

Abstract
The most common form of genetic heart disease is hypertrophic cardiomyopathy (HCM), which is caused by variants in cardiac sarcomeric genes and leads to abnormal heart muscle thickening. Complications of HCM include heart failure, arrhythmia and sudden cardiac death. The dominant-negative c.1208G>A (p.R403Q) pathogenic variant (PV) in β-myosin (MYH7) is a common and well-studied PV that leads to increased cardiac contractility and HCM onset. In this study we identify an adenine base editor and single-guide RNA system that can efficiently correct this human PV with minimal bystander editing and off-target editing at selected sites. We show that delivery of base editing components rescues pathological manifestations of HCM in induced pluripotent stem cell cardiomyocytes derived from patients with HCM and in a humanized mouse model of HCM. Our findings demonstrate the potential of base editing to treat inherited cardiac diseases and prompt the further development of adenine base editor-based therapies to correct monogenic variants causing cardiac disease.
Hypertrophic cardiomyopathy (HCM), a disease of abnormal heart muscle thickening, is the most common form of genetic heart disease in the USA, affecting upwards of 1 in 200 people1,2. Clinical complications of HCM include heart failure, arrhythmia and sudden cardiac death. There is no cure for HCM, aside from heart transplant, which presents its own complications and requires lifetime immunosuppression. Although HCM-causing variants are found in various sarcomeric protein-encoding genes, over one-third of all HCM-causing variants occur in the myosin heavy chain 7 (MYH7) gene, which encodes β-myosin heavy chain, a motor ATPase that incorporates into the thick filament of cardiac muscle and plays a major role in cardiac contraction. These pathogenic variants (PVs) are generally autosomal dominant missense variants, which allow the incorporation of pathogenic myosin heads into cardiac sarcomeres and lead to increased energy consumption, hypercontractility and disease progression of HCM3.
The heterozygous MYH7 c.1208G>A (p.R403Q) pathogenic missense variant causes severe HCM with early-onset and progressive myocardial dysfunction and has a high incidence of early sudden cardiac death, with 50% of patients dying by 40 years of age4,5. It was the first MYH7 variant linked to HCM, leading to the discovery of multiple other missense variants in cardiac sarcomere genes and generating numerous studies of its function. The R403Q missense variant, located on the myosin mesa of the myosin head, results in a loss of positive charge on the myosin head, weakening its interaction with myosin-binding protein C, a molecular brake, and leading to an increase in the number of functionally accessible myosin heads available for contraction that pathologically augments sarcomere contractility6. Incorporation of mutant sarcomeric peptides into myofibrils results in a dominant-negative disease that requires direct correction or ablation of the pathogenic allele.
Base editing has emerged as an attractive method to correct and potentially cure genetically based diseases, especially single nucleotide variants. Base editors are fusion proteins of a deaminase protein and either Cas9 nickase or deactivated Cas9, which allow base-pair edits without double-strand breaks within a defined editing window in relation to the protospacer adjacent motif (PAM) site of a single-guide RNA (sgRNA)7,8. Adenine base editors (ABEs) use deoxyadenosine deaminase to convert DNA A•T base pairs to G•C base pairs via an inosine intermediate and have been previously shown to function in many postmitotic cells in vivo and in vitro9–11.
In this study, we and colleagues who authored the accompanying manuscript (Reichart, Newby, Wakimoto et al.)12 report the use of different gene editing strategies to correct the MYH7 c.1208G>A (p.R403Q) pathogenic missense variant. We develop an ABE-mediated strategy that efficiently corrects and rescues pathological phenotypes of HCM in patient-derived cells. Furthermore, we generate a humanized mouse model containing the pathogenic missense variant with sequence complementarity to the human sgRNA and show that postnatal ABE correction of this mouse model prevents HCM onset.
Results
ABE editing of the R403Q variant in human iPSCs
To screen various ABEs for their efficiencies, we first inserted the MYH7 c.1208G>A (p.R403Q) pathogenic missense variant using CRISPR–Cas9-based homology-directed repair (HDR) into a human induced pluripotent stem cell (iPSC) line derived from a healthy donor (HDWT). We isolated an isogenic heterozygous clone (HD403/+) that mirrors the heterozygous genotype found in patients4 and an isogenic homozygous clone (HD403/403) that has not been previously described in patients. Sequencing confirmed no mutations on the highly homologous MYH6 gene during generation of these clones and these iPSCs readily differentiated into cardiomyocytes (CMs) (Extended Data Fig. 1).
Because ABEs have an optimal activity window in protospacer positions 14–17 (counting the first nucleotide immediately 5′ of the PAM sequence as protospacer position 1), we chose an sgRNA with an NGA PAM that places the MYH7 c.1208G>A PV in protospacer position 16 (h403_sgRNA) (Fig. 1a). To identify an optimal ABE capable of efficiently correcting the pathogenic nucleotide back to the wildtype (WT) nucleotide without introducing any bystander edits, we tested various engineered deaminases, including ABEmax13, which is an optimized, narrow-windowed ABE7.10 variant, and ABE8e14, which is a highly processive, wide-windowed, evolved ABE7.10 variant. Each engineered deaminase variant was fused to engineered SpCas9 variants including SpRY, which targets NRN PAMs15; SpG, which targets NGN PAMs15; SpCas9-NG, which targets NG PAMs16; and SpCas9-VRQR, which targets NGA PAMs17. We then screened these ABEs for their efficiency of correction in our HD403/403 iPSC line via transient transfection with h403_sgRNA (Fig. 1b). We selected the HD403/403 iPSC line for screening to ensure that all G nucleotide Sanger sequencing reads at position c.1208 are due to ABE editing. Similar editing efficiency of the pathogenic adenine was achieved with all ABEmax-SpCas9 variants tested, ranging from 26 ± 2.3% with ABEmax-SpRY to 34 ± 2.5% with ABEmax-VRQR, with minimal bystander editing of neighboring adenines (the average across three bystanders was 2.6 ± 1.7%). ABE8e-SpCas9 variants achieved higher editing efficiencies, ranging from 27 ± 2.6% with ABE8e-SpRY to 37 ± 1.5% with ABE8e-SpG, with slightly increased bystander editing of neighboring adenines (the average across three bystanders was 4.0 ± 2.0%) (Fig. 1c). These bystander edits are predicted to result in K405E, K405R or K405G variants in β-myosin heavy chain, depending on the combination of edits, which may be deleterious due to the high intolerance of MYH7 for missense variants18, although the consequences of these variants on β-myosin heavy chain function have not been described. For subsequent experiments, we opted to use the more narrow-windowed ABEmax to reduce potential bystander edits and the SpCas9-VRQR variant with its more stringent PAM requirements to reduce potential Cas-dependent off-target editing.
Fig. 1 |. In vitro optimization of the ABE system to correct a pathogenic MYH7 variant.
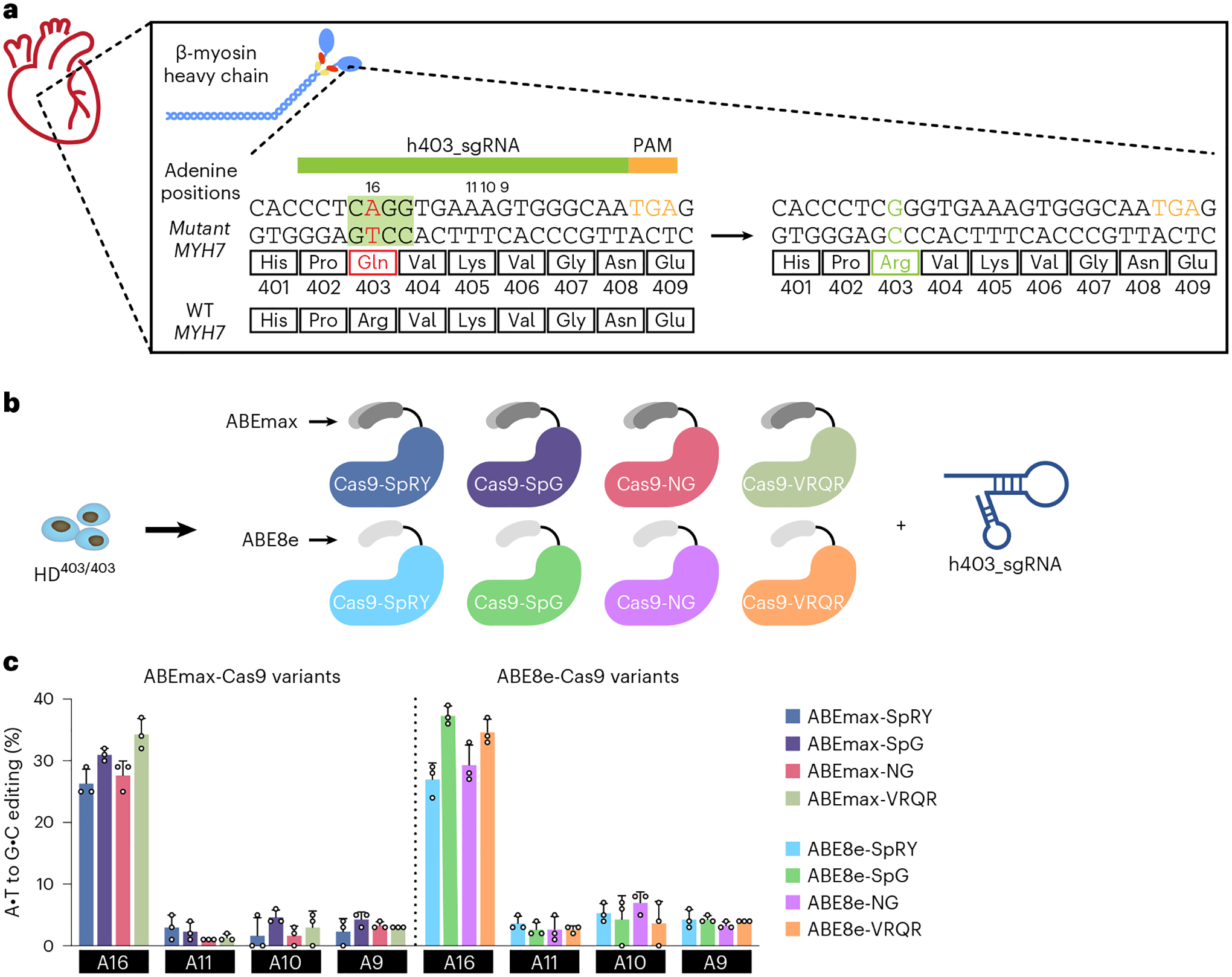
a, A candidate sgRNA, h403_sgRNA, for base editing to correct the MYH7 c.1208G>A (p.R403Q) missense variant. Base editing could convert the neutrally charged glutamine PV back to a positively charged arginine, restoring proper function of the myosin head. The nucleotide sequence highlighted in green corresponds to the adenine base editing window in positions 14–17. b, Eight candidate base editor variants were screened for their efficiencies in correcting the pathogenic adenine to a guanine using the candidate h403_sgRNA within a homozygous MYH7 c.1208G>A iPSC line (HD403/403). c, DNA editing efficiency of all adenines within the target protospacer in HD403/403 iPSCs 72 h posttransfection with candidate base editors. Data are mean ± s.d. across three technical replicates. Numbering is with the first base 5′ of the PAM as 1; target pathogenic adenine is position A16.
ABE correction efficiency in iPSCs derived from patients with HCM
To apply our ABEmax-VRQR and h403_sgRNA system to a disease model, we derived human iPSCs from two patients with HCM and the MYH7403/+ PV (HCM1403/+ and HCM2403/+). We corrected the MYH7403/+ variant via plasmid nucleofection of ABEmax-VRQR-P2a-EGFP and h403_sgRNA and fluorescence-activated cell sorting of GFP+ cells (Fig. 2a). By high-throughput sequencing (HTS), despite 98–99% on-target editing, we observed minimal to no off-target DNA editing (0.12% or less) at all 58 adenine bases for eight tested candidate off-target loci, which were identified using the bioinformatic tool CRISPOR19 (Fig. 2b and Extended Data Fig. 2). A low frequency (0.03–0.48%) of bystander editing was observed at the three bystander adenines for amino acid 505 (K505) of β-myosin. We isolated corrected clonal lines of the iPSCs derived from patients with HCM (HCM1WT and HCM2WT) containing no bystander edits or editing of the highly homologous MYH6 gene, ensuring that subsequent characterizations are due to correction of the pathogenic nucleotide. These results suggest that h403_sgRNA with ABEmax-VRQR can efficiently and specifically correct the target pathogenic missense variant with minimal bystander editing and little to no DNA editing at tested off-target sites.
Fig. 2 |. Analyses of the function of iPSC-CM derived from patients with HCM on base editing correction.
