

Abstract
Introduction
Cyanide exposure can occur in various settings such as industry and metallurgy. The primary mechanism of injury is cellular hypoxia from Complex IV (CIV) inhibition. This leads to decreased ATP production and increased reactive oxygen species production. The brain and the heart are the organs most affected due to their high metabolic demand. While the cardiac effects of cyanide are well known, the cerebral effects on cellular function are less well described. We investigated cerebral metabolism with a combination of brain respirometry, microdialysis, and western blotting using a rodent model of sub-lethal cyanide poisoning.
Methods
Twenty rodents were divided into two groups: control (n = 10) and sub-lethal cyanide (n = 10). Cerebral microdialysis was performed during a 2 mg/kg/h cyanide exposure to obtain real-time measurements of cerebral metabolic status. At the end of the exposure (90 min), brain-isolated mitochondria were measured for mitochondrial respiration. Brain tissue ATP concentrations, acyl-Coenzyme A thioesters, and mitochondrial content were also measured.
Results
The cyanide group showed significantly increased lactate and decreased hypotension with decreased cerebral CIV-linked mitochondrial respiration. There was also a significant decrease in cerebral ATP concentration in the cyanide group and a significantly higher cerebral lactate-to-pyruvate ratio (LPR). In addition, we also found decreased expression of Complex III and IV protein expression in brain tissue from the cyanide group. Finally, there was no change in acyl-coenzyme A thioesters between the two groups.
Conclusions
The key finding demonstrates mitochondrial dysfunction in brain tissue that corresponds with a decrease in mitochondrial function, ATP concentrations, and an elevated LPR indicating brain dysfunction at a sub-lethal dose of cyanide.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13181-022-00928-w.
Keywords: Mitochondria, Cyanide, Basic science, Cerebral metabolism
Introduction
Cyanide is a potent cellular poison with a dose of as little as 50 mg being fatal [1]. It is rapidly acting and can exist either in a salt or gaseous state [2]. Cyanide exposure can occur in a wide range of occupational settings such as in industry, manufacturing plants, and metallurgy. Exposure can also occur from the combustion of plastic and other synthetic material [3, 4]. Cyanide is listed as one of the highest chemical threats due to its potential use as a mass casualty weapon [2]. Finally, while less common, cyanide poisoning may also occur in certain improperly processed foods such as the cassava root, a major source of food for millions of people in the tropics [5].
Cyanide has multiple mechanisms of action, but its primary mechanism of cellular injury involves cellular hypoxia from binding to Complex IV (CIV) of the electron transport system (ETS) [6]. The normal function of the ETS is critical to produce ATP but as cyanide inhibits CIV of the ETS, the movement of electrons across the ETS is hindered and mitochondria are unable to maintain a proper proton gradient across the inner mitochondrial membrane [6–8]. In addition to the reduction in the bioenergetic efficiency of the mitochondria, cyanide will also result in increased production of reactive oxygen species (ROS) due to the inhibition of superoxide dismutase (SOD) and possible increase in reverse electron transport (RET) from CIV inhibition [9].
Cyanide primarily affects the brain and the heart which have a heavy reliance on the production of ATP given their high metabolic demand. Substantial cyanide poisoning manifests in a wide range of signs and symptoms such as seizures, tachycardia, and hypotension [10, 11]. The most common cause of death is cardiovascular shock where patients often have the triad of unresponsiveness, high lactate, and cardiovascular collapse. Despite the well-known effects of cyanide on the heart, an understudied area is the neurologic effects of cyanide [12]. We utilized a comprehensive rodent platform using in vivo and ex vivo measures to investigate the effect of sub-lethal cyanide poisoning on the physiological and molecular levels specifically in the brain.
Material and Methods
Perioperative Procedures and Hemodynamic Monitoring
All procedures have been approved by the Institutional Animal Care and Use Committee at the Children’s Hospital of Philadelphia and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Sprague–Dawley (Charles River Laboratories, Horsham, PA, USA) rats P60 (postnatal day) equivalent to a young human adult (~ 250–300 g) were used for this study of equal sexes. All rodents were placed in an induction chamber with 5% isoflurane exposure for 1–2 min, placed on a SurgiSuite Multi-Functional Surgical Platform (Kent Scientific, Torrington, CT, USA) with warming capacity, and maintained in a temperature range of 36–38 °C, monitored with a rectal probe for the duration of the experiment. After induction, 2–3% isoflurane was delivered by a rodent cone to maintain general anesthesia (tail and toe pinch used to assess the depth of anesthesia). Immediate tracheostomy was performed; the animals were placed on a small animal ventilator (VentElite Small Animal Ventilator from Harvard Apparatus (Holliston, MA, USA)); isoflurane was then weaned down to 1–1.5%, and continuous measurement of oxygen saturation was recorded during the entire experiment. Ventilator settings were as follows on pressure support: respiratory rate of 50–55 breaths/minute and a pressure of 8–10 cm H2O to maintain a partial venous CO2 of 35–45 mmHg confirmed by iStat (Abbott Laboratories, IL, USA) using 100 µL of venous blood every 30 min after the insertion of a 23 g IV catheter placed in the right femoral vein. The right carotid artery, internal jugular vein, and vagal nerve were then dissected out under 3 × magnification. Upon isolation of the carotid artery, a Millar SPR-869 Rat PV Catheter (2F, 4E, 6 mm, 15 cm, PI) was advanced into the left ventricle of the heart and pressure–volume loop analysis was obtained with an ADInstruments PowerLab 8/35 (Sydney, Australia) and Millar (Houston, TX, USA). All data were recorded with PowerLab LabChart 8 Pro software from ADInstruments (Sydney, Australia). The animals were randomized to 2 different groups: sham (n = 10) and sub-lethal cyanide (n = 10).
