664 have a worldwide prevalence of 18%.29,30 Both variants are most common in East Asians (31%) and Europeans (23%), and rare among Africans (5%). Finally, rs62305723 is the least common risk-reducing variant, with the global prevalence of 2%.32 Although current data are indicative, further independent validation in larger multiethnic cohorts is required to understand the risk-reducing impact of HSD17B13 polymorphism on NAFLD progression in different populations.
The role of HSD17B13 in the natural history of NAFLD
Clinical and histological spectrum of NAFLD ranges from simple steatosis to NASH. NASH is characterized as the presence of ≥5% steatosis and lobular inflammation with ballooning, with or without perisinusoidal fibrosis.82 NASH is the potentially progressive subtype of NAFLD that can lead to cirrhosis and HCC, as well as liver-related mortality.83
HSD17B13 and simple steatosis
The exact mechanism underlying the protective effect of HSD17B13 polymorphism on NAFLD development and severity is the subject of ongoing investigation. Hepatic expression of HSD17B13 wild type was 5.9-fold higher in NAFLD patients (n=43) compared to healthy controls (n=14).23 Overexpression of HSD17B13 increased LD sizes and number of cultured human hepatocytes.36 However, studies on the association between HSD17B13 protein and steatosis in murine models have provided conflicting results. Adenovirus-mediated overexpression of HSD17B13 wild type induced fatty liver in mice due to increased lipogenesis.36 In contrast, other studies showed that HSD17B13 knockout mice on a normal chow diet also developed hepatic steatosis and gained body weight.41,84 In essence, HSD17B13 overexpression and knockout have both been linked to steatosis in mice. The human studies produced more consistent data, where inactivating variants of HSD17B13 have been found to have no association with altered hepatic fat content.22,23,25,79 Two large-scale GWAS found no significant association between HSD17B13 protective alleles rs72613567, rs9992651, rs13118664 and simple steatosis in a cohort of NAFLD.22,25 Similarly, Pirola et al. examined the impact of HSD17B13 rs72613567 minor allele in 609 individuals of European ancestry and demonstrated that the protective effect of the variant against hepatic steatosis was not statistically significant when BMI was included in the logistic regression analysis.79 Another HSD17B13 variant rs143404524 was also not associated with the liver fat content in general population derived from Dallas Heart Study.24 All in all, these results suggest that HSD17B13 protein may not have a direct correlation with hepatic steatosis and the protective effect conferred by the lack of HSD17B13 function may be unrelated to hepatic fat accumulation. Further studies are needed to address the interspecies differences in HSD17B13 gene and elucidate the role of HSD17B13 protein in simple steatosis.
Studies that described an association between HSD17B13 SNP and reduced severity of NAFLD
The link between HSD17B13 SNPs and the reduced progression of NAFLD has been evaluated in several independent cohorts (Table 1). The rs72613567 is the most studied variant of HSD17B13, with established influence on the NAFLD progression. The seminal exome-sequence data conducted in 46,544 obese individuals of European descent showed that HSD17B13 rs72613567 variant was associated with a reduced risk of NAFLD and NASH cirrhosis by 17% (95% CI 8–25) and 26% (95% CI 7–40) in heterozygotes, and by 30% (95% CI 13–43) and 49% (95% CI 15–69) in homozygotes, respectively.22 In patients who underwent bariatric surgery, the rs72613567 variant was associated with lower odds of NASH and fibrosis in an allele dose-dependent manner, as compared with simple steatosis. Two independent biopsy-confirmed cohorts from North America and Argentina reported similar findings.23,79 In both studies, rs72613567 variant was associated with decreased histological spectrum of NASH – inflammation, ballooning degeneration, Mallory-Denk bodies and fibrosis – and lower serum levels of ALT and AST among obese and morbidly obese patients with biopsy-proven NAFLD. Gellert-Kristensen et al. tested the hepatoprotective effect of HSD17B13 rs72613567 in 111,612 individuals from the Danish general population and found that each minor allele decreased the risk of cirrhosis and cirrhosis-associated mortality by 15% (95% CI 0.74–0.98) and 49% (95% CI 0.32–0.81), respectively.26
Table 1.
Selected studies reporting the association between HSD17B13 variants and NAFLD
| Author, publication year |
Country | Ethnicity | Study population | Diagnostic criteria for NAFLD | Variant | Findings |
|---|---|---|---|---|---|---|
| Anstee, 2020 | UK, Switzerland, Belgium, France, Sweden, Germany, Italy | European | GWAS cohort: 1,483 NAFLD cases, 17,781 controls; mean age 50, 53% male, median BMI 35 kg/m2 Replication cohort: 559 NAFLD cases, 945 controls; mean age 52, 69% male, median BMI 28 kg/m2 |
Liver biopsy | rs9992651 (A>T) rs13118664 (G>A) |
Associated with decreased risk of NAFLD and NASH. |
| Ajmera, 2021 | US | European, Hispanic | N=264, 122 NAFLD cases; mean age 53, 37% male, mean BMI 29 kg/m2 | MRI-PDFF | rs72613567 (T>TA) | Associated with decrease in liver stiffness on MRE multivariable analysis. |
| Ma, 2019 | US | European | 768 NAFLD cases; mean age 49, 37% male, mean BMI 34.6 kg/m2 | Liver biopsy | rs6834314 (A>G) rs72613567 (T>TA) rs62305723 (G>A) |
Associated with decreased inflammation, ballooning, Mallory-Denk bodies, and liver enzyme levels. Associated with decreased ballooning and inflammation. |
| Paternostro, 2021 | Austria, Switzerland, Germany |
Not specified | 703 NAFLD cases; mean age 47, 45% male, median BMI 43 kg/m2 | Liver biopsy | rs72613567 (T>TA) | Associated with a lower probability of NAS ≥ 5. |
| Pirola, 2019 | Argentina | European | 429 NAFLD cases, 180 controls | Liver biopsy | rs72613567 (T>TA) | Protected against NASH, ballooning, lobular inflammation, and fibrosis. |