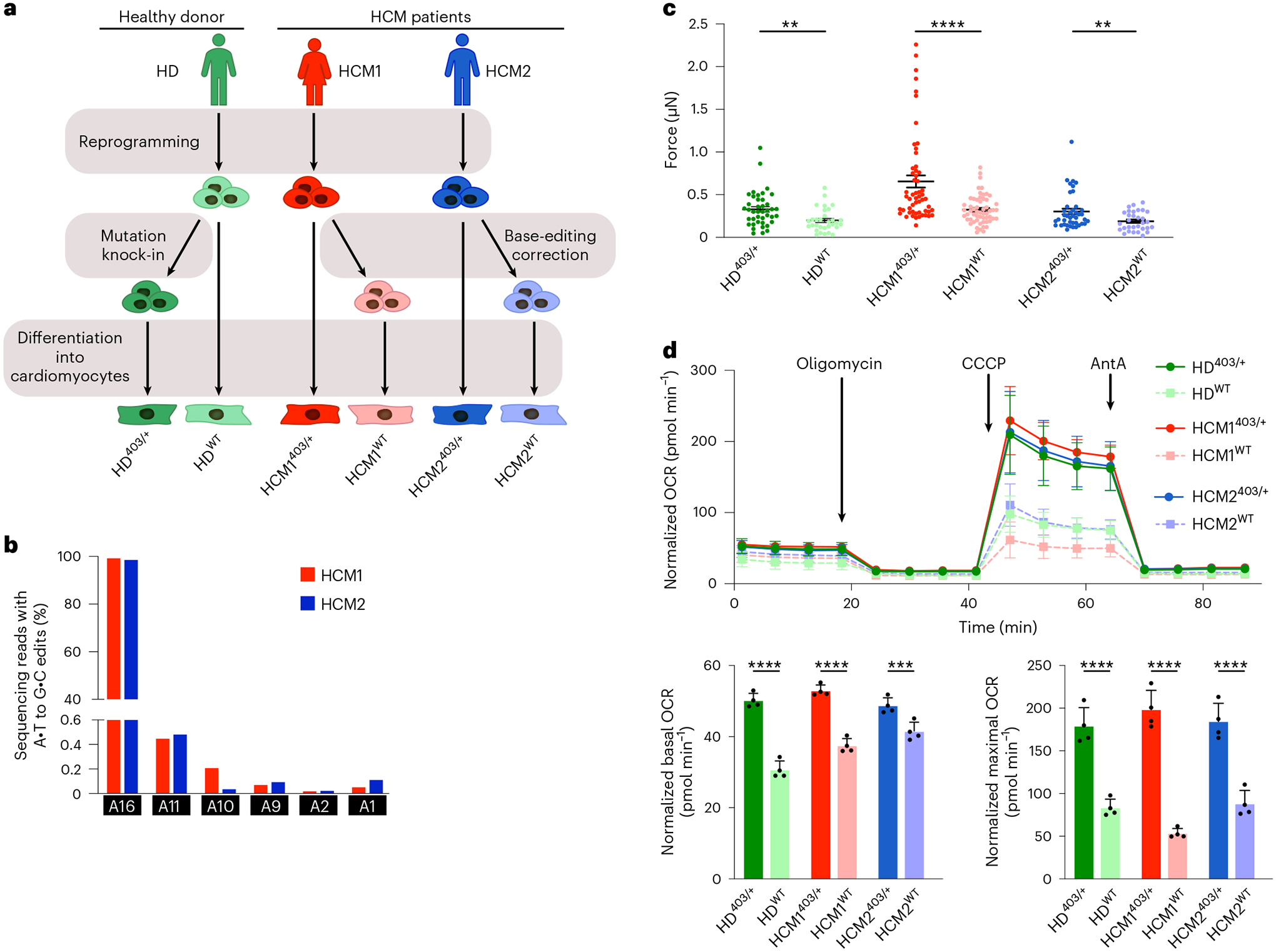
a, Workflow for reprogramming iPSCs from a healthy donor (HD) and two patients with HCM (HCM1 and HCM2), followed by PV knock-in for the HD line and base editing correction for the HCM1 and HCM2 lines. Isogenic clonal lines were then isolated and differentiated into CMs for downstream analyses of iPSC-CM function. b, HTS to measure editing for all adenines within the on-target protospacer. Target pathogenic adenine is A16. HTS was performed for ABE-treated HCM1403/+ and HCM2403/+ iPSCs. c, Quantification of peak systolic force of iPSC-CMs for indicated cell lines. (n = 41 for HD403/+; 31 for HDWT; 55 for HCM1403/+; 57 for HCM1WT; 41 for HCM2403/+; and 31 for HCM2WT). Data are mean ± s.e.m. across three separate differentiations. HD403/+ iPSC-CMs had a 1.7-fold increase in peak systolic force compared with HDWT iPSC-CMs (P = 0.0029); HCM1WT a 2.0-fold decrease compared with HCM1403/+ (P = 0.000013); and HCM2WT a 1.6-fold decrease compared with HCM1403/+ (P = 0.0085). d, Oxygen consumption rate (OCR) as a function of time in indicated cell lines after exposure to the electron transport chain complex inhibitors oligomycin, carbonyl cyanide m-chlorophenyl hydrazone (CCCP) and antimycin A (AntA) (top), and mean and distribution of values across four timepoints for basal OCR (bottom left) and maximal OCR (bottom right) for indicated cell lines. HD403/+ iPSC-CMs had a 1.6-fold increase in basal OCR compared with HDWT iPSC-CMs (P = 0.000018); HCM1WT a 1.4-fold decrease compared with HCM1403/+ (P = 0.000019); and HCM2WT a 1.2-fold decrease compared with HCM1403/+ (P = 0.0056). For maximum OCR, HD403/+ iPSC-CMs had a 2.1-fold increase compared with HDWT iPSC-CMs (P = 0.00020); HCM1WT a 3.7-fold decrease compared with HCM1403/+ (P = 0.000016); and HCM2WT a 2.1-fold decrease compared with HCM1403/+ (P = 0.00034). Data are mean ± s.d. across three separate differentiations. Each data point is from 14–16 Seahorse assay wells. **P < 0.01, ***P < 0.001, ****P < 0.0001 by Student’s unpaired two-sided t-test.
Functional analyses of ABE-edited patient-derived iPSC-CMs
In individual CMs, the MYH7 p.R403Q PV increases the number of functional myosin heads available for contraction, which leads to increased force generation and greater ATP consumption. Increases in both force generation and energy consumption have been previously shown in isogenic iPSC-CMs engineered to have the R403Q PV from a WT line20–22. To determine if these previously validated changes are rescued in our corrected iPSC-CMs derived from patients with HCM, we differentiated both MYH7403/+ pathogenic and MYH7WT healthy clonal lines for all three patient-derived lines (HD, HCM1 and HCM2) into CMs (Fig. 2a).
To investigate whether gene editing correction could reduce hypercontractile force generation in our lines derived from patients with HCM, we plated iPSC-CMs at a single-cell density on soft polydimethylsiloxane surfaces, recorded high frame-rate videos of contracting CMs and calculated the peak systolic force. The HD403/+ iPSC-CMs showed a 1.7-fold increase in peak systolic force compared with HDWT iPSC-CMs originally derived from an HD, consistent with reports of a single MYH7 p.R403Q variant being sufficient to lead to hypercontractility6,22. In contrast, corrected HCM1WT and HCM2WT CMs showed a 2.0-fold and 1.6-fold decrease in peak systolic force, respectively, compared with their isogenic HCM1403/+ and HCM2403/+ counterparts (Fig. 2c).
Because previous studies have shown the p.R403Q HCM variant leads to increased ATP consumption and altered cellular metabolism23, we next assessed changes in cellular energetics via metabolic flux assays after gene editing correction. Basal OCRs were increased 1.6-fold in HD403/+ iPSC-CMs compared with HDWT iPSC-CMs, and HD403/+ iPSC-CMs had a 2.1-fold increase in maximum OCR compared with HDWT iPSC-CMs. Corrected HCM1WT and HCM2WT CMs showed a 1.4-fold and 1.2-fold reduction in basal OCR, respectively, and a 3.7-fold and 2.1-fold reduction in maximum OCR, respectively, compared with isogenic HCM1403/+ and HCM2403/+ CMs (Fig. 2d). These data demonstrate that correction of the PV in human HCM CMs is sufficient to rescue the hypercontractility phenotype and restore normal cellular energetics.
Development of a humanized mouse model of HCM biology
We next sought to apply base editing to a mouse model of HCM. Although β-myosin heavy chain is the dominant myosin isoform found in adult human hearts, the highly homologous α-myosin heavy chain is the dominant myosin isoform expressed in adult mouse hearts and is encoded by the Myh6 gene24. Consequently, previously described mouse models for HCM have placed the corresponding human MYH7 variant on the mouse Myh6 gene to account for these expression differences. Although the 35 amino acids around R403 are 100% identical between human MYH7 and mouse Myh6, the DNA sequence encoding this region of the protein is not identical (Extended Data Fig. 3). Thus, sgRNAs and editing strategies developed for the human genome would not be directly applicable to a mouse model.
To perform preclinical studies using our human sequence-specific base editing strategy, we generated a humanized mouse model containing the MYH7 c.1208G>A (p.R403Q) human missense variant within the mouse Myh6 gene that also has a human DNA sequence identity of at least 21 nucleotides upstream and downstream from the PV to allow testing of human genome specific CRISPR strategies (Fig. 3a). The other Myh6 allele contained the unmodified mouse genomic sequence. This humanized mouse model (Myh6h403/+) mirrors the phenotype of previously described Myh6 p.R403Q mouse models25,26. Most notably, homozygous mice (Myh6h403/h403) have enlarged atria, extensive interstitial fibrosis and die within the first week of life (Fig. 3b). At 9 months old, Myh6h403/+ mice have developed cardiomyopathy with substantial ventricular hypertrophy, myocyte disarray and fibrosis (Fig. 3c).
Fig. 3 |. Generation of a humanized HCM mouse model.
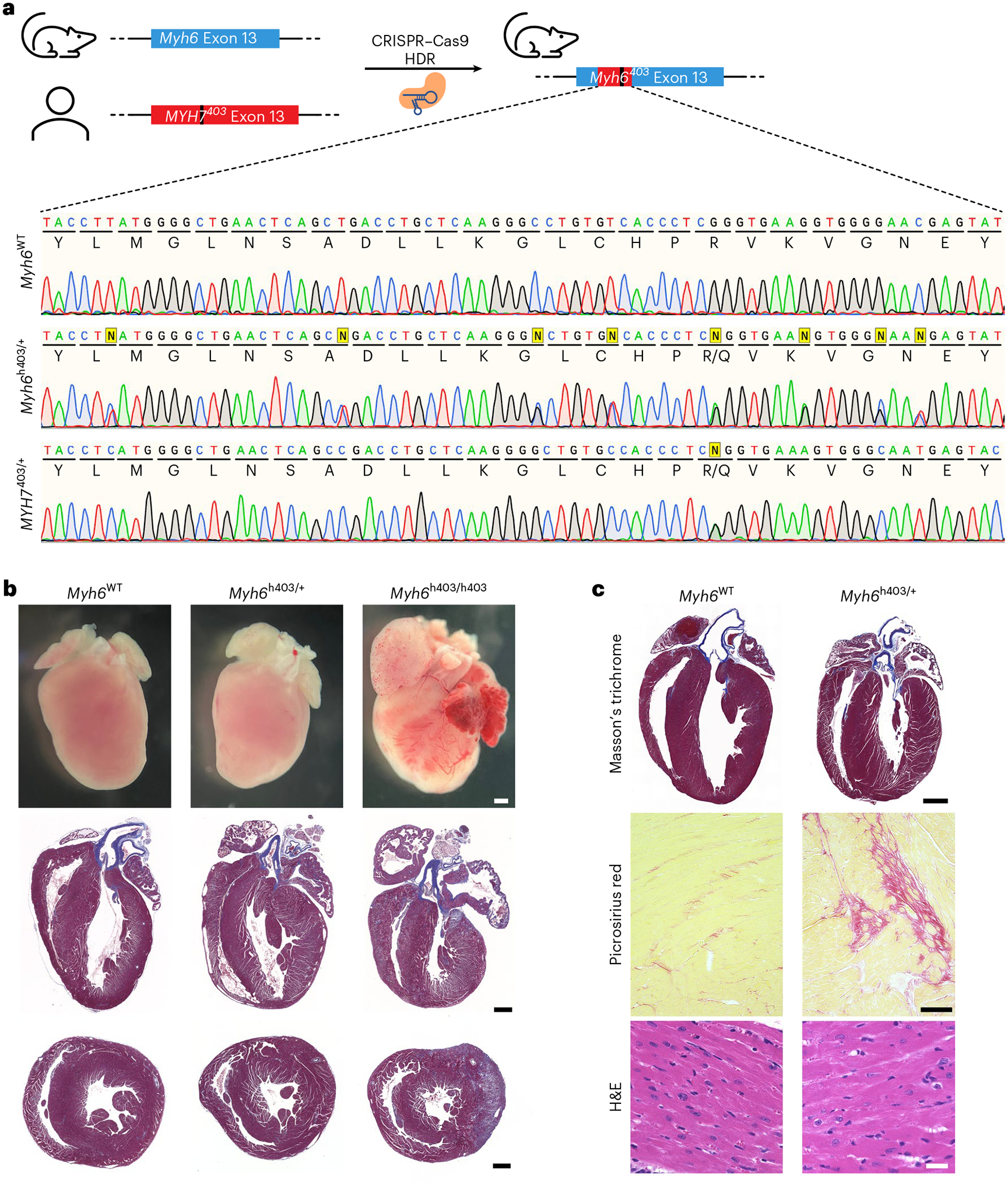
a, A humanized HCM mouse model was generated by replacing part of the native murine Myh6 genomic sequence with the human MYH7 sequence containing the p.R403Q variant via CRISPR–Cas9 HDR. Sanger sequencing chromatograms show the native Myh6WT sequence (top), the humanized Myh6h403/+ mouse model sequence (middle) and the human MYH7 sequence from an HCM patient (bottom). Yellow squares indicate knocked-in single nucleotide polymorphisms (SNPs); Sanger sequencing shows double nucleotide peaks for SNPs. b, Gross histology (top), and Masson’s trichrome staining of coronal (four chamber) (middle) and transverse (bottom) sections of the humanized mouse model for the WT (left), heterozygous (middle) and homozygous (right) genotypes at postnatal day 8. Scale bar, 1 mm. c, Representative Masson’s trichrome, Picrosirius red, and hematoxylin and eosin (H&E) staining of heart sections of the humanized mouse model for the WT (left) and heterozygous (right) genotypes at 9 months of age. Scale bar, 1 mm for ×10 images top, 100 μm for ×10 images middle, 25 μm for ×40 images bottom. Similar results were found in at least three different male mice for each genotype.
In vivo ABE treatment of a mouse model of human HCM
We packaged ABEmax-VRQR and h403_sgRNA within adeno-associated virus (AAV), a US Food and Drug Administration-approved viral delivery method. We chose to use the AAV9 capsid due to its high cardiac transduction and use in clinical trials27. As the full-length base editor (approximately 5.4 kb) exceeds the packaging limit of a single AAV9 (approximately 4.7 kb), we split the base editor across two AAV9s and used trans-splicing inteins to reconstitute the full-length base editor in cells upon protein expression28. Because AAV9 contains broad tissue tropism, we utilized a cardiac troponin T promoter to limit expression of the base editor to CMs29. For this dual AAV9 system, each AAV9 also contained a single copy of an expression cassette encoding h403_sgRNA (Fig. 4a).
Fig. 4 |. Prevention of HCM by dual AAV9 ABE editing of Myh6h403/+ mice.
