

Abstract

Glycosaminoglycans (GAGs) are complex polysaccharides exhibiting a vast structural diversity and fulfilling various functions mediated by thousands of interactions in the extracellular matrix, at the cell surface, and within the cells where they have been detected in the nucleus. It is known that the chemical groups attached to GAGs and GAG conformations comprise “glycocodes” that are not yet fully deciphered. The molecular context also matters for GAG structures and functions, and the influence of the structure and functions of the proteoglycan core proteins on sulfated GAGs and vice versa warrants further investigation. The lack of dedicated bioinformatic tools for mining GAG data sets contributes to a partial characterization of the structural and functional landscape and interactions of GAGs. These pending issues will benefit from the development of new approaches reviewed here, namely (i) the synthesis of GAG oligosaccharides to build large and diverse GAG libraries, (ii) GAG analysis and sequencing by mass spectrometry (e.g., ion mobility-mass spectrometry), gas-phase infrared spectroscopy, recognition tunnelling nanopores, and molecular modeling to identify bioactive GAG sequences, biophysical methods to investigate binding interfaces, and to expand our knowledge and understanding of glycocodes governing GAG molecular recognition, and (iii) artificial intelligence for in-depth investigation of GAGomic data sets and their integration with proteomics.
Keywords: glycosaminoglycans, chondroitin sulfate, dermatan sulfate, heparan sulfate, heparin, keratan sulfate heparosan, hyaluronan
1. Introduction
Glycosaminoglycans (GAGs) are a family of linear, highly negatively charged polydisperse polysaccharides, some variably sulfated but others not, and expressed ubiquitously and abundantly on the cell surface and in the extracellular matrix. GAGs are also found in invertebrates and prokaryotes. Upon their discovery, GAGs were considered to play a minor role, forming an inert “glue” surrounding the cells and thus set aside from “cutting-edge” research efforts. However, during the last decades, the field of GAG research has made giant steps forward, and these macromolecules have emerged as essential players in critical biological processes regulating cellular properties, tissue development and remodeling, homeostasis, and disease progression.
The extraordinary structural diversity of GAGs translates into highly diverse functions not accessible to high-order structures and allows them to modulate interactions with various biological molecules. For example, GAGs participate in extracellular matrix assembly, cell–matrix and cell–cell interactions, ligand–receptor binding, and downstream cellular signaling. They control chemokine and cytokine activities and growth factor sequestration. They regulate multiple biological processes in a physiological context, but they also participate in the progression of many diseases. Besides their biological roles, their physical properties are of major interest for biomaterials and tissue engineering applications.
The chemical and structural complexity of GAGs embodies most of the challenges we can find in glycoscience research, combining the heterogeneity of glycans with the difficulties inherent to charged (“polyelectrolyte”) polysaccharides. Numerous research areas have focused on deciphering the structure-to-function relationships underlying the diversity of functions and properties of GAGs. Significant and decisive advances have been achieved in the past decade, mainly through interdisciplinary collaborations, demonstrating the tremendous potential of future discoveries in health and diseases. Nevertheless, several methodological and biological difficulties still hinder progress in GAGs research and must be overcome to access the full spectrum of analytical, physicochemical, chemical, biophysical, and biochemical investigation capabilities to characterize GAGs and their interactions with proteins. Although these difficulties are usually well identified in each discipline, a global, interdisciplinary approach is needed to identify the major challenges.
Within the context of a European initiative, we asked the GAG research community at large what remains to be solved to fully understand the GAG structure and function. In parallel, we also asked the involved scientists to identify the unmet needs which should be addressed in order for them to acquire further knowledge and perform additional activities in their fields of expertise. Such an approach will allow the scientists to have both a disciplinary and an integrative view of the GAG research field. This endeavor is aimed to create a shared vision for all GAG researchers, from basic research to technological applications, and the present Perspective is a translation of such findings and discussions. More specifically, this work deals with issues relevant to the broad research areas of GAG chemistry, biophysics and biochemistry, and concentrates on the different disciplines within these research fields. A companion publication will address the issues related to the diverse biological functions of GAGs.
2. Glycosaminoglycans
GAGs constitute a structurally heterogeneous class of complex carbohydrates. They are linear, negatively charged polysaccharides characterized by sequences of disaccharide repeating units composed of an (occasionally deacetylated) N-acetylhexosamine alternating with hexuronic acids (glucuronic or iduronic acid) or galactose. Different families of GAGs can be distinguished based on the nature of these disaccharide blocks, namely chondroitin sulfate (CS), dermatan sulfate (DS), heparan sulfate (HS), heparin (HP), and keratan sulfate (KS). Heparosan (HN) is the unsulfated precursor of heparin and heparan sulfate (Figure 1).
Figure 1.

Principle constituent disaccharide units: HS, heparan sulfate: -4-D-GlcNAc-α1,4-D-GlcA-β1-. HP, heparin: -4-D-GlcN, NS, 6S-α1,4-L-IdoA-2S- α1-. HN, heparosan: -4-D-GlcNAc-α1,4-D-GlcA-β1-. HA, hyaluronan: 4-D-GlcA-β1-3-D-GlcNAc-β1. CS, (4S)-chondroitin 4-sulfate: 4-D-GlcA-β1-3-D-GalNAc, 4S-β1. CS, (6S)-chondroitin 6-sulfate: 4-D-GlcA-β1-3-D-GalNAc, 6S-β1. DS, dermatan sulfate: -4-L-IdoA-α1-3-D-GalNAc,4S-β1-. KS, keratan sulfate: -4-D-GlcNAc, 6S-β1-3-D-Gal-β1-. Color displayed in the monosaccharide units follows the SNFG recommendations.
These GAGs are linked to proteins, thus forming proteoglycans (PGs) via a xylose-containing tetrasaccharide and synthesized in the Golgi apparatus. While the synthesis of the core proteins follows a template-driven process, the biosynthesis of the GAG chains is nontemplate-driven. GAGs undergo several chemical modifications, e.g., sulfation, de-N-acetylation, and epimerization, which lead to a large structural diversity. Within this context, hyaluronan (traditionally also called hyaluronic acid, HA) represents a stand-alone case as it is not sulfated and not protein linked. Furthermore, its molecular structure, which may consist of several thousand disaccharide units, is not modified by epimerization.
2.1. Nomenclature
GAGs, as linear and complex polysaccharides, are made of repeating disaccharide units comprised of a hexuronic acid (or galactose in keratan sulfate) and a hexosamine throughout a regular alternation of 1–4 and 1–3 glycosidic linkages.
Complexity stems from the many aspects of GAGs structures: a high degree of polymerization combined with size polydispersity, sequence microheterogeneity, high negative charge density, and the potentially isomeric building blocks. Being highly polydisperse, the length of GAG chains found on a certain proteoglycan at a given position is typically not uniform.
Because of this characteristic microheterogeneity, GAGs cannot be represented by a single, well-defined sequence.
2.1.1. Monosaccharides
The structural information currently encoded in the Symbol Nomenclature for Glycans (SNFG) representation of glycans1 is insufficient for fully describing, building, and handling three-dimensional structures. A proposed extension of the SNFG cartoons allows to represent the nature of the absolute (D or L) and anomeric configurations and to include (α or β)-O-esters and ethers, with labels attached to symbols with a number, e.g., 3S for 3-O-sulfate groups.2 Another proposed extension indicates sulfates within the SNFG representation as red dots attached to the symbols.3 In the D configuration, all pyranoses are assumed to be in the 4C1 chair conformation, whereas those in the L configuration have the 1C4 chair conformation. The descriptors of the ring conformations adopted by idopyranoses (1C4, 4C1, and 2S0) were included within the monosaccharide symbol.
2.1.2. Disaccharides and Higher Oligosaccharides
The proposed extension of the SNFG discussed above led to 202 unique GAG disaccharides.2 All representations must inform the nature of the glycosidic linkage between two consecutive monosaccharides. Higher oligosaccharides are constructed by the sequential addition of a monosaccharide and the nature of its glycosidic linkage to the preceding monosaccharide. All chemical compounds are described with IUPAC, Simplified Molecular Input Line Entry Specification syntax (SMILES), and InChi encodings that are readable by the vast majority of chemo-informatics tools. All glycans are encoded in GlycoCT,4 WURCS (Web3 Unique Representation of Carbohydrate Structures) is the main representation used by the repository,5 and LINUCS (LInear Notation for Unique description of Carbohydrate Sequences).6 The GlycoCT format is used by the major glycan repository GlyTouCan,7 Glyco3D,8 and SugarBind.9 The GlycoCT format describes the residue entities in the RESsection, the bonds in the LIN-section, and the number of repeating units in the REP-section. The GlycanBuilder software automatically converts GlycoCT-encoded GAG sequences into SNFG images. Alternatively, the conversion can be done manually with GlycoWorkBench, using the GlycanBuilder library.10
2.2. Extraction and Purification of Natural GAGs
2.2.1. Extraction, Depolymerization, and Enzymatic Digestion
Cartilage and connective tissues contain significant amounts of proteoglycans, composed of glycosaminoglycans covalently linked to a core protein. GAGs are most commonly sourced from farmed animal tissue as byproducts of the food industry, for example, isolated from the trachea, cartilage, rooster combs, and intestinal mucosa from pigs, cattle, and poultry. The extraction process involves the breakdown of the surrounding tissues and core proteins by alkaline hydrolysis to release the GAGs, which are then subjected to downstream processing for purification and recovery. Increasingly, bacterial production is also deployed (e.g., for HA), and nonfarmed animals can be important GAG sources (e.g., squid for CS).
Chemical Depolymerization
While displaying important versatility, the chemical depolymerization of GAGs mainly occurs by β-elimination and reductive deamination. Throughout a two-step reaction mechanism, the process of β elimination introduces a double bond at the nonreducing ends of each cleaved GAG fragment. The deamination process results in a loss of nitrogen and sulfate but does not alter the stereochemistry of the hexuronic acid. Peroxyl radical cleavage catalyzed by metal ions or gamma irradiation depolymerizes GAGs. Such a method may not be suitable for producing structurally well-defined oligosaccharides.
Enzymatic Depolymerization
Due to the sheer size of full-length GAG polysaccharides, (partial) chemical or enzymatic depolymerization into smaller oligosaccharides is crucial for characterization. Depolymerization combined with chromatographic and electrophoretic separations establishes the link between the shorter oligosaccharides (dp < 12) and the full-length GAG chains (Figure 2). Both approaches aim at tackling challenges arising from dense sulfation and isomerism, two aspects that complicate the analysis of GAGs, yet are amenable through state-of-the-art technologies, such as chromatography, MS, and NMR.
Figure 2.

Common glycosaminoglycan depolymerization strategies are illustrated through the example of heparan sulfate/heparin. (A, left) When glycosidases are used to depolymerize GAG chains, the resulting cleavage preserves the hexuronic acid stereochemistry. Enzymes with endolytic activity are required to obtain oligosaccharides covering the full sequence. As for moderately sulfated HS/heparin chains, using heparanase as endo β-glucuronidases cleaves at the reducing end of GlcA residues (A, right). Prokaryotic lyases, such as heparinase I–III, follow a β-eliminative mechanism, resulting in Δ4,5-unsaturated uronic acid residues at the new nonreducing end. Consequently, stereochemical information is lost in the process (B, left). Benzyl esterification with alkaline β-elimination mimics lyase activity and creates Δ4,5-unsaturated uronic acid residues at the new nonreducing end (B, right). While preserving hexuronic acid stereochemical information at the cleavage site, deamination cleavage alters the structure of the glucosamine through the formation of 2,5-anhydromannose. N-Acetyl groups on glucosamines block the reaction, making prior deacetylation necessary. Reproduced from ref (3). Copyright 2022, American Chemical Society.
Enzymatic Digestion
GAG-degrading enzymes belong to the hydrolase or endo-or exolytic lyase families. They can be of mammalian or bacterial origin. Via an eliminative mechanism, the action of lyases results in a 4,5-unsaturation, i.e., leading to a double bond on the uronosyl residue. This chromophore absorbs at a wavelength of 232 nm.
Albeit more or less specific, HP and HS can be depolymerized from polysaccharides into disaccharides using different classes of bacterial heparinases I, II, and III.11 Heparin oligosaccharides generated from heparinase I digestion will feature sulfated extremities. In contrast, the digestion of HS with heparinase III will display nonsulfated terminal saccharide units. Other heparin-degrading enzymes yield the production of oligosaccharides with distinct structural features. The bacteriophage K5 lyase exclusively cleaves HS nonsulfated disaccharides, thereby generating large fragments.12
A limited subset of enzymes drives the digestion of CS/DS. Chondroitinase ABC catalyzes the complete depolymerization of CS/DS chains into disaccharides. Chondroitinase AC-II and chondroitinase B also degrade CS/DS, with a complementary substrate specificity for GlcA- and IdoA-containing disaccharides, respectively. These yield DS-rich or CS-rich oligosaccharides from CS/DS mixed polymers, respectively.13
The enzymatic degradation of nonsulfated HA is achieved by bacterial lyases rather than extraction from animal tissues, using naturally producing or genetically engineered microbial strains, and used as a starting material through digestion with hyaluronidases.14 Partial digestion with mammalian hyaluronidases generates a wide range of HA oligosaccharide lengths. Mammalian hyaluronidases yield tetra/hexasaccharides because these enzymes display hydrolytic and transglycosylation activities simultaneously.
Perspectives
GAG lyases and hydrolases represent valuable tools for studying GAGs, as exemplified by bacterial heparinases, whose complementary substrate specificities have been proven critical for defining the fine molecular feature of HS chains (composition, size, and domain organization) and for the generation of structurally defined oligosaccharides. However, because of the tremendous structural heterogeneity of GAGs, further progress will require increasing the panel of accessible enzymes by identifying and characterizing new enzymes with alternative and/or higher cleavage specificities. It is also particularly true for HA and CS/DS, for which the number of fully characterized, available enzymes remains limited. One striking example is the absence of commercial HA depolymerase after hyaluronidase from Streptococcus dysgalactiae has been discontinued. Bacteria, with highly diverse activities, represent an immense and poorly explored source of GAG degrading enzymes.(15)In particular, increasing interest may arise from bacterial polysaccharide utilization locus (PUL) encoding new GAG degrading enzymes as invaluable tools for GAG analysis or further understanding of Host/microbiota interactions.
2.2.2. Purification of Natural GAGs Oligosaccharides
Following depolymerization, isolation of GAGs oligosaccharides follows two strategies, based on either physical criteria (size or charge) and/or ligand binding properties.
Affinity Chromatography
Affinity chromatography uses protein-functionalized columns. Alternative techniques, such as filter binding assays, use radiolabeled or biotinylated GAGs. Oligosaccharides bound to the protein are eluted using a NaCl gradient, the ionic strength required for elution indicating the affinity of the interaction. However, these techniques are designed for analytical purposes rather than upscaling oligosaccharides’ production. Furthermore, because of the high electrostatic charge and structural redundancy of GAGs, affinity-based separations are unlikely to lead to pure oligosaccharides. Protein immobilization on columns, usually achieved through amine coupling of lysine residues, could affect GAGs’ binding properties.
Size-Exclusion Chromatography
Size-exclusion chromatography can be used to separate GAG fragments based on properties, such as length, net electrostatic charge, and sulfation pattern, and yields size-defined oligosaccharides. Low-pressure liquid chromatography with commercial resins, such as Bio-Gels (BioRad) and Sepharose gels (Cytiva), yields efficient size separation of HS fragments. According to published calibration curves, Sepharose (CL-4B and CL-6B) resins have been used for estimating the size of HS chains or large fragments.16 Bio-Gel P10 allows the resolution of fragments ranging from di- to octadecasaccharides, commonly used to analyze nitrous acid and heparinase degradation patterns and prepare size-defined oligosaccharides. High-pressure size-exclusion columns separate GAG oligosaccharides ranging from di- to decasaccharides, but these columns are not suited for preparative purposes and, except for short oligosaccharides (di- to tetrasaccharides), do not match the resolution achieved by low-pressure size-exclusion columns.
Anion-Exchange Chromatography
Because of the polyanionic nature of GAGs, ion-exchange chromatography is another method of choice for purifying oligosaccharides (for a review, see ref (17)). Low-pressure, weak ion-exchange chromatography, with resins such as DEAE or Q-Sepharose, has been widely used to purify HSPGs from crude cell extract. However, the resolution of GAG oligosaccharides with fine structural variations requires strong anion exchange and high-performance liquid chromatography. Commercial analytical and preparative columns enable the separation of oligosaccharides primarily according to their charge and, to a certain extent, their sugar content (particularly regarding the IdoA/GlcA epimer ratio) and sulfation pattern. With size-exclusion chromatography, SAX-HPLC remains the reference method for preparing HS oligosaccharide libraries. This technique has also been commonly used for GAG structural characterization by disaccharide analysis until being progressively replaced by separation techniques compatible with MS coupling or disaccharide fluorescent derivatization.18
Gel Electrophoresis
Agarose gel electrophoresis has been applied to the analysis of GAG polysaccharide mixtures from tissue extracts or body fluids and, to some extent, to low molecular weight oligomers. Linear or gradient polyacrylamide gel electrophoresis (PAGE) enables high-resolution GAG and oligosaccharide species analysis. It has been used for oligosaccharide mapping and for purifying small quantities of purified oligosaccharides. Staining with cationic dyes allows the visualization of GAGs, which permits the detection of microgram quantities of material. Silver staining can also improve detection sensitivity to the nanogram level, enabling the dosage of GAGs in biological fluids. After gel electrophoresis, methods for oligosaccharide blotting onto the membrane have also been established. Interestingly, PAGE separation properties, which rely on oligosaccharide size, charge, and shape, significantly differ from size exclusion and SAX chromatography. When combined with these two techniques, PAGE enabled the preparation of oligosaccharides with a very high degree of purity.19
Other Separation Techniques
Finally, the separation of disaccharides/oligosaccharides for analytical purposes can be achieved using a large panel of separation techniques. These include capillary electrophoresis, reversed-phase ion-pairing high-performance liquid chromatography (RPIP-HPLC) and hydrophilic interaction liquid chromatography (HILIC).20 However, these techniques have been developed for direct coupling to mass spectrometry fluorescent derivatization and are only suitable for the resolution of minute amounts of material.
The difficulty of sequencing GAGs from chemical and enzymatic fragmentations results in the many oligosaccharides that originate from all parts of the full length polymer. There exists a strategy that takes advantage of the potential of labeling, via bio-orthogonal groups, the reducing end of GAG chains liberated from proteoglycans by β-elimination. Following an enzymatic fragmentation and a size-separation by PAGE, the labeled fragments are separated from the unlabeled and blotted to paper functionalized via click-chemistry with the bio-orthogonal partner. The sequence can be read by establishing the nature of the nonreducing terminal disaccharide in each band (via HPLC, MS, specific antibodies, etc.). The bioavailability of the labeled fragments allows the study of sequence-defined interactions with biomolecules such as proteins and cells.21
Perspectives
During the last two decades, considerable progress has been made in the structural analysis of GAGs, either purified or from biological samples. However, while these advances have improved our knowledge of GAG structure, access to highly pure, naturally occurring GAG oligosaccharides in semipreparative quantities remains a critical bottleneck for functional studies in biological assays. The only available techniques for preparing structurally defined saccharide libraries remain those used for the past 30–40 years, with major limitations in terms of resolution and processing time. Because of this lack of modern tools and the natural heterogeneity of GAGs, achieving preparative purification to a single species of oligosaccharides beyond the size of a hexasaccharide is still highly challenging. Most protein binding domains involve saccharides of eight sugar units and above, and slight changes in sulfation patterns may dramatically affect biological properties. Therefore, there is a great need for new separation techniques with improved resolutive properties to prepare highly pure GAG oligosaccharide structures.
2.3. Preparation of Synthetic GAGs
Together with native GAG depolymerization, chemical synthesis is one of the most powerful tools for producing well-defined, structurally homogeneous GAG oligosaccharide sequences. Since the pioneering work on the chemical synthesis of the heparin pentasaccharide sequence responsible for the anticoagulant activity of this polysaccharide, multiple total syntheses of GAG oligosaccharides have been reported.22
2.3.1. Solution-Phase Synthesis
The solution-phase synthesis of GAGs oligosaccharides first involves the preparation of conveniently functionalized building blocks, generally mono- or disaccharide units. The coupling of these building blocks leads to the fully protected oligosaccharide chain analogues, and the deprotection–sulfation steps deliver the target GAG oligosaccharides. GAG chemical synthesis is challenging because of the inherent difficulties of oligosaccharide synthesis, namely the control of the regio- and stereochemistry of the glycosidic bonds and the introduction of sulfate functions at specific positions. Furthermore, carefully designed protecting group strategies are required. Uronic acid moieties are usually identified as low reactive sugars in coupling reactions due to the electron-withdrawing effect of the carboxylate functions. In recent years, we have witnessed impressive advances in GAG oligosaccharide synthesis.22,23 However, only a limited number of structures with specific sulfate group distributions is available, a process that initiated the preparation of libraries containing differently sulfated sequences. Developing an intelligent modular strategy based on orthogonally protected disaccharides allowed for generating a library of heparan sulfate tetrasaccharides with different sulfation patterns.24
Pesrpectives
Despite all these impressive contributions, the GAG oligosaccharides synthesized to date only cover a small part of the chemical space, especially when considering sequences longer than tetramers. This lack of more comprehensive GAG collections arises from the difficulties of the solution-phase synthesis of these molecules, which requires column chromatography after each reaction step, making the synthetic process extremely time-intensive.