| Satapathy, 2021 | US | European | 66 LT recipients with NASH; mean age 57, 50% male, BMI>30 kg/m2 in 59% of patients | Liver biopsy | rs6834314 (A>G) | Donor HSD17B13 rs6834314 variant is associated with reduced risk of moderate to severe NAFLD recurrence at 1year post-LT. |
| Seko, 2020 | Japan | Japanese | 290 NAFLD cases; median age 59, 47% male, median BMI 27 kg/m2 | Liver biopsy | rs6834314(A>G) | Associated with lower prevalence of severe inflammation and ballooning. Attenuated the effect of the PNPLA3 rs738409 (I148M) variant on advanced hepatic fibrosis. |
| Seko, 2021 | Japan | Japanese | 140 NAFLD cases; median age 55, 51% male, median BMI 28 kg/m2 | Liver biopsy | rs6834314(A>G) | Associated with significantly lower serum levels of AST, ALT, and FIB-4 index. Associated with the reduction in LSM after 1 year diet therapy. |
| Serper, 2020 | US | European, AA, Hispanic |
Million Veterans Program: 60,542 NAFLD cases, 132,074 controls; mean age 62, 90% male, BMI>30 kg/m2 in 57% of patients | ALT | rs72613567 (T) rs6834314 (A) |
Associated with advanced fibrosis. |
| Ting, 2021 | Malaysia | Malay, Chinese, Indian |
N=428, 223 NAFLD cases, 205 controls; mean age 56, 52% male, mean BMI 29 kg/m2 | Liver biopsy | rs72613567 (T>TA) rs6834314 (A>G) | Associated with lower odds of NASH in the overall cohort and among ethnic Chinese. Associated with a lower incidence of liver-related complications*. Associated with lower grade of hepatocyte ballooning among the ethnic Chinese. |
| Wang, 2021 | US | Multi-ethnic | Multi-ethnic cohort (MEC): 1,232 NAFLD cases without cirrhosis, 8,444 controls; mean age 66, 41% male, mean BMI 26 kg/m2 | ICD (International Classification of Diseases) | rs9992651 (G>A) rs13118664 (A>T) |
Decreased risk of NAFLD among Japanese Americans. |
Liver-related complications: ascites, hepatic encephalopathy, spontaneous bacterial peritonitis, history of gastro-esophageal varices or variceal bleeding, HCC, and hepatorenal syndrome.
Numerous observational studies described an inverse relationship between HSD17B13 rs72613567 SNP and non-invasive markers of fibrosis.78,80,85 For example, in a recent data from 264 patients phenotyped by magnetic resonance elastography (MRE), rs72613567 protective allele was associated with a −0.41 kPa (95% CI −0.76 to −0.05) decrease in liver stiffness.80 Another multi-ethnic large biobank study of 60,542 US Veterans with NAFLD demonstrated that HSD17B13 wild type exhibited strong positive correlation with FIB-4 score (OR 1.06, 95% CI 1.02–1.09; P=9.7×10−4) and a negative association with platelet count (P=2.2×10−16).78 In summary, the protective effect of HSD17B13 rs72613567 variant on the progression of NAFLD has now been independently replicated in numerous studies in both adult85,86,87 and pediatric22,54,88 cohorts across diverse ethnicities, including Hispanic, Chinese, Japanese American and European populations.
HSD17B13 variants rs9992651and rs13118664 were also linked to reduced development of NAFLD with an OR of 0.74 (95% CI 0.671–0.826 and 95% CI 0.667–0.821, respectively) in a large GWAS analysis of 1,483 histologically-confirmed NAFLD patients.25 The results have been replicated in a sample of 838 Japanese Americans (OR 0.87, 95% CI 0.77–0.99).86 Although located in non-coding regions of the HSD17B13 gene, both SNPs exhibit strong linkage disequilibrium with rs72613567, which generates splice variants that are devoid of enzymatic function.
Another SNP highly linked to HSD17B13 rs72613567 is the downstream variant rs6834314, which has a well-established inverse association with NAFLD.23,35 Carriage of minor G allele of HSD17B14 rs6834314 is associated with reduced inflammation, fibrosis67 and liver-related complications86 in patients with NAFLD. Satapathy et al. studied the effect of HSD17B13 rs6834314 variant on prevalence of recurrent NAFLD in NASH transplant recipients.89 Intriguingly, the results demonstrated that the presence of donor rs6834314 variant was associated with significantly decreased risk of moderate to severe NAFLD recurrence (OR 0.11, 95% CI 0.01–0.88; P=0.036) one year after liver transplantation. Lastly, rs62305723 is a low-frequency missense variant that encodes substitution of proline to serine at position 260, which generates loss-of-function mutation and is associated with decreased ballooning and inflammation in patients with NAFLD.23
It is noteworthy to mention the HSD17B13 rs143404524 loss-of-function variant, which is more prevalent among African Americans (18.7%), in contrast to Hispanics (2.4%) and whites (0.2%).24 Kozlitina and colleagues observed that the variant was significantly less frequent among African American adults (OR 0.24, 95% CI 0.07–0.76) and Hispanic children (OR 0.10, 95% CI 0.01–0.79) with chronic liver disease compared to controls. Nevertheless, future research to explore the role of rs143404524 variant in NAFLD progression is warranted.
Impact of HSD17B13 variants on the development of NAFLD-related HCC
With the rise in the rates of obesity and metabolic syndrome, the incidence of NAFLD-related HCC is rapidly increasing.90 In fact, NAFLD is the fastest growing etiology of HCC in the US and parts of Europe, and is projected to rise by 82, 117, and 122% from 2016 to 2030 in China, France, and the USA, respectively.11,14,91,92 Since HSD17B13 truncation and therefore reduced activity were originally reported to be protective against advanced fibrosis and cirrhosis, it is possible that this protective effect would be applicable to hepatocarcinogenesis as well. Indeed, initial findings from a case-control cohort of 44 patients of European descent determined that the HSD17B13 rs72613567 variant was associated with a lower odds of HCC.22 Each TA-allele in the variant was found to reduce the risk of HCC by 28% in the Danish general population.26 Case-control studies conducted in heavy alcohol consumers93,94 and HCV-infected patients identified that HSD17B13 loss-of-function variant attenuated the risk of HCC development, even when the carriage of PNPLA3 I148M mutation status was taken into account.69,94 However, further data from large, prospective cohorts of patients with NAFLD are required to confirm the impact of HSD17B13 variants on HCC incidence.