Cyanide Experimental Protocol
A sub-lethal dose of cyanide (2 mg/kg/h) prepared in saline was administered through a femoral line with a Harvard Apparatus 11 Elite Syringe pump (Holliston, MA, USA). The dose was based on existing literature for dosing of cyanide through various routes, such as oral and intravenous, and in line with what would be considered sub-lethal [13]. All cyanide was prepared on the day of the experiment and placed on ice in a sealed container to avoid off gassing prior to infusion. Animals received their assigned cyanide infusion for 90 min. Animals in the sham group received 90 min of 0.9% normal saline. The volume of fluids was similar between the two groups.
Tissue and Whole Blood Extraction and Preparation
Isolated Brain Mitochondria
Upon completion of the exposure, all subject animals were euthanized (decapitation) and the brain was immediately obtained. Brain tissue immediately underwent rapid but gentle dissection and was immediately transferred to an ice-cold isolation buffer solution (320 mM sucrose, 2 mM EGTA, 10 mM Trizma base, pH 7.4). Next, brain tissue was minced, manually homogenized in ice-cold brain buffer (225 mM D-Mannitol, 75 mM Sucrose, 5 mM HEPES, 1 mM EGTA, and 0.5 L of double deionized water, pH 7.4) containing 0.2% BSA buffer (catalog A6003), and centrifuged at 1300 g and 4 °C to discard cell debris. Subsequently, the supernatant was centrifuged for 10 min at 21,000 g to extract the mitochondrial pellet. Brain mitochondria were further isolated from the derived pellet by differential centrifugation and application of density gradients using 15%, 23%, and 40% Percoll (GE Healthcare cat. no. 17089101), washed with brain buffer, and centrifuged to collect the isolated mitochondrial pellet. Protein count for isolated mitochondria was obtained with a Pierce BCA Protein Assay kit (catalog 23,227) from Thermo Fisher Scientific (Waltham, MA, USA). The full methods for isolated mitochondria from brain tissue are based on our previous works [13, 14]. Snap-frozen brain tissue was stored at -80 °C for CoA measurements, ATP concentrations, and western blotting.
Measurement of Mitochondrial Respiration
Mitochondrial respiratory function was analyzed using Oroboros O2k-FluoRespirometer (Oroboros Instruments, Innsbruck, Austria) with a substrate–uncoupler–inhibitor titration (SUIT) protocol as previously described in our work [13, 15]. The SUIT protocol measures oxidative phosphorylation capacity with electron flow through Complex I (CI) as well as the convergent electron input through CI and Complex II (CI + CII) using the nicotinamide adenine dinucleotide-linked substrates, malate and pyruvate, and glutamate, as well as the flavin adenine dinucleotide-linked substrate succinate, both in the presence of adenosine diphosphate. Oxidative phosphorylation produces adenosine triphosphate (ATP), which is the primary fuel for performing the basic cellular function. Oligomycin, an inhibitor of the ATP synthase, uncouples respiration from ATP-synthase activity to measure respiration where the O2 consumption is dependent on the leakiness of the mitochondrial membrane and back-flux of protons into the mitochondrial matrix that is independent of the ATP synthase (LEAKCI+CII). If LEAK respiration is increased, the electrochemical gradient across the mitochondrial membrane is uncoupled, resulting in inadequate ATP production. Maximal convergent non-phosphorylating respiration of ETSCI+CII is evaluated by titrating the protonophore, carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone. ETSCI+CII is considered a stress test for mitochondria as a marker of mitochondrial respiratory reserve. Non-phosphorylating respiration, specifically through CII (ETSCII), is achieved through the addition of rotenone, an inhibitor of CI. The Complex III (CIII) inhibitor antimycin-A is added to measure the residual non-mitochondrial oxygen consumption, and this value was subtracted from each of the measured respiratory states to provide only mitochondrial respiration. Complex IV (CIV) respiration was measured by the addition of N,N,N,N-tetramethyl-phenylenediamine together with ascorbate. The CIV inhibitor sodium azide was added to reveal the chemical background that is subtracted from the N,N,N,N-tetramethyl-phenylenediamine-induced oxygen consumption rate. All data were acquired using DatLab 7 (Oroboros Instruments, Innsbruck, Austria), and respiration value was normalized to the protein count of the isolated mitochondria obtained from brain tissue also based on our previous works [13, 14, 16].
ATP Fluorometry
ATP concentrations were obtained from the snap-frozen brain using an ATP fluorometric assay kit (Sigma MAK190). Tissue was electrically homogenized in a 1.5 mL centrifuge tube to obtain a concentration of 10 mg weight tissue/100 µL ATP assay buffer per well. A 0.1 mM ATP standard was created with an ATP concentration range of 0 to 1000 pmol/µL; 50 µL of the homogenized sample and standards were loaded in duplicates in a 96-well opaque well plate. The loaded plate was briefly mixed in a shaker and incubated at room temperature for 30 min. ATP fluorometry was measured at excitation and emission values of 535 and 587 nm, respectively. All values were corrected for background and glycerol phosphate (no addition of ATP converter) [13].
