

Abstract
Aims
Recovery of blood flow is a therapeutic approach for myocardial infarction but paradoxically induces injury to the myocardium. Exosomes (exos) are pivotal mediators for intercellular communication that can be released by different cells and are involved in cardiovascular diseases. This study aimed to explore the possible effects and mechanisms of miR‐155‐5p loaded by serum‐derived exos in myocardial infarction reperfusion injury (MIRI).
Methods and results
Exos were isolated from mouse serum after induction of ischaemia reperfusion (I/R) and injected into I/R‐treated mice to assess cardiac function, infarction size, and cardiomyocyte apoptosis. Primary cardiomyocytes were transfected with miR‐155‐5p inhibitor before treatment with oxygen–glucose deprivation and re‐oxygenation (OGD/R) and exos derived from the serum of I/R‐treated mice (I/R‐Exos), in which Bcl‐2, Bax, and cleaved‐caspase‐3 levels were detected. The interactions among miR‐155‐5p, NEDD4, and CypD were evaluated. miR‐155‐5p level was evidently increased in I/R‐Exos than in exos from the serum of sham‐operated mice (P < 0.05). In comparison with the I/R group, the I/R‐Exos + I/R group had increased infarct size, elevated miR‐155‐5p expression, and boosted apoptotic rate in mouse myocardium (P < 0.05). In mice treated with I/R‐Exos and I/R, miR‐155‐5p inhibition reduced cardiac infarct size and apoptosis (P < 0.05). NEDD4 was a target gene of miR‐155‐5p and promoted CypD ubiquitination. Cardiomyocyte apoptosis was markedly increased in the miR‐155‐5p inhibitor + shNEDD4 + OGD/R group versus the miR‐155‐5p inhibitor + OGD/R group (P < 0.05), but decreased in the miR‐155‐5p inhibitor + shNEDD4 + shCypD + OGD/R group than in the miR‐155‐5p inhibitor + shNEDD4 + OGD/R group (P < 0.05).
Conclusions
miR‐155‐5p in I/R‐Exos may facilitate MIRI by inhibiting CypD ubiquitination via targeting NEDD4.
Keywords: Serum‐derived exosome, miR‐155‐5p, NEDD4/CypD axis, Ubiquitination, Myocardial ischaemia–reperfusion injury
Introduction
Myocardial infraction (MI) is generally attributed to increased myocardial oxygen demand and/or reduced supply to the heart. 1 Myocardial ischaemia–reperfusion injury (MIRI) is a common complication of reperfusion therapy for MI during restoration of blood flow to ischaemic myocardium. 2 Despite that great efforts have been made, no effective therapeutic strategy can attenuate MIRI following MI. Therefore, more researches have put their attention on understanding its molecular mechanism.
Recent studies highlighted the involvement of microRNAs (miRs) in cardiac ischaemia reperfusion (I/R). 3 , 4 Multiple miRs have been shown to regulate cardiomyocyte apoptosis and myocardial oxidative stress. 5 , 6 miR‐155 has been reported as a detrimental gene for cardiomyocyte proliferation in a cellular model of MIRI. 7 Inhibition miR‐155‐5p in human mesenchymal stem cells was found to improve cardiomyocyte survival and angiogenesis in infarcted mouse hearts. 8 Exosomes (exos) or microvesicles are an emerging type of diagnostic signs for cell‐free therapeutic application in cardiovascular disorders, and pose an important role for transmitting remote signals for cardioprotection after ischaemic preconditioning. 9 A previous study demonstrated that circulating exos released after MI can carry miRs to influence the angiogenesis of cardiac endothelial cells. 10 Importantly, miR‐155‐5p was highly enriched in extracellular vesicles released from I/R‐insulted heats (I/R‐EVs), and miR‐155‐5p in I/R‐EVs boosted macrophage M1 polarization, contributing to local inflammation in the heart and even systemic inflammation in distant organs. 11 Available evidence has indicated the significance of miR‐155‐5p and circulating exos in MIRI. Therefore, it is intriguing to explore the possibility of serum exos serving as carriers of miR‐155‐5p to affect MIRI and investigate the possible mechanisms.
NEDD4 family is homologous with the carboxyl end of E6AP (HECT) ubiquitin E3 ligases which catalyse the covalent binding of ubiquitin to the protein substrate via the WW domain to facilitate ubiquitination. 12 NEDD4‐1 is cardioprotective against I/R‐induced cardiomyocyte apoptosis, whose expression is reduced in the slate stage of MIRI. 13 NEDD4‐family interacting protein 1 (NDFIP1) was recognized as a direct target of miR‐155 and repressed cell proliferation in uveal melanoma. 14 In a previous study, miR‐454 has been demonstrated to suppress cardiomyocyte apoptosis and attenuate myocardial damage by targeting and downregulating NEDD4‐2. 15 Nevertheless, no evidence identified the possible interaction between miR‐155‐5p and NEDD4 in MIRI. Online bioinformatics prediction in our study showed a possible binding site between miR‐155‐5p and NEDD4, and we therefore speculated that miR‐155‐5p in serum‐derived exos could regulate MIRI by mediating NEDD4 expression. This study was conducted with the aim to elucidate the possible mechanism of serum‐derived exos loaded with miR‐155‐5p in regulating MIRI.
Materials and methods
Myocardial ischaemia reperfusion (I/R) mouse models
Male C57BL/6 mice (n = 60, 6–8 weeks old, 22–25 g) and suckling mice (n = 3, 1–3 days old) from Laboratory Animal Resources, Chinese Academy of Sciences were housed with standard diets and had free access to water. The animal experiments were conducted with the approval of the ethical committee of Nanchang University.
The 6‐ to 8‐week‐old mice were anaesthetised by intraperitoneal injection of pentobarbital sodium (60 mg/kg) and placed in supine positions on an operating table. After endotracheal intubation, the cannula was connected with a small animal ventilator for assisted ventilation, and the anterior chest of mice was exposed for thoracotomy. For establishment of an I/R model, the left anterior descending artery (LAD) of mice was ligated for 30 min and then reperfused for 2 h. The chest of mice was sutured after the surgery. Serum‐derived exos from I/R‐treated mice (I/R‐Exos) were injected into the left ventricular anterior wall and apex of mice (0.5 mg/kg for each injection site). Thirty minutes after the injection, LAD ligation was performed.
Sixty mice were randomly divided into sham, I/R, I/R‐Exos + I/R, I/R‐Exos+ negative control (NC) inhibitor + I/R, and I/R‐Exos + miR‐155‐5p inhibitor + I/R groups (n = 12 for each group). Sham‐operated mice underwent thoracotomy but were free from LAD ligation. Mice in the I/R‐Exos + I/R group were subjected to intramyocardial injection of 1 mg/kg I/R‐Exos 30 min before I/R treatment. Mice in the I/R‐Exos+ NC inhibitor + I/R and the I/R‐Exos + miR‐155‐5p inhibitor + I/R groups were injected with NC inhibitor or miR‐155‐5p inhibitor (100 nm) plus I/R‐Exos (1 mg/kg) to their myocardium 30 min before I/R treatment. Fourteen days after operation, cardiac functions of mice in each group were assessed using the Vevo 2100 Cardiac imaging system (VisualSonics, Canada). Then the mice were euthanized and their hearts were obtained for histological analysis. For each group, six mice were randomly selected for triphenyltetrazolium chloride (TTC) staining, and the others were used for other experiments.
Histological analysis
After ultrasonic inspection, mice were euthanized by high‐concentration carbon dioxide inhalation combined with cervical dislocation. Mice in the sham and I/R groups underwent blood sampling from their orbits and their cardiac tissues were obtained. The cardiac tissues of six mice in each group were made into 2‐mm‐thick sections. The sections were placed in 1% TTC stain at 37°C in the dark for 20 min. Later, excess stain was washed away with phosphate‐buffered saline (PBS). The percentage of MI was calculated using the Image‐Pro Plus 6.0 software. Normal tissues were red and infarcted tissues were grey‐white.