HSD17B13 as a potential prognostic biomarker for NAFLD
In the last few years, human genetic studies underscored the importance of genetic variants on susceptibility and progression of NAFLD.95,96,97As the quantity of NAFLD-associated genetic variants are increasing, integration of numerous SNPs into polygenic risk scores may be utilized to develop personalized risk stratification algorithms. For example, combination of HSD17B13, PNPLA3 and TM6SF2 risk variants into a polygenic risk score was associated with a 12-fold increase in risk of cirrhosis and up to 29-fold increase in risk of HCC in 110,761 individuals from the Danish general population and 334,691 individuals from the UK Biobank.98 Additionally, an alternative polygenic risk score of well-established risk variants (PNPLA3, TM6SF2, GCKR and MBOAT7), adjusted for the presence of HSD17B13 polymorphism (rs72613567) was predictive of HCC development among a NAFLD cohort (n=2,566) from the UK Biobank, independent of the presence of severe fibrosis.99 In general population arm (n=364,048) of the same UK Biobank study, a polygenic risk score detected HCC with ~90% specificity both in cirrhotic (P <0.05) and non-cirrhotic (P <10−5) cohorts. In line with recent data, these findings suggest that genetic variants influencing hepatic fat content facilitate hepatocarcinogenesis. Thus, polygenic risk scoring might be an effective diagnostic tool for predicting HCC in NAFLD patients without advanced fibrosis. Most recently, Wang et al. generated a polygenic risk score comprising 11 NAFLD-associated SNPs (including HSD17B13) in a nested case-control study of multi-ethnic cohort (African Americans, Japanese Americans, Latinos, Native Hawaiians, and Whites).86 Their findings validated the significant association between weighted polygenic risk score and NAFLD progression among diverse ethnicities, taken collectively or individually. It is important to note that the susceptibility to NAFLD conferred by multiple risk genes (PNPLA3, TM6SF2, MBOAT7, HSD17B13, and MARC1) appears to be through impaired hepatic mitochondrial function.103 This mechanism underlying the pathogenesis of NAFLD is fundamentally distinct from that of metabolic component of the disease, which characterized by hepatic surplus of substrates, such as sugars, lipids and amino acids. Therefore, integrating polygenic risk scores into a risk prediction model might be helpful in identifying NAFLD individuals at high risk for disease progression.
Therapeutic strategies for targeting HSD17B13 for treatment of NASH
To date, there is no Food and Drug Administration nor European Medicines Agency approved therapy for NASH. 5 In vivo and in vitro studies indicate that suppression of HSD17B13 expression has favorable effects on NAFLD and represents a novel therapeutic target. In the meantime, lifestyle modifications remain the cornerstone approach to NAFLD treatment. Large prospective studies evaluating the long-term effects of the HSD17B13 SNPs on NAFLD are lacking. Weight loss appears to be beneficial in carriers of the variant allele, according to the results of the first pilot studies. In order to explore the effect of HSD17B13 polymorphism upon the link between the change in liver stiffness and body weight, 140 Japanese patients with biopsy-proven NAFLD were administered diet therapy for the duration of 1 year.104 Interestingly, the change in liver stiffness measurement was independently associated with the reduction in body weight in patients carrying the rs6834314 protective variant only. However, larger studies are required to determine the influence of HSD17B13 polymorphism on the impact of weight loss.
On the other hand, protection conferred by HSD17B13 variants upon the risk of NAFLD seems to be greater among individuals with higher BMI, per genome-wide and exome-wide studies.22,105 More recently, cross-sectional analysis comprising 1,153 non-Hispanic whites with biopsy-proven NAFLD showed that the protective effect of HSD17B13 rs72613567 variant on the risk of NASH and fibrosis was only significant among patients with the traditional risk factors associated with NAFLD progression; including women (β coeff −0.18; P<0.001), patients of age ≥45 years (β coeff −0.18; P<0.001), BMI ≥35 kg/m2 (β coeff −0.17; P<0.001), and diabetes mellitus (β coeff −0.18; P=0.02).106 Moderation analyses were used to explore whether the effect of HSD17B13 rs72613567 on the risk of NASH and fibrosis varies between premenopausal and postmenopausal women. The protective effect of HSD17B13 rs72613567 on risk of NASH was stronger in women aged 51 years or older; the median age of menopause among non-Hispanic white women in the US. In addition, data conducted in Danish cohort found that the protective effect of the same variant was enhanced by the presence of additional risk factors for fatty liver disease; including obesity, alcohol intake, and steatogenic alleles in PNPLA3 and TM6SF2.26 Taken together, evidence suggests that subgroups of populations with higher risk factors for NAFLD progression and associated comorbidities might benefit from the therapeutic targeting of HSD17B13 the most.
Blocking HSD17B13 expression using RNA interference (RNAi) therapeutic modalities
RNAi is a natural cellular process in which double-stranded RNAs inhibit the translation or induce sequence-specific degradation of messenger RNAs of identical sequence, leading to gene silencing. Therapeutics based on RNAi (using small interfering RNAs (siRNAs)) are becoming powerful techniques for identifying potent inhibitors of disease-specific genes. Phase I clinical trials employing approaches that reduce HSD17B13 expression through N-Acetylgalactosamine (GalNAc)-conjugated siRNA are underway (NCT04202354, NCT04565717)33,34. These compounds mimic the protective loss-off-function variant of the HSD17B13 protein. ARO-HSD is a pioneer investigational siRNA therapeutic developed to reduce the expression of HSD17B13 in hepatocytes (NCT04202354). Interim data analysis of a double-blind, placebo-controlled phase I/IIa clinical trial of ARO-HSD was recently presented at The Liver Meeting, the Annual Meeting of the American Association for the Study of Liver Disease.107 The study investigators (ARROWHEAD) recruited 8 healthy volunteers (19–52 years old) and 18 patients (32–61 years old) with suspected NASH, 4 of whom had confirmed NASH. At doses of up to 200 mg administered subcutaneously, hepatic HSD17B13 mRNA and protein levels were reduced by up to 93.4% and 82.7%, respectively. Dose-dependent decreases in ALT and AST were observed with mean reductions of up to 42% and 28%, respectively. Fifty percent of patients had a decline in MRI-PDFF and a third had improvement in liver stiffness (kPa) on transient elastography. The compound was well-tolerated with no reported treatment-related serious adverse events. ALN-HSD is the second siRNA therapeutic that has been developed by Alnylam Pharmaceuticals to knockdown the expression of HSD17B13.34 Preliminary results showed that ALN-HSD suppresses HSD17B13 in vivo and in vitro (rodents and healthy and obese nonhuman primates).108,109 It has also been demonstrated to have no pre-clinical toxicity, with high safety margins and durable pharmacodynamic properties. ALN-HSD is currently being evaluated in a double-blind, placebo-controlled phase I clinical trial in healthy volunteers and patients with NASH (NCT04565717).
