

Abstract
Well-established animal models of depression have described a proximal relationship between stress and central nervous system (CNS) inflammation – a relationship mirrored in the peripheral inflammatory biomarkers of individuals with depression. Evidence also suggests that stress-induced proinflammatory states can contribute to the neurobiology of treatment-resistant depression. Interestingly, ketamine, a rapid-acting antidepressant, can partially exert its therapeutic effects via anti-inflammatory actions on the hypothalamic-pituitary-adrenal (HPA) axis, the kynurenine pathway or by cytokine suppression. Further investigations into the relationship between ketamine, inflammation and stress could provide insight into ketamine’s unique therapeutic mechanisms and stimulate efforts to develop rapid-acting, anti-inflammatory-based antidepressants.
Keywords: Ketamine, depression, stress, inflammation, treatment-resistant depression, anhedonia
Introduction
Depression is the leading cause of disability worldwide, affecting 322 million people.1 In the USA, research suggests that approximately one-third of sufferers have treatment-resistant depression (TRD), broadly defined as non-response to conventional antidepressants.2 One of the primary obstacles to understanding depression is its characteristic heterogeneity in the course of illness, biomarkers, treatment response and genetic polymorphisms. As such, recent efforts within psychiatry have sought to establish clinically relevant biomarker and symptom-based subgroups under the umbrella of depressive disorders.
For over three decades, researchers have studied the relationship between depressive symptoms and inflammatory states.3 This interest began with the observation that chronic administration of interferon (IFN)-α – a proinflammatory cytokine used to treat hepatitis C and other malignancies – precipitated depressive symptoms that responded to standard antidepressant interventions.4 As first introduced by Dantzer and colleagues,5 the concept of ‘sickness-behavior’ has linked inflammation and depression through the hypothesis that constant activation of the peripheral immune system leads to circulating proinflammatory cytokines that increase immune signaling in the brain and subsequently worsen sickness behavior, predisposing a person to depression. Preclinical studies also found that peripheral immune system activation via systemic administration of lipopolysaccharide (LPS), an endotoxin, reliably triggered ‘depressive-like’ behaviors in rodents.6 Acute and chronic stressors that play an integral part in the etiology of depression7 also reliably trigger inflammatory responses.8
Indeed, emerging evidence from population-based studies supports the notion that chronic, low-grade inflammation – although not present in all individuals with depression – could nevertheless play a key part in the pathophysiology of depression for a subset of patients.9 Data from longitudinal studies suggest that dysregulation of the inflammatory response is associated with a more severe course of illness, higher recurrence of depressive symptoms and worse outcomes, including impaired brain connectivity within motivation and reward circuits,10 increased suicidality11 and, notably, greater resistance to conventional therapies.12 Reward circuits can also impact a hallmark symptom of TRD: anhedonia, which has also been consistently linked to inflammation.13 Other factors such as obesity and other conditions associated with chronic inflammation also appear to increase the development of inflammation-associated sickness and depressive symptoms, as well as their persistence.5
Subanesthetic doses of the glutamatergic modulator racemic ketamine, as well as its enantiomers, have consistently been shown to exert rapid-acting antidepressant effects in patients with TRD and treatment-resistant bipolar depression (reviewed in 14). Ketamine has also been found to successfully treat traditionally treatment-refractive symptom domains such as anhedonia, suicidality and amotivation.15,16 Within the context of this review, it is important to note that, although researchers primarily attribute ketamine’s therapeutic effects to upregulated neuroplasticity induced via glutamatergic modulation,17 growing evidence suggests that it might also regulate acute and chronic inflammatory reactions and restore immune homeostasis.18,19
This review of previous and emerging research discusses the links between depression, stress and inflammation, particularly inflammation as an potential indicator of TRD, and summarizes the preclinical and clinical evidence for ketamine’s anti-inflammatory and immunomodulatory properties in the context of its antidepressant effects. Potential mediators of the process – including the kynurenine pathway and the hypothalamic-pituitary-adrenal (HPA) axis – are also discussed, as is the hypothesis that ketamine’s unique ability to reduce depressive symptoms in TRD could in fact be caused by its ability to reduce stress-induced inflammation.
Chronic stress, depression and inflammation: an overview
Stress is an inherent physiologically or emotionally coordinated response that activates processes in the body to maintain homeostasis after threatening stimuli or, under acute stress conditions, helps anticipate challenges or response to dangerous situations. Chronic stress is loosely defined as a sustained threat lasting at least several weeks that is accompanied by a resulting negative emotional state and deleterious effects on body systems. Under chronic, prolonged stress conditions, the brain and body lose their ability to restore homeostasis. The link between inflammation and chronic stress probably results from an evolutionary adaptation.8 In prehistoric environments, this connection between the perception of danger and the risk of subsequent tissue injury or pathogen exposure was believed to be so reliable that evolution favored anticipatory inflammatory responses to many environmental stressors, including psychosocial stressors. In the context of the present review, chronic stress is known to be a major risk factor for depression.7
The relationship between chronic stress and depression holds true in preclinical models, where chronic stress protocols [e.g., social defeat, unpredictable mild stress and chronic corticosterone (CORT) administration] are the gold standard for producing depressive-like behaviors in animals, including symptom profiles such as learned helplessness and anhedonia.20 In animal models, the upregulation of stress hormones was found to robustly increase inflammatory markers such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β.21 Preclinical studies also found that chronic stress induces central nervous system (CNS) inflammation characterized by the secretion of cytokines and neuroinflammation.22 Interestingly, one study found that chronic social instability stress did not alter hippocampal proinflammatory cytokines; however, the study was conducted in females, suggesting potential gender differences in the links between chronic stress and inflammation.23
Multiple clinical studies have reported elevated levels of proinflammatory cytokines in individuals with depression. For instance, meta-analyses found that elevated levels of C-reactive protein (CRP) – a common marker of inflammation – predicted subsequent depressive symptoms24 and were strongly associated with a diagnosis of depression.25 Another recent meta-analysis of individuals with depression found that a quarter of participants had low-grade inflammation (CRP >3 mg/l), and half had elevated CRP levels (CRP >1 mg/l).26 Other meta-analyses reported cerebrospinal fluid (CSF) and peripheral elevations of other proinflammatory markers such as IL-6, IL-8 and TNF-α.27,28 Supporting the notion that higher levels of inflammation play a causative part in depression, one longitudinal study found that participants with elevated IL-6 and CRP levels at age nine were more likely to be evaluated as depressed at age 18.29 Nevertheless, many other studies have found no such association between increased levels of CRP and IL-6 (reviewed in 30), suggesting that any putative relationship between depression and inflammation remains unclear. Such differences could arise from disparities in the chronicity of MDD, given that some evidence suggests that the early phases of MDD might not present with heightened inflammation, which can predict poor response to the initial prescription of antidepressants.31,32 Baseline low-grade inflammation has been shown to mediate endothelial dysfunction, which in turn can predict persistent depressive symptoms and impact the chronicity of MDD.33 In a sample from The Netherlands, higher baseline inflammatory markers predicted a subsequent chronic course of illness in women, and depressive severity predicted subsequent higher levels of IL-6 in men and women.34 Thus, the presence of increased inflammatory markers could represent a distinct subgroup within the depressive diagnostic label, and the mixed results warrant further investigation.
Genetic differences and epigenetic changes are also major drivers of response to chronic stress. Genetically, studies have determined that ~40% of MDD is heritable.35 The past two decades of research have also determined that chronic stress can produce significant epigenetic changes that contribute to the pathophysiology of depression.36 As the major connection between genetics and the environment, epigenetic changes affect the availability of DNA to transcription factors through DNA methylation and histone modifications, including methylation, acetylation, phosphorylation and more.37 Major findings on inflammation-promoting epigenetic changes after chronic stress include histone deacetylase inhibitors, which have been shown to ameliorate depressive-like behavior and microglial activation.38–40 In addition, chronic corticosterone-induced expression of the gene Nfkb1 was shown to be accompanied by upregulation of TNF-α and IL-1β.41 Although an in-depth discussion of epigenetic mechanisms is beyond the scope of this review, these mechanisms were recently reviewed extensively (see 42).
It is also important to acknowledge the potential role of gender in the interplay between chronic stress, depression and inflammation. Rates of depression have consistently been found to be two-to-three-fold higher in females,43 and clinical research also suggests that women might be particularly vulnerable to the effects inflammation on depressive symptomatology.44 By contrast, some preclinical models found that males were more vulnerable to developing depressive-like behaviors after inflammatory insult.45 Throughout this review, gender differences will be discussed wherever possible.
The relationship between inflammation and TRD
As noted above, evidence suggests that individuals with MDD with heightened inflammatory markers can constitute a subpopulation uniquely associated with treatment-refractory symptoms.46 Notably, the ability to model TRD in animals is essential for future contributions to preclinical research exploring the underlying mechanisms of inflammation in treatment-resistance; but this field is still in its infancy.47 Proposed solutions include animal models that receive cyclic exposure to chronic stress and/or using animals that do not respond to conventional antidepressants, such as Wistar–Kyoto rats.48 Another potential proxy would be the measurement of behaviors known to be more present in TRD, such as anhedonia, anergia and motivation.
In clinical research, body mass index (BMI)-corrected serum CRP levels were recently found to be significantly elevated in TRD participants relative to treatment-responsive MDD participants, unmedicated MDD participants and healthy volunteers.49 These findings are complemented by two randomized, controlled trials that found that baseline CRP levels predicted lack of response to conventional antidepressants.50,51 Another study found distinct results in whole-blood samples, with a significant upregulation in mRNA-indicated inflammasome activation and glucocorticoid resistance in the MDD population (untreated versus treatment-responsive versus TRD). Of the mRNAs identified, six (P2RX7, IL-1β, IL-6, TNF-α, CXCL12 and GR) differentiated between TRD and treatment-responsive subgroups.52 By contrast, another study found no evidence of large inflammatory differences in the peripheral blood mononuclear cells (PBMCs) of healthy volunteers versus MDD patients (untreated versus treatment-responsive versus TRD) but did find strong evidence of increased biological aging in the MDD sample.53
One study of unmedicated MDD participants found that those who, on average, had failed to respond to three or more antidepressant trials had significantly higher levels of CRP, IL-6, TNF receptor 2 (sTNF-R2) and TNF-α than those who, on average, had failed to respond to less than one trial.46 A meta-analysis found that higher baseline levels of inflammatory markers in general were associated with poor treatment response, and that high TNF-α levels in particular were associated with TRD.54 An analysis of participants with MDD and bipolar depression who participated in a randomized, controlled trial of escitalopram versus nortriptyline found that cutoffs for absolute mRNA levels of IL-1β and macrophage migration inhibitory factor (MIF) in blood accurately predicted 100% of the non-responders in their study.55 Interestingly, a randomized, controlled trial of the anti-inflammatory agent infliximab found that its antidepressant effects were specific to a subset of TRD participants with elevated baseline plasma CRP levels >5mg/l56; because this impact was not consistent with results observed in individuals with bipolar I and II depression, it suggests a potential unique efficacy for TRD.57 Finally, adjunctive use of the anti-glucocorticoid therapeutic metyrapone actually increased IL-6 levels in individuals diagnosed with TRD, an increase associated with poorer outcomes to treatment; this finding was hypothesized to result from potential glucocorticoid system overcompensation.58
Imaging studies are also beginning to confirm that this peripheral inflammation is mirrored in the brain itself. Positron emission tomography (PET) studies of translocator protein 18 kDa (TSPO) – a biomarker of neuroinflammation – have typically reported greater TSPO binding in the prefrontal cortex (PFC) and anterior cingulate cortex (ACC) of individuals experiencing a major depressive episode.59 In an open-label trial of TRD participants who received the anti-inflammatory agent celecoxib, investigators plotted the reduction in Hamilton Depression Rating Scale (HAM-D) score against baseline TSPO volume in the PFC and ACC and found that HAM-D scores rapidly dropped post-treatment as baseline TSPO distribution volume decreased.60 A recent parallel study measured the impact of minocycline, a tetracycline antibiotic with anti-inflammatory properties, on TRD participants experiencing a major depressive episode; minocycline did not significantly impact TSPO binding,61 although another study found that it significantly decreased HAM-D scores in participants with elevated CRP levels (CRP ≥3mg/ml).62 Other studies investigating minocycline as an adjunctive treatment for TRD found no significant change in depressive symptoms.63 Finally, increases in immune factors after ex vivo LPS stimulation of PBMCs were associated with reduced reward anticipation in the ventral striatum, as measured via functional magnetic resonance imaging (fMRI).64 This builds on previous research that found that endotoxin administration to healthy volunteers significantly increased depressed mood over time and reduced ventral striatum responses to reward.65 This effect could also be gender-dependent, given that females demonstrated greater reductions in ventral striatum activity in response to reward.66 Inflammation can mediate motivational behavioral responses by dampening dopamine activity within reward circuits, resulting in disrupted frontostriatal functional connectivity.67 Inflammatory processes are therefore well-situated to influence the neural circuits underlying motivational symptoms related to anhedonia. This is particularly important because behavioral responses to reward and social stimuli in patients with anhedonia have been associated with suicidality68 and treatment resistance.69 Interestingly, depressive symptoms such as reduced motivation and anhedonia correlate significantly with central IL-6 soluble receptor (IL-6sr)70 as well as peripheral CRP levels.10,70 A resting-state fMRI study of depressed participants found that plasma CRP levels correlated with decreased connectivity between the ventral striatum and ventromedial PFC (vmPFC), and that this change in connectivity was itself correlated with the severity of anhedonia.10 Consistent with this finding, administration of IFN-α for 4–6 weeks in 14 individuals with hepatitis C not only induced anhedonia but also reduced bilateral activation of the ventral striatum in an fMRI reward task71; change in striatal activity again correlated with anhedonia scores.
