

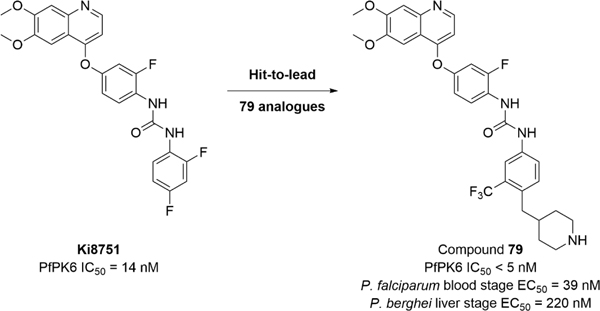
Abstract
Malaria is a devastating disease that causes significant global morbidity and mortality. The rise of drug resistance against artemisinin-based combination therapy demonstrates the necessity to develop alternative antimalarials with novel mechanisms of action. We report the discovery of Ki8751 as an inhibitor of essential kinase PfPK6. 79 derivatives were designed, synthesized and evaluated for PfPK6 inhibition and antiplasmodial activity. Using group efficiency analyses, we established the importance of key groups on the scaffold consistent with a type II inhibitor pharmacophore. We highlight modifications on the tail group that contribute to antiplasmodial activity, cumulating in the discovery of compound 67, a PfPK6 inhibitor (IC50 = 13 nM) active against the P. falciparum blood stage (EC50 = 160 nM), and compound 79, a PfPK6 inhibitor (IC50 < 5 nM) with dual-stage antiplasmodial activity against P. falciparum blood stage (EC50 = 39 nM) and against P. berghei liver stage (EC50 = 220 nM).
Keywords: Malaria, Antiplasmodial, Plasmodium falciparum Protein Kinase 6 (PfPK6), Kinase Inhibitor, Group Efficiency, Structure-Activity Relationship Study
Graphical Abstract
Introduction
Malaria is a devastating infectious disease causing 241 million infections and 627,000 deaths worldwide in 20201. The causative agents of malaria are unicellular eukaryotic parasites from the Plasmodium genus. Of the five known species that infect humans, P. falciparum is associated with the bulk of malaria-associated morbidity and mortality1,2. While there has been a favorable trend of declining mortality rates since the 2000s due to the effectiveness of artemisinin-based combination therapy (ACT), the frontline treatment for malaria globally, progress has stalled in recent years1. Drug resistance remains a challenge in eliminating malaria. Ever since the first report of artemisinin resistance amongst P. falciparum strains isolated from Cambodia in 20093, artemisinin resistance has reached high prevalence and is a worrying trend in the Greater Mekong Subregion of South East Asia1. In the past two years, reports of independent emergence of artemisinin resistance in clinical isolates of parasites from Rwanda4,5 and Uganda6 may indicate the start of a foreboding trend in Africa. This demonstrates the need to expand our arsenal of antimalarial treatments by the development of new drugs with novel mechanisms of action7.
Plasmodium protein kinases present an untapped opportunity for drug development8–10. Of the 85–99 protein kinases identified from the P. falciparum genome11,12, reverse genetics13 and saturation mutagenesis14 studies have identified 36 and 40 kinases, respectively, to be essential for P. falciparum asexual blood stage proliferation. Furthermore, kinases are well-established to be a druggable pharmacological target class, as evident from the 71 kinase inhibitors approved by the FDA for various indications up to May 202115. With an abundance of literature on kinase inhibition to design inhibitors, knowledge of the specific scaffolds can be cautiously extrapolated for inhibition of Plasmodium kinases8. The Plasmodium kinome is significantly divergent from the human kinome, and many Plasmodium kinases have no clear human orthologue, hence the design of a selective Plasmodium kinase inhibitor should be possible10,11. Moreover, no antimalarial drug currently in the market or in clinical trials is known to target any Plasmodium protein kinase7, while only a few investigational drugs, e.g. MMV39004816, target P. falciparum phosphatidylinositol-4-OH kinase (PfPI4K), a lipid kinase. We thus believe that there has been little or no selection pressure for resistance towards kinase inhibitors currently amongst the Plasmodium population. These factors combined support the hypothesis for targeting Plasmodium protein kinases.
Despite the attractiveness of Plasmodium protein kinases as drug targets, few kinases have been pharmacologically validated as targets. While inhibitors of PfPKG17–19 and PfCLK320 have shown promise as new antimalarial preclinical compounds, most other Plasmodium kinases have not had medicinal chemistry campaigns initiated against them. As a result, most Plasmodium kinases have no chemical probes or even tool compounds that could facilitate the investigation of their biological function or possible druggability. To bridge this gap, the overall goal of our research is to discover hits against lesser-studied Plasmodium kinases and develop them into lead compounds potent both against a specific kinase target and against Plasmodium proliferation. The molecules could support biological studies on kinase function as well as future drug development efforts.
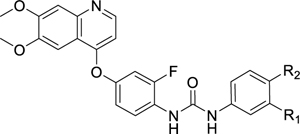
In this report, we have focused on P. falciparum Protein Kinase 6 (PfPK6) (PlasmoDB ID: PF3D7_1337100), a kinase in the CMGC group21 genetically validated to be essential for blood stage proliferation of P. falciparum13,14. The precise function of PfPK6 is not well defined, but it is postulated to be involved in the cell cycle of the Plasmodium parasite in a cyclin-independent manner by virtue of its homology with human CDK2 and its expression profile in late ring, trophozoites and early schizonts21. Two known inhibitors of PfPK6 are the purines roscovitine and olomoucine, which inhibit PfPK6 at biochemical IC50s of 30 μM and 180 μM respectively21. A series of compounds based on the 1-(4-((2-aminopyrimidin-4-yl)(methyl)amino)phenyl)urea scaffold, labelled “Scaffold J”, have also been identified as PfPK6 inhibitors with biochemical IC50s between 138 and 917 nM from the Tres Cantos Antimalarial Set (TCAMS)22 (Figure 1). To the best of our knowledge, these scaffolds were not subject to further development for PfPK6 inhibition. To discover hits against PfPK6, we have screened 110 kinase inhibitors against a panel of 11 P. falciparum kinases using the KinaseSeeker assay, an in vitro assay based on split-luciferase technology we had previously developed for human kinases23. Briefly, displacement of the chemical inducer of dimerization (CID) from the kinase active site by the test compound leads to disassembly of the split luciferase, which results in loss of luminescence. Using this assay, we found Ki8751 to be a potent inhibitor of PfPK6 with an IC50 of 14 nM (Luceome internal data). Ki8751 was initially developed as a VEGFR2 inhibitor24, with several other off-targets identified to be PDGFRα24, c-KIT24, FGFR224, AXL25, AURKB26, AURKC26, and ABL27. Notably, these kinases belong either in the TK group or Aurora family of kinases.
Figure 1.
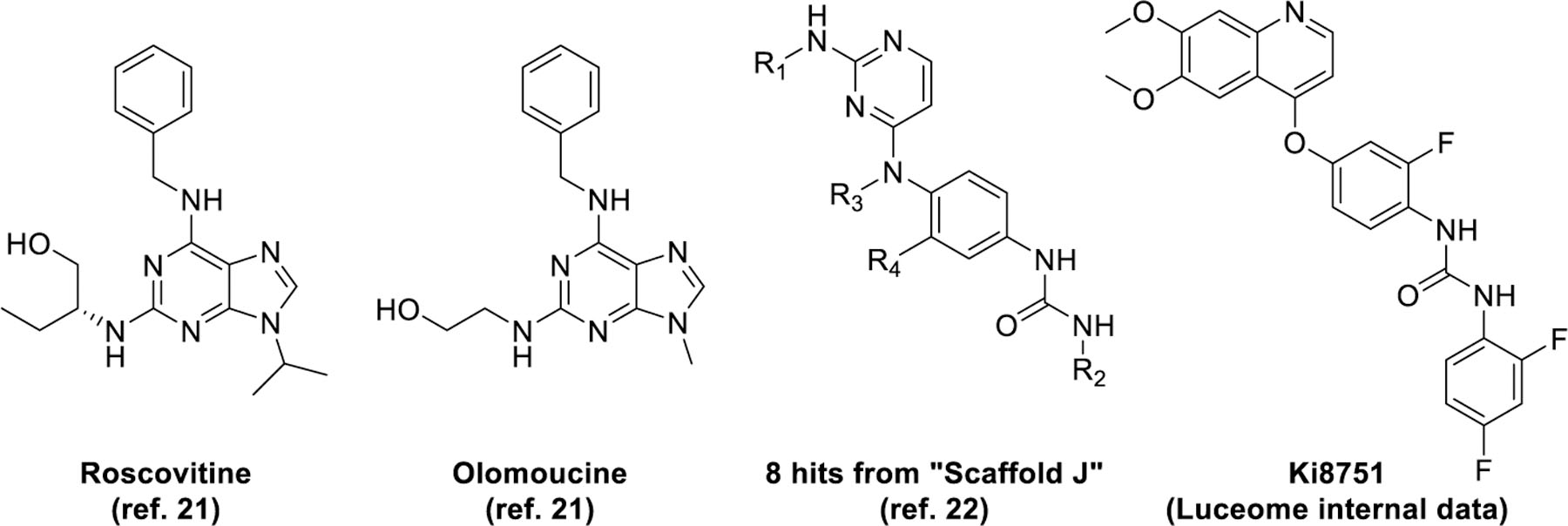
Known inhibitors of PfPK6.
Herein we report results from our early hit-to-lead medicinal chemistry project. We established a structure-activity-relationship (SAR) study of this scaffold, utilizing a group efficiency approach to guide SAR, and the discovery of 79, a potent PfPK6 inhibitor with dual-stage antiplasmodial activity against the P. falciparum asexual blood stage and the P. berghei liver stage.
Results and Discussion
Preliminary Characterization and Group Efficiency.
Considering that known targets of Ki8751 are in the TK group and Aurora family of kinases24–27, it was especially intriguing to find Ki8751 as a hit against PfPK6, a member of the CMGC group. To investigate the kinome-wide inhibitory potential of this scaffold, we screened Ki8751 against 97 human kinases using a thermal shift assay (Figure S1 and Table S2). We found only three kinases (BRAF, LOK, ABL1) with a >10°C shift in melting point, and only seven kinases (PLK4, CHK2, EphA2, EphB3, SLK, CAMKK2, CLK1) with a 5–10°C shift in melting point. This suggests that Ki8751 is not overly promiscuous across the kinome despite its ability to inhibit kinases from diverse groups, and is thus a suitable starting point for further hit-to-lead development.
To the best of our knowledge, Ki8751 is the most potent PfPK6 inhibitor reported to date. With little known about the SAR on this series, we adopted a group efficiency (GE) approach to investigate which substituents on Ki8751 contribute to its potency. GE is a metric commonly used in fragment-based drug discovery to determine which substituents are most efficient in enhancing binding energy per heavy atom28. We hypothesized that through the comparison of the potencies of truncated analogues of Ki8751, we would be able to determine the efficiency of each group, and use this information to inform further optimization.
Removal of the 6-methoxy group (1) on the quinoline ring led to a 3-fold drop in potency against PfPK6 as compared to Ki8751 (Table 1), whereas removal of the 7-methoxy group (2) led to a drastic 15-fold drop in potency. Removal of both methoxy groups (3) resulted in a 34-fold drop in potency relative to Ki8751. These results demonstrate that both methoxy groups are important for PfPK6 inhibition, and the 7-methoxy group is especially efficient, with a GE of 0.81 ± 0.04 (Figure 2 and Figure S2). Interestingly, removal of the annulated phenyl ring from the quinoline of 3 to give pyridine 4 led to a less than 2-fold difference in potency against PfPK6. This suggests that the annulated phenyl ring is a low-contributor to PfPK6 inhibition, which is reflected in the low GE of 0.08 ± 0.03 for the four atoms that comprise the annulated phenyl ring. Compounds 5 and 6, where the pyridine of 4 was removed, were completely inactive against PfPK6, suggesting that the pyridine ring of Ki8751 is essential.
Table 1.
Truncated Analogues of Ki8751
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | PfPK6 activity remaining at 1 μM (%)a | PfPK6 IC50 (nM)b |
| Ki8751 |
|
F |
|
n.d. | 14 ± 1 |
| 1 |
|
F |
|
11 | 40 ± 9 |
| 2 |
|
F |
|
24 | 216 ± 21 |
| 3 |
|
F |
|
40 | 470 ± 80 |
| 4 |
|
F |
|
50 | 830 ± 90 |
| 5 | H | F |
|
100 | n.d. |
| 6 | Me | F |
|
100 | n.d. |
| 7 |
|
F |
|
1 | 1.3 ± 0.13c |
| 8 |
|
F |
|
1 | 2.0 ± 0.12c |
| 9 |
|
F |
|
1 | 1.3 ± 0.24c |
| 10 |
|
H |
|
6 | 18 ± 1.3 |
| 11 |
|
H |
|
4 | 9.0 ± 0.5 |
| 12 |
|
F |
|
28 | 310 ± 15 |
| 13 |
|
F |
|
26 | 240 ± 13 |
| 14 |
|
F | NH2 | 100 | n.d. |
| 15 |
|
H | NH2 | 95 | n.d. |
| 16 |
|
F | H | 84 | 4290 ± 420 |
Results presented as mean values from experiments using the KinaseSeeker assay performed in duplicate. Variability between individual values is less than 10%.
IC50 values were determined using the KinaseSeeker assay with 5-fold dilutions, presented as mean ± s.e.m. values of two experiments performed in duplicate.
IC50 values approach the kinase concentration in the assay. IC50s < 5 nM treated as equipotent. n.d. = not determined.
Figure 2. Group Efficiency (GE) Analysis of Ki8751 on PfPK6 inhibitiona.
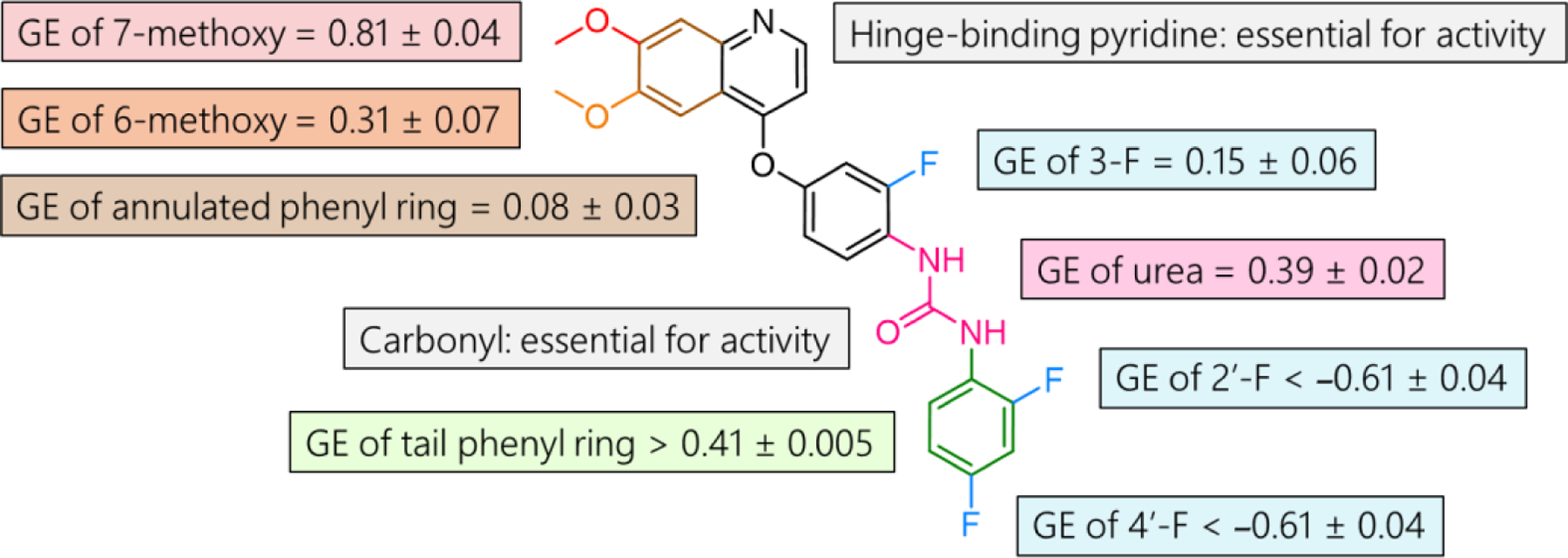
aGE = −ΔΔG / Δ(no. of heavy atoms)
We next investigated the roles of the three fluorine atoms on Ki8751. Removal of either or both of the ortho- or para-substituted fluorine atom on the terminal “tail” phenyl ring (7, 8, and 9) yielded compounds with at least a 3-fold increase in potency as compared to Ki8751. Since the IC50s determined for the most potent compounds approached the kinase concentration in the assay, it would not be prudent to quantitatively discriminate between IC50 values less than ~5 nM. Nevertheless, our results show that both fluorine atoms on the tail phenyl ring were detrimental towards activity against PfPK6. In comparison with the SAR on VEGFR2 and PDGFRα reported, the removal of both fluorine atoms afforded a 3.5-fold increase in potency on VEGFR2 and no change in potency on PDGFRα24. While replacement of the fluorine atom on the middle ring of Ki8751 with a hydrogen atom (10) led to a less than 2-fold difference in PfPK6 inhibition, the corresponding replacement on 9 to give 11 clearly decreased PfPK6 inhibition. We thus show that the fluorine atom in the middle ring is a modest contributor towards activity against PfPK6. In stark contrast, the removal of this fluorine atom did not change VEGFR2 potency and in fact increased PDGFRα potency 20-fold24. To improve selectivity over PDGFRα, we believe that this fluorine atom should be retained on the molecule.
Removal of the entire tail phenyl ring (12 and 13) increased the IC50 against PfPK6 by at least 2 orders of magnitude relative to 9, which demonstrates the importance of the tail phenyl ring, with a GE of >0.41 ± 0.005. Further removal of the carbonyl group of the urea (14 and 15) resulted in a complete loss of activity against PfPK6, which identifies the carbonyl group of Ki8751 as a second essential pharmacophoric element. Removal of the entire urea (16) gave a compound with micromolar activity against PfPK6, and we thus determined the GE of the entire urea group to be 0.39 ± 0.02.
While no crystal structure of PfPK6 is available, our results are consistent with a type II (DFG-out) binding mode to PfPK6. Ki8751 contains the essential pharmacophoric elements of a type II inhibitor29–31: a hydrogen bond acceptor to bind in the hinge region (the quinoline N1 atom), a hydrogen bond donor and acceptor pair 3–5 chemical bond lengths away (the urea) to bridge the cavity formed by the conserved αC-helix Glu and the DFG backbone NH, and lastly a hydrophobic group (the terminal phenyl ring) to occupy the back pocket created by the DFG-out flip. While it would be ideal to confirm the binding mode through structural studies, we were unable to perform docking studies in absence of a crystal structure of PfPK6. While structure prediction by AlphaFold is available, the structure predicted is of an active (DFG-in) state and is thus also unsuitable for docking. Future efforts to obtain a high-resolution crystal structure of PfPK6 in an inactive (DFG-out) state would be necessary to confirm this binding mode.
In summary, key contributors towards the potency of Ki8751 have been identified as both methoxy groups on the quinoline ring, the pyridine component of the quinoline ring, the urea, and the terminal “tail” phenyl ring. Our strategy to improve the potency of this scaffold would be to replace the two underperforming groups (Figure 3). The first group is the inefficient annulated phenyl ring on the pyridine, whereas the second group consists of the two fluorine atoms on the terminal phenyl ring detrimental for activity.
Figure 3.
SAR Plan
SAR of Hinge-Binding Region.
Based on an understanding of the type II pharmacophore, we hypothesize that the two methoxy groups are pointing towards the solvent-exposed region of the kinase ATP-binding pocket. Extension of the two methoxy groups to give a 6,7-bis(ethoxymethoxy)quinoline (19) demonstrated potency equivalent to Ki8751 (Table 2). This is consistent with our hypothesis and shows that this region of the quinoline is amendable for attachment of solubilizing substituents. To better understand the potency-enhancing effect of the two methoxy groups, analogues where the two methoxy groups were cyclized to a 5-membered ring (20) or a 6-membered ring (21) were synthesized and screened. Surprisingly, both 20 and 21 were found to be approximately 40-to-50-fold less potent than Ki8751. Their potencies were comparable to the potency of 3, where both methoxy groups were removed. This eliminates the possibility that the potency-enhancing effects of the two methoxy groups are due simply to their electron-donating effects, instead suggesting that specific interactions are formed between the methoxy groups and the residues surround the ATP-binding pocket. These results also suggest that, within the kinase pocket, the two methyl groups on the methoxy groups are either pointed away from each other in the plane of the quinoline ring, or pointed out of the plane of the quinoline ring, such that when constrained into the plane of the ring they lose their potency-enhancing effect. Our attempt at replacement of the highly-efficient 7-methoxy group with a 7-chloro group (17) was unsuccessful in maintaining its potency, demonstrating that the alkoxy groups at this position may be privileged groups for PfPK6 inhibition.
Table 2.
SAR of Hinge-Binding Heterocycles
| |||
|---|---|---|---|
| Compound | Ar | PfPK6 activity remaining at 1 μM (%)a | PfPK6 IC50 (nM)b |
| Ki8751 |
|
n.d. | 14 ± 1 |
| 3 |
|
40 | 470 ± 80 |
| 17 |
|
38 | 510 ± 160 |
| 18 |
|
86 | n.d. |
| 19 |
|
2 | 10 ± 0.5 |
| 20 |
|
28 | 680 ± 240 |
| 21 |
|
30 | 540 ± 80 |
| 22 |
|
6 | 19 ± 2 |
| 23 |
|
83 | n.d. |
| 24 |
|
100 | n.d. |
| 25 |
|
34 | 327 ± 17 |
| 26 |
|
61 | 1970 ± 240 |
| 27 |
|
43 | 800 ± 100 |
| 28 |
|
80 | n.d |
| 29 |
|
14 | 124 ± 12 |
| 30 |
|
54 | n.d |
| 4 |
|
50 | 830 ± 9 |
| 31 |
|
84 | n.d |
| 32 |
|
92 | n.d |
| 33 |
|
96 | n.d |
| 34 |
|
98 | n.d |
| 35 |
|
60 | n.d |
Results presented as mean values from experiments using the KinaseSeeker assay performed in duplicate. Variability between individual values is less than 10%.
IC50 values were determined using the KinaseSeeker assay with 5-fold dilutions, presented as mean ± s.e.m. values of two experiments performed in duplicate. n.d. = not determined.
We have previously established that the pyridine component of the quinoline ring is essential for PfPK6 inhibition. We thus aim to investigate other heterocycles, maintaining the relative position of the N1 atom of the quinoline ring in Ki8751. Isosteric replacements of the quinoline with thieno[3,2-b]pyridine (25) was tolerated by PfPK6, but the thieno[2,3-b]pyridine isomer (27) was less preferred. We hypothesized that the efficiency of the annulated ring may be enhanced by the formation of an additional hydrogen bond with the carbonyl group of the outer hinge residue of the kinase (hinge.48, based on numbering in KLIFS31). Indeed, replacing the quinoline with a 7-azaindole (29) led to a 4-fold improvement in potency, but this was still an order of magnitude less potent than the 6,7-dimethoxyquinoline group of Ki8751.
We next investigated the effect of introduction of additional nitrogen atoms on the pyridine ring. Introduction of a nitrogen atom at the 2-position, forming a cinnoline (24), led to a complete loss of potency, possibly due to disruption of a CH–O weak hydrogen bond with the backbone carbonyl of the inner hinge residue (hinge.46, based on numbering in KLIFS31) well-recognized in kinase inhibitors32,33. An alternate explanation would be the poorer hydrogen-bond accepting capability of the N1 atom in a cinnoline ring, as estimated by its lower pKa34. One consistent trend we have also observed was that addition of a 3-position nitrogen atom was generally detrimental towards PfPK6 inhibition. Introduction of a nitrogen atom at the 3-position of the quinoline, forming a quinazoline (23), or the addition of a nitrile group at the same position (18) both led to a drastic drop in potency. In some kinases, a hydrogen bond acceptor at this position is known to form a hydrogen bond with either the gatekeeper residue or with a water molecule present, and a nitrile at this position may displace a water molecule the nitrogen atom is hydrogen bonded to35, but this is evidently not applicable to PfPK6. Our results are consistent with the knowledge that the gatekeeper of PfPK6 is a Phe residue. Thus, this region of the ATP-binding site may be lipophilic and the lack of productive hydrogen bonding partners was expected. Interestingly, the 6,7-dimethoxyquinazoline (22), with an IC50 of 19 nM, also possesses comparable potency to Ki8751. This observation further highlights the importance of the two methoxy groups for PfPK6 inhibition, which could almost fully compensate for the detrimental 3-position N atom. We also note that the 6,7-dimethoxyquinoline hinge-binding group led to a ≤2-fold change in potency in VEGFR2 and PDGFRα24, suggesting that selectivity for PfPK6 over the VEGFR2 and PDGFRα cannot be attained with this substitution. Similar to 23, incorporation of a nitrogen atom at the equivalent positions of thieno[3,2-b]pyridine 25, thieno[2,3-b]pyridine 27, 7-azaindole 29 and pyridine 4, to form compounds thieno[3,2-d]pyrimidine 26, thieno[2,3-d]pyrimidine 28, pyrrolo[2,3-d]pyrimidine 30, and pyrimidine 31 respectively, was similarly detrimental towards activity against PfPK6.
Additionally, attempts at the removal of the annulated phenyl ring completely and replacing it with a chloro substituent (32, 33 and 34) were found not to be tolerated by PfPK6, while addition of a benzylamino substituent on the 6-position of the pyrimidine (35) resulted in only a modest improvement in PfPK6 potency over 31.
Our group has previously shown that the configuration of atoms on heterocycles bearing the hinge-binding N atom influences its pKa, which is paramount for effective ligand-kinase molecular recognition36. Using density functional theory (DFT)37–39, we predicted the pKa for each hinge-binding N atom on heterocycles tested (Table S3). As expected, we observed a general trend where increasing pKa of the hinge-binding N atom is correlated with increasing potency against PfPK6 (Figure S3). This is exemplified by the electron-withdrawing 3-position nitrile group of 18, which was predicted to decrease the pKa of the hinge-binding N1 atom to 1.20, a notable decrease as compared to the quinoline group of 3, with a predicted pKa of 5.54. This offers another explanation to the decreased potency of 18. There are a few outliers to this general trend, suggesting that the pKa of the hinge-binding N atom is not the sole determinant of potency on PfPK6; interactions between the kinase active site residues and substituents on the heterocycle, such as the above-discussed two methoxy groups, also contribute to their activity. Nevertheless, our results show that the basicity of the hinge-binding N atom is an important contributor to PfPK6 inhibition.
To sum up the SAR for the hinge-binding region, we have observed modest improvements in potency as compared to quinoline 3 by attempting to replace the annulated phenyl ring. We have also found that incorporation of a 3-position nitrogen atom or nitrile group is disfavored. Comparing analogues varying the methoxy groups have also led us to conclude that the two methoxy groups are essential for activity and the specific 3-dimensional orientation of the methoxy groups are critical for PfPK6 inhibition. With this in mind, we find that the 6,7-dimethoxyquinoline group of Ki8751 appears to be the optimal hinge binder for this chemotype, and would be used in our further optimization process.
Incorporation of CF3 and Piperazines at the Tail Group.
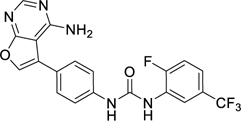
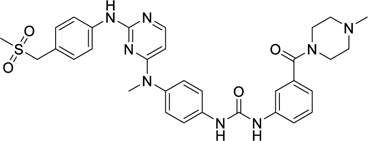
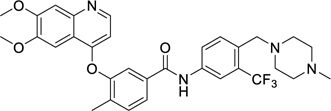
When developing Ki8751 for VEGFR2 inhibition, it was noted that substituents on the tail phenyl ring generally hardly had any influence on VEGFR2 inhibition, although only modest changes were attempted24. To inform our decisions regarding suitable replacements of the two detrimental fluorine atoms on Ki8751 for PfPK6 inhibition, we drew inspiration from three other type II inhibitors in our screen (Table 3). Compounds 36 and 37 were from the Kinase Chemogenomics Set (KCGS), a set of narrow-spectrum kinase inhibitors our group has developed and publicly-released40, while compound 38 was a member of an internal library of type II inhibitors developed based on literature compounds41,42. Compounds 36, 37, and 38 were active against PfPK6 with IC50 values between 5–80 nM. These compounds also showed antiplasmodial activity against the asexual blood stage proliferation of P. falciparum 3D7 in a SYBR Green I-based fluorescence assay43 with EC50s between 390–500 nM. To benchmark the EC50s we have obtained, we compared them to the EC50s of known antimalarial drugs (none of which are known to be type II kinase inhibitors) we have measured here and previously reported using the same assay44. Compounds 36-38 were at least an order of magnitude less potent than dihydroartemisinin (DHA) (EC50 = 0.14 nM) (Table 3), pyrimethamine (EC50 = 27 nM)44 and quinacrine (EC50 = 33 nM)44, which is unsurprising considering these were unoptimized hits. Our assay results for DHA are consistent with literature values45.
Table 3.
Other inhibitors with a Type II pharmacophore benchmarked against dihydroartemisinin (DHA)
| Compound | Structure | PfPK6 IC50 (nM)a | Pf3D7 blood stage EC50 (nM)b |
|---|---|---|---|
| DHA |
|
n.d. | 0.14 ± 0.04 |
| 36 |
|
5.3 ± 0.2 | 500 ± 68 |
| 37 |
|
8.5 ± 0.2 | 390 ± 10 |
| 38 |
|
76 ± 4 | 410 ± 28 |
IC50 values were determined using the KinaseSeeker assay with 5-fold dilutions, presented as mean ± s.e.m. values of two experiments performed in duplicate.
EC50 values were determined using the SYBR Green I-based assay with 2-fold dilutions, presented as mean ± s.e.m. values performed in triplicate. n.d. = not determined
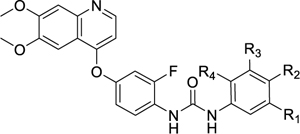
While these three type II inhibitors have different hinge-binding moieties and linker region to their tail group, they possess CF3 (36, 38) or piperazine substituents (37, 38) on the tail group. The CF3 substituent is found at the meta-position (relative to the urea or amide group of the linker) which we hypothesize to be binding in BP-III (using the KLIFS notation31). The piperazine may be found at the meta- or para-positions, linked with a methylene or carbonyl linker, which we hypothesize to bind in BP-V (using the KLIFS notation31). It is clear that these substituents are accepted by PfPK6. We thus took inspiration from these three compounds and incorporated the CF3 and piperazine substituents onto the 1-phenyl-3-(4-(quinolin-4-yloxy)phenyl)urea scaffold of Ki8751. These analogues were then screened for both PfPK6 inhibition and antiplasmodial activity against P. falciparum asexual blood stage parasites (Table 4).
Table 4.
Incorporating CF3 and piperazines to tail group of Ki8751
| ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | PfPK6 IC50 (nM)a | Pf3D7 blood stage EC50 (nM)b |
| Ki8751 | H | F | H | F | 14 ± 1 | n.d. |
| 39 | CF3 | H | H | H | 0.84 ± 0.04c | 540 ± 24 |
| 40 | CF3 | H | H | F | 1.2 ± 0.07c | Inactived |
| 41 | H |
|
H | H | 13 ± 0.5 | 580 ± 65 |
| 42 | H | H |
|
H | 23 ± 1 | Inactivee |
| 43 | H |
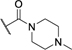
|
H | H | 321 ± 21 | Inactivee |
| 44 | H | H |
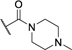
|
H | 68 ± 3 | Inactivee |
| 45 | CF3 |
|
H | H | 1.9 ± 0.1c | 110 ± 4 |
| 46 | CF3 | H |
|
H | 0.81 ± 0.02c | 410 ± 23 |
| 47 | CF3 |
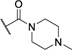
|
H | H | 21 ± 1 | Inactivee |
| 48 | CF3 | H |
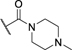
|
H | 0.29 ± 0.02c | Inactivee |
IC50 values were determined using the KinaseSeeker assay with 5-fold dilutions, presented as mean ± s.e.m. values of two experiments performed in duplicate.
EC50 values were determined using the SYBR Green I-based assay with 2-fold dilutions, presented as mean ± s.e.m. values performed in triplicate.
IC50 values approach the kinase concentration in the assay. IC50s < 5 nM treated as equipotent.
No inhibition when tested up to 5 μM in two experiments, testing in dose-response manner.
No inhibition when tested in triplicate at a single concentration of 1 μM. n.d. = not determined.
The incorporation of a CF3 at the meta-position was successful and we obtained compounds with IC50s < 5 nM against PfPK6 (39, 40). Since these compounds have IC50 values approaching the kinase concentration in the assay, we could not quantitatively compare their potency against PfPK6, and hence we elected to assess them by their antiplasmodial activity. In parasite assays, compound 39 was active against P. falciparum blood stage parasites with an EC50 of 540 nM while 40 did not show any inhibition up to 5 μM, suggesting that the incorporation of an ortho-fluorine group is not preferred.
Incorporation of a piperazine with a methylene linker at the para-position afforded compound 41, which was as potent as Ki8751 on PfPK6 and possesses activity against the P. falciparum blood stage with an EC50 of 580 nM. Shifting the piperazine to the meta-position (42) resulted in a less than 2-fold change in PfPK6 inhibition but loss of P. falciparum blood stage activity when tested at 1 μM. Replacement of the methylene with a carbonyl linker to either the para- (43) or meta-positions (44) was less well-tolerated, with a 5-to-25-fold drop in activity against PfPK6 as compared to 41 and, consistent with the reduction in PfPK6 inhibitory potency, we did not observe antiplasmodial activity at 1 μM for either of these compounds. It appears that the para-substituted piperazine with a methylene linker is preferred. The sensitivity of PfPK6 towards this linker merits further investigation (vide infra).
We next combined the meta-CF3 group with the piperazine-containing analogues. It was striking to observe an additive effect of both groups on both PfPK6 inhibition as well as P. falciparum blood stage inhibition. Combining elements of compounds 39 and 41, compound 45 saw an improvement of potency against PfPK6 by an order of magnitude relative to 41, as well as improved potency against P. falciparum blood stage parasites from both 39 and 41 by 5-fold to an EC50 of 110 nM. This one-order-of-magnitude improvement in PfPK6 inhibition was also observed upon addition of a meta-CF3 group to the other piperazine-containing analogues (46, 47 and 48, comparing with 42, 43, and 44, respectively). 46 has also demonstrated antiplasmodial activity with an EC50 of 410 nM, an improvement over 42.
The antiplasmodial activity of compound 45 encouraged us to investigate its activity against the other ten P. falciparum kinases in our KinaseSeeker assay panel (Figure 4). Inhibition of four other P. falciparum kinases, PfCDPK5, PfGSK3, PfNEK3, and PfPKB with IC50 values between 40 and 144 nM were observed. Of these targets, both PfCDPK5 and PfGSK3 have been determined to be essential for the asexual blood stage of P. falciparum through both reverse genetics and saturation mutagenesis studies13,14, whereas PfNEK3 was shown to be non-essential13 or could be disrupted, albeit with a fitness penalty14. Interestingly, while the reverse genetics study found PfPKB to be essential13, the study using saturation mutagenesis found that it could be disrupted without fitness penalty14. Our results demonstrate that 45 likely exerts its antiplasmodial activity through inhibition of multiple essential P. falciparum kinases. Polypharmacology has many advantages for antimalarial drug development, as it reduces the likelihood of resistance, provides opportunities for multistage intervention, and may inhibit enzymes of bypass pathways that would otherwise be able to compensate for inhibition of a single target9,46.
Figure 4. Inhibition of compound 45 on the P. falciparum Kinase Panel.
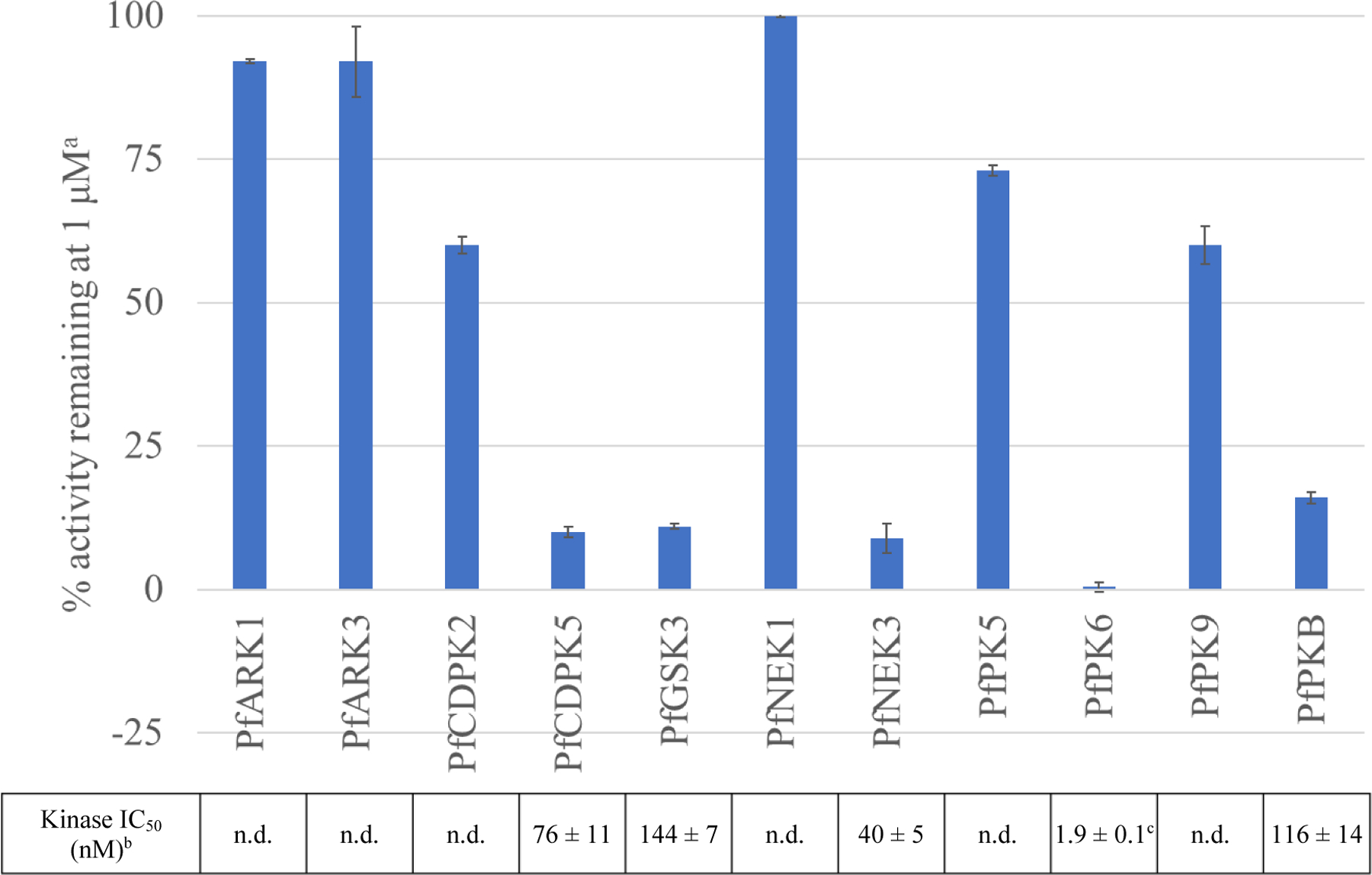
aResults presented as mean ± s.d. values from experiments using the KinaseSeeker assay performed in duplicate. bIC50 values were determined using the KinaseSeeker assay with 5-fold dilutions, presented as mean ± s.e.m. values of two experiments performed in duplicate. cIC50 values approach the kinase concentration in the assay. IC50s < 5 nM treated as equipotent. n.d. = not determined.
However, being able to inhibit five out of the 11 kinases in our P. falciparum kinase panel, across kinases from diverse groups and families, suggests that this compound may be promiscuous and has substantial risk of off-target effects in the human kinome. We thus screened compound 45 at 1 μM against a panel of 468 kinases (403 non-mutant kinases) in the DiscoverX KINOMEscan® profiling service. As expected, 45 potently inhibited 134 non-mutant kinases with ≤1% control, with a selectivity score S1 (1 μM) of 0.33 (Table S4 and Figure S4). Additionally, from the DiscoverX KINOMEscan® profiling, we have found that 45 binds to PfCDPK1 (1.6% control at 1 μM), another essential kinase for P. falciparum blood stage proliferation13,14 that 45 could possibly exert antiplasmodial activity through its inhibition. This finding demonstrates that the rules for improving inhibition against human kinases can be leveraged for inhibition against Plasmodium kinases, at least within this chemotype, adding to the body of evidence that Plasmodium kinases are a druggable target class9. However, like designing kinase inhibitors as treatments for other indications, selectivity between kinases still remains a significant challenge.
We evaluated 45 and other analogues active against P. falciparum blood stage proliferation for cytotoxicity against HepG2 cells, a human hepatoma cell line, using a commercially available CellTiter-Glo assay (Promega) (Table 8). This assay is a commonly used proxy for cytotoxicity against human cells, and has been previously used to evaluate cytotoxicity amongst the antimalarial hits from the TCAMS47. 45 demonstrates only a modest cytotoxic effect against HepG2 cells with a cytotoxicity CC50 of 1.00 μM. This only affords a suboptimal 9-fold window between the antiplasmodial EC50 and the cytotoxicity CC50. This is a promising start for optimization, where we aim to improve the cytotoxicity window of 45.
Table 8.
Antiparasitic Effect on P. berghei Liver Stage Parasite Load, HepG2 Cytotoxicity, and Pharmacokinetic Parameters of Key Compounds
| Compound | HepG2 cytotoxicity CellTiter-Glo CC50 (μM)a | P. berghei liver stage EC50 (μM)b | HepG2 cytotoxicity CellTiter-Fluor CC50 (μM)c | Solubility (μM)d | PAMPA Papp (10−6 cm/s)e | Human Liver Microsomal Stability (% remaining after 30 min)f | Mouse Liver Microsomal Stability (% remaining after 30 min)f |
|---|---|---|---|---|---|---|---|
| Ki8751 | n.d. | n.d. | n.d. | 0.7 | 24.2 | n.d. | n.d. |
| 45 | 1.00 ± 0.24 | n.d. | n.d. | 2.1 | 2.57 | n.d. | n.d. |
| 67 | 4.39 ± 0.17 | inactiveg | 3.29 ± 0.28 | 10.9 | <0.669h | 95.9 | 85.8 |
| 68 | 2.57 ± 0.05 | inactiveg | 6.62 ± 1.32 | 7.4 | <0.709h | 88.6 | 86.7 |
| 75 | 1.00 ± 1.00 | inactiveg | 0.41 ± 0.18 | 1.5 | <0.513h | 88.6 | 69.7 |
| 79 | 1.29 ± 1.34 | 0.22 ± 0.05 | 1.29 ± 0.49 | 2.1 | 0.734 | 98.1 | 88.9 |
CC50 values were presented as mean ± s.d. values from 2–3 experiments using the CellTiter-Glo assay, each performed in quadruplicate.
EC50 values were presented as mean ± s.d. values from two experiments, each performed in triplicate.
CC50 values were presented as mean ± s.d. values from two experiments using the CellTiter-Fluor assay, each performed in triplicate.
Kinetic solubility analysis was carried out in phosphate buffered saline solution (PBS) at pH 7.4 from 10 mM DMSO stock solutions.
Permeability analysis was performed using a chamber separated by an immobilized artificial phospholipid membrane layer into a donor and an acceptor well.
Microsomal stability analysis were performed using a test compound concentration of 5 μM, microsomal protein concentration of 0.5 mg/mL, NADPH concentration of 7–9 mM.
EC50 not reported because we observed a <5-fold difference between observed EC50 and cytotoxicity in host HepG2 cells using the CellTiter-Fluor assay.
Below limit of quantitation in acceptor well. n.d. = not determined.
SAR on the Tail Phenyl Ring.
In an attempt to discover compounds which are less promiscuous but still maintained activity against PfPK6 and against P. falciparum blood stage proliferation, we next investigated if the meta-CF3 group of 39 and the para-piperazine group of 41 could be replaced with alternative substituents. We hypothesized that the additive effect on PfPK6 inhibition and inhibition of P. falciparum blood stage proliferation observed for the meta-CF3 and para-piperazine would be similarly observed for the optimized substituents. Hence, we investigated these alternative substituents at the meta- and para-positions separately before combining the most promising meta- and para-position substituents onto the scaffold.
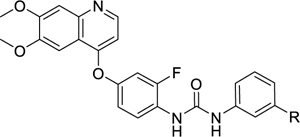
In the optimization of the meta-position substituent, we note that the introduction of meta-CF3, chloro, methyl and methoxy groups have been attempted previously for VEGFR2 and PDGFRα inhibition24,48. As compared to the unsubstituted tail phenyl ring, the meta-methyl and methoxy groups generally maintained potency, but the meta-CF3 led to a 2.5-fold drop in PDGFRα inhibition48 while the meta-chloro group led to a 7-to-8-fold drop in potency against both kinases24. In attempts to replace the meta-CF3 of 39 with halogens, nitrile, alkyl, and alkoxy groups of different sizes (49–58), we found this entire set of analogues to have IC50 < 5 nM for PfPK6 inhibition. Under our assay conditions, we were unable to distinguish which, if any, substituent is preferred (Table 5). This suggests that this pocket of PfPK6 is non-discriminatory and tolerates both small aliphatic hydrophobic groups and groups containing hydrogen bond acceptors reasonably. The two substituents with a submicromolar EC50 against the P. falciparum blood stage were the CF3 (39) and OCF3 (57) groups. Substitution of the CF3 with a chloro (49), methyl (51) or tert-butyl substituent (53) led to a 4-to-6-fold drop in potency against P. falciparum blood stage parasites. Compounds with other substituents were inactive against P. falciparum blood stage parasites when screened at 1 μM. The finding that both the CF3 and OCF3 groups were the most promising substituents suggests that some unique property of the trifluoromethyl group is responsible for activity.
Table 5.
SAR of the meta-position substituent at the tail group
| |||
|---|---|---|---|
| Compound | R | PfPK6 IC50 (nM)a | Pf3D7 blood stage EC50 (nM)b |
| 39 | CF3 | 0.84 ± 0.04c | 540 ± 24 |
| 49 | Cl | 1.0 ± 0.08c | 2190 ± 201 |
| 50 | Br | 1.1 ± 0.03c | Inactived |
| 51 | Me | 1.2 ± 0.07c | 3010 ± 234 |
| 52 | iPr | 1.1 ± 0.07c | Inactived |
| 53 | tBu | 2.1 ± 0.08c | 2520 ± 106 |
| 54 | OMe | 1.7 ± 0.13c | Inactived |
| 55 | OiPr | 1.5 ± 0.12c | Inactived |
| 56 | OCHF2 | 0.44 ± 0.05c | Inactived |
| 57 | OCF3 | 1.2 ± 0.06c | 800 ± 15 |
| 58 | CN | 1.8 ± 0.07c | Inactived |
IC50 values were determined using the KinaseSeeker assay with 5-fold dilutions, presented as mean ± s.e.m. values of two experiments performed in duplicate.
EC50 values were determined using the SYBR Green I-based assay with 2-fold dilutions, presented as mean ± s.e.m. values performed in triplicate.
IC50 values approach the kinase concentration in the assay. IC50s < 5 nM treated as equipotent.
No inhibition when tested in triplicate at a single concentration of 1 μM.
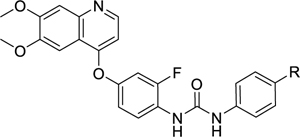
Removal of the methylene linker between the tail phenyl ring and the piperazine of 41 (compound 59) led to a 30-fold drop in potency against PfPK6 (Table 6), suggesting that the linker atom is crucial to position the piperazine in a suitable orientation within the binding site. Surprisingly, the antiplasmodial activity of 59 was similar to 41. To better understand which basic nitrogen atom of the piperazine group contributes to the activity of 41, we first removed the tertiary amine distal to the tail phenyl ring by replacing the N-methylpiperazine of 41 with a morpholine (60), piperidine (61), pyrrolidine (62), or diethylamine (63). These changes led to a 10-to-25-fold drop in potency against PfPK6, which informs us that the basic nitrogen distal to the tail phenyl ring is important for PfPK6 inhibition. In contrast, we observed a <2-fold change in antiplasmodial activity with the piperidine (61), pyrrolidine (62), and diethylamine (63) analogues, but the analogue with a morpholine (60) was inactive at 1 μM. Because the pKa of the tertiary amine in 60 would be lower due to the electron-withdrawing oxygen of the morpholine, this suggests that having at least one sufficiently-basic nitrogen atom in this ring is important for inhibition of the P. falciparum blood stage.
Table 6.
SAR of the para-position substituent at the tail group
| |||
|---|---|---|---|
| Compound | R | PfPK6 IC50 (nM)a | Pf3D7 blood stage EC50 (nM)b |
| 41 |
|
13 ± 0.5 | 580 ± 65 |
| 59 |
|
390 ± 30 | 630 ± 13 |
| 60 |
|
135 ± 8 | Inactivec |
| 61 |
|
310 ± 13 | 250 ± 7 |
| 62 |
|
220 ± 40 | 270 ± 41 |
| 63 |
|
138 ± 14 | 320 ± 13 |
| 64 |
|
26 ± 2 | 330 ± 11 |
| 65 |
|
150 ± 30 | 660 ± 15 |
| 66 |
|
17 ± 3 | 330 ± 5 |
| 67 |
|
13 ± 0.9 | 160 ± 2 |
| 68 |
|
29 ± 3 | 140 ± 4 |
IC50 values were determined using the KinaseSeeker assay with 5-fold dilutions, presented as mean ± s.e.m. values of two experiments performed in duplicate.
EC50 values were determined using the SYBR Green I-based assay with 2-fold dilutions, presented as mean ± s.e.m. values performed in triplicate.
No inhibition when tested in triplicate at a single concentration of 1 μM.
When comparing compounds 41 and 43, the reasons for a preference for a methylene linker over a carbonyl linker to the piperazine were not clear. Possible reasons include steric clash of the carbonyl oxygen atom with the kinase residues around the binding pocket, the incompatibility of hydrogen bond acceptors at this position, conformational restriction of the piperazine ring, or a change from a positively-charged tertiary amine to a neutral amide group. To deconvolute these factors, we replaced the piperazine in 41 with piperidine analogues with alternative linkers (64–68). While oxygen (64), N-methyl (66), and carbon linkers (67, 68) were tolerated by PfPK6, the NH linker (65) was less preferred and led to a 12-fold decrease in inhibition. This suggests that the binding pocket of PfPK6 in the vicinity of this linker is hydrophobic and is unable to compensate for the loss of solvation energy of hydrophilic linkers upon ligand-target binding. Taken together, this suggests that the loss of potency against PfPK6 observed with the carbonyl linker previously (43) was due to the increase in polarity at this position. The argument for steric clash of the carbonyl oxygen atom was ruled out based on the acceptable potency with the similarly-sized N-methyl linker (66) while conformational restriction of the piperazine ring was shown to be tolerable in the case of the isosteric alkene linker (68). The antiplasmodial activity of 64, 65 and 66 are within a 2-fold difference of 41, whereas 67 and 68 possess an approximately 4-fold improvement in P. falciparum blood stage inhibitory activity, with EC50s of 160 and 140 nM respectively. These analogues (64–68) also demonstrate that the basic amine proximal to the tail phenyl ring is neither essential for PfPK6 inhibition nor P. falciparum blood stage inhibition, as long as there is a second basic amine distal to the tail phenyl ring.
Finally, we have combined the meta- and para-position groups of most active compounds against the P. falciparum blood stage (Table 7). Gratifyingly, all compounds were active against PfPK6 and against P. falciparum blood stage proliferation. Compounds 69–74, where either a CF3 or a OCF3 group was added to compounds 61–63, saw an 8-to-26-fold improvement in PfPK6 inhibition but were equally potent with their parent compounds against the P. falciparum blood stage. In contrast to compounds 69-74, an additive effect against both PfPK6 inhibition and P. falciparum blood stage inhibition was instead observed for the other compounds with a basic amine distal to the phenyl ring (75-82). All these compounds are extremely potent against PfPK6, with IC50 values < 5 nM. This approached the kinase concentration in the assay and thus we were unable to provide an exact measurement for the degree of improvement in PfPK6 activity. Nevertheless, at minimum, a 3-to-6-fold improvement in PfPK6 inhibition was observed in all cases. With the addition of a CF3 group onto 64, an 8-fold improvement in potency against P. falciparum blood stage inhibition was additionally observed for 75, affording a potent antimalarial with an EC50 of 40 nM. The OCF3 addition (76) afforded a more modest 4-fold improvement. Similarly, addition of a CF3 group to 67 yielded 79, which demonstrates potent antiplasmodial activity with an EC50 of 39 nM, a 4-fold improvement over 67. The OCF3 analogue (80) only led to a 2-fold improvement. Addition of either a CF3 or OCF3 group to 68 afforded a 2-fold improvement in activity (81, 82). Similarly, the addition of a CF3 group to 66 gave a modest 2-fold improvement in antiplasmodial activity (77), but addition of an OCF3 group only maintained potency (78). Overall, our hypothesis on additive effects between the meta- and para-position substituents was fruitful in finding compounds that possessed enhanced activity against PfPK6 and against P. falciparum blood stage parasites.
Table 7.