2.3.2. Automated Solid-Phase Synthesis
Automated solid-phase synthesis25 offers a promising alternative to address some of the bottlenecks of GAG oligosaccharide synthesis. A solid support equipped with a linker is used in solid-phase synthesis to successfully couple the building blocks and assemble a growing chain of oligomers. The monomers have a temporary protecting group removed from the resin-bound oligomer to allow further chain growth in the next coupling cycle. After each reaction step, the desired product is purified by washing the resin, avoiding multiple chromatography steps.
For oligosaccharide assembly, the regio- and stereochemistry of the coupling must be controlled. It is secured by a suitable selection of orthogonally protected monosaccharide building blocks conveying an ad-hoc combination of temporary and permanent protecting groups.
Due to the straightforward elimination of side products, on-resin reactions can be driven to completion using an excess of reagents or running several reaction cycles.
Solid-phase approaches yielded the preparation of nonsulfated oligosaccharides. Nevertheless, the preparation of long sulfated GAGs oligosaccharides remains difficult. GAG oligosaccharide precursors were prepared on a fully automated computer-controlled synthesizer.26 The sulfated, partially protected intermediates were released from the resin by photocleavage of the linker moiety. However, several additional solution-phase deprotection steps were required to reach the final deprotected GAG oligosaccharides. These final transformations are far from trivial due to the lability of sulfate groups. Furthermore, the high polarity of sulfated compounds complicates purification. Considering the lower scalability of solid-phase synthesis, the difficulties encountered in the final off-resin deprotection steps can limit the utility of these approaches.
Perspectives
Improved solid-phase strategies, including optimized deprotection/sulfation procedures, are in demand. Novel and more efficient on-resin sulfation and deprotection protocols facilitated access to HS disaccharides(27)and sulfated, non-GAG glycans,(28)minimizing the manipulations required after release from the solid support.
Turning automated solid-phase synthesis into a routine operation to prepare GAG oligosaccharides faces some issues. Usually, a large excess (5–15 equiv) of glycosyl donor building blocks is required to complete the resin glycosylation reactions. These units are of high value, containing a complex protecting group distribution that depends on the glycosidic bond sequence and sulfation pattern of the target GAG. Their synthesis usually involves a high number of (solution-phase) reaction steps, and, in general, they are not commercially available. One way to address this problem could be the development of new glycosylation protocols to achieve highly efficient coupling reactions on the solid support, avoiding the use of a large excess of sugar building blocks, for example, by careful control of the reaction temperature.29 Novel approaches for rapidly accessing crucial building blocks required for assembling GAG oligomers are also highly desirable. A good alternative is the production of the needed disaccharide building blocks by the controlled acid hydrolysis of the naturally occurring GAG polysaccharides. Recently, the preparation of core disaccharide building blocks through the controlled acid hydrolysis of heparin and heparosan has been reported.30 Noteworthy, automated solution-phase synthesis of oligosaccharides relying upon preactivation-based, multicomponent one-pot glycosylation sequences was very recently proposed and demonstrated to allow the synthesis of fondaparinux pentasaccharide at gram scale in short times and higher yield with respect to manual one-pot synthesis (Figure 3).31
Figure 3.
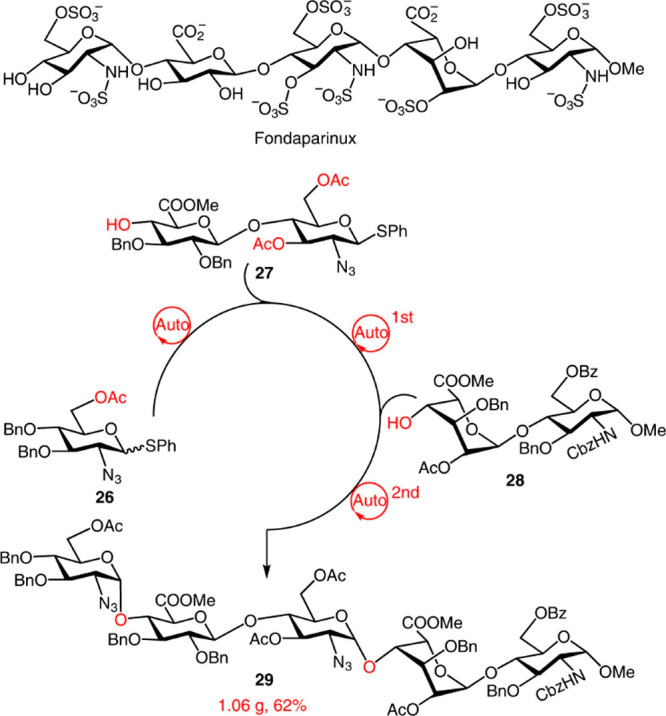
Gram-scale synthesis of a protected fondaparinux pentasaccharide using a dual-mode automated solution-phase glycan synthesizer. A library of oligosaccharides covering various glycoforms and glycosidic linkages assembles rapidly, either in a general promoter-activation mode or light-induced-activation mode. The synthesis used thioglycoside d-glucuronic acid containing disaccharide with 3,6-di-O-acetyl groups for the α-directing glycosylation and l-iduronic-acid-containing disaccharide as building blocks. All compounds are readily obtained from commercially available monosaccharide or disaccharide intermediates. Reproduced with permission form ref (31). Copyright 2022, Springer Nature.
These preparations involved half of the chemical steps usually required for the traditional synthesis of these precursors from commercially available monosaccharides. Controllable enzymatic degradation of CS using bovine testicular hyaluronidase allowed the straightforward isolation of pure tetra- and hexamer intermediates that facilitated access to size-defined fucosylated CS oligosaccharides.32 Despite the growing interest in the peculiar biological and biomedical features of fucosylated CS, synthetic access to pure oligosaccharides higher than a nonasaccharide (being an octasaccharide, the minimum structural unit able to confer remarkable activities) and/or with an isomeric distribution of fucosyl branches is still missing. Synthetic efforts toward these targets are foreseen in the coming years.
Besides solid-phase strategies, alternative methodologies speed up GAG oligosaccharide synthesis by minimizing purification processes associated with repetitive deprotection/glycosylation steps. For instance, despite rapidly growing employment in many organic synthesis fields, the application of continuous flow systems has been poorly investigated for GAG synthesis and could be a topic for interesting developments shortly. Conversely, a programmable one-pot approach has already been successfully employed to synthesize protected heparin pentasaccharides that were selectively deprotected and sulfated to afford sequences with well-defined 6-O sulfation patterns.33 This methodology uses a series of glycosyl donors with different relative reactivity values that can be sequentially activated in “one-pot” to rapidly generate the target oligosaccharide chain without workup and purification of intermediates. Another possibility is the application of fluorous-assisted strategies.34 In this case, a highly fluorinated tag is usually attached to the sugar building block that will constitute the reducing end of the oligosaccharide, similar to the connection to the resin in solid-phase synthesis. Iterative glycosylations and acceptor hydroxyl group deprotections will generate the sugar chain. Since molecules bearing the perfluorinated tag can be easily separated from nonfluorinated compounds by simple fluorous solid-phase extraction, the assembly process is greatly facilitated. Compared to solid-phase approaches, the reactions are run in solution. This has two positive consequences: standard analytical techniques can monitor the reaction course, and less glycosyl donor is required to complete the coupling steps since the reactivity of carbohydrate building blocks in solution is generally higher than that of solid-supported sugars. These new procedures for the GAG oligosaccharide chemical synthesis will expedite the production of more extensive collections of GAG sequences, with a longer length and more diverse sulfation patterns, to cover the wide structural variety found in GAG polysaccharides. Advances in techniques for the structural characterization of these molecules (MS, NMR) will also positively impact GAG synthesis. Undoubtedly, these improvements will increase the information derived from screening glycomic technologies like GAG microarrays.
2.3.3. Enzymatic and Microbial Cell Factory Synthesis of GAG Oligo- and Polysaccharides
Enzymatic and chemoenzymatic approaches have been successfully applied to produce homogeneous GAG oligosaccharides.35 Enzymes catalyze the glycosidic bond formation with exquisite stereo- and regioselectivity and the specific positioning of sulfate groups without the complex protecting group designs required in chemical synthesis. The enzymatic preparation of GAG oligosaccharides first involves glycosyltransferases to build the sugar backbone. Then, different sulfotransferases install the sulfate functions to the designated positions. In the case of IdoA-containing sequences, a C5-epimerase further converts GlcA residues to IdoA units. Glycosyltransferases catalyze the linkage between activated uridine diphosphate (UDP) sugar donors and the corresponding acceptors. UDP sugars are usually expensive, and enzymatic cascades have recently been developed to facilitate access to these substrates.36 Non-natural UDP sugars have been developed to produce specific sulfation patterns. For instance, a non-natural UDP-GlcNTFA donor (NTFA = N-trifluoroacetyl) have been used to introduce an N-sulfate group at a particular position of the oligosaccharide chain by a chemoenzymatic approach.35,37 Other advances in the field have improved accessibility to GAG enzymes, lower production costs, and higher conversion efficiency of 3′-phosphoadenosine-5′phosphosulfate (PAPS), the universal sulfate donor in sulfotransferase-catalyzed reactions. The generation of gram-scale quantities of a wide variety of GAG oligosaccharides is greatly improved. The access to new engineered and GAG biosynthetic enzymes at high expression levels and the development of novel, chemically modified, non-natural acceptors and nucleotide sugar donors will expand the repertoire of GAG oligosaccharides that enzymatic and chemoenzymatic strategies can prepare.
Apart from synthesizing GAG oligosaccharides from monosaccharide primers, enzymes can also be useful in obtaining semisynthetic GAG polysaccharides from microbial-sourced polymeric substrates. Unsulfated heparosan and chondroitin can be obtained as capsular polysaccharides from bacteria such as Escherichia coli, Pasteurella multocida, Streptococcus spp., Yersinia enterocolitica, or Bacillus subtilis (subjected to metabolic engineering strategies and/or tailor-made fermentation processes to improve the yield of the polysaccharides).38 They have been exploited as starting materials for synthesizing heparin and chondroitin sulfate polysaccharides through enzymatic steps catalyzed by N-deacetylase, epimerase, N-sulfotransferase, and several different O-sulfotransferases, respectively. In vitro-enzymatic synthesis can be deployed to produce size-defined polysaccharides, which are not accessible from animal or bacterial sources. These are commercially available for HA and established for other GAGs such as heparosan and chondroitin.39
A key outcome of all the recent advances in the enzymatic synthesis of GAGs has been the direct microbial biosynthesis of structurally homogeneous sulfated GAGs. Suitably metabolically engineered microbial cell factories have been demonstrated to produce sulfated GAGs, employing only methanol as a carbon source for polysaccharide skeleton construction.40 The microbial cell factory approach offers several applications, including biosynthetic access to unnatural GAG polysaccharides and derivatives.
Under the name of GAGOme, a library of isogenic cell lines that differentially display distinct GAGs features has been constructed. The library was engineered from a large panel of Chinese hamster ovary cells with knock-out or knock-in of the genes encoding most of the enzymes involved in GAG biosynthesis. This library can be used for cell-based binding assays, recombinant expression of proteoglycans displaying distinct GAG structures, and the production of distinct GAG chains on metabolic primers. They can be used for the assembly of GAG glycan microarrays.41
Perspectives
Despite the numerous efforts through the different approaches discussed above, preparing a large library of GAG oligosaccharides (and even less so polysaccharides) with defined sulfation patterns remains challenging. A minimal collection of structurally defined GAGs have become commercially available, and this restricts fundamental studies in further understanding of GAG functions
2.3.4. Structural Modification of GAGs
Native GAGs can be functionalized with specific target functional groups or labels.42,43 Most of the structural modifications are chemoselective. They involve, for example, the derivatization of the carboxylate functionalities into amides and hydrazides carrying specific labels (e.g., fluorescent tags, bioactive moieties) or functional groups (e.g., double bonds, thiols, o-quinones etc.) for different applications in controlled drug delivery and tissue engineering.44 To this end, chemoselective derivatizations of alcohol moieties, vicinal diols and acetamido groups have also been achieved. It is worth noting that most of these modifications are randomly distributed along the polysaccharide backbone, with no control of the positions subjected to derivatization. Only a limited number of regioselective modifications have been reported that mainly involve introducing sulfate groups at specific hydroxyl positions within the repeating units of microbial-sourced unsulfated GAG biopolymers.45
Single-site functionalizations aim to derivatize GAGs in a single point of the polymeric structure to introduce a label on the polysaccharide without altering its natural behavior. These functionalizations are currently limited to the (pseudo)reducing end of the polysaccharide chain. They typically exploit the unique reactivity of the hemiacetal moiety present at the reducing end. Alternatively, the possibility to isolate, from proteoglycan proteinase digest, the GAG polysaccharide still carrying a single serine at its pseudoreducing end allows the site-specific derivatization of its amine group.
Perspectives
The regioselective derivatization of GAG polysaccharides remains a challenge. The investigation of tailored chemical methods—i.e., direct regioselective reactions or multistep sequences relying upon suitable protecting groups—is mandatory to avoid random or poorly controlled derivatizations that introduce additional heterogeneity to GAG polysaccharide structures. Despite some accomplishments in the last years, there is still a great deal to be done in the field. Being able to label GAG polysaccharides at defined locations on the chain is even more challenging. Selective modification of the reducing end is relatively straightforward,46and nonreducing end modification of enzyme-digested (but not native) chains is also established.47Site-specific modifications of single sulfation motifs or single positions anywhere along the polysaccharide chain apart from the reducing end, are missing and appear as a distant goal. Recent years have witnessed progress in this area, thanks to automated28and/or enzyme-assisted48syntheses and a limited collection of defined GAGs that have become commercially available. However, the library is still limited and restricts fundamental studies in understanding GAG functions.
2.3.5. Chemical Synthesis of GAG Mimetics
From all the above, it is clear that synthesizing GAG oligosaccharides remains a highly sophisticated and complex task. Therefore, developing novel GAG mimetics, more easily accessible than GAG oligomers, represents a promising line of work. GAG mimetic compounds can be synthesized to imitate the structure and biological functions of naturally occurring GAGs while improving the pharmacological properties of the native oligosaccharides, thus increasing the therapeutic applications of GAG-like molecules.
Several sulfated non-GAG oligosaccharides have been synthesized as GAG mimetics, displaying a simplified chemical structure compared to natural products. For instance, pixatimod (PG545) is a 2,3,6-O-sulfated glucose tetrasaccharide carrying a cholestanol moiety at the reducing end. This clinical-stage HS mimetic has potent anticancer and anti-inflammatory activities, and recently it has been demonstrated that it also inhibits the interaction between the SARS-Cov2 Spike protein and the ACE2 receptor.49 Starting from maltotetraose, the preparation of this derivative is less complex than the synthesis of an HS tetrasaccharide. For compounds with clinical applications, the feasibility of their multigram-scale production is an important point.
A library of IdoA homo-oligosaccharides with different sulfation patterns and chain lengths has also been synthesized as HS mimetics.50,51 These compounds showed the typical conformational plasticity of IdoA-containing molecules and exhibited binding to chemokines and potential applications for cancer treatment. Linear polyglycerol sulfates exhibit good heparin mimetism.52 Other types of GAG mimetics are aromatic ring systems, such as polyphenols, decorated by sulfate groups. These small, structurally homogeneous nonsaccharide mimetics can interact with diverse GAG-binding proteins offering promising opportunities as, for example, antiviral drugs.53 In these compounds, the aromatic backbone can establish additional contacts with hydrophobic regions of the protein receptor, giving additional possibilities for optimizing molecular recognition. The synthesis of multivalent systems, where GAG sequences are introduced as pendant ligands in a nonsaccharide scaffold, is another attractive alternative to access well-defined GAG-like molecules easily.54 Generally, carbohydrate ligands are short synthetic oligosaccharides displaying the characteristic disaccharide repeating unit of a particular GAG and an orthogonal functional group for further conjugation. These fragments are then attached to a dendrimeric or polymeric backbone to afford the corresponding multivalent systems, usually after one single chemical step. Such compounds presenting multiple copies of GAG ligands can be easily produced.
Polymeric GAG mimetics have been mainly obtained from natural, non-GAG polysaccharides by regioselective sulfation through direct or multistep approaches.55 Such engineered sulfated polysaccharides can be produced in large quantities at a low cost from renewable raw materials (e.g., plants, algae, fungi) or microbial fermentations as a more ethical, environmentally and economically sustainable alternative to the isolation of GAGs from animal tissues.56 Furthermore, they may exhibit improved properties compared with natural GAGs, which can be tuned appropriately by additional structural modifications such as the insertion of functional groups for compartmentalization, in vivo biodegradability, hydrogel formation, cross-linking, 3D-printing, etc. An alternative approach to address these challenges in the frame of polymeric GAG mimetics is the synthesis of sulfated glycopolymers. They are obtained by polymerizing sulfated glyco-monomers to yield polymers showing well-defined structures and often closely controlled molecular weights and narrow chain length distributions.57
Perspectives
In GAG-mimetics synthesis, it would be interesting to find a way to construct block copolysaccharides by linking together two or even more structurally diverse GAGs or mimetics thereof, each characterized by a different sulfate content and/or sulfation pattern. The design and synthesis of block copolysaccharides or, more generally, polysaccharide-containing block copolymers is a growing field. Nonetheless, no block copolysaccharides composed of GAG polymer fragments have been reported yet apart from a very recent paper describing the chemo-enzymatic synthesis of some differently sulfated HS hexa- to hexadecasaccharides that were then linked together through CuAAC click reactions to give multidomain structures up to a 28-mer species.58The achievement of this goal is interesting not only for obtaining newly designed, synthetic GAG materials with potential interesting bioactivities but also to have a powerful tool to tackle an almost unexplored issue of GAG structure–activity relationships. It is the effect on GAG biological roles of the clustering of differently sulfated disaccharide subunits in a series of complex regions or domains with variable sulfation patterns along GAG backbones.
GAG mimetics showing negative charges on groups different from sulfates also represent an area of novel development. For example, by comparing sulfate and phosphate groups, their differences in size, polarity, and acid–base properties could lend unreported, interesting properties to phosphorylated GAGs. A theoretical study indicated distinct differences between natural sulfated GAGs and phosphorylated mimetics regarding structural flexibility and intra- and intermolecular interaction patterns.59Phosphorylated glycopolymers with a well-defined structure have been reported.60Conversely, robust, synthetic access to phosphorylated species having the same polysaccharide backbone of natural sulfated GAGs is still lacking, as polysaccharide phosphorylation is a very challenging reaction, requiring rather harsh conditions and generally giving products difficult to characterize, with low yields and degrees of derivatization. Therefore, significant advances in this field are awaiting. Overall, developing novel mimetics that attain similar 3D structures and protein-binding properties to GAGs will provide new tools to control GAG-mediated biological processes, paving the way to new applications in medicine and biotechnology.
2.4. GAG Analysis and Sequencing
Sulfated GAGs are among the most challenging biopolymers in nature to characterize. Obtaining information on the sequence of even the simplest full-length chains is a formidable task. Complexity and the associated challenges stem from the aspects of the GAGs structure described in section 2.1. The dense sulfation of GAGs complicates MS analysis due to Coulomb repulsion, sulfate loss, and the formation of multiple adducts. The occurrence of sulfation and epimerization at various positions generates many isomeric building blocks, which are difficult to distinguish using MS-based methods. There may exist a relationship between the sulfation pattern to specific biological functions. Therefore, elucidating such a “sulfation code” of bioactive sequences involved in protein binding adds to the analytical challenges of all sulfated GAGs. (Figure 4)
Figure 4.

General structures of GAGs with their monosaccharide components. Overview of the characteristic monosaccharide components, N- and O-sulfation motifs, and linkages displayed per the SNFG representation. Repetitive hyaluronan chains (HA) are not modified by sulfation and epimerization. Chondroitin sulfate (CS), dermatan (DS), and keratan sulfate (KS) sulfate display a variety of sulfation motifs. Heparin and heparan sulfate represent the most diverse family of GAGs.
2.4.1. Ion Mobility Spectrometry (IMS)
Online separations are essential for resolving complex mixtures into components before MS analysis. A relatively recent technique to disentangle GAGs mixtures is ion mobility spectrometry (IMS), a technique in which (bio)molecular ions are separated by their mass, charge, size, and shape. A weak electric field guides the analyte ions through a cell filled with inert neutral gas (He, N2). Compact ions collide less frequently with the inert gas than larger ions and traverse the cell faster. Over the last years, several IMS systems have become commercially available, usually in combination with MS as IM-MS. All commercial solutions provide a fast separation; however, the underlying methods are vendor specific and can differ significantly in the electric field, duty cycle, and peak-to-peak resolution. IMS can separate isomeric GAGs and even diastereomers. For reducing adduct formation and the complexity of the analysis, IMS separations are often performed using direct infusion.61−63 However, due to the fast millisecond separation, a direct hyphenation to liquid chromatography is possible, leading to information-rich multidimensional data sets.20
Various IMS techniques were used to characterize GAGs and complex GAG mixtures. Despite the potential of IMS, not all isomers can be quickly resolved, and it is not straightforward to predict a particular separation’s success (or failure). A comprehensive analysis of GAG oligosaccharides, including all structural features, usually requires combining several orthogonal techniques. However, the peak-to-peak resolution in IMS is increasing rapidly, and structurally closely related isomers such as those originating from epimerization can be resolved today.20 As a result, the ability to accurately measure ion mobility-derived collision cross sections for the structural annotation of unknowns rather than the resolution itself may be the major bottleneck for IMS in the future.