Relatedly, reduced motivation has been correlated with central levels of TNF-α.70 Anhedonia, anergia and amotivation all fall under the symptom interest–activity dimension of depression; in the large Genome-Based Therapeutic Drugs for Depression (GENDEP) (n = 811) and Sequenced Treatment Alternatives to Relieve Depression (STAR*D) (n = 3637) studies, this dimension was shown to best predict poor antidepressant response.72
Glutamate can also modulate the interplay between inflammation and depression. Higher glutamate release from microglial cells appears to increase concentrations of extracellular glutamate, promote maladaptive glutamate metabolism, contribute to loss of synaptic fidelity and decrease the specificity of neurotransmission – all of which can worsen depressive-like behaviors and increase circuit dysfunction.73 Although most research in this area has focused on chronic stress, acute traumatic stress can similarly provoke glutamatergic signaling dysfunction.74 Administration of the proinflammatory cytokine IFN-α increased glutamate concentrations in the dorsal anterior cingulate cortex and basal ganglia.75,76 Notably, individuals with depression who also had high concentrations of plasma CRP and high levels of basal ganglia glutamate were significantly more likely to have more-severe symptom presentations of anhedonia and cognitive slowing.77 In analyses of postmortem tissue, glutamate was also found to be increased in the frontal cortex of those diagnosed with MDD or bipolar disorder.78 Increased mGluR2/3 expression was also found in the PFC of individuals with MDD, a finding paralleled in a Rhesus monkey model of depression.79 By contrast, mGluR5 expression was found to be decreased in the PFC of postmortem MDD tissue, and participants with MDD had significantly lower levels of mGluR5 binding in multiple regions, as ascertained through PET imaging.80 mRNA alterations related to glutamate signaling pathways have also been found in the locus coeruleus and hippocampus of individuals with MDD.81,82
Another important indicator of glutamatergic activity implicated in depression is the glutamate–glutamine cycle. Glutamate is synthesized from glutamine in neurons and, after release into the synapse, is taken up by sodium-dependent glutamate transporters (EAAT1 and EEAT2) located on astrocytes. This scavenged glutamate is then converted back to glutamine and transported back to neurons by neutral amino acid transporters (SNAT1 and SNAT2), continuing the cycle. In the CSF, glutamine levels have been found to be upregulated in individuals with MDD.83 In addition, increases in the ratio of glutamine to glutamate have been found in the CSF of individuals with MDD compared with healthy volunteers – a ratio that correlated with the severity of depression in a three-year follow-up.84 Changes in this cycle have also been implicated in suicide; specifically, differential changes in neuronal and astrocytic components of the cycle were observed in postmortem tissue obtained from healthy volunteers, individuals with MDD and those who died by suicide.85
Despite these intriguing findings, the role of glutamate in depression remains unclear. As an example, magnetic resonance spectroscopy (MRS)86 studies found decreased levels of glutamate, or no differences at all,87 in individuals with depression. Other studies found that the glutamatergic neurons of individuals with depression exhibited decreased mitochondrial energy production.88 One important caveat is that glutamate is often measured using Glx – a composite measure that includes glutamate and glutamine. At least one study that separated these measures found no significant differences in glutamate levels in participants with depression.89
Preliminary research is also investigating the response of inflammatory proteins to psychological therapy. In one study, poor response to treatment was associated with higher baseline levels of TNF-α, IL-6 and soluble intracellular adhesion molecule-1 and with higher post-therapeutic levels of CRP, thymus and activation-regulated chemokine, and macrophage chemoattractant protein-4.90 At least one review of the literature also reported a general reduction in inflammation after cognitive behavioral therapy for depression.91
Taken together, inflammatory markers seem to cause bona fide alterations in brain network activity that can, in turn, cause depressive symptoms. Thus, the evidence suggests that inflammation contributes to depressive pathology in at least some cases and that determining potential mediators of the stress response can inform the development of therapeutic interventions.
Potential mediators between depression and inflammation
HPA axis
HPA axis hyperactivity is one of the most consistent findings in studies exploring the underlying pathophysiology of depression. In healthy states, the HPA axis is activated by acute stress, stimulating the release of corticotropin-releasing hormone (CRH) and vasopressin (AVP) from the hypothalamus. This, in turn, stimulates the release of adrenocorticotrophic hormone (ACTH) and glucocorticoids (primarily cortisol in humans and corticosterone in rodents). After an acute stressor, these glucocorticoids interact with their widely expressed receptors (either mineralocorticoid or glucocorticoid receptors), some of which interact with the hypothalamus to form a negative feedback loop to shut off HPA axis activity. Chronic stress disrupts this feedback loop, causing a downregulation of glucocorticoid receptors that impairs the ability to shut off the HPA axis, leading to dysfunctional hyperactivity.92
Many individuals with depression exhibit HPA axis dysfunction, such as continuously elevated levels of cortisol and CRH.93 This hypersecretion can cause hypercortisolism and, as a result, decreased dopaminergic reward-system responsivity.94 In females, increased hair cortisol concentrations were associated with poor performance on measures of cognition and memory, an association that appeared to be mediated by CRP levels.95 Early-life adversity has also been shown to increase vulnerability to acute social stress, an effect mediated by HPA-axis and immune activation,96 and this was also found to impact later diurnal HPA axis functioning in adulthood.97
One of the most compelling theories regarding the clinical relevance of inflammation in depression is that inflammation can differentiate depressive subtypes and mediate specific symptoms. For example, a recent study found that biomarkers of HPA axis activity and subsequent inflammation (such as cortisol and CRP) were more strongly associated with the presence of somatic symptoms rather than cognitive-affective symptoms.98 For instance, a recent review found that cancer patients – who are significantly more likely to have depressive symptoms and a worsened symptom profile – can exhibit increased depressive-like behaviors owing to hyperactivity of the HPA axis caused by cancer and anticancer treatments.99 In a CORT-injected mouse model, the antidepressant-like effects of catalpol, an iridoid glucoside, also appeared to be mediated through the HPA axis, suppressing levels of CORT, ACTH and CRH.100
The kynurenine pathway
One hypothesis of inflammation-mediated depressive pathogenesis is that stress and inflammatory cytokines promote kynurenine pathway signaling.101 Tryptophan, a precursor for serotonin synthesis, is competitively consumed by the kynurenine pathway. One of the rate-limiting enzymes of this pathway: indoleamine-2,3-dioxygenase (IDO), is expressed mainly in immune and neuronal cells and induced by cytokines, cortisol and LPS, generally indicating a proinflammatory state.102 Tryptophan-2,3-dioxygenase (TDO), the other main enzyme in the kynurenine pathway that catalyzes tryptophan catabolism, is also induced under proinflammatory states.103 Thus, increased cytokine and cortisol levels can reduce serotonin levels via tryptophan depletion, a process that has been experimentally shown to induce depressive symptoms in vulnerable persons,104 although these findings have not always been consistent.105 Tryptophan–kynurenine metabolism can also provide a link between the gut–brain axis and inflammatory bowel disease and depression, two disorders that are strongly associated with one another.106 Acute and chronic stress also impact the rate-limiting enzymes involved in the tryptophan–kynurenine balance (Figure 1). For example, IFN-γ and IL-1β are potent inducers of IDO and TDO, which are highly impacted by immune activation in the brain. In addition, stress-induced corticosterone release and the consequent cascading activation of hepatic TDO to tryptophan metabolism ultimately lead to the production of kynurenine, which provokes a depression-related behavioral phenotype.107 IDO and TDO can therefore represent promising targets for the treatment of depression associated with stress-related disorders marked by kynurenine pathway activation (Figure 1).
Figure 1.
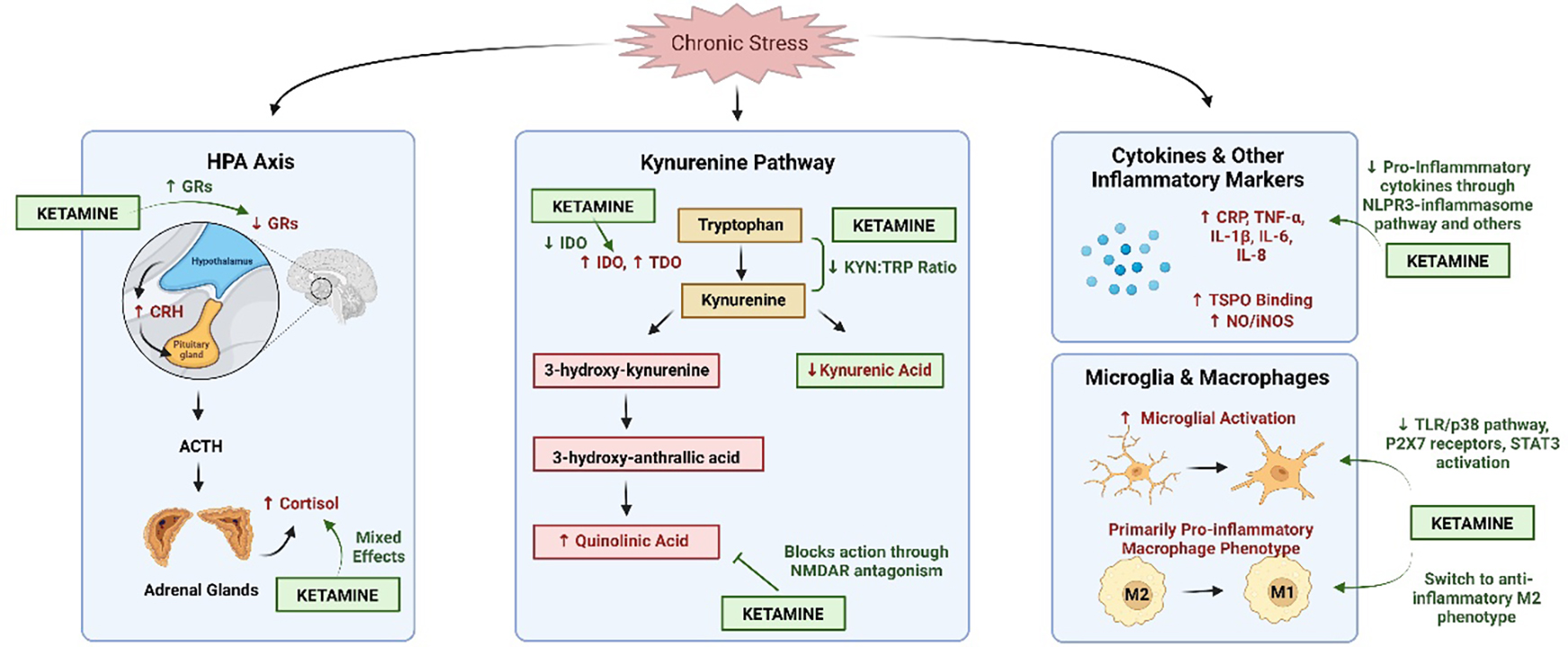
The hypothesized impact of ketamine on stress and inflammatory pathways. Chronic stress leads to overactivation of the hypothalamic-pituitary-adrenal (HPA) axis, which increases levels of corticotropin-releasing hormone (CRH) and cortisol while decreasing expression of glucocorticoid receptors (GRs). This decrease in GR expression prevents the shut-off of the HPA axis, leading to prolonged activation that can have negative consequences. Ketamine, an N-methyl-d-aspartate receptor (NMDAR) antagonist, appears to mediate this stress response by increasing the number of GRs. Studies examining ketamine’s effect on cortisol levels have yielded mixed results. Under chronic stress conditions, the kynurenine pathway, another potential mediator between stress and inflammation, demonstrates increased levels of indoleamine-2,3-dioxygenase (IDO), tryptophan-2,3-dioxygenase (TDO) and quinolinic acid, as well as decreased levels of kynurenic acid. Ketamine decreases IDO levels and the ratio of kynurenine:tryptophan through indirect mechanisms while blocking the action of quinolinic acid through direct NMDAR antagonism. Ketamine also decreases proinflammatory cytokine levels (increased by chronic stress) through the NLPR3-inflammasome pathway, decreasing microglial activation via TLR/p38 signaling, P2X7 receptors and signal transducer and activator of transcription 3 (STAT3) activation, as well as switching macrophages to the anti-inflammatory M2 phenotype. Figure created using Biorender.
