

Abstract
Diabetes mellitus (DM) is a serious epidemic around the globe, and cardiovascular diseases account for the majority of deaths in patients with DM. Diabetic cardiomyopathy (DCM) is defined as a cardiac dysfunction derived from DM without the presence of coronary artery diseases and hypertension. Patients with either type 1 or type 2 DM are at high risk of developing DCM and even heart failure. Metabolic disorders of obesity and insulin resistance in type 2 diabetic environments result in dyslipidaemia and subsequent lipid‐induced toxicity (lipotoxicity) in organs including the heart. Although various mechanisms have been proposed underlying DCM, it remains incompletely understood how lipotoxicity alters cardiac function and how DM induces clinical heart syndrome. With recent progress, we here summarize the latest discoveries on lipid‐induced cardiac toxicity in diabetic hearts and discuss the underlying therapies and controversies in clinical DCM.
Keywords: Diabetic cardiomyopathy, Lipotoxicity, Cardiovascular diseases, Diabetes mellitus
Introduction
Along with lifestyle changes, diabetes mellitus (DM), which is characterized by a disproportional increase in blood glucose, has become a global epidemic with an increased financial burden on individuals, families, and governments. Statistically, the prevalence of DM has risen to 10% in countries such as China and India by the year 2019. 1 According to the World Health Organization (WHO), there are nearly 1.5 million deaths directly caused by DM. Defined by the physiological mechanism of glucose increase, DM can be categorized into type 1 (T1DM) and type 2 (T2DM), with the latter representing the majority. More importantly, DM is intimately associated with cardiovascular diseases, which have been documented to be the leading cause of death among all complications induced by DM. 2 Since Rubler et al. first introduced it in 1972, 3 diabetic cardiomyopathy (DCM) has been used to define a loss of cardiac function in diabetic patients, which is independent of coronary artery disease (CAD), hypertension, or other primary cardiac diseases. 4 According to a survey of over 2000 individuals, the prevalence of DCM was ~16.9% in diabetic patients and 1.1% in the community population. 5 Considering the population diversity and the genetic susceptibility among patients, the epidemic prevalence may be higher than estimated. The pathogenesis and clinical features of DCM differ in types of DM. For instance, systolic dysfunction is more commonly reported in T1DM than in T2DM, whereas diastolic dysfunction can be found in both groups. 6 In terms of pathogenesis, both T1DM and T2DM show metabolic alterations and impaired mitochondrial oxidative capacity, whereas genetic type 1 diabetic mice (Akita mice) do not exhibit cardiac efficiency impairment or mitochondrial uncoupling. 7 In both T1DM and T2DM, a lack of insulin and insulin resistance prompt a metabolic shift wherein the fatty acid (FA) metabolism increases to compensate for the inefficient energy production. Because early findings confirmed an increased lipid content in the hearts of T2DM patients, researchers have developed lipid‐induced toxicity (lipotoxicity) that impairs cardiac function via a cluster of metabolic signalling pathways. 8 , 9 The precise relationship between lipotoxicity and DCM and their underlying molecules remain controversial. In this review, we summarize the most recent findings in lipotoxicity and the pathogenesis of DCM to provide a comprehensive view that could be helpful in scientific research and clinical recognition of DCM.
Metabolic alterations in diabetic heart
Substrate alterations
Type 2 DM is characterized by elevated blood glucose levels and insulin resistance. For cardiomyocytes, ~60–70% of their energy is accounted for by the oxidation of long‐chain FAs, whereas most of the rest derives from glucose metabolism including glycolysis and glucose oxidation. 10 , 11 Insulin resistance and the increased plasma FA concentration in DM cause a shift of metabolic substrate from glucose to FAs, which is associated with excessive FA oxidation (FAO). Studies found a 52% decrease in glycolysis and an 84% decrease in glucose oxidation in states of diabetes, compared with a marked increase in FA uptake and palmitate (a saturated FA) oxidation. 12 , 13 This metabolic inclination to FAs, along with subsequent compensations such as the accumulation of lipid droplets (LDs) and increased mitochondrial activity, plays important role in the lipid‐induced toxicity in diabetic hearts.
Glucose metabolism in diabetes mellitus
The glucose transporter (GLUT), a complex expressed tissue, specifically, was discovered to be the dominant protein governing glucose entry into cells. 14 Of all the GLUT subtypes, GLUT1 and GLUT4 have been studied more thoroughly than the others. GLUT1 activation typically exists in a basal condition, whereas GLUT4 is insulin stimulated, and it expresses in tissues such as adipose, skeletal muscle, and cardiac muscle. 15 , 16 In T2DM, the insulin‐regulated GLUT4 is inefficient to redistribute to cell membranes due to insulin resistance. 17 Wright et al. found reduced GLUT content in mice after only 2 weeks of high‐fat diet (HFD), 18 and that concomitantly impacts the subsequent glucose oxidation and glycolysis. However, the residual glucose uptake that is mediated by the GLUT4‐independent sodium–glucose‐linked transporter (SGLT) has been confirmed. 19 Although the regulation in molecules attempts to maintain glucose homeostasis, un‐oxidative glucose was still observed accumulating by evidence from models, and thus, a deficiency in pyruvate decarboxylation was believed to be partially responsible. 12 , 20
Pyruvate is transported to mitochondria in aerobic oxidation for the tricarboxylic acid cycle (TCA), and it is later dehydrogenated into acetyl‐CoA by pyruvate dehydrogenase (PDH) to assist in energy production. The transport of pyruvate to mitochondria, which is a rate‐limiting factor in pyruvate oxidation, is reduced in a diabetic model. 21 Moreover, the suppressed PDH activity consequent to the regulation of peroxisome proliferator‐activated receptor (PPAR) and FAO also plays a part in the decreased glucose metabolism. 22
Aside from the changes mentioned above, glycolysis deficiency caused by high FA concentration further hampers glucose metabolism. 23 Given the importance of glucose metabolisms, one may hypothesize that disrupted glucose utilization is a possible initiator of DCM. And that was evidenced by impaired GLUT4 translocation and reduced GLUT content preceding FA changes in HFD‐fed mice. 18 More research into glucose metabolism is still required.
Fatty acid utilization increases in diabetic heart
Several mechanisms have been identified that may contribute to the altered FA metabolism in diabetic hearts. The PPAR, a transcription factor activated by specific ligands, was at the centre of investigation. PPAR has been identified into four subtypes, PPAR‐α, PPAR‐β, PPAR‐γ, and PPAR‐δ. According to a study, when PPAR‐α is overexpressed genetically in mice hearts, metabolic changes are similar to those of the diabetic heart. 24 And the activity of carnitine palmitoyltransferase‐1 (CPT‐1), an enzyme responsible for mitochondrial FAO, was found to be increased in this study. All of these suggest that PPARs may be strongly associated with the metabolic alterations in diabetic heart.
