

Abstract
Metabolic reprogramming is a major hallmark of malignant transformation in cancer, and part of the so-called Warburg effect, in which the upregulation of glutamine catabolism plays a major role. The glutaminase enzymes convert glutamine to glutamate, which initiates this pathway. Inhibition of different forms of glutaminase (KGA, GAC, or LGA) demonstrated potential as an emerging anti-cancer therapeutic strategy. The regulation of these enzymes, and the molecular basis for their inhibition, have been the focus of much recent research. This review will explore the recent progress in understanding the molecular basis for activation and inhibition of different forms of glutaminase, as well as the recent focus on combination therapies of glutaminase inhibitors with other anti-cancer drugs.
Keywords: anti-cancer drugs, cancer, cell metabolism, crystallography, glutaminase, medicinal chemistry, structure-function, Warburg effect
Plain language summary
Many strategies exist to inhibit cancer progression, from chemotherapy to more targeted therapies that exploit differences between tumors and healthy tissue. One such targeted strategy involves inhibition of the enzyme glutaminase, which converts glutamine obtained from the bloodstream into nutrients that fuel tumor growth. Research into glutaminase is ongoing, with regulation of the enzyme, and novel molecular approaches to inhibit its activity, being key focus areas. Here, we review recent progress on targeting glutaminase enzymes for anti-cancer therapy, including several approaches in which glutaminase inhibitors are combined with inhibitors of other cancer-relevant targets, to increase the overall effectiveness of the treatment.
Tweetable abstract
Inhibition of the enzyme glutaminase is a promising approach to cancer therapy, which draws on unique metabolic differences between tumors and healthy tissues. This review examines recent progress developing such inhibitors.
Cancer cells frequently undergo metabolic reprogramming to satisfy the increased demand for ATP and biosynthetic precursors characteristic of malignant growth. This phenomenon, termed the ‘Warburg effect’, results in enhanced glucose consumption, with the sugar being predominantly converted to lactate regardless of the availability of oxygen, i.e. ‘aerobic glycolysis’. This effectively uncouples glycolysis from the tricarboxylic acid (TCA) cycle. Despite this uncoupling, cancer cells require the TCA cycle to turn, both to produce synthetic intermediates and to generate ‘high-energy’ molecules, and thus require an alternative source of synthetic material as input. Isotope tracing [1–5] showed that in many aggressive cancers including glioblastoma, breast cancer, pancreatic cancer, and liver cancer, glutamine catabolism is the major contributor to the TCA cycle instead of glucose. These cancer cells in effect become addicted to glutamine consumption to replenish metabolic intermediates for energy production and biogenesis. One of the simplest pathways by which glutamine is introduced to the TCA cycle runs through glutaminase. Glutaminase (EC 3.5.1.2), or L-glutamine amidohydrolase, is a mitochondrial enzyme that initiates glutamine catabolism by catalyzing the conversion of glutamine into glutamate and ammonia (Figure 1A), with glutamate then being converted to the TCA cycle intermediate α-ketoglutarate by glutamate dehydrogenase, or by a variety of reversible transaminases [6]. Glutaminase can thus serve as an essential gateway enzyme to support glutamine addiction. The roles of glutaminase in various cancers have been extensively reviewed in the last few years [7–9]. In this review, we will instead focus on a how this class of enzymes is regulated, along with recent strategies being deployed to inhibit this key onco-supportive enzyme.
Figure 1. . Glutaminase family of enzymes share high degree of homology in sequence and catalytic mechanism.
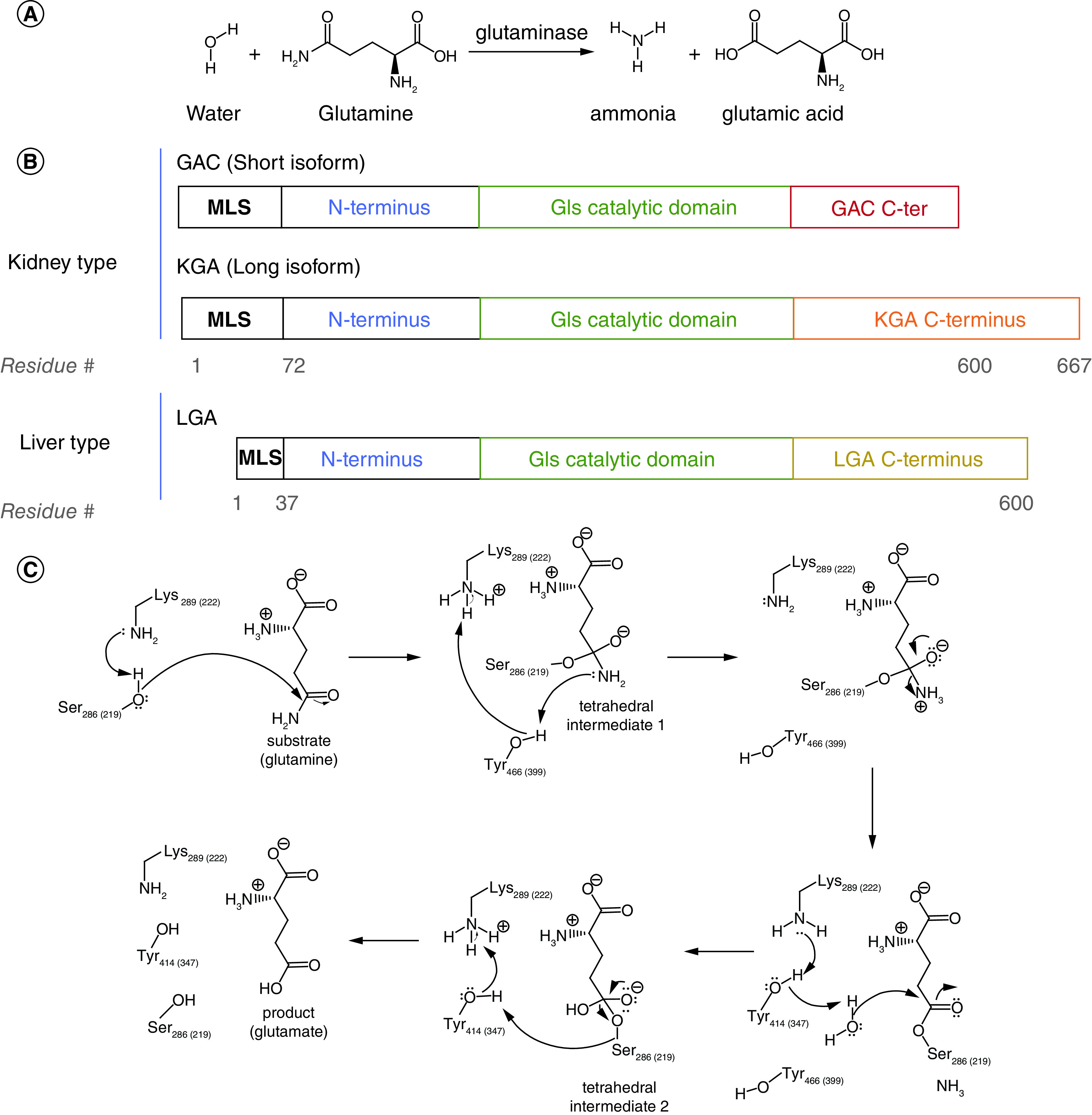
(A) Glutamine hydrolysis as catalyzed by glutaminase enzymes. (B) Glutaminase enzymes are encoded by two independent genes, GLS and GLS2. (C) Mechanism of the hydrolytic reaction of the substrate, glutamine, as catalyzed by the active site residues in glutaminase enzymes. All three active forms of glutaminase share the same active site residues, which are numbered according to human GAC (and human LGA).
C-ter: C-terminus; MLS: Mitochondrial localization sequence.
Two different genes, encoding for different isozymes of glutaminase, exist in mammals (Figure 1B). The two genes, GLS and GLS2, were initially identified in different tissues based on their varying responses to changes in pH [10]. Both GLS and GLS2 play critical roles in the altered metabolism observed in cancer cells, and their differential upregulation in various physiological contexts has been a topic of interest for many researchers. The GLS gene, located on chromosome 2, is predominantly expressed in kidney, brain, small intestine, and is expressed at lower levels in at least 20 other tissues [11]. Through alternate splicing, GLS gives rise to the isoform KGA and its truncated C-terminal variant GAC. The difference in the C termini results in a higher specific activity for GAC, which might explain why it is more frequently overexpressed in aggressive cancers [12,13]. The GLS2 gene, from which the isozyme LGA is derived, is located on chromosome 12 and is expressed mainly in the liver [11]. The glutaminase enzymes all have significant structural homology and have an identical mechanism of enzymatic hydrolysis (Figure 1C). However, the isozymes are not identical in their biological activities, and research is ongoing to examine the differences between GLS and GLS2, which we will outline below.
Metabolic reprogramming has been well established as a major hallmark of cancer cells, and efforts to inhibit glutaminase activity in the past decade have proven to be potentially effective strategies in halting malignant growth [14,15]. Several classes of small molecules that target glutaminase enzymes, ranging from substrate mimetics to allosteric inhibitors, have been identified and optimized. Extensive structure-function characterization efforts, mainly using x-ray crystallography, have revealed several important structural elements of glutaminase that regulate the activity of the enzyme, providing insights into the mechanism of inhibition. In this review, we will discuss the recent progress in understanding the molecular basis for activation and inhibition of different forms of glutaminase. In addition, we will address new research efforts in using glutaminase inhibitors, especially the recent trend toward combination therapies with other anti-cancer drugs.
Controlling the differential expression of glutaminases in cancer cells
Oncogenes regulating glutaminase expression
The basis for the tissue distribution of the different forms of glutaminase is a topic of continuing study. Glutaminase expression can be radically different from one tumor to another (Figure 2 [16,17]A). The choice between whether a cell expresses GLS and GLS2 can have important consequences for cancer progression [18], and so a great deal of effort has been put into understanding the different regulation pathways for either isozyme. The preferential expression of specific forms of glutaminase has been found to be significantly influenced by a number of different factors (Figure 2B). In the hormone-sensitive prostate cancer cell lines LNCaP and LAPC4, KGA expression is promoted by the androgen receptor, and the expression of KGA is reduced when androgen receptors are inhibited during hormone therapy, reducing the cells' ability to satisfy their glutamine addiction. However, glutamine metabolism is ultimately restored through the upregulation of GAC expression [13]. The switch to the GAC isoform, which has higher catalytic activity, results in increased glutaminolysis and hyperproliferation that are independent of the androgen receptors, thus allowing the development of therapeutic resistance for androgen deprivation therapy. This expression of GAC was in fact driven by the oncogenic transcription factor c-Myc, suggesting that c-myc-dependent cancers might also preferentially promote the expression of GAC and become sensitive to glutamine inhibitors.
Figure 2. . Regulated expression of different glutaminase isozymes.
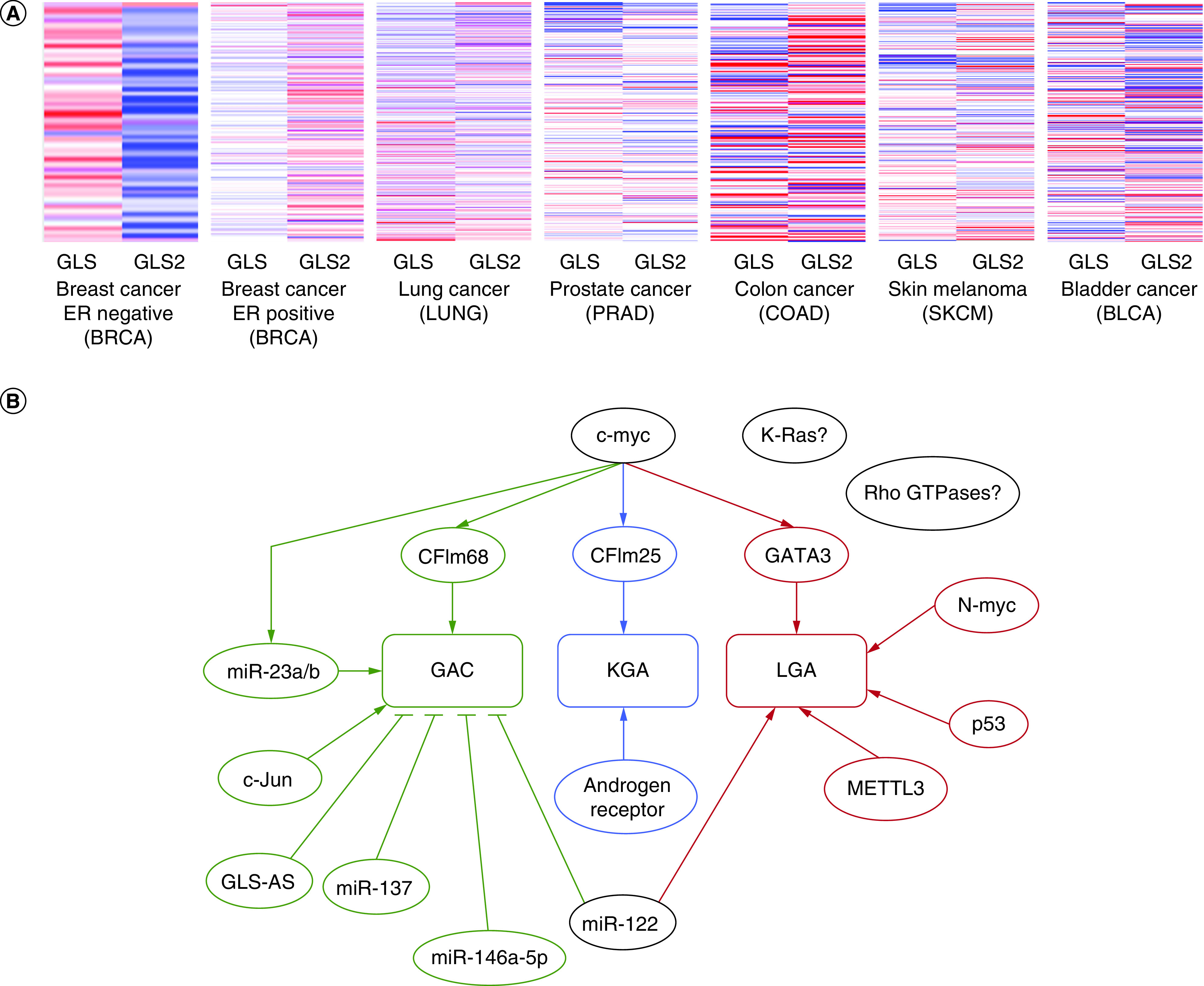
(A) Heat map for differential expression levels of GLS and GLS2 in the most prevalent cancers in the USA [16]. Expression levels are normalized to the respective cancer type study. Relative increase in gene expression is indicated by the red color and relative decreases are in blue. The results shown are based wholly or in part upon data generated by the TCGA Research Network: www.cancer.gov/tcga, and the heat maps were prepared using Xena Browser, analyzed online on 17 September 2022, https://xenabrowser.net/. (B) Many oncogenic factors and transcriptional factors are involved in controlling the differential expression of specific forms of glutaminase in different types of cancer. Whether the regulation is activating, or inhibiting, is indicated by different arrowheads.