Kynurenine pathway products are biologically active. Kynurenic acid (KA) is considered to be neuroprotective108 and a potential therapeutic target for drug development in mood disorders. Another product, quinolinic acid (QA), is an endogenous neurotoxin that generates free radicals109 and causes excitotoxicity by inducing the release and inhibiting the reuptake of glutamate.110 One major component of the kynurenine pathway is its ability to affect the glutamatergic system, where it directly and indirectly influences ionotropic and metabotropic glutamate receptors and vesicular glutamate transport.111 These effects are hypothesized to act as a main link between chronic stress, depression and inflammation.112 For instance, QA directly activates N-methyl-d-aspartate receptors (NMDARs), increases synaptosomal glutamate release and inhibits glutamate uptake, making it uniquely placed to mediate interactions between ketamine and inflammation.113 KA and QA are metabolized from kynurenine by astrocytes and microglia, respectively, and evidence suggests that individuals with MDD have reduced astrocyte density114 and function115 along with increased microglial activation and number.116
Supporting the clinical relevance of this pathway, studies have reported higher ratios of kynurenine to tryptophan levels,117,118 lower levels of KA119 and lower KA:QA ratios120 at baseline in MDD participants. In addition, QA elevations were found in the CSF of recent suicide attempters,121 and more QA-positive cells were found in the brain of suicide decedents.122 Finally, altered peripheral ratios of KA:QA levels were shown to correlate with increased anhedonia in MDD participants123 as well as with depression and fatigue in cancer patients.124
Inflammation-mediated tryptophan metabolism in the gut microbiome has also been implicated in depression.125 Intestinal inflammation, particularly via increased IFN-γ, was able to induce IDO, shifting tryptophan metabolism toward the production of kynurenine rather than serotonin.126,127 Kynurenine is able to cross the blood–brain barrier,128 suggesting that alterations from the gut microbiome could enter the blood circulation and impact levels of kynurenine and kynurenic metabolites in the brain. Fecal microbiota transplantation from participants with depression into a rat model induced depressive-like behaviors that were associated with increased levels of inflammatory markers (IL-6, TNF-α, CRP) as well as an increased kynurenine:tryptophan ratio.129 This finding was paralleled in another fecal microbiota transplant from chronically stressed mice to control mice that increased IDO1 expression and proinflammatory cytokine levels.130 For a recent summary of studies exploring tryptophan metabolism, gut microbiota and depression, we refer the interested reader to a recent summary by Lukic and colleagues.131 Taken together, this evidence suggests that the kynurenine pathway could play a key part in mediating the links between inflammation and depression.
Ketamine and inflammation
The NMDAR antagonist ketamine is uniquely effective for treating TRD, with a response rate ranging from 25% to 85% at 24 h post-infusion.132 It has also been shown to effectively reduce suicidal ideation and anhedonia,15,16 as well as fatigue and amotivation symptoms.133,134 Recent clinical and preclinical evidence indicates that, at antidepressant doses, ketamine can exert these unique therapeutic effects in part by modulating inflammation.19,117 It is important to note that most of the studies described below reflect acute, not chronic, ketamine administration, which could affect the interpretation of results.
Although considerable volumes of research, and clinical studies in particular, have focused on the (S)-ketamine enantiomer, significant preclinical work has begun to explore the mechanisms behind ketamine’s (R)-enantiomer and various ketamine metabolites. Briefly, (R)-ketamine binds with around fourfold less affinity or potency to inhibit NMDARs than the (S)-enantiomer and has shown promise in terms of its antidepressant effects and reduced adverse side effect profile in animal models for depression135 and in early clinical trials.136 In addition, a recent open-label study found that (R)-ketamine had rapid and sustained antidepressant effects,137 and further clinical trials are currently underway by multiple companies. Two major ketamine metabolites [norketamine and hydroxynorketamine (HNK)], which have different binding capacities to NMDARs,138 have also recently been studied in animal models of depression and early-phase clinical trials. Thus far, these metabolites appear to have potential antidepressant-like effects when administered directly.139–141 In addition, varying levels of ketamine metabolites in plasma and CSF have been correlated with clinical response to ketamine.142 Although an in-depth discussion of the hypothesized distinct mechanisms underlying the effects of ketamine, its enantiomers and its metabolites is beyond the scope of this review, these have all recently been reviewed extensively.143 In particular, differences between enantiomers and metabolites should be considered when defining the role that ketamine has in inflammatory processes.
Preclinical evidence of ketamine’s anti-inflammatory effects
Substantial preclinical evidence suggests that ketamine reduces inflammation by regulating the immune system. In vitro ketamine application to rodent glial cells144 and macrophages145 attenuated markers and mediators of LPS-induced inflammatory responses, such as TNF-α, IL-1β, high mobility group box 1 (HMGB1), nitric oxide (NO), inducible nitric oxide synthase (iNOS) and prostaglandin E-2.
In Wistar–Kyoto rats, a proposed model of TRD,146 ketamine showed rapid-acting antidepressant effects by inhibiting the NLR family pyrin domain containing 3 (NLRP3) inflammasome pathway, an effect blocked by application of an autophagy inhibitor.147 In addition, low-dose ketamine effectively mediated the gut microbiome bacterial population associated with inflammation in Wistar–Kyoto rats.148
In other animal models, administration of intraperitoneal ketamine had prophylactic effects against LPS- and chronic-stress-induced depressive behaviors,149–151 an effect that appears unique to ketamine versus other NMDAR antagonists.152 For example, ketamine administered one week before stressor application most effectively reduced freezing behavior in a contextual fear conditioning task153,154 and improved performance on the tail suspension and splash tests.155
Ketamine’s prophylactic effects appear to be, at least in part, mediated via inflammatory signaling, such as the NLRP3 inflammasome pathway after LPS or TNF-α adminstration.156 Cyclooxygenase-2 (COX-2) expression was also shown to be reduced in a gender-specific manner after ketamine administration.153 (R)-Ketamine has also been shown to have prophylactic effects in mice exposed to chronic stress, improving behavioral and biological outcomes, such as altered gene expression of Bdnf and Mecp2 mediated through miR-132-5p activity.157 Interestingly, this prophylactic effect did not generalize to its metabolites [(2R,6R)-HNK and (R)-norketamine]; however, the prophylactic effects of different metabolites could be gender-specific, because ovarian hormones are required for the protective effects of ketamine and (2R,6R)-HNK in female mice.158 These prophylactic effects of (R)-ketamine in chronic stress and inflammation-based models appear to be mediated through microRNA-149 and nuclear factor of activated T cells 4 (NFATc4), which play a part in mediating cytokine expression.159,160 Together, these results emphasize the importance of enantiomer-specific research when discussing the impact of ketamine on inflammatory signaling.
Concurrent with the aforementioned behavioral changes, ketamine also attenuated plasma cytokine elevations161 and cytokine expression in rodent tissue samples from the PFC, hippocampus, cerebellum and spinal cord.151,162 Differences in ketamine’s prophylactic efficacy can arise from different experimental paradigms, such as timing of ketamine administration, which should be considered carefully when designing future experiments. For instance, most, but not all,150 studies focusing on ketamine’s prophylactic effects have administered this agent one week before the stressor to ascertain more long-term effects rather than immediate effects. Age and gender should also be considered in future clinical applications, given that some preclinical research found greater prophylactic efficacy in adolescents153 as well as differences in female response.158
Interestingly, ketamine appears to act directly on immune cells. For instance, S-ketamine – the S-enantiomer of racemic ketamine – decreased microglial activity levels in the CNS after chronic stress exposure.151 An immunohistochemistry study performed on rodent hippocampus samples found that ketamine reversed stress-induced activation of microglia caused by chronic restraint by downregulating Toll-like receptor (TLR)/p38 pathway activation and P2X7 receptors.162 In chronically stressed mice, pharmacological inhibition of TGF-β1 signaling in microglia eliminated (R)-ketamine’s antidepressant effects.163 In addition, ketamine and its antidepressant metabolites altered the localization of signal transducer and activation of transcription 3 (STAT3) in human microglial cells to regulate the ‘response to interferon I’ inflammatory pathway.164 Lastly, a recent study found that the antidepressant-like effects of (R)-ketamine were blocked by microglial depletion in chronic-stress-sensitive mice. (R)-Ketamine was also able to induce brain-derived neurotrophic factor (BDNF) transcription by inhibiting MeCP2 and increasing the expression of nuclear-receptor-binding protein 1 in microglial cultures.165
More recently, acute intraperitoneal ketamine administration in mice was shown to skew the distribution of macrophage populations away from proinflammatory and cytokine-inducing M1 phenotypes toward tissue-supporting M2 phenotypes.166 This finding was also observed in vitro in human monocyte cultures, where the effect could be abolished by inhibiting mammalian target of rapamycin (mTOR), a key protein implicated in ketamine’s antidepressant effects.167 PBMC samples collected from healthy volunteers and participants with MDD after a suicide attempt or with active suicidal ideation found that macrophages in MDD participants also skewed toward the inflammatory M1 phenotype.166
Some of ketamine’s anti-inflammatory effects can also be mediated via apoptosis of various cell types. In neuronal cell types, anesthetic concentrations of (S)-ketamine attenuated expression of Bax, a proapoptotic protein, after cerebral ischemia.168 By contrast, lower concentrations of ketamine have been shown to promote apoptosis of T lymphocytes via mitochondrial signaling in various cell lines, thus potentially inhibiting downstream cytokine production.169 Accumulation of Th17 cells (a proinflammatory T cell subtype) and an imbalance of Th17:Treg cells have also been associated with depression,170 and ketamine was able to suppress differentiation of this cell subtype, although this process was not mediated by apoptosis.171
Ketamine can also indirectly affect inflammation by mediating HPA axis function. In chronically stressed mice, acute ketamine administration restored hippocampal glucocorticoid receptor expression, counteracting the negative feedback associated with HPA overactivation.172 In mice injected with LPS, ketamine significantly reduced corticosterone and ACTH production six hours later.173 Similarly, single and repeated 7-day ketamine administration reduced corticosterone and ACTH levels in mice that had undergone 40 days of chronic mild stress.174
Ketamine and kynurenine appear to converge during stress conditions to affect brain and behavior. One study found that, although ketamine did not affect QA production after LPS administration, it mediated the effects of QA by blocking NMDARs, where QA generally binds to contribute to inflammation.113,150 In a chronic unpredictable mild stress model, ketamine decreased the KYN:tryptophan ratio in addition to other measures of inflammation.175 To more closely mimic TRD, future studies with preclinical models should assess the impact of ketamine on inflammation after multiple cycles of chronic stress.
