

Abstract
Aims
Several patients with heart failure and reduced ejection fraction (HFrEF) do not receive renin–angiotensin–aldosterone system (RAAS) inhibitors at the recommended dose or at all, frequently due to actual or feared hyperkalaemia. Sodium zirconium cyclosilicate (SZC) is an orally administered non‐absorbed intestinal potassium binder proven to lower serum potassium concentrations.
Methods and results
PRIORITIZE‐HF was an international, multicentre, parallel‐group, randomized, double‐blind, placebo‐controlled study to evaluate the benefits and risks of using SZC to intensify RAAS inhibitor therapy. Patients with symptomatic HFrEF were eligible and randomly assigned to receive SZC 5 g or placebo once daily for 12 weeks. Doses of study medication and RAAS inhibitors were titrated during the treatment period. The primary endpoint was the proportion of patients at 12 weeks in the following categories: (i) any RAAS inhibitor at less than target dose, and no MRA; (ii) any RAAS inhibitor at target dose and no MRA; (ii) MRA at less than target dose; and (iv) MRA at target dose. Due to challenges in participant management related to the COVID‐19 pandemic, the study was prematurely terminated with 182 randomized patients. There was no statistically significant difference in the distribution of patients by RAAS inhibitor treatment categories at 3 months (P = 0.43). The proportion of patients at target MRA dose was numerically higher in the SZC group (56.4%) compared with the placebo group (47.0%). Overall, SZC was well tolerated.
Conclusions
PRIORITIZE‐HF was terminated prematurely due to COVID‐19 and did not demonstrate a statistically significant increase in the intensity of RAAS inhibitor therapies with the potassium‐reducing agent SZC compared with placebo.
Keywords: Heart failure with reduced ejection fraction, RAAS inhibitors, Guideline‐directed medical therapy, Hyperkalaemia, Sodium zirconium cyclosilicate
Introduction
Heart failure with reduced ejection fraction (HFrEF) continues to be a major cause of mortality, hospitalization, and impaired quality of life despite advances in its management. These issues are compounded by the increase in prevalence and incidence of heart failure globally. 1 Renin–angiotensin–aldosterone system (RAAS) inhibitors, including angiotensin‐converting enzyme inhibitors (ACEi), angiotensin receptor blockers (ARB), and mineralocorticoid receptor antagonists (MRA) unequivocally reduce the risk of death and hospitalization in patients with HFrEF. Treatment guidelines recommend the administration of an ACEi, ARB or angiotensin receptor, and neprilysin inhibitor (ARNI), as well as an MRA, to patients with heart failure and a left ventricular ejection fraction (LVEF) less than 40%, and more recently, this recommendation has been extended to patients with a mildly reduced ejection fraction between 40% and 50%. 2 , 3 Unfortunately, a sizeable proportion of patients do not receive RAAS inhibitors at the recommended dose or at all, frequently due to actual or feared hyperkalaemia. 4 , 5
Renal function often declines with heart failure, and the ensuing decrease in glomerular filtration rate leads to a reduction in potassium excretion. In addition, many commonly used treatments for HFrEF, including ACEi, ARB, and MRA, predispose patients to hyperkalaemia by decreasing aldosterone production or interfering with its effect, which reduces potassium excretion. Hyperkalaemia occurs in at least 12% of patients treated for heart failure, and the risk is higher in those with comorbidities such as advanced chronic kidney disease and diabetes. 6 Hyperkalaemia is associated with a higher risk of cardiovascular death in patients with heart failure, including the risk of sudden death. Even mild elevations of serum potassium are associated with higher mortality, but a causal relation remains to be established. 7 , 8
Sodium zirconium cyclosilicate (SZC) is an orally administered non‐absorbed intestinal potassium binder proven to lower serum potassium concentrations. 9 SZC exchanges potassium for sodium and hydrogen in the intestinal lumen. As SZC is an efficacious treatment of hyperkalaemia, it may facilitate treatment with RAAS inhibitors in patients unable to receive these agents (or where there is concern about prescribing them) and allow dose optimization. We conducted the Potassium Reduction Initiative to Optimize RAAS Inhibition Therapy With Sodium Zirconium Cyclosilicate in Heart Failure (PRIORITIZE‐HF) study to assess whether SZC would allow RAAS inhibitors to be initiated (in the case of an MRA) and safely uptitrated to target doses without inducing clinically significant hyperkalaemia.
Methods
Trial design
This was an international, multicentre, parallel‐group, randomized, double‐blind, placebo‐controlled Phase 2 study to evaluate the benefits and risks of using SZC to intensify RAAS inhibitor therapy in patients with heart failure under‐treated with an ACEi, ARB or ARNI, and MRA, without inducing clinically significant hyperkalaemia. The trial protocol was designed by the study executive committee in conjunction with the sponsor, AstraZeneca, who funded the trial. The protocol was approved by the institutional review board at all centres involved in the nine countries that participated in the trial (NCT03532009).
The sponsor was responsible for all study support activities including project coordination, data management, site monitoring, and statistical analyses. All patients provided written informed consent. The randomization list was computer‐generated by an unblinded biostatistician and uploaded in the interactive response system. The database was a validated electronic data capture system (eCRF) using InForm 6.0 provided by Oracle. All eCRF users were trained as per completion guidelines, and the data entry was performed by the study staff. The data cleaning activities were performed as per the data management plan. The trial was overseen by a data monitoring committee of independent experts. The study medication and matching placebo were provided by the study sponsor.
The first author (J. C. T.) prepared the first draft of the manuscript; all members of the executive committee had full access to the generated statistical analyses and made the decision to submit the manuscript for publication. All authors assume responsibility for the accuracy and completeness of the data and analyses and for the fidelity to the protocol.
