

Abstract
BACKGROUND:
Our understanding of the pathophysiology underlying Alzheimer’s disease (AD) has benefited from genomic analyses, including those that leverage polygenic risk score (PRS) models of disease. The use of functional annotation has been able to improve the power of genomic models.
OBJECTIVE:
We sought to leverage genomic functional annotations to build tissue-specific AD PRS models and study their relationship with AD and its biomarkers.
METHODS:
We built 13 tissue-specific AD PRS and studied the scores’ relationships with AD diagnosis, cerebrospinal fluid (CSF) amyloid, CSF tau, and other CSF biomarkers in two longitudinal cohort studies of AD.
RESULTS:
The AD PRS model that was most predictive of AD diagnosis (even without APOE) was the liver AD PRS: n = 1,115; odds ratio = 2.15 (1.67–2.78), P = 3.62 × 10−9. The liver AD PRS was also statistically significantly associated with cerebrospinal fluid biomarker evidence of amyloid-β (Aβ42:Aβ40 ratio, P = 3.53 × 10−6) and the phosphorylated tau:amyloid-β ratio (P = 1.45 × 10−5).
DISCUSSION:
These findings provide further evidence of the role of the liver-functional genome in AD and the benefits of incorporating functional annotation into genomic research.
Keywords: Alzheimer’s disease, polygenic risk score, functional annotation, liver, CSF biomarkers
INTRODUCTION
Despite advances in our understanding of Alzheimer’s disease (AD), the causal mechanisms are not fully understood. Since the hallmarks of AD have long been its neuropathological findings [1], the brain has historically been the primary focus of investigations into AD etiology. However, recent research has implicated other systems in AD, including the immune system and processes of inflammation [2], the cardiovascular system [3], and the liver and metabolism [4]. The lack of clarity over the roles of these mechanisms and risk factors in AD hampers our ability to identify novel therapeutic targets and ultimately effective treatments [5].
Genomic research has provided a valuable tool for understanding upstream risk factors in AD. The importance of amyloid-β (Aβ), long known to aggregate and subsequently accumulate in plaques in the brain, has been underscored by the knowledge that familial AD can be driven by genetic mutations in genes directly impacting Aβ processing, like APP, PSEN1, and PSEN2 [6]. In late-onset AD, genomic studies have repeatedly identified risk factors in a number of genes, like APOE, CR1, and ABCA7, expanding our knowledge of the molecular systems likely to be contributing to the development of AD [7–9]. For instance, the discovery of TREM2 as a genetic risk locus is now being expounded in follow-up experimental research on soluble TREM2 levels, highlighting the role of microglia in AD [10–12].
One particular application of genomic research that can help tease apart the mechanisms contributing to a disease involves polygenic risk scores (PRS), which are a measure of risk composed of the contributions of many single nucleotide polymorphisms (SNPs). Genome-wide PRS models and related methods have already been useful in the study of AD, including predicting AD and age of onset [13–15], examining genetic risk beyond the APOE locus [16], identifying PRS-environment associations [17,18], and comparing the genetic basis of related forms of AD [19]. PRS models can further be enhanced by the incorporation of genome functional annotation, which can boost the prediction accuracy of a PRS for disease risk [17,20,21]. One of the benefits of incorporating functional annotation is that it can introduce information about tissues and cell types and their potential relevance to the genomics underlying a particular trait. In AD, where there is growing genetic evidence for the role of different tissue and cell types [22,23], such annotation can provide important information about the full spectrum of biology involved in AD.
Here, we leveraged cell-type-specific genomic functional annotations derived from epigenetic data [23–25] to create tissue-specific PRS models for AD. We estimated tissue-specific genetic risk scores for participants in two longitudinal cohort studies of AD and analyzed the association between each tissue-specific PRS and AD diagnosis. Further, given that AD pathophysiology like brain amyloidosis and tau pathology can be identified using cerebrospinal fluid (CSF) biomarkers, we examined the possible associations between these tissue-specific PRS models and different pathologies in AD using CSF biomarkers of neurodegeneration and inflammation.
MATERIALS AND METHODS
Study participants
Data from two longitudinal AD cohorts focusing on middle and older aged adults were used for this study. The first was the Wisconsin Registry for Alzheimer’s Prevention (WRAP) study, described previously [26]. Briefly, participants were between the ages of 40 and 65, fluent in English, able to perform neuropsychological testing, without a diagnosis or evidence of dementia at baseline, and without any health conditions that might prevent participation in the study.
The second cohort was the Wisconsin Alzheimer’s Disease Research Center (WADRC) study, described previously [27]. WADRC participants were categorized into one of six subgroups: 1) mild late-onset AD; 2) mild cognitive impairment (MCI); 3) age-matched healthy older controls (age > 65); 4) middle-aged adults with a positive parental history of AD; 5) middle-aged adults with a negative parental history of AD; and 6) middle-aged adults with indeterminate parental history of AD. The clinical diagnoses for these groups were based on the National Institute of Neurological and Communicative Disorders and Stroke and Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) [28] and National Institute on Aging and Alzheimer’s Association (NIA-AA) [29]. Briefly, participants were over the age of 45, able to fast from food and drink for 12 hours, with decisional capacity, and without a history of certain medical conditions (like congestive heart failure or major neurologic disorders other than dementia) or any contraindication to biomarker procedures.
This study was performed as part of the GeneRations Of WRAP (GROW) study, which was approved by the University of Wisconsin Health Sciences Institutional Review Board. Participants in the WADRC and WRAP studies provided written informed consent. STREGA reporting guidelines [30] were used in the description of the results.
Clinical diagnoses
AD, MCI, and other diagnoses of cognitive status for both WRAP and WADRC were made by a consensus review committee comprising an expert panel of dementia-specialist physicians, neuropsychologists, and nurse practitioners [26]. CSF biomarker status was not used in the process of making the clinical diagnoses.
CSF biomarkers
Details on the collection of CSF for biomarker analyses have been previously described [31]. Briefly, CSF samples were acquired using a uniform preanalytical protocol and the same staff between 2010 and 2018 for both studies, improving the comparability of the data between the two cohorts. Samples were collected in the morning using a Sprotte 24- or 25-gauge atraumatic spinal needle, and 22 mL of fluid was collected via gentle extraction into polypropylene syringes and combined into a single 30 mL polypropylene tube. After gentle mixing, samples were centrifuged to remove red blood cells and other debris. Then, 0.5 mL CSF was aliquoted into 1.5 mL polypropylene tubes and stored at −80 degrees Celsius within 30 minutes of collection.
All CSF samples were assayed between March 2019 and January 2020 at the Clinical Neurochemistry Laboratory at the University of Gothenburg. CSF biomarkers were assayed using the NeuroToolKit (Roche Diagnostics International Ltd., Rotkreuz, Switzerland), a panel of automated robust prototype immunoassays designed to generate reliable biomarker data that can be compared across cohorts. Measurements with the following immunoassays were performed on a cobas e 601 analyzer: Elecsys® β-amyloid (1–42) CSF (Aβ42), Elecsys Phospho-Tau (181P) CSF, and Elecsys Total-Tau CSF, β-amyloid (1–40) CSF (Aβ40), and interleukin-6 (IL-6). The remaining NTK panel was assayed on a cobas e 411 analyzer, including markers of synaptic damage and neuronal degeneration (neurogranin, neurofilament light protein [NFL], and α-synuclein) and markers of glial activation (chitinase-3-like protein 1 [YKL-40] and soluble triggering receptor expressed on myeloid cells 2 [sTREM2]).
A total of 9 CSF biomarkers were analyzed in this study: the Aβ42/Aβ40 ratio, phosphorylated tau 181 (ptau), the ptau/Aβ42 ratio, NFL, α-synuclein, neurogranin, YKL-40, sTREM2, and IL-6. Details on the relationship of these biomarkers with the stages of AD progression have been previously reported [31]. Since the CSF biomarker measurements were to be used as outcomes, each biomarker was assessed for skewness using the skewness function of the R package moments (version 0.14) [32]. Any biomarker with a skewness ≥ 2 was transformed with a log10-transformation to better meet the normality assumption of regression. The outcomes that were log10-transformed were ptau, the ptau/Aβ42 ratio, NFL, and IL-6.
Genomic data
The genomic data collection in the WRAP and WADRC cohorts has been described previously [33]. The WRAP samples were genotyped using DNA from whole blood samples and the Illumina Multi-Ethnic Genotyping Array at the University of Wisconsin Biotechnology Center [34]. Samples and variants with high missingness (> 5%) or inconsistent genetic and self-reported sex were removed. Samples from individuals of European descent (determined by PCA with the 1000 Genomes Project data using KING [35] and PC-AiR [36], described previously [34]) were then imputed using the Michigan Imputation Server [37] and the Haplotype Reference Consortium (HRC) reference panel [38], with low quality variants again removed (R2 < 0.8) post-imputation. A total of 1,198 samples with 10,499,994 SNPs were present at the end of quality control.
Whole blood samples from the WADRC were genotyped by the Alzheimer’s Disease Genetics Consortium (ADGC) at the National Alzheimer’s Coordinating Center (NACC) using the Illumina HumanOmniExpress-12v1_A, Infinium HumanOmniExpressExome-8 v1-2a, or Infinium Global Screening Array v1-0 (GSAMD-24v1-0_20011747_A1) BeadChip assay. Each chip’s genomic data were initially processed separately. After strict quality control that removed variants or samples with high missingness (> 2%), out of Hardy-Weinberg equilibrium (HWE) (P < 1×10−6), or with inconsistent genetic and self-reported sex, samples were imputed with the Michigan Imputation Server where they were phased using Eagle2 [39] and imputed to the HRC reference panel. Low quality variants (R2 < 0.8) or out of HWE were removed. After imputation, the data sets from the different chips were merged together, leaving a data set with 376 samples of European descent (determined in the same manner as in the WRAP cohort) and 7,049,703 SNPs.
To prevent variant overlap issues, all genomic data sets used in this study were harmonized, including the WADRC, WRAP, International Genomics of Alzheimer’s Project (IGAP) 2019 GWAS of AD [7], and the 1000 Genomes Utah residents with Northern and Western European ancestry (CEU) [40] data sets. The GRCh37 genome build was used, all ambiguous SNPs were removed, and all SNPs were aligned to have strand and allele orientations consistent with the WADRC data set. Only the subset of SNPs successfully harmonized and present across all four data sets was used to build the PRS. To avoid sample overlap between participants in the WADRC and IGAP, 165 overlapping participants were removed, leaving a total of 1,409 participants (211 WADRC, 1,198 WRAP) with 5,631,405 SNPs before any functional annotations were considered.
As an additional measure to ensure the effect of APOE was removed, each sample was additionally assigned an APOE genotype based on the participant’s combination of the ε2, ε3, and ε4 alleles for APOE from a separate set of genotyping. DNA was extracted from whole blood samples, which was then genotyped for the APOE alleles using competitive allele-specific PCR-based KASP genotyping for rs429358 and rs7412 [21]. The count of APOE ε4 alleles was correlated with each tissue-specific PRS without APOE to determine if APOE was still correlated with the PRS.
Tissue-specific functional SNP sets
To categorize the genome into tissue-specific functional regions, annotations from GenoSkyline-PLUS [23–25] (v 1.0.0) were used. GenoSkyline is an unsupervised framework that uses epigenetic data sets from the Roadmap Epigenomics Project [41] to predict tissue-specific functional regions of the genome. For each region of the genome and for each cell type, GenoSkyline predicts a value between 0 and 1 that represents whether that region is likely to be functional for that cell type, with 1 indicating functional and 0 non-functional. Due to the bimodal distribution of these scores, values ≥ 0.5 were considered functional and values < 0.5 were considered non-functional in this study. GenoSkyline-PLUS annotations are available for a variety of cell types, with each cell type labeled as part of a larger tissue type. The full list of GenoSkyline-PLUS tissues and included cell types can be found in Supplementary Table 1. For each tissue, the union of all functional genomic regions from all included cell types was defined as the functional genomic region for that tissue, and all SNPs falling within that region were included in that tissue’s set of tissue-specific functional SNPs. A total of 13 tissue-specific SNP sets were defined in this manner, with a 14th SNP set comprising all SNPs regardless of tissue functionality or annotation to create a non-tissue-specific, non-annotated genome-wide PRS for comparison. For all of the PRS models, the effect size estimate for each SNP came from the 2019 IGAP GWAS results[7]. The nearest protein-coding gene to each SNP was also added using GENCODE (version 19) annotations [42].
Since certain genotypes in the APOE locus are known to be strongly associated with AD, we sought to examine whether the PRS were associated with AD beyond the effect of the APOE locus. To do so, we built tissue-specific PRS using the same procedure as above with the exception that all SNPs in a window around the APOE genomic region were removed (defined as between the PVR and GEMIN7 genes: chromosome 19, base pairs 45,147,098–45,594,782; 932 SNPs were removed in this way). Some SNPs from the APOE locus were considered functional for each tissue, so the PRS scores changed for all tissues when the APOE locus was removed.