Clinical evidence of ketamine’s anti-inflammatory effects
Multiple inflammatory markers have been linked to ketamine’s clinical therapeutic efficacy. In a recent randomized, controlled trial, subanesthetic-dose ketamine (0.5 mg/kg) acutely decreased TNF-α levels in TRD patients, and these decreases correlated with reductions in Montgomery–Åsberg Depression Rating Scale (MADRS) scores.18 A smaller study of individuals with TRD similarly found that higher baseline levels of IL-6 were associated with antidepressant response to ketamine.176 In an open-label trial, ketamine robustly reduced peripheral levels of multiple cytokines elevated at baseline in TRD participants but these levels returned to baseline within 24 h and did not correlate with antidepressant response.177 In addition, a recent study in remitted depressed participants found significant decreases and time x treatment interactions for multiple cytokines.178 However, other studies obtained mixed results. For example, a post hoc analysis of three ketamine randomized, controlled trials of participants with TRD and treatment-resistant bipolar depression found that ketamine decreased levels of soluble tumor necrosis factor receptor 1 (sTNFR1) but increased peripheral levels of IL-6 and TNF-α.179 Interestingly, a recent open-label ketamine trial found that IL-8 did not predict antidepressant response to ketamine but that there was a trend toward prediction in females, suggesting a potential gender-specific effect.180 Rapamycin, an mTORC1 inhibitor, was also found to prolong ketamine’s antidepressant effects, which could be at least partly due to its immunosuppressive actions.181
The effects of ketamine on the HPA system are less clear. One case study found that cortisol levels – as measured by the dexamethasone suppression test – normalized in a TRD participant who received three standard ketamine infusions; cortisol levels rose to baseline a week later as depressive symptoms returned.182 By contrast, a randomized, controlled trial of 12 healthy volunteers who received two back-to-back ketamine infusions (0.29 mg/kg for 1 h, then 0.57 mg/kg for 1 h) reported doubled plasma cortisol levels 200 min later.183 Furthermore, another randomized, placebo-controlled trial of healthy volunteers found that the post-ketamine increase in cortisol was specific to ketamine, because the NMDA antagonist memantine caused no such effect.184 For now, the dearth of properly-powered studies examining potential HPA biomarkers post-ketamine treatment in TRD participants makes it difficult to draw firm conclusions.
Echoing preclinical findings, modulation of the kynurenine pathway might be involved in ketamine’s anti-inflammatory effects. A randomized, controlled trial of TRD participants found that those who responded to ketamine had significantly lower plasma kynurenine:tryptophan ratios as well as lower kynurenine levels 230 min and 24 h post-ketamine administration.117 Furthermore, among participants with TRD and treatment-resistant bipolar depression who received six ketamine infusions over 12 days, those who responded had higher levels of serum KA, both absolute and relative to kynurenine, on Days 1 and 13.118 Moreover, at 24 h, both of these metrics correlated with MADRS score reductions at Days 1, 13 and 26. Finally, a recent randomized, controlled trial of individuals with bipolar depression reported that one ketamine infusion increased KA levels one and three days later and decreased IDO levels from 230 min post-infusion to three days later.185 Despite these promising findings, it should be noted that another study found only trend-level decreases in serum kynurenine after repeated ketamine infusions and no change in cortisol-awakening response.119
There is also indirect evidence of ketamine’s anti-inflammatory effects. One post hoc analysis of four randomized, controlled trials (n = 108) found that greater BMI predicted antidepressant response to ketamine in individuals with MDD or bipolar depression,186 which could be linked to the finding that proinflammatory agents are often deposited in adipose tissue.187 Subsequently, researchers examined adipokine levels and found that ketamine reduced plasma levels of resistin, and that low baseline levels of adiponectin predicted antidepressant response.188 These findings are congruent with anti-inflammatory effects; resistin is a potent proinflammatory agent189 associated with obesity, whereas adiponectin is an anti-inflammatory molecule.190 Another study of medication-free TRD participants found that ketamine decreased the expression of receptor activator of nuclear factor kappa-B ligand (RANKL), a downstream inflammatory mediator.191 In TRD participants, gene expression signatures related to IFN signaling pathway activation were upregulated in comparison to healthy volunteers; but this did not mediate a response to ketamine.192
Despite these promising findings, it is clear that more research is necessary to clarify ketamine’s effects on inflammation in general and on clinical depression subtypes linked to inflammation in particular. The mixed results suggest that future studies should compare acute versus chronic ketamine administration as well as the short- and long-term effects of ketamine, given that some of the aforementioned studies observed an immediate increase in inflammatory indicators post-ketamine administration that decreased with time. It is also important to note that some clinical studies used ketamine adjunctively with current antidepressant therapies, and that administration of concomitant medications could directly impact results on inflammation in comparison to ketamine administered alone. Nevertheless, promising preclinical evidence and strong associations between TRD and inflammation warrant further investigation into the mechanisms by which ketamine can either directly or indirectly mediate an inflammatory response.
Concluding remarks
In this era of personalized medicine, the quest to identify subpopulations of individuals with MDD based on pathophysiology, symptom dimensions and prognostic biomarkers of treatment efficacy holds considerable promise for improving the thus far inadequate therapeutic response associated with many currently available pharmacotherapies. This review presents evidence that chronic-stress-induced, systemic, proinflammatory states can constitute a pathogenic factor that can negatively impact treatment-responsiveness in depression. Meanwhile, preliminary but growing evidence suggests that ketamine’s unique efficacy in treating these same treatment-refractory symptoms could partly be the result of its anti-inflammatory effects, perhaps by directly counteracting the inflammatory consequences of chronic stress; these unique effects are not associated with conventional antidepressants. These effects can occur via some combination of cytokine suppression, alteration of the kynurenine pathway or HPA axis, direct actions on microglia and other monocytes, and additional mechanisms not discussed here. In addition, research that differentiates between the impact of ketamine’s enantiomers [(R)- and (S)-ketamine) and its metabolites [e.g., (2R,6R)-HNK and norketamine] is essential for properly outlining ketamine’s anti-inflammatory effects. Although promising preclinical and early-phase clinical research has been conducted with (R)-ketamine and (2R,6R)-HNK, further clinical studies are needed to verify these initial results.
In this context, the need to verify ketamine’s anti-inflammatory properties with rigorous, prospective, clinical research is clear, as is the need to use preclinical models to elucidate the molecular and cellular basis underlying these effects. The effects of gender must also be considered, given the mixed results regarding gender differences in inflammation. This is particularly important because few preclinical or clinical studies have explored the links between TRD, ketamine and inflammation in a female population. Even less attention has been paid to the effects of race on the links between chronic stress, inflammation and therapeutic response, despite initial correlational studies showing different relationships between inflammation and depression by race.193–195 In addition, the generalizability of ketamine’s therapeutic efficacy has not been determined for multiple ethnic groups. Further research should prioritize determining the effects of race and ethnicity on inflammation-related outcomes and on ketamine’s therapeutic effects. Nevertheless, future research efforts in this area are likely to be complicated by several challenges. First, depressive symptoms that can derive from inflammation and respond to ketamine are neither universal nor specific to any one diagnostic category. Thus, advances in psychiatric nosology are probably needed to replicate research with greater inter-study validity. For example, one crucial issue is a fuller differentiation between unipolar and bipolar depression, because ketamine has been successfully used to treat both these neuropsychiatric disorders despite the fact that the underlying inflammation-mediated mechanisms are probably different. Second, immune system dysregulation has a multitude of other consequences that span multiple systems and that could be further confounded by other factors such as gender and BMI. A more complete understanding of these complex interactions, combined with improved identification of the heterogeneous etiologies of depressive symptoms, are needed to move this field forward.
Regardless, further systematic research into the connections between inflammation, treatment-resistant symptom severity and response to ketamine is warranted. Ideally, such investigations should measure central levels of inflammatory markers and products of related pathways such as the HPA and kynurenine pathways and correlate these with suicidal ideation, anhedonia and other hallmark symptoms of TRD.
Highlights:
Chronic stress, depression and inflammation have been consistently linked
Stress-induced inflammatory states can contribute to treatment resistance
Ketamine could be uniquely placed to target inflammation
Ketamine’s effects can be mediated through the HPA axis or kynurenine pathway
Anti-inflammatory antidepressant therapies could improve treatment resistance
Acknowledgments
The authors thank the 7SE research unit and staff for their support.
Funding
Funding for this work was provided by the Intramural Research Program at the National Institute of Mental Health, National Institutes of Health (IRP-NIMH-NIH; ZIAMH002857). Additional funding was provided by NARSAD Young Investigator Award to Dr Kadriu. The NIMH and NARSAD had no further role in study design; in the collection, analysis or interpretation of data; in the writing of the report; or in the decision to submit the paper for publication. The work was completed as part of the authors’ official duties as Government employees. The views expressed do not necessarily reflect the views of the NIH, the Department of Health and Human Services or the US Government.
Biographies

Jenessa Johnston
Jenessa Johnston is a PhD student at the University of Victoria and a Pre-Doctoral Fellow at the National Institute of Mental Health (NIMH). Her research focuses on the underlying molecular mechanisms of novel antidepressants as well as the discovery of diagnostic and therapeutic biomarkers for various neuropsychiatric disorders.

Max Greenwald
Max Greenwald is an MD/PhD student specializing in neuroscience and psychiatry at the Yale School of Medicine. His research focuses on psychoactive drugs and their therapeutic pathophysiological mechanisms in psychological disorders

Bashkim Kadriu
Bashkim Kadriu, MD, is a board-certified psychiatrist. He is currently Executive Director of Translational and Experimental Medicine, Neuroscience, Jazz Pharmaceuticals. He previously worked as a Clinical Leader at Janssen Pharmaceuticals and as a neuroscientist at the Experimental Therapeutics and Pathophysiology Branch (ETPB), NIMH. His research interests include early drug development as well as the neurobiological correlates of mood disorders, with a particular emphasis on discovering biosignatures that guide novel therapies and their mechanisms of action.
Footnotes
Conflicts of interest
Dr Zarate is listed as a co-inventor on a patent for the use of ketamine in major depression and suicidal ideation; as a co-inventor on a patent for the use of (2R,6R)-hydroxynorketamine, (S)-dehydronorketamine and other stereoisomeric dehydroxylated and hydroxylated metabolites of (R,S)-ketamine metabolites in the treatment of depression and neuropathic pain; and as a co-inventor on a patent application for the use of (2R,6R)-hydroxynorketamine and (2S,6S)-hydroxynorketamine in the treatment of depression, anxiety, anhedonia, suicidal ideation and post-traumatic stress disorders. He has assigned his patent rights to the US Government but will share a percentage of any royalties that might be received by the government. All other authors have no conflicts of interest to disclose, financial or otherwise.