2.4.2. Tandem Mass Spectrometry Techniques
The activation methods most commonly used for GAG characterization via mass spectrometry are collision induced dissociation (CID), electron detachment dissociation (EDD), electron induced dissociation (EID), negative electron transfer dissociation (NETD), infrared multiphoton dissociation (IRMPD), and ultraviolet photodissociation (UVPD). The analysis method selection influences the abundance of cross-ring versus glycosidic cleavage products and the subsequent level of structural information.3
Collision-Induced Dissociation (CID)
In collision-induced dissociation , a kinetically excited precursor molecule that collides with buffer gas gradually imparts enough internal energy to cleave the most labile bonds. In glycans, these are usually the glycosidic linkages. In GAGs, however, the situation is different. Many research groups have applied CID to GAG analysis and found that this method is rather disadvantageous. Especially in highly sulfated GAGs, the sulfates are usually lost first, with the consequent loss of precious structural information.
Due to the sulfate and carboxylic acid groups, GAGs ionize well in negative ion mode. The most suited ion activation methods for negative ions include electron detachment dissociation (EDD) and negative electron transfer dissociation (NETD). EDD, which operates by irradiating multiply charged negative ions with 15–20 eV, has also been highly valuable for studying GAGs and is widely used for analyzing chains. Electron-induced dissociation (EID), which irradiates singly charged anions with 6–20 eV electrons, activates ions by electronic excitation. Without going through the process of cross-ring fragmentation, EID produces similar fragmentation to EDD.
Negative Electron Transfer Dissociation (NETD)
Negative electron transfer dissociation is the desired fragmentation approach to study highly sulfated GAGs as the dissociation of the precursor is accomplished at a faster speed with minimal sulfate loss. In addition, the short reaction time for NETD allows it to be paired with online separation techniques such as high-performance liquid chromatography (HPLC) and capillary-zone-electrophoresis (CZE).
Ultraviolet Photodissociation (UVPD)
Ultraviolet photodissociation helps determine modification sites within a GAG chain.3 UVPD uses an ultraviolet laser to rapidly raise the internal energy of trapped ions by electronic excitation, resulting in fragmentation. A single UV photon can raise the precursor ion into a dissociative state. It favors informative cross-ring fragments and yielded electron photodetachment, along with the corresponding charge-reduced neutral loss products. UVPD, at either 193 or 213 nm, produced both glycosidic and cross-ring fragmentation in GAG standards ionized in negative mode while maintaining sulfate modifications. UVPD does not require a fully ionized precursor to produce informative fragmentation.
Perspectives
Many challenges remain in analyzing GAGs. Recent advances and research in MS of complex GAGs are paving the way for faster and more complete analysis. The evolution of MS/MS methods has led to more detailed structural characterization for this class of carbohydrates. Promising developments address the elucidation of structures of GAG chains with meaningful lengths. Structural modifications can be determined by MS/MS, especially when using electron-based methods. Recent advances in GAG analysis software lead to a faster analysis process and a simplified way to identify unknown sample structures. The variety of separation techniques coupled with MS allows more complex samples to be explored on a reasonable time scale to determine composition and sequence information. GAG analysis has focused chiefly on shorter chains, but in some instances, the sequencing of intact GAG chains demonstrates the capabilities of MS analysis. Future developments will integrate the isolation of biologically relevant regions of GAG chains with MS analysis addressing significant and relevant biology and medical problems.
2.4.3. Gas-Phase Infrared Spectroscopy
The combination of mass spectrometry and gas-phase spectroscopy augments the range of tools for GAG sequencing. The better availability of tunable benchtop lasers, which can cover a broad range of wavelengths, leads to increasing interest in applying gas-phase IR spectroscopy for various classes of biomolecules,64 including sugars.65
Most conventional approaches are action spectroscopy techniques65 in which a photon-mediated “action” such as dissociation or fragmentation is monitored as a function of the wavelength. InfraRed multiple photon dissociation (IRMPD) spectroscopy is based on the sequential absorption of multiple photons. After each absorption, the energy is redistributed within the molecule. This slow heating continues until the internal energy of the ion exceeds the dissociation threshold, and fragments are formed. Plotting the fragmentation yield as a function of the wavelength yields vibrational fingerprints from which valuable structural information can be deduced. In the context of GAGs, IRMPD spectroscopy was successfully applied to determine the stereochemistry of the HexNAc (GlcNAc versus GalNac) and hexuronic acid (GlcA versus IdoA), the presence of HexN, the regiochemistry of the linkages within the oligosaccharide, and the regiochemistry of sulfation.66,67 IRMPD spectroscopy is usually limited to oligomers with a relatively low degree of polymerization as the vibrational spectra become more congested for larger ions and cannot be deconvoluted.68 This problem can be overcome by cryogenic gas-phase IR spectroscopy in which the ions are cooled prior to irradiation, either in a cold trap with subsequent messenger tagging69 or by encapsulation in superfluid helium nanodroplets70 (Figure 5). Even though the underlying principles and the temperature are different in both techniques,65 the spectra are generally comparable: they exhibit narrow and well-resolved vibrational bands that are diagnostic to minute structural details. In combination with sophisticated molecular calculations at the density functional theory (DFT) level, the resulting spectra can be used to obtain detailed structural models of the investigated ions.
Figure 5.
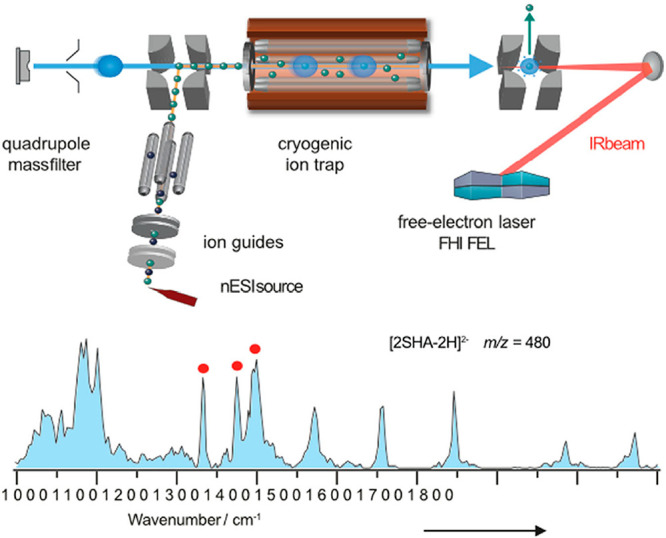
Schematic diagram of an IRMS instrument. The fragment ion charge ratio is measured via time-of-flight analysis. Helium droplets pick up trapped ions, which are immediately cooled to 0.37 K. There is subsequent irradiation of the droplets with monochromatic, high-intensity IR radiation, for example, using a free-electron laser (FEL). Reproduced with permission from ref (71). Copyright 2021, John Wiley and Sons.
Perspectives
The accumulation of experimental data has revealed that closely related isomers have distinct IR fingerprints. Such unique IR fingerprints of well-characterized standards could be organized in a database and used to identify structural features such as the sulfation pattern and perhaps the entire sequence of unknown GAG oligosaccharides in the future. However, already at the level of oligosaccharides, the chemical space of GAGs is too large to be fully covered by synthetic molecules. Theoretical spectra computed from DFT structures may be required to bridge this gap and annotate structures that are not accessible via synthesis. In addition, the combination of experimental and theoretical spectra will help to gain detailed information on the folding behavior and conformational landscape of GAGs.
The biggest technical challenge is undoubtedly the access to instrumentation. Gas-phase spectroscopy techniques require specialized light sources and sophisticated instruments, constraining their application to a few laboratories worldwide. MS technology is developing rapidly, and tunable lasers are becoming commercially available. Both are crucial aspects of transforming gas-phase infrared spectroscopy from a physicist’s toy into an easy-to-use instrument nonspecialists can operate.
2.4.4. Recognition Tunnelling Nanopores
A few reports of the successful sequencing of GAGs using recognition tunnelling nanopores via a single molecule method circumvents the need to obtain homogeneous samples to analyze intact GAG chains or use the complex sequences of analytical techniques mentioned previously. As a device, a recognition tunnelling nanopore provides a sequential reading of a mono- or disaccharide unit when the GAG chains translocate the nanopore.72 The formation of a transient complex between the translocated units and the molecules attached to two tunnelling electrodes generates an electric signal specific to individual monosaccharide units’ structure. The representations of the nanopore data signals of four synthetic GAGs of known composition revealed unambiguously clear differences. A machine-learning algorithm processed the results, distinguished the four different patterns, and identified each variant via image recognition software (Figure 6).73
Figure 6.

Nanopore sequencing. Illustration of the organization of the translocation device showing the insertion of the aerolysin nanopore within the membrane and the elusive depiction of a chondroitin sulfate glycosaminoglycan passing through the channel of aerolysin. Nanopore experiments use a horizontal lipid bilayer Teflon device. The setup comprises the cis and trans chambers connected by a sub-millimeter inner diameter capillary. The lipid bilayer is formed by depositing a film of 1,2-diphytanoyl-sn-glycero-3-phosphocholine over a conical aperture of 20–30 μmin diameter that separates the cis and trans chambers. Two Ag–AgCl electrodes are installed in the cis and trans chambers filled with 100 μL of a buffer allowing the application of a fixed voltage and measurement of the ionic current. The entire setup is placed within a grounded Faraday cage to shield it from electromagnetic interference electrically.74 Upon its translocation, the polysaccharide inside the channel blocks the current. For each translocation event, the block current, the open pore current and the duration of the event are recorded for further identification and statistical analysis. Adapted from ref (75) and is licensed under CC BY 4.0, https://figshare.com/articles/figure/Nanopores_GAGs_sequencing/19391822.
The characterization of GAGs oligosaccharides having various sulfate patterns, epimers of uronic acid residues, and glycosidic bonds can be achieved using a wildtype aerolysin nanopore. Not only can the size from tetra- to icosaccharides from heparin, DS, and CS be discriminated, but the different contents and distributions of sulfate groups as well. The detection of differences in the α versus β anomeric configuration at the 1–4, 1–3 glycosidic linkage highlights the performance of the sequencing.74
Perspectives
Following the proof of concepts that recognition tunnelling signals from disaccharide building blocks of GAGs possess unique signatures that can be used in distinguishing different stereoisomers, many developments remain. The speed of translocation needs to be reduced to the record of sufficient electrical signals for accuracy improvement, and a reference database for recognition tunnelling sequencing of GAGs is needed too.
2.4.5. NMR Analytical Methods
NMR methods are attractive for GAG analysis as they are nondestructive and do not require derivatization. Over the years, the arsenal of NMR methods developed for the structural determination of biomolecules has been applied to GAGs. The most common multidimensional methods involve homonuclear spectroscopy (1H–1H correlation spectroscopy, (COSY), total correlation spectroscopy (TOCSY), nuclear Overhauser effect spectroscopy (NOESY), rotating frame Overhauser effect spectroscopy (ROESY), heteronuclear spectroscopy (1H–13C heteronuclear single quantum coherence (HSQC), and heteronuclear multiple bond coherence (HMBC)). At present, the de novo elucidation of an unknown sample is limited to an octadecasaccharide.
A relatively low sensitivity limits their applications as milligrams of pure samples are usually required for the structural investigation or sulfate distribution through compositional analysis. A 1H–13C 2D NMR-based approach has been developed, directly performed on HS isolated from 13C-labeled cells. Integrating the peak volumes measured at different chemical shifts allows this nondestructive analysis to determine the polysaccharide’s sulfation and the iduronic/glucuronic profiles.76
In 2008, the adulteration of raw heparin with oversulfated CS spawned a global crisis prompting the FDA to revise the old specifications and recommend the development of physicochemical methods for improving the related critical quality attributes of heparin such as identity, purity, and potency assays.77 At the same time, the emerging enoxaparin biosimilars led to the need for thorough similarity proofs not conceivable by the old analytical procedures. Bidimensional NMR, particularly heteronuclear correlation spectroscopy, has become the technology of choice both to detect a variety of potential polysaccharide contaminants and to provide multiple quality attributes regarding the monosaccharide substitution in GAG sequences. The need for quantifying the composition of heparin in more detail, including minor features associated with specific biological activities or specific animal/organ origin of GAGs or to compare production batches, led to extending the use of HSQC for quantitative purposes.
The heterogeneity of heparin and GAGs requires each sample to be characterized by composition in differently substituted disaccharides in their sequence, mean molecular weight, and chain length dispersion. However, these parameters show batch-to-batch differences not only in products of different sources but also in different processes, from the same animal source and even in the same process. Therefore, the composition of a given batch does not bear quality information if it is not compared with large analytical result databases, representing the structural variability of heparin. Statistical methods, such as principal component analysis (PCA), have been used to compare test samples against mono- and bidimensional spectral libraries of heparin of different animal and organ origins. PCA extracts from a high number of variables, which are highly correlated and challenging for interpretation of a small number of orthogonal variables, which are more useful for sample profiling. PCA proved effective in clustering GAGs according to their origin or manufacturing and differentiating different crude heparins; a very complex mixture of GAGs considered the starting material of the active pharmaceutical ingredient (API) heparin production. Novel chemometric techniques, such as spectral filtering, have been applied to HSQC spectral databases to search for unknown features in heparin, whether due to contaminants or manufacturing failures.78
Recently, the FDA alerted industries regarding the potential risk of heparin contamination with nonporcine ruminant material contaminants, suggesting the application of physicochemical methods to ensure the safety of drugs and protect public health. The application of multivariate classification approaches to heparin 1H NMR spectra was a rapid and reliable tool for detecting contaminants. Partial least squares discriminant analysis (PLS-DA) provided the best discrimination of contaminated batches, enabling the detection of samples contaminated by heparin from other animal species at 5%.79
Perspectives
NMR integrated with statistical analysis is a valid quality tool for heparin in the entire production process and should be concurrently used with complementary techniques like SAX-HPLC and disaccharide analysis, but setting acceptance criteria requires the knowledge of the design space of normal processes of different animal source and characteristics of the main anomalies. It will require building large sample databases, supervised by regulatory authorities, and designing analytical procedures with evaluation ranges for results as simple as possible. Moreover, whereas the destination to routine pharmaceutical work usually limits NMR methods to medium field spectrometers (500–600 MHz), the larger availability of high field NMR instruments coupled with high sensitivity probes allows for increasing the sensitivity of these techniques that can also be applied on biological field, where a limited amount of sample is usually available.
2.5. 3D Conformations
Once the composition and sequences are established, a series of methods allow for determining the 3D structural and dynamical features of GAGs. The use of several spectroscopic methods, such as NMR, with appropriate temporal or spatial resolution, provides invaluable experimental data that require the contribution of molecular modeling to be fully interpreted. Structural elucidations of GAGs cover a range of descriptions from local to global properties.
2.5.1. Diffraction Methods
In contrast to other macromolecules, X-ray diffraction of polysaccharides does not provide sufficient experimental information for an unambiguous resolution of 3D structure; therefore, computer modeling techniques are needed to complement the lack of experimental data. The process of structural elucidation combines the calculation of diffraction intensities from various low-energy models with those intensities collected on X-ray diffractograms. In this context, it is even most appropriate to use the term “model” instead of “structure”. Within uniaxially oriented fibers, GAGs chains are extended. X-ray fiber diffraction studies of GAGs have demonstrated that they delineate the boundaries of the possible conformations of their secondary structures and the modes of associations of water molecules and mono- and divalent cations. Frequently, the fibers are embedded in small crystallites, where they make orderly lateral interactions with one another. Such homotypic organizations are artificial, but the observed secondary structures might help illuminate some states of GAGs in solutions and tissues. Those reports with X-ray diffraction and other techniques had proposed HA self-association through interchain hydrophobic interactions and hydrogen bonds, but this view has since been refuted. At physiologically relevant ranges of solvent pH and ionic composition, there is no evidence for interchain association of HA, as demonstrated in the solution phase80,81 and in films of surface-grafted HA chains.82
What Remains To Be Solved
40 years after elucidating such structural features, one may expect the new possibilities offered by X-ray synchrotron sources to investigate polycrystalline materials and explore different levels of structural organizations by microdiffraction would yield significant advances.
2.5.2. Structural NMR
NMR being sensitive to conformational and dynamics changes allows detailed insight into the secondary structure of GAGs and their molecular structures and dynamics in solution. Such analysis requires the correct interpretation of spectral data by applying sufficiently accurate computational approaches. The assignments of the 1H and 13C spectra of heparin, de-N-sulfated, and re-N-acetylated heparin and the measurements of the 1H–1H nuclear Overhauser enhancements and 3J coupling constants provided sufficient experimental data to generate a series of low energy molecular models which oscillate around a conformation similar to that determined by X-ray fiber diffraction.
Although scalar coupling constants usually have a more straightforward interpretation than NOEs, the analysis of spin–spin coupling constants in GAG molecules showed that this might not always be the case. In some instances, magnitudes of proton–proton three-bond coupling constants (3JH–H) have considerably different values as one would expect according to their dependence upon torsion angles, and a simple interpretation could lead to incorrect conclusions. Detailed theoretical analyses in GAG oligosaccharides showed that the magnitudes of the Fermi contributions to 3JH–H depend upon electronic structures proximal to (or neighboring) the coupled nuclei. The presence of oxygen atoms (even quite distant) with lone pairs causes changes in electron densities in the vicinity of the coupled protons and differs for different positions in atoms in various GAG residues. Furthermore, the magnitudes of paramagnetic (PSO) and diamagnetic (DSO) spin–orbit contributions in GAG residues were surprisingly large and caused the Fermi-contact contribution to no longer dominate. As the DSO contributions also alter the locations of the atoms in the molecule, DSO terms can considerably influence the 3JH–H magnitudes. These analyses indicate that the influences of oxygen atom lone pairs, PSO and DSO on the coupling constants magnitudes are rather complex in sulfated GAGs.83 Nevertheless, quantum chemical (QM) methods can provide the first-principle rationale for the effects in detail, allowing for the correct interpretation of spin–spin coupling constants.
Due to the high concentration of negatively charged sulfate and carboxylate groups, GAGs exhibit a high binding affinity to positively charged metal ions. Heparin binds to monovalent cations (Na+, K+), divalent ions (Ca2+ and Mg2+) and trivalent cations such as Al3+. Such bindings can induce structural changes in the three-dimensional structures and modulate their biomolecular interactions. Several techniques have been used to study the binding of metal ions, such as infrared spectroscopy, optical calorimetry, circular dichroism, and potentiometric titration, yielding inconsistent results. Recently, the metal binding to sodium heparin was monitored through a 23Na NMR-based competition assay.84,85 The results of the experiments demonstrate the occurrence of at least two metal-binding sites with different affinities, potentially undergoing dynamic exchange.
Perspectives
Despite significant advances in computational chemistry methods, further development of quantum chemistry approaches, including calculations of NMR parameters such as chemical shifts and spin–spin coupling constants, is desirable. In addition, it is necessary to test other methods of calculating the influence of water and counterions.
2.5.3. Computational Modeling
Once the composition and sequence of GAGs are established, determining the corresponding three-dimensional structural and dynamical features leads to understanding the molecular basis underlying their properties and functions. The range of the relevant computational methods capable of addressing such issues goes from quantum chemistry to mesoscale modeling throughout molecular dynamics and mechanics and coarse-grained and docking calculations.86,87 The structural and physicochemical features of GAGs pose a significant restriction to probing their 3D conformation experimentally. Computational modeling techniques based on classical mechanics are a powerful tool to characterize the statistical ensembles of GAG molecules in solution. Size and structural heterogeneity require multiscale modeling, which can be addressed to GAG fragments, starting from monosaccharides to longer polysaccharides (Figure 7).88,89
Figure 7.

From local to global. From quantum mechanics to coarse-grained simulation, computational methods yield continuous descriptions of the structural features occurring over a wide range of dimensions. Local properties include (1) the description of the monosaccharide low energy conformation characterized by the ring puckers; (2) the potential energy surface computed as a function of the values of the glycosidic torsion angles displaying the occurrence of the low energy conformers and the conformational pathways between them; (3) the interactions of the GAGs chains with ions and water molecules, and the occurrence of the several low energy helical structures of the GAG chain; and (4) the monitoring of fluctuations in lengths and volumes in disordered states and characterization of the radius of gyration and persistence lengths.
The term molecular modeling encompasses approaches at different levels of complexity of molecular description.89 Quantum mechanical (QM) methods allow us to determine molecules’ structural, energetic, and spectroscopic properties from the first-principles of electronic structure theory. Even in their better scaling form, such calculations are too computationally expensive to allow for the routine handling of a system counting over 200 atoms, which requires more approximate representations of matter, often achieved through all-atom (AA) additive or polarizable force fields within a classical mechanics framework. Despite their simplicity, these approximations are remarkably successful and allow for the study within a dynamic context of systems of biological relevance in terms of sufficient size and complexity, enabling routine sampling in the microseconds time scale. As an ultimate level of approximation, coarse-grained (CG) methods allow the study of the structure and dynamics of very large (up to several million atoms) and heterogeneous systems by reducing the complexity of their molecular representation while retaining their fundamental physicochemical characteristics. CG and supra-CG descriptions can be back-mapped to all-atom representation, thus back-tracing important molecular features. The investigations of GAGs primarily exploit such approaches, even if physical issues of these polysaccharides can often limit their application.