PRS calculation
Each SNP set as defined above was then used to construct the corresponding tissue-specific PRS. Each PRS was constructed with PRSice [43] (v 2.2.4) using the Kunkle et al. 2019 IGAP summary statistics [7] as the base data set and the 1000 Genomes CEU samples to estimate linkage disequilibrium (LD). An R2 of 0.5 was used for clumping SNPs and a P threshold of 0.0025 based on an early prototype of the PUMAS method[44] was used for the inclusion of SNPs, leaving a maximum of 649,987 SNPs remaining for PRS construction depending on the SNP set used. The risk score for each tissue was calculated for each participant in the combined WADRC/WRAP data set using the default “average” PRS equation that divides the weighted sum of the alleles by the total count of alleles used (“--score avg” option for PRSice), and then each tissue-specific PRS was standardized to a mean of 0 and variance of 1.
PRS-AD diagnosis associations
Among the WADRC/WRAP data set, all 1,409 participants had at least one study visit with a consensus conference diagnosis. The most recent visit for each participant with an AD, MCI, or cognitively unimpaired diagnosis was kept. Then, among related individuals (defined by estimated genetic relationships in WADRC or self-defined families in WRAP) only one participant was kept per family (chosen arbitrarily), leaving 1,163 unrelated participants. The association between each PRS and AD diagnosis (compared to cognitively unimpaired individuals; n = 78 cases with AD and n = 1,037 cognitively unimpaired controls; MCI cases excluded) was estimated with logistic regression using the R [45] glm function, controlling for sex and age at the time of diagnostic assessment by the consensus review committee. A Bonferroni correction for the number of PRS tested (P = 0.05 / 14 = 0.0036) was used in reporting significant results.
As a follow-up analysis, a comparison of the top-performing risk score between AD, MCI, and cognitively unimpaired participants was made using a box plot, with both ANOVA and pairwise t-tests used to compare the risk score distributions among the groups. Participants were then divided into 5 groups based on the PRS quantiles, and the distributions of the AD diagnoses across these PRS quantiles were compared.
Though the two cohort populations were similar geographically and demographically, the possibility of confounding by cohort or population substructure was assessed. The PRS-diagnosis associations were repeated as above but using only the participants from WADRC and additionally controlling for the first 5 principal components (PCs) of genetics. WADRC was used for this sensitivity analysis because it had a more balanced distribution of clinical diagnoses than WRAP.
To assess whether the effect of APOE was solely driving the associations of the PRS with AD diagnosis, another sensitivity analysis was conducted with the set of tissue-specific PRS constructed without the APOE locus. The associations between these PRS without APOE and AD diagnosis were estimated with logistic regression as before and compared to the original models with APOE included in the PRS.
To assess whether the top-performing PRS was substantially better than the rest of the genome in predicting AD diagnosis, a sensitivity analysis was conducted where only non-tissue-functional SNPs were used to construct a PRS. To build this PRS (referred to as an inverse PRS), the same procedure as before was used for the top-performing tissue PRS except that only SNPs in the non-functional regions for that tissue were included. The association of this PRS with AD diagnosis was estimated in a logistic regression as before and compared to that of the tissue-functional PRS, both with and without the APOE region included.
Finally, to assess whether the top-performing PRS was significantly different from the non-tissue-specific PRS (labeled “all”), we estimated the difference in the odds ratios (OR) of the top-performing PRS and the non-tissue-specific PRS across 1,000 bootstrap samples and calculated the significance of that OR difference. The Z score of the OR difference was calculated as the OR difference in the original data divided by the standard deviation of the odds ratio difference across the bootstrap samples [46].
PRS-CSF biomarker associations
To investigate the intermediate biological pathways driving the association between the PRS most strongly associated with AD diagnosis, the relationship between this PRS (both with and without APOE) and the CSF biomarkers was assessed using the longitudinal data set of unrelated WADRC and WRAP participants with biomarker measurements available (up to 250 visits from 167 individuals). A linear mixed-effects model was used to test each liver PRS-biomarker association, controlling for age at CSF collection and sex and including a random intercept for the individual to account for the longitudinal CSF measurements (R package lme4, version 1.1–26). A Bonferroni correction for the number of biomarkers tested (P = 0.05 / 9 = 0.0056) was used for reporting significant associations. The marginal pseudo-R2 was calculated using the R package MuMIn (version 1.43.17) [47] for each model as well as the model with the PRS term removed in order to quantify the impact of the PRS in prediction.
To assess the robustness of the linear mixed model results, the CSF biomarker analyses were rerun using only the first visit available from each participant (n up to 167). The results were checked against those from the original linear mixed model.
RESULTS
PRS creation
A total of 14 PRS were initially generated based on the GenoSkyline-PLUS functionality annotation: one genome-wide PRS (labeled “all”) and 13 tissue-specific PRS (blood, thymus, and spleen; brain; breast; connective tissue; fat; gastrointestinal; heart; liver; lung; muscle; ovary; pancreas; and skin) (Supplementary Figure 1). The proportion of the 5,631,405 SNPs considered functional for each tissue ranged from 2.5% (ovary) to 25.3% (blood, thymus, and spleen) (Supplementary Figure 2). Among the 1,163 unrelated participants, the genome-wide and tissue-specific PRS values for the WADRC/WRAP cohort were all roughly normally distributed whether the APOE region was included or excluded from the PRS (Supplementary Figures 3–4). The PRS were generally correlated with each other with a median pairwise correlation of 0.872 (APOE included) and 0.754 (APOE excluded) (Supplementary Figures 5–6 and Supplementary Table 2). The correlations of the PRS with APOE excluded with APOE ε4 allele count were all low, ranging from 0.028 to 0.090.
PRS-AD diagnosis associations
The 1,163 unrelated participants were considered for the PRS-AD analyses (Table 1). The majority (1,037, 89.2%) of these participants were cognitively unimpaired at their most recent visit, with the AD and MCI diagnosis participants older and less often female. For the analysis of the PRS-AD diagnosis associations, only the 1,115 AD and cognitively unimpaired participants were used.
Table 1.
AD diagnosis cohort demographics
| By diagnosis | Cohort | ||||
|---|---|---|---|---|---|
| Diagnosis | n | Age (mean, SD) | Female (n, %) | WADRC (n, %) | WRAP (n, %) |
| AD | 78 | 76 (9.1) | 34 (43.6%) | 69 (88.5%) | 9 (11.5%) |
| MCI | 48 | 75.1 (8.2) | 20 (41.7%) | 28 (58.3%) | 20 (41.7%) |
| Cognitively unimpaired | 1037 | 66.2 (6.6) | 716 (69.0%) | 107 (10.3%) | 930 (89.7%) |
| By cohort | |||||
| Cohort | n | Age (mean, SD) | Female (n, %) | ||
| WADRC | 204 | 71.5 (8.8) | 105 (51.5%) | ||
| WRAP | 959 | 66.4 (6.8) | 665 (69.3%) | ||
The association of each PRS with AD diagnosis (versus cognitively unimpaired) is shown in Figure 1 (full regression results and model areas under the curve [AUC] in Supplementary Table 3). All PRS were individually and statistically significantly associated with AD diagnosis after Bonferroni correction for multiple testing, with all PRS showing an increase of polygenic risk associated with a diagnosis of AD relative to cognitively unimpaired and AUCs between 0.839 and 0.865. Three tissue PRS (liver, ovary, and brain) had greater estimated effects on AD diagnosis than the genome-wide PRS, with an odds ratio of having AD relative to being cognitively impaired (OR) (and 95% confidence intervals [CI] and P values) of 2.15 per standard deviation (SD) increase in the PRS (95% CI = 1.67–2.78, P = 3.62 × 10−9), 2.05 (1.58–2.65, P = 5.72 × 10−8), and 1.99 (1.53–2.60, P = 3.84 × 10−7), respectively, compared to the genome-wide PRS with OR of 1.99 (1.53–2.60, P = 3.85 × 10−7).
Figure 1. Association of tissue-specific PRS with AD diagnosis (APOE included).
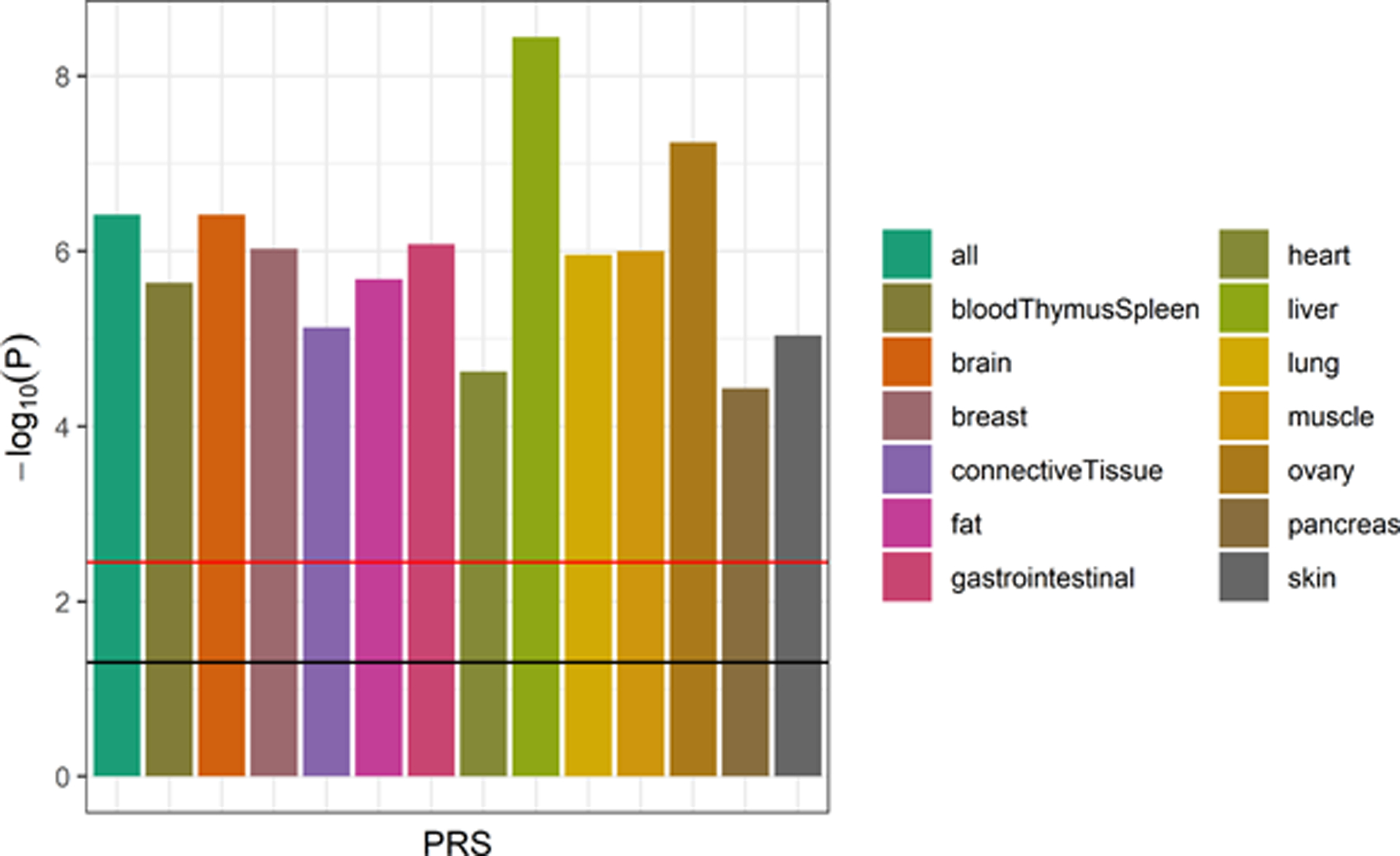
The strength of association of each PRS (with the APOE locus included) with AD diagnosis (relative to cognitively unimpaired participants; MCI cases excluded) from the logistic regression models is shown (n = 1,115). The horizontal lines indicate thresholds for significance, with the black line indicating the nominal threshold of P = 0.05 and the red line indicating the Bonferroni-corrected threshold of P = 0.0036. All PRS scores were statistically significantly associated with AD diagnosis, with the liver PRS being the most strongly associated score.
The genetic risk scores for the liver PRS (with APOE) increased with increasing severity across the three cognitive diagnoses from a mean value of −0.02 (SD = 0.96) for cognitively unimpaired individuals to 0.59 (SD = 1.10) for participants with AD (Figure 2). The mean liver score was different across the three groups (ANOVA P = 3.4 × 10−7) and significantly different between the unimpaired and AD participants (t-test P = 8.4 × 10−6). When the participants were stratified into 5 liver PRS risk quantiles, the highest risk (5th quantile) and lowest risk (1st quantile) showed a corresponding enrichment of AD and MCI participants at the highest PRS risk (19.0%) relative to the lowest PRS risk (6.4%) (Supplementary Figure 7).
Figure 2. Distribution of liver PRS by diagnosis group (APOE included).
