

Abstract
Aims
A causal link between non‐ischaemic heart failure (HF) and humoral autoimmunity against G‐protein‐coupled receptors (GPCR) remains unclear except for Chagas' cardiomyopathy. Uncertainty arises from ambiguous reports on incidences of GPCR autoantibodies, spurious correlations of autoantibody levels with disease activity, and lack of standardization and validation of measuring procedures for putatively cardio‐pathogenic GPCR autoantibodies. Here, we use validated and certified immune assays presenting native receptors as binding targets. We compared candidate GPCR autoantibody species between HF patients and healthy controls and tested associations of serum autoantibody levels with serological, haemodynamic, metabolic, and functional parameters in HF.
Methods
Ninety‐five non‐ischaemic HF patients undergoing transcatheter endomyocardial biopsy and 60 healthy controls were included. GPCR autoantibodies were determined in serum by IgG binding to native receptors or a cyclic peptide (for β1AR autoantibodies). In patients, cardiac function, volumes, and myocardial structural properties were assessed by cardiac magnetic resonance imaging; right heart catheterization served for determination of cardiac haemodynamics; endomyocardial biopsies were used for histological assessment of cardiomyopathy and determination of cardiac mitochondrial oxidative function by high‐resolution respirometry.
Results
Autoantibodies against β1 adrenergic (β1AR), M5‐muscarinic (M5AR), and angiotensin II type 2 receptors (AT2R) were increased in HF (all P < 0.001). Autoantibodies against α1‐adrenergic (α1AR) and angiotensin II type 1 receptors (AT1R) were decreased in HF (all P < 0.001). Correlation of alterations of GPCR autoantibodies with markers of cardiac or systemic inflammation or cardiac damage, haemodynamics, myocardial histology, or left ventricular inflammation (judged by T2 mapping) were weak, even when corrected for total IgG. β1AR autoantibodies were related inversely to markers of left ventricular fibrosis indicated by T1 mapping (r = −0.362, P < 0.05) and global longitudinal strain (r = −0.323, P < 0.05). AT2R autoantibodies were associated with improved myocardial mitochondrial coupling as measured by high‐resolution respirometry in myocardial biopsies (r = −0.352, P < 0.05). In insulin‐resistant HF patients, AT2R autoantibodies were decreased (r = −.240, P < 0.05), and AT1R autoantibodies were increased (r = 0.212, P < 0.05).
Conclusions
GPCR autoantibodies are markedly altered in HF. However, they are correlated poorly or even inversely to haemodynamic, metabolic, and functional markers of disease severity, myocardial histology, and myocardial mitochondrial efficiency. These observations do not hint towards a specific cardio‐pathogenic role of GPCR autoantibodies and suggest that further investigations are required before specific therapies directed at GPCR autoantibodies can be clinically tested in non‐ischaemic HF.
Keywords: Heart failure, Pathophysiology, Autoimmunity, Chronic non‐ischaemic heart failure
Introduction
Chronic heart failure (CHF) is a main cause for hospitalization and death in Western countries. Roughly one‐third of the cases is due to non‐ischaemic myocardial damage caused by genetic predisposition, metabolic stress, and, most notably, myocardial inflammation. 1 Inflammatory CHF is associated with abnormal or misled immune responses and with humoral autoimmunity against various antigens involved in energy metabolism and cardiovascular regulation. 2 , 3 , 4 Heart‐reactive autoantibodies potentially contribute to cardio‐pathogenesis because CHF‐like symptoms can be transferred from patients to mice via B lymphocytes. 5 Consequently, B cells and autoantibodies are emerging targets in CHF therapy. 6 , 7 , 8 , 9 , 10
Among various heart‐reactive autoantibodies discussed in the above context, autoantibodies against cardiac G‐protein‐coupled receptors (GPCRs) are candidates that have been studied earlier. 2 , 11 , 12 CHF‐associated GPCR autoantibodies are intriguing candidates because they impact receptor‐mediated autonomous heart regulation and thereby damage the heart. 13 Evidences of GPCR‐directed/GPCR‐mediated humoral autoimmune cardio‐pathogenesis have been obtained ex vivo and by experimental immunization. These include aberrant activation of signalling pathways, 14 , 15 receptor de‐/hyper‐sensitization, 16 induction of cardiomyocyte apoptosis, 17 and pro‐fibrotic stimulation of cardiac fibroblasts. 13 , 18 Clinical studies of non‐ischaemic CHF patients have shown that activating GPCR autoantibodies (targeting most notably β1AR) are associated with a poorer cardiac function, 19 a poorer disease outcome, 20 a higher incidence of atrial fibrillation, 21 , 22 and a higher incidence of sudden heart death. 23 In one case, appearance of such autoantibodies even preceded clinical symptoms by several years. 22 Moreover, removal 24 , 25 or neutralization of IgG 26 can improve haemodynamics in CHF patients.
However, in clinical medicine, a pathogenic role of humoral autoimmunity against β1AR and other GPCR in non‐ischaemic, inflammatory CHF is not unequivocally accepted for several reasons: (i) GPCR autoantibodies are also associated with a variety of human diseases not involving CHF or cardiac injury 27 , 28 ; (ii) in certain autoimmune diseases, GPCR autoantibodies are thought to play a physiological or even protective role 29 ; (iii) incidence and circulating levels of CHF‐associated GPCR autoantibodies do not stringently correlate with biomarkers of cardiac injury 30 and (iv) it remains unclear which autoantibody species is targeted by the beneficial effects of IgG removal or neutralization in CHF. 31 , 32 , 33
Hesitations regarding the assumption of a specific pathogenetic role of GPCR autoantibodies for the heart is in part be due to an analytical issue: Only GPCR autoantibodies that stimulate the receptors or otherwise modulate their function 13 , 16 are believed to exert the pathogenic effects. 2 , 34 However, assessment of such functional effects is impractical in the setting of clinical studies because it requires functional tests or immune assays employing native receptors or other immunological targets faithfully mimicking the pathogenic conformational receptor autoepitope. 35 , 36 , 37 Consequently, many clinical studies published so far on GPCR autoantibodies in CHF and other diseases have instead assessed GPCR autoantibody via IgG‐binding to linear immobilized peptides, a procedure demonstrated to be unsuitable to detect functional GPCR autoantibodies. 35 Thus, it is quite possible that many published clinical studies available to date may have altogether overlooked the pathogenetically relevant species of GPCR autoantibodies. 36 , 37
Meanwhile, assays appropriate for the determination of antibodies targeting presumably cardio‐pathogenic conformational epitopes have been certified for clinical use. 38 Our study is one of the first making use of these assays on a broad basis. It encompasses all species of GPCR autoantibodies that are currently suspected to play a role in CHF and cardiovascular dysfunction. 2 , 27 Furthermore, to better understand a putative pathophysiological role of GPCR autoantibodies, we have investigated their association with histological, haemodynamic, and metabolic parameters of disease activity obtained by deep phenotyping of the patients with non‐ischaemic CHF. We compare circulating levels of presumably disease‐relevant GPCR autoantibodies with a plethora of patient data on inflammation, cardiac injury, cardiac fibrosis, haemodynamics, metabolism, and myocardial mitochondrial function, which comprehensively delineate disease activity, progression, and prognosis of non‐ischaemic HF.
Methods
Study protocol
These registered clinical trials' protocols were approved by the local ethics board of Heinrich‐Heine University Düsseldorf (study number 5263R and 3786) and registered at ClinicalTrials.gov (NCT03386864). The investigation conforms with the principles outlined in the World's Medical Association Declaration of Helsinki. Before inclusion, all participants have given written informed consent.
Patient enrolment
We included 95 patients with recently diagnosed heart failure (HF) of unknown origin at the University Hospital Düsseldorf. Patients scheduled for transcatheter endomyocardial biopsy were invited to participate in the study. Patient surveillance in the course of routine clinical healthcare included transthoracic echocardiography, cardiac magnetic resonance imaging (whenever feasible), coronary angiography, and right heart catheterization with endomyocardial biopsy.