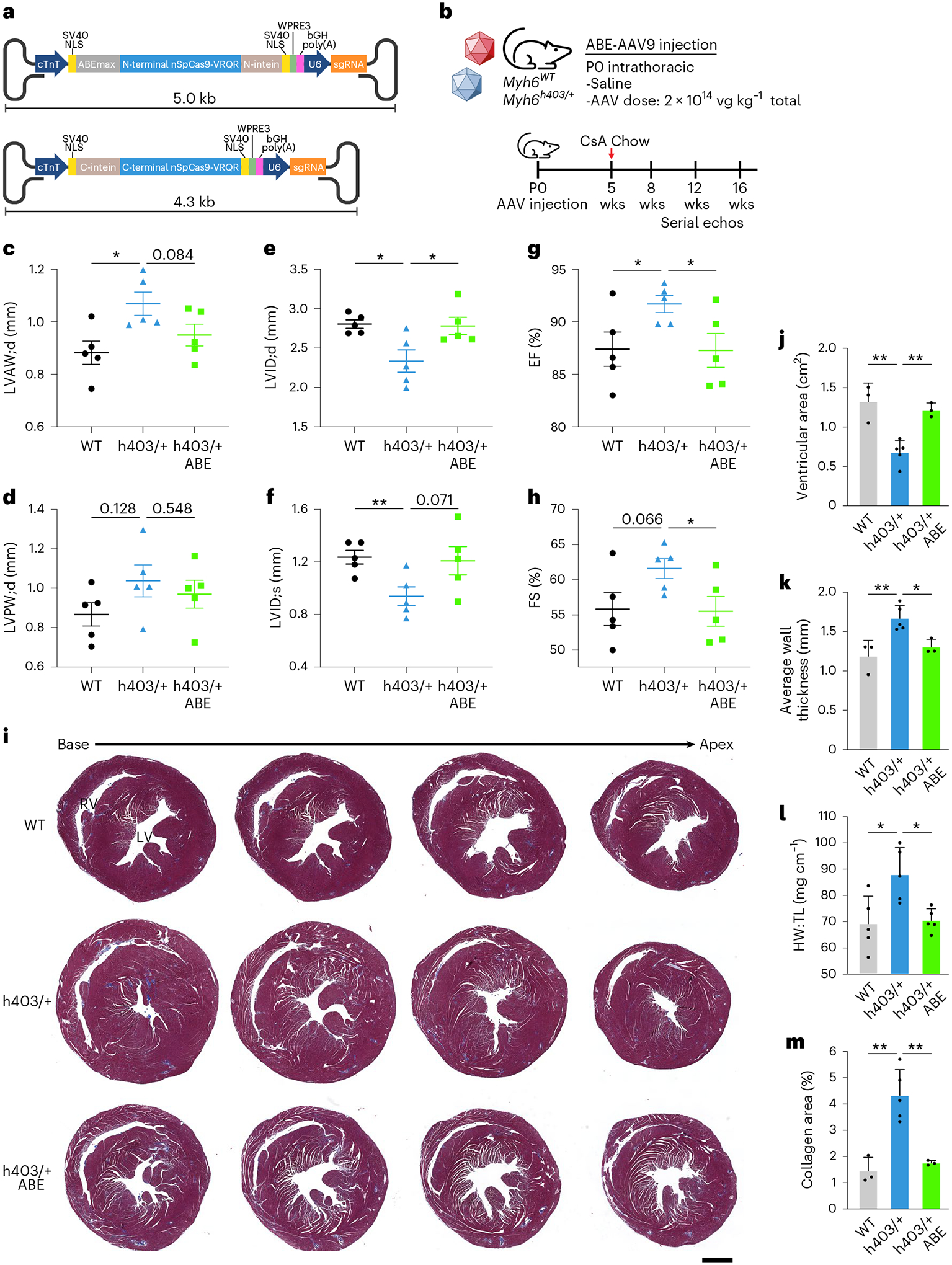
a, Schematic of dual AAV9 ABE system encoding ABEmax-VRQR base editor halves under the cardiac troponin T promoter (cTnT) and h403_sgRNA to target the human MYH7 p.R403Q variant. b, Experimental outline for intrathoracic injection of Myh6WT or Myh6h403/+ mice with saline or dual AAV9 ABE at P0 followed by serial echocardiograms. Chow diet supplemented with 0.1% cyclosporine A (CsA) was given at 5 weeks of age for 11 weeks. c–h, Left ventricular anterior wall thickness at diastole (LVAW;d) (c), left ventricular posterior wall thickness at diastole (LVPW;d) (d), left ventricular internal diameter at diastole (LVID;d) (e) and systole (LVID;s) (f), ejection fraction (EF) (g) and fractional shortening (FS) (h), of Myh6WT mice, Myh6h403/+ mice and ABE-treated Myh6h403/+ mice at 16 weeks of age. n = 5 male mice for each group. Data are mean ± s.e.m. Exact P values can be found in Table 1. i, Representative Masson’s trichrome staining of serial (500-μm interval) transverse sections for Myh6WT mice, Myh6h403/+ mice or ABE-treated Myh6h403/+ mice at 16 weeks of age. Scale bar, 1 mm. j, Ventricular cross-sectional area measurements from n = 3 for Myh6WT male mice; 5, Myh6h403/+ male mice; 3, ABE-treated Myh6h403/+ male mice. Myh6h403/+ mice had significantly decreased ventricular cross-sectional area (0.679 cm2) compared with Myh6WT mice (1.32 cm2, P = 0.0032) and ABE-treated Myh6h403/+ mice (1.22 cm2, P = 0.0016). Data are mean ± s.d. k, Average wall thickness measurements from n = 3 for Myh6WT male mice; 5, Myh6h403/+ male mice; 3, ABE-treated Myh6h403/+ male mice. Data are mean ± s.d. Myh6h403/+ mice had significantly increased average wall thickness (1.67 mm) compared with Myh6WT mice (1.19 mm, P = 0.0094) and ABE-treated Myh6h403/+ mice (1.31 cm2, P = 0.013). l, Heart weight (HW) to tibia length (TL) measurements from n = 5 male mice for each experimental group. Data are mean ± s.d. Myh6h403/+ mice had significantly increased HW and TL (87.95 mg cm−1) compared with Myh6WT mice (69.23 mg cm−1, P = 0.022) and ABE-treated Myh6h403/+ mice (70.48 mg cm−1, P = 0.0082). m, Percentage of collagen area from n = 3 for Myh6WT male mice; 5, Myh6h403/+ male mice; 3, ABE-treated Myh6h403/+ male mice. Myh6h403/+ mice had significantly increased collagen area (4.33%) compared with Myh6WT mice (1.45%, P = 0.022) and ABE-treated Myh6h403/+ mice (1.75%, P = 0.0047). Data are mean ± s.d. *P < 0.05, **P < 0.01 by Student’s unpaired two-sided t-test. LV, left ventricle; RV, right ventricle.
We first sought to validate the efficiency of our dual AAV9 ABE system by trying to rescue Myh6h403/h403 mice, which die within the first week of life. Notably, no human patients have been reported to have the homozygous genotype. We injected P0 (postnatal day 0) Myh6h403/h403 pups intrathoracically with either saline, a low dose (4 × 1013vg kg−1) or a high dose (1.5 × 1014vg kg−1) of each AAV9 (total of 8 × 1013vg kg−1 for low and 3 × 1014vg kg−1 for high) and monitored their development (Extended Data Fig. 4a). The 3 × 1014vg kg−1 high dose is the highest dose administered in clinical trials30. The Myh6h403/+ and Myh6WT mice survived past weaning and well into adulthood. The median survival of saline-injected Myh6h403/h403 mice was 7 days, whereas that of low-dose ABE-treated Myh6h403/h403 mice was increased to 9 days (1.3-fold longer, P < 0.05 by Mantel–Cox test). The median survival of high-dose ABE-treated Myh6h403/h403 mice was increased to 15 days (2.1-fold longer, P < 0.01 by Mantel–Cox test) (Extended Data Fig. 4b). Sanger sequencing of complementary DNA (cDNA) of the heart from a high-dose mouse indicated 35% correction of the pathogenic nucleotide at the transcript level, suggesting that our dual AAV9 ABE system enabled editing in the heart (Extended Data Fig. 4).
Because the MYH7 p.R403Q variant only exists in a heterozygous form in human patients, we next deployed our dual AAV9 ABE system to prevent HCM disease onset in Myh6h403/+ mice. We injected Myh6h403/+ P0 pups intrathoracically with either saline or 1 × 1014vg kg−1 of each AAV9 (2 × 1014vg kg−1 total) and their littermate Myh6WT control pups with saline. At 5 weeks of age, the mice were put on a chow diet containing 0.1% cyclosporine A, which has previously been shown to accelerate the onset of HCM in mouse models of sarcomere PVs31. Serial echocardiograms were conducted at 8, 12 and 16 weeks of age to monitor disease progression (Fig. 4b). We found that Myh6h403/+ mice had increased features of HCM compared with Myh6WT controls, including increased left ventricular anterior wall thickness at diastole (LVAW;d) (1.07 ± 0.0443 mm versus 0.883 ± 0.0441 mm, P = 0.017) and increased left ventricular posterior wall thickness at diastole (LVPW;d) (1.04 ± 0.0809 mm versus 0.867 ± 0.0590 mm, P = 0.128). These mice also had decreased left ventricular internal diameter at diastole (LVID;d) (2.34 ± 0.142 mm versus 2.81 ± 0.0540 mm, P = 0.015) and systole (LVID;s) (0.940 ± 0.0713 mm versus 1.24 ± 0.0520, P = 0.010), with increased ejection fraction (EF) and fractional shortening (FS) (Fig. 4c–h). The increased ventricular wall thickness and a concomitant decrease in ventricular diameter of Myh6h403/+ mice, along with high-normal FS is consistent with the clinical progression in human patients1.
In contrast, ABE-treated Myh6h403/+ mice had reduced features of HCM with similar echocardiographic measurements to Myh6WT control mice, suggesting that gene correction of the pathogenic nucleotide was sufficient to prevent the onset of HCM (Fig. 4c–h, Table 1 and Extended Data Fig. 5). Histological analysis also revealed increased cardiac wall thickness and decreased ventricular cross-sectional area in Myh6h403/+ mice compared with Myh6WT control mice, whereas ABE-treated Myh6h403/+ mice had similar cardiac dimensions to Myh6WT control mice (Fig. 4i–k). When normalized to tibia length, Myh6h403/+ mice had 1.3-fold larger hearts by heart weight compared with Myh6WT control mice, whereas ABE-treated Myh6h403/+ mice had no significant difference in heart weight compared with Myh6WT mice (Fig. 4l). As a measure of fibrosis, hearts from Myh6h403/+ mice had threefold more collagen area compared with Myh6WT control mice, whereas ABE-treated Myh6h403/+ mice had no significant difference in collagen area compared with Myh6WT mice (Fig. 4m). These data suggest that dual AAV9 ABE treatment was sufficient to prevent the onset of HCM-mediated pathological remodeling of the heart.
Table 1 |.
Summary of echocardiographic measurements for humanized mice
| Echocardiography measurements | ||||||
|---|---|---|---|---|---|---|
| LVAW;d (mm) | P value | |||||
| +/+ | h403/+ | ABE h403/+ | h403/+ vs +/+ | ABE h403/+ vs +/+ | ABE h403/+ vs h403/+ | |
| 8 weeks | 0.796 ± 0.0453 | 0.908 ± 0.0283 | 0.775 ± 0.0510 | 0.069 | 0.757 | 0.051 |
| 12 weeks | 0.908 ± 0.0434 | 1.07 ± 0.0428 | 0.829 ± 0.0243 | 0.023 1 | 0.154 | 0.001 |
| 16 weeks | 0.883 ± 0.0441 | 1.07 ± 0.0443 | 0.950 ± 0.0414 | 0.017 | 0.299 | 0.084 |
| LVID;d (mm) | P value | |||||
| +/+ | h403/+ | ABE h403/+ | h403/+ vs +/+ | ABE h403/+ vs +/+ | ABE h403/+ vs h403/+ | |
| 8 weeks | 2.83 ± 0.126 | 2.54 ± 0.110 | 2.64 ± 0.0749 | 0.069 | 0.757 | 0.051 |
| 12 weeks | 2.90 ± 0.0983 | 2.45 ± 0.0986 | 2.84 ± 0.149 | 0.013 | 0.763 | 0.060 |
| 16 weeks | 2.81 ± 0.0540 | 2.34 ± 0.142 | 2.78 ± 0.110 | 0.015 | 0.844 | 0.038 |
| LVID;s (mm) | P value | |||||
| +/+ | h403/+ | ABE h403/+ | h403/+ vs +/+ | ABE h403/+ vs +/+ | ABE h403/+ vs h403/+ | |
| 8 weeks | 1.27 ± 0.109 | 1.05 ± 0.0580 | 1.20 ± 0.0988 | 0.107 | 0.638 | 0.222 |
| 12 weeks | 1.37 ± 0.0729 | 1.00 ± 0.0544 | 1.16 ± 0.0794 | 0.004 | 0.085 | 0.147 |
| 16 weeks | 1.24 ± 0.0520 | 0.940 ± 0.0713 | 1.21 ± 0.108 | 0.010 | 0.829 | 0.071 |
| LVPW;d (mm) | P value | |||||
| +/+ | h403/+ | ABE h403/+ | h403/+ vs +/+ | ABE h403/+ vs +/+ | ABE h403/+ vs h403/+ | |
| 8 weeks | 0.850 ± 0.0349 | 0.899 ± 0.0262 | 0.771 ± 0.0695 | 0.300 | 0.335 | 0.123 |
| 12 weeks | 0.910 ± 0.0471 | 1.00 ± 0.0605 | 0.807 ± 0.0546 | 0.264 | 0.192 | 0.044 |
| 16 weeks | 0.867 ± 0.0590 | 1.04 ± 0.0809 | 0.970 ± 0.0709 | 0.128 | 0.299 | 0.548 |
| EF (%) | P value | |||||
| +/+ | h403/+ | ABE h403/+ | h403/+ vs +/+ | ABE h403/+ vs +/+ | ABE h403/+ vs h403/+ | |
| 8 weeks | 87.0 ± 1.79 | 88.6 ± 0.606 | 86.8 ± 1.86 | 0.428 | 0.922 | 0.374 |
| 12 weeks | 85.8 ± 2.01 | 89.4 ± 1.40 | 88.2 ± 2.13 | 0.188 | 0.448 | 0.652 |
| 16 weeks | 87.4 ± 1.63 | 91.7 ± 0.807 | 87.3 ± 1.61 | 0.045 | 0.965 | 0.039 |
| FS (%) | P value | |||||
| +/+ | h403/+ | ABE h403/+ | h403/+ vs +/+ | ABE h403/+ vs +/+ | ABE h403/+ vs h403/+ | |
| 8 weeks | 55.4 ± 2.19 | 56.7 ± 0.894 | 52.6 ± 3.30 | 0.592 | 0.509 | 0.269 |
| 12 weeks | 53.9 ± 2.40 | 58.1 ± 2.25 | 57.1 ± 2.79 | 0.238 | 0.404 | 0.798 |
| 16 weeks | 55.8 ± 2.33 | 61.6 ± 1.40 | 55.5 ± 2.12 | 0.066 | 0.928 | 0.043 |
P values < 0.05 are in bold. Echocardiographic measurements in Myh6WT mice, Myh6h403/+ mice and ABE-treated Myh6h403/+ mice (8–16 weeks, n = 5 male mice) for changes in LVAW, LVID;d, LVID;s, LVPW, EF and FS. P values are calculated by Student’s unpaired two-sided t-test for given comparisons. Data are mean ± s.e.m.
Genomic and transcriptomic analyses of ABE-treated mice
To determine tissue-level viral transduction and gene editing efficiency, we collected the heart, lung, liver, spleen and quadriceps muscle from saline-treated Myh6h403/+ mice and ABE-treated Myh6h403/+ mice at 18 weeks of age. The heart was further dissected into the right atrium, right ventricle, left atrium and left ventricle and each chamber was separately analyzed. We conducted a viral copy number assay and found similar viral transduction in all four chambers of the heart, ranging from five to nine copies of each viral half per diploid genome. The liver had the highest viral transduction of the dissected tissues, with about ten copies of each viral half per diploid genome, whereas the spleen had the least, with about 0.2 copies of each viral half per diploid genome (Extended Data Fig. 6a). Despite similar levels of viral copy numbers among tissues, we found low levels of DNA editing in the liver, lung, spleen and quadriceps (< 0.12%) compared with DNA editing in each of the heart chambers (5.5–8.0%), confirming the cardiac specificity of the cardiac troponin T promoter (Extended Data Fig. 6b). Because CMs make up around 30% of cells32 in the heart but are the only cells to express Myh6, we next looked at cDNA efficiency. We found that although the left atria had less correction of pathogenic transcripts compared with the ventricles, there was similar editing efficiency across the four chambers (12.9–26.7%) (Extended Data Fig. 6c). We further confirmed no changes in protein expression of key sarcomere proteins (Extended Data Fig. 6d).