Trial population
Adult women (practicing adequate contraception and with a negative pregnancy test at baseline, or without child‐bearing potential) or men were eligible if they had a documented diagnosis of symptomatic HFrEF [New York Heart Association (NYHA) functional Class II to IV] that had been present for at least three months, a LVEF equal to or less than 40% within the past 12 months, were receiving background standard of care and treated according to locally recognized guidelines with both drugs and devices as appropriate. Treatment with RAAS inhibitors and a beta‐blocker had to be stable for at least four weeks before randomization. Patients needed to be treated with an ACEi, ARB or ARNI to be eligible for enrollment. Participants could either be taking a low dose of spironolactone or eplerenone (less than or equal to 12.5 mg once daily or 25 mg every other day) or not receiving an MRA. If patients were taking a low dose of MRA, the rationale had to be that the patient could not tolerate a higher dose due to documented hyperkalaemia observed at higher doses.
At baseline, patients were required to have mild hyperkalaemia or be at risk of developing hyperkalaemia during the study defined as follows: estimated glomerular filtration rate (eGFR) 20 to 44 mL/min/1.73 m2 [calculated using the Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) equation] and serum potassium concentration between 4.0 and 5.5 mmol/L, or eGFR 45 to 59 mL/min/1.73 m2 and serum potassium between 5.1 and 5.5 mmol/L, or eGFR 45 to 59 mL/min/1.73 m2 and serum potassium concentration between 4.0 and 5.0 mmol/L, and prior documented serum potassium concentration higher than 5.0 mmol/L attributed to use of a RAAS inhibitor.
Patients were excluded if they had heart failure due to restrictive cardiomyopathy, active myocarditis, constrictive pericarditis, obstructive hypertrophic cardiomyopathy, or uncorrected primary valvular disease; acute decompensated heart failure within 4 weeks prior to enrolment; acute coronary syndrome, stroke, or transient ischaemic attack within 12 weeks prior to enrolment; coronary revascularization or cardiac valvular intervention performed within 12 weeks prior to enrolment or planned after randomization; implantation of a cardiac resynchronization therapy device or cardiac defibrillator within 12 weeks prior to enrolment or intent to perform such procedure; previous cardiac transplantation or implantation of a ventricular assist device, or transplantation or implantation expected after randomization; symptomatic bradycardia or second‐degree (Mobitz Type 2) or third‐degree heart block without a pacemaker; symptomatic hypotension or systolic blood pressure less than 95 mmHg on two consecutive measurements; receiving dialysis or anticipated to require such therapy during the study; prior history of hypersensitivity to a RAAS inhibitor; hypoaldosteronism; active malignancy requiring treatment; any condition outside cardiovascular and renal diseases with a life expectancy of less than 2 years; treatment with any potassium binding resin within 7 days prior to the first dose of study medication; treatment with potassium supplements within 7 days prior to randomization; family history of long QT syndrome, presence of cardiac arrhythmias or conduction defects that require immediate treatment, or a QTc interval of 550 ms or more.
Trial procedures
Blinded randomization was centralized and performed electronically through an automated interactive response system. The allocation sequence was computer‐generated in a 1:1 ratio scheme without stratification. Patients were randomly assigned to receive SZC 5 g or placebo once daily for 12 weeks. Those with serum potassium concentration greater than 5.0 mmol/L at the last assessment before randomization received SZC 10 g or placebo three times daily for the first 2 days according to their random study group assignment. All staff involved, including study investigators and nurses, and patients were blinded to the treatment received. Clinical evaluations and measurements of serum electrolytes and creatinine were performed at 1, 2, 3, 5, 7, 9, and 13 weeks. Doses of study medication (SZC or placebo, Supporting information, Table S1 ) and RAAS inhibitor(s) (Table S2 ) were titrated during the study treatment period up to the 12 week visit, directing investigators to titrate the latter to target doses.
The study was paused from May to October 2019 and went through a major protocol amendment, which included updates to the inclusion criteria (patients with low‐dose MRA and/or lower serum potassium values could be enrolled) as well as guidance on treatment uptitration starting with MRA followed by ACEi, ARB, or ARNI.
Endpoints
The primary efficacy endpoint was the proportion of patients at 12 weeks in the following categories: (i) any of ACEi, ARB, or ARNI at less than target dose (including no treatment), and no MRA; (ii) any of ACEi, ARB, or ARNI at target dose and no MRA; (iii) MRA at less than target dose; and (4) MRA at target dose. Exploratory endpoints were the proportions of patients in the same categories according to serum potassium concentrations above 5.0 mmol/L, and 5.0 mmol/L or less at the time of randomization; the proportion of patients treated with MRA and those at target dose at 12 weeks; the proportion of patients on higher doses of RAAS inhibitors compared with baseline; a dose intensity score 10 ; changes from baseline in blood pressure, NYHA class, Patient Global Impression of Change Questionnaire, the overall summary score of the Kansas City Cardiomyopathy Questionnaire; NT‐pro‐BNP and troponin; and urine albumin to creatinine ratio in patients with albuminuria at baseline. Safety endpoints included adverse events, serious adverse events, changes on the electrocardiogram, serum creatinine, other laboratory parameters, and the numbers of patients and events with abnormal serum kalaemia concentrations (greater than 5.5 and 6.0 mmol/L and less than 3.5 and 3.0 mmol/L).
Statistical analysis
A sample size of 140 patients per group provided at least 90% power to detect a between‐groups difference of 20% in the proportion of patients treated with an MRA at 3 months, at a significance level of 5%, using a χ 2 test. Due in large part to challenges in participant management related to the COVID‐19 pandemic, the study was prematurely terminated with a total sample size of 182 randomized patients.
All efficacy analyses were based on all randomized patients. The analysis of the primary efficacy endpoint was a test of the equality of distributions, with the null hypothesis that there was no difference between treatments in the distribution of proportion of patients in the four RAAS inhibitor treatment categories. The hypothesis was tested at a two‐sided significance level of 5%. To mitigate the impact of missing data caused by, among other factors, study termination due to COVID‐19, treatment categories at 3 months for patients where the information regarding renin–angiotensin–aldosterone inhibitor dosage was missing were imputed using multiple imputation. The imputation model included RAAS inhibitor category at 3 months as the outcome variable and the following covariates: RAAS inhibitor treatment category, serum potassium, eGFR, and systolic blood pressure measured at the last visit before 3 months when all the covariate data were available. For each of the imputed datasets, a χ 2 test was performed with the corresponding test statistics pooled and used in an F‐test. 11
Sensitivity analyses of the primary efficacy endpoint were performed by (i) using only available non‐imputed RAAS inhibitor dose data at 3 months, (ii) using the worse of the last two dose observations carried forward, and (iii) removing the observations potentially affected by COVID‐19, defined as those obtained during visits on dates later than 11 March 2020.
