

Abstract

Herein, we report a strategy for the formation of isotopically labeled carboxylic esters from boronic esters/acids using a readily accessible palladium carboxylate complex as an organometallic source of isotopically labeled functional groups. The reaction allows access to either unlabeled or full 13C- or 14C-isotopically labeled carboxylic esters, and the method is characterized by its operational simplicity, mild conditions, and general substrate scope. Our protocol is further extended to a carbon isotope replacement strategy, involving an initial decarbonylative borylation procedure. Such an approach allows access to isotopically labeled compounds directly from the unlabeled pharmaceutical, which can have implications for drug discovery programs.
Keywords: isotope labeling, carbon-14, pharmaceuticals, organic acids, palladium
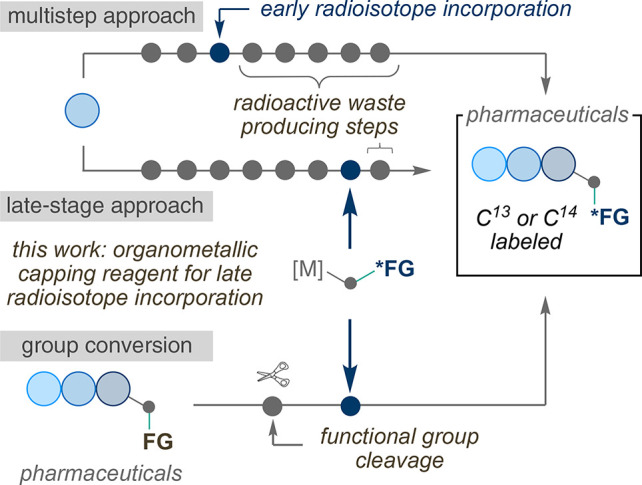
Carboxylic acids and esters are a central motif in drug discovery programs and constitute versatile synthetic intermediates en route to active pharmaceutical ingredients (APIs).1 Pharmacokinetic studies of these APIs, however, are costly to perform, as they require isotope enrichment of the API for adsorption, metabolism, elimination (ADME) studies.2 Often, staple synthetic techniques used in process or medicinal chemistry to access carboxylic acids and esters do not transfer to isotope labeling programs due to stringent limitations in efficiently incorporating the valuable radiolabel. The synthesis of isotopically labeled compounds differs from traditional organic synthesis due to (1) the scarcity of isotopically labeled reagents, (2) the high cost of these reagents, and (3) the production of dangerous radioactive waste, which therefore requires new synthetic routes to be devised.3 Radiochemists therefore aim to incorporate the radiolabel in high radiochemical yields at the latest stage possible in a synthetic route in order to minimize cost and hazardous waste.
Of the radiolabeled motifs, accessing carbonyl containing compounds are among the most valuable with three carbonyl containing functional groups (amides, carboxylic acids, and esters) among the most prevalent in bioactive molecules.4 Furthermore, these methods often utilize feedstock radiolabels that originate from Ba14CO3, which is the source of all radioactive carbon-14 containing compounds and liberates isotopically labeled carbon dioxide (Scheme 1).5
Scheme 1. Pharmaceuticals with Aryl Carboxylic Acid Functionalities and Traditional Synthetic Routes for Isotope Incorporation.

