

Abstract
Peroxisome proliferator-activated receptor γ (PPARγ) belongs to a family of nuclear receptors that could serve as lipid sensors. PPARγ is the target of a group of insulin sensitizers called thiazolidinediones (TZD) which regulate the expression of genes involved in glucose and lipid metabolism, as well as adipokines that regulate metabolic function in other tissues. Non-alcoholic fatty liver disease (NAFLD) has a high prevalence worldwide and is even higher in patients with obesity and insulin resistance. TZD-mediated activation of PPARγ could serve as a good treatment for NAFLD because TZD have shown anti-fibrogenic and anti-inflammatory effects in vitro, and increase insulin sensitivity in peripheral tissues which improves liver pathology. However, mechanistic studies in mouse models suggest that the activation of PPARγ in hepatocytes might reduce or limit the therapeutic potential of TZD against NAFLD. In this review, we briefly describe the short history of PPAR isoforms, the relevance of their expression in different tissues, as well as the pathogenesis and potential therapeutics for NAFLD. We also discuss some evidence derived from mouse models that could be useful for endocrinologists to assess tissue-specific roles of PPARs, complement reverse endocrinology approaches, and understand the direct role that PPARγ has in hepatocytes and non-parenchymal cells.
Introduction.
Peroxisome proliferator-activated receptors (PPAR) are a group of nuclear receptors that were initially identified as targets of compounds that increase peroxisome proliferation. Three PPAR genes have been identified: NR1C1, NR1C2, and NR1C3 which are commonly named PPARα, PPARβ/δ, and PPARγ, respectively. These receptors are widely expressed throughout the organism at different levels, could serve as lipid sensors, and are the targets of drugs with positive effects on metabolism. As such, extensive research has been performed on them to develop pharmacological therapies to treat diseases such as non-alcoholic fatty liver disease (NAFLD). The activation of these receptors with exogenous ligands, however, may evoke side effects that reduce their use in clinic. In this review, we discuss the cell-specific contribution of PPARγ in in hepatic pathophysiology by using mechanistic mouse studies.
PPAR were identified as orphan receptors and tested with reverse endocrinology.
The first PPAR was identified in 1990 from a cDNA library from mouse liver, and was described as a target of hepatocarcinogens that increase the number of peroxisomes, fatty acid oxidation, and reduce plasma lipid levels (Issemann et al. 1990). The highest expression of this mouse PPAR (mPPAR) was detected in the liver, kidney, and heart with lower expression in other tissues. Later, three sequences with high similarity to mPPAR were identified from a cDNA library of the ovary of Xanepous laevis (African clawed toad) (Dreyer et al. 1992). Xanepous PPARα (xPPARα) showed the highest similarity to mPPAR (77% in amino acid sequence), whereas xPPARβ and xPPARγ had reduced similarity with mPPAR (50–60%). In humans, the first sequence with 62% similarity to mPPAR was isolated using a cDNA library from osteosarcoma cells (Schmidt et al. 1992) and would later be identified as PPARδ. Finally, the three genes of PPARα, PPARβ/δ, and PPARγ were identified in mouse and humans (Issemann et al. 1990, Schmidt et al. 1992, Zhu et al. 1993, Kliewer et al. 1994, Mukherjee et al. 1994, Greene et al. 1995). Of note, the PPARγ gene produces two isoforms: PPARγ1 and PPARγ2, where PPARγ2 has 30 additional amino acids on the N-terminal domain as compared to PPARγ1 (Zhu et al. 1995).
The proteins encoded by PPARα, PPARβ/δ, and PPARγ genes in mice and humans show a high interspecies similarity (93–98%) suggesting similar structures and functions (Table 1). Although each PPAR isoforms have a high similarity in their ligand-binding domain (60–70%), the 3D structural analysis indicates that each PPAR could hold different hydrophobic compounds (Xu et al. 2001). The PPARs are orphan receptors, but analyses of the hydrophobic ligand-binding pocket suggests that fatty acids and their derivates are potent activators of PPARs (Lemberger et al. 1996, Kliewer et al. 1999, Grygiel-Gorniak 2014). To date, it remains unclear which primary endogenous ligands activate PPARs, but their activation (endogenously or exogenously) causes their dimerization with retinoid X receptor (RXR) which then binds to the PPAR response elements in the DNA. The heterodimer complex of PPARs/RXR binds the DNA and controls a diverse range of genes (Lemberger et al. 1996, Scholtes et al. 2022), by acting as direct activator (ligand-dependent transactivation) or inhibitor of gene expression with ligand-dependent transrepression or ligand-independent repression (Ricote et al. 2007).
Table 1.
Similarity of mouse and human PPARα (NR1C1), PPARβ/δ, (NR1C2), and PPARγ (NR1C3) proteins.
| PPARα | PPAR β/δ | PPARγ1 | PPARγ2 | ||
|---|---|---|---|---|---|
| Mouse | Gene ID | 19013 | 19015 | 19016 | 19016 |
| # AA | 468 | 440 | 475 | 505 | |
| Human | Gene ID | 5465 | 5467 | 5468 | 5468 |
| # AA | 468 | 441 | 475 | 505 | |
| Similarity between mouse and human | 97% | 93% | 98% | 96% | |
Similarity was calculated by comparing mouse and human protein sequences in BLAST protein of the National Center for Biotechnology Information.
To determine the functional roles of PPARs, reverse endocrinology was used with potential agonists (Kliewer et al. 1999). To date, the functional role of PPARs are widely known thanks to the use of exogenous ligands rather than by their endogenous activation. However, using reverse endocrinology to assess PPAR functions has risks because these compounds may not recapitulate the same response triggered by endogenous ligands. Additionally, administration of these exogenous ligands could activate PPARs in multiple tissues simultaneously, thus offsetting some of the physiological effects of PPAR activation in a specific tissue of interest. For instance, fibrates are exogenous ligands of PPARα that lower triglyceride levels and regulate genes involved in lipoprotein and fatty acid oxidation (Lemberger et al. 1996, Grygiel-Gorniak 2014). While it would be expected that fibrate-mediated activation of PPARα in the liver reduces fat accumulation, PPARα activation with fibrates increases liver weight, the expression of genes involved in fatty acid oxidation and synthesis, and failed to reduce hepatic fat accumulation in mice (Rajamoorthi et al. 2017). In a pilot trial, 48 weeks of fenofibrate had minimal effects on liver histology (including steatosis) in patients with NAFLD (Fernandez-Miranda et al. 2008). Other examples would be thiazolidinediones (TZD), a class of drugs that are exogenous ligands of PPARγ. TZD activate PPARγ in adipose tissue, muscle, and liver. In adipose tissue, TZD increase the expression of genes involved in insulin signaling, glucose uptake, and the lipid and fatty acid metabolism. This is sufficient for a complete insulin sensitization and control of glucose levels (Sugii et al. 2009, Soccio et al. 2014). Moreover, TZD increase the expression of adiponectin in adipocytes (Iwaki et al. 2003) that could indirectly control glucose and lipid metabolism in liver and muscle by the regulation of AMP-activated protein kinase (AMPK) signaling (Nawrocki et al. 2006). Furthermore, TZD directly activate PPARγ in liver and muscle to reduce insulin resistance (Chao et al. 2000, Hevener et al. 2003, Norris et al. 2003). This insulin sensitizing effect of TZD is used to reduce hepatic fat content in humans with NASH and insulin resistance. Indeed, three major clinical trials have shown the anti-steatogenic effects of TZD (Ratziu et al. 2010, Sanyal et al. 2010, Cusi et al. 2016), as we will briefly describe in this review. However, when TZD are used in mice, model- or study-dependent differences in the anti-steatogenic effects of TZD arise. In fact, TZD enhance liver steatosis in mouse models with NAFLD: diet-induced obese wild-type mice (Gao et al. 2016, Lee et al. 2021a), lipodystrophic mice (Gavrilova et al. 2003), and ob/ob mice (Matsusue et al. 2003). These potential steatogenic effects of TZD in mouse liver have been proved to be hepatocyte-specific PPARγ-dependent as we will describe below in more detail. In addition, TZD evoke undesired effects potentially due to the activation of PPARγ in multiple tissues simultaneously (Ahmadian et al. 2013). Overall, the use of reverse endocrinology is highly informative and helps to describe major roles of orphan receptors. However, it may not reveal all the insights and functions of PPARs in different tissues. Therefore, it is important to use additional methods to study the tissue-specific actions of these PPAR isoforms, and their activation by their respective endogenous ligands to reveal the physiological relevance of PPARs.
Lessons from tissue-specific knock out of PPARα and PPARγ.
PPARα is abundantly expressed in the liver, PPARγ in adipose tissue, and PPARβ/δ in nearly all tissues (Lemberger et al. 1996, Kliewer et al. 1999). Certainly, new RNA-seq data from mouse and human tissues confirms the differential expression of PPARs in different tissues. As shown by Figure 1, the liver predominantly expresses PPARα, while the heart and kidney express high levels of both PPARα and PPARβ/δ. The lung, brain, stomach, spleen, adrenal, testis, and ovary express high levels of PPARβ/δ while the bladder and placenta express high levels of PPARβ/δ and PPARγ. Finally, adipose tissue expresses high levels of PPARγ. Figure 1 does not include all the tissues analyzed in these RNA-seq studies (Bioprojects #PRJNA66167 and #PRJEB4337) (Fagerberg et al. 2014), and we chose the tissues that were included in both mouse and human analyses. Nonetheless, this figure clearly indicates that PPAR isoforms are ubiquitously expressed throughout multiple tissues in basal conditions in mice and humans where they may regulate physiological functions. From a simplistic point of view, it could be assumed that each isoform would have physiological effects only in tissues where they have high expression, and where exogenous ligands easily activate them. However, low expression of each PPAR isoform may have a significant contribution to the tissue physiology as well. To define the tissue-specific roles of these receptors, animal models need to be used to knock out the expression of PPARs in specific tissues. This tissue- or cell-specific knockout of a gene can be achieved with the cell-specific promoter-driven Cre-LoxP recombination system (Kim et al. 2018). Below, we will briefly review some examples of these models to study tissue-specific PPAR physiology.
Figure 1. Tissue-specific expression level of PPARα, PPARβ/δ, and PPARγ.