Teaser: This review investigates the role of inflammation in chronic stress and depression, as well as how the rapid-acting antidepressant ketamine can exert some of its therapeutic effects via anti-inflammatory mechanisms.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Friedrich MJ. Depression is the leading cause of disability around the world. JAMA. 2017;317(15):1517-. [DOI] [PubMed] [Google Scholar]
- 2.Gaynes BN, Warden D, Trivedi MH, Wisniewski SR, Fava M, Rush AJ. What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Psychiatr Serv. 2009;60(11):1439–45. [DOI] [PubMed] [Google Scholar]
- 3.Zunszain PA, Hepgul N, Pariante CM. Inflammation and depression. Curr Top Behav Neurosci. 2013;14:135–51. [DOI] [PubMed] [Google Scholar]
- 4.Renault PF, Hoofnagle JH, Park Y, Mullen KD, Peters M, Jones DB, et al. Psychiatric complications of long-term interferon alfa therapy. Arch Intern Med. 1987;147(9):1577–80. [PubMed] [Google Scholar]
- 5.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yirmiya R Endotoxin produces a depressive-like episode in rats. Brain Res. 1996;711(1):163–74. [DOI] [PubMed] [Google Scholar]
- 7.Hammen C Stress and depression. Annu Rev Clin Psychol. 2004;1(1):293–319. [DOI] [PubMed] [Google Scholar]
- 8.Slavich GM, Irwin MR. From stress to inflammation and major depressive disorder: a social signal transduction theory of depression. Psychol Bull. 2014;140(3):774–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leonard BE. Inflammation and depression: a causal or coincidental link to the pathophysiology? Acta Neuropsychiatr. 2018;30:1–16. [DOI] [PubMed] [Google Scholar]
- 10.Felger JC, Li Z, Haroon E, Woolwine BJ, Jung MY, Hu X, et al. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry. 2016;21(10):1358–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Enache D, Pariante CM, Mondelli V. Markers of central inflammation in major depressive disorder: A systematic review and meta-analysis of studies examining cerebrospinal fluid, positron emission tomography and post-mortem brain tissue. Brain Behav Immun. 2019;81:24–40. [DOI] [PubMed] [Google Scholar]
- 12.Arteaga-Henríquez G, Simon MS, Burger B, Weidinger E, Wijkhuijs A, Arolt V, et al. Low-grade inflammation as a predictor of antidepressant and anti-inflammatory therapy response in MDD patients: a systematic review of the literature in combination with an analysis of experimental data collected in the EU-MOODINFLAME consortium. Front Psychiatry. 2019;10:458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly CA, Freeman KB, Schumacher JA. Treatment-resistant depression with anhedonia: Integrating clinical and preclinical approaches to investigate distinct phenotypes. Neurosci Biobehav Rev. 2022;136:104578. [DOI] [PubMed] [Google Scholar]
- 14.Kraus C, Rabl U, Vanicek T, Carlberg L, Popovic A, Spies M, et al. Administration of ketamine for unipolar and bipolar depression. Int J Psychiatry Clin Pract. 2017;21:2–12. [DOI] [PubMed] [Google Scholar]
- 15.Lally N, Nugent AC, Luckenbaugh DA, Ameli R, Roiser JP, Zarate CA. Anti-anhedonic effect of ketamine and its neural correlates in treatment-resistant bipolar depression. Transl Psychiatry. 2014;4:e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilkinson ST, Ballard ED, Bloch MH, Mathew SJ, Murrough JW, Feder A, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2018;175:150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kadriu B, Musazzi L, Henter ID, Graves M, Popoli M, Zarate CA Jr.. Glutamatergic neurotransmission: pathway to developing novel rapid-acting antidepressant treatments. Int J Neuropsychopharmacol. 2019;22(2):119–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen M-H, Li C-T, Lin W-C, Hong C-J, Tu P-C, Bai Y-M, et al. Rapid inflammation modulation and antidepressant efficacy of a low-dose ketamine infusion in treatment-resistant depression: a randomized, double-blind control study. Psychiatry Res. 2018;269:207–11. [DOI] [PubMed] [Google Scholar]
- 19.Zhan Y, Zhou Y, Zheng W, Liu W, Wang C, Lan X, et al. Alterations of multiple peripheral inflammatory cytokine levels after repeated ketamine infusions in major depressive disorder. Transl Psychiatry. 2020;10:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q, Timberlake MA, Prall K, Dwivedi Y. The recent progress in animal models of depression. Prog Neuropsychopharmacol Biol Psych. 2017;77:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kubera M, Obuchowicz E, Goehler L, Brzeszcz J, Maes M. In animal models, psychosocial stress-induced (neuro)inflammation, apoptosis and reduced neurogenesis are associated to the onset of depression. Prog Neuro-Psychopharmacol Biol Psychiatry. 2011;35:744–59. [DOI] [PubMed] [Google Scholar]
- 22.Reader BF, Jarrett BL, McKim DB, Wohleb ES, Godbout JP, Sheridan JF. Peripheral and central effects of repeated social defeat stress: monocyte trafficking, microglial activation, and anxiety. Neuroscience. 2015;289:429–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Labaka A, Gómez-Lázaro E, Vegas O, Pérez-Tejada J, Arregi A, Garmendia L. Reduced hippocampal IL-10 expression, altered monoaminergic activity and anxiety and depressive-like behavior in female mice subjected to chronic social instability stress. Behav Brain Res. 2017;335:8–18. [DOI] [PubMed] [Google Scholar]
- 24.Valkanova V, Ebmeier KP, Allan CL. CRP, IL-6 and depression: A systematic review and meta-analysis of longitudinal studies. J Affect Disord. 2013;150(3):736–44. [DOI] [PubMed] [Google Scholar]
- 25.Horn SR, Long MM, Nelson BW, Allen NB, Fisher PA, Byrne ML. Replication and reproducibility issues in the relationship between C-reactive protein and depression: A systematic review and focused meta-analysis. Brain Behav Immun. 2018;73:85–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osimo EF, Baxter LJ, Lewis G, Jones PB, Khandaker GM. Prevalence of low-grade inflammation in depression: a systematic review and meta-analysis of CRP levels. Psychol Med. 2019;49:1958–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haapakoski R, Mathieu J, Ebmeier KP, Alenius H, Kivimäki M. Cumulative meta-analysis of interleukins 6 and 1β, tumour necrosis factor α and C-reactive protein in patients with major depressive disorder. Brain Behav Immun. 2015;49:206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Köhler CA, Freitas TH, Maes M, Andrade NQd, Liu CS, Fernandes BS, et al. Peripheral cytokine and chemokine alterations in depression: a meta-analysis of 82 studies. Acta Psychiatr Scand. 2017;135(5):373–87. [DOI] [PubMed] [Google Scholar]
- 29.Khandaker GM, Pearson RM, Zammit S, Lewis G, Jones PB. Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: a population-based longitudinal study. JAMA Psychiatry. 2014;71(10):1121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mac Giollabhui N, Ng TH, Ellman LM, Alloy LB. The longitudinal associations of inflammatory biomarkers and depression revisited: systematic review, meta-analysis, and meta-regression. Mol Psychiatry. 2021;26:3302–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vogelzangs N, Beekman AT, van Reedt Dortland AK, Schoevers RA, Giltay EJ, De Jonge P, et al. Inflammatory and metabolic dysregulation and the 2-year course of depressive disorders in antidepressant users. Neuropsychopharmacology. 2014;39:1624–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson G, Berk M, Dean O, Moylan S, Maes M. Role of immune-inflammatory and oxidative and nitrosative stress pathways in the etiology of depression: therapeutic implications. CNS Drugs. 2014;28:1–10. [DOI] [PubMed] [Google Scholar]
- 33.Janssen EPCJ, Köhler S, Geraets AFJ, Stehouwer CDA, Schaper NC, Sep SJS, et al. Low-grade inflammation and endothelial dysfunction predict four-year risk and course of depressive symptoms: The Maastricht study. Brain Behav Immun. 2021;97:61–7. [DOI] [PubMed] [Google Scholar]
- 34.Lamers F, Milaneschi Y, Smit JH, Schoevers RA, Wittenberg G, Penninx BW. Longitudinal association between depression and inflammatory markers: results from the Netherlands study of depression and anxiety. Biol Psychiatry. 2019;85:829–37. [DOI] [PubMed] [Google Scholar]
- 35.Geschwind DH, Flint J. Genetics and genomics of psychiatric disease. Science. 2015;349:1489–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scarpa JR, Fatma M, Loh Y-HE, Traore SR, Stefan T, Chen TH, et al. Shared transcriptional signatures in major depressive disorder and mouse chronic stress models. Biol Psychiatry. 2020;88:159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500. [DOI] [PubMed] [Google Scholar]
- 38.Yamawaki Y, Yoshioka N, Nozaki K, Ito H, Oda K, Harada K, et al. Sodium butyrate abolishes lipopolysaccharide-induced depression-like behaviors and hippocampal microglial activation in mice. Brain Res. 2018;1680:13–38. [DOI] [PubMed] [Google Scholar]
- 39.Faraco G, Pittelli M, Cavone L, Fossati S, Porcu M, Mascagni P, et al. Histone deacetylase (HDAC) inhibitors reduce the glial inflammatory response in vitro and in vivo. Neurobiol Dis. 2009;36:269–79. [DOI] [PubMed] [Google Scholar]
- 40.Kannan V, Brouwer N, Hanisch U-W, Regen T, Eggen BJL, Boddeke HWGM. Histone deacetylase inhibitors suppress immune activation in primary mouse microglia. J Neurosci Res. 2013;91:1133–42. [DOI] [PubMed] [Google Scholar]
- 41.Kv A, Madhana RM, Js IC, Lahkar M, Sinha S, Naidu VGM. Antidepressant activity of vorinostat is associated with amelioration of oxidative stress and inflammation in a corticosterone-induced chronic stress model in mice. Behav Brain Res. 2018;344:73–84. [DOI] [PubMed] [Google Scholar]
- 42.de Carvalho LM, Chen W-Y, Lasek AW. Epigenetic mechanisms underlying stress-induced depression. Int Rev Neurobiol. 2021;156:87–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eid RS, Gobinath AR, Galea LA. Sex differences in depression: insights from clinical and preclinical studies. Prog Neurobiol. 2019;176:86–102. [DOI] [PubMed] [Google Scholar]
- 44.Lasselin J, Lekander M, Axelsson J, Karshikoff B. Sex differences in how inflammation affects behavior: what we can learn from experimental inflammatory models in humans. Front Neuroendocrinol. 2018;50:91–106. [DOI] [PubMed] [Google Scholar]
- 45.Ma L, Xu Y, Wang G, Li R. What do we know about sex differences in depression: a review of animal models and potential mechanisms. Prog Neuro-Psychopharmacol Biol Psychiatry. 2019;89:48–56. [DOI] [PubMed] [Google Scholar]
- 46.Haroon E, Daguanno AW, Woolwine BJ, Goldsmith DR, Baer WM, Wommack EC, et al. Antidepressant treatment resistance is associated with increased inflammatory markers in patients with major depressive disorder. Psychoneuroendocrinology. 2018;95:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Papp M, Cubala WJ, Swiecicki L, Newman-Tancredi A, Willner P. Perspectives for therapy of treatment-resistant depression. Br J Pharmacol. 2022;179:4181–200. [DOI] [PubMed] [Google Scholar]
- 48.Planchez B, Surget A, Belzung C. Animal models of major depression: drawbacks and challenges. J Neural Transm (Vienna). 2019;126:1383–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chamberlain SR, Cavanagh J, de Boer P, Mondelli V, Jones DNC, Drevets WC, et al. Treatment-resistant depression and peripheral C-reactive protein. Br J Psychiatry. 2019;214(1):11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jha MK, Minhajuddin A, Gadad B, Greer T, Grannemann B, Soyombo A, et al. Can C-reactive protein inform antidepressant medication selection in depressed outpatients? Findings from the CO-MED trial. Psychoneuroendocrinology. 2017;78:105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uher R, Tansey KE, Dew T, Maier W, Mors O, Hauser J, et al. An inflammatory biomarker as a differential predictor of outcome of depression treatment with escitalopram and nortriptyline. Am J Psychiatry. 2014;171(12):1278–86. [DOI] [PubMed] [Google Scholar]