From this stage the CO2 can be used directly or reduced to afford a variety of one carbon synthons such as CO. Most commonly utilized strategies for the synthesis of isotopically labeled carboxylic acids include nitrile substitution followed by hydrolysis or direct carboxylation from organomagnesium or -lithium reagents.6−8 However, both of these approaches suffer from harsh reaction conditions and poor functional group compatibility, which limits their applicability en route to functional group dense molecules often found within drug discovery programs. Modern carboxylation or alkoxycarbonylation strategies relevant to isotope labeling have been developed from aryl halides,9−11 pseudohalides,12,13 and boronic esters derivatives14−17 using palladium, nickel, or copper catalyzed/mediated processes with superior substrate compatibility. However, these methods require the use of gaseous reagents as radiolabels, which have specific drawbacks, including the requirement for specialized equipment and safety considerations. Furthermore, the radiolabel is often used in significant excess, which produces challenging to handle gaseous radioactive waste and storage of gaseous radiolabeled CO gas may undergo radiolysis. Alternative approaches that take advantage of functional group exchange have also been developed with recent dynamic carbon isotope exchange strategies developed by Audisio18 and Lundgren19 utilizing aryl carboxylate salts and by Baran20 and Martin21 using redox active alkyl esters. These dynamic exchange methods have emerged as atom efficient routes for isotope labeling, though have a generalized drawback with incomplete isotope incorporation occurring due to the presence of an equilibrium of labeled to unlabeled compound.
Considering these challenges, our group has aimed to develop isotope labeling reagents that can readily incorporate carbonyl motifs and be compatible with late-stage functionalization and functional group interconversion technologies (Scheme 2). Imagining general and robust reagents, we envisioned an efficient synthesis toward an organometallic species bearing an isotopically labeled carbonyl functional group may serve as “organometallic capping reagents”. In line with this objective, our group recently reported a procedure for the carbon isotope labeling of aliphatic esters from alkyl iodides using a nickel mediated approach.22 We initially designed this approach through the in situ formation of an isotopically labeled nickel(II) carboxylate species (NiII-*COOMe), which could react with an open-shell alkyl species. However, our mechanistic investigations suggested an alternative pathway was operative, involving alkyl-radical addition to a metal ligated CO species to generate a nickel-acyl complex en route to the aliphatic ester. As such, we re-evaluated this approach and targeted well-defined complexes that could be accessed in a single, efficient step in high yields, and be isolated as an air-stable and readily weighed solid. If realized, this reagent could offer a complementary method to our previous report without the need for gaseous reagents or specialized equipment, and it would provide an operationally simple and robust method for the synthesis of isotopically labeled carboxylic esters and acids. We targeted our approach toward related, well-defined metallacarboxylate complexes, coupling these reagents to transmetalation reactions from boronic esters/acids offered an ideal scenario due to its robust nature and the stability of these reagents.23,24 Boronic esters/acids are particularly attractive precursors due to the ability of boron to be installed at late-stages in a synthetic route, which merges well within the context of late-stage isotope incorporation.25 Herein, we describe our efforts toward this goal using an isotopically labeled Pd-carboxylate complex.
Scheme 2. Approaches to Radiolabeling and Carboxylate Labeling Reagent.
We began our investigations by synthesizing metal carboxylate complexes, which could be accessed from simple commercially available starting materials, in high yielding and utilize low equivalents of CO or CO2 as the carbon isotope label source. The resultant complex should also be easy to purify, store, and utilize in subsequent reactions. Surveying the literature of complexes that fit these criteria, we initially examined nickellacarboxylate complexes isolated utilizing low equivalence of one carbon synthons, (CO or CO2) amenable to isotope labeling.26 However, due to their multistep synthesis and the formation of reactive species en route to the final Ni(II)-COOR compounds, we began to investigate alternatives. The utilization of cobalt-carboxylates such as (salen)Co(III)-*COOMe (salen = N,N′-Ethylenebis(salicylimine)) was initially promising as they could be accessed directly from (salen)Co(II), CO, and MeOH under oxidative conditions,27,28 but we found these organometallic complexes were unreactive to standard coupling reagents.