Cerebral Microdialysis
We performed cMD that allows semicontinuous monitoring of brain extracellular fluid. The cMD system consisted of a CMA 402 Syringe Pump with 1 mL microsyringe, a CMA 142 Microfraction Collector with 300 uL plastic vials, and a CMA 12 Probe with FEP tubing and tubing adapters from CMA, a division of Harvard Biosciences, Inc. (Holliston, MA, USA). The probe was placed in the left frontal cortex at a depth of 0.5–1 cm in the brain parenchyma. Sterile saline was perfused at 1 µl/minute, and after a 30-min equilibration period, samples were collected in 30-min intervals throughout the cyanide exposure. Samples were immediately frozen at -80 °C. Pyruvate, lactate, glycerol, and glucose concentrations were analyzed in a blinded fashion using the automated ISCUS FlexTM Microdialysis Analyzer, and data were processed using the ICUpilot software from mDialysis (Stockholm, Sweden) [13].
Acyl-Coenzyme A (CoA) Thioesters
Flash-frozen brain tissue samples were kept on dry ice throughout processing. To measure acyl-coenzyme A (CoA) thioesters, the frozen tissues were cut on a frozen tile (kept on dry ice) and added to 10% (w/v) trichloroacetic acid (Sigma cat. #T6399) in water then were sonicated for 12 × 0.5 s pulses, the protein was pelleted by centrifugation at 17,000 × g from 10 min at 4 °C. The supernatant was purified by solid-phase extraction using Oasis HLB 1 cc (30 mg) SPE columns (Waters). The wash step was with 1 mL methanol and the equilibration with 1 mL water. The supernatant was loaded, desalted with 1 mL water, and eluted with 1 mL methanol containing 25 mM ammonium acetate. The elutes were evaporated to dryness under nitrogen and then resuspended in 100 μL 5% (w/v) 5-sulfosalicylic acid in water. Acyl-CoAs were measured by liquid chromatography-high resolution mass spectrometry. Briefly, 5 μL of resuspended samples in 5% SSA were injected on an Ultimate 3000 UHPLC coupled to a Q Exactive-HF (Thermo Scientific) mass spectrometer in positive ESI mode using the settings described previously [17]. Quantification of acyl-CoAs was via their MS1. Data were integrated using Xcalibur 4.2 (Thermo Scientific) software. The standard curve generation can be seen in Supplementary Table 2.
Western Blot Protocol
Western blot was performed on brain tissue with all reagents and antibodies purchased from Invitrogen (Carlsbad, CA, USA) unless otherwise noted. Brain tissues were incubated on ice in RIPA lysis and extraction buffer (catalog 89,900) containing Pierce Protease inhibitor (catalog A32963) for 5 min. Tissues were homogenized with the electric homogenizer and then subjected to centrifugation at 14,000 g at 4 °C for 10 min with the collection of the supernatant. Protein concentrations of the supernatant were quantified using a Pierce BCA Protein Assay Kit from Thermo Fisher Scientific (Waltham, MA, USA). We have published specifics of our protein quantification [18]. The equal protein content of 20 µg was then loaded into each well of 4–12% Bis–Tris gel and separated by sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Gel proteins were transferred onto a nitrocellulose membrane (Catalog IB23001) and then incubated with a citrate synthase (CS) recombinant rabbit monoclonal antibody (catalog #703,361) with a dilution factor of 1:4000 in the iBind solution (Catalog SLF1020). Primary rabbit monoclonal anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody conjugated to horseradish peroxidase (HRP) (#3683 Cell Signaling Technology; 1:1000) was used as an internal control. CS levels were detected using goat anti-rabbit IgG secondary antibody conjugated to HRP (catalog A16096; 1:400) and a chemiluminescent substrate reagent kit.
The same method was performed to obtain protein quantification of components of the OXPHOS system using an OXPHOS cocktail (CI-V). Gel proteins were transferred onto a PVDF membrane (catalog IB24001) and then incubated with an OXPHOS monoclonal antibody cocktail (catalog 458,099) with a dilution factor of 1:1600 in iBind solution (Catalog SLF1020). Primary mouse monoclonal anti-GAPDH antibody (GA1R) conjugated to HRP (#3683 Cell Signaling Technology; 1:4000) was used as an internal control. OXPHOS levels were detected using rabbit anti-mouse IgG secondary antibody conjugated to HRP (catalog A16160, 1:1600) and a chemiluminescent substrate reagent kit.
Immunoblotting steps used the iBind Western Device with images captured with the iBright CL1000, and iBright Analysis Software (Thermo Scientific) was used in the quantification and densitometric analysis of the blots. All experiments were performed in duplicates, and the local background corrected density values were normalized against respective GAPDH (CS) and GA1R (OXPHOS) values.