To identify genomic and transcriptomic changes more deeply in CMs after base editing, we isolated CM nuclei from the ventricles of saline-treated Myh6WT control mice, saline-treated Myh6h403/+ mice and ABE-treated Myh6h403/+ mice at 18 weeks of age (Fig. 5a). We first evaluated on-target editing efficiencies after dual AAV9 ABE treatment. In ABE-treated Myh6h403/+ mice, DNA editing efficiency of the target pathogenic adenine was 32.3 ± 2.87%, resulting in a 33.1 ± 9.08% reduction in pathogenic transcripts compared with Myh6h403/+ mice (Fig. 5b,c), which is comparable with other in vivo studies using base editing26 or RNAi-based knockdown of pathogenic transcripts33. Furthermore, there was no detectable bystander editing in ABE-treated Myh6h403/+ mice (Fig. 5d). We then assessed potential off-target RNA editing via transcriptome-wide RNA sequencing (RNA-seq), because ABEmax contains deaminase activity. RNA-seq analysis revealed no significant increase in the average frequency of A-to-I editing in the transcriptome of ABE-treated mice compared with that of saline-treated mice (Fig. 5e). This finding suggests that in vivo treatment with our dual AAV9 ABE system does not increase RNA deamination above background levels of endogenous cellular deaminase activity.
Fig. 5 |. Genomic and transcriptomic changes following dual AAV9 ABE injection in mice.
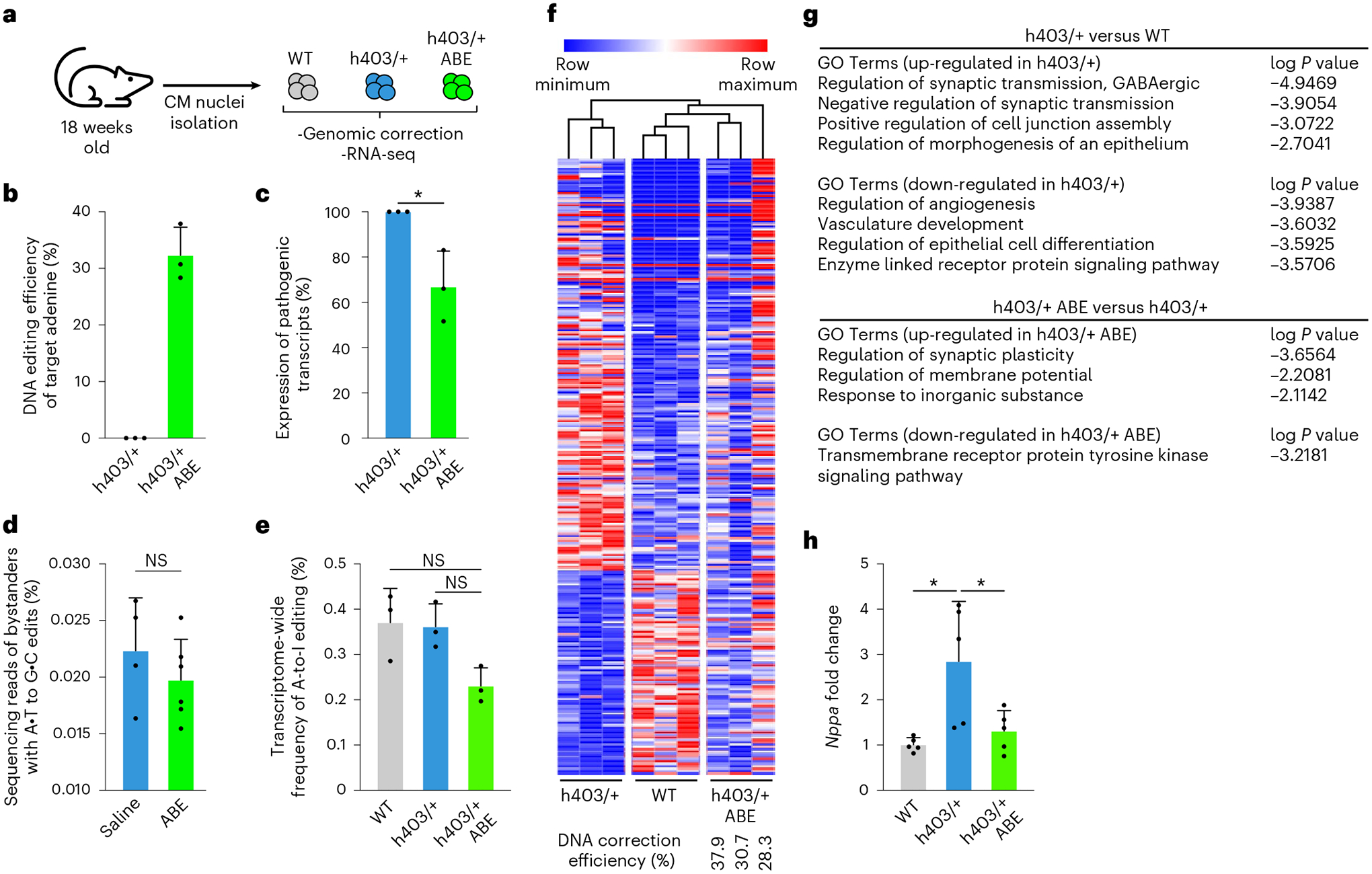
a, CM nuclei were isolated from the ventricles of 18-week-old Myh6WT mice, Myh6h403/+ mice or ABE-treated Myh6h403/+ mice to assess genomic correction and transcriptomic changes. b, DNA editing efficiency for correcting the pathogenic adenine nucleotide was higher in ABE-treated Myh6h403/+ mice (32.3%) than Myh6h403/+ mice (0.15%, P = 0.000359). Data are mean ± s.d. c, Percentage of expressed pathogenic transcripts was lower in ABE-treated Myh6h403/+ mice (66.9%) compared with Myh6h403/+ mice (100.0%, P = 0.0216). Data are mean ± s.d. d, Bystander editing in ABE-treated Myh6h403/+ mice (0.0224%) was not significantly different from saline-treated mice (0.0197%, P = 0.347). Data are mean ± s.d. e, Transcriptome-wide nuclear levels of A-to-I RNA editing was lower in ABE-treated Myh6h403/+ mice (0.231%) compared with Myh6WT mice (0.371%, P = 0.0464) and Myh6h403/+ mice (0.361%, P = 0.0247). Data are mean ± s.d. f, Heat map of 257 differentially expressed genes amongst Myh6WT or Myh6h403/+ mice and ABE-treated Myh6h403/+ mice. Samples and genes are ordered by hierarchical clustering. Data were scaled by the sum of each row and are displayed as row minimum and row maximum. ABE-treated Myh6h403/+ mice cluster with Myh6WT mice. Editing efficiency for each ABE-treated Myh6h403/+ mouse is indicated. g, Top gene ontology (GO) terms associated with the differentially expressed genes in the comparison of h403/+ versus WT (top) and h403/+ ABE-treated versus h403/+ (bottom). P values were obtained using Metascape (Methods). h, Fold-change of Nppa mRNA expression was higher for Myh6h403/+ mice (2.85) than for ABE-treated Myh6h403/+ mice (1.31, P = 0.0395) normalized to Myh6WT mice (1.01, P = 0.0151). Data from RNA-seq and qPCR. Data are mean ± s.d. For all: *P < 0.05 by Student’s unpaired two-sided t-test, n = 3 male mice for each group. NS, not significant.
We next evaluated transcriptome-wide changes in ABE-treated Myh6h403/+ mice via RNA-seq. We first identified 257 differentially regulated genes between Myh6WT mice and Myh6h403/+ mice. Heat maps showed that ABE-treated Myh6h403/+ mice had transcriptome profiles more similar to Myh6WT mice than to Myh6h403/+ mice in a correction efficiency-dependent response (Fig. 5f, and Extended Data Fig. 7). Gene ontology analyses of differentially regulated genes between Myh6h403/+ mice and Myh6WT mice indicated dysregulation of intercellular signaling and angiogenesis, whereas intercellular signaling was dysregulated between Myh6h403/+ mice and ABE-treated Myh6h403/+ mice (Fig. 5g). Additionally, expression of the prototypic hypertrophic marker Nppa was 2.8-fold higher in Myh6h403/+ mice compared with Myh6WT mice, whereas expression of Nppa in the ABE-treated Myh6h403/+ mice was not significantly different from Myh6WT mice (Fig. 5h). Taken together, these data suggest our dual AAV9 ABE system can efficiently correct the pathogenic nucleotide in genomic DNA and prevent transcriptomic dysregulation.
Discussion
In this proof-of-concept study, we show that an ABE can directly correct a common and well-studied PV that leads to HCM, serving as a step towards clinical translation of base editing in patients with HCM. In iPSCs derived from patients with clinically diagnosed HCM, we identified the optimal base editor as ABEmax-VRQR, because it corrected the target pathogenic adenine at 98–99% efficiency in iPSCs sorted to contain all editing components, with minimal bystander editing of neighboring adenines and low DNA off-target editing at tested sites within the human genome. These corrected patient-derived iPSCs, when differentiated into CMs, demonstrated normalization of contractile force and rescued cellular energetics compared with uncorrected patient-derived CMs, suggesting that correction of this PV is sufficient to reduce the abnormalities of HCM.
To extend this work to a clinically relevant animal model, we generated a humanized mouse model of HCM. This humanized mouse model demonstrates similar HCM onset to other previously described models, but contains DNA sequence complementarity to the human allele, thus allowing testing of human-specific sgRNAs. Injection of a single dose of dual AAV9 encoding ABEmax-VRQR and the h403_sgRNA in clinically relevant heterozygous postnatal Myh6h403/+ mice resulted in correction of the pathogenic allele, reduction of pathogenic transcripts and reduction in cardiac hypertrophy and histopathologic remodeling. Our study, along with that of our colleagues12, marks the first demonstration, to our knowledge, of efficient single nucleotide gene correction in vivo in postnatal mammalian CMs. Future studies will seek to determine whether this rescue is sufficient to prevent HCM onset for the lifespan of the mouse.
Base editing in Myh6h403/h403 homozygous mice could double their lifespan, but ultimately could not prevent their death before weaning. This is predictable, because only the heterozygous form of the MYH7 p.R403Q variant exists in human patients and, most likely, the homozygous form is nonviable. In mice, Myh7 protein is partially expressed within the ventricles for the first week of life, which likely protects the heart against the pathogenic Myh6 protein24, because these mice die around 7 days after birth, coincident with isoform switching in the ventricles. Although we could achieve approximately 35% correction of transcripts at the highest dual AAV9 dose, the lag between healthy protein production after correction and myosin turnover and replacement of the pathogenic protein was likely too long to prevent death.
Because HCM is an autosomal dominant disease, there is a strong founder effect bias in patient frequency; the initial study linking the c.1208G>A PV to HCM was based on genetic mapping of a large French-Canadian kindred (>80 members) over five generations34. Nonetheless, this PV is likely found worldwide because it was also reported in a Korean family35. Based on data from the Sarcomeric Human Cardiomyopathy Registry36, which performed genetic testing on patients with HCM across eight different institutions in three different continents, out of all patients with identified MYH7 gene variants, 2.4% (15 out of 613) of them had the c.1208G>A PV, making it the fifth most frequent PV out of 229 MYH7 variants detected. For all sarcomere gene variants identified, 0.5% (15 out of 2,763) of patients had the c.1208G>A PV. The World Bank estimates that there are 5.1 billion people worldwide aged 15–64 (https://databank.worldbank.org/source/population-estimates-and-projections). It is conservatively estimated that one in 500 people have HCM, of which 50% have a known genetic cause in a sarcomere gene. From these data, we can estimate that perhaps 25,000 patients exist with the specific c.1208G>A p.R403Q PV and would be amenable to adenine base editing correction.
Although we demonstrate base editing for a single pathogenic missense variant in MYH7, we envision that this base editing approach can be applied to the hundreds of other documented missense variants in MYH7 and to other cardiac sarcomeric proteins in which dominant-negative variants lead to HCM18. Base editing is an ideal tool to correct the many dominant-negative cardiac diseases, because gene therapy is precluded in dominant-negative diseases and CRISPR–Cas9 single-cutting may edit the healthy WT allele, which can have deleterious effects for essential genes such as MYH737. In base editing, careful selection of an sgRNA and a base editor with an appropriate editing window and activity can minimize off-target binding of the sgRNA to the healthy WT allele and minimize DNA off-target and bystander editing. As a clinical treatment, we envision that although the initial base editing trials in the heart will be rigorously evaluated before clinical translation, these and other initial studies will establish a pipeline for gene editing of HCM. In the future, treatment of other MYH7 PVs may then simply involve changing the Cas9 variant and gRNA sequence and evaluating potential gRNA-specific off-target editing for each patient’s genome.
Although our study showed that gene editing of newborn mice can prevent the onset of HCM caused by the highly penetrant p.R403Q variant, some patients are genotype positive for an HCM-causing variant but are phenotype negative by not demonstrating LV hypertrophy at any point in the clinical course38. These patients would be unlikely to need gene editing correction unless clinical HCM develops. Future studies will seek to determine whether in vivo gene editing in established HCM can either revert or limit the progression of the disease. Previous studies in a mouse39 and cat model40 of HCM have shown that mavacamten, a small molecule myosin inhibitor, can cause partial regression of hypertrophy in adult animals with established HCM, suggesting a potentially wide therapeutic window for other treatments such as gene editing. Use of adult animals with larger hearts could also allow testing of other delivery methods not explored in our study, such as coronary injection41, which could then allow testing of other delivery modalities for base editors, including lipid nanoparticles42 or virus-like particles43. Our study also did not explore optimization in viral dosage, use of a nontargeting sgRNA as a control treatment in place of saline administration, or use of other cardiotropic AAV capsid variants44,45. Finally, future studies will need to demonstrate correction in larger animal models that endogenously express β-myosin heavy chain predominantly within the heart, such as rabbits, cats and pigs46, before ultimately treating patients with HCM. Myosin-binding drugs that can modulate cardiac function, such as mavacamten39, hold great promise in treating HCM; clinical trials for mavacamten have met their primary endpoints and it was recently approved by the Food and Drug Adminstration to treat symptomatic obstructive HCM47,48. Nevertheless, there is a continued need for new therapeutic modalities, especially considering the on-label warning of heart failure of mavacamten49. Our study suggests that base editing can provide an alternative one-time treatment modality to directly and permanently correct pathogenic HCM-causing variants and prevent HCM onset, especially in cardiac genes for which no drug has been identified, or for patients with symptomatic nonobstructive HCM, for which mavacamten did not significantly improve exercise capacity or symptoms compared with placebo50.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-022-02176-5.