No formal statistical inference was planned for exploratory endpoints; therefore, no correction for multiplicity was applied. As such, confidence intervals provided for any of the exploratory variables should be interpreted with caution. Except for subgroup analyses of the primary endpoint, exploratory variables were based on available data at the visits in question.
Safety analyses included all randomized patients who took at least one dose of study medication (SZC or placebo). Adverse events and serious adverse events were summarized by intensity and relatedness. Number and percentage of patients with events related to fluid overload or oedema were also evaluated and defined by the following terms: fluid overload, fluid retention, generalized oedema, hypervolemia, localized oedema, oedema, and oedema peripheral and peripheral swelling. Summaries of clinical chemistry and haematology variables were based on samples analysed at the central laboratory. Number and percentage of patients with serum potassium less than 3.5, 3.0, and 2.5 mmol/L, as well as those with values greater than 5.0, 5.5, 6.0, and 6.5 mmol/L, were also determined.
To assess the degree of missingness related to the COVID‐19 pandemic, the number and proportion of patients attending the different study visits and those remaining on treatment were tabulated for visits occurring after 11 March 2020.
Results
Patients
A total of 182 patients were randomly assigned to the SZC group (92 patients) or the placebo group (90 patients). The majority of patients completed the study [SZC: 90 patients (97.8%); placebo: 86 patients (95.6%)]. Patients who completed the end‐of‐treatment visit early due to the decision to prematurely terminate the study because of the COVID‐19 pandemic were considered to have completed. More than half of randomized patients completed the intended 3 months of treatment [SZC: 57/92 (62.0%); placebo: 51/90 patients (56.7%)]. The most common reason for premature discontinuation of treatment was the decision to prematurely terminate the study due to the pandemic [SZC: 19 patients (20.7%); placebo: 26 patients (28.9%)].
Baseline patient characteristics are shown in Table 1 . The mean age of patients was 71.9 years, the majority were male (59.3%), White (98.4%), and in NYHA Class II (64.8%). Mean LVEF was 33.8%. Most patients (62.1%) had an eGFR between 20 and 45 mL/min/1.73 m2, and mean serum potassium concentration was 4.86 mmol/L at baseline. Of the patients, 73% were normokalaemic at baseline. Duration of exposure to the study medication was similar between the SZC and placebo groups (median duration of 80.0 and 77.5 days, respectively). The mean average daily dose of study medication was 6.9 g for the SZC group and 8.2 g for the placebo group.
Table 1.
Characteristics of the patients at baseline
| SZC (N = 92) | Placebo (N = 90) | ||
|---|---|---|---|
| Age (years) | Mean ± SD | 72.9 ± 8.8 | 71.0 ± 8.1 |
| Sex n (%) | Male | 51 (55.4) | 57 (63.3) |
| Race n (%) | White | 90 (97.8) | 89 (98.9) |
| Body‐mass index (kg/m2) | Mean ± SD | 29.6 ± 5.6 | 30.2 ± 5.6 |
| NYHA class n (%) | II | 61 (66.3) | 57 (63.3) |
| III | 31 (33.7) | 33 (36.7) | |
| LV ejection fraction (%) | Mean ± SD | 33.8 ± 5.8 | 33.9 ± 6.1 |
| NT‐proBNP (pmol/L) | Mean ± SD | 319.4 ± 428.3 | 288.5 ± 399.6 |
| Potassium (mmol/L)a | Mean ± SD | 4.85 ± 0.37 | 4.87 ± 0.33 |
| Potassium group (mmol/L)a | ≤5.0 | 65 (70.7) | 67 (74.4) |
| >5.0 | 27 (29.3) | 23 (25.6) | |
| eGFR (mL/min/1.73 m2)b | Mean ± SD | 40.0 ± 11.0 | 42.7 ± 11.5 |
| eGFR group (mL/min/1.73 m2)b | ≤20 | 2 (2.2) | 1 (1.1) |
| >20–≤45 | 60 (65.2) | 53 (58.9) | |
| >45–<60 | 26 (28.3) | 31 (34.4) | |
| ≥60 | 4 (4.3) | 5 (5.6) | |
| Diabetes mellitus n (%) | 40 (43.5) | 42 (46.7) | |
| Atrial fibrillation n (%) | 42 (45.7) | 42 (46.7) | |
| Hypertension n (%) | 86 (93.5) | 85 (94.4) | |
| ACE inhibitor n (%) | 43 (46.7) | 45 (50.0) | |
| Angiotensin II receptor blocker n (%) | 34 (37.0) | 28 (31.1) | |
| Sacubitril‐valsartan n (%) | 14 (15.2) | 16 (17.8) | |
| Mineralocorticoid antagonist n (%) | 16 (17.4) | 18 (20.0) | |
| Diuretic n (%) | 78 (84.8) | 77 (85.6) | |
| Beta‐blocker n (%) | 83 (90.2) | 83 (92.2) | |
Abbreviations: ACE, angiotensin‐converting enzyme; eGFR, estimated glomerular filtration rate; NT‐proBNP, N‐terminal pro–B‐type natriuretic peptide; NYHA, New York Heart Association.
Efficacy endpoints
Table 2 shows results for the primary efficacy analysis. There was no statistically significant difference in the distribution of patients by RAAS inhibitor treatment categories at 3 months (P = 0.43). The proportion of patients at target MRA dose was numerically higher in the SZC group compared with the placebo group (56.4% and 47.0%, respectively).
Table 2.