One mechanism by which c-Myc drives glutaminase expression works through microRNAs (miRNAs), a class of short, non-coding RNAs. For example, c-Myc has been shown to select for the expression of GLS over GLS2 in P493-6 lymphoma cancer cells and PC3 prostate cancer cells by repressing the microRNAs miR-23a and miR-23b [19]. These microRNAs bind to the GLS gene, but not the GLS2 gene, and thus decrease GLS transcription. Expression of another transcription factor in the same family, N-Myc, correlates with GLS2 mRNA expression in neuroblastoma cells. Mirroring this finding, glutamine deprivation was more damaging to MYCN amplified Kelly cells than to MYCN nonamplified SHEP cells, and siRNA silencing of n-Myc in the Kelly cells reduced the sensitivity to glutamine withdrawal [20].
In some cases, radically different genes drive isozyme preference in different categories of cancer effecting the same tissue. For example, glutaminase isozyme expression is distinct for luminal versus basal breast cancer [4]. GLS2 is expressed in luminal-subtype breast cancer cell lines (MDA-MB-453, T-47D, BT-474, MCF7 cells), and its expression is driven by the luminal-transcription factor GATA3. However, in basal-subtype breast cancers, such as the MDA-MD-231, TSE, Hs 578T, and HCC38 cell lines, the GLS2 promoter is methylated and therefore protein expression is suppressed. These cancer cells instead upregulate GLS expression by utilizing the c-Jun oncogenic transcription factor. The reverse was observed in liver tumors where GLS2 expression was increased upon knockout of GLS [2].
One interesting pathway by which some oncogenes regulate glutaminase expression is by preventing its degradation. For example, GLS2 transcript levels can be protected from degradation, resulting in higher GLS2 expression compared with GLS. Investigation of TE13 and TE1 esophageal squamous cell carcinoma cells revealed that the RNA-methylating enzyme METTL3 enhances the stability of the GLS2 transcript in a YTHDF1/IGF2BP2-mediated m6A-dependent manner [21]. GLS is regulated in a similar matter by Sirtuin 5 (Sirt5). In MDA-MB-231 or MDA-MB-468 breast cancer cells, and in A549 lung adenocarcinoma cells, silencing of SIRT5 resulted in a significant decrease in GLS expression [22]. It was determined that this occurs because SIRT5 desuccynylates GLS at residue Lys164. This, in turn, prevents ubiquitination of GLS at Lys158, resulting in reduced ubiquitination-driven protein degradation.
There are many other cases where oncogenic signals triggered by Rho-GTPases [14], K-Ras [5,23], and c-Myc [24,25] upregulate glutamine metabolism, but little is known about the specific glutaminase isoforms or isozymes involved in these signaling pathways. Such studies tend to rely upon examining the effects of glutamine deprivation in cell culture models, tracking the fate of isotope-labeled glutamine, or measuring ammonia production (a by-product of glutamine hydrolysis by glutaminase). Clearly, more work is required to both fully understand the signaling pathways which lead to glutaminase expression and degradation, and to characterize which form of glutaminase is most involved in any given instance of glutamine dependence.
Role of non-coding RNAs
As mentioned above, miRNA could help oncogenes such as c-myc control the expression of specific isozyme of glutaminase at the transcript level. Besides miR-23a/b, another direct binder to the 3′-UTR of glutaminase mRNAs is miR-122, the most abundant miRNA in the liver. This miRNA suppresses GLS level while increasing GLS2 level [26], as shown through western blot analysis of GLS and GLS2 expression. A clear reduction in GLS expression and an increase in GLS2 level was observed in the liver of liver-tumor-bearing mice, as well as in human primary hepatocellular carcinoma cell line EC4 when miR-122 is introduced. miR-137 plays a similar role in melanoma by targeting GLS and thus acts as a tumor suppressor, as shown in A375 and SK-MEL-1 cells and accompanying mouse xenograft model [27]. Other miRNAs have been found to regulate glutaminase expression through different modes. For example, in chronic myeloid leukemia, miR-146a-5p can target ubiquitin-specific protease 6 (USP6), which in turn affects the level of ubiquitination and hence degradation of GLS using K562-R cells [28]. A more comprehensive screen using glioblastoma cell lines (LN229 and T98G) also highlights other miRNAs and their specific association with either GLS or GLS2 in order to select for specific expression of each isozyme [29].
In addition, long non-coding RNAs (lncRNAs) could also influence the expression of the GLS gene. For example, the alternative splicing of GLS or the GAC:KGA ratio can be controlled at the pre-transcriptional level by long non-coding RNA (CCAT2) and polyadenylation factors (CFIm25 and CFIm68) [12]. The long non-coding RNA CCAT2 has two alleles G and T. The G allele preferentially binds to CFIm25, and the complex can then associate with the poly(A) site within intron 14 of the GLS pre-mRNA to selectively direct splicing toward generating the GAC transcript. The T allele of CCAT2 on the other hand binds with higher affinity to CFIm68 and selects for the production of the KGA transcript. In colorectal cancer, the G allele of CCAT2 is overexpressed [12], and thus upregulates GAC expression to promote metastases and cell proliferation. However, GAC is not always the ultimate choice to support malignant growth in aggressive cancers. Triple-negative breast cancer cells (MDA-MB-231) utilize both GAC and KGA [14,30] and removal of either isoform is sufficient to reduce glutamine catabolism [30]. In pancreatic cancer, it has been found that GLS expression is regulated by a nuclear-enriched antisense lncRNA (GLS-AS) at the post-transcriptional level [31]. Specifically, GLS-AS can inhibit GLS transcription by ADAR1/Dicer-dependent RNA interference, and hence pancreatic cancer cells such as BxPC-3 or PANC-1 cells show low expression of GLS-AS as these cells are dependent on glutaminase activity and glutamine catabolism.
Localization regulation
The localization of each form of glutaminase might also provide insights into their preferential expression. Prediction algorithms, such as TPpred3 [32] and TargetP 2.0 [33], when performed on KGA, GAC, and LGA have consistently predicted that each localizes to the mitochondria. Specifically, when analyzing the full-length cytosolic precursors of the glutaminase isoforms, these servers predict that GAC and KGA share a cleavable mitochondrial localization signal peptide at the start of the N-terminus based on matching cleavage site motifs (i.e. cleavage after the initial 45 amino acids – TPpred3, and 72 amino acids – TargetP 2.0). LGA is also predicted to have an N-terminal signal peptide (cleavage after the initial 21 amino acids – TPpred3, and 40 amino acids – TargetP 2.0). Predictions using the entire sequence (BAR 3.0 [34] and ESLPred [35]) also show similar results. Interestingly, one experimental study provided evidence showing that GAC localizes to the mitochondria while KGA remains in the cytosol in MDA-MB-231 and SKBR3 breast cancer cells, PC3 and DU145 prostate tumor cells and A549 lung cancer tumor cells, using cell fractionation approach, western blot analysis, and immunofluorescence [36]. Similarly, another study that examined LGA expression in human hepatocellular carcinoma, glioblastoma, neuroblastoma cells, and monkey COS-7 cells using immunocytochemistry and subcellular fractionation also suggested that LGA can also localize to the nucleus besides the mitochondria [37]. Therefore, experimental approaches beyond sequence analyses of the localization of the various glutaminases for different contexts of cancer progression might be important in offering an alternative explanation for their differential regulation in various types of cancer cells.
Molecular basis for activation of glutaminase
The glutaminase enzymes have a number of key features that relate to their activity. The N- and C-termini are poorly resolved in crystal structures to date, but are believed to contribute to the regulation of these enzymes, possibly by interacting with binding partners, or by facilitating the formation of large multimeric structures [38]. By comparison, in-depth structural data is available for the bifurcated catalytic domain, many features of which are well conserved among the glutaminase isozymes. Considering GLS and GLS2, sequence alignment with BLAST shows 88% similarity for the catalytic domains from Ile221 to Arg544 in GAC/KGA and Ile154 to Arg477 in LGA. Structure alignment of the catalytic domains of KGA (PDB: 3czd) and LGA (PDB: 4bqm) is shown in Figure 3A. Specifically, all three glutaminases are highly similar in their catalytic sites, with each containing a Ser-Lys-Tyr catalytic triad (Figures 1C & 3B). GAC, KGA, and LGA have all been found to self-associate to form dimers and tetramers in solution (Figure 4A). The dimers are held together mainly by the salt bridges between Asp386 and Lys396. Important structural elements such as the ‘activation loop’ (Gly315 to Glu325 in GAC/KGA and Gly248 to Glu258 in LGA) [39] and the ‘lid region’ (Tyr249 to Lys255 in GAC/KGA and Tyr182 to Lys188 in LGA) [40,41] have been implicated in regulating the activation of GAC and LGA, and as discussed below are also implicated in small-molecule inhibitory mechanisms (Figure 4B). Despite the close similarity between the various isozymes, several small molecules have been found which can discriminate between GLS and GLS2, as discussed further below.
Figure 3. . GLS and GLS2 share highly conserved active sites.

(A) Alignment of the catalytic domains of KGA (green, PDB: 3czd) and LGA (cyan, PDB: 4bqm). The few differences are highlighted in magenta. (B) A closer look at the substrate-binding pocket, with the catalytically active residues shown as sticks.
Figure 4. . Important structural elements of glutaminase.

(A) Glutaminase monomers assemble into dimers and tetramers, with the crystallographic assembly shown. Different important structural units are highlighted in different colors (PDB: 7sbm). (B) Binding sites for glutaminase inhibitors are shown, all determined by crystallography except for 968 and AV-1 (which were determined by molecular modeling).
The transition from dimers to tetramers increases the affinity of glutaminase enzymes for glutamine [36,40,41], which correlates with increased catalytic activity. All three forms of glutaminase exhibit maximal catalytic activity in the presence of the allosteric activator inorganic phosphate (Pi). The activation by phosphate [39] differs markedly between GLS and GLS2, as shown in Table 1. The efficient catalytic turnover of GAC [13] is thought to be a driving force for the preferred expression of this form by many cancer cells, and thus its mechanism of activation has been extensively studied. Phosphate drives the formation of GAC tetramers in vitro [36], and the formation of an active tetramer is essential for maximal catalytic activity [42,43]. It has recently been suggested that the presence of phosphate activates GAC through a conformational shift in the ‘lid region’ of the enzyme. Specifically, residue Tyr249 forms a ‘lid’ which is able to cover the glutamine binding site of the enzyme, and thus converts from a ‘closed’ conformation which covers the active site and blocks substrate entry or product exit, to an open conformation where the residue projects away from the site. The conversion between the two positions of Tyr249 is essential for high affinity glutamine binding and maximal catalytic turnover in the presence of phosphate [41]. The tetramer is generally thought to be the biologically meaningful glutaminase multimer, but addition of large amounts of phosphate can lead to even higher-order structures [36,38]. It remains unclear however why phosphate induces the formation of these higher-order oligomers of GAC in solution, and how this impacts enzyme activation [38,44]. Enhanced catalytic activity can also be achieved by mutating Lys320 in the activation loop of GAC to Ala, which yields a constitutively active enzyme with activity matching that of GAC (wt) in the presence of phosphate, and also results in oligomer formation [43]. In contrast, KGA and LGA do not show the same capability for forming higher order oligomers [38]. In fact, LGA only forms tetramers in the presence of phosphate [40].