Cardiac tissues were dehydrated using alcohol gradient for 1 min each time, cleared twice with xylene for 5 min each time, and embedded in paraffin. The paraffin‐embedded tissues were continuously sliced (5 μm), and the tissue sections were placed in an oven (80°C) for 1 h. The sections were cooled, hydrated using alcohol gradient, cleared with xylene, and rinsed. Subsequently, the sections were stained with haematoxylin (H8070‐5g, Solarbio, Beijing, China) for 4 min and differentiated for 10 s with alcohol hydrochloride. Following staining for 2 min with eosin (PT001, Shanghai Bogoo Biotechnology Co., Ltd., Shanghai, China), the sections were dehydrated using alcohol gradient for 1 min each time, cleared twice with xylene for 1 min each time, and blocked with neutral balsam. The histopathological changes of the tissues were observed under an optical microscope (DMM‐300D, Shanghai Caikon Optical Instrument Co., Ltd., Shanghai, China).
Transferase‐mediated deoxyuridine triphosphate‐biotin nick end labelling (TUNEL) staining
Paraffin‐embedded cardiac tissue sections were dried in an oven (<60°C) for 30 min, dewaxed, and hydrated. After that, the sections were immersed in citrate buffer solution for boiling. After cooling, the sections were washed thrice in PBS and stained using a fluorescein‐conjugated TUNEL in situ cell death detection kit (Roche Diagnostics, Mannheim, Germany). Fluorescence micrographs were obtained under an Olympus IX‐73 fluorescence (Olympus, Tokyo, Japan). Data were obtained from five randomly selected fields and averaged, and the apoptotic rate (%) was calculated as positive cells/total cells × 100.
Isolation and identification of exos
According to the protocol of the ExoQuick ULTRA extraction kit (System Biosciences), exos were isolated from the serum of mice in the sham and I/R groups. ExoQuick ULTRA was mixed with mouse serum (250 μL) for overnight incubation (4°C). Afterwards, the serum was centrifuged for 10 min (3000 g) and the supernatant was removed. After centrifugation for 5 min (1500 g), the upper liquid was discarded and the exos were collected after resuspension. Protein quantification of exos was performed using BCA kits (Beyotime, Jiangsu, China). The morphology of the exos was observed under a transmission electron microscope (TEM, Hitachi, Japan). Then 20 μL of exos were added to a copper grid, dried using filter paper, and added to one drop of 1% uranyl acetate. After 1 min, the exos were dried using filter paper. The exos were observed under the TEM. After that, 0.5 mL of exos were diluted in 4.5 mL ultrapure water and filtered through a 0.22‐μm filter membrane. The size of the exos was measured using nanoparticle tracking analysis (NTA). The expression levels of exo biomarkers [CD9 (ab92726, 1:1000, Abcam, Cambridge, MA, USA), CD63 (ab217345, 1:1000, Abcam), ALIX (ab275377, 1:1000, Abcam), and Calnexin (ab133615, 1:2000, Abcam)] were measured using western blotting.
Exos labelling
Exos were labelled with PKH26 (Sigma‐Aldrich, St. Louis, MO, USA) at 37°C, based on the instructions of the manufacturer. Exos (20 μg) were resuspended in 100 μL of PBS and added to 0.5 mL of dilution buffer C, followed by the addition of the mixture of 4 μL PKH26 and 0.5 mL dilution buffer C. After the reaction was terminated, the solution was wholly transferred to a new tube, filtered through a 0.22‐μm membrane, and centrifuged for 2 h (120 000 g, and 4°C).
Isolation and culture of primary cardiomyocytes
Three suckling mice were euthanized, and their left ventricles were collected for isolation of primary cardiomyocytes. The tissues were immersed in Hank's solution, the vessels were removed, and the cardiac tissues were cut into 1–2 mm2 pieces using ophthalmic scissors. The tissues were vibrated and digested in Hank's solution containing 0.125% trypsin and 150 U/mL type II collagenase (37°C, 10 min). The disassociation solution was isolated, added with fetal bovine serum (FBS) to terminate the digestion, and centrifuged for 5 min at 1200 rpm. The supernatant was removed and the cell pellets were resuspended in 10% FBS‐Dulbecco's Modified Eagle Medium (DMEM). The residual tissues were repeatedly digested for three to five times till the tissue blocks were generally digested. The cell suspension was mixed, flicked, and centrifuged at 1200 rpm for 3 min. The supernatant was removed, and the cells were resuspended in 10% FBS‐DMEM, inoculated to a culture flask, and cultured at 37°C with 5% CO2.
Immunofluorescence staining
Cardiomyocytes were seeded onto 6‐well plates (3 × 104 cells/well) and then incubated in 4% paraformaldehyde (dissolved in PBS, pH 7.4) for 10 min at room temperature. Following thrice ice‐cold PBS washing, the cells were treated with primary antibodies against α‐actin (ab179467, 1:50, Abcam) and cTnT (PA5‐102358, 1:100, Cell Signalling Technology, Danvers, MA, USA) overnight at 4°C. After thrice washing in PBS, the cells were incubated for 1 h with goat‐anti rabbit IgG (1:200, Beijing ComWin Biotech Co., Ltd., Beijing, China). After PBS washing, the cells were added with 4′,6‐diamidino‐2‐phenylindole 2hci (DAPI) for nuclear staining. Staining was visualized by a fluorescence microscope (TCSSP2, Leica, Germany) and percentage of positive cells was calculated as positive cells/total cells × 100%.
Plasmid construction and cell transfection
miR‐155‐5p mimic, miR‐155‐5p inhibitor, mimic NC, or inhibitor NC (Guangzhou RiboBio Co., Ltd., China) as well as shRNAs for NEDD4 and Cyclophilin‐D (CypD) (50 nM) (Invitrogen, Carlsbad, CA, USA) were transduced into primary cardiomyocytes using Lipofectamine 3000 (Thermo Fishier Scientific) in accordance with the instructions. Cell experiments were initiated 48 h after cell transfection.
Oxygen and glucose deprivation/reoxygenation (OGD/R) cell model
Primary cardiomyocytes were cultured in DMEM (Gibco, Grand Island, NY, USA) containing 10% FBS and 1% penicillin/streptomycin at 37°C with 5% CO2. The OGD/R model was conducted based on the protocols of Yi et al. 16 : Cardiomyocytes were exposed to a hypoxic atmosphere (94% N2, 5% CO2, and 1% O2) in sugar‐free medium for 6 h, and transferred to normal medium and incubated for 6 h at 37°C with 5% CO2.
Cell grouping was as follows: control group: untreated cardiomyocytes; OGD/R groupI/R‐Exos + OGD/R group: OGD/R‐induced cardiomyocytes were co‐cultured with I/R‐Exos; I/R‐Exos + NC inhibitor + OGD/R group: cardiomyocytes transfected with NC inhibitor were subjected to OGD/R induction and co‐cultured with I/R‐Exos; I/R‐Exos + miR‐155‐5p inhibitor + OGD/R group: cardiomyocytes transfected with miR‐155‐5p inhibitor were stimulated with OGD/R and co‐cultured with I/R‐Exos; mimic NC + OGD/R group: cardiomyocytes transfected with NC mimic were induced by OGD/R; miR‐155‐5p mimic + OGD/R group: cardiomyocytes transfected with miR‐155‐5p mimic were induced by OGD/R; NC inhibitor + OGD/R group: cardiomyocytes transfected with NC inhibitor were induced by OGD/R; miR‐155‐5p inhibitor + OGD/R group: cardiomyocytes transfected with miR‐155‐5p inhibitor were induced by OGD/R; miR‐155‐5p inhibitor + sh NEDD4 + OGD/R group: cardiomyocytes transfected with miR‐155‐5p inhibitor and sh NEDD4 were induced by OGD/R; miR‐155‐5p inhibitor + sh NEDD4 + sh CypD + OGD/R group: cardiomyocytes transfected with miR‐155‐5p inhibitor, sh NEDD4, and sh CypD were induced by OGD/R.