A cohort of 60 healthy humans served as controls in this study. Exclusion criteria included systemic antibiotic treatment within 6 months prior to study enrolment, history of endocarditis, bleeding disorders, organ transplantation, dialysis, pregnancy, and lactation. Absence of HF at the time of blood drawing was in addition confirmed by determination of serum levels of N‐terminal pro‐brain natriuretic peptide (pBNP) at <125 ng/L and troponin T (TpT) at <4 ng/L. Absence of chronic systemic inflammation was ascertained by determination of serum levels of C‐reactive protein (CRP) at <5 mg/L and interleukin 6 (IL‐6) at <7 ng/L.
None of the study participants (patients or controls) had a history of Chagas' disease and/or had lived in areas where Chagas' disease was endemic within the past 20 years.
Cardiac magnetic resonance imaging
In 39 participants, cardiac magnetic resonance (CMR) was conducted on a 1.5‐T scanner (Achieva, Philips, Best, Netherlands). In addition to standard protocols for assessment of ventricular volumes and function, including global longitudinal strain (GLS) and diastolic strain rate, left ventricular T2 relaxation time reflecting myocardial oedema and inflammation was quantified using the Gradient and SpinEcho (GraSE) sequence as described previously. 39 T1 relaxation time reflecting diffuse myocardial fibrosis was quantified according to current recommendations. 40
Catheterization procedures and endomyocardial biopsies
Right heart catheterization was performed to measure right atrial pressure (RAP), pulmonary artery pressure (PAP), and pulmonary capillary wedge pressure (PCWP). Afterwards, endomyocardial biopsies were taken from the interventricular septum. One additional biopsy (1–2 mg) was taken for respirometry.
High‐resolution respirometry
Respirometry was carried out using the Oxygraph‐2k (OROBOROS Instruments, Innsbruck, Austria) as recently established and described in detail. 41 Oxidative phosphorylation capacity (OXPHOS) was measured for octanyl‐carnitine, glutamate, and succinate. The leak control ratio was calculated as the ratio of the LEAK state induced by addition of oligomycin divided by electron transfer system capacity. 42 , 43 Integrity of the outer mitochondrial membrane was confirmed by assessing respiration increase after cytochrome C addition by less than 10%.
Measurement of GPCR antibodies in serum samples
Antibodies against α1‐ and β1‐adrenergic receptors, the muscarinic acetylcholine receptor 5, the receptor for complement C5a, angiotensin II receptor types 1 and 2, endothelin receptors A and B, and ACE were measured with commercially available immunoassays (CellTrend GmbH, Luckenwalde, Germany) according to the instructions of the manufacturer. These assays provide native receptors presented in their physiological membrane environment as immunogenic targets for autoantibodies and were calibrated with polyclonal standard sera, yielding quantitative values of autoantibody levels expressed in arbitrary units/mL. Antibodies against β1‐adrenergic receptors (β1AR‐Aabs, the most prominent candidate) were determined by IgG binding to a cyclic peptide providing a valid representation of the conformational epitope within the second extracellular loop of the receptor associated with the active receptor conformation. It has been demonstrated that pre‐absorption with this peptide neutralizes the cardio‐pathogenic potency of stimulatory receptor antibodies in immunized mice. 38 The cyclic peptide was coated onto microtitre plates by established procedures, and these plates were processed in a similar manner as the above commercial immunoassays. It was calibrated with a humanized mouse monoclonal antibody against the autoepitope (kindly provided by Drs Holthoff and. Ungerer, AdvanceCor, Munich, Germany), allowing expression of autoantibody levels in ng IgG/mL. The two assays for β1AR‐Aabs exhibited a reasonable correlation (Figure S1 ) with a few outliers, which are explained by the presence of β1AR‐Aabs not directed against the second extracellular loop of the receptor. It should be noted, however, that some correlations with corroborative clinical data such as left ventricular T1‐relaxation times and myocardial mitochondrial leak control ratio were only seen with levels of β1AR autoantibodies determined by the cyclopeptide assay (Table S1 ).
Statistical analyses
Quantitative measurements of receptor autoantibodies were optionally normalized to total IgG levels of the respective patients' serum by a simple division and subsequent (log + 1) transformation. The influence of confounders was tested for each antibody species in a separate general linear model. All statistical analyses were carried out using GraphPad Prism version 9.1.2 (GraphPad Software, San Diego, CA, USA) and SPSS Statistics 25.0.0.2 (International Business Machines Corporation, Armonk, NY, USA). The statistical significance threshold was alpha = 0.05. The homeostasis model assessment was used to estimate the participants' insulin resistance (HOMA‐IR) based on fasting insulin and glucose concentrations. 43
Results
Basal characteristics
Characteristics of the two groups are summarized in Table 1 . A total of 155 participants were recruited for the study. Ninety‐five of these were HF patients (HF group) undergoing endomyocardial biopsy. Advanced left‐sided HF within the HF group was established by cardiac index (2.17 ± 0.62 L/min/m2), left ventricular ejection fraction (28.95 ± 10.26%), and elevated pulmonary artery pressures (18.71 ± 8.55 mmHg). The remaining 60 were healthy humans, who tended to be slightly older and comprised more females. In the HF group, supra‐normal serum levels of CRP and IL‐6 indicated evidence for mostly subclinical or low‐grade systemic inflammation, whereas in the control group, total serum IgG was higher. Of note, the groups also differed in biomarkers of kidney function and cardiac injury.
Table 1.
Patient characteristics
| Heart failure | Control | Total | P | |
|---|---|---|---|---|
| Overall, n | 95 | 60 | 155 | |
| Anthropometry | ||||
| Male, % | 74 | 47 | 63 | 0.001 |
| Age, y | 56.7 ± 12.1 | 59.1 ± 13.6 | 57.6 ± 12.7 | 0.069 |
| Cardiac measurements | ||||
| Cardiac index, L/min/m2 | 2.17 ± 0.62 | ‐ | ‐ | ‐ |
| EF, % | 28.95 ± 10.26 | ‐ | ‐ | ‐ |
| RA (M), mmHg | 7.87 ± 5.64 | ‐ | ‐ | ‐ |
| PA (M), mmHg | 27.64 ± 10.68 | ‐ | ‐ | ‐ |
| PA (sys), mmHg | 42.10 ± 14.63 | ‐ | ‐ | ‐ |
| PA (dia), mmHg | 17.93 ± 8.50 | ‐ | ‐ | ‐ |
| PCWP, mmHg | 18.71 ± 8.55 | ‐ | ‐ | ‐ |
| Cardiac MRI | ||||
| T1 relaxation time, msec | 1058 ± 74.16 | ‐ | ‐ | ‐ |
| T2 relaxation time, msec | 58.80 ± 10.90 | ‐ | ‐ | ‐ |
| Laboratory results | ||||
| Creatinine (serum), mg/dL | 1.22 ± 1.03 | 0.78 ± 0.15 | 1.04 ± 0.84 | <0.001 |
| Creatine kinase, U/L | 75.74 ± 58.17 | 84.85 ± 74.93 | 79.31 ± 65.17 | 0.0330 |
| Total protein, g/dL | 14.27 ± 75.87 | 7.39 ± 0.63 | 11.58 ± 59.12 | <0.001 |
| Total IgG, mg/dL | 955.0 ± 306.3 | 1139 ± 230.2 | 1028 ± 292.0 | <0.001 |
| Troponin, ng/L | 64.38 ± 59.14 | 3.64 ± 4.03 | 40.87 ± 54.97 | <0.001 |
| nt‐proBNP, pg/mL | 8075 ± 36 169 | 44.54 ± 34.01 | 4966 ± 28 529 | < 0.001 |
| IL‐6, pg/mL | 15.97 ± 44.89 | 1.71 ± 0.75 | 10.45 ± 35.76 | < 0.001 |
| Cystatin C, mg/L | 1.40 ± 0.81 | 0.80 ± 0.12 | 1.16 ± 0.70 | < 0.001 |
| CRP, mg/dL | 1.96 ± 5.94 | 0.19 ± 0.40 | 1.27 ± 4.72 | < 0.001 |
| Medication | ||||
| Beta‐blockers, % | 94 | ‐ | ‐ | ‐ |
| AT1‐receptor antagonists, % | 12 | ‐ | ‐ | ‐ |
| Angiotensin receptor‐neprilysin inhibitor, % | 24 | ‐ | ‐ | ‐ |
Patient characteristics of the heart failure and the control group. Data given as mean ± SD, P calculated using unpaired t test or Mann–Whitney test.