Continuing our investigation, we synthesized (PPh3)2Pd(Cl)(CO2Me) Pd-1 from an adapted procedure originally reported by Yamamoto et al.,29 with Pd-1 being accessed in a single step from commercial (PPh3)4Pd. After some optimization, a final procedure was developed that reacted (PPh3)4Pd with LiCl and O2 along with near stochiometric CO released from SilaCOgen, to afford Pd-1 as a white powder in a satisfactory 96% isolated yield (Scheme 3). The structure of Pd-1 could be unambiguously determined by XRD as a square planar complex with trans-oriented PPh3 ligands. We note the synthesis of Pd-1 is in stark contrast to the synthesis from similar Pd(II) species (PPh3)2PdCl2, which required pressures of 30 bar CO to proceed toward the carboxylate formation.29 As anticipated, isotopically labeled analogs of Pd-1 could be accessed using 13CO or 14CO applying either COgen (10% 14C-labeled) or SilaCOgen reagents.30 Importantly the 14C-radiolabeled complex was found to be stable over a storage period of 6 months as both a solid and as a solution in dichloromethane at −32 °C seeing no loss in radioactivity. In a similar nature to our desired reactivity, palladium(0) sources have been been utilized under oxidative conditions for the direct synthesis of esters from boronic ester/acids under atmospheric pressures of CO and were later applied to 11C isotope labeling.31−33
Scheme 3. Synthesis of Palladium-Carboxylate Complex.

12C- or 13C- SilaCOgen (1.5 equiv.).
14C–COgen (3 equiv.).
With Pd-1 in hand, we turned to evaluating its reaction with boronic esters/acids (Table 1A). After some optimization, the reaction between the aryl neopentyl glycol ester and Pd-1 at room temperature with KF and Na2CO3 in a dioxane/water mixture afforded the 4-phenyl–phenylboronic ester 1 in excellent yields (91%). Boronic acids showed similar reactivity to boronic esters (entry 2), and of the bases evaluated, the inorganic base Na2CO3 performed optimally (entries 3 and 4). Lower yields were found by replacing dioxane (entries 5 and 6), while comparable yields were found using H2O or MeOH or heating the reaction (entries 7 and 8). The reaction components KF, H2O and Na2CO3 were all found to be beneficial for the reaction outcome (entries 9–11). Furthermore, performing the reaction in air rather than an inert atmosphere only resulted in a slight reduction in yield (85%), highlighting the simplicity of the experimental setup.
Table 1. Optimization and Hammett Analysis of the Reaction Conditionsa.

Pd-1 (0.1 mmol), 4-phenyl–phenylboronic ester (0.1 mmol), KF (0.1 mmol), Na2CO3 (0.2 mmol), dioxane/H2O (5.4 mL/0.6 mL), r.t., 16 h.
isolated yield
Pd-1 (0.025 mmol), boronic acid (0.25 mmol), KF (0.025 mmol), Na2CO3 (0.05 mmol), dioxane/H2O (1.35 mL/0.15 mL), r.t..
To gain further information on the reaction, we monitored the standard reaction and performed a Hammett analysis (Table 1B). Monitoring the reaction of 4-biphenyl boronic acid with Pd-1 under standard conditions, we observed full conversion within 4 h, while performing a Hammett analysis of phenyl boronic acids bearing p-OMe, p-tBu, p-Cl, and p-CN groups observed a σ value of −0.64, which supports the reaction kinetics being relatively insensitive to the electronic nature of the boronic acid.
Next, we turned to studying the generality of the protocol (Table 2). Gratifyingly, a range of electron-donating or electron-withdrawing aryl boronic esters delivered the targeted methyl esters 1–3 (R = OMe, tBu, CN). Substrates containing free N–H or O–H bonds (4, 5, 6, and 9) and those containing acrylates (8) and phenothiazine (10) groups were also amenable to the reaction conditions. Aryl bromide (7) could also be tolerated with the addition of PhIOAc2 as an external oxidant which reacts with the Pd(0) species formed following transmetalation and reductive elimination. Derivatives of drug compounds clofibrate (11) and indomethacin (12) were smoothly synthesized under the standard conditions. The synthesis of 14C-labeled methyl esters was carried out using 14C labeled material diluted with unlabeled 12C material (approximately 10% 14C, see the Supporting Information for details). The synthesis proceeded smoothly, with the standard biphenyl substrate (13) being synthesized in a 93% yield with >98% radiochemical purity and a radiochemical yield of 98%. 14C-Labeling was extended to functionalities such as oxathianes (14), amides (15), or a drug derivative of fenofibrate containing ester and ketone functionalities (16), which were well tolerated in this method.