SAR combining optimal meta- and para-position substituents
| ||||
|---|---|---|---|---|
| Compound | R1 | R2 | PfPK6 IC50 (nM)a | Pf3D7 blood stage EC50 (nM)b |
| 69 | CF3 |
|
12 ± 1 | 490 ± 15 |
| 70 | OCF3 | 14 ± 2 | 310 ± 14 | |
| 71 | CF3 |
|
27 ± 3 | 330 ± 13 |
| 72 | OCF3 | 12 ± 1 | 230 ± 5 | |
| 73 | CF3 |
|
8.1 ± 0.8 | 300 ± 3 |
| 74 | OCF3 | 14 ± 2 | 270 ± 7 | |
| 75 | CF3 |
|
1.9 ± 0.09c | 40 ± 0.7 |
| 76 | OCF3 | 3.1 ± 0.1c | 75 ± 1.8 | |
| 77 | CF3 |
|
1.9 ± 0.1c | 150 ± 12 |
| 78 | OCF3 | 1.8 ± 0.1c | 250 ± 11 | |
| 79 | CF3 |
|
1.6 ± 0.3c | 39 ± 0.3 |
| 80 | OCF3 | 2.8 ± 0.3c | 82 ± 0.4 | |
| 81 | CF3 |
|
2.0 ± 0.2c | 84 ± 1.6 |
| 82 | OCF3 | 2.7 ± 0.2c | 84 ± 0.3 | |
IC50 values were determined using the KinaseSeeker assay with 5-fold dilutions, presented as mean ± s.e.m. values of two experiments performed in duplicate.
EC50 values were determined using the SYBR Green I-based assay with 2-fold dilutions, presented as mean ± s.e.m. values performed in triplicate.
cIC50 values approach the kinase concentration in the assay. IC50s < 5 nM treated as equipotent.
Characterization of 67, 68, 75, and 79.
With potent antimalarials (75 and 79) on hand, we next sought to measure the cytotoxicity of these compounds and evaluate if we have improved the therapeutic window compared to 45. We screened compounds that have antiplasmodial effect for cytotoxicity of HepG2 cells using the CellTiter-Glo assay (Table S1), and found that compounds 75 and 79 exhibit cytotoxic effects with CC50s values of 1.00 μM and 1.29 μM respectively (Table 8), demonstrating a 25-fold and 33-fold selectivity window, respectively, for antiplasmodial activity against the P. falciparum asexual blood stage over HepG2 cytotoxicity. In our cytotoxicity screen, two additional compounds stood out with a good balance of antiplasmodial activity and HepG2 cytotoxicity. Compound 67 and 68 have HepG2 cytotoxicity CC50 values of 4.39 μM and 2.57 μM, respectively, while also having good antiplasmodial activity of 160 and 140 nM, respectively. This affords 67 a 27-fold therapeutic window and 68 an 18-fold therapeutic window. While 67 has a similar therapeutic window as compared to 75 and 79, 68 has a slightly lower therapeutic window than 75 and 79. Both 67 and 68 are still considerable improvements over 45. Our results demonstrate that this scaffold has potential to be optimized for specificity against Plasmodium over human targets.
Ideally, an antimalarial drug would fulfill multiple target candidate profiles (TCPs) as outlined by the Medicines for Malaria Venture (MMV)49. We thus investigated if our compounds demonstrate antiproliferative activity against other life stages of the Plasmodium life cycle. To this end, we tested 67, 68, 75, and 79 for activity using a common liver stage model system. Specifically, compounds were tested for activity against P. berghei ANKA parasite load in HepG2 cells50. Due to high sequence similarity between PfPK6 and P. berghei PK6 (81.9% similarity), we hypothesized that these compounds could demonstrate antiproliferative effects against P. berghei. As a counter-screen for host cell cytotoxicity, we screened these compounds in parallel in a cell viability assay in HepG2 cells with the CellTiter-Fluor assay. Our results from the CellTiter-Fluor assay generally agree with the results from our CellTiter-Glo assay, with 75 exhibiting a slightly greater cytotoxic response in the CellTiter-Fluor assay. For 67, 68 and 75, we observed some antiplasmodial effect against P. berghei liver stage, but with only a 1.4-, 1.9- and 1.9-fold difference between the P. berghei EC50 and the CellTiter-Fluor CC50 respectively. Therefore, they likely exert their antiplasmodial effect via host cell cytotoxicity. In contrast, compound 79 was observed to be active against P. berghei liver stage with an EC50 of 0.22 μM (Table 8) and possesses a 5.9-fold window of selectivity between the P. berghei EC50 and the CellTiter-Fluor CC50, suggesting that its antiplasmodial activity may be attributed to parasite-specific toxicity. Compound 79 is thus a promising antiplasmodial lead compound with dual-stage activity against the P. falciparum blood stage and P. berghei liver stage.
We next obtained in vitro pharmacokinetic parameters for key compounds (Table 8). With our modifications to the tail group, 67 possesses the greatest solubility of the compounds tested (10.9 μM), a noteworthy improvement over Ki8751 (0.7 μM) and 45 (2.1 μM) but still leaves room for improvement. This increase in solubility came with a trade-off of decreased permeability. While the PAMPA assay we used does not fully model permeability into the parasite, the low permeability of these compounds of this series across lipid bilayers could be a factor for the disconnect between the in vitro kinase inhibitory activity and the activity against the parasite, especially considering that three membrane barriers (host cell membrane, parasitophorous vacuolar membrane, parasite plasma membrane) lay between the extracellular environment and the parasite cytoplasm. We hence envision that improvement in the permeability of compounds of this series during future optimization might be the key to improve cellular activity. Compounds 67, 68, 75, and 79 also demonstrate good microsomal stability in human liver microsomes (88.6%−98.1% remaining after 30 min of incubation) and mouse liver microsomes (69.7%−88.9% remaining after 30 min of incubation). These pharmacokinetic data support further development of these compounds into novel antimalarial lead compounds, although future efforts would be required to address the permeability liabilities.
To establish the extent of possible off-targets in the human kinome, we screened compounds 67 and 79 in the DiscoverX KINOMEscan® profiling service. Compound 67 and 79 inhibited 22 and 97 non-mutant kinases with ≤1% control, respectively, with a selectivity score S1 (1 μM) of 0.055 and 0.24 respectively (Table S4, Figures S5 and S6). As a point of reference, we also profiled Ki8751 in the DiscoverX KINOMEscan® profiling service and found Ki8751 to inhibit 16 non-mutant kinases with ≤1% control, with a selectivity score S1 (1 μM) of 0.040 (Table S4, Figure S7). This demonstrates that 67 has maintained a similar degree of promiscuity of Ki8751 in the human kinome, while being a considerable improvement over 45. 79 also demonstrates a slight improvement over 45.
From the KINOMEscan® profiling, PfCDPK1 was also identified as a target for 79 (1.1% control at 1 μM) (Table 9), and thus PfCDPK1 may be one other target through which 79 may exert its potent antiplasmodial activity. To investigate alternative targets in the Plasmodium kinome that could contribute to the antimalarial efficacy of compounds 67, 68, 75, and 79, we screened them against the other ten P. falciparum kinases in our kinase panel in the KinaseSeeker assay (Table 9). Notably, consistent with our results from the DiscoverX KINOMEscan® profiling, 67 and 79 do not inhibit PfPK5 in both orthogonal assay formats. In our kinase panel, apart from PfPK6, 67 and 68 only weakly inhibits PfPKB, suggesting that their antiplasmodial activity is primarily exerted through inhibition of PfPK6, although this does not rule out inhibition of other targets beyond those screened by our efforts. On the other hand, compounds 75 and 79 demonstrate inhibition of PfCDPK5, PfGSK3, PfNEK3, and PfPKB, similar to 45. The potency of 75 and 79 against the P. falciparum blood stage could thus be attributed to polypharmacology. We also acknowledge the possibility that these compounds exert their antiplasmodial activity through other targets beyond those screened by our efforts. Further studies such as the evaluation of our compounds in inducible PfPK6 knockdown parasite lines or cellular thermal shift assays would be required to validate if PfPK6 is the primary target of these compounds in living parasites.
Table 9.
Inhibition of compound 67, 68, 75, and 79 on other P. falciparum kinases
| % Activity remaining at 1 μM (IC50)a | % control at 1 μMb | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PfARK1 | PfARK3 | PfCDPK2 | PfCDPK5 | PfGSK3 | PfNEK1 | PfNEK3 | PfPK5 | PfPK6 | PfPK9 | PfPKB | PfCDPK1 | PfPK5 | |
| 67 | 97 | 100 | 88 | 91 | 97 | 100 | 82 | 97 | 2 (13 nM) | 99 | 30 | 51 | 84 |
| 68 | 98 | 100 | 96 | 98 | 100 | 100 | 84 | 100 | 1 (29 nM) | 100 | 38 | n.d. | n.d. |
| 75 | 94 | 97 | 78 | 25 | 30 | 100 | 6 | 94 | 0 (1.9 nM)c | 79 | 20 | n.d. | n.d. |
| 79 | 100 | 87 | 64 | 27 | 34 | 100 | 7 | 94 | 1 (1.6 nM)c | 89 | 21 | 1.1 | 80 |
Results presented as mean values from experiments using the KinaseSeeker assay performed in duplicate. Variability between individual values is less than 10%. IC50 values were determined using the KinaseSeeker assay with 5-fold dilutions, presented as mean values of two experiments performed in duplicate.
Results from DiscoverX KINOMEscan® profiling service.
IC50 values approach the kinase concentration in the assay. IC50s < 5 nM treated as equipotent. n.d. = not determined.
In general, we observe some disconnect between the SAR on PfPK6 inhibition and antiplasmodial activity for these analogues. This may be due to a multitude of factors. Low solubility could hinder detection of activity in cellular assays as poorly-soluble compounds could fail to achieve sufficient concentration for activity. Low permeability could result in insufficient intracellular concentration for target inhibition. The different physiochemical environment of PfPK6 and its binding partners in our in vitro kinase assay and in living parasites, which are not known, may be another factor for this disconnect. While we demonstrate that lead compound 67 primarily inhibits PfPK6 amongst the kinases screened, lead compound 79 additionally inhibits PfCDPK1, PfCDPK5, PfGSK3, PfNEK3, and PfPKB. It is possible that 67 and 79, as well as other analogues in this series, target other kinases beyond these screened by our efforts and could have additional mechanisms of action. It is noteworthy that the most active compounds of this series against the P. falciparum blood stage have PfPK6 IC50s < 5 nM (Figure S8), which suggests that that PfPK6 is at least one of the targets they act upon. Possibly, different extents of compound retention within intracellular compartments may also play a role in the disconnect of SAR observed. A sufficiently-basic amine in these analogues may be important for retention of the compound inside acidic intracellular compartments of the parasite such as the digestive vacuole. pH-trapping was established to be important for the antimalarial drug chloroquine and analogues51–53, which inhibits hemozoin formation within the digestive vacuole54. Perhaps the targets of these compounds may be localized in or associated with the digestive vacuole. We speculate that pH-trapping may also allow for a slow release of the compound from within acidic intracellular compartments for sustained inhibition of its target(s) in live P. falciparum cells. Further experiments would be necessary to investigate these hypotheses.
Conclusions
In summary, we have synthesized 79 analogues based on the 1-phenyl-3-(4-(quinolin-4-yloxy)phenyl)urea scaffold of the initial hit Ki8751, focusing on diverse hinge-binding groups and tail groups, and we report the SAR of this scaffold on PfPK6 inhibition and inhibition of P. falciparum asexual blood stage parasites. Compound 67 was identified to be a potent PfPK6 inhibitor (IC50 = 13 nM) active against the P. falciparum blood stage (EC50 = 160 nM) while compound 79 was identified with excellent PfPK6 inhibition (IC50 < 5 nM) and dual-stage antiplasmodial activity against P. falciparum at the blood stage (EC50 = 39 nM) and against P. berghei in the liver stage (EC50 = 0.22 μM). Both compounds were evaluated against other P. falciparum kinases. While 67 was found to primarily inhibit PfPK6, 79 likely exerts its antiplasmodial effects via polypharmacology. In conclusion, our results provide a good starting point for further lead optimization of compounds 67 and 79. Further optimization would need to address key concerns found in this study regarding selectivity against human kinase targets, solubility and permeability, as well as demonstrate activity against Plasmodium in vivo, before these compounds could be considered suitable antimalarial drug candidates.
Chemistry
All synthetic procedures are described in detail in the Experimental Section. Truncated analogues of Ki8751 were synthesized by a divergent synthetic strategy (Scheme 1). Nucleophilic aromatic substitution reactions with the appropriate phenol on 4-chloro-6,7-dimethoxyquinoline (83) under neutral or basic conditions afforded compounds 16, 15, and 84. 15 was reacted with 2,4-difluoro-1-isocyanatobenzene to afford the urea 10, and added to 1,1’-carbonyl diimidazole (CDI) and aniline to afford urea 11. Reduction of the nitro group on 84 using iron and ammonium chloride in ethanol and water afforded 14 in good yields, which was then coupled with the appropriate aniline with CDI to form ureas 7, 8, and 9. The experiments with CDI also revealed the highly-varied, substrate-dependent reactivity of the two aniline groups. We observed that 14 preferred to form its symmetrical urea by-product even when reacted with super-stoichiometric amounts of CDI, and low yields of the desired unsymmetrical urea were obtained after addition of the second aniline. Reactions of the latter anilines with CDI before addition of 14 were generally higher-yielding, although symmetrical urea by-products of both anilines were observed in most cases. Addition of acetyl chloride with DIPEA to 14 afforded 13 in excellent yields, while addition of aqueous sodium cyanate to 14 in acetic acid afforded the urea 12, although trace amounts of 13 were also observed due to a side reaction with acetic acid.
Scheme 1. Synthesis of truncated analogues of Ki8751a.
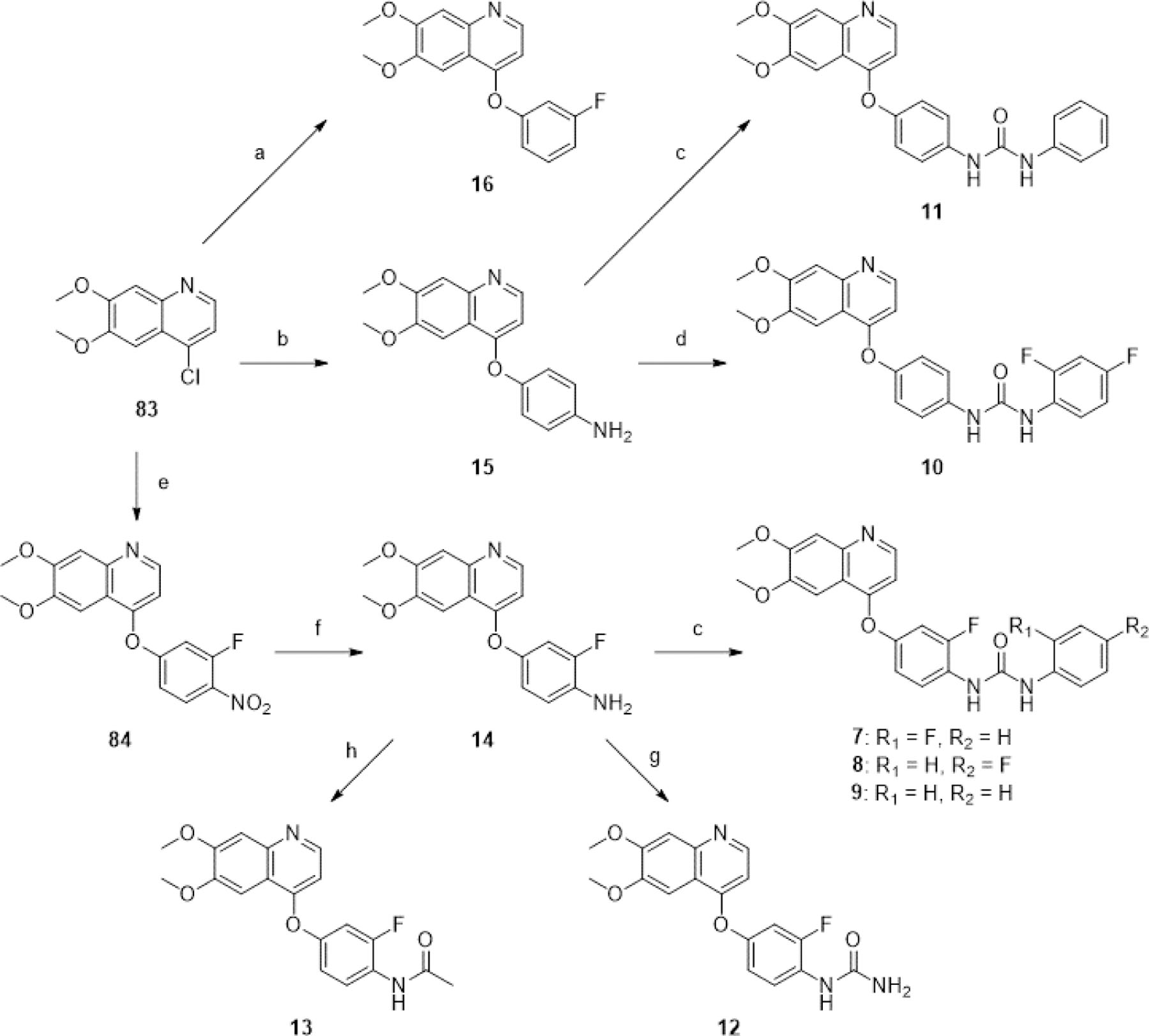
aReagents and conditions: (a) 3-fluorophenol, neat, 170°C, 20 min, 34%; (b) 4-aminophenol, NaOtBu, DMF, 0–110°C, 4 h, 25%; (c) 2-fluoroaniline, 4-fluoroaniline or aniline, CDI, THF or DMSO, rt, [1–2.5 h], then [45–50°C], [1.5–4 h], [7 (5%), 8 (11%), 9 (34%), 11 (71%)]; (d) 2,4-difluoro-1-isocyanatobenzene, EtOH, 0°C-rt, 1 h, 8%; (e) 3-fluoro-4-nitrophenol (87), o-xylene, 135°C, 16 h, 76%; (f) Fe, NH4Cl, EtOH/H2O (3:1), 78°C, 5 h, 78%; (g) NaOCN, AcOH/H2O (5:4), rt, 2 h, 61%; (h) acetyl chloride, DIPEA, 1,4-dioxane, 0°C-rt, 2.5 h, 92%.
We first attempted the synthesis of analogues varying the hinge-binding moiety by a tail-to-head strategy (Scheme 2), planning for diversification at the final step to expedite synthesis. Urea formation between 85 and 86 with 2,4-difluoro-1-isocyanatobenzene afforded 5 and 6 respectively. 5 was then subjected to nucleophilic aromatic substitution reactions under basic conditions with suitable aryl chlorides to afford 3, 17, 22, and 26, and with pyridine-4-yl triflate to afford 4. Yields of these reactions were difficult to optimize as the urea group of 5 was unexpectedly labile under harsher reaction conditions. We thus adopted a head-to-tail synthetic strategy for the remaining compounds (Scheme 3). Refluxing 87 with aryl chlorides in o-xylene overnight afforded compounds 88-95, in varying yields, depending on electrophilicity of the aryl chloride and ease of isolation. For less electrophilic aryl chlorides 4-chloro-1H-pyrrolo[2,3-b]pyridine and 4-chloro-7H-pyrrolo[2,3-d]pyrimidine, reaction with 87 under neat conditions in the microwave reactor or under basic conditions in NMP with 2,6-lutidine afforded 96 and 97 respectively in low yields. For more electron-deficient dichloropyrimidines, a milder reaction condition using potassium carbonate as a base in DMF with mild heating afforded 98 and 99 with minimal overreaction. Reduction of the nitro group using iron and ammonium chloride proceeded with moderate to excellent yields to afford anilines 100-105, 108, 109, 111, 112, 115, 116. Anilines 106, 107, 110, 113, and 114 were obtained from nucleophilic aromatic substitution of 85 with the appropriate aryl chloride using potassium tert-butoxide as the base in DMF or DMA. These anilines were then coupled with 2,4-difluoroaniline using CDI to afford ureas 1, 2, 18-21, 23-25, 27-34. An additional nucleophilic aromatic substitution reaction between the 6-chloropyrimidine group of 33 and benzyl amine under basic conditions in refluxing THF afforded 35 with moderate yields.
Scheme 2. Tail-to-head synthesis of analogues varying hinge-binding groupa.
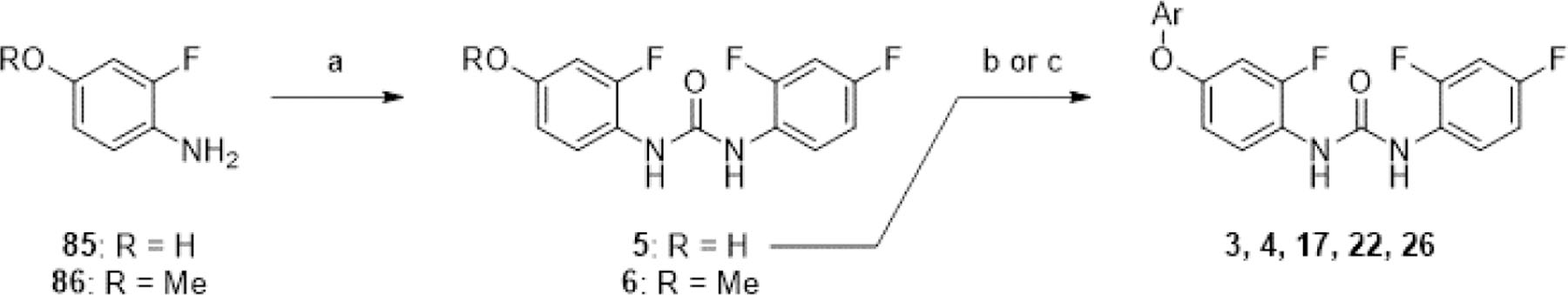
aReagents and conditions: (a) 2,4-difluoro-1-isocyanatobenzene, MeCN, rt, 30 min, 32–36%; (b) ArCl or ArOTf, DIPEA, NMP or DMF, [90–120°C], [1.5–18 h], 3–10%; (c) (for 4) pyridin-4-yl trifluoromethanesulfonate, DIPEA, DMF, μW, 130°C, 3 h, 6%.
Scheme 3. Head-to-tail synthesis of analogues varying hinge-binding groupa.
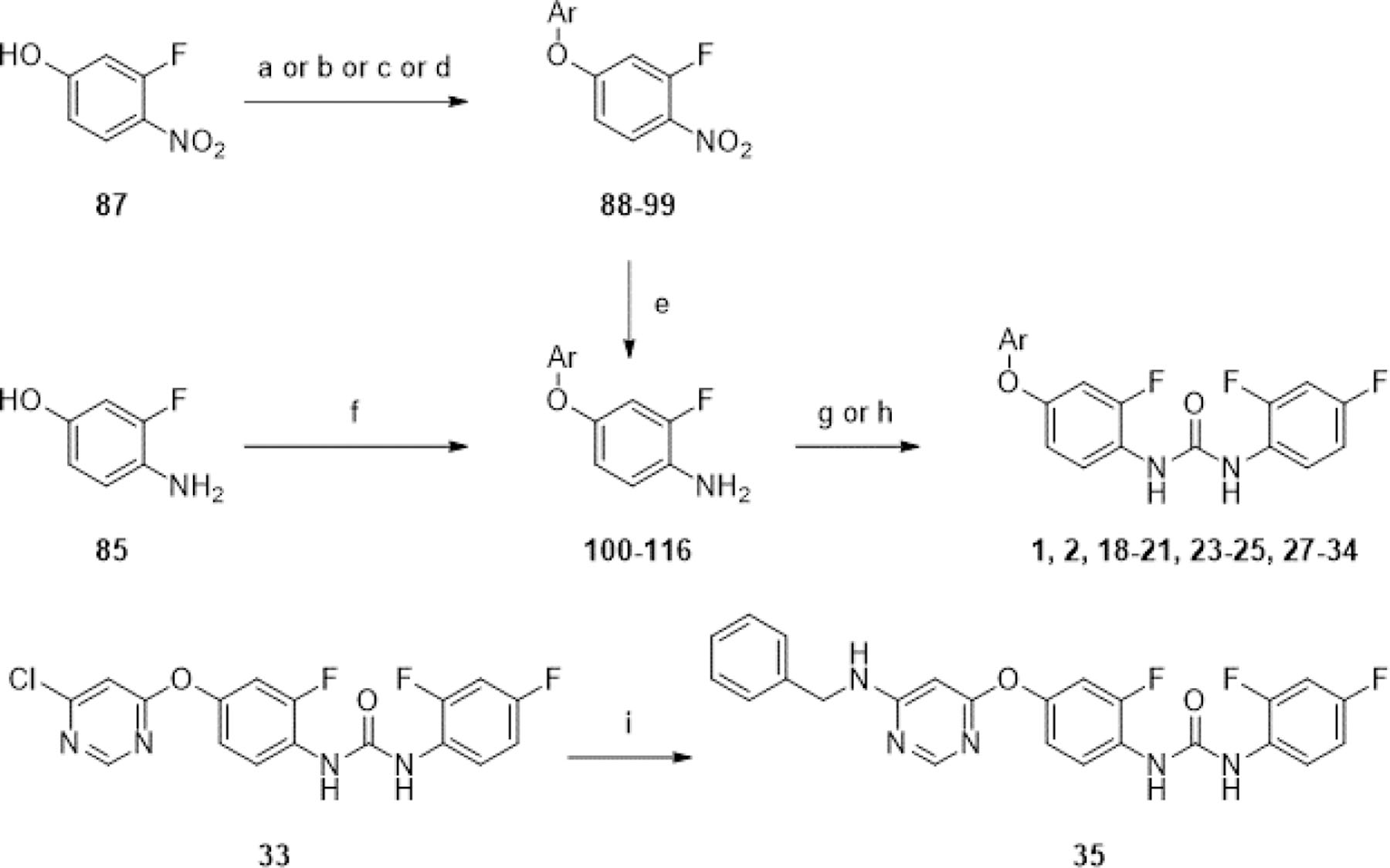
aReagents and conditions: (a) ArCl, o-xylene, 135°C, [14–60 h], 18–76%; (b) (for 96) 4-chloro-1H-pyrrolo[2,3-b]pyridine, neat, μW, 150°C, 20 min, 12%; (c) (for 97) 4-chloro-7H-pyrrolo[2,3-d]pyrimidine, 2,6-lutidine, NMP, 130°C, 22 h, 28%; (d) (for 98 and 99) 4,6-dichloropyrimidine or 2,4-dichloropyrimidine, K2CO3, DMF, 70°C, 16 h, 54–61%; (e) Fe, NH4Cl, EtOH/H2O (3:1), [31–78°C], [2–6 h], 58–95%; (f) ArCl, KOtBu, DMF or DMA, [0–110°C or rt-80°C], [4–16 h], 11–58%; (g) 2,4-difluoro-1-isocyanatobenzene, EtOH, 0°C-rt, [0.5–24 h], 5–58%; (h) 2,4-difluoroaniline, CDI, DCM or DMSO, 0°C or rt, [0.5–2 h], then [35–40°C], [2.5–4 h], 4–67%; (i) benzylamine, Et3N, THF, 66°C, 10 h, 52%.
The majority of aryl chlorides used above were commercially available. The non-commercially available aryl chlorides 118, 119, 122, 124, and 127 were synthesized as described (Scheme 4). Removal of the methyl groups from 83 using boron tribromide in DCM afforded diphenol 117, which reacted with diiodomethane and 1,2-dibromoethane to yield 118 and 119 respectively. Treatment of 120 with DMF-DMA afforded the formamidine intermediate, which was reacted with n-butyllithium-deprotonated acetonitrile to yield compound 121. Condensation of 125 with formamide under refluxing conditions afforded 126. 121, 126, and the commercially available 123 were subject to POCl3 to form aryl chlorides 122, 127 and 124, respectively, suitable for the above nucleophilic aromatic substitution reactions.
Scheme 4. Preparation of aryl chlorides 118, 119, 122, 124, 127a.
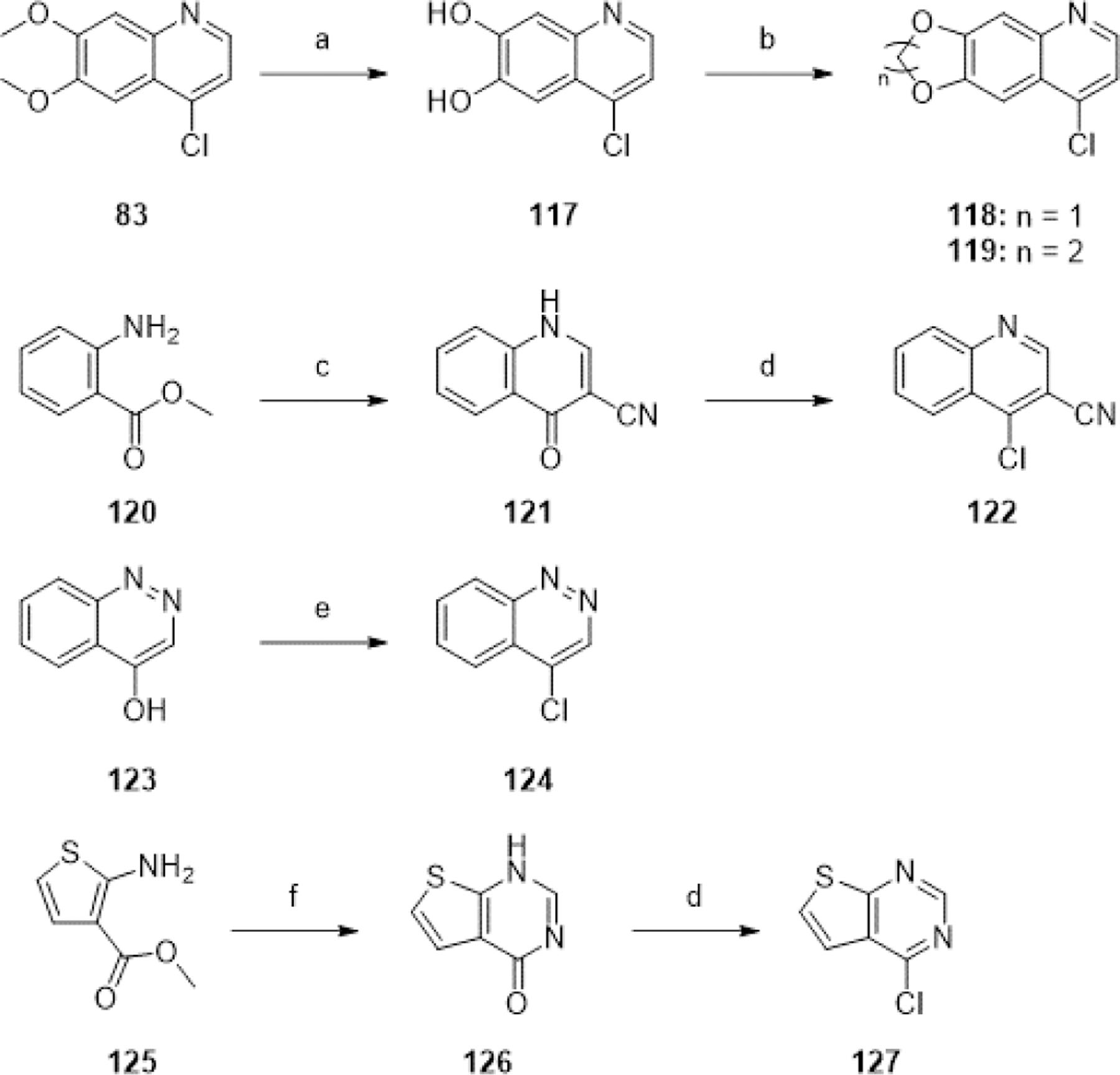
aReagents and conditions: (a) BBr3, DCM, 0°C-rt, 18 h, 42%; (b) diiodomethane or 1,2-dibromoethane, K2CO3, DMF, 60°C, 2 h, 53–66%; (c) (i) DMF-DMA, DMF, 150°C, 3 h, (ii) MeCN, nBuLi, THF, −78°C, 1 h, 80% (over two steps); (d) POCl3, neat, 100°C, 2 h, 50–95%; (e) POCl3, THF, 66°C, 1 h, 86%; (f) formamide, neat, 160°C, 24 h, 52%.
To synthesize analogues with diverse tail group substituents, key intermediate 14 was coupled with the appropriate commercially available aniline using CDI to form ureas 40-43, 45, 49-60, and 63 in poor-to-moderate yields (Scheme 5). In cases where the available aniline was available as a hydrochloride salt, one equivalent of DIPEA or Et3N was additionally used for neutralization. 39 was synthesized by coupling 14 with 1-isocyanato-3-(trifluoromethyl)benzene.
Scheme 5. One-step diversification at the tail groupa.
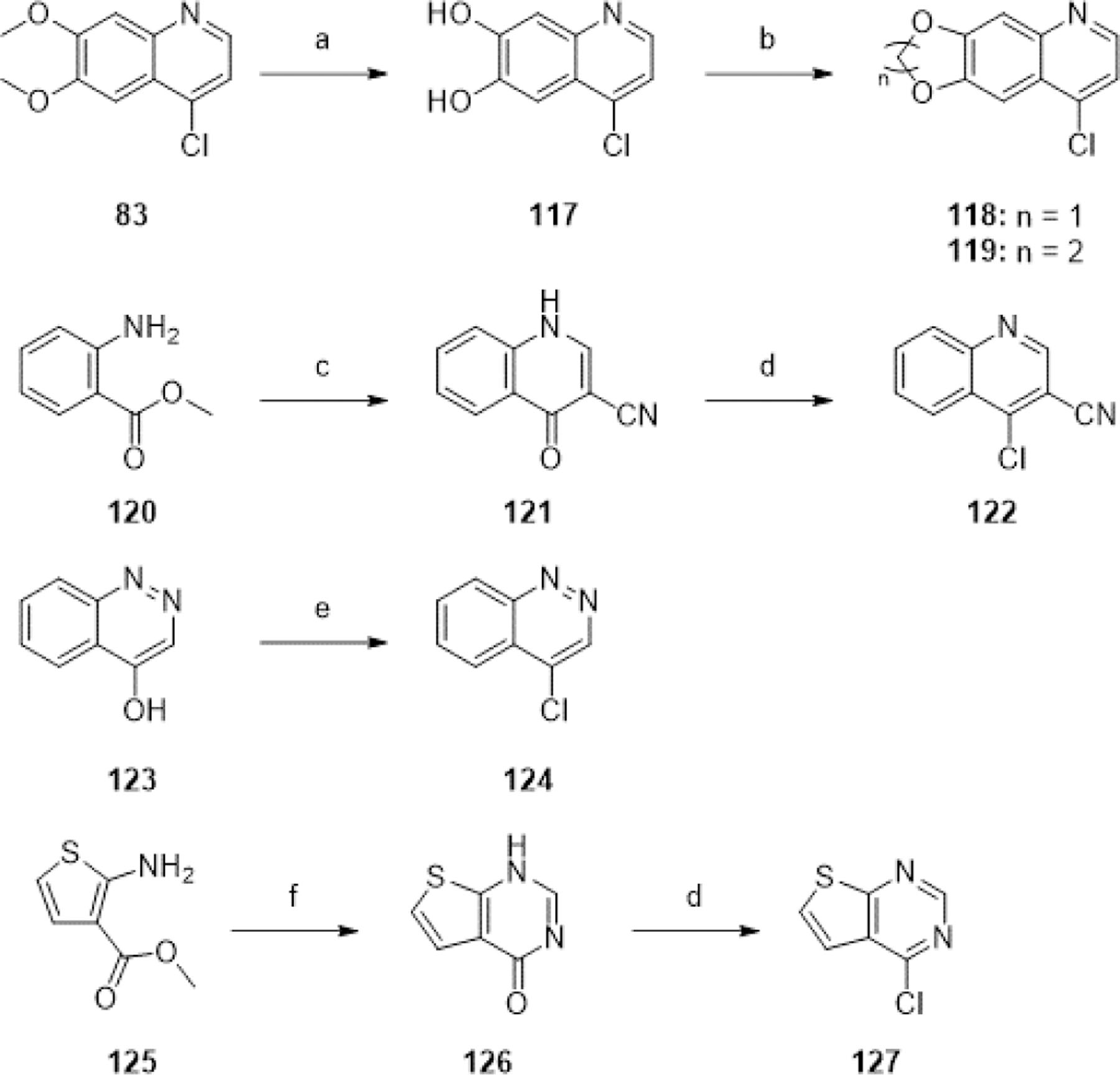
aReagents and conditions: (a) ArNH2, CDI, DMSO, [rt or rt-40°C], [5 min-3.5 h], then 60°C, [2–5 h], 15–55%; (b) (for 39) 1-isocyanato-3-(trifluoromethyl)benzene, EtOH/DCM (2:1), rt, 16 h, 58%; (c) (for 51) m-toluidine hydrochloride, DIPEA, CDI, DMSO, rt, 45 min, then 60 °C, 4 h, 35%; (d) (for 63) 4-((diethylamino)methyl)aniline hydrochloride, Et3N, CDI, DMSO, rt, 1 h, then 60 °C, 2 h, 60%.
With complex substituents where a suitable aniline or isocyanate was not commercially available, the appropriate aniline of the tail group was synthesized before coupling with 14 (Scheme 6). The coupling of acyl chloride 128 with 1-methylpiperazine followed by reduction of the nitro group using iron and ammonium chloride afforded 129. The EDC-mediated amide coupling between 1-methylpiperazine and 130 followed by simultaneous reduction of both the nitro group and the resultant amide using LiAlH4 yielded 131. Similarly, EDC-mediated amide coupling of 1-methylpiperazine with 132 and 134 afforded anilines 133 and 135 respectively. 129, 131, 133, and 135 were then reacted with CDI and 14 to yield 44, 46, 47, and 48 respectively.
Scheme 6. Synthesis of tail group anilines and urea formationa.
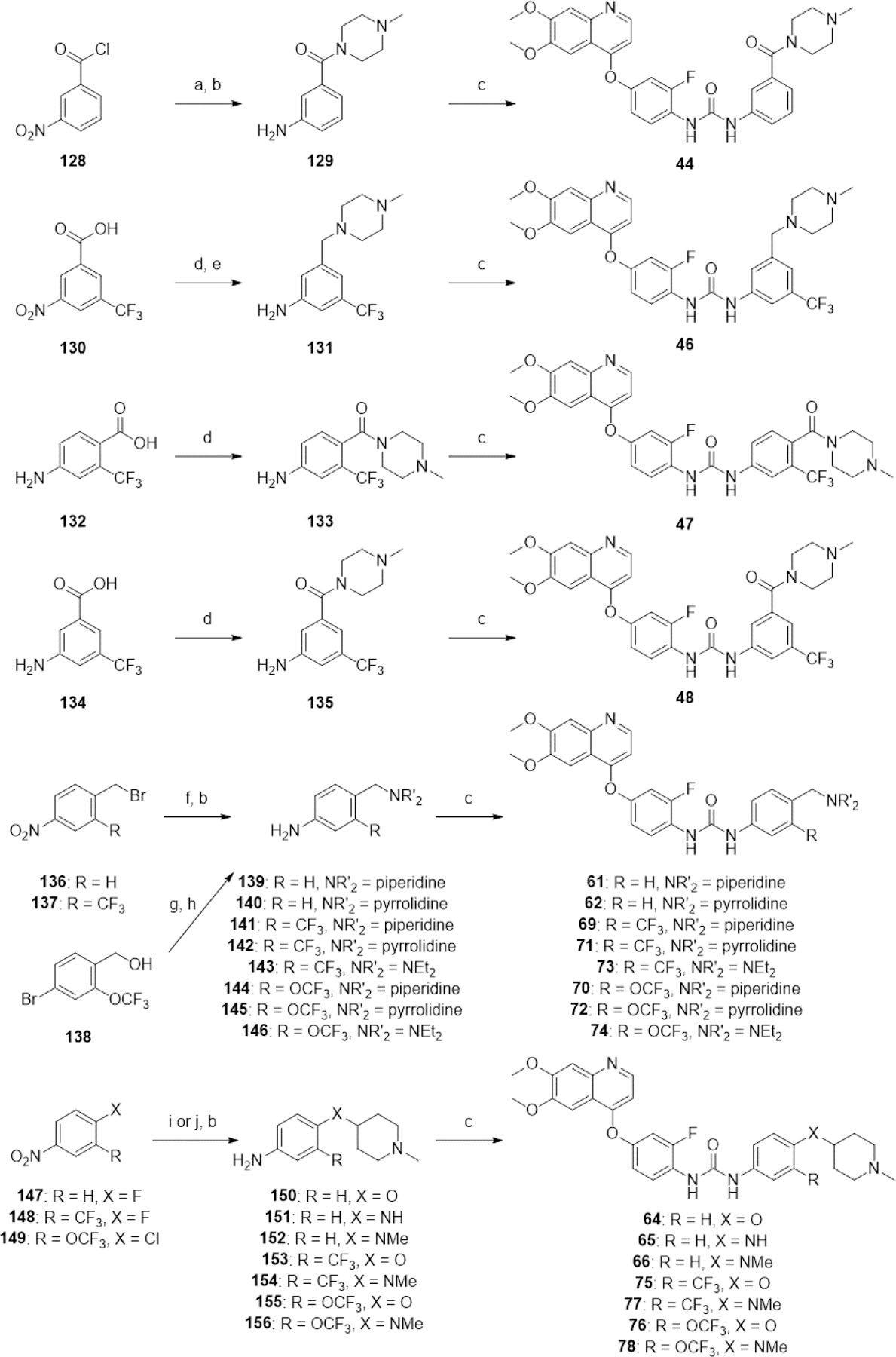
aReagents and conditions: (a) 1-methylpiperazine, THF, 54%; (b) Fe, NH4Cl, EtOH/H2O (3:1), [31–78°C], [5–24 h], 53–100%; (c) 14, CDI, DMSO, [rt-40°C], [0.5–3 h], then 60°C, [2–5 h], 15–69%; (d) 1-methylpiperazine, EDC, DCM, rt, 18 h, 46–79%; (e) LiAlH4, THF, 66°C, 5 h, 28%; (f) piperidine, pyrrolidine, or Et2NH, KI, K2CO3, MeCN, rt, [1.5–3 h], 54–92%; (g) (i) MsCl, Et3N, DCM, 0°C-rt, 2 h, (ii) piperidine, pyrrolidine, or Et2NH, Et3N, rt, 16 h, 75–89% (over two steps); (h) (i) LiHMDS, Pd(tBu3P)2, toluene, rt, 16 h, (ii), HCl (aq), rt, 10 min, 60–87% (over two steps); (i) 1-methylpiperidin-4-ol, KOtBu, DMSO, rt, 16 h, 28–58%; (j) 1-methylpiperidin-4-amine or N,1-dimethylpiperidin-4-amine, Et3N, DMSO, [90–100°C], [16–20 h], 48–82%.
The potassium iodide-catalyzed substitution of the benzyl bromide group of 136 and 137 with piperidine, pyrrolidine or diethylamine using potassium carbonate as a base, followed by nitro reduction using iron and ammonium chloride afforded anilines 139-143. The benzyl alcohol of aryl bromide 138 was converted to a mesylate using mesyl chloride and triethylamine in DCM, followed by one-pot substitution by piperidine, pyrrolidine or diethylamine. The aryl bromides were then converted to anilines 144-146 in excellent yields using a procedure modified from Lee et al., 200155 using Pd(tBu3P)2 as the catalyst and LiHMDS as the nitrogen source. The anilines 139, 140, 141, 142, 143, 144, 145, and 146 were then successfully coupled with 14 to form ureas 61, 62, 69, 71, 73, 70, 72 and 74 respectively.
Nucleophilic aryl substitutions of aryl fluorides 147, 148 and aryl chloride 149 with 1-methylpiperidin-4-ol, using potassium tert- butoxide as the base in DMSO at room temperature, and with 1-methylpiperidin-4-amine or N,1-dimethylpiperidin-4-amine using triethylamine as a base in DMSO at 90°C or 100°C were successful. The nitro groups of these substitution products were then reduced by iron and ammonium chloride to afford anilines 150-156. Intriguingly, where there is a trifluoromethoxy group present ortho to the aryl chloride, a minor product was observed during the nucleophilic aromatic substitution step, where displacement of the nitro group occurred instead of the chloro group. This minor product was only separable after the subsequent nitro reduction step. Anilines 150, 151, 152, 153, 154, 155, and 156 were then coupled with CDI and 14 to afford ureas 64, 65, 66, 75, 77, 76, and 78 respectively.
The synthesis of analogues with carbon linkers between the terminal phenyl ring and the piperidine ring required a different approach (Scheme 7). Heck coupling between 157 and 158 using Pd(dppf)Cl2 and triethylamine in DMF heated at 100°C overnight afforded 159 as a minor product; the major product was not identified. Ester saponification proceeded with excellent yields to yield the corresponding benzoic acid, which was subjected to a one-pot microwave-assisted Curtius rearrangement using triethylamine and diphenylphosphoryl azide (DPPA), trapping the isocyanate using the aniline 14, following the procedure by Kulkarni et al., 201756, to yield 160 in excellent yields. 160 was subjected to 20% TFA in DCM to remove the Boc group, yielding 68. Hydrogenation of the alkene of 68 afforded 67 in moderate yields.
Scheme 7. Synthesis of compounds 67, 68, 79–82a.
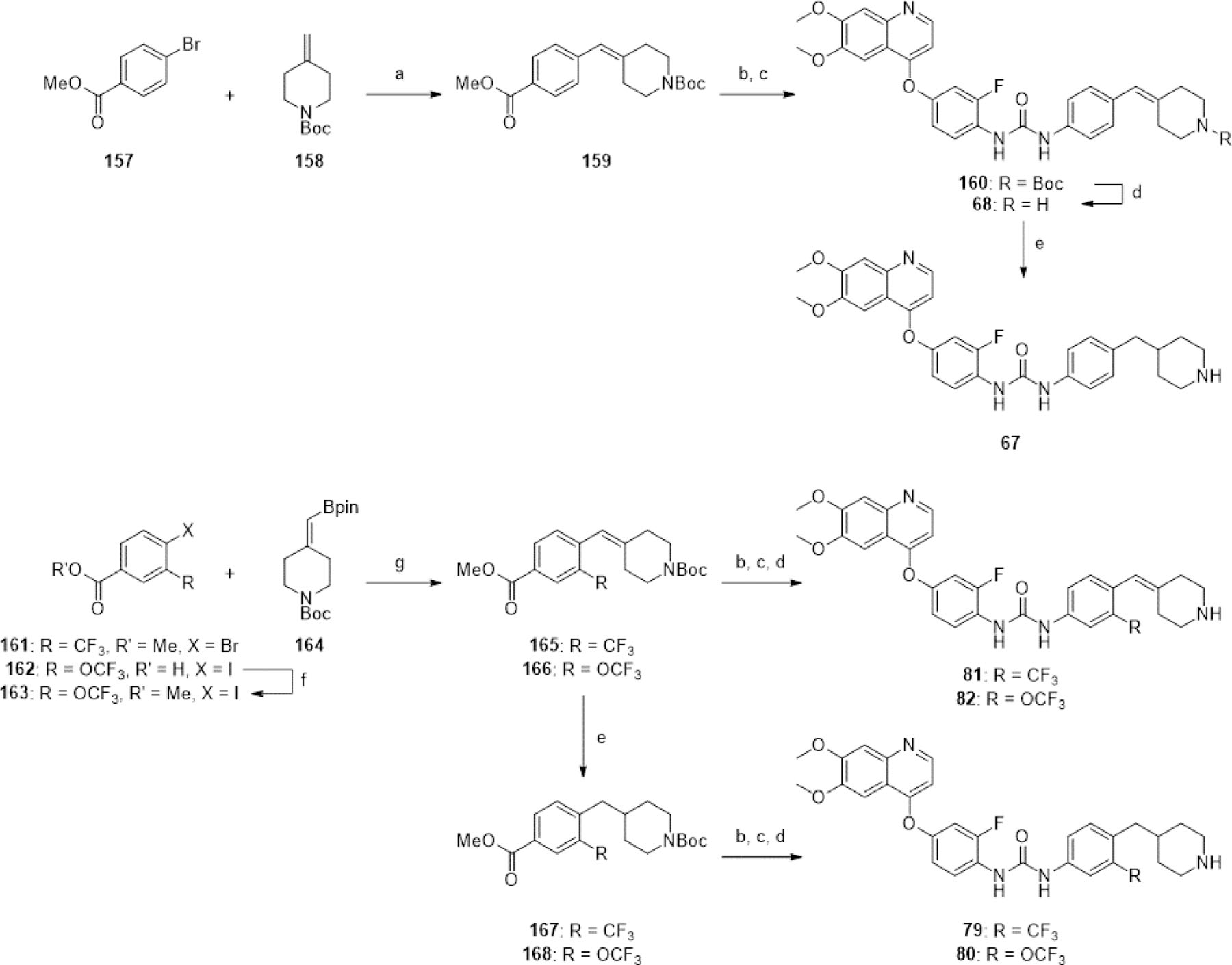
aReagents and conditions: (a) Et3N, Pd(dppf)Cl2, DMF, 100°C, 16 h, 18%; (b) LiOH, THF/H2O (4:1), 50°C, [4–6 h], 88–94%; (c) 14, DPPA, Et3N, toluene, μW, 100°C, 10 min, [160 (82%)]; (d) TFA/DCM (1:4), rt, 1 h, [68 (79%), 79 (63% over two steps), 80 (71% over two steps), 81 (63% over two steps), 82 (69% over two steps)]; (e) H2, Pd/C, MeOH, rt, [18–24 h], 55–88%; (f) MeI, K2CO3, DMSO, rt, 6 h, 95%; (g) Pd2(dba)3, Xphos, K3PO4, 1,4-dioxane/H2O (9:1), 100°C, 1 h, 67–83%.
Attempts at the analogous Heck coupling between 161 and 158 were unsuccessful, thus 165 was instead prepared by Suzuki coupling between 161 and 164, using Pd2(dba)3 and Xphos as the catalytic system, potassium phosphate as the base, in 1,4-dioxane and water at 100°C for 1 h. Methylation of 162 with methyl iodide and potassium carbonate in DMSO afforded 163, which was subjected to the same Suzuki coupling conditions to afford 166. Hydrogenation of 165 and 166 yielded 167 and 168 in excellent yields. The methyl esters 165, 166, 167, and 168 were saponified, next reacted with 14 in the abovementioned one-pot Curtius rearrangement, and finally subject to Boc deprotection by 20% TFA in DCM to afford 81, 82, 79 and 80 respectively in good yields.
Experimental Section
KinaseSeeker Assay.
Stock solutions of compounds were serially diluted in DMSO to make assay stocks. Prior to initiating a profiling campaign, the compounds were evaluated for false positive against split-luciferase. The compounds were then screened in duplicate against each of the kinases. For kinase assays, each Cfluc-Kinase was translated along with Fos-Nfluc using a cell-free system (cell lysate) at 30°C for 90 min. 24 µL aliquot of this lysate containing either 1 µL of DMSO (for no-inhibitor control) or compound solution in DMSO (final concentration: 1 µM) was incubated for 2 hours at room temperature in presence of a kinase specific probe. 80 µL of luciferin assay reagent was added to each solution and luminescence was immediately measured on a luminometer.
The % inhibition was calculated using the following equation:
P. falciparum Asexual Blood Stage Culture and Viability Assay.
P. falciparum 3D7 parasites were continuously cultured in vitro in complete medium (10.44 g/L RPMI 1640 (ThermoFisher Scientific), 25 mM HEPES, pH 7.2 (ThermoFisher Scientific), 0.37 mM hypoxanthine (Sigma), 24 mM sodium bicarbonate (Sigma), 0.5% (wt/vol) AlbuMAX II (ThermoFisher Scientific), 25 μg/mL gentamicin (Sigma)) supplemented with freshly washed human erythrocytes (Gulf Coast Regional Blood Center, Houston, TX) approximately every 48 h. The parasite cultures were maintained at 2– 10% parasitemia with 1% hematocrit at 37°C in a 3% O2, 5% CO2, 92% N2 atmosphere. Highly synchronized cultures were generated by treatment with 25 volumes of 5% (wt/vol) D-sorbitol (Sigma) at 37°C for 10 min during the early ring stage.
Prior to the assays, P. falciparum 3D7 parasites were synchronized as described above and adjusted to 2% parasitemia and 2% hematocrit. Compounds were initially assayed for parasite inhibition at 1 μM. Dose response curves for select compounds were generated by dispensing 100 μL of the culture into each well of a 96-well black microplate (Corning), followed by administration of 1 μL serial diluted compounds in triplicate. Final compound concentrations ranged from 0 to 5 μM. Quinacrine at 125 nM was employed as the positive control and 0.5% DMSO as the negative control. Plates were incubated at 37°C in a 3% O2, 5% CO2, 92% N2 atmosphere before and after drug administration. At 34 h post-reinvasion (i.e. 72 h after drug administration), 40 μL lysis solution (20 mM Tris- HCl, pH 7.5 (Fisher Chemical), 5 mM EDTA dipotassium salt dihydrate (Fisher Chemical), 0.16% (wt/vol) saponin (Sigma), 1.6% (vol/vol) Triton X-100 (Fisher Chemical)) containing fresh 10x SYBR Green I (ThermoFisher Scientific) was added to each well and incubated in the dark at room temperature for 24 h. The fluorescent signals were measured at 535 nm with excitation at 485 nm using an EnVision plate reader (PerkinElmer). Data was normalized to the negative and positive controls to obtain the relative percent parasite load. EC50 values were determined by fitting data to a standard dose response equation (GraphPad Prism). The Z-factor ranged from 0.5–0.9.