CRP, C‐reactive protein; EF, ejection fraction; IgG, immunoglobulin G; IL‐6, interleukin 6; nt‐proBNP, n‐terminal pro‐brain natriuretic peptide; PA, pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure; RA, right atrial pressure.
HF‐associated differences in circulating levels of GPCR autoantibodies
Out of all GPCR autoantibodies here examined, significant differences between the HF group and the control group were observed for autoantibodies against β1AR, M5R, α1AR, AT1R, and AT2R, but not for antibodies against component 5a receptor, angiotensin converting enzyme II receptor, Endothelin A receptor or Endothelin B receptor. As illustrated in Figure 1 , levels of β1AR, M5R, and AT2R autoantibodies were higher in the HF group than in the control group, whereas levels of α1AR and AT1R autoantibodies were lower in the HF group than in the control group. All these differences were highly significant at P < 0.0001. The above findings could be replicated in each of the following settings: (i) in non‐transformed raw data; (ii) in IgG‐normalized data; (iii) in IgG‐normalized and log‐transformed data; and (iv) in IgG‐normalized and log‐transformed data corrected for age and gender in linear regression models.
Figure 1.
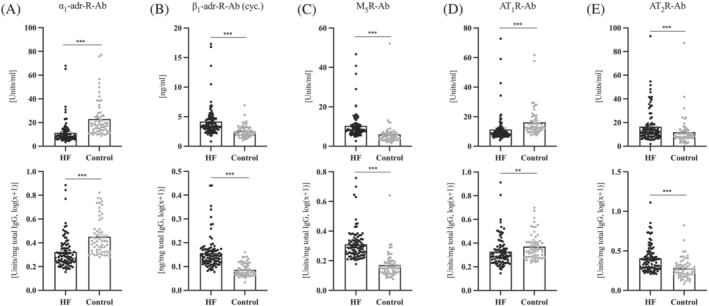
Levels of GPCR‐Abs in the heart failure group and the control group. Top row: Native data. Bottom row: Data normalized to total IgG and log(x + 1) transformed. HF, heart failure; IgG, immunoglobulin G. (A) α1‐Adrenergic receptor antibodies. (B) β1‐Adrenergic receptor antibodies, measured via cyclic peptide assay. (C) Muscarinic receptor M5 antibodies. (D) Angiotensin II receptor type 1 antibodies. (E) Angiotensin II receptor type 2 antibodies. **P ≤ 0.01, ***P ≤ 0.001 vs. control group. P calculated using Mann–Whitney test. n = 95–91 vs. n = 60.
Subsequently, we divided the HF cohort according to histopathology into subgroups of myocarditis (n = 30), dilated cardiomyopathy (n = 29), other cardiomyopathy (n = 25), and no evidence of pathology (n = 11). Neither the increases in β1AR, M5R, and AT2R autoantibodies nor the decreases of α1AR and AT1R autoantibodies were found clustered to any of these histopathological categories (Table S2 ).
Association of GPCR autoantibodies with biomarkers of cardiac injury and inflammation
GPCR autoantibody levels were quantitatively compared with serum markers of inflammation (IL‐6 and CRP) or cardiac injury (pBNP and TpT). β1AR, M5R, and AT2R autoantibodies exhibited a positive correlation with pBNP, α1AR and AT1R autoantibodies exhibiting an inverse correlation with pBNP when we analysed the complete cohort consisting of HF and healthy patients (Figure 2 ). However, within the HF group, the above associations of GPCR autoantibody levels with pBNP or high‐sensitive TpT were not detectable. Moreover, within the HF group, an apparent association of β1AR autoantibody levels with IL‐6 and CRP disappeared upon normalization to total IgG content (Table 2 ).
Figure 2.
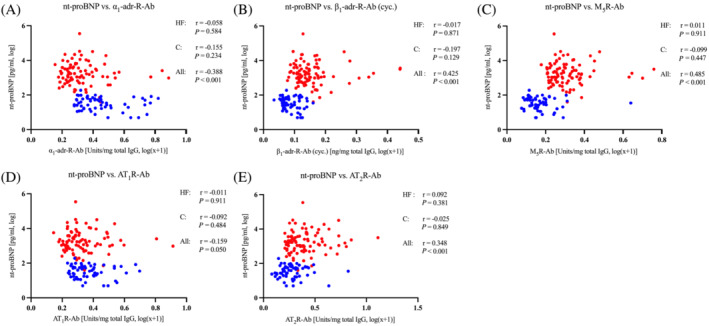
Association of GPCR‐Abs and nt‐proBNP; GPCR‐Abs were normalized to total IgG and logarithmized. Red: Heart failure group. Blue: Control group. C, control; HF, heart failure; IgG, immunoglobulin G. Data given as Spearman rho. (A) α1‐adrenergic antibodies. (B) β1‐Adrenergic receptor antibodies, measured via cyclic peptide assay. (C) Muscarinic receptor M5 antibodies. (D) Angiotensin II receptor type 1 antibodies. (E) Angiotensin II receptor type 2 antibodies. n = 91 vs. n = 60.
Table 2.
GPCR antibodies and cardiac und inflammatory markers
| IL‐6 | CRP | Troponin | nt‐proBNP | |
|---|---|---|---|---|
| Panel A | ||||
| α1‐adr‐R‐Ab | 0.194 | 0.059 | 0.095 | −0.026 |
| β1‐adr‐R‐Ab (cyc.) | 0.327* | 0.226* | 0.084 | −0.052 |
| M5R‐Ab | 0.171 | 0.017 | 0.004 | 0.017 |
| AT1R‐Ab | 0.174 | −0.000 | 0.043 | 0.013 |
| AT2R‐Ab | 0.139 | −0.127 | −0.029 | 0.105 |
| Panel B | ||||
| α1‐adr‐R‐Ab | 0.053 | −0.034 | 0.070 | −0.021 |
| β1‐adr‐R‐Ab (cyc.) | 0.184 | 0.116 | 0.185 | −0.094 |
| M5R‐Ab | −0.009 | −0.164 | 0.076 | −0.003 |
| AT1R‐Ab | 0.020 | −0.125 | 0.030 | −0.008 |
| AT2R‐Ab | 0.054 | −0.198 | −0.014 | 0.095 |
Association of GPCR antibodies with IL‐6, CRP, troponin, and BNP within the heart failure group. Panel A: Native data. Panel B: Antibodies normalized to total IgG and logarithmized. Data given as Spearman rho with *P ≤ 0.05. n = 95–90.
CRP, C‐reactive protein; IL‐6, interleukin 6; nt‐proBNP, n‐terminal pro‐brain natriuretic peptide.
Association of GPCR autoantibodies with parameters of impaired haemodynamics
Within the HF group, we observed an inverse correlation between pulmonary vascular resistance and α1AR autoantibodies, which, however, disappeared upon normalization of the autoantibody levels to total IgG (Table S3 ). Apart from that, we failed to find any significant association of GPCR autoantibodies (unprocessed or IgG‐normalized serum levels) with haemodynamic parameters of the patients including ejection fraction, cardiac index, PCWP, PAP, or RAP of the HF patients studied.
Associations of GPCR autoantibodies with parameters of cardiac structure and contractility
We compared serum levels of GPCR autoantibodies in the HF patient group with myocardial T2 mapping reflecting myocardial oedema and inflammation 39 and myocardial T1 mapping considered a marker of tissue fibrosis as well as lipid and iron accumulation. 44 These results are summarized in Table S4 . Global left ventricular T1‐relaxation times were significantly correlated to serum levels of M5R and β1AR autoantibodies. The inverse correlation of T1‐mapping data with β1AR autoantibodies was retained after normalization to total IgG, but the correlation with M5R autoantibody levels disappeared upon normalization to total IgG. We failed to observe any correlations between the various GPCR autoantibody levels and T2‐mapping data of the patients.