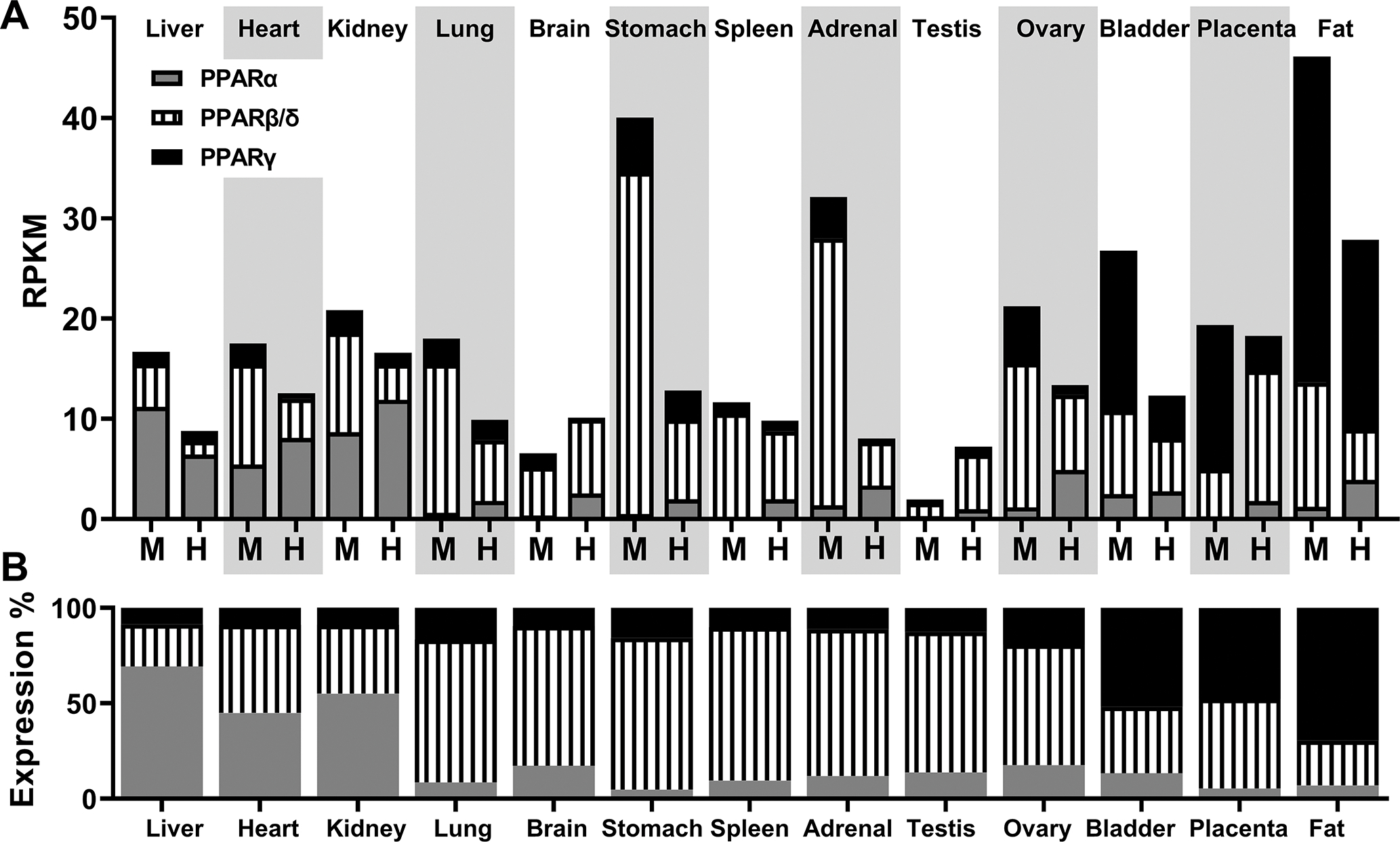
A) Normalized gene expression data: reads per kilobase of transcripts, per million mapped reads (RPKM) of PPARα, PPARβ/δ, and PPARγ in mouse (M) and human (H) tissues. Values were obtained from Gene database of the National Center for Biotechnology Information: Gene IDs 19013, 19015, 19016, 5465, 5467, 5468, using expression data of Bioproject #PRJNA66167 (RNA profiling data sets generated by the Mouse ENCODE project) and #PRJEB4337 (RNA-seq of tissue samples from 95 human individuals). B) Representation of percentage expression level of mouse and human PPAR isoforms by tissue.
Hepatocyte-specific PPARα knockout mice have been generated by crossbreeding PPARα-floxed mice with albumin (Alb) promoter-driven Cre recombinase mice (Montagner et al. 2016, Regnier et al. 2020). These mice show that PPARα serves as a hepatocyte-specific lipid sensor during fasting, it can be activated by increased lipolytic activity of adipose tissue and increases the expression of genes involved in fatty acid oxidation to prevent fat accumulation in hepatocytes (Montagner et al. 2016). Interestingly, when hepatocyte-specific PPARα knockout mice are challenged with a high-fat diet (60% Kcal from fat) they show evidence of non-alcoholic steatohepatitis (NASH) as shown by increased steatosis, inflammation, and plasma alanine transaminase (ALT) levels that were not developed in wild-type mice (Regnier et al. 2020). This suggests that high expression of PPARα in hepatocytes prevents lipid accumulation and the development of inflammation, thereby reducing NAFLD.
Adipocyte-specific PPARγ knockout mice show that PPARγ serves as an essential gene involved in the differentiation and maintenance of adipose tissue. Two interesting studies knocked out the expression of PPARγ in adipocytes by crossbreeding PPARγ-floxed mice with the fatty acid binding protein 4 (aP2) promoter-driven Cre mouse model (He et al. 2003) or with the adiponectin promoter-driven Cre mouse model (Wang et al. 2013). Although these mouse lines are specifically selected to target adipose tissue, the phenotype of the adipose tissue-specific PPARγ knockout mice significantly differs. Specifically, He et al. showed a modest effect on lipodystrophy in the adipose-tissue specific PPARγ knockout mice (aP2-driven Cre) which was associated with a reduced effect on glucose homeostasis when the mice were fed a high fat diet (He et al. 2003). By contrast, Wang et al. showed severe lipodystrophy in the adipose-tissue specific PPARγ knockout mice (adiponectin-driven Cre) associated with fatty liver, glucose intolerance and severe insulin resistance (Wang et al. 2013). In both studies, loxP sites were located upstream and downstream the exons 1 and 2 of the gene, and thereby the adipose tissue-specific PPARγ knockout mice lose both PPARγ isoforms. However, it should be known that aP2 is not only expressed in adipocytes but also in macrophages (Furuhashi et al. 2008). The non-specificity of the aP2-Cre line, together with the high expression of PPARγ in macrophages, might explain the differences in the phenotype of adipose tissue-specific PPARγ knockout mice. These differences highlight the importance of selecting the appropriate method to knockout the gene to be studied as the model may impact the phenotype obtained in the knockout mouse model, and the understanding of the physiological relevance of the gene studied. Therefore, it should be necessary to assess the tissue-specific effects of PPARs with several Cre-LoxP-based methods (ie. different Cre lines, congenital vs inducible Cre-mediated recombination) to dissect out the real function of PPARs in a specific tissue.
The previous two examples highlight the importance of highly expressed PPARs in the liver or adipose tissue. However, it is also important to consider the relevance of PPAR isoforms with low expression which are presumed to have a reduced physiological relevance. Adipose tissue-specific PPARα knock out mice obtained by crossbreeding PPARα-floxed mice with adiponectin promoter-driven Cre mice show that PPARα controls adipocyte metabolism by reducing lipogenesis, inflammation, and cholesterol ester accumulation to prevent adiposity (Hinds et al. 2021). Another example would be the cardiomyocyte-specific PPARγ knockout mice obtained by crossbreeding PPARγ floxed mice with α-myosin heavy-chain promoter-driven Cre mouse model. These mice show that cardiomyocyte PPARγ suppresses cardiac growth and embryonic gene expression and inhibits nuclear factor kB (Duan et al. 2005). Importantly, this study shows that the effect of rosiglitazone (a U.S Food and Drug Administration-approved TZD) on cardiac hypertrophy was independent of cardiomyocyte-specific PPARγ expression and may be due to the actions of the rosiglitazone on other cell types or tissues.
Overall, these examples show us that alteration (cell-specific knockout) of PPAR isoforms, independent of their expression level, may reveal novel actions regulated by these nuclear receptors. Moreover, it should be considered that certain physiological (ie. fasting) or pathological (i.e obesity and insulin resistance) conditions may alter the tissue-specific expression of PPAR isoforms thereby altering their potential tissue-specific relevance. Finally, the combination of cell-specific PPARs knockout mouse models and the treatment with exogenous ligands in normal and pathophysiological conditions will help to reveal the true nature of the tissue-specific actions of each PPAR isoform.
NAFLD onset and potential therapeutics.
Non-alcoholic fatty liver disease (NAFLD) has emerged as the leading global cause of chronic liver disease with a worldwide prevalence of 33% which increases to 70% in overweight individuals (Younossi et al. 2016, Quek et al. 2022). The hallmark of this disease is the accumulation of fat in >5% of hepatic parenchyma (in hepatocytes), which is known as steatosis. Hepatocytes are the main cell type of the liver accounting for ~60% of the hepatic cell population and ~80% of hepatic volume. The non-parenchymal cells represent ~40% of hepatic cells but account for a reduced amount of hepatic mass: endothelial cells (~3% volume), Kupffer cells (~2% volume), and hepatic stellate cells (HSCs) (~1.5% volume). Cholangiocytes are also an important cell type of the liver and account for 1–5% of liver cells (Blouin et al. 1977, Nagy et al. 2020). The development of steatosis is largely attributed to obesity and type 2 diabetes, where insulin resistance increases lipolysis in adipose tissue and allows the mobilization of fatty acids to hepatocytes. Furthermore, increased insulin levels and availability of glucose in insulin resistant individuals promotes de novo lipogenesis (DNL) in hepatocytes. Also, excess dietary fat and recycled lipoproteins in obese and insulin resistant individuals increase the availability of fatty acids to hepatocytes (Figure 2). The hepatocytes use the fatty acids as source of energy in the mitochondrial β-oxidation or mostly re-esterify them with glycerol to generate phospholipids and triglycerides (TG). These re-esterified fatty acids can be exported to the circulation as very-low density lipoproteins (VLDL) or stored as lipid droplets in the cytosol of hepatocytes (Figure 2). The imbalance between synthesis, uptake, re-esterification of fatty acids, storage of TG, and lipid oxidation and export can lead to the development of steatosis. Livers with steatosis can then progress to NASH which is characterized by enhanced steatosis, hepatocyte ballooning, and inflammation with the possibility of developing fibrosis (Figure 2) (Takahashi et al. 2014, Friedman et al. 2018). This progression to NASH could be accelerated by enhanced DNL, and reduction in lipid oxidation after short periods of fructose consumption (Schwarz et al. 2015). Furthermore, the type of dietary fat influences NASH, as saturated fatty acids, cholesterol, and trans fats all increase oxidative stress, lipid peroxidation and the production of ceramides (Luukkonen et al. 2016, Luukkonen et al. 2018, Rosqvist et al. 2019). The accumulation of lipids, combined with oxidative and lipid peroxidative stress, promotes hepatocyte ballooning and triggers cell death. Consequently, endogenous proteins and lipids known as damaged associated molecular patterns (DAMPs) are released into the extracellular environment and activate non-parenchymal cells: Kupffer cells, immune cells, HSC, and endothelial cells (An et al. 2020) (Figure 2). In addition, these DAMPs and the chemokines released by non-parenchymal cells attract circulating inflammatory cells such as lymphocytes, neutrophils, and eosinophils to the liver. The combination of activated immune/inflammatory cells and HSC promote inflammation and fibrosis, respectively, that is observed in the progression of NASH (Figure 2). Specifically, both activated immune cells and HSC play a pivotal role in the inflammatory and fibrogenic phenotype of NAFLD (Carter et al. 2022, Huby et al. 2022).
Figure 2. Representation of the changes in the hepatic phenotype during the progression of obesity-related nonalcoholic fatty liver disease (NAFLD).