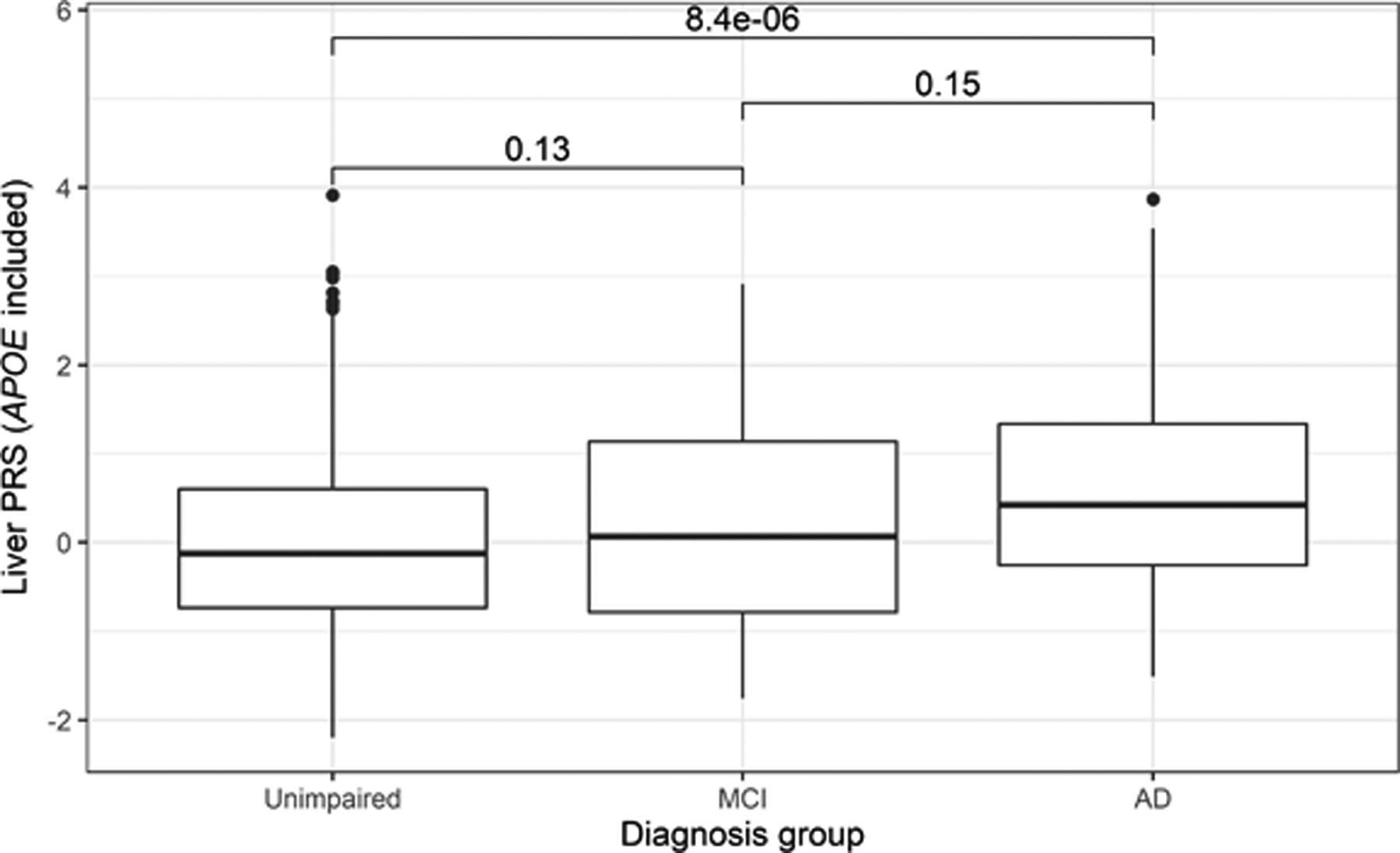
The distribution of the liver PRS with the APOE region included across the three clinical stages of AD is shown (n = 1,409). Pairwise t-tests of the diagnosis group means (P values shown above each box plot pair) revealed a statistically significant difference between the liver PRS scores of the cognitively unimpaired and AD groups. An ANOVA test similarly identified a difference in means across all three groups (P = 3.4 × 10−7).
When these PRS-AD diagnosis analyses were repeated using just the WADRC cohort (n = 176) and including the first 5 genetic PCs as additional covariates, the results were similar but with weaker associations. All PRS with APOE included were statistically significantly associated with AD diagnosis with the liver PRS having the largest estimated OR of 2.19 (95% CI: 1.49–3.23, P = 6.68 × 10−5). When APOE was excluded, only the liver PRS remained statistically significantly associated with an OR of 2.04 (1.29–3.24, P = 0.00227), though its P value was just below the Bonferroni-corrected significance threshold.
To rule out the possibility that the PRS associations with AD diagnosis were solely driven by the effect of the APOE locus, the PRS-AD diagnosis associations were recalculated using the PRS that excluded the APOE region. In this sensitivity analysis, only the liver and ovary PRS remained significantly associated with AD diagnosis after Bonferroni correction for the number of PRS tested with an OR of 1.58 (95% CI: 1.20–2.06, P = 9.01 × 10−4) and an OR of 1.46 (1.13–1.88, P = 3.47 × 10−3), respectively, (Figure 3), although the AUCs remained similar across PRS models (range: 0.814–0.826; Supplementary Table 3). A similar increasing genetic risk score across the diagnosis groups from cognitively unimpaired to AD was seen with the liver PRS without the APOE locus. A statistically significant difference in the genetic risk scores for the liver PRS between cognitively unimpaired (mean score = −0.001, SD = 0.97) and AD (mean score = 0.35, SD = 0.99) groups was observed with a t-test P = 0.0034, and the proportion of AD and MCI participants among the highest risk quantile was 14.7% compared to 6.9% in the lowest risk quantile.
Figure 3. Association of tissue-specific PRS with AD diagnosis (APOE excluded).
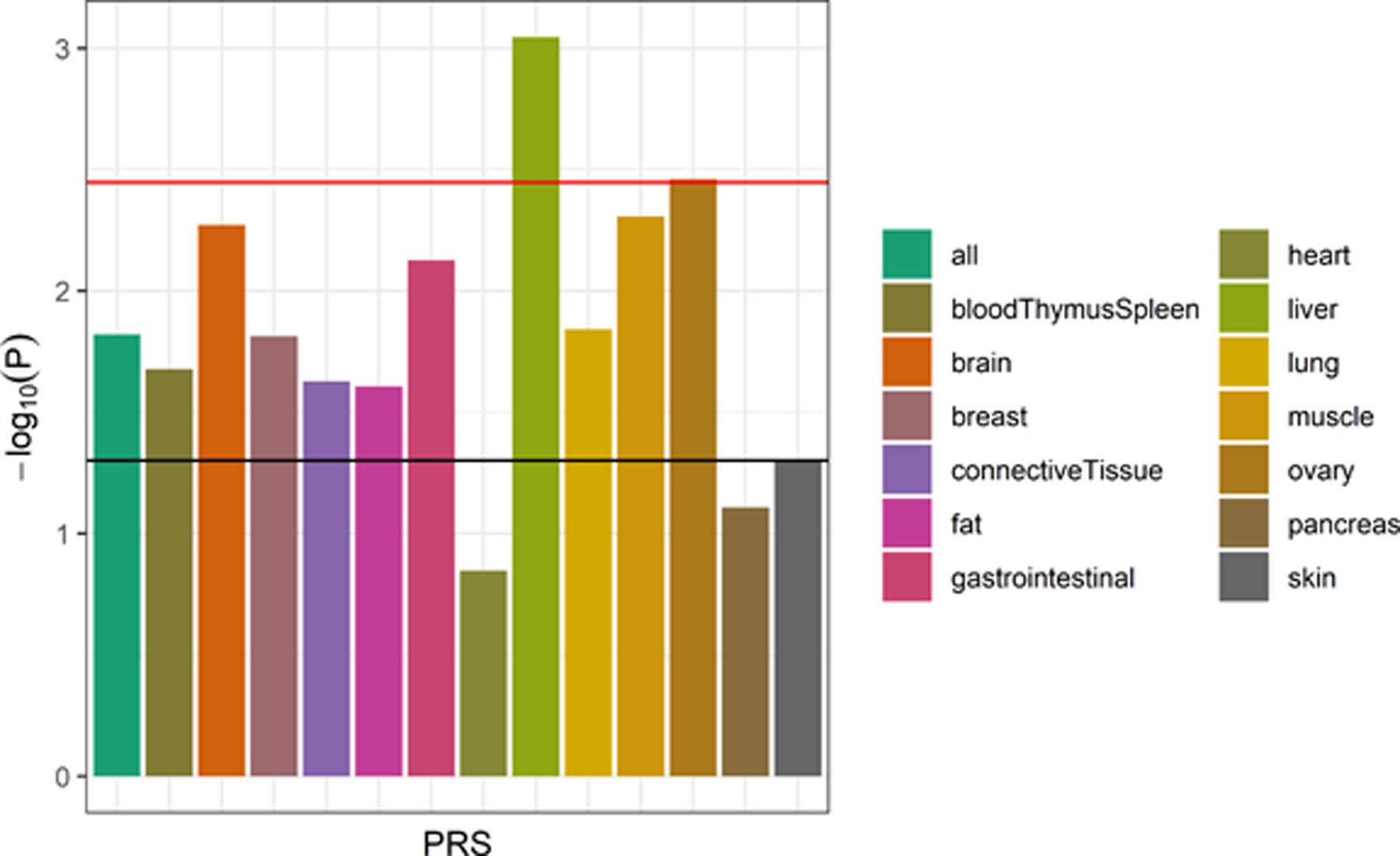
The strength of association of each PRS (with the APOE locus excluded) with AD diagnosis (relative to cognitively unimpaired participants; MCI cases excluded) from the logistic regression models is shown (n = 1,115). The horizontal lines indicate thresholds for significance, with the black line indicating the nominal threshold of P = 0.05 and the red line indicating the Bonferroni-corrected threshold of P = 0.0036. Only the liver and ovary PRS remained statistically significantly associated with AD diagnosis after multiple testing correction.
To explore whether the liver-functional genome was indeed more predictive of AD diagnosis than the remaining genome, the performance of the liver PRS was compared to the PRS constructed using every part of the genome except for the liver-functional genome (i.e., the “liver inverse” PRS), both with and without the APOE region included. Regardless of the inclusion of the APOE region, the liver-functional genome PRS had a greater estimated effect on AD diagnosis (vs. cognitively unimpaired) than the liver inverse PRS, although all PRS were nominally associated. With APOE included, the liver PRS’ OR was 2.15 (95% CI = 1.67–2.78, P = 3.62 × 10−9) compared to the liver inverse PRS with an OR of 1.86 (95% CI = 1.43–2.43, P = 4.30 × 10−6). Without APOE, the liver PRS OR was 1.58 (95% CI = 1.20–2.06, P = 0.0009) compared to the liver inverse PRS with an OR of 1.36 (95% CI = 1.05–1.75, P = 0.018) (Supplementary Figure 8).
Finally, we compared the liver PRS to the non-tissue-specific PRS (“all” PRS) to see if the annotation-stratified liver PRS was statistically significantly different in terms of its association with AD diagnosis than a typical AD PRS (both scores including APOE). The mean OR difference (liver PRS OR minus all PRS OR) across the bootstrap resampling procedure was 0.17 (95% CI = −0.17–0.51, P = 0.37).
PRS-CSF biomarker associations
The longitudinal data set for the CSF biomarkers included all available WADRC/WRAP visits for participants where CSF biomarker and genetic data were available, which ranged from 1 to 5 visits per participant (median average time between visits for those with multiple visits = 1.75 years). The total number of participants included per biomarker analysis ranged from 164–167, comprising 245–250 total visits (Table 2). The mean age at visit across all included visits was 64.1 (SD 7.1) with 64.0% of the visits from female participants.
Table 2.
CSF biomarker data set description
| Outcome | Individuals | Visits | Outcome mean | Outcome SD |
|---|---|---|---|---|
| α-synuclein (pg/mL) | 167 | 250 | 183.87 | 83.43 |
| Aβ42:Aβ40 ratio | 166 | 247 | 0.060 | 0.020 |
| IL-6 (pg/mL) | 164 | 245 | 4.48 | 2.62 |
| Neurogranin (pg/mL) | 167 | 250 | 849.62 | 354.44 |
| NFL (pg/mL) | 167 | 250 | 115.78 | 84.49 |
| ptau (pg/mL) | 166 | 247 | 21.59 | 12.55 |
| ptau:Aβ42 ratio | 166 | 247 | 0.034 | 0.034 |
| sTREM2 (ng/mL) | 167 | 250 | 8.74 | 2.82 |
| YKL-40 (ng/mL) | 167 | 250 | 160.05 | 63.76 |
The results of the linear mixed-effects models that regressed each outcome on the liver PRS (controlling for age at visit and sex) are summarized in Figure 4 (full regression results in Supplementary Table 4). After Bonferroni correction, the liver PRS was statistically significantly associated with three outcomes: the Aβ42/Aβ40 ratio, ptau, and the ptau/Aβ42 ratio. The liver PRS was nominally associated with three other outcomes: NFL, neurogranin, and α-synuclein. The remaining three outcomes (YKL-40, sTREM2, and IL-6) were not associated with the liver PRS. The increase in marginal pseudo-R2 between the models with and without the PRS term ranged from 0.001 (sTREM2) to 0.101 (ptau/Aβ42) and 0.112 (Aβ42/Aβ40).