Table 1. . GAC, KGA, and GAC show distinct enzymatic activity profiles in the presence of phosphate.
| Isoform | Ka for Pi (mM) | kcat (s-1) | KM for glutamine (mM) | Cooperativity |
|---|---|---|---|---|
| GAC | 80 ± 8 | 22 ± 2 | 1.4 ± 0.4 | Michaelis-Menten kinetics |
| KGA | 76 ± 7 | 10 ± 1 | 1.9 ± 0.4 | Michaelis-Menten kinetics |
| LGA | 8 ± 1 | 2.5 ± 0.3 | 4.0 ± 0.2 | Positive cooperativity |
Table adapted from DeLaBarre et al. [39].
As an interesting contrast, at higher enzyme concentrations, LGA shows positive cooperativity for glutamine catalysis while both GAC and KGA show classical Michaelis-Menten kinetics [40]. The positive cooperativity indicates that LGA binds glutamine at reduced affinity at low concentrations of substrate, only to undergo a switch to a higher affinity state that is driven by an increase in substrate concentration, resulting in a lower overall KM (Table 1). The different responses to substrate binding and catalysis might account for the differences in the mechanism of activation for these enzymes, offering a basis for the preferential expression of each glutaminase in varying contexts. As a result, it may be possible that new competitive inhibitors can be developed to better compete with glutamine in targeting LGA, given the isozyme's reduced affinity for the substrate at low concentration. Further mechanistic investigation of substrate binding in LGA can provide a potential avenue for the development of effective competitive inhibitors in LGA.
Glutaminase can also be activated by several other bivalent anions such as sulfate [42], though to a lesser extent. Isolation of intact mitochondria also shows an enhanced rate of glutamine hydrolysis in the presence of ADP, ATP, citrate, and tricarboxylic acid [45,46]; however, these effects have not been investigated with recombinant enzymes. Determination of the local concentration of inorganic phosphate at the sites where the glutaminase enzymes localize, along with further studies investigating the metabolic profiles in cancer cells with enhanced glutaminase activities, will help to shed light on the physiological mechanisms of glutaminase activation.
Molecular basis for glutaminase inhibition
Competitive inhibition by glutamine analogs
Efforts targeting glutaminase enzymes, mainly GAC and KGA, have found success using both substrate mimetics and small-molecule allosteric inhibitors [47,48]. Glutamine analogs share structural similarities with the substrate but typically contain other functional groups in place of the amide moiety, and they bind directly to the catalytic active site (Figure 4B). 6-diazo-5-oxo-L-norleucine (DON) and its prodrugs JHU-083 and DRP-104, azaserine, chloroketone, acivicin, and their derivatives function as competitive inhibitors versus the substrate, either through an irreversible or reversible binding mechanism (Figure 5 [48]A). Among these compounds, DON exhibits the lowest IC50 value with the recombinant glutaminase enzymes compared with the other analogs [49], and thus has been the focal point for optimization. The hydrolysis of glutamine catalyzed by GAC/KGA involves a nucleophilic attack on the acyl carbon by Ser286 in the enzyme active site, and subsequent proton transfer from Tyr466 facilitates the deamidation/hydrolysis reaction. The same nucleophilic attack occurs at the acyl carbon in DON, but without subsequent hydrolysis. Instead, the diazo group is released as Ser286 forms a covalent bond with the remainder of the small molecule, locking in the suicide inhibition. Co-crystallization with the enzyme (PDB: 4O7D) shows that this glutamine mimetic binds in a highly specific manner to the active site, making hydrogen bonds with Tyr249, Gln285, Ser286, Asn335, Glu381, Asn388, Tyr414, Tyr466 and Val484 [49]. Aside from DON, there have not been any direct structural characterizations for glutaminase and other glutamine mimetics. However, a closely related enzyme, γ-glutamyltransferase, which also catalyzes glutamine hydrolysis, has been crystallized with DON [50], azaserine [51], and acivin [51] in three independent structures (PDB: 5Y9B, 2Z8I, and 2Z8K respectively). The similarities in these structures suggest that the glutamine mimetics are all binding to γ-glutamyltransferase in a similar manner (Figure 5B), and presumably the same would be true for GLS. Unfortunately, these structures also demonstrate how glutamine analogs have significant off-target effects. Beyond GLS and γ-glutamyltransferase, glutamine mimetics inhibit many other enzymes that hydrolyze glutamine [52], including but not limited to asparagine synthetase, CTP synthetase, NAD+ synthetase, and carbamyl phosphate synthetase.
Figure 5. . Glutamine analogs and their conserved mechanism of action at the glutamine-binding site.
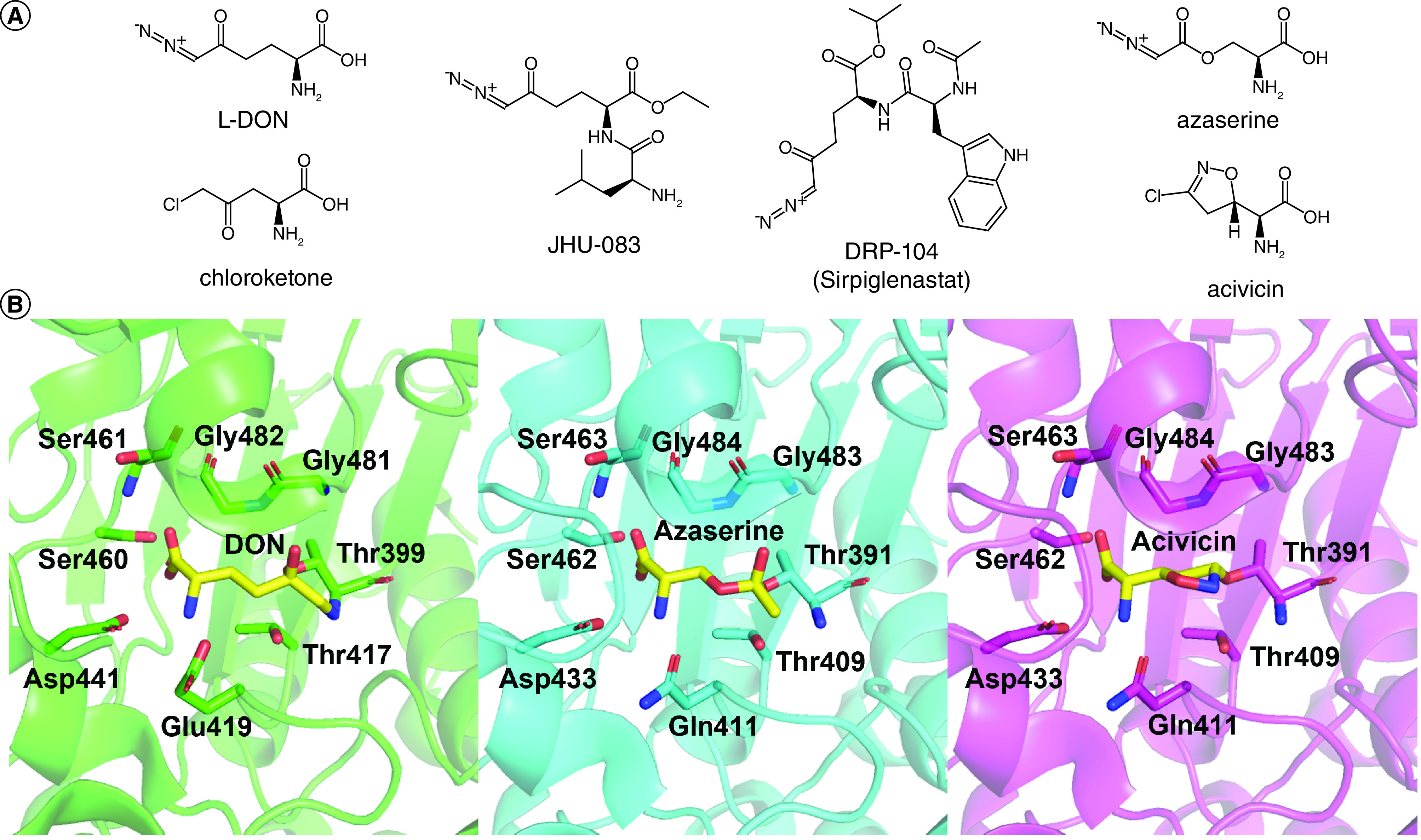
(A) Structures of various glutamine analogs have shown inhibition of glutaminase activity (B) Alignment of the active sites of γ-glutamyltranspeptidase when bound to different glutamine mimetics showed highly similar interactions between the analogs and the catalytically active residues. From left to right is the structure of Bacillus licheniformis γ-glutamyltranspeptidase with DON (green, PDB: 5Y9B), Escherichia coli γ-glutamyltranspeptidase with azaserine (cyan, PDB: 5Z8I), Escherichia coli γ-glutamyltranspeptidase with acivicin (magenta, PDB: 2Z8K).
Despite its anti-tumor effects [15,52], DON showed high levels of gastrointestinal toxicity and was removed from clinical trials after phase II as patients experienced severe nausea and vomiting [52]. For this reason, efforts have been made to create prodrugs based on DON that might bypass the gastrointestinal tract by adding protecting groups on both the amine and carboxylate functional groups [53]. With this strategy, the prodrugs showed increased stability in human plasma, swine liver and intestinal homogenates. Further, one of the better studied such prodrugs, JHU-083, has been shown to be effective in models of brain cancer [54], with 10 μM JHU-083 significantly slowing the growth of several different MYC-driven medulloblastoma cell lines, including one derived from a PDX tumor. Similar results were obtained with another pro-drug, JHU-395 [55]. These compounds also showed improved human tumor cell-to-plasma partitioning and are better directed to the central nervous system. The compound DRP-104, currently in a clinical trial, has shown promising results with lower toxicity compared with DON, and sustained glutaminase inhibition in CT26 murine colon carcinoma models [56].
Inhibition by allosteric inhibitors
The ability to specifically inhibit the glutaminase enzymes greatly advanced upon the development of small-molecule allosteric inhibitors for GAC/KGA. These compounds are also more potent than the glutamine analogs, and mainly target two distinct allosteric sites on the enzyme: the dimer-dimer interface and the monomer-monomer interface. Kinetics studies thus far suggest that the allosteric inhibitors of glutaminase are either uncompetitive or noncompetitive [57,58].
BPTES class
The first class of inhibitors are all derived from the initial hit compound bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl) ethyl sulfide (BPTES) (Figure 6A). Among these is CB-839, which was optimized for better potency from BPTES [59], and is currently the lead compound for this class, being in phase II clinical trials (Table 2). A second BPTES derivative, IPN60090 [60], has also entered clinical trials (Supplementary Table 1, see Glutaminase inhibitors and conventional therapies), but is considerably less well studied.
Figure 6. . Structures of the small-molecule allosteric inhibitors that target glutaminase enzymes.
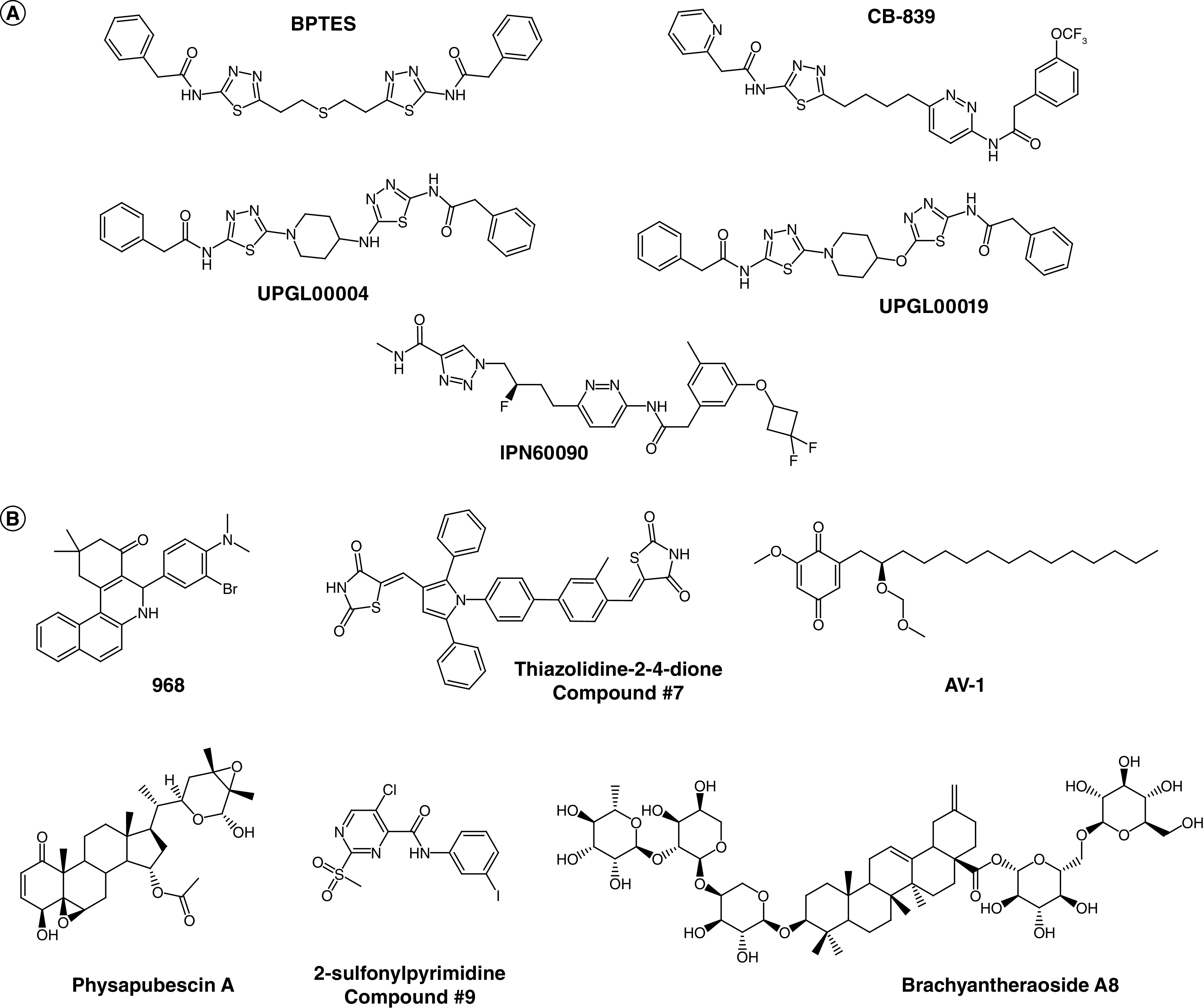
(A) Structures of representative compounds from the BPTES class that specifically target GLS. (B) Structures of other allosteric inhibitors that can target GLS and GLS2.