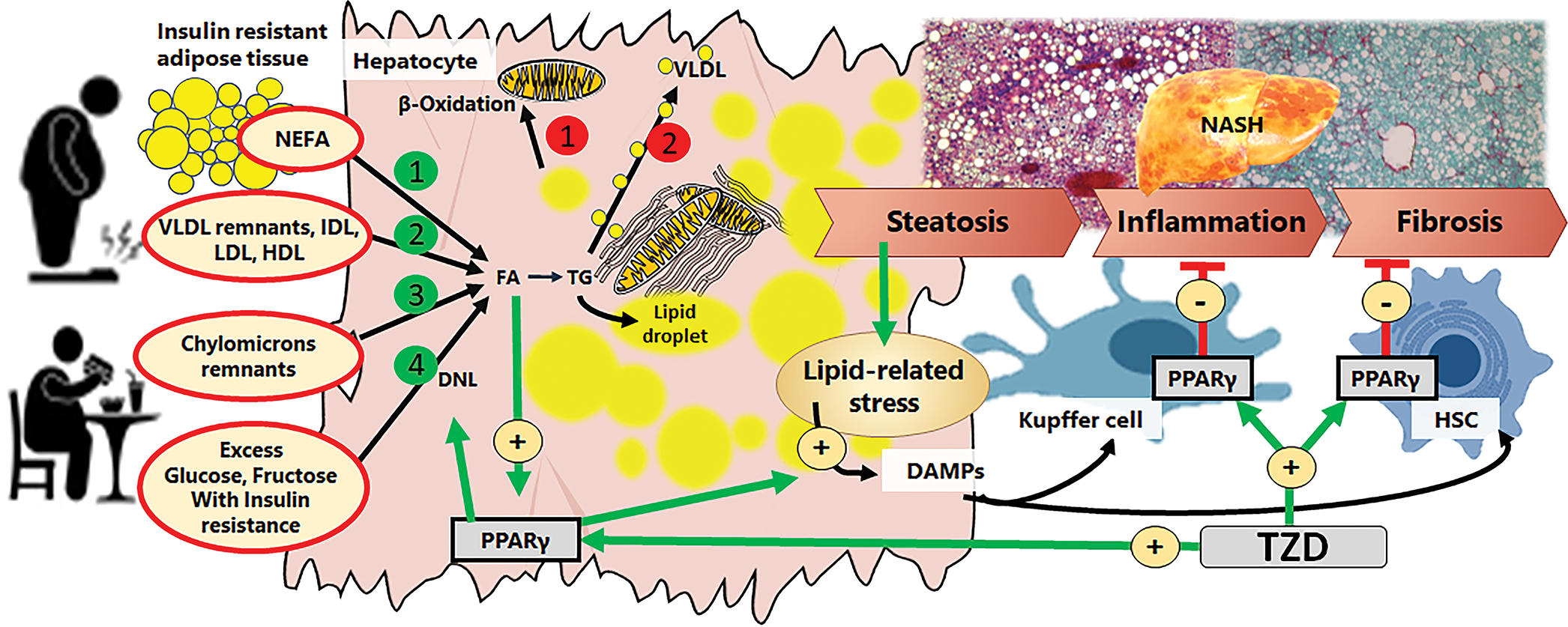
In obese individuals, insulin resistance increases adipose tissue lipolysis resulting in increased availability of non-esterified fatty acids (1, green), and recycled lipoproteins (2, green). In addition, increased food intake and content of carbohydrates and fat in the diet, increases availability of chylomicrons remnants (3, green) and carbohydrates that will fuel de novo lipogenesis (DNL, 4, green). These mechanisms increase the production of triglycerides (TG) and store of fat as lipid droplets in the cytosol. Also, increased fat accumulation in hepatocytes could be associated with impaired fatty acid β-oxidation (1, red) and reduced production of very low density lipoprotein release (VLDL, 2, red). Excess accumulation of fat in hepatocytes leads to steatosis, and promotes lipid-related stress that promotes the generation of damage-associated molecular patterns (DAMPs). These DAMP will be released by hepatocytes to activate the non-parenchymal cells: Kupffer cells and hepatic stellate cells (HSC), which promote the development of inflammation and fibrosis, respectively, and the progression of NAFLD to non-alcoholic steatohepatitis (NASH). TZD activate PPARγ in hepatocytes, Kupffer cells, and HSC. In hepatocytes, PPARγ is also activated by incoming fatty acids and accumulated lipids. PPARγ activates steatogenic mechanisms (green circles 1–4), and may increase the production of DAMPs to activate non-parenchymal cells. In Kupffer cells, PPARγ acts as an anti-inflammatory agent. In HSC, PPARγ acts as an anti-fibrogenic agent.
Lifestyle modifications such as diet and exercise, improve systemic insulin sensitivity and liver histology. In fact, as little as a 10% weight loss leads to significant improvement of liver histology in NAFLD patients (Vilar-Gomez et al. 2015, Babu et al. 2021). However, since lifestyle interventions have proven to be difficult to sustain, clinical trials are testing multiple drugs to treat NAFLD. Some potential candidates could directly target hepatocyte fat and carbohydrate metabolism to reduce NAFLD. Thyroid hormone receptor beta agonists (Resmestriom) could promote fatty acid oxidation and reduce steatosis (Harrison et al. 2019). Also, inhibitors of DNL enzymes (GS-0976, PF-05221304, aramchol), and ketohexokinase (PF-06835919) may reduce the levels of hepatic lipids (Loomba et al. 2018, Calle et al. 2021, Kazierad et al. 2021, Ratziu et al. 2021). Other drugs could target non-parenchymal cells to reduce inflammation and fibrosis such as the inhibitors of galectin 3 (Belapectin) or CCR2/CCR5 (Cenicriviroc) (Chalasani et al. 2020, Ratziu et al. 2020). As indicated above, improved systemic insulin sensitivity could indirectly improve liver histology. Thus, TZD reduce steatosis and inflammation but they increase body weight (Ratziu et al. 2010, Sanyal et al. 2010, Cusi et al. 2016), whereas GLP-1 receptor agonists (liraglutide or semaglutide) reduce steatosis and inflammation associated with body weight loss (Armstrong et al. 2016, Newsome et al. 2021). Agonists of FXR (obeticholic acid) improve insulin sensitivity and reduce NASH and fibrosis (Younossi et al. 2019). These are only a few of the drugs and therapeutic mechanisms being explored to reduce NAFLD. To date, we do not have a specific pharmacological treatment for NAFLD. Although many clinical trials have tested multiple drugs, they cannot reduce NAFLD/NASH efficiently (Vuppalanchi et al. 2021), and it is possible that some of these drugs are excellent candidates but they evoke unknown functions in specific cell-types that reduce or limit their therapeutic effects.
Thiazolidinediones as a therapy for NAFLD
TZD are a group of drugs with strong insulin sensitizing properties that were developed in the 1980s and later were found to be agonists for PPARγ (Fujita et al. 1983, Lehmann et al. 1995). Rosiglitazone (Avandia, 4–8 mg/day) and Pioglitazone (Actos, 15–30 mg/day) are the current U.S Food and Drug Administration-approved TZD that can be used as a second-line treatment for diabetes due to their potent insulin-sensitizing effects (Soccio et al. 2014). However, side-effects of TZD such as weight gain, edema, bone loss, and heart problems have reduced and limited their use in clinic (Soccio et al. 2014). Briefly, TZD activate PPARγ in white adipose tissue to increase insulin sensitivity and promote adipogenesis allowing the safe storage of plasma lipids into newly formed adipocytes, thus reducing hyperglycemia, insulin levels, and dyslipidemia (Soccio et al. 2014). Of note, TZD increase the release of adiponectin (Iwaki et al. 2003), an adipokine known to be reduced in obese and diabetic patients (Bugianesi et al. 2005), that improves glucose and lipid metabolism in liver and skeletal muscle. Adiponectin signaling activates AMPK signaling which phosphorylates acetyl-CoA carboxylase to reduce DNL and promote fatty acid oxidation (Yu et al. 2002, LeBrasseur et al. 2006). Therefore, TZD could reduce insulin resistance, dyslipidemia, hepatic lipid synthesis and increase fatty acid oxidation indirectly in patients with NAFLD to reduce liver steatosis.
TZD have shown anti-fibrogenic and anti-inflammatory properties in vitro as well. TZDs suppress the activation of isolated human HSC and prevent the release of transforming growth factor β1 (Galli et al. 2000, Galli et al. 2002). Furthermore, it has been previously observed that oral administration of TZD reduced fibrosis in various rodent models of liver fibrosis (Galli et al. 2002). Moreover, TZD switch the pro-inflammatory M1 to an anti-inflammatory M2 phenotype in macrophages of adipose tissue (Prieur et al. 2011). Interestingly, pioglitazone increased the expression of M2 macrophages markers in obese patients (Satoh et al. 2010) while rosiglitazone reduced inflammatory markers in blood (Mohanty et al. 2004). Based on these strong anti-NAFLD/NASH properties of TZD, these drugs must be excellent therapeutic tools to reduce inflammation, and fibrosis in patients with NASH.
The effects of TZD on humans with NASH were recently reviewed (Ferguson et al. 2021, Lange et al. 2022). Three long-term major clinical trials have described the positive effects of TZD (Rosiglitazone and Pioglitazone) on NASH, but some effects on histological improvement slightly differ between studies (Ratziu et al. 2010, Sanyal et al. 2010, Cusi et al. 2016). Nonetheless, it is well appreciated that TZD therapies increase insulin sensitivity which improves liver histology, and have been recommended for NASH patients. In fact, the positive effects of TZD on NAFLD are currently being tested in another clinical trial that uses pioglitazone (Clinical Trial # NCT04501406). Nonetheless, pioglitazone but not rosiglitazone improves fibrosis (Musso et al. 2017), and TZD “therapy beyond 18 months does not offer significant additional histological benefit” (Musso et al. 2017) for both rosiglitazone (Ratziu et al. 2010), and pioglitazone ((Cusi et al. 2016), appendix figure 1 - 36 months results). It should be noted that pioglitazone is a weaker PPARγ agonist than rosiglitazone, and activates PPARα to a lesser extent which could promote fatty acid oxidation in hepatocytes (Orasanu et al. 2008). Therefore, the stronger positive effects of pioglitazone might in part be due to a weak PPARγ agonism, or to its PPARγ-independent activity. Thus, it is not unusual to find that additional PPAR agonists with ability to bind PPARα and PPARβ/δ, (elafibranor), PPARα, PPARβ/δ, and PPARγ (lanifibranor), or the second generation TZD (MSDC-0620K) with reduced ability to activate PPARγ are potential therapeutics to treat NAFLD (Ratziu et al. 2016, Harrison et al. 2020, Francque et al. 2021). In addition, the limitation of continued improvements in liver histology in the long-term treatment with TZD, and specifically the “exhaustion” of the anti-steatogenic effect of rosiglitazone, may be suggesting an incomplete understanding about the potential therapeutic effects of TZD in liver histology. Whether a direct PPARγ agonism with exogenous ligands on hepatic cells compromises the significant benefits of TZD-mediated insulin sensitization remains to be determined. To unveil the effects of PPARγ agonism in the liver, that cannot be deciphered in descriptive clinical trials, mouse models can be used where we can tease apart the cell-specific actions of PPARγ and their impact on the effects of TZD therapies on the liver.
Cell-specific expression of PPARγ and potential cell-specific effect of TZD in the liver of mouse models
PPARγ is expressed in both hepatocytes and non-parenchymal cells (Kupffer cells, immune cells, and HSC) in the liver. As indicated above, low-level expression of a PPAR isoform could be important in the control of tissue physiology, and the cells could be sensitive to TZD-mediated PPARγ activation. Also, the expression level of PPARγ in pathological conditions may differ from that at basal stage (Figure 1) and alter the cell-specific actions of TZD. Liver diseases activate HSC, which is associated with reduced expression of PPARγ (likely PPARγ1) in isolated HSC (Marra et al. 2000, Miyahara et al. 2000). However, since the percentage of HSC in the liver is reduced as compared to other cell types, the activation of HSC may not account for significant changes in total hepatic PPARγ expression. Furthermore, liver diseases increase the activation and infiltration of immune cells (including macrophages) that express PPARγ which can also be activated with TZD (Gautier et al. 2012). The development of fatty liver disease alters these immune cells that develop an inflammatory phenotype, but the percentage of immune cells in the liver may not account for a significant change of total PPARγ expression. The accumulation of lipids in hepatocytes in different liver diseases may increase the expression and/or activity of PPARγ because this could serve as a sensor for hydrophobic molecules (Figure 2). It is well known that mice fed a high fat diet show increased expression of hepatic PPARγ (Inoue et al. 2005), and the activator protein 1 (AP-1, Fos/Jun) complex activate the expression of PPARγ2 in hepatocytes of mice fed a high fat diet (Hasenfuss et al. 2014). This would be in line with early studies by Vidal-Puig et al that showed a dietary (high-fat diet)-mediated upregulation of PPARγ2 in adipose tissue (Vidal-Puig et al. 1996, Vidal-Puig et al. 1997), as well as our recent study that shows how high-fat diet increases PPARγ2 levels in male and female mice (Lee et al. 2023). Thereby, although PPARγ is expressed at low levels in the livers of lean patients (Vidal-Puig et al. 1997), its expression (or the pathways controlled by PPARγ in the liver) is significantly increased in patients with NAFLD/NASH (Nakamuta et al. 2005, Lima-Cabello et al. 2011, Pettinelli et al. 2011, Jia et al. 2019, Namjou et al. 2019, Frohlich et al. 2020, Lee et al. 2023). Specifically, we have measured the expression of PPARγ in a cohort of 102 patients with severe obesity and found that obese patients with NAFLD/NASH (70% of the cohort) have increased expression of PPARγ and PPARγ-target genes in the liver (Lee et al. 2023). Similarly, the expression of PPARγ is also increased in the livers of mouse models with NAFLD, suggesting that PPARγ is a steatogenic factor that contributes to the progression of this disease (Gavrilova et al. 2003, Matsusue et al. 2003, Inoue et al. 2005, Morán-Salvador et al. 2011, Gao et al. 2016, Wolf Greenstein et al. 2017, de Conti et al. 2020, Lee et al. 2021a, Lee et al. 2021b). To assess the cell-specific relevance of PPARγ expression in NAFLD/NASH, different mouse models were used to knockout PPARγ in specific cell types with the cell-specific promoter-driven Cre-LoxP recombination. Below, we will describe some of these studies, and assess the potential effect of PPARγ in macrophages, HSC, and hepatocytes in the progression of NAFLD.