Table 2. Accessing Radiolabeled Methyl Estersa,b.

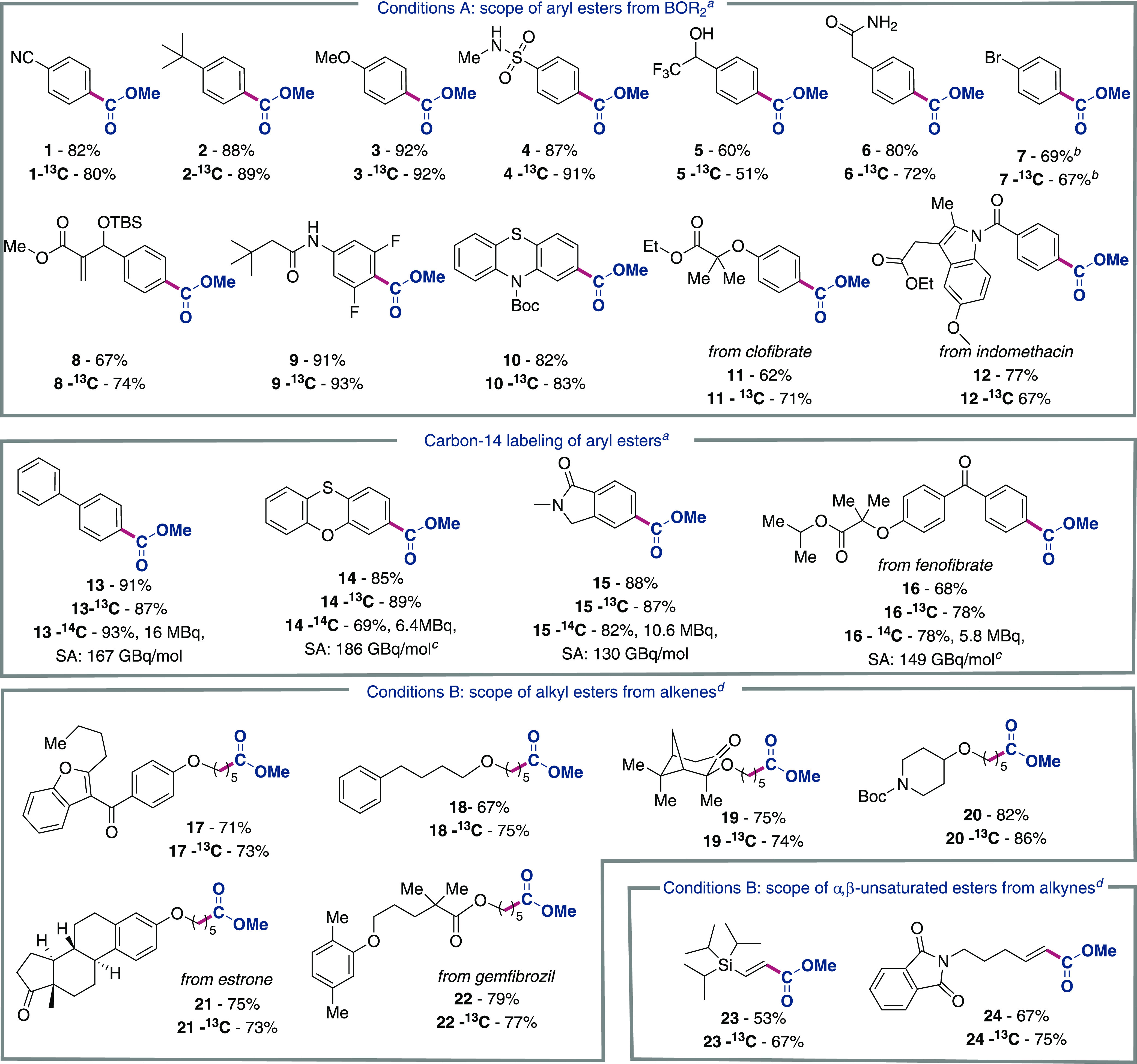
Encouraged by these results, we wondered if this protocol could be extended to alkyl and alkenyl boronic ester derivatives. After additional optimization, a modified procedure was developed in which alkenes or alkynes underwent hydroboration to afford alkyl or alkenyl 9-BBN derivatives that could be coupled to our standard radiolabeling conditions using the Pd-carboxylate labeling reagent. As shown, substrates containing furans (17), enolizable sites (19), phthalimides (24), or drug derivatives of the hormone estrone (21) and pharmaceutical gemfibrozil (22) were able to be isolated in high yields. Comparing this protocol to Cu-catalyzed carboxylation reactions34 or Pd-catalyzed alkoxycarbonylation protocols of aryl boronic esters,33 the corresponding reactions were performed under modified conditions limiting the isotopically labeled CO or CO2 equivalence released to 1.5 equivalents from BaCO3 or SilaCOgen, respectively. Under the Cu-catalyzed conditions, we observed significantly reduced yields (1–19%) for electronically varied substrates (1, 2, and 13) while the Pd-catalyzed conditions accessed these substrates in moderate yields (24–54% yield), supporting the notion that our approach offers complementary reactivity to existing protocols in the context of radiolabeling (see SI).
Lastly, we aimed to apply the developed reaction conditions for the carbon isotope labeling of carboxylic acids and pharmaceutically relevant compounds (Scheme 4). Building on a report by the Sanford group, aryl carboxylic acids can be transformed into boronic acids in a single step.35 We speculated this report could be coupled with Pd-1 as a radiolabeling reagent for a carbon isotope replacement strategy. Gratifyingly, reacting pharmaceuticals bearing carboxylic acid moieties (Bexarotene, Adapalene, Probenecid) with the borylation conditions