Primary endpoint of renin–angiotensin–aldosterone system inhibitor treatment categories at 3 months
| (%) of subjects a | Global test of no difference | |||||
|---|---|---|---|---|---|---|
| Group | n | No ACEi/ARB/ARNI or at less than target dose and no MRA | ACEi/ARB/ARNI at target dose and no MRA | MRA at less than target dose b | MRA at target dose b (50 mg daily) | Pooled P value |
| A—Primary analysis in the intent‐to‐treat population | ||||||
| SZC (N = 92) | 89 | 14.7 | 14.7 | 14.2 | 56.4 | 0.43 |
| Placebo (N = 90) | 87 | 13.5 | 15.1 | 24.5 | 47.0 | |
| B—Sensitivity analysis without imputation in the intent‐to‐treat population | ||||||
| SZC (N = 92) | 58 | 8 (13.8) | 9 (15.5) | 4 (6.9) | 37 (63.8) | 0.35 |
| Placebo (N = 90) | 49 | 6 (12.2) | 10 (20.4) | 8 (16.3) | 25 (51.0) | |
| C—Sensitivity analysis with last value carry forward in the intent‐to‐treat population | ||||||
| SZC (N = 92) | 92 | 16 (17.4) | 15 (16.3) | 16 (17.4) | 45 (48.9) | 0.42 |
| Placebo (N = 90) | 90 | 16 (17.8) | 15 (16.7) | 24 (26.7) | 35 (38.9) | |
| D—Sensitivity analysis related to COVID‐19 (prior to 11 March 2020, ITT population) | ||||||
| SZC (N = 92) | 76 | 14.4 | 15.9 | 15.8 | 53.9 | 0.62 |
| Placebo (N = 90) | 81 | 13.3 | 16.3 | 24.9 | 45.6 | |
Abbreviations: ACEi, angiotensin‐converting enzyme inhibitors; ARB, angiotensin receptor blockers; ARNI, angiotensin receptor and neprolysin inhibitors; MRA, mineralocorticoid receptor antagonist; n, number of subjects in analysis.
Multiple imputation technique was used to impute missing values at 3 months. Pooled P value was obtained from F distribution with combined results from χ 2 tests on imputed individual data sets. Proportion of subjects in each category was obtained by pooling/averaging the proportion from individual data sets, each calculated using n as denominator.
Irrespective of ACEi, ARB, or ARNi dose.
The results of the sensitivity analyses of the primary endpoint using only available non‐imputed RAAS inhibitor dose data at 3 months (complete case analysis) and using the worse of the last two dose observations carried forward are presented in Table 2 . Consistent with the primary analysis, the sensitivity analyses did not show a statistically significant difference between treatment groups, although the SZC group had a numerically higher proportion of patients at target MRA dose compared with the placebo group. A sensitivity analysis was performed restricting results to only those obtained prior to 11 March 2020 (in relation to the COVID‐19 pandemic), which were consistent with the primary analysis.
As post‐hoc subgroup analyses, sensitivity analyses of the primary endpoint with no imputation are presented by protocol version (date of randomization earlier than 12 June 2019 or on or after 12 June 2019). Consistent with the full analysis, when analysed based on available data, there was no statistically significant difference between treatment groups for any of the subgroups. For patients randomized earlier than 12 June 2019, the difference between the two treatment groups in the proportion of patients at MRA target dose at 3 months (in favour of SZC) was larger than in the full analysis (SZC: 58.6%; placebo: 30.4%). For patients randomized on or after 12 June 2019, there was no notable difference between the treatment groups in the proportion of patients at target MRA dose (SZC: 69.0%; placebo: 69.2%).
Subgroup analyses were performed considering the serum potassium concentration before randomization. For patients with a baseline potassium concentration higher than 5.0 mmol/L, the proportions at target MRA dose were 36.5% in the SZC group and 24.5% in the placebo group. For those with a baseline potassium equal to or less than 5.0 mmol/L, the proportions of patients at target MRA dose were 65.6% and 55.0% in the SZC and placebo groups, respectively.
A summary of RAAS inhibitor dose intensity scores during the study is shown in Table 3 . The majority of patients in both treatment groups were not taking MRA at baseline [score value 0; SZC: 78 patients (84.8%); placebo: 74 patients (82.2%)]. At 3 months, the proportions of patients who had titrated to MRA target dose (score value: 3) were 63.8% in the SZC group and 51.0% in the placebo group.
Table 3.
Renin–angiotensin–aldosterone system inhibitor therapy intensity at 3 months (intent‐to‐treat population)
| Number (%) of subjects | ||||||
|---|---|---|---|---|---|---|
| SZC (N = 92) | Placebo (N = 90) | |||||
| n = 58 | n = 49 | |||||
| Ranking of RAASi therapy intensity | ACEi/ARB/ARNI | MRA | ||||
| 0 | No dose | No dose | 2 | (3.4) | 2 | (4.1) |
| 1 | Some | No dose | 6 | (10.3) | 4 | (8.2) |
| 2 | No dose | Some | 0 | (0.0) | 0 | (0.0) |
| 3 | Target | No dose | 9 | (15.5) | 10 | (20.4) |
| 4 | Some | Some | 2 | (3.4) | 5 | (10.2) |
| 5 | No dose | Target | 1 | (1.7) | 0 | (0.0) |
| 6 | Target | Some | 2 | (3.4) | 3 | (6.1) |
| 7 | Some | Target | 14 | (24.1) | 8 | (16.3) |
| 8 | Target | Target | 22 | (37.9) | 17 | (34.7) |
Mean serum potassium concentrations during the treatment period were numerically lower in patients randomized to the SZC group than those in the placebo group (Figure 1 ). At end of treatment, mean serum potassium was 4.65 and 4.90 mmol/L in the SZC and placebo groups, respectively. The proportions of normokalaemic patients were numerically higher across all post‐baseline on‐treatment time points in the SZC group compared with the placebo group. At end of treatment, 80.4% of patients in the SZC group were normokalaemic compared with 63.6% of patients in the placebo group.
Figure 1.