Methods
Study design and approval
All mouse experiments complied with all relevant ethical regulations and were performed according to protocols approved by the Institutional Animal Care and Use Committees at the University of Texas (UT) Southwestern Medical Center. UT Southwestern uses the Guide for the Care and Use of Laboratory Animals when establishing animal research standards. All mice used in this study were housed at the pathogen-free Animal Resource Center at the UT Southwestern Medical Center. All animals were bred inside a specific pathogen-free facility with 12 h light:dark cycles with a temperature of 18–24 °C and humidity of 35–60% and monitored daily with no health problems. All animals were housed in groups of a maximum five per cage with ad libitum access to food and water. Informed consent was obtained from all donors of peripheral blood mononuclear cells for use of their cells as patient-derived iPSCs. Stem-cell work was conducted under the oversight of the UT Southwestern Stem Cell Research Oversight Committee.
The objective of this study was to determine whether base editing correction of a pathogenic HCM-causing variant could prevent the onset of HCM pathological features in human CMs and a humanized mouse model. In human CMs, this was done by base editing correction of HCM patient-derived iPSCs and measuring changes in characteristic CM function. In a humanized mouse model, a dual AAV9 system was used to deliver the base editing components to CMs and changes in heart function, dimensions and transcriptomics were measured. For all experiments, the number of replicates, type of replicates and statistical test used are reported in the figure legends. For in vitro CM experiments, data are collected from three separate differentiations and no outliers or other data points were excluded.
For in vivo experiments, male and female mice were assigned to treatment based on genotype. Male mice were used for heterozygous experiments because female mice have better cardio-protection than male mice51 and did not display the phenotype as readily as males, which has been described in other mouse studies of HCM26,33. Both male and female mice were used for homozygous experiments because the phenotype is highly penetrant for both sexes. Echocardiographic measurements were conducted in a blinded fashion. Runt mice with reduced body weight more than two s.d. from the mean were excluded. Endpoints were guided by changes in echocardiographic measurements.
Plasmids and vector construction
The pSpCas9(BB)-2A-GFP (PX458) plasmid was a gift from Feng Zhang (Addgene plasmid no. 48138)52 and was used as the primary scaffold to clone in the following base editors and SpCas9 nickases: ABE8e, a gift from David Liu (Addgene plasmid no. 138489)14; VRQR-ABEmax, a gift from David Liu (Addgene plasmid no. 119811)53; NG-ABEmax, a gift from David Liu (Addgene plasmid no. 124163)53; pCMV-T7-SpG-HF1-P2A-EGFP (RTW5000), a gift from Benjamin Kleinstiver (Addgene plasmid no. 139996)15; and pCMV-T7-SpRY-HF1-P2A-EGFP (RTW5008), a gift from Benjamin Kleinstiver (Addgene plasmid no. 139997)15. The N-terminal ABE and C-terminal ABE constructs were adapted from Cbh_v5 AAV-ABE N terminus (Addgene plasmid no. 137177)54 and Cbh_v5 AAV-ABE C terminus (Addgene plasmid no. 137178)54 and synthesized by Twist Bioscience. Polymerase chain reaction (PCR) amplification of select plasmids was done using PrimeStar GXL Polymerase (Takara) and cloning was done using NEBuilder HiFi DNA Assembly (NEB) into restriction enzyme-digested destination vectors.
Generation of patient-derived iPSCs and isogenic lines
Peripheral blood mononuclear cells from two patients with the MYH7 c.1208G>A (p.R403Q) PV were reprogrammed to iPSCs (HCM1 and HCM2) using Sendai virus. The HCM1 line was derived from a 56-year-old woman with extensive family history of HCM and nonobstructive HCM with a history of reduced left ventricular EF and low maximal oxygen uptake (VO2 max). A biventricular pacemaker was placed for a complete heart block. The HCM2 line was derived from a 32-year-old man with a history of HCM, an implantable cardioverter-defibrillator and a strong family history of HCM. He has a dilated left atrium but has improved VO2 max, metabolic equivalent and no evidence of atrial fibrillation by cardiopulmonary exercise testing. These two human iPSC lines were obtained from Joseph C. Wu, MD, PhD at the Stanford Cardiovascular Institute funded by National Institutes of Health grant R24 HL117756. Peripheral blood mononuclear cells from a male HD were reprogrammed to iPSCs at the UT Southwestern Wellstone Myoediting Core using Sendai virus (CytoTune 2.0 Sendai Reprogramming Kit, Thermo Fisher Scientific). To generate isogenic iPSCs containing the MYH7 c.1208G>A (p.R403Q) variant via homology-directed repair, HD iPSCs were nucleofected using the P3 Primary Cell 4D-Nucleofector X Kit (Lonza) with a single-stranded oligodeoxynucleotide template (Integrated DNA Technologies, IDT) including the PV, and the PX458 plasmid encoding SpCas9-P2a-EGFP and a sgRNA targeting MYH7. For base editing correction of HCM1 and HCM2 patient-derived lines, iPSCs were nucleofected with a single plasmid encoding ABEmax-VRQR-P2a-EGFP and h403_sgRNA. After 48 h, GFP + iPSCs were collected by fluorescence-activated cell sorting, clonally expanded, and genotyped by Sanger sequencing.
iPSC maintenance and differentiation
iPSC culture and differentiation were performed as previously described11. Briefly, iPSCs were cultured on Matrigel (Corning)-coated tissue culture polystyrene plates and maintained in mTeSR1 media (STEMCELL) and passaged at 70–80% confluency using Versene. iPSCs were differentiated into CMs at 70–80% confluency by treatment with CHIR99021 (Selleckchem) in RPMI supplemented with ascorbic acid (50 μg ml−1) and B27 without insulin (RPMI/B27-) for 24 h (from day 0 to day 1). At day 1, media were replaced with RPMI/B27-. At day 3, cells were treated with RPMI/B27- supplemented with WNT-C59 (Selleckchem). At day 5, media were refreshed with RPMI/B27-. From day 7 onwards, iPSC-CMs were maintained in RPMI supplemented with ascorbic acid (50 μg ml−1) and B27 (RPMI/B27) with media refreshed every 3–4 days. Metabolic selection of CMs was performed for 6 days starting at day 10 by culturing cells in RPMI without glucose and supplemented with 5 mM sodium dl-lactate and CDM3 supplement (500 μg ml−1 Oryza sativa-derived recombinant human albumin, A0237, Sigma-Aldrich; and 213 μg ml−1 ʟ-ascorbic acid 2-phosphate, Sigma-Aldrich). To induce their maturation, iPSC-CMs were maintained in RPMI without glucose supplemented with B27, 50 μmol palmitic acid, 100 μmol oleic acid, 10 mmol galactose and 1 mmol glutamine (Sigma-Aldrich)55,56. All CM functional studies were done at days 40–50.
Plasmid transfection and editing efficiency analysis
iPSCs were seeded on a 48-well plate 24 h before transfection. At around 20% confluency, cells were transiently transfected with 0.5 μg of plasmid encoding a base editor and the h403_sgRNA using 1 μl of Lipo-fectamine Stem Transfection Reagent (Thermo Fisher Scientific) per well. After 48 h posttransfection, cells were lysed in Direct PCR Lysis Reagent (Cell) (Viagen). PCR amplification of target sites was done using PrimeStar GXL Polymerase (Takara) and PCR cleanup was done using ExoSap-IT Express (Thermo Fisher Scientific) before Sanger sequencing on a ABI 3730XL Genetic Analyzer. Chromatograms were analyzed using EditR to determine base editing efficiencies57.
Contractility analyses of iPSC-CMs
iPSC-CMs were plated at single-cell density on flexible polydimethylsiloxane 527 substrates (Young’s modulus = 5 kPa) prepared according to a previously established protocol58. Recordings of contracting iPSC-CMs were captured at 37 °C using a Nikon A1R + confocal system at 59 frames per second in resonance scanning mode. Contractile force generation of iPSC-CMs was quantified using a previously established method. In brief, recordings were analyzed using Fiji to measure maximum and minimum cell lengths and cell widths during contraction. A previously published customized MATLAB code was used to calculate peak systolic forces59.
Extracellular flux analyses of iPSC-CMs
iPSC-CMs were plated at 40,000 cells per well in Seahorse XFe96 V3 PS Cell Culture Microplates (Agilent) coated with Matrigel. Then 1 week post-plating, cells were washed three times with prewarmed assay media (pyruvate-free DMEM (Sigma D5030) supplemented with 2 mM ʟ-glutamine, 1 mM sodium pyruvate and 10 mM glucose, pH 7.4) and incubated at 37 °C for 60 min in a non-CO2 incubator. OCR was measured in a Seahorse XFe96 instrument using consecutive cycles of 2 min of measurement, 10 s of waiting and 3 min of mixing. Mitochondrial stress testing was performed by injecting oligomycin (final concentration 2 μM), carbonyl cyanide m-chlorophenyl hydrazone (final concentration 1 μM) and antimycin A (final concentration 1 μM) at indicated time intervals. Data were analyzed using the WAVE software (Agilent).
Immunofluorescence staining
iPSC-CMs were plated on glass surfaces and fixed with 4% paraformaldehyde for 10 min, followed by blocking with 5% goat serum/0.1% Tween-20 (Sigma-Aldrich) for 1 h. Primary and secondary antibodies were diluted in blocking buffer and added to cells for 2 h and 1 h, respectively. Nuclei were counterstained using DAPI. Antibodies used included rabbit anti-troponin-I (H-170 sc-15368, Santa Cruz Biotechnology, 1:200) and fluorescein-conjugated donkey anti-rabbit IgG (711–095-152, Jackson ImmunoResearch, 1:50). Confocal images were acquired using a Zeiss LSM 800 and Zeiss Zen-2.
Off-target analyses
Candidate off-target sites were identified with CRISPOR and the top eight sites by cutting frequency determination score, for which PCR products were successfully obtained, were selected19,60. Genomic DNA was isolated using a DNeasy Blood & Tissue Kit (Qiagen) from HCM1 and HCM2 cell lines that had been nucleofected with plasmids encoding ABEmax-VRQR-P2a-EGFP and h403_sgRNA and sorted for GFP + cells. Target sites were PCR amplified using PrimeStar GXL Polymerase (Takara), and a second round of PCR was used to add Illumina flow cell binding sequences and barcodes. PCR products were purified with AMPure XP Beads (Beckman Coulter), analyzed for integrity on a 2200 TapeStation System (Agilent) and quantified by QuBit dsDNA high-sensitivity assay (Invitrogen) before pooling and loading on an Illumina MiSeq. After demultiplexing, the resulting reads were analyzed with CRISPResso2 for editing frequency61.
Generation of AAVs
Recombinant AAV9 (rAAV9) viruses were made at the University of Michigan Vector Core using ultracentrifugation through an iodixanol gradient. rAAV9s were washed three times with phosphate buffered saline (PBS) using Amicon Ultra centrifugal filter units (Millipore) and resuspended in PBS + 0.001% Pluronic F68. Titers were assessed by quantitative PCR (qPCR). rAAV9 was stored in 25 μl aliquots at −80 °C.
Mice
Mice were maintained on standard chow (2916 Teklad Global). The humanized Myh6h403/+ PV was introduced via microinjection of zygotes with Cas9 mRNA (50 ng μl−1) (TriLink Biotechnologies), a sgRNA (20 ng μl−1) (IDT) and a single-stranded oligodeoxynucleotide donor template (15 ng μl−1) (IDT) following a modified protocol62. Mice were bred into the C57BL/6 background. Genotyping was performed using a custom TaqMan SNP Genotyping Assay (Assay ID ANPRZE6) (Thermo Fisher Scientific). To accelerate the onset of HCM, mice were treated with a custom chow (2916 Teklad Global base) containing cyclosporine A (Alfa Aesar) at 1 g kg−1 and blue food dye at 0.2 g kg−1. For injections, mice were genotyped at P0 and received either saline or a AAV9 dose via a single 40 μl bolus using a 31 G insulin syringe through the diaphragm by a subxiphoid approach into the inferior mediastinum, avoiding the heart and the lung.
Transthoracic echocardiography
Cardiac function on conscious mice was evaluated by two-dimensional transthoracic echocardiography using a VisualSonics Vevo2100 imaging system. M-mode tracings were used to measure LVAW;d, LVPW;d, LVID;d and LVID;s. FS was calculated according to the following formula: FS (%) = ((LVID;d − LVID;s)/LVID;d) × 100. EF was calculated according to the following formula: EF (%) = ((LVEDV − LVESV)/LVEDV) × 100. All measurements were performed by an experienced operator blinded to the study.
Histology
Mouse hearts were dissected out and submerged in PBS with cardio-plegic 0.2 M KCl for 5 min before fixation in 4% paraformaldehyde in PBS overnight, followed by dehydration in 70% ethanol and paraffin embedding. Serial transverse cross-sections at 500-μm intervals were cut and mounted on slides, followed by hematoxylin and eosin staining, picrosirius red or Masson’s trichrome staining. Images were captured on a BZ-X800 all-in-one microscope (Keyence) at ×10 or ×40 magnification. Analyses were performed with Fiji and Adobe Photoshop.
Viral copy number assay
Genomic DNA was isolated from mouse tissue using the DNeasy Blood & Tissue Kit (Qiagen). AAV viral copy number was determined by qPCR using custom-designed primers and Taqman probes (IDT) (Supplementary Table 1) and the Applied Biosystems TaqMan Fast Advanced Master Mix (Applied Biosystems). The primers and probes anneal to the N-terminal and C-terminal Cas9 genes. A copy number standard curve of the AAV plasmids used for packaging was used to determine copy number for each cycle threshold. The 18S ribosomal RNA gene was used as a reference to calculate genomic DNA quantity.
Cardiac myofibril isolation and analysis
Cardiac myofibrils were isolated from hearts using a previously reported protocol, with minor modifications63. Collected hearts were homogenized using CK28-R hard tissue homogenizing tubes in the Percellys homogenizer (Bertin Instruments) on the hard tissue setting. After myofibril isolation using various buffers, protein amounts were quantified by Pierce BCA Protein Assay (Thermo Fisher Scientific). For each sample, 10 μg protein was run on a 4–20% polyacrylamide gel, then stained with Coomassie G-250.
CM nuclei isolation
For each nuclear sample, ventricular heart tissue was isolated. CM nuclei were isolated as previously described using various buffers and immunolabeled cell sorting64. Isolated nuclei were immediately used for downstream processing. RNA was isolated from nuclei using the RNeasy Micro Kit (Qiagen). For qPCR and cDNA HTS, RNA was reverse transcribed using iScript cDNA Synthesis Kit (Bio-Rad). For DNA sequencing, nuclei were lysed in Direct PCR Lysis Reagent (Cell) (Viagen).
Mouse DNA and cDNA sequencing and analysis
On-target DNA and cDNA sites were PCR amplified using PrimeStar GXL Polymerase (Takara) and a second round of PCR was used to add Illumina flow cell binding sequences and barcodes. PCR products were purified with AMPure XP Beads (Beckman Coulter), analyzed for integrity on a 2200 TapeStation System (Agilent) and quantified by QuBit dsDNA high-sensitivity assay (Invitrogen) before pooling and loading on an Illumina MiSeq. After demultiplexing, resulting reads were analyzed with CRISPResso2 for editing frequency61.