- 52.Cattaneo A, Ferrari C, Turner L, Mariani N, Enache D, Hastings C, et al. Whole-blood expression of inflammasome-and glucocorticoid-related mRNAs correctly separates treatment-resistant depressed patients from drug-free and responsive patients in the BIODEP study. Transl Psychiatry. 2020;10:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cole JJ, McColl A, Shaw R, Lynall M-E, Cowen PJ, de Boer P, et al. No evidence for differential gene expression in major depressive disorder PBMCs, but robust evidence of elevated biological ageing. Transl Psychiatry. 2021;11:404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Strawbridge R, Arnone D, Danese A, Papadopoulos A, Herane Vives A, Cleare AJ. Inflammation and clinical response to treatment in depression: A meta-analysis. Eur Neuropsychopharmacol. 2015;25(10):1532–43. [DOI] [PubMed] [Google Scholar]
- 55.Cattaneo A, Ferrari C, Uher R, Bocchio-Chiavetto L, Riva MA, Pariante CM. Absolute measurements of macrophage migration inhibitory factor and interleukin-1-β mRNA levels accurately predict treatment response in depressed patients. Int J Neuropsychopharmacol. 2016;19(10):pyw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McIntyre RS, Subramaniapillai M, Lee Y, Pan Z, Carmona NE, Shekotikhina M, et al. Efficacy of adjunctive infliximab vs placebo in the treatment of adults with bipolar I/II depression: a randomized clinical trial. JAMA Psychiatry. 2019;76:783–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strawbridge R, Jamieson A, Hodsoll J, Ferrier IN, McAllister-Williams RH, Powell TR, et al. The role of inflammatory proteins in anti-glucocorticoid therapy for treatment-resistant depression. J Clin Med. 2021;10:784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meyer JH, Cervenka S, Kim M-J, Kreisl WC, Henter ID, Innis RB. Neuroinflammation in psychiatric disorders: PET imaging and promising new targets. Lancet Psychiatry. 2020;7:1064–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Attwells S, Setiawan E, Rusjan PM, Xu C, Hutton C, Rafiei D, et al. Translocator protein distribution volume predicts reduction of symptoms during open-label trial of celecoxib in major depressive disorder. Biol Psychiatry. 2020;88:649–56. [DOI] [PubMed] [Google Scholar]
- 61.Attwells S, Setiawan E, Rusjan PM, Xu C, Kish SJ, Vasdev N, et al. A double-blind placebo-controlled trial of minocycline on translocator protein distribution volume in treatment-resistant major depressive disorder. Transl Psychiatry. 2021;11:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nettis MA, Lombardo G, Hastings C, Zajkowska Z, Mariani N, Nikkheslat N, et al. Augmentation therapy with minocycline in treatment-resistant depression patients with low-grade peripheral inflammation: results from a double-blind randomised clinical trial. Neuropsychopharmacology. 2021;46:939–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Husain MI, Cullen C, Umer M, Carvalho AF, Kloiber S, Meyer JH, et al. Minocycline as adjunctive treatment for treatment-resistant depression: study protocol for a double blind, placebo-controlled, randomized trial (MINDEP2). BMC Psychiatry. 2020;20:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Costi S, Morris LS, Collins A, Fernandez NF, Patel M, Xie H, et al. Peripheral immune cell reactivity and neural response to reward in patients with depression and anhedonia. Transl Psychiatry. 2021;11:565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry. 2010;68:748–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moieni M, Tan KM, Inagaki TK, Muscatell KA, Dutcher JM, Jevtic I, et al. Sex differences in the relationship between inflammation and reward sensitivity: a randomized controlled trial of endotoxin. Biol Psychiatry Cogn Neurosci Neuroimaging. 2019;4:619–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Felger JC, Treadway MT. Inflammation effects on motivation and motor activity: role of dopamine. Neuropsychopharmacology. 2017;42(1):216–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fawcett J, Scheftner WA, Fogg L, Clark DC, Young MA, Hedeker D, et al. Time-related predictors of suicide in major affective disorder. Am J Psychiatry. 1990;147(9):1189–94. [DOI] [PubMed] [Google Scholar]
- 69.Nierenberg AA, Keefe BR, Leslie VC, Alpert JE, Pava JA, Worthington JJ, et al. Residual symptoms in depressed patients who respond acutely to fluoxetine. J Clin Psychiatry. 1999;60(4):221–5. [DOI] [PubMed] [Google Scholar]
- 70.Felger JC, Haroon E, Patel TA, Goldsmith DR, Wommack EC, Woolwine BJ, et al. What does plasma CRP tell us about peripheral and central inflammation in depression? Mol Psychiatry. 2018;25:1301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Capuron L, Pagnoni G, Drake DF, Woolwine BJ, Spivey JR, Crowe RJ, et al. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry. 2012;69(10):1044–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Uher R, Perlis RH, Henigsberg N, Zobel A, Rietschel M, Mors O, et al. Depression symptom dimensions as predictors of antidepressant treatment outcome: replicable evidence for interest-activity symptoms. Psychological Med. 2012;42(5):967–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haroon E, Miller AH, Sanacora G. Inflammation, glutamate, and glia: a trio of trouble in mood disorders. Neuropsychopharmacology. 2017;42:193–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sanacora G, Yan Z, Popoli M. The stressed synapse 2.0: pathophysiological mechanisms in stress-related neuropsychiatric disorders. Nat Rev Neurosci. 2022;23:86–103. [DOI] [PubMed] [Google Scholar]
- 75.Capuron L, Pagnoni G, Demetrashvili MF, Lawson DH, Fornwalt FB, Woolwine B, et al. Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology. 2007;32:2384–92. [DOI] [PubMed] [Google Scholar]
- 76.Capuron L, Pagnoni G, Demetrashvili M, Woolwine BJ, Nemeroff CB, Berns GS, et al. Anterior cingulate activation and error processing during interferon-alpha treatment. Biol Psychiatry. 2005;58:190–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Haroon E, Fleischer CC, Felger JC, Chen X, Woolwine BJ, Patel T, et al. Conceptual convergence: increased inflammation is associated with increased basal ganglia glutamate in patients with major depression. Mol Psychiatry. 2016;21:1351–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hashimoto K, Sawa A, Iyo M. Increased levels of glutamate in brains from patients with mood disorders. Biol Psychiatry. 2007;62:1310–6. [DOI] [PubMed] [Google Scholar]
- 79.Feyissa A, Woolverton WL, Miguel-Hidalgo JJ, Wang Z, Kyle PB, Hasler G, et al. Elevated level of metabotropic glutamate receptor 2/3 in the prefrontal cortex in major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Deschwanden A, Karolewicz B, Feyissa AM, Treyer V, Ametamey SM, Johayem A, et al. Reduced metabotropic glutamate receptor 5 density in major depression determined by [11C] ABP688 PET and postmortem study. Am J Psychiatry. 2011;168:727–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bernard R, Kerman IA, Thompson RC, Jones EG, Bunney WE, Barchas JD, et al. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol Psychiatry. 2011;16:634–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Duric V, Banasr M, Stockmeier CA, Simen AA, Newton SS, Overholser JC, et al. Altered expression of synapse and glutamate related genes in post-mortem hippocampus of depressed subjects. Int J Neuropsychopharmacol. 2013;16:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Levine J, Panchalingam K, Rapoport A, Gershon S, McClure RJ, Pettegrew JW. Increased cerebrospinal fluid glutamine levels in depressed patients. Biol Psychiatry. 2000;47:586–93. [DOI] [PubMed] [Google Scholar]
- 84.Hashimoto K, Bruno D, Nierenberg J, Marmar CR, Zetterberg H, Blennow K, et al. Abnormality in glutamine–glutamate cycle in the cerebrospinal fluid of cognitively intact elderly individuals with major depressive disorder: a 3-year follow-up study. Transl Psychiatry. 2016;6:e744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhao J, Verwer RWH, van Wamelen DJ, Qi X-R, Gao S-F, Lucassen PJ, et al. Prefrontal changes in the glutamate-glutamine cycle and neuronal/glial glutamate transporters in depression with and without suicide. J Psychiatr Res. 2016;82:8–15. [DOI] [PubMed] [Google Scholar]
- 86.Luykx JJ, Laban KG, Van Den Heuvel MP, Boks MPM, Mandl RCW, Kahn RS, et al. Region and state specific glutamate downregulation in major depressive disorder: a meta-analysis of 1H-MRS findings. Neurosci Biobehav Rev. 2012;36:198–205. [DOI] [PubMed] [Google Scholar]
- 87.Truong V, Cheng PZ, Lee H-C, Lane TJ, Hsu T-Y, Duncan NW. Occipital gamma-aminobutyric acid and glutamate-glutamine alterations in major depressive disorder: An MRS study and meta-analysis. Psychiatry Res Neuroimaging. 2021;308:111238. [DOI] [PubMed] [Google Scholar]
- 88.Abdallah CG, Jiang L, De Feyter HM, Fasula M, Krystal JH, Rothman DL, et al. Glutamate metabolism in major depressive disorder. Am J Psychiatry. 2014;171:1320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Godlewska RB, Masaki C, Sharpley AL, Cowen PJ, Emir UE. Brain glutamate in medication-free depressed patients: a proton MRS study at 7 Tesla. Psychol Med. 2018;48:1731–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Strawbridge R, Marwood L, King S, Young AH, Pariante CM, Colasanti A, et al. Inflammatory proteins and clinical response to psychological therapy in patients with depression: an exploratory study. J Clin Med. 2020;9:3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lopresti AL. Cognitive behaviour therapy and inflammation: A systematic review of its relationship and the potential implications for the treatment of depression. Aust N Z J Psychiatry. 2017;51:565–82. [DOI] [PubMed] [Google Scholar]
- 92.Gadek-Michalska A, Spyrka J, Rachwalska P, Tadeusz J, Bugajski J. Influence of chronic stress on brain corticosteroid receptors and HPA axis activity. Pharmacol Rep. 2013;65:1163–75. [DOI] [PubMed] [Google Scholar]
- 93.Stetler C, Miller GE. Depression and hypothalamic-pituitary-adrenal activation: a quantitative summary of four decades of research. Psychosom Med. 2011;73:114–26. [DOI] [PubMed] [Google Scholar]
- 94.Gold PW, Chrousos GP. Organization of the stress system and its dysregulation in melancholic and atypical depression: high vs low CRH/NE states. Mol Psychiatry. 2002;7(3):254–75. [DOI] [PubMed] [Google Scholar]
- 95.van den Heuvel LL, Suliman S, Bröcker E, Kilian S, Stalder T, Kirschbaum C, et al. The association between hair cortisol levels, inflammation and cognitive functioning in females. Psychoneuroendocrinology. 2022;136:105619. [DOI] [PubMed] [Google Scholar]
- 96.Kuhlman KR, Cole SW, Craske MG, Fuligni AJ, Irwin MR, Bower JE. Enhanced immune activation following acute social stress among adolescents with early-life adversity. BP:GOS. 2022;March 12 [online ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Maier BCL, Zillich L, Streit F, Wildenberg K, Rietschel M, Hammes H-P, et al. Adverse childhood experiences and late-life diurnal HPA axis activity: associations of different childhood adversity types and interaction with timing in a sample of older East Prussian World War II refugees. Psychoneuroendocrinology. 2022;139:105717. [DOI] [PubMed] [Google Scholar]
- 98.Iob E, Kirschbaum C, Steptoe A. Persistent depressive symptoms, HPA-axis hyperactivity, and inflammation: the role of cognitive-affective and somatic symptoms. Mol Psychiatry. 2020;25:1130–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ahmad MH, Rizvi MA, Fatima M, Mondal AC. Pathophysiological implications of neuroinflammation mediated HPA axis dysregulation in the prognosis of cancer and depression. Mol Cell Endocrinol. 2021;520:111093. [DOI] [PubMed] [Google Scholar]