CMR‐based left ventricular strain analyses indicated a weak association between elevated β1AR autoantibodies and impaired systolic GLS, as a marker of myocardial deformation (Table S4 ). Diastolic strain rate and left ventricular ejection fraction were not related to circulating autoantibodies.
Association of GPCR autoantibodies with serum markers of metabolic alterations
Within the HF group, 26 participants were normoglycaemic, 47 participants had prediabetes, and 22 participants had type 2 diabetes mellitus. Among the HF‐associated GPCR autoantibodies, AT1R–AT2R autoantibodies exhibited associations with certain markers of metabolic alterations: AT1R autoantibody levels were correlated to circulating non‐esterified fatty acids, and AT2R autoantibody levels were inversely related to insulin resistance, as assessed according to the homoeostasis model (HOMA‐IR). Furthermore, higher serum cholesterol and triacylglycerol related inversely to serum AT2R autoantibody levels. These associations were robust in as much as they were retained following normalization to total IgG (Table S5 ).
GPCR autoantibodies and myocardial mitochondrial function
Impaired mitochondrial function has been shown to play an important role in the development of HF. 41 , 45 To follow up on possible interrelations between myocardial mitochondrial function and GPCR autoimmunity, we compared serum levels of HF‐associated GPCR autoantibodies to data from high‐resolution respirometry performed in corresponding myocardial specimen. There was no direct association of the receptor autoantibodies with myocardial mitochondrial oxidative phosphorylation activity and electron transport capacity (Table S6 ). However, the leak control ratio, which is a marker of mitochondrial uncoupling, was related inversely to the IgG‐normalized circulating antibodies of AT2R, M5R, and β1AR. Without IgG‐normalization, the correlation of leak control ratio with M5R and β1AR autoantibody levels disappeared, whereas the correlation with AT2R autoantibody remained significant.
Discussion
The salient findings of this study are:
GPCR autoantibodies for α1AR, β1AR, M5R, AT1R, and AT2R differ between patients with non‐ischaemic HF and healthy controls independent of total IgG when assessed with state‐of‐the‐art assays.
In patients with non‐ischaemic HF, increases in these autoantibodies do neither relate to histological diagnosis nor to cardiac haemodynamics, adverse prognostic parameters, or cardiac structural properties.
In patients with non‐ischaemic HF, AT2R autoantibodies relate inversely to systemic insulin resistance (HOMA‐IR) and myocardial mitochondrial uncoupling. M5R autoantibodies also relate to myocardial mitochondrial uncoupling but not to insulin resistance.
Key questions
Autoantibodies against adrenergic and muscarinic receptors are discussed as pathogenetic principle in chronic HF since more than four decades. 46 , 47 , 48 , 49 , 50 The basic pathogenetic mechanism has been extensively demonstrated by ex vivo studies and animal models, 13 , 15 and we know of a human disease (Chagas' cardiomyopathy) based thereon. 51 However, published evidence regarding a putative role of humoral GPCR autoimmunity in human chronic HF other than Chagas' cardiomyopathy is inconsistent, and the measurements employed in most available clinical studies to determine potentially cardio‐pathogenic GPCR autoantibodies are either impractical, not certified, and/or poorly reproducible 52 , 53 or invalid. 35 , 36 , 37
Here, we aimed at a comprehensive re‐assessment of the above issue in a cohort of thoroughly characterized, manifest HF patients of established aetiology (excluding Chagas' cardiomyopathy) using validated/certified and reproducible high‐throughput assays 38 for a panel of GPCR autoantibodies currently suggested to play a role in HF. 2 Results thus obtained allow us to address three key questions: (i) Are any of the tested GPCR autoantibodies associated with HF? (ii) Can serological patterns of such HF‐associated GPCR autoantibodies distinguish non‐ischaemic HF or even sub‐entities thereof from the healthy condition? (iii) Are serum levels of HF‐associated GPCR autoantibodies quantitatively correlated with established serological, haemodynamic, histological, radiological, and biochemical parameters of disease activity?
Association of imbalances of GPCR autoantibodies with non‐ischaemic HF
We observed that circulating levels of autoantibodies against α1AR, β1AR, M5R, AT1R, and AT2R differed significantly between HF patients and healthy controls. These differences remained significant upon correction for total IgG. Among a broad panel of GPCR autoantibodies analysed simultaneously by comparable assays, only these five GPCR autoantibody species were altered in the patients. The above alterations were equally distributed between histological sub‐entities of non‐ischaemic HF classified as myocarditis, dilated cardiomyopathy, other cardiomyopathy, and no histological correlate. Thus, our data support the notion that alterations of the above five GPCR autoantibody species are associated with non‐Chagas' and non‐ischaemic HF in a significant and possibly meaningful manner, but a distinction of histological sub‐entities does not show a distinct GPCR autoantibody immunity profile. Interestingly, not all the observed HF‐associated alterations of GPCR autoantibody levels pointed in the same direction: β1AR, M5R, and AT2R autoantibodies were increased, whereas α1AR and AT1R autoantibodies were decreased in the HF patients, and component 5a receptor, angiotensin‐converting enzyme II receptor, and Endothelin A receptor or Endothelin B receptor autoantibodies were not significantly altered.
HF‐associated increases in GPCR autoantibodies: The increase in β1AR autoantibodies conforms to a plethora of published data suggesting a specific association of that autoantibody species with various cardiovascular diseases, most notably with idiopathic dilated cardiomyopathy. 2 The increase in M5R autoantibodies observed here is reminiscent of a similar phenomenon recently observed in elderly persons subjected to invasive periodontal therapy, which, however, was correlated with the intensity and time course of periodontal surgery but not with cardiac markers. 30 The increase in AT2R autoantibodies finally, which we observed in HF patients, remains to be evaluated with respect to its possible relevance for non‐ischaemic HF. To our knowledge, a similar humoral alteration has not been observed before in any human disease or syndrome. However, the role of the AT2R itself has been thoroughly investigated in HF. Recent research suggests that AT2R activation plays a protective role in HF. 54 The demonstrated HF‐associated increase in circulating AT2R antibodies might therefore indicate a novel compensatory mechanism after HF onset, provided the measured antibodies exert a stimulating effect on the AT2R, which still needs to be demonstrated.
HF‐associated decreases of GPCR autoantibodies: α1AR, AT1R, ETA/BR, and ACEII autoantibodies were included in this study, because increased levels thereof play an established role in vascular allograft rejection and blood pressure dysregulation. 27 However, levels of ETA/BR and ACEII autoantibodies did not significantly differ between patients and controls, whereas α1AR and AT1R autoantibody levels were lower in HF patients. Decreases in GPCR autoantibodies could be just as relevant as increases given current pathogenetic concepts proposing a physiological regulatory role of GPCR antibodies 15 that becomes imbalanced in autoimmune diseases. 29 α1AR and AT1R autoantibodies are known to upregulate blood pressure in various human pathologies, 27 and AT2R autoantibodies can counteract these effects in immunized rabbits. 55 The observed HF‐associated alterations of AT1R, α1AR, and AT2R autoantibodies mirror the known effects of corresponding receptor activations in cardiovascular regulation 54 , 56 and, thus, could be relevant for HF due to an impact on blood pressure regulation.
Association of GPCR autoantibodies with disease activity and myocardial damage
We have performed an extensive screen for associations or quantitative correlations between HF‐associated alterations of GPCR autoantibodies and clinical and experimental parameters of disease activity, prognosis parameters, or myocardial damage. However, we have not detected any association with serum markers of myocardial damage or haemodynamic parameters of impaired systolic function. Neither have we observed any association with myocardial oedema and inflammation as judged by T2 mapping 39 or biopsy histology or inflammation serum markers.