2.5.3.1. Quantum Mechanical Simulations
The quantum mechanical (QM) description is widely adopted to investigate GAG systems’ hydrogen bonding and coordination interactions and calculate spectroscopic properties needed to complement or interpret experimental data (see the section on IRMPD above).89,90 Due to the hefty computational cost required, even the better-scaling QM methods are regularly used to study monosaccharides and disaccharides, with rare applications to longer oligosaccharides.
The QM simulations in the gas phase complement the gas-phase IR spectroscopy experiments to decipher a complex experimental spectrum. Due to their high flexibility, GAGs populate multiple conformations at room temperature, not only in a solvent but also in the gas phase, which is relevant to the back-calculation of IR spectroscopy data. Even though the accessibility to different conformers, and thus the rate of conformational interchange, decreases with temperature, GAGs retain a significant degree of structural flexibility even in cryogenic conditions. The complex spectrum is still needed for structural annotation to decompose into components from different conformational states and species. The original spectrum is compared with the calculated spectra of the conformers likely to be present in the mixture. The IR spectra calculations of representative conformers follow an exhaustive conformational sampling of the molecule of interest. Optimizing the structure at a high level with the density functional theory (DFT) formalism, such as PBE0+D3/def2-TZVP, provides a reliable computed IR spectrum. Certain absorption bands are strongly anharmonic, so calculating anharmonic frequencies may be required, albeit computationally expensive.
At a different level of complexity, the water molecules are considered. Because of some additional degrees of freedom arising from water molecules and the low energy cost of their conformational transitions, a long time is required to minimize energy. The B3LYP functional and the 6-311++G(d,p) or 6-311++G(2d,2p) basis set provide experimentally relevant geometries, as demonstrated for tetra- and pentaheparin fragments91 The discrete nature of the explicit water model in the calculations enables the analysis of water positions located at hydration sites in GAGs. One can expect significant hydrogen bond interactions between oxygen atoms in GAG molecules (especially those oxygens in sulfate and carboxylate groups) and water molecules. DFT data showed that bifurcated, donor, and acceptor hydrogen bonds occur between water molecules and oxygens from the sulfate groups, an arrangement influenced by the structure of the first hydration shell in the vicinity of the sulfate and carboxylate groups. Theoretical analysis indicates that the strength of intermolecular hydrogen bonds between carboxylates in GAGs and water molecules is weaker than in the carboxylic acid–water complex.
DFT yields reliable NMR spectral parameters, such as spin–spin coupling constants. The measurements and interpretations of spin–spin coupling constants are the most accurate and accessible approach for determining molecular structures in solution. This approach was applied to the analysis of various GAG mono- and oligosaccharides, including the analysis of pseudorotation of the 2-O-sulfated L-iduronic acid (IdoA2S) pyranose ring. The application of DFT calculations enabled the calculation of accurate three-bond proton–proton (3JH–H) spin–spin coupling constants which agree well with experimental data.91,92
Other challenges arise from the strong polyelectrolyte nature of the sulfated GAGs. The GAGs’ Coulombic interactions with counterions exhibit site-specific coordination among sulfates, carboxylates, and positively charged counterions. Location and coordination depend upon the counterion type. DFT analysis indicated that sodium ions tend to form 6-fold coordination with oxygens from sulfates and water molecules, which occurs independently of the pyranose ring conformations. The coordination of bivalent calcium ions showed a tendency to form a pentagonal bipyramid. The DFT-derived structure and the computed spin–spin coupling constants suggest that the formation of the bipyramid is more appropriate in the chair form, which agrees with the published experimental data.93
Small cations (e.g., Na+) can strongly influence the first hydration shell of sulfated GAGs. Ionic interactions are generally considered stronger than the intra- and intermolecular hydrogen bonds; consequently, they play a significant role in shaping the 3D GAG structures. DFT calculations showed that the energetically more stable conformer is stabilized mainly by ionic interactions rather than by intramolecular hydrogen bonds (see the review by Nagarajan et al.88). For heparin fragments, the first hydration shell is strongly influenced by ion–ion and ion–dipole interactions between cations, sulfates, carboxyl, and hydroxyl groups, stabilizing the spatial structure. The counterions bound to sulfate groups stabilize some transient local chain conformations that can be probed experimentally by FTIR spectroscopy since the electron density of the sulfate group redistributes according to the strength of cation binding.
2.5.3.2. QM or Molecular Mechanics
QM or molecular mechanics methods throughout the all-atom (AA) molecular description are applied to sample GAG conformation and GAG-biomolecular interactions in solution. GAGs fragment length in such computer experiments is usually limited mainly due to the GAG or GAG-biomolecular starting structure. This level characterizes physicochemical interactions of GAGs and other biomolecules; it describes direct and allosteric mechanisms induced by GAGs. In this approximation, it is not trivial to investigate the contribution of solvation and counterions to GAG conformations due to the static point description of the charges in sulfate groups, water, and ions.
GAGs have benefitted from the development and application of molecular dynamics (MD) simulations to probe the conformational dynamics, inter- and intramolecular interactions, energetics of complexation, and atomistic structure–function for both free and protein-bound GAGs.94 Most investigations used the TIP3P water model for all-atom simulations specifying a 3-site rigid molecule with charges and Lennard-Jones parameters assigned to each of the three atoms. The orientation of two lone pairs of oxygen atoms required for the cation coordination is omitted. The application of TIP5P or polarizable water models can tackle this problem at the cost of significantly increasing computational time. Another approximation is the representation of cations as a single-point charge, albeit the coordination number is different. Ca2+ interacts with many anionic polysaccharides and is represented as a single point, but its coordination number spans from 6 to 8. Some ion models account for total charge distribution by introducing dummy centers that mimic the coordination features to minimize the approximation.
MD simulations revealed a wealth of atomic (e.g., torsions, puckering, hydrogen bonding, bridging waters), molecular (e.g., global shape), and thermodynamic (e.g., potential energy) information. Potential energy surfaces are also visually appealing for GAGs because they directly convey favored glycosidic torsions (Φ and Ψ), thereby quickly revealing local similarities and differences. Whereas a collection of such classical potential energy surfaces is freely available, new tools have been developed to help their visualizations. NMR studies have also supported these results, which is an independent approach to understanding GAG structure.51 MD and NMR provide a synergistic approach to understanding preferred conformational states for GAGs in solution.90 Conventional MD simulations may fail to adequately sample the free energy landscape, grasping only the conformations around the equilibrium. For this reason, replica-exchange MD and Gaussian-accelerated MD accelerate the conformational sampling of GAGs, increasing the sampling of conformational landscapes.
Perspectives
(1) A well-curated nonredundant, systematically designed data set containing structural and binding data for protein–GAG interactions should be created to test, calibrate, and further develop MD-based approaches for protein–GAG containing systems. (2) Novel MD-based techniques mainly designed to improve conformational sampling should be further adapted and applied for highly flexible GAG-containing molecular systems. (3) More focus on proper modeling solvent, ions, and glycosylation should be put in the MD-based studies of GAGs and their complexes.
2.5.3.3. Coarse-Grained Models
The coarse-grained (CG) description mainly applies to complex system evolution and molecular mechanisms or conformational changes occurring in long time-scale ranges. The approximation relies on representing groups of atoms as one pseudoatom (usually called a “bead”). This approximation results in decreasing the degrees of freedom in the molecular system and flattening the energy landscape, which allows us to speed up the calculations by some orders of magnitude. The speed increase depends on the type of mapping and the size of the grains adopted. The general drawback is the loss of stereoisomers description and the directionality of hydrogen bonds, which can be indirectly tuned via the mapping type and nonbonded interaction parameters. Several GAG CG models exist.95 Most of them, like very broadly used MARTINI,96 are based on the spherical pseudoatom parametrization, which could be potentially valuable in describing some purely electrostatics-driven processes physically inappropriate to reflect the sulfation code and, therefore, the specificity of GAG interactions. Physically based models using a nonspherical representation of pseudoatoms would be highly recommended instead of using empirical or machine learning-based approaches, whose underlying physics of the intermolecular interactions remains obscure.
Computational modeling of GAGs allows for describing GAG conformational space, achieved via the adiabatic approximation or its extension. The former approach implies that the rotations of adjacent glycosidic linkages in a linear polysaccharide are independent. The constituent disaccharides are exhaustively sampled, yielding a conformational map as a function of potential energy versus two coordinates, Φ and Ψ angles of the glycosidic linkage. The most energetically favorable regular configurations can be generated using the (Φ, Ψ) values of the ground state. The most representative conformation adopted by GAG is a helix; for HA, numerous helical conformations, both left- and right-handed, have comparable energies. This approach allows the generation of long polysaccharide chains with statistically relevant torsion angle values distribution along the chain. The latter approach characterizes the evolution of a long GAG chain in solution using usually all-atom molecular dynamics.
Perspectives
Even when the MD simulations describe GAGs or GAG–biomolecule complexes, the length of these sugar chains is far from the biological conditions since natural GAGs can reach up to 200 monosaccharide units (and much longer for HA). Furthermore, these methods lack a description of the dynamics of biologically relevant dimensions. Each approach has its strengths and limitations, and the most appropriate method must be selected based on the question that needs to be addressed.
2.6. Physical Properties: From Free-Floating to Bio-interfaces
2.6.1. Free-Floating GAG Polysaccharides
GAG polysaccharides intrinsically lack defined secondary or higher order structures at physiological pH and ionic strength. Instead, they dynamically sample a wide range of low-energy conformations. Characterization of the physical properties of GAGs thus falls within the scope of physicochemical methods to measure the molecular mass and the (average) size of GAG molecules in solution.
GAGs from biological sources are heterogeneous in their mass. The most common way to quantify the mass distribution of a GAG population is through the weight-average molecular mass (Mw), the number-averaged molecular mass (Mn), and the mass dispersity (Đ = Mw/Mn). The mass of sulfated GAGs (CS, DS, HS, including heparin and KS) in vertebrates is typically 10–50 kDa. HA mass varies much more widely and can reach values of many MDa. Well-established methods to quantify GAG mass distribution are available.97 Multiangle light scattering (MALS), when coupled with size-exclusion chromatography (SEC) or field flow fractionation (FFF), provides Mw, Mn, and Đ without the need for a size standard but requires relatively large amounts of sample (typically many μg).
Gas-phase electrophoretic mobility molecular analysis (GEMMA)98 and, more recently, solid-state nanopore sensors99,100 are well suited to quantify HA mass. These techniques analyze one molecule at a time and thus provide the mass distribution of HA samples with exquisite detail beyond what is encompassed by the average mass and mass dispersity values. They also need much less samples than MALS. However, they do require a set of mass standards for analysis and measurements are affected by GAG charge in addition to mass, which limits the use of these methods for samples of sulfated GAGs with unknown or varying charge distribution.
Gel electrophoresis (GE) is a simple way to estimate GAG mass which can be readily implemented in any biochemistry laboratory. It can analyze GAGs of any size, from the smallest oligosaccharides to the largest polysaccharides. Drawbacks, like for GEMMA and nanopores sensors, are that it requires mass standards and does not readily separate mass from charge effects.
Light scattering methods can determine the size of GAG molecules in solution. MALS provides the radius of gyration (Rg), i.e., the average distance of the constituent monosaccharides from the molecule’s center of mass. Quasi-elastic light scattering yields hydrodynamic radii (Rh) from the rate of molecular diffusion and is sensitive to smaller molecule sizes than MALS.
The persistence length (Lp) measures the flexibility of GAG chains. It defines the contour length range over which a linear polymer retains a stiff appearance and provides a link between the total contour length (Lc) and the in-solution size (Rg) of GAG molecules. The total contour length of GAG polysaccharides is readily estimated from the GAG mass, considering that each disaccharide unit has a contour length of 1.0 nm and a mass of 400 Da (for unsulfated GAGs) or approximately 500 Da (for sulfated GAGs, with minor variations depending on their degree of sulfation). The persistence length is 4 nm for HA at physiological pH and ionic strength and is likely similar for other GAGs (although computer simulations suggest that this depends on their degree of sulfation). This value is larger than most other biological and synthetic polymers (e.g., unfolded polypeptides, polyethylene glycol) and implies that GAG polysaccharides pervade comparatively more space and are easier to stretch.101
Novel methods based on nonlinear spectroscopy describe the molecular interactions that mediate critical mechanical properties of HA, such as the pH-induced gelation of HA, which undergoes a transition from a viscous to an elastic state in a narrow pH range around 2.5102 or the molecular mechanism underlying the condensation of Ca2+ and the subsequent influence of the chain flexibility.103
Perspectives
Methods are needed to quantify the size of small and variably sulfated GAG polysaccharides. Gold standard methods such as SEC-MALS work well for larger polysaccharides but have limited sensitivity for small polysaccharides, and all other methods suffer from difficulties in separating mass from charge effects. In particular, methods are needed to analyze GAG mass in the often minute amounts extracted from small tissues or tissue sections. An open question is also how much the persistence length varies across GAG types and as a function of GAG sulfation and environmental changes (e.g., ion types in saline solution). Experimental methods are needed to probe this directly and complement the predictions of computational methods.104
2.6.2. GAG Polysaccharides at Bio-interfaces
In many biological settings and biomaterials applications, GAGs are displayed on surfaces or other scaffolding structures, thus forming films of varying thicknesses. For example, HA can be retained on the cell surface through one end or multiple attachment points along the chain contour, and sulfated GAGs are typically tethered to their core protein via their reducing end. Characterizing the mass and conformation of end-attached or side-attached GAGs in situ remains challenging. Conventionally, GAGs are stripped off surfaces and scaffolds for subsequent solution-phase mass analysis, though this multistep processing is time-consuming and may entail artifacts.
A recently developed method105 based on the quartz crystal microbalance with dissipation monitoring (QCM-D) exploits that the softness and thickness of films of end-grafted HA increase monotonically with HA contour length. The results establish a quantitative method to determine the mass of end-attached HA, and sulfated GAGs from 1 to 500 kDa, with a resolution of better than 10%. A workflow recapitulates the main steps involved in GAG sizing on surfaces using this method, thereby offering quality control of GAG-based surface coatings.
Of all biomolecules known, sulfated GAGs have the highest charge density. The charge density of GAGs entails a high osmotic swelling pressure owing to counterions and endows GAG matrices with exceptional mechanical properties. Among the physiologically relevant properties of GAG matrices are their ability to lubricate (e.g., synovial fluid of joints) and bear dynamic loads (e.g., in cartilage). The rheological properties of semidilute HA solutions are well documented, with a particular emphasis on the influence of temperature, concentration, and ionic strength on dynamic mechanical properties. Like other flexible and well-solvated polymers, HA solutions display a viscoelastic behavior exhibiting a transition from viscous to elastic state with increasing deformation frequency. An increase in HA concentration and molecular mass yields a more elastic behavior. Overlapping time–temperature, time–concentration, and time–ionic strength superpositions demonstrate that temperature, concentration, and ionic strength do not alter the nature of the relaxation process but rather the relaxation time scale.
Perspectives
In situ analyses (and mapping) of GAG molecular mass, GAG chain conformation, and GAG concentration in GAG-rich matrices remain a distant goal. However, if accessible, they would provide a much richer view, focusing on the organization of GAGs and their associated molecules, rather than their mere presence, to better understand the mechanisms of GAG function in cells and tissues and for the design and quality control of GAG-rich biomaterials.
2.7. Data Management and Deep Learning Methods
Glycoscience is heavily rooted in multidisciplinary approaches, often implemented orthogonally to compensate for the absence of genetic determinants for glycosylation. These approaches all produce rich catalogues of data, each not sufficient to determine glycan types of glycosylation sites/populations but necessary when put into context with additional complementary sets. It is essentially the very definition of a data set on which Deep Learning algorithms can be trained, tested, and used for prediction or classification. Glycan sequencing requires orthogonal protocols involving multiple combinations of different liquid chromatography (LC), mass spectrometry (MS), NMR, and IR, each producing heaps of data, usually analyzed separately by highly trained scientists. Through the compilation of well-designed and curated data sets, Artificial Intelligence would have the potential to substitute human intervention, providing consistent data interpretation and, ultimately, rapid glycoanalytics. Toward this goal, the role of glycobioinformatics in the setup and curation of glycomics/glycoanalytics data sets is a notable example of Unicarb-DB.
In terms of 3D structure determination, the limitations glycoscience currently suffers from are similar to the difficulties inherent to the structural determination of intrinsically disordered proteins (IDPs) or regions (IDRs), for which AlphaFold2 is not greatly useful. As it stands, the contribution of glycan builders, such as glycam.org, CHARMM-GUI, GAG-Builder, and databases of equilibrated MD structures in combination with adequate conformational sampling, can address most queries. Nevertheless, limitations of the empirical additive force field formalism remain problematic in some cases and work toward developing machine learning force fields for glycans is one of the highly anticipated upcoming innovations in the field.
Perspectives
The use of Machine Learning algorithms to advance glycomics and glycoanalytics is in its infancy as this article is being written, with notable initial contributions from Daniel Bojar and colleagues in the prediction of lectin binding specificities and glycan-mediated host–pathogen interactions(106,107)in the development of tools to load and extract information from glycan data sets.108The use of Machine Learning methods to predict and understand GAGs structure to functions relationships from existing data sets and under-development databases is one of the promising upcoming fields of GAGs research soon.
3.0. Protein-Glycosaminoglycan Interactions
3.1. Analytical Tools to Identify GAG-Mediated Interactions
The identification and characterization of GAG–protein interactions rely on low- and high-throughput methods. The most commonly used are affinity chromatography, analytical ultracentrifugation, electrophoretic mobility shift assays, and real-time, label-free methods such as surface plasmon resonance and biolayer interferometry, which yield kinetic data (i.e., association and dissociation rates). In contrast, thermodynamic data (enthalpy changes, changes in entropy, changes in molar heat capacity) can be calculated using isothermal titration calorimetry.109 Microarrays and affinity proteomics have been developed as high-throughput methods to identify GAG-binding proteins and can be performed with GAG oligosaccharides of defined size, length, and charge prepared and fractionated as described in section 2 or with full-length, physiological, GAG chains. GAG and protein arrays have also been used to characterize the GAG repertoire of a protein and the protein repertoire of a GAG, respectively. The number of GAGs and/or GAG oligosaccharides spotted on these microarrays is generally limited (<100) partly because the purification and synthesis of GAGs are still challenging. The GAG arrays developed in the past few years have been recently reviewed.109 Although these GAG microarrays are not routinely available to the glycobiology community, neo-glycolipid-based microarrays have been developed. They contain a subset of GAGs and GAG oligosaccharides and are available for screening analysis at an affordable cost (https://glycosciences.med.ic.ac.uk/glycanLibraryIndex.html). GAG and protein arrays require purified GAGs, oligosaccharides, and proteins, whereas affinity proteomics can capture GAG-binding proteins from crude biological samples (e.g., cell or tissue lysates, culture supernatants, and biological fluids). Although GAG-binding proteins identified by affinity proteomics may contribute to the biological and structural functions of GAGs, their direct binding to GAGs and the biological or structural relevance of these interactions remain to be demonstrated.
In vitro binding measurements are not likely to reflect the biological context of the extracellular matrix, cell surface, or interior and nucleus. Cell or tissue experiments are needed to assess their biological significance by monitoring the interaction’s biological response. Chemical or enzymatic treatments globally or specifically altering GAG chemical groups and/or selective deletion of GAGs at the cell surface by knocking down the expression of GAG biosynthetic enzymes provide essential information. In addition, methods should be developed to study the ability of various GAGs to assemble multimeric protein complexes and follow the motions of GAGs and GAG-binding proteins throughout the extracellular matrix and the glycocalyx to reach and bind to their partners (e.g., cell-surface receptors).
3.2. Structure of GAG–Protein Complexes
3.2.1. Experimental Techniques: X-ray, NMR, and SAXS
X-ray Diffraction
The Protein Data Base comprehensively records more than 120 structures of GAGs protein complexes and 15 structures of long-chain GAGs s established by X-ray fiber diffractometry, X-ray scattering or by solution NMR. The size of GAGs bound to proteins ranges from disaccharides to full length polysaccharide, including the following degrees of polymerization (DP): DP2 (34), DP 3 (1), DP 4 (18), DP 5 (13), DP 6 (15), DP 7 (7), DP 8 (8), and DP 9 (1). More than 80% of the GAGs involved in the complexes are heparin and hyaluronic acid oligosaccharides, which does not reflect the diversity of GAGs.
The GAG-DB database (https://gagdb.glycopedia.eu/) provides the architecture and navigation tools to query the Protein Data Bank, UniProtKB, and GlyTouCan (the universal glycan repository) identifiers. Special attention was devoted to describing the bound glycan ligands using simple graphical representation and numerical format for cross-referencing other databases in glycoscience and functional data. GAG-DB provides detailed information on GAGs, their bound protein ligands, and interaction features using several open-access applications. The binding energy calculated using the Poisson Boltzmann Surface Area method and the evaluation of quaternary structure are also displayed.110 This work confirmed the lack of counterion effect in the interaction between GAGs, the identification of amino acids preferentially bringing the electrostatic neutrality of the interaction, and the lack of influence of the sulfate groups on the glycosidic torsion angles.