Quantitative reverse transcription polymerase chain reaction (qRT‐PCR)
Total RNA in cardiac tissues and cardiomyocytes were extracted using TRIzol reagent and 5 μL of total RNA was diluted 20 fold using RNA‐free ultrapure water. The absorbance at 260 nm and 280 nm was measured using an ultraviolet spectrophotometer to determine RNA purity and concentration. OD260/OD280 ratios within the range of 1.7–2.1 suggest high purity. RNA was reverse transcribed into cDNA templates using a PCR instrument. Real‐time quantitative RT‐PCR was performed using an ABI7500 quantitative PCR instrument (pre‐denaturation at 95°C for 10 min, and 40 cycles of denaturation at 95°C for 10 s, annealing at 60°C for 20 s, and extension at 72°C for 34 s). The data were analysed using the 2−ΔΔCt method: ΔΔCt = [Ct(target gene) − Ct(internal gene)]experimental group − [Ct(target gene) − Ct(internal gene)]control group. Three biological replicates were set for each test and the primer sequences are listed in Table 1 .
Table 1.
Primer sequences
| Name of primer | Sequences |
|---|---|
| miR‐155‐5p‐F | TTAATGCTAATTGTGAT |
| miR‐155‐5p‐R | GCAGGGTCCGAGGTATTC |
| U6‐F | TCGCTTCGGCAGCACATATAC |
| U6‐R | GCGTGTCATCCTTGCGCAG |
| NEDD4‐F | GAGTGGAATCCTTACCAGCGTG |
| NEDD4‐R | AGAATGCGGTGTCGCTGTGGAA |
| CypD‐F | GGACGCTTGAAAATGTAGAGGTG |
| CypD‐R | GGATGACTGTCACCAGAGCCAT |
| β‐actin‐F | CATTGCTGACAGGATGCAGAAGG |
| β‐actin ‐R | TGCTGGAAGGTGGACAGTGAGG |
Note: F, forward; R, reverse.
Flow cytometry
Primary cardiomyocytes at logarithmic phases were collected and the concentration of cell suspension was adjusted into 1 × 106 cells/mL. The cardiomyocytes were plated onto a 6‐well plate for cell culture in an incubator at 37°C with 5% CO2. About 48 h after cell transfection, the cells were disassociated in trypsin and collected for Annexin V‐FITC and PI staining in a darkroom for 15 min based on the instructions of the Annexin V‐FITC apoptosis assay kit (BD Biosciences, USA). Cell apoptotic rate was measured by a flow cytometer.
Western blotting
Cardiomyocytes and cardiac tissues were washed thrice in pre‐cold PBS before cell lysis buffer in a 100 μL/50 mL culture disk was added for cell lysis on ice for 30 min. The cell lysates were centrifuged at 12000 rpm and 4°C for 10 min, and the supernatant was collected for determination of protein concentration using BCA method. The proteins were added with loading buffer for 5 min of boiling and then 30 μg of proteins were collected for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and electroblotted onto a PVDF membrane. The unspecific reaction was blocked using skimmed milk powder for 2 h before the primary antibodies of Bcl‐2 (ab196495, Abcam), Bax (ab32503, Abcam), cleaved caspase 3 (ab49822, Abcam), NEDD4 (sc‐25508, Santa Cruz), and CypD (Mitosciences) were incubated with the membrane at 4°C for overnight. The membrane was washed thrice with TBST for 10 min each before incubation with horseradish peroxidase‐labelled IgG at room temperature for 2 h, followed by thrice TBST washes for 10 min each. The membrane was then subjected to ECL colour development and the grey value was analysed using Image J software, using β‐actin as an internal control.
Dual luciferase reporter assay
The binding sites of NEDD4 and miR‐155‐5p were predicted using Starbase. The mutant and wide sequences of the binding site were accordingly designed and cloned into reporter vectors (Promega, Madison, WI, USA). Cells were seeded onto a culture plate and transduced with miR‐155‐5p mimic or mimic NC plus reporter vectors loading with the mutant or wild sequence. The luciferase activities in each group were measured using the dual luciferase reporter gene detection system (Promega, United States).
Co‐immunoprecipitation (co‐IP)
Cells were lysed on ice with 0.6 mL of lysis buffer and protease inhibitor (Roche) for 40 min, followed by 15 min of centrifugation (12 000 g). The lysates were incubated with 1 μg specific antibody at 4°C for overnight. After that, 30 μL of protein A agarose (Invitrogen) or protein G agarose (Santa Cruz) beads were washed and incubated with the antigen–antibody complex for 3 h. Then the complex was washed with cell lysis buffer for 4 times and boiled with loading buffer for 3 min before SDS‐PAGE and western blotting.
Statistical analysis
Data were analysed using GraphPad Prism 5.0 (GraphPad Software Inc.). Measuring data were expressed as mean ± standard deviation. Comparisons between two groups were analysed using the t test whereas those among multiple groups were determined using one‐way analysis of variance, with Tukey's multiple comparisons test as the post hoc analysis. P value <0.05 was considered to have significant difference.
Results
Isolation and identification of serum‐derived exos
Increasing evidence has shown exos can be derived from stem cells, macrophage, and serum. To explore the effects of exos on I/R‐induced myocardial injury, exos were isolated from the serum of mice in the sham and I/R groups. Under the TEM, exos had membranous structures (Figure 1 A ). The particle size of the exos was analysed by NTA tests. Each sample was examined for three times and the results represented the average of the three NTA tests. Exos were approximately 100 nm in diameters, with an average of 104.26 nm in the sham group and 109.75 nm in the I/R group (Figure 1 B ). Western blots showed that the exos in the sham and I/R groups expressed CD9, CD63, and ALIX but did not express Calnexin (Figure 1 C ). Collectively, those results suggested exos were successfully isolated. In addition, there were no significant differences in the particle number and protein amount per millilitre of exos as well as the protein amount per million particles in the I/R group versus the sham group (Figure 1 D–F ).
Figure 1.
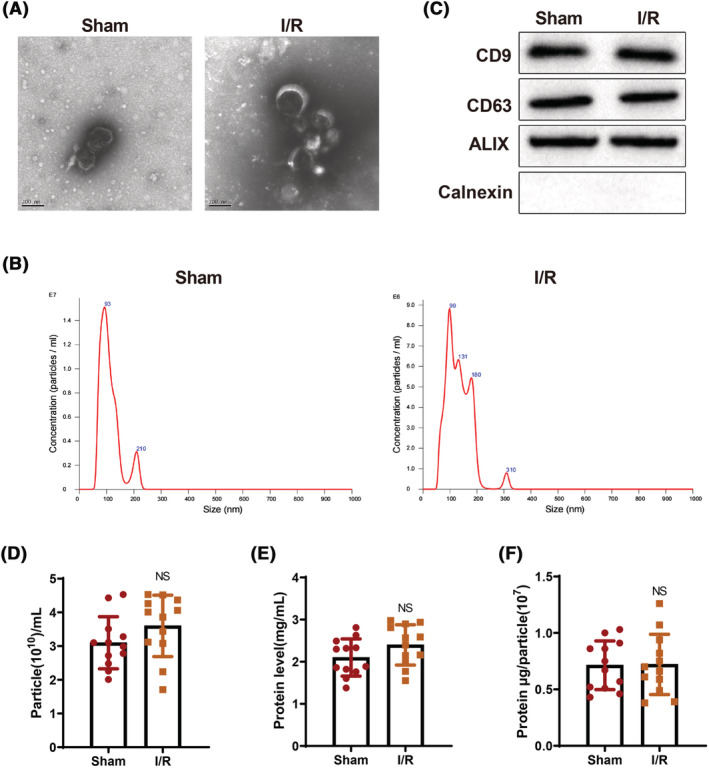
Isolation and identification of exos derived from serum of mice in the sham and I/R groups. The morphology of exos was observed under the TEM (A); the size of serum‐derived exos was measured by nanoparticle tracking analysis (B); the expression of CD9, CD63, ALIX, and Calnexin in exos was detected by western blotting (C). The experiment was set with three biological replicates. The particle number and protein amount per millilitre of exos were examined (D, E); the protein amount per million particles was detected (F). N = 12. TEM, transmission electron microscope; I/R, ischaemia–reperfusion; exos, exosomes.