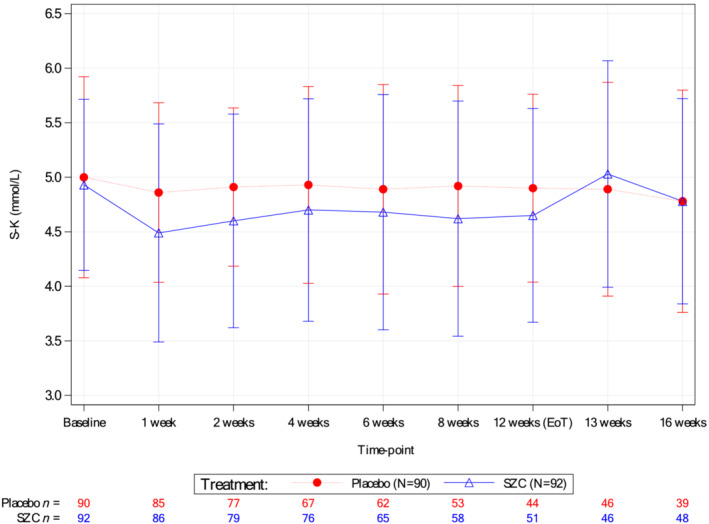
Mean serum potassium concentrations over time (intent‐to‐treat population).
Mean NT‐pro‐BNP at baseline was 2707 and 2445 ng/L for the SZC and placebo groups, respectively. There were no clinically meaningful differences in NT‐pro‐BNP and troponin biomarkers over time between the treatment groups (data not shown). No differences were detected between the SZC and placebo groups in the estimated GFR and urine albumin to creatinine ratio at 3 months (data not shown).
Most patients remained in the same NYHA class at baseline and 3 months. Only one patient worsened NYHA class in each group (both from Class II to III). Table 4 shows a summary of patient global impression of change in heart failure symptoms at baseline and end‐of‐study. Table 5 shows corresponding overall summary scores, total symptom scores, and clinical summary scores from the Kansas City Cardiomyopathy questionnaire. The mean scores showed a numerically greater improvement from baseline to 3 months in the SZC group compared with the placebo group [difference in means for overall summary score: 5.20, 95% CI (−0.17, 10.57); for TSS: 3.28, 95% CI (−3.59, 10.15); and for CSS: 6.04, 95% CI (0.21, 11.88)]. Results were similar for patients randomized before or after 12 June 2019.
Table 4.
Patient global impression of change at 3 months
| PGIC class | Number (%) of subjects | |||
|---|---|---|---|---|
| SZC (N = 92) | Placebo (N = 90) | |||
| Total | 53 | 47 | ||
| Subject evaluation of change in heart failure symptoms | ||||
| Much better | 10 | (18.9) | 4 | (8.5) |
| Moderately better | 8 | (15.1) | 8 | (17.0) |
| A little better | 22 | (41.5) | 11 | (23.4) |
| About the same | 12 | (22.6) | 23 | (48.9) |
| A little worse | 1 | (1.9) | 1 | (2.1) |
| Moderately worse | 0 | (0) | 0 | (0) |
| Much worse | 0 | (0) | 0 | (0) |
Abbreviations: N, number of patients in treatment group; PGIC, Patient Global Impression of Change.
Note: The percentages are calculated using the total number of subjects with non‐missing data as denominator.
Table 5.
Change in Kansas City cardiomyopathy questionnaire from baseline to 3 months
| Group | Baseline | 3 month change from baseline | |||||
|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | Difference in means | 95% CI | ||
| Overall summary score | SZC | 90 | 57.69 (20.00) | 53 | 9.94 (14.62) | 5.20 | (−0.17, 10.57) |
| Placebo | 87 | 60.78 (21.10) | 47 | 4.74 (12.12) | |||
| Total symptom score | SZC | 90 | 61.21 (23.31) | 53 | 10.36 (16.84) | 3.28 | (−3.59, 10.15) |
| Placebo | 87 | 63.68 (23.11) | 47 | 7.08 (17.77) | |||
| Clinical summary score | SZC | 90 | 57.56 (21.66) | 53 | 9.54 (14.13) | 6.04 | (0.21, 11.88) |
| Placebo | 87 | 62.72 (21.93) | 47 | 3.50 (15.27) | |||
Abbreviation: CI, confidence interval.
Safety endpoints
One patient (1.1%) in each treatment group died during the study, both events were cardiac disorders not considered related to study medication. Overall, SZC was well tolerated during the study (Table 6 ). The incidence of adverse events was 47.3% in the SZC group and 52.2% in the placebo group. The most commonly reported adverse were chronic cardiac failure, chronic kidney disease, and viral upper respiratory tract infection in both groups. Most adverse events were mild or moderate in intensity.
Table 6.
Adverse events (safety population) a
| Number (%) of patients [a] | ||
|---|---|---|
| SZC (N = 91) | Placebo (N = 90) | |
| Any adverse event | 43 (47.3) | 47 (52.2) |
| Acute cardiac failure | 2 (2.2) | 1 (1.1) |
| Chronic cardiac failure | 9 (9.9) | 4 (4.4) |
| Chronic kidney disease | 6 (6.6) | 4 (4.4) |
| Viral upper respiratory tract infection | 6 (6.6) | 3 (3.3) |
| Hypotension | 4 (4.4) | 1 (1.1) |
| Nasopharyngitis | 4 (4.4) | 3 (3.3) |
| Constipation | 3 (3.3) | 2 (2.2) |
| Any serious adverse event (including events resulting in death) | 14 (15.4) | 10 (11.1) |
| Death | 1 (1.1) | 1 (1.1) |
| Infection | 2 (2.2) | 2 (2.2) |
| Cardiac disorders | 8 (8.8) | 5 (5.6) |
| Acute myocardial infarction | 2 (2.2) | 1 (1.1) |
| Unstable angina | 0 (0.0) | 1 (1.1) |
| Atrial fibrillation | 1 (1.1) | 0 (0.0) |
| Acute cardiac failure | 2 (2.2) | 1 (1.1) |
| Chronic cardiac failure | 2 (2.2) | 2 (2.2) |
| Myocardial fibrosis | 1 (1.1) | 0 (0.0) |
Number of patients with adverse events that occurred during the study, on and off treatment, among patients who received at least one dose of the study medication.