Gaining knowledge from accumulating high-quality results largely depends on the structural diversity of the investigated samples. An overview of the literature indicates that almost half of the studies involving GAGs considered heparin in isolation or in complex with proteins. It is primarily due to the vast usage of heparin in medical applications and the search for heparin mimics for therapeutic purposes. Its structural similarity to HS makes it a substitute or model for HS in biochemical and simulation studies. Heparin is used as a model of GAGs because it is available in good purity at an acceptable price. The number of protein–heparin experimental structures is far higher than the number available for other GAGs (https://gagdb.glycopedia.eu/). There is a long list of “orphan GAGs” for which the role, involvement, and mechanisms of action are only partially taken into account.
NMR Spectroscopy
NMR of Protein–GAGS Interactions
Nuclear magnetic resonance is widely used to study the conformation of GAGs alone or in complexes with proteins.111 In NMR, the behavior of two interacting molecules depends on the relative lifetimes of the species in the equilibrium and the strength of the magnetic field. Then, we can distinguish three different chemical exchange kinetics for protein–GAGs interactions: slow, medium, and fast in the NMR time scale. In the case of slow exchange interactions, the signals appear individually in two sets (one for the complex and the other for the free components). A single signal appears averaged in the chemical shift time scale between the complex and the free species for fast interactions. There is an intermediate exchange regime with a characteristic point, denominated coalescence, where the shape of the signals cannot be ascribed to a single or multiple signal behavior. The performance of a compound at a given temperature depends on the magnetic field strength used, as coalescence depends on how the chemical exchange kinetics compares with the separation between the signals in Hertz.
In most cases, for the GAG length typically used in solution NMR studies, the kinetics of the GAG–protein interactions fall primarily on the fast exchange regime. Then, to study them from the whole complex perspective, it is necessary to solve the 3D structure of the protein and stoichiometric complex using protein NMR techniques, complemented with a deep knowledge of the GAG NMR properties. Such investigations use standard protein NMR methods, commonly with double-labeled 13C and 15N proteins. Provided that the protein is fully assigned and its 3D structure determined, a 15N heteronuclear single quantum correlation (HSQC) titration provides the location of the ligand. This experiment uses the primary assignment from the free protein and translates it to the complex. Then the regions with larger chemical shift perturbations (CSP) can be located, indicating the protein regions with more significant interaction with the ligand. Typically, the results are displayed as heat maps of the CSP on the protein structure that are very informative about the regions involved in the interaction. When this method compares several complexes, special care must be taken to quantify the affinity. The 15N-HSQC titration method can be used together with a complete analysis of KD per residue. KD, CSP, and stoichiometry per residue can be obtained from this analysis. In this case, the precise location of the ligand in the complex can be easily found, along with the relative importance of a given residue in various complexes. Stoichiometry can also be deduced from this type of analysis. This approach has the benefit of observing two frequencies, 15N and 1H. Still, the disadvantage is that the protein assignment must be previously done and that the information comes only from backbone or side chain 15N atoms. Also, observing stoichiometric complexes avoids the potential interference of other stoichiometries with larger proportions of ligands.112,113
An additional analysis, based on using filtered (13C and/or 15N) experiments, facilitates the assignment and evaluation of the restrictions of the ligand signals and eventually locates the position of the GAG binding site. In this case, the set of experiments allows the extraction of ligand-to-ligand, ligand-to-receptor, receptor-to-ligand, and receptor-to-receptor homonuclear correlations to assign and characterize through bond connections and through space contacts via NOE nuclear Overhauser effect (NOE). From the NOEs, intermolecular distances can be extracted as a key structural magnitude. By using filtered experiments, it can also be analyzed if the binding has any influence on the 3D structure of the ligand.
Ligand-Observed NMR Techniques
Several challenges can complicate the protein NMR approach, for example, undesirable binding-induced protein aggregation or precipitation using long GAGs. Besides, commercial long GAGs or those resulting from enzymatic digestions show an intrinsic heterogeneity that poses difficulties in interpreting 1H,15N HSQC binding studies, e.g., what specific sulfation pattern and oligosaccharide length are involved in the interaction with the receptor. These issues have encouraged using shorter GAG oligosaccharides, which typically show weak affinity. Fortunately, a low binding affinity indicates that the molecular recognition process occurs with a fast exchange in the NMR relaxation time scale so that an excess of the GAG over the receptor facilitates an efficient “transfer of information” from the bound to the free state. This feature forms the basis of the “ligand-observed NMR techniques” to study protein–GAG interactions. With an excess of GAG, the analysis focuses on the free GAG signals, while the fast exchange efficiently transfers properties of the bound state (exchange broadening, large negative NOEs) onto the observable signals of the free GAG.
Among ligand-observed NMR techniques, transverse relaxation-based approaches (CPMG, 1H-T1ρ), and NOE-based approaches (water-ligand observed via gradient spectroscopy (waterLOGSY), exchange-transferred NOESY, and saturation transfer difference STD NMR) are of direct applicability to study small/medium-sized GAGs binding to protein receptors.114 From them, only STD NMR and exchange-transferred NOESY are appropriate to gain structural information in atomic detail about the binding mode and bioactive conformation of the ligands in the complexes.
Exchange-transferred NOESY experiments are important to characterize the bioactive conformations of GAGs containing iduronate rings. Not only the global GAG bound conformation can be determined, but the diagnostic H2–H5 NOE allows detection of the presence of the 2SO skew-boat conformation,115 a unique feature of the iduronate ring, as well as to determine changes in the population ratios of the 1C4/2SO/4C1 ring conformers between the free and bound states.116
STD NMR experiments identify the GAG oligosaccharide regions making close contact with the protein surface in the bound state (the so-called ligand binding epitope mapping). Except for long GAGs with regular repetitive sequences (e.g., heparin), this allows for determining the binding mode and what residues most likely constitute the minimum structural requirement for binding. It is a relevant information to understand the molecular recognition of GAGs by receptors and eventually design potential ligands to interfere in the process. Additionally, novel multifrequency STD NMR approaches provide information on the relative orientation of the ligand within the protein binding pocket. The so-called differential epitopes (DEEP-STD NMR) by comparison of STD NMR experiments in H2O and D2O allow identifying of contacts of the GAG oligosaccharide with arginine side chains commonly found in the binding pocket of GAG-binding proteins.117−120
Perspectives
Future progress in applying the novel multifrequency STD NMR approaches along with other ligand-observed NMR techniques, like exchange-transferred NOESY, will strongly benefit from the efficient implementation of STD NMR-derived restraints into docking calculations to generate 3D molecular models of GAG–protein complexes. These models can be validated by applying a complete conformational exchange matrix approach (CORCEMA-ST), which can still be time-consuming. In that regard, faster quantitative approaches dealing with the whole network of dipole–dipole coupled protons at the interface of the protein–ligand interaction are needed to be able to implement efficient binding epitope prediction and validation along long molecular dynamics simulations (MD), as well as future inclusion of STD NMR-derived restraints into MD simulations to generate a validated model of the GAG–protein complex including structure and dynamics of the molecular recognition process. Isotopically labeled GAGs, from chemoenzymatic synthesis from bacterial polysaccharide precursors (e.g., heparosan from E. coli K5) on the application of novel automated synthetic technologies are also highly desirable, as they will alleviate the difficulties related to the challenging narrow1H chemical shift dispersion shown by GAGs.
Computational Prediction of the 3D Structure of GAG–Protein Complexes
When applied to GAG–protein complexes, computational approaches aim to characterize the molecular nature of the interactions (e.g., the noncovalent bonds, the GAG chemical groups, and the amino acids involved), their mechanism of action, the role of basic domain motions, and the influence of possible mutations.121,122
Such knowledge is crucial to understanding their biological function, relevance in pathological alteration, and developing new therapeutics. Methods to accurately elucidate these questions are in high demand. Experimental methods described in the previous section have been developed to determine the structures of proteins and have subsequently been applied to study GAG and GAG–protein complex structures. However, due to the structural features of GAGs, such as their length, flexibility, and periodicity, and their tendency to assume a wide distribution of conformational states, these methods often failed to describe the structure of long GAGs alone or in complex with other biomolecules.123,124 At the same time, these methods could successfully characterize individual proteins complexed with GAG oligosaccharides of defined length up to DP8 according to the available experimental structures. Nevertheless, they cannot reproduce processes, such as the creation of protein gradients or GAG-mediated protein multimerization, involving natural GAGs up to 200 monosaccharide units. The description of the dynamics of these systems is progressing.
Molecular modeling and simulations are “computational microscopes” that aim to provide real-time visualization of biological phenomena by describing their dynamics. For GAGs, about 12% of all the investigations focused on GAG alone, 82% on GAG–protein complexes, and 6% on GAG–drug, GAG–lipid/membrane, or GAG–GAG interactions see review.123 Studies on GAGs alone mainly describe the dynamics of polysaccharide chains to investigate ring puckering and conformation, linear and bent conformations, and the role of sulfation on GAG monosaccharides. The past decade witnessed the implementation of multiple computational protocols based on molecular docking or advanced MD techniques aiming at modeling long GAGs chains alone or bound to proteins. However, the possible misinterpretations of the strength and stability of the complexes are not frequently discussed. Gaining accuracy requires more complex and time-consuming MD simulations to get additional insights into the binding stability, the role of nonionic and polar residues, and bridging water’s influence on GAG conformations.125,126 Binding energies can be calculated from MD simulations following either the linear interaction energy approximation or the Poisson–Boltzmann Surface(PBSA)/generalized Born, umbrella sampling, and Potential of Mean Force (PMF) methods and compared with experimental data. Free-energy-end point methods are not even close to chemical accuracy for tight binding of drugs, let alone for highly flexible multicontact-based interactions of low affinity. The poor documentation regarding the origin of the GAG samples investigated and their heterogeneity (molecular weight, sulfation) can primarily impair the comparison with experimental data
About 90% of the studies have been performed with short GAG oligosaccharides (< DP5). It represents a substantial limitation in translating the results of the computational prediction to biological processes at larger scales.127 Experimentally, this limitation is mainly due to the lack of available well-characterized longer oligosaccharides and full-length GAGs and the subsequent limitation of 3D structures of GAG–proteins complexes (currently more than 120 PDB entries, including less than 40 nonredundant nonenzymatic complexes).110 Computational methods to tackle long polysaccharide chains are deficient because running MD simulations for many sequences remains challenging, especially at a high-throughput level. As a result, biological phenomena induced by long GAG chains, such as protein homo-oligomerization, the formation of multimeric biomolecule complexes and cooperative effects, are underexplored.128
Despite all these difficulties, the accumulation of MD results, combined with docking studies, suggests that the patterns of recognition of GAGs by proteins follow a continuum. From a highly selective pattern to the unselective one, there exists a continuum of intermediate levels of selectivity, including moderately selective and plastic (Figure 8).88
Figure 8.
GAG-binding to proteins: From “high selectivity” to “non-selectivity”,88 a continuous spectrum of interactions intermediate cases exists. (a) The panel displays a system in which unique interactions between sulfate groups of heparin and basic residues of antithrombin result from a highly selective recognition. (b) The GAG–protein mode of interactions exhibits a “moderate selectivity” where several binding modes occur throughout the “chain polarity” in terms of the directionality of binding. (c) Illustrates a “plastic” type of interaction, where the GAG chain slides along the protein binding site while maintaining the directionality of the chain. (d) The structure on the right panel shows no preference for the protein’s GAG sequence, as the several binding poses are fully nonselective.
The quest for bioactivity and drugability still drives most of the research to which computational modeling might contribute. Novel and foreseeable developments concern the screening of combinatorial virtual libraries hoping to uncover bioactive GAG sequences before implementing molecular dynamics simulations. Identifying highly selective systems (both from the protein and GAG side) is still pending.129 The fundamental biological role of GAGs in regulating such responses as chemotaxis, cell signaling, nuclear translocation, and viral invasion throughout complex yet poorly characterized plasticity remains lagging behind the quest for druggability.
Perspectives
Crucial questions regarding the interaction with highly glycosylated protein receptors,(130−134)ions,(135)and solvent(122)need appropriate and thorough MD treatments. Even if their progressive uses have not yet significantly affected the percentage of the overall literature concerning GAGs, they are paving the way to unveil GAG-induced mechanistic and allosteric effects on proteins.
3.3. GAG Interactomes
As key biomolecular players, GAGs organize the pericellular and extracellular matrix, contributing to extracellular matrix architecture, cell–matrix interactions, and subsequent cell signaling. Advances in GAG sequencing allow the characterization of GAG sequences binding to proteins, and GAG-mediated pull-down proteomics can identify GAG-binding proteins on a proteome-wide scale in various biological and clinical samples. The availability of these data led to the building of GAGs interaction networks that help decipher the molecular and cellular mechanisms underlying GAGs functions mediated by their binding to proteins. All GAGs, except for hyaluronan, are covalently attached to core proteins to form proteoglycans, and GAG interactions occur in vivo in the molecular context of proteoglycans. Consequently, GAG interactomes should be contextualized by integrating proteoglycan interactions, transcriptomics, and/or proteomics data to generate cell-, tissue-, or disease-specific interactomes. Additional GAG interactomes can be built based on a molecular function, a biological pathway, or a cellular location. It should aid in deciphering the mechanisms associated with GAG interactions in various physio-pathological contexts.
However, the integration of these data is focused on GAG-binding proteins. The development of high-throughput assays measuring the number of individual GAGs in biological samples would allow the collection of GAGomic data sets, which could then be integrated into GAG networks to refine their specificity.136 The setup of high-trough-put binding assays based on surface plasmon resonance imaging using commercially available instruments will be helpful in calculating the kinetic and affinity parameters of an increasing number of GAG interactions. The integration of these parameters in GAG networks would be crucial to determine if a specific range of kinetic and affinity values are associated with specific locations, amount of disorder, and molecular functions of GAG-binding proteins or with the biological processes they are involved in as previously shown for a subset of heparin-binding proteins.137
Large-scale, high-throughput binding assays, including GAG microarrays and affinity proteomics, have identified hundreds of GAG-binding proteins. They have been used to generate GAG interactomes.138−140 At the time of writing, the most comprehensive GAG interactome available includes 4290 interactions and 3464 unique GAG-binding proteins.141 (Figure 9). These large data sets provide the opportunity to investigate the location, molecular function, biological process and biological pathways associated with the protein partners of a specific GAG. The integration in these networks of the chemical features (e.g., sulfation) and size of GAGs and of the amino acid sequences and domains of GAG-binding proteins would be very valuable to explore the relationships, if any, between these features and the biological or structural roles of GAG-binding proteins.
Figure 9.
Interaction network of the six natural glycosaminoglycans. Glycosaminoglycan partners are represented as symbols, and interactions as edges: CS, chondroitin sulfate (green); DS, dermatan sulfate (pink); HA, hyaluronan (dark yellow); HP/HS, heparin/heparan sulfate (blue); KS, keratan sulfate (light red). Partners shared by two or more GAGs are in light gray. Reproduced with permission from ref (141). Copyright 2022, American Physiology Society.
GAG interaction data are freely available in public databases such as MatrixDB (http://matrixdb.univ-lyon1.fr/) and GAGDB (https://gagdb.glycopedia.eu/), which have recently been reviewed.109 The development of standards in data sharing (e.g., the FAIR principle) and storage repositories and databases have markedly changed access to large data sets and their retrieval and (re)use. “Big data” have become a precious resource, and its exploration will benefit from recent advances in artificial Intelligence (AI). This term covers numerous approaches that constitute the algorithmic framework needed to interpret and make sense of big data, whatever they are. The AI revolution has impacted structural biology and life sciences by allowing the development of the AlphaFold algorithm used to predict the 3D structure of proteins from their amino acid sequences. It is still not a substitute for experimental structural biology and will probably never be, yet it will undoubtedly expand the knowledge in structural biology and speed up the experimental determination of 3D structures. It can also be applied to glycoproteins by grafting glycosylations from a library of glycan blocks onto AlphaFold protein models.142
Perspectives
The ability of Deep Learning algorithms to extract valuable and meaningful information from big data is likely to provide the long-awaited step change in glycoscience discovery, i.e., bring glycomics on par with proteomics and genomics. The challenges still slowing down glycoscience discovery are directly linked to the lack of routine, easy-to-perform sequencing of bioactive GAGs and GAG oligosaccharides, which might be addressed by nanopore sequencing and infrared spectroscopy in the future, and by the difficulty of studying the 3D structure of these flexible and dynamic molecules, and capturing their conformational states required for or stabilized by their binding to proteins. The structural characterization of GAGs is a huge task due to their high structural diversity (e.g., 48 theoretical disaccharides for HS), further complicated by their template-independent biosynthesis. Thinking outside the box, where the “box” contains glycomic/glycoanalytic data sets and the “thinking” is AI algorithms, is undoubtedly a promising way to solve the above issues, to mine glycomic data sets, and promote significant achievements in glycoscience. The Glycompare tool recently developed to analyze glycomics data sets using artificial intelligence(143)is an excellent example of this approach.
3.4. Self-Organization of GAGS and GAG-Binding Proteins
Extracellular GAG organization plays critical roles in GAG function, influencing cell-matrix, cell–cell recognition, cell adhesion, and migration. GAGs, however, do not self-associate on their own under physiological conditions. Instead, the self-organization of GAGs is intimately related to the proteins they bind and the core proteins to which they are attached. Methods to analyze how proteins cross-link GAGs and how such cross-linking defines the organization and physical properties (i.e., mechanics and permeability) of GAG-rich matrices’ organization are underdeveloped.
3.4.1. GAG Cross-Linking
The functional importance of GAG cross-linking is firmly established for a range of tissues. An early example was the extended HA/aggrecan complexes (cross-linked by link proteins) that confer load-bearing properties to cartilage.144 Perineuronal nets, GAG-rich coats on neurons with a reticular morphology that modulate synapse formation and neuronal plasticity, are a molecular network made from HA, CS proteoglycan, and their cross-linking proteins.101 Another example is the expanded and ultrasoft yet elastic matrix that surrounds mammalian oocytes during ovulation. It regulates transport and fertilization in the oviduct, which requires HA, a set of HA-cross-linking proteins (TSG-6, pentraxin 3, and inter-α-inhibitor), and also contains proteoglycans such as versican.145 Despite their functional importance, little is currently known about the structure and dynamics of the cross-linking nodes in all these matrices. We also do not know how the degree of cross-linking is regulated to achieve matrices with the desired morphology and biophysical properties (e.g., elasticity and permeability).
Intercellular signaling molecules (e.g., morphogens/growth factors for tissue development/repair and chemokines for immune cell trafficking) rely on GAGs for their precise distribution across the extracellular space. In this context, control over the presentation of intercellular signaling proteins to the cognate cell surface receptors (and the downstream intracellular signaling process) has long been thought to be the primary function of GAGs. Emerging evidence, however, suggests that many signaling proteins are also capable of cross-linking GAGs.146,147 These proteins, thus, may also exert their functions independently from cognate receptors by dynamically reorganizing GAG-rich extracellular matrix and modulating matrix morphology and biophysical properties. Such a promising new area of investigation will require new tools and ways of thinking.
3.4.2. Response of GAG–Protein Bonds to Mechanical Strain
Pericellular coats and extracellular matrices in which GAGs reside are exposed to varying mechanical strains, and such strains will impact the flexible GAGs and their interactions. This dimension of GAG–protein interactions has historically received very little attention but can now be explored. Single-molecule force spectroscopy (SMFS) methods are sensitive to probing the mechanics of individual GAG–protein bonds.148 They have already revealed specialized interactions such as “catch” bonds (i.e., bonds that strengthen under mechanical force, whereas most bonds weaken under force) between HS and the cell surface sulfatase Sulf1.149 However, these methods are not widespread and must be expanded to more protein/GAG interactions. For example, catch bonds have been proposed to occur between HA and its cell-surface receptor CD44.150 Such an interaction is required for the migration of activated leukocytes from the blood vasculature into the interstitial tissue. However, current data are inconclusive as to whether HA and CD44 are indeed forming catch bonds.148
3.4.3. Multivalent Interactions and Super Selective Binding
GAG polysaccharides can engage with several binding partners simultaneously. Such multivalent binding impacts cells’ recognition of GAGs and their extracellular complexes. A case in point is HA, which exploits multivalent binding to discriminate its receptors (e.g., CD44, LYVE-1) sharply based on their comparative surface densities in contrast to mere affinity.151,152 Such “super selective” binding may explain the long-standing controversy of how high and low molecular mass HA exert radically different biological effects (e.g., anti- vs pro-inflammatory) and, more generally, how multivalent GAG interactions modulate the communication of cells with their environment.
3.4.4. Topology-Dependent Recognition
There is a high level of complexity in how GAGs interact with proteins. The linear GAGs allow for distinct topology-dependent binding behaviors, such as one-dimensional sliding, or selective binding to chain ends (i.e., the nonreducing or the reducing end), or sulfation motifs. For example, some GAG degrading enzymes and other proteins153 selectively operate at the nonreducing end of GAG chains, chemokines may “slide along” GAG chains, and GAG chains may organize multiprotein functional complexes. Revealing these effects requires bespoke biophysical tools, which are currently underdeveloped.