RNA-seq library preparation, sequencing and analysis
RNA-seq libraries were generated using the SMARTer Stranded Total RNA-Seq Kit v2-Pico Input Mammalian kit (Takara), containing Illumina sequencing adapters. Libraries were visualized on a 2200 TapeStation System (Agilent) and quantified by QuBit dsDNA high-sensitivity assay (Invitrogen) before pooling and loading on an Illumina NextSeq 500. FastQC tool (v.0.11.8) was used for quality control of RNA-seq data to determine low-quality or adapter portions of the reads for trimming. Read trimming was performed using Trimmomatic (v.0.39) and strandness was determined using RSeQC (v.4.0.0), then reads were aligned to the mm10 reference genome using HiSAT2 (v.2.1.0) with default settings and -rna-strandness R. Aligned reads were counted using feature-Counts (v.1.6.2). Differential gene expression analysis was performed using R package DESeq (v.1.38.0). Genes with fold-change over two and P value < 0.01 were designated as differentially expressed genes between sample group comparisons. To calculate the average percentage of A-to-I editing among adenosines sequenced in transcriptome-wide sequencing analysis, we adopted a previous strategy9. In brief, REDItools2 was used to quantify the percentage editing in each sample. Nucleotides except adenosines were removed and remaining adenosines with read coverage less than ten or read quality score below 25 were also filtered to avoid errors due to low sampling or low sequencing quality. We then calculated the number of A-to-I conversion in each sample and divided this by the total number of adenosines in our dataset after filtering to get the percentage of A-to-I editing in the transcriptome.
Quantitative real-time PCR analysis
qPCR reactions were assembled using Applied Biosystems TaqMan Fast Advanced Master Mix (Applied Biosystems). Assays were performed using Applied Biosystems QuantStudio 5 Real-Time PCR System (Applied Biosystems). Expression values were normalized to 18S mRNA and represented as fold-change.
Statistics
All data are presented as means ± s.e.m. or means ± s.d. as indicated. Unpaired two-tailed Student’s t-tests were performed for comparison between the respective two groups as indicated in the figures. Kaplan–Meier analysis and log-rank (Mantel–Cox) test were used to evaluate the difference in survival between different genotypes. Data analyses were performed with statistical software (GraphPad Prism Software v.9.4.0). P values < 0.05 were considered statistically significant. For differentially expressed genes sets, P adjust values are calculated by two-tailed Wilcoxon rank-sum test. Enrichment analysis was performed with Metascape by supplying upregulated or downregulated differentially expressed genes65.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Generation of isogenic HD403/+ and HD403/403 iPSCs by homology-directed repair.
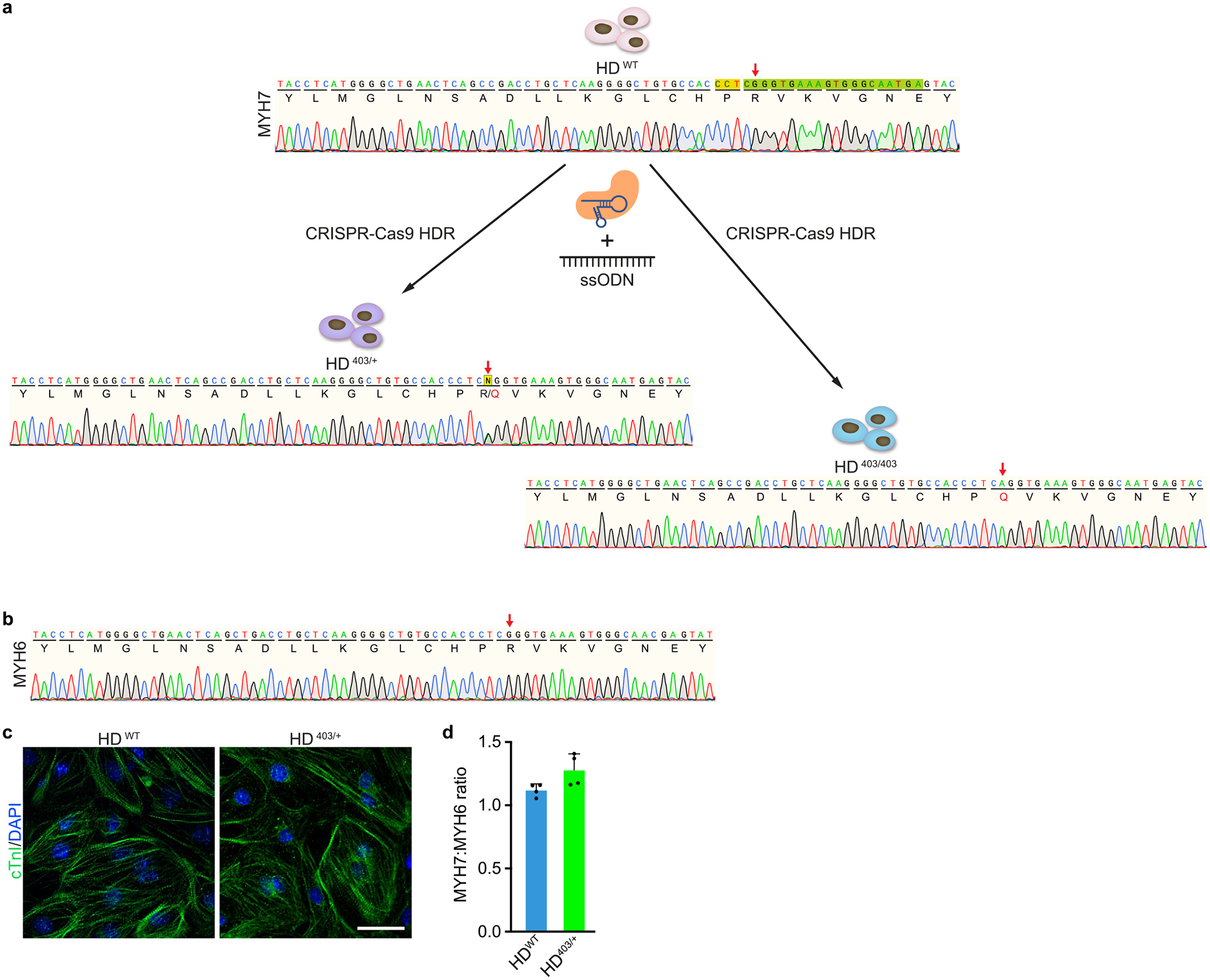
a, Using iPSCs derived from a healthy donor (HDWT), the MYH7 p.R403Q (c.1208 G > A) variant was introduced by CRISPRCas9-based homology-directed repair (HDR) using SpCas9, a sgRNA (spacer sequence colored in green, PAM sequence colored in gold), and a single-stranded oligodeoxynucleotide (ssODN) donor template containing the PV. A heterozygous genotype (HD403/+) and homozygous genotype (HD403/403) were isolated. Chromatograms highlighting insertion of the PV and corresponding amino acid changes are shown for indicated genotypes. Red arrows indicate coding nucleotide 1208 and amino acid 403. b, Sanger sequencing chromatogram showing no insertion of the PV on the highly homologous MYH6 gene. Red arrow indicates coding nucleotide 1211 and amino acid 404. c, HDWT and HD403/+ iPSCs readily differentiate into CMs. Cardiac troponin I (cTnI, green) highlights CMs; nuclei (blue) are marked by DAPI (4′,6-diamidino-2-phenylindole). Scale bar, 25 μm. Similar ability for iPSCs to differentiate into CMs was found in at least three separate differentiations for each genotype. d, Ratio of MYH7 to MYH6 gene expression in HDWT and HD403/+ iPSC-CMs as measured by quantitative PCR. Data are mean ± s.d. across four separate differentiations.
Extended Data Fig. 2 |. Computationally determined off-target sites for h403_sgRNA with ABEmax-VRQR.
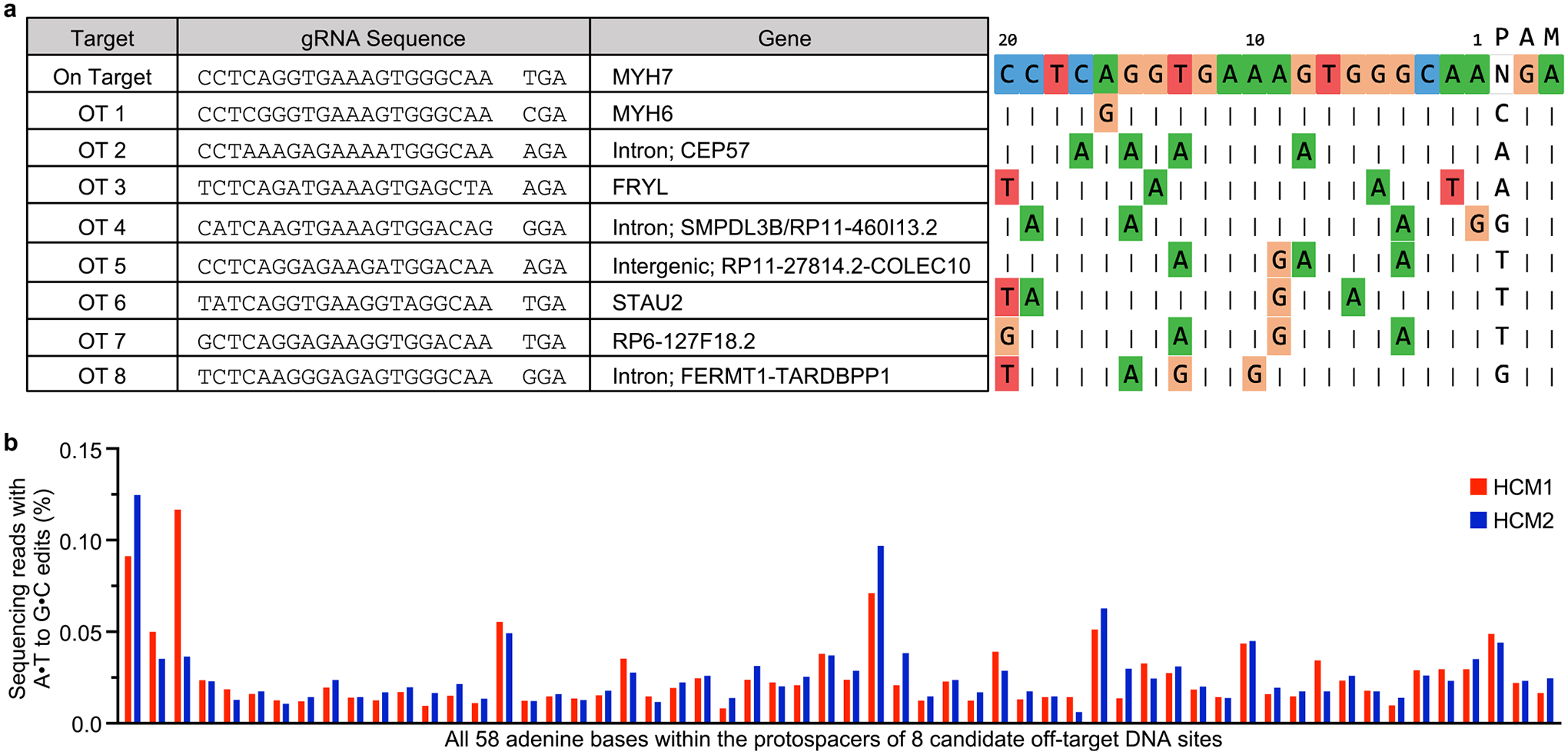
a, Genomic loci of eight candidate off-target (OT) sites (left) and alignments of eight candidate off-target sequences to the on-target protospacer (right). Nucleotides that match the protospacer are indicated with a vertical dash. Nucleotides that differ are shown for each site. Numbering of nucleotides in protospacer starts with the nucleotide immediately 5′ of the PAM as nucleotide 1. b, HTS to measure editing for all 58 adenines within the protospacers of the top 8 CRISPOR-identified candidate off-target loci. HTS was performed for ABE-treated MYH7403/+ HCM1 and MYH7403/+ HCM2 iPSCs.
Extended Data Fig. 3 |. Comparison of predominantly expressed mouse and human myosin heavy chain sequences.
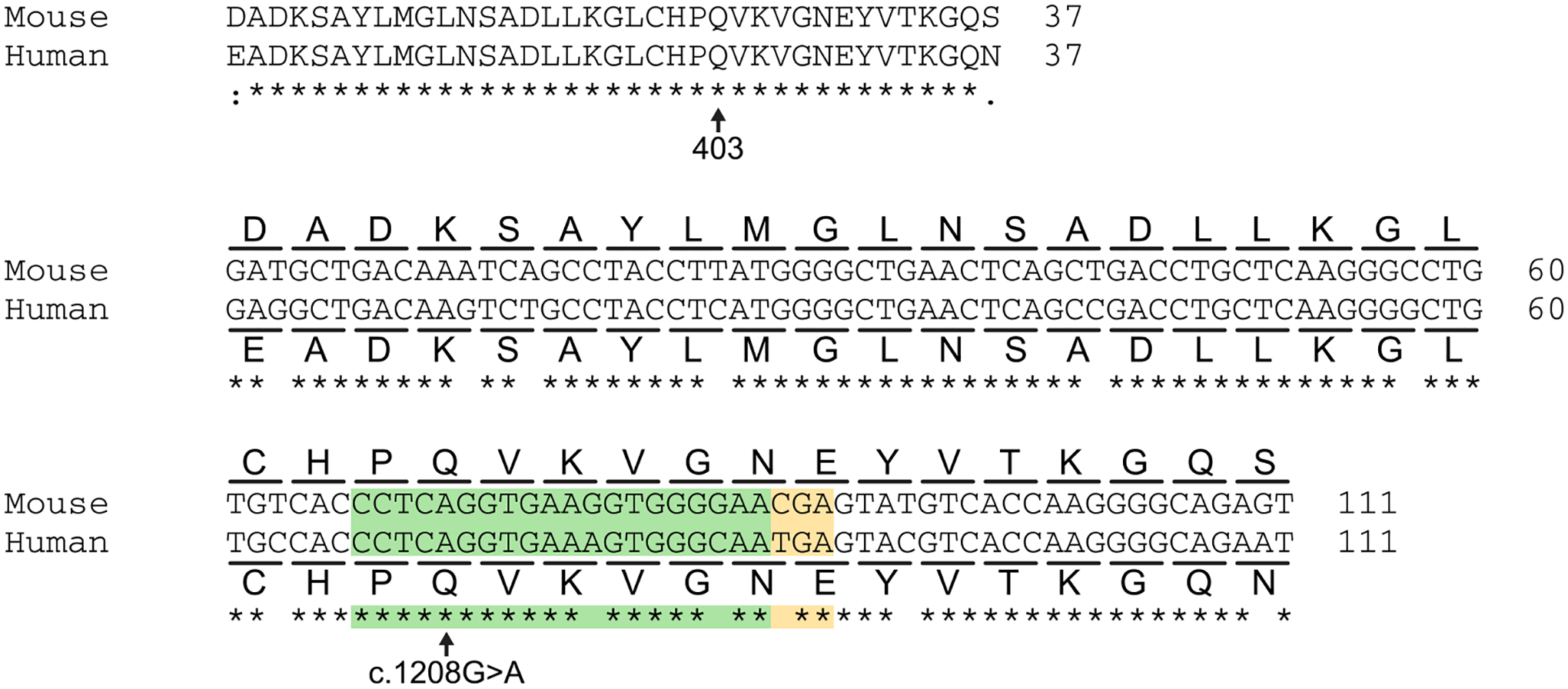
Homology comparison for mouse α-myosin heavy chain (Myh6) and human β-myosin heavy chain (MYH7) at the amino acid level (top) and DNA sequence level (bottom) around glutamine 403. The h403_sgRNA is illustrated in green and the PAM sequence is illustrated in yellow. The pathogenic c.1208 G > A variant is located at position 16 within the canonical base editing window of positions 14–17, counting the adenine nucleotide immediately 5′ of the PAM as position 1.