Abbreviation: SZC, sodium zirconium cyclosilicate.
The incidence of serious adverse events was 15.4% in the SZC group and 11.1% in the placebo group. One serious adverse event (generalized oedema) in the SZC group was considered related to study medication and occurred along with a concurrent adverse event of chronic cardiac failure following an accidental overdose of SZC and led to its discontinuation. The only other adverse event (nausea) causing discontinuation of study medication was considered related to study medication in one patient of the SZC group.
An oedema‐related adverse event occurred in 3 patients (3.3%) in the SZC group and 1 patient (1.1%) in the placebo group. There were numerically more patients who developed worsening heart failure in the SZC group [11 patients (12.1%), of which 9 patients (9.9%) reported events while on treatment] compared with the placebo group [5 patients (5.6%), of which 4 patients (4.4%) reported events while on treatment]. A similar proportions of patients in both treatment groups were hospitalized and received an intravenous diuretic (4 and 3 patients in the SZC and placebo groups, respectively, of which 2 patients in each treatment group were on treatment). Serum potassium values greater than 6.0 mmol/L were reported in 3.3% and 4.4% of patients in the SZC and placebo groups, respectively. Seven patients (7.7%) in the SZC group had a serum potassium concentration less than 3.5 mmol/L compared with no patient in the placebo group, and no value less than 3.0 mmol/L was reported in either group.
Discussion
In this exploratory, randomized, double‐blind, placebo‐controlled trial, we aimed to assess if a treatment regimen containing SZC would allow RAAS inhibitors to be uptitrated to target doses in patients with heart failure and elevated serum potassium concentration or at high risk of developing elevated serum potassium.
The World Health Organization characterized COVID‐19 as a pandemic in the first quarter of 2020. To minimize the risk of COVID‐19 infection in this vulnerable high‐risk population who required multiple study visits (including for monitoring of potassium), enrolment was stopped in March 2020, treatment with the study medication was ended early for all patients as of 1 April 2020, and the study was prematurely terminated. A total of 182 patients were randomized, far below the 280 patients originally planned and for which the primary efficacy endpoint was powered. More than half of the patients completed treatment, with 20.7% and 28.9% of patients in the SZC and placebo groups discontinuing treatment due to the COVID‐19 pandemic. There were no COVID‐19‐related concerns about the overall quality of the data collected during the study, and no patients were diagnosed with COVID‐19 during the study.
This study did not show evidence of a difference between the SZC and placebo groups in the primary endpoint of distribution of patient proportions in RAAS inhibitor treatment categories at 3 months. Sensitivity analyses using only available non‐imputed RAAS inhibitor dose data at 3 months, the worse of the last two dose observations carried forward, and results restricted to only those obtained before the onset of the COVID‐19 pandemic broadly supported the conclusions of the main analysis.
A possible reason for the less than desired facilitation of RAAS inhibitors during the study is that the protocol encouraged but did not mandate uptitration of agents. Other contributing factors may have included concerns of investigators (who did not know if patients were receiving SZC or placebo) about hyperkalaemia, kidney function, and hypotension related to initiation or uptitration of MRAs and/or uptitration of ACEi/ARB/ARNIs. Approximately 30% of patients did not have their dose of ACEi/ARB/ARNI increased, and approximately 14% of patients did not have an MRA initiated or the dose increased during the trial. This was reflected in investigators' reported reasons for not uptitrating RAASi, where concerns of hyperkalaemia tended to be more common in the placebo group while concerns of hypotension were more prominent in the SZC group. Furthermore, the smaller than expected difference between the two treatment groups in the proportion of patients reaching the target dose of MRA at 3 months (SZC: 56.4%; placebo: 47.0%) may have been due to a lower than predicted risk of developing hyperkalaemia, reflected by this high percentage of patients in the placebo group being able to reach guidelines‐recommended MRA dose without developing overt hyperkalaemia.
The overall efficacy results may also relate, at least in part, to the major changes in the study protocol made in 2019, as described above. In patients randomized before 12 June 2019, a notably higher proportion achieved MRA target dose at 3 months in the SZC group compared with the placebo group (52.7% and 29.6%, respectively), while the difference between the two groups was minimal in patients randomized on or after the date of the implementation of the updated protocol (SZC: 64.0%, placebo: 61.7%). In light of the high proportion of placebo‐treated patients enrolled under the modified protocol who were able to titrate to target MRA dose, this may indicate that the study population was not at a particularly high risk of developing hyperkalaemia. The observation that RAASi doses could be uptitrated in patients on placebo in the study suggests that more aggressive uptitration of RAASi in patients at risk of hyperkalaemia could be considered, provided that potassium levels are adequately monitored.
PRIORITIZE‐HF is one of several trials to examine the use of intestinal potassium binders to facilitate management of other disease states treated with inhibitors of the RAAS. Results from three randomized placebo‐controlled trials were published in 2014–2015, demonstrating that the intestinal potassium binders patiromer sorbitex calcium and SZC could control hyperkalaemia in patients with impaired kidney function, the majority of whom were treated with inhibitors of the RAAS to slow progression of CKD. 12 , 13 , 14 Another trial of 295 patients with resistant hypertension treated with spironolactone showed that those randomized to patiromer sorbitex calcium were significantly more likely to remain on the MRA at 12 weeks than those randomized to placebo (86% vs. 66%). 15 Patiromer is a potassium binder that exchanges potassium for calcium in the intestinal lumen.
The DIAMOND trial was intended to be a multinational, multicentre, double‐blind, placebo‐controlled, randomized withdrawal, parallel‐group study including approximately 2400 patients with HFrEF and hyperkalaemia (serum potassium higher than 5.0 mmol/L) on guideline‐directed therapy including ACEi/ARB/ARNI and MRA, with time to the first event of cardiovascular death or cardiovascular hospitalization as the primary composite endpoint. 16 That trial was modified owing to lower‐than‐anticipated enrollment and incidence of cardiovascular events due to the impact of the COVID‐19 pandemic. The updated primary endpoint in DIAMOND was the adjusted mean change in serum potassium level, which was 0.03 mmol/L in the patiromer group and 0.13 mmol/L with placebo (P < 0.001). 17 The REALIZE‐K trial is a prospective, randomized, double‐blind, placebo‐controlled, parallel‐group, multicentre Phase 4 study, which will evaluate SZC for the management of hyperkalaemia in patients with symptomatic HFrEF and who are taking optimized spironolactone guideline‐directed therapy (ClinicalTrials.gov Identifier: NCT04676646).