Perspectives
Our understanding of the molecular and physical mechanisms underpinning GAG functions under the influence of proteins is still in its infancy, not the least because we lack the biophysical tools to probe the complexity of molecular interactions involving GAGs. Biochemical tools have established a plethora of protein binding partners for GAGs and basic interaction parameters (e.g., affinity, the composition of interaction complexes). However, they fall short of mechanistically explaining the complexity of GAG–protein interactions. More and different biophysical methods are needed to resolve the structure and dynamics (including under mechanical force) of GAG cross-linking nodes and to probe protein binding modes unique to polymer chains, such as sliding interactions and selective chain-end recognition. In addition to analytical tools, we also need new reagents and methods to reconstitute multipartner GAG–protein interactions and protein-mediated GAG self-organization in vitro. Such molecularly defined environments will enable new studies of structure/property/function interrelationships(154)that are impossible with the more complex, less defined, and less tunable matrices produced by cells and tissues alone.
4.0. Future Directions: “Be FAIR to GAGs.”
The previous sections highlighted the accomplishments and progress achieved using the full spectrum of current analytical, chemical, physicochemical, structural, biophysical, and biochemical methods devoted to GAGs. The wealth and the amount of research data produced represent a gold mine for the discipline. Nevertheless, further progress could benefit from methods that help understand the extent and complexity of the accumulated data set. Future research directions aiming at serving the whole community are presented along several lines, which are analyzed in light of the most recent development.
Update on Standards
A first condition would require that the experimental and computational data are comprehensively characterized, made publicly available, and organized. In proteomics and genomics, bioinformatics has generated powerful methods and software tools to apprehend the extent and complexity of data sets. It is not yet the case for the field of glycomics, and this particularly challenges the inclusion of GAGs in physiological and pathological contexts. Most omics applications rely on robust analytical technology and strong bioinformatics support to extract as much information as possible from large data sets generated by automated analytical methods. The sparsity and heterogeneity of GAG data and the variety of techniques used for their study still limit the automation of analytical procedures.
The lack of common glycomics standards has hindered progress in representing information stored in databases and hampered data exchange and interoperability of such databases. Several cooperative initiatives for representing and collecting glycomic data have been launched in response to this situation. An international glycan structure repository named GlyTouCan was released to become a community resource.7 As a first step in adopting a standard view on glycans, it relies on the depiction of 2D structures in the SNFG notation increasingly shared within and outside glycoscience and applicable to representing GAGs. Furthermore, semantic Web technology, commonly used in bioinformatics via Resource Description Framework (RDF) data modeling, is implemented as a new standard for representing and storing knowledge in various databases.155 This trend was introduced with the GlycoRDF model of glycans156 that was subsequently implemented in GlyTouCan but should be refined to become readily usable with GAGs. Recently, GlySTreeM, another RDF-based model designed to tolerate the frequent ambiguity of glycan structures (e.g., missing linkage information and loosely defined monosaccharides), was proposed.157 It could be helpful to capture uncertain sulfation patterns in GAGs.
On top of the expected bioinformatic development, GAG data management and exchange require consolidation and compliance to standards, as initiated through the Minimum Information Data Required for Glycomics (MIRAGE) initiative of the Beilstein Institute (https://www.beilstein-institut.de/en/projects/mirage/). At this point, only a few glycoinformatics resources are collecting raw data for further availability to the community. While channelling the structural data (mass spectrometry (MS), NMR, etc.) through a pipeline is essential, only partial consideration has been given and mainly focused on N- and O-linked glycans.158 GlycoPOST is the first implementation of a working MS data repository159 but remains rather confidential, especially in the GAG community. Nonetheless, a pipeline to translate GAGs sequences into a conventional computer-readable format and generate corresponding 3D models was first attempted to move the field forward.2 Despite the long way to go, the cited initiatives are not attempted in isolation. However, they can be integrated within the global alliance named GlySpace,160 formed to step up the definition and adoption of good glycoinformatics practices and to coordinate research data processing.
FAIR and TRUST Principles
In the context of open science, four guiding principles, Findability, Accessibility, Interoperability and Reusability (FAIR),161 underline the practice of systematic data sharing to chart new horizons. Findability (F) is indispensable because data search is a frequent task that should be easy for the largest community of life scientists. It primarily requires that data and related metadata (information supplementing data) are associated with a unique and persistent identifier and, secondarily, readability by humans and computers. Accessibility (A) involves retrieval using these identifiers with a standardized protocol such as hypertext transfer protocol (HTTP). Interoperability (I) is a crucial constraint in attempts to merge or integrate data from different sources. Data must be described with standard languages reflecting knowledge representations, commonly known as ontologies, otherwise also qualified as controlled vocabularies. Reusability (R) can be achieved through well-described metadata, data provenance, and community standards. The precise recording and depiction of the heterogeneous experimental and theoretical information is a definite challenge. Besides, it should be reckoned that in contemporaneous purpose-focused research, only a limited fraction (approximately 10%) of the data leading to the results are reported in the published studies. While many data are not fully characterized, the lack of information on the metadata (explaining and characterizing the measured or computed data), the ontologies (the relationships in metadata), and the workflow of different research groups are difficult to adjust. As a result, most research data are neither findable nor interoperable.
Nonetheless, to boost expected changes and ease programmatic access, all members of the GlySpace Alliance have developed Application Programming Interfaces (APIs) in their respective resources, enabling data exposure and sharing for any user in compliance with FAIR principles. When data and metadata are fully and unambiguously characterized (and results reproducible), they can be combined with different studies, utilized in different contexts, and used for deeper analysis and data mining. FAIR was recently reinforced by TRUST (Transparency, Responsibility, User focus, Sustainability, Technology) principles that mainly account for the previously missing time component and emphasize the difficulty of maintenance in the longer term.162 If FAIR has gained recognition in the past decade and shaped many bioinformatics initiatives, TRUST is just beginning to unveil more delicate issues of maintaining resources while coping with the constant evolution of technology and respecting user needs.
Cross-Referencing
Within the disciplines covered in the present article, not all comply with one or several FAIR principles. A large proportion of research and the vast majority of data is not utilized in chemistry. The existing digital ecosystem surrounding scholarly data publication prevents the maximum extraction that would benefit from research investments. On the whole, the chemistry community, at large, does not have an inherent FAIR culture. It should go beyond conventional Open Access to journal articles and provide access to chemistry-specific data, such as molecules and properties, in a form that can be validated and reused. Organic synthesis is often based on personal experience and tricks, which are not readily shared with others.
Nevertheless, many important data sets emerge from traditional, low-throughput bench science. These data sets are no less critical concerning integrative research, reproducibility, and reuse. Linking those experimental, theoretical, and biological data using common schemes and ontology will generate a new level of the science of GAGs.
Data Modeling
The use of computational methods receives ever-growing attention due to hardware, algorithmic data, and software evolution. Such development allows for capturing three-dimensional structures over several temporal and spatial orders of magnitude. Implementing multiscale data faces many challenges due to the heterogeneity of simulation setups, force fields, etc. as well as the heterogeneity of the meaning of the produced data. Related to this heterogeneity is accuracy, as error bars are usually missing, which impacts the interoperability of the data with experimental results. Concerning the data volume in molecular dynamics calculations, it is hardly possible to store all the information, i.e., the detailed time evolution of the positions of all the atoms, as they are several thousand. Selection and compression strategies must be developed to deal with the type and amount of data.
Big Data and AI Approach
Ultimately, standardized, structured, and well-annotated data can accumulate and provide opportunities to train models and improve the prediction of GAG behavior in definite contexts or environments. Recent impressive progress in using Deep Learning methods such as AlfaFold for predicting protein 3D structure163 demonstrates the worth of collecting well-characterized data shaped over a long period. Closer to GAGomics, Deep Learning approaches have also mushroomed lately for predicting glycan protein interactions (LectinOracle, GlyNet, ref of D Mattox) thanks to the availability of large amounts of glycan array data. This trend will likely expand in the coming years, emphasizing all points made earlier in this section, leading to deciphering the many unknowns embedded in GAGs.
Acknowledgments
This article is based upon work from COST Action INNOGLY CA 18103, supported by COST (European Cooperation in Science and Technology).
Author Contributions
S.P., D.N., S.R.-B. created the general concept and led the planning of the separate written contributions provided by all the authors. S.P. organized the article’s structure and wrote the article from the submitted contributions. O.M. processed the manuscript and managed all bibliographic references. All authors, whose names appear in alphabetic order, carefully read and edited the whole article. CRediT: Serge Perez conceptualization, writing-original draft, writing-review & editing; Olga Mashkakova writin-original draft, software, visualization and editing; Jesus Angulo writing-original draft; Emiliano Bedini writing-original draft; Antonella Bisio writing-original draft; José L. de Paz writing-original draft; Elisa Fadda writing-original draft, writing-review & editing; Marco Guerrini writing-original draft; Michal Hricovini writing-original draft; Milos Hricovini writing-original draft; Frederique Lisacek writing-original draft; Pedro M Nieto writing-original draft; Kevin Pagel visualization, writing-original draft; Giulia Paiardi writing-original draft; Ralf P. Richter writing-original draft, writing-review & editing; Sergey A. Samsonov writing-original draft; Romain R Vives writing-original draft; Dragana Nikitovic conceptualization, supervision; Sylvie Ricard-Blum conceptualization, writing-original draft, writing-review & editing.
The authors declare no competing financial interest.
This paper was published ASAP on March 2, 2023, with an error in an author’s name. The corrected version reposted March 13, 2023.
References
- Varki A.; Cummings R. D.; Aebi M.; Packer N. H.; Seeberger P. H.; Esko J. D.; Stanley P.; Hart G.; Darvill A.; Kinoshita T.; Prestegard J. J.; Schnaar R. L.; Freeze H. H.; Marth J. D.; Bertozzi C. R.; Etzler M. E.; Frank M.; Vliegenthart J. F.; Lutteke T.; Perez S.; Bolton E.; Rudd P.; Paulson J.; Kanehisa M.; Toukach P.; Aoki-Kinoshita K. F.; Dell A.; Narimatsu H.; York W.; Taniguchi N.; Kornfeld S. Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology 2015, 25 (12), 1323–4. 10.1093/glycob/cwv091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc O.; Mariethoz J.; Rivet A.; Lisacek F.; Perez S.; Ricard-Blum S. A pipeline to translate glycosaminoglycan sequences into 3D models. Application to the exploration of glycosaminoglycan conformational space. Glycobiology 2019, 29 (1), 36–44. 10.1093/glycob/cwy084. [DOI] [PubMed] [Google Scholar]
- Grabarics M.; Lettow M.; Kirschbaum C.; Greis K.; Manz C.; Pagel K. Mass Spectrometry-Based Techniques to Elucidate the Sugar Code. Chem. Rev. 2022, 122 (8), 7840–7908. 10.1021/acs.chemrev.1c00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herget S.; Ranzinger R.; Maass K.; Lieth C. W. GlycoCT-a unifying sequence format for carbohydrates. Carbohydr. Res. 2008, 343 (12), 2162–71. 10.1016/j.carres.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Matsubara M.; Aoki-Kinoshita K. F.; Aoki N. P.; Yamada I.; Narimatsu H. WURCS 2.0 Update To Encapsulate Ambiguous Carbohydrate Structures. J. Chem. Inf Model 2017, 57 (4), 632–637. 10.1021/acs.jcim.6b00650. [DOI] [PubMed] [Google Scholar]
- Bohne-Lang A.; Lang E.; Forster T.; von der Lieth C. W. LINUCS: linear notation for unique description of carbohydrate sequences. Carbohydr. Res. 2001, 336 (1), 1–11. 10.1016/S0008-6215(01)00230-0. [DOI] [PubMed] [Google Scholar]
- Tiemeyer M.; Aoki K.; Paulson J.; Cummings R. D.; York W. S.; Karlsson N. G.; Lisacek F.; Packer N. H.; Campbell M. P.; Aoki N. P.; Fujita A.; Matsubara M.; Shinmachi D.; Tsuchiya S.; Yamada I.; Pierce M.; Ranzinger R.; Narimatsu H.; Aoki-Kinoshita K. F. GlyTouCan: an accessible glycan structure repository. Glycobiology 2017, 27 (10), 915–919. 10.1093/glycob/cwx066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez S.; Sarkar A.; Rivet A.; Breton C.; Imberty A. Glyco3D: a portal for structural glycosciences. Methods Mol. Biol. 2015, 1273, 241–58. 10.1007/978-1-4939-2343-4_18. [DOI] [PubMed] [Google Scholar]
- Mariethoz J.; Khatib K.; Alocci D.; Campbell M. P.; Karlsson N. G.; Packer N. H.; Mullen E. H.; Lisacek F. SugarBindDB, a resource of glycan-mediated host-pathogen interactions. Nucleic Acids Res. 2016, 44 (D1), D1243–50. 10.1093/nar/gkv1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damerell D.; Ceroni A.; Maass K.; Ranzinger R.; Dell A.; Haslam S. M. The GlycanBuilder and GlycoWorkbench glycoinformatics tools: updates and new developments. Biol. Chem. 2012, 393 (11), 1357–62. 10.1515/hsz-2012-0135. [DOI] [PubMed] [Google Scholar]
- Boyce A.; Walsh G. Production, characteristics and applications of microbial heparinases. Biochimie 2022, 198, 109–140. 10.1016/j.biochi.2022.03.011. [DOI] [PubMed] [Google Scholar]
- Murphy K. J.; Merry C. L.; Lyon M.; Thompson J. E.; Roberts I. S.; Gallagher J. T. A new model for the domain structure of heparan sulfate based on the novel specificity of K5 lyase. J. Biol. Chem. 2004, 279 (26), 27239–45. 10.1074/jbc.M401774200. [DOI] [PubMed] [Google Scholar]
- Wang W.; Wang J.; Li F. Hyaluronidase and Chondroitinase. Adv. Exp. Med. Biol. 2016, 925, 75–87. 10.1007/5584_2016_54. [DOI] [PubMed] [Google Scholar]
- Zhang Y. S.; Gong J. S.; Yao Z. Y.; Jiang J. Y.; Su C.; Li H.; Kang C. L.; Liu L.; Xu Z. H.; Shi J. S. Insights into the source, mechanism and biotechnological applications of hyaluronidases. Biotechnol Adv. 2022, 60, 108018. 10.1016/j.biotechadv.2022.108018. [DOI] [PubMed] [Google Scholar]
- Ndeh D.; Basle A.; Strahl H.; Yates E. A.; McClurgg U. L.; Henrissat B.; Terrapon N.; Cartmell A. Metabolism of multiple glycosaminoglycans by Bacteroides thetaiotaomicron is orchestrated by a versatile core genetic locus. Nat. Commun. 2020, 11 (1), 646. 10.1038/s41467-020-18097-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasteson A. A method for the determination of the molecular weight and molecular-weight distribution of chondroitin sulphate. J. Chromatogr. 1971, 59 (1), 87–97. 10.1016/S0021-9673(01)80009-1. [DOI] [PubMed] [Google Scholar]
- Fasciano J. M.; Danielson N. D. Ion chromatography for the separation of heparin and structurally related glycosaminoglycans: A review. J. Sep Sci. 2016, 39 (6), 1118–29. 10.1002/jssc.201500664. [DOI] [PubMed] [Google Scholar]
- Laguri C.; Sadir R.; Gout E.; Vives R. R.; Lortat-Jacob H. Preparation and Characterization of Heparan Sulfate-Derived Oligosaccharides to Investigate Protein-GAG Interaction and HS Biosynthesis Enzyme Activity. Methods Mol. Biol. 2022, 2303, 121–137. 10.1007/978-1-0716-1398-6_11. [DOI] [PubMed] [Google Scholar]
- Vivès R. R.; Goodger S.; Pye D. A. Combined strong anion-exchange HPLC and PAGE approach for the purification of heparan sulphate oligosaccharides. Biochem. J. 2001, 354 (Pt 1), 141–7. 10.1042/bj3540141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallero G. J.; Zaia J. Resolving Heparan Sulfate Oligosaccharide Positional Isomers Using Hydrophilic Interaction Liquid Chromatography-Cyclic Ion Mobility Mass Spectrometry. Anal. Chem. 2022, 94 (5), 2366–2374. 10.1021/acs.analchem.1c03543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kuppevelt T. H.; Oosterhof A.; Versteeg E. M. M.; Podhumljak E.; van de Westerlo E. M. A.; Daamen W. F. Sequencing of glycosaminoglycans with potential to interrogate sequence-specific interactions. Sci. Rep 2017, 7 (1), 14785. 10.1038/s41598-017-15009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mende M.; Bednarek C.; Wawryszyn M.; Sauter P.; Biskup M. B.; Schepers U.; Brase S. Chemical Synthesis of Glycosaminoglycans. Chem. Rev. 2016, 116 (14), 8193–255. 10.1021/acs.chemrev.6b00010. [DOI] [PubMed] [Google Scholar]
- Pongener I.; O’Shea C.; Wootton H.; Watkinson M.; Miller G. J. Developments in the Chemical Synthesis of Heparin and Heparan Sulfate. Chem. Rec 2021, 21 (11), 3238–3255. 10.1002/tcr.202100173. [DOI] [PubMed] [Google Scholar]
- Zong C.; Venot A.; Li X.; Lu W.; Xiao W.; Wilkes J. L.; Salanga C. L.; Handel T. M.; Wang L.; Wolfert M. A.; Boons G. J. Heparan Sulfate Microarray Reveals That Heparan Sulfate-Protein Binding Exhibits Different Ligand Requirements. J. Am. Chem. Soc. 2017, 139 (28), 9534–9543. 10.1021/jacs.7b01399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph A. A.; Pardo-Vargas A.; Seeberger P. H. Total Synthesis of Polysaccharides by Automated Glycan Assembly. J. Am. Chem. Soc. 2020, 142 (19), 8561–8564. 10.1021/jacs.0c00751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eller S.; Collot M.; Yin J.; Hahm H. S.; Seeberger P. H. Automated solid-phase synthesis of chondroitin sulfate glycosaminoglycans. Angew. Chem., Int. Ed. Engl. 2013, 52 (22), 5858–61. 10.1002/anie.201210132. [DOI] [PubMed] [Google Scholar]