developed by Sanford, followed by our conditions with Pd-1 after only filtration and no intermediate purification steps smoothly transformed these APIs into the labeled methyl carboxylic esters 25–27 (Scheme 4). Isolation of the neopentyl boronic ester intermediate of 27 resulted in an elevated yield of 79% for the single step reaction.
Scheme 4. Applications to Carbon Isotope Replacement.

aryl carboxylic acid (0.1 mmol), TFFH (0.1 mmol), proton sponge (0.1 mmol), THF (0.2 mL), 15 min, r.t., then PCy3 (10 mol %), Ni(COD)2 (5 mol %), B2nep2 (2.0 equiv), 115 °C, 24 h, and then reaction conditions as Table 1, entry 1.
In summary, we have developed a simple synthesis of an organometallic capping reagent that can be accessed from commercial reagents and is effectively adapted to its radiolabeled analogues. This reagent is used in a mild and selective protocol to access valuable radiolabeled methyl esters from readily accessible alkyl, alkenyl and aryl boronic esters/acids in high radiochemical yields. The reaction is further characterized by its operational simplicity, broad substrate scope, and applicability to pharmaceutically relevant scaffolds. The potential of this capping reagent is also showcased in a carbon isotope replacement strategy from drugs containing carboxylic acids.
Acknowledgments
We are highly appreciative of the financial support from the Danish National Research Foundation (grant no. DNRF118), NordForsk (grant no. 85378), and Aarhus University. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 862179 and the Marie Sklodowska-Curie grant agreement No. 859910. This publication reflects the views only of the authors, and the Commission cannot be held responsible for any use which may be made of the information contained therein. The authors are grateful to Clemens Kaussler for assistance with the X-ray crystallographic analysis.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.2c00708.