Despite the lack of a statistically significant difference in RAAS inhibitor treatment categories between the study groups, there is much to learn from the PRIORITIZE‐HF trial. The SZC group had numerically higher proportions of patients at target MRA dose at 3 months compared with the placebo group, and mean serum potassium concentrations were lower during the treatment period despite the fact that a higher proportion of SZC‐treated patients were receiving MRAs at target dose. Both patient‐reported outcomes (the Patient Global Impression of Change and Kansas City Cardiomyopathy Questionnaire) showed numerical improvements for SZC compared with placebo. For example, an improvement in heart failure symptoms as assessed by the patient global impression of change at 3 months was observed in 75.5% of patients in the SZC group and 48.9% of those in the placebo group. Of note, this study was the first controlled trial that examined the effect of SZC on patient‐reported outcomes in heart failure patients with elevated serum potassium concentrations or at high risk of developing elevated serum potassium.
The incidence of adverse events and serious adverse events was comparable between the treatment groups. oedema‐related events were consistent with the known safety profile of SZC. Numerically more patients in the SZC group developed worsening of heart failure during the study compared with the placebo group. Of note, these cases of worsening heart failure were based on investigators' assessment and were not stringently defined or adjudicated, were mostly mild or moderate in intensity, and resulted in similar proportions of patients in both treatment groups being hospitalized (4 and 3 patients in the SZC and placebo groups, respectively, of which 2 patients in each treatment group were on treatment). Even without an increased risk of hospitalization, such episodes if confirmed in future studies could affect the quality of life of patients. They might be related to the release of sodium in the intestinal lumen in exchange of potassium being captured by SZC. In conclusion, the PRIORITIZE‐HF study was terminated prematurely due to COVID‐19 and did not demonstrate a statistically significant increase in the intensity of RAAS inhibitor therapies with the potassium‐reducing agent SZC compared with placebo.
Funding
This work was supported by AstraZeneca.
Conflict of interest
Dr. Tardid reports grants and personal fees from AstraZeneca during the conduct of the study; grants from Amarin; grants from AstraZeneca; grants from Ceapro; grants, personal fees, and minor equity interest from Dalcor; grants from Esperion; personal fees from HLS Pharmaceuticals; grants from Ionis; grants from Novartis; personal fees from Pendopharm; grants from Pfizer; grants from RegenXBio, outside the submitted work; in addition, Dr. Tardif has a patent on Genetic markers for predicting responsiveness to therapy with HDL‐raising or HDL mimicking agent pending, a patent on Methods for using low‐dose colchicine after MI pending assigned to the Montreal Heart Institute, and a patent on Methods of treating a coronavirus infection using Colchicine pending assigned to the Montreal Heart Institute. Dr. Tardif has waived his rights in the colchicine patents and does not stand to gain financially.
Dr. Rouleau reports fees from AstraZeneca, Novartis, Bayer, BMS (consultancy). Dr. Chertow served on the Steering Committee of the PRIORITIZE‐HF trial. Drs. Al‐Shurbaji, Lisovskaja and Gustavson are full time employees and shareholders of AstraZeneca. Dr. Zhao is a full time employee of AstraZeneca. Dr. Bouabdallaoui reports fees from AstraZeneca (2019, consultancy). Dr. Desai reports research grant support to institution from Abbott, AstraZeneca, Alnylam, Bayer, Novartis, and Consulting Fees/Honoraria from Abbott, Alnylam, Amgen, AstraZeneca, Avidity, Bayer, Biofourmis, Boston Scientific, Cytokinetics, Dalcor Pharma, Lupin Pharma, Merck, Novartis, Relypsa, Regeneron, Sun Pharma, Verily. Dr. Chernyavskiy reports participating in the study as the site's principal investigator, PRIORITIZE‐HF6. Dr. Evsina reports no conflicts of interest. Dr. Merkely reports personal payment from AstraZeneca, Boehringer Ingelheim, and Novartis. Dr. McMurray declares payments from his employer, Glasgow University, for his work on clinical trials, consulting, and other activities: Alnylam, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, BMS, Cardurion, Cytokinetics, Dal‐Cor, GSK, Ionis, KBP Biosciences, Novartis, Pfizer, and Theracos; personal lecture fees: Abbott, Alkem Metabolics, AstraZeneca, Eris Lifesciences, Hikma, Lupin, Sun Pharmaceuticals, Medscape/Heart.Org, ProAdWise Communications, S & L Solutions Event Management Inc, Radcliffe Cardiology, Servier, the Corpus, Translational Medical Academy, Web MD, and (as Director) the Global Clinical Trial Partners Ltd (GCTP). Dr. Pfeffer reports research grant support from the following institutions: Novartis; consultant to: AstraZeneca, Boehringer Ingelheim, and Eli Lilly Alliance, Corvidia, DalCor, GlaxoSmithKline, Lexicon, NHLBI CONNECTs (Master Protocol Committee), Novartis, Novo Nordisk, Peerbridge, and Sanofi; and has equity in DalCor.
Supporting information
Table S1. Study medications (IP) SZC/placebo dose titration table.
Table S2. RAASi dose titration instructions.
Tardif, J.‐C. , Rouleau, J. , Chertow, G. M. , Al‐Shurbaji, A. , Lisovskaja, V. , Gustavson, S. , Zhao, Y. , Bouabdallaoui, N. , Desai, A. S. , Chernyavskiy, A. , Evsina, M. , Merkely, B. , McMurray, J. J. V. , and Pfeffer, M. A. (2023) Potassium reduction with sodium zirconium cyclosilicate in patients with heart failure. ESC Heart Failure, 10: 1066–1076. 10.1002/ehf2.14268.