I/R‐exos aggravates myocardial injury
Ischaemia reperfusion can cause myocardial infarction and a decrease in left ventricular ejection fraction (LVEF). TTC staining is the gold standard for detecting infarct size, 17 and LVEF is an important indicator for cardiac function. 18 The detection of heart function by ultrasonic cardiogram showed that mice in the sham group had normal heart function; the heart function of mice in the I/R group was damaged compared with that in the sham group; the severity of heart injury in the I/R‐Exos + I/R group was more obvious than in the I/R group (P < 0.05, Figure 2 A ). TTC staining suggested that the infarct size of mice was increased in the I/R group than in the sham group. The infarct size of mice in the I/R‐Exos + I/R group was further increased than that of the I/R group (P < 0.05, Figure 2 B ). H&E staining in Figure 2 C demonstrated that the cardiomyocytes in the sham group arranged neatly, and had normal morphology, centred nuclei, and no obvious inflammatory cell infiltration. However, inflammatory infiltration was obviously noticed in the I/R group, and the worst inflammatory infiltration was found in the I/R‐Exos +I/R group versus the I/R group. Furthermore, TUNEL staining showed that cardiomyocyte apoptosis in the I/R group was elevated compared with that in the sham group, and further aggravated in the I/R‐Exos + I/R group compared with the I/R group (P < 0.05, Figure 2 D ). Bax and caspase‐3 are pro‐apoptotic proteins, 19 , 20 and Bcl‐2 shows an anti‐apoptotic activity. 21 Western blots indicated increased Bax and cleaved caspase3 protein expression and decreased Bcl‐2 protein expression in the I/R group, and the expression trends of these proteins were stronger in the I/R‐Exos + I/R group compared with those in the I/R group (P < 0.05, Figure 2 E ). Above results showed I/R‐Exos aggravated myocardial injury in I/R‐treated mice.
Figure 2.
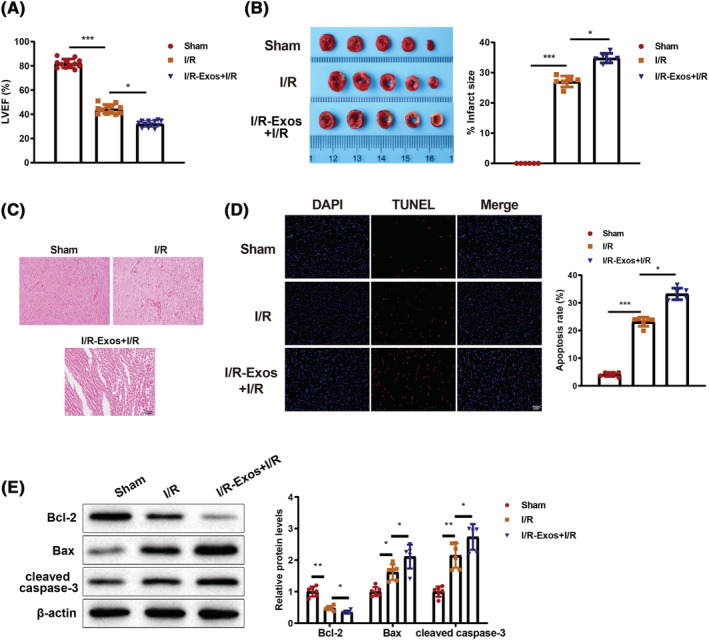
Exos derived from serum of I/R‐treated mice deteriorated myocardial injury in mice. The heart functions of mice in each group were assessed by detecting left ventricular ejection fraction using ultrasonic cardiogram (A). The infarction area in each group was detected by TCC staining (B). Histological changes of cardiac tissues were observed after H&E staining (C). Cell apoptosis in myocardium was assessed after TUNEL staining (D). Western blot was used to detect Bcl‐2, Bax, and cleaved caspase3 protein expression in myocardium (E). N = 6 except N = 12 in panel (A); *P < 0.05; **P < 0.01; ***P < 0.001. Data were exhibited in the form of mean ± standard deviation, and comparisons among multiple groups were analysed using one‐way analysis of variance, followed by Tukey's multiple comparisons test. I/R, ischaemia–reperfusion; exos, exosomes.
miR‐155‐5p inhibition attenuates I/R‐exos‐induced myocardial injury in mice
Because I/R‐Exos were found to impel myocardial injury in I/R‐treated mice, we speculated the miRs carried by I/R‐Exos may play pivotal roles in MIRI. In addition, downregulation of miR‐155 may be involved in sevoflurane‐mediated cardioprotection through mediating SIRT1. 22 Therefore, we detected the level of miR‐155‐5p in the sham‐Exos and I/R‐Exos and the results showed that miR‐155‐5p level was elevated in the I/R‐Exos group compared with the sham‐Exos group (P < 0.05, Figure 3 A ). The expression level of miR‐155‐5p in mouse myocardium was measured after injection of I/R‐Exos. qRT‐PCR demonstrated that miR‐155‐5p expression level in the I/R group was increased compared with the sham group (P < 0.05), and further elevated in the I/R‐Exos + I/R group compared with the I/R group (P < 0.05, Figure 3 B ). After the exos were dissolved in PBS and added with RNase or/and TritonX‐100, the level of miR‐155‐5p was detected. Co‐treatment by RNase and TritonX‐100 resulted in reduced miR‐155‐5p expression level (P < 0.05, Figure 3 C ), whereas RNase treatment alone did not alter the level of miR‐155‐5p, suggesting miR‐155‐5p was packaged by membrane and may have critical functions in myocardial injury in I/R‐treated mice. To validate the impacts of exo‐loaded miR‐155‐5p in myocardial injury, I/R‐Exos and miR‐155‐5p inhibitor were injected into mice. Compared with the I/R‐Exos + NC inhibitor + I/R group, the expression level of miR‐155‐5p in the I/R‐Exos + miR‐155‐5p inhibitor + I/R group was suppressed (P < 0.05, Figure 3 D ). TTC staining (Figure 3 E ) and H&E staining (Figure 3 F ) demonstrated increased infraction area in the I/R‐Exos + NC inhibitor + I/R group and decreased infarction area in the I/R‐Exos + miR‐155‐5p inhibitor + I/R group (P < 0.05). TUNEL staining showed that miR‐155‐5p inhibitor suppressed ischaemia‐induced apoptosis of cardiomyocytes (P < 0.05, Figure 3 G ). The findings of western blotting indicated that decreased Bax and cleaved caspase3 protein expression and increased Bcl‐2 protein expression in the I/R‐Exos + miR‐155‐5p inhibitor + I/R group versus the I/R‐Exos + NC inhibitor + I/R group (P < 0.05, Figure 3 H ). Above results showed inhibition of miR‐155‐5p expression level attenuated I/R‐Exos‐induced myocardial injury.
Figure 3.