Figure 4. Association of the liver PRS with CSF biomarkers (APOE included).
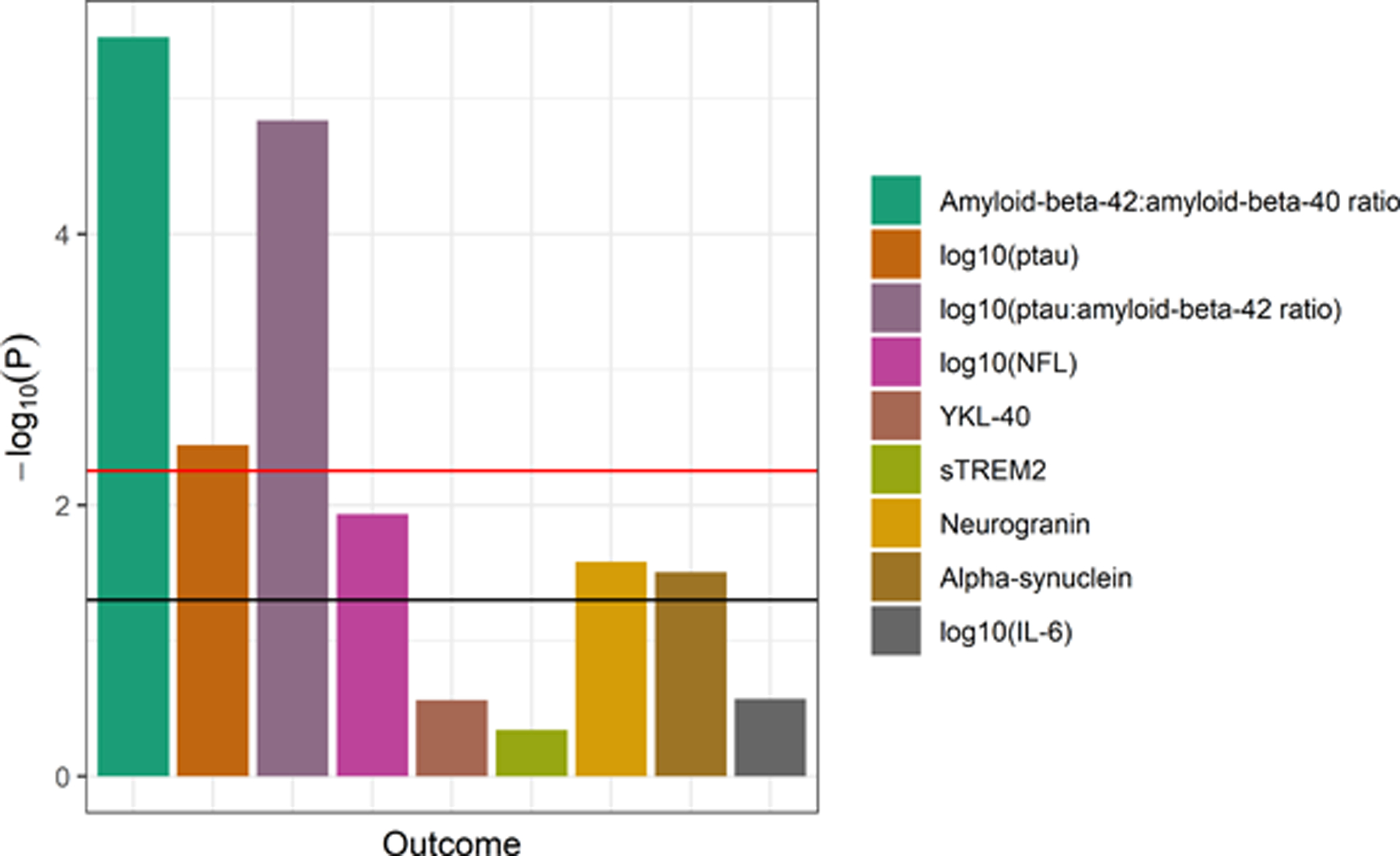
The strength of association of the liver PRS (with the APOE locus included) with each CSF biomarker from the linear mixed effects regression models is shown (n range = 245–250 visits). The horizontal lines indicate thresholds for significance, with the black line indicating the nominal threshold of P = 0.05 and the red line indicating the Bonferroni-corrected threshold of P = 0.0056. The liver PRS was statistically significantly associated with the measures of amyloid and tau but not with the other biomarkers.
When these analyses were repeated with the APOE region removed from the liver PRS, the majority of the association signal was lost: no outcome was statistically significantly associated with the liver PRS without APOE after Bonferroni correction, although the PRS was nominally associated with sTREM2 (Supplementary Figure 9). The difference in marginal pseudo-R2 was negligible except for sTREM2, which had an increase of 0.018 from the model without the PRS term to the model with the PRS term. Furthermore, when these analyses were rerun using only the first visit available per participant, the results did not substantively change, indicating that the results were robust to whether a linear model or linear mixed model were used: the liver PRS with APOE was still statistically significantly associated with just the Aβ42/Aβ40 ratio, ptau, and the ptau/Aβ42 ratio while being nominally associated with NFL, neurogranin, and α-synuclein, and the liver PRS without APOE was only nominally associated with sTREM2.
DISCUSSION
The main result from the analysis of the association between tissue-specific PRS and AD diagnosis was that the liver PRS showed the greatest effect size and area under the curve (AUC), although the differences in AUC were subtle and the difference between the liver PRS and the typical, non-tissue-specific PRS was not statistically significant. When the effect of APOE was mitigated by excluding all SNPs in the APOE region, the liver PRS was the only PRS to remain statistically significant following multiple testing correction in its association to AD diagnosis and be significantly associated in just the WADRC cohort alone. The importance of the APOE region in driving much of the association signal for the PRS models, including the liver PRS, was expected, as the APOE locus has long been known to be strongly associated with AD risk [48,49], especially among a population predominantly of European ancestry [50], as was the case here. However, the liver PRS was associated with AD diagnosis beyond the impact of the APOE locus.
Interpreting the meaning of the liver PRS’ relationship with AD was aided by the follow-up analysis with the CSF biomarkers. The liver PRS was most strongly associated with the core biomarkers of AD, amyloid and tau (CSF Aβ42/Aβ40, ptau, and ptau/Aβ42), suggesting that the PRS was more directly capturing these features of AD pathology rather than some of the other processes of neuroinflammation and neurodegeneration. However, these associations with amyloid and tau were removed when the APOE locus was removed from the liver PRS, leaving only a nominal association with sTREM2. Weaker association signals among the CSF biomarker data set could be attributed in part to the much smaller sample size available with CSF biomarker data compared to that with AD diagnosis data (n = 250 vs n = 1,115). Still, the reason for the liver PRS without APOE being associated with AD diagnosis but not any of the CSF biomarkers was unclear.
Whether the liver PRS’ association with AD risk indicates a role for the liver organ itself remains an open question. The liver PRS here may be associated with AD due to some role of the liver itself or simply through the genes that happen to be functional in the liver but are not uniquely expressed in the liver. Across the 560 SNPs that were part of the liver PRS (APOE excluded), many of the major AD loci were represented, including CLU, BIN1, PICALM, SPI1, ABCA7, SORL1, and others (Supplementary Table 5), many of which are expressed in brain as well where their connection to AD may be most relevant [51]. Previous studies that looked to aggregate AD genetic associations to the roles of tissues have at times indicated an enrichment of AD-associated genes expressed in the liver [8], though gene-set based tissue-enrichment approaches have tended to identify the spleen as an enriched tissue [8,52]. Nevertheless, there is mounting evidence pointing to metabolic dysregulation and the liver as relevant to AD. Several metabolic traits, including dyslipidemia, metabolic syndrome, obesity, and type 2 diabetes, appear to be risk factors for AD [53,54]. More specific to the liver, Neuner et al. suggest that cholesterol regulation may be a point of common ground between the liver, APOE, neurons, and AD [6]. Recent evidence has also indicated an association between measures of liver function, including blood levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), and AD diagnosis, amyloid, tau, and neurodegeneration [4]. Perhaps most intriguing was a recent study in mice showed additional evidence for the liver, highlighting how Aβ produced specifically by the liver could lead to neurodegeneration and cognitive decline [55]. Our findings here provide potential further evidence of the relevance of the liver and metabolism in relation to AD.
More generally, this study reinforces the benefit of using functional annotation to improve genomic prediction as the PRS’ performance was improved, albeit subtly, by incorporating predicted functional information. Among the PRS models with APOE excluded, 6 of the 13 tissue-specific PRS were more strongly associated with AD diagnosis than the genome-wide PRS in association. This increased strength of association is likely the result of improved filtering of the included SNPs to just those that are more likely to be causal due to their predicted functionality. This finding would support a general theme among the functional annotation literature that suggests that functional annotation can improve genomic analyses of disease. Early work demonstrated that genomic functional annotation could be used to filter down a set of SNPs to those more likely to be causal for both dominant and recessive Mendelian traits [56]. Recent approaches have used functional annotation to boost GWAS power in identifying SNP associations [57], stratify heritability of complex disease by functional annotation [58], and improve genetic risk prediction for disease [20,59]. Our work further demonstrates the potential utility of incorporating functional annotation in genetic risk prediction, though additional work is needed to quantify whether the improvement in this case is reproducible and enough to be clinically relevant.
Limitations of this study included the limited sample size. In the study of PRS-AD diagnosis associations, the sample was predominantly cognitively unimpaired with only 78 individuals diagnosed with AD, and in the follow-up analysis of CSF biomarker data, only 167 unique individuals were available with data. However, even among these smaller sample sizes, detectable association signals were still observed. As these cohort studies continue to grow so too will our capability to investigate genetic associations with AD pathology. We note too that there is potential for selection bias in the underlying cohorts, for example with WRAP due to its explicit recruitment of individuals without dementia at baseline. While our sensitivity analysis using just the WADRC cohort alone showed results consistent with the full analysis with the WRAP cohort included, additional sensitivity analyses and external replication would be helpful to further rule out potential biases. Also, here, we focused on tissue-specific functional annotation, but information at the cell type level would also be informative, so an application of our framework to individual cell types could be useful. We also use tissue functionality models that did not incorporate some data sets like DNA conservation and single-cell transcriptomics, both of which may be useful in further improving these models. It is also worth noting that this method of choosing SNPs for a PRS based on predicted functionality could theoretically include SNPs that tag causal SNPs further away that are not functional for the tissue for which the tagging SNP is functional. Given the fact that individual SNPs tend to have weak effects on AD risk (with the exception of APOE) and that the simple annotation-stratified approach we used here is known to effectively quantify heritability enrichment at the genome-wide scale for complex traits, including AD [23], we expect that the SNPs used here in the tissue-specific PRS models would be strongly enriched for causal variants that overlap with functional genomic regions in that tissue. The correlation between the different tissue-specific PRS also merits caution, as the differences in the predictiveness of tissue-specific PRS were subtle and the OR difference between the liver PRS and the typical, non-tissue-specific PRS was not statistically significant with bootstrap resampling. This overlap in PRS across tissue types may reflect, in part, SNPs that are functional in multiple tissues, similar to the observed sharing of genetic effects across tissues in GTEx [60]. Larger sample sizes and different strategies for annotating SNPs with tissue types that would minimize overlap between the PRS would be beneficial in further determining differences in predictiveness between tissue-specific PRS and more common non-tissue-specific PRS approaches. Another limitation was the population of study, which was limited to European ancestry due to lack of sample size in other populations in the data set at the time the data were pulled. Further studies will be needed to better understand the transferability of these tissue-specific PRS findings to other populations.
In conclusion, we leveraged genome functional annotation to create tissue-specific PRS for AD, identifying the liver PRS as being associated with AD diagnosis. Follow-up analysis of the liver PRS with CSF biomarkers of AD, neurodegeneration, and neuroinflammation revealed potential intermediate pathways related to the role of the liver-functional genome in AD, but the limited sample size of the biomarker data set and the apparent role of APOE in driving these biomarker results merit further study. Altogether, these findings provide further evidence for the role of the liver-functional genome in AD and highlight the benefit of incorporating genomic functional annotation into genetic research of complex disease.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank WRAP and WADRC participants and the Wisconsin Alzheimer’s Institute (WAI) and WADRC staff for their contributions to the WRAP and WADRC studies. Without their efforts this research would not be possible. ELECSYS, COBAS and COBAS E are trademarks of Roche. The Roche NeuroToolKit robust prototype assays are for investigational purposes only and are not approved for clinical use. We thank the University of Wisconsin Madison Biotechnology Center Gene Expression Center for providing Illumina Infinium genotyping services. We thank Dr. Brian Kunkle for his help with discussions on the sample overlap issue of genomic cohorts. We also thank Jiacheng Miao for his advice on testing the difference of two PRS models.