References
- 1. Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Kichtman JH, Longenecker CT, Shane Loop M, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, Marma Perak A, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah AH, Sahy C, Spartano NL, Stokes A, Tirschwell DL, Van Wagner LB, Tsao CW, American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee . Heart Disease and Stroke Statistics–2020 update: a report from the American Heart Association. Circulation 2020; 141: e139‐e596. [DOI] [PubMed] [Google Scholar]
- 2. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, Drazner MH, Filippatos GS, Fonarow GC, Givertz MM, Hollenberg SM, Lindenfeld J, Masoudi FA, McBride PE, Peterson PN, Stevenson LW, Westlake C. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2017; 136: e137–e161. [DOI] [PubMed] [Google Scholar]
- 3. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Bohm M, Burri H, Butler J, Čelutkienė J, Chioncel O, Cleland JGF, Coats AJS, Crespo‐Leiro MG, Farmakis D, Gilard M, Heymans S, Hoes AW, Jaarsma T, Jankowska EA, Lainscak M, Lam CSP, Lyon AR, McMurray JJV, Mebazaa A, Mindham R, Muneretto C, Piepoli MF, Price S, Rosano GMC, Ruschitzka F, Skibelund AK, ESC Scientific Document Group . 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2021; 42: 3599–3726. [DOI] [PubMed] [Google Scholar]
- 4. Epstein M, Alvarez PJ, Reaven NL, Funk SE, McGaughey KJ, Brenner MS, Benton W, Golestaneh L. Evaluation of clinical outcomes and costs based on prescribed dose level of renin–angiotensin–aldosterone system inhibitors. Am J Manag Care. 2016; 22: s311–s324. [PubMed] [Google Scholar]
- 5. Epstein M, Pecoits‐Filho R, Clase CM, Sood MM, Kovesdy CP. Hyperkalemia with mineralocorticoid receptor antagonist use in people with CKD: understanding and mitigating the risks. Clin J Am Soc Nephrol. 2021; 17: CJN.13541021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kovesdy CP. Management of hyperkalemia: an update for the internist. Am J Med. 2015; 128: 1281–1287. [DOI] [PubMed] [Google Scholar]
- 7. Nakhoul GN, Huang H, Arrigain S, Jolly SE, Schold JD, Nally JV Jr, Navaneethan SD. Serum potassium, end‐stage renal disease and mortality in chronic kidney disease. Am J Nephrol. 2015; 41: 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Einhorn LM, Zhan M, Hsu VD, Walker LD, Moen MF, Seliger SL, Weir MR, Fink JC. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Intern Med. 2009; 169: 1156–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hoy SM. Sodium zirconium Cyclosilicate: a review in hyperkalaemia. Drugs. 2018; 78: 1605–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vasudevan A, Jazi HH, Won JI, Ball T, Patankar GR, Sarmast SA, Shin HJ, McCullough PA. Personalized treatment of heart failure with biomarker guidance using a novel disease severity score. Proc (Bayl Univ Med Cent). 2017; 30: 139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ratitch B, O'Kelly M, Tosiello R. Missing data in clinical trials: from clinical assumptions to statistical analysis using pattern mixture models. Pharm Stat. 2013; 12: 337–347. [DOI] [PubMed] [Google Scholar]
- 12. Kosiborod M, Rasmussen HS, Lavin P, Qunibi WY, Spinowitz B, Packham D, Roger SD, Yang A, Lerma E, Singh B. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia: the HARMONIZE randomized clinical trial. JAMA. 2014; 312: 2223–2233. [DOI] [PubMed] [Google Scholar]
- 13. Packham DK, Rasmussen HS, Lavin PT, El‐Shahawy MA, Roger SD, Block G, Qunibi W, Pergola P, Singh B. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med. 2015; 372: 222–231. [DOI] [PubMed] [Google Scholar]
- 14. Weir MR, Bakris GL, Bushinsky DA, Mayo MR, Garza D, Stasiv Y, Wittes J, Christ‐Schmidt H, Berman L, Pitt B, Investigators OPAL‐HK. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med. 2015; 372: 211–221. [DOI] [PubMed] [Google Scholar]
- 15. Agarwal R, Rossignol P, Romero A, Garza D, Mayo MR, Warren S, Ma J, White WB, Williams B. Patiromer versus placebo to enable spironolactone use in patients with resistant hypertension and chronic kidney disease (AMBER): a phase 2, randomised, double‐blind, placebo‐controlled trial. Lancet. 2019; 394: 1540–1550. [DOI] [PubMed] [Google Scholar]
- 16. Butler J, Anker SD, Siddiqi TJ, Coats AJS, Dorigotti F, Filippatos G, Friede T, Göhring UM, Kosiborod MN, Lund LH, Metra M, MorenoQuinn C, Pina IL, Pinto FJ, Rossignol P, Szecödy P, Van Der Meer P, Weir M, Pitt B. Patiromer for the management of hyperkalaemia in patients receiving renin–angiotensin–aldosterone system inhibitors for heart failure: design and rationale of the DIAMOND trial. Eur J Heart Fail. 2022; 24: 230–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Butler J, Anker SD, Lund LH, Coats AJS, Filippatos G, Siddiqi TJ, Friede T, Fabien V, Kosiborod M, Metra M, Pina IL, Pinto F, Rossignol P, van deer Meer P, Bahit C, Belohavek J, Bohm M, Brugts JJ, Cleland JGF, Ezekowitz J, Bayes‐Genis A, Gotsman I, Goudev A, Khintibidze I, Lindenfeld J, Mentz RJ, Merkely B, Castro Montes E, Mullens W, Nicolau JC, Parkhomenko A, Ponikowski P, Seferovic PM, Senni M, Shlyakhto E, Cohen‐Solal A, Szecsody P, Jensen K, Dorigotti F, Weir MR, Pitt B. Patiromer for the management of hyperkalemia in heart failure with reduced ejection fraction: the DIAMOND trial. Eur Heart J. 2022; 43: 4362–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Study medications (IP) SZC/placebo dose titration table.
Table S2. RAASi dose titration instructions.