Together, these data suggest that inhibition of HSD17B13 expression using siRNA modalities may be a potential therapeutic approach to prevent NAFLD/NASH progression (Figure 4).
Figure 4. siRNA-based inhibition of HSD17B13 expression.
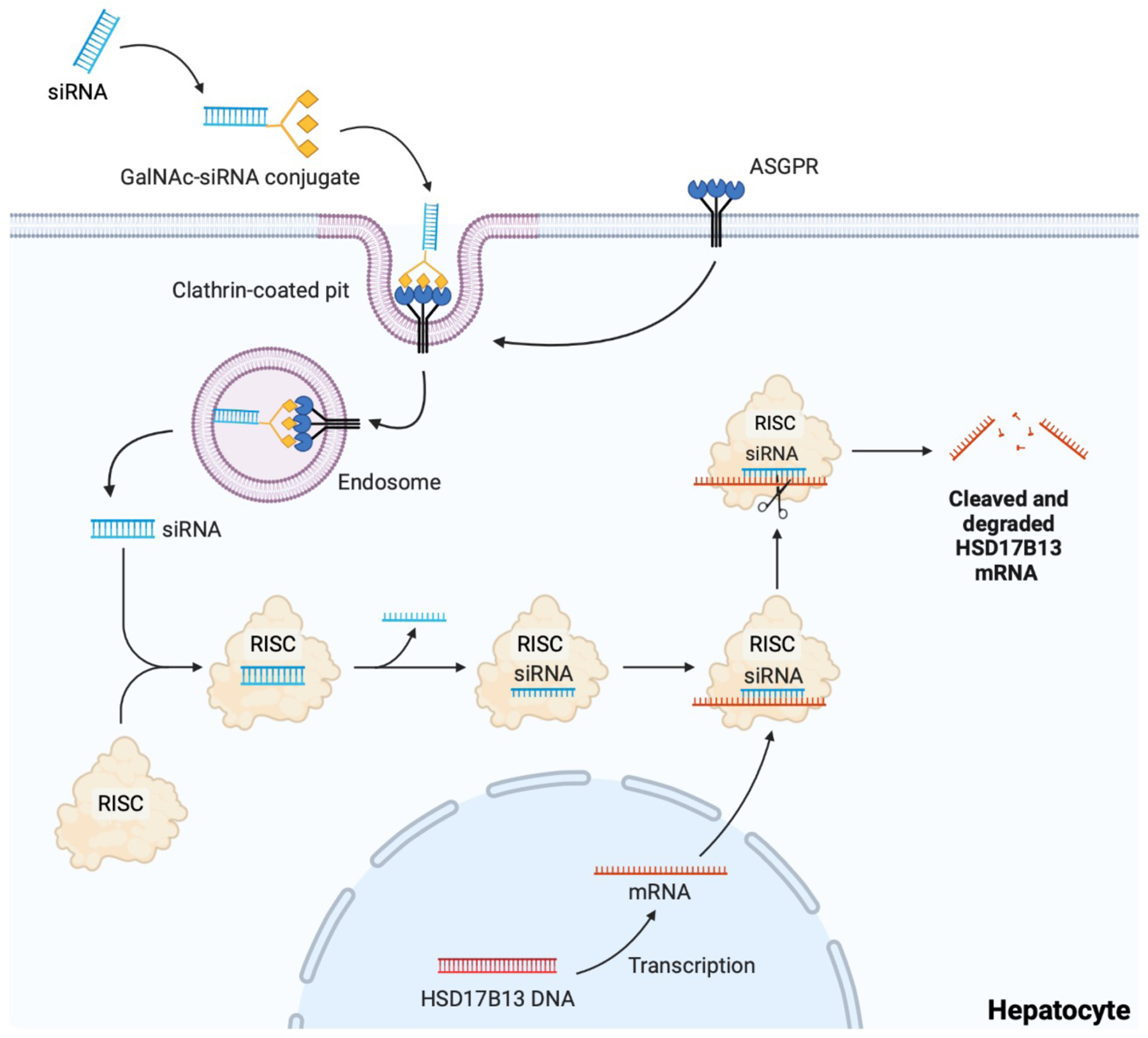
The small interfering RNA (siRNA) targeting HSD17B13 mRNA is conjugated with a triantennary N-Acetylgalactosamine (GalNAc). GalNAc binds to the asialoglycoprotein receptor (ASGPR), which is highly expressed on hepatocytes, thus targeting siRNA to the liver. The siRNA-ASGPR complex is then taken into hepatocytes by clathrin-mediated endocytosis, where the siRNA causes HSD17B13 mRNA destruction through the RNA-induced silencing complex (RISC) in the cytoplasm.
Small molecule inhibitors targeting HSD17B13 protein
Owing to their compact size, small molecule inhibitors can infiltrate the cell membrane easily to target proteins present inside a cell. As anticipated, small molecule inhibitors of HSD17B13 have been of interest to pharmaceutical industry for the last several years. More recently, inno.N developed a potent inhibitor of human HSD17B13, first-in-class oral potent molecule Compound A.110 Results of a preclinical study demonstrated that Compound A successfully inhibits HSD17B13 and decreases profibrogenic marker alpha-SMA mRNA level. Moreover, in C57BL/6 mice fed choline-deficient, L-amino acid-defined high-fat diet (HFD), Compound A decreased ALT, NAFLD activity score (NAS), lactate dehydrogenase and liver to body weight ratio, and improved plasma lipid profile.