For FA uptake, CD36 is believed to be pivotal in guiding FAs through the cell membrane. 25 Researchers found an up‐regulation of cardiac CD36 in HFD mice, and CD36 is believed to be important in myocardial FA uptake in human. 26 , 27 As a receptor known as FA translocase (FAT), CD36 was originally thought to be enabled by PPAR‐α, but it was later discovered to be PPAR‐γ responsible as well. 28 , 29 In fact, PPARs may be multifunctional in activating elements of FA metabolisms (MCAD and CD36) as well as glucose metabolisms (GLUT1). 30 For example, PPAR‐γ accounts for both glucose uptake and lipid genesis. 31 PPAR‐δ deficiency impaired FA metabolism, glucose metabolism, and even cardiac efficiency by suppressing metabolic proteins and antioxidants. 32 According to the findings stated above, it has been suggested that the imbalanced expression between PPAR‐α and PPAR‐γ may be one of the causes of metabolic dysregulation in diabetic heart. 33 And that remains elucidated as PPARs regulate inflammatory and vascular homeostasis‐related pathways. 34
Imbalance of fatty acid metabolism was assumed
In contrast to the metabolic inclination to glucose and the reduced FAO in heart failure (HF), diabetic environment up‐regulates oxidation‐associated proteins. Robust data now support the idea that increased FAO is generally associated with impaired mitochondrial function, increased reactive oxygen species (ROS) synthesis, and even cardiac ineffectiveness in db/db or ob/ob mice. 23 , 35 Paradoxically, when FAO was manually promoted by reducing malonyl CoA or deleting acetyl‐CoA carboxylase 2 (ACC2), no cardiac dysfunction was observed. 36 , 37 Following this, a lengthy debate ensued as to whether the increased FAO impairs cardiac function. 38 , 39
Recently, a hypothesis proposed by Shao et al. concerning the imbalance between FA absorption and FAO has amply explained such conflicts. 36 That theory was similar to a previous concept of triglyceride (TAG) accumulation resulting from a mismatch between FA uptake and oxidation, but it was more detailed. 40 In brief, lipolysis in adipose tissue is detected in the context of insulin resistance and other metabolic disorders in T2DM, and that gives rise to a high level of FAs in circulation. Cardiomyocytes exposed to this dyslipidaemia have increased FA absorption and FAO following regulations by PPARs and other molecules. 22 , 41 , 42 , 43 However, the enhanced uptake of FAs exceeds the ability of the cardiomyocytes to perform FAO, which accumulates unprocessed FAs and is considered to be toxic to the cell itself (lipotoxicity). 36 From this, the inadequate settlement of FAs, rather than FAO alone, is seen as contributing to the weakening of diabetic heart. That theory may explain some of the proposed controversies, whereas more cellular components in this metabolic homeostasis, such as LDs and autophagosomes, need to be taken into account.
Lipid droplets may serve as a compensation
Following incomplete processing, FAs are stored into LDs in the form of TAGs. Cholesterol ester and ether lipid monoalkenyl diacylglycerol (DAG) can also be found within LDs. 44
Functions of LDs remain to be elucidated, but recent studies inclined to the view that they are components to buffer FA toxicity by avoiding productions of toxic molecules such as ceramides and ROS. Furthermore, as they include special elements known as LDs resident proteins, LDs may trigger lipid synthesis, lipid hydrolysis, and even signal transduction. 45 In addition to these, it has also been found that LDs interact with mitochondria to facilitate the exchange of FAs, which helps to maintain the intracellular energy homeostasis.
Moreover, we may notice that these metabolic alterations in diabetic cardiomyocytes are quite similar to those of the non‐alcoholic fatty liver disease (NAFLD). Acquisition of lipids in NAFLD occurs when the activities of absorption and lipogenesis [including de novo lipogenesis (DNL)] overwhelmed the ability of disposal by oxidations and exporting. 46 Their major difference with DCM is the limitation for cardiomyocytes to carry out lipogenesis. Although the reason for this is still unclear, a study found that some of the proteins involved in lipogenesis may play another role in cardiac calcium control and the corresponding arrhythmia. 47
Lipophagy may play a role in lipid accumulation
Autophagy was first described in the 1960s as a process of degrading cellular components via interaction with lysosomes to meet physiological demands, and it differs in process and activation when dealing with different components such as mitochondria (mitophagy) and LDs (lipophagy). 48 By detecting levels of LC3 to assess autophagy flux, a rate of autophagosome formation or degradation, Tong et al. observed an early‐stage rise of LC3, which peaked at about the sixth week after the start of HFD and returned to baseline at the second month. 49 , 50 That phenomenon strongly implies that autophagy may be activated in states of lipid overload to diminish certain organelles or complexes in order to support cellular function.
Lipophagy is majorly investigated in hepatocytes or corresponding liver diseases for lipid metabolism. Scientists found that suppressing lipophagy in hepatocytes promotes lipid accumulation, and lipid accumulation further weakens lipophagy. 51 , 52 However, it remains unknown whether hallmarkers exist in hepatocytes‐related lipophagy and whether these understandings of lipophagy can be applied to cardiomyocytes and even HFD‐fed diabetic heart. 49 , 52 A research by Mardani et al. suggests that plin2 is involved in lipophagy and cardiac lipid accumulation, but the specifics of this regulation need to be further confirmed and explored. 53 Furthermore, a study revealed that lipophagy and lipases may be complementary and cooperative in the lipolysis process. 54 These findings would allow one to consider the possibility of activating lipophagy to counteract excessive FAs or LDs in diabetic cardiomyocytes.
The pathogenesis of lipid toxicity in diabetic cardiomyopathy
Energy production and mitochondrial dysfunction
In aerobic oxidation, cellular glucose undergoes glycolysis and TCA to produce adenosine triphosphate (ATP), whereas FAs undergo β‐oxidation and TCA. Experts measured a higher level of oxygen consumption rate (MVO2) in ob/ob mice, which is accompanied by attenuated cardiac function. 23 Due to their differences in molecules, FAs are less efficient than glucose in energy production, as they consume more oxygen to produce ATPs. For that reason, substrate alteration occurs in ischaemia to facilitate recovery, as well as in diabetes. 55 , 56 However, inefficient energy production by FAs is inadequate to explain the observed failing heart in diabetes. The role of mitochondria has to be taken into account.
Mitochondria are vulnerable to disturbances from specific molecules or medications. Because the absorbed FAs are transported through the mitochondrial membranes to be oxidized, abundant FAs burden mitochondria. This excessive handling of FAs, along with multifaceted downstream impairments such as detrimental ROS synthesis, toxic lipid intermediates, and mitochondrial uncoupling, constructs the lipotoxicity network. Before delving into these in detail, it is worth noting that dynamic mitochondrial fission, mitochondrial fusion, and mitophagy are crucial in mitochondrial quality control. 57
Mitochondrial fusion, fission, and remodelling
Fusion is initiated when mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) facilitate the outer mitochondrial membrane (OMM) whereas the optic atrophy 1 (Opa1) gene facilitates the inner mitochondrial membrane (IMM). In a model overexpressing long‐chain acyl‐CoA synthetase 1 to mimic lipotoxicity by Tsushima et al., increased mitochondrial fission and decreased fusion have been confirmed by detecting dynamin‐related protein 1 (Drp1) (fission‐associated protein) and Opa1. 58 After the deletion of fusion and fission‐associated regulators, developed cardiomyopathy has been observed, suggesting that the dysregulated balance between mitochondrial fission and fusion is one of the causes of cardiac dysfunction. 59 , 60 The transition from fusion to fission, along with the increased mitochondrial fragmentation, is also believed to be related to detrimental ROS synthesis. 61 , 62 From the heart of a diabetic mouse model, experts found a decrease in the number of ordered‐cristae mitochondria and an imbalance between the mitochondrial fusion–fission axis, which supports the aforementioned hypothesis. 63 They subsequently suggested that this kind of imbalance, in concert with stalled mitophagy, induces mitochondrial remodelling events and functional changes.
