

Abstract
Heterogeneous nuclear ribonucleoproteins (hnRNPs) are RNA binding proteins, which aid in maturation, stabilization and transport of mRNA. They have a significant role in cellular nucleic acid metabolism. The hnRNPs alter gene expression and are linked to various neurodegenerative disorders and cancers. Previously, six unrelated girls with developmental delay, intellectual disability and hypotonia were found to have de novo heterozygous pathogenic missense variants in HNRNPH2, located on the X chromosome. A gain-of-function effect was proposed for the variant and it was thought to be lethal in males as no surviving males were identified. We describe a family with two affected siblings, one male and one female, with a known pathogenic variant in HNRNPH2, possibly due to maternal germline mosaicism.
Keywords: Heterogeneous nuclear ribonucleoproteins, developmental delay, intellectual disability, hypotonia, X-linked dominant, HNRNPH2
1. INTRODUCTION
Heterogeneous nuclear ribonucleoproteins (hnRNPs) are RNA binding proteins, which play a significant role in nucleic acid metabolism by influencing alternative splicing, maturation, stabilization and transport of RNAs (Dreyfuss, Kim & Kataoka, 2002; Dreyfuss, Matunis, Pinol-Roma & Burd, 1993; Geuens, Bouhy & Timmerman, 2016). There are twenty types of hnRNPs named alphabetically from A1 to U (Geuens, Bouhy & Timmerman, 2016). Altered expression of hnRNPs has been associated with many types of cancer (Singh & Cooper, 2012). Misregulation of hnRNPs also causes aberrant splicing and has been implicated in the pathogenesis of several neurological disorders such as spinal muscular atrophy, amyotropic lateral sclerosis and fragile X-associated tremor/ataxia syndrome (Feng & Xie, 2013).
X-linked syndromic mental retardation, Bain type (MRXSB, MIM # 300986) was first described among six unrelated females with intellectual disability, developmental delay and hypotonia with de novo missense variants in HNRNPH2 (Bain et al., 2016). Here, we report two siblings, one male and one female, born to a consanguineous Indian couple, with features of MRXSB and a known pathogenic variant in HNRNPH2, identified by whole exome sequencing.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
The study was approved by the Institutional Ethics Committee. Written informed consents were obtained for the use of photographs and research findings.
2.2. Clinical report
Proband 1 (P1), the first-born boy of a third degree consanguineous couple was evaluated at 8-year-9 months, for global developmental delay, intellectual disability and seizures. He was born by normal vaginal delivery at term with a birth weight of 1.7 kg (−3 to −4 SD). He had delayed cry and was admitted to neonatal intensive unit. He achieved head control at 10 months, rolling over at 12 months and sitting with support at 18 months. He reached out to objects at age 12 months and transferred objects at 18 months. He had generalized tonic-clonic seizures at 18 months of age following which he lost all his previously attained milestones. Myoclonic jerks were noted at 8 years of age. On examination, his weight was 19 kg (−3 SD), height was 115cm (−3 to −4 SD) and his head circumference was 48 cm (−2 SD). He had hypertelorism, medial flaring of eyebrows, bilateral ptosis, thick vermillion of upper and lower lips, short philtrum and high arched palate. He also had mild scoliosis, bilateral 5th finger clinodactyly. Skin hyperextensibility and significant joint hypermobility were present (Figure 1A–1D). He had abnormal flapping of hands and tremors, generalized hypotonia and preserved deep tendon reflexes. Karyotype, chromosomal microarray, tandem mass spectrometry, ophthalmic evaluation, electroencephalogram, magnetic resonance imaging (MRI) and computed tomography of brain revealed normal results.