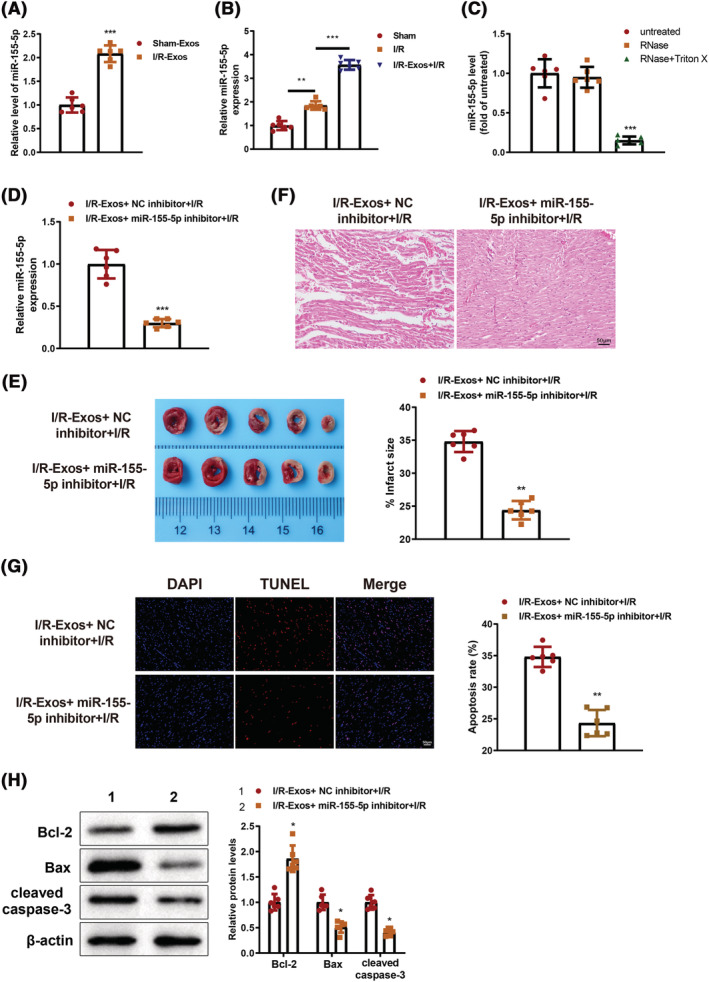
Inhibition of miR‐155‐5p exerts cardioprotection on I/R‐Exos‐induced myocardial injury in I/R‐treated mice. The level of miR‐155‐5p in exos‐derived from mouse serum (A) and myocardium (B) was detected by qRT‐PCR. After treatment by RNase A or/and Triton X‐100, the level of miR‐155‐5p in exos was measured by qRT‐PCR (C). The efficiency of miR‐155‐5p inhibitor was assessed by qRT‐PCR. (D). Infarction, morphology, and cell apoptosis in mouse myocardium were determined by TTC (E), H&E (F), and TUNEL staining (G), respectively. Western blotting was used to detect Bcl‐2, Bax, and cleaved caspase3 protein expression in myocardium (H). N = 6; *P < 0.05; **P < 0.01; ***P < 0.001. Data were exhibited in the form of mean ± standard deviation. Comparisons between two groups were analysed using the t test whereas that among multiples was determined using one‐way analysis of variance combined with Tukey's multiple comparisons test. I/R, ischaemia–reperfusion; exos, exosomes.
miR‐155‐5p inhibition reverses IR‐exos‐induced cardiomyocyte injury
To verify the role of I/R‐Exos on cardiomyocyte injury, we isolated primary cardiomyocytes from myocardium of suckling mice. Spontaneous contraction was observed in individual cardiomyocytes under an inverted microscope 24 h after cell culture, and the cells were then gradually spread out, projecting pseudopodia to the surround and forming irregular star shapes. On the 3rd to 4th day of culture, the cells contacted and interweaved each other into a network, forming a cell monolayer or cell clusters. Reportedly, α‐actin and cTnT are specific antigens of cardiomyocytes. 23 The expression of α‐actin and cTnT was detected by immunofluorescence (Figure 4 A ). The results showed that α‐actin and cTnT were highly expressed in 90% of the isolated cardiomyocytes. Next, cardiomyocytes were exposed to OGD/R to induce injury. The isolated cells were co‐cultured with exos and the uptake of Exos was observed by a fluorescence microscope. After co‐culture with PKH26‐labelled exos, the cardiomyocytes exhibited red fluorescence (Figure 4 B ). qRT‐PCR detection showed elevated miR‐155‐5p expression level in the OGD/R group compared with that in the control group (P < 0.05), whereas further elevated expression of miR‐155‐5p was observed in the I/R‐Exos + OGD/R group versus the OGD/R group, and miR‐155‐5p inhibitor evidently repressed miR‐155‐5p expression level (P < 0.05, Figure 4 C ). Relatedly, the effect of miR‐155‐5p on apoptosis of OGD/R treated‐cardiomyocytes was evaluated, which showed boosted cell apoptosis in the OGD/R and I/R‐Exos + OGD/R groups compared with their controls, and suppressed apoptotic rate in the I/R‐Exos + miR‐155‐5p inhibitor + OGD/R group compared with the I/R‐Exos + NC inhibitor + OGD/R group (P < 0.05, Figure 4 D ). Additionally, upregulated Bax and cleaved caspase3 expression levels and downregulated Bcl‐2 expression level were found in the OGD/R and I/R‐Exos + OGD/R groups than in their controls (P < 0.05). Further treatment with miR‐155‐5p inhibitor reversed the expression patterns of these proteins (P < 0.05, Figure 4 E ). In vitro data showed that inhibition of miR‐155‐5p reversed exos‐induced cardiomyocyte injury.
Figure 4.
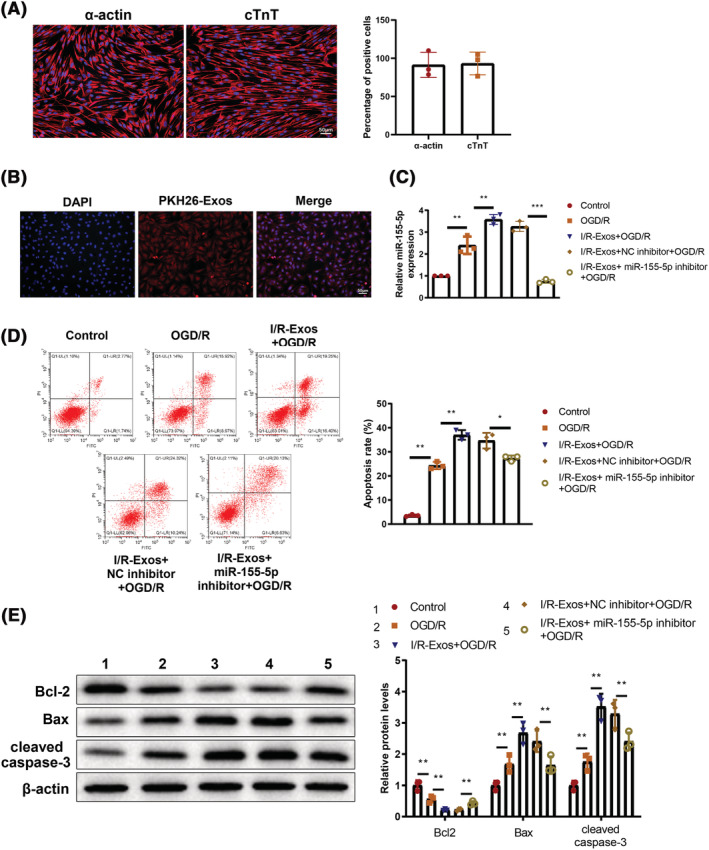
Inhibition of miR‐155‐5p expression in OGD/R‐treated cardiomyocytes reverses exos‐induced cardiomyocyte injury. The expression of α‐actin and cTnT in isolated cardiomyocytes was detected by immunofluorescence staining (A). The uptake of exos by cardiomyocytes was observed under a fluorescence microscope (B). The expression level of miR‐155‐5p was detected by qRT‐PCR (C). Cell apoptosis was determined by flow cytometry (D). The expression level of Bcl‐2, Bax, and cleaved caspase3 in each group was measured by western blot (E). The experiment was set with three biological replicates; *P < 0.05; **P < 0.01; ***P < 0.001. Data were expressed as mean ± standard deviation, and comparisons among multiple groups were analysed using one‐way analysis of variance combined with Tukey's multiple comparisons test. OGD/R, oxygen–glucose deprivation and re‐oxygenation; exos, exosomes.
miR‐155‐5p targets NEDD4
The target genes of miR‐155‐5p were predicted by Starbase and the results showed that miR‐155‐5p could bind NEDD4 in the 3′‐untranslated region (UTR) (Figure 5 A ). Measurement on luciferase activities showed that cells co‐transfected with miR‐155‐5p mimic and wild plasmid containing the NEDD4 3’UTR had suppressed luciferase activity (P < 0.05), whereas co‐transfection with mutant plasmid had no significant change in luciferase activity (P > 0.05, Figure 5 B ). Additionally, qRT‐PCR analysis demonstrated that knockdown of miR‐155‐5p in cardiomyocytes before OGD/R treatment resulted in elevated expression levels of NEDD4 mRNA and protein compared with the NC inhibitor + OGD/R group (P < 0.05, Figure 5 C,D ). To determine the effect of miR‐155‐5p on NEDD4 in mouse myocardium, we injected I/R‐Exos and miR‐155‐5p inhibitor into mice and induced the I/R model. In comparison to the I/R‐Exos + NC inhibitor + I/R group, NEDD4 protein expression was increased in the I/R‐Exos + miR‐155‐5p inhibitor + I/R group (P < 0.05, Figure 5 E ). Collectively, miR‐155‐5p can bind and negatively regulate NEDD4.