FUNDING
This research is supported by National Institutes of Health (NIH) grants R01AG27161 (Wisconsin Registry for Alzheimer Prevention: Biomarkers of Preclinical AD), R01AG054047 (Genomic and Metabolomic Data Integration in a Longitudinal Cohort at Risk for Alzheimer’s Disease), R21AG067092 (Identifying Metabolomic Risk Factors in Plasma and Cerebrospinal Fluid for Alzheimer’s Disease), R01AG037639 (White Matter Degeneration: Biomarkers in Preclinical Alzheimer’s Disease), P30AG017266 (Center for Demography of Health and Aging), and P50AG033514 and P30AG062715 (Wisconsin Alzheimer’s Disease Research Center Grant), the Helen Bader Foundation, Northwestern Mutual Foundation, Extendicare Foundation, State of Wisconsin, the Clinical and Translational Science Award (CTSA) program through the NIH National Center for Advancing Translational Sciences (NCATS) grant UL1TR000427, and the University of Wisconsin-Madison Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation. This research was supported in part by the Intramural Research Program of the National Institute on Aging. Computational resources were supported by a core grant to the Center for Demography and Ecology at the University of Wisconsin-Madison (P2CHD047873). We also acknowledge use of the facilities of the Center for Demography of Health and Aging at the University of Wisconsin-Madison, funded by NIA Center grant P30AG017266. Author DJP was supported by NLM training grants to the Bio-Data Science Training Program (T32LM012413) and the Interdisciplinary Training Program in Cardiovascular and Pulmonary Biostatistics (5T32HL83806). Author BFD was supported by an NLM training grant to the Computation and Informatics in Biology and Medicine Training Program (NLM 5T15LM007359). Author YKD was supported by a training grant from the National Institute on Aging (T32AG000213). Author HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018-02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG-720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), the AD Strategic Fund and the Alzheimer’s Association (#ADSF-21-831376-C, #ADSF-21-831381-C and #ADSF-21-831377-C), the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2019-0228), the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 860197 (MIRIADE), and the UK Dementia Research Institute at UCL. Author KB was supported by the Swedish Research Council (#2017-00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809-2016615), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017-0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986), the European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236), and the NIH, USA, (grant #1R01AG068398-01).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
CONFLICTS OF INTEREST
Author HZ has served at scientific advisory boards and/or as a consultant for Abbvie, Alector, Annexon, AZTherapies, CogRx, Denali, Eisai, Nervgen, Pinteon Therapeutics, Red Abbey Labs, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure and Biogen, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. Author KB has served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, Biogen, JOMDD/Shimadzu. Julius Clinical, Lilly, MagQu, Novartis, Roche Diagnostics, and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. Author GK is a full-time employee of Roche Diagnostics GmbH. Author IS is a full-time employee and shareholder of Roche Diagnostics International Ltd. Author SCJ served as a consultant to Roche Diagnostics in 2018. Other authors have no competing interests to declare.
DATA AVAILABILITY
The data sets analyzed in this study may be requested from the WADRC at https://www.adrc.wisc.edu/apply-resources.
REFERENCES
- [1].Vinters HV (2015) Emerging Concepts in Alzheimer’s Disease. Annu Rev Pathol Mech Dis 10, 291–319. [DOI] [PubMed] [Google Scholar]
- [2].Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WST, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata–Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss–Coray T (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21, 383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Luchsinger JA, Mayeux R (2004) Cardiovascular risk factors and Alzheimer’s disease. Curr Atheroscler Rep 6, 261–266. [DOI] [PubMed] [Google Scholar]
- [4].Nho K, Kueider-Paisley A, Ahmad S, MahmoudianDehkordi S, Arnold M, Risacher SL, Louie G, Blach C, Baillie R, Han X, Kastenmüller G, Trojanowski JQ, Shaw LM, Weiner MW, Doraiswamy PM, van Duijn C, Saykin AJ, Kaddurah-Daouk R (2019) Association of Altered Liver Enzymes With Alzheimer Disease Diagnosis, Cognition, Neuroimaging Measures, and Cerebrospinal Fluid Biomarkers. JAMA Netw Open 2,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kumar A, Singh A, Ekavali (2015) A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol Rep 67, 195–203. [DOI] [PubMed] [Google Scholar]
- [6].Neuner SM, Tcw J, Goate AM (2020) Genetic architecture of Alzheimer’s disease. Neurobiol Dis 143, 104976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, Bellenguez C, Frizatti A, Chouraki V, Martin ER, Sleegers K, Badarinarayan N, Jakobsdottir J, Hamilton-Nelson KL, Moreno-Grau S, Olaso R, Raybould R, Chen Y, Kuzma AB, Hiltunen M, Morgan T, Ahmad S, Vardarajan BN, Epelbaum J, Hoffmann P, Boada M, Beecham GW, Garnier J-G, Harold D, Fitzpatrick AL, Valladares O, Moutet M-L, Gerrish A, Smith AV, Qu L, Bacq D, Denning N, Jian X, Zhao Y, Zompo MD, Fox NC, Choi S-H, Mateo I, Hughes JT, Adams HH, Malamon J, Sanchez-Garcia F, Patel Y, Brody JA, Dombroski BA, Naranjo MCD, Daniilidou M, Eiriksdottir G, Mukherjee S, Wallon D, Uphill J, Aspelund T, Cantwell LB, Garzia F, Galimberti D, Hofer E, Butkiewicz M, Fin B, Scarpini E, Sarnowski C, Bush WS, Meslage S, Kornhuber J, White CC, Song Y, Barber RC, Engelborghs S, Sordon S, Voijnovic D, Adams PM, Vandenberghe R, Mayhaus M, Cupples LA, Albert MS, Deyn PPD, Gu W, Himali JJ, Beekly D, Squassina A, Hartmann AM, Orellana A, Blacker D, Rodriguez-Rodriguez E, Lovestone S, Garcia ME, Doody RS, Munoz-Fernadez C, Sussams R, Lin H, Fairchild TJ, Benito YA, Holmes C, Karamujić-Čomić H, Frosch MP, Thonberg H, Maier W, Roschupkin G, Ghetti B, Giedraitis V, Kawalia A, Li S, Huebinger RM, Kilander L, Moebus S, Hernández I, Kamboh MI, Brundin R, Turton J, Yang Q, Katz MJ, Concari L, Lord J, Beiser AS, Keene CD, Helisalmi S, Kloszewska I, Kukull WA, Koivisto AM, Lynch A, Tarraga L, Larson EB, Haapasalo A, Lawlor B, Mosley TH, Lipton RB, Solfrizzi V, Gill M, Longstreth WT, Montine TJ, Frisardi V, Diez-Fairen M, Rivadeneira F, Petersen RC, Deramecourt V, Alvarez I, Salani F, Ciaramella A, Boerwinkle E, Reiman EM, Fievet N, Rotter JI, Reisch JS, Hanon O, Cupidi C, Uitterlinden AGA, Royall DR, Dufouil C, Maletta RG, Rojas I de, Sano M, Brice A, Cecchetti R, George-Hyslop PS, Ritchie K, Tsolaki M, Tsuang DW, Dubois B, Craig D, Wu C-K, Soininen H, Avramidou D, Albin RL, Fratiglioni L, Germanou A, Apostolova LG, Keller L, Koutroumani M, Arnold SE, Panza F, Gkatzima O, Asthana S, Hannequin D, Whitehead P, Atwood CS, Caffarra P, Hampel H, Quintela I, Carracedo Á, Lannfelt L, Rubinsztein DC, Barnes LL, Pasquier F, Frölich L, Barral S, McGuinness B, Beach TG, Johnston JA, Becker JT, Passmore P, Bigio EH, Schott JM, Bird TD, Warren JD, Boeve BF, Lupton MK, Bowen JD, Proitsi P, Boxer A, Powell JF, Burke JR, Kauwe JSK, Burns JM, Mancuso M, Buxbaum JD, Bonuccelli U, Cairns NJ, McQuillin A, Cao C, Livingston G, Carlson CS, Bass NJ, Carlsson CM, Hardy J, Carney RM, Bras J, Carrasquillo MM, Guerreiro R, Allen M, Chui HC, Fisher E, Masullo C, Crocco EA, DeCarli C, Bisceglio G, Dick M, Ma L, Duara R, Graff-Radford NR, Evans DA, Hodges A, Faber KM, Scherer M, Fallon KB, Riemenschneider M, Fardo DW, Heun R, Farlow MR, Kölsch H, Ferris S, Leber M, Foroud TM, Heuser I, Galasko DR, Giegling I, Gearing M, Hüll M, Geschwind DH, Gilbert JR, Morris J, Green RC, Mayo K, Growdon JH, Feulner T, Hamilton RL, Harrell LE, Drichel D, Honig LS, Cushion TD, Huentelman MJ, Hollingworth P, Hulette CM, Hyman BT, Marshall R, Jarvik GP, Meggy A, Abner E, Menzies GE, Jin L-W, Leonenko G, Real LM, Jun GR, Baldwin CT, Grozeva D, Karydas A, Russo G, Kaye JA, Kim R, Jessen F, Kowall NW, Vellas B, Kramer JH, Vardy E, LaFerla FM, Jöckel K-H, Lah JJ, Dichgans M, Leverenz JB, Mann D, Levey AI, Pickering-Brown S, Lieberman AP, Klopp N, Lunetta KL, Wichmann H-E, Lyketsos CG, Morgan K, Marson DC, Brown K, Martiniuk F, Medway C, Mash DC, Nöthen MM, Masliah E, Hooper NM, McCormick WC, Daniele A, McCurry SM, Bayer A, McDavid AN, Gallacher J, McKee AC, van den Bussche H, Mesulam M, Brayne C, Miller BL, Riedel-Heller S, Miller CA, Miller JW, Al-Chalabi A, Morris JC, Shaw CE, Myers AJ, Wiltfang J, O’Bryant S, Olichney JM, Alvarez V, Parisi JE, Singleton AB, Paulson HL, Collinge J, Perry WR, Mead S, Peskind E, Cribbs DH, Rossor M, Pierce A, Ryan NS, Poon WW, Nacmias B, Potter H, Sorbi S, Quinn JF, Sacchinelli E, Raj A, Spalletta G, Raskind M, Caltagirone C, Bossù P, Orfei MD, Reisberg B, Clarke R, Reitz C, Smith AD, Ringman JM, Warden D, Roberson ED, Wilcock G, Rogaeva E, Bruni AC, Rosen HJ, Gallo M, Rosenberg RN, Ben-Shlomo Y, Sager MA, Mecocci P, Saykin AJ, Pastor P, Cuccaro ML, Vance JM, Schneider JA, Schneider LS, Slifer S, Seeley WW, Smith AG, Sonnen JA, Spina S, Stern RA, Swerdlow RH, Tang M, Tanzi RE, Trojanowski JQ, Troncoso JC, Deerlin VMV, Eldik LJV, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Wilhelmsen KC, Williamson J, Wingo TS, Woltjer RL, Wright CB, Yu C-E, Yu L, Saba Y, Pilotto A, Bullido MJ, Peters O, Crane PK, Bennett D, Bosco P, Coto E, Boccardi V, Jager PLD, Lleo A, Warner N, Lopez OL, Ingelsson M, Deloukas P, Cruchaga C, Graff C, Gwilliam R, Fornage M, Goate AM, Sanchez-Juan P, Kehoe PG, Amin N, Ertekin-Taner N, Berr C, Debette S, Love S, Launer LJ, Younkin SG, Dartigues J-F, Corcoran C, Ikram MA, Dickson DW, Nicolas G, Campion D, Tschanz J, Schmidt H, Hakonarson H, Clarimon J, Munger R, Schmidt R, Farrer LA, Broeckhoven CV, O’Donovan MC, DeStefano AL, Jones L, Haines JL, Deleuze J-F, Owen MJ, Gudnason V, Mayeux R, Escott-Price V, Psaty BM, Ramirez A, Wang L-S, Ruiz A, van Duijn CM, Holmans PA, Seshadri S, Williams J, Amouyel P, Schellenberg GD, Lambert J-C, Pericak-Vance MA (2019) Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 51, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hägg S, Athanasiu L, Voyle N, Proitsi P, Witoelar A, Stringer S, Aarsland D, Almdahl IS, Andersen F, Bergh S, Bettella F, Bjornsson S, Brækhus A, Bråthen G, Leeuw C de, Desikan RS, Djurovic S, Dumitrescu L, Fladby T, Hohman TJ, Jonsson PV, Kiddle SJ, Rongve A, Saltvedt I, Sando SB, Selbæk G, Shoai M, Skene NG, Snaedal J, Stordal E, Ulstein ID, Wang Y, White LR, Hardy J, Hjerling-Leffler J, Sullivan PF, van der Flier WM, Dobson R, Davis LK, Stefansson H, Stefansson K, Pedersen NL, Ripke S, Andreassen OA, Posthuma D (2019) Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51, 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, Naj AC, Campos-Martin R, Grenier-Boley B, Andrade V, Holmans PA, Boland A, Damotte V, van der Lee SJ, Costa MR, Kuulasmaa T, Yang Q, de Rojas I, Bis JC, Yaqub A, Prokic I, Chapuis J, Ahmad S, Giedraitis V, Aarsland D, Garcia-Gonzalez P, Abdelnour C, Alarcón-Martín E, Alcolea D, Alegret M, Alvarez I, Álvarez V, Armstrong NJ, Tsolaki A, Antúnez C, Appollonio I, Arcaro M, Archetti S, Pastor AA, Arosio B, Athanasiu L, Bailly H, Banaj N, Baquero M, Barral S, Beiser A, Pastor AB, Below JE, Benchek P, Benussi L, Berr C, Besse C, Bessi V, Binetti G, Bizarro A, Blesa R, Boada M, Boerwinkle E, Borroni