- Ramadan S.; Su G.; Baryal K.; Hsieh-Wilson L. C.; Liu J.; Huang X. Automated solid phase assisted synthesis of a heparan sulfate disaccharide library. Organic Chemistry Frontiers 2022, 9 (11), 2910–2920. 10.1039/D2QO00439A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrikos-Ergas T.; Sletten E. T.; Huang J. Y.; Seeberger P. H.; Delbianco M. On resin synthesis of sulfated oligosaccharides. Chem. Sci. 2022, 13 (7), 2115–2120. 10.1039/D1SC06063E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuck O. T.; Sletten E. T.; Danglad-Flores J.; Seeberger P. H. Towards a Systematic Understanding of the Influence of Temperature on Glycosylation Reactions. Angew. Chem., Int. Ed. Engl. 2022, 61 (15), e202115433 10.1002/anie.202115433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar N. J.; Wang L.; Higo T.; Bhattacharya C.; Kancharla P. K.; Zhang F.; Baryal K.; Huo C. X.; Liu J.; Linhardt R. J.; Huang X.; Hsieh-Wilson L. C. Expedient Synthesis of Core Disaccharide Building Blocks from Natural Polysaccharides for Heparan Sulfate Oligosaccharide Assembly. Angew. Chem., Int. Ed. Engl. 2019, 58 (51), 18577–18583. 10.1002/anie.201908805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao W.; Xiong D.-C.; Yang Y.; Geng C.; Cong Z.; Li F.; Li B.-H.; Qin X.; Wang L.-N.; Xue W.-Y.; Yu N.; Zhang H.; Wu X.; Liu M.; Ye X.-S. Automated solution-phase multiplicative synthesis of complex glycans up to a 1,080-mer. Nature Synthesis 2022, 1, 854. 10.1038/s44160-022-00171-9. [DOI] [Google Scholar]
- Zhang X.; Liu H.; Lin L.; Yao W.; Zhao J.; Wu M.; Li Z. Synthesis of Fucosylated Chondroitin Sulfate Nonasaccharide as a Novel Anticoagulant Targeting Intrinsic Factor Xase Complex. Angew. Chem., Int. Ed. Engl. 2018, 57 (39), 12880–12885. 10.1002/anie.201807546. [DOI] [PubMed] [Google Scholar]
- Dey S.; Wong C. H. Programmable one-pot synthesis of heparin pentasaccharides enabling access to regiodefined sulfate derivatives. Chem. Sci. 2018, 9 (32), 6685–6691. 10.1039/C8SC01743C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Paz J. L.; Nieto P. M. Fluorous-Tag-Assisted Synthesis of GAG-Like Oligosaccharides. Methods Mol. Biol. 2022, 2303, 37–47. 10.1007/978-1-0716-1398-6_4. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Lin L.; Huang H.; Linhardt R. J. Chemoenzymatic Synthesis of Glycosaminoglycans. Acc. Chem. Res. 2020, 53 (2), 335–346. 10.1021/acs.accounts.9b00420. [DOI] [PubMed] [Google Scholar]
- Gottschalk J.; Elling L. Current state on the enzymatic synthesis of glycosaminoglycans. Curr. Opin Chem. Biol. 2021, 61, 71–80. 10.1016/j.cbpa.2020.09.008. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Arnold K.; Dhurandhare V. M.; Xu Y.; Liu J. Investigation of the biological functions of heparan sulfate using a chemoenzymatic synthetic approach. RSC Chem. Biol. 2021, 2 (3), 702–712. 10.1039/D0CB00199F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini D.; Restaino O. F.; Schiraldi C. Microbial production and metabolic engineering of chondroitin and chondroitin sulfate. Emerg Top Life Sci. 2018, 2 (3), 349–361. 10.1042/ETLS20180006. [DOI] [PubMed] [Google Scholar]
- Jing W.; DeAngelis P. L. Synchronized chemoenzymatic synthesis of monodisperse hyaluronan polymers. J. Biol. Chem. 2004, 279 (40), 42345–42349. 10.1074/jbc.M402744200. [DOI] [PubMed] [Google Scholar]
- Jin X.; Zhang W.; Wang Y.; Sheng J.; Xu R.; Li J.; Du G.; Kang Z. Biosynthesis of non-animal chondroitin sulfate from methanol using genetically engineered Pichia pastoris. Green Chem. 2021, 23 (12), 4365–4374. 10.1039/D1GC00260K. [DOI] [Google Scholar]
- Chen Y. H.; Narimatsu Y.; Clausen T. M.; Gomes C.; Karlsson R.; Steentoft C.; Spliid C. B.; Gustavsson T.; Salanti A.; Persson A.; Malmstrom A.; Willen D.; Ellervik U.; Bennett E. P.; Mao Y.; Clausen H.; Yang Z. The GAGOme: a cell-based library of displayed glycosaminoglycans. Nat. Methods 2018, 15 (11), 881–888. 10.1038/s41592-018-0086-z. [DOI] [PubMed] [Google Scholar]
- Hintze V.; Schnabelrauch M.; Rother S. Chemical Modification of Hyaluronan and Their Biomedical Applications. Front Chem. 2022, 10, 830671. 10.3389/fchem.2022.830671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedini E.; Laezza A.; Iadonisi A. Chemical Derivatization of Sulfated Glycosaminoglycans. Eur. J. Org. Chem. 2016, 2016 (18), 3018–3042. 10.1002/ejoc.201600108. [DOI] [Google Scholar]
- Sodhi H.; Panitch A. Glycosaminoglycans in Tissue Engineering: A Review. Biomolecules 2021, 11 (1), 29. 10.3390/biom11010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessella G.; Traboni S.; Cimini D.; Iadonisi A.; Schiraldi C.; Bedini E. Development of Semisynthetic, Regioselective Pathways for Accessing the Missing Sulfation Patterns of Chondroitin Sulfate. Biomacromolecules 2019, 20 (8), 3021–3030. 10.1021/acs.biomac.9b00590. [DOI] [PubMed] [Google Scholar]
- Thakar D.; Migliorini E.; Coche-Guerente L.; Sadir R.; Lortat-Jacob H.; Boturyn D.; Renaudet O.; Labbe P.; Richter R. P. A quartz crystal microbalance method to study the terminal functionalization of glycosaminoglycans. Chem. Commun. (Camb) 2014, 50 (96), 15148–51. 10.1039/C4CC06905F. [DOI] [PubMed] [Google Scholar]
- Przybylski C.; Bonnet V.; Vives R. R. A microscale double labelling of GAG oligosaccharides compatible with enzymatic treatment and mass spectrometry. Chem. Commun. (Camb) 2019, 55 (29), 4182–4185. 10.1039/C9CC00254E. [DOI] [PubMed] [Google Scholar]
- Sterner E.; Masuko S.; Li G.; Li L.; Green D. E.; Otto N. J.; Xu Y.; DeAngelis P. L.; Liu J.; Dordick J. S.; Linhardt R. J. Fibroblast growth factor-based signaling through synthetic heparan sulfate blocks copolymers studied using high cell density three-dimensional cell printing. J. Biol. Chem. 2014, 289 (14), 9754–65. 10.1074/jbc.M113.546937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimond S. E.; Mycroft-West C. J.; Gandhi N. S.; Tree J. A.; Le T. T.; Spalluto C. M.; Humbert M. V.; Buttigieg K. R.; Coombes N.; Elmore M. J.; Wand M.; Nystrom K.; Said J.; Setoh Y. X.; Amarilla A. A.; Modhiran N.; Sng J. D. J.; Chhabra M.; Young P. R.; Rawle D. J.; Lima M. A.; Yates E. A.; Karlsson R.; Miller R. L.; Chen Y. H.; Bagdonaite I.; Yang Z.; Stewart J.; Nguyen D.; Laidlaw S.; Hammond E.; Dredge K.; Wilkinson T. M. A.; Watterson D.; Khromykh A. A.; Suhrbier A.; Carroll M. W.; Trybala E.; Bergstrom T.; Ferro V.; Skidmore M. A.; Turnbull J. E. Synthetic Heparan Sulfate Mimetic Pixatimod (PG545) Potently Inhibits SARS-CoV-2 by Disrupting the Spike-ACE2 Interaction. ACS Cent Sci. 2022, 8 (5), 527–545. 10.1021/acscentsci.1c01293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanthamurthy C. D.; Gimeno A.; Leviatan Ben-Arye S.; Kumar N. V.; Jain P.; Padler-Karavani V.; Jimenez-Barbero J.; Kikkeri R. Sulfation Code and Conformational Plasticity of l-Iduronic Acid Homo-Oligosaccharides Mimic the Biological Functions of Heparan Sulfate. ACS Chem. Biol. 2021, 16 (11), 2481–2489. 10.1021/acschembio.1c00582. [DOI] [PubMed] [Google Scholar]
- Shanthamurthy C. D.; Leviatan Ben-Arye S.; Kumar N. V.; Yehuda S.; Amon R.; Woods R. J.; Padler-Karavani V.; Kikkeri R. Heparan Sulfate Mimetics Differentially Affect Homologous Chemokines and Attenuate Cancer Development. J. Med. Chem. 2021, 64 (6), 3367–3380. 10.1021/acs.jmedchem.0c01800. [DOI] [PubMed] [Google Scholar]
- Nie C.; Pouyan P.; Lauster D.; Trimpert J.; Kerkhoff Y.; Szekeres G. P.; Wallert M.; Block S.; Sahoo A. K.; Dernedde J.; Pagel K.; Kaufer B. B.; Netz R. R.; Ballauff M.; Haag R. Polysulfates Block SARS-CoV-2 Uptake through Electrostatic Interactions*. Angew. Chem., Int. Ed. Engl. 2021, 60 (29), 15870–15878. 10.1002/anie.202102717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangji R. N.; Sankaranarayanan N. V.; Elste J.; Al-Horani R. A.; Afosah D. K.; Joshi R.; Tiwari V.; Desai U. R. Inhibition of Herpes Simplex Virus-1 Entry into Human Cells by Nonsaccharide Glycosaminoglycan Mimetics. ACS Med. Chem. Lett. 2018, 9 (8), 797–802. 10.1021/acsmedchemlett.7b00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo J.; Nieto P. M.; de Paz J. L. GAG Multivalent Systems to Interact with Langerin. Curr. Med. Chem. 2022, 29 (7), 1173–1192. 10.2174/0929867328666210705143102. [DOI] [PubMed] [Google Scholar]
- Bedini E.; Laezza A.; Parrilli M.; Iadonisi A. A review of chemical methods for the selective sulfation and desulfation of polysaccharides. Carbohydr. Polym. 2017, 174, 1224–1239. 10.1016/j.carbpol.2017.07.017. [DOI] [PubMed] [Google Scholar]
- Arlov Ø.; Rütsche D.; Asadi Korayem M.; Öztürk E.; Zenobi-Wong M. Engineered Sulfated Polysaccharides for Biomedical Applications. Adv. Funct. Mater. 2021, 31 (19), 2010732. 10.1002/adfm.202010732. [DOI] [Google Scholar]
- Liu Q.; Chen G.; Chen H. Chemical synthesis of glycosaminoglycan-mimetic polymers. Polym. Chem. 2019, 10 (2), 164–171. 10.1039/C8PY01338A. [DOI] [Google Scholar]
- Sun L.; Chopra P.; Boons G. J. Chemoenzymatic Synthesis of Heparan Sulfate Oligosaccharides having a Domain Structure. Angew. Chem., Int. Ed. Engl. 2022, 61 (47), e202211112 10.1002/anie.202211112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojarski K. K.; Becher J.; Riemer T.; Lemmnitzer K.; Möller S.; Schiller J.; Schnabelrauch M.; Samsonov S. A. Synthesis and in silico characterization of artificially phosphorylated glycosaminoglycans. J. Mol. Struct. 2019, 1197, 401–416. 10.1016/j.molstruc.2019.07.064. [DOI] [Google Scholar]
- Varghese M.; Haque F.; Lu W.; Grinstaff M. W. Synthesis and Characterization of Regioselectively Functionalized Mono-Sulfated and -Phosphorylated Anionic Poly-Amido-Saccharides. Biomacromolecules 2022, 23 (5), 2075–2088. 10.1021/acs.biomac.2c00086. [DOI] [PubMed] [Google Scholar]
- Sarbu M.; Ica R.; Sharon E.; Clemmer D. E.; Zamfir A. D. Identification and Structural Characterization of Novel Chondroitin/Dermatan Sulfate Hexassacharide Domains in Human Decorin by Ion Mobility Tandem Mass Spectrometry. Molecules 2022, 27 (18), 6026. 10.3390/molecules27186026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmnitzer K.; Kohling S.; Freyse J.; Rademann J.; Schiller J. Characterization of defined sulfated heparin-like oligosaccharides by electrospray ionization ion trap mass spectrometry. J. Mass Spectrom 2021, 56 (2), e4692 10.1002/jms.4692. [DOI] [PubMed] [Google Scholar]
- Miller R. L.; Guimond S. E.; Schworer R.; Zubkova O. V.; Tyler P. C.; Xu Y.; Liu J.; Chopra P.; Boons G. J.; Grabarics M.; Manz C.; Hofmann J.; Karlsson N. G.; Turnbull J. E.; Struwe W. B.; Pagel K. Shotgun ion mobility mass spectrometry sequencing of heparan sulfate saccharides. Nat. Commun. 2020, 11 (1), 1481. 10.1038/s41467-020-15284-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens J.; Berden G.; Bentlage H.; Coene K. L. M.; Engelke U. F.; Wishart D.; van Scherpenzeel M.; Kluijtmans L. A. J.; Wevers R. A.; Oomens J. Unraveling the unknown areas of the human metabolome: the role of infrared ion spectroscopy. J. Inherit Metab Dis 2018, 41 (3), 367–377. 10.1007/s10545-018-0161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greis K.; Kirschbaum C.; von Helden G.; Pagel K. Gas-phase infrared spectroscopy of glycans and glycoconjugates. Curr. Opin Struct Biol. 2022, 72, 194–202. 10.1016/j.sbi.2021.11.006. [DOI] [PubMed] [Google Scholar]
- Renois-Predelus G.; Schindler B.; Compagnon I. Analysis of Sulfate Patterns in Glycosaminoglycan Oligosaccharides by MS(n) Coupled to Infrared Ion Spectroscopy: the Case of GalNAc4S and GalNAc6S. J. Am. Soc. Mass Spectrom. 2018, 29 (6), 1242–1249. 10.1007/s13361-018-1955-5. [DOI] [PubMed] [Google Scholar]
- Schindler B.; Renois-Predelus G.; Bagdadi N.; Melizi S.; Barnes L.; Chambert S.; Allouche A. R.; Compagnon I. MS/IR, a new MS-based hyphenated method for analysis of hexuronic acid epimers in glycosaminoglycans. Glycoconj J. 2017, 34 (3), 421–425. 10.1007/s10719-016-9741-8. [DOI] [PubMed] [Google Scholar]
- Mucha E.; Stuckmann A.; Marianski M.; Struwe W. B.; Meijer G.; Pagel K. In-depth structural analysis of glycans in the gas phase. Chemical Science 2019, 10 (5), 1272–1284. 10.1039/C8SC05426F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanal N.; Masellis C.; Kamrath M. Z.; Clemmer D. E.; Rizzo T. R. Glycosaminoglycan Analysis by Cryogenic Messenger-Tagging IR Spectroscopy Combined with IMS-MS. Anal. Chem. 2017, 89 (14), 7601–7606. 10.1021/acs.analchem.7b01467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettow M.; Grabarics M.; Mucha E.; Thomas D. A.; Polewski L.; Freyse J.; Rademann J.; Meijer G.; von Helden G.; Pagel K. IR action spectroscopy of glycosaminoglycan oligosaccharides. Anal Bioanal Chem. 2020, 412 (3), 533–537. 10.1007/s00216-019-02327-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappe A.; Miller R. L.; Struwe W. B.; Pagel K. State-of-the-art glycosaminoglycan characterization. Mass Spectrom Rev. 2022, 41 (6), 1040–1071. 10.1002/mas.21737. [DOI] [PubMed] [Google Scholar]
- Im J.; Lindsay S.; Wang X.; Zhang P. Single Molecule Identification and Quantification of Glycosaminoglycans Using Solid-State Nanopores. ACS Nano 2019, 13 (6), 6308–6318. 10.1021/acsnano.9b00618. [DOI] [PubMed] [Google Scholar]
- Xia K.; Hagan J. T.; Fu L.; Sheetz B. S.; Bhattacharya S.; Zhang F.; Dwyer J. R.; Linhardt R. J. Synthetic heparan sulfate standards and machine learning facilitate the development of solid-state nanopore analysis. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (11), e2022806118 10.1073/pnas.2022806118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayat P.; Rambaud C.; Priem B.; Bourderioux M.; Bilong M.; Poyer S.; Pastoriza-Gallego M.; Oukhaled A.; Mathe J.; Daniel R. Comprehensive structural assignment of glycosaminoglycan oligo- and polysaccharides by protein nanopore. Nat. Commun. 2022, 13 (1), 5113. 10.1038/s41467-022-32800-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez S.Nanopore GAGs sequencing 2022https://figshare.com/articles/figure/Nanopores_GAGs_sequencing/19391822, CC BY 4.0, accessed: 1 Dec 2022. Perez S..
- Pegeot M.; Sadir R.; Eriksson I.; Kjellen L.; Simorre J. P.; Gans P.; Lortat-Jacob H. Profiling sulfation/epimerization pattern of full-length heparan sulfate by NMR following cell culture 13C-glucose metabolic labeling. Glycobiology 2015, 25 (2), 151–6. 10.1093/glycob/cwu114. [DOI] [PubMed] [Google Scholar]
- Rudd T. R.; Yates E. A.; Guerrini M., CHAPTER 14 New Methods for the Analysis of Heterogeneous Polysaccharides - Lessons Learned from the Heparin Crisis. In NMR in Glycoscience and Glycotechnology; The Royal Society of Chemistry, 2017; pp 305–334. [Google Scholar]
- Rudd T. R.; Mauri L.; Marinozzi M.; Stancanelli E.; Yates E. A.; Naggi A.; Guerrini M. Multivariate analysis applied to complex biological medicines. Faraday Discuss. 2019, 218 (0), 303–316. 10.1039/C9FD00009G. [DOI] [PubMed] [Google Scholar]
- Colombo E.; Mauri L.; Marinozzi M.; Rudd T. R.; Yates E. A.; Ballabio D.; Guerrini M. NMR spectroscopy and chemometric models to detect a specific non-porcine ruminant contaminant in pharmaceutical heparin. J. Pharm. Biomed Anal 2022, 214, 114724. 10.1016/j.jpba.2022.114724. [DOI] [PubMed] [Google Scholar]
- Gribbon P.; Heng B. C.; Hardingham T. E. The molecular basis of the solution properties of hyaluronan investigated by confocal fluorescence recovery after photobleaching. Biophys. J. 1999, 77 (4), 2210–6. 10.1016/S0006-3495(99)77061-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribbon P.; Heng B. C.; Hardingham T. E. The analysis of intermolecular interactions in concentrated hyaluronan solutions suggest no evidence for chain-chain association. Biochem. J. 2000, 350 (Part 1), 329–35. 10.1042/bj3500329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attili S.; Borisov O. V.; Richter R. P. Films of end-grafted hyaluronan are a prototype of a brush of a strongly charged, semiflexible polyelectrolyte with intrinsic excluded volume. Biomacromolecules 2012, 13 (5), 1466–77. 10.1021/bm3001759. [DOI] [PubMed] [Google Scholar]
- Hricovini M.; Driguez P. A.; Malkina O. L. NMR and DFT analysis of trisaccharide from heparin repeating sequence. J. Phys. Chem. B 2014, 118 (41), 11931–42. 10.1021/jp508045n. [DOI] [PubMed] [Google Scholar]
- Sieme D.; Griesinger C. N.; Rezaei-Ghaleh N. Metal Binding to Sodium Heparin Monitored by Quadrupolar NMR. Int. J. Mol. Sci. 2022, 23 (21), 13185. 10.3390/ijms232113185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauri L.; Marinozzi M.; Phatak N.; Karfunkle M.; St Ange K.; Guerrini M.; Keire D. A.; Linhardt R. J. 1D and 2D-HSQC NMR: Two Methods to Distinguish and Characterize Heparin From Different Animal and Tissue Sources. Front Med. (Lausanne) 2019, 6, 142. 10.3389/fmed.2019.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadda E. Molecular simulations of complex carbohydrates and glycoconjugates. Curr. Opin Chem. Biol. 2022, 69, 102175. 10.1016/j.cbpa.2022.102175. [DOI] [PubMed] [Google Scholar]
- Perez S.; Makshakova O. Multifaceted Computational Modeling in Glycoscience. Chem. Rev. 2022, 122, 15914. 10.1021/acs.chemrev.2c00060. [DOI] [PubMed] [Google Scholar]
- Nagarajan B.; Holmes S. G.; Sankaranarayanan N. V.; Desai U. R. Molecular dynamics simulations to understand glycosaminoglycan interactions in the free- and protein-bound states. Curr. Opin Struct Biol. 2022, 74, 102356. 10.1016/j.sbi.2022.102356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almond A. Multiscale modeling of glycosaminoglycan structure and dynamics: current methods and challenges. Curr. Opin Struct Biol. 2018, 50, 58–64. 10.1016/j.sbi.2017.11.008. [DOI] [PubMed] [Google Scholar]
- Kunze G.; Huster D.; Samsonov S. A. Investigation of the structure of regulatory proteins interacting with glycosaminoglycans by combining NMR spectroscopy and molecular modeling - the beginning of a wonderful friendship. Biol. Chem. 2021, 402 (11), 1337–1355. 10.1515/hsz-2021-0119. [DOI] [PubMed] [Google Scholar]
- Hricovini M. Solution Structure of Heparin Pentasaccharide: NMR and DFT Analysis. J. Phys. Chem. B 2015, 119 (38), 12397–409. 10.1021/acs.jpcb.5b07046. [DOI] [PubMed] [Google Scholar]
- Hricovini M.; Hricovini M. Solution Conformation of Heparin Tetrasaccharide. DFT Analysis of Structure and Spin(−)Spin Coupling Constants. Molecules 2018, 23 (11), 3042. 10.3390/molecules23113042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hricovini M. Effect of solvent and counterions upon structure and NMR spin-spin coupling constants in heparin disaccharide. J. Phys. Chem. B 2011, 115 (6), 1503–11. 10.1021/jp1098552. [DOI] [PubMed] [Google Scholar]
- Singh A.; Tessier M. B.; Pederson K.; Wang X.; Venot A. P.; Boons G. J.; Prestegard J. H.; Woods R. J. Extension and validation of the GLYCAM force field parameters for modeling glycosaminoglycans. Can. J. Chem. 2016, 94 (11), 927–935. 10.1139/cjc-2015-0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsonov S. A.; Bichmann L.; Pisabarro M. T. Coarse-Grained Model of Glycosaminoglycans. J. Chem. Inf. Model. 2015, 55 (1), 114–124. 10.1021/ci500669w. [DOI] [PubMed] [Google Scholar]
- Souza P. C. T.; Alessandri R.; Barnoud J.; Thallmair S.; Faustino I.; Grunewald F.; Patmanidis I.; Abdizadeh H.; Bruininks B. M. H.; Wassenaar T. A.; Kroon P. C.; Melcr J.; Nieto V.; Corradi V.; Khan H. M.; Domanski J.; Javanainen M.; Martinez-Seara H.; Reuter N.; Best R. B.; Vattulainen I.; Monticelli L.; Periole X.; Tieleman D. P.; de Vries A. H.; Marrink S. J. Martini 3: a general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18 (4), 382–388. 10.1038/s41592-021-01098-3. [DOI] [PubMed] [Google Scholar]