Figure 1:
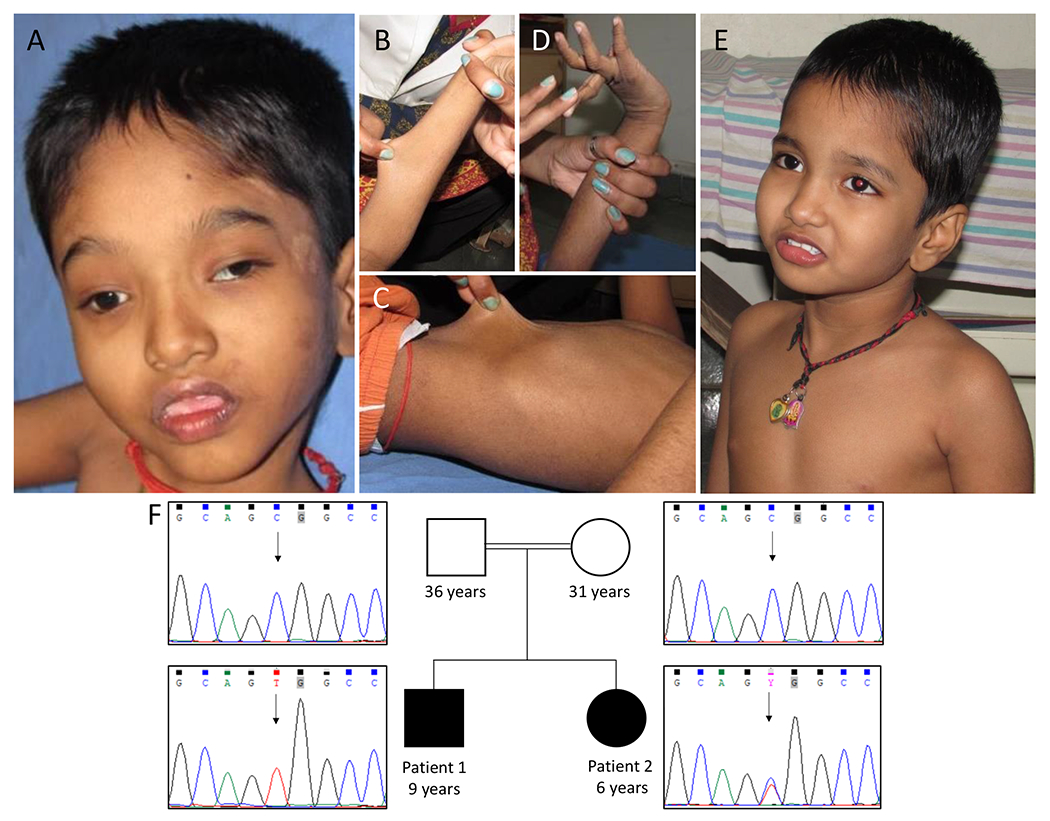
Photographs of affected individuals. Patient 1 shows hypertelorism, medial flaring of eyebrows, ptosis, short philtrum, thick vermillion of upper and lower lips (A), skin hyperextensibility (B and C), severe joint hypermobility (D). Patient 2 with no facial dysmorphism (E). Pedigree of the family and electropherograms show c.616C>T in HNRNPH2 in patient 1 and patient 2 in hemizygous and heterozygous state respectively. Parents had wild type allele (F).
Proband 2 (P2), the younger sibling of patient 1, a 5-year-9-months-old girl, also presented with global developmental delay, intellectual disability and neuroregression after 2 years of age. She was born at term with a birth weight of 2.75 kg (−1 to −2 SD). She achieved head control at 5 months, sat with support at 7 months and spoke monosyllables at 2 years. She did not have seizures. On examination her weight was 14.5 kg (−2 to −3 SD), height was 103 cm (−2 to −3 SD) and head circumference was 48 cm (−1 to −2 SD). No facial dysmorphism was noted in her (Figure 1E). Her fingers were long and slender. She had skin hyperextensibility and joint hypermobility, but of lesser severity when compared to her brother. She had generalized hypotonia and normal deep tendon reflexes. She also had involuntary movements of her hands. Karyotype was normal. MRI of brain at three years of age, showed a hyperintense lesion in the inferior part of splenium of corpus callosum, suggestive of lipoma of corpus callosum. Parents were clinically normal.
Genomic analysis
Genomic DNA was isolated by standard phenol-chloroform method from EDTA blood samples. Whole exome sequencing (WES) was performed using Illumina’s Nextera Rapid Capture Exome Kit and NextSeq500 Sequencer with NextSeq™ 500 High Output Kit (Illumina, Inc., San Diego, CA, USA) with a coverage of >95% of all amplicons at minimum of 20X depth for proband 1 and 2 as previously described (Shukla et al., 2017). Additionally, OMIM phenotypes, HPO terms and in-house allele frequency from exome data of 583 individuals of Indian origin were integrated into the WES data. Filtering strategy used to identify the candidate variant is provided in supplementary table 1. Validation and segregation analysis of the variant was done by Sanger sequencing.