These negative findings stand in contrast to some previous studies, which may be attributable to several factors. Firstly, most other studies have been based on quantitative determinations of circulating GPCR antibodies by IgG binding to linear peptides. However, such measurements do not necessarily reflect the impact of the antibodies on receptor function. 35 , 36 , 37 , 53 Other previous studies have been exclusively based on functional readouts, which are only loosely related to circulating levels of the antibodies. 52 , 57 Here, we employed a type of assay that to the best of our knowledge combines both approaches as it measures IgG binding to the native receptor or a circular peptide faithfully reflecting the conformational autoepitope related to the active receptor conformation. 38 , 58 Another source of divergence may be a different representation of receptor polymorphisms in the study population. At least for the β1AR, it has been demonstrated that certain frequent polymorphisms have a significant influence on binding as well as functional effects of GPCR autoantibodies. 59 Moreover, to our knowledge, in none of the previous studies, measurements of circulating levels of GPCR autoantibodies have been normalized to total IgG. Consequently, these studies cannot exclude confounding effects of global inflammatory phenomena on the circulating levels of certain GPCR autoantibodies. Such confounding effects have previously demonstrated in periodontitis patients 30 and were here observed in HF patients. Finally, all patients in the HF group of this study exhibited an advanced HF phenotype justifying endomyocardial biopsy. Thus, in contrast to most previous studies, not all existing phenotypes and earlier stages of non‐ischaemic HF were included in our study population.
The only notable links between GPCR autoantibodies and indicators of HF disease activity found in this study were a correlation between circulating levels β1AR autoantibodies and global left ventricular T1‐relaxation time as a marker of myocardial fibrosis as well as lipid and iron accumulation, 44 as well as left ventricular GLS as a marker of cardiac contractile function. These correlations were robustly conserved following normalization to total IgG. However, both correlations were inverse, which is inconsistent with data obtained by experimental immunization and transfer of immune components to animals suggesting that β1AR autoantibodies stimulate myocardial fibrosis and may impair cardiac contractile function. 5 , 60 , 61 , 62 However, it is conceivable that humoral β1AR autoimmunity disappears again once myocardial fibrosis has established, which would be the case in the advanced state of disease of the HF patients included in our study. Recent research also hints towards a compensatory increase of β‐adrenergic receptor autoantibodies in paediatric myocarditis. 63 As only cross‐sectional non‐causal correlations are provided in our study, future studies of the timing of events linking cardiac fibrosis and functional impairment to humoral β1AR autoimmunity are required to address a presumed biphasic time course of β1AR autoantibodies.
Association of GPCR autoantibodies with metabolic disease
Our data demonstrate an inverse correlation of AT2R autoantibodies with insulin resistance, whereas an elevation in circulating free fatty acids, as often found in the insulin resistant state, 64 was associated with higher AT1R autoantibody titres. As known from previous studies, AT2R stimulation exhibits protective effects in heart failure as well as diabetes‐associated atherosclerosis. 54 , 65 AT2R activation through angiotensin II increases capillary blood flow and raises insulin‐mediated glucose usage. In contrast, AT1R activation leads to reduced nitric oxide bioavailability, impaired insulin signalling, vasoconstriction, and insulin resistance. 66
Furthermore, we find that increased levels of AT2R autoantibodies are correlated to increased mitochondrial efficiency as estimated via leak control ratio. 41 , 67 These findings could support the hypothesis of GPCR antibodies having a physiological regulatory role. 29 In this case, a role of AT2R autoantibodies in the adaptation of mitochondrial metabolism would further corroborate the concept of protective effects of AT2R on the myocardium in HF.
On the one hand, our data could indicate that low levels of AT2R autoantibodies might promote insulin resistance, which in turn is a key factor for a developing type 2 diabetes mellitus. 68 , 69 , 70 On the other hand, an effect of insulin resistance on AT2R activity and their cardioprotective effects via circulating AT2R autoantibodies cannot be excluded. 71 Therefore, it is tempting to speculate that the link between AT2R autoantibodies and insulin resistance here observed in HF patients may be mechanistically involved in diabetes‐related heart disease or in the worse outcomes of HF patients with diabetes mellitus or both. 71 , 72 , 73 , 74 , 75
Limitations
This study comes with several limitations. Firstly, and most importantly, the observational and cross‐sectional design of the study and the correlations shown in this study do not allow to infer causation or timing of events and should therefore be regarded as hypothesis‐generating. Moreover, we were not able to fully phenotype the control group in the same fashion as the HF group. Finally, our groups differed in age and sex distribution, which we accounted for by adjusting for those parameters in linear regression analysis in the main group comparisons of GPCR autoantibodies in HF and controls.
Concluding remarks
Our data clearly show a link between non‐ischaemic HF and alterations of GPCR autoantibody‐mediated immunity. Although the associations shown in this hypothesis‐generating study alone cannot imply or preclude causality, the consistency of these alterations among different causes of non‐ischaemic HF does not hint towards a direct and global cardio‐pathogenic effect of GPCR autoantibodies as demonstrated for other anti‐heart autoantibodies. 76 , 77 On the contrary, correlations of GPCR autoantibodies with cardiac metabolic or structural parameters lend support to the concepts of protective rather than pathogenic properties of specific autoantibodies and reactive alterations of GPCR autoantibodies in HF. Therefore, it seems counterintuitive that removal or neutralization of HF‐associated GPCR autoantibodies (as opposed to unselective removal total IgG or certain IgG subclasses) should improve human HF in the same manner as it does in certain animal models. 7 Overall, our study stresses the importance to further differentiate the effects of GPCR autoantibodies within their class prior to establishing GPCR autoantibody‐regulating therapies.
Conflict of interest
None of the authors have a conflict of interest with respect to the content of this manuscript to declare.
Funding
This work was supported by funding from the German Research Foundation (SFB1116), a grant provided by the research commission of the Medical Faculty, Heinrich‐Heine‐University, Düsseldorf, Germany, the German Center for Diabetes Research (DZD e.V.) and the German Diabetes Center, which is funded by the German Federal Ministry of Health (Berlin, Germany) and the Ministry of Innovation, Science, Research, and Technology of the state North Rhine Westphalia (Düsseldorf, Germany).
Supporting information
Table S1. Comparison of immune assays.
Table S2. Histopathological classification.
Table S3. GPCR‐Antibodies and cardiac parameters.
Table S4. GPCR‐Antibodies and cardiac MRI.
Table S5. GPCR‐Antibodies and metabolic markers.
Table S6. GPCR‐Antibodies and mitochondrial parameters.
Figure S1. Correlation between β1‐adr‐R‐Antibodies measured via cyclopeptide or membrane essay within the heart failure group; GPCR‐Antibodies were normalized to total IgG and logarithmized. IgG immunoglobin G. Data given as Spearman rho. n = 91.
Acknowledgements
Open Access funding enabled and organized by Projekt DEAL.
Zweck, E. , Karschnia, M. , Scheiber, D. , Heidecke, H. , Dechend, R. , Barthuber, C. , Kaufmann, S. , Kelm, M. , Roden, M. , Westenfeld, R. , Szendrödi, J. , and Boege, F. (2023) Receptor autoantibodies: Associations with cardiac markers, histology, and function in human non‐ischaemic heart failure. ESC Heart Failure, 10: 1258–1269. 10.1002/ehf2.14293.
Elric Zweck and Maximilian Karschnia contributed equally to this article.