Statistics and Data Analysis
With 10 rodents per group, the study was powered to detect an effect size of 1.3 or larger for all measurements with 80% power and alpha set at 0.05. To determine differences between control and cyanide groups for blood gasses, hemodynamics, ATP, citrate synthase, and acyl-CoAs, Wilcoxon rank sum tests were used. Data for these analyses are presented as median and interquartile range (IQR). To adjust for multiple direct comparisons (n = 7), a more stringent p-value of 0.007 was used. To assess differences between groups over time for lactate-to-pyruvate ratio and mitochondrial respiration, an analysis of variance in repeated measures was used. To adjust for multiple comparisons between groups and time or respiratory states, post-hoc pairwise comparisons using Tukey Kramer t-tests were performed. All analyses were performed using SAS statistical software (version 9.4, SAS Institute, Cary, NC). Figures were created using GraphPad Prism (version 9.3.1, GraphPad Software, San Diego, CA, USA).
Result
Baseline Characteristics, Blood Gas Chemistry, and Hemodynamics Variables
The baseline weight and blood gasses obtained at the start of the experiment were similar between animals (data not shown). Serial venous blood gasses were obtained every 30 min, along with continuous vital sign monitoring throughout the experiment. There was a significant difference between the two groups at the end of the experiment, where the cyanide group had significantly lower heart rate and mean arterial pressure along with higher blood lactate (Table 1). The blood oxygen saturation was over 95% for both groups throughout the entire experiment.
Table 1.
Subject animal characteristics.
| Group characteristics | |||
|---|---|---|---|
| Variable | Sham (n = 10) |
Cyanide (n = 10) |
P-value* |
| Blood gasses | |||
| pH | 7.41 (7.4–7.42) | 7 (6.9–7.1) | 0.0014 |
| PaCO2 (mmHg) | 41 (40–41) | 20.5 (19–22) | 0.0013 |
| Lactate (mmol/L) | 1.1 (1.1–1.2) | 6.25 (5.1–7.3) | 0.0013 |
| Hemodynamics | |||
| Heart rate (bpm) | 305.5 (299–312) | 210.5 (206–220) | 0.0014 |
| Mean arterial pressure (mmHg) | 71.5 (70–73) | 39 (37–42) | 0.0013 |
The variables reported are at the 90-min time point. At the 90-min time point, the venous blood gas and basic hemodynamics are presented as well as their respective P-values obtained with a 2-sample t-test. All values are represented as median (IQR)
*P-values derived from Wilcoxon rank sum tests
Cerebral Microdialysis
Cerebral microdialysis (cMD) samples were taken at 30-min time points during the experimental procedure, starting with baseline measurements. The following substrates were measured to gauge cerebral metabolism: lactate, pyruvate, glycerol, and glucose. With the exception of baseline measurement prior to the exposure, LPR was significantly higher for the cyanide group compared to the control group over 30-min intervals (P < 0.0001 for all time points beyond the baseline, Fig. 1).
Fig. 1.
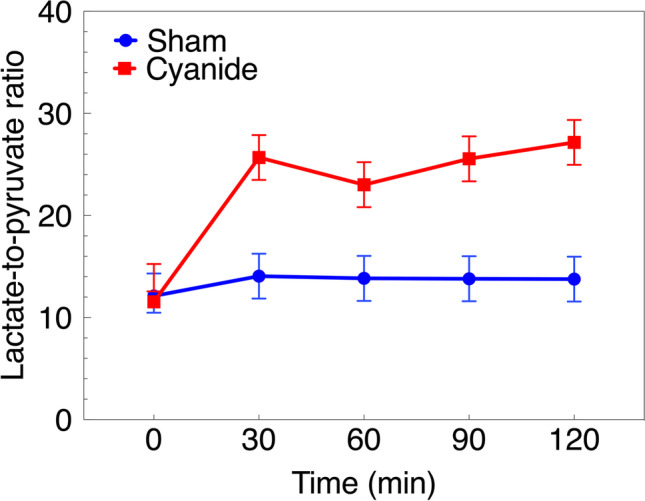
Cerebral microdialysis (cMD) measurements during cyanide exposure. Values expressed as mean ± 95%CI. Sham vs. cyanide groups significantly different at all time points (p < 0.0001 for all). CI: confidence interval.
Mitochondrial Respiration
The oxygen consumption of isolated mitochondria from cortical brain tissue was measured ex vivo at the conclusion of the experiment. CIV-linked respiration as well as all other respiratory states were significantly lower in the cyanide group when compared to the sham group (P < 0.0001 for all respiratory states, Fig. 2 and Supplementary Table 1).
Fig. 2.
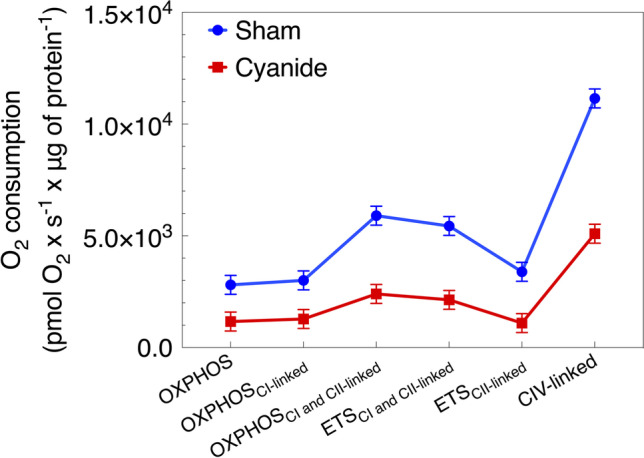
Isolated mitochondrial respiration of brain (cortical) tissue. Respiration states of isolated mitochondria from brain tissue between the sham and cyanide group. Values expressed as mean ± 95% CI. Sham vs. cyanide groups significantly different for all key respiratory states (p < 0.0001 for all). OXPHOS: oxidative phosphorylation; ETS electron transport chain; C: Complex; CI: confidence interval.