3. RESULTS
WES of the siblings identified a known missense pathogenic variant, c.616C>T [p.(Arg206Trp), rs886039763] in HNRNPH2 (NM_001032393.2). Patient 1 was hemizygous and patient 2 was heterozygous for the variant. Parents were found to have wild type allele (Figure 1F). This variant was not found in population databases such as gnomAD, 1000 Genome Project and our in-house WES data of 583 individuals of Indian origin. The variant is conserved across species with GERP score of 3.06. In-silico prediction tools such as MutationTaster, SIFT and CADD Phred predicted the variant to be damaging to the protein function.
4. DISCUSSION
We identified a known missense pathogenic variant, c.616C>T [p.(Arg206Trp)] in HNRNPH2 in hemizygous state in P1 and heterozygous state in P2. Bain et al., previously described this de novo missense heterozygous variant in four unrelated females with developmental delay and intellectual disability. Two other de novo missense heterozygous variants, c.617 G>A [p. (Arg206Gln)] and c.626C>T [p. (Pro209Leu)] in HNRNPH2 were identified in two other girls with a similar phenotype (Bain et al., 2016).
All the females described by Bain et al., had developmental delay, intellectual disability and hypotonia. These features were consistently seen in the siblings we describe. Neuroregression was not a constant feature and was observed only in three of the six females described by Bain et al. The two subjects in our study had regression of milestones. P1 was more severely affected than P2. P1 had seizures, which were not seen in P2. Seizures were observed in three of the six females previously described. Two individuals described by Bain et al had acquired microcephaly. The head circumference of both probands in our study was within normal range. Speech was either absent or severely delayed in five of the six girls. The siblings we describe had absent speech. All the previously described individuals had variable facial dysmorphic features like hypertelorism, thick vermillion of lower lips, concave eyebrows and almond shaped eyes. Though P1 had facial dysmorphism, P2 did not show any dysmorphic features. Three reported individuals had features of autistic spectrum disorder and two of the older individuals had behavioral issues like attention deficit hyperactivity disorder and obsessive-compulsive disorders. Though both P1 and P2 had repetitive hand flapping and involuntary movements, formal assessment for autism was not carried out. Skin hyperextensibility and joint hypermobility were prominent findings in both probands. These findings were observed only in one individual in the previously described cohort. A detailed comparison of clinical features is given in Table 1.
Table 1:
Clinical features of proband 1 and proband 2 in comparison to the previously reported patients.
| Current study | JM Bain et al., 2016 | Harmsen et al., 2019 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Proband | 1 | 2 | 1 | 2 | 3 | 4 | 5 | 6 | 1 |
| Gender | Male | Female | Female | Female | Female | Female | Female | Female | Male |
| Age (years) | 9 | 6 | 34 | 8 | 4 | 6 | 21 | 2 | 3 |
| Variant | c.616 C>T | c.616C>T | c.616C>T | c.616C>T | c.617G>A | c.626C>T | c.616C>T | c.616C>T | c. 617G>A |
| Birth weight | 1.7 kg (−3 to −4 SD) | 2.75 kg (−1 to −2 SD) | Not available | 2.6 kg (−1 to −2 SD) | 2.7 kg (−1 to −2 SD) | 2.9 kg (−1 SD) | 3.9 kg (+1 to +2 SD) | Not available | 3.92 kg (+1 to +2 SD) |
| Weight | 19 kg (−3 SD) | 14.5 kg (−2 to −3 SD) | 56 kg (0 to −1 SD) | 20 kg at 6 years (normal) | Not available | 11.8 kg (−4 to −5 SD) | 42.5 kg at 19 years (−2 to −3 SD) | Not available | Not available |
| Length | 115cm (−3 to −4 SD) | 103 cm (−2 to −3 SD) | 154 cm (−1 to −2 SD) | Not available | Not available | 104 cm (−2 to −3 SD) | 159.8 cm at 19 years (−1 SD) | Not available | Not available |
| Head circumference | 48 cm (−2 SD) | 48 cm (−1 to −2 SD) | 48 cm at 7 years (−1 to −2 SD) | 51 cm at 6.5 years (normal) | Not available | 46.5 cm (−2 to −3 SD) | 55 cm at 19 years (normal) | 46 cm (−1 to 2 SD) | 45.6 cm (−2 to 3 SD) |
| Developmental delay | + | + | + | + | + | + | + | + | + |
| Intellectual disability | + | + | + | + | + | + | + | + | + |
| Regression | + | + | + | − | + | + | − | − | − |
| Autism spectrum disorder | − | − | + | + | − | − | + | − | − |