References
- 1. Tschöpe C, Ammirati E, Bozkurt B, Caforio ALP, Cooper LT, Felix SB, Hare JM, Heidecker B, Heymans S, Hübner N, Kelle S, Klingel K, Maatz H, Parwani AS, Spillmann F, Starling RC, Tsutsui H, Seferovic P, van Linthout S. Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat Rev Cardiol. 2021; 18: 169–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boivin‐Jahns V, Jahns R. GPCR‐autoantibodies in chronic heart failure. Front Biosci (Landmark Ed). 2018; 23: 2065–2081. [DOI] [PubMed] [Google Scholar]
- 3. Müller AM, Bockstahler M, Hristov G, Weiß C, Fischer A, Korkmaz‐Icöz S, Giannitsis E, Poller W, Schultheiss HP, Katus HA, Kaya Z. Identification of novel antigens contributing to autoimmunity in cardiovascular diseases. Clin Immunol. 2016; 173: 64–75. [DOI] [PubMed] [Google Scholar]
- 4. Kallwellis‐Opara A, Dörner A, Poller WC, Noutsias M, Kühl U, Schultheiss HP, Pauschinger M. Autoimmunological features in inflammatory cardiomyopathy. Clin Res Cardiol. 2007; 96: 469–480. [DOI] [PubMed] [Google Scholar]
- 5. Omerovic E, Bollano E, Andersson B, Kujacic V, Schulze W, Almarson AH, Waagstein F, Fu M. Induction of cardiomyopathy in severe combined immunodeficiency mice by transfer of lymphocytes from patients with idiopathic dilated cardiomyopathy. Autoimmunity. 2000; 32: 271–280. [DOI] [PubMed] [Google Scholar]
- 6. García‐Rivas G, Castillo EC, Gonzalez‐Gil AM, Maravillas‐Montero JL, Brunck M, Torres‐Quintanilla A, Elizondo‐Montemayor L, Torre‐Amione G. The role of B cells in heart failure and implications for future immunomodulatory treatment strategies. ESC Heart Fail. 2020; 7: 1387–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Werner S, Wallukat G, Becker NP, Wenzel K, Müller J, Schimke I, Wess G. The aptamer BC 007 for treatment of dilated cardiomyopathy: evaluation in Doberman pinschers of efficacy and outcomes. ESC Heart Fail. 2020; 7: 844–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boivin‐Jahns V, Uhland K, Holthoff HP, Beyersdorf N, Kocoski V, Kerkau T, Münch G, Lohse MJ, Ungerer M, Jahns R. Cyclopeptide COR‐1 to treat beta1‐adrenergic receptor antibody‐induced heart failure. PLoS ONE. 2018; 13: e0201160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seferović PM, Polovina M, Bauersachs J, Arad M, Ben Gal T, Lund LH, Felix SB, Arbustini E, Caforio ALP, Farmakis D, Filippatos GS, Gialafos E, Kanjuh V, Krljanac G, Limongelli G, Linhart A, Lyon AR, Maksimović R, Miličić D, Milinković I, Noutsias M, Oto A, Oto Ö, Pavlović SU, Piepoli MF, Ristić AD, Rosano GMC, Seggewiss H, Ašanin M, Seferović JP, Ruschitzka F, Čelutkiene J, Jaarsma T, Mueller C, Moura B, Hill L, Volterrani M, Lopatin Y, Metra M, Backs J, Mullens W, Chioncel O, de Boer RA, Anker S, Rapezzi C, Coats AJS, Tschöpe C. Heart failure in cardiomyopathies: a position paper from the heart failure Association of the European Society of cardiology. Eur J Heart Fail. 2019; 21: 553–576. [DOI] [PubMed] [Google Scholar]
- 10. Elsanhoury A, Tschope C, Van Linthout S. A toolbox of potential immune‐related therapies for inflammatory cardiomyopathy. J Cardiovasc Transl Res. 2021; 14: 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Düngen HD, Dordevic A, Felix SB, Pieske B, Voors AA, McMurray JJV, Butler J. beta1‐Adrenoreceptor autoantibodies in heart failure: physiology and therapeutic implications. Circ Heart Fail. 2020; 13: e006155. [DOI] [PubMed] [Google Scholar]
- 12. Patel PA, Hernandez AF. Targeting anti‐beta‐1‐adrenergic receptor antibodies for dilated cardiomyopathy. Eur J Heart Fail. 2013; 15: 724–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boivin‐Jahns V, Jahns R, Boege F. Relevant effects of beta1‐adrenoceptor autoantibodies in chronic heart failure. Front Biosci (Landmark Ed). 2018; 23: 2146–2156. [DOI] [PubMed] [Google Scholar]
- 14. Stavrakis S, Kem DC, Patterson E, Lozano P, Huang S, Szabo B, Cunningham MW, Lazzara R, Yu X. Opposing cardiac effects of autoantibody activation of beta‐adrenergic and M2 muscarinic receptors in cardiac‐related diseases. Int J Cardiol. 2011; 148: 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Skiba MA, Kruse AC. Autoantibodies as endogenous modulators of GPCR signaling. Trends Pharmacol Sci. 2021; 42: 135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Herda LR, Felix SB, Boege F. Drug‐like actions of autoantibodies against receptors of the autonomous nervous system and their impact on human heart function. Br J Pharmacol. 2012; 166: 847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jane‐wit D, Altuntas CZ, Johnson JM, Yong S, Wickley PJ, Clark P, Wang Q, Popović ZB, Penn MS, Damron DS, Perez DM, Tuohy VK. Beta 1‐adrenergic receptor autoantibodies mediate dilated cardiomyopathy by agonistically inducing cardiomyocyte apoptosis. Circulation. 2007; 116: 399–410. [DOI] [PubMed] [Google Scholar]
- 18. Liu J, Mao W, Iwai C, Fukuoka S, Vulapalli R, Huang H, Wang T, Sharma VK, Sheu SS, Fu M, Liang CS. Adoptive passive transfer of rabbit beta1‐adrenoceptor peptide immune cardiomyopathy into the Rag2−/− mouse: participation of the ER stress. J Mol Cell Cardiol. 2008; 44: 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jahns R, Boivin V, Siegmund C, Inselmann G, Lohse MJ, Boege F. Autoantibodies activating human beta1‐adrenergic receptors are associated with reduced cardiac function in chronic heart failure. Circulation. 1999; 99: 649–654. [DOI] [PubMed] [Google Scholar]
- 20. Störk S, Boivin V, Horf R, Hein L, Lohse MJ, Angermann CE, Jahns R. Stimulating autoantibodies directed against the cardiac beta1‐adrenergic receptor predict increased mortality in idiopathic cardiomyopathy. Am Heart J. 2006; 152: 697–704. [DOI] [PubMed] [Google Scholar]
- 21. Stavrakis S, Yu X, Patterson E, Huang S, Hamlett SR, Chalmers L, Pappy R, Cunningham MW, Morshed SA, Davies TF, Lazzara R, Kem DC. Activating autoantibodies to the beta‐1 adrenergic and m2 muscarinic receptors facilitate atrial fibrillation in patients with Graves' hyperthyroidism. J Am Coll Cardiol. 2009; 54: 1309–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu X, Patterson E, Stavrakis S, Huang S, de Aos I, Hamlett S, Cunningham MW, Lazarra R, Kem DC. Development of cardiomyopathy and atrial tachyarrhythmias associated with activating autoantibodies to beta‐adrenergic and muscarinic receptors. J Am Soc Hypertens. 2009; 3: 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pei J, Li N, Chen J, Li X, Zhang Y, Wang Z, Zhang P, Cao K, Pu J. The predictive values of beta1‐adrenergic and M2 muscarinic receptor autoantibodies for sudden cardiac death in patients with chronic heart failure. Eur J Heart Fail. 2012; 14: 887–894. [DOI] [PubMed] [Google Scholar]
- 24. Schimke I, Müller J, Dandel M, Gremmels HD, Bayer W, Wallukat B, Wallukat G, Hetzer R. Reduced oxidative stress in parallel to improved cardiac performance one year after selective removal of anti‐beta 1‐adrenoreceptor autoantibodies in patients with idiopathic dilated cardiomyopathy: data of a preliminary study. J Clin Apher. 2005; 20: 137–142. [DOI] [PubMed] [Google Scholar]