Contribution of myeloid cell-specific PPARγ to the phenotype of the liver in NAFLD.
To test the actions regulated by PPARγ in Kupffer and other immune (ie. infiltrating macrophages) cells, macrophage (myeloid)-specific PPARγ knockout mice have been generated using the myeloid cell-specific lysozyme 2 (LysM)-promoter driven Cre recombinase mouse model (Greenhalgh et al. 2015) (Table 2). The knockout of PPARγ in macrophages (PpargΔMac) of Balb/c mice reveals a critical role of PPARγ in the regulation of macrophage phenotype and in the reversion of the anti-inflammatory response. Furthermore, PpargΔMac increases adiposity, glucose intolerance and insulin resistance in Balb/c mice fed a high fat diet (Odegaard et al. 2007). Similarly, in a different study, PpargΔMac increased glucose intolerance in chow-fed mice, and increased dyslipidemia and ALT in C57Bl/6J mice fed a high-fat diet. However, in this latter study PpargΔMac did not impact obesity, glucose homeostasis, nor inflammation (Morán-Salvador et al. 2011). The differences in the effect of PpargΔMac could be due to the PPARγ floxed lines (Akiyama et al. 2002, He et al. 2003) and/or the different strains (Balb/c vs C57Bl/6J). In addition, PpargΔMac increases inflammatory damage in mice fed a methionine and choline-deficient diet (Ni et al. 2022), and makes the liver more susceptible to liver damage and fibrosis in a model of hepatoxicity induced by carbon tetrachloride (Moran-Salvador et al. 2013). In mice, rosiglitazone reduces the pro-inflammatory phenotype associated with liver steatosis in PPARγ-intact mice fed a high-fat diet (Luo et al. 2017), and promotes the anti-inflammatory M2 phenotype of adipose-tissue macrophages (Prieur et al. 2011). Therefore, expression and/or activation of PPARγ in macrophages may prevent steatosis and inflammation directly or indirectly when associated with improved systemic insulin sensitivity. In fact, these anti-inflammatory effects are also described in humans (Mohanty et al. 2004, Satoh et al. 2010). However, most of these mouse studies used a model of high-fat diet that resembles early stages of NAFLD where hepatic inflammation is not dramatically increased as in advance stages of NASH. Additional studies using PpargΔMac mice reinforce the role of PPARγ as an anti-inflammatory factor in macrophages but question the role of PPARγ in the glucocorticoid-mediated suppression of pro-inflammatory cytokine production (Heming et al. 2018). Of note, long-term treatment of rosiglitazone in patients with NASH resulted in increased expression of pro-inflammatory genes such as suppressor of cytokine signaling 3, toll like receptor 4 and monocyte chemoattractant protein-1 (Lemoine et al. 2014), and it is possible that this adverse effect is due to a pro-inflammatory role of rosiglitazone in hepatocytes (Rogue et al. 2011). Although TZD have clear anti-inflammatory properties (Figure 2), their actions may be limited in the liver when inflammation is already developed and/or is derived from myeloid and parenchymal cells.
Table 2.
Summary of studies that used cell-specific PPARγ knockout mouse models to assess the contribution of PPARγ in macrophages, HSC and hepatocytes in different models of fatty liver disease.
| Year [Ref] | Model | Hepatic PPARγ expression | Cre, mouse line. | Results of PpargΔ Mac | Results of TZD treatment (Macrophage-PPARγ dependent) |
| 2007 (Odegaard et al. 2007) | Balb/c HFD (18 weeks) | ns | LysM-Cre, 1 | Insulin resistance | NA |
| 2011 (Morán-Salvador et al. 2011) | HFD (12 weeks) | ↑ PPARγ1 and 2 | LysM-Cre, 2 | Glucose intolerance (chow) and ↓ steatosis (HFD) | NA |
| 2013 (Moran-Salvador et al. 2013) | CCl4 (8 weeks) | ↑ PPARγ2 | LysM-Cre, 2 | ↑ Liver damage and fibrosis | NA |
| 2018 (Heming et al. 2018) | Bone marrow cells | ns | LysM-Cre, ns | Pro-inflammatory phenotype. | NA |
| 2022 (Ni et al. 2022) | MCD (4weeks) | ns | LysM-Cre, 3 | ↑ inflammation and fibrosis | NA |
| Ref. | Model | Hepatic PPARγ expression | Cre, mouse line. | Results of PpargΔHSC | Results of TZD treatment (HSC-PPARγ dependent), dose |
| 2013 (Moran-Salvador et al. 2013) | CCl4 (8 weeks) | ↑ PPARγ2 | aP2-Cre, 2 | ↑ Liver damage and fibrosis | NA |
| 2020 (Liu et al. 2020) | CCl4 + recovery | ns | Lrat-Cre, 2 | ↑ fibrosis | Accelerated regression of liver fibrosis. |
| Ref. | Model | Hepatic PPARγ expression | Cre, mouse line. | Results of PpargΔHep | Results of TZD treatment [dose] (Hepatocyte-PPARγ dependent) |
| 2003 (Matsusue et al. 2003) | ob/ob | ↑ PPARγ | Alb-Cre, 1 | ↓ steatosis. Impaired glucose homeostasis, and chylomicrons remnants clearance. | ↑ steatosis by rosiglitazone [~3 mg/kg/day, 3 weeks] in PPARγ-intact ob/ob mice. |
| 2003 (Gavrilova et al. 2003) | A/ZIP | ↑ PPARγ2 | Alb-Cre, 1 | ↓ steatosis. Impaired triglyceride clearance. | ↑ steatosis by rosiglitazone [~3 mg/kg/day, 5 weeks] in PPARγ-intact A/ZIP mice. |
| 2011 (Morán-Salvador et al. 2011) | HFD (12 weeks) | ↑ PPARγ1 and 2 | Alb-Cre, 2 | ↓ steatosis. Improved glucose homeostasis. | ↑ steatosis by rosiglitazone [10μM] in PPARγ-intact PCLS |
| 2013 (Moran-Salvador et al. 2013) | CCl4 (8 weeks) | ↑ PPARγ2 | Alb-Cre, 2 | ↑ hepatic IL1B, TIMP1 expression. | NA |
| 2016 (Zhang et al. 2016) | Alcohol (8 weeks) | ↑t PARγ2 | Alb-Cre, 2 | ↓ steatosis and inflammation | NA |
| 2017 (Wang et al. 2017) | HFD-Alcohol (3 months) | ↑ PPARγ | Alb-Cre, 2 | ↓ steatosis and fibrosis, but ↑ neutrophil infiltration | NA |
| 2017 (Wolf Greenstein et al. 2017) | HFD (14 weeks) | ↑ PPARγ | AAV8-TBG-Cre, 2 | ↓ steatosis | NA |
| 2020 (Cordoba-Chacon 2020) | MCD (3 weeks) | ↑ PPARγ | AAV8-TBG-Cre, 2 | ↓ fibrosis | NA |
| 2020 (Kulkarni et al. 2020) | HFD (3 months) | ↑ PPARγ | Alb-Cre | ↓ steatosis | ↑ steatosis by pioglitazone [100 mg/Kg diet, 3 months] in PPARγ-intact mice. |
| 2021 (Lee et al. 2021a) | HFD (23 weeks) | ↑ PPARγ2 | AAV8-TBG-Cre, 2 | ↓ steatosis in severe obese mice, and ↑ adiposity | ↑ steatosis by rosiglitazone [70 mg/Kg diet, 6 weeks] in PPARγ-intact severe obese mice. |
| 2021 (Lee et al. 2021b) | HFCF (24/34 weeks) | ↑ PPARγ in male mice | AAV8-TBG-Cre, 2 | ↓ steatosis and NASH in male mice | Rosiglitazone [50 mg/Kg diet, 8 weeks] efficiently decreases NASH in PpargΔHepmice |
| 2022 (Lee et al. 2023) | HFD-HFCF (18–16 weeks) | ↑ PPARγ2 | AAV8-TBG-Cre, 2 | ↓ steatosis in HFD-fed and NASH in HFCF-fed male and female mice | NA |
Models: ob/ob, leptin-deficient hyperphagic mouse model with severe obesity; A/ZIP, lipodystrophie mouse model; HFD, high-fat diet-induced obese mouse model; CCl4, carbon tetrachloride-induced hepatoxicity; MCD: methionine and choline-deficient diet; Alcohol, alcoholic liver disease- induced by Lieber-deCarli diet; HFD-Alcohol, high-fat diet plus binge ethanol mouse model; MCD, methionine and choline-deficient diet-induced steatohepatitis; HFCF, high-fat, cholesterol, and fructose diet-induced non-alcoholic steatohepatitis. ↑, increased; ↓, decreased. Ns, not shown or described. NA, not applicable to this study. IL1B: Interleukin 1B, TIMP1: Tissue inhibitor of metalloproteinase 1. PCLS: Precision-cut liver slices. PPARγ floxed mouse line: 1, mouse line developed by Akiyama et al (Akiyama et al. 2002); 2, mouse line developed by He et al (He et al. 2003) (Jackson Laboratories Strain #:004584); 3, mouse line obtained in GemPharmatech (Nanjing, China).
Contribution of HSC-specific PPARγ to the phenotype of the liver in NAFLD
To test the contribution of PPARγ in HSC, PPARγ has been knocked out using the aP2 promoter-driven Cre recombinase model (Moran-Salvador et al. 2013). The cell-specific deletion of PPARγ using this model is questionable because aP2 is expressed in parenchymal and non-parenchymal cells of the liver, as well as in the adipose tissue. Regardless, aP2-Cre/PPARγ-floxed mice show increased liver damage and fibrogenesis induced by carbon tetrachloride-mediated hepatoxicity (Moran-Salvador et al. 2013) (Table 2). A recent study used the lecithin:retinol acyltransferase (Lrat) promoter-driven Cre mouse model (Greenhalgh et al. 2015) to knock out PPARγ in HSC (PpargΔHSC). PpargΔHSC increases the susceptibility to develop fibrosis induced by carbon tetrachloride and impairs the resolution of fibrosis (Liu et al. 2020) (Table 2). These studies support the well-known effect of TZD as anti-fibrogenic agents and support that expression of PPARγ in the HSC is required to maintain a quiescent phenotype (Galli et al. 2000, Galli et al. 2002) (Figure 2). However, these studies did not explore the role of PPARγ in the activation of HSC in models of NAFLD/NASH. In fact, the effect of TZD in the reversion of fibrosis in patients with NASH did not achieve a clear and significant result (Ratziu et al. 2010, Sanyal et al. 2010, Cusi et al. 2016). Therefore, it is possible that in NASH, DAMPs and cytokines released by parenchymal and non-parenchymal cells reduce the efficiency of TZD in the reversion of fibrosis.
Contribution of hepatocyte-specific PPARγ to the phenotype of the liver in NAFLD.