Figure 5.
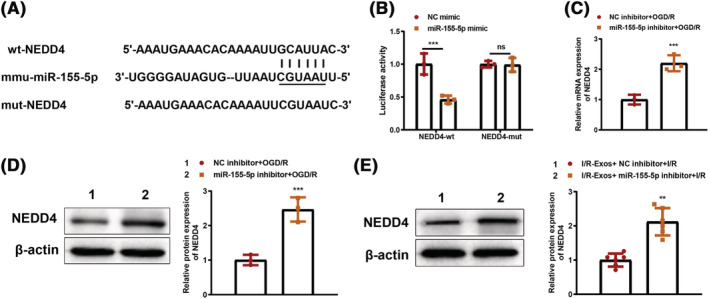
miR‐155‐5p negatively regulates NEDD4 expression. The binding site of miR‐155‐5p in the 3′‐UTR of NEDD4 (A). The luciferase activities were detected after co‐transfection of miR‐155‐5p mimic plus NEDD4‐wt or NEDD4‐mut (B). NEDD4 mRNA expression was measured by qRT‐PCR (C). NEDD4 protein expression in cardiomyocytes (D) or myocardial tissues (E) was determined by western blot. In panel E, N = 6. The rest panels were cell experiments, and the experiment was set with three biological replicates; *P < 0.05; **P < 0.01; ***P < 0.001. Data were expressed as mean ± standard deviation, and comparisons between two groups were analysed using the t test.
miR‐155‐5p inhibits CypD ubiquitination degradation through NEDD4 expression
NEDD4 family of carboxyl end of E6AP (HECT) ubiquitin E3 ligases can catalyse ubiquitination by recognizing different protein substrates via the WW domain. 12 Previously, Parkin was found to regulate the ubiquitination of CypD and thus mitigate programed necrosis and MIRI. 24 Apoptosis repressor with caspase recruitment domain (ARC) can inhibit mPTP expression by mediating CypD (a principal regulatory factor of mPTP), thereby reducing cardiomyocyte apoptosis. 25 To explore the mechanism of NEDD4‐mediated cardiomyocyte injury, in vitro experiments were conducted. Overexpression of miR‐155‐5p in cardiomyocytes before OGD/R treatment had no effect on CypD mRNA expression and resulted in elevated CypD protein expression, whereas downregulation of miR‐155‐5p reduced CypD protein expression (P < 0.05, Figure 6 A–C ). Cells were treated by cycloheximide (100 μg/mL) and half‐life of CypD was extended in response to miR‐155‐5p overexpression (P < 0.05, Figure 6 D ), suggesting miR‐155‐5p may post‐translationally regulate CypD expression. Co‐IP assay was used to detect the ubiquitination of CypD, which demonstrated that miR‐155‐5p suppressed the ubiquitination of CypD (P < 0.05, Figure 6 E ). Taken together, miR‐155‐5p targeted and downregulated NEDD4 expression to inhibit CypD ubiquitination degradation.
Figure 6.
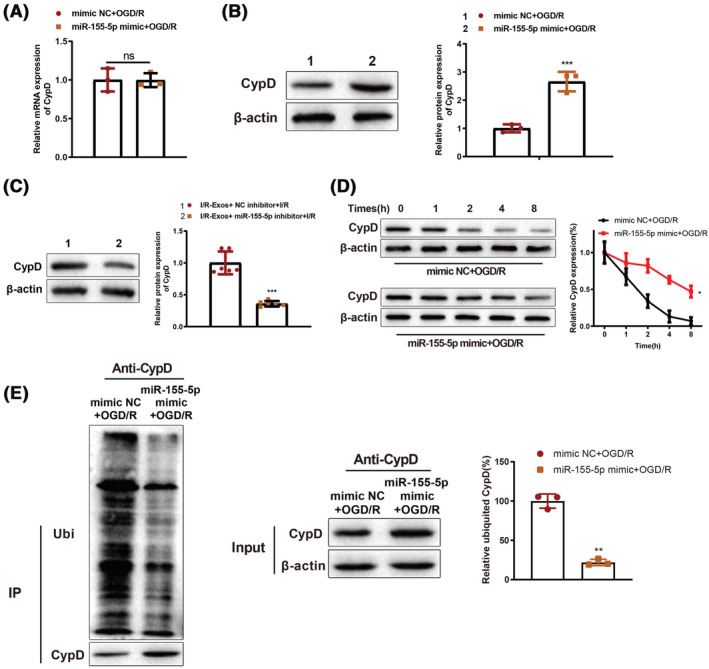
miR‐155‐5p inhibits CypD ubiquitination degradation by binding NEDD4. RIP assay was used to detect the binding of miR‐155‐5p with CypD (A). NEDD4 protein expression levels in vitro (B) and in vivo (C) were determined by western blot. Western blot was used to detect the stability of CypD protein in cardiomyocytes (D). The CypD ubiquitination in cardiomyocytes was determined by Co‐IP assay (E). N = 6 in panel C. The rest were cell experiments and had three biological replicates; *P < 0.05; **P < 0.01; ***P < 0.001. Data were shown as mean ± standard deviation, and comparisons between two groups were analysed using the t test.
miR‐155‐5p promotes OGD/R‐induced cardiomyocyte injury through the NEDD4/CypD axis
Cardiomyocytes were co‐transfected with miR‐155‐5p inhibitor, sh NEDD4, and sh CypD and theapoptotic rate was evaluated to further identify the effects of miR‐155‐5p/NEDD4/CypD signalling axis on cardiomyocyte injury. The results showed that knockdown of miR‐155‐5p significantly inhibited OGD/R‐induced cell apoptosis (P < 0.05), and further inhibition of NEDD4 resulted in elevated cell apoptosis (P < 0.05). CypD shRNA treatment in the presence of miR‐155‐5p inhibitor and sh NEDD4 substantially blocked cell apoptosis (P < 0.05, Figure 7 A ). Detection of apoptosis‐related proteins indicated that miR‐155‐5p knockdown resulted in suppressed Bax and cleaved caspase 3 expression and increased Bcl‐2 expression in OGD/R‐induced cardiomyocytes (P < 0.05), whose expression patterns were reversed by further transfection of sh NEDD4. Co‐transfection with miR‐155‐5p inhibitor, sh NEDD4, and sh CypD led to suppressed Bax and cleaved caspase 3 expression and increased Bcl‐2 expression in OGD/R‐induced cardiomyocytes compared with cells co‐transfected with miR‐155‐5p inhibitor and sh NEDD4 (P < 0.05, Figure 7 B ). Those results demonstrated that miR‐155‐5p exacerbated OGD/R‐induced cardiomyocyte injury by regulating the NEDD4/CypD axis.
Figure 7.