B, Boschi S, Bossù P, Bråthen G, Bressler J, Bresner C, Brodaty H, Brookes KJ, Brusco LI, Buiza-Rueda D, Bûrger K, Burholt V, Bush WS, Calero M, Cantwell LB, Chene G, Chung J, Cuccaro ML, Carracedo Á, Cecchetti R, Cervera-Carles L, Charbonnier C, Chen H-H, Chillotti C, Ciccone S, Claassen JAHR, Clark C, Conti E, Corma-Gómez A, Costantini E, Custodero C, Daian D, Dalmasso MC, Daniele A, Dardiotis E, Dartigues J-F, de Deyn PP, de Paiva Lopes K, de Witte LD, Debette S, Deckert J, Del Ser T, Denning N, DeStefano A, Dichgans M, Diehl-Schmid J, Diez-Fairen M, Rossi PD, Djurovic S, Duron E, Düzel E, Dufouil C, Eiriksdottir G, Engelborghs S, Escott-Price V, Espinosa A, Ewers M, Faber KM, Fabrizio T, Nielsen SF, Fardo DW, Farotti L, Fenoglio C, Fernández-Fuertes M, Ferrari R, Ferreira CB, Ferri E, Fin B, Fischer P, Fladby T, Fließbach K, Fongang B, Fornage M, Fortea J, Foroud TM, Fostinelli S, Fox NC, Franco-Macías E, Bullido MJ, Frank-García A, Froelich L, Fulton-Howard B, Galimberti D, García-Alberca JM, García-González P, Garcia-Madrona S, Garcia-Ribas G, Ghidoni R, Giegling I, Giorgio G, Goate AM, Goldhardt O, Gomez-Fonseca D, González-Pérez A, Graff C, Grande G, Green E, Grimmer T, Grünblatt E, Grunin M, Gudnason V, Guetta-Baranes T, Haapasalo A, Hadjigeorgiou G, Haines JL, Hamilton-Nelson KL, Hampel H, Hanon O, Hardy J, Hartmann AM, Hausner L, Harwood J, Heilmann-Heimbach S, Helisalmi S, Heneka MT, Hernández I, Herrmann MJ, Hoffmann P, Holmes C, Holstege H, Vilas RH, Hulsman M, Humphrey J, Biessels GJ, Jian X, Johansson C, Jun GR, Kastumata Y, Kauwe J, Kehoe PG, Kilander L, Ståhlbom AK, Kivipelto M, Koivisto A, Kornhuber J, Kosmidis MH, Kukull WA, Kuksa PP, Kunkle BW, Kuzma AB, Lage C, Laukka EJ, Launer L, Lauria A, Lee C-Y, Lehtisalo J, Lerch O, Lleó A, Longstreth W, Lopez O, de Munain AL, Love S, Löwemark M, Luckcuck L, Lunetta KL, Ma Y, Macías J, MacLeod CA, Maier W, Mangialasche F, Spallazzi M, Marquié M, Marshall R, Martin ER, Montes AM, Rodríguez CM, Masullo C, Mayeux R, Mead S, Mecocci P, Medina M, Meggy A, Mehrabian S, Mendoza S, Menéndez-González M, Mir P, Moebus S, Mol M, Molina-Porcel L, Montrreal L, Morelli L, Moreno F, Morgan K, Mosley T, Nöthen MM, Muchnik C, Mukherjee S, Nacmias B, Ngandu T, Nicolas G, Nordestgaard BG, Olaso R, Orellana A, Orsini M, Ortega G, Padovani A, Paolo C, Papenberg G, Parnetti L, Pasquier F, Pastor P, Peloso G, Pérez-Cordón A, Pérez-Tur J, Pericard P, Peters O, Pijnenburg YAL, Pineda JA, Piñol-Ripoll G, Pisanu C, Polak T, Popp J, Posthuma D, Priller J, Puerta R, Quenez O, Quintela I, Thomassen JQ, Rábano A, Rainero I, Rajabli F, Ramakers I, Real LM, Reinders MJT, Reitz C, Reyes-Dumeyer D, Ridge P, Riedel-Heller S, Riederer P, Roberto N, Rodriguez-Rodriguez E, Rongve A, Allende IR, Rosende-Roca M, Royo JL, Rubino E, Rujescu D, Sáez ME, Sakka P, Saltvedt I, Sanabria Á, Sánchez-Arjona MB, Sanchez-Garcia F, Juan PS, Sánchez-Valle R, Sando SB, Sarnowski C, Satizabal CL, Scamosci M, Scarmeas N, Scarpini E, Scheltens P, Scherbaum N, Scherer M, Schmid M, Schneider A, Schott JM, Selbæk G, Seripa D, Serrano M, Sha J, Shadrin AA, Skrobot O, Slifer S, Snijders GJL, Soininen H, Solfrizzi V, Solomon A, Song Y, Sorbi S, Sotolongo-Grau O, Spalletta G, Spottke A, Squassina A, Stordal E, Tartan JP, Tárraga L, Tesí N, Thalamuthu A, Thomas T, Tosto G, Traykov L, Tremolizzo L, Tybjærg-Hansen A, Uitterlinden A, Ullgren A, Ulstein I, Valero S, Valladares O, Broeckhoven CV, Vance J, Vardarajan BN, van der Lugt A, Dongen JV, van Rooij J, van Swieten J, Vandenberghe R, Verhey F, Vidal J-S, Vogelgsang J, Vyhnalek M, Wagner M, Wallon D, Wang L-S, Wang R, Weinhold L, Wiltfang J, Windle G, Woods B, Yannakoulia M, Zare H, Zhao Y, Zhang X, Zhu C, Zulaica M, EADB, GR@ACE, DEGESCO, EADI, GERAD, Demgene, FinnGen, ADGC, CHARGE, Farrer LA, Psaty BM, Ghanbari M, Raj T, Sachdev P, Mather K, Jessen F, Ikram MA, de Mendonça A, Hort J, Tsolaki M, Pericak-Vance MA, Amouyel P, Williams J, Frikke-Schmidt R, Clarimon J, Deleuze J-F, Rossi G, Seshadri S, Andreassen OA, Ingelsson M, Hiltunen M, Sleegers K, Schellenberg GD, van Duijn CM, Sims R, van der Flier WM, Ruiz A, Ramirez A, Lambert J-C (2022) New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet 54, 412–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K (2013) Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N Engl J Med 368, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Deming Y, Filipello F, Cignarella F, Cantoni C, Hsu S, Mikesell R, Li Z, Del-Aguila JL, Dube U, Farias FG, Bradley J, Budde J, Ibanez L, Fernandez MV, Blennow K, Zetterberg H, Heslegrave A, Johansson PM, Svensson J, Nellgård B, Lleo A, Alcolea D, Clarimon J, Rami L, Molinuevo JL, Suárez-Calvet M, Morenas-Rodríguez E, Kleinberger G, Ewers M, Harari O, Haass C, Brett TJ, Benitez BA, Karch CM, Piccio L, Cruchaga C (2019) The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci Transl Med 11,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ewers M, Franzmeier N, Suárez-Calvet M, Morenas-Rodriguez E, Caballero MAA, Kleinberger G, Piccio L, Cruchaga C, Deming Y, Dichgans M, Trojanowski JQ, Shaw LM, Weiner MW, Haass C, Initiative ADN (2019) Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci Transl Med 11,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Escott-Price V, Sims R, Bannister C, Harold D, Vronskaya M, Majounie E, Badarinarayan N, Morgan K, Passmore P, Holmes C, Powell J, Brayne C, Gill M, Mead S, Goate A, Cruchaga C, Lambert J-C, van Duijn C, Maier W, Ramirez A, Holmans P, Jones L, Hardy J, Seshadri S, Schellenberg GD, Amouyel P, Williams J (2015) Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain 138, 3673–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Escott-Price V, Shoai M, Pither R, Williams J, Hardy J (2017) Polygenic score prediction captures nearly all common genetic risk for Alzheimer’s disease. Neurobiol Aging 49, 214.e7–214.e11. [DOI] [PubMed] [Google Scholar]
- [15].Desikan RS, Fan CC, Wang Y, Schork AJ, Cabral HJ, Cupples LA, Thompson WK, Besser L, Kukull WA, Holland D, Chen C-H, Brewer JB, Karow DS, Kauppi K, Witoelar A, Karch CM, Bonham LW, Yokoyama JS, Rosen HJ, Miller BL, Dillon WP, Wilson DM, Hess CP, Pericak-Vance M, Haines JL, Farrer LA, Mayeux R, Hardy J, Goate AM, Hyman BT, Schellenberg GD, McEvoy LK, Andreassen OA, Dale AM (2017) Genetic assessment of age-associated Alzheimer disease risk: Development and validation of a polygenic hazard score. PLOS Med 14, e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Escott-Price V, Myers A, Huentelman M, Shoai M, Hardy J (2019) Polygenic Risk Score Analysis of Alzheimer’s Disease in Cases without APOE4 or APOE2 Alleles. J Prev Alzheimers Dis 6, 16–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schultz SA, Boots EA, Darst BF, Zetterberg H, Blennow K, Edwards DF, Koscik RL, Carlsson CM, Gallagher CL, Bendlin BB, Asthana S, Sager MA, Hogan KJ, Hermann BP, Cook DB, Johnson SC, Engelman CD, Okonkwo OC (2017) Cardiorespiratory fitness alters the influence of a polygenic risk score on biomarkers of AD. Neurology 88, 1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Matloff WJ, Zhao L, Ning K, Conti DV, Toga AW (2020) Interaction effect of alcohol consumption and Alzheimer disease polygenic risk score on the brain cortical thickness of cognitively normal subjects. Alcohol 85, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cruchaga C, Del-Aguila JL, Saef B, Black K, Fernandez MV, Budde J, Ibanez L, Deming Y, Kapoor M, Tosto G, Mayeux RP, Holtzman DM, Fagan AM, Morris JC, Bateman RJ, Goate AM, Harari O (2018) Polygenic risk score of sporadic late-onset Alzheimer’s disease reveals a shared architecture with the familial and early-onset forms. Alzheimers Dement 14, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hu Y, Lu Q, Powles R, Yao X, Yang C, Fang F, Xu X, Zhao H (2017) Leveraging functional annotations in genetic risk prediction for human complex diseases. PLOS Comput Biol 13, e1005589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Darst BF, Koscik RL, Racine AM, Oh JM, Krause RA, Carlsson CM, Zetterberg H, Blennow K, Christian BT, Bendlin BB, Okonkwo OC, Hogan KJ, Hermann BP, Sager MA, Asthana S, Johnson SC, Engelman CD (2017) Pathway-Specific Polygenic Risk Scores as Predictors of Amyloid-β Deposition and Cognitive Function in a Sample at Increased Risk for Alzheimer’s Disease. J Alzheimers Dis 55, 473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Efthymiou AG, Goate AM (2017) Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener 12, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lu Q, Powles RL, Abdallah S, Ou D, Wang Q, Hu Y, Lu Y, Liu W, Li B, Mukherjee S, Crane PK, Zhao H (2017) Systematic tissue-specific functional annotation of the human genome highlights immune-related DNA elements for late-onset Alzheimer’s disease. PLoS Genet 13, e1006933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lu Q, Powles RL, Wang Q, He BJ, Zhao H (2016) Integrative Tissue-Specific Functional Annotations in the Human Genome Provide Novel Insights on Many Complex Traits and Improve Signal Prioritization in Genome Wide Association Studies. PLoS Genet 12, e1005947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lu Q, Yao X, Hu Y, Zhao H (2016) GenoWAP: GWAS signal prioritization through integrated analysis of genomic functional annotation. Bioinformatics 32, 542–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Johnson SC, Koscik RL, Jonaitis EM, Clark LR, Mueller KD, Berman SE, Bendlin BB, Engelman CD, Okonkwo OC, Hogan KJ, Asthana S, Carlsson CM, Hermann BP, Sager MA (2017) The Wisconsin Registry for Alzheimer’s Prevention: A review of findings and current directions. Alzheimers Dement (Amst) 10, 130–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Melah KE, Lu SY-F, Hoscheidt SM, Alexander AL, Adluru N, Destiche DJ, Carlsson CM, Zetterberg H, Blennow K, Okonkwo OC, Gleason CE, Dowling NM, Bratzke LC, Rowley HA, Sager MA, Asthana S, Johnson SC, Bendlin BB (2016) CSF markers of Alzheimer’s pathology and microglial activation are associated with altered white matter microstructure in asymptomatic adults at risk for Alzheimer’s disease. J Alzheimers Dis 50, 873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944. [DOI] [PubMed] [Google Scholar]
- [29].Jack CR, Albert M, Knopman DS, McKhann GM, Sperling RA, Carillo M, Thies W, Phelps CH (2011) Introduction to Revised Criteria for the Diagnosis of Alzheimer’s Disease: National Institute on Aging and the Alzheimer Association Workgroups. Alzheimers Dement 7, 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Little J, Higgins JPT, Ioannidis JPA, Moher D, Gagnon F, von Elm E, Khoury MJ, Cohen B, Davey-Smith G, Grimshaw J, Scheet P, Gwinn M, Williamson RE, Zou GY, Hutchings K, Johnson CY, Tait V, Wiens M, Golding J, van Duijn C, McLaughlin J, Paterson A, Wells G, Fortier I, Freedman M, Zecevic M, King R, Infante-Rivard C, Stewart A, Birkett N (2009) STrengthening the REporting of Genetic Association Studies (STREGA)--an extension of the STROBE statement. Genet Epidemiol 33, 581–598. [DOI] [PubMed] [Google Scholar]
- [31].Van Hulle CA, Jonaitis EM, Betthauser TJ, Batrla R, Wild N, Kollmorgen G, Andreasson U, Okonkwo O, Bendlin BB, Asthana S, Carlsson CM, Johnson SC, Zetterberg H, Blennow K (2021) An examination of a novel multipanel of CSF biomarkers in the Alzheimer’s disease clinical and pathological continuum. Alzheimers Dement 17, 431–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Komsta L, Novomestky F (2015) moments: Moments, cumulants, skewness, kurtosis and related tests.