To test the contribution of PPARγ in hepatocytes, PPARγ has been mostly knocked out using the Alb promoter-driven Cre recombinase: PpargΔHep (Gavrilova et al. 2003, Matsusue et al. 2003, Morán-Salvador et al. 2011, Moran-Salvador et al. 2013, Zhang et al. 2016, Wang et al. 2017, Kulkarni et al. 2020). Our research group has used adeno-associated virus serotype 8 to deliver a thyroxin binding globulin (TBG) promoter-driven Cre recombinase (AAV8-TBG-Cre) to generate PpargΔHep in adult mice (Wolf Greenstein et al. 2017, Cordoba-Chacon 2020, Lee et al. 2021a, Lee et al. 2021b, Lee et al. 2023) (Table 2). The generation of PpargΔHep mice clearly shows that PPARγ serves as a steatogenic factor in hepatocytes. PPARγ controls the expression of genes involved in lipid metabolism: DNL, fatty acid re-esterification, and lipid storage (Gavrilova et al. 2003, Matsusue et al. 2003, Morán-Salvador et al. 2011, Wolf Greenstein et al. 2017, Lee et al. 2021a). Of note, the steatogenic genes are upregulated in the liver of mice that overexpress PPARγ1 or PPARγ2 (Yu et al. 2003, Lee et al. 2012). Moreover, the development of liver steatosis associated with hyperphagia and obesity in ob/ob mice, with lipodystrophy in A/ZIP mice, with high-fat diet-induced obesity, or with alcohol-mediated steatosis or carbon tetrachloride-mediated hepatoxicity is associated with increased expression of hepatic PPARγ (Table 2). In fact, PPARγ2 is mostly induced in these models (Table 2), and PpargΔHep mice show that this isoform is hepatocyte-specific and regulated by high-fat diets (Gavrilova et al. 2003, Matsusue et al. 2003, Morán-Salvador et al. 2011, Wolf Greenstein et al. 2017, Lee et al. 2021a, Lee et al. 2023), as previously described in adipose tissue (Vidal-Puig et al. 1996, Vidal-Puig et al. 1997).
Rosiglitazone or pioglitazone treatments in PPARγ-intact mice result in an TZD-mediated increase of liver steatosis (Table 2, Figure 2) (Gavrilova et al. 2003, Matsusue et al. 2003, Morán-Salvador et al. 2011, Gao et al. 2016, Kulkarni et al. 2020, Lee et al. 2021a). However, this steatogenic effect of TZD is not commonly observed in mice with mild or reduced obesity, nor in low-fat fed mice where TZD decreases hepatic fat accumulation (Gao et al. 2016, Lee et al. 2021a). Pioglitazone may activate PPARα as well and directly increase fatty acid oxidation, whereas rosiglitazone is a powerful and selective agonist of PPARγ. Therefore, it is common to see that rosiglitazone, rather than pioglitazone, increases hepatic fat accumulation in a hepatocyte-PPARγ dependent manner in obese mice (Table 2). Overall, these results indicate that if TZD are provided to mice with obesity and insulin resistance, they will likely increase liver steatosis in a hepatocyte-specific PPARγ dependent manner despite the increase in whole-body insulin sensitivity. Of course, when compared to human studies, it is important to understand that TZD-treated mice do not follow lifestyle modifications (diet and exercise) and mice are kept in the same obesogenic diet while treated with TZD. Also, it should be noted that the studies included in this review mostly provide the TZD treatment (in diet, Table 2) after liver steatosis has developed by the genetic background or diet-induced obesity. Therefore, increased expression of PPARγ in the liver (that likely is derived from increased fat accumulation in hepatocytes) may limit the therapeutic effects of TZD on liver steatosis which mostly is the consequence of their insulin sensitizing properties in peripheral tissues.
As described above, TZD show PPARγ-dependent anti-fibrotic and anti-inflammatory properties. However, the anti-inflammatory effects of TZD in NASH patients are unclear, and the anti-fibrotic effects of TZD on NASH patients are questionable. Our lab has performed the first studies that assess the role of hepatocyte-specific PPARγ in the progression of diet-induced NASH and in the response to rosiglitazone treatment (initiated after development of NASH). Male PpargΔHep mice showed reduced progression of NASH induced with a high fat, cholesterol, and fructose diet (Lee et al. 2021b). This PpargΔHep-mediated protection was associated with reduced expression of inflammatory and fibrogenic genes in the liver and with reduced progression liver histology: steatosis, inflammation, and fibrosis. This PpargΔHep-mediated improvement in liver histology is independent of improved peripheral metabolism or adiposity of male mice (Lee et al. 2021b). Furthermore, these results suggest that the protection against diet-induced NASH by PpargΔHep mice is due to biological processes regulated by PPARγ in hepatocytes. In addition, the induction of PpargΔHep after the development of diet-induced NASH increased the therapeutic effect of TZD in the liver. Specifically, rosiglitazone reduced steatosis, liver injury, and expression of hepatic genes involved in fibrosis in PpargΔHep mice but not in PPARγ-intact mice when these mice were maintained in a high fat, cholesterol and fructose diet (Lee et al. 2021b). This dramatic effect of rosiglitazone in the liver of PpargΔHep mice with NASH was associated with an enhanced effects of TZD on adipose tissue: increased TZD-mediated adiposity, adiponectin production, and phosphorylation of hepatic AMPK (Lee et al. 2021b). The enhanced effect of rosiglitazone on adipose tissue was also found in severe obese mice where PpargΔHep was induced after the development of obesity (Lee et al. 2021a). This suggests that hepatocyte-specific PPARγ may reduce effects of TZD on the adipose tissue and the indirect benefits on the liver.
Although the protective effect of PpargΔHep on fibrosis and inflammation is not expected, it has been previously shown that increased expression of PPARγ in the liver (likely hepatocyte-specific) could be associated with development of inflammation and fibrosis (Lemoine et al. 2014, Bhushan et al. 2019, Cordoba-Chacon 2020). Our latest study further supports the role of hepatocyte-specific PPARγ in the development of fibrosis, where PpargΔHep was induced after the development of diet-induced obesity and before diet-induced NASH. This study suggests that PPARγ expression in hepatocytes contributes to the development of fibrosis in male mice, and that effect could be due to a negative effect of PPARγ in the metabolism of methionine. Specifically, we observed that PPARγ expression was positively associated with the levels of homocysteine which could serve as a DAMP, promote NASH (Tripathi et al. 2022) (Figure 2), and activate HSC and the development of fibrosis (Lee et al. 2021b, Lee et al. 2023).
Role of hepatic PPARγ in NAFLD beyond expression.
The regulation of hepatic PPARγ expression, activity, and protein stability has been reported by multiple studies that describe a multilayer and complex regulatory network which regulates hepatic PPARγ actions. This section will briefly cover novel mechanisms that could regulate hepatic PPARγ activity. For instance, long non-coding RNA (lncRNA) H19 has been reported to control hepatic metabolism by epigenetic mechanisms, and it is upregulated by fatty acids and diet-induced steatosis. lncRNA-H19 increases hepatic lipogenesis by upregulating the expression of PPARγ in hepatocytes via downregulation of miR-130a, which negatively regulates PPARγ through binding onto the 3′-UTR region of PPARγ mRNA (Liu et al. 2019). Another example, carbohydrate response element binding protein promotes the secretion of hepatocyte growth factor activator (a protease) from hepatocytes which subsequently activates hepatocyte growth factor (HGF). Then, HGF activates c-Met receptor tyrosine kinase in hepatocytes and increases PPARγ expression and steatogenic genes (Sargsyan et al. 2022). Over nutrition reduces the expression and activity of DNA 6mA demethylase AlkB homolog 1 (ALKBH1) which is an epigenetic regulator in hepatocytes. As a consequence, ALKBH1 cannot demethylate 6mA in intergenic regions and introns of PPARγ and its target genes (i.e Cd36) which favors the expression of PPARγ-regulated steatogenic program (Luo et al. 2022). Another important consideration is whether post-translation modification of PPARγ influences its activity. For example, PPARγ is acetylated in multiple sites, and TZD deacetylate lysine 268 and 293 in a SIRT1-dependent manner to promote browning of white adipose tissue. A mouse model bearing deacetylation-mimetic mutations of lysine 268 and 293 to arginine showed enhanced effects of rosiglitazone in diet-induced obese mice where rosiglitazone was able to reduce liver weight and steatosis (Kraakman et al. 2018). This suggests that acetylation may also promote the steatogenic effects of PPARγ. Furthermore, the activity of PPARγ in the liver may be negatively regulated by high mobility group box 1 (HMGB1) which may suppress direct PPARγ transcriptional activity to prevent the development of NAFLD (Personnaz et al. 2022). Also, a synthetic derivate of xanthohumol named tetrahydroxanthohumol could directly bind to PPARγ and work as an antagonist of PPARγ to reduce fat accumulation in hepatocytes of mice fed a high fat diet (Zhang et al. 2021). Moreover, nutrition-mediated suppression of AMPK-TBC1D1 signaling increases the levels of GTP-bound Rab2A and the stability of PPARγ in hepatocytes that promotes NAFLD. Of note, GTP-bound Rab2A binds the AF-2 domain of PPARγ and inhibits its proteasomal degradation (Chen et al. 2022). Furthermore, ubiquitin-specific protease 22 (USP22) stabilizes PPARγ via K48-linked deubiquitination. USP22 may interact with the DNA-binding domain of PPARγ and stabilizes its function and transcriptional activity to promote NAFLD (Ning et al. 2022). These are recent examples that highlight the critical role of hepatic PPARγ as a steatogenic factor. Further research needs to be performed in preclinical mouse models of diet-induced NASH to assess the cell-specific contribution of these mechanisms that regulate PPARγ activity in hepatocytes to the development and progression of NAFLD.
Conclusions.
PPARγ belongs to a family of nuclear receptors that serve as lipid sensors and contribute to multiple functions in several tissues. Since PPARγ is the target of TZD, massive attention to the therapeutic effects of TZD have been drawn in the last decades. Cell-specific knockout mouse models have become a valuable tool to decipher the contribution of PPARγ in liver pathology. Most of these studies indicate that hepatocyte-specific expression of PPARγ alters hepatic metabolism and the activation of non-parenchymal cells. Also, these studies show that while TZD increase steatosis in a hepatocyte-PPARγ-dependent manner, they can also reduce inflammation and fibrosis by activating PPARγ in non-parenchymal cells. The cell-specific effects of TZD on the liver might limit the therapeutic benefits derived from TZD treatments in mouse models with obesity/insulin resistance and diet-induced NASH. Therefore, in obese and insulin resistant individuals, the cell-specific activation of PPARγ: hepatocytes vs non-parenchymal hepatic cells, should be considered to enhance the positive effects of TZD on NASH, and to design future therapeutical approaches to treat NAFLD and NASH.
Grant Support:
National Institutes of Health K01DK125525, R01DK131038, R03DK129419 [JCC].
Footnotes
Conflict of interest: Authors do not have any conflict of interest.