Table 2. . Current clinical trials with CB-839.
| Indication | Trial No. (NCT___) | Phase | Status (Number of patients) | Ref. |
|---|---|---|---|---|
| NF1 positive tumors & nerve sheath tumors | 03872427 | II | Active – Recruiting (108) | |
| Hematological tumors | 02071888 | I and II | Completed (25) | |
| CB-839 was well tolerated at and above effective doses Dosing reduction from TID to BID with food improved the PK profile and resolved LFT elevations |
[61] | |||
| Healthy patients (PK/formulation study) | 02944435 | I | Completed (14) | |
| Leukemia | 02071927 | I | Completed (43) | |
| Observations included CB-839 being generally well tolerated for prolonged periods of BID dosing with food, and reductions in marrow and peripheral blast counts in acute leukemia populations | [62] | |||
| Solid tumors, multiple indications | 02071862 | I | Completed (210) | |
| Rapid absorption and clearance with a half-life of about 4 hours, inhibition was targeted in tumors. | [63] | |||
Additional details on the trials are provided when available (trials marked with *).
BID: Twice a day; LFT: Liver function test; PK: Pharmacokinetics; TID: Three-times a day.
Despite being the lead compound in clinical trials, CB-839 shows higher calculated logP (a measure of lipophilicity) and lower lipophilic efficiency (LipE, a measure of inhibitory potency relative to lipophilicity, and often used as a predictor of long-term clinical success) than BPTES [64], and also requires an as-yet unexplained long pre-incubation times to achieve maximum potency [57]. Thus, other derivatives are being developed to improve the pharmacokinetics of the compounds in this class, including for example the UPGL series of allosteric inhibitors or IPN60090 [60], which substitute a heterocyclic ring in place of the flexible core of BPTES or CB-839 in order to constrain the conformational space of the molecules and reduce the entropic penalty for binding, while simultaneously improving metabolic stability and increasing LipE [65]. Moreover, improvement in the delivery strategy for BPTES, such as encapsulation of the compound in dense poly(ethylene glycol) nanoparticles, also showed enhanced drug exposure and lower liver toxicity in mice bearing orthotopic pancreatic tumors [66,67].
The BPTES class of compounds are highly selective for GLS [39]. The difference in potency seems to be dictated by the activation loops of GAC/KGA and LGA. Extensive structural characterizations using x-ray crystallography have been carried out for compounds in this class. It has been found that these compounds occupy the space between two adjacent activation loops of GAC and KGA, which span from residues Gly315 to Glu325. Important hydrogen bonds that stabilize the enzyme-drug interactions involve the backbones of Leu323, Phe322, and Tyr394. By binding near the dimer-dimer interface of the glutaminase tetramers, these compounds can stabilize an inactive tetrameric conformation of the enzyme [42]. However, despite the knowledge of this general mechanism, traditional cryo-cooled x-ray crystallography has not been able to explain the differences in potency among the compounds in the BPTES class.
Serial room-temperature x-ray crystallography, which circumvents the cryo-cooling step traditionally used in x-ray crystallography in order to examine molecules in a more natural conformation, has helped to provide important insights into the chemical determinants underlying the differences in potency between BPTES-type molecules by capturing distinct conformations of the inhibitors [64]. When comparing BPTES (IC50 = 371 nM) with the more potent UPGL00004 compound (IC50 = 29 nM), it was observed that BPTES forms a more extended, linear conformation compared with the cup-shaped conformation of UPGL00004. This linear conformation demonstrates that even when bound to the enzyme, the less potent inhibitor molecule is more flexible and undergoes additional conformational orientations which do not make optimal contacts with the protein binding site, before eventually settling into an energetically favored conformation that resembles that of the more potent inhibitors. Moreover, these two compounds share a number of hydrogen bonds with the activation loop, but the average B-factor for BPTES is much higher than that of UPGL00004, further indicating that the interactions between BPTES and glutaminase are less stable. Indeed, an effort in which macrocyclic molecules were prepared that maintained the cup-like orientation even with a flexible molecular core did show improvements in the IC50 compared with parent compound BPTES, though the addition of the various alkyl linkers also worsens the pharmacological properties [68].
The development of fluorescence spectroscopic read-outs for the binding of inhibitor molecules to the glutaminase enzymes have also contributed greatly to our understanding of their mechanisms of action. For example, the substitution of a tryptophan residue for Phe327 at the activation loop showed that the BPTES-class of compounds compete with inorganic phosphate in modulating the activation loop conformation [42]. This tryptophan mutant, together with another tryptophan substitution for Tyr466 in the active site of GAC, has provided valuable tools for assaying the binding affinities of different compounds in this class of inhibitors [42,64]. Additional mechanistic insights into how different amino acids in the activation loop contribute to the binding of the BPTES-class of inhibitors have also been obtained using these fluorescence readouts. For example, they have shown that mutating Lys320 to an alanine abolished the binding of the UPGL00019 to GAC, indicating that the lysine side chain interacts with the terminal phenyl rings of the inhibitor. This is further support for the previously identified high B-factors for Lys320 and these terminal rings in the co-crystal structure of GAC and e.g. UPGL00019 or CB-839, and strongly suggests that one important role for the terminal aromatic rings in these molecules is to flexibly maintain a binding contact with a mobile Lys320.
This class of allosteric compound has been shown to be effective in a number of different cancers. The compounds have been most heavily studied in the context of triple-negative breast cancer, where potent compounds such as CB-839 [57] or UPGL00004 [69] can inhibit the growth of models such as MDA-MB-231 or Hs578T with nanomolar potency. However, CB-839 especially has been widely studied in other contexts, and has been demonstrated to be effective against e.g. A549 and H460 lung tumor cells [70], Cal-27 tongue cancer cells [71], and U226 Bzr multiple myeloma cells [72]. These are only some of the tumor classes that have been successfully inhibited in vitro by BPTES-class allosteric regulators of GLS, and it is likely that more will be discovered in the coming years.
Benzophenanthridinone class
Another group of allosteric inhibitors is comprised of molecules derived from benzophenanthridinone, with the lead compound being 968 (Figure 6B). This compound was first used to show the importance of GAC activity for oncogenic transformation in cell culture, as well as for the growth and survival of cancer cell lines and tumor growth in mice [14], and it has since been shown to exhibit a slight selectivity (<= twofold) for inhibiting LGA, compared with GAC [4]. 968 and its optimized analogs all contain a ‘hotspot ring’ (generally an ortho-bromo, N-methyl-amino-phenyl moiety or a structurally similar group). This so-called hotspot ring was used to precipitate its target, GAC, from oncogenic transformed NIH 3T3 cell lysates [14]. Using GAC molecules labeled with fluorescent reporter groups, it was shown by FRET that 968 uses an inhibition mechanism distinct from that of BPTES. Specifically, 968 does not interfere with the dimer-dimer interaction within the glutaminase tetramers. Instead, it was reported to preferentially bind to the monomeric form of GAC, with the requirement that it associates with the enzyme prior to the binding of activators, as it engages and stabilizes an inactive state. 968 is also able to inhibit the catalytic activity of the LGA isozyme though the mechanism of inhibition has not been characterized in detail as with GAC [4].
Structure-activity relationship studies were carried out for the benzophenanthridinone class of inhibitors. A docking study suggested that 968 binds in a hydrophobic pocket between the monomer-monomer interface of GAC, at the junction where the N- and C-termini meet the catalytic domain [58]. This binding position was supported by site-directed mutagenesis that caused a corresponding reduction in 968 potency, but has not been unambiguously verified e.g. by x-ray crystallographic structural determinations, as the low solubility of this compound and its preference for binding to the enzyme monomer (which exists only at relatively low protein concentrations) has hindered crystallization efforts. Other structural approaches have been complicated by the intrinsic absorbance of 968, which shares the same wavelength as tryptophan, rendering biophysical studies similar to those conducted on the BPTES-class compounds challenging. In addition, as the binding affinities of these compounds are reduced with increasing enzyme concentration, characterization methods which require high protein concentrations, such as isothermal titration calorimetry, or surface plasmon resonance, are not ideal for studying this class of glutaminase inhibitors. A newer technique, microscale thermophoresis, might offer an alternative as it requires less protein for measurement [73], but to the best of our knowledge no such study has yet been attempted.
Like in the case of the BPTES-class of inhibitors, 968 has been used to inhibit the growth of a number of different cancers. When originally discovered, 968 was shown to have potency against MDA-MB-231 and SKBR3 breast cancer cells [14]. However, in more recent years, 968 has shown efficacy against HEY, SKOV3, and IGROV-1 ovarian cancer cell lines [74], A549, Spc-A1, H23, H292, and H1299 non-small cell lung cancer cells [75], and HepG2, 7402, and LM3 hepatocellular carcinoma cells [76]. Crucially, in these studies, 968 was shown to be relatively ineffective at blocking the growth of e.g. human mammary epithelial cells [14], bronchial epithelial cells [75], and EVC304 non-malignant endothelial cells [76], helping to demonstrate the potential therapeutic window for clinical glutaminase inhibitor use.
Other classes of allosteric inhibitors
There are several additional lead compounds from other classes of inhibitors that target glutaminase enzymes, many of which were identified through high-throughput screens (Figure 6B). Thiazolidine-2,4-dione derivatives [77] possess unique structures from the previous classes of molecules, and can inhibit both GLS and GLS2. Molecular modeling and site-directed mutagenesis studies with KGA suggest that these compounds occupy the active sites of the glutaminase enzymes, likely engaging in a hydrophobic interaction with Arg387, and thus might explain the low selectivity toward the different enzyme forms. A recent effort to create high throughput assays for glutaminase inhibitors using a fluorescence enzyme-coupled read-out identified novel scaffolds with 2-sulfonylpyrimidine/2-thiopyrimidine derivatives for inhibitor design [78]. These scaffolds were found to engage in noncompetitive inhibition versus substrate and to bind and stabilize GAC in a tetrameric state. Structural modeling suggested that the lead compound C9 might bind to the activation loops of GAC similarly to BPTES. However, while BPTES decreases polydispersity of the enzyme upon binding (i.e. caused the enzyme to reduce aggregation), C9 did not exhibit such an effect. Natural-product inhibitors have also been identified, including physapubescin [73] and brachyantheraoside A8 [79]. Interestingly, physapubescin shares structural similarity with the benzophenathridinone class of inhibitors, displaying even lower IC50. However, the interactions of these compounds with the glutaminase enzymes have not been well characterized. Further structural characterizations and structure-activity relationship studies across different classes of compounds might provide new insights into their mechanism of inhibition.
In addition, since the role of LGA in cancer progression has not gained significant attention until recently, inhibitor development specific for this isozyme also warrants additional efforts. Currently, no other molecule besides AV-1 [80], a natural alkyl benzoquinone, can selectively inhibit LGA over GAC/KGA (almost tenfold selectivity against LGA, IC50 being 0.28 μM vs IC50 being 2.1 μM for KGA). Molecular docking also suggested that this compound binds at a distinct site from DON, BPTES, or 968, but as is the case for most of these allosteric compounds, a definitive binding site has yet to be identified by structural approaches.
Combinational therapies with glutaminase inhibitors & other anti-cancer treatments
Combining the effects of small molecules that target glutaminase
The expression of the different glutaminase isozymes is highly dependent on the type of cancer cells, and upstream regulations could influence the switch from one isozyme to another, as discussed above. Moreover, GLS2 has been found to compensate for loss of GLS in certain cases [2], and in such cases, it may be beneficial to simultaneously inhibit all major glutaminase isozymes at once. The effect of simultaneously inhibiting GLS and GLS2 is most clearly demonstrated with the use of compound 968, an inhibitor that has been shown to inhibit all forms glutaminase (KGA, GAC, and LGA). However, it is important to also note the alternate role of GLS2 as a tumor suppressor in some contexts, and that we still do not have a clear understanding on the role of glutamine catabolism in these cases. Interestingly, it has also been shown that combining 968 and BPTES can have an additive or synergistic effect in inhibiting the growth of triple-negative breast cancer cells which predominantly express GAC, suggesting that the binding of one class of inhibitors to GAC may enhance the binding of the other class of compounds [58]. Future studies examining the combination of substrate mimics like DON and the BPTES-family of inhibitors might also result in synergistic effects. Such combination approaches could eventually contribute to lowering the treatment dose of each drug and limit off-target effects.