- [33].Panyard DJ, Kim KM, Darst BF, Deming YK, Zhong X, Wu Y, Kang H, Carlsson CM, Johnson SC, Asthana S, Engelman CD, Lu Q (2021) Cerebrospinal fluid metabolomics identifies 19 brain-related phenotype associations. Commun Biol 4,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Darst BF, Lu Q, Johnson SC, Engelman CD (2019) Integrated analysis of genomics, longitudinal metabolomics, and Alzheimer’s risk factors among 1,111 cohort participants. Genet Epidemiol 43, 657–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen W-M (2010) Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Conomos MP, Miller MB, Thornton TA (2015) Robust Inference of Population Structure for Ancestry Prediction and Correction of Stratification in the Presence of Relatedness. Genet Epidemiol 39, 276–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, Schlessinger D, Stambolian D, Loh P-R, Iacono WG, Swaroop A, Scott LJ, Cucca F, Kronenberg F, Boehnke M, Abecasis GR, Fuchsberger C (2016) Next-generation genotype imputation service and methods. Nat Genet 48, 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, Luo Y, Sidore C, Kwong A, Timpson N, Koskinen S, Vrieze S, Scott LJ, Zhang H, Mahajan A, Veldink J, Peters U, Pato C, van Duijn CM, Gillies CE, Gandin I, Mezzavilla M, Gilly A, Cocca M, Traglia M, Angius A, Barrett JC, Boomsma D, Branham K, Breen G, Brummett CM, Busonero F, Campbell H, Chan A, Chen S, Chew E, Collins FS, Corbin LJ, Smith GD, Dedoussis G, Dorr M, Farmaki A-E, Ferrucci L, Forer L, Fraser RM, Gabriel S, Levy S, Groop L, Harrison T, Hattersley A, Holmen OL, Hveem K, Kretzler M, Lee JC, McGue M, Meitinger T, Melzer D, Min JL, Mohlke KL, Vincent JB, Nauck M, Nickerson D, Palotie A, Pato M, Pirastu N, McInnis M, Richards JB, Sala C, Salomaa V, Schlessinger D, Schoenherr S, Slagboom PE, Small K, Spector T, Stambolian D, Tuke M, Tuomilehto J, Van den Berg LH, Van Rheenen W, Volker U, Wijmenga C, Toniolo D, Zeggini E, Gasparini P, Sampson MG, Wilson JF, Frayling T, de Bakker PIW, Swertz MA, McCarroll S, Kooperberg C, Dekker A, Altshuler D, Willer C, Iacono W, Ripatti S, Soranzo N, Walter K, Swaroop A, Cucca F, Anderson CA, Myers RM, Boehnke M, McCarthy MI, Durbin R, Abecasis G, Marchini J, the Haplotype Reference Consortium (2016) A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 48, 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Loh P-R, Danecek P, Palamara PF, Fuchsberger C, Reshef YA, Finucane HK, Schoenherr S, Forer L, McCarthy S, Abecasis GR, Durbin R, Price AL (2016) Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet 48, 1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, Flicek P, Gabriel SB, Gibbs RA, Green ED, Hurles ME, Knoppers BM, Korbel JO, Lander ES, Lee C, Lehrach H, Mardis ER, Marth GT, McVean GA, Nickerson DA, Schmidt JP, Sherry ST, Wang J, Wilson RK, Gibbs RA, Boerwinkle E, Doddapaneni H, Han Y, Korchina V, Kovar C, Lee S, Muzny D, Reid JG, Zhu Y, Wang J, Chang Y, Feng Q, Fang X, Guo X, Jian M, Jiang H, Jin X, Lan T, Li G, Li J, Li Y, Liu S, Liu X, Lu Y, Ma X, Tang M, Wang B, Wang G, Wu H, Wu R, Xu X, Yin Y, Zhang D, Zhang W, Zhao J, Zhao M, Zheng X, Lander ES, Altshuler DM, Gabriel SB, Gupta N, Gharani N, Toji LH, Gerry NP, Resch AM, Flicek P, Barker J, Clarke L, Gil L, Hunt SE, Kelman G, Kulesha E, Leinonen R, McLaren WM, Radhakrishnan R, Roa A, Smirnov D, Smith RE, Streeter I, Thormann A, Toneva I, Vaughan B, Zheng-Bradley X, Bentley DR, Grocock R, Humphray S, James T, Kingsbury Z, Lehrach H, Sudbrak R, Albrecht MW, Amstislavskiy VS, Borodina TA, Lienhard M, Mertes F, Sultan M, Timmermann B, Yaspo M-L, Mardis ER, Wilson RK, Fulton L, Fulton R, Sherry ST, Ananiev V, Belaia Z, Beloslyudtsev D, Bouk N, Chen C, Church D, Cohen R, Cook C, Garner J, Hefferon T, Kimelman M, Liu C, Lopez J, Meric P, O’Sullivan C, Ostapchuk Y, Phan L, Ponomarov S, Schneider V, Shekhtman E, Sirotkin K, Slotta D, Zhang H, McVean GA, Durbin RM, Balasubramaniam S, Burton J, Danecek P, Keane TM, Kolb-Kokocinski A, McCarthy S, Stalker J, Quail M, Schmidt JP, Davies CJ, Gollub J, Webster T, Wong B, Zhan Y, Auton A, Campbell CL, Kong Y, Marcketta A, Gibbs RA, Yu F, Antunes L, Bainbridge M, Muzny D, Sabo A, Huang Z, Wang J, Coin LJM, Fang L, Guo X, Jin X, Li G, Li Q, Li Y, Li Z, Lin H, Liu B, Luo R, Shao H, Xie Y, Ye C, Yu C, Zhang F, Zheng H, Zhu H, Alkan C, Dal E, Kahveci F, Marth GT, Garrison EP, Kural D, Lee W-P, Fung Leong W, Stromberg M, Ward AN, Wu J, Zhang M, Daly MJ, DePristo MA, Handsaker RE, Altshuler DM, Banks E, Bhatia G, del Angel G, Gabriel SB, Genovese G, Gupta N, Li H, Kashin S, Lander ES, McCarroll SA, Nemesh JC, Poplin RE, Yoon SC, Lihm J, Makarov V, Clark AG, Gottipati S, Keinan A, Rodriguez-Flores JL, Korbel JO, Rausch T, Fritz MH, Stütz AM, Flicek P, Beal K, Clarke L, Datta A, Herrero J, McLaren WM, Ritchie GRS, Smith RE, Zerbino D, Zheng-Bradley X, Sabeti PC, Shlyakhter I, Schaffner SF, Vitti J, Cooper DN, Ball EV, Stenson PD, Bentley DR, Barnes B, Bauer M, Keira Cheetham R, Cox A, Eberle M, Humphray S, Kahn S, Murray L, Peden J, Shaw R, Kenny EE, Batzer MA, Konkel MK, Walker JA, MacArthur DG, Lek M, Sudbrak R, Amstislavskiy VS, Herwig R, Mardis ER, Ding L, Koboldt DC, Larson D, Ye K, Gravel S, The 1000 Genomes Project Consortium, Corresponding authors, Steering committee, Production group, Baylor College of Medicine, BGI-Shenzhen, Broad Institute of MIT and Harvard, Coriell Institute for Medical Research, European Molecular Biology Laboratory EBI, Illumina, Max Planck Institute for Molecular Genetics, McDonnell Genome Institute at Washington University, US National Institutes of Health, University of Oxford, Wellcome Trust Sanger Institute, Analysis group, Affymetrix, Albert Einstein College of Medicine, Bilkent University, Boston College, Cold Spring Harbor Laboratory, Cornell University, European Molecular Biology Laboratory, Harvard University, Human Gene Mutation Database, Icahn School of Medicine at Mount Sinai, Louisiana State University, Massachusetts General Hospital, McGill University, National Eye Institute N (2015) A global reference for human genetic variation. Nature 526, 68–74.26432245 [Google Scholar]
- [41].Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu Y-C, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh K-H, Feizi S, Karlic R, Kim A-R, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJM, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai L-H, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M (2015) Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, Barnes I, Berry A, Bignell A, Carbonell Sala S, Chrast J, Cunningham F, Di Domenico T, Donaldson S, Fiddes IT, García Girón C, Gonzalez JM, Grego T, Hardy M, Hourlier T, Hunt T, Izuogu OG, Lagarde J, Martin FJ, Martínez L, Mohanan S, Muir P, Navarro FCP, Parker A, Pei B, Pozo F, Ruffier M, Schmitt BM, Stapleton E, Suner M-M, Sycheva I, Uszczynska-Ratajczak B, Xu J, Yates A, Zerbino D, Zhang Y, Aken B, Choudhary JS, Gerstein M, Guigó R, Hubbard TJP, Kellis M, Paten B, Reymond A, Tress ML, Flicek P (2019) GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Euesden J, Lewis CM, O’Reilly PF (2015) PRSice: Polygenic Risk Score software. Bioinformatics 31, 1466–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhao Z, Yi Y, Song J, Wu Y, Zhong X, Lin Y, Hohman TJ, Fletcher J, Lu Q (2021) PUMAS: fine-tuning polygenic risk scores with GWAS summary statistics. Genome Biol 22, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].R Core Team (2019) R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- [46].Spence JP, Sinnott-Armstrong N, Assimes TL, Pritchard JK (2022) A flexible modeling and inference framework for estimating variant effect sizes from GWAS summary statistics. bioRxiv 2022.04.18.488696. [Google Scholar]
- [47].Barton K (2020) MuMIn: Multi-Model Inference.