References
- Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M and Evans RM (2013). “PPARgamma signaling and metabolism: the good, the bad and the future.” Nat Med 19(5): 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama TE, Sakai S, Lambert G, Nicol CJ, Matsusue K, Pimprale S, Lee YH, Ricote M, Glass CK, Brewer HB Jr. and Gonzalez FJ (2002). “Conditional disruption of the peroxisome proliferator-activated receptor gamma gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux.” Mol Cell Biol 22(8): 2607–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An P, Wei LL, Zhao S, Sverdlov DY, Vaid KA, Miyamoto M, Kuramitsu K, Lai M and Popov YV (2020). “Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis.” Nat Commun 11(1): 2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, Hazlehurst JM, Guo K, team L. t., Abouda G, Aldersley MA, Stocken D, Gough SC, Tomlinson JW, Brown RM, Hubscher SG and Newsome (2016). “Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study.” Lancet 387(10019): 679–690. [DOI] [PubMed] [Google Scholar]
- Babu AF, Csader S, Lok J, Gomez-Gallego C, Hanhineva K, El-Nezami H and Schwab U (2021). “Positive Effects of Exercise Intervention without Weight Loss and Dietary Changes in NAFLD-Related Clinical Parameters: A Systematic Review and Meta-Analysis.” Nutrients 13(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan B, Banerjee S, Paranjpe S, Koral K, Mars WM, Stoops JW, Orr A, Bowen WC, Locker J and Michalopoulos GK (2019). “Pharmacologic Inhibition of Epidermal Growth Factor Receptor Suppresses Nonalcoholic Fatty Liver Disease in a Murine Fast-Food Diet Model.” Hepatology 70(5): 1546–1563. [DOI] [PubMed] [Google Scholar]
- Blouin A, Bolender RP and Weibel ER (1977). “Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study.” J Cell Biol 72(2): 441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugianesi E, Pagotto U, Manini R, Vanni E, Gastaldelli A, de Iasio R, Gentilcore E, Natale S, Cassader M, Rizzetto M, Pasquali R and Marchesini G (2005). “Plasma adiponectin in nonalcoholic fatty liver is related to hepatic insulin resistance and hepatic fat content, not to liver disease severity.” J Clin Endocrinol Metab 90(6): 3498–3504. [DOI] [PubMed] [Google Scholar]
- Calle RA, Amin NB, Carvajal-Gonzalez S, Ross TT, Bergman A, Aggarwal S, Crowley C, Rinaldi A, Mancuso J, Aggarwal N, Somayaji V, Inglot M, Tuthill TA, Kou K, Boucher M, Tesz G, Dullea R, Bence KK, Kim AM, Pfefferkorn JA and Esler WP (2021). “ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials.” Nat Med 27(10): 1836–1848. [DOI] [PubMed] [Google Scholar]
- Carter JK and Friedman SL (2022). “Hepatic Stellate Cell-Immune Interactions in NASH.” Front Endocrinol (Lausanne) 13: 867940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, Noureddin M, Pyko M, Shiffman M, Sanyal A, Allgood A, Shlevin H, Horton R, Zomer E, Irish W, Goodman Z, Harrison SA, Traber PG and Belapectin Study I (2020). “Effects of Belapectin, an Inhibitor of Galectin-3, in Patients With Nonalcoholic Steatohepatitis With Cirrhosis and Portal Hypertension.” Gastroenterology 158(5): 1334–1345 e1335. [DOI] [PubMed] [Google Scholar]
- Chao L, Marcus-Samuels B, Mason MM, Moitra J, Vinson C, Arioglu E, Gavrilova O and Reitman ML (2000). “Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones.” J Clin Invest 106(10): 1221–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Sun YT, Wang ZM, Hong J, Xu M, Zhang FT, Zhou XQ, Rong P, Wang Q, Wang HY, Wang H, Chen S and Chen L (2022). “Rab2A regulates the progression of nonalcoholic fatty liver disease downstream of AMPK-TBC1D1 axis by stabilizing PPARgamma.” PLoS Biol 20(1): e3001522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordoba-Chacon J (2020). “Loss of Hepatocyte-Specific PPARgamma Expression Ameliorates Early Events of Steatohepatitis in Mice Fed the Methionine and Choline-Deficient Diet.” PPAR Res 2020: 9735083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, Tio F, Hardies J, Darland C, Musi N, Webb A and Portillo-Sanchez P (2016). “Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial.” Ann Intern Med 165(5): 305–315. [DOI] [PubMed] [Google Scholar]
- de Conti A, Tryndyak V, Willett RA, Borowa-Mazgaj B, Watson A, Patton R, Khare S, Muskhelishvili L, Olson GR, Avigan MI, Cerniglia CE, Ross SA, Sanyal AJ, Beland FA, Rusyn I and Pogribny IP (2020). “Characterization of the variability in the extent of nonalcoholic fatty liver induced by a high-fat diet in the genetically diverse Collaborative Cross mouse model.” FASEB J 34(6): 7773–7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer C, Krey G, Keller H, Givel F, Helftenbein G and Wahli W (1992). “Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors.” Cell 68(5): 879–887. [DOI] [PubMed] [Google Scholar]
- Duan SZ, Ivashchenko CY, Russell MW, Milstone DS and Mortensen RM (2005). “Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-gamma both induce cardiac hypertrophy in mice.” Circ Res 97(4): 372–379. [DOI] [PubMed] [Google Scholar]
- Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, Asplund A, Sjostedt E, Lundberg E, Szigyarto CA, Skogs M, Takanen JO, Berling H, Tegel H, Mulder J, Nilsson P, Schwenk JM, Lindskog C, Danielsson F, Mardinoglu A, Sivertsson A, von Feilitzen K, Forsberg M, Zwahlen M, Olsson I, Navani S, Huss M, Nielsen J, Ponten F and Uhlen M (2014). “Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics.” Mol Cell Proteomics 13(2): 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson D and Finck BN (2021). “Emerging therapeutic approaches for the treatment of NAFLD and type 2 diabetes mellitus.” Nature Reviews Endocrinology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Miranda C, Perez-Carreras M, Colina F, Lopez-Alonso G, Vargas C and Solis-Herruzo JA (2008). “A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease.” Dig Liver Dis 40(3): 200–205. [DOI] [PubMed] [Google Scholar]
- Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, Loomba R, Harrison SA, Balabanska R, Mateva L, Lanthier N, Alkhouri N, Moreno C, Schattenberg JM, Stefanova-Petrova D, Vonghia L, Rouzier R, Guillaume M, Hodge A, Romero-Gomez M, Huot-Marchand P, Baudin M, Richard MP, Abitbol JL, Broqua P, Junien JL, Abdelmalek MF and Group NS (2021). “A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH.” N Engl J Med 385(17): 1547–1558. [DOI] [PubMed] [Google Scholar]
- Friedman SL, Neuschwander-Tetri BA, Rinella M and Sanyal AJ (2018). “Mechanisms of NAFLD development and therapeutic strategies.” Nat Med 24(7): 908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich J, Kovacovicova K, Mazza T, Emma MR, Cabibi D, Foti M, Sobolewski C, Oben JA, Peyrou M, Villarroya F, Soresi M, Rezzani R, Cervello M, Bonomini F, Alisi A and Vinciguerra M (2020). “GDF11 induces mild hepatic fibrosis independent of metabolic health.” Aging (Albany NY) 12(20): 20024–20046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T, Sugiyama Y, Taketomi S, Sohda T, Kawamatsu Y, Iwatsuka H and Suzuoki Z (1983). “Reduction of insulin resistance in obese and/or diabetic animals by 5-[4-(1-methylcyclohexylmethoxy)benzyl]-thiazolidine-2,4-dione (ADD-3878, U-63,287, ciglitazone), a new antidiabetic agent.” Diabetes 32(9): 804–810. [DOI] [PubMed] [Google Scholar]
- Furuhashi M, Fucho R, Gorgun CZ, Tuncman G, Cao H and Hotamisligil GS (2008). “Adipocyte/macrophage fatty acid-binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice.” J Clin Invest 118(7): 2640–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli A, Crabb D, Price D, Ceni E, Salzano R, Surrenti C and Casini A (2000). “Peroxisome proliferator-activated receptor gamma transcriptional regulation is involved in platelet-derived growth factor-induced proliferation of human hepatic stellate cells.” Hepatology 31(1): 101–108. [DOI] [PubMed] [Google Scholar]
- Galli A, Crabb DW, Ceni E, Salzano R, Mello T, Svegliati-Baroni G, Ridolfi F, Trozzi L, Surrenti C and Casini A (2002). “Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro.” Gastroenterology 122(7): 1924–1940. [DOI] [PubMed] [Google Scholar]
- Gao M, Ma Y, Alsaggar M and Liu D (2016). “Dual Outcomes of Rosiglitazone Treatment on Fatty Liver.” AAPS J 18(4): 1023–1031. [DOI] [PubMed] [Google Scholar]
- Gautier EL, Chow A, Spanbroek R, Marcelin G, Greter M, Jakubzick C, Bogunovic M, Leboeuf M, van Rooijen N, Habenicht AJ, Merad M and Randolph GJ (2012). “Systemic analysis of PPARgamma in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity.” J Immunol 189(5): 2614–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, Nicol CJ, Vinson C, Gonzalez FJ and Reitman ML (2003). “Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass.” J Biol Chem 278(36): 34268–34276. [DOI] [PubMed] [Google Scholar]
- Greene ME, Blumberg B, McBride OW, Yi HF, Kronquist K, Kwan K, Hsieh L, Greene G and Nimer SD (1995). “Isolation of the human peroxisome proliferator activated receptor gamma cDNA: expression in hematopoietic cells and chromosomal mapping.” Gene Expr 4(4–5): 281–299. [PMC free article] [PubMed] [Google Scholar]
- Greenhalgh SN, Conroy KP and Henderson NC (2015). “Cre-ativity in the liver: transgenic approaches to targeting hepatic nonparenchymal cells.” Hepatology 61(6): 2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grygiel-Gorniak B (2014). “Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications--a review.” Nutr J 13: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SA, Alkhouri N, Davison BA, Sanyal A, Edwards C, Colca JR, Lee BH, Loomba R, Cusi K, Kolterman O, Cotter G and Dittrich HC (2020). “Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase IIb study.” J Hepatol 72(4): 613–626. [DOI] [PubMed] [Google Scholar]
- Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, Alkhouri N, Bansal MB, Baum S, Neuschwander-Tetri BA, Taub R and Moussa SE (2019). “Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial.” Lancet 394(10213): 2012–2024. [DOI] [PubMed] [Google Scholar]
- Hasenfuss SC, Bakiri L, Thomsen MK, Williams EG, Auwerx J and Wagner EF (2014). “Regulation of steatohepatitis and PPARgamma signaling by distinct AP-1 dimers.” Cell Metab 19(1): 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Barak Y, Hevener A, Olson P, Liao D, Le J, Nelson M, Ong E, Olefsky JM and Evans RM (2003). “Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle.” Proc Natl Acad Sci U S A 100(26): 15712–15717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heming M, Gran S, Jauch SL, Fischer-Riepe L, Russo A, Klotz L, Hermann S, Schafers M, Roth J and Barczyk-Kahlert K (2018). “Peroxisome Proliferator-Activated Receptor-gamma Modulates the Response of Macrophages to Lipopolysaccharide and Glucocorticoids.” Front Immunol 9: 893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevener AL, He W, Barak Y, Le J, Bandyopadhyay G, Olson P, Wilkes J, Evans RM and Olefsky J (2003). “Muscle-specific Pparg deletion causes insulin resistance.” Nat Med 9(12): 1491–1497. [DOI] [PubMed] [Google Scholar]
- Hinds TD Jr., Kipp ZA, Xu M, Yiannikouris FB, Morris AJ, Stec DF, Wahli W and Stec DE (2021). “Adipose-Specific PPARalpha Knockout Mice Have Increased Lipogenesis by PASK-SREBP1 Signaling and a Polarity Shift to Inflammatory Macrophages in White Adipose Tissue.” Cells 11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huby T and Gautier EL (2022). “Immune cell-mediated features of non-alcoholic steatohepatitis.” Nat Rev Immunol 22(7): 429–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Ohtake T, Motomura W, Takahashi N, Hosoki Y, Miyoshi S, Suzuki Y, Saito H, Kohgo Y and Okumura T (2005). “Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice.” Biochem Biophys Res Commun 336(1): 215–222. [DOI] [PubMed] [Google Scholar]
- Issemann I and Green S (1990). “Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators.” Nature 347(6294): 645–650. [DOI] [PubMed] [Google Scholar]