Recently, another small molecule inhibitor of HSD17B13 INI-678 developed by Inipharm has been tested in a human liver cell-based 3D “liver-on-a-chip” model of NASH.111 INI-678 has been shown to decrease biomarkers of fibrosis including α-SMA (35.4±7.5%; P<0.0001) and collagen type 1 (42.5±6.4 %; P<0.0001). Moreover, INI-678 inhibited HSD17B13-catalyzed oxidation of retinol, estradiol and LTB3 which was accompanied by a notable trend towards decrease in levels of TG, bile acids and IL-6. The latter result is consistent with the findings of a clinical study, where the carriers of HSD17B13 inactive variant had lower plasma concentrations of IL-6 compared with noncarriers (9.0 ± 0.5 vs. 10.2 ± 0.5 pg/ml; P<0.05).55
Conclusions
HSD17B13 is a novel LD-associated protein that is principally expressed in hepatocytes. Truncation of the HSD17B13 protein may be associated with reduced risk of NASH, fibrosis, cirrhosis, and HCC in patients with NAFLD. The underlying mechanism is likely to be the loss of HSD17B13 enzymatic activity against retinol and proinflammatory mediators. The frequency of the protective minor alleles differs across different ethnic populations, which might contribute to the interethnic variabilities in the prevalence and severity of NAFLD. Therapeutic studies targeting HSD17B13 through RNAi or small molecule inhibitors are underway.
Acknowledgments
Figures created with BioRender.com
Declaration of personal interests: RLserves as a consultant to Aardvark Therapeutics, Altimmune, Anylam/Regeneron, Amgen, Arrowhead Pharmaceuticals, AstraZeneca, Bristol-Myer Squibb, CohBar, Eli Lilly, Galmed, Gilead, Glympse bio, Hightide, Inipharma, Intercept, Inventiva, Ionis, Janssen Inc., Madrigal, Metacrine, Inc., NGM Biopharmaceuticals, Novartis, Novo Nordisk, Merck, Pfizer, Sagimet, Theratechnologies, 89 bio, Terns Pharmaceuticals and Viking Therapeutics. In addition, his institutions received research grants from Arrowhead Pharmaceuticals, Astrazeneca, Boehringer-Ingelheim, Bristol-Myers Squibb, Eli Lilly, Galectin Therapeutics, Galmed Pharmaceuticals, Gilead, Intercept, Hanmi, Intercept, Inventiva, Ionis, Janssen, Madrigal Pharmaceuticals, Merck, NGM Biopharmaceuticals, Novo Nordisk, Merck, Pfizer, Sonic Incytes and Terns Pharmaceuticals. Co-founder of LipoNexus Inc. D.Q.H. receives funding support from Singapore Ministry of Health’s National Medical Research Council under its NMRC Research Training Fellowship (MOH- 000595–01). In addition, he has served as an advisory board member for Eisai.
Funding Information
RL received funding support from NIEHS (5P42ES010337), NCATS (5UL1TR001442), DOD PRCRP (W81XWH-18–2-0026), NIDDK (U01DK061734, R01DK106419, R01DK121378, R01DK124318, P30DK120515), NHLBI (P01HL147835) and NIAAA (U01AA029019).
References
- 1.Loomba R & Sanyal AJ The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol 10, 686–690 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Younossi ZM et al. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Chalasani N et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases: Hepatology, Vol. XX, No. X, 2017. Hepatology 67, 328–357 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Loomba R, Friedman SL & Shulman GI Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184, 2537–2564 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Tan DJH et al. Global burden of liver cancer in males and females: Changing etiological basis and the growing contribution of NASH. Hepatol. Baltim. Md (2022). [DOI] [PubMed]
- 6.Marchesini G et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 37, 917–923 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Adams LA, Waters OR, Knuiman MW, Elliott RR & Olynyk JK NAFLD as a Risk Factor for the Development of Diabetes and the Metabolic Syndrome: An Eleven-Year Follow-up Study. Off. J. Am. Coll. Gastroenterol. ACG 104, 861–867 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Ye Q et al. Global prevalence, incidence, and outcomes of non-obese or lean non-alcoholic fatty liver disease: a systematic review and meta-analysis. Lancet Gastroenterol. Hepatol 5, 739–752 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Angulo P et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 149, 389–397.e10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wree A, Broderick L, Canbay A, Hoffman HM & Feldstein AE From NAFLD to NASH to cirrhosis—new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol 10, 627–636 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Huang DQ, El-Serag HB & Loomba R Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol 18, 223–238 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orci LA et al. Incidence of Hepatocellular Carcinoma in Patients With Nonalcoholic Fatty Liver Disease: A Systematic Review, Meta-analysis, and Meta-regression. Clin. Gastroenterol. Hepatol 20, 283–292.e10 (2022). [DOI] [PubMed] [Google Scholar]
- 13.Tan DJH et al. Clinical characteristics, surveillance, treatment allocation, and outcomes of non-alcoholic fatty liver disease-related hepatocellular carcinoma: a systematic review and meta-analysis. Lancet Oncol (2022). [DOI] [PMC free article] [PubMed]
- 14.Younossi Z et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc 17, 748–755.e3 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Eslam M & George J Genetic contributions to NAFLD: leveraging shared genetics to uncover systems biology. Nat. Rev. Gastroenterol. Hepatol 17, 40–52 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Romeo S et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet 40, 1461–1465 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kozlitina J et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet 46, 352–356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mancina RM et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 150, 1219–1230.e6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teo K et al. rs641738C>T near MBOAT7 is associated with liver fat, ALT and fibrosis in NAFLD: A meta-analysis. J. Hepatol 74, 20–30 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan H-L et al. Association of glucokinase regulatory gene polymorphisms with risk and severity of non-alcoholic fatty liver disease: an interaction study with adiponutrin gene. J. Gastroenterol 49, 1056–1064 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Eslam M, Valenti L & Romeo S Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol 68, 268–279 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Abul-Husn NS et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med 378, 1096–1106 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma Y et al. 17‐Beta Hydroxysteroid Dehydrogenase 13 Is a Hepatic Retinol Dehydrogenase Associated With Histological Features of Nonalcoholic Fatty Liver Disease. Hepatology 69, 1504–1519 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kozlitina J, Stender S, Hobbs HH & Cohen JC HSD17B13 and Chronic Liver Disease in Blacks and Hispanics. N. Engl. J. Med 379, 1876–1877 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Anstee QM et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort☆. J. Hepatol 73, 505–515 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Gellert‐Kristensen H, Nordestgaard BG, Tybjærg‐Hansen A & Stender S High Risk of Fatty Liver Disease Amplifies the Alanine Transaminase–Lowering Effect of a HSD17B13 Variant. Hepatology 71, 56–66 (2020). [DOI] [PubMed] [Google Scholar]
- 27.rs6834314 (SNP) - Population genetics - Homo_sapiens - Ensembl genome browser 105. https://uswest.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=4:87292156-87293156;v=rs6834314;vdb=variation;vf=93467431.