P. berghei Liver Stage Parasite Load Assay and HepG2 CellTiter-Fluor Cytotoxicity Assay.
HepG2 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) with L-glutamine (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS) (v/v) (Sigma-Aldrich) and 1% antibiotic-antimycotic (Thermo Fisher Scientific) in a standard tissue culture incubator (37°C, 5% CO2). P. berghei ANKA sporozoites used for liver stage experiments were isolated from freshly dissected salivary glands of infected mosquitoes (University of Georgia SporoCore). Dose response curves were generated for select compounds by assessing P. berghei parasite load in hepatocytes as previously described50. Briefly, HepG2 (8,000 cells/well) were seeded into 384-well white microplates (Corning). After 24 hours, compounds (0–100 µM) were added (HP D300 Digital Dispenser) before infection with P. berghei ANKA sporozoites (4,000 spz/well). DMSO (1% v/v) was added as the negative control. All samples were evaluated in triplicate and had a final DMSO concentration of 1%. After 44 hours post-infection, HepG2 cell viability and parasite load were assessed using CellTiter-Fluor (Promega) and Bright-Glo (Promega) reagents, respectively, according to manufacturer’s protocols. Relative fluorescence and luminescence signal was measured using an EnVision plate reader (PerkinElmer). The signal intensity of each well was normalized to the negative control (1% DMSO) to assess relative viability. Dose response analysis was performed with GraphPad Prism.
HepG2 CellTiter-Glo Cytotoxicity Assay.
HepG2 cells were maintained in DMEM (Gibco) supplemented with 10% FBS, 1% NEAA, 1% L-glutamine. No antibiotics were used. Cells were plated at 4000 cells/well in 384-well plate (Costar) and incubated overnight (37°C, 5% CO2) before adding compound. Compounds were added in quadruplicate and incubated for 48 hrs. DMSO percentage was constant across all concentrations of compound. Cell viability was measured using CellTiter-Glo2 (Promega) and luminescence signal was read on a GloMax plate reader (Promega). Dose response analysis was performed using GraphPad Prism.
DSF-based selectivity screening against a curated kinase library.
The assay was performed as previously described57,58. Briefly, recombinant protein kinase domains at a concentration of 2 μM were mixed with 10 μM compound in a buffer containing 20 mM HEPES, pH 7.5, and 500 mM NaCl. SYPRO Orange (5000×, Invitrogen) was added as a fluorescence probe (1 µL per mL). Subsequently, temperature-dependent protein unfolding profiles were measured using the QuantStudio™ 5 realtime PCR machine (Thermo Fisher). Excitation and emission filters were set to 465 nm and 590 nm, respectively. The temperature was raised with a step rate of 3°C per minute. Data points were analyzed with the internal software (Thermal Shift SoftwareTM Version 1.4, Thermo Fisher) using the Boltzmann equation to determine the inflection point of the transition curve.
pKa Calculation.
The pKa value of PfPK6 inhibitors was simulated by the computational method proposed by one of the coauthors using the molecular electrostatic potential (MEP) on the acidic/basic nucleus and valence natural atomic orbital (NAO) energies as the equivalent descriptors37–39,59–61. All calculations were carried out with Gaussian 16 package version C0162 with tight self-consistent-field convergence and ultrafine integration grids. The density functional theory B3LYP63,64 approximate exchange-correlation functional was employed for all calculations with Pople’s 6–311+G(d) basis set65. MEP and NAO descriptors were obtained by the natural population analysis66 available from the Gaussian package. The reference pKa values for structural analogues were from the reference67.
General Chemistry Methods.
All reagents and solvents were used directly as received from commercial suppliers without further purification. Reactions were run under nitrogen or argon atmosphere unless otherwise noted. Solvents were degassed with argon for cross-coupling reactions. All microwave (μW) reactions were carried out in a Biotage Initiator EXP US 400W microwave synthesizer. Thin-layer chromatography (TLC) analyses were performed using aluminum-backed 200 μm pre-coated silica gel Sorbtech 254 nm-fluorescent TLC plates, and spots were visualized using UV light (254/365 nm). Flash chromatography was performed with RediSep 40–63 μm irregular silica prepacked cartridges or RediSep 20–40 μm spherical C18-coated silica prepacked cartridges on Biotage Isolera One or Prime instruments, or with a Phenomenex Luna Phenyl-Hexyl (5 μm particle size, 100 Å pore size, 75 × 30 mm) column on an Agilent 1100 Series preparatory high-performance liquid chromatography (prep-HPLC) instrument equipped with an Agilent G1315B diode array detector measured at 220/254 nm.
Nuclear magnetic resonance (NMR) spectra were recorded in DMSO-d6, Methanol-d4, or CDCl3 on Varian Inova 400 MHz, Bruker Avance 700 MHz or Bruker Avance 850 MHz instruments. Chemical shifts are reported in parts per million (ppm, δ), with residual solvent peaks referenced as the internal standard. Coupling constants are reported in Hz. Spin multiplicities are described as s (singlet), br s (broad singlet), d (doublet), t (triplet), q (quartet), p (pentet), and m (multiplet). Data were processed using MestReNova.
Yields are reported for pure material after isolation. All compounds tested for biological activity were ≥95% pure based on HPLC. High-resolution mass spectrometry samples were analyzed with a ThermoFisher Q Exactive HF-X (ThermoFisher, Bremen, Germany) mass spectrometer coupled with a Waters Acquity H-class liquid chromatograph system. Samples were introduced via a heated electrospray source (HESI) at a flow rate of 0.3 mL/min. Electrospray source conditions were set as: spray voltage 3.0 kV, sheath gas (nitrogen) 60 arb, auxillary gas (nitrogen) 20 arb, sweep gas (nitrogen) 0 arb, nebulizer temperature 375°C, capillary temperature 380°C, RF funnel 45 V. The mass range was set to 150–2000 m/z. All measurements were recorded at a resolution setting of 120,000. Separations were conducted on a Waters Acquity UPLC BEH C18 column (2.1 × 50 mm, 1.7 um particle size). LC conditions were set at 95 % water with 0.1% formic acid (A) ramped linearly over 5.0 mins to 100% acetonitrile with 0.1% formic acid (B) and held until 6.0 mins. At 7.0 mins the gradient was switched back to 95% A and allowed to re-equilibrate until 9.0 mins. Injection volume for all samples was 3 μL. Xcalibur (ThermoFisher, Breman, Germany) was used to analyze the data. Solutions were analyzed at 0.1 mg/mL or less based on responsiveness to the ESI mechanism. Molecular formula assignments were determined with Molecular Formula Calculator (v 1.2.3). All observed species were singly charged, as verified by unit m/z separation between mass spectral peaks corresponding to the 12C and 13C12Cc-1 isotope for each elemental composition.
Experimental Procedures in Scheme 1.
4-(3-fluorophenoxy)-6,7-dimethoxyquinoline (16).
A mixture of 4-chloro-6,7-dimethoxyquinoline (83) (50 mg, 0.22 mmol, 1 eq) and 3-fluorophenol (75 mg, 0.67 mmol, 3 eq) was heated at 170°C for 20 min. The reaction mixture was cooled to rt, and saturated aqueous NaHCO3 solution (20 mL) was added. The aqueous was extracted with EtOAc (30 mL x 3). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (5–40% EtOAc in Hexanes on silica gel column) to afford 16 (23 mg, 34% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.52 (d, J = 5.2 Hz, 1H), 7.55 (td, J = 8.3, 6.8 Hz, 1H), 7.46 (s, 1H), 7.41 (s, 1H), 7.24 (dt, J = 10.0, 2.4 Hz, 1H), 7.18 (tdd, J = 8.5, 2.5, 0.9 Hz, 1H), 7.12 (ddd, J = 8.2, 2.5, 0.9 Hz, 1H), 6.60 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.92 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −110.22 (ddd, J = 9.6, 8.2, 6.9 Hz). 13C NMR (101 MHz, DMSO-d6) δ 162.89 (d, J = 245.7 Hz), 158.92, 155.47 (d, J = 11.0 Hz), 152.65, 149.49, 148.89, 146.62, 131.73 (d, J = 9.6 Hz), 116.82 (d, J = 3.1 Hz), 115.32, 112.37 (d, J = 21.1 Hz), 108.61 (d, J = 24.1 Hz), 107.90, 104.15, 98.98, 55.76, 55.71. HRMS: calcd for C17H15FNO3 [M + H]+ m/z, 300.1036; found m/z, 300.1021.
4-((6,7-dimethoxyquinolin-4-yl)oxy)aniline (15).
To a solution of 4-chloro-6,7-dimethoxyquinoline (83) (500 mg, 2.24 mmol, 1 eq) and 4-aminophenol (342 mg, 3.13 mmol, 1.4 eq) in DMF (10 mL) was added a suspension of sodium tert-butoxide (301 mg, 3.13 mmol, 1.4 eq) in DMF (10 mL) dropwise at 0°C. The reaction mixture was stirred at 110°C for 4 h. The reaction mixture was cooled to 0°C using an ice bath, water (60 mL) was added and stirred overnight. The solid was collected by filtration, washed with water, and dried. The crude material was dissolved in a mixture of DCM and MeOH, concentrated onto Celite and purified by flash chromatography (0–6% MeOH in DCM on silica gel column) to afford 15 (166 mg, 25% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (d, J = 5.2 Hz, 1H), 7.50 (s, 1H), 7.36 (s, 1H), 6.92 (d, J = 8.8 Hz, 2H), 6.66 (d, J = 8.7 Hz, 2H), 6.37 (d, J = 5.2 Hz, 1H), 5.15 (s, 2H), 3.93 (s, 3H), 3.93 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 160.95, 152.40, 149.10, 148.84, 146.65, 146.24, 143.40, 121.77, 115.00, 114.86, 107.79, 102.28, 99.19, 55.67, 55.65. HRMS: calcd for C17H17N2O3 [M + H]+ m/z, 297.1239; found m/z, 297.1224.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)phenyl)-3-phenylurea (11).
To a solution of aniline (14 mg, 0.15 mmol, 1.3 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (27 mg, 0.17 mmol, 1.4 eq). The reaction mixture was stirred at rt for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)aniline (15) (35 mg, 0.12 mmol, 1 eq) in DMSO (1 mL). The reaction mixture was stirred at 50°C for 4 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–3% MeOH in EtOAc on silica gel column) to afford 11 (35 mg, 71% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.81 (s, 1H), 8.69 (s, 1H), 8.47 (d, J = 5.3 Hz, 1H), 7.63 – 7.56 (m, 2H), 7.52 (s, 1H), 7.50 – 7.44 (m, 2H), 7.39 (s, 1H), 7.29 (dd, J = 8.5, 7.3 Hz, 2H), 7.24 – 7.17 (m, 2H), 6.98 (t, J = 7.3 Hz, 1H), 6.44 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.94 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 160.16, 152.58, 152.53, 149.28, 148.86, 148.13, 146.42, 139.65, 137.33, 128.81, 121.89, 121.56, 119.83, 118.25, 115.10, 107.85, 102.88, 99.12, 55.71, 55.69. HRMS: calcd for C24H22N3O4 [M + H]+ m/z, 416.1610; found m/z, 416.1591.
1-(2,4-difluorophenyl)-3-(4-((6,7-dimethoxyquinolin-4-yl)oxy)phenyl)urea (10).
To a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)aniline (15) (60 mg, 0.20 mmol, 1 eq) in ethanol (0.6 mL) cooled to 0°C using an ice bath was added 2,4-difluoro-1-isocyanatobenzene (31 mg, 0.20 mmol, 1 eq) dropwise. The reaction mixture was warmed to rt and stirred at rt for 1 h. The reaction mixture was concentrated onto Celite and purified by flash chromatography (0–5% MeOH in EtOAc on silica gel column) to afford 10 (7.7 mg, 8% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.16 (s, 1H), 8.54 (d, J = 2.3 Hz, 1H), 8.47 (d, J = 5.2 Hz, 1H), 8.08 (td, J = 9.2, 6.1 Hz, 1H), 7.62 – 7.55 (m, 2H), 7.52 (s, 1H), 7.39 (s, 1H), 7.32 (ddd, J = 11.6, 8.9, 2.9 Hz, 1H), 7.25 – 7.18 (m, 2H), 7.11 – 7.01 (m, 1H), 6.44 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.94 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −117.62 – −118.43 (m), −124.67 (t, J = 10.8 Hz). 13C NMR (101 MHz, DMSO-d6) δ 160.13, 156.88 (dd, J = 241.7, 11.9 Hz), 152.54, 152.37, 152.28 (dd, J = 243.8, 12.3 Hz), 149.30, 148.86, 148.34, 146.43, 137.02, 124.03 (dd, J = 11.1, 3.5 Hz), 122.10 (dd, J = 9.1, 3.2 Hz), 121.65, 119.80, 115.11, 111.91 – 110.58 (m), 107.85, 103.82 (dd, J = 26.8, 23.8 Hz), 102.92, 99.13, 55.72, 55.70. HRMS: calcd for C24H20F2N3O4 [M + H]+ m/z, 452.1422; found m/z, 452.1402.
4-(3-fluoro-4-nitrophenoxy)-6,7-dimethoxyquinoline (84).
A suspension of 4-chloro-6,7-dimethoxyquinoline (83) (2.50 g, 11.2 mmol, 1 eq) and 3-fluoro-4-nitrophenol (87) (3.16 g, 20.1 mmol, 1.8 eq) in o-xylene (24 mL) was stirred at 135°C for 16 h. The reaction mixture was cooled to rt. EtOAc (60 mL) was added and the mixture was stirred for 1 h at rt. The solid formed was collected by filtration, washed with EtOAc and dried. The solid was added to an aqueous NaOH solution (1 M, 60 mL) and stirred for 1 h at rt. The solids were collected by filtration, washed with water, and dried to afford 84 (2.936 g, 76% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.64 (d, J = 5.1 Hz, 1H), 8.27 (t, J = 9.0 Hz, 1H), 7.56 (dd, J = 12.4, 2.6 Hz, 1H), 7.46 (s, 1H), 7.33 (s, 1H), 7.20 (ddd, J = 9.1, 2.6, 1.1 Hz, 1H), 6.98 (d, J = 5.1 Hz, 1H), 3.96 (s, 3H), 3.88 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −113.95 (dd, J = 12.3, 8.9 Hz). 13C NMR (101 MHz, DMSO-d6) δ 160.85 (d, J = 11.3 Hz), 156.82, 156.29 (d, J = 263.3 Hz), 152.88, 149.92, 148.90, 147.04, 133.14 (d, J = 7.1 Hz), 128.51 (d, J = 1.8 Hz), 115.81, 115.44 (d, J = 3.4 Hz), 109.34 (d, J = 24.1 Hz), 108.01, 107.03, 98.68, 55.81, 55.75.
4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14).
A suspension of 4-(3-fluoro-4-nitrophenoxy)-6,7-dimethoxyquinoline (84) (2.90 g, 8.42 mmol, 1 eq), iron (2.35 g, 42.1 mmol, 5 eq), and ammonium chloride (3.60 g, 67.4 mmol, 8 eq) in ethanol (40 mL) and water (13 mL) was heated at 78°C for 5 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between DCM (100 mL) and aqueous NaOH solution (1 M, 100 mL). The aqueous layer was extracted with DCM (2 × 100 mL). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–10% MeOH in DCM on silica gel column) to afford 14 (2.064 g, 78% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.45 (d, J = 5.3 Hz, 1H), 7.48 (s, 1H), 7.37 (s, 1H), 7.05 (dd, J = 12.1, 2.3 Hz, 1H), 6.93 – 6.76 (m, 2H), 6.42 (d, J = 5.2 Hz, 1H), 5.20 (s, 2H), 3.94 (s, 3H), 3.93 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −131.32 (dd, J = 11.8, 8.9 Hz). 13C NMR (101 MHz, DMSO-d6) δ 160.52, 152.46, 150.18 (d, J = 239.8 Hz), 149.19, 148.86, 146.31, 142.83 (d, J = 9.3 Hz), 134.45 (d, J = 12.9 Hz), 117.48 (d, J = 3.1 Hz), 116.43 (d, J = 5.8 Hz), 114.91, 109.15 (d, J = 21.0 Hz), 107.81, 102.49, 99.11, 55.69, 55.68. HRMS: calcd for C17H16FN2O3 [M + H]+ m/z, 315.1145; found m/z, 315.1131.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(2-fluorophenyl)urea (7).
To a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (60 mg, 0.19 mmol, 1 eq) in THF (1 mL) was added 1,1′- carbonyldiimidazole (40 mg, 0.25 mmol, 1.3 eq). The reaction mixture was stirred at rt for 2.5 h, followed by stirring at 45°C for 1.5 h, then 60°C for 1 h, monitoring reaction progress by TLC. A solution of 2-fluoroaniline (25 mg, 0.23 mmol, 1.2 eq) in THF (1 mL) was added. The reaction mixture was stirred at 45°C for 2 h, then cooled to rt. Water (10 mL) was added to precipitate a white solid. The solid was collected by filtration, washed with water and dried. The crude material was dissolved in a mixture of DCM and MeOH, concentrated onto Celite and purified by flash chromatography (0–10% MeOH in EtOAc on silica gel column) to afford 7 (4.4 mg, 5% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.29 (d, J = 2.4 Hz, 1H), 8.22 (d, J = 2.6 Hz, 1H), 7.67 (d, J = 5.2 Hz, 1H), 7.45 (t, J = 9.1 Hz, 1H), 7.35 (td, J = 8.2, 1.6 Hz, 1H), 6.67 (s, 1H), 6.58 (s, 1H), 6.53 (dd, J = 11.8, 2.7 Hz, 1H), 6.43 (ddd, J = 11.7, 8.1, 1.3 Hz, 1H), 6.33 (t, J = 7.8 Hz, 1H), 6.28 (ddd, J = 9.1, 2.6, 1.2 Hz, 1H), 6.25 – 6.16 (m, 1H), 5.72 (d, J = 5.2 Hz, 1H), 3.12 (s, 3H), 3.11 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −125.74 (t, J = 10.7 Hz), −129.71 – −129.91 (m). 13C NMR (214 MHz, DMSO-d6) δ 159.76, 152.74, 152.38 (d, J = 245.3 Hz), 152.19, 152.15 (d, J = 241.9 Hz), 149.52, 149.01, 148.54 (d, J = 10.2 Hz), 146.54, 127.36 (d, J = 10.1 Hz), 125.08 (d, J = 10.6 Hz), 124.71 (d, J = 3.7 Hz), 122.93 (d, J = 7.4 Hz), 121.77 (d, J = 2.9 Hz), 120.73, 117.24 (d, J = 3.1 Hz), 115.22, 115.18 (d, J = 19.9 Hz), 109.14 (d, J = 21.9 Hz), 107.89, 103.49, 99.16, 55.86, 55.84. HRMS: calcd for C24H20F2N3O4 [M + H]+ m/z, 452.1422; found m/z, 452.1403.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-fluorophenyl)urea (8).
To a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (60 mg, 0.19 mmol, 1 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (40 mg, 0.25 mmol, 1.3 eq). The reaction mixture was stirred at rt for 2.5 h, followed by stirring at 45°C for 1.5 h, monitoring reaction progress by TLC. A solution of 4-fluoroaniline (25 mg, 0.23 mmol, 1.2 eq) in DMSO (1 mL) was added. The reaction mixture was stirred at 45°C for 3 h, then cooled to rt. Water (10 mL) was added to precipitate a white solid. The solid was collected by filtration, washed with water and dried. The crude material was dissolved in a mixture of DCM and MeOH, concentrated onto Celite and purified by flash chromatography (0–10% MeOH in EtOAc on silica gel column) to afford 8 (9.9 mg, 11% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.10 (s, 1H), 8.60 (d, J = 2.4 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.22 (t, J = 9.1 Hz, 1H), 7.56 – 7.43 (m, 3H), 7.40 (s, 1H), 7.34 (dd, J = 11.8, 2.7 Hz, 1H), 7.18 – 7.07 (m, 3H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −121.14 (ddd, J = 13.6, 8.9, 4.9 Hz), −125.79 (t, J = 10.6 Hz). 13C NMR (101 MHz, DMSO-d6) δ 159.63, 157.48 (d, J = 238.2 Hz), 152.59, 152.32, 152.31 (d, J = 244.3 Hz), 149.39, 148.88, 148.31 (d, J = 10.3 Hz), 146.49, 135.72 (d, J = 2.3 Hz), 125.16 (d, J = 10.7 Hz), 121.71 (d, J = 3.2 Hz), 119.90 (d, J = 7.6 Hz), 117.10, 115.42 (d, J = 22.2 Hz), 115.08, 109.02 (d, J = 21.9 Hz), 107.87, 103.32, 99.03, 55.74, 55.72. HRMS: calcd for C24H20F2N3O4 [M + H]+ m/z, 452.1422; found m/z, 452.1403.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-phenylurea (9).
To a solution of aniline (14 mg, 0.15 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (27 mg, 0.17 mmol, 1.3 eq). The reaction mixture was stirred at rt for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (40 mg, 0.13 mmol, 1 eq) in DMSO (1 mL). The reaction mixture was stirred at 45°C for 3 h, followed by stirring at 60°C for 2 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–4% MeOH in EtOAc on silica gel column) to afford 9 (18.5 mg, 34% yield) as a light purple solid. 1H NMR (400 MHz, DMSO-d6) δ 9.07 (s, 1H), 8.62 (d, J = 2.5 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.25 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.47 (dd, J = 8.6, 1.2 Hz, 2H), 7.40 (s, 1H), 7.35 (dd, J = 11.8, 2.7 Hz, 1H), 7.30 (dd, J = 8.5, 7.3 Hz, 2H), 7.10 (ddd, J = 8.9, 2.7, 1.2 Hz, 1H), 7.00 (tt, J = 7.3, 1.2 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −125.93 (dd, J = 10.4, 9.5 Hz). 13C NMR (101 MHz, DMSO-d6) δ 160.09, 153.03, 152.67 (d, J = 244.4 Hz), 152.66, 149.82, 149.31, 148.66 (d, J = 10.4 Hz), 146.93, 139.83, 129.34, 125.67 (d, J = 10.6 Hz), 122.57, 122.02 (d, J = 2.8 Hz), 118.57, 117.55 (d, J = 3.1 Hz), 115.51, 109.45 (d, J = 22.1 Hz), 108.31, 103.74, 99.47, 56.18, 56.15. HRMS: calcd for C24H21FN3O4 [M + H]+ m/z, 434.1516; found m/z, 434.1498.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)urea (12).
To a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (40 mg, 0.13 mmol, 1 eq) in acetic acid (0.25 mL) was added a solution of sodium cyanate (12 mg, 0.19 mmol, 1.5 eq) in water (0.2 mL). The reaction mixture was stirred at rt for 2 h, then concentrated under reduced pressure. Saturated aqueous NaHCO3 solution was added, adjusting pH to 8. The solid formed was collected by filtration, washed with water and dried. The crude material was dissolved in a mixture of DCM and MeOH, concentrated onto Celite and purified by flash chromatography (0–6% MeOH in DCM on silica gel column) to afford 12 (28 mg, 61% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.48 (d, J = 5.3 Hz, 1H), 8.42 (d, J = 2.4 Hz, 1H), 8.23 (t, J = 9.2 Hz, 1H), 7.49 (s, 1H), 7.39 (s, 1H), 7.27 (dd, J = 11.9, 2.7 Hz, 1H), 7.03 (ddd, J = 8.9, 2.7, 1.3 Hz, 1H), 6.50 (d, J = 5.2 Hz, 1H), 6.21 (s, 2H), 3.94 (s, 3H), 3.93 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −126.54 (t, J = 10.9 Hz). 13C NMR (101 MHz, DMSO-d6) δ 159.77, 155.67, 152.56, 151.82 (d, J = 243.8 Hz), 149.34, 148.86, 147.45 (d, J = 10.4 Hz), 146.45, 126.08 (d, J = 10.6 Hz), 121.31 (d, J = 3.4 Hz), 116.98 (d, J = 3.1 Hz), 115.02, 108.84 (d, J = 22.2 Hz), 107.85, 103.13, 99.03, 55.72, 55.70. HRMS: calcd for C18H17FN3O4 [M + H]+ m/z, 358.1203; found m/z, 358.1186.
N-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)acetamide (13).
To a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (25 mg, 0.080 mmol, 1 eq) and diisopropylethylamine (30 mg, 0.23 mmol, 2.9 eq) in 1,4-dioxane (0.5 mL) cooled to 0°C using an ice bath was added a solution of acetyl chloride (18 mg, 0.22 mmol, 2.8 eq) in 1,4-dioxane (0.5 mL). The reaction mixture was warmed to rt and stirred at rt for 2.5 h. The reaction mixture was concentrated onto Celite and purified by flash chromatography (0–3% MeOH in EtOAc on silica gel column) to afford 13 (26 mg, 92% yield) as a pink solid. 1H NMR (400 MHz, DMSO-d6) δ 9.81 (s, 1H), 8.50 (d, J = 5.2 Hz, 1H), 7.97 (t, J = 8.9 Hz, 1H), 7.47 (s, 1H), 7.40 (s, 1H), 7.32 (dd, J = 11.4, 2.7 Hz, 1H), 7.08 (ddd, J = 8.9, 2.7, 1.2 Hz, 1H), 6.56 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.92 (s, 3H), 2.11 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −120.69 (dd, J = 10.3, 9.5 Hz). 13C NMR (101 MHz, DMSO-d6) δ 168.72, 159.30, 153.94 (d, J = 247.7 Hz), 152.62, 150.41 (d, J = 10.4 Hz), 149.43, 148.88, 146.54, 125.32, 123.76 (d, J = 11.6 Hz), 116.67 (d, J = 3.4 Hz), 115.16, 109.06 (d, J = 22.7 Hz), 107.88, 103.67, 99.00, 55.75, 55.71, 23.41. HRMS: calcd for C19H18FN2O4 [M + H]+ m/z, 357.1251; found m/z, 357.1233.
Experimental Procedures in Scheme 2.
1-(2,4-difluorophenyl)-3-(2-fluoro-4-methoxyphenyl)urea (6).
To a solution of 2-fluoro-4-methoxyaniline (86) (70 mg, 0.50 mmol, 1 eq) in acetonitrile (0.8 mL) at room temperature was added 2,4-difluoro-1-isocyanatobenzene (77 mg, 0.50 mmol, 1 eq) dropwise. The resultant solution was stirred at room temperature for 30 minutes. Water (10 mL) was added and the solid was collected by filtration, washed with water and dried. The crude material was dissolved in a mixture of DCM and MeOH, concentrated onto Celite and purified by flash chromatography (10% EtOAc in Hexanes on silica gel column) to afford 6 (46 mg, 32% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.80 (d, J = 1.8 Hz, 1H), 8.69 (s, 1H), 8.10 (td, J = 9.2, 6.1 Hz, 1H), 7.89 (t, J = 9.2 Hz, 1H), 7.30 (ddd, J = 11.7, 8.8, 2.9 Hz, 1H), 7.04 (t, J = 8.8 Hz, 1H), 6.90 (dd, J = 13.0, 2.8 Hz, 1H), 6.75 (dd, J = 8.6, 1.8 Hz, 1H), 3.74 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −118.16 – −119.01 (m), −125.08 (t, J = 10.5 Hz), −126.21 (t, J = 10.9 Hz). 13C NMR (101 MHz, DMSO-d6) δ 156.67 (dd, J = 241.3, 11.8 Hz), 155.33 (d, J = 10.2 Hz), 153.24 (d, J = 242.2 Hz), 152.35, 151.94 (dd, J = 244.4, 11.7 Hz), 124.12 (dd, J = 10.7, 3.7 Hz), 122.62 (d, J = 3.1 Hz), 121.66 (dd, J = 9.2, 3.2 Hz), 119.83 (d, J = 11.3 Hz), 111.02 (dd, J = 21.6, 3.4 Hz), 109.70 (d, J = 2.9 Hz), 103.76 (dd, J = 27.0, 23.7 Hz), 101.72 (d, J = 22.9 Hz), 55.62. HRMS: calcd for C14H12F3N2O2 [M + H]+ m/z, 297.0851; found m/z, 297.0842.
1-(2,4-difluorophenyl)-3-(2-fluoro-4-hydroxyphenyl)urea (5).
To a solution of 4-amino-3-fluorophenol (85) (250 mg, 1.97 mmol, 1 eq) in acetonitrile (0.8 mL) at room temperature was added 2,4-difluoro-1-isocyanatobenzene (305 mg, 1.97 mmol, 1 eq) dropwise. The resultant solution was stirred at room temperature for 30 minutes. Water (10 mL) was added and the solid was collected by filtration, washed with water and dried. The crude material was dissolved in a mixture of DCM and MeOH, concentrated onto Celite and purified by flash chromatography (0–80% EtOAc in Hexanes on silica gel column) to afford 5 (197 mg, 36% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.59 (s, 1H), 8.74 (d, J = 2.3 Hz, 1H), 8.55 (s, 1H), 8.14 – 8.04 (m, 1H), 7.71 (t, J = 9.2 Hz, 1H), 7.29 (ddd, J = 11.7, 8.9, 2.9 Hz, 1H), 7.03 (t, J = 2.3 Hz, 0H), 6.62 (dd, J = 12.7, 2.6 Hz, 1H), 6.56 (ddd, J = 8.9, 2.7, 1.1 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −118.50 – −118.65 (m), −125.18 (t, J = 10.7 Hz), −126.41 (dd, J = 11.1, 10.5 Hz). 13C NMR (101 MHz, DMSO-d6) δ 156.59 (dd, J = 241.0, 11.5 Hz), 153.80 (d, J = 11.0 Hz), 153.58 (d, J = 241.8 Hz), 152.46, 151.88 (dd, J = 244.5, 12.4 Hz), 124.24 (dd, J = 10.6, 3.5 Hz), 123.35 (d, J = 3.2 Hz), 121.57 (dd, J = 9.1, 3.0 Hz), 118.12 (d, J = 11.4 Hz), 110.99 (dd, J = 21.8, 3.3 Hz), 110.92 (d, J = 3.0 Hz), 103.72 (dd, J = 26.9, 23.6 Hz), 102.59 (d, J = 21.7 Hz). HRMS: calcd for C13H10F3N2O2 [M + H]+ m/z, 283.0694; found m/z, 283.0681.
1-(2,4-difluorophenyl)-3-(2-fluoro-4-(quinolin-4-yloxy)phenyl)urea (3).
To a solution of 1-(2,4-difluorophenyl)-3-(2-fluoro-4-hydroxyphenyl)urea (5) (85 mg, 0.30 mmol, 1.1 eq) and 4-chloroquinoline (45 mg, 0.28 mmol, 1 eq) in NMP (2 mL) was added DIPEA (71 mg, 0.55 mmol, 2 eq). The solution was heated at 90°C for 1.5 h. The reaction mixture was cooled to rt and partitioned between EtOAc (30 mL) and water (20 mL). The aqueous layer was extracted with EtOAc (30 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (10–70% EtOAc in Hexanes on silica gel column) to afford 3 (11 mg, 10% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.07 (d, J = 2.4 Hz, 1H), 9.00 (d, J = 2.3 Hz, 1H), 8.71 (d, J = 5.1 Hz, 1H), 8.34 – 8.23 (m, 2H), 8.13 (td, J = 9.2, 6.1 Hz, 1H), 8.04 (d, J = 8.5 Hz, 1H), 7.83 (ddd, J = 8.5, 6.8, 1.5 Hz, 1H), 7.67 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 7.40 (dd, J = 11.8, 2.7 Hz, 1H), 7.33 (ddd, J = 11.7, 8.9, 2.9 Hz, 1H), 7.14 (ddd, J = 9.0, 2.7, 1.3 Hz, 1H), 7.10 – 7.01 (m, 1H), 6.69 (d, J = 5.1 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −117.88 – −118.03 (m), −124.82 (t, J = 10.1 Hz), −125.61 (t, J = 10.5 Hz). 13C NMR (101 MHz, DMSO-d6) δ 160.88, 156.87 (dd, J = 241.5, 11.6 Hz), 152.25 (d, J = 244.7 Hz), 152.12, 152.07 (dd, J = 244.8, 12.3 Hz), 151.49, 149.24, 148.17 (d, J = 10.5 Hz), 130.31, 128.82, 126.45, 125.19 (d, J = 10.7 Hz), 123.87 (dd, J = 10.8, 3.5 Hz), 121.79 (dd, J = 9.1, 3.0 Hz), 121.62 (d, J = 2.9 Hz), 121.43, 120.52, 117.17 (d, J = 3.3 Hz), 111.12 (dd, J = 21.5, 3.5 Hz), 109.11 (d, J = 22.1 Hz), 104.43, 103.85 (dd, J = 26.9, 23.6 Hz). HRMS: calcd for C22H15F3N3O2 [M + H]+ m/z, 410.1116; found m/z, 410.1098.
1-(2,4-difluorophenyl)-3-(2-fluoro-4-(pyridin-4-yloxy)phenyl)urea (4).
To a solution of 1-(2,4-difluorophenyl)-3-(2-fluoro-4-hydroxyphenyl)urea (5) (48 mg, 0.17 mmol, 1.1 eq) and pyridin-4-yl trifluoromethanesulfonate (35 mg, 0.15 mmol, 1 eq) in DMF (1 mL) was added DIPEA (40 mg, 0.31 mmol, 2 eq). The solution was irradiated in a microwave reactor at 130°C for 3 h. The reaction mixture was cooled to rt and partitioned between EtOAc (30 mL) and water (20 mL). The aqueous layer was extracted with EtOAc (30 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–80% EtOAc in Hexanes on silica gel column) to afford 4 (3.3 mg, 6% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.04 (s, 1H), 8.99 (s, 1H), 8.49 – 8.44 (m, 2H), 8.21 (t, J = 9.1 Hz, 1H), 8.12 (td, J = 9.3, 6.1 Hz, 1H), 7.33 (ddd, J = 11.7, 8.9, 2.9 Hz, 1H), 7.26 (dd, J = 11.9, 2.7 Hz, 1H), 7.09 – 7.04 (m, 1H), 7.04 – 6.99 (m, 1H), 6.97 – 6.93 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −117.97 (tdd, J = 8.9, 6.0, 3.6 Hz), −124.79 (t, J = 9.6 Hz), −125.59 (t, J = 10.5 Hz). 13C NMR (214 MHz, DMSO-d6) δ 151.12, 143.95 (dd, J = 242.4, 12.0 Hz), 139.33 (d, J = 245.1 Hz), 139.19, 139.17 (dd, J = 245.1, 12.1 Hz), 138.53, 135.01 (d, J = 10.1 Hz), 111.93 (d, J = 10.9 Hz), 110.82 (dd, J = 10.8, 3.2 Hz), 108.96 (dd, J = 8.3, 3.3 Hz), 108.81 (dd, J = 5.8, 1.4 Hz), 103.92 (d, J = 2.9 Hz), 98.90, 98.15 (dd, J = 21.5, 3.2 Hz), 95.86 (d, J = 21.9 Hz), 90.87 (dd, J = 26.8, 23.6 Hz). HRMS: calcd for C18H13F3N3O2 [M + H]+ m/z, 360.0960; found m/z, 360.0944.
1-(4-((7-chloroquinolin-4-yl)oxy)-2-fluorophenyl)-3-(2,4-difluorophenyl)urea (17).
To a solution of 1-(2,4-difluorophenyl)-3-(2-fluoro-4-hydroxyphenyl)urea (5) (71 mg, 0.25 mmol, 1.1 eq) and 4,7-dichloroquinoline (45 mg, 0.23 mmol, 1 eq) in DMF (2 mL) was added DIPEA (59 mg, 0.45 mmol, 2 eq). The solution was heated at 120°C for 18 h. The reaction mixture was cooled to rt and partitioned between EtOAc (30 mL) and water (20 mL). The aqueous layer was extracted with EtOAc (30 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–80% EtOAc in Hexanes on silica gel column) to afford 17 (3.4 mg, 3% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.08 (dd, J = 29.9, 1.6 Hz, 1H), 9.00 (d, J = 1.5 Hz, 1H), 8.74 (d, J = 5.2 Hz, 1H), 8.33 (d, J = 8.9 Hz, 1H), 8.27 (t, J = 9.1 Hz, 1H), 8.17 – 8.08 (m, 2H), 7.70 (dd, J = 8.9, 2.2 Hz, 1H), 7.42 (dd, J = 11.8, 2.7 Hz, 1H), 7.33 (ddd, J = 11.7, 8.8, 2.9 Hz, 1H), 7.16 (dt, J = 8.8, 1.5 Hz, 1H), 7.07 (t, J = 8.9 Hz, 1H), 6.72 (d, J = 5.2 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −117.92 (tdd, J = 8.9, 6.1, 3.6 Hz), −124.79 (t, J = 10.5 Hz), −125.53 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 161.17, 157.01 (dd, J = 241.4, 11.5 Hz), 153.13, 152.33 (d, J = 245.2 Hz), 152.22, 152.21 (dd, J = 244.6, 12.2 Hz), 149.70, 147.94 (d, J = 10.1 Hz), 135.11, 127.51, 127.15, 125.45 (d, J = 10.3 Hz), 123.91 (d, J = 2.8 Hz), 123.86, 121.99 (dd, J = 9.0, 2.4 Hz), 121.76 (d, J = 1.8 Hz), 119.28, 117.31, 111.21 (dd, J = 21.4, 3.0 Hz), 109.25 (d, J = 22.2 Hz), 104.87, 103.94 (dd, J = 26.8, 23.7 Hz). HRMS: calcd for C22H14ClF3N3O2 [M + H]+ m/z, 444.0727; found m/z, 444.0708.
1-(2,4-difluorophenyl)-3-(4-((6,7-dimethoxyquinazolin-4-yl)oxy)-2-fluorophenyl)urea (22).
To a solution of 1-(2,4-difluorophenyl)-3-(2-fluoro-4-hydroxyphenyl)urea (5) (62 mg, 0.22 mmol, 1.1 eq) and 4-chloro-6,7-dimethoxyquinazoline (45 mg, 0.20 mmol, 1 eq) in DMF (2 mL) was added DIPEA (52 mg, 0.40 mmol, 2 eq). The solution was heated at 110°C for 18 h. The reaction mixture was cooled to rt and partitioned between EtOAc (30 mL) and water (20 mL). The aqueous layer was extracted with EtOAc (30 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (10–100% EtOAc in Hexanes on silica gel column) to afford 22 (6.7 mg, 7% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.02 (d, J = 2.3 Hz, 1H), 8.99 (d, J = 2.2 Hz, 1H), 8.57 (s, 1H), 8.20 (t, J = 9.1 Hz, 1H), 8.14 (td, J = 9.2, 6.1 Hz, 1H), 7.55 (s, 1H), 7.43 – 7.38 (m, 2H), 7.33 (ddd, J = 11.6, 8.8, 2.8 Hz, 1H), 7.14 (d, J = 8.7 Hz, 1H), 7.10 – 7.02 (m, 1H), 3.99 (s, 3H), 3.98 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −117.52 – −119.07 (m), −124.83 (t, J = 10.2 Hz), −126.88 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 165.14, 157.40 (dd, J = 242.2, 11.6 Hz), 156.23, 152.68, 152.65 (dd, J = 245.5, 12.4 Hz), 152.53, 152.48 (d, J = 244.0 Hz), 150.51, 149.12, 147.29 (d, J = 10.9 Hz), 125.16 (d, J = 10.8 Hz), 124.04 (dd, J = 10.3, 3.2 Hz), 122.58 (dd, J = 8.5, 2.4 Hz), 121.77 (d, J = 2.4 Hz), 118.57 (d, J = 2.6 Hz), 111.50 (dd, J = 21.8, 3.4 Hz), 110.54 (d, J = 22.1 Hz), 110.04, 106.90, 104.20 (dd, J = 27.0, 23.4 Hz), 101.08, 56.55, 56.39. HRMS: calcd for C23H18F3N4O4 [M + H]+ m/z, 471.1280; found m/z, 471.1262.
1-(2,4-difluorophenyl)-3-(2-fluoro-4-(thieno[3,2-d]pyrimidin-4-yloxy)phenyl)urea (26).
To a solution of 1-(2,4-difluorophenyl)-3-(2-fluoro-4-hydroxyphenyl)urea (5) (64 mg, 0.23 mmol, 1.1 eq) and 4-chloro-6,7-dimethoxyquinazoline (35 mg, 0.21 mmol, 1 eq) in DMF (1.5 mL) was added DIPEA (53 mg, 0.41 mmol, 2 eq). The solution was heated at 120°C for 18 h. The reaction mixture was cooled to rt and partitioned between EtOAc (30 mL) and water (20 mL). The aqueous layer was extracted with EtOAc (30 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–50% EtOAc in Hexanes on silica gel column) to afford 26 (4.4 mg, 5% yield) as an orange-brown solid. 1H NMR (400 MHz, DMSO-d6) δ 9.04 (d, J = 2.4 Hz, 1H), 8.99 (d, J = 2.2 Hz, 1H), 8.73 (s, 1H), 8.49 (d, J = 5.4 Hz, 1H), 8.20 (t, J = 9.1 Hz, 1H), 8.13 (td, J = 9.2, 6.1 Hz, 1H), 7.69 (d, J = 5.4 Hz, 1H), 7.45 (dd, J = 11.8, 2.7 Hz, 1H), 7.33 (ddd, J = 11.6, 8.8, 2.9 Hz, 1H), 7.17 (dq, J = 9.0, 1.2 Hz, 1H), 7.07 (t, J = 8.8 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −116.75 – −120.17 (m), −124.82 (t, J = 10.4 Hz), −126.65 (dd, J = 11.8, 9.2 Hz). 13C NMR (101 MHz, DMSO-d6) δ 163.60, 163.19, 154.11, 152.14, 152.06 (dd, J = 244.6, 11.7 Hz), 151.85 (d, J = 243.7 Hz), 146.02 (d, J = 10.6 Hz), 137.44, 125.36 (d, J = 10.7 Hz), 124.29, 123.89 (d, J = 7.0 Hz), 121.78 (d, J = 10.7 Hz), 121.00, 118.15 (d, J = 3.3 Hz), 116.81, 111.10 (d, J = 21.9 Hz), 110.18 (d, J = 22.6 Hz), 103.84. (only one doublet of the dd was observed at 158.06 ppm (d, J = 11.2 Hz), the other doublet was not observed due to signal to noise). HRMS: calcd for C19H12F3N4O2S [M + H]+ m/z, 417.0633; found m/z, 417.0616.
Experimental Procedures in Schemes 3 and 4.
4-(3-fluoro-4-nitrophenoxy)-7-methoxyquinoline (88).
A suspension of 4-chloro-7-methoxyquinoline (250 mg, 1.29 mmol, 1 eq) and 3-fluoro-4-nitrophenol (87) (406 mg, 2.58 mmol, 2 eq) in o-xylene (3.5 mL) was stirred at 135°C for 16 h. The reaction mixture was cooled to rt. EtOAc (15 mL) was added and the mixture was stirred for 1 h at rt. The solid formed was collected by filtration, washed with EtOAc and dried. The solid was added to an aqueous NaOH solution (1 M, 20 mL) and stirred for 1 h at rt. The solids were collected by filtration, washed with water, and dried under vacuum to afford 88 (265 mg, 65% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.78 (d, J = 5.1 Hz, 1H), 8.27 (t, J = 8.9 Hz, 1H), 8.03 (d, J = 9.2 Hz, 1H), 7.59 (dd, J = 12.3, 2.6 Hz, 1H), 7.48 (d, J = 2.5 Hz, 1H), 7.31 (dd, J = 9.2, 2.5 Hz, 1H), 7.21 (ddd, J = 9.2, 2.7, 1.2 Hz, 1H), 6.98 (d, J = 5.1 Hz, 1H), 3.95 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −113.92 (dd, J = 12.3, 8.8 Hz).
2-fluoro-4-((7-methoxyquinolin-4-yl)oxy)aniline (100).
A suspension of 4-(3-fluoro-4-nitrophenoxy)-7-methoxyquinoline (88) (263 mg, 0.84 mmol, 1 eq), iron (234 mg, 4.2 mmol, 5 eq), and ammonium chloride (358 mg, 6.7 mmol, 8 eq) in ethanol (4.5 mL) and water (15 mL) was heated at 78°C for 6 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between DCM (30 mL) and aqueous NaOH solution (1 M, 30 mL). The aqueous layer was extracted with DCM (2 × 30 mL). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–6% MeOH in DCM on silica gel column) to afford 100 (203 mg, 85% yield) as an orange solid. 1H NMR (400 MHz, DMSO-d6) δ 8.58 (d, J = 5.2 Hz, 1H), 8.18 (d, J = 9.1 Hz, 1H), 7.38 (d, J = 2.5 Hz, 1H), 7.26 (dd, J = 9.2, 2.6 Hz, 1H), 7.07 (dd, J = 12.2, 2.1 Hz, 1H), 6.96 – 6.78 (m, 2H), 6.43 (d, J = 5.2 Hz, 1H), 5.20 (s, 2H), 3.93 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −131.33 (dd, J = 11.9, 8.5 Hz). 13C NMR (214 MHz, DMSO-d6) δ 162.14, 160.94, 151.71, 150.81, 150.26 (d, J = 239.8 Hz), 142.75 (d, J = 9.5 Hz), 134.67 (d, J = 12.5 Hz), 122.99, 118.75, 117.50 (d, J = 2.9 Hz), 116.56 (d, J = 5.6 Hz), 115.13, 109.17 (d, J = 21.0 Hz), 107.07, 102.26, 55.61.
1-(2,4-difluorophenyl)-3-(2-fluoro-4-((7-methoxyquinolin-4-yl)oxy)phenyl)urea (1).
To a solution of 2-fluoro-4-((7-methoxyquinolin-4-yl)oxy)aniline (100) (50 mg, 0.18 mmol, 1 eq) in ethanol (0.5 mL) cooled to 0°C using an ice bath was added 2,4-difluoro-1-isocyanatobenzene (27 mg, 0.18 mmol, 1 eq) dropwise. The reaction mixture was warmed to rt and stirred at rt for 1 h. The reaction mixture was concentrated onto Celite and purified by flash chromatography (0–3% MeOH in DCM on silica gel column) to afford 1 (24 mg, 31% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.06 (d, J = 2.4 Hz, 1H), 8.99 (d, J = 2.3 Hz, 1H), 8.63 (d, J = 5.2 Hz, 1H), 8.25 (t, J = 9.1 Hz, 1H), 8.18 (d, J = 9.1 Hz, 1H), 8.13 (td, J = 9.2, 6.1 Hz, 1H), 7.41 (d, J = 2.6 Hz, 1H), 7.40 – 7.26 (m, 3H), 7.15 – 7.09 (m, 1H), 7.06 (t, J = 8.1 Hz, 1H), 3.94 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −117.92 – −118.01 (m), −124.83 (t, J = 9.7 Hz), −125.67 (t, J = 10.7 Hz). 13C NMR (101 MHz, DMSO-d6) δ 160.90, 160.81, 156.90 (dd, J = 241.6, 11.5 Hz), 152.27 (d, J = 244.6 Hz), 152.15, 152.10 (dd, J = 245.0, 12.2 Hz), 151.88, 151.30, 148.27 (d, J = 10.3 Hz), 125.11 (d, J = 10.8 Hz), 123.89 (dd, J = 10.7, 3.5 Hz), 122.78, 121.83 (dd, J = 9.1, 2.6 Hz), 121.64 (d, J = 2.8 Hz), 118.85, 117.13 (d, J = 3.2 Hz), 115.16, 111.14 (dd, J = 21.6, 3.5 Hz), 109.07 (d, J = 22.1 Hz), 107.43, 103.87 (dd, J = 27.1, 23.6 Hz), 102.95, 55.55. HRMS: calcd for C23H17F3N3O3 [M + H]+ m/z, 440.1222; found m/z, 440.1204.
4-(3-fluoro-4-nitrophenoxy)-6-methoxyquinoline hydrochloride (89).
A suspension of 4-chloro-6-methoxyquinoline (250 mg, 1.25 mmol, 1 eq) and 3-fluoro-4-nitrophenol (87) (406 mg, 2.58 mmol, 2 eq) in o-xylene (3.5 mL) was stirred at 135°C for 16 h. The reaction mixture was cooled to rt. EtOAc (15 mL) was added and the mixture was stirred for 1 h at rt. The solid formed was collected by filtration, washed with EtOAc and dried. The solid was added to an aqueous NaOH solution (1 M, 20 mL) and stirred for 1 h at rt. The solids were collected by filtration, washed with water, and dried. The residue was then transferred to an aqueous HCl solution (2 M, 5 mL) and the aqueous solution was decanted. EtOAc was added to the remaining residue, and decanted. The remaining residue was dried under vacuum to afford 89 (83 mg, 20% yield) as a light brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.97 (d, J = 6.1 Hz, 1H), 8.41 (t, J = 8.9 Hz, 1H), 8.28 (d, J = 9.3 Hz, 1H), 7.90 – 7.75 (m, 2H), 7.69 (d, J = 2.7 Hz, 1H), 7.57 – 7.43 (m, 1H), 7.33 (d, J = 6.1 Hz, 1H), 3.99 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −113.59 (dd, J = 11.9, 8.6 Hz).
2-fluoro-4-((6-methoxyquinolin-4-yl)oxy)aniline (101).
A suspension of 4-(3-fluoro-4-nitrophenoxy)-6-methoxyquinoline (89) (83 mg, 0.26 mmol, 1 eq), iron (74 mg, 1.3 mmol, 5 eq), and ammonium chloride (110 mg, 2.1 mmol, 8 eq) in ethanol (1.5 mL) and water (0.5 mL) was heated at 78°C for 5 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between DCM (30 mL) and aqueous NaOH solution (1 M, 30 mL). The aqueous layer was extracted with DCM (2 × 30 mL). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–6% MeOH in DCM on silica gel column) to afford 101 (55 mg, 73% yield) as a yellow solid. 1H NMR (400 MHz, Methanol-d4) δ 8.38 (d, J = 5.3 Hz, 1H), 7.84 (d, J = 9.3 Hz, 1H), 7.57 (d, J = 2.8 Hz, 1H), 7.37 (dd, J = 9.2, 2.8 Hz, 1H), 6.98 – 6.87 (m, 2H), 6.80 (ddd, J = 8.6, 2.6, 1.2 Hz, 1H), 6.53 (d, J = 5.3 Hz, 1H), 3.89 (s, 3H). 19F NMR (376 MHz, Methanol-d4) δ −132.99 (t, J = 10.7 Hz).
1-(2,4-difluorophenyl)-3-(2-fluoro-4-((6-methoxyquinolin-4-yl)oxy)phenyl)urea (2).
To a solution of 2-fluoro-4-((6-methoxyquinolin-4-yl)oxy)aniline (101) (53 mg, 0.19 mmol, 1 eq) in ethanol (0.55 mL) cooled to 0°C using an ice bath was added 2,4-difluoro-1-isocyanatobenzene (29 mg, 0.19 mmol, 1 eq) dropwise. The reaction mixture was warmed to rt and stirred at rt for 1 h. The reaction mixture was concentrated onto Celite and purified by flash chromatography (0–10% MeOH in DCM on silica gel column) to afford 2 (40 mg, 49% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.07 (d, J = 2.4 Hz, 1H), 9.00 (d, J = 2.2 Hz, 1H), 8.56 (d, J = 5.1 Hz, 1H), 8.27 (t, J = 9.1 Hz, 1H), 8.13 (td, J = 9.2, 6.1 Hz, 1H), 7.95 (d, J = 9.2 Hz, 1H), 7.55 (d, J = 2.9 Hz, 1H), 7.47 (dd, J = 9.2, 2.9 Hz, 1H), 7.38 (dd, J = 11.8, 2.7 Hz, 1H), 7.33 (ddd, J = 11.7, 8.5, 2.5 Hz, 1H), 7.13 (ddd, J = 9.1, 2.7, 1.3 Hz, 1H), 7.10 – 7.03 (m, 1H), 6.66 (d, J = 5.1 Hz, 1H), 3.93 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −117.96 (tdd, J = 8.9, 6.0, 3.6 Hz), −124.81 (t, J = 10.7 Hz), −125.67 (t, J = 10.6 Hz). 13C NMR (101 MHz, DMSO-d6) δ 159.96, 157.35, 156.87 (dd, J = 241.1, 11.6 Hz), 152.23 (d, J = 244.7 Hz), 152.12, 152.07 (dd, J = 245.2, 11.8 Hz), 148.73, 148.28 (d, J = 10.4 Hz), 145.27, 130.53, 125.10 (d, J = 10.7 Hz), 123.87 (dd, J = 10.7, 3.8 Hz), 122.63, 121.80 (dd, J = 9.1, 3.0 Hz), 121.58 (d, J = 2.6 Hz), 121.33, 117.19 (d, J = 2.9 Hz), 111.12 (dd, J = 21.5, 3.6 Hz), 109.11 (d, J = 22.0 Hz), 104.76, 103.85 (dd, J = 26.9, 23.9 Hz), 99.13, 55.60. HRMS: calcd for C23H17F3N3O3 [M + H]+ m/z, 440.1222; found m/z, 440.1214.
4-oxo-1,4-dihydroquinoline-3-carbonitrile (121).
To a solution of methyl 2-aminobenzoate (120) (200 mg, 1.3 mmol, 1 eq) in DMF (1.5 mL) was added N,N-dimethylformamide dimethyl acetal (473 mg, 4.0 mmol, 3 eq). The reaction mixture was refluxed at 150°C for 3 h, then cooled to rt. Repeatedly, toluene was added and volatiles were removed in vacuo until the crude formamidine was obtained as a dark purple liquid. Separately, to a degassed round-bottom-flask with anhydrous THF (2 mL) cooled to −78°C using a dry-ice/acetone bath was added n-butyllithium (2.5 M in hexanes, 1.3 mL, 2.5 eq), a solution of anhydrous MeCN (136 mg, 0.17 mL, 2.5 eq) in anhydrous THF (2.6 mL) dropwise, and the crude formamidine dissolved in anhydrous THF (2.6 mL) dropwise. The reaction mixture was stirred at −78°C for 1 h, then quenched by dropwise addition of water (0.5 mL) and acetic acid (0.3 mL), warmed to rt and concentrated in vacuo. Water was added, and the solids were collected by filtration, washed with water and chloroform, and dried in vacuo to afford 121 (180 mg, 80% yield) as an off- white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.81 (s, 1H), 8.74 (s, 1H), 8.13 (dd, J = 8.1, 1.5 Hz, 1H), 7.78 (ddd, J = 8.5, 7.0, 1.5 Hz, 1H), 7.64 (d, J = 8.3 Hz, 1H), 7.48 (ddd, J = 8.1, 7.1, 1.1 Hz, 1H). 13C NMR (214 MHz, DMSO-d6) δ 177.66, 149.81, 142.21, 136.49, 128.74, 128.23, 128.14, 122.36, 119.93, 96.68.