Combining inhibitors of glutaminase & other metabolic enzymes
In cancer cells that are addicted to glutamine, glucose uptake is also frequently highly upregulated to support subsequent aerobic glycolysis, producing high levels of lactate. Inhibition of glycolysis has the potential to synergize with the inhibition of glutamine metabolism. Piperazine-2-one (Glutor), an inhibitor of glucose transporters GLUT-1/-3, has been found to sensitize colon cancer cells (HCT 116) to treatment with CB-839 [81]. The presence of 1 μM Glutor resulted in a GI50 value of less than 50 nM for CB839, compared with a GI50 value of 12 μM in the absence of Glutor. Conversely, the presence of 5 μM CB-839 reduced the GI50 for Glutor by 40-fold to a value of 10 nM compared with a GI50 of 428 nM in the absence of CB-839. In addition to blocking glucose uptake, the inhibition of other glycolytic enzymes represents a potential strategy worth exploring. For example, it has been found that under conditions of glutamine deprivation, or upon silencing of GLS, glioblastoma becomes dependent on pyruvate carboxylase, which allows cells to utilize glucose-derived pyruvate as an energy source instead of glutamine [82]. Thus, dual targeting of both pyruvate carboxylase and glutaminase could be a viable therapeutic strategy when a tumor acquires resistance to therapies targeting glutamine metabolism.
mTOR is generally considered to be a master regulator of cell function, and hyper-activation of mTOR has been linked to a number of cancers. Inhibition of the mTOR complexes mTORC1 and mTORC2 has proven to be an important synergistic strategy together with glutaminase inhibition, as it was found that inhibition of mTOR resulted in a dependence on glutamine metabolism in lung squamous cell carcinoma [83], renal cell carcinoma [84], and ovarian cancer [85]. In a lung squamous cell carcinoma model that became resistant to the mTOR inhibitor MLN128, the GSK3a/b pathway was activated to upregulate c-MYC and c-JUN expression, which in turn increased the expression of glutaminase enzymes. In ovarian cancer cell lines, GLS promoted resistance through STAT3 signaling [85]. The combined use of CB-839 together with an mTOR inhibitor (i.e. MLN128, PP242, or rapamycin) overcame this resistance. Similarly, inhibition of glutaminase sensitized glioblastoma multiforme cells derived from patients and ovarian cancer cell lines to treatments that target mTOR, and the combination of both treatments blocked tumor growth in xenograft models [86]. The combinational therapy might even offer synergistic effects, as shown in renal cell carcinoma cell lines, with the combination index being less than 1, i.e. indicative of synergy [84]. Furthermore, treatment with an Hsp90 inhibitor, in addition to inhibiting mTOR and GLS, was shown to induce apoptosis in cell xenograft tumors with tuberous sclerosis complex deficiency [86].
Blocking amino acid uptake has also been found to be an effective strategy to overcome resistance and insufficiency when inhibiting glutamine catabolism alone. A recent study showed that compound V-9302, which inhibits the amino acid transporter ASCT2, sensitizes glutamine-addicted liver cancer cells to CB-839 treatment [87]. Specifically, using a hepatocellular carcinoma xenograft mouse model, the combination treatment resulted in a reduction of glutathione levels, inducing cytotoxic effects and apoptosis in the liver cancer cells, and hindered xenograft growth in vivo. Blocking amino acid synthase can also provide an additional level of inhibition when glutamine catabolism is reduced. Dual siRNA-knockdowns of GLS in cancer-associated fibroblasts and glutamine synthetase in neighboring stromal cells resulted in a reduction in tumor weight, as well as in the number of tumor nodules, and metastases in a tumor-bearing orthotopic mouse model for ovarian carcinoma [88]. In addition, blocking asparagine catabolism has also been shown to synergize with glutaminase inhibition. This is because under conditions of glutamine deprivation, asparagine can restore glutamine biosynthesis by upregulating glutamine synthetase as well as activate mTORC1 to support glutamine-independent cell growth [89]. Inhibition of asparagine usage can block these processes. For example, treatment with recombinant L-asparaginase, together with the glutaminase inhibitor DON, showed a synergistic reduction of cell proliferation, induced apoptosis, and autophagy in several glioblastoma cell lines (U251, U87, and SF767) [90].
Inhibiting lipid metabolism together with glutaminase activity is another potential combination strategy. Triple-negative breast cancer cells, when treated with 968, underwent further metabolic reprogramming to increase lipid catabolism over time [91]. These cells, upon becoming resistant to glutaminase inhibition, showed increased CPT1 activity levels, an enzyme involved in lipid processing [92]. Therefore, targeting enzymes important for lipid metabolism offers promising potential in overcoming resistance in breast cancer cells.
Combining glutaminase inhibitors with checkpoint inhibitors
One mechanism by which cancer cells survive is by evading the immune system. They frequently do so by targeting receptors on immune cells that send kill-signals to those cells, activating immune checkpoints. Targeting the immune checkpoint via blockade therapy in cancer has gained tremendous interest in the past decade, and clinical trials involving combination therapy between immune checkpoint inhibitors and other cancer treatments have become a preferred strategy due to the enhanced efficacy versus checkpoint inhibitors alone [93]. One such combination approach centers on glutaminase inhibition. Upon activation, T-cells undergo metabolic reprogramming, switching from oxidative phosphorylation to aerobic glycolysis to support the rapid growth and proliferation necessary for an effective immune response [94]. To evade the immune system, cancer cells express PD-L1, which upon interaction with PD-1 on activated T cells impairs glycolysis and glutaminolysis in these cells, and promotes apoptosis [95]. Since immune cells compete with cancer cells in the tumor microenvironment and decrease the tumor-accessible supply of nutrients and oxygen [96], glutamine metabolism becomes an attractive target for combination therapy with immunotherapy. In fact, inhibition of the PD-1/PD-L1 axis shows synergistic effects when combined with inhibitors of glutamine metabolism in restoring the immune response by T cells on patient-derived melanoma, as well as in a colon carcinoma model, and in clear cell ovarian carcinoma [3,97,98].
In addition to immune checkpoint inhibitors, cell cycle checkpoints have also proven to be promising combinational therapeutic targets to use together with glutaminase inhibitors. Upon inhibition or depletion of the key cell cycle regulators CDK4 or CDK6 in colorectal cancer cells, there is an increase in myc expression, leading to an enhancement in glycolysis and mitochondrial glutamine metabolism [99]. Moreover, inhibiting glutaminase activity by CB-839 overcame the acquired resistance of esophageal squamous cell carcinoma to CDK4/6 inhibition [100]. Synergistic reduction of cell growth and tumor burden were observed when using both treatments simultaneously. The dual inhibition of CDK4/6 and glutaminase has now entered clinical trials in combination with CB-839 (Supplementary Table 1, see Glutaminase inhibitors and conventional therapies). Given that cell cycle progression and glutamine metabolism are interconnected, therapeutic strategies that combine cell cycle checkpoint and glutaminase inhibitors offer promising research potential [101].
One particular difficulty in combining glutaminase and immune checkpoint inhibitors, however, is that glutaminase also plays a role in T cell function. For example, in freshly activated T cells [102], c-Myc was correlated with the expression of GLS2, but not GLS, during the cells growth phase prior to initiation of proliferation. Similarly, when CD8+ T cells in a lung cancer model were activated by anti-PD1 therapy, GLS inhibition with CB-839 was found to inhibit clonal expansion of the activated cells [103]. This mirrors similar findings in which CD4+ T cell expansion shows dependence on glutamine and the activity of GLS [104]. However, competing research suggests that T cells can adapt to glutamine denial, and that the GLS inhibitor JHU-083 in fact leads to metabolic reprogramming resulting in enhanced growth, proliferation and survival [3]. This in turn aligns with research showing that inhibition of CDK4 and/or CDK6 [105] decreased T cell proliferation, but enhanced overall T cell activity and tumor infiltration. Given the many similarities between cancer and immune cell metabolism, these various and sometimes conflicting reports are not entirely surprising. These findings also highlight the importance of selecting the appropriate model systems that encompass glutamine catabolism in both tumor and immune cells. Immune-competent models are particularly important when studying checkpoint inhibitors. But this also limits the studies to highly artificial systems in which human immune systems are implanted into mice, or fully murine systems, which do not necessarily replicate a human cancer. In the end, many complications need to be overcome to allow any combination metabolic & checkpoint therapy to be used in the clinic.
Glutaminase inhibitors & conventional therapies
Many different combinations of traditional therapeutic treatments of cancer have been examined for synergistic effects with glutaminase inhibitors (Supplementary Table 1). Current clinical trials that use a combinatorial approach with glutaminase inhibition include chemotherapies targeting DNA, topoisomerase, proteasome inhibition, PARP inhibition, and growth receptors' inhibition. Enhanced outcomes have also been observed when combining radiotherapy with CB-839 in head and neck squamous cell carcinoma cell lines and xenograft mouse models [71]. For a subset of non-small cell lung cancer having STK11/LKB1 mutations, GLS inhibition was able to overcome resistance to standard radiation [106].
Metformin, a conventional therapy for diabetes, has been found to have synergistic effects with glutaminase inhibitors. Metformin is widely known to primarily limit cellular glucose uptake in diabetic patients [107], but has recently entered phase III clinical cancer trials. A recent study showed that metformin strongly synergized with CB-839 in inhibiting osteosarcoma cell growth and metastasis [108]. Metabolic profiling using [13C]glucose and [13C15N]glutamine with orthotopic tumors derived from patients with pancreatic ductal adenocarcinoma showed that a combination of BPTES-nanoparticles and metformin is efficient in reducing tumor cell growth as they complement each other in inhibiting glutamine and glucose metabolism, respectively. Similarly, by promoting aerobic glycolysis and lactate production, metformin also causes prostate cancer cells to be more susceptible to glutaminase inhibition by 968 and BPTES [109]; in a reciprocal fashion, glutamine deprivation sensitizes these cells to metformin treatment.
Complications arising from glutaminase inhibition
Despite the general promise shown to date by glutaminase inhibitors, both when used on cell culture models, animal models, and clinical patients, a number of complications may arise when glutaminase inhibitors are employed. Perhaps preeminent among these is that, in some cases, glutaminase (and particularly GLS2) has been suggested to be a tumor suppressor, rather than a tumor promoter. For example, GLS2 has been shown to be promoted in some cases by the tumor suppressing transcription factor p53 [110,111]. In hepatocellular carcinomas (HCC), the GLS2 promoter was found to be hypermethylated in the majority of samples. Reduced expression of GLS2 in HCC compared with healthy tissue was verified by staining tissue microarrays for protein, and by using real-time PCR to detect GLS2 mRNA. Overexpression of GLS2 in Huh1 and Huh7 cells was shown to block PI3K/AKT activation, although the mechanism causing that reduction in activity was not determined [112]. Largely similar effects were demonstrated in T98G glioblastoma cells, which have naturally low levels of GLS2, and experienced reduced growth rate upon transient transfection of GLS2 [113]. GLS2 has also been shown to promote ferroptosis, an iron-dependent form of programmed cell death in which lipidic peroxides accumulate within cells, in GLS2-enriched mouse embryonic stem cells, STAM HCC cells [114], and mouse embryonic fibroblasts [115].
Other challenges include inhibiting compensatory pathways which allow cells in the tumor microenvironment to obtain α-ketoglutarate, or other TCA cycle intermediates, without the involvement of glutaminase [67,116–118]. One such input involves importing pyruvate to form oxaloacetate. This was recently demonstrated in triple-negative/basal-subtype breast cancers, which normally respond well to GLS inhibitors [117]. Here, a panel of breast cancer cell lines including MDA-MB-231, MDA-MB 436, and BT549 cells were treated with CB-839 or BPTES in a variety of different culture media. The drug IC50 values were lowest in RPMI-1640 medium, and considerably higher in α-MEM or DMEM media, both of which have a much higher concentration of pyruvic acid. Introduction of sodium pyruvate to RPMI-1640 provided resistance to the drugs. This change in sensitivity was found to depend upon pyruvate carboxylase, which converts pyruvate to oxaloacetate. One could imagine similar effects if cells were provided with other common anaplerotic inputs to enhance formation of e.g. succinyl-CoA or fumarate.