- 100.Song L, Wu X, Wang J, Guan Y, Zhang Y, Gong M, et al. Antidepressant effect of catalpol on corticosterone-induced depressive-like behavior involves the inhibition of HPA axis hyperactivity, central inflammation and oxidative damage probably via dual regulation of NF-κB and Nrf2. Brain Res Bull. 2021;177:81–91. [DOI] [PubMed] [Google Scholar]
- 101.Kowalczyk M, Szemraj J, Bliźniewska K, Maes M, Berk M, Su K-P, et al. An immune gate of depression – Early neuroimmune development in the formation of the underlying depressive disorder. Pharmacol Rep. 2019;71(6):1299–307. [DOI] [PubMed] [Google Scholar]
- 102.O’Connor JC, Lawson MA, André C, Moreau M, Lestage J, Castanon N, et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry. 2009;14(5):511–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen Y, Guillemin GJ. Kynurenine pathway metabolites in humans: disease and healthy states. Int J Tryptophan Res. 2009;2:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Young SN. The effect of raising and lowering tryptophan levels on human mood and social behaviour. Philos Trans R Soc Lond, B, Biol Sci. 2013;368(1615):20110375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wichers MC, Koek GH, Robaeys G, Verkerk R, Scharpe S, Maes MJMP. IDO and interferon-alpha-induced depressive symptoms: a shift in hypothesis from tryptophan depletion to neurotoxicity. Mol Psychiatry. 2005;10:538–44. [DOI] [PubMed] [Google Scholar]
- 106.Chen LM, Bao CH, Wu Y, Liang SH, Wang D, Wu LY, et al. Tryptophan-kynurenine metabolism: a link between the gut and brain for depression in inflammatory bowel disease. J Neuroinflammation. 2021;18:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gibney SM, Fagan EM, Waldron A-M, O’Byrne J, Connor TJ, Harkin A. Inhibition of stress-induced hepatic tryptophan 2,3-dioxygenase exhibits antidepressant activity in an animal model of depressive behaviour. Int J Neuropsychopharmacol. 2014;17:917–28. [DOI] [PubMed] [Google Scholar]
- 108.Foster AC, Vezzani A, French ED, Schwarcz R. Kynurenic acid blocks neurotoxicity and seizures induced in rats by the related brain metabolite quinolinic acid. Neuroscience Letters. 1984;48(3):273–8. [DOI] [PubMed] [Google Scholar]
- 109.Lugo-Huitrón R, Ugalde Muñiz P, Pineda B, Pedraza-Chaverrí J, Ríos C, Pérez-de la Cruz V. Quinolinic acid: an endogenous neurotoxin with multiple targets. Oxid Med Cell Longev. 2013;2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tavares RG, Tasca CI, Santos CES, Alves LB, Porciúncula LO, Emanuelli T, et al. Quinolinic acid stimulates synaptosomal glutamate release and inhibits glutamate uptake into astrocytes. Neurochem Int. 2002;40:621–7. [DOI] [PubMed] [Google Scholar]
- 111.Kynurenines Schwarcz R. and glutamate: multiple links and therapeutic implications. Adv Pharmacol. 2016;76:13–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dantzer R, Walker AK. Is there a role for glutamate-mediated excitotoxicity in inflammation-induced depression? J Neural Transm. 2014;121:925–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Miller AH. Conceptual confluence: the kynurenine pathway as a common target for ketamine and the convergence of the inflammation and glutamate hypotheses of depression. Neuropsychopharmacology. 2013;38:1607–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, et al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45(9):1085–98. [DOI] [PubMed] [Google Scholar]
- 115.Rajkowska G, Stockmeier CA. Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr Drug Targets. 2013;14(11):1225–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry. 2015;72(3):268–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Moaddel R, Shardell M, Khadeer M, Lovett J, Kadriu B, Ravichandran S, et al. Plasma metabolomic profiling of a ketamine and placebo crossover trial of major depressive disorder and healthy control subjects. Psychopharmacology (Berl). 2018;235(10):3017–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhou Y, Zheng W, Liu W, Wang C, Zhan Y, Li H, et al. Antidepressant effect of repeated ketamine administration on kynurenine pathway metabolites in patients with unipolar and bipolar depression. Brain Behav Immun. 2018;74:205–12. [DOI] [PubMed] [Google Scholar]
- 119.Allen AP, Naughton M, Dowling J, Walsh A, O’Shea R, Shorten G, et al. Kynurenine pathway metabolism and the neurobiology of treatment-resistant depression: Comparison of multiple ketamine infusions and electroconvulsive therapy. J Psychiatr Res. 2018;100:24–32. [DOI] [PubMed] [Google Scholar]
- 120.Doolin K, Allers KA, Pleiner S, Liesener A, Farrell C, Tozzi L, et al. Altered tryptophan catabolite concentrations in major depressive disorder and associated changes in hippocampal subfield volumes. Psychoneuroendocrinology. 2018;95:8–17. [DOI] [PubMed] [Google Scholar]
- 121.Erhardt S, Lim CK, Linderholm KR, Janelidze S, Lindqvist D, Samuelsson M, et al. Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology. 2013;38:743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein H-G, Sarnyai Z, et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: Evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflammation. 2011;8:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Savitz J, Drevets WC, Wurfel BE, Ford BN, Bellgowan PSF, Victor TA, et al. Reduction of kynurenic acid to quinolinic acid ratio in both the depressed and remitted phases of major depressive disorder. Brain Behav Immun. 2015;46:55–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lanser L, Kink P, Egger EM, Willenbacher W, Fuchs D, Weiss G, et al. Inflammation-induced tryptophan breakdown is related with anemia, fatigue, and depression in cancer. Front Immunol. 2020;2020:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Waclawiková B, El Aidy S. Role of microbiota and tryptophan metabolites in the remote effect of intestinal inflammation on brain and depression. Pharmaceuticals (Basel). 2018;11:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yeung AWS, Terentis AC, King NJC, Thomas SR. Role of indoleamine 2,3-dioxygenase in health and disease. Clin Sci. 2015;129:601–72. [DOI] [PubMed] [Google Scholar]
- 127.Dantzer R Role of the kynurenine metabolism pathway in inflammation-induced depression: preclinical approaches. In: Dantzer R, Capuron L, eds. Inflammation-Associated Depression: Evidence, Mechanisms and Implications. Current Topics in Behavioral Neurosciences. Cham, Switzerland: Springer; 2016:117–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fukui S, Schwarcz R, Rapoport SI, Takada Y, Smith QR. Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem. 1991;56:2007–17. [DOI] [PubMed] [Google Scholar]
- 129.Kelly JR, Borre Y, O’Brien C, Patterson E, El Aidy S, Deane J, et al. Transferring the blues: depression-associated gut microbiota induces neurobehavioural changes in the rat. J Psychiatr Res. 2016;82:109–18. [DOI] [PubMed] [Google Scholar]
- 130.Li N, Wang Q, Wang Y, Sun A, Lin Y, Jin Y, et al. Fecal microbiota transplantation from chronic unpredictable mild stress mice donors affects anxiety-like and depression-like behavior in recipient mice via the gut microbiota-inflammation-brain axis. Stress. 2019;22:592–602. [DOI] [PubMed] [Google Scholar]
- 131.Lukić I, Ivković S, Mitić M, Adžić M. Tryptophan metabolites in depression: Modulation by gut microbiota. Front Behav Neurosci. 2022;16:987697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hashimoto K Rapid-acting antidepressant ketamine, its metabolites and other candidates: A historical overview and future perspective. Psychiatry Clin Neurosci. 2019;73:613–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ballard ED, Yarrington J, Farmer CA, Lener MS, Kadriu B, Lally N, et al. Parsing the heterogeneity of depression: An exploratory factor analysis across commonly used depression rating scales. J Affect Disord. 2018;231:51–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Saligan LN, Farmer C, Ballard ED, Kadriu B, Zarate CA. Disentangling the association of depression on the anti-fatigue effects of ketamine. J Affect Disord. 2019;244:42–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fukumoto K, Toki H, Iijima M, Hashihayata T, Yamaguchi J-I, Hashimoto K, et al. Antidepressant potential of (R)-ketamine in rodent models: comparison with (S)-ketamine. J Pharmacol Exp Ther. 2017;361:9–16. [DOI] [PubMed] [Google Scholar]
- 136.Zhang J-C, Yao W, Hashimoto K. Arketamine, a new rapid-acting antidepressant: a historical review and future directions. Neuropharmacology. 2022;218:109219. [DOI] [PubMed] [Google Scholar]
- 137.Leal GC, Bandeira ID, Correia-Melo FS, Telles M, Mello RP, Vieira F, et al. Intravenous arketamine for treatment-resistant depression: open-label pilot study. Eur Arch Psychiatr Clin Neurosci. 2021;271:577–82. [DOI] [PubMed] [Google Scholar]
- 138.Suzuki K, Nosyreva E, Hunt KW, Kavalali ET, Monteggia LM. Effects of a ketamine metabolite on synaptic NMDAR function. Nature. 2017;546:E1–E3. [DOI] [PubMed] [Google Scholar]
- 139.Pham TH, Defaix C, Xu X, Deng SX, Fabresse N, Alvarez JC, et al. Common neurotransmission recruited in (R,S)-ketamine and (2R,6R)-hydroxynorketamine-induced sustained antidepressant-like effects. Biol Psychiatry. 2018;84(1):e3–e6. [DOI] [PubMed] [Google Scholar]
- 140.Fukumoto K, Fogaca MV, Liu RJ, Duman C, Kato T, Li XY, et al. Activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2R,6R)-hydroxynorketamine. Proc Natl Acad Sci U S A. 2019;116(1):297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yang C, Kobayashi S, Nakao K, Dong C, Han M, Qu Y, et al. AMPA receptor activation-independent antidepressant actions of ketamine metabolite (S)-norketamine. Biol Psychiatry. 2018;84:591–600. [DOI] [PubMed] [Google Scholar]
- 142.Zarate CA Jr,., Brutsche N, Laje G, Luckenbaugh DA, Venkata SL, Ramamoorthy A, et al. Relationship of ketamine’s plasma metabolites with response, diagnosis, and side effects in major depression. Biol Psychiatry. 2012;72(4):331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yang C, Yang J, Luo A, Hashimoto K. Molecular and cellular mechanisms underlying the antidepressant effects of ketamine enantiomers and its metabolites. Transl Psychiatry. 2019;9:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shibakawa YS, Sasaki Y, Goshima Y, Echigo N, Kamiya Y, Kurahashi K, et al. Effects of ketamine and propofol on inflammatory responses of primary glial cell cultures stimulated with lipopolysaccharide. Br J Anaesth. 2005;95(6):803–10. [DOI] [PubMed] [Google Scholar]
- 145.Tan Y, Wang Q, She Y, Bi X, Zhao B. Ketamine reduces LPS-induced HMGB1 via activation of the Nrf2/HO-1 pathway and NF-κB suppression. J Trauma Acute Care Surg. 2015;78(4):784–92. [DOI] [PubMed] [Google Scholar]
- 146.Willner P, Belzung C. Treatment-resistant depression: are animal models of depression fit for purpose? Psychopharmacology. 2015;232:3473–95. [DOI] [PubMed] [Google Scholar]
- 147.Lyu D, Wang F, Zhang M, Yang W, Huang H, Huang Q, et al. Ketamine induces rapid antidepressant effects via the autophagy-NLRP3 inflammasome pathway. Psychopharmacology (Berl). 2022;239:3201–12. [DOI] [PubMed] [Google Scholar]
- 148.Getachew B, Aubee JI, Schottenfeld RS, Csoka AB, Thompson KM, Tizabi Y. Ketamine interactions with gut-microbiota in rats: relevance to its antidepressant and anti-inflammatory properties. BMC Microbiol 2018;18(1):222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mastrodonato A, Cohensedgh O, LaGamma CT, McGowan JC, Hunsberger HC, Denny CA. Prophylactic (R,S)-ketamine selectively protects against inflammatory stressors. Behav Brain Res. 2019;378:112238. [DOI] [PubMed] [Google Scholar]