4-chloroquinoline-3-carbonitrile (122).
A mixture of 4-oxo-1,4-dihydroquinoline-3-carbonitrile (121) (175 mg, 1.03 mmol, 1 eq) in phosphoryl trichloride (2 mL) was refluxed at 100°C for 2 h. The reaction was cooled to rt, and concentrated in vacuo. The mixture was then cooled to 0°C using an ice bath. Water (20 mL) and dichloromethane (20 mL) was carefully added to the mixture while stirring, followed by careful addition of solid potassium carbonate until the pH was between 8–9. The mixture was left to stir for 30 min at rt. The organic layer was separated, washed with water, then brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give 122 (185 mg, 95% yield) as a light brown solid. 1H NMR (400 MHz, DMSO-d6) δ 9.21 (s, 1H), 8.34 (dd, J = 8.5, 1.4 Hz, 1H), 8.21 (dd, J = 8.5, 1.2 Hz, 1H), 8.08 (ddd, J = 8.5, 6.9, 1.4 Hz, 1H), 7.93 (ddd, J = 8.3, 6.9, 1.2 Hz, 1H). 13C NMR (214 MHz, DMSO-d6) δ 174.50, 146.64, 139.14, 133.30, 125.56, 125.13, 125.00, 119.26, 116.83, 93.55.
4-(3-fluoro-4-nitrophenoxy)quinoline-3-carbonitrile (90).
A suspension of 4-chloroquinoline-3-carbonitrile (122) (175 mg, 0.928 mmol, 1 eq) and 3-fluoro-4-nitrophenol (87) (262 mg, 1.67 mmol, 1.8 eq) in o-xylene (2.3 mL) was stirred at 135°C for 40 h. The reaction mixture was cooled to rt. EtOAc (15 mL) was added and the mixture was stirred for 1 h at rt. The solid formed was collected by filtration, washed with EtOAc and dried. The solid was added to an aqueous NaOH solution (1 M, 20 mL) and stirred for 1 h at rt. The solids were collected by filtration, washed with water, and dried under vacuum. The crude material was dissolved in a mixture of DCM and MeOH, concentrated onto Celite and purified by flash chromatography (0–35% EtOAc in Hexanes on silica gel column) to afford 90 (118 mg, 41% yield) as a light yellow-brown solid. 1H NMR (400 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.29 – 8.20 (m, 2H), 8.10 – 8.01 (m, 2H), 7.79 (ddd, J = 8.3, 6.9, 1.2 Hz, 1H), 7.62 (dd, J = 12.3, 2.7 Hz, 1H), 7.22 (ddd, J = 9.3, 2.7, 1.2 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −113.55 (dd, J = 12.4, 8.7 Hz). 13C NMR (214 MHz, DMSO-d6) δ 161.20 (d, J = 11.0 Hz), 160.21, 156.31 (d, J = 263.6 Hz), 151.74, 150.76, 133.83, 133.21 (d, J = 7.1 Hz), 129.73, 129.14, 128.73, 122.28, 120.52, 114.01, 113.35 (d, J = 3.2 Hz), 107.26 (d, J = 25.3 Hz), 98.88.
4-(4-amino-3-fluorophenoxy)quinoline-3-carbonitrile (102).
A suspension of 4-(3-fluoro-4-nitrophenoxy)quinoline-3-carbonitrile (90) (113 mg, 0.365 mmol, 1 eq), iron (102 mg, 1.83 mmol, 5 eq), and ammonium chloride (156 mg, 2.92 mmol, 8 eq) in ethanol (1.0 mL) and water (0.35 mL) was heated at 31°C for 4 h. The reaction mixture was cooled to rt and filtered through Celite. The filtrate was concentrated onto Celite, and purified by flash chromatography (0–40% EtOAc in Hexanes on silica gel column) to afford 102 (59 mg, 58% yield) as a yellow-brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.97 (s, 1H), 8.28 – 8.24 (m, 1H), 8.12 (dt, J = 8.5, 1.0 Hz, 1H), 7.99 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.75 (ddd, J = 8.3, 6.9, 1.2 Hz, 1H), 7.15 (dd, J = 11.8, 2.7 Hz, 1H), 6.88 – 6.75 (m, 2H), 5.21 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −131.65 (dd, J = 11.9, 9.7 Hz). 13C NMR (214 MHz, DMSO-d6) δ 163.43, 152.49, 150.02 (d, J = 239.8 Hz), 149.92, 145.01 (d, J = 9.7 Hz), 134.84 (d, J = 13.2 Hz), 133.26, 129.18, 128.16, 122.73, 120.44, 116.08 (d, J = 5.7 Hz), 115.78 (d, J = 3.0 Hz), 114.24, 107.83 (d, J = 22.4 Hz), 93.91.
1-(4-((3-cyanoquinolin-4-yl)oxy)-2-fluorophenyl)-3-(2,4-difluorophenyl)urea (18).
To a solution of 2,4-difluoroaniline (28 mg, 0.21 mmol, 2 eq) in DCM (1 mL) cooled to 0°C using an ice bath was added 1,1′-carbonyldiimidazole (39 mg, 0.24 mmol, 2.25 eq). The reaction mixture was stirred at 0°C for 30 min, monitoring reaction progress by TLC. This was added to a solution of 4-(4-amino-3-fluorophenoxy)quinoline-3-carbonitrile (102) (30 mg, 0.11 mmol, 1 eq) in DCM (0.5 mL). The reaction mixture was stirred at 35°C for 4 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–40% EtOAc in Hexanes on silica gel column) to afford 18 (18.3 mg, 40% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.09 (s, 1H), 9.02 (d, J = 2.4 Hz, 1H), 8.96 (d, J = 2.2 Hz, 1H), 8.23 – 8.14 (m, 3H), 8.10 (td, J = 9.2, 6.1 Hz, 1H), 8.02 (ddd, J = 8.5, 7.0, 1.4 Hz, 1H), 7.77 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 7.41 (dd, J = 11.8, 2.8 Hz, 1H), 7.32 (ddd, J = 11.6, 8.9, 2.9 Hz, 1H), 7.11 – 7.01 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −117.74 – −118.49 (m), −124.75 – −124.85 (m), −125.84 (ddt, J = 11.0, 9.3, 1.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 162.27, 156.89 (dd, J = 241.5, 11.6 Hz), 152.16, 152.15 (d, J = 244.8 Hz), 152.10 (dd, J = 245.0, 12.2 Hz), 152.09, 150.57 (d, J = 10.3 Hz), 150.29, 133.47, 129.40, 128.51, 124.79 (d, J = 10.5 Hz), 123.83 (dd, J = 10.8, 3.6 Hz), 122.61, 121.86 (dd, J = 9.1, 2.4 Hz), 121.37 (d, J = 2.2 Hz), 120.65, 114.43 (d, J = 3.2 Hz), 114.20, 111.10 (dd, J = 21.7, 3.5 Hz), 106.84 (d, J = 23.4 Hz), 103.84 (dd, J = 27.0, 23.6 Hz), 95.97. HRMS: calcd for C23H14F3N4O2 [M + H]+ m/z, 435.1069; found m/z, 435.1059.
4-(3-fluoro-4-nitrophenoxy)-6,7-bis(2-methoxyethoxy)quinoline (91).
A suspension of 4-chloro-6,7-bis(2-methoxyethoxy)quinoline (350 mg, 1.12 mmol, 1 eq) and 3-fluoro-4-nitrophenol (87) (353 mg, 2.25 mmol, 2 eq) in o-xylene (2.8 mL) was stirred at 135°C for 14 h. The reaction mixture was cooled to rt. EtOAc (10 mL) was added and the mixture was stirred for 1 h at rt. The solid formed was collected by filtration, washed with EtOAc and dried. The solid was added to an aqueous NaOH solution (1 M, 15 mL) and stirred for 1 h at rt. The solids were collected by filtration, washed with water, and dried under vacuum to afford 91 (290 mg, 60% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.64 (d, J = 5.1 Hz, 1H), 8.27 (t, J = 9.0 Hz, 1H), 7.55 (dd, J = 12.4, 2.6 Hz, 1H), 7.49 (s, 1H), 7.37 (s, 1H), 7.18 (ddd, J = 9.2, 2.7, 1.1 Hz, 1H), 6.98 (d, J = 5.1 Hz, 1H), 4.34 – 4.28 (m, 2H), 4.25 – 4.18 (m, 2H), 3.80 – 3.73 (m, 2H), 3.72 – 3.69 (m, 2H), 3.36 (s, 3H), 3.32 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −114.00 (dd, J = 12.4, 8.8 Hz). 13C NMR (214 MHz, DMSO-d6) δ 160.92 (d, J = 11.2 Hz), 156.75, 156.27 (d, J = 263.3 Hz), 152.18, 149.19, 149.05, 146.99, 133.13 (d, J = 6.9 Hz), 128.52, 115.87, 115.35 (d, J = 3.0 Hz), 109.27 (d, J = 24.1 Hz), 109.09, 107.24, 100.12, 70.07, 68.17, 68.11, 58.35, 58.30. (1 aliphatic carbon was not observed, probably due to overlap at the extraordinarily intense peak at 70.07 ppm).
4-((6,7-bis(2-methoxyethoxy)quinolin-4-yl)oxy)-2-fluoroaniline (103).
A suspension of 4-(3-fluoro-4-nitrophenoxy)-6,7-bis(2-methoxyethoxy)quinoline (91). (274 mg, 0.634 mmol, 1 eq), iron (177 mg, 3.17 mmol, 5 eq), and ammonium chloride (271 mg, 5.07 mmol, 8 eq) in ethanol (1.7 mL) and water (0.55 mL) was heated at 31°C for 4 h. The reaction mixture was cooled to rt and filtered through Celite. The filtrate was concentrated onto Celite, and purified by flash chromatography (0–10% MeOH in DCM on silica gel column) to afford 103 (226 mg, 89% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.53 (d, J = 5.6 Hz, 1H), 7.58 (s, 1H), 7.45 (s, 1H), 7.13 – 7.02 (m, 1H), 6.94 – 6.81 (m, 2H), 6.54 (d, J = 5.6 Hz, 1H), 5.32 (br s, 2H), 4.34 – 4.26 (m, 4H), 3.81 – 3.72 (m, 4H), 3.36 (s, 3H), 3.35 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −131.12 – −131.28 (m). 13C NMR (214 MHz, DMSO-d6) δ 161.91, 152.63, 150.12 (d, J = 240.2 Hz), 148.92, 147.51, 143.91, 142.46 (d, J = 9.1 Hz), 134.74 (d, J = 12.5 Hz), 117.42 (d, J = 3.2 Hz), 116.41 (d, J = 5.9 Hz), 115.02, 109.10 (d, J = 21.3 Hz), 106.82, 102.66, 100.74, 70.10, 70.04, 68.24, 68.19, 58.38, 58.33.
1-(4-((6,7-bis(2-methoxyethoxy)quinolin-4-yl)oxy)-2-fluorophenyl)-3-(2,4-difluorophenyl)urea (19).
To a solution of 2,4-difluoroaniline (32 mg, 0.25 mmol, 2 eq) in DCM (1.2 mL) cooled to 0°C using an ice bath was added 1,1′-carbonyldiimidazole (51 mg, 0.31 mmol, 2.5 eq). The reaction mixture was stirred at 0°C for 30 min. This was added to a solution of 4-((6,7-bis(2-methoxyethoxy)quinolin-4-yl)oxy)-2-fluoroaniline (103) (55 mg, 0.13 mmol, 1 eq) in DCM (0.7 mL). The reaction mixture was stirred at 40°C for 2.5 h. Separately, another solution of 2,4-difluoroaniline (22 mg, 0.17 mmol, 1.4 eq) in DCM (0.8 mL) cooled to 0°C was added 1,1′-carbonyldiimidazole (34 mg, 0.21 mmol, 1.7 eq) and stirred at 0°C for 30 min, followed by addition to the former reaction mixture. The reaction mixture was stirred at 40°C for 1 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (50–100% EtOAc in Hexanes on silica gel column) to afford 19 (45 mg, 64% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.05 (d, J = 2.4 Hz, 1H), 8.99 (d, J = 2.3 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.25 (t, J = 9.1 Hz, 1H), 8.13 (td, J = 9.2, 6.1 Hz, 1H), 7.52 (s, 1H), 7.43 (s, 1H), 7.40 – 7.26 (m, 2H), 7.18 – 6.98 (m, 2H), 6.54 (d, J = 5.2 Hz, 1H), 4.32 – 4.24 (m, 4H), 3.79 – 3.72 (m, 4H), 3.36 (s, 3H), 3.35 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −117.97 (tdd, J = 8.9, 6.1, 3.6 Hz), −124.78 – −124.86 (m), −125.75 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.57, 156.87 (dd, J = 241.6, 11.8 Hz), 152.22 (d, J = 244.5 Hz), 152.12, 152.07 (dd, J = 244.8, 12.3 Hz), 151.89, 148.97, 148.66, 148.44 (d, J = 10.4 Hz), 146.45, 124.94 (d, J = 10.8 Hz), 123.87 (dd, J = 10.9, 3.8 Hz), 121.80 (dd, J = 9.0, 2.4 Hz), 121.57, 117.08 (d, J = 3.1 Hz), 115.15, 111.10 (dd, J = 21.8, 3.3 Hz), 109.05, 108.95, 103.84 (dd, J = 26.9, 23.6 Hz), 103.39, 100.44, 70.14, 70.12, 68.12, 68.00, 58.36, 58.33. HRMS: calcd for C28H27F3N3O6 [M + H]+ m/z, 558.1852; found m/z, 558.1842.
4-chloroquinoline-6,7-diol (117).
To a solution of 4-chloro-6,7-dimethoxyquinoline (83) (1.40 g, 6.26 mmol, 1 eq) in DCM (45 mL) cooled to 0°C using an ice bath was added boron tribromide (4.23 g, 16.9 mL, 2.7 eq) dissolved in DCM (15 mL) dropwise. The solution was stirred at 0°C for 2 h, then allowed to warm slowly to rt and stir for 16 h. Water (70 mL) was added to quench the reaction and the pH was adjusted to pH 6 by addition of solid NaOH. 23 mmol, 1 eq) in DCM (1.1 mL). The reaction mixture was stirred at 40°C for 2.5 h, then cooled to rt. The aqueous layer was extracted with EtOAc (100 mL x 3). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4 and filtered. The solvent was removed in vacuo to afford 117 (520 mg, 42% yield) as a bright yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.33 (s, 2H), 8.48 (d, J = 4.9 Hz, 1H), 7.42 (d, J = 4.8 Hz, 1H), 7.37 (s, 1H), 7.30 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 151.16, 148.79, 146.65, 145.19, 138.28, 120.84, 118.54, 111.00, 104.51.
8-chloro-[1,3]dioxolo[4,5-g]quinoline (118).
To a solution of 4-chloroquinoline-6,7-diol (117) (342 mg, 1.75 mmol, 1 eq) in DMF (12 mL) was added potassium carbonate (906 mg, 6.56 mmol, 3.75 eq) and diiodomethane (1.87 g, 6.99 mmol, 4 eq). The reaction mixture was stirred at 60°C for 2 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (60 mL) and water (60 mL). The aqueous layer was extracted with EtOAc (60 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (10–50% EtOAc on Hexanes on silica gel column to afford 118 (193 mg, 53% yield) as a light green solid. 1H NMR (400 MHz, DMSO-d6) δ 8.59 (d, J = 4.9 Hz, 1H), 7.57 (d, J = 4.8 Hz, 1H), 7.48 (s, 1H), 7.43 (s, 1H), 6.28 (s, 2H). 13C NMR (214 MHz, DMSO-d6) δ 151.36, 149.12, 147.97, 147.13, 139.57, 122.59, 119.91, 105.50, 102.67, 98.74.
8-(3-fluoro-4-nitrophenoxy)-[1,3]dioxolo[4,5-g]quinoline (92).
A suspension of 8-chloro-[1,3]dioxolo[4,5-g]quinoline (118) (190 mg, 0.92 mmol, 1 eq) and 3-fluoro-4-nitrophenol (87) (288 mg, 1.83 mmol, 2 eq) in o-xylene (2.3 mL) was stirred at 135°C for 16 h. The reaction mixture was cooled to rt. EtOAc (20 mL) was added and the mixture was stirred for 1 h at rt. Aqueous NaOH solution (1 M, 20 mL) was added. The aqueous layer was extracted with EtOAc (30 mL x 2). The combined organic extracts were washed with aqueous NaOH (1 M), brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (10–60% EtOAc on Hexanes on silica gel column to afford 92 (228 mg, 76% yield) as a beige solid. 1H NMR (400 MHz, DMSO-d6) δ 8.64 (d, J = 5.1 Hz, 1H), 8.25 (t, J = 9.0 Hz, 1H), 7.50 (dd, J = 12.4, 2.6 Hz, 1H), 7.45 (s, 1H), 7.36 (s, 1H), 7.13 (ddd, J = 9.2, 2.6, 1.1 Hz, 1H), 7.05 (d, J = 5.1 Hz, 1H), 6.24 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −113.95 (dd, J = 12.7, 8.9 Hz). 13C NMR (214 MHz, DMSO-d6) δ 161.04 (d, J = 10.9 Hz), 157.09, 156.29 (d, J = 263.3 Hz), 151.32, 149.17, 148.33, 148.26, 133.04 (d, J = 6.7 Hz), 128.54, 117.49, 114.93 (d, J = 3.2 Hz), 108.90 (d, J = 24.2 Hz), 108.18, 105.40, 102.41, 96.42.
4-([1,3]dioxolo[4,5-g]quinolin-8-yloxy)-2-fluoroaniline (104).
A suspension of 8-(3-fluoro-4-nitrophenoxy)-[1,3]dioxolo[4,5-g]quinoline (92) (225 mg, 0.68 mmol, 1 eq), iron (189 mg, 3.4 mmol, 5 eq), and ammonium chloride (290 mg, 5.4 mmol, 8 eq) in ethanol (1.8 mL) and water (0.6 mL) was heated at 60°C for 3 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between DCM (30 mL) and aqueous NaOH solution (1 M, 30 mL). The aqueous layer was extracted with DCM (2 × 30 mL). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–5% MeOH in DCM on silica gel column) to afford 104 (190 mg, 94% yield) as a beige solid. 1H NMR (400 MHz, DMSO-d6) δ 8.43 (d, J = 5.3 Hz, 1H), 7.51 (s, 1H), 7.34 (s, 1H), 7.03 (dd, J = 11.8, 2.5 Hz, 1H), 6.89 – 6.75 (m, 2H), 6.46 (d, J = 5.2 Hz, 1H), 6.22 (s, 2H), 5.19 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −131.34 (dd, J = 11.8, 9.5 Hz). 13C NMR (214 MHz, DMSO-d6) δ 160.98, 150.82, 150.16 (d, J = 239.8 Hz), 149.02, 147.51, 147.43, 142.89 (d, J = 9.3 Hz), 134.44 (d, J = 12.7 Hz), 117.33 (d, J = 3.0 Hz), 116.41 (d, J = 5.8 Hz), 116.38, 109.02 (d, J = 21.0 Hz), 105.12, 103.04, 102.08, 96.77.
1-(4-([1,3]dioxolo[4,5-g]quinolin-8-yloxy)-2-fluorophenyl)-3-(2,4-difluorophenyl)urea (20).
To a solution of 1,1′-carbonyldiimidazole (65 mg, 0.40 mmol, 3 eq) in DCM (0.6 mL) cooled to 0°C using an ice bath was added a solution of 2,4-difluoroaniline (52 mg, 0.40 mmol, 3 eq) in DCM (0.6 mL) dropwise. The reaction mixture was stirred at 0°C for 30 min, monitoring reaction progress by TLC. This was added to a solution of 4-([1,3]dioxolo[4,5-g]quinolin-8-yloxy)-2-fluoroaniline (104) (40 mg, 0.13 mmol, 1 eq) in DCM (0.7 mL). The reaction mixture was stirred at 40°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (20–80% EtOAc in Hexanes on silica gel column) to afford 20 (41 mg, 67% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.05 (s, 1H), 9.00 (s, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.23 (t, J = 9.1 Hz, 1H), 8.12 (td, J = 9.2, 6.1 Hz, 1H), 7.51 (s, 1H), 7.38 (s, 1H), 7.41 – 7.23 (m, 2H), 7.12 – 6.91 (m, 2H), 6.59 (d, J = 5.3 Hz, 1H), 6.24 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −117.98 (tdd, J = 8.9, 6.1, 3.7 Hz), −124.80 (t, J = 9.8 Hz), −125.67 (t, J = 10.4 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.99, 156.86 (dd, J = 241.8, 11.5 Hz), 152.26 (d, J = 244.6 Hz), 152.13, 152.07 (dd, J = 244.9, 12.1 Hz), 150.97, 149.03, 148.54 (d, J = 10.1 Hz), 147.72, 147.65, 124.89 (d, J = 10.5 Hz), 123.88 (dd, J = 10.9, 3.4 Hz), 121.80 (dd, J = 8.7, 2.4 Hz), 121.64 (d, J = 2.6 Hz), 116.84 (d, J = 2.9 Hz), 116.62, 111.09 (dd, J = 21.4, 3.6 Hz), 108.81 (d, J = 22.0 Hz), 105.19, 104.06, 103.83 (dd, J = 26.8, 23.4 Hz), 102.17, 96.70. HRMS: calcd for C23H15F3N3O4 [M + H]+ m/z, 454.1015; found m/z, 454.1004.
9-chloro-2,3-dihydro-[1,4]dioxino[2,3-g]quinoline (119).
To a solution of 4-chloroquinoline-6,7-diol (117) (311 mg, 1.59 mmol, 1 eq) in DMF (17 mL) was added potassium carbonate (1.21 g, 8.75 mmol, 5.5 eq) and 1,2-dibromoethane (2.24 g, 11.9 mmol, 7.5 eq). The reaction mixture was stirred at 60°C for 2 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (100 mL) and water (100 mL). The aqueous layer was extracted with EtOAc (100 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (10–50% EtOAc on Hexanes on silica gel column to afford 119 (234 mg, 66% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.58 (d, J = 4.7 Hz, 1H), 7.62 (s, 1H), 7.55 (s, 1H), 7.32 (d, J = 4.7 Hz, 1H), 4.40 (s, 4H). 13C NMR (214 MHz, CDCl3) δ 148.43, 147.69, 145.78, 145.41, 140.88, 122.77, 119.69, 114.78, 109.13, 64.56, 64.56.
9-(3-fluoro-4-nitrophenoxy)-2,3-dihydro-[1,4]dioxino[2,3-g]quinoline (93).
9-chloro-2,3-dihydro-[1,4]dioxino[2,3-g]quinoline (119) (234 mg, 1.06 mmol, 1 eq) and 3-fluoro-4-nitrophenol (87) (333 mg, 2.12 mmol, 2 eq) in o-xylene (2.6 mL) was stirred at 135°C for 16 h. The reaction mixture was cooled to rt. EtOAc (20 mL) was added and the mixture was stirred for 1 h at rt. Aqueous NaOH solution (1 M, 20 mL) was added. The aqueous layer was extracted with EtOAc (30 mL x 2). The combined organic extracts were washed with aqueous NaOH (1 M), brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (10–60% EtOAc on Hexanes on silica gel column to afford 93 (78 mg, 22% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.65 (d, J = 5.0 Hz, 1H), 8.25 (t, J = 9.0 Hz, 1H), 7.52 (dd, J = 12.4, 2.6 Hz, 1H), 7.49 (s, 1H), 7.39 (s, 1H), 7.14 (ddd, J = 9.2, 2.6, 1.2 Hz, 1H), 6.99 (d, J = 5.0 Hz, 1H), 4.43 – 4.36 (m, 4H). 19F NMR (376 MHz, DMSO-d6) δ −113.92 (dd, J = 12.4, 8.8 Hz). 13C NMR (214 MHz, DMSO-d6) δ 160.95 (d, J = 11.3 Hz), 156.72, 156.28 (d, J = 263.6 Hz), 149.76, 147.56, 146.34, 144.72, 133.11 (d, J = 6.9 Hz), 128.56, 116.59, 115.04 (d, J = 2.9 Hz), 113.69, 109.01 (d, J = 24.1 Hz), 107.44, 105.39, 64.30, 64.25.
4-((2,3-dihydro-[1,4]dioxino[2,3-g]quinolin-9-yl)oxy)-2-fluoroaniline (105).
A suspension of 9-(3-fluoro-4-nitrophenoxy)-2,3-dihydro-[1,4]dioxino[2,3-g]quinoline (93) (73 mg, 0.21 mmol, 1 eq), iron (59 mg, 1.1 mmol, 5 eq), and ammonium chloride (91 mg, 1.7 mmol, 8 eq) in ethanol (0.6 mL) and water (0.2 mL) was heated at 45°C for 4 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between DCM (30 mL) and aqueous NaOH solution (1 M, 30 mL). The aqueous layer was extracted with DCM (2 × 30 mL). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–5% MeOH in DCM on silica gel column) to afford 105 (47 mg, 71% yield) as a light purple solid. 1H NMR (400 MHz, DMSO-d6) δ 8.44 (d, J = 5.1 Hz, 1H), 7.55 (s, 1H), 7.38 (s, 1H), 7.03 (dd, J = 11.8, 2.5 Hz, 1H), 6.91 – 6.76 (m, 2H), 6.39 (d, J = 5.2 Hz, 1H), 5.19 (s, 2H), 4.42 – 4.37 (m, 4H). 19F NMR (376 MHz, DMSO-d6) δ −131.36 (dd, J = 11.9, 9.3 Hz). 13C NMR (214 MHz, DMSO-d6) δ 160.61, 150.16 (d, J = 239.8 Hz), 149.67, 147.11, 145.71, 143.94, 142.84 (d, J = 9.3 Hz), 134.45 (d, J = 13.1 Hz), 117.33 (d, J = 3.0 Hz), 116.40 (d, J = 5.5 Hz), 115.88, 113.28, 109.02 (d, J = 21.0 Hz), 105.77, 102.21, 64.31, 64.20.
1-(2,4-difluorophenyl)-3-(4-((2,3-dihydro-[1,4]dioxino[2,3-g]quinolin-9-yl)oxy)-2-fluorophenyl)urea (21).
To a solution of 1,1′-carbonyldiimidazole (65 mg, 0.40 mmol, 3 eq) in DCM (0.6 mL) cooled to 0°C using an ice bath was added a solution of 2,4-difluoroaniline (52 mg, 0.40 mmol, 3 eq) in DCM (0.6 mL) dropwise. The reaction mixture was stirred at 0°C for 30 min, monitoring reaction progress by TLC. This was added to a solution of 4-((2,3-dihydro-[1,4]dioxino[2,3-g]quinolin-9-yl)oxy)-2-fluoroaniline (105) (43 mg, 0.13 mmol, 1 eq) in DCM (0.7 mL). The reaction mixture was stirred at 40°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (20–80% EtOAc in Hexanes on silica gel column) to afford 21 (39 mg, 62% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.06 (d, J = 2.2 Hz, 1H), 9.00 (d, J = 2.1 Hz, 1H), 8.49 (d, J = 5.1 Hz, 1H), 8.23 (t, J = 9.1 Hz, 1H), 8.12 (td, J = 9.2, 6.1 Hz, 1H), 7.55 (s, 1H), 7.41 (s, 1H), 7.38 – 7.28 (m, 2H), 7.10 – 7.02 (m, 2H), 6.52 (d, J = 5.1 Hz, 1H), 4.42 – 4.38 (m, 4H). 19F NMR (376 MHz, DMSO-d6) δ −117.98 (tdd, J = 8.9, 6.1, 3.6 Hz), −124.80 (t, J = 10.0 Hz), −125.67 (t, J = 10.7 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.62, 156.86 (dd, J = 241.8, 11.6 Hz), 152.25 (d, J = 244.5 Hz), 152.13, 152.07 (dd, J = 244.9, 12.2 Hz), 149.66, 148.48 (d, J = 10.3 Hz), 147.24, 145.87, 144.14, 124.92 (d, J = 10.4 Hz), 123.88 (dd, J = 10.8, 3.5 Hz), 121.81 (dd, J = 8.9, 2.6 Hz), 121.63 (d, J = 2.4 Hz), 116.88 (d, J = 3.1 Hz), 116.02, 113.38, 111.09 (dd, J = 21.6, 3.6 Hz), 108.84 (d, J = 21.9 Hz), 105.70, 103.83 (dd, J = 27.1, 23.5 Hz), 103.23, 64.31, 64.22. HRMS: calcd for C24H17F3N3O4 [M + H]+ m/z, 468.1171; found m/z, 468.1160.
2-fluoro-4-(quinazolin-4-yloxy)aniline (106).
To a solution of 4-chloroquinazoline (350 mg, 2.13 mmol, 1 eq) and 4-amino-3-fluorophenol (85) (378 mg, 2.98 mmol, 1.4 eq) in DMF (8 mL) cooled to 0°C was added potassium tert-butoxide (334 mg, 2.98 mmol, 1.4 eq). The reaction mixture was stirred at 110°C for 4 h, then cooled to rt. The reaction mixture was partitioned between DCM (50 mL) and water (50 mL). The aqueous layer was extracted with DCM (50 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–6% MeOH in DCM on silica gel column) to afford 106 (58 mg, 11% yield) as a light brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.72 (s, 1H), 8.33 (ddd, J = 8.2, 1.4, 0.7 Hz, 1H), 8.05 – 7.95 (m, 2H), 7.76 (ddd, J = 8.2, 6.7, 1.5 Hz, 1H), 7.11 (dd, J = 11.9, 2.4 Hz, 1H), 6.95 – 6.78 (m, 2H), 5.15 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −132.56 (dd, J = 11.9, 9.3 Hz).
1-(2,4-difluorophenyl)-3-(2-fluoro-4-(quinazolin-4-yloxy)phenyl)urea (23).
To a solution of 2-fluoro-4-(quinazolin-4-yloxy)aniline (106) (50 mg, 0.20 mmol, 1 eq) in ethanol (0.5 mL) cooled to 0°C using an ice bath was added 2,4-difluoro-1-isocyanatobenzene (30 mg, 0.20 mmol, 1 eq) dropwise. The reaction mixture was warmed to rt and stirred at rt for 30 min. The reaction mixture was concentrated onto Celite and purified by flash chromatography (0–10% MeOH in EtOAc on silica gel column then 0–100% EtOAc in Hexanes on silica gel) to afford 23 (13.7 mg, 17% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.04 (s, 1H), 9.00 (s, 1H), 8.76 (s, 1H), 8.40 – 8.36 (m, 1H), 8.21 (t, J = 9.5 Hz, 1H), 8.16 – 8.11 (m, 1H), 8.10 – 7.99 (m, 2H), 7.80 (q, J = 6.4 Hz, 1H), 7.51 – 7.40 (m, 1H), 7.40 – 7.28 (m, 1H), 7.24 – 7.14 (m, 1H), 7.14 – 7.02 (m, 1H). 19F NMR (376 MHz, DMSO-d6) δ −118.43 – −118.57 (m), −125.31 (t, J = 10.7 Hz), −127.16 – −127.26 (m). 13C NMR (101 MHz, DMSO-d6) δ 166.44, 156.91 (dd, J = 241.5, 11.8 Hz), 153.88, 152.21, 152.12 (dd, J = 244.9, 12.3 Hz), 152.00 (d, J = 243.6 Hz), 151.19, 146.59 (d, J = 10.7 Hz), 134.79, 128.23, 127.58, 125.20 (d, J = 10.7 Hz), 123.92 (dd, J = 10.8, 3.5 Hz), 123.44, 121.86 (dd, J = 9.0, 3.1 Hz), 121.14, 118.19 (d, J = 3.3 Hz), 115.54, 111.15 (dd, J = 21.5, 3.6 Hz), 110.23 (d, J = 22.3 Hz), 103.88 (dd, J = 27.0, 23.7 Hz). HRMS: calcd for C21H14F3N4O2 [M + H]+ m/z, 411.1069; found m/z, 411.1060.
4-chlorocinnoline (124).
To a suspension of cinnolin-4-ol (123) (200 mg, 1.37 mmol, 1 eq) in anhydrous THF (14 mL) was added phosphoryl trichloride (629 mg, 4.11 mmol, 3 eq). The reaction mixture was refluxed at 66°C for 1 h, then cooled to 0°C using an ice bath. Saturated NaHCO3 solution (24 mL) was added dropwise and the reaction mixture was allowed to warm to rt and stirred at rt for 1 h. The reaction mixture was partitioned between DCM (50 mL) and water (50 mL). The aqueous layer was extracted with DCM (50 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated in vacuo to afford 124 (193 mg, 86% yield) as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 9.60 (s, 1H), 8.62 – 8.53 (m, 1H), 8.28 – 8.19 (m, 1H), 8.14 – 8.02 (m, 2H).
4-(cinnolin-4-yloxy)-2-fluoroaniline (107).
To a solution of 4-chlorocinnoline (124) (190 mg, 1.15 mmol, 1 eq) and 4-amino-3-fluorophenol (85) (205 mg, 1.62 mmol, 1.4 eq) in DMF (3.8 mL) cooled to 0°C was added potassium tert-butoxide (181 mg, 1.62 mmol, 1.4 eq). The reaction mixture was stirred at 110°C for 4 h, then cooled to rt. Water (60 mL) was added and the solid formed was collected by filtration, washed with water and dried. The crude material was dissolved in a mixture of DCM and MeOH, concentrated onto Celite and purified by flash chromatography (0-s8% MeOH in DCM on silica gel column) to afford 107 (100 mg, 34% yield) as a yellow-orange solid. 1H NMR (400 MHz, DMSO-d6) δ 8.64 (s, 1H), 8.46 (dt, J = 8.5, 1.0 Hz, 1H), 8.33 (ddd, J = 8.3, 1.4, 0.7 Hz, 1H), 8.01 (ddd, J = 8.5, 6.8, 1.4 Hz, 1H), 7.92 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 7.20 (dd, J = 11.8, 2.5 Hz, 1H), 6.99 – 6.85 (m, 2H), 5.29 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −130.95 (dd, J = 12.0, 9.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 152.94, 150.53, 150.22 (d, J = 240.4 Hz), 142.20 (d, J = 9.5 Hz), 135.08 (d, J = 12.9 Hz), 131.72, 131.45, 130.89, 128.45, 120.75, 117.85, 117.16 (d, J = 3.0 Hz), 116.58 (d, J = 5.6 Hz), 108.93 (d, J = 21.3 Hz).
1-(4-(cinnolin-4-yloxy)-2-fluorophenyl)-3-(2,4-difluorophenyl)urea (24).
To a solution of 4-(cinnolin-4-yloxy)-2-fluoroaniline (107) (98 mg, 0.38 mmol, 1 eq) in ethanol (1 mL) cooled to 0°C using an ice bath was added 2,4-difluoro-1-isocyanatobenzene (60 mg, 0.38 mmol, 1 eq) dropwise. The reaction mixture was warmed to rt and stirred at rt for 1 h. The reaction mixture was concentrated onto Celite and purified by flash chromatography (0–10% MeOH in DCM on silica gel column then 0–10% MeOH in EtOAc on silica gel) to afford 24 (10.1 mg, 6% yield) as a beige solid. 1H NMR (400 MHz, DMSO-d6) δ 9.11 (d, J = 2.4 Hz, 1H), 9.03 (d, J = 2.2 Hz, 1H), 8.78 (s, 1H), 8.49 (dt, J = 8.6, 1.0 Hz, 1H), 8.34 (d, J = 7.9 Hz, 1H), 8.30 (t, J = 9.1 Hz, 1H), 8.13 (td, J = 9.2, 6.1 Hz, 1H), 8.04 (ddd, J = 8.5, 6.9, 1.4 Hz, 1H), 7.94 (ddd, J = 8.2, 6.8, 1.2 Hz, 1H), 7.50 (dd, J = 11.7, 2.7 Hz, 1H), 7.34 (ddd, J = 11.7, 8.9, 2.9 Hz, 1H), 7.21 (ddd, J = 9.3, 2.5, 1.3 Hz, 1H), 7.07 (tq, J = 8.7, 1.8 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −117.90 (tdd, J = 8.9, 6.1, 3.7 Hz), −124.72 (t, J = 10.6 Hz), −125.27 (t, J = 10.6 Hz). 13C NMR (101 MHz, DMSO-d6) δ 156.90 (dd, J = 241.3, 11.9 Hz), 152.21 (d, J = 245.1 Hz), 152.12, 152.11 (dd, J = 244.8, 12.8 Hz), 152.11, 150.69, 147.64 (d, J = 10.6 Hz), 131.97, 131.74, 131.05, 128.53, 125.62 (d, J = 10.7 Hz), 123.84 (dd, J = 10.8, 3.6 Hz), 121.86 (dd, J = 9.1, 2.9 Hz), 121.58 (d, J = 3.1 Hz), 120.65, 117.93, 116.85 (d, J = 3.3 Hz), 111.12 (dd, J = 21.9, 3.7 Hz), 108.90 (d, J = 22.5 Hz), 103.86 (dd, J = 26.8, 23.6 Hz). HRMS: calcd for C21H14F3N4O2 [M + H]+ m/z, 411.1069; found m/z, 411.1058.
7-(3-fluoro-4-nitrophenoxy)thieno[3,2-b]pyridine hydrochloride (94).
A suspension of 7-chlorothieno[3,2-b]pyridine (300 mg, 1.77 mmol, 1 eq) and 3-fluoro-4-nitrophenol (87) (556 mg, 3.54 mmol, 2 eq) in o-xylene (4 mL) was stirred at 135°C for 16 h. The reaction mixture was cooled to rt. EtOAc (15 mL) was added and the mixture was stirred for 1 h at rt. The solid formed was collected by filtration, washed with EtOAc and dried. The solid was added to an aqueous NaOH solution (1 M, 20 mL) and stirred for 1 h at rt. The solids were collected by filtration, acidified with HCl solution (2 M, 20 mL). The solids formed were washed with water, then EtOAc and dried under vacuum to afford 94 (104 mg, 18% yield) as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.77 (dd, J = 5.8, 1.1 Hz, 1H), 8.37 (dd, J = 5.5, 1.5 Hz, 1H), 8.33 (t, J = 8.9 Hz, 1H), 7.77 – 7.71 (m, 2H), 7.37 (ddd, J = 9.0, 2.5, 1.2 Hz, 1H), 7.19 (d, J = 5.7 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −113.73 (dd, J = 12.2, 8.8 Hz).
2-fluoro-4-(thieno[3,2-b]pyridin-7-yloxy)aniline (108).
A suspension of 7-(3-fluoro-4-nitrophenoxy)thieno[3,2-b]pyridine hydrochloride (94) (100 mg, 0.306 mmol, 1 eq), iron (85.5 mg, 1.53 mmol, 5 eq), and ammonium chloride (131 mg, 2.45 mmol, 8 eq) in ethanol (2 mL) and water (0.65 mL) was heated at 78°C for 5 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between DCM (30 mL) and aqueous NaOH solution (1M, 30 mL). The aqueous layer was extracted with DCM (2 × 30 mL). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–5% MeOH in DCM on silica gel column) to afford 108 (63 mg, 79% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.48 (d, J = 5.4 Hz, 1H), 8.12 (d, J = 5.4 Hz, 1H), 7.57 (d, J = 5.4 Hz, 1H), 7.14 – 7.02 (m, 1H), 6.91 – 6.79 (m, 2H), 6.59 (d, J = 5.4 Hz, 1H), 5.24 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −131.27 – −131.39 (m).
1-(2,4-difluorophenyl)-3-(2-fluoro-4-(thieno[3,2-b]pyridin-7-yloxy)phenyl)urea (25).
To a solution of 2-fluoro-4-(thieno[3,2-b]pyridin-7-yloxy)aniline (108) (60 mg, 0.23 mmol, 1 eq) in ethanol (0.6 mL) cooled to 0°C using an ice bath was added 2,4-difluoro-1-isocyanatobenzene (36 mg, 0.23 mmol, 1 eq) dropwise. The reaction mixture was warmed to rt and stirred at rt for 45 min. The reaction mixture was concentrated onto Celite and purified by flash chromatography (0–10% MeOH in DCM on silica gel column) to afford 25 (55 mg, 58% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.09 (d, J = 2.4 Hz, 1H), 9.02 (d, J = 2.3 Hz, 1H), 8.54 (d, J = 5.4 Hz, 1H), 8.25 (t, J = 9.1 Hz, 1H), 8.17 (d, J = 5.4 Hz, 1H), 8.13 (td, J = 9.2, 6.1 Hz, 1H), 7.61 (d, J = 5.4 Hz, 1H), 7.41 (dd, J = 11.7, 2.7 Hz, 1H), 7.33 (ddd, J = 11.6, 8.9, 2.9 Hz, 1H), 7.14 (ddd, J = 9.1, 2.8, 1.3 Hz, 1H), 7.10 – 7.03 (m, 1H), 6.72 (d, J = 5.4 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −117.26 – −118.41 (m), −124.76 (t, J = 10.4 Hz), −125.61 (t, J = 10.5 Hz). 13C NMR (101 MHz, DMSO-d6) δ 159.59, 158.64, 156.88 (dd, J = 241.5, 11.5 Hz), 152.10, 152.08 (dd, J = 245.0, 12.2 Hz), 152.07 (d, J = 244.8 Hz), 149.38, 147.46 (d, J = 10.3 Hz), 132.29, 125.44 (d, J = 10.6 Hz), 124.82, 123.84 (dd, J = 10.7, 3.5 Hz), 121.81 (dd, J = 9.0, 2.9 Hz), 121.51, 121.42 (d, J = 2.9 Hz), 117.13 (d, J = 3.4 Hz), 111.11 (dd, J = 21.6, 3.5 Hz), 109.09 (d, J = 22.2 Hz), 104.30, 104.14 – 103.53 (m). HRMS: calcd for C20H13F3N3O2S [M + H]+ m/z, 416.0681; found m/z, 416.0671.
4-(3-fluoro-4-nitrophenoxy)thieno[2,3-b]pyridine (95).
A suspension of 4-chlorothieno[2,3-b]pyridine (350 mg, 2.06 mmol, 1 eq) and 3-fluoro-4-nitrophenol (87) (583 mg, 3.71 mmol, 1.8 eq) in o-xylene (4.5 mL) was stirred at 135°C for 60 h. The reaction mixture was cooled to rt and partitioned between EtOAc (50 mL) and aqueous NaOH solution (1 M, 50 mL). The aqueous layer was extracted with EtOAc (50 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated into celite, and purified by flash chromatography (0–35% EtOAc in Hexanes on silica gel column) to afford 95 (267 mg, 45% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.57 (d, J = 5.3 Hz, 1H), 8.27 (t, J = 9.0 Hz, 1H), 7.93 (d, J = 6.1 Hz, 1H), 7.58 (dd, J = 12.4, 2.6 Hz, 1H), 7.40 (d, J = 6.1 Hz, 1H), 7.21 (ddd, J = 9.2, 2.6, 1.2 Hz, 1H), 7.12 (d, J = 5.3 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −113.86 (dd, J = 12.3, 8.8 Hz). 13C NMR (214 MHz, DMSO-d6) δ 163.74, 160.32 (d, J = 10.9 Hz), 156.47, 156.26 (d, J = 263.7 Hz), 148.79, 128.57, 128.15, 124.24, 117.82, 115.47 (d, J = 3.2 Hz), 109.39 (d, J = 24.1 Hz), 108.53. (missing aromatic carbon is probably due to overlap at the intense peak at 128.15 ppm.)
2-fluoro-4-(thieno[2,3-b]pyridin-4-yloxy)aniline (109).
A suspension of 4-(3-fluoro-4-nitrophenoxy)thieno[2,3-b]pyridine (95) (264 mg, 0.909 mmol, 1 eq), iron (254 mg, 4.55 mmol, 5 eq), and ammonium chloride (389 mg, 7.28 mmol, 8 eq) in ethanol (2.7 mL) and water (0.9 mL) was heated at 31°C for 4 h. The reaction mixture was cooled to rt and filtered through Celite. The filtrate was concentrated onto Celite, and purified by flash chromatography (0–40% EtOAc in Hexanes on silica gel column) to afford 109 (184 mg, 78% yield) as an orange solid. 1H NMR (400 MHz, DMSO-d6) δ 8.37 (d, J = 5.5 Hz, 1H), 7.82 (d, J = 6.0 Hz, 1H), 7.50 (d, J = 6.0 Hz, 1H), 7.11 – 7.03 (m, 1H), 6.92 – 6.78 (m, 2H), 6.56 (d, J = 5.4 Hz, 1H), 5.21 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −131.24 (dd, J = 11.9, 8.2 Hz). 13C NMR (214 MHz, DMSO-d6) δ 163.04, 160.39, 150.13 (d, J = 239.8 Hz), 148.57, 142.57 (d, J = 9.3 Hz), 134.62 (d, J = 12.6 Hz), 126.30, 122.94, 118.15, 117.28 (d, J = 3.3 Hz), 116.41 (d, J = 6.0 Hz), 108.98 (d, J = 21.0 Hz), 104.27.
1-(2,4-difluorophenyl)-3-(2-fluoro-4-(thieno[2,3-b]pyridin-4-yloxy)phenyl)urea (27).
To a solution of 2,4-difluoroaniline (39 mg, 0.30 mmol, 1.3 eq) in DMSO (1.2 mL) was added 1,1′-carbonyldiimidazole (56 mg, 0.35 mmol, 1.5 eq). The reaction mixture was stirred at rt for 2 h, monitoring reaction progress by TLC. This was added to a solution of 2-fluoro-4-(thieno[2,3-b]pyridin-4-yloxy)aniline (109) (60 mg, 0.23 mmol, 1 eq) in DMSO (1.2 mL). The reaction mixture was stirred at 40°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–40% EtOAc in Hexanes on silica gel column) to afford 27 (23 mg, 24% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.06 (d, J = 2.5 Hz, 1H), 8.99 (d, J = 2.3 Hz, 1H), 8.43 (d, J = 5.4 Hz, 1H), 8.25 (t, J = 9.1 Hz, 1H), 8.13 (td, J = 9.2, 6.1 Hz, 1H), 7.86 (d, J = 6.0 Hz, 1H), 7.51 (d, J = 6.0 Hz, 1H), 7.45 – 7.25 (m, 2H), 7.19 – 7.03 (m, 2H), 6.69 (d, J = 5.5 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −117.70 – −118.08 (m), −124.82 (t, J = 10.8 Hz), −125.60 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 163.22, 159.39, 158.07 – 156.03 (m), 152.19 (d, J = 244.9 Hz), 152.10, 152.06 (dd, J = 245.0, 12.3 Hz), 148.62, 148.08 (d, J = 10.3 Hz), 126.77, 125.14 (d, J = 10.7 Hz), 123.85 (dd, J = 10.8, 3.3 Hz), 123.21, 121.79 (dd, J = 9.0, 2.3 Hz), 121.56 (d, J = 2.8 Hz), 118.06, 116.92 (d, J = 3.1 Hz), 111.11 (dd, J = 21.4, 3.4 Hz), 108.89 (d, J = 22.0 Hz), 105.08, 103.84 (dd, J = 27.1, 23.4 Hz). HRMS: calcd for C20H13F3N3O2S [M + H]+ m/z, 416.0681; found m/z, 416.0670.
thieno[2,3-d]pyrimidin-4(1H)-one (126).
A solution of methyl 2-aminothiophene-3-carboxylate (125) (1.0 g, 6.3 mmol, 1 eq) in formamide (5 mL, 127 mmol, 20 eq) was stirred at 160°C for 24 h, then cooled to rt. Water was added, and the solid formed was collected by filtration, washed with water, and dried in vacuo to afford 126 (507 mg, 52% yield) as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.12 (s, 1H), 7.58 (d, J = 5.8 Hz, 1H), 7.39 (d, J = 5.8 Hz, 1H). 13C NMR (214 MHz, DMSO-d6) δ 164.26, 157.54, 145.63, 124.64, 123.88, 121.66.
4-chlorothieno[2,3-d]pyrimidine (127).
A solution of thieno[2,3-d]pyrimidin-4(1H)-one (126) (627 mg, 4.123 mmol) in phosphoryl trichloride (6 mL) was refluxed at 100°C for 2 h. The reaction mixture was cooled to rt and an ice-cold saturated aqueous NaHCO3 solution was added dropwise. The solid was collected by filtration, washed with water and dried. The crude material was dissolved in a mixture of DCM and MeOH, concentrated onto Celite and purified by flash chromatography (0–25% EtOAc in Hexanes on silica gel column) to afford 127 (349 mg, 50% yield) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.87 (s, 1H), 7.64 (d, J = 6.1 Hz, 1H), 7.46 (d, J = 6.1 Hz, 1H).
2-fluoro-4-(thieno[2,3-d]pyrimidin-4-yloxy)aniline (110).
To a solution of 4-chlorothieno[2,3-d]pyrimidine (127) (340 mg, 1.99 mmol, 1 eq) and 4-amino-3-fluorophenol (85) (355 mg, 2.79 mmol, 1.4 eq) in DMF (1.5 mL) cooled to 0°C was added a solution of potassium tert-butoxide (313 mg, 2.79 mmol, 1.4 eq) in DMF (5 mL) dropwise. The reaction mixture was stirred at 110°C for 4 h, then cooled to rt. Water (100 mL) was added. The solid formed was collected by filtration, washed with water and dried. The crude material was dissolved in a mixture of DCM and MeOH, concentrated onto Celite and purified by flash chromatography (0–5% MeOH in DCM on silica gel column then 0–60% EtOAc in Hexanes on silica gel column) to afford 110 (238 mg, 46% yield) as a light brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.61 (s, 1H), 7.94 (d, J = 5.9 Hz, 1H), 7.61 (d, J = 5.9 Hz, 1H), 7.17 – 7.04 (m, 1H), 6.88 – 6.75 (m, 2H), 5.14 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −132.50 (dd, J = 11.9, 8.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 168.88, 163.59, 153.05, 149.95 (d, J = 238.9 Hz), 141.34 (d, J = 9.8 Hz), 134.32 (d, J = 12.9 Hz), 127.22, 118.55, 118.54, 117.93 (d, J = 3.1 Hz), 115.90 (d, J = 5.4 Hz), 109.80 (d, J = 21.6 Hz).
1-(2,4-difluorophenyl)-3-(2-fluoro-4-(thieno[2,3-d]pyrimidin-4-yloxy)phenyl)urea (28).
To a solution of 2-fluoro-4-(thieno[2,3-d]pyrimidin-4-yloxy)aniline (110) (92 mg, 0.352 mmol, 1 eq) in ethanol (1.0 mL) cooled to 0°C using an ice bath was added 2,4-difluoro-1-isocyanatobenzene (55 mg, 0.35 mmol, 1 eq) dropwise. The reaction mixture was warmed to rt and stirred at rt for 1.5 h. 2,4-difluoro-1-isocyanatobenzene (55 mg, 0.35 mmol, 1 eq) was added dropwise and the reaction mixture was stirred at rt for 45 min. The reaction mixture was concentrated onto Celite and purified by flash chromatography (0–8% MeOH in DCM on silica gel column then 30–100% MeCN in water with 0.1% formic acid on C18 column) to afford 28 (7.9 mg, 5% yield) as a light pink solid. 1H NMR (400 MHz, DMSO-d6) δ 9.05 (d, J = 2.1 Hz, 1H), 9.00 (s, 1H), 8.64 (s, 1H), 8.19 (t, J = 9.1 Hz, 1H), 8.13 (td, J = 9.2, 6.1 Hz, 1H), 7.98 (d, J = 6.0 Hz, 1H), 7.67 (d, J = 6.0 Hz, 1H), 7.41 (dd, J = 11.8, 2.6 Hz, 1H), 7.33 (ddd, J = 11.7, 8.9, 2.9 Hz, 1H), 7.15 (ddd, J = 9.0, 2.7, 1.3 Hz, 1H), 7.12 – 7.03 (m, 1H). 19F NMR (376 MHz, DMSO-d6) δ −117.61 – −118.37 (m), −124.76 (t, J = 9.3 Hz), −126.61 (t, J = 10.4 Hz). 13C NMR (101 MHz, DMSO-d6) δ 169.08, 163.11, 156.86 (dd, J = 242.2, 11.5 Hz), 152.93, 152.16, 152.07 (dd, J = 244.9, 12.0 Hz), 151.93 (d, J = 243.8 Hz), 146.38 (d, J = 10.9 Hz), 127.54, 125.15 (d, J = 10.8 Hz), 123.90 (dd, J = 10.7, 3.7 Hz), 121.80 (dd, J = 9.1, 2.9 Hz), 121.10 (d, J = 2.7 Hz), 118.54, 118.46, 118.06 (d, J = 3.3 Hz), 111.11 (dd, J = 21.9, 3.7 Hz), 110.10 (d, J = 22.4 Hz), 103.84 (dd, J = 26.8, 23.5 Hz). HRMS: calcd for C19H12F3N4O2S [M + H]+ m/z, 417.0633; found m/z, 417.0624.
4-(3-fluoro-4-nitrophenoxy)-1H-pyrrolo[2,3-b]pyridine (96).