Experimental procedures and spectral and crystallographic data (PDF)
Author Contributions
† S.J.T. and A.K.R. contributed equally to this work. CRediT: Stephanie J. Ton conceptualization, data curation, formal analysis, investigation, methodology, writing-original draft, writing-review & editing; Anne Ravn data curation, formal analysis, investigation, methodology; Daniel Hoffmann data curation, formal analysis; Craig S. Day data curation, formal analysis, investigation, methodology; Lee Kingston data curation, formal analysis; Charles S. Elmore formal analysis, project administration, supervision, writing-review & editing; Troels Skrydstrup conceptualization, formal analysis, funding acquisition, project administration, supervision, writing-review & editing.
The authors declare the following competing financial interest(s): T.S. is co-owner of SyTracks A/S, which commercializes the two-chamber system (COware) and SilaCOgen.
Supplementary Material
References
- Maag H.Prodrugs of Carboxylic Acids. In Prodrugs: Challenges and Rewards Part 1; Stella V. J., Borchardt R. T., Hageman M. J., Oliyai R., Maag H., Tilley J. W., Eds.; Springer, 2007; pp 703–729. [Google Scholar]
- Marathe H. P.; Shyu C. W.; Humphreys G. W. The Use of Radiolabeled Compounds for ADME Studies in Discovery and Exploratory Development. Curr. Pharm. Des. 2004, 10 (24), 2991–3008. 10.2174/1381612043383494. [DOI] [PubMed] [Google Scholar]
- Babin V.; Taran F.; Audisio D. Late-Stage Carbon-14 Labeling and Isotope Exchange: Emerging Opportunities and Future Challenges. JACS Au 2022, 2 (6), 1234–1251. 10.1021/jacsau.2c00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertl P.; Altmann E.; McKenna J. M. The Most Common Functional Groups in Bioactive Molecules and How Their Popularity Has Evolved over Time. J. Med. Chem. 2020, 63 (15), 8408–8418. 10.1021/acs.jmedchem.0c00754. [DOI] [PubMed] [Google Scholar]
- Voges R.; Heys J. R.; Moenius T.. Preparation of Compounds Labeled with Tritium and Carbon-14; Wiley, 2009; p 688. [Google Scholar]
- Derdau V. New trends and applications in cyanation isotope chemistry. J. Labelled Comp Radiopharm 2018, 61 (14), 1012–1023. 10.1002/jlcr.3630. [DOI] [PubMed] [Google Scholar]
- Bragg R. A.; Sardana M.; Artelsmair M.; Elmore C. S. New trends and applications in carboxylation for isotope chemistry. J. Labelled Comp Radiopharm 2018, 61 (13), 934–948. 10.1002/jlcr.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauben W.; Reid J.; Yankwich P. Techniques in Using Carbon 14. Anal. Chem. 1947, 19 (11), 828–832. 10.1021/ac60011a003. [DOI] [Google Scholar]
- Correa A.; Martín R. Palladium-Catalyzed Direct Carboxylation of Aryl Bromides with Carbon Dioxide. J. Am. Chem. Soc. 2009, 131 (44), 15974–15975. 10.1021/ja905264a. [DOI] [PubMed] [Google Scholar]