Mitophagy
In the cardiac domain, mitophagy is more comprehensively studied than lipophagy. Literally, mitophagy is a process of eliminating dysfunctional mitochondria by autophagosome to maintain intracellular homeostasis. 64
There are several ways to induce mitophagy: PTEN‐inducible kinase (PINK)/Parkin pathway, FUN14 domain‐containing (FUNDC1) pathway, and BCL2/adenovirus E1B 19 kDa‐interacting protein 3 (BNIP3)/NIX pathway, the first two of which are ubiquitin‐dependent. 65 , 66 This processing of mitochondria is believed to disrupt the homeostasis of ATP synthesis and ROS production. When PINK1 is depleted, oxidative stress, apoptosis, and fibrosis are promoted. 67 The elevated ROS level in turn triggers mitophagy by mitochondrial depolarization, and it damages cellular components by superoxide anions and other oxidative molecules. 65 In this way, mitophagy is regarded as a protective mechanism for preserving mitochondrial quality and intracellular homeostasis.
Using transgenic mice expressing cardiac‐specific Mito‐Keima, increased level of mitophagy has been confirmed in the early stages of a diabetic model, which lasts continuously even after 2 months of HFD when it begins to decrease. 49 Although recently, Tong et al. proposed and confirmed the existence of an LC3‐independent alternative mitophagy mediated by the Ulk1/Rab9 pathway, which culminates at approximately the fifth month of HFD. 68 Cardiac function was impaired in these diabetic models as well as in transgenic mice with knockout of mitophagy‐associated genes (Parkin and Ulk1). 49 , 68 We have known that autophagy flux rises in the fifth week and decreases in the second month, and their differences with mitophagy have raised questions about how upstream mechanisms control these alterations and how these alterations exert an influence on cardiac function. Moreover, the differences between a periodic activation of mitophagy and a gradual deterioration of DCM allow one to hypothesize that the increased mitophagy was inefficient in restoring mitochondrial quality or that different types of mitophagy may differ in efficiency. And if mitophagy is not sufficient to protect cells from lipotoxicity, whether there is a point at which mitophagy begins to impair cells, known as lethal mitophagy, should be discussed.
Metabolic intermediates
Ceramide
Quantities of molecules are produced when FAs undergo oxidation, including ceramide, DAGs, and acylcarnitine. As a metabolic intermediate, ceramide can be produced by different synthases (ceramide synthase 1–6) and pathways in different locations. 69 Those pathways, including the de novo synthetic pathway in the endoplasmic reticulum (ER), produce ceramide of different fatty acyl chain lengths and functions. 70 In the heart of a Zucker diabetes fatty (ZDF) rat and human, a significant rise in the intracellular concentration of ceramide was detected and that was believed to be partly responsible for the development of DCM. 71
Ceramide, as a toxic intermediate, may induce apoptosis, 72 insulin resistance, 73 autophagy and mitochondrial dysfunction. In terms of mitochondrial dysfunction, Di Paola et al. reported an inhibition of electron transport chain (ETC) complex I using C2‐ceramide. 74 Later, Raichur et al. confirmed that C16‐ceramide may also interrupt ETC while acting on different complexes. 75 Nevertheless, these types of inhibition do not apply to C24‐ceramide. 76 Recently, some experts suggested that apoptosis may be involved in this induction of mitochondrial dysfunction, whereas details should be confirmed. 77
Ceramide has also been shown to play a role in autophagy and even mitophagy. 78 Studies of ceramide‐induced autophagy have largely focused on cancer, and that results in cell death and tumour suppression (lethal autophagy). 70 Besides lethal autophagy, non‐lethal autophagy can also be induced by ceramide. 79 However, it is still unclear whether these differences in cellular outcomes are attributed to different types or levels of ceramides. When it comes to mitophagy, it is revealed that ceramide, specifically C18‐ceramide, targets autophagosome to mitochondria and then suppresses ATP production and tumour growth, a process known as lethal mitophagy. 70 Conversely, no conclusions have been drawn about ceramide‐induced pro‐survival mitophagy. Given the correlation between increased ceramide level and functional deterioration, the effect of ceramide‐induced cardiac mitophagy should be further investigated.
Diacylglycerols
Diacylglycerols are metabolic intermediates formed when FAs are attached to glycerol‐3‐phosphate in the composition of TAGs. 80 For investigations on the adverse impacts of DAGs, the DAG–protein kinase C (PKC) pathways are consistently addressed. PKC, a Ca2+, oxidant, or lipid‐activated element, is believed to be associated with calcium handling, fibrosis, cardiomyopathy, and even HF. 81 , 82 Also, ceramide may enable PKCs independent of DAGs to induce contractility dysfunction. 83 In conclusion, the research on the effects of ceramides, DAGs, and acylcarnitine will be promising in studies of DCM.
Reactive oxygen species and oxidative stress
Reactive oxygen species are groups of molecules with unmatched electrons outside the orbit of proton O, which commonly exist in the form of H2O2, O2−, or ·OH. Mitochondria produce the vast majority of ROS, whereas some of the rests are generated by the xanthine oxidation or the nicotinamide adenine dinucleotide phosphate (NADPH) oxidation. 84 Normally, ~1–2% of the imported electrons leak out of the ETC, which is one of the mechanisms underlying mitochondrial ROS synthesis. 85 Hyperglycaemia, palmitate, and lipotoxicity‐related mitochondrial impairments are the most well‐known causes of the rising ROS levels in DCM. 86
Due to their short half‐lives, ROS are easily driven to damage nearby organelles, even mitochondria themselves. 87 Detrimental effects of ROS include oxidative damage to proteins (such as carbonylation), lipid peroxidation product generations, and deoxyribonucleic acid (DNA) strand breakage. 86 Because dysfunctional mitochondria are more likely to produce ROS, which causes their own morphological alterations such as swelling, cristae disruption, and fragmentation, a vicious cycle was therefore established. 88 , 89 For functionality, the tendency of ROS to combine sarcomere proteins increases cellular stiffness and leads to impaired contractility. 90 Besides, lipid accumulation may also be exacerbated by the mitochondrial inadequacy in handling FAs due to ROS impairment. Despite the presence of anti‐oxidative components such as superoxide dismutase (SOD) and reduced glutathione (GSH), the interference of ROS on mitochondria cannot be balanced, leading to oxidative stress. 91 Given such detriments, therapeutic applications targeting ROS could be prominently efficient.
Mitochondrial uncoupling
In mitochondria, successive electron transfers to oxygen produce H2O, from which ATPs are produced by cooperating with an electrochemical gradient. Any decoupling of these procedures may prevent mitochondria from producing ATPs, and this is known as mitochondrial uncoupling. Major mediators of mitochondrial uncoupling are uncoupling proteins (UCPs), and most research is focused on UCP2 or UCP3.
According to a previous study, UCPs and plasma FA levels are favourably correlated, suggesting that UCPs may play a role in lipid stress and energy deficiency in HF. 92 Later, UCPs were believed to be involved in mitochondrial ROS synthesis by studies. 93 Under these conditions of oxidative stress and up‐regulated PPARs, the expression of UCPs is activated. 30 Recent findings in hearts of diabetic animals suggest that the property of ameliorating ROS damage by UCPs is strongly correlated with improved cardiac function. 94 , 95 More details on UCPs should be explored by experiments.