Extended Data Fig. 4 |. Validation of a dual AAV9 ABE system in mice.
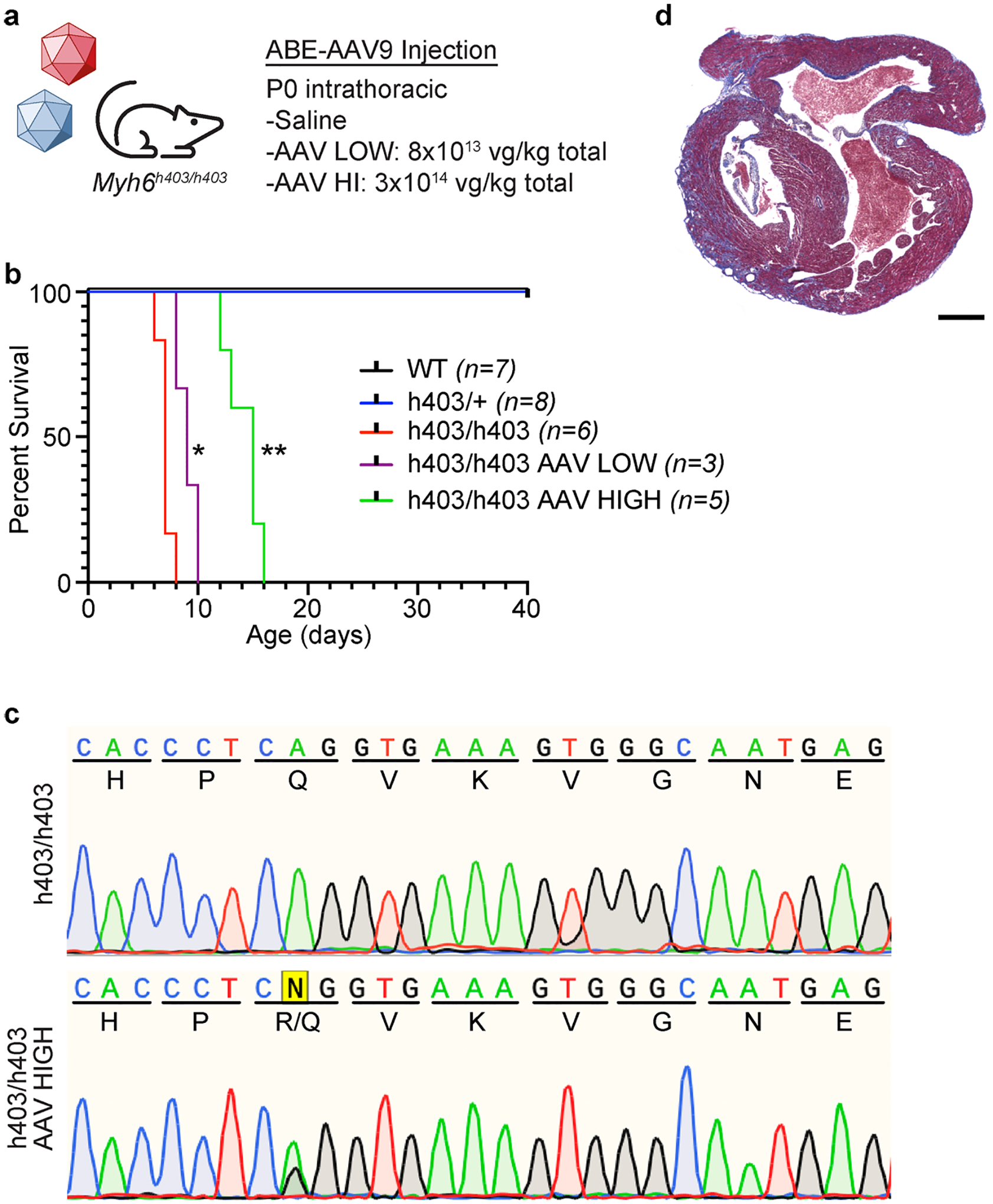
a, Injection details for treating Myh6h403/h403 mice with ABE-AAV9 or saline. b, Kaplan-Meier curve for Myh6WT mice (n = 7; 4 male, 3 female), Myh6h403/+ mice (n = 8; 2 male, 6 female), Myh6h403/h403 mice (n = 6; 1 male, 5 female), and ABE-treated Myh6h403/h403 mice at a low (AAV LOW, n = 3; 1 male, 2 female) or high dose (AAV HIGH, n = 5; 4 male, 1 female). Median lifespans: Myh6WT and Myh6h403/+ mice, >40 days; Myh6h403/h403 mice, 7 days; AAV LOW Myh6h403/h403 mice, 9 days (1.3-fold longer, P = 0.0201); AAV HIGH Myh6h403/h403 mice, 15 days (2.1-fold longer, P = 0.0014). *P < 0.05, **P < 0.01 by log-rank (Mantel–Cox) test for AAV LOW Myh6h403/h403 mice and AAV HIGH Myh6h403/h403 mice, each, compared to Myh6h403/h403 mice. c, Sanger sequencing chromatograms for a Myh6h403/h403 mouse and a AAV HIGH Myh6h403/h403 mouse showing 35% on-target editing of the target pathogenic adenine at the cDNA level. d, Four-chamber sectioning and Masson’s trichrome staining of a AAV HIGH Myh6h403/h403 male mouse at 15 days of age. No other replications were possible as all other pups (5 total) were cannibalized before hearts could be collected.
Extended Data Fig. 5 |. Serial echocardiograms following dual AAV9 ABE editing of Myh6h403/+ mice.
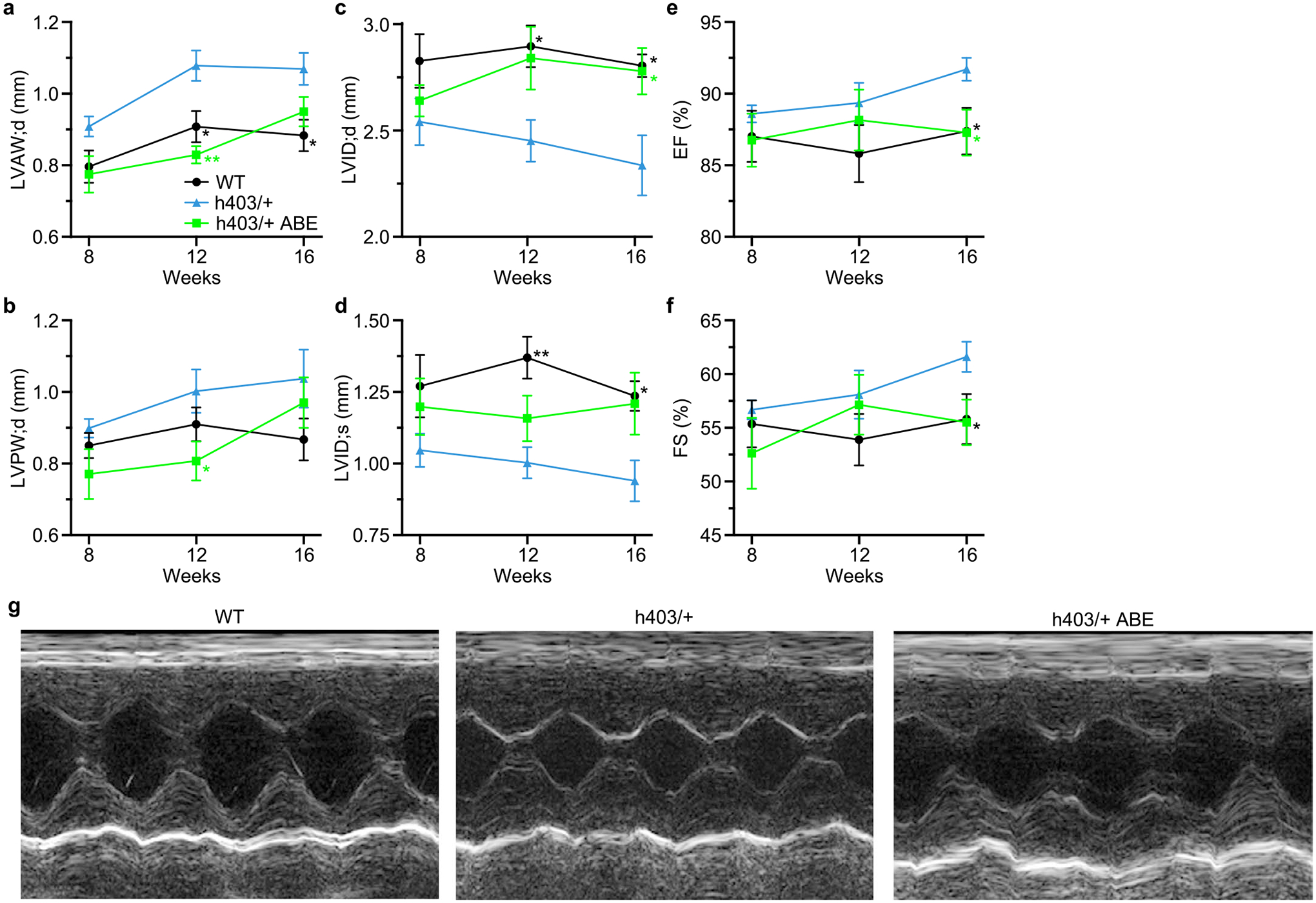
a–f, Left ventricular anterior wall thickness at diastole (a) left ventricular posterior wall thickness at diastole (b), left ventricular internal diameter at diastole (c) and systole (d), ejection fraction (e), and fractional shortening (f), of Myh6WT mice, Myh6h403/+ mice, or ABE-treated Myh6h403/+ mice from 8–16 weeks of age. n = 5 male mice for each group. Exact P values can be found in Table 1. Data are mean ± s.e.m. *P < 0.05, **P < 0.01 by Student’s unpaired two-sided t-test for Myh6WT mice compared to Myh6h403/+ mice (black) and ABE-treated Myh6h403/+ mice compared to Myh6h403/+ mice (green). g, Representative M-mode images for Myh6WT mice, Myh6h403/+ mice, and ABE-treated Myh6h403/+ mice at 16 weeks of age.
Extended Data Fig. 6 |. Genomic and proteomic analysis of select tissues following dual AAV9 ABE editing.
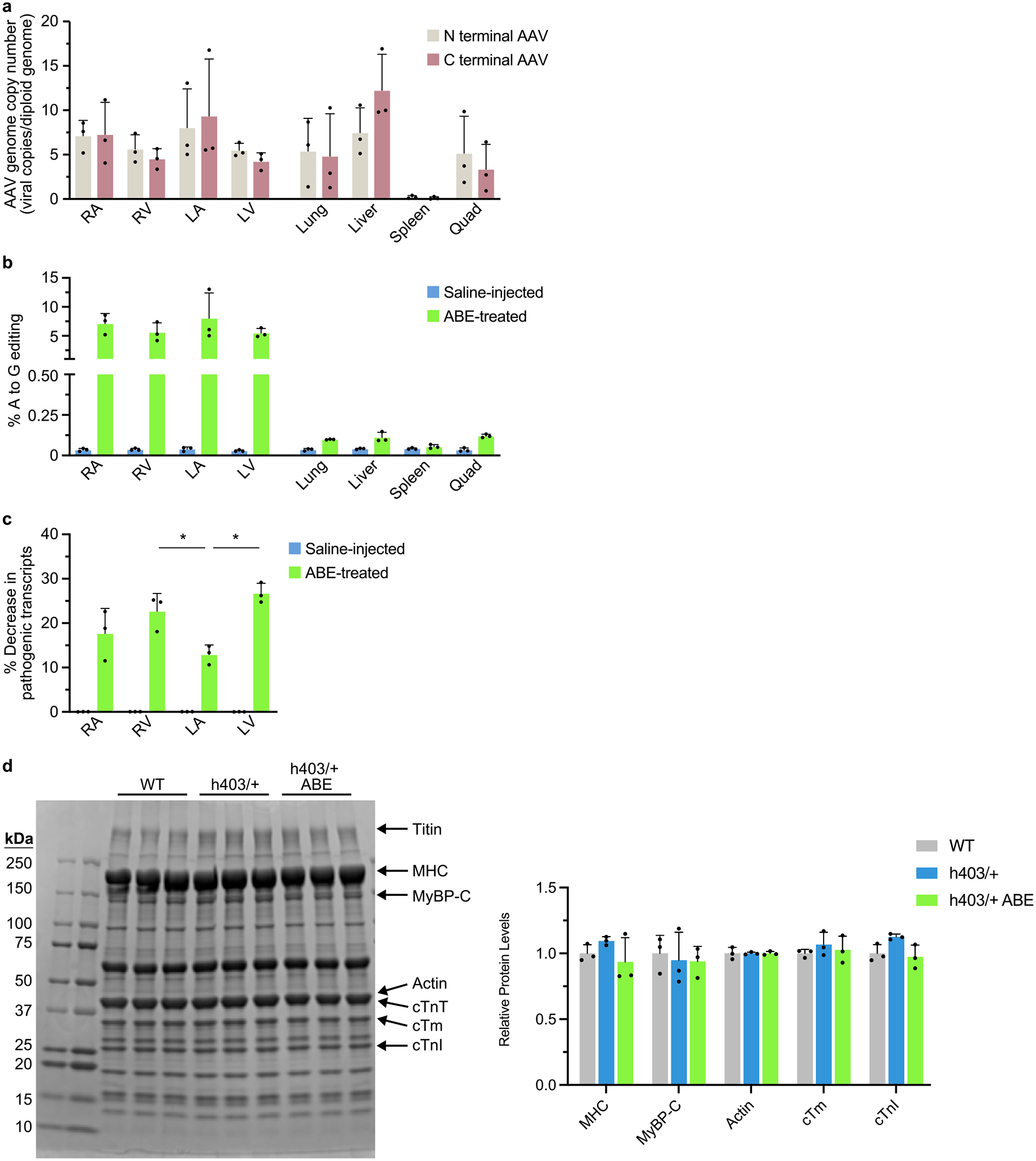
a, Viral copy numbers for the N terminal AAV and C terminal AAV were quantified from the right atrium (RA), right ventricle (RV), left atrium (LA), left ventricle (LV), lung, liver, spleen, and quadriceps muscle (Quad) from ABE-treated Myh6h403/+ mice at 16 weeks of age. b, The percentage of A to G editing was determined by HTS of genomic DNA in the RA, RV, LA, LV, lung, liver, spleen and Quad from ABE-treated and saline-injected Myh6h403/+ mice. c, The percentage decrease in mutant transcripts in the RA, RV, LA, and LV was determined by HTS of cDNA from ABE-treated and saline-injected Myh6h403/+ mice. The percentage decrease was greater in the RV (22.7%, P = 0.0202) and the LV (26.7%, P = 0.00157) compared to the LA (12.9%). d, Cardiac myofibrils were isolated from Myh6WT mice, Myh6h403/+ mice, and ABE-treated Myh6h403/+ mice, run on a 4–20% polyacrylamide gel, and stained with Coomassie G-250. Key sarcomeric proteins are marked, including titin, myosin heavy chain (MHC), myosin binding protein C (MyBP-C), actin, cardiac troponin T (cTnT), cardiac tropomyosin (cTm), and cardiac troponin I (cTnI). Sizes for ladder markings are in kDa. Relative protein amounts for each key sarcomeric protein are normalized to WT. Data are mean ± s.d. *P < 0.05 by Student’s unpaired two-sided t-test, n = 3 male mice for each group.