- 25. Dandel M, Englert A, Wallukat G, Riese A, Knosalla C, Stein J, Hetzer R. Immunoadsorption can improve cardiac function in transplant candidates with non‐ischemic dilated cardiomyopathy associated with diabetes mellitus. Atheroscler Suppl. 2015; 18: 124–133. [DOI] [PubMed] [Google Scholar]
- 26. Becker NP, Haberland A, Wenzel K, Göttel P, Wallukat G, Davideit H, Schulze‐Rothe S, Hönicke AS, Schimke I, Bartel S, Grossmann M, Sinn A, Iavarone L, Boergermann JH, Prilliman K, Golor G, Müller J, Becker S. A three‐part, randomised study to investigate the safety, tolerability, pharmacokinetics and mode of action of BC 007, neutraliser of pathogenic autoantibodies against G‐protein coupled receptors in healthy, young and elderly subjects. Clin Drug Investig. 2020; 40: 433–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Meyer C, Heidecke H. Antibodies against GPCR. Front Biosci (Landmark Ed). 2018; 23: 2177–2194. [DOI] [PubMed] [Google Scholar]
- 28. Loebel M, Grabowski P, Heidecke H, Bauer S, Hanitsch LG, Wittke K, Meisel C, Reinke P, Volk HD, Fluge Ø, Mella O, Scheibenbogen C. Antibodies to beta adrenergic and muscarinic cholinergic receptors in patients with chronic fatigue syndrome. Brain Behav Immun. 2016; 52: 32–39. [DOI] [PubMed] [Google Scholar]
- 29. Cabral‐Marques O, Marques A, Giil LM, de Vito R, Rademacher J, Günther J, Lange T, Humrich JY, Klapa S, Schinke S, Schimke LF, Marschner G, Pitann S, Adler S, Dechend R, Müller DN, Braicu I, Sehouli J, Schulze‐Forster K, Trippel T, Scheibenbogen C, Staff A, Mertens PR, Löbel M, Mastroianni J, Plattfaut C, Gieseler F, Dragun D, Engelhardt BE, Fernandez‐Cabezudo MJ, Ochs HD, al‐Ramadi BK, Lamprecht P, Mueller A, Heidecke H, Riemekasten G. GPCR‐specific autoantibody signatures are associated with physiological and pathological immune homeostasis. Nat Commun. 2018; 9: 5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scherbaum I, Heidecke H, Bunte K, Peters U, Beikler T, Boege F. Autoantibodies against M5‐muscarinic and beta1‐adrenergic receptors in periodontitis patients. Aging (Albany NY). 2020; 12: 16609–16620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trimpert C, Herda LR, Eckerle LG, Pohle S, Müller C, Landsberger M, Felix SB, Staudt A. Immunoadsorption in dilated cardiomyopathy: long‐term reduction of cardiodepressant antibodies. Eur J Clin Invest. 2010; 40: 685–691. [DOI] [PubMed] [Google Scholar]
- 32. Staudt A, Herda LR, Trimpert C, Lubenow L, Landsberger M, Dörr M, Hummel A, Eckerle LG, Beug D, Müller C, Hoffmann W, Weitmann K, Klingel K, Kandolf R, Kroemer HK, Greinacher A, Felix SB. Fcgamma‐receptor IIa polymorphism and the role of immunoadsorption in cardiac dysfunction in patients with dilated cardiomyopathy. Clin Pharmacol Ther. 2010; 87: 452–458. [DOI] [PubMed] [Google Scholar]
- 33. Eckerle LG, Felix SB, Herda LR. Measurement of antibody effects on cellular function of isolated cardiomyocytes. J Vis Exp: JoVE. 2013; 73: e4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luft FC. Activating autoantibodies and cardiovascular disease. Physiology (Bethesda). 2013; 28: 254–261. [DOI] [PubMed] [Google Scholar]
- 35. Bornholz B, Hanzen B, Reinke Y, Felix SB, Jahns R, Schimke I, Wallukat G, Boege F. Detection of DCM‐associated beta1‐adrenergic receptor autoantibodies requires functional readouts or native human beta1‐receptors as targets. Int J Cardiol. 2016; 202: 728–730. [DOI] [PubMed] [Google Scholar]
- 36. Boege F, Westenfeld R, Jahns R. beta1AAb determined by peptide ELISA: a signal in the noise? J Am Coll Cardiol. 2017; 70: 807–808. [DOI] [PubMed] [Google Scholar]
- 37. Jahns R, Boege F. Questionable validity of petide‐based ELISA strategies in the diagnostics of cadrdiopathogenic autoantibodies that activate G‐protein‐coupled receptors. Cardiology. 2015; 131: 149–150. [DOI] [PubMed] [Google Scholar]
- 38. Wölfel A, Sättele M, Zechmeister C, Nikolaev VO, Lohse MJ, Boege F, Jahns R, Boivin‐Jahns V. Unmasking features of the auto‐epitope essential for beta1 ‐adrenoceptor activation by autoantibodies in chronic heart failure. ESC Heart Fail. 2020; 7: 1830–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Spieker M, Katsianos E, Gastl M, Behm P, Horn P, Jacoby C, Schnackenburg B, Reinecke P, Kelm M, Westenfeld R, Bönner F. T2 mapping cardiovascular magnetic resonance identifies the presence of myocardial inflammation in patients with dilated cardiomyopathy as compared to endomyocardial biopsy. Eur Heart J Cardiovasc Imaging. 2018; 19: 574–582. [DOI] [PubMed] [Google Scholar]
- 40. Messroghli DR, Moon JC, Ferreira VM, Grosse‐Wortmann L, He T, Kellman P, Mascherbauer J, Nezafat R, Salerno M, Schelbert EB, Taylor AJ, Thompson R, Ugander M, van Heeswijk R, Friedrich MG. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: a consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J Cardiovasc Magn Reson. 2017; 19: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scheiber D, Jelenik T, Zweck E, Horn P, Schultheiss HP, Lassner D, Boeken U, Saeed D, Kelm M, Roden M, Westenfeld R, Szendroedi J. High‐resolution respirometry in human endomyocardial biopsies shows reduced ventricular oxidative capacity related to heart failure. Exp Mol Med. 2019; 51: 16–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scheiber D, Zweck E, Jelenik T, Horn P, Albermann S, Masyuk M, Boeken U, Saeed D, Kelm M, Roden M, Szendroedi J, Westenfeld R. Reduced myocardial mitochondrial ROS production in mechanically unloaded hearts. J Cardiovasc Transl Res. 2018; 12: 107–115. [DOI] [PubMed] [Google Scholar]
- 43. Jelenik T, Flögel U, Álvarez‐Hernández E, Scheiber D, Zweck E, Ding Z, Rothe M, Mastrototaro L, Kohlhaas V, Kotzka J, Knebel B, Müller‐Wieland D, Moellendorf S, Gödecke A, Kelm M, Westenfeld R, Roden M, Szendroedi J. Insulin resistance and vulnerability to cardiac ischemia. Diabetes. 2018; 67: 2695–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Puntmann VO, Peker E, Chandrashekhar Y, Nagel E. T1 mapping in characterizing myocardial disease: a comprehensive review. Circ Res. 2016; 119: 277–299. [DOI] [PubMed] [Google Scholar]
- 45. Neubauer S. The failing heart — an engine out of fuel. N Engl J Med. 2007; 356: 1140–1151. [DOI] [PubMed] [Google Scholar]
- 46. Sterin‐Borda L, Cossio PM, Gimeno MF, Gimeno AL, Diez C, Laguens RP, Meckert PC, Arana RM. Effect of chagasic sera on the rat isolated atrial preparation: immunological, morphological and function aspects. Cardiovasc Res. 1976; 10: 613–622. [DOI] [PubMed] [Google Scholar]
- 47. Fraser C, Venter J. Anti‐receptor antibodies in human disease. J Allergy Clin Immunol. 1984; 74: 661–673. [DOI] [PubMed] [Google Scholar]
- 48. Wallukat G, Wollenberger A. Effects of the serum gamma globulin fraction of patients with allergic asthma and dilated cardiomyopathy on chronotropic beta adrenoceptor function in cultured neonatal rat heart myocytes. Biomed Biochim Acta. 1987; 46: S634–S639. [PubMed] [Google Scholar]
- 49. Limas CJ, Goldenberg IF, Limas C. Autoantibodies against beta‐adrenoceptors in human idiopathic dilated cardiomyopathy. Circ Res. 1989; 64: 97–103. [DOI] [PubMed] [Google Scholar]