ATP Concentration
ATP concentrations were obtained from snap-frozen brain cortical tissue using a commercially available kit. There was a significant decrease in the ATP concentration in brain tissue obtained from the sham group when compared to the cyanide group (median = 3,011,393 vs. 1,155,689, respectively; p = 0.0002, Fig. 3).
Fig. 3.
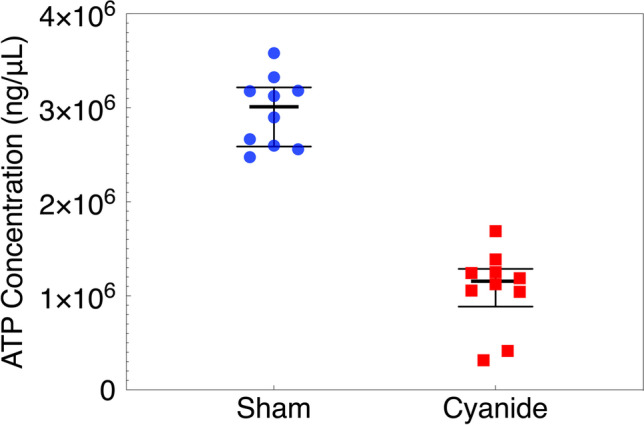
Adenosine triphosphate (ATP) concentrations in brain tissue. ATP concentrations were obtained from snap-frozen brain tissue performed in duplicates from the sham and cyanide group. Error bars are expressed as median ± IQR. ATP: adenosine triphosphate; IQR: interquartile range. Sham and cyanide group ATP significantly different (median = 3,011,393 vs. 1,155,689, respectively; p = 0.0002).
Western Blot for Mitochondrial Content
The relative density of citrate synthase (CS) and OXPHOS complexes in snap-frozen cortical brain tissue were compared in the two groups. Results show no significant difference in the relative density value for both CS and OXPHOS complex in the two groups except for CIII and CIV (*p = 0.003 and **p = 0.0002, respectively, Fig. 4a,b). Supplementary Fig. 1 displays representative images used for western blot analysis of both CS and OXPHOS complexes. The ~ 45 kDa band corresponds to the molecular weight of CS protein which was detected using a polyclonal secondary antibody against a rabbit monoclonal anti-CS primary that is known to bind human CS. A second band was observed at ~ 37 kDa using a monoclonal rabbit anti-GAPDH antibody known to detect human GAPDH.
Fig. 4.

A(a) Citrate synthase relative density value for brain tissue. Relative density value for citrate synthase corrected for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) used as a loading control. Error bars are expressed as median ± IQR. Statistical comparison between the sham and cyanide group. No significant statistical comparison between the sham group and the cyanide group. (b) Mitochondrial respiratory chain complex densities in brain tissue. Relative density value for the oxidative phosphorylation (OXPHOS) complexes corrected for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) used as a loading control obtained in snap-frozen brain performed in duplicates from the sham and cyanide group. Error bars are expressed as median ± IQR. Significant differences between sham and cyanide for CIII and CIV (*p = 0.003 and **p = 0.0002). C: Complex.
Acyl-Coenzyme A (CoA) Thioesters
Acetyl-CoA, Succinyl-CoA, HMG-CoA, B-Hydroxybutyrate (BHB)-CoA, and Malate-CoA were also compared between the sham and cyanide group. There was no significant difference between the two groups for any of the five metabolites (p > 0.05 for all comparisons, Fig. 5).
Fig. 5.

Coenzyme A for brain tissue. Data shown are median ± IQR. No statistically significant differences found between the sham and cyanide group (p > 0.05 for all comparisons). AcCoA: Acetyl-CoA; Succ-CoA: Succinyl-CoA; HMG-CoA: β-Hydroxy β-methylglutaryl-CoA; BHB-CoA: β-hydroxybutyrate CoA; Mal-CoA: Malonyl-CoA.
Discussion
The objective of this study was to investigate alterations in cerebral metabolism in a rodent model of sub-lethal cyanide poisoning. To achieve this, we employed measurements of cerebral metabolism and mitochondrial function to study the metabolic effects of cyanide poisoning in the brain. We also measured mitochondrial respiration in the brain to evaluate the effects of CIV inhibition on other components of the electron transport system [9, 19]. Our key findings from this study include (1) changes in the metabolic response of the brain that were characterized by a significant decrease in mitochondrial function (most prominently in, but not restricted to, CIV respiration) and no changes in mitochondrial content or respiratory chain complexes protein abundance; (2) substantial decrease in cerebral tissue ATP concentrations; and (3) significant increase in cerebral LPR as seen by cMD, corroborating the diminished mitochondrial function in the brain imposed by cyanide.