Endoplasmic reticulum stress
Endoplasmic reticulum stress, also known as unfolded protein response (UPR), has been proposed to be associated with a variety of metabolic processes or diseases such as diabetes. Previously, ER stress was found to be correlated with the establishment of diabetes in studies of β‐cell function and insulin signalling. 96 , 97 By palmitate incubation, an altered ER morphology, but not an up‐regulated UPR, was observed. 98 , 99 Other research on the hypothalamus and liver still backs up the role of ER stress in diabetes. 97
Endoplasmic reticulum stress may interrupt several metabolic processes by misfolding proteins, for example, PDH kinase 4 (PDK4) of glucose metabolisms, Parkin and Beclin1 of mitophagy, and Drp1 of mitochondrial fission. 100 Under ER stress conditions, ER–mitochondria contacts to enhance ATP production can be triggered. And conversely, depletion of mitochondria‐associated ER membranes (MAMs) affects ER stress by interrupting ER–mitochondria communication. 100 , 101 These interactive processes between mitochondria–ER and ER stress provide promising targets for future DCM research. Specific mechanisms, however, are still unknown.
Impaired calcium handling
Calcium handling, including cytosolic calcium handling and mitochondrial calcium handling, plays a pivotal role in driving contraction, as well as in the development of cellular dysfunction. The homeostasis of mitochondrial calcium is maintained by the dynamic uptake and release of calcium. 102 In a diabetic model, down‐regulated mitochondrial calcium handling was detected and was thought to be the consequence of the permeability transition. 103 Later, ceramide and disturbed protein level in mitochondrial calcium uniporter (MCU) and sarcoplasmic reticulum (SR)–mitochondria microdomains were identified as causes of dysregulated mitochondrial calcium handling in diabetes. 30 , 102 This dysregulated calcium handling can be rescued by restoring calcium to the mitochondria, which is followed by improved mitochondrial activity and energy production. Those improvements provide a new target for the treatment of DCM.
Lipid droplets–mitochondria interaction
Lipid droplets–mitochondria interaction refers to a process of lipid trafficking and FA transfer between LDs and mitochondria. Using electronic microscopy, a physical interaction and even membrane conjunction were seen in vitro and in vivo. 104 It has been speculated that this interaction can be distinguished into two types: stable contact and dynamic contact, which differ in conjunctional structures to facilitate transfer under different conditions. 45
Lipid droplets serve to store FAs when mitochondria are inefficient at handling excessive FAs. When extra oxidation or storage of FAs is required, LDs contact mitochondria to transfer the essential FAs. 105 This process prevents the production of toxic intermediates (Figure 1 ) and may consequently improve mitochondrial functions.
Figure 1.
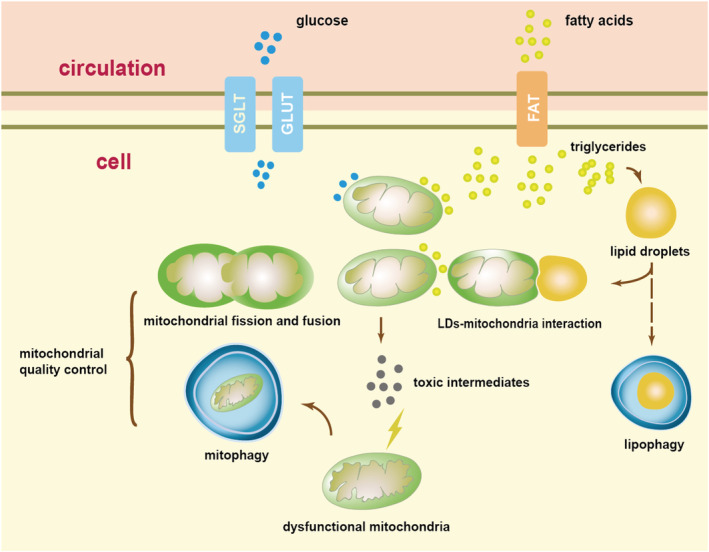
Mismatch between fatty acid (FA) and glucose metabolism results in a series of pathophysiological processes, compensations, and finally dysfunctional mitochondria. FAs and glucose in circulation were absorbed into cells by protein complexes. An increase uptake of FAs followed by an increase oxidation. Excessive FAs were stored into lipid droplets (LDs) in the form of triglycerides (TAGs), and LDs interact with mitochondria to exchange FAs when necessary. Elevated FA and glucose metabolism resulted in an elevation of reactive oxygen species (ROS) and toxic intermediates, and these molecules damage mitochondria when mitophagy, mitochondrial fission, and mitochondrial fusion were not enough to compensate this impairment. The undetermined lipophagy may also be important in lipid stress. The arrows refer to the intracellular signalling and process. The dotted arrow refers to undetermined elements. FAT, fatty acid translocase; GLUT, glucose transporter; SGLT, sodium–glucose‐linked transporter.
As a result, the size and quantity of LDs intracellularly may not be positively correlated with lipid toxicity and may be inappropriate for assessing metabolic or functional impairment in diabetic cardiomyocytes. Moreover, detecting ceramide and ROS for assessment of lipid toxicity will be insufficiently specific. In this regard, more research into the precise evaluation of lipid toxicity should be addressed.
Additional mechanisms underlying diabetic cardiomyopathy
We have concluded that various mechanisms are responsible for mitochondrial dysfunction. And dysfunctional mitochondria may also be the reason for these detriments in the pathogenesis of DCM (Figure 2 ). From that perspective, mitochondrial dysfunction can be regarded as an indispensable element of the lipotoxicity network. To better understand mitochondrial dysfunction and lipotoxicity, there should be investigations on mitochondria‐independent detriments.
Figure 2.
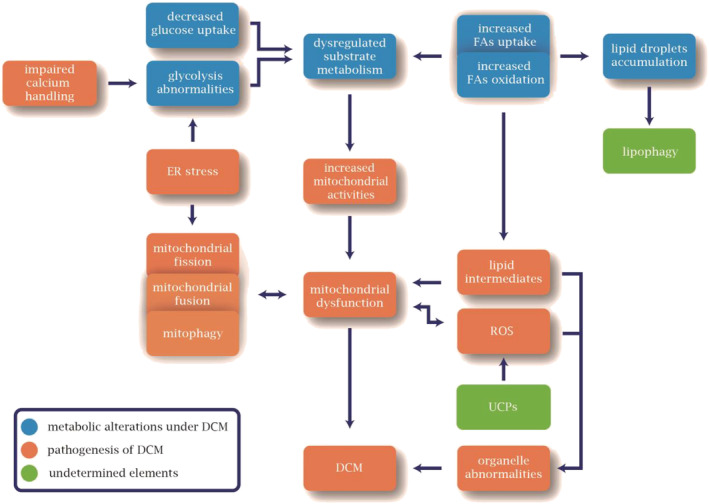
Metabolic alterations and pathophysiological process in diabetic cardiomyopathy (DCM). Metabolic alterations of elevated fatty acid (FA) metabolism and down‐regulated glucose metabolism result in increased mitochondrial activity, generation of lipid intermediates, and accumulation of lipid droplets. Later, mitochondrial dysfunction happened owing to detrimental reactive oxygen species (ROS) synthesis, dysregulated mitochondrial fission, mitochondrial fusion, and mitophagy, and lipid intermediates. Besides mitochondrial dysfunction, organelle dysfunction induced by lipid intermediates and ROS also underlies the pathogenesis of DCM. In this process, endoplasmic reticulum (ER) stress and impaired calcium handling are also believed to be contributing, whereas the role of lipophagy and uncoupling proteins (UCPs) remains undetermined.