In a microwave vial charged with a stir bar was added 4-chloro-1H-pyrrolo[2,3-b]pyridine (500 mg, 3.28 mmol, 1 eq) and 3-fluoro-4-nitrophenol (87) (618 mg, 3.93 mmol, 1.2 eq). The microwave vial was sealed and irradiated at 150°C for 20 min. The crude material was dissolved in EtOAc (3 mL) and was partitioned between EtOAc (50 mL) and aqueous NaOH solution (1 M, 50 mL). The aqueous layer was extracted with EtOAc (50 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–5% MeOH in DCM on silica gel column then 0–50% EtOAc in Hexanes on silica gel column) to afford 96 (103 mg, 12% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 11.96 (s, 1H), 8.33 – 8.13 (m, 2H), 7.47 (dd, J = 3.5, 2.5 Hz, 1H), 7.40 (dd, J = 12.6, 2.6 Hz, 1H), 7.06 (ddd, J = 9.2, 2.6, 1.2 Hz, 1H), 6.86 (d, J = 5.3 Hz, 1H), 6.21 (dd, J = 3.5, 1.9 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −114.11 (dd, J = 12.6, 8.9 Hz). 13C NMR (214 MHz, DMSO-d6) δ 161.36 (d, J = 11.2 Hz), 156.29 (d, J = 263.2 Hz), 153.56, 151.47, 144.39, 132.54 (d, J = 7.0 Hz), 128.41, 126.26, 114.34 (d, J = 3.1 Hz), 111.15, 108.16 (d, J = 24.1 Hz), 105.50, 96.75.
4-((1H-pyrrolo[2,3-b]pyridin-4-yl)oxy)-2-fluoroaniline (111).
A suspension of 4-(3-fluoro-4-nitrophenoxy)-1H-pyrrolo[2,3-b]pyridine (96) (105 mg, 0.384 mmol, 1 eq), iron (107 mg, 1.92 mmol, 5 eq), and ammonium chloride (164 mg, 3.07 mmol, 8 eq) in ethanol (1 mL) and water (0.3 mL) was heated at 31°C for 4 h. The reaction mixture was cooled to rt and filtered through Celite. The filtrate was concentrated onto Celite, and purified by flash chromatography (20–70% EtOAc in Hexanes on silica gel column) to afford 111 (83 mg, 88% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 7.21 (d, J = 5.5 Hz, 1H), 6.50 (dd, J = 3.5, 2.5 Hz, 1H), 6.15 (dd, J = 11.9, 2.5 Hz, 1H), 6.06 – 5.90 (m, 2H), 5.51 (d, J = 5.4 Hz, 1H), 5.38 (dd, J = 3.5, 2.0 Hz, 1H), 4.31 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −131.62 (dd, J = 12.1, 9.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 158.19, 151.02, 150.10 (d, J = 239.6 Hz), 144.17, 143.68 (d, J = 8.9 Hz), 134.01 (d, J = 13.0 Hz), 124.32, 117.13 (d, J = 3.1 Hz), 116.30 (d, J = 5.5 Hz), 109.65, 108.78 (d, J = 20.9 Hz), 101.12, 97.11.
1-(4-((1H-pyrrolo[2,3-b]pyridin-4-yl)oxy)-2-fluorophenyl)-3-(2,4-difluorophenyl)urea (29).
To a solution of 2,4-difluoroaniline (59 mg, 0.45 mmol, 2 eq) in DCM (2.2 mL) cooled to 0°C was added 1,1′-carbonyldiimidazole (92 mg, 0.57 mmol, 2.5 eq). The reaction mixture was stirred at 0°C for 30 min. This was added to a solution of 4-((1H-pyrrolo[2,3-b]pyridin-4-yl)oxy)-2-fluoroaniline (111) (60 mg, 0.23 mmol, 1 eq) in DCM (1.1 mL). The reaction mixture was stirred at 40°C for 2.5 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–60% EtOAc on Hexanes on silica gel column then 20–50% MeCN in water with 0.1% formic acid on C18 column) to afford 29 (3.3 mg, 4% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.76 (s, 1H), 9.08 (s, 1H), 9.05 (s, 1H), 8.29 – 8.01 (m, 3H), 7.37 (s, 1H), 7.32 (t, J = 10.2 Hz, 1H), 7.24 (d, J = 11.8 Hz, 1H), 7.14 – 6.96 (m, 2H), 6.47 (d, J = 5.4 Hz, 1H), 6.22 (s, 1H). 19F NMR (376 MHz, DMSO-d6) δ −115.96 – −119.80 (m), −124.65 (t, J = 10.3 Hz), −125.77 (t, J = 11.0 Hz). 13C NMR (214 MHz, DMSO-d6) δ 156.95, 156.85 (dd, J = 241.6, 11.6 Hz), 152.28 (d, J = 244.4 Hz), 152.19, 152.11 (dd, J = 245.0, 12.1 Hz), 151.15, 149.31 (d, J = 10.0 Hz), 144.23, 124.85, 124.34 (d, J = 10.6 Hz), 123.92 (dd, J = 10.7, 3.3 Hz), 121.87 (dd, J = 8.7, 2.6 Hz), 121.65 (d, J = 2.1 Hz), 116.41 (d, J = 3.0 Hz), 111.08 (dd, J = 21.6, 3.5 Hz), 110.06, 108.33 (d, J = 22.1 Hz), 103.83 (dd, J = 26.7, 23.5 Hz), 102.18, 97.01. HRMS: calcd for C20H14F3N4O2 [M + H]+ m/z, 399.1069; found m/z, 399.1059.
4-(3-fluoro-4-nitrophenoxy)-7H-pyrrolo[2,3-d]pyrimidine (97).
To a solution of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (405 mg, 2.64 mmol, 1 eq) and 3-fluoro-4-nitrophenol (87) (539 mg, 3.43 mmol, 1.3 eq) in N-methyl-2-pyrrolidone (1.5 mL) was added 2,6-lutidine (339 mg, 3.16 mmol, 1.2 eq). The reaction mixture was stirred at 130°C for 22 h. then cooled to rt and poured into ice water (30 mL). The solid was collected by filtration, washed with water and diethyl ether, then re-suspended in isopropanol (5 mL) and collected by filtration, washed with isopropanol and dried under vacuum to afford 97 (202 mg, 28% yield) as a light grey solid. 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 8.39 (s, 1H), 8.28 (t, J = 8.9 Hz, 1H), 7.72 (dd, J = 12.4, 2.5 Hz, 1H), 7.57 (dd, J = 3.5, 2.4 Hz, 1H), 7.40 (ddd, J = 9.1, 2.5, 1.2 Hz, 1H), 6.62 (dd, J = 3.5, 1.8 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −115.28 (dd, J = 12.5, 8.8 Hz). 13C NMR (214 MHz, DMSO-d6) δ 160.30, 158.24 (d, J = 11.0 Hz), 155.76 (d, J = 262.4 Hz), 154.11, 150.03, 134.04 (d, J = 7.5 Hz), 127.80, 126.44, 118.67 (d, J = 3.4 Hz), 112.20 (d, J = 23.5 Hz), 105.17, 98.01.
4-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)-2-fluoroaniline (112).
A suspension of 4-(3-fluoro-4-nitrophenoxy)-7H-pyrrolo[2,3-d]pyrimidine (97) (80 mg, 0.29 mmol, 1 eq), iron (81 mg, 1.5 mmol, 5 eq), and ammonium chloride (120 mg, 2.3 mmol, 8 eq) in ethanol (0.9 mL) and water (0.3 mL) was heated at 78°C for 5 h. The reaction mixture was cooled to rt and filtered through Celite. The filtrate was concentrated onto Celite, and purified by flash chromatography (0– 60% EtOAc in Hexanes on silica gel column) to afford 112 (62 mg, 87% yield) as a light pink solid. 1H NMR (400 MHz, DMSO-d6) δ 12.15 (s, 1H), 8.28 (s, 1H), 7.42 (d, J = 3.5 Hz, 1H), 7.14 – 6.96 (m, 1H), 6.85 – 6.75 (m, 2H), 6.35 (d, J = 3.4 Hz, 1H), 5.09 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −131.17 – −138.43 (m). 13C NMR (214 MHz, DMSO-d6) δ 162.08, 153.46, 150.20, 149.94 (d, J = 238.3 Hz), 142.03 (d, J = 9.5 Hz), 134.03 (d, J = 12.6 Hz), 124.96, 117.98 (d, J = 3.2 Hz), 115.84 (d, J = 5.5 Hz), 109.79 (d, J = 21.1 Hz), 104.39, 98.08.
1-(4-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)-2-fluorophenyl)-3-(2,4-difluorophenyl)urea (30).
To a solution of 4-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)-2-fluoroaniline (112) (58 mg, 0.24 mmol, 1 eq) in ethanol (0.7 mL) cooled to 0°C using an ice bath was added 2,4-difluoro-1-isocyanatobenzene (37 mg, 0.24 mmol, 1 eq) dropwise. The reaction mixture was warmed to rt and stirred at rt for 1 h. 2,4-difluoro-1-isocyanatobenzene (48 mg, 0.31 mmol, 1.3 eq) was added over the course of 1.5 h portionwise. The reaction mixture was stirred at rt for another 1 h. The reaction mixture was concentrated onto Celite and purified by flash chromatography (0–100% EtOAc in Hexanes on silica gel column then 100% MeCN in water with 0.1% formic acid on C18 column) to afford 30 (33 mg, 35% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.24 (s, 1H), 9.02 (d, J = 2.3 Hz, 1H), 9.00 (d, J = 1.3 Hz, 1H), 8.32 (s, 1H), 8.19 – 8.09 (m, 2H), 7.48 (dd, J = 3.6, 1.6 Hz, 1H), 7.39 – 7.27 (m, 2H), 7.15 – 7.02 (m, 2H), 6.50 (d, J = 3.5 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −117.46 – −118.53 (m), −124.82 (t, J = 10.2 Hz), −126.82 (t, J = 10.6 Hz). 13C NMR (101 MHz, DMSO-d6) δ 161.48, 156.83 (dd, J = 241.3, 11.3 Hz), 153.57, 152.19, 152.05 (dd, J = 241.3, 12.3 Hz), 152.02 (d, J = 243.3 Hz), 150.08, 147.16 (d, J = 10.7 Hz), 125.38, 124.65 (d, J = 10.6 Hz), 123.94 (dd, J = 10.6, 3.5 Hz), 121.79 (dd, J = 9.0, 2.9 Hz), 121.16 (d, J = 2.6 Hz), 118.03 (d, J = 3.2 Hz), 111.10 (dd, J = 21.1, 3.5 Hz), 110.01 (d, J = 22.0 Hz), 104.54, 103.83 (dd, J = 26.7, 23.7 Hz), 97.96. HRMS: calcd for C19H13F3N5O2 [M + H]+ m/z, 400.1021; found m/z, 400.1013.
2-fluoro-4-(pyrimidin-4-yloxy)aniline (113).
To a solution of 4-chloropyrimidine hydrochloride (250 mg, 1.66 mmol, 1 eq) and 4-amino-3-fluorophenol (85) (295 mg, 2.32 mmol, 1.4 eq) in DMF (10 mL) cooled to 0°C using an ice bath was added a suspension of potassium tert-butoxide (446 mg, 3.97 mmol, 2.4 eq) in DMF (5 mL) dropwise. The reaction mixture was stirred at 110°C for 4 h. The reaction mixture was cooled to 0°C using an ice bath, water (50 mL) was added. The aqueous layer was extracted with DCM (50 mL x 3). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–5% MeOH in DCM on silica gel column) to afford 113 (183 mg, 54% yield) as a beige solid. 1H NMR (400 MHz, DMSO-d6) δ 8.75 (dd, J = 1.2, 0.6 Hz, 1H), 8.63 (dd, J = 5.8, 0.7 Hz, 1H), 7.03 (dd, J = 5.8, 1.2 Hz, 1H), 6.99 (dd, J = 11.9, 2.4 Hz, 1H), 6.84 – 6.74 (m, 2H), 5.13 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −132.21 (dd, J = 12.0, 9.5 Hz). 13C NMR (101 MHz, DMSO-d6) δ 169.20, 158.93, 158.40, 149.92 (d, J = 239.1 Hz), 141.15 (d, J = 9.6 Hz), 134.44 (d, J = 13.0 Hz), 117.64 (d, J = 3.2 Hz), 115.93 (d, J = 5.7 Hz), 109.45 (d, J = 21.3 Hz), 108.07.
1-(2,4-difluorophenyl)-3-(2-fluoro-4-(pyrimidin-4-yloxy)phenyl)urea (31).
To a solution of 2-fluoro-4-(pyrimidin-4-yloxy)aniline (113) (64 mg, 0.31 mmol, 1 eq) in ethanol (0.8 mL) cooled to 0°C using an ice bath was added 2,4-difluoro-1-isocyanatobenzene (49 mg, 0.31 mmol, 1 eq) dropwise. The reaction mixture was warmed to rt and stirred at rt for 1 h. The reaction mixture was concentrated onto Celite and purified by flash chromatography (0–8% MeOH in DCM on silica gel column then 0–80% EtOAc in Hexanes on silica gel column) to afford 31 (18.1 mg, 16% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.01 (s, 1H), 8.98 (s, 1H), 8.78 (dd, J = 1.2, 0.6 Hz, 1H), 8.69 (dd, J = 5.8, 0.6 Hz, 1H), 8.17 (t, J = 9.1 Hz, 1H), 8.12 (td, J = 9.2, 6.1 Hz, 1H), 7.41 – 7.27 (m, 2H), 7.16 (dd, J = 5.8, 1.2 Hz, 1H), 7.09 – 7.01 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −118.02 (tdd, J = 8.8, 5.8, 3.5 Hz), −124.85 (t, J = 10.7 Hz), −126.49 (t, J = 10.7 Hz). 13C NMR (101 MHz, DMSO-d6) δ 168.64, 159.21, 158.36, 156.87 (dd, J = 241.3, 11.8 Hz), 152.15, 152.07 (dd, J = 244.9, 12.2 Hz), 152.02 (d, J = 243.7 Hz), 146.41 (d, J = 10.6 Hz), 125.07 (d, J = 10.7 Hz), 123.90 (dd, J = 10.7, 3.6 Hz), 121.80 (dd, J = 9.1, 3.0 Hz), 121.26 (d, J = 2.8 Hz), 117.82 (d, J = 3.3 Hz), 111.12 (dd, J = 21.7, 3.5 Hz), 109.81 (d, J = 22.2 Hz), 108.56, 103.85 (dd, J = 27.0, 23.6 Hz). HRMS: calcd for C17H12F3N4O2 [M + H]+ m/z, 361.0912; found m/z, 361.0902.
4-((2-chloropyridin-4-yl)oxy)-2-fluoroaniline (114).
To a solution of 4-amino-3-fluorophenol (85) (1.00 g, 7.9 mmol, 1.17 eq) in DMA (13.5 mL) was added potassium tert-butoxide (910 mg, 8.1 mmol, 1.2 eq) and stirred at rt for 30 min. 2,4-dichloropyridine (1.00 g, 6.76 mmol, 1 eq) was added and the reaction mixture was stirred at 80°C for 16 h. The reaction mixture was partitioned between EtOAc (100 mL) and water (100 mL). The aqueous layer was extracted with EtOAc (100 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–35% EtOAc in Hexanes on silica gel column) to afford 114 (940 mg, 58% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.25 (dd, J = 5.7, 0.6 Hz, 1H), 7.02 (dd, J = 11.8, 2.5 Hz, 1H), 6.93 – 6.88 (m, 2H), 6.87 – 6.75 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −131.09 (dd, J = 12.0, 9.7 Hz). 13C NMR (214 MHz, DMSO-d6) δ 166.99, 151.46, 151.15, 150.06 (d, J = 240.0 Hz), 141.95 (d, J = 9.3 Hz), 134.84 (d, J = 13.0 Hz), 117.17 (d, J = 3.1 Hz), 116.40 (d, J = 5.6 Hz), 111.16, 111.02, 108.89 (d, J = 21.1 Hz).
1-(4-((2-chloropyridin-4-yl)oxy)-2-fluorophenyl)-3-(2,4-difluorophenyl)urea (32).
To a solution of 1,1′-carbonyldiimidazole (469 mg, 2.89 mmol, 2.5 eq) in DCM (5 mL) cooled to 0°C using an ice bath was added 2,4-difluoroaniline (299 mg, 2.31 mmol, 2 eq) in DCM (7 mL) dropwise over 10 min. The reaction mixture was stirred at 0°C for 30 min, monitoring reaction progress by TLC. This was added to a solution of 4-((2-chloropyridin-4-yl)oxy)-2-fluoroaniline (114) (300 mg, 1.16 mmol, 1 eq) in DCM (3 mL). The reaction mixture was stirred at 40°C for 2 h. Separately, to another solution of 1,1′-carbonyldiimidazole (469 mg, 2.89 mmol, 2.5 eq) in DCM (5 mL) cooled to 0°C using an ice bath was added 2,4-difluoroaniline (299 mg, 2.31 mmol, 2 eq) in DCM (5 mL) dropwise and stirred at 0°C for 10 min. The reaction mixtures were combined and stirred at 40°C for 2 h. The crude reaction mixture was partitioned between EtOAc (60 mL) and water (50 mL). The aqueous layer was extracted with EtOAc (50 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (5–30% EtOAc in Hexanes on silica gel column) to afford 32 (285 mg, 63% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.05 (d, J = 2.4 Hz, 1H), 8.99 (d, J = 2.2 Hz, 1H), 8.30 (d, J = 5.8 Hz, 1H), 8.23 (t, J = 9.1 Hz, 1H), 8.12 (td, J = 9.3, 6.1 Hz, 1H), 7.32 (dtd, J = 11.7, 4.4, 2.9 Hz, 2H), 7.12 – 7.02 (m, 3H), 6.98 (dd, J = 5.7, 2.3 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −117.77 – −118.13 (m), −124.81 (t, J = 10.9 Hz), −125.39 (t, J = 10.5 Hz). 13C NMR (214 MHz, DMSO-d6) δ 166.13, 156.88 (dd, J = 241.7, 11.5 Hz), 152.15 (d, J = 245.8 Hz), 152.08, 152.07 (dd, J = 245.0, 12.2 Hz), 151.59, 151.32, 147.34 (d, J = 10.4 Hz), 125.46 (d, J = 10.7 Hz), 123.83 (dd, J = 10.9, 3.5 Hz), 121.79 (dd, J = 9.2, 2.2 Hz), 121.57 (d, J = 2.8 Hz), 116.98 (d, J = 3.3 Hz), 111.62, 111.57, 111.11 (dd, J = 21.8, 3.3 Hz), 108.97 (d, J = 22.0 Hz), 103.84 (dd, J = 26.8, 23.6 Hz). HRMS: calcd for C18H12ClF3N3O2 [M + H]+ m/z, 394.0570; found m/z, 394.0561.
4-chloro-6-(3-fluoro-4-nitrophenoxy)pyrimidine (98).
To a solution of 4,6-dichloropyrimidine (500 mg, 3.36 mmol, 1 eq) and 3-fluoro-4-nitrophenol (87) (527 mg, 3.36 mmol, 1 eq) in DMF (11 mL) was added potassium carbonate (464 mg, 3.36 mmol, 1 eq). The reaction mixture was stirred at 70°C for 16 h, then cooled to rt and was partitioned between EtOAc (60 mL) and water (60 mL). The aqueous layer was extracted with EtOAc (60 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–35% EtOAc in Hexanes on silica gel column) to afford 98 (554 mg, 61% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.73 (d, J = 0.9 Hz, 1H), 8.30 (t, J = 8.9 Hz, 1H), 7.72 (dd, J = 12.1, 2.5 Hz, 1H), 7.63 (d, J = 0.9 Hz, 1H), 7.40 (ddd, J = 9.1, 2.5, 1.3 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −114.92 (dd, J = 12.1, 8.7 Hz). 13C NMR (214 MHz, DMSO-d6) δ 168.84, 161.44, 158.52, 156.94 (d, J = 11.0 Hz), 155.54 (d, J = 263.1 Hz), 134.60 (d, J = 7.0 Hz), 127.90, 118.59 (d, J = 3.4 Hz), 112.28 (d, J = 24.0 Hz), 109.11.
4-((6-chloropyrimidin-4-yl)oxy)-2-fluoroaniline (115).
A suspension of 4-chloro-6-(3-fluoro-4-nitrophenoxy)pyrimidine (98) (365 mg, 1.35 mmol, 1 eq), iron (378 mg, 6.77 mmol, 5 eq), and ammonium chloride (579 mg, 10.8 mmol, 8 eq) in ethanol (4 mL) and water (1.35 mL) was heated at 31°C for 4 h. The reaction mixture was cooled to rt and filtered through Celite. The filtrate was concentrated onto Celite, and purified by flash chromatography (0–40% EtOAc in Hexanes on silica gel column) to afford 115 (307 mg, 95% yield) as a beige solid. 1H NMR (400 MHz, DMSO-d6) δ 8.64 (d, J = 0.9 Hz, 1H), 7.25 (d, J = 0.9 Hz, 1H), 7.07 – 6.97 (m, 1H), 6.89 – 6.72 (m, 2H), 5.17 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −132.12 (dd, J = 11.9, 8.1 Hz). 13C NMR (214 MHz, DMSO-d6) δ 170.54, 160.77, 158.61, 149.86 (d, J = 239.1 Hz), 140.99 (d, J = 9.5 Hz), 134.67 (d, J = 12.8 Hz), 117.50 (d, J = 3.3 Hz), 115.90 (d, J = 5.7 Hz), 109.34 (d, J = 21.4 Hz), 107.62.
1-(4-((6-chloropyrimidin-4-yl)oxy)-2-fluorophenyl)-3-(2,4-difluorophenyl)urea (33).
To a solution of 4-((6-chloropyrimidin-4-yl)oxy)-2-fluoroaniline (115) (448 mg, 1.87 mmol, 1 eq) in ethanol (4.5 mL) was added 2,4-difluoro-1-isocyanatobenzene (1.45 g, 9.35 mmol, 5 eq) portionwise over 10 h while stirring at rt, monitoring reaction progress by TLC. The reaction mixture was stirred at rt for 14 h, then concentrated onto Celite, and purified by flash chromatography (0–30% EtOAc in Hexanes on silica gel column then 25%−100% DCM in Hexanes followed by 0–10% MeOH in DCM on silica gel column) to afford 33 (81 mg, 11% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.03 (d, J = 2.3 Hz, 1H), 8.99 (d, J = 2.2 Hz, 1H), 8.67 (d, J = 0.9 Hz, 1H), 8.22 – 8.08 (m, 2H), 7.42 (d, J = 0.9 Hz, 1H), 7.34 (dd, J = 11.7, 2.7 Hz, 1H), 7.32 (dd, J = 11.5, 2.9 Hz, 1H), 7.11 – 7.02 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −117.98 (tdd, J = 8.9, 6.1, 3.6 Hz), −124.82 (t, J = 10.9 Hz), −126.34 (t, J = 10.8 Hz). 13C NMR (214 MHz, DMSO-d6) δ 169.95, 160.95, 158.56, 156.86 (dd, J = 241.6, 11.2 Hz), 152.12, 152.06 (dd, J = 245.4, 12.2 Hz), 151.93 (d, J = 244.4 Hz), 146.18 (d, J = 10.2 Hz), 125.36 (d, J = 10.9 Hz), 124.30 – 123.34 (m), 121.78 (dd, J = 9.1, 2.5 Hz), 121.17, 117.89 – 117.04 (m), 111.22 – 110.62 (m), 109.69 (d, J = 22.4 Hz), 108.13, 103.84 (dd, J = 27.2, 23.6 Hz). HRMS: calcd for C17H11ClF3N4O2 [M + H]+ m/z, 395.0523; found m/z, 395.0512.
2-chloro-4-(3-fluoro-4-nitrophenoxy)pyrimidine (99).
To a solution of 2,4-dichloropyrimidine (800 mg, 5.37 mmol, 1 eq) and 3-fluoro-4-nitrophenol (87) (844 mg, 5.37 mmol, 1 eq) in DMF (15 mL) was added potassium carbonate (742 mg, 5.37 mmol, 1 eq). The reaction mixture was stirred at 70°C for 16 h, then cooled to rt and was partitioned between EtOAc (60 mL) and water (60 mL). The aqueous layer was extracted with EtOAc (60 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (20–100% DCM in Hexanes on silica gel column) to afford 99 (788 mg, 54% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.73 (d, J = 5.7 Hz, 1H), 8.31 (t, J = 8.9 Hz, 1H), 7.75 (dt, J = 12.0, 2.1 Hz, 1H), 7.43 (ddd, J = 9.1, 2.5, 1.3 Hz, 1H), 7.37 (dd, J = 5.7, 1.1 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −114.62 – −114.74 (m). 13C NMR (214 MHz, DMSO-d6) δ 168.87, 162.15, 158.75, 156.51 (d, J = 11.4 Hz), 155.51 (d, J = 263.2 Hz), 134.72 (d, J = 6.9 Hz), 127.95, 118.53 (d, J = 3.5 Hz), 112.20 (d, J = 23.8 Hz), 108.36.
4-((2-chloropyrimidin-4-yl)oxy)-2-fluoroaniline (116).
A suspension of 2-chloro-4-(3-fluoro-4-nitrophenoxy)pyrimidine (99) (770 mg, 2.86 mmol, 1 eq), iron (798 mg, 14.3 mmol, 5 eq), and ammonium chloride (1.22 g, 22.8 mmol, 8 eq) in ethanol (7.5 mL) and water (2.5 mL) was heated at 40°C for 5 h. The reaction mixture was cooled to rt and filtered through Celite. The filtrate was concentrated onto Celite, and purified by flash chromatography (0–40% EtOAc in Hexanes on silica gel column) to afford 116 (625 mg, 91% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.57 (d, J = 5.8 Hz, 1H), 7.12 – 6.97 (m, 2H), 6.87 – 6.76 (m, 2H), 5.21 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −128.17 – −134.17 (m). 13C NMR (214 MHz, DMSO-d6) δ 170.73, 161.32, 159.05, 149.83 (d, J = 239.5 Hz), 140.73 (d, J = 9.5 Hz), 134.82 (d, J = 12.8 Hz), 117.44 (d, J = 3.1 Hz), 115.96 (d, J = 5.5 Hz), 109.25 (d, J = 21.7 Hz), 107.13.
1-(4-((2-chloropyrimidin-4-yl)oxy)-2-fluorophenyl)-3-(2,4-difluorophenyl)urea (34).
To a solution of 1,1′-carbonyldiimidazole (761 mg, 4.69 mmol, 2.25 eq) in DCM (10 mL) cooled to 0°C using an ice bath was added 2,4-difluoroaniline (539 mg, 4.17 mmol, 2 eq) in DCM (10 mL) dropwise over 10 min. The reaction mixture was stirred at 0°C for 50 min, monitoring reaction progress by TLC. This was added to a solution of 4-((2-chloropyrimidin-4-yl)oxy)-2-fluoroaniline (116) (500 mg, 2.09 mmol, 1 eq) in DCM (5 mL). The reaction mixture was stirred at 40°C for 3 h. The crude reaction mixture was partitioned between EtOAc (60 mL) and water (50 mL). The aqueous layer was extracted with EtOAc (50 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (5–35% EtOAc in Hexanes on silica gel column) to afford 34 (443 mg, 54% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.04 (d, J = 2.3 Hz, 1H), 9.00 (d, J = 2.1 Hz, 1H), 8.63 (d, J = 5.7 Hz, 1H), 8.20 (t, J = 9.1 Hz, 1H), 8.12 (td, J = 9.2, 6.1 Hz, 1H), 7.44 – 7.29 (m, 2H), 7.19 (d, J = 5.7 Hz, 1H), 7.14 – 6.99 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −117.96 (tdd, J = 8.9, 6.1, 3.6 Hz), −124.81 (t, J = 10.2 Hz), −126.15 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 170.08, 161.54, 158.95, 156.87 (dd, J = 241.7, 11.5 Hz), 152.11, 152.06 (dd, J = 245.1, 12.3 Hz), 151.88 (d, J = 244.3 Hz), 145.83 (d, J = 10.3 Hz), 125.55 (d, J = 10.3 Hz), 123.85 (dd, J = 11.0, 3.5 Hz), 121.79 (dd, J = 8.7, 2.8 Hz), 121.18 (d, J = 2.5 Hz), 117.62 (d, J = 3.1 Hz), 111.10 (dd, J = 21.6, 3.4 Hz), 109.60 (d, J = 22.5 Hz), 107.58, 103.84 (dd, J = 26.7, 23.5 Hz). HRMS: calcd for C17H11ClF3N4O2 [M + H]+ m/z, 395.0523; found m/z, 395.0515.
1-(4-((6-(benzylamino)pyrimidin-4-yl)oxy)-2-fluorophenyl)-3-(2,4-difluorophenyl)urea (35).
To a solution of 1-(4-((6-chloropyrimidin-4-yl)oxy)-2-fluorophenyl)-3-(2,4-difluorophenyl)urea (33) (61 mg, 0.15 mmol, 1 eq) in THF (0.6 mL) was added triethylamine (23 mg, 1.5 eq, 0.23 mmol) and benzylamine (100 mg, 0.93 mmol, 6 eq). The reaction mixture was stirred at 66°C for 10 h, then concentrated onto Celite, and purified by flash chromatography (0–4% MeOH in DCM on silica gel column then 25–100% MeCN in water with 0.1% formic acid on C18 column) to afford 35 (37 mg, 52% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.99 (s, 1H), 8.98 (s, 1H), 8.20 – 8.06 (m, 3H), 7.90 (s, 1H), 7.38 – 7.28 (m, 5H), 7.27 – 7.22 (m, 1H), 7.18 (d, J = 10.7 Hz, 1H), 7.05 (t, J = 8.7 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 5.87 (s, 1H), 4.50 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −118.02 – −118.12 (m), −124.83 (t, J = 9.7 Hz), −126.57. 13C NMR (214 MHz, DMSO-d6) δ 168.79, 164.59, 158.09, 156.81 (dd, J = 241.5, 11.6 Hz), 152.15, 152.04 (d, J = 243.9 Hz), 152.04 (dd, J = 244.9, 12.2 Hz), 147.31 (d, J = 10.3 Hz), 139.55, 128.35, 127.22, 126.85, 124.44 (d, J = 11.7 Hz), 123.92 (dd, J = 10.8, 3.7 Hz), 121.76 (dd, J = 9.0, 2.0 Hz), 121.30, 117.62 (d, J = 2.4 Hz), 111.08 (dd, J = 21.6, 3.4 Hz), 109.54 (d, J = 21.8 Hz), 103.82 (dd, J = 27.1, 23.5 Hz), 87.21, 43.64. HRMS: calc for C24H19F3N5O2 [M + H]+ m/z, 466.1491; found m/z, 466.1482.
Experimental Procedures in Schemes 5 and 6.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(3-(trifluoromethyl)phenyl)urea (39).
To a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (80 mg, 0.25 mmol, 1 eq) in DCM (0.3 mL) and ethanol (0.6 mL) was added 1-isocyanato-3-(trifluoromethyl)benzene (144 mg, 0.75 mmol, 3 eq). The reaction mixture was stirred at rt for 16 h, then concentrated onto Celite, and purified by flash chromatography (1–5% MeOH in DCM on silica gel column) to afford 39 (75 mg, 58% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.42 (s, 1H), 8.72 (d, J = 2.3 Hz, 1H), 8.50 (d, J = 5.3 Hz, 1H), 8.21 (t, J = 9.1 Hz, 1H), 8.04 (d, J = 2.2 Hz, 1H), 7.59 – 7.51 (m, 2H), 7.49 (s, 1H), 7.41 (s, 1H), 7.39 – 7.31 (m, 2H), 7.11 (ddd, J = 8.9, 2.8, 1.3 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −61.35 (s, 3F), −125.21 (t, J = 10.6 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.58, 152.62 (d, J = 244.8 Hz), 152.62, 152.28, 149.41, 148.87, 148.77 (d, J = 10.2 Hz), 146.52, 140.28, 130.06, 129.63 (q, J = 31.5 Hz), 124.75 (d, J = 10.8 Hz), 124.19 (q, J = 271.8 Hz), 122.20 (d, J = 2.2 Hz), 121.79, 118.39 (q, J = 3.6 Hz), 117.09 (d, J = 3.1 Hz), 115.11, 114.04 (q, J = 3.9 Hz), 109.05 (d, J = 21.9 Hz), 107.87, 103.39, 99.04, 55.74, 55.72. HRMS: calcd for C25H20F4N3O4 [M + H]+ m/z, 502.1390; found m/z, 502.1380.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(2-fluoro-5-(trifluoromethyl)phenyl)urea (40).
To a solution of 2-fluoro-5-(trifluoromethyl)aniline (51 mg, 0.29 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (50 mg, 0.31 mmol, 1.3 eq). The reaction mixture was stirred at rt for 1 h then warmed to 40°C and stirred for 2.5 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (75 mg, 0.24 mmol, 1 eq) in DMSO (1 mL). The reaction mixture was stirred at 60°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (40–100% EtOAc in Hexanes on silica gel column) to afford 40 (47 mg, 38% yield) as a light pink solid. 1H NMR (400 MHz, DMSO-d6) δ 9.38 (d, J = 2.8 Hz, 1H), 9.23 (d, J = 2.3 Hz, 1H), 8.65 (dd, J = 7.2, 2.3 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.27 (t, J = 9.1 Hz, 1H), 7.52 (dd, J = 10.8, 8.7 Hz, 1H), 7.49 (s, 1H), 7.44 – 7.40 (m, 1H), 7.41 (s, 1H), 7.38 (dd, J = 11.8, 2.7 Hz, 1H), 7.12 (ddd, J = 9.0, 2.8, 1.2 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −60.74 (s, 3F), −123.98 (t, J = 8.3 Hz, 1F), −125.46 (t, J = 10.7 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.55, 153.47 (d, J = 247.8 Hz), 152.61, 152.37 (d, J = 245.1 Hz), 152.00, 149.41, 148.87, 148.76 (d, J = 10.2 Hz), 146.51, 128.50 (d, J = 11.3 Hz), 125.42 (qd, J = 31.9, 2.2 Hz), 124.55 (d, J = 10.8 Hz), 123.90 (q, J = 271.8 Hz), 121.84 (d, J = 2.5 Hz), 119.55 (dq, J = 7.7, 3.9 Hz), 117.13 (d, J = 3.1 Hz), 116.57 (dq, J = 4.0, 3.2 Hz), 116.17 (d, J = 20.4 Hz), 115.10, 109.07 (d, J = 21.9 Hz), 107.87, 103.40, 99.03, 55.74, 55.72. HRMS: calcd for C25H19F5N3O4 [M + H]+ m/z, 520.1296; found m/z, 520.1287.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-((4-methylpiperazin-1-yl)methyl)phenyl)urea (41).
To a solution of 4-((4-methylpiperazin-1-yl)methyl)aniline (55 mg, 0.27 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (47 mg, 0.29 mmol, 1.3 eq). The reaction mixture was stirred at rt for 2 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (70 mg, 0.22 mmol, 1 eq) in DMSO (1 mL). The reaction mixture was stirred at 60°C for 2 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (5–15% MeOH in DCM on silica gel column) to afford 41 (55 mg, 45% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.21 (s, 1H), 8.68 (d, J = 2.5 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.24 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.44 – 7.39 (m, 3H), 7.34 (dd, J = 11.8, 2.7 Hz, 1H), 7.21 (d, J = 8.5 Hz, 2H), 7.09 (ddd, J = 9.0, 2.7, 1.3 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.40 (s, 2H), 2.38 (br s, 8H), 2.21 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −125.74 (t, J = 10.7 Hz). 13C NMR (101 MHz, DMSO-d6) δ 160.11, 153.03, 152.77, 152.70 (d, J = 244.6 Hz), 149.82, 149.32, 148.61 (d, J = 10.3 Hz), 146.93, 138.79, 132.01, 129.89, 125.75 (d, J = 10.6 Hz), 122.07 (d, J = 2.8 Hz), 118.36, 117.50 (d, J = 2.9 Hz), 115.52, 109.44 (d, J = 22.2 Hz), 108.31, 103.74, 99.49, 61.95, 56.18, 56.16, 54.91, 52.52, 45.78. HRMS: calcd for C30H33FN5O4 [M + H]+ m/z, 546.2517; found m/z, 546.2511.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(3-((4-methylpiperazin-1-yl)methyl)phenyl)urea diformate (42).
To a solution of 3-((4-methylpiperazin-1-yl)methyl)aniline (55 mg, 0.27 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (47 mg, 0.29 mmol, 1.2 eq). The reaction mixture was stirred at rt for 2 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (70 mg, 0.22 mmol, 1 eq) in DMSO (1 mL). The reaction mixture was stirred at 60°C for 2 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (5–15% MeOH in DCM on silica gel column then 10% MeCN in water with 0.1% formic acid on C18 column) to afford 42 (78 mg, 55% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.13 (s, 1H), 8.63 (d, J = 2.6 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.24 (t, J = 9.1 Hz, 1H), 8.16 (s, 2H), 7.49 (s, 1H), 7.43 (t, J = 1.9 Hz, 1H), 7.40 (s, 1H), 7.36 (d, J = 2.5 Hz, 1H), 7.35 – 7.31 (m, 1H), 7.24 (t, J = 7.8 Hz, 1H), 7.10 (ddd, J = 9.3, 2.5, 1.3 Hz, 1H), 6.92 (d, J = 7.5 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.44 (s, 2H), 2.42 (br s, 8H), 2.23 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −125.80 (t, J = 10.9 Hz). 13C NMR (101 MHz, DMSO-d6) δ 163.52, 159.65, 152.59, 152.33 (d, J = 244.5 Hz), 152.25, 149.39, 148.86, 148.28 (d, J = 10.3 Hz), 146.49, 139.43, 138.76, 128.67, 125.23 (d, J = 10.5 Hz), 122.65, 121.75 (d, J = 3.0 Hz), 118.43, 117.07 (d, J = 3.2 Hz), 116.84, 115.08, 109.01 (d, J = 22.1 Hz), 107.86, 103.31, 99.03, 61.87, 55.73, 55.71, 54.19, 51.78, 44.89. HRMS: calcd for C30H34FN5O4 [M + 2H]2+ m/z, 273.6298; found m/z, 273.6284.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(4-methylpiperazine-1-carbonyl)phenyl)urea (43).
To a solution of (4-aminophenyl)(4-methylpiperazin-1-yl)methanone (59 mg, 0.27 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (47 mg, 0.29 mmol, 1.3 eq). The reaction mixture was stirred at rt for 2 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (70 mg, 0.22 mmol, 1 eq) in DMSO (1 mL). The reaction mixture was stirred at 60°C for 2 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–12% MeOH in DCM on silica gel column) to afford 43 (57 mg, 45% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.33 (s, 1H), 8.71 (d, J = 2.5 Hz, 1H), 8.50 (d, J = 5.3 Hz, 1H), 8.24 (t, J = 9.1 Hz, 1H), 7.55 – 7.51 (m, 2H), 7.49 (s, 1H), 7.40 (s, 1H), 7.38 – 7.33 (m, 3H), 7.11 (ddd, J = 8.9, 2.8, 1.3 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.49 (br s, 4H), 2.32 (br s, 4H), 2.20 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −125.57 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 168.87, 159.61, 152.60, 152.38 (d, J = 244.5 Hz), 152.14, 149.40, 148.88, 148.47 (d, J = 9.7 Hz), 146.50, 140.70, 129.13, 128.28, 125.01 (d, J = 10.2 Hz), 121.77, 117.45, 117.20 – 116.90 (m), 115.09, 109.04 (d, J = 22.1 Hz), 107.87, 103.35, 99.04, 55.74, 55.72, 55.67 (br s), 54.49 (br s), 45.60. HRMS: calcd for C30H31FN5O5 [M + H]+ m/z, 560.2309; found m/z, 560.2303.
(4-methylpiperazin-1-yl)(3-nitrophenyl)methanone (169).
To a solution of 3-nitrobenzoyl chloride (128) (335 mg, 1.81 mmol, 1 eq) in THF (2.25 mL) was added 1-methylpiperazine (181 mg, 1.81 mmol, 1 eq). The reaction mixture was stirred at rt for 30 min, then partitioned between EtOAc (30 mL) and aqueous NaOH solution (1 M, 30 mL). The aqueous layer was extracted with EtOAc (30 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and filtered to afford 169 (243 mg, 54% yield) as an orange liquid. 1H NMR (400 MHz, DMSO-d6) δ 8.30 (ddd, J = 8.2, 2.4, 1.1 Hz, 1H), 8.18 (t, J = 1.9 Hz, 1H), 7.85 (dt, J = 7.7, 1.3 Hz, 1H), 7.74 (t, J = 7.9 Hz, 1H), 3.64 (s, 2H), 3.30 (s, 2H), 2.38 (s, 2H), 2.27 (s, 2H), 2.20 (s, 3H). 13C NMR (214 MHz, DMSO-d6) δ 166.70, 147.70, 137.44, 133.35, 130.29, 124.24, 121.79, 54.55 (br s), 54.05 (br s), 47.04 (br s), 45.54, 41.57 (br s).
(3-aminophenyl)(4-methylpiperazin-1-yl)methanone (129).
A suspension of (4-methylpiperazin-1-yl)(3-nitrophenyl)methanone (169) (240 mg, 0.96 mmol, 1 eq), iron (269 mg, 4.82 mmol, 5 eq), and ammonium chloride (412 mg, 7.71 mmol, 8 eq) in ethanol (3 mL) and water (1 mL) was stirred at 31°C for 4 h then warmed to 40°C and stirred for 1 h. The reaction mixture was cooled to rt and filtered through Celite. The filtrate was concentrated onto Celite, and purified by flash chromatography (3–10% MeOH in DCM on silica gel column) to afford 129 (142 mg, 67% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 7.04 (t, J = 7.7 Hz, 1H), 6.59 (ddd, J = 8.1, 2.3, 1.0 Hz, 1H), 6.52 (t, J = 1.9 Hz, 1H), 6.43 (dt, J = 7.4, 1.3 Hz, 1H), 5.24 (s, 2H), 3.54 (br s, 2H), 3.35 (br s, 2H), 2.29 (br s, 4H), 2.19 (s, 3H). 13C NMR (214 MHz, DMSO-d6) δ 172.75, 151.86, 139.75, 131.97, 117.78, 116.86, 115.05, 57.90 (br s), 57.46 (br s), 50.02 (br s), 48.69, 44.34 (br s).
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(3-(4-methylpiperazine-1-carbonyl)phenyl)urea (44).
To a solution of (3-aminophenyl)(4-methylpiperazin-1-yl)methanone (129) (59 mg, 0.27 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (47 mg, 0.29 mmol, 1.3 eq). The reaction mixture was stirred at rt for 2 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (70 mg, 0.22 mmol, 1 eq) in DMSO (1 mL). The reaction mixture was stirred at 60°C for 2 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–12% MeOH in DCM on silica gel column) to afford 44 (58 mg, 47% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.26 (s, 1H), 8.68 (d, J = 2.5 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.22 (t, J = 9.1 Hz, 1H), 7.58 (t, J = 1.9 Hz, 1H), 7.49 (s, 1H), 7.46 – 7.40 (m, 2H), 7.39 – 7.32 (m, 2H), 7.11 (ddd, J = 8.9, 2.8, 1.3 Hz, 1H), 6.99 (dt, J = 7.5, 1.4 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.61 (br s, 2H), 3.36 (br s, 2H), 2.33 (br s, 4H), 2.20 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −125.49 (t, J = 10.5 Hz). 13C NMR (214 MHz, DMSO-d6) δ 168.79, 159.61, 152.60, 152.43 (d, J = 244.8 Hz), 152.25, 149.40, 148.87, 148.49 (d, J = 9.6 Hz), 146.50, 139.50, 136.62, 129.08, 125.00 (d, J = 10.8 Hz), 121.88, 120.41, 118.99, 117.10, 116.31, 115.09, 109.02 (d, J = 22.1 Hz), 107.87, 103.35, 99.03, 55.73, 55.71, 54.75 (br s), 54.26 (br s), 47.02 (br s), 45.58, 41.38 (br s). HRMS: calcd for C30H31FN5O5 [M + H]+ m/z, 560.2309; found m/z, 560.2302.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)urea diformate (45).
To a solution of 4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)aniline (79 mg, 0.29 mmol, 1.3 eq) in DMSO (1.3 mL) was added 1,1′-carbonyldiimidazole (65 mg, 0.40 mmol, 1.8 eq). The reaction mixture was stirred at rt for 3 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (70 mg, 0.22 mmol, 1 eq) in DMSO (1 mL). The reaction mixture was stirred at 60°C for 2 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (3–15% MeOH in DCM on silica gel column then 8–15% MeCN in water with 0.1% formic acid on C18 column) to afford 45 (66 mg, 42% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.63 (s, 1H), 8.91 (s, 1H), 8.50 (d, J = 5.3 Hz, 1H), 8.26 – 8.14 (m, 3H), 8.00 (s, 1H), 7.69 – 7.55 (m, 2H), 7.49 (s, 1H), 7.40 (s, 1H), 7.38 – 7.31 (m, 1H), 7.16 – 7.04 (m, 1H), 6.54 (d, J = 5.3 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.55 (s, 2H), 2.55 (br s, 4H), 2.44 (br s, 4H), 2.31 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −58.03 (s, 3F), −124.88 (t, J = 10.7 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 163.51, 159.58, 152.67 (d, J = 245.1 Hz), 152.60, 152.37, 149.40, 148.86, 148.72 (d, J = 10.1 Hz), 146.50, 138.73, 131.52, 130.01, 127.65 (q, J = 29.6 Hz), 124.85 (d, J = 10.8 Hz), 124.35 (q, J = 274.2 Hz), 122.32 (d, J = 2.2 Hz), 121.51, 117.05 (d, J = 3.2 Hz), 115.10, 114.92 (q, J = 6.1 Hz), 109.04 (d, J = 22.0 Hz), 107.86, 103.37, 99.03, 57.20, 55.73, 55.71, 54.27, 51.94, 44.90. HRMS: calcd for C31H33F4N5O4 [M + 2H]2+ m/z, 307.6234; found m/z, 307.6220.
(4-methylpiperazin-1-yl)(3-nitro-5-(trifluoromethyl)phenyl)methanone (170).
To a solution of 3-nitro-5-(trifluoromethyl)benzoic acid (130) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (856 mg, 4.47 mmol, 1.5 eq) in DCM (6 mL) was added 1-methylpiperazine (358 mg, 3.57 mmol, 1.2 eq). The reaction mixture was stirred at rt for 18 h, then was partitioned between DCM (50 mL) and saturated aqueous NaHCO3 solution (50 mL). The aqueous layer was extracted with DCM (50 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–4% MeOH in DCM on silica gel column) to afford 170 (744 mg, 79% yield) as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 8.55 (t, J = 2.1 Hz, 1H), 8.48 (t, J = 1.8 Hz, 1H), 8.26 (t, J = 1.7 Hz, 1H), 3.65 (s, 2H), 3.29 (s, 2H), 2.40 (s, 2H), 2.27 (s, 2H), 2.20 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −61.30. 13C NMR (214 MHz, DMSO-d6) δ 165.28, 148.22, 139.03, 130.70 (q, J = 33.7 Hz), 129.80 (q, J = 3.7 Hz), 125.70, 122.74 (q, J = 273.2 Hz), 121.15 (q, J = 3.9 Hz), 54.35, 53.87, 46.96, 45.53, 41.64.
3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)aniline (131).
To a solution of lithium aluminium hydride (299 mg, 7.9 mmol, 10 eq) in THF (8.5 mL) was added a solution of (4-methylpiperazin-1-yl)(3-nitro-5-(trifluoromethyl)phenyl)methanone (170) (250 mg, 0.79 mmol, 1 eq) in THF (1.5 mL). The reaction mixture was stirred at 66°C for 5 h, then cooled to 0°C using an ice bath. Water (0.3 mL) was added dropwise to the reaction mixture, followed by aqueous NaOH solution (15%, 0.3 mL) then water (0.9 mL). The reaction mixture was warmed to rt and stirred at rt for 2 h and the resultant grey suspension was filtered through Celite. The filtrate was concentrated onto Celite and purified by flash chromatography (1–20% MeOH in DCM on silica gel column) to afford 131 (60 mg, 28% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 6.76 (s, 1H), 6.71 (s, 1H), 6.68 (s, 1H), 5.52 (s, 2H), 3.35 (s, 2H), 2.33 (br s, 8H), 2.14 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −61.31.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)urea (46).
To a solution of 3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)aniline (131) (55 mg, 0.20 mmol, 1 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (59 mg, 0.36 mmol, 1.8 eq). The reaction mixture was stirred at rt for 2 h then warmed to 40°C and stirred for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (89 mg, 0.28 mmol, 1.4 eq) in DMSO (1 mL). The reaction mixture was stirred at 60°C for 2 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–15% MeOH in DCM on silica gel column) to afford 46 (71 mg, 58% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.48 (s, 1H), 8.68 (d, J = 2.4 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.20 (t, J = 9.1 Hz, 1H), 7.93 (t, J = 2.0 Hz, 1H), 7.51 (s, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.36 (dd, J = 11.7, 2.7 Hz, 1H), 7.23 (s, 1H), 7.10 (ddd, J = 9.0, 2.8, 1.3 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.52 (s, 2H), 2.39 (br s, 4H), 2.35 (br s, 4H), 2.16 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −61.26 (s, 3F), −125.21 (t, J = 10.5 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.57, 152.61 (d, J = 245.1 Hz), 152.60, 152.25, 149.40, 148.86, 148.76 (d, J = 10.2 Hz), 146.51, 140.99, 140.30, 129.53 (q, J = 31.2 Hz), 124.74 (d, J = 10.6 Hz), 124.21 (q, J = 272.4 Hz), 122.21 (d, J = 2.0 Hz), 121.52, 118.33 (q, J = 3.5 Hz), 117.07 (d, J = 3.1 Hz), 115.10, 112.69 (q, J = 3.9 Hz), 109.04 (d, J = 22.1 Hz), 107.87, 103.38, 99.03, 61.19, 55.73, 55.71, 54.71, 52.46, 45.70. HRMS: calcd for C31H32F4N5O4 [M + H]+ m/z, 614.2390; found m/z, 614.2381.
(4-amino-2-(trifluoromethyl)phenyl)(4-methylpiperazin-1-yl)methanone (133).
To a solution of 4-amino-2-(trifluoromethyl)benzoic acid (132) (250 mg, 1.2 mmol, 1 eq) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (350 mg, 1.8 mmol, 1.5 eq) in DCM (2.4 mL) was added 1-methylpiperazine (134 mg, 1.3 mmol, 1.1 eq). The reaction mixture was stirred at rt for 18 h, then was partitioned between DCM (30 mL) and saturated aqueous NaHCO3 solution (30 mL). The aqueous layer was extracted with DCM (30 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–10% MeOH in DCM on silica gel column) to afford 133 (216 mg, 62% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.01 (d, J = 8.3 Hz, 1H), 6.88 (d, J = 2.3 Hz, 1H), 6.78 (dd, J = 8.4, 2.3 Hz, 1H), 5.77 (s, 2H), 3.56 (br s, 2H), 3.13 (br s, 1H), 3.08 (br s, 1H), 2.36 – 2.09 (m, 7H). 19F NMR (376 MHz, DMSO-d6) δ −58.80. 13C NMR (214 MHz, DMSO-d6) δ 170.31, 152.62, 131.61, 129.52 (q, J = 30.7 Hz), 127.11 (q, J = 273.8 Hz), 124.23 (q, J = 2.2 Hz), 119.57, 113.40 (q, J = 5.0 Hz), 57.40, 57.26, 49.77, 48.75, 44.16.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(4-methylpiperazine-1-carbonyl)-3-(trifluoromethyl)phenyl)urea (47).
To a solution of (4-amino-2-(trifluoromethyl)phenyl)(4-methylpiperazin-1-yl)methanone (133) (90 mg, 0.31 mmol, 1.4 eq) in DMSO (1.3 mL) was added 1,1′-carbonyldiimidazole (54 mg, 0.33 mmol, 1.5 eq). The reaction mixture was stirred at rt for 2 h, then warmed to 40°C and stirred for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (70 mg, 0.22 mmol, 1 eq) in DMSO (1 mL). The reaction mixture was stirred at 60°C for 2 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–20% MeOH in DCM on silica gel column) to afford 47 (26 mg, 19% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.56 (s, 1H), 8.77 (d, J = 2.4 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.19 (t, J = 9.1 Hz, 1H), 8.08 (d, J = 2.1 Hz, 1H), 7.62 (dd, J = 8.5, 2.2 Hz, 1H), 7.49 (s, 1H), 7.41 (s, 1H), 7.39 – 7.34 (m, 2H), 7.11 (ddd, J = 8.9, 2.8, 1.2 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.70 – 3.56 (m, 2H), 3.21 – 2.94 (m, 2H), 2.45 – 2.12 (m, 7H). 19F NMR (376 MHz, DMSO-d6) δ −58.84 (s, 3F), −124.95 (t, J = 10.6 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 166.10, 159.55, 152.73 (d, J = 245.4 Hz), 152.62, 152.24, 149.42, 148.94 (d, J = 10.3 Hz), 148.88, 146.52, 140.35, 128.46, 128.01, 126.04 (q, J = 29.6 Hz), 124.60 (d, J = 10.8 Hz), 123.62 (q, J = 274.9 Hz), 122.39, 121.51, 117.10, 115.11, 115.09, 109.08 (d, J = 22.5 Hz), 107.88, 103.42, 99.03, 55.75, 55.72, 54.14, 54.02, 46.55, 45.55, 41.02. HRMS: calcd for C31H30F4N5O5 [M + H]+ m/z, 628.2183; found m/z, 628.2175.
(3-amino-5-(trifluoromethyl)phenyl)(4-methylpiperazin-1-yl)methanone (135).