Another compensatory pathway has been shown in a recent study in medulloblastoma where possible input to the TCA cycle from the glutamine transaminase KYAT1 was examined [118]. In this study, KYAT1 was found to transfer a nitrogen group directly from glutamine to α-ketoglutarate, to form α-ketoglutaramate and glutamate. The α-ketoglutaramate was then hydrolyzed to α-ketoglutarate by the omega-amidase, NIT2, for a net conversion of glutamine to glutamate. This set of reaction is sometimes referred to as the ‘glutamine transaminase–amidase’ pathway [67,116,119]. The authors treated D425MED cells with uniformly labeled (m + 7) glutamine for two hours, then examined the molecular weight of glutamate in cell lysates. In this case, they found a relatively even amount of m + 6 glutamate (presumably from glutaminase activity, which would remove one heavy atom), and of m + 1 and m + 5 glutamate (the former coming from conversion of KYAT1-formed α-ketoglutarate to glutamate, and the latter from the conversion of α-ketoglutaramate to α-ketoglutarate, then to glutamate). However, when a similar experiment was conducted in mice with flank injections of tumor cells, the m + 5 signal almost entirely disappeared, and the m + 6 signal decreased. This effect was even more pronounced in orthotopic models, which lost both signals, and had only an m + 1 signal, and the authors concluded that this represented a dominance of glutamine consumption by KYAT1. Even though the elimination of the m + 5 signal might suggest that the labeled glutamate may have simply arisen from carbon that had gone through the TCA cycle several times, this study does highlight how changing the tumor microenvironment can change its metabolic patterns. Those interested in a more detailed examination of this complicated system, and opinions pertaining to how involved it may be in cancer, are encouraged to examine recent reviews dedicated to the topic [116,119]. Given the multiple ways glutamine is utilized in biological systems, compensatory metabolic pathways such as those described above might interfere with glutaminase inhibition in complex ways, and the potential exists that inhibiting multiple glutaminase-adjacent pathways simultaneously may eventually be shown to have beneficial effects in cancer treatment.
Glutaminase inhibitors to date are, largely, less than ideal in terms of aqueous solubility. CB-839, the best-studied clinical candidate, is for instance dosed in mice at an incredible 200 mg/kg BID (oral administration). This particular formulation of the drug relied upon solubilization with 25% w/v hydroxypropyl–cyclodextrin [57]. Another study examined injected UPGL-00004 (1 mg/kg mouse body weight), in a carrier agent consisting of RPMI-1640, 5% DMSO, 5% Cremophor EL, and 5% ethanol [69]. While this was a reduced amount of a comparable drug, it was also in a relatively aggressive injection carrier, depending upon Cremophor EL, a non-ionic surfactant which, despite its long history of pharmaceutical use to solubilize hydrophobic drugs, also has several identified negative health effects associated with it [120–122]. One popular technique to address these problems, briefly mentioned above, is to encapsulate drugs of this class in nanoparticle carriers. Several approaches have been taken here. Elgogary has encapsulated BPTES in biodegradable poly (lactic-co-glycolic acid) (PLGA) nanoparticles coated in a dense polyethylene glycol (PEG) shell, and found that this improved BPTES potency to be nearly identical to CB-839, while preventing the elevation in liver enzymes which accompanies CB-839 treatment, and can be indicative of toxic side effects [66]. Choi and coworkers took a second approach, creating the core of their nanoparticles from pH-sensing peptides, which precipitate as nanoparticles at pH >7, and solubilize to release their cargo at pH <6.9. When BPTES was enclosed in these pH-peptide nanoparticles, the growth of HCC1806 xenografted tumors was reduced significantly more-so than in the case of treatment with BPTES alone [123]. Despite its comparatively enhanced potency, similar approaches have been taken to CB-839 itself as well. Jian and coworkers used a zeolitic imidazolate framework (ZIF) to form nanoparticles which could deliver both CB-839 and iron, thus inhibiting GLS while activating ferroptotic pathways [124]. These ZIF nanoparticles were able to inhibit the growth of MDA-MB-231 breast cancer cells both in vitro and in vitro in xenograft models. Yet another approach involved encapsulating CB-839 in gold nanoparticles coated with CD133, to direct the nanoparticles to cancer stem cells [125]. Like previous nanoparticles, these were found to be more effective in inhibiting the growth of cancer stem cells compared with the individual components used to synthesize them. These sorts of studies help to show the importance of formulation chemistry, and the improvements to treatment that can be made when solubility problems are overcome.
A final complication is that glutamine hydrolysis by glutaminase also produces ammonia as a byproduct (Figure 1A). Ammonia is both necessary for cell survival and toxic at high concentrations. This is further complicated by metabolic changes cells experience upon hypoxia, which is frequently encountered in tumors, where for example MCF7, HeLa, and 4T1 cells under hypoxic conditions increase storage of nitrogen in orotate and dihydroorotate [126]. Conceivably, any inhibition of glutaminase (a major producer of ammonia) could influence these tightly regulated pathways and result in either cell death or enhanced cell survival, and thus the fate of nitrogen from glutaminase activity is worth monitoring.
Conclusion
The glutaminase family enzymes, which initiate glutamine catabolism in order to satisfy the “glutamine addiction” experienced by many cancer cells as part of the Warburg effect, have shown to be promising targets for cancer therapeutics development. Regulation of these enzymes appears to be complex, and glutamine metabolism feeds into several pathways implicated in various cancer types. As a result, glutaminase inhibition, either alone or as part of combination therapies, can be particularly effective in some cancers. This understanding has driven the development of glutaminase inhibitors in recent years, which has led to several promising clinical trials. Further progress toward the development of potent allosteric inhibitors of glutaminase, with improved efficacy and pharmacokinetics compared to current generation molecules, and ongoing development of potent inhibitors capable of modulating the specific activities of the different glutaminase isozymes, have the potential to greatly improve prognostic outlook for patients with glutamine-addicted cancers.
Future perspective
Targeting glutamine catabolism has provided a promising approach for cancer therapeutics and developing small molecule inhibitors of glutaminase has proven to be an effective strategy. Modulating the expression and activity of the different forms of glutaminase is critical to tuning metabolic pathways in various forms of cancer. Further work optimizing small-molecule inhibitors for glutaminase will be beneficial in being able to selectively inhibit each glutaminase isozyme. A focus on newer structural approaches, such as serial room-temperature crystallography [64] and time-resolved x-ray crystallography [127], may offer new insights into the interactions of various inhibitors with the glutaminase enzymes and further help the development of potent drug candidates. This may be of particular value in the case of allosteric inhibitors, where the inhibitory mechanisms remain less clear, and new opportunities for inhibitor optimization may arise as these mechanisms come into clearer focus
In addition, exciting possibilities exist regarding the synergistic benefits of combining glutaminase inhibitors with other cancer therapeutics. We suspect that the most important advance here will be due to improvement of model systems, ranging from cell lines, xenografts, to mouse models [128], in which such synergies are studied. Much of the metabolic nature of a tumor model is dependent upon its microenvironment, and changing that microenvironment, from using different cell culture medium to moving from cell culture to whole animal approaches, can greatly affect the potency of metabolic inhibitors such as those discussed here. Similarly, particularly promising approaches, such as combining glutaminase inhibitors with immune checkpoint inhibitors, currently can only be properly investigated in highly complex immune-competent murine model systems. Such models are incredibly powerful, but are also incredibly expensive to deploy, and similarly include ethical complications in their use. As research advances in the generation of in vitro models which can more fully reconstitute such systems, studies of glutaminase inhibition will become both easier, more accurate, and more accessible to scientists globally. In addition, an agreement in the field on standardized methods of pathway analysis to study glutamine catabolism, such as 13C/15N-glutamine tracing studies and ammonia formation detection, would also unify the various approaches to study the modulatory effects of inhibiting glutaminase in different biological processes.
Given the important roles of glutaminase enzymes in such systems, these enzymes have also been found to have important implications in other diseases related to cancer. For example, as cancer is frequently an age-related impairment, the role of glutamine catabolism is also being studied in senescence. A recent report showed that KGA expression in response to lysosomal membrane damage is essential for maintaining senescence [129]. Glutaminolysis has also been found to modulate cell cycle arrest in human endothelial cells [130]. Future work on controlling cellular fate through metabolic reprogramming with glutaminase inhibition may offer new strategies for regulating senescence [131]. One caveat here, however, is that while glutaminase monotherapy has been generally shown to have a wide therapeutic window between e.g. cancer and non-cancer cells, these studies do show that non-cancer cells may still require intact glutaminolysis in some cases. This becomes doubly problematic when examining synergies between glutaminase inhibitors and other compounds. Indeed, the vast majority of the studies we examined which detail drug synergies did not test these synergies against healthy cell lines, and it is likely that such testing in the future will help to indicate the most promising combination therapies going forward.
Targeting glutamine catabolism might also offer potential therapeutic strategies against viral infection, an interesting possibility in light of the COVID-19 pandemic. Virus infection often results in the metabolic reprogramming of host-infected cells with the upregulation of glutamine catabolism to satisfy the requirements of viral replication. GLS inhibition has been found to halt the replication of adenovirus, herpes simplex virus 1 and influenza A in cultured primary cells [132], while α-ketoglutarate was shown to rescue human cytomegalovirus propagation upon glutamine deprivation [133]. Glutamine addiction was also observed in host cells upon infection with hepatitis C, rhinovirus, as well as several other common viruses [134]. Targeting glutamine catabolism in virus-infected host cells might eventually offer an alternative to the current FDA-approved antiviral drugs, which primarily inhibit different components of the virus machinery [135]. The important role of glutamine catabolism is also being established in other therapeutic areas, including allograft acceptance [136], acute lung injury [137], and neural tube defects [138]. And, given the central role of cellular metabolism in biology, we expect that even more disease states will be found to be at least partially driven by glutaminase by the end of the decade. With the importance of glutaminase enzymes, both in cancer and other diseases, and the ever-improving nature of model systems in which to study glutaminase biologically, and technical systems by which to study glutaminase chemically, we anticipate that the development of glutaminase inhibitors, and more generally studies upon the glutaminase enzymes, will be thriving scientific fields for many years to come.
Executive summary.
Controlling the differential expression of glutaminases in cancer cells
GAC, KGA, and LGA are three distinct glutaminase enzymes that are regulated by various oncogenes for differential expression depending on specific types of cancer. Glutaminases share similar catalytic domains but can differ in their mechanisms of allosteric activation and inhibition.
Major mechanisms that regulate glutaminase expression and activity patterns include transcription factors, long non-coding RNAs, and localization.
Glutamine catabolism has been established as potentially important therapeutic target. Inhibition of glutaminase is an effective mechanism for inhibiting glutamine catabolism.
Molecular Basis for activation of glutaminase
Different forms of glutaminase have different binding potency for glutamine and for phosphate, and different turnover rate.
The detailed basis of glutamine hydrolysis is well understood, but much research remains before there is an understanding of how activating signals are received and turned into enzymatic efficiency.
Molecular basis for glutaminase inhibition
Glutamine analogs can target glutaminase as competitive inhibitors, and prodrugs are being developed to enhance therapeutic index and reduce systematic exposure.
Allosteric inhibitors for glutaminase such as the BPTES-class and 968-class of compounds show better specificity and potency than competitive inhibitors, and thus have undergone extensive structure-activity analysis.
Combinational therapies with glutaminase inhibitors & other anti-cancer treatments
The emergence of combination therapies using glutaminase inhibitors together with other anti-cancer drugs have shown to be an effective strategy in targeting the metabolic programs of different types of cancer. This includes combining small-molecule inhibitors of glutaminase together, with other metabolic inhibitors, checkpoint inhibitors, and with additional conventional cancer therapies.
Complications arising from glutaminase inhibition
Several alternate biological pathways exist which either consume glutamine or produce glutamate.
The role of GLS2 is not fully understood, and it may act as a tumor suppressor in some situations.
Current glutaminase inhibitors have some poor pharmacological properties, particularly low aqueous solubility, which are being addressed via formulation chemistry.
Future perspective
Efforts focusing on optimizing allosteric inhibitors specific for each form of glutaminase will be important in modulating the specific catalytic activities and functions of these enzymes in various types of cancer. These may be advanced via e.g. new approaches to protein structure determination, and more relevant biological model systems.
Targeting glutamine catabolism might also offer new therapeutic strategies in non-cancer related treatments involving diseases linked to aging, viral infection, and injury recovery.
Supplementary Material
Acknowledgments
T-T T Nguyen was responsible for conception of the review and prioritization of topics for consideration. All authors helped to write, edit, and finalize the manuscript. We would also like to thank M Antonyak for his insightful comments and Wendy Sweeny for her assistance in assembling and submitting the final manuscript.
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.future-science.com/doi/suppl/10.4155/fdd-2022-0011
Financial & competing interests disclosure
RA Cerione acknowledges funding from the National Institute of General Medical Sciences (NIGMS, 5R35GM122575-05). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Yang C, Ko B, Hensley CT et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 56(3), 414–424 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Méndez-Lucas A, Lin W, Driscoll PC et al. Identifying strategies to target the metabolic flexibility of tumours. Nat. Metab. 2(4), 335–350 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leone RD, Zhao L, Englert JM et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366(6468), 1013–1021 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lukey MJ, Cluntun AA, Katt WP et al. Liver-Type Glutaminase GLS2 Is a Druggable Metabolic Node in Luminal-Subtype Breast Cancer. Cell Rep. 29(1), 76–88; e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Son J, Lyssiotis CA, Ying H et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496(7443), 101–105 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lukey MJ, Katt WP, Cerione RA. Targeting amino acid metabolism for cancer therapy. Drug Discov. Today 22(5), 796–804 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study was among the first to identify GLS2 as necessary for the survival of a cancer, in this case receptor-positive breast cancers.