Finally, even though lipid overload has been confirmed in diabetic animal models and human heart biopsies, it is still insufficient to induce DCM on its own. Renin–angiotensin system (RAS), insulin resistance, advanced glycation products (AGEs), and more mechanisms should be highlighted. 20 And these mechanisms, including lipotoxicity, appear to have varying relevance for different DCM phenotypes. 106
Clinical implications and underlying therapies
Clinical diabetic cardiomyopathy
It takes a long time to progress from pathophysiological alterations to pathological alterations and even morphological alterations to clinical DCM. The majority of the mechanisms described above are manifestations of an accepted model (ZDF rat, ob/ob mice, db/db mice, or HFD mice), but whether these models accurately reflect the characteristics of human DCM is debatable. Confounding factors, such as coronary microvascular dysfunction (CMVD), may impede the diagnosis or our recognition of DCM. Moreover, parameters in echocardiography may be heterogenetic when they are used to diagnose DCM. 107 For these reasons, it remains inconclusive whether DCM is a clinical entity and whether diabetes alone may ultimately lead to clinical heart syndrome. 108
As early as the 1970s, diabetes was considered as a discrete risk factor for HF in the Framingham study. 109 Subsequent trials adjusted for CAD, hypertension, and even intercurrent myocardial infarction to support that conclusion. 110 , 111 In recent years, investigations into metabolic syndrome have brought down the boundaries between metabolic and cardiovascular diseases. Even though these findings did not confirm the existing entity of DCM, they still provide critical information for the prevention and management of diabetes‐related HF. Recently, a clinical update of DCM held the opinion that there are two independently evolved phenotypes of DCM, which are the restrictive/HF with preserved ejection fraction (HFpEF) phenotype and the dilated/HF with reduced ejection fraction (HFrEF) phenotype. 106 That was in contrast to the conventional view that the dilated/HFrEF phenotype is the successive stage of the restrictive/HFpEF phenotype. Although their relationship is still controversial, these two phenotypes are both aetiologies of cardiac remodelling. And most importantly, regardless of whether diabetes alone or other risk factors lead to diabetes‐related HF, cardioprotective therapies should be advised aside from glucose‐lowering drugs.
Currently, treatment regimens for diabetes include blood glucose‐lowering therapies (insulin stimulating and excretion stimulating) and appetite‐reducing therapies, as well as lifestyle‐improving recommendations. And the development of glucose‐lowering drugs with cardioprotective benefits will definitely facilitate the prevention of diabetes‐related HF. Finally, for the recognition and prevention of severe HF in diabetic patients, the staging system described by Maisch et al. is clinically applicable, 112 and treatment for the two phenotypes of DCM (dilated or restricted) should be respectively considered.
Promising approaches to detect diabetic cardiomyopathy
Echocardiography, including conventional Doppler, tissue Doppler, and intravenous contrast echocardiography, is the first choice for measuring cardiac function in clinical diagnosis and trial enrolment. Decreased E/A, increased E/E′, or decreased integrated backscatter (IB) can be reported in different phenotypes or phases of DCM. 113 Additionally, magnetic resonance (MR) imaging (MRI) and nuclear imaging are occasionally used. Despite the widespread application of these techniques, they still have a cluster of drawbacks, including a lack of specificity and an inability to identify cardiac metabolism, TAG content, and corresponding lipotoxicity. From these, Rijzewijk et al. described a technique for measuring myocardial TAG content using 1H‐MR spectroscopy (MRS). 114 They subsequently found that increased TAGs in the heart tissue of T2DM patients are correlated with impaired left ventricular diastolic function as measured by MRI. Later, in a study using 13C MRS to measure cardiac metabolism (PDH, lactate dehydrogenase, and alanine transaminase) and 31P MRS to assess cardiac energy, Rider et al. discovered a down‐regulation of metabolic flux and an impairment of myocardial energetics in T2DM patients. 115 These methods, along with T1 mapping to assess cardiac fibrosis, may provide promising approaches for the precise detection of DCM and lipotoxicity in the future.
Peroxisome proliferator‐activated receptors
Drugs that target PPARs to normalize glucose include single PPAR agonists, dual agonists, and pan agonists, with fibrates and thiazolidinediones (TZDs) being the most well known and widely used. Fibrates are single PPAR‐α agonists that benefit patients by stabilizing serum triacylglycerol levels and inhibiting excessive lipid metabolism. However, the increased creatine level after prescribing fibrates has cast doubt on the possibility of deteriorating renal function, whereas subsequent evidence has challenged that viewpoint. 116 Aside from fibrates, there are TZDs targeting PPAR‐γ, which act as insulin sensitizers. TZDs were initially proven to be beneficial, offering long‐term glycaemic control and preventing the progression of pre‐diabetes, whereas their limited efficiency compared with fibrates and the adverse effects (mild oedema, body weight gain, fatigue, and fracture) constrained their wide application. 117 , 118 However, further evidence revealed that the glucose‐lowering benefits of TZDs were independent of PPAR‐γ, which is the cause of side effects. 119 As a result, the drug MSDC‐0602 and its subtypes were developed, with low intimation for PPAR‐γ and similar effect of improving diabetes. 118 Most importantly, by fixing the imbalanced activities of osteoclasts and osteoblasts, no evidence of fracture was documented. 118 Although the molecular mechanisms of TZDs or MSDC‐0602 remain uncertain, drugs targeting PPARs would be promising in diabetic treatment.
Sodium–glucose‐linked transporter
Sodium–glucose‐linked transporter inhibitors are well known for their efficiency in the treatment of diabetes and a low risk of hypoglycaemia. 120 SGLT1 and SGLT2 vary by organ, with the former facilitating the intestinal glucose absorption and the latter assisting reabsorption of glucose filtered by the kidney. Text above suggested that SGLT is responsible for the residual glucose uptake in insulin resistance. This function is different from the mechanisms of the currently available SGLT2 inhibitors, which target glucose absorptions and excretions, thereby alleviating ‘glucotoxicity’.
A growing body of clinical evidence supports the cardiovascular benefit of SGLT2 inhibitors. The CANVAS Program and the EMPA‐REG OUTCOME trial demonstrated the cardioprotective effect of two SGLT2 inhibitors: canagliflozin and empagliflozin. 121 , 122 Both of them showed a lower incidence of cardiovascular events, including deaths from cardiovascular causes, stroke, and cardiac infarction. Additionally, SGLT2 inhibitors benefit those at risk of atherosclerotic cardiovascular disease (the DECLARE–TIMI 58 trial). 123 Regarding HF, numerous trials have been conducted recently. For HFrEF, both the DAPA‐HF trial and the EMPEROR‐Reduced trial reported lower cardiovascular death by taking SGLT2 inhibitors. 124 , 125 For HFpEF, the EMPEROR‐Preserved trial found that the combined risk of cardiovascular death or hospitalization for HF is reduced, even in those without diabetes. 126 And the PRESERVED‐HF trial in HFpEF patients demonstrated that dapagliflozin improve patient‐reported symptoms and physical limitations. 127 Furthermore, cardioprotective conclusions are supported even in patients with decompensated HF (the SOLOIST‐WHF trial). 128
All these trials highlight the urgent need for understanding the cardioprotective mechanisms of SGLT2 inhibitors. 129 First of all, as a mild diuretic drug facilitating glucose excretion, a blood pressure‐lowering effect can be expected by taking SGLT2 inhibitors, and these drugs have low risks of developing hypotension. 130 Some researchers reported an amelioration of oxidative stress in the heart tissue and diabetic models. 131 Besides, attenuated cardiac fibrosis, improved cardiac metabolisms, ameliorated inflammation, and autophagy may be contributing. 132 , 133 , 134 However, whether these mechanisms underlie the benefits on DCM should be thereafter discussed.