To a solution of 3-amino-5-(trifluoromethyl)benzoic acid (134) (250 mg, 1.2 mmol, 1 eq) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (350 mg, 1.8 mmol, 1.5 eq) in DCM (2.4 mL) was added 1-methylpiperazine (134 mg, 1.3 mmol, 1.1 eq). The reaction mixture was stirred at rt for 18 h, then was partitioned between DCM (30 mL) and saturated aqueous NaHCO3 solution (30 mL). The aqueous layer was extracted with DCM (30 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–12% MeOH in EtOAc on silica gel column) to afford 135 (161 mg, 46% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 6.89 (t, J = 2.0 Hz, 1H), 6.76 (t, J = 1.8 Hz, 1H), 6.69 (s, 1H), 5.79 (s, 2H), 3.57 (br s, 2H), 3.29 (br s, 2H), 2.32 (br s, 2H), 2.28 (br s, 2H), 2.19 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −61.63. 13C NMR (214 MHz, DMSO-d6) δ 168.12, 149.63, 137.74, 129.96 (q, J = 31.3 Hz), 124.13 (q, J = 272.4 Hz), 115.01, 110.10 (q, J = 3.7 Hz), 109.46 (q, J = 4.2, 3.7 Hz), 54.71 (br s), 54.21 (br s), 46.95 (br s), 45.59, 41.39 (br s).
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(3-(4-methylpiperazine-1-carbonyl)-5-(trifluoromethyl)phenyl)urea formate (48).
To a solution of (3-amino-5-(trifluoromethyl)phenyl)(4-methylpiperazin-1-yl)methanone (135) (80 mg, 0.28 mmol, 1.25 eq) in DMSO (1.2 mL) was added 1,1′-carbonyldiimidazole (54 mg, 0.33 mmol, 1.5 eq). The reaction mixture was stirred at rt for 2 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (70 mg, 0.22 mmol, 1 eq) in DMSO (1 mL). The reaction mixture was stirred at 60°C for 2 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–12% MeOH in DCM on silica gel column then 8–20% MeCN in water with 0.1% formic acid on C18 column) to afford 48 (38 mg, 26% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.70 (s, 1H), 8.93 (s, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.19 – 8.10 (m, 2H), 8.01 (s, 1H), 7.66 (s, 1H), 7.49 (s, 1H), 7.41 (s, 1H), 7.36 (dd, J = 11.6, 2.7 Hz, 1H), 7.31 (s, 1H), 7.16 – 7.08 (m, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.63 (br s, 2H), 3.33 (br s, 2H), 2.39 (br s, 2H), 2.32 (br s, 2H), 2.21 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −61.47 (s, 3F), −124.36 (t, J = 10.4 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 167.24, 163.40, 159.52, 152.97 (d, J = 245.2 Hz), 152.61, 152.37, 149.41, 149.10 (d, J = 10.9 Hz), 148.86, 140.63, 137.78, 129.92 (q, J = 32.5 Hz), 124.52 (d, J = 10.9 Hz), 123.76 (q, J = 273.2 Hz), 122.80, 119.76, 117.04, 116.56, 115.12, 114.92, 109.08 (d, J = 22.6 Hz), 107.87, 103.44, 99.03, 55.74, 55.71, 54.58 (br s), 54.07 (br s), 46.96 (br s), 45.49, 41.47 (br s). HRMS: calcd for C31H31F4N5O5 [M + 2H]2+ m/z, 314.6131; found m/z, 314.6117.
1-(3-chlorophenyl)-3-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)urea (49).
To a solution of 3-chloroaniline (24 mg, 0.19 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (34 mg, 0.21 mmol, 1.3 eq). The reaction mixture was stirred at rt for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 4 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (20–100% EtOAc in Hexanes on silica gel column then 1–4% MeOH in DCM on silica gel column) to afford 49 (24 mg, 32% yield) as a light orange solid. 1H NMR (400 MHz, DMSO-d6) δ 9.26 (s, 1H), 8.68 (d, J = 2.4 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.20 (t, J = 9.1 Hz, 1H), 7.74 (t, J = 2.0 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.35 (dd, J = 11.8, 2.7 Hz, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.26 (ddd, J = 8.2, 2.1, 1.1 Hz, 1H), 7.11 (ddd, J = 9.0, 2.8, 1.3 Hz, 1H), 7.05 (ddd, J = 7.8, 2.1, 1.1 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −125.38 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.63, 152.64, 152.52 (d, J = 245.0 Hz), 152.15, 149.43, 148.87, 148.65 (d, J = 10.6 Hz), 146.47, 140.94, 133.31, 130.55, 124.85 (d, J = 10.7 Hz), 122.01 (d, J = 2.8 Hz), 121.80, 117.52, 117.12 (d, J = 2.5 Hz), 116.64, 115.11, 109.06 (d, J = 22.3 Hz), 107.84, 103.40, 99.06, 55.76, 55.74. HRMS: calcd for C24H20ClFN3O4 [M + H]+ m/z, 468.1126; found m/z, 468.1115.
1-(3-bromophenyl)-3-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)urea (50).
To a solution of 3-bromoaniline (33 mg, 0.19 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (34 mg, 0.21 mmol, 1.3 eq). The reaction mixture was stirred at rt for 30 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 5 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (20–70% EtOAc in Hexanes on silica gel column) to afford 50 (17 mg, 21% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.25 (s, 1H), 8.69 (d, J = 2.5 Hz, 1H), 8.49 (s, 0H), 8.20 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.38 – 7.33 (m, 1H), 7.30 (dt, J = 8.2, 1.7 Hz, 1H), 7.26 (t, J = 7.8 Hz, 1H), 7.18 (dt, J = 7.5, 1.7 Hz, 1H), 7.11 (ddd, J = 8.9, 2.8, 1.3 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −125.36 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.57, 152.60, 152.50 (d, J = 244.7 Hz), 152.11, 149.40, 148.87, 148.63 (d, J = 10.3 Hz), 146.50, 141.07, 130.83, 124.83 (d, J = 10.3 Hz), 124.67, 121.98 (d, J = 2.0 Hz), 121.81, 120.35, 117.09 (d, J = 3.0 Hz), 117.00, 115.09, 109.04 (d, J = 21.9 Hz), 107.87, 103.38, 99.03, 55.74, 55.71. HRMS: calcd for C24H20BrFN3O4 [M + H]+ m/z, 512.0621; found m/z, 512.0611.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(m-tolyl)urea (51).
To a solution of m-toluidine hydrochloride (27 mg, 0.19 mmol, 1.2 eq) in DMSO (1 mL) was added DIPEA (25 mg, 0.19 mmol, 1.2 eq) and 1,1′-carbonyldiimidazole (34 mg, 0.21 mmol, 1.3 eq). The reaction mixture was stirred at rt for 45 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 4 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (20–75% EtOAc in Hexanes on silica gel column) to afford 51 (25 mg, 35% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.00 (s, 1H), 8.61 (d, J = 2.5 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.25 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.34 (dd, J = 11.8, 2.7 Hz, 1H), 7.31 (t, J = 1.9 Hz, 1H), 7.27 – 7.22 (m, 1H), 7.18 (t, J = 7.7 Hz, 1H), 7.09 (ddd, J = 8.9, 2.8, 1.3 Hz, 1H), 6.82 (dt, J = 7.4, 1.7 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 2.29 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −125.95 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.64, 152.59, 152.21 (d, J = 244.1 Hz), 152.20, 149.38, 148.87, 148.19 (d, J = 10.2 Hz), 146.49, 139.31, 138.08, 128.73, 125.26 (d, J = 10.3 Hz), 122.86, 121.54 (d, J = 2.6 Hz), 118.65, 117.08 (d, J = 3.0 Hz), 115.32, 115.08, 108.99 (d, J = 22.0 Hz), 107.87, 103.31, 99.04, 55.73, 55.71, 21.22. HRMS: calcd for C25H23FN3O4 [M + H]+ m/z, 448.1673; found m/z, 448.1661.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(3-isopropylphenyl)urea (52).
To a solution of 3-isopropylaniline (26 mg, 0.19 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (34 mg, 0.21 mmol, 1.3 eq). The reaction mixture was stirred at rt for 20 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 4 h then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (20–70% EtOAc in Hexanes on silica gel column) to afford 52 (39 mg, 52% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.03 (s, 1H), 8.59 (d, J = 2.5 Hz, 1H), 8.50 (d, J = 5.3 Hz, 1H), 8.25 (t, J = 9.1 Hz, 1H), 7.50 (s, 1H), 7.40 (s, 1H), 7.37 – 7.31 (m, 2H), 7.27 (ddd, J = 8.1, 2.2, 1.2 Hz, 1H), 7.21 (t, J = 7.7 Hz, 1H), 7.09 (ddd, J = 9.0, 2.7, 1.2 Hz, 1H), 6.88 (dt, J = 7.4, 1.5 Hz, 1H), 6.54 (d, J = 5.3 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 2.86 (hept, J = 7.0 Hz, 1H), 1.20 (d, J = 6.9 Hz, 6H). 19F NMR (376 MHz, DMSO-d6) δ −125.97 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.65, 152.59, 152.24, 152.23 (d, J = 244.4 Hz), 149.38, 149.14, 148.87, 148.20 (d, J = 10.2 Hz), 146.49, 139.36, 128.79, 125.26 (d, J = 10.6 Hz), 121.62 (d, J = 1.9 Hz), 120.22, 117.09 (d, J = 3.0 Hz), 116.08, 115.74, 115.07, 108.99 (d, J = 21.9 Hz), 107.87, 103.29, 99.03, 55.73, 55.71, 33.47, 23.86. HRMS: calcd for C27H27FN3O4 [M + H]+ m/z, 476.1986; found m/z, 476.1973.
1-(3-(tert-butyl)phenyl)-3-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)urea (53).
To a solution of 3-(tert-butyl)aniline (28 mg, 0.19 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (34 mg, 0.21 mmol, 1.3 eq). The reaction mixture was stirred at rt for 30 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 5 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (20–75% EtOAc in Hexanes on silica gel column) to afford 53 (42 mg, 54% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.05 (s, 1H), 8.57 (d, J = 2.5 Hz, 1H), 8.49 (d, J = 5.3 Hz, 1H), 8.25 (t, J = 9.1 Hz, 1H), 7.50 (s, 1H), 7.47 (t, J = 2.0 Hz, 1H), 7.40 (s, 1H), 7.34 (dd, J = 11.8, 2.7 Hz, 1H), 7.30 (ddd, J = 8.1, 2.1, 1.1 Hz, 1H), 7.22 (t, J = 7.9 Hz, 1H), 7.09 (ddd, J = 9.1, 2.8, 1.3 Hz, 1H), 7.04 (ddd, J = 7.8, 1.9, 1.1 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.94 (s, 3H), 1.28 (s, 9H). 19F NMR (376 MHz, DMSO-d6) δ −126.01 (t, J = 10.4 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.66, 152.59, 152.27, 152.23 (d, J = 244.3 Hz), 151.39, 149.38, 148.87, 148.18 (d, J = 10.3 Hz), 146.49, 139.12, 128.54, 125.27 (d, J = 10.4 Hz), 121.66 (d, J = 2.6 Hz), 119.16, 117.10 (d, J = 3.0 Hz), 115.40, 115.11, 115.07, 108.99 (d, J = 22.2 Hz), 107.87, 103.28, 99.03, 55.73, 55.71, 34.40, 31.10. HRMS: calcd for C28H29FN3O4 [M + H]+ m/z, 490.2142; found m/z, 490.2129.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(3-methoxyphenyl)urea (54).
To a solution of 3-methoxyaniline (24 mg, 0.19 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (34 mg, 0.21 mmol, 1.3 eq). The reaction mixture was stirred at rt for 5 min. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 4 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (20–70% EtOAc in Hexanes on silica gel column) to afford 54 (11 mg, 15% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.09 (s, 1H), 8.61 (d, J = 2.5 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.24 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.35 (dd, J = 11.8, 2.7 Hz, 1H), 7.24 – 7.16 (m, 2H), 7.10 (ddd, J = 8.9, 2.8, 1.3 Hz, 1H), 6.93 (ddd, J = 8.1, 2.0, 0.9 Hz, 1H), 6.58 (ddd, J = 8.3, 2.5, 0.9 Hz, 1H), 6.54 (d, J = 5.3 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.74 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −125.85 (t, J = 10.5 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.74, 159.63, 152.59, 152.27 (d, J = 244.4 Hz), 152.16, 149.38, 148.87, 148.29 (d, J = 10.5 Hz), 146.49, 140.60, 129.67, 125.14 (d, J = 10.6 Hz), 121.68 (d, J = 2.6 Hz), 117.10 (d, J = 2.3 Hz), 115.07, 110.43, 109.00 (d, J = 21.8 Hz), 107.87, 107.53, 103.90, 103.30, 99.03, 55.73, 55.71, 54.94. HRMS: calcd for C25H23FN3O5 [M + H]+ m/z, 464.1622; found m/z, 464.1611.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(3-isopropoxyphenyl)urea (55).
To a solution of 3-isopropoxyaniline (29 mg, 0.19 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (34 mg, 0.21 mmol, 1.3 eq). The reaction mixture was stirred at rt for 20 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 4 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (20–70% EtOAc in Hexanes on silica gel column) to afford 55 (38 mg, 47% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.05 (s, 1H), 8.61 (d, J = 2.5 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.24 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.34 (dd, J = 11.8, 2.7 Hz, 1H), 7.21 – 7.14 (m, 2H), 7.10 (ddd, J = 9.0, 2.7, 1.3 Hz, 1H), 6.88 (ddd, J = 8.2, 1.9, 0.9 Hz, 1H), 6.57 – 6.52 (m, 2H), 4.56 (hept, J = 6.0 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 1.28 (s, 3H), 1.26 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −125.84 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.64, 157.93, 152.59, 152.27 (d, J = 244.4 Hz), 152.18, 149.38, 148.87, 148.27 (d, J = 10.3 Hz), 146.49, 140.62, 129.65, 125.16 (d, J = 10.3 Hz), 121.66 (d, J = 2.4 Hz), 117.10 (d, J = 3.1 Hz), 115.07, 110.26, 109.29, 109.00 (d, J = 22.1 Hz), 107.87, 105.56, 103.30, 99.03, 69.09, 55.73, 55.71, 21.86. HRMS: calcd for C27H27FN3O5 [M + H]+ m/z, 492.1935; found m/z, 492.1920.
1-(3-(difluoromethoxy)phenyl)-3-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)urea (56).
To a solution of 3-(difluoromethoxy)aniline (30 mg, 0.19 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (34 mg, 0.19 mmol, 1.2 eq). The reaction mixture was stirred at rt for 10 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 4 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (20–70% EtOAc in Hexanes on silica gel column) to afford 56 (19 mg, 24% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.28 (s, 1H), 8.67 (d, J = 2.5 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.22 (t, J = 9.1 Hz, 1H), 7.52 – 7.47 (m, 2H), 7.40 (s, 1H), 7.38 – 7.29 (m, 2H), 7.21 (t, J = 74.1 Hz, 1H), 7.19 (ddd, J = 8.3, 2.1, 1.0 Hz, 1H), 7.11 (ddd, J = 9.0, 2.9, 1.3 Hz, 1H), 6.80 (dd, J = 8.3, 2.4 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −81.53 (d, J = 74.2 Hz, 2F), −125.51 (t, J = 10.7 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.59, 152.60, 152.44 (d, J = 244.9 Hz), 152.14, 151.42 (t, J = 3.2 Hz), 149.39, 148.87, 148.56 (d, J = 10.3 Hz), 146.50, 140.99, 130.25, 124.89 (d, J = 10.7 Hz), 121.92 (d, J = 2.6 Hz), 117.10 (d, J = 3.2 Hz), 116.39 (t, J = 256.3 Hz), 115.08, 114.69, 111.91, 109.03 (d, J = 21.8 Hz), 108.34, 107.87, 103.35, 99.03, 55.74, 55.71. HRMS: calcd for C25H21F3N3O5 [M + H]+ m/z, 500.1433; found m/z, 500.1425.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(3-(trifluoromethoxy)phenyl)urea (57).
To a solution of 3-(trifluoromethoxy)aniline (34 mg, 0.19 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (34 mg, 0.21 mmol, 1.3 eq). The reaction mixture was stirred at rt for 20 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 4 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (20–70% EtOAc in Hexanes on silica gel column) to afford 57 (17 mg, 21% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.37 (s, 1H), 8.69 (d, J = 2.4 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.20 (t, J = 9.1 Hz, 1H), 7.73 (s, 1H), 7.49 (s, 1H), 7.42 (t, J = 8.2 Hz, 1H), 7.41 (s, 1H), 7.36 (dd, J = 11.8, 2.7 Hz, 1H), 7.28 (ddd, J = 8.3, 2.1, 0.9 Hz, 1H), 7.11 (ddd, J = 8.8, 2.8, 1.3 Hz, 1H), 6.97 (ddt, J = 8.1, 2.1, 1.0 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −56.62 (s, 3F), −125.33 (t, J = 10.5 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.57, 152.60, 152.54 (d, J = 244.6 Hz), 152.16, 149.40, 148.87, 148.76 (q, J = 1.6 Hz), 148.70 (d, J = 10.5 Hz), 146.50, 141.17, 130.54, 124.76 (d, J = 10.8 Hz), 122.10 (d, J = 2.3 Hz), 120.10 (q, J = 256.2 Hz), 117.10 (d, J = 2.4 Hz), 116.77, 115.09, 113.99, 110.09, 109.04 (d, J = 21.9 Hz), 107.87, 103.37, 99.03, 55.74, 55.72. HRMS: calcd for C25H20F4N3O5 [M + H]+ m/z, 518.1339; found m/z, 518.1327.
1-(3-cyanophenyl)-3-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)urea (58).
To a solution of 3-aminobenzonitrile (23 mg, 0.19 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (34 mg, 0.21 mmol, 1.3 eq). The reaction mixture was stirred at rt for 15 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 4 h, then cooled to rt. To achieve complete conversion, additional 1,1′-carbonyldiimidazole (68 mg, 0.42 mmol, 2.6 eq) and a solution of 3-aminobenzonitrile (23 mg, 0.19 mmol, 1.2 eq) in DMSO (0.5 mL) was added to the reaction mixture. The reaction mixture was stirred at 60°C for 18 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (20–75% EtOAc in Hexanes on silica gel column) to afford 58 (20 mg, 28% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.41 (s, 1H), 8.78 (d, J = 2.4 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.20 (t, J = 9.1 Hz, 1H), 8.00 (t, J = 1.9 Hz, 1H), 7.68 (ddd, J = 8.5, 2.2, 1.2 Hz, 1H), 7.52 (t, J = 7.9 Hz, 1H), 7.51 – 7.42 (m, 2H), 7.41 (s, 1H), 7.36 (dd, J = 11.7, 2.7 Hz, 1H), 7.12 (ddd, J = 9.0, 2.7, 1.4 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −125.13 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.55, 152.61 (d, J = 245.0 Hz), 152.60, 152.20, 149.40, 148.87, 148.80 (d, J = 10.4 Hz), 146.51, 140.31, 130.31, 125.64, 124.68 (d, J = 10.9 Hz), 122.85, 122.15 (d, J = 2.2 Hz), 120.70, 118.82, 117.10 (d, J = 2.9 Hz), 115.10, 111.71, 109.06 (d, J = 22.4 Hz), 107.87, 103.41, 99.03, 55.74, 55.72. HRMS: calcd for C25H20FN4O4 [M + H]+ m/z, 459.1469; found m/z, 459.1459.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(4-methylpiperazin-1-yl)phenyl)urea (59).
To a solution of 4-(4-methylpiperazin-1-yl)aniline (37 mg, 0.19 mmol, 1.2 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (34 mg, 0.21 mmol, 1.3 eq). The reaction mixture was stirred at rt for 2 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–15% MeOH in DCM on silica gel column) to afford 59 (31.4 mg, 19% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.83 (s, 1H), 8.52 (d, J = 2.6 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.25 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.35 – 7.24 (m, 3H), 7.08 (ddd, J = 8.8, 2.8, 1.2 Hz, 1H), 6.96 – 6.84 (m, 2H), 6.53 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.05 (t, J = 5.0 Hz, 4H), 2.44 (t, J = 5.0 Hz, 4H), 2.21 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −126.20 (t, J = 10.7 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.69, 152.59, 152.35, 152.09 (d, J = 243.9 Hz), 149.38, 148.88, 147.94 (d, J = 10.5 Hz), 146.69, 146.48, 131.31, 125.52 (d, J = 10.4 Hz), 121.39 (d, J = 3.2 Hz), 119.55, 117.09 (d, J = 3.1 Hz), 116.15, 115.07, 108.96 (d, J = 21.9 Hz), 107.86, 103.26, 99.05, 55.74, 55.72, 54.69, 48.80, 45.79. HRMS: calcd for C29H31FN5O4 [M + H]+ m/z, 532.2360; found m/z, 532.2353.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(morpholinomethyl)phenyl)urea (60).
To a solution of 4-(morpholinomethyl)aniline (39 mg, 0.20 mmol, 1.4 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (35 mg, 0.21 mmol, 1.5 eq). The reaction mixture was stirred at rt for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (45 mg, 0.14 mmol, 1 eq) in DMSO (0.7 mL). The reaction mixture was stirred at 60°C for 2.5 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–10% MeOH in DCM on silica gel column) to afford 60 (28.3 mg, 37% yield) as a white solid. 1H NMR (850 MHz, DMSO-d6) δ 9.09 (s, 1H), 8.64 (d, J = 2.6 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.24 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.43 – 7.41 (m, 2H), 7.40 (s, 1H), 7.34 (dd, J = 11.7, 2.7 Hz, 1H), 7.24 – 7.19 (m, 2H), 7.09 (ddd, J = 8.9, 2.7, 1.1 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.56 (t, J = 4.7 Hz, 4H), 3.39 (s, 2H), 2.33 (br s, 4H). 19F NMR (376 MHz, DMSO-d6) δ −125.90 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.64, 152.58, 152.23, 152.22 (d, J = 244.4 Hz), 149.38, 148.86, 148.19 (d, J = 10.1 Hz), 146.48, 138.26, 131.36, 129.52, 125.26 (d, J = 10.4 Hz), 121.56 (d, J = 2.6 Hz), 117.97, 117.09 (d, J = 3.2 Hz), 115.07, 108.99 (d, J = 21.9 Hz), 107.86, 103.30, 99.03, 66.20, 62.01, 55.73, 55.71, 53.10. HRMS: calcd for C29H30FN4O5 [M + H]+ m/z, 533.2200; found m/z, 533.2194.
1-(4-nitrobenzyl)piperidine (171).
To a solution of 1-(bromomethyl)-4-nitrobenzene (136) (500 mg, 2.3 mmol, 1 eq) in MeCN (5 mL) was added potassium carbonate (640 mg, 4.6 mmol, 2 eq), potassium iodide (38 mg, 0.23 mmol, 0.1 eq), and piperidine (217 mg, 2.6 mmol, 1.1 eq). The reaction mixture was stirred at rt for 3 h, then partitioned between EtOAc (50 mL) and water (50 mL). The aqueous layer was extracted with EtOAc (50 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and filtered to afford 171 (470 mg, 92% yield) as an orange oil. 1H NMR (400 MHz, DMSO-d6) δ 8.28 – 8.11 (m, 2H), 7.67 – 7.47 (m, 2H), 3.55 (s, 2H), 2.33 (t, J = 5.3 Hz, 4H), 1.50 (p, J = 5.4 Hz, 4H), 1.39 (q, J = 6.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 147.27 (br s), 146.49, 129.60, 123.33, 61.84, 53.95, 25.55, 23.85.
4-(piperidin-1-ylmethyl)aniline (139).
A suspension of 1-(4-nitrobenzyl)piperidine (171) (467 mg, 2.1 mmol, 1 eq), iron (593 mg, 10.6 mmol, 5 eq), and ammonium chloride (908 mg, 17.0 mmol, 8 eq) in ethanol (6.3 mL) and water (2.1 mL) was heated at 45°C for 5 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between EtOAc (30 mL) and aqueous NaOH solution (1 M, 30 mL). The aqueous layer was extracted with EtOAc (30 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–15% MeOH in DCM on silica gel column) to afford 139 (385 mg, 95% yield) as a yellow-orange solid. 1H NMR (400 MHz, DMSO-d6) δ 7.01 – 6.82 (m, 2H), 6.62 – 6.38 (m, 2H), 4.90 (s, 2H), 3.20 (s, 2H), 2.24 (t, J = 6.0 Hz, 4H), 1.45 (p, J = 5.5 Hz, 4H), 1.35 (q, J = 9.3, 4.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 147.41, 129.70, 125.28, 113.58, 62.79, 53.71, 25.57, 24.18.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(piperidin-1-ylmethyl)phenyl)urea bis(2,2,2-trifluoroacetate) (61).
To a solution of 4-(piperidin-1-ylmethyl)aniline (139) (35 mg, 0.19 mmol, 1.3 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (37 mg, 0.23 mmol, 1.6 eq). The reaction mixture was stirred at rt for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (45 mg, 0.14 mmol, 1 eq) in DMSO (0.7 mL). The reaction mixture was stirred at 60°C for 2 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–12% MeOH in DCM on silica gel column then 10–100% MeOH in water with 0.05% TFA on reverse-phase column in prep-HPLC) to afford 61 (29.7 mg, 27% yield) as a light grey solid. 1H NMR (850 MHz, DMSO-d6) δ 9.45 (s, 1H), 9.39 (s, 1H), 8.91 (d, J = 2.9 Hz, 1H), 8.78 (d, J = 6.3 Hz, 1H), 8.30 (t, J = 9.0 Hz, 1H), 7.69 (s, 1H), 7.60 – 7.53 (m, 3H), 7.50 (dd, J = 11.4, 2.7 Hz, 1H), 7.42 (dd, J = 8.8, 2.2 Hz, 2H), 7.25 – 7.21 (m, 1H), 6.89 (d, J = 6.2 Hz, 1H), 4.22 (d, J = 5.1 Hz, 2H), 4.03 (s, 3H), 4.02 (s, 4H), 3.32 (d, J = 12.2 Hz, 2H), 2.89 – 2.80 (m, 2H), 1.82 (dt, J = 15.1, 3.4 Hz, 2H), 1.69 (dt, J = 13.3, 3.7 Hz, 1H), 1.66 – 1.57 (m, 2H), 1.35 (qt, J = 12.7, 3.8 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ −73.96 (s, 6H), −124.80 (t, J = 10.0 Hz, 1H). 13C NMR (214 MHz, DMSO-d6) δ 163.98 (br s), 158.17 (q, J = 32.7 Hz), 155.30, 152.33 (d, J = 245.3 Hz), 152.17, 150.75, 147.08 (d, J = 8.8 Hz), 144.63 (br s), 140.58, 132.13, 126.13 (d, J = 9.8 Hz), 122.88, 121.87 (d, J = 2.1 Hz), 118.08, 117.36 (d, J = 2.6 Hz), 116.76 (q, J = 297.9 Hz), 115.27, 109.36 (d, J = 22.7 Hz), 103.49, 99.98, 58.73, 56.42, 56.30, 51.54, 22.37, 21.33. (two expected aromatic singlets are not observed, probably due to overlap or signal-to-noise.) HRMS: calcd for C30H32FN4O4 [M + H]+ m/z, 531.2408; found m/z, 531.2400.
1-(4-nitrobenzyl)pyrrolidine (172).
To a solution of 1-(bromomethyl)-4-nitrobenzene (136) (500 mg, 2.3 mmol, 1 eq) in MeCN (5 mL) was added potassium carbonate (640 mg, 4.6 mmol, 2 eq), potassium iodide (38 mg, 0.23 mmol, 0.1 eq), and pyrrolidine (181 mg, 2.6 mmol, 1.1 eq). The reaction mixture was stirred at rt for 3 h, then partitioned between EtOAc (50 mL) and water (50 mL). The aqueous layer was extracted with EtOAc (50 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and filtered to afford 172 (259 mg, 54% yield) as a yellow-orange oil. 1H NMR (850 MHz, DMSO-d6) δ 8.22 – 8.15 (m, 2H), 7.64 – 7.53 (m, 2H), 3.72 (s, 2H), 2.46 (t, J = 6.0 Hz, 4H), 1.71 (p, J = 3.1 Hz, 4H). 13C NMR (214 MHz, DMSO-d6) δ 146.46, 129.42, 123.38, 58.70, 53.52, 23.19. (missing carbon signal is likely due to overlap at the extraordinarily intense peak at 123.38 ppm.)
4-(pyrrolidin-1-ylmethyl)aniline (140).
A suspension of 1-(4-nitrobenzyl)pyrrolidine (172) (255 mg, 1.2 mmol, 1 eq), iron (346 mg, 6.2 mmol, 5 eq), and ammonium chloride (530 mg, 9.9 mmol, 8 eq) in ethanol (3.75 mL) and water (1.25 mL) was heated at 45°C for 7 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between EtOAc (30 mL) and aqueous NaOH solution (1 M, 30 mL). The aqueous layer was extracted with EtOAc (30 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–15% MeOH in DCM on silica gel column) to afford 140 (218 mg, 100% yield) as a yellow-orange solid. 1H NMR (400 MHz, DMSO-d6) δ 7.04 – 6.83 (m, 2H), 6.54 – 6.42 (m, 2H), 4.90 (s, 2H), 3.36 (s, 2H), 2.41 – 2.29 (m, 4H), 1.65 (p, J = 3.0 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 147.37, 129.30, 126.28, 113.62, 59.37, 53.29, 23.04.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(pyrrolidin-1-ylmethyl)phenyl)urea (62).
To a solution of 4-(pyrrolidin-1-ylmethyl)aniline (140) (36 mg, 0.21 mmol, 1.3 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (44 mg, 0.27 mmol, 1.7 eq). The reaction mixture was stirred at rt for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 4 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–15% MeOH in DCM on silica gel column) to afford 62 (51 mg, 63% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.05 (s, 1H), 8.61 (d, J = 2.6 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.25 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.40 (dd, J = 6.3, 2.2 Hz, 3H), 7.34 (dd, J = 11.8, 2.7 Hz, 1H), 7.25 – 7.18 (m, 2H), 7.09 (ddd, J = 8.9, 2.8, 1.3 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.50 (s, 2H), 2.44 – 2.33 (m, 4H), 1.68 (p, J = 3.0 Hz, 4H). 19F NMR (376 MHz, DMSO-d6) δ −125.98 (t, J = 10.7 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.65, 152.59, 152.23, 152.20 (d, J = 244.3 Hz), 149.38, 148.87, 148.16 (d, J = 10.2 Hz), 146.49, 137.98, 133.20, 128.99, 125.29 (d, J = 10.6 Hz), 121.52 (d, J = 2.6 Hz), 117.96, 117.09 (d, J = 3.0 Hz), 115.07, 108.98 (d, J = 21.9 Hz), 107.87, 103.30, 99.04, 59.14, 55.73, 55.71, 53.40, 23.09. HRMS: calcd for C29H30FN4O4 [M + H]+ m/z, 517.2251; found m/z, 517.2244.
1-(4-((diethylamino)methyl)phenyl)-3-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)urea (63).
To a solution of 4-((diethylamino)methyl)aniline hydrochloride (44 mg, 0.21 mmol, 1.3 eq) in DMSO (1 mL) was added triethylamine (21 mg, 0.21 mmol, 1.3 eq) and 1,1′-carbonyldiimidazole (44 mg, 0.27 mmol, 1.7 eq). The reaction mixture was stirred at rt for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 2 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–12% MeOH in DCM on silica gel column) to afford 63 (50 mg, 60% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.04 (s, 1H), 8.61 (d, J = 2.6 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.25 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.43 – 7.37 (m, 3H), 7.34 (dd, J = 11.8, 2.7 Hz, 1H), 7.25 – 7.18 (m, 2H), 7.09 (ddd, J = 9.0, 2.8, 1.3 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.46 (s, 2H), 2.44 (q, J = 7.1 Hz, 4H), 0.97 (t, J = 7.1 Hz, 6H). 19F NMR (376 MHz, DMSO-d6) δ −126.01 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.65, 152.59, 152.23, 152.18 (d, J = 244.4 Hz), 149.38, 148.87, 148.15 (d, J = 10.1 Hz), 146.48, 137.89, 133.51, 129.06, 125.30 (d, J = 10.3 Hz), 121.50 (d, J = 1.9 Hz), 117.97, 117.09 (d, J = 3.1 Hz), 115.07, 108.98 (d, J = 22.2 Hz), 107.86, 103.29, 99.03, 56.38, 55.73, 55.71, 45.95, 11.64. HRMS: calcd for C29H32FN4O4 [M + H]+ m/z, 519.2408; found m/z, 519.2400.
1-methyl-4-(4-nitrophenoxy)piperidine (173).
To a solution of 1-fluoro-4-nitrobenzene (147) (404 mg, 2.9 mmol, 1.1 eq) in DMSO (10 mL) in a water bath was added 1-methylpiperidin-4-ol (300 mg, 2.6 mmol, 1 eq) and potassium tert-butoxide (351 mg, 3.1 mmol, 1.2 eq) portionwise. The reaction mixture was stirred at rt for 16 h, then poured into ice water. The solid precipitated was collected by filtration, washed with water and dried under vacuum to afford 173 (219 mg, 36% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.21 – 8.14 (m, 2H), 7.20 – 7.12 (m, 2H), 4.58 (tt, J = 8.2, 4.0 Hz, 1H), 2.65 – 2.55 (m, 2H), 2.22 – 2.14 (m, 5H), 2.01 – 1.89 (m, 2H), 1.66 (dtd, J = 12.7, 8.8, 3.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 162.73, 140.55, 125.96, 115.84, 73.10, 52.20, 45.78, 30.31.
4-((1-methylpiperidin-4-yl)oxy)aniline (150).
A suspension of 1-methyl-4-(4-nitrophenoxy)piperidine (173) (215 mg, 0.91 mmol, 1 eq), iron (254 mg, 4.6 mmol, 5 eq), and ammonium chloride (389 mg, 7.3 mmol, 8 eq) in ethanol (2.7 mL) and water (0.9 mL) was heated at 45°C for 4 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between EtOAc (30 mL) and aqueous NaOH solution (1 M, 30 mL). The aqueous layer was extracted with EtOAc (30 mL x2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–15% MeOH in DCM on silica gel column) to afford 173 (152 mg, 81% yield) as a yellow-orange solid. 1H NMR (850 MHz, DMSO-d6) δ 6.70 – 6.53 (m, 2H), 6.53 – 6.37 (m, 2H), 4.60 (s, 2H), 4.03 (tt, J = 8.8, 4.1 Hz, 1H), 2.61 – 2.57 (m, 2H), 2.16 (s, 3H), 2.10 (t, J = 10.9 Hz, 2H), 1.86 – 1.80 (m, 2H), 1.55 (dtd, J = 12.9, 8.9, 3.6 Hz, 2H). 13C NMR (214 MHz, DMSO-d6) δ 147.98, 142.79, 117.77, 114.90, 73.06 (br s), 52.51, 45.80, 30.81.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-((1-methylpiperidin-4-yl)oxy)phenyl)urea (64).
To a solution of 4-((1-methylpiperidin-4-yl)oxy)aniline (150) (43 mg, 0.21 mmol, 1.3 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (44 mg, 0.27 mmol, 1.7 eq). The reaction mixture was stirred at rt for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–15% MeOH in DCM on silica gel column) to afford 64 (57 mg, 66% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (s, 1H), 8.54 (d, J = 2.6 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.24 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.37 – 7.30 (m, 3H), 7.09 (ddd, J = 8.9, 2.7, 1.3 Hz, 1H), 6.96 – 6.85 (m, 2H), 6.53 (d, J = 5.2 Hz, 1H), 4.26 (tt, J = 8.4, 3.8 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 2.67 – 2.57 (m, 2H), 2.20 – 2.09 (m, 5H), 1.90 (d, J = 12.5 Hz, 2H), 1.60 (dtd, J = 12.7, 9.0, 3.6 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −126.12 (t, J = 10.7 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.68, 152.59, 152.36, 152.34, 152.14 (d, J = 244.3 Hz), 149.38, 148.87, 148.03 (d, J = 10.0 Hz), 146.48, 132.51, 125.43 (d, J = 10.6 Hz), 121.45 (d, J = 2.5 Hz), 119.94, 117.09 (d, J = 3.1 Hz), 116.47, 115.07, 108.97 (d, J = 22.2 Hz), 107.86, 103.27, 99.04, 72.37 (br s), 55.73, 55.71, 52.45, 45.85, 30.68. HRMS: calcd for C30H32FN4O5 [M + H]+ m/z, 547.2357; found m/z, 547.2348.
1-methyl-N-(4-nitrophenyl)piperidin-4-amine (174).
To a solution of 1-fluoro-4-nitrobenzene (147) (435 mg, 3.1 mmol, 1 eq) in DMSO (6 mL) was added triethylamine (936 mg, 9.3 mmol, 3 eq) and 1-methylpiperidin-4-amine (387 mg, 3.4 mmol, 1.1 eq). The reaction mixture was stirred at 90°C for 16 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (50 mL) and water (50 mL). The aqueous layer was extracted with EtOAc (50 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–10% MeOH in DCM on silica gel column) to afford 174 (596 mg, 82% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.00 – 7.94 (m, 2H), 7.18 (d, J = 7.7 Hz, 1H), 6.70 – 6.60 (m, 2H), 3.43 – 3.24 (m, 1H), 2.73 (dt, J = 12.6, 3.5 Hz, 2H), 2.16 (s, 3H), 2.01 (td, J = 11.5, 2.5 Hz, 2H), 1.92 – 1.80 (m, 2H), 1.51 – 1.36 (m, 2H). 13C NMR (214 MHz, DMSO-d6) δ 153.71, 135.37, 126.33, 111.03 (br s), 53.91, 48.64, 45.97, 31.29.
N1-(1-methylpiperidin-4-yl)benzene-1,4-diamine (151).
A suspension of 1-methyl-N-(4-nitrophenyl)piperidin-4-amine (174) (384 mg, 1.6 mmol, 1 eq), iron (456 mg, 8.2 mmol, 5 eq), and ammonium chloride (698 mg, 13.1 mmol, 8 eq) in ethanol (4.8 mL) and water (1.6 mL) was heated at 60°C for 7 h, then cooled to rt. Additional iron (912 mg, 16.4 mmol, 10 eq) and ammonium chloride (1.40 g, 26.2 mmol, 16 eq) were added and the reaction mixture was stirred at 78°C for 26 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between EtOAc (50 mL) and aqueous NaOH solution (1 M, 50 mL). The aqueous layer was extracted with EtOAc (50 mL x2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (5–15% MeOH in DCM on silica gel column) to afford 151 (177 mg, 53% yield) as a dull rose solid, which was stored under vacuum or under an argon atmosphere to prevent oxidation by atmospheric oxygen. 1H NMR (850 MHz, DMSO-d6) δ 6.41 – 6.38 (m, 2H), 6.38 – 6.35 (m, 2H), 4.36 (s, 1H), 4.23 (s, 2H), 2.97 (tt, J = 10.2, 3.7 Hz, 1H), 2.70 (d, J = 10.9 Hz, 2H), 2.14 (s, 3H), 1.94 (t, J = 10.9 Hz, 2H), 1.83 – 1.80 (m, 2H), 1.33 – 1.26 (m, 2H). 13C NMR (214 MHz, DMSO-d6) δ 139.08, 138.92, 115.48, 114.55, 54.37, 49.82 (br s), 46.01, 32.05.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-((1-methylpiperidin-4-yl)amino)phenyl)urea (65).
To a solution of N1-(1-methylpiperidin-4-yl)benzene-1,4-diamine (151) (42 mg, 0.21 mmol, 1.3 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (44 mg, 0.27 mmol, 1.7 eq). The reaction mixture was stirred at rt for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 5 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–18% MeOH in DCM on silica gel column then 10–100% MeOH in water with 0.05% TFA on reverse-phase column on prep-HPLC) to afford the desired compound as a tris-(2,2,2-trifluoroacetate) salt. The salt was partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 65 (30 mg, 34% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.60 (s, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.44 (d, J = 2.6 Hz, 1H), 8.25 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.31 (dd, J = 11.9, 2.7 Hz, 1H), 7.18 – 7.10 (m, 2H), 7.07 (ddd, J = 8.9, 2.8, 1.3 Hz, 1H), 6.54 (d, J = 6.2 Hz, 2H), 6.52 (d, J = 2.5 Hz, 1H), 5.16 (d, J = 8.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.17 – 3.02 (m, 1H), 2.72 (d, J = 11.5 Hz, 2H), 2.16 (s, 3H), 1.99 (t, J = 11.3 Hz, 2H), 1.86 (d, J = 12.4 Hz, 2H), 1.45 – 1.28 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −126.48 (t, J = 10.8 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.72, 152.57, 152.49, 151.95 (d, J = 243.7 Hz), 149.36, 148.87, 147.70 (d, J = 10.0 Hz), 146.47, 143.84, 127.95, 125.73 (d, J = 10.5 Hz), 121.19 (d, J = 2.5 Hz), 120.72, 117.07 (d, J = 3.1 Hz), 115.05, 112.78, 108.92 (d, J = 21.8 Hz), 107.86, 103.21, 99.04, 55.73, 55.71, 54.27, 48.93, 46.01, 31.79. HRMS: calcd for C30H33FN5O4 [M + H]+ m/z, 546.2517; found m/z, 546.2511.
N,1-dimethyl-N-(4-nitrophenyl)piperidin-4-amine (175).
To a solution of 1-fluoro-4-nitrobenzene (147) (400 mg, 2.8 mmol, 1 eq) in DMSO (5.7 mL) was added triethylamine (717 mg, 7.1 mmol, 2.5 eq) and N,1-dimethylpiperidin-4-amine (400 mg, 3.1 mmol, 1.1 eq). The reaction mixture was stirred at 90°C for 16 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (50 mL) and water (50 mL). The aqueous layer was extracted with EtOAc (50 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–10% MeOH in DCM on silica gel column) to afford 175 (536 mg, 76% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.15 – 7.84 (m, 2H), 7.03 – 6.65 (m, 2H), 3.80 (tt, J = 11.7, 4.0 Hz, 1H), 2.89 (s, 3H), 2.87 – 2.78 (m, 2H), 2.18 (s, 3H), 2.05 (td, J = 11.8, 2.4 Hz, 2H), 1.80 (qd, J = 12.1, 3.9 Hz, 2H), 1.65 – 1.53 (m, 2H). 13C NMR (214 MHz, DMSO-d6) δ 154.08, 135.53, 125.92, 111.08, 55.17, 54.55, 45.84, 31.43, 28.45.
N1-methyl-N1-(1-methylpiperidin-4-yl)benzene-1,4-diamine (152).
A suspension of N,1-dimethyl-N-(4-nitrophenyl)piperidin-4-amine (175) (221 mg, 0.89 mmol, 1 eq), iron (346 mg, 6.2 mmol, 7 eq), and ammonium chloride (474 mg, 8.9 mmol, 10 eq) in ethanol (2.7 mL) and water (0.9 mL) was heated at 78°C for 16 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between EtOAc (40 mL) and aqueous NaOH solution (1 M, 40 mL). The aqueous layer was extracted with EtOAc (50 mL x2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (5–15% MeOH in DCM on silica gel column) to afford 152 (177 mg, 91% yield) as a brown oil, which was stored under vacuum or under an argon atmosphere to prevent oxidation by atmospheric oxygen. 1H NMR (400 MHz, DMSO-d6) δ 6.68 – 6.59 (m, 2H), 6.53 – 6.42 (m, 2H), 4.50 (s, 2H), 3.17 – 2.99 (m, 1H), 2.84 – 2.71 (m, 2H), 2.54 (s, 3H), 2.12 (s, 3H), 1.97 – 1.81 (m, 2H), 1.60 – 1.45 (m, 4H). 13C NMR (214 MHz, DMSO-d6) δ 141.56, 141.41, 118.83, 114.83, 58.54, 55.06, 45.91, 33.87, 28.39.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(methyl(1-methylpiperidin-4-yl)amino)phenyl)urea (66).
To a solution of N1-methyl-N1-(1-methylpiperidin-4-yl)benzene-1,4-diamine (152) (45 mg, 0.21 mmol, 1.3 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (44 mg, 0.27 mmol, 1.7 eq). The reaction mixture was stirred at rt for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (3–15% MeOH in DCM on silica gel column) to afford 66 (62 mg, 69% yield) as a light grey solid. 1H NMR (400 MHz, DMSO-d6) δ 8.77 (s, 1H), 8.51 (d, J = 2.6 Hz, 1H), 8.49 (d, J = 5.3 Hz, 1H), 8.25 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.32 (dd, J = 11.8, 2.7 Hz, 1H), 7.29 – 7.24 (m, 2H), 7.08 (ddd, J = 9.1, 2.7, 1.3 Hz, 1H), 6.82 – 6.74 (m, 2H), 6.53 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 2.87 – 2.74 (m, 3H), 2.67 (s, 3H), 2.16 (s, 3H), 1.97 (td, J = 11.7, 2.4 Hz, 2H), 1.69 (qd, J = 12.1, 3.9 Hz, 2H), 1.58 – 1.48 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −126.32 (t, J = 10.7 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.71, 152.58, 152.42, 152.02 (d, J = 243.7 Hz), 149.37, 148.87, 147.81 (d, J = 10.1 Hz), 146.47, 145.86, 129.29, 125.64 (d, J = 10.7 Hz), 121.31 (d, J = 2.7 Hz), 120.11, 117.07 (d, J = 3.1 Hz), 115.06, 114.47, 108.94 (d, J = 21.8 Hz), 107.86, 103.23, 99.04, 56.37, 55.73, 55.71, 55.09, 45.93, 31.60, 28.27. HRMS: calcd for C31H35FN5O4 [M + H]+ m/z, 560.2673; found m/z, 560.2665.
1-(4-nitro-2-(trifluoromethyl)benzyl)piperidine (177).
To a solution of 1-(bromomethyl)-4-nitro-2-(trifluoromethyl)benzene (137) (250 mg, 0.88 mmol, 1 eq) in acetonitrile (1.8 mL) was added potassium carbonate (243 mg, 1.8 mmol, 2 eq), potassium iodide (15 mg, 0.088 mmol, 0.1 eq), and piperidine (90 mg, 1.1 mmol, 1.2 eq). The reaction mixture was stirred at rt for 1.5 h. The crude reaction mixture was partitioned between EtOAc (40 mL) and water (40 mL). The aqueous layer was extracted with EtOAc (40 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–15% EtOAc in Hexanes on silica gel column) to afford 177 (233 mg, 92% yield) as a yellow liquid. 1H NMR (400 MHz, DMSO-d6) δ 7.70 (dd, J = 8.6, 2.4 Hz, 1H), 7.59 (d, J = 2.4 Hz, 1H), 7.29 (d, J = 8.6 Hz, 1H), 2.86 (s, 2H), 1.56 (t, J = 5.0 Hz, 4H), 0.72 (p, J = 5.6 Hz, 4H), 0.60 (p, J = 5.7 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −58.97. 13C NMR (214 MHz, DMSO-d6) δ 146.19, 145.99, 132.02, 127.99 (q, J = 31.4 Hz), 127.20, 123.20 (q, J = 274.7 Hz), 120.99 (q, J = 6.2 Hz), 58.03, 54.09, 25.56, 23.66.
4-(piperidin-1-ylmethyl)-3-(trifluoromethyl)aniline (141).
A suspension of 1-(4-nitro-2-(trifluoromethyl)benzyl)piperidine (177) (233 mg, 0.81 mmol, 1 eq), iron (226 mg, 4.0 mmol, 5 eq), and ammonium chloride (346 mg, 6.5 mmol, 8 eq) in ethanol (2.4 mL) and water (0.8 mL) was heated at 40°C for 3 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between EtOAc (30 mL) and aqueous NaOH solution (1 M, 30 mL). The aqueous layer was extracted with EtOAc (30 mL x2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–10% MeOH in DCM on silica gel column) to afford 141 (196 mg, 94% yield) as a yellow liquid. 1H NMR (400 MHz, DMSO-d6) δ 7.30 (d, J = 8.3 Hz, 1H), 6.84 (d, J = 2.4 Hz, 1H), 6.75 (dd, J = 8.4, 2.4 Hz, 1H), 5.39 (s, 2H), 3.34 (s, 2H), 2.28 (t, J = 5.3 Hz, 4H), 1.47 (p, J = 5.4 Hz, 4H), 1.43 – 1.32 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −57.85. 13C NMR (214 MHz, DMSO-d6) δ 147.65, 131.60, 127.68 (q, J = 28.8 Hz), 124.77 (q, J = 274.2 Hz), 123.32, 116.90, 110.21 (q, J = 5.8 Hz), 58.27, 53.99, 25.63, 24.04.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(piperidin-1-ylmethyl)-3-(trifluoromethyl)phenyl)urea (69).
To a solution of 4-(piperidin-1-ylmethyl)-3-(trifluoromethyl)aniline (141) (53 mg, 0.21 mmol, 1.3 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (44 mg, 0.27 mmol, 0.16 eq). The reaction mixture was stirred at rt for 30 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–10% MeOH in DCM on silica gel column) to afford 69 (30 mg, 32% yield) as a light pink solid. 1H NMR (400 MHz, DMSO-d6) δ 9.36 (s, 1H), 8.67 (d, J = 2.4 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.21 (t, J = 9.1 Hz, 1H), 7.99 (d, J = 2.2 Hz, 1H), 7.67 (d, J = 8.5 Hz, 1H), 7.55 (dd, J = 8.5, 2.3 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.35 (dd, J = 11.7, 2.7 Hz, 1H), 7.10 (ddd, J = 9.0, 2.8, 1.3 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.49 (s, 2H), 2.33 (t, J = 5.4 Hz, 4H), 1.51 (p, J = 5.5 Hz, 4H), 1.46 – 1.36 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −58.10 (s, 3F), −125.40 (t, J = 10.6 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.71, 152.63, 152.52 (d, J = 244.7 Hz), 152.32, 149.42, 148.96, 148.57 (d, J = 10.3 Hz), 146.53, 138.43, 131.34, 130.97, 127.54 (q, J = 29.6 Hz), 124.97 (d, J = 10.7 Hz), 124.45 (q, J = 274.0 Hz), 122.02, 121.62, 117.27 (d, J = 3.2 Hz), 115.09, 114.81 (q, J = 5.8 Hz), 109.22 (d, J = 22.2 Hz), 107.89, 103.33, 99.03, 58.14, 55.80, 55.77, 54.16, 25.69, 23.99. HRMS: calcd for C31H31F4N4O4 [M + H]+ m/z, 599.2281; found m/z, 599.2272.
1-(4-bromo-2-(trifluoromethoxy)benzyl)piperidine (178).
To a solution of (4-bromo-2-(trifluoromethoxy)phenyl)methanol (138) (300 mg, 1.1 mmol, 1 eq) in DCM (7.4 mL) was added triethylamine (224 mg, 2.2 mmol, 2 eq). The reaction solution was cooled to 0°C using an ice bath and methanesulfonyl chloride (165 mg, 1.4 mmol, 1.3 eq) was added dropwise. The reaction mixture was warmed slowly to rt and stirred at rt for 2 h. Triethylamine (112 mg, 1.1 mmol, 1 eq) and pyrrolidine (315 mg, 4.4 mmol, 4 eq) were added. The reaction mixture was stirred at rt for 16 h, then concentrated onto Celite, and purified by flash chromatography (0–15% EtOAc in Hexanes on silica gel column) to afford 178 (333 mg, 89% yield) as a colorless liquid. 1H NMR (400 MHz, DMSO-d6) δ 7.61 (dd, J = 8.3, 1.9 Hz, 1H), 7.56 (dq, J = 2.0, 1.4 Hz, 1H), 7.51 (d, J = 8.3 Hz, 1H), 3.43 (s, 2H), 2.32 (t, J = 5.3 Hz, 4H), 1.48 (p, J = 5.5 Hz, 4H), 1.37 (p, J = 5.6 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −56.23. 13C NMR (101 MHz, DMSO-d6) δ 147.22 (q, J = 1.4 Hz), 132.70, 131.24, 130.57, 123.71 (q, J = 1.2 Hz), 120.03 (q, J = 257.4 Hz), 119.94, 55.60, 53.83, 25.55, 23.77.
4-(piperidin-1-ylmethyl)-3-(trifluoromethoxy)aniline (144).
To a solution of 1-(4-bromo-2-(trifluoromethoxy)benzyl)piperidine (178) (328 mg, 0.97 mmol, 1 eq) in toluene (2.4 mL) was added bis(tri-tert-butylphosphine)palladium(0) (50 mg, 0.10 mmol, 0.1 eq) and lithium bis(trimethylsilyl)amide (1.5 M in THF, 0.97 mL, 1.5 mmol, 1.5 eq). The reaction mixture was stirred at rt for 16 h, then aqueous HCl (1 M, 2 mL) was added. The reaction mixture was stirred at rt for 10 min, then partitioned between EtOAc (20 mL) and aqueous NaOH (1 M, 15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated in vacuo, and purified by flash chromatography (1–10% MeOH in DCM on silica gel column) to afford 144 (233 mg, 87% yield) as a yellow liquid. 1H NMR (400 MHz, DMSO-d6) δ 7.08 (d, J = 8.1 Hz, 1H), 6.57 – 6.43 (m, 2H), 5.39 (s, 2H), 3.26 (2H), 2.27 (br s, 4H), 1.45 (p, J = 5.4 Hz, 4H), 1.40 – 1.33 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −55.49. 13C NMR (214 MHz, DMSO-d6) δ 149.27, 148.01, 131.75, 120.23 (q, J = 255.3 Hz), 116.64, 112.49, 104.78, 55.97, 53.65, 25.57, 24.00.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(piperidin-1-ylmethyl)-3-(trifluoromethoxy)phenyl)urea (70).