- 7.Masisi BK, El Ansari R, Alfarsi L, Rakha EA, Green AR, Craze ML. The role of glutaminase in cancer. Histopathology 76(4), 498–508 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Katt WP, Cerione RA. Glutaminase regulation in cancer cells: a druggable chain of events. Drug Discov. Today 19(4), 450–457 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matés JM, Campos-Sandoval JA, Santos-Jiménez J de L, Márquez J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett. 467, 29–39 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Katt WP, Lukey MJ, Cerione RA. A tale of two glutaminases: homologous enzymes with distinct roles in tumorigenesis. Future Med. Chem. 9(2), 223–243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ardlie KG, DeLuca DS, Segrè AV et al. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348(6235), 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Redis RS, Vela LE, Lu W et al. Allele-Specific Reprogramming of Cancer Metabolism by the Long Non-coding RNA CCAT2. Mol. Cell 61(4), 520–534 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu L, Yin Y, Li Y et al. A glutaminase isoform switch drives therapeutic resistance and disease progression of prostate cancer. Proc. Natl Acad. Sci. USA 118(13), 1–11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang JB, Erickson JW, Fuji R et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18(3), 207–219 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; • First description of compound 968 as a glutaminase inhibitor.
- 15.Shen YA, Chen CL, Huang YH et al. Inhibition of glutaminolysis in combination with other therapies to improve cancer treatment. Curr. Opin. Chem. Biol. 62(March), 64–81 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.NIH NCI. Cancer Statistics. (2020). www.cancer.gov/about-cancer/understanding/statistics#:~:text=The most common cancers (listed,endometrial cancer%2C leukemia%2C pancreatic cancer
- 17.Goldman MJ, Craft B, Hastie M et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 38(6), 675–678 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saha SK, Riazul Islam SM, Abdullah-Al-Wadud M, Islam S, Ali F, Park KS. Multiomics analysis reveals that GLS and GLS2 differentially modulate the clinical outcomes of cancer. J. Clin. Med. 8(3), 1–28 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao P, Tchernyshyov I, Chang T et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458(April), (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; • One of the earliest studies to find a link between the transcription c-Myc and glutaminase expression.
- 20.Qing G, Li B, Vu A et al. ATF4 Regulates MYC-Mediated Neuroblastoma Cell Death upon Glutamine Deprivation. Cancer Cell 22(5), 631–644 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X, Huang L, Yang T et al. METTL3 Promotes Esophageal Squamous Cell Carcinoma Metastasis Through Enhancing GLS2 Expression. Front. Oncol. 11(May), 1–13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greene KS, Lukey MJ, Wang X et al. SIRT5 stabilizes mitochondrial glutaminase and supports breast cancer tumorigenesis. Proc. Natl Acad. Sci. USA 116(52), 26625–26632 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaglio D, Metallo CM, Gameiro PA et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 7(523), 1–15 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 178(1), 93–105 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wise DR, Deberardinis RJ, Mancuso A et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl Acad. Sci. USA 105(48), 18782–18787 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sengupta D, Cassel T, Teng K-Y et al. Regulation of hepatic glutamine metabolism by miR-122. Mol. Metab. 34(January), 174–186 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luan W, Zhou Z, Zhu Y, Xia Y, Wang J, Xu B. miR-137 inhibits glutamine catabolism and growth of malignant melanoma by targeting glutaminase. Biochem. Biophys. Res. Commun. 495(1), 46–52 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Chen X, Chen Y, Zhang M et al. HucMSC exosomes promoted imatinib-induced apoptosis in K562-R cells via a miR-145a-5p/USP6/GLS1 axis. Cell Death Dis. 13(1), 1–11 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de los Santos-Jiménez J, Campos-Sandoval JA, Márquez-Torres C et al. Glutaminase isoforms expression switches microRNA levels and oxidative status in glioblastoma cells. J. Biomed. Sci. 28(1), 1–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lampa M, Arlt H, He T et al. Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLOS ONE 12(9), (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng SJ, Chen HY, Zeng Z et al. Nutrient stress-dysregulated antisense lncRNA GLS-As impairs GLS-mediated metabolism and represses pancreatic cancer progression. Cancer Res. 79(7), 1398–1412 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Savojardo C, Martelli PL, Fariselli P, Casadio R. TPpred3 detects and discriminates mitochondrial and chloroplastic targeting peptides in eukaryotic proteins. Bioinformatics 31(20), 3269–3275 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Armenteros JJA, Salvatore M, Emanuelsson O et al. Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance 2(5), 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Profiti G, Martelli PL, Casadio R. The Bologna Annotation Resource (BAR 3.0): improving protein functional annotation. Nucleic Acids Res. 45(W1), W285–W290 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhasin M, Raghava GPS. ESLpred: SVM-based method for subcellular localization of eukaryotic proteins using dipeptide composition and PSI-BLAST. Nucleic Acids Res. 32(WEB SERVER ISS.), 414–419 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cassago A, Ferreira APS, Ferreira IM et al. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc. Natl Acad. Sci. USA 109(4), 1092–1097 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.López de la Oliva AR, Campos-Sandoval JA, Gómez-García MC et al. Nuclear Translocation of Glutaminase GLS2 in Human Cancer Cells Associates with Proliferation Arrest and Differentiation. Sci. Rep. 10(1), 1–17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferreira APS, Cassago A, De Almeida Gonçalves K et al. Active glutaminase C self-assembles into a supratetrameric oligomer that can be disrupted by an allosteric inhibitor. J. Biol. Chem. 288(39), 28009–28020 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delabarre B, Gross S, Fang C et al. Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 50(50), 10764–10770 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Ferreira IM, Quesñay JEN, Bastos AC et al. Structure and activation mechanism of the human liver-type glutaminase GLS2. Biochimie 185, 96–104 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen T-TT, Ramachandran S, Hill MJ, Cerione RA. High-resolution structures of mitochondrial glutaminase C tetramers indicate conformational changes upon phosphate binding. J. Biol. Chem. 298(2), 101564 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Offers some of the most important current structural insights into how inorganic phosphate stimulates enzymatic activity in glutaminase enzymes.
- 42.Stalnecker CA, Erickson JW, Cerione RA. Conformational changes in the activation loop of mitochondrial glutaminase C: a direct fluorescence readout that distinguishes the binding of allosteric inhibitors from activators. J. Biol. Chem. 292(15), 6095–6107 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, Erickson JW, Stalnecker CA et al. Mechanistic basis of glutaminase activation: a key enzyme that promotes glutamine metabolism in cancer cells. J. Biol. Chem. 291(40), 20900–20910 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Møller M, Nielsen SS, Ramachandran S et al. Small Angle X-Ray Scattering Studies of Mitochondrial Glutaminase C Reveal Extended Flexible Regions, and Link Oligomeric State with Enzyme Activity. PLOS ONE 8(9), 1–13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masola B, Ngubane NP. The activity of phosphate-dependent glutaminase from the rat small intestine is modulated by ADP and is dependent on integrity of mitochondria. Arch. Biochem. Biophys. 504(2), 197–203 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Szweda LI, Atkinson DE. Response of rat liver glutaminase to pH, ammonium, and citrate. Possible regulatory role of glutaminase in ureagenesis. J. Biol. Chem. 265(34), 20869–20873 (1990). [PubMed] [Google Scholar]
- 47.Wu CR, Chen LX, Jin S, Li H. Glutaminase inhibitors: a patent review. Expert Opin. Ther. Pat. 28(11), 823–835 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Xu X, Meng Y, Li L et al. Overview of the Development of Glutaminase Inhibitors: Achievements and Future Directions. J. Med. Chem. 62(3), 1096–1115 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Thangavelu K, Chong QY, Low BC, Sivaraman J. Structural Basis for the Active Site Inhibition Mechanism of Human Kidney-Type Glutaminase (KGA). Sci. Rep. 4, 1–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goel M, Kumari S, Pal R, Gupta R. Crystal structure of Bacillus licheniformis Gamma glutamyl transpeptidase with DON. (2018). www.rcsb.org/structure/5Y9B
- 51.Wada K, Hiratake J, Irie M et al. Crystal Structures of Escherichia coli γ-Glutamyltranspeptidase in Complex with Azaserine and Acivicin: Novel Mechanistic Implication for Inhibition by Glutamine Antagonists. J. Mol. Biol. 380(2), 361–372 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Lemberg KM, Vornov JJ, Rais R, Slusher BS. We're not ‘don’ yet: optimal dosing and prodrug delivery of 6-diazo-5-oxo-L-norleucine. Mol. Cancer Ther. 17(9), 1824–1832 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu X, Nedelcovych MT, Thomas AG et al. JHU-083 selectively blocks glutaminase activity in brain CD11b+ cells and prevents depression-associated behaviors induced by chronic social defeat stress. Neuropsychopharmacology 44(4), 683–694 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hanaford AR, Alt J, Rais R et al. Orally bioavailable glutamine antagonist prodrug JHU-083 penetrates mouse brain and suppresses the growth of MYC-driven medulloblastoma. Transl. Oncol. 12(10), 1314–1322 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Reports the discovery of pro-drug variants of DON, derivatives of which are currently in clinical trials.
- 55.Pham K, Maxwell MJ, Sweeney H et al. Novel Glutamine Antagonist JHU395 Suppresses MYC-Driven Medulloblastoma Growth and Induces Apoptosis. J. Neuropathol. Exp. Neurol. 80(4), 336–344 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.34th Annual Meeting & Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2019): Part 1. National Harbor, MD, USA: (2019). [Google Scholar]
- 57.Gross MI, Demo SD, Dennison JB et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 13(4), 890–901 (2014). [DOI] [PubMed] [Google Scholar]; •• Describes the discovery of CB-839/teleglenastat, currently the most clinically relevant glutaminase inhibitor.