- 50. Fu MLX. Anti‐M2 muscarinic receptor autoantibodies and idiopathic dilated cardiomyopathy. Int J Cardiol. 1996; 54: 127–135. [DOI] [PubMed] [Google Scholar]
- 51. Munoz‐Saravia SG, Haberland A, Wallukat G, Schimke I. Chronic Chagas' heart disease: a disease on its way to becoming a worldwide health problem: epidemiology, etiopathology, treatment, pathogenesis and laboratory medicine. Heart Fail Rev. 2012; 17: 45–64. [DOI] [PubMed] [Google Scholar]
- 52. Wallukat G, Wenzel K, Schimke I. Analytics of functional autoantibodies in patients with Chagas disease. Methods Mol Biol. 2019; 1955: 247–261. [DOI] [PubMed] [Google Scholar]
- 53. Bornholz B, Benninghaus T, Reinke Y, Felix SB, Roggenbuck D, Jahns‐Boivin V, Jahns R, Boege F. A standardised FACS assay based on native, receptor transfected cells for the clinical diagnosis and monitoring of beta1‐adrenergic receptor autoantibodies in human heart disease. Clin Chem Lab Med. 2016; 54: 683–691. [DOI] [PubMed] [Google Scholar]
- 54. Kaschina E, Namsolleck P, Unger T. AT2 receptors in cardiovascular and renal diseases. Pharmacol Res. 2017; 125: 39–47. [DOI] [PubMed] [Google Scholar]
- 55. Liles C, Li H, Veitla V, Liles JT, Murphy TA, Cunningham MW, Yu X, Kem DC. AT2R autoantibodies block angiotensin II and AT1R autoantibody‐induced vasoconstriction. Hypertension. 2015; 66: 830–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kawai T, Forrester SJ, O'Brien S, Baggett A, Rizzo V, Eguchi S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol Res. 2017; 125: 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Becker NP, Goettel P, Mueller J, Wallukat G, Schimke I. Functional autoantibody diseases: basics and treatment related to cardiomyopathies. Front Biosci (Landmark Ed). 2019; 24: 48–95. [DOI] [PubMed] [Google Scholar]
- 58. Bornholz B, Weidtkamp‐Peters S, Schmitmeier S, Seidel CA, Herda LR, Felix SB, Lemoine H, Hescheler J, Nguemo F, Schäfer C, Christensen MO, Mielke C, Boege F. Impact of human autoantibodies on beta1‐adrenergic receptor conformation, activity, and internalization. Cardiovasc Res. 2013; 97: 472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bornholz B, Hanzen B, Reinke Y, Felix SB, Boege F. Impact of common beta‐adrenergic receptor polymorphisms on the interaction with agonistic autoantibodies in dilated cardiomyopathy. Int J Cardiol. 2016; 214: 83–85. [DOI] [PubMed] [Google Scholar]
- 60. Lv T, Du Y, Cao N, Zhang S, Gong Y, Bai Y, Wang W, Liu H. Proliferation in cardiac fibroblasts induced by beta1‐adrenoceptor autoantibody and the underlying mechanisms. Sci Rep. 2016; 6: 32430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gurses KM, Yalcin MU, Kocyigit D, Kesikli SA, Canpolat U, Yorgun H, Sahiner ML, Kaya EB, Hazirolan T, Ozer N, Oto MA, Guc D, Aytemir K. M2‐muscarinic acetylcholine receptor autoantibody levels predict left atrial fibrosis severity in paroxysmal lone atrial fibrillation patients undergoing cryoablation. EP Europace. 2015; 17: 239–246. [DOI] [PubMed] [Google Scholar]
- 62. Matsui S, Fu ML, Hayase M, Katsuda S, Yamaguchi N, Teraoka K, Kurihara T, Takekoshi N. Active immunization of combined beta1‐adrenoceptor and M2‐muscarinic receptor peptides induces cardiac hypertrophy in rabbits. J Card Fail. 1999; 5: 246–254. [DOI] [PubMed] [Google Scholar]
- 63. Seidel F, Scheibenbogen C, Heidecke H, Opgen‐Rhein B, Pickardt T, Klingel K, Berger F, Messroghli D, Schubert S. Compensatory upregulation of anti‐beta‐adrenergic receptor antibody levels might prevent heart failure presentation in pediatric myocarditis. Front Pediatr. 2022; 10: 881208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Boden G. Free fatty acids, insulin resistance, and type 2 diabetes mellitus. Proc Assoc Am Physicians. 1999; 111: 241–248. [DOI] [PubMed] [Google Scholar]
- 65. Chow BS, Koulis C, Krishnaswamy P, Steckelings UM, Unger T, Cooper ME, Jandeleit‐Dahm KA, Allen TJ. The angiotensin II type 2 receptor agonist compound 21 is protective in experimental diabetes‐associated atherosclerosis. Diabetologia. 2016; 59: 1778–1790. [DOI] [PubMed] [Google Scholar]
- 66. Muniyappa R, Yavuz S. Metabolic actions of angiotensin II and insulin: a microvascular endothelial balancing act. Mol Cell Endocrinol. 2013; 378: 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pesta D, Gnaiger E. High‐resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol Biol. 2012; 810: 25–58. [DOI] [PubMed] [Google Scholar]
- 68. Lebovitz HE. Insulin resistance: definition and consequences. Exp Clin Endocrinol Diabetes. 2001; 109: S135–S148. [DOI] [PubMed] [Google Scholar]
- 69. Roden M, Shulman GI. The integrative biology of type 2 diabetes. Nature. 2019; 576: 51–60. [DOI] [PubMed] [Google Scholar]
- 70. Ohshima K, Mogi M, Jing F, Iwanami J, Tsukuda K, Min LJ, Ogimoto A, Dahlöf B, Steckelings UM, Unger T, Higaki J, Horiuchi M. Direct angiotensin II type 2 receptor stimulation ameliorates insulin resistance in type 2 diabetes mice with PPARgamma activation. PLoS ONE. 2012; 7: e48387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bernardi S, Michelli A, Zuolo G, Candido R, Fabris B. Update on RAAS modulation for the treatment of diabetic cardiovascular disease. J Diabetes Res. 2016; 2016: 8917578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zweck E, Scheiber D, Jelenik T, Bönner F, Horn P, Pesta D, Schultheiss HP, Boeken U, Akhyari P, Lichtenberg A, Kelm M, Roden M, Westenfeld R, Szendroedi J. Exposure to type 2 diabetes provokes mitochondrial impairment in apparently healthy human hearts. Diabetes Care. 2021; 44: e82–e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. 2018; 122: 624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rawshani A, Rawshani A, Franzén S, Eliasson B, Svensson AM, Miftaraj M, McGuire DK, Sattar N, Rosengren A, Gudbjörnsdottir S. Mortality and cardiovascular disease in type 1 and type 2 diabetes. N Engl J Med. 2017; 376: 1407–1418. [DOI] [PubMed] [Google Scholar]
- 75. McMurray JJV, Gerstein HC, Holman RR, Pfeffer MA. Heart failure: a cardiovascular outcome in diabetes that can no longer be ignored. Lancet Diabetes Endocrinol. 2014; 2: 843–851. [DOI] [PubMed] [Google Scholar]
- 76. Caforio AL, Tona F, Bottaro S, Vinci A, Dequal G, Daliento L, Thiene G, Iliceto S. Clinical implications of anti‐heart autoantibodies in myocarditis and dilated cardiomyopathy. Autoimmunity. 2008; 41: 35–45. [DOI] [PubMed] [Google Scholar]
- 77. Caforio AL, Daliento L, Angelini A, Bottaro S, Vinci A, Dequal G, Tona F, Iliceto S, Thiene G, McKenna WJ. Autoimmune myocarditis and dilated cardiomyopathy: focus on cardiac autoantibodies. Lupus. 2005; 14: 652–655. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Comparison of immune assays.
Table S2. Histopathological classification.
Table S3. GPCR‐Antibodies and cardiac parameters.
Table S4. GPCR‐Antibodies and cardiac MRI.
Table S5. GPCR‐Antibodies and metabolic markers.
Table S6. GPCR‐Antibodies and mitochondrial parameters.
Figure S1. Correlation between β1‐adr‐R‐Antibodies measured via cyclopeptide or membrane essay within the heart failure group; GPCR‐Antibodies were normalized to total IgG and logarithmized. IgG immunoglobin G. Data given as Spearman rho. n = 91.