One of the strengths of our study was the ability to investigate the effect of cyanide on cerebral metabolism using cMD in conjunction with our other measures such as mitochondrial respiration of the brain [20, 21]. We measured concentrations of glucose, glycerol, lactate, and pyruvate across the exposure period to examine changes in cerebral metabolism related to the CIV inhibition caused by cyanide. While there were no significant differences in both the glucose and glycerol concentrations, there was a substantial difference in the lactate/pyruvate ratio, which is an indication of dysfunction within the electron transport system. This result attests to the implications of cyanide on cerebral metabolism leading to high LPR in the cyanide group compared to the control group. In general, an LPR of greater than 20 often indicates cellular dysfunction [22]. For our study, the LPR in the cyanide group was above 20 during the entire exposure period with a normal baseline for both groups, which further supports the mitochondrial dysfunction observed in the brain. cMD is a translational method that is used for clinical application in traumatic brain injury (TBI), so cerebral LPR may serve as a potential translational biomarker for future clinical trials. cMD also has other important applications for future studies in this area that include delivery of therapeutics and measurements of other important substrates that can lead to further identification of other critical pathways amendable to therapy. It is important to note that while cMD has been used clinically in the TBI field, it is considered invasive and has not shown clear benefit over other markers of injury or prognosis in TBI.
We also performed an analysis of mitochondrial respiration to evaluate the effects of sub-lethal cyanide poisoning on cellular respiration in a rodent model. While cyanide poisoning can present clinically with seizures, tachycardia, and hypotension, the physiologic response and its effect on cerebral function have been less studied. The results of our study showed a decrease in mitochondrial function and oxidative phosphorylation, with the most significant inhibition in CIV-linked respiration. Furthermore, we saw a significant decrease in non-phosphorylating respiration of through Complex I and II (ETSCI+CII.) Maximal convergent non-phosphorylating respiration of ETSCI+CII is evaluated by titrating the protonophore, carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone. ETSCI+CII is considered a stress test for mitochondria, as a marker of mitochondrial respiratory reserve. We specifically test the whole pathway of CI + II to CIII to CIV, and that the reduction or respiration here likely is an effect of the downstream blockade at CIV [23].
Furthermore, oxidative phosphorylation produces ATP which is the primary fuel for performing the basic function. In times of distress or injury, the ability to produce ATP may be reduced. To characterize this relationship, we measured ATP concentrations in snap-frozen tissue with the use of fluorometric analysis to better investigate the consequence of altered cellular respiration. Since the primary function of the mitochondria is to generate ATP for cellular bioenergetic function, having the ability to measure this critical molecule in relation to respiration will be important for future studies. The significant decrease in ATP concentration in the cyanide group compared to the sham group corroborates the depleting effect of cyanide poisoning on the mitochondrial function which consequently implicates ATP production. Low levels of ATP in other states of critical illness have also been linked with increased mortality and morbidity [19, 24, 25].
In addition to respiration, we also measured acyl-coenzyme A thioesters (acyl-CoAs). CoAs are metabolites involved in multiple metabolic pathways that involve the mitochondria [26]. Acetyl-CoA, the most abundant acyl-CoA, is the product of several catabolic processes in the mitochondria as well as serving an important function in the formation of citrate in the TCA cycle. It is the primary substrate for anabolic processes in the cytosol such as fatty acid synthesis. Our group developed a Stable Isotope Labeling of Essential nutrients in cell Culture (SILEC) approach using 15N113C3–vitamin B5 (pantothenate) to generate cell lines highly enriched for 15N113C3–labeled acyl-CoAs [27]. SILEC applies SILEC-labeled cells as rigorous isotope-labeled analogs used as internal standard controls introduced prior to quantify acyl-CoA in cells and tissues. The metabolic sequelae of cyanide poisoning include lactic acidosis with increased glycolysis to compensate for impaired mitochondrial respiration. The lactate acidosis leads to impaired hepatic and pyruvate uptake as well as impaired gluconeogenesis supported by elevated concentrations of gluconeogenic precursors such as alanine and glycerol with some indication that there is the activation of fatty acyl-CoA with cyanide poisoning. While our study did not find a significant difference in the CoA measured, larger studies or varying doses of cyanide may result in measurable changes in CoA or longer exposures.
We also investigated if cyanide resulted in alterations in the protein expression of CS and structural OXPHOS proteins using Western blotting. CS is a pace-maker enzyme in the Krebs cycle with a molecular weight of 51,709 Da and is localized in the mitochondrial matrix [28–30]. The OXPHOS mitochondrial antibody cocktail represents the five mitochondrial complexes with different band densities and is used to examine the expression of mitochondrial proteins. The OXPHOS cocktail was used to determine if the protein quantification of components of the OXPHOS system changed due to cyanide poisoning. One of the possible factors in changes in both respiration and ROS generation may be related to a change in the number of mitochondria or content. We demonstrated that there was decreased protein expression of CIII and CIV, but otherwise, no other proteins were significantly different in the two conditions.
One of the limitations of our study was using a fixed dose of cyanide exposure. Future studies may implement varying doses of cyanide to evaluate the dose-dependent effect on cellular function [31]. For future studies, it will be useful to determine a range of doses that will allow us to evaluate for changes in cellular function that may be predictive of toxicity before any clinical manifestations. Another limitation of our study was that we did not specifically measure cyanide concentrations as a confirmatory test. Cyanide poisoning is considered a clinical diagnosis, and our animals exposed to cyanide demonstrated high lactates, hypotension, and tachycardia. Furthermore, in our recent ex vivo study, we demonstrated a dose–response relationship between cyanide and mitochondrial function using blood cells, without confirmatory levels [9]. Another limitation was that we did not measure metabolites in plasma, and hence, we cannot relate the values found in the brain to those in plasma. In general, there is an equilibrium of lactate through the blood–brain barrier but not clear if this is the case with pyruvate as that typically requires active transport and has an important role in the pyruvate and malate shuttle. We also did not perform any behavioral assessments as this was a non-survivor study. Future survivor studies in this area should ideally examine mitochondrial physiology to gauge any persistent cellular dysfunction after cyanide exposure has stopped, as well as neurobehavioral outcomes. Finally, our rodent model used mechanical ventilation to avoid the potential for hypoventilation related to sedation and hence does not strictly represent what may occur clinically, but this controlled experiment allows us to better isolate the effects of cyanide on our proposed metrics.