- 28.rs72613567 (INDEL) - Population genetics - Homo_sapiens - Ensembl genome browser 105. http://uswest.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=4:87309741-87310741;v=rs72613567;vdb=variation;vf=100585521.
- 29.rs9992651 (SNP) - Population genetics - Homo_sapiens - Ensembl genome browser 105. https://uswest.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=4:87310858-87311858;v=rs9992651;vdb=variation;vf=94574947.
- 30.rs13118664 (SNP) - Population genetics - Homo_sapiens - Ensembl genome browser 105. https://uswest.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=4:87317957-87318957;v=rs13118664;vdb=variation;vf=96366017.
- 31.rs80182459 (INDEL) - Population genetics - Homo_sapiens - Ensembl genome browser 105. http://uswest.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=4:87313445-87314445;v=rs80182459;vdb=variation;vf=94017030#population_freq_AMR.
- 32.rs62305723 (SNP) - Population genetics - Homo_sapiens - Ensembl genome browser 105. https://uswest.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=4:87309777-87310777;v=rs62305723;vdb=variation;vf=99892100.
- 33.Arrowhead Pharmaceuticals. A Phase 1/2a Single and Multiple Dose-Escalating Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamic Effects of ARO-HSD in Normal Healthy Volunteers as Well as in Patients With NASH or Suspected NASH https://clinicaltrials.gov/ct2/show/NCT04202354 (2021).
- 34.Alnylam Pharmaceuticals. A Phase 1, Randomized, Double-Blind, Placebo-Controlled, 2-Part Study of the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Single Dose ALN-HSD in Healthy Adult Subjects and Multiple Dose ALN-HSD in Adult Patients With Nonalcoholic Steatohepatitis (NASH) https://clinicaltrials.gov/ct2/show/NCT04565717 (2022).
- 35.Chambers et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat. Genet 43, 1131–1138 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su W et al. Comparative proteomic study reveals 17 -HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci 111, 11437–11442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luu-The V, Bélanger A & Labrie F Androgen biosynthetic pathways in the human prostate. Best Pract. Res. Clin. Endocrinol. Metab 22, 207–221 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Saloniemi T, Jokela H, Strauss L, Pakarinen P & Poutanen M The diversity of sex steroid action: novel functions of hydroxysteroid (17β) dehydrogenases as revealed by genetically modified mouse models. J. Endocrinol 212, 27–40 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Liu S et al. Molecular cloning and expression analysis of a new gene for short-chain dehydrogenase/reductase 9. Acta Biochim. Pol 54, 213–218 (2007). [PubMed] [Google Scholar]
- 40.Horiguchi Y, Araki M & Motojima K 17β-Hydroxysteroid dehydrogenase type 13 is a liver-specific lipid droplet-associated protein. Biochem. Biophys. Res. Commun 370, 235–238 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Ma Y et al. 17‐Beta Hydroxysteroid Dehydrogenase 13 Deficiency Does Not Protect Mice From Obesogenic Diet Injury. Hepatology 73, 1701–1716 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma Y et al. Characterization of essential domains in HSD17B13 for cellular localization and enzymatic activity. J. Lipid Res 61, 1400–1409 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rotinen M, Villar J, Celay J & Encío I Type 10 17β-hydroxysteroid dehydrogenase expression is regulated by C/EBPβ in HepG2 cells. J. Steroid Biochem. Mol. Biol 122, 164–171 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Su W et al. Liver X receptor α induces 17β-hydroxysteroid dehydrogenase-13 expression through SREBP-1c. Am. J. Physiol.-Endocrinol. Metab 312, E357–E367 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Schultz JR et al. Role of LXRs in control of lipogenesis. Genes Dev 14, 2831–2838 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beaven SW et al. Reciprocal Regulation of Hepatic and Adipose Lipogenesis by Liver X Receptors in Obesity and Insulin Resistance. Cell Metab 18, 106–117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Higuchi N et al. Liver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatol. Res. Off. J. Jpn. Soc. Hepatol 38, 1122–1129 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Rotroff DM et al. Genetic Variants in HSD17B3 , SMAD3 , and IPO11 Impact Circulating Lipids in Response to Fenofibrate in Individuals With Type 2 Diabetes. Clin. Pharmacol. Ther 103, 712–721 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tailleux A, Wouters K & Staels B Roles of PPARs in NAFLD: Potential therapeutic targets. Biochim. Biophys. Acta BBA - Mol. Cell Biol. Lipids 1821, 809–818 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Gluchowski NL, Becuwe M, Walther TC & Farese RV Lipid droplets and liver disease: from basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol 14, 343–355 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carr RM & Ahima RS Pathophysiology of lipid droplet proteins in liver diseases. Exp. Cell Res 340, 187–192 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang M et al. Down-Regulating the High Level of 17-Beta-Hydroxysteroid Dehydrogenase 13 Plays a Therapeutic Role for Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci 23, 5544 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carleton M, Florio V, Odingo J, Anandan S, Swaminathan S, Ramaiah S, Rohil AN, Guenigault G, Silva R, Kostrzewski T, Hsu HK. POTENT AND SELECTIVE SMALL MOLECULE HSD17B13 INHIBITOR INI-678 DECREASES FIBROTIC MARKERS IN NASH LIVER-ON-A-CHIP (2021).