To a solution of 4-(piperidin-1-ylmethyl)-3-(trifluoromethoxy)aniline (144) (68 mg, 0.25 mmol, 1.3 eq) in DMSO (1.3 mL) was added 1,1′-carbonyldiimidazole (53 mg, 0.32 mmol, 1.7 eq). The reaction mixture was stirred at rt for 45 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (60 mg, 0.19 mmol, 1 eq) in DMSO (1 mL). The reaction mixture was stirred at 60°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–12% MeOH in DCM on silica gel column then 10–100% MeOH in water with 0.05% TFA on reverse-phase column on prep-HPLC). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 70 (42 mg, 35% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.35 (s, 1H), 8.66 (d, J = 2.4 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.21 (t, J = 9.1 Hz, 1H), 7.75 (p, J = 1.7 Hz, 1H), 7.49 (s, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.40 (s, 1H), 7.35 (dd, J = 11.7, 2.7 Hz, 1H), 7.23 (dd, J = 8.4, 2.1 Hz, 1H), 7.10 (ddd, J = 9.0, 2.8, 1.3 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.41 (s, 2H), 2.33 (br s, 4H), 1.49 (p, J = 5.4 Hz, 4H), 1.38 (td, J = 9.8, 4.2 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −55.79 (s, 3F), −125.46 (t, J = 10.7 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.58, 152.60, 152.49 (d, J = 244.6 Hz), 152.17, 149.40, 148.85, 148.62 (d, J = 10.2 Hz), 147.21, 146.50, 139.60, 131.44, 124.82 (d, J = 10.4 Hz), 124.13 (br s), 122.05 (d, J = 2.3 Hz), 120.22 (q, J = 256.2 Hz), 117.10 (d, J = 3.1 Hz), 116.62, 115.08, 109.67, 109.03 (d, J = 22.1 Hz), 107.87, 103.34, 99.02, 55.79, 55.73, 55.71, 53.78, 25.56, 23.88. HRMS: calcd for C31H31F4N4O5 [M + H]+ m/z, 615.2231; found m/z, 615.2226.
1-(4-nitro-2-(trifluoromethyl)benzyl)pyrrolidine (179).
To a solution of 1-(bromomethyl)-4-nitro-2-(trifluoromethyl)benzene (137) (250 mg, 0.88 mmol, 1 eq) in acetonitrile (1.8 mL) was added potassium carbonate (243 mg, 1.8 mmol, 2 eq), potassium iodide (15 mg, 0.088 mmol, 0.1 eq), and pyrrolidine (75 mg, 1.1 mmol, 1.2 eq). The reaction mixture was stirred at rt for 1.5 h. The crude reaction mixture was partitioned between EtOAc (40 mL) and water (40 mL). The aqueous layer was extracted with EtOAc (40 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–20% EtOAc in Hexanes on silica gel column) to afford 179 (187 mg, 78% yield) as a yellow liquid. 1H NMR (400 MHz, DMSO-d6) δ 8.50 (dd, J = 8.6, 2.4 Hz, 1H), 8.40 (d, J = 2.4 Hz, 1H), 8.09 (d, J = 8.6 Hz, 1H), 3.86 (s, 2H), 2.54 – 2.51 (m, 4H), 1.80 – 1.69 (m, 4H). 19F NMR (376 MHz, DMSO-d6) δ −59.02. 13C NMR (214 MHz, DMSO-d6) δ 146.14, 132.10, 127.54 (q, J = 31.7 Hz), 127.26, 123.20 (q, J = 274.7 Hz), 120.95 (q, J = 6.0 Hz), 55.05, 53.60, 23.29. (missing aromatic carbon peak is probably due to overlap at 146.14 ppm.)
4-(pyrrolidin-1-ylmethyl)-3-(trifluoromethyl)aniline (142).
A suspension of 1-(4-nitro-2-(trifluoromethyl)benzyl)pyrrolidine (179). (184 mg, 0.67 mmol, 1 eq), iron (187 mg, 3.4 mmol, 5 eq), and ammonium chloride (287 mg, 5.4 mmol, 8 eq) in ethanol (2 mL) and water (0.65 mL) was heated at 40°C for 3 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between EtOAc (30 mL) and aqueous NaOH solution (1 M, 30 mL). The aqueous layer was extracted with EtOAc (30 mL x2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–10% MeOH in DCM on silica gel column) to afford 142 (149 mg, 91% yield) as a yellow liquid. 1H NMR (400 MHz, DMSO-d6) δ 7.31 (d, J = 8.3 Hz, 1H), 6.84 (d, J = 2.4 Hz, 1H), 6.75 (dd, J = 8.3, 2.4 Hz, 1H), 5.39 (s, 2H), 3.52 (s, 2H), 2.41 (t, J = 5.9 Hz, 4H), 1.73 – 1.63 (m, 4H). 19F NMR (376 MHz, DMSO-d6) δ −57.88. 13C NMR (214 MHz, DMSO-d6) δ 147.61, 131.56, 127.22 (q, J = 28.5, 27.8 Hz), 124.78 (q, J = 274.1 Hz), 123.93, 116.90, 110.19 (q, J = 5.9 Hz), 55.09, 53.52, 23.13.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(pyrrolidin-1-ylmethyl)-3-(trifluoromethyl)phenyl)urea (71).
To a solution of 4-(pyrrolidin-1-ylmethyl)-3-(trifluoromethyl)aniline (142) (51 mg, 0.21 mmol, 1.3 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (52 mg, 0.32 mmol, 2 eq). The reaction mixture was stirred at rt for 45 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–12% MeOH in DCM on silica gel column then 10–100% MeOH in water with 0.05% TFA on reverse-phase column on prep-HPLC). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 71 (40 mg, 43% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.36 (s, 1H), 8.67 (d, J = 2.4 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.21 (t, J = 9.1 Hz, 1H), 7.98 (d, J = 2.3 Hz, 1H), 7.66 (d, J = 8.5 Hz, 1H), 7.56 (dd, J = 8.5, 2.3 Hz, 1H), 7.49 (s, 1H), 7.41 (s, 1H), 7.35 (dd, J = 11.7, 2.7 Hz, 1H), 7.10 (ddd, J = 8.9, 2.8, 1.3 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.68 (s, 2H), 2.46 (d, J = 6.0 Hz, 4H), 1.76 – 1.68 (m, 4H). 19F NMR (376 MHz, DMSO-d6) δ −58.12 (s, 3F), −125.38 (t, J = 10.4 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.71, 152.63, 152.54 (d, J = 244.8 Hz), 152.32, 149.42, 148.95, 148.59 (d, J = 10.2 Hz), 146.53, 138.41, 131.40, 127.09 (q, J = 29.0, 28.4 Hz), 124.96 (d, J = 10.5 Hz), 124.45 (q, J = 274.1 Hz), 122.05, 121.62, 117.27 (d, J = 3.0 Hz), 115.10, 114.78 (q, J = 5.5 Hz), 109.22 (d, J = 21.9 Hz), 107.89, 103.33, 99.03, 55.80, 55.77, 55.04, 53.69, 23.25. (missing aromatic carbon is probably due to overlap of signals at 131.40 ppm) HRMS: calcd for C30H29F4N4O4 [M + H]+ m/z, 585.2125; found m/z 585.2115.
1-(4-bromo-2-(trifluoromethoxy)benzyl)pyrrolidine (180).
To a solution of (4-bromo-2-(trifluoromethoxy)phenyl)methanol (138) (300 mg, 1.1 mmol, 1 eq) in DCM (7.4 mL) was added triethylamine (224 mg, 2.2 mmol, 2 eq). The reaction solution was cooled to 0°C using an ice bath and methanesulfonyl chloride (165 mg, 1.4 mmol, 1.3 eq) was added dropwise. The reaction mixture was warmed slowly to rt and stirred at rt for 2 h. Triethylamine (112 mg, 1.1 mmol, 1 eq) and pyrrolidine (315 mg, 4.4 mmol, 4 eq) were added. The reaction mixture was stirred at rt for 16 h, then concentrated onto Celite, and purified by flash chromatography (0–15% EtOAc in Hexanes on silica gel column) to afford 180 (310 mg, 86% yield) as a colorless liquid. 1H NMR (400 MHz, DMSO-d6) δ 7.60 (dd, J = 8.3, 1.9 Hz, 1H), 7.56 (dq, J = 1.9, 1.5 Hz, 1H), 7.53 (d, J = 8.3 Hz, 1H), 3.60 (s, 2H), 2.46 – 2.41 (m, 4H), 1.69 (p, J = 3.0 Hz, 4H). 19F NMR (376 MHz, DMSO-d6) δ −56.34 (d, J = 1.3 Hz). 13C NMR (101 MHz, DMSO-d6) δ 146.76 (q, J = 1.6 Hz), 132.63, 131.80, 130.62, 123.65 (q, J = 1.4 Hz), 120.03 (q, J = 257.5 Hz), 119.87, 53.40, 52.37, 23.16.
4-(pyrrolidin-1-ylmethyl)-3-(trifluoromethoxy)aniline (145).
To a solution of 1-(4-bromo-2-(trifluoromethoxy)benzyl)pyrrolidine (180) (303 mg, 0.94 mmol, 1 eq) in toluene (2.3 mL) was added bis(tri-tert-butylphosphine)palladium(0) (48 mg, 0.09 mmol, 0.1 eq) and lithium bis(trimethylsilyl)amide (1.5 M in THF, 0.94 mL, 1.4 mmol, 1.5 eq). The reaction mixture was stirred at rt for 16 h, then aqueous HCl (1 M, 2 mL) was added. The reaction mixture was stirred at rt for 10 min, then partitioned between EtOAc (20 mL) and aqueous NaOH (1 M, 15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated in vacuo, and purified by flash chromatography (1–10% MeOH in DCM on silica gel column) to afford 145 (147 mg, 60% yield) as a yellow liquid. 1H NMR (400 MHz, DMSO-d6) δ 7.10 (d, J = 8.2 Hz, 1H), 6.54 – 6.45 (m, 2H), 5.39 (s, 2H), 3.43 (s, 2H), 2.39 (t, J = 5.7 Hz, 4H), 1.72 – 1.60 (m, 4H). 19F NMR (376 MHz, DMSO-d6) δ −55.61. 13C NMR (214 MHz, DMSO-d6) δ 149.24, 147.55, 131.58, 120.23 (q, J = 255.4 Hz), 117.37 (br s), 112.55, 104.75, 53.23, 52.46, 23.05.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(pyrrolidin-1-ylmethyl)-3-(trifluoromethoxy)phenyl)urea (72).
To a solution of 4-(pyrrolidin-1-ylmethyl)-3-(trifluoromethoxy)aniline (145) (65 mg, 0.25 mmol, 1.3 eq) in DMSO (1.3 mL) was added 1,1′-carbonyldiimidazole (53 mg, 0.32 mmol, 1.7 eq). The reaction mixture was stirred at rt for 45 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (60 mg, 0.19 mmol, 1 eq) in DMSO (1 mL). The reaction mixture was stirred at 60°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–12% MeOH in DCM on silica gel column then 10–100% MeOH in water with 0.05% TFA on reverse-phase column on prep-HPLC). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 72 (38 mg, 33% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.36 (s, 1H), 8.66 (d, J = 2.5 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.21 (t, J = 9.1 Hz, 1H), 7.75 (t, J = 1.8 Hz, 1H), 7.49 (s, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.41 (s, 1H), 7.35 (dd, J = 11.7, 2.7 Hz, 1H), 7.24 (dd, J = 8.4, 2.1 Hz, 1H), 7.10 (ddd, J = 9.3, 2.7, 1.4 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.58 (s, 2H), 2.45 (t, J = 5.6 Hz, 4H), 1.77 – 1.64 (m, 4H). 19F NMR (376 MHz, DMSO-d6) δ −55.89 (s, 3F), −125.42 (t, J = 10.6 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.58, 152.60, 152.50 (d, J = 244.6 Hz), 152.18, 149.40, 148.86, 148.63 (d, J = 10.1 Hz), 146.75, 146.50, 139.59 (br s), 131.35 (br s), 124.81 (d, J = 10.4 Hz), 124.81 (br s), 122.07 (d, J = 2.0 Hz), 120.22 (q, J = 256.4 Hz), 117.09 (d, J = 3.1 Hz), 116.66, 115.09, 109.62, 109.03 (d, J = 22.1 Hz), 107.87, 103.34, 99.02, 55.73, 55.71, 53.38, 52.41 (br s), 23.12. HRMS: calcd for C30H29F4N4O5 [M + H]+ m/z, 601.2074; found m/z, 601.2070.
N-ethyl-N-(4-nitro-2-(trifluoromethyl)benzyl)ethanamine (181).
To a solution of 1-(bromomethyl)-4-nitro-2-(trifluoromethyl)benzene (137) (250 mg, 0.88 mmol, 1 eq) in acetonitrile (1.8 mL) was added potassium carbonate (243 mg, 1.8 mmol, 2 eq), potassium iodide (15 mg, 0.088 mmol, 0.1 eq), and diethylamine (77.3 mg, 1.0 mmol, 1.2 eq). The reaction mixture was stirred at rt for 3 h, then another portion of diethylamine (77.3 mg, 1.0 mmol, 1.2 eq) was added. The reaction mixture was stirred at 50°C for 4 h, then another portion of diethylamine (77.3 mg, 1.0 mmol, 1.2 eq) was added to drive reaction to completion. The reaction mixture was stirred at 50°C for 16 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (40 mL) and water (40 mL). The aqueous layer was extracted with EtOAc (40 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–25% EtOAc in Hexanes on silica gel column) to afford 181 (154 mg, 64% yield) as a yellow liquid. 1H NMR (400 MHz, DMSO-d6) δ 8.51 (dd, J = 8.7, 2.4 Hz, 1H), 8.39 (d, J = 2.4 Hz, 1H), 8.17 (d, J = 8.6 Hz, 1H), 3.78 (s, 2H), 2.53 (t, J = 7.1 Hz, 4H), 0.98 (t, J = 7.1 Hz, 6H). 19F NMR (376 MHz, DMSO-d6) δ −59.18. 13C NMR (214 MHz, DMSO-d6) δ 147.66, 146.10, 131.98, 127.60 (q, J = 31.4 Hz), 127.14, 123.25 (q, J = 274.3 Hz), 120.88 (q, J = 6.2 Hz), 53.11, 46.97, 11.79.
4-((diethylamino)methyl)-3-(trifluoromethyl)aniline (143).
A suspension of N-ethyl-N-(4-nitro-2-(trifluoromethyl)benzyl)ethanamine (181). (151 mg, 0.55 mmol, 1 eq), iron (214 mg, 3.8 mmol, 7 eq), and ammonium chloride (292 mg, 5.5 mmol, 10 eq) in ethanol (1.7 mL) and water (0.55 mL) was heated at 50°C for 3 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between EtOAc (30 mL) and aqueous NaOH solution (1 M, 30 mL). The aqueous layer was extracted with EtOAc (30 mL x2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–10% MeOH in DCM on silica gel column) to afford 143 (89 mg, 66% yield) as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 7.37 (d, J = 8.4 Hz, 1H), 6.84 (d, J = 2.4 Hz, 1H), 6.75 (dd, J = 8.5, 2.4 Hz, 1H), 5.38 (s, 2H), 3.45 (s, 2H), 2.42 (q, J = 7.1 Hz, 4H), 0.94 (t, J = 7.1 Hz, 6H). 19F NMR (376 MHz, DMSO-d6) δ −58.06. 13C NMR (214 MHz, DMSO-d6) δ 147.51, 131.46, 127.39 (q, J = 29.2 Hz), 124.81 (q, J = 274.1 Hz), 124.61, 116.94, 110.19 (q, J = 5.8 Hz), 52.85, 46.25, 11.66.
1-(4-((diethylamino)methyl)-3-(trifluoromethyl)phenyl)-3-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)urea (73).
To a solution of 4-((diethylamino)methyl)-3-(trifluoromethyl)aniline (143) (51 mg, 0.21 mmol, 1.3 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (44 mg, 0.27 mmol, 1.7 eq). The reaction mixture was stirred at 40°C for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–12% MeOH in DCM on silica gel column then 10–100% MeOH in water with 0.05% TFA on reverse-phase column on prep-HPLC). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 73 (14 mg, 15% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.35 (s, 1H), 8.67 (d, J = 2.5 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.21 (t, J = 9.1 Hz, 1H), 7.99 (d, J = 2.3 Hz, 1H), 7.74 (d, J = 8.5 Hz, 1H), 7.59 – 7.51 (m, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.35 (dd, J = 11.7, 2.7 Hz, 1H), 7.14 – 7.07 (m, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.60 (s, 2H), 2.47 (q, J = 7.0 Hz, 4H), 0.97 (t, J = 7.1 Hz, 6H). 19F NMR (376 MHz, DMSO-d6) δ −58.32 (s, 3F), −125.42 (t, J = 10.8 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.58, 152.60, 152.51 (d, J = 244.7 Hz), 152.27, 149.40, 148.86, 148.62 (d, J = 10.2 Hz), 146.50, 138.23, 132.33, 131.19, 127.18 (q, J = 29.5 Hz), 124.85 (d, J = 10.7 Hz), 124.42 (q, J = 274.2 Hz), 122.04 (d, J = 2.5 Hz), 121.55, 117.09 (d, J = 3.0 Hz), 115.09, 114.76 (q, J = 6.5, 6.0 Hz), 109.04 (d, J = 22.0 Hz), 107.87, 103.36, 99.03, 55.73, 55.71, 52.83, 46.55, 11.73. HRMS: calcd for C30H31F4N4O4 [M + H]+ m/z, 587.2281; found m/z, 587.2271.
N-(4-bromo-2-(trifluoromethoxy)benzyl)-N-ethylethanamine (182).
To a solution of (4-bromo-2-(trifluoromethoxy)phenyl)methanol (138) (250 mg, 0.92 mmol, 1 eq) in DCM (6 mL) was added triethylamine (187 mg, 1.8 mmol, 2 eq). The reaction solution was cooled to 0°C using an ice bath and methanesulfonyl chloride (137 mg, 1.2 mmol, 1.3 eq) was added dropwise. The reaction mixture was warmed slowly to rt and stirred at rt for 2 h. Triethylamine (93 mg, 0.92 mmol, 1 eq) and diethylamine (337 mg, 4.6 mmol, 5 eq) were added. The reaction mixture was stirred at rt for 16 h, then concentrated onto Celite, and purified by flash chromatography (0–15% EtOAc in Hexanes on silica gel column) to afford 182 (226 mg, 75% yield) as a colorless liquid. 1H NMR (400 MHz, DMSO-d6) δ 7.61 (dd, J = 8.3, 1.9 Hz, 1H), 7.58 – 7.53 (m, 2H), 3.52 (s, 2H), 2.45 (q, J = 7.1 Hz, 4H), 0.96 (t, J = 7.1 Hz, 6H). 19F NMR (376 MHz, DMSO-d6) δ −56.29. 13C NMR (176 MHz, DMSO-d6) δ 147.10, 132.72, 132.66, 130.52, 123.63, 120.07 (q, J = 257.5 Hz), 119.76, 50.31, 46.50, 11.70.
4-((diethylamino)methyl)-3-(trifluoromethoxy)aniline (146).
To a solution of N-(4-bromo-2-(trifluoromethoxy)benzyl)-N-ethylethanamine (182) (103 mg, 0.32 mmol, 1 eq) in toluene (0.8 mL) was added bis(tri-tert-butylphosphine)palladium(0) (16 mg, 0.03 mmol, 0.1 eq) and lithium bis(trimethylsilyl)amide (1.5 M in THF, 0.32 mL, 0.47 mmol, 1.5 eq). The reaction mixture was stirred at rt for 16 h, then aqueous HCl (1 M, 2 mL) was added. The reaction mixture was stirred at rt for 10 min, then partitioned between EtOAc (20 mL) and aqueous NaOH (1 M, 15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated in vacuo, and purified by flash chromatography (1–10% MeOH in DCM on silica gel column) to afford 146 (69 mg, 83% yield) as a yellow liquid. 1H NMR (400 MHz, DMSO-d6) δ 7.11 (d, J = 8.2 Hz, 1H), 6.53 – 6.46 (m, 2H), 5.38 (s, 2H), 3.37 (s, 2H), 2.41 (q, J = 7.1 Hz, 4H), 0.95 (t, J = 7.1 Hz, 6H). 19F NMR (376 MHz, DMSO-d6) δ −55.57 (d, J = 1.8 Hz). 13C NMR (214 MHz, DMSO-d6) δ 149.34, 148.03, 131.87, 120.33 (q, J = 255.2 Hz), 112.53, 104.85, 50.26, 46.00, 11.59. (missing aromatic carbon peak is probably due to overlap at 112.53 ppm.)
1-(4-((diethylamino)methyl)-3-(trifluoromethoxy)phenyl)-3-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)urea (74).
To a solution of 4-((diethylamino)methyl)-3-(trifluoromethoxy)aniline (146) (66 mg, 0.25 mmol, 1.3 eq) in DMSO (1.3 mL) was added 1,1′-carbonyldiimidazole (53 mg, 0.33 mmol, 1.7 eq). The reaction mixture was stirred at rt for 45 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (61 mg, 0.19 mmol, 1 eq) in DMSO (1 mL). The reaction mixture was stirred at 60°C for 4 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–12% MeOH in DCM on silica gel column then 10–100% MeOH in water with 0.05% TFA on reverse-phase column on prep-HPLC). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 74 (31 mg, 27% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.39 (s, 1H), 8.68 (s, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.21 (t, J = 9.1 Hz, 1H), 7.76 (s, 1H), 7.49 (s, 1H), 7.47 (d, J = 8.4 Hz, 1H), 7.41 (s, 1H), 7.35 (dd, J = 11.7, 2.7 Hz, 1H), 7.23 (dd, J = 8.6, 2.1 Hz, 1H), 7.10 (ddd, J = 9.0, 2.7, 1.3 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.51 (s, 2H), 2.46 (br s, 4H), 0.98 (t, J = 7.3 Hz, 6H). 19F NMR (376 MHz, DMSO-d6) δ −55.84 (s, 3F), −125.42 (t, J = 10.8 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.58, 152.60, 152.50 (d, J = 244.8 Hz), 152.19, 149.39, 148.85, 148.61 (d, J = 9.9 Hz), 147.11 (br s), 146.49, 139.47 (br s), 131.40 (br s), 125.40 (br s), 124.82 (d, J = 10.5 Hz), 122.06, 120.24 (q, J = 256.5 Hz), 117.09 (d, J = 3.1 Hz), 116.56, 115.08, 109.64 (br s), 109.03 (d, J = 22.1 Hz), 107.86, 103.34, 99.02, 55.73, 55.71, 50.28 (br s), 46.28, 11.67 (br s). HRMS: calcd for C30H31F4N4O5 [M + H]+ m/z, 603.2231; found m/z, 603.2229.
1-methyl-4-(4-nitro-2-(trifluoromethyl)phenoxy)piperidine (183).
To a solution of 1-fluoro-4-nitro-2-(trifluoromethyl)benzene (148) (629 mg, 3.0 mmol, 1.1 eq) in DMSO (10 mL) in a water bath was added 1-methylpiperidin-4-ol (315 mg, 2.7 mmol, 1 eq) and potassium tert-butoxide (368 mg, 3.3 mmol, 1.2 eq) portionwise. The reaction mixture was stirred at rt for 16 h, then was partitioned between EtOAc (60 mL) and water (60 mL). The aqueous layer was extracted with EtOAc (60 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (2–10% MeOH in DCM on silica gel column) to afford 183 (485 mg, 58% yield) as an orange solid. 1H NMR (400 MHz, DMSO-d6) δ 8.47 (dd, J = 9.3, 2.9 Hz, 1H), 8.37 (d, J = 2.8 Hz, 1H), 7.56 (d, J = 9.3 Hz, 1H), 4.87 (tt, J = 6.8, 3.5 Hz, 1H), 2.49 – 2.41 (m, 2H), 2.39 – 2.25 (m, 2H), 2.17 (s, 3H), 1.95 (ddt, J = 12.1, 7.7, 3.7 Hz, 2H), 1.74 (dtd, J = 13.6, 7.0, 3.5 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −61.97. 13C NMR (214 MHz, DMSO-d6) δ 160.08, 139.70, 129.93, 123.17 (q, J = 5.3 Hz), 122.52 (q, J = 272.2 Hz), 117.90 (q, J = 31.5 Hz), 115.21, 73.72, 51.24, 45.86, 29.76.
4-((1-methylpiperidin-4-yl)oxy)-3-(trifluoromethyl)aniline (153).
A suspension of 1-methyl-4-(4-nitro-2-(trifluoromethyl)phenoxy)piperidine (183) (473 mg, 1.6 mmol, 1 eq), iron (434 mg, 7.8 mmol, 5 eq), and ammonium chloride (665 mg, 12.4 mmol, 8 eq) in ethanol (4.65 mL) and water (1.55 mL) was heated at 45°C for 16 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between EtOAc (50 mL) and aqueous NaOH solution (1 M, 50 mL). The aqueous layer was extracted with EtOAc (50 mL x2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (3–15% MeOH in DCM on silica gel column) to afford 153 (379 mg, 89% yield) as a yellow solid, which was stored under vacuum or under an argon atmosphere to prevent oxidation by atmospheric oxygen. 1H NMR (400 MHz, DMSO-d6) δ 6.97 (d, J = 8.8 Hz, 1H), 6.80 (d, J = 2.8 Hz, 1H), 6.75 (dd, J = 8.8, 2.8 Hz, 1H), 5.01 (s, 2H), 4.27 (tt, J = 8.0, 3.8 Hz, 1H), 2.59 – 2.52 (m, 2H), 2.21 – 2.07 (m, 5H), 1.96 – 1.77 (m, 2H), 1.61 (dtd, J = 12.3, 8.3, 3.5 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −60.05. 13C NMR (214 MHz, DMSO-d6) δ 145.35, 142.41, 123.98 (q, J = 272.5 Hz), 118.65 (q, J = 29.3 Hz), 118.52, 117.15, 111.42 (q, J = 5.3 Hz), 73.02 (br s), 52.04, 45.83, 30.50.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-((1-methylpiperidin-4-yl)oxy)-3-(trifluoromethyl)phenyl)urea (75).
To a solution of 4-((1-methylpiperidin-4-yl)oxy)-3-(trifluoromethyl)aniline (153) (57 mg, 0.21 mmol, 1.3 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (44 mg, 0.27 mmol, 1.7 eq). The reaction mixture was stirred at rt for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–20% MeOH in DCM on silica gel column then 10–100% MeOH in water with 0.05% TFA on reverse-phase column on prep-HPLC). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 75 (36 mg, 37% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.16 (s, 1H), 8.60 (d, J = 2.3 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.20 (t, J = 9.1 Hz, 1H), 7.85 (d, J = 2.7 Hz, 1H), 7.52 (dd, J = 9.0, 2.7 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.34 (dd, J = 11.7, 2.7 Hz, 1H), 7.26 (d, J = 9.1 Hz, 1H), 7.09 (ddd, J = 9.0, 2.7, 1.3 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 4.52 (tt, J = 7.5, 3.6 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 2.54 (dd,J = 9.4, 5.7 Hz, 2H), 2.27 – 2.19 (m, 2H), 2.17 (s, 3H), 1.90 (tt, J = 10.7, 3.5 Hz, 2H), 1.74 – 1.61 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −60.62 (s, 3F), −125.49 (t, J = 10.5 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.60, 152.59, 152.45 (d, J = 244.6 Hz), 152.42, 149.95, 149.38, 148.85, 148.46 (d, J = 10.3 Hz), 146.49, 132.14, 125.02 (d, J = 10.7 Hz), 123.75, 123.63 (q, J = 272.5 Hz), 121.99 (d, J = 2.5 Hz), 117.92 (q, J = 29.5 Hz), 117.06 (d, J = 3.1 Hz), 116.83 (q, J = 5.2 Hz), 115.79, 115.08, 109.01 (d, J = 21.9 Hz), 107.86, 103.32, 99.02, 72.45 (br s), 55.73, 55.70, 51.72, 45.87, 30.19. HRMS: calcd for C31H31F4N4O5 [M + H]+ m/z, 615.2231; found m/z, 615.2219.
1-methyl-4-(4-nitro-2-(trifluoromethoxy)phenoxy)piperidine (184).
To a solution of 1-chloro-4-nitro-2-(trifluoromethoxy)benzene (149) (300 mg, 1.24 mmol, 1 eq) in DMSO (5 mL) in a water bath was added 1-methylpiperidin-4-ol (143 mg, 1.24 mmol, 1 eq) and potassium tert-butoxide (167 mg, 1.2 mmol) portionwise. The reaction mixture was stirred at rt for 18 h, then was partitioned between EtOAc (30 mL) and aqueous NaOH (1M, 30 mL). The aqueous layer was extracted with EtOAc (30 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (2–10% MeOH in DCM on silica gel column) to afford 184 (111 mg, 28% yield) as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.26 (dd, J = 9.2, 2.8 Hz, 1H), 8.23 – 8.21 (m, 1H), 7.55 (d, J = 9.4 Hz, 1H), 4.76 (tt, J = 7.4, 3.9 Hz, 1H), 2.55 – 2.51 (m, 2H), 2.27 (ddd, J = 11.4, 8.0, 3.3 Hz, 2H), 2.17 (s, 3H), 2.01 – 1.91 (m, 2H), 1.71 (dtd, J = 12.1, 7.9, 3.7 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −57.37. 13C NMR (176 MHz, DMSO-d6) δ 155.40, 139.79, 136.51, 124.99, 120.10 (q, J = 258.2 Hz), 119.44, 115.22, 73.95 (br s), 51.60, 45.82, 29.93.
4-((1-methylpiperidin-4-yl)oxy)-3-(trifluoromethoxy)aniline (155).
A suspension of 1-methyl-4-(4-nitro-2-(trifluoromethoxy)phenoxy)piperidine (184) (108 mg, 0.34 mmol, 1 eq), iron (94 mg, 1.7 mmol, 5 eq), and ammonium chloride (144 mg, 2.7 mmol, 8 eq) in ethanol (1 mL) and water (0.35 mL) was heated at 60°C for 3 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between DCM (30 mL) and aqueous NaOH solution (1 M, 30 mL). The aqueous layer was extracted with DCM (30 mL x2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (3–12% MeOH in DCM on silica gel column) to afford 155 (87 mg, 90% yield) as a yellow oil, which was stored under vacuum or under an argon atmosphere to prevent oxidation by atmospheric oxygen. 1H NMR (400 MHz, DMSO-d6) δ 6.91 (d, J = 8.7 Hz, 1H), 6.52 (dq, J = 2.8, 1.4 Hz, 1H), 6.48 (dd, J = 8.7, 2.7 Hz, 1H), 5.04 (s, 2H), 4.05 (tt, J = 7.8, 3.8 Hz, 1H), 2.59 – 2.52 (m, 2H), 2.14 (s, 3H), 2.14 – 2.06 (m, 2H), 1.90 – 1.76 (m, 2H), 1.59 (dtd, J = 12.4, 8.4, 3.6 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −56.40. 13C NMR (214 MHz, DMSO-d6) δ 144.11, 139.49, 139.17, 120.32 (q, J = 255.5 Hz), 119.82, 113.23, 107.70, 74.64 (br s), 52.24 (br s), 45.96, 30.73.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-((1-methylpiperidin-4-yl)oxy)-3-(trifluoromethoxy)phenyl)urea (76).
To a solution of 4-((1-methylpiperidin-4-yl)oxy)-3-(trifluoromethoxy)aniline (155) (86 mg, 0.30 mmol, 1.3 eq) in DMSO (1.5 mL) was added 1,1′-carbonyldiimidazole (63 mg, 0.39 mmol, 1.7 eq). The reaction mixture was stirred at rt for 45 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (72 mg, 0.23 mmol, 1 eq) in DMSO (1 mL). The reaction mixture was stirred at 60°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–15% MeOH in DCM on silica gel column then 10–100% MeOH in water with 0.05% TFA on reverse-phase column on prep-HPLC). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 76 (81 mg, 56% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.15 (s, 1H), 8.59 (d, J = 2.5 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.20 (t, J = 9.1 Hz, 1H), 7.68 (q, J = 1.4 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.34 (dd, J = 11.8, 2.7 Hz, 1H), 7.22 (d, J = 1.5 Hz, 2H), 7.09 (ddd, J = 8.9, 2.8, 1.3 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 4.36 (tt, J = 7.6, 3.5 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 2.60 – 2.52 (m, 2H), 2.24 – 2.15 (m, 5H), 1.93 – 1.82 (m, 2H), 1.65 (dtd, J = 12.1, 8.0, 3.6 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −56.76 (s, 3F), −125.57 (dd, J = 12.2, 9.1 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.61, 152.59, 152.43 (d, J = 244.6 Hz), 152.29, 149.39, 148.86, 148.47 (d, J = 10.3 Hz), 146.49, 144.20, 137.90, 133.10, 124.99 (d, J = 10.6 Hz), 121.97 (d, J = 2.0 Hz), 120.28 (q, J = 255.8 Hz), 118.08, 117.40, 117.08 (d, J = 3.0 Hz), 115.08, 113.17, 109.01 (d, J = 22.1 Hz), 107.87, 103.32, 99.03, 73.48 (br s), 55.73, 55.71, 51.96, 45.87, 30.40. HRMS: calcd for C31H31F4N4O6 [M + H]+ m/z, 631.2180; found m/z, 631.2169.
N,1-dimethyl-N-(4-nitro-2-(trifluoromethyl)phenyl)piperidin-4-amine (185).
To a solution of 1-fluoro-4-nitro-2-(trifluoromethyl)benzene (148) (500 mg, 2.4 mmol, 1 eq) in DMSO (4.8 mL) was added triethylamine (605 mg, 6.0 mmol, 2.5 eq) and N,1-dimethylpiperidin-4-amine (337 mg, 2.6 mmol, 1.1 eq). The reaction mixture was stirred at 90°C for 20 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (50 mL) and water (50 mL). The aqueous layer was extracted with EtOAc (50 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (2–10% MeOH in DCM on silica gel column) to afford 185 (567 mg, 75% yield) as a reddish orange oil. 1H NMR (400 MHz, DMSO-d6) δ 8.37 (d, J = 2.8 Hz, 1H), 8.33 (dd, J = 9.2, 2.8 Hz, 1H), 7.53 (d, J = 9.2 Hz, 1H), 3.23 (tt, J = 11.0, 4.0 Hz, 1H), 2.82 – 2.76 (m, 5H), 2.14 (s, 3H), 1.88 (td, J = 11.7, 2.5 Hz, 2H), 1.72 (qd, J = 11.8, 3.7 Hz, 2H), 1.66 – 1.59 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −57.84. 13C NMR (214 MHz, DMSO-d6) δ 156.85, 140.18, 127.80, 124.42 (q, J = 5.9 Hz), 123.87, 123.33 (q, J = 273.3 Hz), 120.64 (q, J = 30.8 Hz), 60.92, 54.48, 45.72, 36.28, 28.55.
N1-methyl-N1-(1-methylpiperidin-4-yl)-2-(trifluoromethyl)benzene-1,4-diamine (154).
A suspension of N,1-dimethyl-N-(4-nitro-2-(trifluoromethyl)phenyl)piperidin-4-amine (185) (561 mg, 1.8 mmol, 1 eq), iron (691 mg, 12.4 mmol, 7 eq), and ammonium chloride (946 mg, 17.7 mmol, 10 eq) in ethanol (5.25 mL) and water (1.75 mL) was heated at 78°C for 16 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between DCM (30 mL) and aqueous NaOH solution (1 M, 30 mL). The aqueous layer was extracted with DCM (30 mL x2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (5–15% MeOH in DCM on silica gel column) to afford 154 (371 mg, 73% yield) as a reddish oil, which was stored under vacuum or under an argon atmosphere to prevent oxidation by atmospheric oxygen. 1H NMR (400 MHz, DMSO-d6) δ 7.18 (d, J = 8.5 Hz, 1H), 6.79 (d, J = 2.7 Hz, 1H), 6.76 (dd, J = 8.5, 2.7 Hz, 1H), 5.31 (s, 2H), 2.70 (d, J = 11.3 Hz, 2H), 2.56 (t, J = 9.9 Hz, 1H), 2.46 (s, 3H), 2.09 (s, 3H), 1.75 (td, J = 12.0, 2.3 Hz, 2H), 1.62 (d, J = 12.3 Hz, 2H), 1.30 (qd, J = 12.2, 3.8 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −58.66. 13C NMR (214 MHz, DMSO-d6) δ 146.24, 139.65, 127.61 (q, J = 27.3 Hz), 127.18, 124.19 (q, J = 273.3 Hz), 117.80, 110.46 (q, J = 5.5 Hz), 60.19, 54.45, 45.81, 41.23, 30.09.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(methyl(1-methylpiperidin-4-yl)amino)-3-(trifluoromethyl)phenyl)urea (77).
To a solution of N1-methyl-N1-(1-methylpiperidin-4-yl)-2-(trifluoromethyl)benzene-1,4-diamine (154) (59 mg, 0.21 mmol, 1.3 eq) in DMSO (1 mL) was added 1,1′-carbonyldiimidazole (44 mg, 0.27 mmol, 1.7 eq). The reaction mixture was stirred at rt for 1 h, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (50 mg, 0.16 mmol, 1 eq) in DMSO (0.8 mL). The reaction mixture was stirred at 60°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (1–18% MeOH in DCM on silica gel column then 10–100% MeOH in water with 0.05% TFA on reverse-phase column on prep-HPLC). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 77 (31 mg, 31% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.34 (s, 1H), 8.66 (d, J = 2.4 Hz, 1H), 8.50 (d, J = 5.3 Hz, 1H), 8.21 (t, J = 9.1 Hz, 1H), 7.91 (d, J = 2.5 Hz, 1H), 7.58 (dd, J = 8.8, 2.6 Hz, 1H), 7.50 (d, J = 9.5 Hz, 2H), 7.41 (s, 1H), 7.34 (dd, J = 11.7, 2.7 Hz, 1H), 7.10 (ddd, J = 8.9, 2.8, 1.3 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 2.72 (t, J = 10.2 Hz, 3H), 2.54 (s, 3H), 2.12 (s, 3H), 1.81 (t, J = 11.7 Hz, 2H), 1.65 (d, J = 11.6 Hz, 2H), 1.38 (qd, J = 12.0, 3.8 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −58.77 (s, 3F), −125.39 (t, J = 10.6 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.59, 152.59, 152.49 (d, J = 245.1 Hz), 152.28, 149.39, 148.86, 148.59 (d, J = 10.2 Hz), 146.50, 145.97, 136.67, 127.49, 127.40 (q, J = 27.2 Hz), 124.89 (d, J = 10.6 Hz), 123.82 (q, J = 273.3 Hz), 122.62, 122.03 (d, J = 2.4 Hz), 117.09 (d, J = 3.2 Hz), 115.71 (q, J = 5.6 Hz), 115.08, 109.03 (d, J = 22.0 Hz), 107.87, 103.34, 99.02, 60.02, 55.73, 55.71, 54.32, 45.71, 40.78, 29.83. HRMS: calcd for C32H34F4N5O4 [M + H]+ m/z, 628.2547; found m/z, 628.2535.
N,1-dimethyl-N-(4-nitro-2-(trifluoromethoxy)phenyl)piperidin-4-amine (186).
To a solution of 1-chloro-4-nitro-2-(trifluoromethoxy)benzene (149) (300 mg, 1.24 mmol, 1 eq) in DMSO (2.5 mL) was added N,1-dimethylpiperidin-4-amine (510 mg, 4.0 mmol, 3.2 eq) and potassium phosphate (527 mg, 2.48 mmol, 2 eq). The reaction mixture was stirred at 100°C for 18 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (30 mL) and water (30 mL). The aqueous layer was extracted with EtOAc (30 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (2–10% MeOH in DCM on silica gel column) to afford 186 (198 mg, 48% yield) as a yellow-brown solid. 1H NMR (400 MHz, DMSO-d6) δ 8.11 (dd, J = 9.3, 2.7 Hz, 1H), 8.04 (dq, J = 2.7, 1.3 Hz, 1H), 7.21 (d, J = 9.3 Hz, 1H), 3.53 (tt, J = 11.2, 3.9 Hz, 1H), 2.91 – 2.81 (m, 5H), 2.17 (s, 3H), 2.01 – 1.80 (m, 4H), 1.62 – 1.49 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −57.18. 13C NMR (214 MHz, DMSO-d6) δ 150.46, 137.63, 136.32, 124.23, 120.22 (q, J = 258.1 Hz), 119.32, 118.70, 59.51, 54.72, 45.76, 33.41, 28.41.
N1-methyl-N1-(1-methylpiperidin-4-yl)-2-(trifluoromethoxy)benzene-1,4-diamine (156).
A suspension of N,1-dimethyl-N-(4-nitro-2-(trifluoromethoxy)phenyl)piperidin-4-amine (186). (194 mg, 0.58 mmol, 1 eq), iron (261 mg, 4.6 mmol, 8 eq), and ammonium chloride (313 mg, 5.8 mmol, 10 eq) in ethanol (1.8 mL) and water (0.6 mL) was heated at 60°C for 24 h. The reaction mixture was cooled to rt and filtered through Celite. Solvent was removed from the filtrate and the residue was partitioned between DCM (30 mL) and aqueous NaOH solution (1 M, 30 mL). The aqueous layer was extracted with DCM (30 mL x2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (3–12% MeOH in DCM on silica gel column) to afford 156 (113 mg, 64% yield) as an orange oil, which was stored under vacuum or under an argon atmosphere to prevent oxidation by atmospheric oxygen. 1H NMR (400 MHz, DMSO-d6) δ 7.04 – 6.90 (m, 1H), 6.56 – 6.42 (m, 2H), 5.16 (s, 2H), 2.78 – 2.70 (m, 2H), 2.63 (tt, J = 11.1, 4.1 Hz, 1H), 2.52 (s, 3H), 2.10 (s, 3H), 1.78 (td, J = 11.7, 2.4 Hz, 2H), 1.64 – 1.53 (m, 2H), 1.43 (qd, J = 11.9, 3.8 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −55.41. 13C NMR (214 MHz, DMSO-d6) δ 146.19, 144.66, 132.92, 125.82, 120.34 (q, J = 255.2 Hz), 112.83, 106.42, 60.25, 54.84, 46.05, 37.16, 28.99.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(methyl(1-methylpiperidin-4-yl)amino)-3-(trifluoromethoxy)phenyl)urea (78).
To a solution of N1-methyl-N1-(1-methylpiperidin-4-yl)-2-(trifluoromethoxy)benzene-1,4-diamine (156) (55 mg, 0.18 mmol, 1.3 eq) in DMSO (0.9 mL) was added 1,1′-carbonyldiimidazole (39 mg, 0.24 mmol, 1.7 eq). The reaction mixture was stirred at rt for 45 min, monitoring reaction progress by TLC. This was added to a solution of 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (44 mg, 0.14 mmol, 1 eq) in DMSO (0.7 mL). The reaction mixture was stirred at 60°C for 3 h, then cooled to rt. The crude reaction mixture was partitioned between EtOAc (15 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (3–15% MeOH in DCM on silica gel column then 10–100% MeOH in water with 0.05% TFA on reverse-phase column on prep-HPLC). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 78 (40 mg, 44% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.19 (s, 1H), 8.60 (d, J = 2.5 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.21 (t, J = 9.1 Hz, 1H), 7.66 (s, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.34 (dd, J = 11.7, 2.7 Hz, 1H), 7.23 – 7.13 (m, 2H), 7.09 (dt, J = 8.9, 1.8 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 2.89 (tt, J = 10.6, 4.7 Hz, 1H), 2.78 (d, J = 11.3 Hz, 2H), 2.61 (s, 3H), 2.12 (s, 3H), 1.83 (td, J = 11.2, 3.4 Hz, 2H), 1.68 – 1.51 (m, 4H). 19F NMR (376 MHz, DMSO-d6) δ −55.86 (s, 3F), −125.64 (t, J = 10.4 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.73, 152.62, 152.40 (d, J = 244.5 Hz), 152.28, 149.41, 148.95, 148.36 (d, J = 10.3 Hz), 146.52, 142.22, 139.31, 134.72, 125.12 (d, J = 10.6 Hz), 123.51, 121.87 (d, J = 2.7 Hz), 120.25 (q, J = 256.2 Hz), 117.31, 117.28 (d, J = 2.4 Hz), 115.07, 111.75, 109.20 (d, J = 21.9 Hz), 107.89, 103.27, 99.03, 59.95, 55.79, 55.76, 54.90, 45.90, 34.76, 28.39. HRMS: calcd for C32H34F4N5O5 [M + H]+ m/z, 644.2496; found m/z, 644.2485.
Experimental Procedures in Scheme 7.
tert-butyl 4-(4-(methoxycarbonyl)benzylidene)piperidine-1-carboxylate (159).
To a solution of methyl 4-bromobenzoate (157) (700 mg, 3.26 mmol, 1 eq) and tert-butyl 4-methylenepiperidine-1-carboxylate (158) (963 mg, 4.88 mmol, 1.5 eq) in DMF (6.5 mL) was added triethylamine (988 mg, 9.77 mmol, 3 eq) and [1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) (238 mg, 0.33 mmol, 0.1 eq). The reaction mixture was stirred at 100°C for 16 h. The reaction mixture was cooled to rt and filtered through Celite. The filtrate was portioned between EtOAc (50 mL) and water (50 mL), and the organic layer was washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–20% EtOAc in Hexanes on silica gel column) to afford 159 (199 mg, 18% yield) as a light green solid. 1H NMR (400 MHz, DMSO-d6) δ 7.91 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.3 Hz, 2H), 6.43 (s, 1H), 3.84 (s, 3H), 3.42 (t, J = 5.8 Hz, 2H), 3.35 (t, J = 5.9 Hz, 2H), 2.42 (t, J = 5.8 Hz, 2H), 2.31 (t, J = 5.5 Hz, 2H), 1.41 (s, 9H). 13C NMR (214 MHz, DMSO-d6) δ 166.01, 153.77, 141.94, 140.87, 129.15, 128.97, 127.31, 123.34, 78.81, 52.04, 44.78 (*), 43.78 (*), 35.66 (br s), 28.89 (br s), 28.06. (*not observed on 1D 13C spectrum, peaks were confirmed by (1H-13C)-HSQC. 1D spectrum shows three very broad singlets at this region probably reflecting chemical exchange)
4-((1-(tert-butoxycarbonyl)piperidin-4-ylidene)methyl)benzoic acid (176).
To a solution of tert-butyl 4-(4-(methoxycarbonyl)benzylidene)piperidine-1-carboxylate (159) (194 mg, 0.58 mmol, 1 eq) in THF (3.6 mL) and water (0.90 mL) was added lithium hydroxide (42 mg, 1.8 mmol, 3 eq). The reaction mixture was stirred at 50°C for 6 h. The reaction mixture was cooled to rt and THF was removed in vacuo, then the reaction mixture was acidified to pH 2 using aqueous HCl (2 M). The solid formed was collected by filtration, washed with water and dried to afford 176 (165 mg, 89% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.89 (s, 1H), 7.97 – 7.83 (m, 2H), 7.34 (d, J = 8.3 Hz, 2H), 6.43 (s, 1H), 3.42 (t, J = 5.8 Hz, 2H), 3.35 (t, J = 5.9 Hz, 2H), 2.42 (t, J = 5.9 Hz, 2H), 2.31 (t, J = 5.7 Hz, 2H), 1.41 (s, 9H). 13C NMR (214 MHz, DMSO-d6) δ 167.10, 153.78, 141.47, 140.52, 129.31, 128.80, 123.47, 78.81, 44.59 (*), 43.63 (*), 35.68 (br s), 28.86 (br s), 28.08. (*not observed on 1D 13C spectrum, peaks were confirmed by (1H-13C)-HSQC. 1D spectrum shows three very broad singlets at this region probably reflecting chemical exchange) (missing aromatic carbon signal is likely due to overlap of peaks)
tert-butyl 4-(4-(3-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)ureido)benzylidene)piperidine-1-carboxylate (160).
In a microwave vial was added 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (107 mg, 0.34 mmol, 1 eq), 4-((1-(tert-butoxycarbonyl)piperidin-4-ylidene)methyl)benzoic acid (176) (180 mg, 0.51 mmol, 1.5 eq), toluene (0.75 mL) and triethylamine (103 mg, 1.02 mmol, 3 eq). The suspension was sonicated until fine particles were obtained, then diphenylphosphoryl azide (140 mg, 0.51 mmol, 1.5 eq) was added and the reaction mixture was irradiated at 100°C for 10 min. The reaction mixture was partitioned between EtOAc (30 mL) and water (20 mL). The aqueous layer was extracted with EtOAc (30 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (40–100% EtOAc in Hexanes on silica gel column) to afford 160 (176 mg, 82% yield) as a beige solid. 1H NMR (400 MHz, DMSO-d6) δ 9.12 (d, J = 2.8 Hz, 1H), 8.64 (s, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.25 (t, J = 9.2 Hz, 1H), 7.49 (s, 1H), 7.44 (d, J = 8.4 Hz, 2H), 7.40 (s, 1H), 7.35 (dd, J = 11.8, 2.7 Hz, 1H), 7.17 (d, J = 8.4 Hz, 2H), 7.10 (dd, J = 9.1, 2.7 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 6.31 (s, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.46 – 3.38 (m, 2H), 3.37 – 3.34 (m, 2H), 2.42 (d, J = 6.1 Hz, 2H), 2.27 (t, J = 5.7 Hz, 2H), 1.42 (s, 9H). 19F NMR (376 MHz, DMSO-d6) δ −125.88 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.64, 153.81, 152.59, 152.24 (d, J = 244.5 Hz), 152.17, 149.38, 148.87, 148.24 (d, J = 10.3 Hz), 146.49, 137.67, 136.96, 130.86, 129.29, 125.22 (d, J = 10.4 Hz), 123.77, 121.58 (d, J = 2.5 Hz), 117.91, 117.11 (d, J = 3.0 Hz), 115.07, 109.00 (d, J = 22.2 Hz), 107.87, 103.30, 99.03, 78.74, 55.73, 55.71, 44.83 (*), 43.64 (*), 35.61 (br s), 28.81 (br s), 28.09. (*not observed on 1D 13C spectrum, peaks were confirmed by (1H-13C)-HSQC. 1D spectrum shows three very broad singlets at this region probably reflecting chemical exchange) HRMS: calcd for C35H38FN4O6 [M + H]+ m/z, 629.2775; found m/z, 629.2766.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(piperidin-4-ylidenemethyl)phenyl)urea (68).
To a solution of tert-butyl 4-(4-(3-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)ureido)benzylidene)piperidine-1-carboxylate (160) (144 mg, 0.23 mmol) in DCM (2 mL) was added TFA (0.5 mL). The reaction mixture was stirred at rt for 1 h, then concentrated in vacuo, and purified by flash chromatography (10–100% MeOH in water with 0.1% TFA on C18 column). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 68 (96 mg, 79% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.12 (s, 1H), 8.65 (s, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.25 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.42 (d, J = 8.7 Hz, 2H), 7.40 (s, 1H), 7.34 (dd, J = 11.8, 2.7 Hz, 1H), 7.14 (d, J = 8.6 Hz, 2H), 7.10 (ddd, J = 9.1, 2.8, 1.3 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 6.18 (s, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 2.77 (t, J = 5.6 Hz, 2H), 2.69 (t, J = 5.6 Hz, 2H), 2.35 (t, J = 5.7 Hz, 2H), 2.21 (t, J = 5.6 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −125.87 (t, J = 10.3 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.65, 152.59, 152.23 (d, J = 244.4 Hz), 152.19, 149.38, 148.87, 148.20 (d, J = 10.2 Hz), 146.49, 139.70, 137.38, 131.31, 129.23, 125.26 (d, J = 10.5 Hz), 121.90, 121.57 (d, J = 2.9 Hz), 117.90, 117.10 (d, J = 3.0 Hz), 115.08, 109.00 (d, J = 21.9 Hz), 107.87, 103.30, 99.04, 55.73, 55.71, 48.13, 47.26, 37.74, 30.63. HRMS: calcd for C30H30FN4O4 [M + H]+ m/z, 529.2251; found m/z, 529.2245.
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(piperidin-4-ylmethyl)phenyl)urea (67).
To a solution of 1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(piperidin-4-ylidenemethyl)phenyl)urea (68) (76 mg, 0.14 mmol, 1 eq) in methanol (1.4 mL) was added palladium on carbon (10% Pd by wt, 15 mg, 0.1 eq). The solution was purged with argon, then hydrogen gas. The reaction mixture was stirred under hydrogen atmosphere at rt for 18 h, then filtered through Celite. The filtrate was concentrated onto Celite and purified by flash chromatography (10–100% MeOH in water with 0.1% TFA on reverse-phase column in prep-HPLC). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 67 (42 mg, 55% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.06 (s, 1H), 8.64 (d, J = 2.6 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.24 (t, J = 9.1 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.38 – 7.30 (m, 3H), 7.12 – 7.04 (m, 3H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 2.94 (dt, J = 12.3, 3.2 Hz, 2H), 2.44 (td, J = 10.9, 9.7, 2.6 Hz, 4H), 1.60 – 1.47 (m, 3H), 1.06 (qd, J = 12.0, 4.0 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −125.91 (t, J = 10.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 159.66, 152.59, 152.27, 152.20 (d, J = 244.4 Hz), 149.38, 148.87, 148.12 (d, J = 10.2 Hz), 146.48, 137.17, 133.88, 129.37, 125.34 (d, J = 10.3 Hz), 121.53 (d, J = 2.4 Hz), 118.12, 117.08 (d, J = 3.2 Hz), 115.07, 108.99 (d, J = 21.9 Hz), 107.86, 103.29, 99.04, 55.73, 55.71, 45.61, 42.23, 37.63, 32.12. HRMS: calcd for C30H32FN4O4 [M + H]+ m/z, 531.2408; found m/z, 531.2401.
tert-butyl 4-(4-(methoxycarbonyl)-2-(trifluoromethyl)benzylidene)piperidine-1-carboxylate (165).