| Seizures | + | − | + | + | − | + | − | − | − |
| Speech | Absent | Absent | Single words and short sentences achieved at 7 years | Absent | Not available | Few words | Speaking in sentences achieved at 8 years | Absent (babbles) | Absent |
| Tone | Hypotonia | Hypotonia | Truncal hypotonia and limbs hypertonia | Hypotonia | Hypotonia | Hypotonia | Hypotonia | Hypotonia | Axial hypotonia |
| Skin hyperextensibility | + | + | − | − | − | − | + | − | − |
| Joint hypermobility | + | + | − | − | − | − | + | − | − |
| Facial features | Hypertelorism, medial flared eyebrow, ,short philtrum, thick vermillion of upper and lower lips, high arched palate, ptosis | Hypertelorism, short philtrum | Hypotelorism, high narrow nasal bridge, wide mouth, full lips | Bilateral epicanthal folds, midface hypoplasia, almond shaped eyes | Short palpebral fissures | Hypertelorism | Mild micrognathia, high arched palate | Concave eyebrows, fissure in the upper lip | Mild facial dysmorphism with almond-shaped eyes |
| Skeletal abnormalities | Scoliosis mild, bilateral 5th digit clinodactyly, proximally placed thumb | Long slender fingers | Lordosis, bilateral femoral osteotomies, arachnodactyly | − | Pes planus | Talus valgus | Thoracolumbar scoliosis, pectus carinatum, pes planus, joint hypermobility, elongated fingers | − | Scoliosis |
| Brain MRI | Normal | Lipoma in Corpus callosum region | Normal | Cerebellar vermis hypoplasia | Not available | Normal | Normal | Distorted cerebellar vermis | Normal |
| Other findings | Stereotypic hand flapping, tremors while trying to hold objects | Involuntary movements | Abnormal gait, ankle clonus, attention deficit hyperactivity disorder, obsessive-compulsive disorder | Ataxia, mild hearing loss, anemia, epistaxis, constipation | Gastroesophageal reflux disease, hand flapping | Incoordination, Failure to thrive, walks with support, feeding difficulty, anxiety, sensory issues, atrial septal defect and mitral valve prolapse, fetal finger pads Incoordination | Attention deficit hyperactivity disorder, mitral valve prolapse, mitral regurgitation, gastroesophageal reflux disease | Dystonia of left hand, Dyspraxia, feeding difficulty, drooling | Lack of independent movement, spasticity of the extremities, feeding difficulties |
HNRNPH2 is located on the Xq22.1and the HNRNPH2 protein has five domains including three homologous quasi-RNA recognition motif (RRMs) and two glycine-rich domains (GRDs). RRMs are involved in binding to G-rich RNA and GRDs are essential for protein-protein interactions and nuclear localization of the protein. GRDs contain highly conserved nuclear localization sequence between amino acid residues 194 to 220, which interacts with transportin 1, a nuclear transport receptor. HNRNPH2 protein is ubiquitously expressed and localizes predominantly in the nucleus as well as cytoplasm. It is mainly involved in alternating splicing either by enhancing or silencing splicing of pre-RNA and nucleo-cytoplasmic shuttling mRNA (Geuens, Bouhy & Timmerman, 2016; Grammatikakis et al., 2016; Van Dusen, Yee, Mcnally & Mcnally, 2010). The variant, p.(Arg206Trp) is located in the highly conserved nuclear localization sequence of GRD of HNRNPH2 protein.
Since no surviving male was identified previously, it was postulated that variants in HNRNPH2 were incompatible with survival in males and only females survived, suggesting an X-linked dominant inheritance with male lethality. Recently, an earlier reported variant, c.617G>A (p.Arg206Gln), has been described in a 3-years-old male with clinical findings similar to those of females with MRXSB (Harmsen, Buchert, Mayatepek, Haack & Distelmaier, 2019). Ascertainment of another male with this condition, though with more severe clinical manifestations indicates that pathogenic variants in HNRNPH2 are compatible with postnatal survival. Germline mosaicism in mother has been previously described in X-linked dominant conditions like periventricular nodular heterotopias due to variants in FLNA gene (Lapointe, Spriggs & Mhanni, 2014). In the present study, both the parents had wild type alleles. The most likely explanation for recurrence in two siblings could be of maternal germline mosaicism. A de novo variant, c.616C>T in HNRNPH1, an autosomal paralogue of HNRNPH2, was reported in a boy with similar phenotypic features of MRXSB (Pilch et al., 2018).