- 54.Hudert CA et al. Variants in MARC1 and HSD17B13 reduce severity of NAFLD in children, perturb phospholipid metabolism, and suppress fibrotic pathways. MedRxiv Prepr. Serv. Health Sci (2020).
- 55.Luukkonen PK et al. Hydroxysteroid 17-β dehydrogenase 13 variant increases phospholipids and protects against fibrosis in nonalcoholic fatty liver disease. JCI Insight 5, e132158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Puri P et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 46, 1081–1090 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Zeisel SH et al. Choline, an essential nutrient for humans. FASEB J 5, 2093–2098 (1991). [PubMed] [Google Scholar]
- 58.Vrablic AS, Albright CD, Craciunescu CN, Salganik RI & Zeisel SH Altered mitochondrial function and overgeneration of reactive oxygen species precede the induction of apoptosis by 1-O-octadecyl-2-methyl-rαc-grycero-3-phosphocholine in p53-defective hepatocytes. FASEB J 15, 1739–1744 (2001). [DOI] [PubMed] [Google Scholar]
- 59.Saeed A, Dullaart RPF, Schreuder TCMA, Blokzijl H & Faber KN Disturbed Vitamin A Metabolism in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 10, E29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blaner WS Vitamin A signaling and homeostasis in obesity, diabetes, and metabolic disorders. Pharmacol. Ther 197, 153–178 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yanagitani A et al. Retinoic acid receptor α dominant negative form causes steatohepatitis and liver tumors in transgenic mice. Hepatology 40, 366–375 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Motomura K, Sakai H, Isobe H & Nawata H Effects of retinoids on the production of tumour necrosis factor-alpha and nitric oxide by lipopolysaccharide-stimulated rat Kupffer cells in vitro: evidence for participation of retinoid X receptor signalling pathway. Cell Biochem. Funct 15, 95–101 (1997). [DOI] [PubMed] [Google Scholar]
- 63.Seifert WF et al. Vitamin A deficiency potentiates carbon tetrachloride-induced liver fibrosis in rats. Hepatology 19, 193–201 (1994). [PubMed] [Google Scholar]
- 64.Pingitore P et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum. Mol. Genet ddw341 (2016). [DOI] [PMC free article] [PubMed]
- 65.Ashla AA et al. Genetic analysis of expression profile involved in retinoid metabolism in non-alcoholic fatty liver disease. Hepatol. Res 40, 594–604 (2010). [DOI] [PubMed] [Google Scholar]
- 66.Saeed A et al. Impaired Hepatic Vitamin A Metabolism in NAFLD Mice Leading to Vitamin A Accumulation in Hepatocytes. Cell. Mol. Gastroenterol. Hepatol 11, 309–325.e3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seko Y et al. Attenuated effect of PNPLA3 on hepatic fibrosis by HSD17B13 in Japanese patients with non‐alcoholic fatty liver disease. Liver Int 40, 1686–1692 (2020). [DOI] [PubMed] [Google Scholar]
- 68.Bellan M et al. Severity of Nonalcoholic Fatty Liver Disease in Type 2 Diabetes Mellitus: Relationship between Nongenetic Factors and PNPLA3/HSD17B13 Polymorphisms. Diabetes Metab. J 43, 700 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.De Benedittis C et al. Interplay of PNPLA3 and HSD17B13 Variants in Modulating the Risk of Hepatocellular Carcinoma among Hepatitis C Patients. Gastroenterol. Res. Pract 2020, 4216451 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pirazzi C et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet 23, 4077–4085 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mondul A et al. PNPLA3 I148M Variant Influences Circulating Retinol in Adults with Nonalcoholic Fatty Liver Disease or Obesity. J. Nutr 145, 1687–1691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bruschi FV et al. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 65, 1875–1890 (2017). [DOI] [PubMed] [Google Scholar]
- 73.Le MH et al. 2019 Global NAFLD Prevalence: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol S1542356521012805 (2021). [DOI] [PubMed]
- 74.Lazo M et al. Prevalence of Nonalcoholic Fatty Liver Disease in the United States: The Third National Health and Nutrition Examination Survey, 1988–1994. Am. J. Epidemiol 178, 38–45 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rich NE et al. Racial and Ethnic Disparities in Nonalcoholic Fatty Liver Disease Prevalence, Severity, and Outcomes in the United States: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol 16, 198–210.e2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ciardullo S & Perseghin G Prevalence of NAFLD, MAFLD and associated advanced fibrosis in the contemporary United States population. Liver Int 41, 1290–1293 (2021). [DOI] [PubMed] [Google Scholar]
- 77.Sun D-Q et al. The HSD17B13 rs72613567 variant is associated with lower levels of albuminuria in patients with biopsy-proven nonalcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis 31, 1822–1831 (2021). [DOI] [PubMed] [Google Scholar]
- 78.Serper M et al. Validating a non-invasive, ALT-based non-alcoholic fatty liver phenotype in the million veteran program. PLOS ONE 15, e0237430 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pirola CJ et al. Splice variant rs72613567 prevents worst histologic outcomes in patients with nonalcoholic fatty liver disease. J. Lipid Res 60, 176–185 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ajmera V et al. The impact of genetic risk on liver fibrosis in non‐alcoholic fatty liver disease as assessed by magnetic resonance elastography. Aliment. Pharmacol. Ther 54, 68–77 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rutledge S, Belbin G, Soper E, Kenny E, Abul-Husn N Loss-of-function variants in HSD17B13 in an ancestrally diverse patient population-implications for liver disease risk. In Hepatology 2021. (Vol. 74, pp. 131A–132A). 111 River St, Hoboken 07030–5774, NJ USA: Wiley. [Google Scholar]
- 82.Kleiner DE et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321 (2005). [DOI] [PubMed] [Google Scholar]
- 83.Dulai PS et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 65, 1557–1565 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Adam M et al. Hydroxysteroid (17β) dehydrogenase 13 deficiency triggers hepatic steatosis and inflammation in mice. FASEB J 32, 3434–3447 (2018). [DOI] [PubMed] [Google Scholar]