Glucagon‐like peptide‐1
Glucagon‐like peptide‐1 (GLP‐1) receptor agonist (GLP‐1 RA), an anti‐diabetes drug that stimulates insulin secretion while suppressing glucagon release, was found to be efficient in diabetic patients with cardiovascular events. As one eats, the gastrointestinal tract is activated to secret incretin, a hormone that favours releasing insulin and reducing appetite, to help digest food. And GLP‐1 is one particular form of incretin.
The clinical effects of GLP‐1 RA are partially beneficial. The LEADER trial and the SUSTAIN‐6 trial confirmed the efficiency of liraglutide and semaglutide in reducing the incidence of cardiovascular death, nonfatal cardiac infarction, and stroke. 135 , 136 However, neither the ELIXA trial nor the EXSCEL trial reported a reduction of major cardiovascular events when compared with the placebo. 137 , 138 Recently, liraglutide have been shown to protect the heart by promoting glucose oxidation and PDH activity. 139 Furthermore, some studies reported that GLP‐1 RA may be a renal‐beneficial drug because it reduces sodium absorption and was followed by a diuretic effect. 140 Improvements in diastolic dysfunction were also demonstrated by assessing E/E′ or E/A. 139 Other benefits of GLP‐1 RA, including the ability to inhibit apoptosis and alleviate oxidative stress, have also been reported. 141 , 142 These benefits and trials on cardiovascular diseases cannot be applied specifically to DCM, but they do suggest a possibility of ameliorating DCM. Clinical follow‐up and trials on DCM are still required.
Dipeptidyl peptidase‐4
An active form of GLP‐1 is degraded by dipeptidyl peptidase‐4 (DPP‐4), which was previously identified as CD26 in the immunological system. DPP‐4 is speculated to be associated with cardiovascular diseases in different researches. 143 , 144 Trials of DPP‐4 inhibitors in diabetic patients with cardiovascular diseases showed conflicting results. 145 , 146 For instance, the TECOS trial in older patients identified sitagliptin as having a neutral effect on cardiovascular risks. 147 The SAVOR‐TIMI 53 trial, however, reported an increased rate of HF hospitalization in those with saxagliptin, even though the overall cardiovascular safety is supported. 148
These conflicts raise questions about its molecular mechanisms. DPP‐4 was found to act through either a GLP‐1‐dependent or a GLP‐1‐independent pathway. The GLP‐1‐dependent pathway stimulates insulin secretion while reducing glucagon secretion, and the GLP‐1‐independent pathway favours peptide generation. 145 Ameliorations in macrovascular or microvascular endothelial damage by targeting inflammatory responses have been reported. 149 In addition to research on mechanisms, White et al. speculated that the adverse effects of increased HF in the SAVOR‐TIMI 53 trial may be independent of the drug‐class effect of DPP‐4 inhibitors. 150 Anyway, the role of DPP‐4 inhibitors in cardiovascular diseases is not fully understood by now.
Novel therapies and biomarkers
Numerous targets have been proposed that may contribute to the development of novel therapies. A recent investigation showed that recombinant human neuregulin‐1 (rhNRG‐1) can improve cardiac function and reverse cardiac remodelling in DCM rats. 151 Huynh et al. administered coenzyme Q10 to diabetic mice and found that it improved both levels of pro‐inflammatory markers and diastolic function. 152 Furthermore, microRNA‐based treatment and modulation of oxidative stress may be promising in future research. 153 , 154
Conclusions
In conclusion, a variety of mechanisms have been proposed that underlie lipotoxicity or DCM, whereas some of them remain controversial. Despite all the uncertainties, research into mechanisms advances our understanding of whether DCM exists as a clinical entity. And we may realize that the stagnation in clinical awareness has not kept pace with its advancing exploration of molecules. One of the reasons for this is that there are no approaches specifically for detecting DCM. Sophisticated techniques, such as 1H‐MRS for measuring lipid content in the heart, could be beneficial for researchers studying lipotoxicity and DCM in biomedicine or for physicians looking to administer therapies to diabetic patients. In light of all the above, the giant number of diabetic patients around the globe entails efficient cardioprotective therapy and precise medicine for DCM. And we believed that with more clinical trials and biomedical research, lipotoxicity would be addressed in developing effective DCM medications.
Conflict of interest
None declared.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Number 82070381, 82270356 and 81670293) and the Project of Shanghai Science and Technology Commission (Grant Number 20ZR1431100).
Ke, J. , Pan, J. , Lin, H. , and Gu, J. (2023) Diabetic cardiomyopathy: a brief summary on lipid toxicity. ESC Heart Failure, 10: 776–790. 10.1002/ehf2.14224.
References
- 1. Cosentino F, Grant PJ, Aboyans V, Bailey CJ, Ceriello A, Delgado V, Federici M, Filippatos G, Grobbee DE, Hansen TB, Huikuri HV, Johansson I, Jüni P, Lettino M, Marx N, Mellbin LG, Östgren CJ, Rocca B, Roffi M, Sattar N, Seferović PM, Sousa‐Uva M, Valensi P, Wheeler DC, ESC Scientific Document Group , Piepoli MF, Birkeland KI, Adamopoulos S, Ajjan R, Avogaro A, Baigent C, Brodmann M, Bueno H, Ceconi C, Chioncel O, Coats A, Collet JP, Collins P, Cosyns B, di Mario C, Fisher M, Fitzsimons D, Halvorsen S, Hansen D, Hoes A, Holt RIG, Home P, Katus HA, Khunti K, Komajda M, Lambrinou E, Landmesser U, Lewis BS, Linde C, Lorusso R, Mach F, Mueller C, Neumann FJ, Persson F, Petersen SE, Petronio AS, Richter DJ, Rosano GMC, Rossing P, Rydén L, Shlyakhto E, Simpson IA, Touyz RM, Wijns W, Wilhelm M, Williams B, Aboyans V, Bailey CJ, Ceriello A, Delgado V, Federici M, Filippatos G, Grobbee DE, Hansen TB, Huikuri HV, Johansson I, Jüni P, Lettino M, Marx N, Mellbin LG, Östgren CJ, Rocca B, Roffi M, Sattar N, Seferović PM, Sousa‐Uva M, Valensi P, Wheeler DC, Windecker S, Aboyans V, Baigent C, Collet JP, Dean V, Delgado V, Fitzsimons D, Gale CP, Grobbee DE, Halvorsen S, Hindricks G, Iung B, Jüni P, Katus HA, Landmesser U, Leclercq C, Lettino M, Lewis BS, Merkely B, Mueller C, Petersen SE, Petronio AS, Richter DJ, Roffi M, Shlyakhto E, Simpson IA, Sousa‐Uva M, Touyz RM, Zelveian PH, Scherr D, Jahangirov T, Lazareva I, Shivalkar B, Naser N, Gruev I, Milicic D, Petrou PM, Linhart A, Hildebrandt P, Hasan‐Ali H, Marandi T, Lehto S, Mansourati J, Kurashvili R, Siasos G, Lengyel C, Thrainsdottir IS, Aronson D, di Lenarda A, Raissova A, Ibrahimi P, Abilova S, Trusinskis K, Saade G, Benlamin H, Petrulioniene Z, Banu C, Magri CJ, David L, Boskovic A, Alami M, Liem AH, Bosevski M, Svingen GFT, Janion M, Gavina C, Vinereanu D, Nedogoda S, Mancini T, Ilic MD, Fabryova L, Fras Z, Jiménez‐Navarro MF, Norhammar A, Lehmann R, Mourali MS, Ural D, Nesukay E, Chowdhury TA. 2019 ESC guidelines on diabetes, pre‐diabetes, and cardiovascular diseases developed in collaboration with the EASD: the Task Force for Diabetes, Pre‐diabetes, and Cardiovascular Diseases of the European Society of Cardiology (ESC) and the European Association for the Study of Diabetes (EASD). Eur Heart J. 2020; 41: 255–323. [DOI] [PubMed] [Google Scholar]