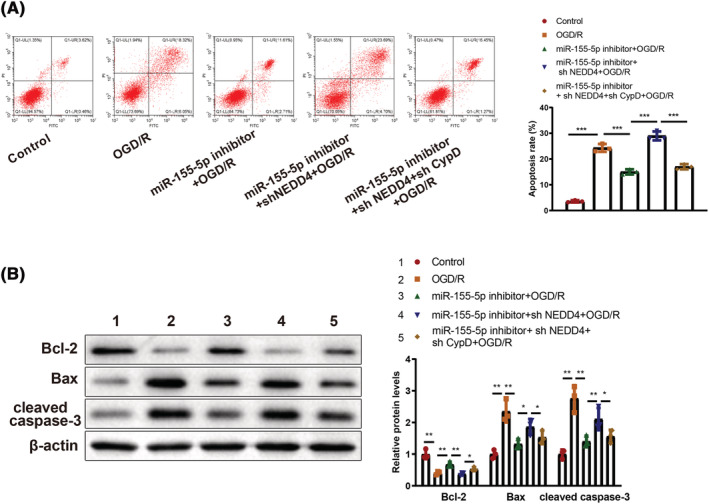
miR‐155‐5p regulates OGD/R‐induced cardiomyocyte injury through the NEDD4/CypD axis. Cardiomyocyte apoptosis was measured by flow cytometry (A). The expression levels of Bcl‐2, Bax, and cleaved caspase3 in cardiomyocytes were determined by western blotting (B). The experiment was set with three biological replicates; *P < 0.05; **P < 0.01;***P < 0.001. Data were reflected as mean ± standard deviation, and comparisons among multiple groups were analysed using one‐way analysis of variance and Tukey's multiple comparisons test. OGD/R, oxygen–glucose deprivation and re‐oxygenation.
Discussion
MIRI occurs when the blood and oxygen supply is reestablished to the ischaemic myocardium following MI, in which the occluded vessels were reopened whereas the previously ischaemic tissues are also damaged. 26 There are no effective approaches that can be taken to prevent this complication. In this case, better understanding on how MIRI is triggered and how to attenuate MIRI progression will certainly benefit those suffering from MIRI. In our study, we identified miR‐155‐5p carried by serum‐derived exos could promote MIRI progression both in vivo and in vitro. This was closely related to its regulation on NEDD4 and CypD ubiquitination degradation.
Exos were extracted from I/R‐treated mice and then injected into mice. The observation on infraction area and inflammation demonstrated that mice injected with I/R‐Exos had increased infraction area, elevated inflammatory infiltration, and cardiomyocyte apoptosis, suggesting the exos derived from serum of I/R‐treated mice can further deteriorate I/R injury. Exos are nanometre‐sized membranous vesicles that can serve as carriers for lipids, proteins, mRNAs, and miRs and are responsible for cell to cell communication. 27 MiRs transferred by Exos have been proposed for modulating cellular functions. 28 In this study, we firstly measured the level of miR‐155‐5p in I/R‐Exos and found elevated level of miR‐155‐5p. Relatedly, the expression level of miR‐155‐5p in I/R‐treated mice was also detected. Unsurprisingly, miR‐155‐5p was highly expressed in I/R‐treated mice than in sham‐operated mice. Further in vivo experiments demonstrated that miR‐155‐5p inhibition could attenuate the I/R‐Exos‐induced myocardial injury in mice. Similar results can be found in a published study which showed that the inhibition on miR‐155 expression can improve acute MI‐induced cardiac rupture and cardiac function. 29 A study has also highlighted the suppressive effects of miR‐155 deficiency on cardiac inflammation and cardiomyocyte apoptosis in injured heart. 30 Consistently, in vitro experiments in our study showed that OGD/R‐induced cardiomyocytes had increased apoptotic rate after co‐culture with I/R‐Exos, whereas the pro‐apoptotic effect of I/R‐Exos was reversed by miR‐155‐5p inhibition. Taken together, in vivo and in vitro data showed that inhibition of miR‐155‐5p could attenuate I/R‐Exos‐exaggerated I/R injury in mice.
Further identification by online software showed that miR‐155‐5p negatively regulated NEDD4 expression. NEDD4 was reported to regulate Phosphatase and Tensin Homologue (PTEN) through polyubiquitination in multiple diseases, including glioma, 31 pancreatic cancer 32 and hepatocellular carcinoma. 33 In melanoma, NEDD4, an E3 ligase, can interact with the PPxY motifs of VDAC2/3 through its WW domain and degrade them. 34 Evidence from a previous study has also supported the implication of NEDD4L in cardiovascular disease. 35 In our study, we identified that miR‐155‐5p bound to NEDD4 to further inhibit CypD ubiquitination degradation. Specifically, miR‐155‐5p post‐translationally regulated CypD expression and decreased CypD ubiquitination. In a previous study by Wang et al., miR‐30b was shown to target CypD to exert cardio‐protective effects after I/R injury. 36 In addition, CypD was also targeted by miR‐1281 in macrophages. 37 In vitro experiments in our study demonstrated that knockdown of miR‐155‐5p suppressed OGD/R‐induced cell apoptosis, whereas the anti‐apoptotic effect of miR‐155‐5p inhibition was counteracted by NEDD4 suppression. However, the synergic effects of miR‐155‐5p inhibitor and NEDD4 shRNA on cardiomyocyte apoptosis were reversed by CypD shRNA. A previous study on cardiac microvascular I/R injury has demonstrated that CypD phosphorylation can be increased in response to increased PGAM5 expression induced by Rip3 activation. 38 In our study, miR‐155‐5p overexpression can extend the half‐life of CypD protein and miR‐155‐5p can suppress CypD ubiquitination in OGD/R‐induced cardiomyocytes. Collectively, miR‐155‐5p in serum‐derived exos was found to enhance OGD/R‐induced cardiomyocyte injury by regulating the NEDD4/CypD axis. However, because a miR could target many downstream genes, we could not exclude the likelihood that miR‐155‐5p also affects MIRI by regulating other target genes. In order to have a better knowledge about the role of miR‐155‐5p in MIRI, more research is needed to see if miR‐155‐5p could regulate other target genes in MIRI.
Conclusions
Taken together, in vivo and in vitro data in this study demonstrate that miR‐155‐5p in serum‐derived exos can aggravate myocardial injury by negatively mediating NEDD4 and further inhibiting CypD ubiquitination. These findings may be helpful for the establishment of a new paradigm in I/R injury, but more data are required to validate the result of this study.
Conflict of interest
None declared.
Funding
Thanks for the grant from the Natural Science Foundation of Guangxi Province (Grant No. 2020GXNSFDA238007) and the Guangxi Health Commission Key Laboratory of Disease Proteomics Research.
Hu, C. , Liao, J. , Huang, R. , Su, Q. , and He, L. (2023) MicroRNA‐155‐5p in serum derived‐exosomes promotes ischaemia–reperfusion injury by reducing CypD ubiquitination by NEDD4. ESC Heart Failure, 10: 1144–1157. 10.1002/ehf2.14279.