In summary, in our sub-lethal rodent model of cyanide poisoning, we demonstrate mitochondrial respiratory dysfunction in brain tissue that corresponds to decreased ATP concentrations and an elevated LPR, indicative of brain dysfunction. This model may allow more comprehensive monitoring of brain function in response to future therapies for cyanide.
Supplementary Information
Below is the link to the electronic supplementary material.
{kind=link}
Funding
1. R21ES031243 (Jang).
2. R03HL154232 (Jang).
3. R01HL141386 (Kilbaugh).
4. R56HL158696 (Jang).
5. P30ES013508 (Mesaros).
This publication was made possible by P30 ES013508 from the National Institute of Environmental Health Sciences, NIH. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIEHS, NIH.
Declarations
Conflicts of Interest
None.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lee SK, Rhee JS, Yum HS. Cyanide poisoning deaths detected at the national forensic service headquarters in Seoul of Korea: a six year survey (2005–2010) Toxicol Res. 2012;28(3):195–199. doi: 10.5487/TR.2012.28.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bebarta VS. Cyanide poisoning and antidotes. Emerg Med Australas. 2012;24(6):680; author reply 1–2. 10.1111/1742-6723.12004 [DOI] [PubMed]
- 3.Anseeuw K, Delvau N, Burillo-Putze G, De Iaco F, Geldner G, Holmstrom P, et al. Cyanide poisoning by fire smoke inhalation: a European expert consensus. Eur J Emergency Med : Off J Eur Soc Emergency Med. 2013;20(1):2–9. doi: 10.1097/MEJ.0b013e328357170b. [DOI] [PubMed] [Google Scholar]
- 4.Birngruber CG, Veit F, Lang J, Verhoff MA. Inhaled cyanide poisoning as a vital sign in a room fire victim. Forensic Sci Int. 2017;281:e16–e18. doi: 10.1016/j.forsciint.2017.10.037. [DOI] [PubMed] [Google Scholar]
- 5.Tshala-Katumbay DD, Ngombe NN, Okitundu D, David L, Westaway SK, Boivin MJ, et al. Cyanide and the human brain: perspectives from a model of food (cassava) poisoning. Ann N Y Acad Sci. 2016;1378(1):50–57. doi: 10.1111/nyas.13159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. IV. The respiratory chain. J Biol Chem. 1955;217(1):429–38. doi: 10.1016/S0021-9258(19)57192-7. [DOI] [PubMed] [Google Scholar]
- 7.Chance B, Williams GR, Holmes WF, Higgins J. Respiratory enzymes in oxidative phosphorylation V. A mechanism for oxidative phosphorylation. J Biol Chem. 1955;217(1):439–51. doi: 10.1016/S0021-9258(19)57193-9. [DOI] [PubMed] [Google Scholar]
- 8.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. VI. The effects of adenosine diphosphate on azide-treated mitochondria. J Biol Chem. 1956;221(1):477–89. doi: 10.1016/S0021-9258(18)65266-4. [DOI] [PubMed] [Google Scholar]
- 9.Jang DH, Shofer FS, Weiss SL, Becker LB. Impairment of mitochondrial respiration following ex vivo cyanide exposure in peripheral blood mononuclear cells. Clin Toxicol (Phila) 2016;54(4):303–307. doi: 10.3109/15563650.2016.1139712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dodds RG, Penney DG, Sutariya BB. Cardiovascular, metabolic and neurologic effects of carbon monoxide and cyanide in the rat. Toxicol Lett. 1992;61(2–3):243–254. doi: 10.1016/0378-4274(92)90151-9. [DOI] [PubMed] [Google Scholar]
- 11.Salkowski AA, Penney DG. Metabolic, cardiovascular, and neurologic aspects of acute cyanide poisoning in the rat. Toxicol Lett. 1995;75(1–3):19–27. doi: 10.1016/0378-4274(94)03152-W. [DOI] [PubMed] [Google Scholar]
- 12.Baud FJ, Borron SW, Megarbane B, Trout H, Lapostolle F, Vicaut E, et al. Value of lactic acidosis in the assessment of the severity of acute cyanide poisoning. Crit Care Med. 2002;30(9):2044–2050. doi: 10.1097/00003246-200209000-00015. [DOI] [PubMed] [Google Scholar]
- 13.Jang DH, Piel S, Greenwood JC, Kelly M, Mazandi VM, Ranganathan A et al. Alterations in cerebral and cardiac mitochondrial function in a porcine model of acute carbon monoxide poisoning. Clin Toxicol (Phila). 2021;59(9):801–809. 10.1080/15563650.2020.1870691. [DOI] [PMC free article] [PubMed]
- 14.Janowska JI, Piel S, Saliba N, Kim CD, Jang DH, Karlsson M, et al. Mitochondrial respiratory chain complex I dysfunction induced by N-methyl carbamate ex vivo can be alleviated with a cell-permeable succinate prodrug. Toxicol in vitro : an Int J Published Assoc BIBRA. 2020;65:104794. doi: 10.1016/j.tiv.2020.104794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jang DH, Orloski CJ, Owiredu S, Shofer FS, Greenwood JC, Eckmann DM. Alterations in mitochondrial function in blood cells obtained from patients with sepsis presenting to an emergency department. Shock. 2018;51(5):580–584. 10.1097/SHK.0000000000001208. [DOI] [PMC free article] [PubMed]