- 150.Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B, et al. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology. 2013;38(9):1609–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Edem EE, Anyanwu C-KC, Nebo KE, Akinluyi ET, Fafure AA, Ishola AO, et al. Ketamine abrogates sensorimotor deficits and cytokine dysregulation in a chronic unpredictable mild stress model of depression. Psychopharmacology (Berl). 2022;239:185–200. [DOI] [PubMed] [Google Scholar]
- 152.Camargo A, Torrá ACNC, Dalmagro AP, Valverde AP, Kouba BR, Fraga DB, et al. Prophylactic efficacy of ketamine, but not the low-trapping NMDA receptor antagonist AZD6765, against stress-induced maladaptive behavior and 4E-BP1-related synaptic protein synthesis impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2022;115:110509. [DOI] [PubMed] [Google Scholar]
- 153.Mastrodonato A, Pavlova I, Kee NC, Pham VA, McGowan JC, Mann JJ, et al. Prophylactic (R, S)-ketamine is effective against stress-induced behaviors in adolescent but not aged mice. Int J Neuropsychopharmacol. 2022;25:512–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.McGowan JC, LaGamma CT, Lim SC, Tsitsiklis M, Neria Y, Brachman RA, et al. Prophylactic ketamine attenuates learned fear. Neuropsychopharmacology. 2017;42:1577–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Camargo A, Dalmagro AP, de Souza MM, Zeni ALB, Rodrigues ALS. Ketamine, but not guanosine, as a prophylactic agent against corticosterone-induced depressive-like behavior: possible role of long-lasting pro-synaptogenic signaling pathway. Exp Neurol. 2020;334:113459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Camargo A, Dalmagro AP, Wolin IAV, Kaster MP, Rodrigues ALS. The resilient phenotype elicited by ketamine against inflammatory stressors-induced depressive-like behavior is associated with NLRP3-driven signaling pathway. J Psychiatr Res. 2021;144:118–28. [DOI] [PubMed] [Google Scholar]
- 157.Ma L, Wang L, Chang L, Shan J, Qu Y, Wang X, et al. A key role of miR-132–5p in the prefrontal cortex for persistent prophylactic actions of (R)-ketamine in mice. Transl Psychiatry. 2022;12:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chen BK, Luna VM, LaGamma CT, Xu X, Deng SX, Suckow RF, et al. Sex-specific neurobiological actions of prophylactic (R,S)-ketamine, (2R,6R)-hydroxynorketamine, and (2S,6S)-hydroxynorketamine. Neuropsychopharmacology. 2020;45(9):1545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ma L, Zhang J, Fujita Y, Qu Y, Shan J, Wan X, et al. Nuclear factor of activated T cells 4 in the prefrontal cortex is required for prophylactic actions of (R)-ketamine. Transl Psychiatry. 2022;12:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ma L, Wang L, Chang L, Shan J, Qu Y, Wang X, et al. A role of microRNA-149 in the prefrontal cortex for prophylactic actions of (R)-ketamine in inflammation model. Neuropharmacology. 2022;219:109250. [DOI] [PubMed] [Google Scholar]
- 161.Clarke M, Razmjou S, Prowse N, Dwyer Z, Litteljohn D, Pentz R, et al. Ketamine modulates hippocampal neurogenesis and pro-inflammatory cytokines but not stressor induced neurochemical changes. Neuropharmacology. 2017;112(Pt A):210–20. [DOI] [PubMed] [Google Scholar]
- 162.Tan S, Wang Y, Chen K, Long Z, Zou J. Ketamine alleviates depressive-like behaviors via down-regulating inflammatory cytokines induced by chronic restraint stress in mice. Biol Pharm Bull. 2017;40(8):1260–7. [DOI] [PubMed] [Google Scholar]
- 163.Zhang K, Yang C, Chang L, Sakamoto A, Suzuki T, Fujita Y, et al. Essential role of microglial transforming growth factor-β1 in antidepressant actions of (R)-ketamine and the novel antidepressant TGF-β1. Transl Psychiatry. 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ho M-F, Zhang C, Zhang L, Li H, Weinshilboum RM. Ketamine and active ketamine metabolites regulate STAT3 and the type I interferon pathway in human microglia: molecular mechanisms linked to the antidepressant effects of ketamine. Front Pharmacol. 2019;10:1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yao W, Cao Q, Luo S, He L, Yang C, Chen J, et al. Microglial ERK-NRBP1-CREB-BDNF signaling in sustained antidepressant actions of (R)-ketamine. Mol Psychiatry. 2022;27:1618–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nowak W, Grendas LN, Sanmarco LM, Estecho IG, Arena AR, Eberhardt N, et al. Pro-inflammatory monocyte profile in patients with major depressive disorder and suicide behaviour and how ketamine induces anti-inflammatory M2 macrophages by NMDAR and mTOR. EBioMedicine. 2019;50:290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zanos P, Gould TD. Mechanisms of ketamine action as an antidepressant. Mol Psychiatry. 2018;23(4):801–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Proescholdt M, Heimann A, Kempski O. Neuroprotection of S(+) ketamine isomer in global forebrain ischemia. Brain Res. 2001;904:245–51. [DOI] [PubMed] [Google Scholar]
- 169.Braun S, Gaza N, Werdehausen R, Hermanns H, Bauer I, Durieux ME, et al. Ketamine induces apoptosis via the mitochondrial pathway in human lymphocytes and neuronal cells. Br J Anaesth. 2010;105:347–54. [DOI] [PubMed] [Google Scholar]
- 170.Cui M, Dai W, Kong J, Chen H. Th17 Cells in depression: are they crucial for the antidepressant effect of ketamine? Front Pharmacol. 2021;12:649144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lee JE, Lee JM, Park YJ, Kim BS, Jeon YT, Chung Y. Inhibition of autoimmune Th17 cell responses by pain killer ketamine. Oncotarget. 2017;8:89475–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang W, Liu L, Yang X, Gao H, Tang Q-K, Yin L-Y, et al. Ketamine improved depressive-like behaviors via hippocampal glucocorticoid receptor in chronic stress induced- susceptible mice. Behav Brain Res. 2019;364:75–84. [DOI] [PubMed] [Google Scholar]
- 173.Besnier E, Clavier T, Tonon M-C, Selim J, Lefevre-Scelles A, Morin F, et al. Ketamine and etomidate down-regulate the hypothalamic-pituitary-adrenal axis in an endotoxemic mouse model. Anesthesiology. 2017;127(2):347–54. [DOI] [PubMed] [Google Scholar]
- 174.Garcia LSB, Comim CM, Valvassori SS, Réus GZ, Stertz L, Kapczinski F, et al. Ketamine treatment reverses behavioral and physiological alterations induced by chronic mild stress in rats. Prog Neuro-Psychopharmacol Biol Psychiatry. 2009;33:450–5. [DOI] [PubMed] [Google Scholar]
- 175.Wang N, Yu H-Y, Shen X-F, Gao Z-Q, Yang C, Yang J-J, et al. The rapid antidepressant effect of ketamine in rats is associated with down-regulation of pro-inflammatory cytokines in the hippocampus. Ups J Med Sci. 2015;120:241–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yang J-j, Wang N, Yang C, Shi J-y, Yu H-y, Hashimoto. Serum interleukin-6 is a predictive biomarker for ketamine’s antidepressant effect in treatment-resistant patients with major depression. Biol Psychiatry. 2015;77(3):e19–e20. [DOI] [PubMed] [Google Scholar]
- 177.Kiraly DD, Horn SR, Van Dam NT, Costi S, Schwartz J, Kim-Schulze S, et al. Altered peripheral immune profiles in treatment-resistant depression: response to ketamine and prediction of treatment outcome. Transl Psychiatry. 2017;7(3):e1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Nikkheslat N, Kotoula V, Mazibuko N, Mondelli V, Mehta M, Pariante CM. Anti-inflammatory effect of ketamine: a double-blind placebo-controlled randomised study. Psychoneuroendocrinology. 2021;131 (Suppl):105505. [Google Scholar]
- 179.Park M, Newman LE, Gold PW, Luckenbaugh DA, Yuan P, Machado-Vieira R, et al. Change in cytokine levels is not associated with rapid antidepressant response to ketamine in treatment-resistant depression. J Psychiatr Res. 2017;84:113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kruse JL, Vasavada MM, Olmstead R, Helleman G, Wade B, Breen EC, et al. Depression treatment response to ketamine: sex-specific role of interleukin-8, but not other inflammatory markers. Transl Psychiatry. 2021;11:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Abdallah CG, Averill LA, Gueorguieva R, Goktas S, Purohit P, Ranganathan M, et al. Modulation of the antidepressant effects of ketamine by the mTORC1 inhibitor rapamycin. Neuropsychopharmacology. 2020;45:990–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ostroff R, Kothari JS. Reversal of non-suppression of cortisol levels in a patient with refractory depression receiving ketamine. Am J Psychiatry. 2015;172(1):95–6. [DOI] [PubMed] [Google Scholar]
- 183.Khalili-Mahani N, Martini CH, Olofsen E, Dahan A, Niesters M. Effect of subanaesthetic ketamine on plasma and saliva cortisol secretion. Br J Anaesth. 2015;115(1):68–75. [DOI] [PubMed] [Google Scholar]
- 184.Hergovich N, Singer E, Agneter E, Eichler HG, Graselli U, Simhandl C, et al. Comparison of the effects of ketamine and memantine on prolactin and cortisol release in men. a randomized, double-blind, placebo-controlled trial. Neuropsychopharmacology. 2001;24:590–3. [DOI] [PubMed] [Google Scholar]
- 185.Kadriu B, Farmer CA, Yuan P, Park LT, Deng Z, Moaddel R, et al. The kynurenine pathway and bipolar disorder: intersection of the monoaminergic and glutamatergic systems and immune response. Mol Psychiatry. 2021;26:4085–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Niciu MJ, Luckenbaugh DA, Ionescu DF, Guevara S, Machado-Vieira R, Richards EM, et al. Clinical predictors of ketamine response in teatment-resistant major depression. J Clin Psychiatry. 2014;75(5):e417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gold PW. The organization of the stress system and its dysregulation in depressive illness. Mol Psychiatry. 2015;20(1):32–47. [DOI] [PubMed] [Google Scholar]
- 188.Machado-Vieira R, Gold PW, Luckenbaugh DA, Ballard ED, Richards EM, Henter ID, et al. The role of adipokines in the rapid antidepressant effects of ketamine. Mol Psychiatry. 2017;22(1):127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Jung HS, Park K-H, Cho YM, Chung SS, Cho HJ, Cho SY, et al. Resistin is secreted from macrophages in atheromas and promotes atherosclerosis. Cardiovasc Res. 2006;69(1):76–85. [DOI] [PubMed] [Google Scholar]
- 190.Ouchi N, Walsh K. Adiponectin as an anti-inflammatory factor. Clin Chim Acta. 2007;380(1–2):24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kadriu B, Gold PW, Luckenbaugh DA, Lener MS, Ballard ED, Niciu MJ, et al. Acute ketamine administration corrects abnormal inflammatory bone markers in major depressive disorder. Mol Psychiatry. 2018;23:1626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cathomas F, Bevilacqua L, Ramakrishnan A, Kronman H, Costi S, Schneider M, et al. Whole blood transcriptional signatures associated with rapid antidepressant response to ketamine in patients with treatment resistant depression. Transl Psychiatry. 2022;12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Morris AA, Zhao L, Ahmed Y, Stoyanova N, De Staercke C, Hooper WC, et al. Association between depression and inflammation–differences by race and sex: the META-Health study. Psychosom Med. 2011;73:462–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Beydoun MA, Obhi H, Weiss J, Canas J, Beydoun HA, Evans MK, et al. Systemic inflammation is associated with depressive symptoms differentially by sex and race: a longitudinal study of urban adults. Mol Psychiatry. 2020;25:1286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Stewart JC. One effect size does not fit all—Is the depression-inflammation link missing in racial/ethnic minority individuals. JAMA Psychiatry. 2016;73:301–2. [DOI] [PubMed] [Google Scholar]