- Tran-Vu H.; Daugulis O. Copper-Catalyzed Carboxylation of Aryl Iodides with Carbon Dioxide. ACS Catal. 2013, 3 (10), 2417. 10.1021/cs400443p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friis S. D.; Andersen T. L.; Skrydstrup T. Palladium-Catalyzed Synthesis of Aromatic Carboxylic Acids with Silacarboxylic Acids. Org. Lett. 2013, 15 (6), 1378–1381. 10.1021/ol4003465. [DOI] [PubMed] [Google Scholar]
- Rebih F.; Andreini M.; Moncomble A.; Harrison-Marchand A.; Maddaluno J.; Durandetti M. Direct Carboxylation of Aryl Tosylates by CO2 Catalyzed by In situ-Generated Ni0. Chem.—Eur. J. 2016, 22 (11), 3758–3763. 10.1002/chem.201503926. [DOI] [PubMed] [Google Scholar]
- Minami H.; Nogi K.; Yorimitsu H. Palladium-Catalyzed Alkoxycarbonylation of Arylsulfoniums. Org. Lett. 2019, 21 (8), 2518–2522. 10.1021/acs.orglett.9b00067. [DOI] [PubMed] [Google Scholar]
- Takaya J.; Tadami S.; Ukai K.; Iwasawa N. Copper(I)-Catalyzed Carboxylation of Aryl- and Alkenylboronic Esters. Org. Lett. 2008, 10 (13), 2697–2700. 10.1021/ol800829q. [DOI] [PubMed] [Google Scholar]
- Ohishi T.; Nishiura M.; Hou Z. Carboxylation of Organoboronic Esters Catalyzed by N-Heterocyclic Carbene Copper(I) Complexes. Angew. Chem., Int. Ed. 2008, 47 (31), 5792–5795. 10.1002/anie.200801857. [DOI] [PubMed] [Google Scholar]
- Riss P. J.; Lu S.; Telu S.; Aigbirhio F. I.; Pike V. W. CuI-Catalyzed 11C Carboxylation of Boronic Acid Esters: A Rapid and Convenient Entry to 11C-Labeled Carboxylic Acids, Esters, and Amides. Angew. Chem., Int. Ed. 2012, 51 (11), 2698–2702. 10.1002/anie.201107263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Li G.; He J.; Liu J.; Li P.; Lei A. Palladium-Catalyzed Aerobic Oxidative Carbonylation of Arylboronate Esters under Mild Conditions. Angew. Chem., Int. Ed. 2010, 49 (19), 3371–3374. 10.1002/anie.201000460. [DOI] [PubMed] [Google Scholar]
- Destro G.; Loreau O.; Marcon E.; Taran F.; Cantat T.; Audisio D. Dynamic Carbon Isotope Exchange of Pharmaceuticals with Labeled CO2. J. Am. Chem. Soc. 2019, 141 (2), 780–784. 10.1021/jacs.8b12140. [DOI] [PubMed] [Google Scholar]
- Kong D.; Munch M.; Qiqige Q.; Cooze C. J. C.; Rotstein B. H.; Lundgren R. J. Fast Carbon Isotope Exchange of Carboxylic Acids Enabled by Organic Photoredox Catalysis. J. Am. Chem. Soc. 2021, 143 (5), 2200–2206. 10.1021/jacs.0c12819. [DOI] [PubMed] [Google Scholar]
- Kingston C.; Wallace M. A.; Allentoff A. J.; deGruyter J. N.; Chen J. S.; Gong S. X.; Bonacorsi S. Jr; Baran P. S. Direct Carbon Isotope Exchange through Decarboxylative Carboxylation. J. Am. Chem. Soc. 2019, 141 (2), 774–779. 10.1021/jacs.8b12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortajada A.; Duan Y.; Sahoo B.; Cong F.; Toupalas G.; Sallustrau A.; Loreau O.; Audisio D.; Martin R. Catalytic Decarboxylation/Carboxylation Platform for Accessing Isotopically Labeled Carboxylic Acids. ACS Catal. 2019, 9 (7), 5897–5901. 10.1021/acscatal.9b01921. [DOI] [Google Scholar]
- Ton S. J.; Neumann K. T.; Nørby P.; Skrydstrup T. Nickel-Mediated Alkoxycarbonylation for Complete Carbon Isotope Replacement. J. Am. Chem. Soc. 2021, 143 (42), 17816–17824. 10.1021/jacs.1c09170. [DOI] [PubMed] [Google Scholar]