- 16.Lewis AT, Forti RM, Alomaja O, Mesaros C, Piel S, Greenwood JC, et al. Preliminary research: application of non-invasive measure of cytochrome c oxidase redox states and mitochondrial function in a porcine model of carbon monoxide poisoning. J Med Toxicol. 2022 doi: 10.1007/s13181-022-00892-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, et al. ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep. 2016;17(4):1037–1052. doi: 10.1016/j.celrep.2016.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Owiredu S, Ranganathan A, Greenwood JC, Piel S, Janowska JI, Eckmann DM, et al. In vitro comparison of hydroxocobalamin (B12a) and the mitochondrial directed therapy by a succinate prodrug in a cellular model of cyanide poisoning. Toxicol Rep. 2020;7:1263–1271. doi: 10.1016/j.toxrep.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jang DH, Greenwood JC, Spyres MB, Eckmann DM. Measurement of mitochondrial respiration and motility in acute care: sepsis, trauma, and poisoning. J Intensive Care Med. 2017;32(1):86–94. doi: 10.1177/0885066616658449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inoue Y, Kaizaki-Mitsumoto A, Numazawa S. Toxicokinetic evaluation during intoxication of psychotropic drugs using brain microdialysis in mice. J Toxicol Sci. 2022;47(3):99–108. doi: 10.2131/jts.47.99. [DOI] [PubMed] [Google Scholar]
- 21.Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011;31(37):13110–13117. doi: 10.1523/JNEUROSCI.2569-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rimachi R, Bruzzi de Carvahlo F, Orellano-Jimenez C, Cotton F, Vincent JL, De Backer D. Lactate/pyruvate ratio as a marker of tissue hypoxia in circulatory and septic shock. Anaesth Intensive Care. 2012;40(3):427–32. doi: 10.1177/0310057X1204000307. [DOI] [PubMed] [Google Scholar]
- 23.Khailova LS, Rokitskaya TI, Kotova EA, Antonenko YN. Effect of Cyanide on mitochondrial membrane depolarization induced by uncouplers. Biochemistry (Mosc) 2017;82(10):1140–1146. doi: 10.1134/S0006297917100066. [DOI] [PubMed] [Google Scholar]
- 24.Callahan LA, Supinski GS. Sepsis induces diaphragm electron transport chain dysfunction and protein depletion. Am J Respir Crit Care Med. 2005;172(7):861–868. doi: 10.1164/rccm.200410-1344OC. [DOI] [PubMed] [Google Scholar]
- 25.d'Avila JC, Santiago AP, Amancio RT, Galina A, Oliveira MF, Bozza FA. Sepsis induces brain mitochondrial dysfunction. Crit Care Med. 2008;36(6):1925–1932. doi: 10.1097/CCM.0b013e3181760c4b. [DOI] [PubMed] [Google Scholar]
- 26.Wang S, Blair IA, Mesaros C. Analytical methods for mass spectrometry-based metabolomics studies. Adv Exp Med Biol. 2019;1140:635–647. doi: 10.1007/978-3-030-15950-4_38. [DOI] [PubMed] [Google Scholar]
- 27.Basu SS, Blair IA. SILEC: a protocol for generating and using isotopically labeled coenzyme A mass spectrometry standards. Nat Protoc. 2011;7(1):1–12. doi: 10.1038/nprot.2011.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ehinger JK, Morota S, Hansson MJ, Paul G, Elmer E. Mitochondrial dysfunction in blood cells from amyotrophic lateral sclerosis patients. J Neurol. 2015;262(6):1493–1503. doi: 10.1007/s00415-015-7737-0. [DOI] [PubMed] [Google Scholar]
- 29.Piel S, Ehinger JK, Elmer E, Hansson MJ. Metformin induces lactate production in peripheral blood mononuclear cells and platelets through specific mitochondrial complex I inhibition. Acta Physiol (Oxf) 2015;213(1):171–180. doi: 10.1111/apha.12311. [DOI] [PubMed] [Google Scholar]
- 30.Weiss SL, Selak MA, Tuluc F, Perales Villarroel J, Nadkarni VM, Deutschman CS, et al. Mitochondrial dysfunction in peripheral blood mononuclear cells in pediatric septic shock. Pediatr Crit Care Med. 2015;16(1):e4–e12. doi: 10.1097/pcc.0000000000000277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cambal LK, Swanson MR, Yuan Q, Weitz AC, Li HH, Pitt BR, Pearce LL, Peterson J. Acute, sublethal cyanide poisoning in mice is ameliorated by nitrite alone: complications arising from concomitant administration of nitrite and thiosulfate as an antidotal combination. Chem Res Toxicol. 2011;24(7):1104–1112. doi: 10.1021/tx2001042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.