- 85.Kallwitz E et al. Association of HSD17B13 rs72613567:TA with non‐alcoholic fatty liver disease in Hispanics/Latinos. Liver Int 40, 889–893 (2020). [DOI] [PubMed] [Google Scholar]
- 86.Ting Y-W et al. Loss-of-function HSD17B13 variants, non-alcoholic steatohepatitis and adverse liver outcomes: Results from a multi-ethnic Asian cohort. Clin. Mol. Hepatol 27, 486–498 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang J et al. Association of Genetic Risk Score With NAFLD in An Ethnically Diverse Cohort. Hepatol. Commun 5, 1689–1703 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Di Sessa A et al. The rs72613567:TA Variant in the Hydroxysteroid 17-beta Dehydrogenase 13 Gene Reduces Liver Damage in Obese Children. J. Pediatr. Gastroenterol. Nutr 70, 371–374 (2020). [DOI] [PubMed] [Google Scholar]
- 89.Satapathy SK et al. Clinical and Genetic Risk Factors of Recurrent Nonalcoholic Fatty Liver Disease After Liver Transplantation. Clin. Transl. Gastroenterol 12, e00302 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Estes C, Razavi H, Loomba R, Younossi Z & Sanyal AJ Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 67, 123–133 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Estes C et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol 69, 896–904 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Huang DQ et al. Comparative efficacy of an optimal exam between ultrasound versus abbreviated MRI for HCC screening in NAFLD cirrhosis: A prospective study. Aliment. Pharmacol. Ther 55, 820–827 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang J et al. A 17‐Beta‐Hydroxysteroid Dehydrogenase 13 Variant Protects From Hepatocellular Carcinoma Development in Alcoholic Liver Disease. Hepatology hep 30623 (2019). [DOI] [PubMed] [Google Scholar]
- 94.Stickel F et al. Genetic Variation in HSD17B13 Reduces the Risk of Developing Cirrhosis and Hepatocellular Carcinoma in Alcohol Misusers. Hepatology 72, 88–102 (2020). [DOI] [PubMed] [Google Scholar]
- 95.Trépo E & Valenti L Update on NAFLD genetics: From new variants to the clinic. J. Hepatol 72, 1196–1209 (2020). [DOI] [PubMed] [Google Scholar]
- 96.Dongiovanni P et al. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol 19, 6969–6978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Speliotes EK et al. Genome-Wide Association Analysis Identifies Variants Associated with Nonalcoholic Fatty Liver Disease That Have Distinct Effects on Metabolic Traits. PLOS Genet 7, e1001324 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gellert-Kristensen H et al. Combined Effect of PNPLA3, TM6SF2, and HSD17B13 Variants on Risk of Cirrhosis and Hepatocellular Carcinoma in the General Population. Hepatology 72, 845–856 (2020). [DOI] [PubMed] [Google Scholar]
- 99.Bianco C et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J. Hepatol 74, 775–782 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li L et al. Differential requirement for de novo lipogenesis in cholangiocarcinoma and hepatocellular carcinoma of mice and humans. Hepatology 63, 1900–1913 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Burlone ME et al. HSD17B13 and other liver fat-modulating genes predict development of hepatocellular carcinoma among HCV-positive cirrhotics with and without viral clearance after DAA treatment. Clin. J. Gastroenterol (2022). [DOI] [PubMed]
- 102.Donati B et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep 7, 4492 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Luukkonen PK et al. Distinct contributions of metabolic dysfunction and genetic risk factors in the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol S0168827821021504 (2021). [DOI] [PMC free article] [PubMed]
- 104.Seko Y et al. The Effect of Genetic Polymorphism in Response to Body Weight Reduction in Japanese Patients with Nonalcoholic Fatty Liver Disease. Genes 12, 628 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gao C et al. Genome‐wide association analysis of serum alanine and aspartate aminotransferase, and the modifying effects of BMI in 388k European individuals. Genet. Epidemiol 45, 664–681 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vilar-Gomez E et al. The Protection Conferred by HSD17B13 rs72613567 Polymorphism on Risk of Steatohepatitis and Fibrosis May Be Limited to Selected Subgroups of Patients With NAFLD. Clin. Transl. Gastroenterol 12, e00400 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Arrowhead Presents Additional Clinical Data on Investigational ARO-HSD Treatment at AASLD Liver Meeting | Arrowhead Pharmaceuticals Inc. https://ir.arrowheadpharma.com/news-releases/news-release-details/arrowhead-presents-additional-clinical-data-investigational-1.
- 108.Goga A & Stoffel M Therapeutic RNA-silencing oligonucleotides in metabolic diseases. Nat. Rev. Drug Discov 1–23 (2022). [DOI] [PubMed]
- 109.OliX Pharmaceuticals, ‘GalNAc-siRNA platform’ competitiveness against HBV-NASH? http://www.biospectator.com/view/news_view.php?varAtcId=14766.
- 110.Choi JW et al. A first-in-class small molecule targeting 17-beta-hydroxysteroid dehydrogenase 13 for the treatment of non-alcoholic steatohepatitis. InJournal of hepatology 2021. Jul 1 (Vol. 75, pp. S263–S263). RADARWEG 29, 1043 NX AMSTERDAM, NETHERLANDS: ELSEVIER. [Google Scholar]
- 111.Carleton M et al. Potent and selective small molecule HSD17B13 inhibitor INI-678 decreases fibrotic markers in NASH liver-on-a-chip. In Hepatology 2021. Oct 1 (Vol. 74, pp. 143A–143A). 111 River St, Hoboken 07030–5774, NJ USA: Wiley. [Google Scholar]
- 112.Paternostro R et al. Combined effects of PNPLA3, TM6SF2 and HSD17B13 variants on severity of biopsy-proven non-alcoholic fatty liver disease. Hepatol. Int 15, 922–933 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Koo BK et al. Development and Validation of a Scoring System, Based on Genetic and Clinical Factors, to Determine Risk of Steatohepatitis in Asian Patients with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol 18, 2592–2599.e10 (2020). [DOI] [PubMed] [Google Scholar]