- 2. Dillmann WH. Diabetic cardiomyopathy: what is it and can it be fixed? Circ Res. 2019; 124: 1160–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972; 30: 595–602. [DOI] [PubMed] [Google Scholar]
- 4. Seferovic PM, Petrie MC, Filippatos GS, Anker SD, Rosano G, Bauersachs J, Paulus WJ, Komatjda M, Cosentino F, de Boer RA, Farmakis D. Type 2 diabetes mellitus and heart failure: a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2018; 20: 853–872. [DOI] [PubMed] [Google Scholar]
- 5. Dandamudi S, Slusser J, Mahoney DW, Redfield MM, Rodeheffer RJ, Chen HH. The prevalence of diabetic cardiomyopathy: a population‐based study in Olmsted County, Minnesota. J Card Fail. 2014; 20: 304–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hölscher ME, Bode C, Bugger H. Diabetic cardiomyopathy: does the type of diabetes matter? Int J Mol Sci. 2016; 17: 2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bugger H, Boudina S, Hu XX, Tuinei J, Zaha VG, Theobald HA, Yun UJ, McQueen AP, Wayment B, Litwin SE, Abel ED. Type 1 diabetic Akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3. Diabetes. 2008; 57: 2924–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van de Weijer T, Schrauwen‐Hinderling VB, Schrauwen P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc Res. 2011; 92: 10–18. [DOI] [PubMed] [Google Scholar]
- 9. Ruberg FL. Myocardial lipid accumulation in the diabetic heart. Circulation. 2007; 116: 1110–1112. [DOI] [PubMed] [Google Scholar]
- 10. Liedtke AJ. Alterations of carbohydrate and lipid metabolism in the acutely ischemic heart. Prog Cardiovasc Dis. 1981; 23: 336. [DOI] [PubMed] [Google Scholar]
- 11. Camici P, Ferrannini E, Opie LH. Myocardial metabolism in ischemic heart disease: basic principles and application to imaging by positron emission tomography. Prog Cardiovasc Dis. 1989; 32: 245. [DOI] [PubMed] [Google Scholar]
- 12. Darrell D, Belke TSL, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2000; 279: E1104–E1113. [DOI] [PubMed] [Google Scholar]
- 13. Rijzewijk LJ, van der Meer RW, Lamb HJ, de Jong HW, Lubberink M, Romijn JA, Bax JJ, de Roos A, Twisk JW, Heine RJ, Lammertsma AA. Altered myocardial substrate metabolism and decreased diastolic function in nonischemic human diabetic cardiomyopathy: studies with cardiac positron emission tomography and magnetic resonance imaging. J Am Coll Cardiol. 2009; 54: 1524–1532. [DOI] [PubMed] [Google Scholar]
- 14. Simmons RA, Flozak AS, Ogata ES. Glucose regulates Glut 1 function and expression in fetal rat lung and muscle in vitro. Endocrinology. 1993; 132: 2312–2318. [DOI] [PubMed] [Google Scholar]
- 15. Simmons RA. Cell glucose transport and glucose handling during fetal and neonatal development. In Polin Richard A., Abman Steven H., Rowitch David H., Benitz William E., Fox William W., eds. Fetal and Neonatal Physiology, 5th ed. Philadelphia, PA: Elsevier; 2017. p428–435.e3. [Google Scholar]
- 16. Abel ED. Glucose transport in the heart. Front Biosci. 2004; 9: 201–215. [DOI] [PubMed] [Google Scholar]
- 17. Uphues I, Kolter T, Goud B, Eckel J. Failure of insulin‐regulated recruitment of the glucose transporter GLUT4 in cardiac muscle of obese Zucker rats is associated with alterations of small‐molecular‐mass GTP‐binding proteins. Biochem J. 1995; 311: 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wright JJ, Kim J, Buchanan J, Boudina S, Sena S, Bakirtzi K, Ilkun O, Theobald HA, Cooksey RC, Kandror KV, Abel ED. Mechanisms for increased myocardial fatty acid utilization following short‐term high‐fat feeding. Cardiovasc Res. 2009; 82: 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Banerjee SK, McGaffin KR, Pastor‐Soler NM, Ahmad F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc Res. 2009; 84: 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zamora M, Villena JA. Contribution of impaired insulin signaling to the pathogenesis of diabetic cardiomyopathy. Int J Mol Sci. 2019; 20: 2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vadvalkar SS, Matsuzaki S, Eyster CA, Giorgione JR, Bockus LB, Kinter CS, Kinter M, Humphries KM. Decreased mitochondrial pyruvate transport activity in the diabetic heart: role of mitochondrial pyruvate carrier 2 (MPC2) acetylation. J Biol Chem. 2017; 292: 4423–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee TW, Bai KJ, Lee TI, Chao TF, Kao YH, Chen YJ. PPARs modulate cardiac metabolism and mitochondrial function in diabetes. J Biomed Sci. 2017; 24: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mazumder PK, O'Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin‐resistant ob/ob mouse hearts. Diabetes. 2004; 53: 2366–2374. [DOI] [PubMed] [Google Scholar]
- 24. Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J Clin Investig. 2002; 109: 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol. 2020; 17: 585–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Greenwalt DE, Scheck SH, Rhinehart‐Jones T. Heart CD36 expression is increased in murine models of diabetes and in mice fed a high fat diet. J Clin Invest. 1995; 96: 1382–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cifarelli V, Abumrad NA. Enterocyte fatty acid handling proteins and chylomicron formation. In Said Hamid M., ed. Physiology of the Gastrointestinal Tract, 6th ed. Cambridge, MA: Academic Press; 2018. p1087–1107. [Google Scholar]
- 28. Yang X, Zhang W, Chen Y, Li Y, Sun L, Liu Y, Liu M, Yu M, Li X, Han J, Duan Y. Activation of peroxisome proliferator‐activated receptor γ (PPARγ) and CD36 protein expression: the dual pathophysiological roles of progesterone. J Biol Chem. 2016; 291: 15108–15118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fliegner D, Westermann D, Riad A, Schubert C, Becher E, Fielitz J, Tschöpe C, Regitz‐Zagrosek V. Up‐regulation of PPARγ in myocardial infarction. Eur J Heart Fail. 2008; 10: 30–38. [DOI] [PubMed] [Google Scholar]
- 30. Schilling JD. The mitochondria in diabetic heart failure: from pathogenesis to therapeutic promise. Antioxid Redox Signal. 2015; 22: 1515–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mirza AZ, Althagafi II, Shamshad H. Role of PPAR receptor in different diseases and their ligands: physiological importance and clinical implications. Eur J Med Chem. 2019; 166: 502–513. [DOI] [PubMed] [Google Scholar]
- 32. Wang P, Liu J, Li Y, Wu S, Luo J, Yang H, Subbiah R, Chatham J, Zhelyabovska O, Yang Q. Peroxisome proliferator‐activated receptor δ is an essential transcriptional regulator for mitochondrial protection and biogenesis in adult heart. Circ Res. 2010; 106: 911–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee TI, Kao YH, Chen YC, Huang JH, Hsiao FC, Chen YJ. Peroxisome proliferator‐activated receptors modulate cardiac dysfunction in diabetic cardiomyopathy. Diabetes Res Clin Pract. 2013; 100: 330–339. [DOI] [PubMed] [Google Scholar]