- Rivas F.; Erxleben D.; Smith I.; Rahbar E.; DeAngelis P. L.; Cowman M. K.; Hall A. R. Methods for isolating and analyzing physiological hyaluronan: a review. Am. J. Physiol Cell Physiol 2022, 322 (4), C674–C687. 10.1152/ajpcell.00019.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm L.; Hellman U.; Larsson G. Size determination of hyaluronan using a gas-phase electrophoretic mobility molecular analysis. Glycobiology 2012, 22 (1), 7–11. 10.1093/glycob/cwr096. [DOI] [PubMed] [Google Scholar]
- Rivas F.; Zahid O. K.; Reesink H. L.; Peal B. T.; Nixon A. J.; DeAngelis P. L.; Skardal A.; Rahbar E.; Hall A. R. Label-free analysis of physiological hyaluronan size distribution with a solid-state nanopore sensor. Nat. Commun. 2018, 9 (1), 1037. 10.1038/s41467-018-03439-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas F.; DeAngelis P. L.; Rahbar E.; Hall A. R. Optimizing the sensitivity and resolution of hyaluronan analysis with solid-state nanopores. Sci. Rep 2022, 12 (1), 4469. 10.1038/s41598-022-08533-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter R. P.; Baranova N. S.; Day A. J.; Kwok J. C. Glycosaminoglycans in extracellular matrix organisation: are concepts from soft matter physics key to understanding the formation of perineuronal nets?. Curr. Opin Struct Biol. 2018, 50, 65–74. 10.1016/j.sbi.2017.12.002. [DOI] [PubMed] [Google Scholar]
- Giubertoni G.; Burla F.; Martinez-Torres C.; Dutta B.; Pletikapic G.; Pelan E.; Rezus Y. L. A.; Koenderink G. H.; Bakker H. J. Molecular Origin of the Elastic State of Aqueous Hyaluronic Acid. J. Phys. Chem. B 2019, 123 (14), 3043–3049. 10.1021/acs.jpcb.9b00982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giubertoni G.; Perez de Alba Ortiz A.; Bano F.; Zhang X.; Linhardt R. J.; Green D. E.; DeAngelis P. L.; Koenderink G. H.; Richter R. P.; Ensing B.; Bakker H. J. Strong Reduction of the Chain Rigidity of Hyaluronan by Selective Binding of Ca(2+) Ions. Macromolecules 2021, 54 (3), 1137–1146. 10.1021/acs.macromol.0c02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innes-Gold S. N.; Jacobson D. R.; Pincus P. A.; Stevens M. J.; Saleh O. A. Flexible, charged biopolymers in monovalent and mixed-valence salt: Regimes of anomalous electrostatic stiffening and of salt insensitivity. Phys. Rev. E 2021, 104 (1–1), 014504. 10.1103/PhysRevE.104.014504. [DOI] [PubMed] [Google Scholar]
- Srimasorn S.; Souter L.; Green D. E.; Djerbal L.; Goodenough A.; Duncan J. A.; Roberts A. R. E.; Zhang X.; Debarre D.; DeAngelis P. L.; Kwok J. C. F.; Richter R. P. A quartz crystal microbalance method to quantify the size of hyaluronan and other glycosaminoglycans on surfaces. Sci. Rep 2022, 12 (1), 10980. 10.1038/s41598-022-14948-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojar D.; Meche L.; Meng G.; Eng W.; Smith D. F.; Cummings R. D.; Mahal L. K. A Useful Guide to Lectin Binding: Machine-Learning Directed Annotation of 57 Unique Lectin Specificities. ACS Chem. Biol. 2022, 17 (11), 2993–3012. 10.1021/acschembio.1c00689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojar D.; Powers R. K.; Camacho D. M.; Collins J. J. Deep-Learning Resources for Studying Glycan-Mediated Host-Microbe Interactions. Cell Host Microbe 2021, 29 (1), 132–144. 10.1016/j.chom.2020.10.004. [DOI] [PubMed] [Google Scholar]
- Thomes L.; Burkholz R.; Bojar D. Glycowork: A Python package for glycan data science and machine learning. Glycobiology 2021, 31 (10), 1240–1244. 10.1093/glycob/cwab067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricard-Blum S.; Perez S. Glycosaminoglycan interaction networks and databases. Curr. Opin Struct Biol. 2022, 74, 102355. 10.1016/j.sbi.2022.102355. [DOI] [PubMed] [Google Scholar]
- Perez S.; Bonnardel F.; Lisacek F.; Imberty A.; Ricard Blum S.; Makshakova O.. GAG-DB, the New Interface of the Three-Dimensional Landscape of Glycosaminoglycans. Biomolecules 2020, 10 ( (12), ).1660. 10.3390/biom10121660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu C.; Jin L. NMR Characterization of the Interactions Between Glycosaminoglycans and Proteins. Front Mol. Biosci 2021, 8, 646808. 10.3389/fmolb.2021.646808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons M.; Millet O. Dynamic NMR Studies of Supramolecular Complexes. ChemInform 2001, 32, 291–291. 10.1002/chin.200129291. [DOI] [Google Scholar]
- Williamson M. P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Pomin V. H.; Wang X. Glycosaminoglycan-Protein Interactions by Nuclear Magnetic Resonance (NMR) Spectroscopy. Molecules 2018, 23 (9), 2314. 10.3390/molecules23092314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Garcia J. C.; Lopez-Prados J.; Angulo J.; Diaz-Contreras I.; Reichardt N.; de Paz J. L.; Martin-Lomas M.; Nieto P. M. Effect of the substituents of the neighboring ring in the conformational equilibrium of iduronate in heparin-like trisaccharides. Chemistry 2012, 18 (51), 16319–31. 10.1002/chem.201202770. [DOI] [PubMed] [Google Scholar]
- Munoz-Garcia J. C.; Chabrol E.; Vives R. R.; Thomas A.; de Paz J. L.; Rojo J.; Imberty A.; Fieschi F.; Nieto P. M.; Angulo J. Langerin-heparin interaction: two binding sites for small and large ligands as revealed by a combination of NMR spectroscopy and cross-linking mapping experiments. J. Am. Chem. Soc. 2015, 137 (12), 4100–10. 10.1021/ja511529x. [DOI] [PubMed] [Google Scholar]
- Monaco S.; Tailford L. E.; Juge N.; Angulo J. Differential Epitope Mapping by STD NMR Spectroscopy To Reveal the Nature of Protein-Ligand Contacts. Angew. Chem., Int. Ed. Engl. 2017, 56 (48), 15289–15293. 10.1002/anie.201707682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco S.; Walpole S.; Doukani H.; Nepravishta R.; Martinez-Bailen M.; Carmona A. T.; Ramos-Soriano J.; Bergstrom M.; Robina I.; Angulo J. Exploring Multi-Subsite Binding Pockets in Proteins: DEEP-STD NMR Fingerprinting and Molecular Dynamics Unveil a Cryptic Subsite at the GM1 Binding Pocket of Cholera Toxin B. Chemistry 2020, 26 (44), 10024–10034. 10.1002/chem.202001723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepravishta R.; Monaco S.; Distefano M.; Rizzo R.; Cescutti P.; Angulo J. Multifrequency STD NMR Unveils the Interactions of Antibiotics With Burkholderia multivorans Biofilm Exopolysaccharide. Front Mol. Biosci 2021, 8, 727980. 10.3389/fmolb.2021.727980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepravishta R.; Walpole S.; Tailford L.; Juge N.; Angulo J. Deriving Ligand Orientation in Weak Protein-Ligand Complexes by DEEP-STD NMR Spectroscopy in the Absence of Protein Chemical-Shift Assignment. Chembiochem 2019, 20 (3), 340–344. 10.1002/cbic.201800568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogut M. M.; Marcisz M.; Samsonov S. A. Modeling glycosaminoglycan-protein complexes. Curr. Opin Struct Biol. 2022, 73, 102332. 10.1016/j.sbi.2022.102332. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan N. V.; Nagarajan B.; Desai U. R. So you think computational approaches to understanding glycosaminoglycan-protein interactions are too dry and too rigid? Think again!. Curr. Opin Struct Biol. 2018, 50, 91–100. 10.1016/j.sbi.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiardi G.; Milanesi M.; Wade R. C.; D’Ursi P.; Rusnati M. A Bittersweet Computational Journey among Glycosaminoglycans. Biomolecules 2021, 11 (5), 739. 10.3390/biom11050739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. H.; Kearns F. L.; Rosenfeld M. A.; Casalino L.; Papanikolas M. J.; Simmerling C.; Amaro R. E.; Freeman R. GlycoGrip: Cell Surface-Inspired Universal Sensor for Betacoronaviruses. ACS Cent Sci. 2022, 8 (1), 22–42. 10.1021/acscentsci.1c01080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R.; Jonniya N. A.; Kar P. Effect of Sulfation on the Conformational Dynamics of Dermatan Sulfate Glycosaminoglycan: A Gaussian Accelerated Molecular Dynamics Study. J. Phys. Chem. B 2022, 126 (21), 3852–3866. 10.1021/acs.jpcb.2c01807. [DOI] [PubMed] [Google Scholar]
- Whitmore E. K.; Martin D.; Guvench O. Constructing 3-Dimensional Atomic-Resolution Models of Nonsulfated Glycosaminoglycans with Arbitrary Lengths Using Conformations from Molecular Dynamics. Int. J. Mol. Sci. 2020, 21 (20), 7699. 10.3390/ijms21207699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcisz M.; Zacharias M.; Samsonov S. A. Modeling Protein-Glycosaminoglycan Complexes: Does the Size Matter?. J. Chem. Inf Model 2021, 61 (9), 4475–4485. 10.1021/acs.jcim.1c00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojarski K. K.; Karczynska A. S.; Samsonov S. A. Role of Glycosaminoglycans in Procathepsin B Maturation: Molecular Mechanism Elucidated by a Computational Study. J. Chem. Inf Model 2020, 60 (4), 2247–2256. 10.1021/acs.jcim.0c00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan N. V.; Desai U. Computerized Molecular Modeling for Discovering Promising Glycosaminoglycan Oligosaccharides that Modulate Protein Function. Methods Mol. Biol. 2022, 2303, 513–537. 10.1007/978-1-0716-1398-6_41. [DOI] [PubMed] [Google Scholar]
- Schuurs Z. P.; Hammond E.; Elli S.; Rudd T. R.; Mycroft-West C. J.; Lima M. A.; Skidmore M. A.; Karlsson R.; Chen Y. H.; Bagdonaite I.; Yang Z.; Ahmed Y. A.; Richard D. J.; Turnbull J.; Ferro V.; Coombe D. R.; Gandhi N. S. Evidence of a putative glycosaminoglycan binding site on the glycosylated SARS-CoV-2 spike protein N-terminal domain. Comput. Struct Biotechnol J. 2021, 19, 2806–2818. 10.1016/j.csbj.2021.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison A. M.; Fogarty C. A.; Phung T. K.; Satheesan A.; Schulz B. L.; Fadda E. Fine-tuning the spike: role of the nature and topology of the glycan shield in the structure and dynamics of the SARS-CoV-2 S. Chem. Sci. 2022, 13 (2), 386–395. 10.1039/D1SC04832E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiardi G.; Richter S.; Oreste P.; Urbinati C.; Rusnati M.; Wade R. C. The binding of heparin to spike glycoprotein inhibits SARS-CoV-2 infection by three mechanisms. J. Biol. Chem. 2022, 298 (2), 101507. 10.1016/j.jbc.2021.101507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugatti A.; Paiardi G.; Urbinati C.; Chiodelli P.; Orro A.; Uggeri M.; Milanesi L.; Caruso A.; Caccuri F.; D’Ursi P.; Rusnati M. Heparin and heparan sulfate proteoglycans promote HIV-1 p17 matrix protein oligomerization: computational, biochemical and biological implications. Sci. Rep 2019, 9 (1), 15768. 10.1038/s41598-019-52201-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittum J. E.; Sankaranarayanan N. V.; O’Hara C. P.; Desai U. R. On the Selectivity of Heparan Sulfate Recognition by SARS-CoV-2 Spike Glycoprotein. ACS Med. Chem. Lett. 2021, 12 (11), 1710–1717. 10.1021/acsmedchemlett.1c00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogut M. M.; Maszota-Zieleniak M.; Marcisz M.; Samsonov S. A. Computational insights into the role of calcium ions in protein-glycosaminoglycan systems. Phys. Chem. Chem. Phys. 2021, 23 (5), 3519–3530. 10.1039/D0CP05438K. [DOI] [PubMed] [Google Scholar]
- Ricard-Blum S.; Lisacek F. Glycosaminoglycanomics: where we are. Glycoconj J. 2017, 34 (3), 339–349. 10.1007/s10719-016-9747-2. [DOI] [PubMed] [Google Scholar]
- Peysselon F.; Ricard-Blum S. Heparin-protein interactions: from affinity and kinetics to biological roles. Application to an interaction network regulating angiogenesis. Matrix Biol. 2014, 35, 73–81. 10.1016/j.matbio.2013.11.001. [DOI] [PubMed] [Google Scholar]
- Ori A.; Wilkinson M. C.; Fernig D. G. A systems biology approach for the investigation of the heparin/heparan sulfate interactome. J. Biol. Chem. 2011, 286 (22), 19892–904. 10.1074/jbc.M111.228114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Toledo A.; Sorrentino J. T.; Sandoval D. R.; Malmstrom J.; Lewis N. E.; Esko J. D. A Systems View of the Heparan Sulfate Interactome. J. Histochem Cytochem 2021, 69 (2), 105–119. 10.1369/0022155420988661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallet S. D.; Clerc O.; Ricard-Blum S. Glycosaminoglycan-Protein Interactions: The First Draft of the Glycosaminoglycan Interactome. J. Histochem Cytochem 2021, 69 (2), 93–104. 10.1369/0022155420946403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallet S. D.; Berthollier C.; Ricard-Blum S. The glycosaminoglycan interactome 2.0. Am. J. Physiol Cell Physiol 2022, 322 (6), C1271–C1278. 10.1152/ajpcell.00095.2022. [DOI] [PubMed] [Google Scholar]
- Bagdonas H.; Fogarty C. A.; Fadda E.; Agirre J. The case for post-predictional modifications in the AlphaFold Protein Structure Database. Nat. Struct Mol. Biol. 2021, 28 (11), 869–870. 10.1038/s41594-021-00680-9. [DOI] [PubMed] [Google Scholar]
- Bao B.; Kellman B. P.; Chiang A. W. T.; Zhang Y.; Sorrentino J. T.; York A. K.; Mohammad M. A.; Haymond M. W.; Bode L.; Lewis N. E. Correcting for sparsity and interdependence in glycomics by accounting for glycan biosynthesis. Nat. Commun. 2021, 12 (1), 4988. 10.1038/s41467-021-25183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seror J.; Merkher Y.; Kampf N.; Collinson L.; Day A. J.; Maroudas A.; Klein J. Articular cartilage proteoglycans as boundary lubricants: structure and frictional interaction of surface-attached hyaluronan and hyaluronan-aggrecan complexes. Biomacromolecules 2011, 12 (10), 3432–43. 10.1021/bm2004912. [DOI] [PubMed] [Google Scholar]
- Russell D. L.; Salustri A. Extracellular matrix of the cumulus-oocyte complex. Semin Reprod Med. 2006, 24 (4), 217–27. 10.1055/s-2006-948551. [DOI] [PubMed] [Google Scholar]
- Dyer D. P.; Migliorini E.; Salanga C. L.; Thakar D.; Handel T. M.; Richter R. P. Differential structural remodelling of heparan sulfate by chemokines: the role of chemokine oligomerization. Open Biol. 2017, 7 (1), 160286. 10.1098/rsob.160286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliorini E.; Thakar D.; Kuhnle J.; Sadir R.; Dyer D. P.; Li Y.; Sun C.; Volkman B. F.; Handel T. M.; Coche-Guerente L.; Fernig D. G.; Lortat-Jacob H.; Richter R. P. Cytokines and growth factors cross-link heparan sulfate. Open Biol. 2015, 5 (8), 150046. 10.1098/rsob.150046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bano F.; Banerji S.; Howarth M.; Jackson D. G.; Richter R. P. A single molecule assay to probe monovalent and multivalent bonds between hyaluronan and its key leukocyte receptor CD44 under force. Sci. Rep. 2016, 6 (1), 34176. 10.1038/srep34176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder A.; Möller A. K.; Milz F.; Neuhaus P.; Walhorn V.; Dierks T.; Anselmetti D. Catch bond interaction between cell-surface sulfatase Sulf1 and glycosaminoglycans. Biophys. J. 2015, 108 (7), 1709–1717. 10.1016/j.bpj.2015.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke-Roos M.; Meseck G. R.; Rosenhahn A. Catch bond interaction allows cells to attach to strongly hydrated interfaces. Biointerphases 2016, 11 (1), 018905. 10.1116/1.4939040. [DOI] [PubMed] [Google Scholar]
- Lawrance W.; Banerji S.; Day A. J.; Bhattacharjee S.; Jackson D. G. Binding of Hyaluronan to the Native Lymphatic Vessel Endothelial Receptor LYVE-1 Is Critically Dependent on Receptor Clustering and Hyaluronan Organization. J. Biol. Chem. 2016, 291 (15), 8014–30. 10.1074/jbc.M115.708305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curk T.; Dubacheva G. V.; Brisson A. R.; Richter R. P. Controlling Superselectivity of Multivalent Interactions with Cofactors and Competitors. J. Am. Chem. Soc. 2022, 144 (38), 17346–17350. 10.1021/jacs.2c06942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz V.; Suflita M.; Liu X.; Zhang X.; Yu Y.; Li L.; Green D. E.; Xu Y.; Zhang F.; DeAngelis P. L.; Liu J.; Linhardt R. J. Heparan Sulfate Domains Required for Fibroblast Growth Factor 1 and 2 Signaling through Fibroblast Growth Factor Receptor 1c. J. Biol. Chem. 2017, 292 (6), 2495–2509. 10.1074/jbc.M116.761585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burla F.; Tauber J.; Dussi S.; van der Gucht J.; Koenderink G. H. Stress management in composite biopolymer networks. Nat. Phys. 2019, 15 (6), 549–553. 10.1038/s41567-019-0443-6. [DOI] [Google Scholar]
- Aoki-Kinoshita K. F.; Campbell M. P.; Lisacek F.; Neelamegham S.; York W. S.; Packer N. H., Glycoinformatics. In Essentials of Glycobiology Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Mohnen D., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., Seeberger P. H., Eds.; Cold Spring Harbor: New York, 2022; pp 705–718. [Google Scholar]
- Ranzinger R.; Aoki-Kinoshita K. F.; Campbell M. P.; Kawano S.; Lutteke T.; Okuda S.; Shinmachi D.; Shikanai T.; Sawaki H.; Toukach P.; Matsubara M.; Yamada I.; Narimatsu H. GlycoRDF: an ontology to standardize glycomics data in RDF. Bioinformatics 2015, 31 (6), 919–25. 10.1093/bioinformatics/btu732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daponte V.; Hayes C.; Mariethoz J.; Lisacek F. Dealing with the Ambiguity of Glycan Substructure Search. Molecules 2022, 27 (1), 65. 10.3390/molecules27010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Macias M. A.; Mariethoz J.; Andersson P.; Jin C.; Venkatakrishnan V.; Aoki N. P.; Shinmachi D.; Ashwood C.; Madunic K.; Zhang T.; Miller R. L.; Horlacher O.; Struwe W. B.; Watanabe Y.; Okuda S.; Levander F.; Kolarich D.; Rudd P. M.; Wuhrer M.; Kettner C.; Packer N. H.; Aoki-Kinoshita K. F.; Lisacek F.; Karlsson N. G. Towards a standardized bioinformatics infrastructure for N- and O-glycomics. Nat. Commun. 2019, 10 (1), 3275. 10.1038/s41467-019-11131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Aoki-Kinoshita K. F.; Ishihama Y.; Okuda S. GlycoPOST realizes FAIR principles for glycomics mass spectrometry data. Nucleic Acids Res. 2021, 49 (D1), D1523–D1528. 10.1093/nar/gkaa1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki-Kinoshita K. F.; Lisacek F.; Mazumder R.; York W. S.; Packer N. H. The GlySpace Alliance: toward a collaborative global glycoinformatics community. Glycobiology 2020, 30 (2), 70–71. 10.1093/glycob/cwz078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson M. D.; Dumontier M.; Aalbersberg I. J.; Appleton G.; Axton M.; Baak A.; Blomberg N.; Boiten J. W.; da Silva Santos L. B.; Bourne P. E.; Bouwman J.; Brookes A. J.; Clark T.; Crosas M.; Dillo I.; Dumon O.; Edmunds S.; Evelo C. T.; Finkers R.; Gonzalez-Beltran A.; Gray A. J.; Groth P.; Goble C.; Grethe J. S.; Heringa J.; t Hoen P. A.; Hooft R.; Kuhn T.; Kok R.; Kok J.; Lusher S. J.; Martone M. E.; Mons A.; Packer A. L.; Persson B.; Rocca-Serra P.; Roos M.; van Schaik R.; Sansone S. A.; Schultes E.; Sengstag T.; Slater T.; Strawn G.; Swertz M. A.; Thompson M.; van der Lei J.; van Mulligen E.; Velterop J.; Waagmeester A.; Wittenburg P.; Wolstencroft K.; Zhao J.; Mons B. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. 10.1038/sdata.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D.; Crabtree J.; Dillo I.; Downs R. R.; Edmunds R.; Giaretta D.; De Giusti M.; L’Hours H.; Hugo W.; Jenkyns R.; Khodiyar V.; Martone M. E.; Mokrane M.; Navale V.; Petters J.; Sierman B.; Sokolova D. V.; Stockhause M.; Westbrook J. The TRUST Principles for digital repositories. Sci. Data 2020, 7 (1), 144. 10.1038/s41597-020-0486-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Zidek A.; Potapenko A.; Bridgland A.; Meyer C.; Kohl S. A. A.; Ballard A. J.; Cowie A.; Romera-Paredes B.; Nikolov S.; Jain R.; Adler J.; Back T.; Petersen S.; Reiman D.; Clancy E.; Zielinski M.; Steinegger M.; Pacholska M.; Berghammer T.; Bodenstein S.; Silver D.; Vinyals O.; Senior A. W.; Kavukcuoglu K.; Kohli P.; Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]