References
- 1. Saleh M, Ambrose JA. Understanding myocardial infarction. F1000Res. 2018; 7: F1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boag SE, Andreano E, Spyridopoulos I. Lymphocyte communication in myocardial ischemia/reperfusion injury. Antioxid Redox Signal. 2017; 26: 660–675. [DOI] [PubMed] [Google Scholar]
- 3. Chen Y, Ye X, Yan F. MicroRNA 3113‐5p is a novel marker for early cardiac ischemia/reperfusion injury. Diagn Pathol. 2019; 14: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhou Y, Chen Q, Lew KS, Richards AM, Wang P. Discovery of potential therapeutic miRNA targets in cardiac ischemia‐reperfusion injury. J Cardiovasc Pharmacol Ther. 2016; 21: 296–309. [DOI] [PubMed] [Google Scholar]
- 5. Liu B, Wei H, Lan M, Jia N, Liu J, Zhang M. MicroRNA‐21 mediates the protective effects of salidroside against hypoxia/reoxygenation‐induced myocardial oxidative stress and inflammatory response. Exp Ther Med. 2020; 19: 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhai CL, Tang GM, Qian G, Hu HL, Wang SJ, Yin D, Zhang S. MicroRNA‐98 attenuates cardiac ischemia‐reperfusion injury through inhibiting DAPK1 expression. IUBMB Life. 2019; 71: 166–176. [DOI] [PubMed] [Google Scholar]
- 7. Xi J, Li QQ, Li BQ, Li N. miR155 inhibition represents a potential valuable regulator in mitigating myocardial hypoxia/reoxygenation injury through targeting BAG5 and MAPK/JNK signaling. Mol Med Rep. 2020; 21: 1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hong Y, He H, Jiang G, Zhang H, Tao W, Ding Y, Yuan D, Liu J, Fan H, Lin F, Liang X, Li X, Zhang Y. miR‐155‐5p inhibition rejuvenates aged mesenchymal stem cells and enhances cardioprotection following infarction. Aging Cell. 2020; 19: e13128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shyu KG, Wang BW, Fang WJ, Pan CM, Lin CM. Hyperbaric oxygen‐induced long non‐coding RNA MALAT1 exosomes suppress MicroRNA‐92a expression in a rat model of acute myocardial infarction. J Cell Mol Med. 2020; 24: 12945–12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Geng T, Song ZY, Xing JX, Wang BX, Dai SP, Xu ZS. Exosome derived from coronary serum of patients with myocardial infarction promotes angiogenesis through the miRNA‐143/IGF‐IR pathway. Int J Nanomedicine. 2020; 15: 2647–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ge X, Meng Q, Wei L, Liu J, Li M, Liang X, Lin F, Zhang Y, Li Y, Liu Z, Fan H, Zhou X. Myocardial ischemia‐reperfusion induced cardiac extracellular vesicles harbour proinflammatory features and aggravate heart injury. J Extracell Vesicles. 2021; 10: e12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Y, Qian H, Wu B, You S, Wu S, Lu S, Wang P, Cao L, Zhang N, Sun Y. E3 ubiquitin ligase NEDD4 familyregulatory network in cardiovascular disease. Int J Biol Sci. 2020; 16: 2727–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu W, Zhang P, Gu J, Yu Q, Zhang D. NEDD4–1 protects against ischaemia/reperfusion‐induced cardiomyocyte apoptosis via the PI3K/Akt pathway. Apoptosis. 2017; 22: 437–448. [DOI] [PubMed] [Google Scholar]
- 14. Peng J, Liu H, Liu C. MiR‐155 promotes uveal melanoma cell proliferation and invasion by regulating NDFIP1 expression. Technol Cancer Res Treat. 2017; 16: 1160–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Y, Pan W, Bai X, Wang X, Wang Y, Yin Y. microRNA‐454‐mediated NEDD4–2/TrkA/cAMP axis in heart failure: mechanisms and cardioprotective implications. J Cell Mol Med. 2021; 25: 5082–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yi Q, Tan FH, Tan JA, Chen XH, Xiao Q, Liu YH, Zhang GP, Luo JD. Minocycline protects against myocardial ischemia/reperfusion injury in rats by upregulating MCPIP1 to inhibit NF‐kappaB activation. Acta Pharmacol Sin. 2019; 40: 1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Basalay MV, Yellon DM, Davidson SM. Targeting myocardial ischaemic injury in the absence of reperfusion. Basic Res Cardiol. 2020; 115: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murphy SP, Ibrahim NE, Januzzi JL Jr. Heart failure with reduced ejection fraction: A review. JAMA. 2020; 324: 488–504. [DOI] [PubMed] [Google Scholar]
- 19. Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in cell death, inflammation, and Pyroptosis. Annu Rev Immunol. 2020; 38: 567–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010; 11: 621–632. [DOI] [PubMed] [Google Scholar]
- 21. Youle RJ, Strasser A. The BCL‐2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008; 9: 47–59. [DOI] [PubMed] [Google Scholar]
- 22. Huang G, Hao F, Hu X. Downregulation of microRNA‐155 stimulates sevoflurane‐mediated cardioprotection against myocardial ischemia/reperfusion injury by binding to SIRT1 in mice. J Cell Biochem. 2019; 120: 15494–15505. [DOI] [PubMed] [Google Scholar]
- 23. Sun Y, Liu J, Xu Z, Lin X, Zhang X, Li L, Li Y. Matrix stiffness regulates myocardial differentiation of human umbilical cord mesenchymal stem cells. Aging (Albany NY). 2021; 13: 2231–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sun T, Ding W, Xu T, Ao X, Yu T, Li M, Liu Y, Zhang X, Hou L, Wang J. Parkin regulates programmed necrosis and myocardial ischemia/reperfusion injury by targeting Cyclophilin‐D. Antioxid Redox Signal. 2019; 31: 1177–1193. [DOI] [PubMed] [Google Scholar]
- 25. Xu T, Ding W, Ao X, Chu X, Wan Q, Wang Y, Xiao D, Yu W, Li M, Yu F, Wang J. ARC regulates programmed necrosis and myocardial ischemia/reperfusion injury through the inhibition of mPTP opening. Redox Biol. 2019; 20: 414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Minutoli L, Puzzolo D, Rinaldi M, Irrera N, Marini H, Arcoraci V, Bitto A, Crea G, Pisani A, Squadrito F, Trichilo V, Bruschetta D, Micali A, Altavilla D. ROS‐mediated NLRP3 Inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid Med Cell Longev. 2016; 2016: 2183026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang K, Yang J, Guo S, Zhao G, Wu H, Deng G. Peripheral circulating exosome‐mediated delivery of miR‐155 as a novel mechanism for acute lung inflammation. Mol Ther. 2019; 27: 1758–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alexander M, Hu R, Runtsch MC, Kagele DA, Mosbruger TL, Tolmachova T, Seabra MC, Round JL, Ward DM, O’Connell RM. Exosome‐delivered microRNAs modulate the inflammatory response to endotoxin. Nat Commun. 2015; 6: 7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang C, Zhang C, Liu L, A X, Chen B, Li Y, Du J. Macrophage‐derived mir‐155‐containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol Ther. 2017; 25: 192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen JG, Xu XM, Ji H, Sun B. Inhibiting miR‐155 protects against myocardial ischemia/reperfusion injury via targeted regulation of HIF‐1alpha in rats. Iran J Basic Med Sci. 2019; 22: 1050–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen WL, Chen HJ, Hou GQ, Zhang XH, Ge JW. LINC01198 promotes proliferation and temozolomide resistance in a NEDD4–1‐dependent manner, repressing PTEN expression in glioma. Aging (Albany NY). 2019; 11: 6053–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weng M, Luo ZL, Wu XL, Zeng WZ. The E3 ubiquitin ligase NEDD4 is translationally upregulated and facilitates pancreatic cancer. Oncotarget. 2017; 8: 20288–20296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang ZJ, Zhu JJ, Yang XY, Biskup E. NEDD4 promotes cell growth and migration via PTEN/PI3K/AKT signaling in hepatocellular carcinoma. Oncol Lett. 2017; 14: 2649–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang Y, Luo M, Zhang K, Zhang J, Gao T, Connell DO’, Yao F, Mu C, Cai B, Shang Y, Chen W. Nedd4 ubiquitylates VDAC2/3 to suppress erastin‐induced ferroptosis in melanoma. Nat Commun. 2020; 11: 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dahlberg J, Sjögren M, Hedblad B, Engström G, Melander O. Genetic variation in NEDD4L, an epithelial sodium channel regulator, is associated with cardiovascular disease and cardiovascular death. J Hypertens. 2014; 32: 294–299. [DOI] [PubMed] [Google Scholar]
- 36. Wang K, An T, Zhou LY, Liu CY, Zhang XJ, Feng C, Li PF. E2F1‐regulated miR‐30b suppresses Cyclophilin D and protects heart from ischemia/reperfusion injury and necrotic cell death. Cell Death Differ. 2015; 22: 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sun Q, Shen X, Wang P, Ma J, Sha W. Targeting cyclophilin‐D by miR‐1281 protects human macrophages from Mycobacterium tuberculosis ‐induced programmed necrosis and apoptosis. Aging (Albany NY). 2019; 11: 12661–12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou H, Li D, Zhu P, Ma Q, Toan S, Wang J, Hu S, Chen Y, Zhang Y. Inhibitory effect of melatonin on necroptosis via repressing the Ripk3‐PGAM5‐CypD‐mPTP pathway attenuates cardiac microvascular ischemia‐reperfusion injury. J Pineal Res. 2018; 65: e12503. [DOI] [PubMed] [Google Scholar]