- Iwaki M, Matsuda M, Maeda N, Funahashi T, Matsuzawa Y, Makishima M and Shimomura I (2003). “Induction of adiponectin, a fat-derived antidiabetic and antiatherogenic factor, by nuclear receptors.” Diabetes 52(7): 1655–1663. [DOI] [PubMed] [Google Scholar]
- Jia X and Zhai T (2019). “Integrated Analysis of Multiple Microarray Studies to Identify Novel Gene Signatures in Non-alcoholic Fatty Liver Disease.” Front Endocrinol (Lausanne) 10: 599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazierad DJ, Chidsey K, Somayaji VR, Bergman AJ, Birnbaum MJ and Calle RA (2021). “Inhibition of ketohexokinase in adults with NAFLD reduces liver fat and inflammatory markers: A randomized phase 2 trial.” Med (N Y) 2(7): 800–813 e803. [DOI] [PubMed] [Google Scholar]
- Kim H, Kim M, Im SK and Fang S (2018). “Mouse Cre-LoxP system: general principles to determine tissue-specific roles of target genes.” Lab Anim Res 34(4): 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, Umesono K and Evans RM (1994). “Differential expression and activation of a family of murine peroxisome proliferator-activated receptors.” Proc Natl Acad Sci U S A 91(15): 7355–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Lehmann JM and Willson TM (1999). “Orphan nuclear receptors: shifting endocrinology into reverse.” Science 284(5415): 757–760. [DOI] [PubMed] [Google Scholar]
- Kraakman MJ, Liu Q, Postigo-Fernandez J, Ji R, Kon N, Larrea D, Namwanje M, Fan L, Chan M, Area-Gomez E, Fu W, Creusot RJ and Qiang L (2018). “PPARgamma deacetylation dissociates thiazolidinedione’s metabolic benefits from its adverse effects.” J Clin Invest 128(6): 2600–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni S, Huang J, Tycksen E, Cliften PF and Rudnick DA (2020). “Diet Modifies Pioglitazone’s Influence on Hepatic PPARγ-Regulated Mitochondrial Gene Expression.” PPAR Research 2020: 3817573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange NF, Graf V, Caussy C and Dufour JF (2022). “PPAR-Targeted Therapies in the Treatment of Non-Alcoholic Fatty Liver Disease in Diabetic Patients.” Int J Mol Sci 23(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBrasseur NK, Kelly M, Tsao TS, Farmer SR, Saha AK, Ruderman NB and Tomas E (2006). “Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues.” Am J Physiol Endocrinol Metab 291(1): E175–181. [DOI] [PubMed] [Google Scholar]
- Lee SM, Muratalla J, Diaz-Ruiz A, Remon-Ruiz P, McCann M, Liew CW, Kineman RD and Cordoba-Chacon J (2021a). “Rosiglitazone Requires Hepatocyte PPARγ Expression to Promote Steatosis in Male Mice With Diet-Induced Obesity.” Endocrinology 162(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Muratalla J, Karimi S, Diaz-Ruiz A, Frutos MD, Guzman G, Ramos-Molina B and Cordoba-Chacon J (2023). “Hepatocyte PPARγ contributes to the progression of non-alcoholic steatohepatitis in male and female obese mice.” Cellular and Molecular Life Sciences In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Pusec CM, Norris GH, De Jesus A, Diaz-Ruiz A, Muratalla J, Sarmento-Cabral A, Guzman G, Layden BT and Cordoba-Chacon J (2021b). “Hepatocyte-Specific Loss of PPARγ Protects Mice From NASH and Increases the Therapeutic Effects of Rosiglitazone in the Liver.” Cell Mol Gastroenterol Hepatol 11(5): 1291–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Ko EH, Kim JE, Kim E, Lee H, Choi H, Yu JH, Kim HJ, Seong JK, Kim KS and Kim JW (2012). “Nuclear receptor PPARgamma-regulated monoacylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis.” Proc Natl Acad Sci U S A 109(34): 13656–13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM and Kliewer SA (1995). “An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma).” J Biol Chem 270(22): 12953–12956. [DOI] [PubMed] [Google Scholar]
- Lemberger T, Desvergne B and Wahli W (1996). “Peroxisome proliferator-activated receptors: a nuclear receptor signaling pathway in lipid physiology.” Annu Rev Cell Dev Biol 12: 335–363. [DOI] [PubMed] [Google Scholar]
- Lemoine M, Serfaty L, Cervera P, Capeau J and Ratziu V (2014). “Hepatic molecular effects of rosiglitazone in human non-alcoholic steatohepatitis suggest long-term pro-inflammatory damage.” Hepatol Res 44(12): 1241–1247. [DOI] [PubMed] [Google Scholar]
- Lima-Cabello E, Garcia-Mediavilla MV, Miquilena-Colina ME, Vargas-Castrillon J, Lozano-Rodriguez T, Fernandez-Bermejo M, Olcoz JL, Gonzalez-Gallego J, Garcia-Monzon C and Sanchez-Campos S (2011). “Enhanced expression of pro-inflammatory mediators and liver X-receptor-regulated lipogenic genes in non-alcoholic fatty liver disease and hepatitis C.” Clin Sci (Lond) 120(6): 239–250. [DOI] [PubMed] [Google Scholar]
- Liu J, Tang T, Wang GD and Liu B (2019). “LncRNA-H19 promotes hepatic lipogenesis by directly regulating miR-130a/PPARgamma axis in non-alcoholic fatty liver disease.” Biosci Rep 39(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Xu J, Rosenthal S, Zhang LJ, McCubbin R, Meshgin N, Shang L, Koyama Y, Ma HY, Sharma S, Heinz S, Glass CK, Benner C, Brenner DA and Kisseleva T (2020). “Identification of Lineage-Specific Transcription Factors That Prevent Activation of Hepatic Stellate Cells and Promote Fibrosis Resolution.” Gastroenterology 158(6): 1728–1744.e1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, Kayali Z, Noureddin M, Ruane P, Lawitz EJ, Bennett M, Wang L, Harting E, Tarrant JM, McColgan BJ, Chung C, Ray AS, Subramanian GM, Myers RP, Middleton MS, Lai M, Charlton M and Harrison SA (2018). “GS-0976 Reduces Hepatic Steatosis and Fibrosis Markers in Patients With Nonalcoholic Fatty Liver Disease.” Gastroenterology 155(5): 1463–1473 e1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Liu Y, Nizigiyimana P, Ye M, Xiao Y, Guo Q, Su T, Luo X, Huang Y and Zhou H (2022). “DNA 6mA Demethylase ALKBH1 Orchestrates Fatty Acid Metabolism and Suppresses Diet-Induced Hepatic Steatosis.” Cell Mol Gastroenterol Hepatol 14(6): 1213–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Xu Q, Wang Q, Wu H and Hua J (2017). “Effect of modulation of PPAR-gamma activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease.” Sci Rep 7: 44612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luukkonen PK, Sädevirta S, Zhou Y, Kayser B, Ali A, Ahonen L, Lallukka S, Pelloux V, Gaggini M, Jian C, Hakkarainen A, Lundbom N, Gylling H, Salonen A, Orešič M, Hyötyläinen T, Orho-Melander M, Rissanen A, Gastaldelli A, Clément K, Hodson L and Yki-Järvinen H (2018). “Saturated Fat Is More Metabolically Harmful for the Human Liver Than Unsaturated Fat or Simple Sugars.” Diabetes Care 41(8): 1732–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luukkonen PK, Zhou Y, Sädevirta S, Leivonen M, Arola J, Orešič M, Hyötyläinen T and Yki-Järvinen H (2016). “Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease.” J Hepatol 64(5): 1167–1175. [DOI] [PubMed] [Google Scholar]
- Marra F, Efsen E, Romanelli RG, Caligiuri A, Pastacaldi S, Batignani G, Bonacchi A, Caporale R, Laffi G, Pinzani M and Gentilini P (2000). “Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells.” Gastroenterology 119(2): 466–478. [DOI] [PubMed] [Google Scholar]
- Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, Brewer B Jr., Reitman ML and Gonzalez FJ (2003). “Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes.” J Clin Invest 111(5): 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyahara T, Schrum L, Rippe R, Xiong S, Yee HF Jr., Motomura K, Anania FA, Willson TM and Tsukamoto H (2000). “Peroxisome proliferator-activated receptors and hepatic stellate cell activation.” J Biol Chem 275(46): 35715–35722. [DOI] [PubMed] [Google Scholar]
- Mohanty P, Aljada A, Ghanim H, Hofmeyer D, Tripathy D, Syed T, Al-Haddad W, Dhindsa S and Dandona P (2004). “Evidence for a potent antiinflammatory effect of rosiglitazone.” J Clin Endocrinol Metab 89(6): 2728–2735. [DOI] [PubMed] [Google Scholar]
- Montagner A, Polizzi A, Fouche E, Ducheix S, Lippi Y, Lasserre F, Barquissau V, Regnier M, Lukowicz C, Benhamed F, Iroz A, Bertrand-Michel J, Al Saati T, Cano P, Mselli-Lakhal L, Mithieux G, Rajas F, Lagarrigue S, Pineau T, Loiseau N, Postic C, Langin D, Wahli W and Guillou H (2016). “Liver PPARalpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD.” Gut 65(7): 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morán-Salvador E, López-Parra M, García-Alonso V, Titos E, Martínez-Clemente M, González-Périz A, López-Vicario C, Barak Y, Arroyo V and Clària J (2011). “Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts.” Faseb j 25(8): 2538–2550. [DOI] [PubMed] [Google Scholar]
- Moran-Salvador E, Titos E, Rius B, Gonzalez-Periz A, Garcia-Alonso V, Lopez-Vicario C, Miquel R, Barak Y, Arroyo V and Claria J (2013). “Cell-specific PPARgamma deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells.” J Hepatol 59(5): 1045–1053. [DOI] [PubMed] [Google Scholar]
- Mukherjee R, Jow L, Noonan D and McDonnell DP (1994). “Human and rat peroxisome proliferator activated receptors (PPARs) demonstrate similar tissue distribution but different responsiveness to PPAR activators.” J Steroid Biochem Mol Biol 51(3–4): 157–166. [DOI] [PubMed] [Google Scholar]
- Musso G, Cassader M, Paschetta E and Gambino R (2017). “Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: A Meta-analysis.” JAMA Intern Med 177(5): 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy P, Thorgeirsson SS and Grisham JW (2020). Organizational Principles of the Liver. The Liver : 1–13. [Google Scholar]
- Nakamuta M, Kohjima M, Morizono S, Kotoh K, Yoshimoto T, Miyagi I and Enjoji M (2005). “Evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease.” Int J Mol Med 16(4): 631–635. [PubMed] [Google Scholar]
- Namjou B, Lingren T, Huang Y, Parameswaran S, Cobb BL, Stanaway IB, Connolly JJ, Mentch FD, Benoit B, Niu X, Wei WQ, Carroll RJ, Pacheco JA, Harley ITW, Divanovic S, Carrell DS, Larson EB, Carey DJ, Verma S, Ritchie MD, Gharavi AG, Murphy S, Williams MS, Crosslin DR, Jarvik GP, Kullo IJ, Hakonarson H, Li R, Xanthakos M. N. e, S. A and Harley JB (2019). “GWAS and enrichment analyses of non-alcoholic fatty liver disease identify new trait-associated genes and pathways across eMERGE Network.” BMC Med 17(1): 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki AR, Rajala MW, Tomas E, Pajvani UB, Saha AK, Trumbauer ME, Pang Z, Chen AS, Ruderman NB, Chen H, Rossetti L and Scherer PE (2006). “Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists.” J Biol Chem 281(5): 2654–2660. [DOI] [PubMed] [Google Scholar]
- Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, Sanyal AJ, Sejling AS, Harrison SA and Investigators NN (2021). “A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis.” N Engl J Med 384(12): 1113–1124. [DOI] [PubMed] [Google Scholar]
- Ni XX, Ji PX, Chen YX, Li XY, Sheng L, Lian M, Guo CJ and Hua J (2022). “Regulation of the macrophage-hepatic stellate cell interaction by targeting macrophage peroxisome proliferator-activated receptor gamma to prevent non-alcoholic steatohepatitis progression in mice.” Liver Int 42(12): 2696–2712. [DOI] [PubMed] [Google Scholar]