5. CONCLUSIONS
The study expands the phenotype of MRXSB and reports a surviving male with a variant in HNRNPH2. The recurrence in the two siblings could be due to gonadal mosaicism of the variant in the mother. This aspect is important while offering counseling to families with apparent de novo variants in HNRNPH2. Also, additional report of subjects with this condition and functional validation of variants would aid in further elucidating the role of HNRNPH2 as a cause of syndromic X-linked intellectual disability.
Supplementary Material
ACKNOWLEDGMENTS
We thank patients and family members for their participation in the study. This study was supported by National Institutes of Health funded project, ‘Genetic diagnosis of heritable neurodevelopmental disorders in India: investigating the use of whole exome sequencing and genetic counseling to address the high burden of neurodevelopmental disorders’ (Grant Number: 1R21NS094047-01).
Footnotes
CONFLICTS OF INTEREST
Authors declare no conflict of interest.
REFERENCE
- Bain JM, Cho MT, Telegrafi A, Wilson A, Brooks S, Botti C, … Chung WK (2016). Variants in HNRNPH2 on the X Chromosome Are Associated with a Neurodevelopmental Disorder in Females. American journal of human genetics, 99(3), 728–734. doi: 10.1016/j.ajhg.2016.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss G, Kim VN, & Kataoka N (2002). Messenger-RNA-binding proteins and the messages they carry. Nature reviews. Molecular cell biology, 3(3), 195–205. doi: 10.1038/nrm760 [DOI] [PubMed] [Google Scholar]
- Dreyfuss G, Matunis MJ, Pinol-Roma S, & Burd CG (1993). hnRNP proteins and the biogenesis of mRNA. Annual review of biochemistry, 62, 289–321. doi: 10.1146/annurev.bi.62.070193.001445 [DOI] [PubMed] [Google Scholar]
- Feng D, & Xie J (2013). Aberrant splicing in neurological diseases. Wiley interdisciplinary reviews. RNA, 4(6), 631–649. doi: 10.1002/wrna.1184 [DOI] [PubMed] [Google Scholar]
- Geuens T, Bouhy D, & Timmerman V (2016). The hnRNP family: insights into their role in health and disease. Human genetics, 135(8), 851–867. doi: 10.1007/s00439-016-1683-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammatikakis I, Zhang P, Panda AC, Kim J, Maudsley S, Abdelmohsen K, … Gorospe M (2016). Alternative Splicing of Neuronal Differentiation Factor TRF2 Regulated by HNRNPH1/H2. Cell reports, 15(5), 926–934. doi: 10.1016/j.celrep.2016.03.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmsen S, Buchert R, Mayatepek E, Haack TB, & Distelmaier F (2019). Bain type of X-linked syndromic mental retardation in boys. Clinical Genetics, 95(6), 734–735. doi: 10.1111/cge.13524 [DOI] [PubMed] [Google Scholar]
- LaPointe MM, Spriggs EL, & Mhanni AA (2014). Germline mosaicism in X-linked periventricular nodular heterotopia. BMC neurology, 14, 125. doi: 10.1186/1471-2377-14-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilch J, Koppolu AA, Walczak A, Pienkowski VAM, Biernacka A, Skiba P, … Ploski R (2018). Evidence for HNRNPH1 being another gene for Bain type syndromic mental retardation. Clinical genetics. doi: 10.1111/cge.13410 [DOI] [PubMed] [Google Scholar]
- Shukla A, Upadhyai P, Shah J, Neethukrishna K, Bielas S, & Girisha KM (2017). Autosomal recessive spinocerebellar ataxia 20: Report of a new patient and review of literature. European journal of medical genetics, 60(2), 118–123. doi: 10.1016/j.ejmg.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, & Cooper TA (2012). Pre-mRNA splicing in disease and therapeutics. Trends in molecular medicine, 18(8), 472–482. doi: 10.1016/j.molmed.2012.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dusen CM, Yee L, McNally LM, & McNally MT (2010). A glycine-rich domain of hnRNP H/F promotes nucleocytoplasmic shuttling and nuclear import through an interaction with transportin 1. Molecular and cellular biology, 30(10), 2552–2562. doi: 10.1128/mcb.00230-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.