To a solution of methyl 4-bromo-3-(trifluoromethyl)benzoate (161) (250 mg, 0.88 mmol, 1 eq) and tert-butyl 4-((4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)methylene)piperidine-1-carboxylate (164) (286 mg, 0.88 mmol, 1 eq) in 1,4-dioxane (5.7 mL) and water (0.6 mL) was added tris(dibenzylideneacetone)dipalladium(0) (61 mg, 0.066 mmol, 0.075 eq), Xphos (63 mg, 0.13 mmol, 0.15 eq) and potassium phosphate (281 mg, 1.3 mmol, 1.5 eq). The reaction mixture was stirred at 100°C for 1 h, then cooled to rt and partitioned between EtOAc (30 mL) and water (20 mL). The aqueous layer was extracted with EtOAc (30 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and filtered. The filtrate was exposed to atmosphere for 2 days to oxidize an otherwise difficult-to-separate impurity, then concentrated onto Celite, and purified by flash chromatography (0–12% EtOAc in Hexanes on silica gel column) to afford 165 (238 mg, 67% yield) as a light yellow liquid. 1H NMR (400 MHz, DMSO-d6) δ 8.24 – 8.13 (m, 2H), 7.53 (d, J = 7.6 Hz, 1H), 6.52 (s, 1H), 3.90 (s, 3H), 3.42 (t, J = 5.8 Hz, 2H), 3.30 (t, J = 5.8 Hz, 2H), 2.33 (t, J = 5.8 Hz, 2H), 2.17 (t, J = 5.8 Hz, 2H), 1.41 (s, 9H). 19F NMR (376 MHz, DMSO-d6) δ −59.91. 13C NMR (214 MHz, DMSO-d6) δ 165.01, 153.81, 142.97, 140.52, 132.82, 132.64, 128.41, 127.56 (q, J = 30.0 Hz), 126.23 (q, J = 6.4, 4.9 Hz), 123.74 (q, J = 273.8 Hz), 119.49, 79.00, 52.68, 44.94 (*), 44.06 (*), 35.71 (br s), 29.09 (br s), 28.11. (*not observed on 1D 13C spectrum, peaks were confirmed by (1H-13C)-HSQC. 1D spectrum shows three very broad singlets at this region probably reflecting chemical exchange)
tert-butyl 4-(4-(methoxycarbonyl)-2-(trifluoromethyl)benzyl)piperidine-1-carboxylate (167).
To a solution of tert-butyl 4-(4-(methoxycarbonyl)-2-(trifluoromethyl)benzylidene)piperidine-1-carboxylate (165) (235 mg, 0.59 mmol, 1 eq) in methanol (3.9 mL) was added palladium on carbon (10% Pd by wt, 63 mg, 0.1 eq). The solution was purged with argon, then hydrogen gas. The reaction mixture was stirred under hydrogen atmosphere at rt for 24 h, then filtered through Celite. The filtrate was concentrated onto Celite and purified by flash chromatography (0–15% EtOAc in Hexanes on silica gel column) to afford 167 (203 mg, 86% yield) as a colorless liquid. 1H NMR (400 MHz, DMSO-d6) δ 8.20 – 8.12 (m, 2H), 7.66 (d, J = 7.9 Hz, 1H), 4.00 – 3.85 (m, 5H), 2.75 (d, J = 7.2 Hz, 2H), 2.62 (s, 3H), 1.78 (dtt, J = 14.5, 7.1, 3.8 Hz, 1H), 1.52 (dd, J = 13.3, 2.9 Hz, 1H), 1.38 (s, 9H), 1.10 (qd, J = 12.4, 4.2 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −58.10. 13C NMR (214 MHz, DMSO-d6) δ 165.09, 153.82, 144.32, 133.05, 132.64, 128.10, 127.89 (q, J = 29.8 Hz), 126.45 (q, J = 5.7 Hz), 124.08 (q, J = 274.1 Hz), 78.58, 52.63, 43.89 (br s), 42.90 (br s), 38.54, 37.09, 31.68 (br s), 31.47 (br s), 28.15.
4-((1-(tert-butoxycarbonyl)piperidin-4-yl)methyl)-3-(trifluoromethyl)benzoic acid (187).
To a solution of tert-butyl 4-(4-(methoxycarbonyl)-2-(trifluoromethyl)benzyl)piperidine-1-carboxylate (167) (200 mg, 0.50 mmol, 1 eq) in THF (3.2 mL) and water (0.8 mL) was added lithium hydroxide (36 mg, 1.5 mmol, 3 eq). The reaction mixture was stirred at 50°C for 4 h then cooled to rt. THF was removed in vacuo and the solution was acidified to pH 2 using aqueous HCl (1 M). The solid was collected by filtration, washed with water and dried in vacuo to afford 187 (173 mg, 90% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.16 (d, J = 1.7 Hz, 1H), 8.08 (dd, J = 8.0, 1.7 Hz, 1H), 7.52 (d, J = 8.0 Hz, 1H), 3.92 (d, J = 13.2 Hz, 2H), 2.71 (d, J = 7.1 Hz, 2H), 2.62 (br s, 2H), 1.83 – 1.68 (m, 1H), 1.52 (d, J = 13.0 Hz, 2H), 1.38 (s, 9H), 1.08 (qd, J = 12.3, 4.3 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −57.76. 13C NMR (214 MHz, DMSO-d6) δ 166.44, 153.79, 141.72 (br s), 133.02 (br s), 132.51, 132.13, 127.12 (q, J = 29.7, 29.0 Hz), 126.47 (q, J = 6.1 Hz), 124.46 (q, J = 274.0 Hz), 78.47, 43.88 (br s), 42.96 (br s), 38.45, 37.04, 31.59 (br s), 28.10. (missing aliphatic carbon peak is probably due to overlap at 31.59 ppm.)
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(piperidin-4-ylmethyl)-3-(trifluoromethyl)phenyl)urea (79).
In a microwave vial was added 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (38 mg, 0.12 mmol, 1 eq), 4-((1-(tert-butoxycarbonyl)piperidin-4-yl)methyl)-3-(trifluoromethyl)benzoic acid (187) (65 mg, 0.17 mmol, 1.4 eq), toluene (0.8 mL) and triethylamine (36 mg, 0.36 mmol, 3 eq). The suspension was sonicated until fine particles were obtained, then diphenylphosphoryl azide (46 mg, 0.17 mmol, 1.4 eq) was added and the reaction mixture was irradiated at 100°C for 10 min. The reaction mixture was partitioned between EtOAc (20 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (50– 100% EtOAc in Hexanes on silica gel column). Solvent was removed in vacuo from fractions containing the desired Boc-protected product. The solid was redissolved in DCM (2 mL) and TFA (0.5 mL) was added. The reaction solution was stirred at rt for 1 h, then concentrated in vacuo and purified by flash chromatography (10–100% MeOH in water with 0.05% TFA on reverse-phase column on prep-HPLC). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 79 (45 mg, 63% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.43 (s, 1H), 8.74 (s, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.20 (t, J = 9.1 Hz, 1H), 7.96 (d, J = 2.3 Hz, 1H), 7.53 (dd, J = 8.5, 2.3 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.38 – 7.32 (m, 2H), 7.10 (dt, J = 8.9, 1.8 Hz, 1H), 6.54 (d, J = 5.2 Hz, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 2.92 (d, J = 12.0 Hz, 2H), 2.58 (d, J = 7.0 Hz, 2H), 2.45 – 2.35 (m, 2H), 1.67 – 1.57 (m, 1H), 1.49 (d, J = 12.6 Hz, 2H), 1.09 (qd, J = 12.0, 3.9 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −57.78 (s, 3F), −125.22 (t, J = 10.2 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.71, 152.62, 152.57 (d, J = 244.9 Hz), 152.39, 149.41, 148.95, 148.56 (d, J = 10.1 Hz), 146.53, 137.86, 132.72, 131.84, 127.63 (q, J = 28.9 Hz), 125.01 (d, J = 10.6 Hz), 124.60 (q, J = 274.1 Hz), 122.11, 121.47, 117.25 (d, J = 3.0 Hz), 115.09, 115.02 (q, J = 6.0 Hz), 109.22 (d, J = 21.8 Hz), 107.89, 103.32, 99.03, 55.80, 55.76, 45.98, 38.74, 37.70, 32.74. HRMS: calcd for C31H31F4N4O4 [M + H]+ m/z, 599.2281; found m/z, 599.2273.
4-((1-(tert-butoxycarbonyl)piperidin-4-ylidene)methyl)-3-(trifluoromethyl)benzoic acid (188).
To a solution of tert-butyl 4-(4-(methoxycarbonyl)-2-(trifluoromethyl)benzylidene)piperidine-1-carboxylate (165) (179 mg, 0.45 mmol, 1 eq) in THF (2.8 mL) and water (0.7 mL) was added lithium hydroxide (32 mg, 1.3 mmol, 3 eq). The reaction mixture was stirred at 50°C for 4 h then cooled to rt. THF was removed in vacuo and the solution was acidified to pH 2 using aqueous HCl (1 M). The solid was collected by filtration, washed with water and dried in vacuo to afford 188 (153 mg, 88% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.18 (d, J = 1.7 Hz, 1H), 8.14 (dd, J = 7.9, 1.7 Hz, 1H), 7.48 (d, J = 8.0 Hz, 1H), 6.51 (s, 1H), 3.42 (t, J = 5.8 Hz, 2H), 3.30 (t, J = 5.9 Hz, 2H), 2.36 – 2.28 (m, 2H), 2.17 (t, J = 5.9 Hz, 2H), 1.41 (s, 9H). 19F NMR (376 MHz, DMSO-d6) δ −59.81 (d, J = 2.4 Hz). 13C NMR (214 MHz, DMSO-d6) δ 166.02, 153.78, 142.44, 139.75, 132.80, 132.28, 130.24 (br s), 127.32 (q, J = 29.6 Hz), 126.28 (q, J = 5.2 Hz), 123.84 (q, J = 273.8 Hz), 119.62, 78.91, 44.92 (*), 43.86 (*), 35.47 (br s), 29.19 (br s), 28.06. (*not observed on 1D 13C spectrum, peaks were confirmed by (1H-13C)-HSQC. 1D spectrum shows three very broad singlets at this region probably reflecting chemical exchange)
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(piperidin-4-ylidenemethyl)-3-(trifluoromethyl)phenyl)urea (81).
In a microwave vial was added 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (35 mg, 0.11 mmol, 1 eq), 4-((1-(tert-butoxycarbonyl)piperidin-4-ylidene)methyl)-3-(trifluoromethyl)benzoic acid (188) (55 mg, 0.14 mmol, 1.3 eq), toluene (0.75 mL) and triethylamine (33 mg, 0.33 mmol, 3 eq). The suspension was sonicated until fine particles were obtained, then diphenylphosphoryl azide (39 mg, 0.14 mmol, 1.3 eq) was added and the reaction mixture was irradiated at 100°C for 10 min. The reaction mixture was partitioned between EtOAc (20 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (50– 100% EtOAc in Hexanes on silica gel column). Solvent was removed in vacuo from fractions containing the desired Boc-protected product. The solid was redissolved in DCM (2 mL) and TFA (0.5 mL) was added. The reaction solution was stirred at rt for 1 h, then concentrated in vacuo and purified by flash chromatography (10–100% MeOH in water with 0.05% TFA on reverse-phase column on prep-HPLC). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 81 (41 mg, 63% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.44 (s, 1H), 8.73 (d, J = 2.4 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.21 (t, J = 9.1 Hz, 1H), 8.01 (d, J = 2.3 Hz, 1H), 7.53 (dd, J = 8.4, 2.3 Hz, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.35 (dd, J = 11.7, 2.7 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.10 (ddd, J = 8.9, 2.8, 1.3 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 6.29 (s, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 2.78 (t, J = 5.5 Hz, 2H), 2.66 (t, J = 5.6 Hz, 2H), 2.24 (t, J = 5.6 Hz, 2H), 2.12 (t, J = 5.7 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −59.65 (d, J = 2.5 Hz, 3F), −125.25 (t, J = 10.5 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.70, 152.62, 152.58 (d, J = 245.0 Hz), 152.33, 149.42, 148.95, 148.61 (d, J = 10.4 Hz), 146.53, 142.68, 138.27, 132.55, 129.32, 127.51 (q, J = 28.4 Hz), 124.95 (d, J = 10.5 Hz), 124.26 (q, J = 273.9 Hz), 122.11, 121.14, 118.10, 117.26 (d, J = 3.3 Hz), 115.09, 114.89 (q, J = 6.0 Hz), 109.23 (d, J = 22.1 Hz), 107.89, 103.33, 99.03, 55.79, 55.76, 48.19, 47.35, 37.50, 30.94. HRMS: calcd for C31H29F4N4O4 [M + H]+ m/z, 597.2125; found m/z, 597.2116.
methyl 4-iodo-3-(trifluoromethoxy)benzoate (163).
To a solution of 4-iodo-3-(trifluoromethoxy)benzoic acid (162) (500 mg, 1.5 mmol, 1 eq) in DMSO (10 mL) was added potassium carbonate (416 mg, 3.0 mmol, 2 eq) and iodomethane (321 mg, 2.3 mmol, 1.5 eq). The reaction mixture was stirred at rt for 6 h, then partitioned between EtOAc (80 mL) and water (80 mL). The aqueous layer was extracted with EtOAc (80 mL x 2). The combined organic extracts were washed with water, brine, dried over anhydrous Na2SO4, filtered, concentrated in vacuo to afford 163 (495 mg, 95% yield) as a light orange liquid. 1H NMR (400 MHz, DMSO-d6) δ 8.18 (d, J = 8.2 Hz, 1H), 7.81 (dq, J = 1.8, 1.4 Hz, 1H), 7.70 (dd, J = 8.2, 1.8 Hz, 1H), 3.88 (s, 3H). 19F NMR (376 MHz, DMSO-d6) δ −56.19 (d, J = 1.4 Hz). 13C NMR (176 MHz, DMSO-d6) δ 164.65, 148.80, 141.05, 131.67, 129.35, 121.05, 120.05 (q, J = 257.6 Hz), 98.20, 52.75.
tert-butyl 4-(4-(methoxycarbonyl)-2-(trifluoromethoxy)benzylidene)piperidine-1-carboxylate (166).
To a solution of methyl 4-iodo-3-(trifluoromethoxy)benzoate (163) (490 mg, 1.4 mmol, 1 eq) and tert-butyl 4-((4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)methylene)piperidine-1-carboxylate (164) (458 mg, 1.4 mmol, 1 eq) in 1,4-dioxane (9 mL) and water (1 mL) was added tris(dibenzylideneacetone)dipalladium(0) (130 mg, 0.14 mmol, 0.1 eq), Xphos (135 mg, 0.28 mmol, 0.2 eq) and potassium phosphate (451 mg, 2.1 mmol, 1.5 eq). The reaction mixture was stirred at 100°C for 1 h, then cooled to rt and partitioned between EtOAc (40 mL) and water (30 mL). The aqueous layer was extracted with EtOAc (40 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (0–12% EtOAc in Hexanes on silica gel column) to afford 166 (490 mg, 83% yield) as an orange-brown liquid. 1H NMR (400 MHz, DMSO-d6) δ 7.94 (dd, J = 8.0, 1.7 Hz, 1H), 7.82 (dq, J = 3.0, 1.4 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 6.37 (s, 1H), 3.88 (s, 3H), 3.42 (t, J = 5.7 Hz, 2H), 3.33 (t, J = 5.7 Hz, 2H), 2.34 (t, J = 5.8 Hz, 2H), 2.25 (t, J = 5.9 Hz, 2H), 1.41 (s, 9H). 19F NMR (376 MHz, DMSO-d6) δ −56.41 (d, J = 5.4 Hz). 13C NMR (214 MHz, DMSO-d6) δ 164.84, 153.77, 145.84, 143.57, 135.36, 132.03, 129.70, 127.97, 121.44, 120.12 (q, J = 257.5 Hz), 116.92, 78.93, 52.59, 44.91 (*), 43.94 (*), 35.55 (br s), 29.23 (br s), 28.06. (*not observed on 1D 13C spectrum, peaks were confirmed by (1H-13C)-HSQC. 1D spectrum shows three very broad singlets at this region probably reflecting chemical exchange)
tert-butyl 4-(4-(methoxycarbonyl)-2-(trifluoromethoxy)benzyl)piperidine-1-carboxylate (168).
To a solution of tert-butyl 4-(4-(methoxycarbonyl)-2-(trifluoromethoxy)benzylidene)piperidine-1-carboxylate (166) (239 mg, 0.58 mmol, 1 eq) in methanol (3.8 mL) was added palladium on carbon (10% Pd by wt, 61 mg, 0.1 eq). The solution was purged with argon, then hydrogen gas. The reaction mixture was stirred under hydrogen atmosphere at rt for 24 h, then filtered through Celite. The filtrate was concentrated onto Celite and purified by flash chromatography (0–15% EtOAc in Hexanes on silica gel column) to afford 168 (211 mg, 88% yield) as a light yellow liquid. 1H NMR (400 MHz, DMSO-d6) δ 7.91 (dd, J = 8.0, 1.6 Hz, 1H), 7.79 (dq, J = 1.8, 1.3 Hz, 1H), 7.57 (d, J = 8.0 Hz, 1H), 3.87 (s, 5H), 2.65 (d, J = 7.2 Hz, 2H), 2.62 (br s, 2H), 1.73 (ttt, J = 11.8, 7.2, 3.1 Hz, 1H), 1.50 (d, J = 14.1 Hz, 2H), 1.38 (s, 9H), 1.06 (qd, J = 12.3, 4.2 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −56.04 (d, J = 8.7 Hz). 13C NMR (214 MHz, DMSO-d6) δ 165.00, 153.82, 146.93, 138.27, 132.70, 129.54, 127.89, 120.51, 120.18 (q, J = 257.8 Hz), 78.56, 52.64, 43.81 (br s), 42.82 (br s), 36.23, 35.97, 31.54 (br s), 31.35 (br s), 28.14.
4-((1-(tert-butoxycarbonyl)piperidin-4-yl)methyl)-3-(trifluoromethoxy)benzoic acid (189).
To a solution of tert-butyl 4-(4-(methoxycarbonyl)-2-(trifluoromethoxy)benzyl)piperidine-1-carboxylate (168) (208 mg, 0.50 mmol, 1 eq) in THF (3.2 mL) and water (0.8 mL) was added lithium hydroxide (36 mg, 1.5 mmol, 3 eq). The reaction mixture was stirred at 50°C for 4 h then cooled to rt. THF was removed in vacuo and the solution was acidified to pH 2 using aqueous HCl (1 M). The solid was collected by filtration, washed with water and dried in vacuo to afford 189 (184 mg, 92% yield) as a gray solid. 1H NMR (400 MHz, DMSO-d6) δ 7.87 (dd, J = 7.9, 1.6 Hz, 1H), 7.77 (dq, J = 1.7, 1.6 Hz, 1H), 7.52 (d, J = 8.0 Hz, 1H), 3.90 (d, J = 13.2 Hz, 2H), 2.72 – 2.60 (m, 4H), 1.80 – 1.65 (m, 1H), 1.50 (dd, J = 13.3, 2.0 Hz, 2H), 1.38 (s, 9H), 1.06 (qd, J = 12.4, 4.2 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −55.98 (d, J = 1.6 Hz). 13C NMR (214 MHz, DMSO-d6) δ 166.04, 153.79, 146.84, 137.26, 132.29, 131.38 (br s), 127.83, 120.44, 120.16 (q, J = 256.9 Hz), 78.49, 43.78 (br s), 42.82 (br s), 36.17, 35.89, 31.41 (br s), 28.09. (missing aliphatic carbon peak is probably due to overlap at 31.41 ppm.)
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(piperidin-4-ylmethyl)-3-(trifluoromethoxy)phenyl)urea (80).
In a microwave vial was added 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (36 mg, 0.12 mmol, 1 eq), 4-((1-(tert-butoxycarbonyl)piperidin-4-yl)methyl)-3-(trifluoromethoxy)benzoic acid (189) (70 mg, 0.17 mmol, 1.5 eq), toluene (0.8 mL) and triethylamine (35 mg, 0.35 mmol, 3 eq). The suspension was sonicated until fine particles were obtained, then diphenylphosphoryl azide (48 mg, 0.17 mmol, 1.5 eq) was added and the reaction mixture was irradiated at 100°C for 10 min. The reaction mixture was partitioned between EtOAc (20 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (40–100% EtOAc in Hexanes on silica gel column). Solvent was removed in vacuo from fractions containing the desired Boc-protected product. The solid was redissolved in DCM (2 mL) and TFA (0.5 mL) was added. The reaction solution was stirred at rt for 1 h, then concentrated in vacuo and purified by flash chromatography (10–100% MeOH in water with 0.05% TFA on reverse-phase column on prep-HPLC). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 80 (50 mg, 71% yield) as a white solid. 1H NMR (850 MHz, DMSO-d6) δ 9.51 (s, 1H), 8.82 (s, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.20 (t, J = 9.0 Hz, 1H), 7.75 (s, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.37 (dd, J = 11.6, 2.7 Hz, 1H), 7.26 (d, J = 8.3 Hz, 1H), 7.19 (dd, J = 8.3, 2.2 Hz, 1H), 7.10 (dd, J = 8.4, 2.4 Hz, 1H), 6.53 (d, J = 5.2 Hz, 1H), 3.94 (s, 3H), 3.93 (s, 3H), 2.92 (dt, J = 12.4, 3.4 Hz, 2H), 2.47 (d, J = 7.2 Hz, 2H), 2.40 (td, J = 12.2, 2.6 Hz, 2H), 1.60 – 1.53 (m, 1H), 1.48 (d, J = 11.7 Hz, 2H), 1.07 (qd, J = 12.2, 4.0 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −55.68 (s, 3F), −125.21 (t, J = 10.9 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.72, 152.62, 152.57 (d, J = 244.8 Hz), 152.33, 149.41, 148.95, 148.53 (d, J = 10.2 Hz), 147.10, 146.53, 139.04, 131.96, 125.43, 125.01 (d, J = 10.6 Hz), 122.14, 120.29 (q, J = 256.3 Hz), 117.25 (d, J = 2.0 Hz), 116.62, 115.08, 109.54, 109.21 (d, J = 21.9 Hz), 107.89, 103.30, 99.03, 55.79, 55.76, 45.78, 36.89, 36.16, 32.40. HRMS: calcd for C31H31F4N4O5 [M + H]+ m/z, 615.2231; found m/z, 615.2228.
4-((1-(tert-butoxycarbonyl)piperidin-4-ylidene)methyl)-3-(trifluoromethoxy)benzoic acid (190).
To a solution of tert-butyl 4-(4-(methoxycarbonyl)-2-(trifluoromethoxy)benzylidene)piperidine-1-carboxylate (166) (226 mg, 0.54 mmol, 1 eq) in THF (3.4 mL) and water (0.85 mL) was added lithium hydroxide (39 mg, 1.6 mmol, 3 eq). The reaction mixture was stirred at 50°C for 4 h then cooled to rt. THF was removed in vacuo and the solution was acidified to pH 2 using aqueous HCl (1 M). The solid was collected by filtration, washed with water and dried in vacuo to afford 190 (205 mg, 94% yield) as a black solid. 1H NMR (400 MHz, DMSO-d6) δ 7.90 (dd, J = 7.9, 1.6 Hz, 1H), 7.80 (dq, J = 1.6, 1.3 Hz, 1H), 7.48 (d, J = 8.0 Hz, 1H), 6.36 (s, 1H), 3.42 (t, J = 5.8 Hz, 2H), 3.32 (t, J = 5.9 Hz, 2H), 2.37 – 2.29 (m, 2H), 2.25 (t, J = 5.9 Hz, 2H), 1.41 (s, 9H). 19F NMR (376 MHz, DMSO-d6) δ −56.35 (d, J = 1.3 Hz). 13C NMR (214 MHz, DMSO-d6) δ 165.95, 153.78, 145.81, 143.11, 134.51, 131.79 (br s), 131.72, 128.00, 121.43, 120.15 (q, J = 256.9 Hz), 117.09, 78.92, 44.81 (*), 43.78 (*), 35.57 (br s), 29.27 (br s), 28.07. (*not observed on 1D 13C spectrum, peaks were confirmed by (1H-13C)-HSQC. 1D spectrum shows three very broad singlets at this region probably reflecting chemical exchange)
1-(4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluorophenyl)-3-(4-(piperidin-4-ylidenemethyl)-3-(trifluoromethoxy)phenyl)urea (82).
In a microwave vial was added 4-((6,7-dimethoxyquinolin-4-yl)oxy)-2-fluoroaniline (14) (38 mg, 0.12 mmol, 1 eq), 4-((1-(tert-butoxycarbonyl)piperidin-4-ylidene)methyl)-3-(trifluoromethoxy)benzoic acid (190) (68 mg, 0.17 mmol, 1.4 eq), toluene (0.8 mL) and triethylamine (37 mg, 0.36 mmol, 3 eq). The suspension was sonicated until fine particles were obtained, then diphenylphosphoryl azide (47 mg, 0.17 mmol, 1.4 eq) was added and the reaction mixture was irradiated at 100°C for 10 min. The reaction mixture was partitioned between EtOAc (20 mL) and water (15 mL). The aqueous layer was extracted with EtOAc (20 mL x 2). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, concentrated onto Celite, and purified by flash chromatography (50–100% EtOAc in Hexanes on silica gel column). Solvent was removed in vacuo from fractions containing the desired Boc-protected product. The solid was redissolved in DCM (2 mL) and TFA (0.5 mL) was added. The reaction solution was stirred at rt for 1 h, then concentrated in vacuo and purified by flash chromatography (10–100% MeOH in water with 0.05% TFA on reverse-phase column on prep-HPLC). Fractions containing the desired product were concentrated in vacuo, then partitioned between EtOAc (15 mL) and saturated aqueous NaHCO3 solution (15 mL). The aqueous layer was extracted with EtOAc (15 mL x 2). The combined organic extracts were washed with saturated aqueous NaHCO3 solution, water, brine, dried over anhydrous Na2SO4, filtered, and dried under vacuum to afford 82 (51 mg, 69% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.42 (s, 1H), 8.71 (s, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.20 (t, J = 9.1 Hz, 1H), 7.78 – 7.74 (m, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.35 (dd, J = 11.7, 2.7 Hz, 1H), 7.23 (d, J = 1.2 Hz, 2H), 7.14 – 7.07 (m, 1H), 6.54 (d, J = 5.2 Hz, 1H), 6.11 (s, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 2.77 (t, J = 5.5 Hz, 2H), 2.67 (t, J = 5.6 Hz, 2H), 2.23 (t, J = 5.5 Hz, 2H), 2.19 (t, J = 5.4 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −56.06 (s, 3F), −125.32 (t, J = 10.7 Hz, 1F). 13C NMR (214 MHz, DMSO-d6) δ 159.71, 152.62, 152.55 (d, J = 244.8 Hz), 152.24, 149.42, 148.95, 148.58 (d, J = 10.2 Hz), 146.53, 146.19, 143.04, 139.35, 131.63, 124.94 (d, J = 10.8 Hz), 123.98, 122.09 (d, J = 2.7 Hz), 120.29 (q, J = 256.2 Hz), 117.27 (d, J = 3.1 Hz), 116.57, 115.42, 115.08, 110.36, 109.22 (d, J = 21.9 Hz), 107.89, 103.31, 99.03, 55.80, 55.76, 48.28, 47.31, 37.67, 31.09. HRMS: calcd for C31H29F4N4O5 [M + H]+ m/z, 613.2074; found m/z, 613.2068.
Supplementary Material
Ki8751, a type II human VEGFR2 inhibitor, was discovered to be a PfPK6 inhibitor
79 analogues were designed, synthesized, and screened for PfPK6 inhibition
Top analogues were screened for antiplasmodial activity
67 inhibits PfPK6 and is active against P. falciparum asexual blood stage
79 inhibits PfPK6 and is active against Plasmodium liver and asexual blood stages
Acknowledgements
This material is based upon work supported by the National Institutes of Health under Grant No. (1R44AI150237–01) and the National Science Foundation under Grant No. (1644868 and CHE-1726291). We thank the University of North Carolina’s Department of Chemistry Mass Spectrometry Core Laboratory, especially Diane Weatherspoon, for their assistance with mass spectrometry analysis.
We are grateful for support by the Structural Genomics Consortium (SGC), a registered charity (no: 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada through Ontario Genomics Institute [OGI-196], EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking [EUbOPEN grant 875510], Janssen, Merck KGaA (aka EMD in Canada and US), Pfizer and Takeda.
Abbreviations Used
- ABL
tyrosine-protein kinase ABL1
- ACT
artemisinin-based combination therapy
- ATP
adenosine triphosphate
- AURKB
aurora kinase B
- AURKC
aurora kinase C
- AXL
tyrosine-protein kinase receptor UFO
- CDI
1,1’-carbonyl diimidazole
- CDK2
cyclin-dependent kinase 2
- c-KIT
mast/stem cell growth factor receptor Kit
- CID
chemical inducer of dimerization
- DCM
dichloromethane
- DFT
density functional theory
- DHA
dihydroartemisinin
- DIPEA
diisopropylethylamine
- DMA
dimethylacetamide
- DMF
dimethylformamide
- DMF-DMA
N,N-dimethylformamide dimethyl acetal
- DMSO
dimethylsulfoxide
- DPPA
diphenylphosphoryl azide
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- FDA
United States Food and Drug Administration
- FGFR2
fibroblast growth factor receptor 2
- GE
group efficiency
- KCGS
Kinase Chemogenomics Set
- LiHMDS
lithium bis(trimethylsilyl)amide
- MEP
molecular electrostatic potential
- MMV
Medicines for Malaria Venture
- NAO
natural atomic orbital
- NMP
N-methylpyrrolidone
- PDGFRα
platelet-derived growth factor receptor alpha
- PfCDPK1
P. falciparum calcium-dependent protein kinase 1
- PfCDPK5
P. falciparum calcium-dependent protein kinase 5
- PfCLK3
P. falciparum cyclin-dependent-like kinase CLK3
- PfGSK3
P. falciparum glycogen synthase kinase 3
- PfNEK3
P. falciparum NIMA related kinase 3
- PfPI4K
P. falciparum phosphatidylinositol 4-kinase beta
- PfPK5
P. falciparum Protein Kinase 5
- PfPK6
P. falciparum Protein Kinase 6
- PfPKB
P. falciparum RAC-beta serine/threonine protein kinase
- PfPKG
P. falciparum cGMP-dependent protein kinase
- SAR
structure-activity-relationship
- TCAMS
Tres Cantos Antimalarial Set
- TCP
target candidate profile
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- VEGFR2
vascular endothelial growth factor receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary Data
The following supporting data to this article is provided:
Activity of Ki8751 on 97 human kinases determined using a thermal shift assay; compound pairs used for group efficiency analysis; correlation between predicted pKa and %activity remaining on PfPK6; activity of compounds 45, 67, and 79 on 403 non-mutant kinases determined from DiscoverX KINOMEscan®; plot of Pf3D7 EC50 values against PfPK6 IC50 values; HepG2 cytotoxicity data determined using the CellTiter-Glo assay; supplemental methods for PK studies; NMR spectra and HPLC chromatograms (DOCX)
Thermal Shift Assay results on 97 human kinases for Ki8751; pKa prediction of Ki8751 and analogues varying hinge-binder; KINOMEscan® results on 468 kinases for compounds 67, 79, 45 and Ki8751 (XLSX)
Molecular formula strings (CSV)
Data availability
Data will be made available on request.
References
- (1).World Health Organization. World Malaria Report 2021; 2021.
- (2).Phillips MA; Burrows JN; Manyando C; van Huijsduijnen RH; Van Voorhis WC; Wells TNC Malaria. Nat. Rev. Dis. Prim 2017, 3, 17050. [DOI] [PubMed] [Google Scholar]
- (3).Dondorp AM; Nosten F; Yi P; Das D; Phyo AP; Tarning J; Lwin KM; Ariey F; Hanpithakpong W; Lee SJ; Ringwald P; Silamut K; Imwong M; Chotivanich K; Lim P; Herdman T; An SS; Yeung S; Singhasivanon P; Day NPJ; Lindegardh N; Socheat D; White NJ Artemisinin Resistance in Plasmodium Falciparum Malaria. N. Engl. J. Med 2009, 361, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Uwimana A; Legrand E; Stokes BH; Ndikumana J-LM; Warsame M; Umulisa N; Ngamije D; Munyaneza T; Mazarati J-B; Munguti K; Campagne P; Criscuolo A; Ariey F; Murindahabi M; Ringwald P; Fidock DA; Mbituyumuremyi A; Menard D Emergence and Clonal Expansion of in Vitro Artemisinin-Resistant Plasmodium Falciparum Kelch13 R561H Mutant Parasites in Rwanda. Nat. Med 2020, 26, 1602–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Uwimana A; Umulisa N; Venkatesan M; Svigel SS; Zhou Z; Munyaneza T; Habimana RM; Rucogoza A; Moriarty LF; Sandford R; Piercefield E; Goldman I; Ezema B; Talundzic E; Pacheco MA; Escalante AA; Ngamije D; Mangala J-LN; Kabera M; Munguti K; Murindahabi M; Brieger W; Musanabaganwa C; Mutesa L; Udhayakumar V; Mbituyumuremyi A; Halsey ES; Lucchi NW Association of Plasmodium Falciparum Kelch13 R561H Genotypes with Delayed Parasite Clearance in Rwanda: An Open-Label, Single-Arm, Multicentre, Therapeutic Efficacy Study. Lancet Infect. Dis 2021, 21, 1120–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Balikagala B; Fukuda N; Ikeda M; Katuro OT; Tachibana S-I; Yamauchi M; Opio W; Emoto S; Anywar DA; Kimura E; Palacpac NMQ; Odongo-Aginya EI; Ogwang M; Horii T; Mita T Evidence of Artemisinin-Resistant Malaria in Africa. N. Engl. J. Med 2021, 385, 1163–1171. [DOI] [PubMed] [Google Scholar]
- (7).Tse EG; Korsik M; Todd MH The Past, Present and Future of Anti-Malarial Medicines. Malar. J 2019, 18, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Brown JR; Drewry D; Gamo F-J; Garcia-Bustos JF Kinase Inhibitors Among Hits from Malaria Cellular Screens. In Protein Phosphorylation in Parasites; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp 261–291. [Google Scholar]
- (9).Lucet IS; Tobin A; Drewry D; Wilks AF; Doerig C Plasmodium Kinases as Targets for New-Generation Antimalarials. Future Med. Chem 2012, 4, 2295–2310. [DOI] [PubMed] [Google Scholar]
- (10).Arendse LB; Wyllie S; Chibale K; Gilbert IH Plasmodium Kinases as Potential Drug Targets for Malaria: Challenges and Opportunities. ACS Infect. Dis 2021, 7, 518–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ward P; Equinet L; Packer J; Doerig C Protein Kinases of the Human Malaria Parasite Plasmodium Falciparum: The Kinome of a Divergent Eukaryote. BMC Genomics 2004, 5, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Anamika; Srinivasan N; Krupa A A Genomic Perspective of Protein Kinases in Plasmodium Falciparum. Proteins Struct. Funct. Bioinforma 2005, 58, 180–189. [DOI] [PubMed] [Google Scholar]
- (13).Solyakov L; Halbert J; Alam MM; Semblat J-P; Dorin-Semblat D; Reininger L; Bottrill AR; Mistry S; Abdi A; Fennell C; Holland Z; Demarta C; Bouza Y; Sicard A; Nivez M-P; Eschenlauer S; Lama T; Thomas DC; Sharma P; Agarwal S; Kern S; Pradel G; Graciotti M; Tobin AB; Doerig C Global Kinomic and Phospho-Proteomic Analyses of the Human Malaria Parasite Plasmodium Falciparum. Nat. Commun 2011, 2, 565. [DOI] [PubMed] [Google Scholar]
- (14).Zhang M; Wang C; Otto TD; Oberstaller J; Liao X; Adapa SR; Udenze K; Bronner IF; Casandra D; Mayho M; Brown J; Li S; Swanson J; Rayner JC; Jiang RHY; Adams JH Uncovering the Essential Genes of the Human Malaria Parasite Plasmodium Falciparum by Saturation Mutagenesis. Science 2018, 360, eaap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Attwood MM; Fabbro D; Sokolov AV; Knapp S; Schiöth HB Trends in Kinase Drug Discovery: Targets, Indications and Inhibitor Design. Nat. Rev. Drug Discov 2021, 20, 839–861. [DOI] [PubMed] [Google Scholar]
- (16).Paquet T; Le Manach C; Cabrera DG; Younis Y; Henrich PP; Abraham TS; Lee MCS; Basak R; Ghidelli-Disse S; Lafuente-Monasterio MJ; Bantscheff M; Ruecker A; Blagborough AM; Zakutansky SE; Zeeman A-M; White KL; Shackleford DM; Mannila J; Morizzi J; Scheurer C; Angulo-Barturen I; Martínez MS; Ferrer S; Sanz LM; Gamo FJ; Reader J; Botha M; Dechering KJ; Sauerwein RW; Tungtaeng A; Vanachayangkul P; Lim CS; Burrows J; Witty MJ; Marsh KC; Bodenreider C; Rochford R; Solapure SM; Jiménez-Díaz MB; Wittlin S; Charman SA; Donini C; Campo B; Birkholtz L-M; Hanson KK; Drewes G; Kocken CHM; Delves MJ; Leroy D; Fidock DA; Waterson D; Street LJ; Chibale K Antimalarial Efficacy of MMV390048, an Inhibitor of Plasmodium Phosphatidylinositol 4-Kinase. Sci. Transl. Med 2017, 9, eaad9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Baker DA; Stewart LB; Large JM; Bowyer PW; Ansell KH; Jiménez-Díaz MB; El Bakkouri M; Birchall K; Dechering KJ; Bouloc NS; Coombs PJ; Whalley D; Harding DJ; Smiljanic-Hurley E; Wheldon MC; Walker EM; Dessens JT; Lafuente MJ; Sanz LM; Gamo F-J; Ferrer SB; Hui R; Bousema T; Angulo-Barturén I; Merritt AT; Croft SL; Gutteridge WE; Kettleborough CA; Osborne SA A Potent Series Targeting the Malarial CGMP-Dependent Protein Kinase Clears Infection and Blocks Transmission. Nat. Commun 2017, 8, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Vanaerschot M; Murithi JM; Pasaje CFA; Ghidelli-Disse S; Dwomoh L; Bird M; Spottiswoode N; Mittal N; Arendse LB; Owen ES; Wicht KJ; Siciliano G; Bösche M; Yeo T; Kumar TRS; Mok S; Carpenter EF; Giddins MJ; Sanz O; Ottilie S; Alano P; Chibale K; Llinás M; Uhlemann A-C; Delves M; Tobin AB; Doerig C; Winzeler EA; Lee MCS; Niles JC; Fidock DA Inhibition of Resistance-Refractory P. Falciparum Kinase PKG Delivers Prophylactic, Blood Stage, and Transmission-Blocking Antiplasmodial Activity. Cell Chem. Biol 2020, 27, 806–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Matralis AN; Malik A; Penzo M; Moreno I; Almela MJ; Camino I; Crespo B; Saadeddin A; Ghidelli-Disse S; Rueda L; Calderon F; Osborne SA; Drewes G; Böesche M; Fernández-Álvaro E; Martin Hernando JI; Baker DA Development of Chemical Entities Endowed with Potent Fast-Killing Properties against Plasmodium Falciparum Malaria Parasites. J. Med. Chem 2019, 62, 9217–9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Alam MM; Sanchez-Azqueta A; Janha O; Flannery EL; Mahindra A; Mapesa K; Char AB; Sriranganadane D; Brancucci NMB; Antonova-Koch Y; Crouch K; Simwela NV; Millar SB; Akinwale J; Mitcheson D; Solyakov L; Dudek K; Jones C; Zapatero C; Doerig C; Nwakanma DC; Vázquez MJ; Colmenarejo G; Lafuente-Monasterio MJ; Leon ML; Godoi PHC; Elkins JM; Waters AP; Jamieson AG; Álvaro EF; Ranford-Cartwright LC; Marti M; Winzeler EA; Gamo FJ; Tobin AB Validation of the Protein Kinase PfCLK3 as a Multistage Cross-Species Malarial Drug Target. Science 2019, 365, eaau1682. [DOI] [PubMed] [Google Scholar]
- (21).Bracchi-Ricard V; Barik S; DelVecchio C; Doerig C; Chakrabarti R; Chakrabarti D PfPK6, a Novel Cyclin-Dependent Kinase/Mitogen-Activated Protein Kinase-Related Protein Kinase from Plasmodium Falciparum. Biochem. J 2000, 347, 255. [PMC free article] [PubMed] [Google Scholar]
- (22).Crowther GJ; Hillesland HK; Keyloun KR; Reid MC; Lafuente-Monasterio MJ; Ghidelli-Disse S; Leonard SE; He P; Jones JC; Krahn MM; Mo JS; Dasari KS; Fox AMW; Boesche M; El Bakkouri M; Rivas KL; Leroy D; Hui R; Drewes G; Maly DJ; Van Voorhis WC; Ojo KK Biochemical Screening of Five Protein Kinases from Plasmodium Falciparum against 14,000 Cell-Active Compounds. PLoS One 2016, 11, e0149996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Jester BW; Cox KJ; Gaj A; Shomin CD; Porter JR; Ghosh I A Coiled-Coil Enabled Split-Luciferase Three-Hybrid System: Applied Toward Profiling Inhibitors of Protein Kinases. J. Am. Chem. Soc 2010, 132, 11727–11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kubo K; Shimizu T; Ohyama S; Murooka H; Iwai A; Nakamura K; Hasegawa K; Kobayashi Y; Takahashi N; Takahashi K; Kato S; Izawa T; Isoe T Novel Potent Orally Active Selective VEGFR-2 Tyrosine Kinase Inhibitors: Synthesis, Structure−Activity Relationships, and Antitumor Activities of N-Phenyl-N ‘-{4-(4-Quinolyloxy)Phenyl}ureas. J. Med. Chem 2005, 48, 1359–1366. [DOI] [PubMed] [Google Scholar]
- (25).Szabadkai I; Torka R; Garamvölgyi R; Baska F; Gyulavári P; Boros S; Illyés E; Choidas A; Ullrich A; Őrfi L Discovery of N-[4-(Quinolin-4-Yloxy)Phenyl]Benzenesulfonamides as Novel AXL Kinase Inhibitors. J. Med. Chem 2018, 61, 6277–6292. [DOI] [PubMed] [Google Scholar]
- (26).Jester BW; Gaj A; Shomin CD; Cox KJ; Ghosh I Testing the Promiscuity of Commercial Kinase Inhibitors Against the AGC Kinase Group Using a Split-Luciferase Screen. J. Med. Chem 2012, 55, 1526–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ogunleye LO; Jester BW; Riemen AJ; Badran AH; Wang P; Ghosh I When Tight Is Too Tight: Dasatinib and Its Lower Affinity Analogue for Profiling Kinase Inhibitors in a Three-Hybrid Split-Luciferase System. Medchemcomm 2014, 5, 328–332. [Google Scholar]
- (28).Hopkins AL; Keserü GM; Leeson PD; Rees DC; Reynolds CH The Role of Ligand Efficiency Metrics in Drug Discovery. Nat. Rev. Drug Discov 2014, 13, 105–121. [DOI] [PubMed] [Google Scholar]
- (29).Liu Y; Gray NS Rational Design of Inhibitors That Bind to Inactive Kinase Conformations. Nat. Chem. Biol 2006, 2, 358–364. [DOI] [PubMed] [Google Scholar]
- (30).Zhao Z; Wu H; Wang L; Liu Y; Knapp S; Liu Q; Gray NS Exploration of Type II Binding Mode: A Privileged Approach for Kinase Inhibitor Focused Drug Discovery? ACS Chem. Biol 2014, 9, 1230–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).van Linden OPJ; Kooistra AJ; Leurs R; de Esch IJP; de Graaf C KLIFS: A Knowledge-Based Structural Database To Navigate Kinase–Ligand Interaction Space. J. Med. Chem 2014, 57, 249–277. [DOI] [PubMed] [Google Scholar]
- (32).Pierce AC; Sandretto KL; Bemis GW Kinase Inhibitors and the Case for CH-O Hydrogen Bonds in Protein-Ligand Binding. Proteins Struct. Funct. Genet 2002, 49, 567–576. [DOI] [PubMed] [Google Scholar]
- (33).Panigrahi SK Strong and Weak Hydrogen Bonds in Protein-Ligand Complexes of Kinases: A Comparative Study. Amino Acids 2008, 34, 617–633. [DOI] [PubMed] [Google Scholar]
- (34).Williams R pKa Values in Water Compilation https://organicchemistrydata.org/hansreich/resources/pka/pka_data/pka-compilation-williams.pdf (accessed 2022-11-09).
- (35).Wissner A; Berger DM; Boschelli DH; Floyd MB; Greenberger LM; Gruber BC; Johnson BD; Mamuya N; Nilakantan R; Reich MF; Shen R; Tsou H-R; Upeslacis E; Wang YF; Wu B; Ye F; Zhang N 4-Anilino-6,7-Dialkoxyquinoline-3-Carbonitrile Inhibitors of Epidermal Growth Factor Receptor Kinase and Their Bioisosteric Relationship to the 4-Anilino-6,7-Dialkoxyquinazoline Inhibitors. J. Med. Chem 2000, 43, 3244–3256. [DOI] [PubMed] [Google Scholar]
- (36).Picado A; Chaikuad A; Wells CI; Shrestha S; Zuercher WJ; Pickett JE; Kwarcinski FE; Sinha P; de Silva CS; Zutshi R; Liu S; Kannan N; Knapp S; Drewry DH; Willson TM A Chemical Probe for Dark Kinase STK17B Derives Its Potency and High Selectivity through a Unique P-Loop Conformation. J. Med. Chem 2020, 63, 14626–14646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Liu S; Pedersen LG Estimation of Molecular Acidity via Electrostatic Potential at the Nucleus and Valence Natural Atomic Orbitals. J. Phys. Chem. A 2009, 113, 3648–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Cao X; Rong C; Zhong A; Lu T; Liu S Molecular Acidity: An Accurate Description with Information-Theoretic Approach in Density Functional Reactivity Theory. J. Comput. Chem 2018, 39, 117–129. [DOI] [PubMed] [Google Scholar]
- (39).Huang Y; Liu L; Liu S Towards Understanding Proton Affinity and Gas-Phase Basicity with Density Functional Reactivity Theory. Chem. Phys. Lett 2012, 527, 73–78. [Google Scholar]
- (40).Wells CI; Al-Ali H; Andrews DM; Asquith CRM; Axtman AD; Dikic I; Ebner D; Ettmayer P; Fischer C; Frederiksen M; Futrell RE; Gray NS; Hatch SB; Knapp S; Lücking U; Michaelides M; Mills CE; Müller S; Owen D; Picado A; Saikatendu KS; Schröder M; Stolz A; Tellechea M; Turunen BJ; Vilar S; Wang J; Zuercher WJ; Willson TM; Drewry DH The Kinase Chemogenomic Set (KCGS): An Open Science Resource for Kinase Vulnerability Identification. Int. J. Mol. Sci 2021, 22, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Tan L; Nomanbhoy T; Gurbani D; Patricelli M; Hunter J; Geng J; Herhaus L; Zhang J; Pauls E; Ham Y; Choi HG; Xie T; Deng X; Buhrlage SJ; Sim T; Cohen P; Sapkota G; Westover KD; Gray NS Discovery of Type II Inhibitors of TGFβ-Activated Kinase 1 (TAK1) and Mitogen-Activated Protein Kinase Kinase Kinase Kinase 2 (MAP4K2). J. Med. Chem 2015, 58, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Weir MC; Shu ST; Patel RK; Hellwig S; Chen L; Tan L; Gray NS; Smithgall TE Selective Inhibition of the Myeloid Src-Family Kinase Fgr Potently Suppresses AML Cell Growth in Vitro and in Vivo. ACS Chem. Biol 2018, 13, 1551–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Smilkstein M; Sriwilaijaroen N; Kelly JX; Wilairat P; Riscoe M Simple and Inexpensive Fluorescence-Based Technique for High-Throughput Antimalarial Drug Screening. Antimicrob. Agents Chemother 2004, 48, 1803–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lu K-Y; Pasaje CFA; Srivastava T; Loiselle DR; Niles JC; Derbyshire E Phosphatidylinositol 3-Phosphate and Hsp70 Protect Plasmodium Falciparum from Heat-Induced Cell Death. Elife 2020, 9, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Duffy S; Avery VM Plasmodium Falciparum in Vitro Continuous Culture Conditions: A Comparison of Parasite Susceptibility and Tolerance to Anti-Malarial Drugs throughout the Asexual Intra-Erythrocytic Life Cycle. Int. J. Parasitol. Drugs Drug Resist 2017, 7, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Forte B; Ottilie S; Plater A; Campo B; Dechering KJ; Gamo FJ; Goldberg DE; Istvan ES; Lee M; Lukens AK; McNamara CW; Niles JC; Okombo J; Pasaje CFA; Siegel MG; Wirth D; Wyllie S; Fidock DA; Baragaña B; Winzeler EA; Gilbert IH Prioritization of Molecular Targets for Antimalarial Drug Discovery. ACS Infect. Dis 2021, 7, 2764–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Gamo F-J; Sanz LM; Vidal J; de Cozar C; Alvarez E; Lavandera J-L; Vanderwall DE; Green DVS; Kumar V; Hasan S; Brown JR; Peishoff CE; Cardon LR; Garcia-Bustos JF Thousands of Chemical Starting Points for Antimalarial Lead Identification. Nature 2010, 465, 305–310. [DOI] [PubMed] [Google Scholar]
- (48).Kubo K; Ohyama S; Shimizu T; Nishitoba T; Kato S; Murooka H; Kobayashi Y Quinoline Derivatives and Quinazoline Derivatives Inhibiting Autophosphorylation of Growth Factor Receptor Originating in Platelet and Phamaceutical Compositions Containing the Same WO9717329A1, 1997.
- (49).Burrows JN; Duparc S; Gutteridge WE; Hooft van Huijsduijnen R; Kaszubska W; Macintyre F; Mazzuri S; Möhrle JJ; Wells, T. N. C. New Developments in Anti-Malarial Target Candidate and Product Profiles. Malar. J 2017, 16, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Derbyshire ER; Prudencio M; Mota MM; Clardy J Liver-Stage Malaria Parasites Vulnerable to Diverse Chemical Scaffolds. Proc. Natl. Acad. Sci 2012, 109, 8511–8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Egan TJ; Hunter R; Kaschula CH; Marques HM; Misplon A; Walden J Structure−Function Relationships in Aminoquinolines: Effect of Amino and Chloro Groups on Quinoline−Hematin Complex Formation, Inhibition of β-Hematin Formation, and Antiplasmodial Activity. J. Med. Chem 2000, 43, 283–291. [DOI] [PubMed] [Google Scholar]
- (52).Natarajan JK; Alumasa JN; Yearick K; Ekoue-Kovi KA; Casabianca LB; de Dios AC; Wolf C; Roepe PD 4- N -, 4- S -, and 4- O -Chloroquine Analogues: Influence of Side Chain Length and Quinolyl Nitrogen p K a on Activity vs Chloroquine Resistant Malaria. J. Med. Chem 2008, 51, 3466–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kaur K; Jain M; Reddy RP; Jain R Quinolines and Structurally Related Heterocycles as Antimalarials. Eur. J. Med. Chem 2010, 45, 3245–3264. [DOI] [PubMed] [Google Scholar]
- (54).Sullivan DJ; Gluzman IY; Russell DG; Goldberg DE On the Molecular Mechanism of Chloroquine’s Antimalarial Action. Proc. Natl. Acad. Sci 1996, 93, 11865–11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lee S; Jørgensen M; Hartwig JF Palladium-Catalyzed Synthesis of Arylamines from Aryl Halides and Lithium Bis(Trimethylsilyl)Amide as an Ammonia Equivalent. Org. Lett 2001, 3, 2729–2732. [DOI] [PubMed] [Google Scholar]
- (56).Kulkarni AR; Garai S; Thakur GA Scalable, One-Pot, Microwave-Accelerated Tandem Synthesis of Unsymmetrical Urea Derivatives. J. Org. Chem 2017, 82, 992–999. [DOI] [PubMed] [Google Scholar]
- (57).Krämer A; Kurz CG; Berger B-T; Celik IE; Tjaden A; Greco FA; Knapp S; Hanke T Optimization of Pyrazolo[1,5-a]Pyrimidines Lead to the Identification of a Highly Selective Casein Kinase 2 Inhibitor. Eur. J. Med. Chem 2020, 208, 112770. [DOI] [PubMed] [Google Scholar]
- (58).Fedorov O; Niesen FH; Knapp S Kinase Inhibitor Selectivity Profiling Using Differential Scanning Fluorimetry In Kinase Inhibitors; 2012; pp 109–118. [DOI] [PubMed] [Google Scholar]
- (59).Liu S; Schauer CK; Pedersen LG Molecular Acidity: A Quantitative Conceptual Density Functional Theory Description. J. Chem. Phys 2009, 131, 164107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Huang Y; Liu L; Liu W; Liu S; Liu S Modeling Molecular Acidity with Electronic Properties and Hammett Constants for Substituted Benzoic Acids. J. Phys. Chem. A 2011, 115, 14697–14707. [DOI] [PubMed] [Google Scholar]
- (61).Xiao X; Cao X; Zhao D; Rong C; Liu S Quantification of Molecular Basicity for Amines: A Combined Conceptual Density Functional Theory and Information-Theoretic Approach Study. Acta Physico-Chimica Sin 2020, 36, 1906030–1906034. [Google Scholar]
- (62).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA,J; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 16, Revision C.01 Gaussian, Inc., Wallingford CT: 2019. [Google Scholar]
- (63).Becke AD Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [DOI] [PubMed] [Google Scholar]
- (64).Lee C; Yang W; Parr RG Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- (65).Ditchfield R; Hehre WJ; Pople JA Self‐Consistent Molecular‐Orbital Methods. IX. An Extended Gaussian‐Type Basis for Molecular‐Orbital Studies of Organic Molecules. J. Chem. Phys 1971, 54, 724–728. [Google Scholar]
- (66).Glendening ED; Landis CR; Weinhold F Natural Bond Orbital Methods. WIREs Comput. Mol. Sci 2012, 2, 1–42. [Google Scholar]
- (67).Williams R pKa Values https://organicchemistrydata.org/hansreich/resources/pka/pka_data/pka-compilation-williams.pdf (accessed 2022-05-09).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.