- Neochoritis C. G.; Shaabani S.; Ahmadianmoghaddam M.; Zarganes-Tzitzikas T.; Gao L.; Novotná M.; Mitríková T.; Romero A. R.; Irianti M. I.; Xu R. Rapid approach to complex boronic acids. Sci. Adv. 2019, 5 (7), aaw4607. 10.1126/sciadv.aaw4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D. G.Structure, Properties, and Preparation of Boronic Acid Derivatives. In Boronic Acids; Wiley, 2011; p 133. [Google Scholar]
- Ni N.; Wang B.. Applications of Boronic Acids in Chemical Biology and Medicinal Chemistry. In Boronic Acids; Wiley, 2011; pp 591–620. [Google Scholar]
- Yoo C.; Kim J.; Lee Y. Synthesis and Reactivity of Nickel(II) Hydroxycarbonyl Species, NiCOOH-κC. Organometallics 2013, 32 (23), 7195–7203. 10.1021/om400881j. [DOI] [Google Scholar]
- Costa G.; Mestroni G. Acyl- and carboxyalkyl—cobalt(III) chelates. Tetrahedron Lett. 1967, 8 (19), 1783–1784. 10.1016/S0040-4039(00)90723-8. [DOI] [Google Scholar]
- Chambers D. R.; Sullivan R. E.; Martin D. B. C. Synthesis and Characterization of Alkoxycarbonyl Cobalt Complexes via Direct Carbonylation Methods. Organometallics 2017, 36 (8), 1630–1639. 10.1021/acs.organomet.7b00186. [DOI] [Google Scholar]
- Izawa Y.; Shimizu I.; Yamamoto A. Palladium-Catalyzed Oxidative Carbonylation of 1-Alkynes into 2-Alkynoates with Molecular Oxygen as Oxidant. Bull. Chem. Soc. Jpn. 2004, 77 (11), 2033–2045. 10.1246/bcsj.77.2033. [DOI] [Google Scholar]
- Ravn A. K.; Johansen M. B.; Skrydstrup T. Controlled Release of Reactive Gases: A Tale of Taming Carbon Monoxide. ChemPlusChem. 2020, 85 (7), 1529–1533. 10.1002/cplu.202000319. [DOI] [PubMed] [Google Scholar]
- Ohe T.; Ohe K.; Uemura S.; Sugita N. Palladium(0)-catalyzed carbonylation of alkenyl- and aryl- borates and boronic acids with carbon monoxide. J. Organomet. Chem. 1988, 344 (1), C5–C7. 10.1016/0022-328X(88)80220-1. [DOI] [Google Scholar]
- Ishii H.; Minegishi K.; Nagatsu K.; Zhang M.-R. Pd(0)-mediated [11C]carbonylation of aryl and heteroaryl boronic acid pinacol esters with [11C]carbon monoxide under ambient conditions and a facile process for the conversion of [carbonyl-11C]esters to [carbonyl-11C]amides. Tetrahedron 2015, 71 (10), 1588–1596. 10.1016/j.tet.2015.01.008. [DOI] [Google Scholar]
- Yamamoto Y. The First General and Selective Palladium(II)-Catalyzed Alkoxycarbonylation of Arylboronates: Interplay among Benzoquinone-Ligated Palladium(0) Complex, Organoboron, and Alcohol Solvent. Adv. Synth. Catal. 2010, 352 (2–3), 478–492. 10.1002/adsc.200900836. [DOI] [Google Scholar]
- Nayal O. S.; Hong J.; Yang Y.; Mo F. Cu-Catalysed carboxylation of aryl boronic acids with CO2. Org. Chem. Front. 2019, 6 (21), 3673–3677. 10.1039/C9QO01023H. [DOI] [Google Scholar]
- Malapit C. A.; Bour J. R.; Laursen S. R.; Sanford M. S. Mechanism and Scope of Nickel-Catalyzed Decarbonylative Borylation of Carboxylic Acid Fluorides. J. Am. Chem. Soc. 2019, 141 (43), 17322–17330. 10.1021/jacs.9b08961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.