- 58.Katt WP, Ramachandran S, Erickson JW, Cerione RA. Dibenzophenanthridines as inhibitors of glutaminase C and cancer cell proliferation. Mol. Cancer Ther. 11(6), 1269–1278 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang W-H, Qiu Y, Stamatatos O, Janowitz T, Lukey MJ. Enhancing the Efficacy of Glutamine Metabolism Inhibitors in Cancer Therapy. Trends in Cancer 7(8), 790–804 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soth MJ, Le K, Di Francesco ME et al. Discovery of IPN60090, a Clinical Stage Selective Glutaminase-1 (GLS-1) Inhibitor with Excellent Pharmacokinetic and Physicochemical Properties. J. Med. Chem. 63(21), 12957–12977 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vogl DT, Younes A, Stewart K et al. Phase 1 Study of CB-839, a First-in-Class, Glutaminase Inhibitor in Patients with Multiple Myeloma and Lymphoma. Blood 126(23), 3059–3059 (2015). [Google Scholar]
- 62.Wang ES, Frankfurt O, Orford KW et al. Phase 1 Study of CB-839, a First-in-Class, Orally Administered Small Molecule Inhibitor of Glutaminase in Patients with Relapsed/Refractory Leukemia. Blood 126(23), 2566–2566 (2015). [Google Scholar]
- 63.Harding JJ, Telli ML, Munster PN et al. Safety and tolerability of increasing doses of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase, in solid tumors. J. Clin. Oncol. 33(Suppl. 15), 2512 (2015). [Google Scholar]
- 64.Milano SK, Huang Q, Nguyen T-TT et al. New insights into the molecular mechanisms of glutaminase C inhibitors in cancer cells using serial room temperature crystallography. J. Biol. Chem. 298(2), 101535 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McDermott LA, Iyer P, Vernetti L et al. Design and evaluation of novel glutaminase inhibitors. Bioorganic Med. Chem. 24(8), 1819–1839 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Elgogary A, Xu Q, Poore B et al. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc. Natl Acad. Sci. USA 113(36), E5328–E5336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Udupa S, Nguyen S, Hoang G et al. Upregulation of the Glutaminase II Pathway Contributes to Glutamate Production upon Glutaminase 1 Inhibition in Pancreatic Cancer. Proteomics 19(21-22), 1–7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu X, Wang J, Wang M et al. Structure-Enabled Discovery of Novel Macrocyclic Inhibitors Targeting Glutaminase 1 Allosteric Binding Site. J. Med. Chem. 64(8), 4588–4611 (2021). [DOI] [PubMed] [Google Scholar]
- 69.Huang Q, Stalnecker C, Zhang C et al. Characterization of the interactions of potent allosteric inhibitors with glutaminase C, a key enzyme in cancer cell glutamine metabolism. J. Biol. Chem. 293(10), 3535–3545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boysen G, Jamshidi-Parsian A, Davis MA et al. Glutaminase inhibitor CB-839 increases radiation sensitivity of lung tumor cells and human lung tumor xenografts in mice. Int. J. Radiat. Biol. 95(4), 436–442 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wicker CA, Hunt BG, Krishnan S et al. Glutaminase inhibition with telaglenastat (CB-839) improves treatment response in combination with ionizing radiation in head and neck squamous cell carcinoma models. Cancer Lett. 502, 180–188 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thompson RM, Dytfeld D, Reyes L et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 8(22), 35863–35876 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang KY, Wu CR, Zheng MZ et al. Physapubescin I from husk tomato suppresses SW1990 cancer cell growth by targeting kidney-type glutaminase. Bioorg. Chem. 92(August), 103186 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Yuan L, Sheng X, Clark LH et al. Glutaminase inhibitor compound 968 inhibits cell proliferation and sensitizes paclitaxel in ovarian cancer. Am. J. Transl. Res. 8(10), 4265–4277 (2016). [PMC free article] [PubMed] [Google Scholar]
- 75.Han T, Guo M, Zhang T, Gan M, Xie C, Wang J-B. A novel glutaminase inhibitor-968 inhibits the migration and proliferation of non-small cell lung cancer cells by targeting EGFR/ERK signaling pathway. Oncotarget 8(17), 28063–28073 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang D, Meng G, Zheng M et al. The glutaminase-1 inhibitor 968 enhances dihydroartemisinin-mediated antitumor efficacy in hepatocellular carcinoma cells. PLOS ONE 11(11), e0166423 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yeh TK, Kuo CC, Lee YZ et al. Design, Synthesis, and Evaluation of Thiazolidine-2,4-dione Derivatives as a Novel Class of Glutaminase Inhibitors. J. Med. Chem. 60(13), 5599–5612 (2017). [DOI] [PubMed] [Google Scholar]
- 78.Renna RK, Rodrigues CT, Jean JC et al. High-Throughput Screening Reveals New Glutaminase Inhibitor Molecules. ACS Pharmacol. Transl. Sci. 4(6), 1849–1866 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li R, Wei P, Wang Y, Liu Y, Liu X, Meng D. Brachyantheraoside A8, a new natural nor-oleanane triterpenoid as a kidney-type glutaminase inhibitor from: stauntonia brachyanthera. RSC Adv. 7(83), 52533–52542 (2017). [Google Scholar]
- 80.Lee YZ, Yang CW, Chang HY et al. Discovery of selective inhibitors of Glutaminase-2, which inhibit mTORC1, activate autophagy and inhibit proliferation in cancer cells. Oncotarget 5(15), 6087–6101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reckzeh ES, Karageorgis G, Schwalfenberg M et al. Inhibition of Glucose Transporters and Glutaminase Synergistically Impairs Tumor Cell Growth. Cell Chem. Biol. 26(9), 1214–1228; e25 (2019). [DOI] [PubMed] [Google Scholar]
- 82.Cheng T, Sudderth J, Yang C et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl Acad. Sci. USA 108(21), 8674–8679 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Momcilovic M, Bailey ST, Lee JT et al. The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma. Cancer Cell 33(5), 905–921; e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Emberley E, Pan A, Chen J et al. The glutaminase inhibitor telaglenastat enhances the antitumor activity of signal transduction inhibitors everolimus and cabozantinib in models of renal cell carcinoma. PLOS ONE 16(11 November), 1–20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guo L, Zhou B, Liu Z et al. Blockage of glutaminolysis enhances the sensitivity of ovarian cancer cells to PI3K/mTOR inhibition involvement of STAT3 signaling. Tumor Biol. 37(8), 11007–11015 (2016). [DOI] [PubMed] [Google Scholar]
- 86.Li J, Csibi A, Yang S et al. Synthetic lethality of combined glutaminase and Hsp90 inhibition in mTORC1-driven tumor cells. Proc. Natl Acad. Sci. USA 112(1), E21–E29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 87.Jin H, Wang S, Zaal EA et al. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. Elife 9, 1–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang L, Achreja A, Yeung TL et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 24(5), 685–700 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pavlova NN, Hui S, Ghergurovich JM et al. As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab. 27(2), 428–438; e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ohba S, Hirose Y. L-Asparaginase and 6-Diazo-5-Oxo-L-Norleucine Synergistically Inhibit the Growth of Glioblastoma Cells. J. Neurooncol. 146(3), 469–475 (2020). [DOI] [PubMed] [Google Scholar]
- 91.Halama A, Kulinski M, Dib SS et al. Accelerated lipid catabolism and autophagy are cancer survival mechanisms under inhibited glutaminolysis. Cancer Lett. 430(May), 133–147 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Dos Reis LM, Adamoski D, Souza ROO et al. Dual inhibition of glutaminase and carnitine palmitoyltransferase decreases growth and migration of glutaminase inhibition-resistant triple-negative breast cancer cells. J. Biol. Chem. 294(24), 9342–9357 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Upadhaya S, Neftelinov ST, Hodge J, Campbell J. Challenges and opportunities in the PD1/PDL1 inhibitor clinical trial landscape. England (2022). [DOI] [PubMed] [Google Scholar]
- 94.Chang CH, Curtis JD, Maggi LB et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153(6), 1239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patsoukis N, Bardhan K, Chatterjee P et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 6, 6692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chang CH, Qiu J, O'Sullivan D et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162(6), 1229–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Varghese S, Pramanik S, Williams LJ et al. The glutaminase inhibitor CB-839 (Telaglenastat) enhances the antimelanoma activity of T-cell-mediated immunotherapies. Mol. Cancer Ther. 20(3), 500–511 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ma G, Li C, Zhang Z et al. Targeted Glucose or Glutamine Metabolic Therapy Combined With PD-1/PD-L1 Checkpoint Blockade Immunotherapy for the Treatment of Tumors – Mechanisms and Strategies. Front. Oncol. 11(July), 1–11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tarrado-Castellarnau M, de Atauri P, Tarragó-Celada J et al. De novo MYC addiction as an adaptive response of cancer cells to CDK 4/6 inhibition. Mol. Syst. Biol. 13(10), 940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Qie S, Yoshida A, Parnham S et al. Targeting glutamine-addiction and overcoming CDK4/6 inhibitor resistance in human esophageal squamous cell carcinoma. Nat. Commun. 10(1), 1–15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Icard P, Fournel L, Wu Z, Alifano M, Lincet H. Interconnection between Metabolism and Cell Cycle in Cancer. Trends Biochem. Sci. 44(6), 490–501 (2019). [DOI] [PubMed] [Google Scholar]
- 102.Wang R, Dillon CP, Shi LZ et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 35(6), 871–882 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Best SA, Gubser PM, Sethumadhavan S et al. Glutaminase inhibition impairs CD8 T cell activation in STK11-/Lkb1-deficient lung cancer. Cell Metab. 34(6), 874–887; e6 (2022). [DOI] [PubMed] [Google Scholar]
- 104.Sener Z, Cederkvist FH, Volchenkov R, Holen HL, Skålhegg BS. T helper cell activation and expansion is sensitive to glutaminase inhibition under both hypoxic and normoxic conditions. PLOS ONE 11(7), 1–18 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Deng J, Wang ES, Jenkins RW et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 8(2), 216–233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sitthideatphaiboon P, Galan-Cobo A, Negrao MV et al. STK11/LKB1 mutations in NSCLC are associated with KEAP1/NRF2-dependent radiotherapy resistance targetable by glutaminase inhibition. Clin. Cancer Res. 27(6), 1720–1733 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kamarudin MNA, Sarker MMR, Zhou JR, Parhar I. Metformin in colorectal cancer: molecular mechanism, preclinical and clinical aspects. J. Exp. Clin. Cancer Res. 38, 491 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ren L, Ruiz-Rodado V, Dowdy T et al. Glutaminase-1 (GLS1) inhibition limits metastatic progression in osteosarcoma. Cancer Metab. 8(1), 1–13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fendt SM, Bell EL, Keibler MA et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 73(14), 4429–4438 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl Acad. Sci. USA 107(16), 7455–7460 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Suzuki S, Tanaka T, Poyurovsky MV et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl Acad. Sci. USA 107(16), 7461–7466 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu J, Zhang C, Lin M et al. Glutaminase 2 negatively regulates the PI3K/AKT signaling and shows tumor suppression activity in human hepatocellular carcinoma. Oncotarget 5(9), 2635–2647 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Szeliga M, Obara-Michlewska M, Matyja E et al. Transfection with liver-type glutaminase cDNA alters gene expression and reduces survival, migration and proliferation of T98G glioma cells. Glia 57(9), 1014–1023 (2009). [DOI] [PubMed] [Google Scholar]
- 114.Suzuki S, Venkatesh D, Kanda H et al. GLS2 is a tumor suppressor and a regulator of ferroptosis in hepatocellular carcinoma. Cancer Res. 82(18), 3209–3222 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 59(2), 298–308 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dorai T, Pinto JT, Denton TT, Krasnikov BF, Cooper AJL. The metabolic importance of the glutaminase II pathway in normal and cancerous cells. Anal. Biochem. 644, 114083 (2022). [DOI] [PubMed] [Google Scholar]
- 117.Singleton DC, Dechaume AL, Murray PM, Katt WP, Baguley BC, Leung EY. Pyruvate anaplerosis is a mechanism of resistance to pharmacological glutaminase inhibition in triple-receptor negative breast cancer. BMC Cancer 20(1), 1–13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pham K, Hanaford AR, Poore BA et al. Comprehensive Metabolic Profiling of MYC-Amplified Medulloblastoma Tumors Reveals Key Dependencies on Amino Acid, Tricarboxylic Acid and Hexosamine Pathways. Cancers (Basel) 14(5), (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cooper AJL, Dorai T, Pinto JT, Denton TT. α-Ketoglutaramate-A key metabolite contributing to glutamine addiction in cancer cells. Front. Med. 13, 1035335 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dorr RT. Pharmacology and toxicology of cremophor el diluent. Ann. Pharmacother. 28, S11–S14 (1994). [DOI] [PubMed] [Google Scholar]
- 121.Campos FC, Victorino VJ, Martins-Pinge MC, Cecchini AL, Panis C, Cecchini R. Systemic toxicity induced by paclitaxel in vivo is associated with the solvent cremophor EL through oxidative stress-driven mechanisms. Food Chem. Toxicol. 68, 78–86 (2014). [DOI] [PubMed] [Google Scholar]
- 122.Kiss L, Walter FR, Bocsik A et al. Kinetic Analysis of the Toxicity of Pharmaceutical Excipients Cremophor EL and RH40 on Endothelial and Epithelial Cells. J. Pharm. Sci. 102(4), 1173–1181 (2013). [DOI] [PubMed] [Google Scholar]
- 123.Choi H, Liu T, Nath K, Zhou R, Chen I-W. Peptide nanoparticle with pH-sensing cargo solubility enhances cancer drug efficiency. Nano Today 13, 15–22 (2017). [Google Scholar]
- 124.Jian H, Zhang Y, Wang J, Chen Z, Wen T. Zeolitic imidazolate framework-based nanoparticles for the cascade enhancement of cancer chemodynamic therapy by targeting glutamine metabolism. Nanoscale 14(24), 8727–8743 (2022). [DOI] [PubMed] [Google Scholar]
- 125.Poonaki E, Nickel A-C, Shafiee Ardestani M et al. CD133-Functionalized Gold Nanoparticles as a Carrier Platform for Telaglenastat (CB-839) against Tumor Stem Cells. Int. J. Mol. Sci. 23(10), 5479 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang Y, Bai C, Ruan Y et al. Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat. Commun. 10, 201 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Calvey GD, Katz AM, Zielinski KA, Dzikovski B, Pollack L. Characterizing Enzyme Reactions in Microcrystals for Effective Mix-and-Inject Experiments using X-ray Free-Electron Lasers. Anal. Chem. 92(20), 13864–13870 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schcolnik-Cabrera A, Dueñas-Gonzalez A. Mouse Model for Efficient Simultaneous Targeting of Glycolysis, Glutaminolysis, and De Novo Synthesis of Fatty Acids in Colon Cancer. In: Cancer Cell Signaling: Methods and Protocols. Robles-Flores M (Ed.). 45–69, Springer US, New York, NY: (2021). [DOI] [PubMed] [Google Scholar]
- 129.Johmura Y, Yamanaka T, Omori S et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 371(6526), 265–270 (2021). [DOI] [PubMed] [Google Scholar]
- 130.Peyton KJ, Liu X-M, Yu Y, Yates B, Behnammanesh G, Durante W. Glutaminase-1 stimulates the proliferation, migration, and survival of human endothelial cells. Biochem. Pharmacol. 156(August), 204–214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhang W, Li J, Duan Y et al. Metabolic Regulation: A Potential Strategy for Rescuing Stem Cell Senescence. Stem Cell Rev. Reports 18(5), 1728–1742 (2022). [DOI] [PubMed] [Google Scholar]
- 132.Thai M, Thaker SK, Feng J et al. MYC-induced reprogramming of glutamine catabolism supports optimal virus replication. Nat. Commun. 6, 1–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chambers JW, Maguire TG, Alwine JC. Glutamine Metabolism Is Essential for Human Cytomegalovirus Infection. J. Virol. 84(4), 1867–1873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hirabara SM, Gorjao R, Levada-Pires AC et al. Host cell glutamine metabolism as a potential antiviral target. Clin. Sci. 135(2), 305–325 (2021). [DOI] [PubMed] [Google Scholar]
- 135.Tompa DR, Immanuel A, Skrikanth S, Kadhirvel S. Trends and strategies to combat viral infections: a review on FDA approved antiviral drugs. Int. J. Biol. Macromol. 172(January), 524–541 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee CF, Cheng CH, Hung HC, Chan KM, Lee WC. Targeting glutamine metabolism as an effective means to promote allograft acceptance while inhibit tumor growth. Transpl. Immunol. 63(September), 101336 (2020). [DOI] [PubMed] [Google Scholar]
- 137.Vigeland CL, Beggs HS, Collins SL et al. Inhibition of glutamine metabolism accelerates resolution of acute lung injury. Physiol. Rep. 7(5), e14019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Benavides-Rivas C, Tovar LM, Zúñiga N et al. Altered Glutaminase 1 Activity During Neurulation and Its Potential Implications in Neural Tube Defects. Front. Pharmacol. 11(June), 1–8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.