- Ning Z, Guo X, Liu X, Lu C, Wang A, Wang X, Wang W, Chen H, Qin W, Liu X, Zhou L, Ma C, Du J, Lin Z, Luo H, Otkur W, Qi H, Chen D, Xia T, Liu J, Tan G, Xu G and Piao HL (2022). “USP22 regulates lipidome accumulation by stabilizing PPARgamma in hepatocellular carcinoma.” Nat Commun 13(1): 2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris AW, Chen L, Fisher SJ, Szanto I, Ristow M, Jozsi AC, Hirshman MF, Rosen ED, Goodyear LJ, Gonzalez FJ, Spiegelman BM and Kahn CR (2003). “Muscle-specific PPARgamma-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones.” J Clin Invest 112(4): 608–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW and Chawla A (2007). “Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance.” Nature 447(7148): 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orasanu G, Ziouzenkova O, Devchand PR, Nehra V, Hamdy O, Horton ES and Plutzky J (2008). “The peroxisome proliferator-activated receptor-gamma agonist pioglitazone represses inflammation in a peroxisome proliferator-activated receptor-alpha-dependent manner in vitro and in vivo in mice.” J Am Coll Cardiol 52(10): 869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Personnaz J, Piccolo E, Dortignac A, Iacovoni JS, Mariette J, Rocher V, Polizzi A, Batut A, Deleruyelle S, Bourdens L, Delos O, Combes-Soia L, Paccoud R, Moreau E, Martins F, Clouaire T, Benhamed F, Montagner A, Wahli W, Schwabe RF, Yart A, Castan-Laurell I, Bertrand-Michel J, Burlet-Schiltz O, Postic C, Denechaud PD, Moro C, Legube G, Lee CH, Guillou H, Valet P, Dray C and Pradere JP (2022). “Nuclear HMGB1 protects from nonalcoholic fatty liver disease through negative regulation of liver X receptor.” Sci Adv 8(12): eabg9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinelli P and Videla LA (2011). “Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction.” J Clin Endocrinol Metab 96(5): 1424–1430. [DOI] [PubMed] [Google Scholar]
- Prieur X, Mok CY, Velagapudi VR, Nunez V, Fuentes L, Montaner D, Ishikawa K, Camacho A, Barbarroja N, O’Rahilly S, Sethi JK, Dopazo J, Oresic M, Ricote M and Vidal-Puig A (2011). “Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice.” Diabetes 60(3): 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quek J, Chan KE, Wong ZY, Tan C, Tan B, Lim WH, Tan DJH, Tang ASP, Tay P, Xiao J, Yong JN, Zeng RW, Chew NWS, Nah B, Kulkarni A, Siddiqui MS, Dan YY, Wong VW, Sanyal AJ, Noureddin M, Muthiah M and Ng CH (2022). “Global prevalence of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in the overweight and obese population: a systematic review and meta-analysis.” Lancet Gastroenterol Hepatol. [DOI] [PubMed] [Google Scholar]
- Rajamoorthi A, Arias N, Basta J, Lee RG and Baldan A (2017). “Amelioration of diet-induced steatohepatitis in mice following combined therapy with ASO-Fsp27 and fenofibrate.” J Lipid Res 58(11): 2127–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratziu V, Charlotte F, Bernhardt C, Giral P, Halbron M, Lenaour G, Hartmann-Heurtier A, Bruckert E and Poynard T (2010). “Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial.” Hepatology 51(2): 445–453. [DOI] [PubMed] [Google Scholar]
- Ratziu V, de Guevara L, Safadi R, Poordad F, Fuster F, Flores-Figueroa J, Arrese M, Fracanzani AL, Ben Bashat D, Lackner K, Gorfine T, Kadosh S, Oren R, Halperin M, Hayardeny L, Loomba R, Friedman S, group A. i. s and Sanyal AJ (2021). “Aramchol in patients with nonalcoholic steatohepatitis: a randomized, double-blind, placebo-controlled phase 2b trial.” Nat Med 27(10): 1825–1835. [DOI] [PubMed] [Google Scholar]
- Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, Romero-Gomez M, Boursier J, Abdelmalek M, Caldwell S, Drenth J, Anstee QM, Hum D, Hanf R, Roudot A, Megnien S, Staels B, Sanyal A and Group G-IS (2016). “Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening.” Gastroenterology 150(5): 1147–1159 e1145. [DOI] [PubMed] [Google Scholar]
- Ratziu V, Sanyal A, Harrison SA, Wong VW, Francque S, Goodman Z, Aithal GP, Kowdley KV, Seyedkazemi S, Fischer L, Loomba R, Abdelmalek MF and Tacke F (2020). “Cenicriviroc Treatment for Adults With Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study.” Hepatology 72(3): 892–905. [DOI] [PubMed] [Google Scholar]
- Regnier M, Polizzi A, Smati S, Lukowicz C, Fougerat A, Lippi Y, Fouche E, Lasserre F, Naylies C, Betoulieres C, Barquissau V, Mouisel E, Bertrand-Michel J, Batut A, Saati TA, Canlet C, Tremblay-Franco M, Ellero-Simatos S, Langin D, Postic C, Wahli W, Loiseau N, Guillou H and Montagner A (2020). “Hepatocyte-specific deletion of Pparalpha promotes NAFLD in the context of obesity.” Sci Rep 10(1): 6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M and Glass CK (2007). “PPARs and molecular mechanisms of transrepression.” Biochim Biophys Acta 1771(8): 926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogue A, Lambert C, Josse R, Antherieu S, Spire C, Claude N and Guillouzo A (2011). “Comparative gene expression profiles induced by PPARgamma and PPARalpha/gamma agonists in human hepatocytes.” PLoS One 6(4): e18816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosqvist F, Kullberg J, Ståhlman M, Cedernaes J, Heurling K, Johansson HE, Iggman D, Wilking H, Larsson A, Eriksson O, Johansson L, Straniero S, Rudling M, Antoni G, Lubberink M, Orho-Melander M, Borén J, Ahlström H and Risérus U (2019). “Overeating Saturated Fat Promotes Fatty Liver and Ceramides Compared With Polyunsaturated Fat: A Randomized Trial.” J Clin Endocrinol Metab 104(12): 6207–6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, Van Natta M, Clark J, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR and Nash CRN (2010). “Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis.” N Engl J Med 362(18): 1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargsyan A, Doridot L, Hannou SA, Tong W, Srinivasan H, Ivison R, Monn R, Kou HH, Haldeman JM, Arlotto M, White PJ, Grimsrud PA, Astapova I, Tsai LT and Herman MA (2022). “HGFAC is a ChREBP regulated hepatokine that enhances glucose and lipid homeostasis.” JCI Insight. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh N, Shimatsu A, Himeno A, Sasaki Y, Yamakage H, Yamada K, Suganami T and Ogawa Y (2010). “Unbalanced M1/M2 phenotype of peripheral blood monocytes in obese diabetic patients: effect of pioglitazone.” Diabetes Care 33(1): e7. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Endo N, Rutledge SJ, Vogel R, Shinar D and Rodan GA (1992). “Identification of a new member of the steroid hormone receptor superfamily that is activated by a peroxisome proliferator and fatty acids.” Mol Endocrinol 6(10): 1634–1641. [DOI] [PubMed] [Google Scholar]
- Scholtes C and Giguere V (2022). “Transcriptional control of energy metabolism by nuclear receptors.” Nat Rev Mol Cell Biol. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Noworolski SM, Wen MJ, Dyachenko A, Prior JL, Weinberg ME, Herraiz LA, Tai VW, Bergeron N, Bersot TP, Rao MN, Schambelan M and Mulligan K (2015). “Effect of a High-Fructose Weight-Maintaining Diet on Lipogenesis and Liver Fat.” J Clin Endocrinol Metab 100(6): 2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soccio RE, Chen ER and Lazar MA (2014). “Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes.” Cell Metab 20(4): 573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugii S, Olson P, Sears DD, Saberi M, Atkins AR, Barish GD, Hong SH, Castro GL, Yin YQ, Nelson MC, Hsiao G, Greaves DR, Downes M, Yu RT, Olefsky JM and Evans RM (2009). “PPAR gamma activation in adipocytes is sufficient for systemic insulin sensitization.” Proceedings of the National Academy of Sciences of the United States of America 106(52): 22504–22509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y and Fukusato T (2014). “Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis.” World J Gastroenterol 20(42): 15539–15548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi M, Singh BK, Zhou J, Tikno K, Widjaja A, Sandireddy R, Arul K, Abdul Ghani SAB, Bee GGB, Wong KA, Pei HJ, Shekeran SG, Sinha RA, Singh MK, Cook SA, Suzuki A, Lim TR, Cheah CC, Wang J, Xiao RP, Zhang X, Chow PKH and Yen PM (2022). “Vitamin B(12) and folate decrease inflammation and fibrosis in NASH by preventing syntaxin 17 homocysteinylation.” J Hepatol 77(5): 1246–1255. [DOI] [PubMed] [Google Scholar]
- Vidal-Puig A, Jimenez-Linan M, Lowell BB, Hamann A, Hu E, Spiegelman B, Flier JS and Moller DE (1996). “Regulation of PPAR gamma gene expression by nutrition and obesity in rodents.” J Clin Invest 97(11): 2553–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Puig AJ, Considine RV, Jimenez-Linan M, Werman A, Pories WJ, Caro JF and Flier JS (1997). “Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids.” J Clin Invest 99(10): 2416–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, Friedman SL, Diago M and Romero-Gomez M (2015). “Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis.” Gastroenterology 149(2): 367–378 e365; quiz e314–365. [DOI] [PubMed] [Google Scholar]
- Vuppalanchi R, Noureddin M, Alkhouri N and Sanyal AJ (2021). “Therapeutic pipeline in nonalcoholic steatohepatitis.” Nat Rev Gastroenterol Hepatol 18(6): 373–392. [DOI] [PubMed] [Google Scholar]
- Wang F, Mullican SE, DiSpirito JR, Peed LC and Lazar MA (2013). “Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARgamma.” Proc Natl Acad Sci U S A 110(46): 18656–18661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xu MJ, Cai Y, Zhou Z, Cao H, Mukhopadhyay P, Pacher P, Zheng S, Gonzalez FJ and Gao B (2017). “Inflammation is independent of steatosis in a murine model of steatohepatitis.” Hepatology 66(1): 108–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Greenstein A, Majumdar N, Yang P, Subbaiah PV, Kineman RD and Cordoba-Chacon J (2017). “Hepatocyte-specific, PPARγ-regulated mechanisms to promote steatosis in adult mice.” J Endocrinol 232(1): 107–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HE, Lambert MH, Montana VG, Plunket KD, Moore LB, Collins JL, Oplinger JA, Kliewer SA, Gampe RT Jr., McKee DD, Moore JT and Willson TM (2001). “Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors.” Proc Natl Acad Sci U S A 98(24): 13919–13924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L and Wymer M (2016). “Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes.” Hepatology 64(1): 73–84. [DOI] [PubMed] [Google Scholar]
- Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, Bedossa P, Geier A, Beckebaum S, Newsome PN, Sheridan D, Sheikh MY, Trotter J, Knapple W, Lawitz E, Abdelmalek MF, Kowdley KV, Montano-Loza AJ, Boursier J, Mathurin P, Bugianesi E, Mazzella G, Olveira A, Cortez-Pinto H, Graupera I, Orr D, Gluud LL, Dufour JF, Shapiro D, Campagna J, Zaru L, MacConell L, Shringarpure R, Harrison S, Sanyal AJ and Investigators RS (2019). “Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial.” Lancet 394(10215): 2184–2196. [DOI] [PubMed] [Google Scholar]
- Yu JG, Javorschi S, Hevener AL, Kruszynska YT, Norman RA, Sinha M and Olefsky JM (2002). “The effect of thiazolidinediones on plasma adiponectin levels in normal, obese, and type 2 diabetic subjects.” Diabetes 51(10): 2968–2974. [DOI] [PubMed] [Google Scholar]
- Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, Rao MS, Gonzalez FJ and Reddy JK (2003). “Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression.” J Biol Chem 278(1): 498–505. [DOI] [PubMed] [Google Scholar]
- Zhang W, Sun Q, Zhong W, Sun X and Zhou Z (2016). “Hepatic Peroxisome Proliferator-Activated Receptor Gamma Signaling Contributes to Alcohol-Induced Hepatic Steatosis and Inflammation in Mice.” Alcohol Clin Exp Res 40(5): 988–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Bobe G, Miranda CL, Lowry MB, Hsu VL, Lohr CV, Wong CP, Jump DB, Robinson MM, Sharpton TJ, Maier CS, Stevens JF and Gombart AF (2021). “Tetrahydroxanthohumol, a xanthohumol derivative, attenuates high-fat diet-induced hepatic steatosis by antagonizing PPARgamma.” Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Alvares K, Huang Q, Rao MS and Reddy JK (1993). “Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver.” J Biol Chem 268(36): 26817–26820. [PubMed] [Google Scholar]
- Zhu Y, Qi C, Korenberg JR, Chen XN, Noya D, Rao MS and Reddy JK (1995). “Structural organization of mouse peroxisome proliferator-activated receptor gamma (mPPAR gamma) gene: alternative promoter use and different splicing yield two mPPAR gamma isoforms.” Proc Natl Acad Sci U S A 92(17): 7921–7925. [DOI] [PMC free article] [PubMed] [Google Scholar]