- [48].Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356. [PubMed] [Google Scholar]
- [49].Bertram L, Lill CM, Tanzi RE (2010) The Genetics of Alzheimer Disease: Back to the Future. Neuron 68, 270–281. [DOI] [PubMed] [Google Scholar]
- [50].Tang M-X, Stern Y, Marder K, Bell K, Gurland B, Lantigua R, Andrews H, Feng L, Tycko B, Mayeux R (1998) The APOE-∊4 Allele and the Risk of Alzheimer Disease Among African Americans, Whites, and Hispanics. JAMA 279, 751–755. [DOI] [PubMed] [Google Scholar]
- [51].Ochoa D, Hercules A, Carmona M, Suveges D, Gonzalez-Uriarte A, Malangone C, Miranda A, Fumis L, Carvalho-Silva D, Spitzer M, Baker J, Ferrer J, Raies A, Razuvayevskaya O, Faulconbridge A, Petsalaki E, Mutowo P, Machlitt-Northen S, Peat G, McAuley E, Ong CK, Mountjoy E, Ghoussaini M, Pierleoni A, Papa E, Pignatelli M, Koscielny G, Karim M, Schwartzentruber J, Hulcoop DG, Dunham I, McDonagh EM (2021) Open Targets Platform: supporting systematic drug–target identification and prioritisation. Nucleic Acids Res 49, D1302–D1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D, Rongve A, Børte S, Winsvold BS, Drange OK, Martinsen AE, Skogholt AH, Willer C, Bråthen G, Bosnes I, Nielsen JB, Fritsche LG, Thomas LF, Pedersen LM, Gabrielsen ME, Johnsen MB, Meisingset TW, Zhou W, Proitsi P, Hodges A, Dobson R, Velayudhan L, Sealock JM, Davis LK, Pedersen NL, Reynolds CA, Karlsson IK, Magnusson S, Stefansson H, Thordardottir S, Jonsson PV, Snaedal J, Zettergren A, Skoog I, Kern S, Waern M, Zetterberg H, Blennow K, Stordal E, Hveem K, Zwart J-A, Athanasiu L, Selnes P, Saltvedt I, Sando SB, Ulstein I, Djurovic S, Fladby T, Aarsland D, Selbæk G, Ripke S, Stefansson K, Andreassen OA, Posthuma D (2021) A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet 53, 1276–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mayeux R, Stern Y (2012) Epidemiology of Alzheimer Disease. Cold Spring Harb Perspect Med 2,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Reitz C, Mayeux R (2014) Alzheimer disease: Epidemiology, Diagnostic Criteria, Risk Factors and Biomarkers. Biochem Pharmacol 88, 640–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lam V, Takechi R, Hackett MJ, Francis R, Bynevelt M, Celliers LM, Nesbit M, Mamsa S, Arfuso F, Das S, Koentgen F, Hagan M, Codd L, Richardson K, O’Mara B, Scharli RK, Morandeau L, Gauntlett J, Leatherday C, Boucek J, Mamo JCL (2021) Synthesis of human amyloid restricted to liver results in an Alzheimer disease–like neurodegenerative phenotype. PLOS Biol 19, e3001358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164–e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Pickrell JK (2014) Joint Analysis of Functional Genomic Data and Genome-wide Association Studies of 18 Human Traits. Am J Hum Genet 94, 559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gusev A, Lee SH, Trynka G, Finucane H, Vilhjálmsson BJ, Xu H, Zang C, Ripke S, Bulik-Sullivan B, Stahl E, Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans PA, Lee P, Bulik-Sullivan B, Collier DA, Huang H, Pers TH, Agartz I, Agerbo E, Albus M, Alexander M, Amin F, Bacanu SA, Begemann M, Belliveau RA, Bene J, Bergen SE, Bevilacqua E, Bigdeli TB, Black DW, Børglum AD, Bruggeman R, Buccola NG, Buckner RL, Byerley W, Cahn W, Cai G, Campion D, Cantor RM, Carr VJ, Carrera N, Catts SV, Chambert KD, Chan RCK, Chen RYL, Chen EYH, Cheng W, Cheung EFC, Chong SA, Cloninger CR, Cohen D, Cohen N, Cormican P, Craddock N, Crowley JJ, Curtis D, Davidson M, Davis KL, Degenhardt F, Del Favero J, DeLisi LE, Demontis D, Dikeos D, Dinan T, Djurovic S, Donohoe G, Drapeau E, Duan J, Dudbridge F, Durmishi N, Eichhammer P, Eriksson J, Escott-Price V, Essioux L, Fanous AH, Farrell MS, Frank J, Franke L, Freedman R, Freimer NB, Friedl M, Friedman JI, Fromer M, Genovese G, Georgieva L, Gershon ES, Giegling I, Giusti-Rodrguez P, Godard S, Goldstein JI, Golimbet V, Gopal S, Gratten J, Grove J, de Haan L, Hammer C, Hamshere ML, Hansen M, Hansen T, Haroutunian V, Hartmann AM, Henskens FA, Herms S, Hirschhorn JN, Hoffmann P, Hofman A, Hollegaard MV, Hougaard DM, Ikeda M, Joa I, Julià A, Kahn RS, Kalaydjieva L, Karachanak-Yankova S, Karjalainen J, Kavanagh D, Keller MC, Kelly BJ, Kennedy JL, Khrunin A, Kim Y, Klovins J, Knowles JA, Konte B, Kucinskas V, Kucinskiene ZA, Kuzelova-Ptackova H, Kähler AK, Laurent C, Keong JLC, Lee SH, Legge SE, Lerer B, Li M, Li T, Liang K-Y, Lieberman J, Limborska S, Loughland CM, Lubinski J, Lnnqvist J, Macek M, Magnusson PKE, Maher BS, Maier W, Mallet J, Marsal S, Mattheisen M, Mattingsdal M, McCarley RW, McDonald C, McIntosh AM, Meier S, Meijer CJ, Melegh B, Melle I, Mesholam-Gately RI, Metspalu A, Michie PT, Milani L, Milanova V, Mokrab Y, Morris DW, Mors O, Mortensen PB, Murphy KC, Murray RM, Myin-Germeys I, Mller-Myhsok B, Nelis M, Nenadic I, Nertney DA, Nestadt G, Nicodemus KK, Nikitina-Zake L, Nisenbaum L, Nordin A, O’Callaghan E, O’Dushlaine C, O’Neill FA, Oh S-Y, Olincy A, Olsen L, Van Os J, Pantelis C, Papadimitriou GN, Papiol S, Parkhomenko E, Pato MT, Paunio T, Pejovic-Milovancevic M, Perkins DO, Pietilinen O, Pimm J, Pocklington AJ, Powell J, Price A, Pulver AE, Purcell SM, Quested D, Rasmussen HB, Reichenberg A, Reimers MA, Richards AL, Roffman JL, Roussos P, Ruderfer DM, Salomaa V, Sanders AR, Schall U, Schubert CR, Schulze TG, Schwab SG, Scolnick EM, Scott RJ, Seidman LJ, Shi J, Sigurdsson E, Silagadze T, Silverman JM, Sim K, Slominsky P, Smoller JW, So H-C, Spencer CCA, Stahl EA, Stefansson H, Steinberg S, Stogmann E, Straub RE, Strengman E, Strohmaier J, Stroup TS, Subramaniam M, Suvisaari J, Svrakic DM, Szatkiewicz JP, Sderman E, Thirumalai S, Toncheva D, Tooney PA, Tosato S, Veijola J, Waddington J, Walsh D, Wang D, Wang Q, Webb BT, Weiser M, Wildenauer DB, Williams NM, Williams S, Witt SH, Wolen AR, Wong EHM, Wormley BK, Wu JQ, Xi HS, Zai CC, Zheng X, Zimprich F, Wray NR, Stefansson K, Visscher PM, Adolfsson R, Andreassen OA, Blackwood DHR, Bramon E, Buxbaum JD, Brglum AD, Cichon S, Darvasi A, Domenici E, Ehrenreich H, Esko T, Gejman PV, Gill M, Gurling H, Hultman CM, Iwata N, Jablensky AV, Jönsson EG, Kendler KS, Kirov G, Knight J, Lencz T, Levinson DF, Li QS, Liu J, Malhotra AK, McCarroll SA, McQuillin A, Moran JL, Mortensen PB, Mowry BJ, Nthen MM, Ophoff RA, Owen MJ, Palotie A, Pato CN, Petryshen TL, Posthuma D, Rietschel M, Riley BP, Rujescu D, Sham PC, Sklar P, St. Clair D, Weinberger DR, Wendland JR, Werge T, Daly MJ, Sullivan PF, O’Donovan MC, Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kähler AK, Akterin S, Bergen S, Magnusson PKE, Neale BM, Ruderfer D, Scolnick E, Purcell S, McCarroll S, Sklar P, Hultman CM, Sullivan PF, Kähler AK, Hultman CM, Purcell SM, McCarroll SA, Daly M, Pasaniuc B, Sullivan PF, Neale BM, Wray NR, Raychaudhuri S, Price AL (2014) Partitioning Heritability of Regulatory and Cell-Type-Specific Variants across 11 Common Diseases. Am J Hum Genet 95, 535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Amariuta T, Ishigaki K, Sugishita H, Ohta T, Koido M, Dey KK, Matsuda K, Murakami Y, Price AL, Kawakami E, Terao C, Raychaudhuri S (2020) Improving the trans-ancestry portability of polygenic risk scores by prioritizing variants in predicted cell-type-specific regulatory elements. Nat Genet 52, 1346–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Aguet F, Brown AA, Castel SE, Davis JR, He Y, Jo B, Mohammadi P, Park Y, Parsana P, Segrè AV, Strober BJ, Zappala Z, Cummings BB, Gelfand ET, Hadley K, Huang KH, Lek M, Li X, Nedzel JL, Nguyen DY, Noble MS, Sullivan TJ, Tukiainen T, MacArthur DG, Getz G, Addington A, Guan P, Koester S, Little AR, Lockhart NC, Moore HM, Rao A, Struewing JP, Volpi S, Brigham LE, Hasz R, Hunter M, Johns C, Johnson M, Kopen G, Leinweber WF, Lonsdale JT, McDonald A, Mestichelli B, Myer K, Roe B, Salvatore M, Shad S, Thomas JA, Walters G, Washington M, Wheeler J, Bridge J, Foster BA, Gillard BM, Karasik E, Kumar R, Miklos M, Moser MT, Jewell SD, Montroy RG, Rohrer DC, Valley D, Mash DC, Davis DA, Sobin L, Barcus ME, Branton PA, Abell NS, Balliu B, Delaneau O, Frésard L, Gamazon ER, Garrido-Martín D, Gewirtz ADH, Gliner G, Gloudemans MJ, Han B, He AZ, Hormozdiari F, Li X, Liu B, Kang EY, McDowell IC, Ongen H, Palowitch JJ, Peterson CB, Quon G, Ripke S, Saha A, Shabalin AA, Shimko TC, Sul JH, Teran NA, Tsang EK, Zhang H, Zhou Y-H, Bustamante CD, Cox NJ, Guigó R, Kellis M, McCarthy MI, Conrad DF, Eskin E, Li G, Nobel AB, Sabatti C, Stranger BE, Wen X, Wright FA, Ardlie KG, Dermitzakis ET, Lappalainen T, Aguet F, Ardlie KG, Cummings BB, Gelfand ET, Getz G, Hadley K, Handsaker RE, Huang KH, Kashin S, Karczewski KJ, Lek M, Li X, MacArthur DG, Nedzel JL, Nguyen DT, Noble MS, Segrè AV, Trowbridge CA, Tukiainen T, Abell NS, Balliu B, Barshir R, Basha O, Battle A, Bogu GK, Brown A, Brown CD, Castel SE, Chen LS, Chiang C, Conrad DF, Cox NJ, Damani FN, Davis JR, Delaneau O, Dermitzakis ET, Engelhardt BE, Eskin E, Ferreira PG, Frésard L, Gamazon ER, Garrido-Martín D, Gewirtz ADH, Gliner G, Gloudemans MJ, Guigo R, Hall IM, Han B, He Y, Hormozdiari F, Howald C, Kyung Im H, Jo B, Yong Kang E, Kim Y, Kim-Hellmuth S, Lappalainen T, Li G, Li X, Liu B, Mangul S, McCarthy MI, McDowell IC, Mohammadi P, Monlong J, Montgomery SB, Muñoz-Aguirre M, Ndungu AW, Nicolae DL, Nobel AB, Oliva M, Ongen H, Palowitch JJ, Panousis N, Papasaikas P, Park Y, Parsana P, Payne AJ, Peterson CB, Quan J, Reverter F, Sabatti C, Saha A, Sammeth M, Scott AJ, Shabalin AA, Sodaei R, Stephens M, Stranger BE, Strober BJ, Sul JH, Tsang EK, Urbut S, van de Bunt M, Wang G, Wen X, Wright FA, Xi HS, Yeger-Lotem E, Zappala Z, Zaugg JB, Zhou Y-H, Akey JM, Bates D, Chan J, Chen LS, Claussnitzer M, Demanelis K, Diegel M, Doherty JA, Feinberg AP, Fernando MS, Halow J, Hansen KD, Haugen E, Hickey PF, Hou L, Jasmine F, Jian R, Jiang L, Johnson A, Kaul R, Kellis M, Kibriya MG, Lee K, Billy Li J, Li Q, Li X, Lin J, Lin S, Linder S, Linke C, Liu Y, Maurano MT, Molinie B, Montgomery SB, Nelson J, Neri FJ, Oliva M, Park Y, Pierce BL, Rinaldi NJ, Rizzardi LF, Sandstrom R, Skol A, Smith KS, Snyder MP, Stamatoyannopoulos J, Stranger BE, Tang H, Tsang EK, Wang L, Wang M, Van Wittenberghe N, Wu F, Zhang R, Nierras CR, Branton PA, Carithers LJ, Guan P, Moore HM, Rao A, Vaught JB, Gould SE, Lockart NC, Martin C, Struewing JP, Volpi S, Addington AM, Koester SE, Little AR, GTEx Consortium, Lead analysts:, Laboratory DA& CC (LDACC):, NIH program management:, Biospecimen collection:, Pathology:, eQTL manuscript working group:, Laboratory DA& CC (LDACC)—Analysis WG, Statistical Methods groups—Analysis Working Group, Enhancing GTEx (eGTEx) groups, NIH Common Fund, NIH/NCI, NIH/NHGRI, NIH/NIMH, NIH/NIDA, Biospecimen Collection Source Site—NDRI (2017) Genetic effects on gene expression across human tissues. Nature 550, 204–213.29022597 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets analyzed in this study may be requested from the WADRC at https://www.adrc.wisc.edu/apply-resources.