Extended Data Fig. 7 |. RNA-sequencing analysis of dual AAV9 ABE editing of Myh6h403/+ mice.
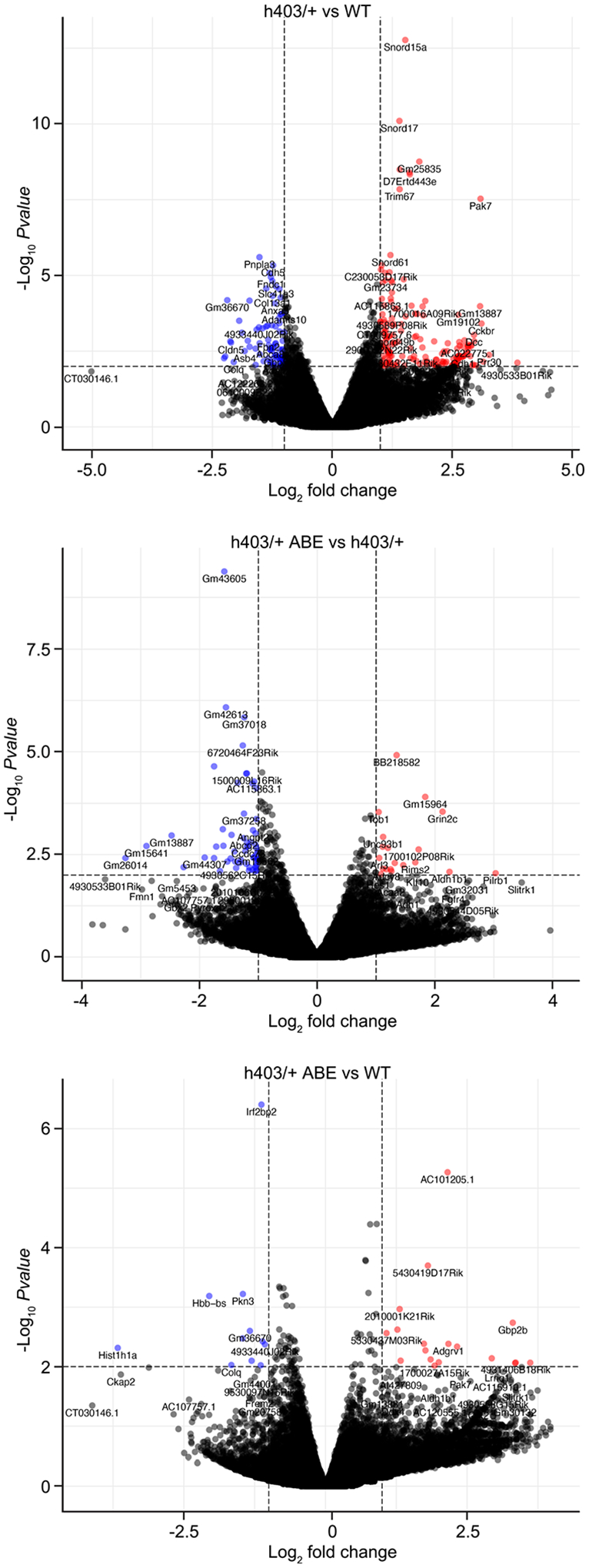
Volcano plot showing fold-change and p-value of genes up-regulated (red) and down-regulated (blue) in Myh6h403/+ mice compared to Myh6WT mice (top), ABE-treated Myh6h403/+ mice compared to Myh6h403/+ mice (middle), and ABE-treated Myh6h403/+ mice compared to Myh6WT mice (bottom). P-adjust < 0.01 and fold-change > 2.0 were used for cutoffs; p-adjust values are calculated by two-tailed Wilcoxon rank-sum test. n = 3 male mice for each group.
Supplementary Material
Acknowledgements
We thank the members of the E.N.O. laboratory for helpful discussions; J. C. Wu and the Stanford Cardiovascular Institute for providing the HCM patient-derived iPSCs; C. Seidman and H. Wakimoto for suggestions on intrathoracic AAV9 delivery; T. Lanigan, H. Kopera and R. Agate from the University of Michigan Vector Core for rAAV9 production; C. Llamas and P. Mishra from the Children’s Medical Center Research Institute for help with Seahorse assays; J. Cabrera and S. Vargas for graphics; D. Martin from Envigo for custom chow consultation; J. Xu and Y. J. Kim from the Children’s Medical Center Research Institute for performing the Illumina NextSeq sequencing; C. Rodriguez-Caycedo for assistance with iPSCs; the UT Southwestern McDermott Center Sanger Sequencing Core; the UT Southwestern McDermott Center Next-Generation Sequencing Core; the UT Southwestern Flow Cytometry Core; and J. Shelton from the Molecular Histopathology Core for help with histology. This work was supported by grants from the National Institutes of Health (R01HL130253, P50HD087351 and R01HL157281 to E.N.O. and R.B.-D.; F30HL163915 to A.C.C.), the American Heart Association (907611 to A.C.C.), the Foundation Leducq Transatlantic Networks of Excellence in Cardiovascular Research and the Robert A. Welch Foundation (grant 1-0025 to E.N.O.). The E.N.O. laboratory is supported by CureHeart, the British Heart Foundation’s Big Beat Challenge Award (BBC/F/21/220106).
Footnotes
Code availability
The MATLAB code used to perform contractile force measurements of iPSC-CMs has been deposited to GitHub: https://github.com/DarisaLLC/Cardio.
Competing interests
E.N.O. is a consultant for Vertex Pharmaceuticals and Tenaya Therapeutics. The other authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41591-022-02176-5.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41591-022-02176-5.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper, extended data and supplementary material. Raw and analyzed RNA-seq data generated during this study are available in the Gene Expression Omnibus repository (http://www.ncbi.nlm.nih.gov/geo/) and are accessible through Gene Expression Omnibus series accession number GSE201755. DNA sequencing files can be accessed at the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA) with accession code PRJNA902011. The mm10 reference genome is available at https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/.
References
- 1.Maron BJ Clinical course and management of hypertrophic cardiomyopathy. N. Engl. J. Med 379, 655–668 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Semsarian C, Ingles J, Maron MS & Maron BJ New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol 65, 1249–1254 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Trivedi DV, Adhikari AS, Sarkar SS, Ruppel KM & Spudich JA Hypertrophic cardiomyopathy and the myosin mesa: viewing an old disease in a new light. Biophys. Rev 10, 27–48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geisterfer-Lowrance AA et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 62, 999–1006 (1990). [DOI] [PubMed] [Google Scholar]
- 5.Tyska MJ et al. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ. Res 86, 737–744 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Sarkar SS et al. The hypertrophic cardiomyopathy mutations R403Q and R663H increase the number of myosin heads available to interact with actin. Sci. Adv 6, eaax0069 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaudelli NM et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koblan LW et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature 589, 608–614 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suh S et al. Restoration of visual function in adult mice with an inherited retinal disease via adenine base editing. Nat. Biomed. Eng 5, 169–178 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chemello F et al. Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci. Adv 7, eabg4910 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reichart D et al. Efficient in vivo genome editing prevents hypertrophic cardiomyopathy in mice. Nat. Med (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koblan LW et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol 36, 843–846 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richter MF et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol 38, 883–891 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walton RT, Christie KA, Whittaker MN & Kleinstiver BP Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 368, 290–296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishimasu H et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361, 1259–1262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kleinstiver BP et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marian AJ & Braunwald E Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ. Res 121, 749–770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Concordet JP & Haeussler M CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic. Acid. Res 46, W242–W245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pua CJ et al. Genetic studies of hypertrophic cardiomyopathy in Singaporeans identify variants in TNNI3 and TNNT2 that are common in Chinese patients. Circ. Genom. Precis. Med 13, 424–434 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toepfer CN et al. Myosin sequestration regulates sarcomere function, cardiomyocyte energetics, and metabolism, informing the pathogenesis of hypertrophic cardiomyopathy. Circulation 141, 828–842 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohn R et al. A contraction stress model of hypertrophic cardiomyopathy due to sarcomere mutations. Stem Cell Rep 12, 71–83 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vakrou S & Abraham MR Hypertrophic cardiomyopathy: a heart in need of an energy bar? Front. Physiol 5, 309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lyons GE, Schiaffino S, Sassoon D, Barton P & Buckingham M Developmental regulation of myosin gene expression in mouse cardiac muscle. J. Cell Biol 111, 2427–2436 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geisterfer-Lowrance AA et al. A mouse model of familial hypertrophic cardiomyopathy. Science 272, 731–734 (1996). [DOI] [PubMed] [Google Scholar]
- 26.Ma S et al. Efficient correction of a hypertrophic cardiomyopathy mutation by ABEmax-NG. Circ. Res 129, 895–908 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Ishikawa K, Weber T & Hajjar RJ Human cardiac gene therapy. Circ. Res 123, 601–613 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zettler J, Schutz V & Mootz HD The naturally split Npu DnaE intein exhibits an extraordinarily high rate in the protein trans-splicing reaction. FEBS Lett 583, 909–914 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Prasad KM, Xu Y, Yang Z, Acton ST & French BA Robust cardiomyocyte-specific gene expression following systemic injection of AAV: in vivo gene delivery follows a Poisson distribution. Gene Ther 18, 43–52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mendell JR et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther 29, 464–488 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teekakirikul P et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J. Clin. Invest 120, 3520–3529 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pinto AR et al. Revisiting cardiac cellular composition. Circ. Res 118, 400–409 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang J, Wakimoto H, Seidman JG & Seidman CE Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science 342, 111–114 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pare JA, Fraser RG, Pirozynski WJ, Shanks JA & Stubington D Hereditary cardiovascular dysplasia. A form of familial cardiomyopathy. Am. J. Med 31, 37–62 (1961). [DOI] [PubMed] [Google Scholar]
- 35.Fananapazir L & Epstein ND Genotype-phenotype correlations in hypertrophic cardiomyopathy. Insights provided by comparisons of kindreds with distinct and identical beta-myosin heavy chain gene mutations. Circulation 89, 22–32 (1994). [DOI] [PubMed] [Google Scholar]
- 36.Ho CY et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 138, 1387–1398 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carroll KJ et al. A mouse model for adult cardiac-specific gene deletion with CRISPR/Cas9. Proc. Natl Acad. Sci. USA 113, 338–343 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maron BJ, Yeates L & Semsarian C Clinical challenges of genotype positive (+)-phenotype negative (−) family members in hypertrophic cardiomyopathy. Am. J. Cardiol 107, 604–608 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Green EM et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 351, 617–621 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stern JA et al. A small molecule inhibitor of sarcomere contractility acutely relieves left ventricular outflow tract obstruction in feline hypertrophic cardiomyopathy. PLoS ONE 11, e0168407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ladage D, Ishikawa K, Tilemann L, Muller-Ehmsen J & Kawase Y Percutaneous methods of vector delivery in preclinical models. Gene Ther 19, 637–641 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Cheng Q et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol 15, 313–320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banskota S et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell 185, 250–265 e216 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tabebordbar M et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 184, 4919–4938 e4922 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weinmann J et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat. Commun 11, 5432 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lompre AM et al. Species- and age-dependent changes in the relative amounts of cardiac myosin isoenzymes in mammals. Dev. Biol 84, 286–290 (1981). [DOI] [PubMed] [Google Scholar]
- 47.Desai MY et al. Study design and rationale of VALOR-HCM: evaluation of mavacamten in adults with symptomatic obstructive hypertrophic cardiomyopathy who are eligible for septal reduction therapy. Am. Heart J 239, 80–89 (2021). [DOI] [PubMed] [Google Scholar]
- 48.Saberi S et al. Mavacamten favorably impacts cardiac structure in obstructive hypertrophic cardiomyopathy: EXPLORER-HCM Cardiac Magnetic Resonance Substudy Analysis. Circulation 143, 606–608 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Keam SJ Mavacamten: First Approval. Drugs 82, 1127–1135 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ho CY et al. Evaluation of mavacamten in symptomatic patients with nonobstructive hypertrophic cardiomyopathy. J. Am. Coll. Cardiol 75, 2649–2660 (2020). [DOI] [PubMed] [Google Scholar]
References
- 51.Murphy E & Steenbergen C Gender-based differences in mechanisms of protection in myocardial ischemia-reperfusion injury. Cardiovasc. Res 75, 478–486 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang TP et al. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat. Biotechnol 37, 626–631 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Levy JM et al. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng 4, 97–110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burridge PW et al. Chemically defined generation of human cardiomyocytes. Nat. Method 11, 855–860 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Correia C et al. Distinct carbon sources affect structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Sci. Rep 7, 8590 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kluesner MG et al. EditR: a method to quantify base editing from Sanger sequencing. CRISPR J 1, 239–250 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Atmanli A et al. Cardiac myoediting attenuates cardiac abnormalities in human and mouse models of Duchenne muscular dystrophy. Circ. Res 129, 602–616 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kijlstra JD et al. Integrated analysis of contractile kinetics, force generation, and electrical activity in single human stem cell-derived cardiomyocytes. Stem Cell Rep 5, 1226–1238 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clement K et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miura H, Quadros RM, Gurumurthy CB & Ohtsuka M Easi-CRISPR for creating knock-in and conditional knockout mouse models using long ssDNA donors. Nat. Protoc 13, 195–215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Creed HA & Tong CW Preparation and identification of cardiac myofibrils from whole heart samples. Methods Mol. Biol 2319, 15–24 (2021). [DOI] [PubMed] [Google Scholar]
- 64.Cui M & Olson EN Protocol for single-nucleus transcriptomics of diploid and tetraploid cardiomyocytes in murine hearts. STAR Protoc 1, 100049 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou Y et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper, extended data and supplementary material. Raw and analyzed RNA-seq data generated during this study are available in the Gene Expression Omnibus repository (http://www.ncbi.nlm.nih.gov/geo/) and are accessible through Gene Expression Omnibus series accession number GSE201755. DNA sequencing files can be accessed at the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA) with accession code PRJNA902011. The mm10 reference genome is available at https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/.