- 34. Szanto A, Nagy L. The many faces of PPARγ: anti‐inflammatory by any means? Immunobiology. 2008; 213: 789–803. [DOI] [PubMed] [Google Scholar]
- 35. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity‐related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007; 56: 2457–2466. [DOI] [PubMed] [Google Scholar]
- 36. Shao D, Kolwicz SC Jr, Wang P, Roe ND, Villet O, Nishi K, Hsu YA, Flint GV, Caudal A, Wang W, Regnier M. Increasing fatty acid oxidation prevents high‐fat diet‐induced cardiomyopathy through regulating parkin‐mediated mitophagy. Circulation. 2020; 142: 983–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kolwicz SC Jr, Olson DP, Marney LC, Garcia‐Menendez L, Synovec RE, Tian R. Cardiac‐specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure‐overload hypertrophy. Circ Res. 2012; 111: 728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sambandam N, Lopaschuk GD, Brownsey RW, Allard MF. Energy metabolism in the hypertrophied heart. Heart Fail Rev. 2002; 7: 161–173. [DOI] [PubMed] [Google Scholar]
- 39. Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, Schaffer JE, Yu YH, Goldberg IJ. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem. 2009; 284: 36312–36323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004; 18: 1692–1700. [DOI] [PubMed] [Google Scholar]
- 41. Yong‐Xu Wang C‐HL, Tiep S, Yu RT, Ham J, Kang H, Evans RM. Peroxisome‐proliferator‐activated receptor δ activates fat metabolism to prevent obesity. Cell. 2003; 113: 159–170. [DOI] [PubMed] [Google Scholar]
- 42. Ravnskjaer K, Frigerio F, Boergesen M, Nielsen T, Maechler P, Mandrup S. PPARδ is a fatty acid sensor that enhances mitochondrial oxidation in insulin‐secreting cells and protects against fatty acid‐induced dysfunction. J Lipid Res. 2010; 51: 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, Nielsen LB. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology. 2003; 144: 3483–3490. [DOI] [PubMed] [Google Scholar]
- 44. Bartz R, Li WH, Venables B, Zehmer JK, Roth MR, Welti R, Anderson RG, Liu P, Chapman KD. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J Lipid Res. 2007; 48: 837–847. [DOI] [PubMed] [Google Scholar]
- 45. Cui L, Liu P. Two types of contact between lipid droplets and mitochondria. Front Cell Dev Biol. 2020; 8: 618322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ipsen DH, Lykkesfeldt J, Tveden‐Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non‐alcoholic fatty liver disease. Cell Mol Life Sci. 2018; 75: 3313–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Razani B, Zhang H, Schulze PC, Schilling JD, Verbsky J, Lodhi IJ, Topkara VK, Feng C, Coleman T, Kovacs A, Kelly DP, Saffitz JE, Dorn GW II, Nichols CG, Semenkovich CF. Fatty acid synthase modulates homeostatic responses to myocardial stress. J Biol Chem. 2011; 286: 30949–30961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011; 13: 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, Sadoshima J. Mitophagy is essential for maintaining cardiac function during high fat diet‐induced diabetic cardiomyopathy. Circ Res. 2019; 124: 1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sheridan M, Ogretmen B. The role of ceramide metabolism and signaling in the regulation of mitophagy and cancer therapy. Cancers (Basel). 2021; 13: 2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martinez‐Lopez N, Singh R. Autophagy and lipid droplets in the liver. Annu Rev Nutr. 2015; 35: 215–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009; 458: 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mardani I, Tomas Dalen K, Drevinge C, Miljanovic A, Stahlman M, Klevstig M, Scharin Tang M, Fogelstrand P, Levin M, Ekstrand M, Nair S. Plin2‐deficiency reduces lipophagy and results in increased lipid accumulation in the heart. Sci Rep. 2019; 9: 6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Martinez‐Lopez N, Garcia‐Macia M, Sahu S, Athonvarangkul D, Liebling E, Merlo P, Cecconi F, Schwartz GJ, Singh R. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. 2016; 23: 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cross HR, Opie LH, Radda GK, Clarke K. Is a high glycogen content beneficial or detrimental to the ischemic rat heart? A controversy resolved. Circ Res. 1996; 78: 482–491. [DOI] [PubMed] [Google Scholar]
- 56. Ishihama S, Yoshida S, Yoshida T, Mori Y, Ouchi N, Eguchi S, Sakaguchi T, Tsuda T, Kato K, Shimizu Y, Ohashi K, Okumura T, Bando YK, Yagyu H, Wettschureck N, Kubota N, Offermanns S, Kadowaki T, Murohara T, Takefuji M. LPL/AQP7/GPD2 promotes glycerol metabolism under hypoxia and prevents cardiac dysfunction during ischemia. FASEB J. 2021; 35: e22048. [DOI] [PubMed] [Google Scholar]
- 57. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders—a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis. 2017; 1863: 1066–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tsushima K, Bugger H, Wende AR, Soto J, Jenson GA, Tor AR, McGlauflin R, Kenny HC, Zhang Y, Souvenir R, Hu XX, Sloan CL, Pereira RO, Lira VA, Spitzer KW, Sharp TL, Shoghi KI, Sparagna GC, Rog‐Zielinska EA, Kohl P, Khalimonchuk O, Schaffer JE, Abel ED. Mitochondrial reactive oxygen species in lipotoxic hearts induce post‐translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission. Circ Res. 2018; 122: 58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015; 116: 264–278. [DOI] [PubMed] [Google Scholar]
- 60. Chen Y, Liu Y, Dorn GW 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. 2011; 109: 1327–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Serasinghe MN, Chipuk JE. Mitochondrial fission in human diseases. Handb Exp Pharmacol. 2017; 240: 159–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006; 103: 2653–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rajab BS, Kassab S, Stonall CD, Daghistani H, Gibbons S, Mamas M, Smith D, Mironov A, AlBalawi Z, Zhang YH, Baudoin F, Zi M, Prehar S, Cartwright EJ, Kitmitto A. Differential remodelling of mitochondrial subpopulations and mitochondrial dysfunction are a feature of early stage diabetes. Sci Rep. 2022; 12: 978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pickles S, Vigie P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018; 28: R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Belousov DM, Mikhaylenko EV, Somasundaram SG, Kirkland CE, Aliev G. The dawn of mitophagy: what do we know by now? Curr Neuropharmacol. 2021; 19: 170–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Luan Y, Luan Y, Feng Q, Chen X, Ren KD, Yang Y. Emerging role of mitophagy in the heart: therapeutic potentials to modulate mitophagy in cardiac diseases. Oxid Med Cell Longev. 2021; 2021: 3259963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN‐inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A. 2011; 108: 9572–9577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, Sadoshima J. Alternative mitophagy protects the heart against obesity‐associated cardiomyopathy. Circ Res. 2021; 129: 1105–1121. [DOI] [PubMed] [Google Scholar]
- 69. Hannun YA, Obeid LM. Many ceramides. J Biol Chem. 2011; 286: 27855–27862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dany M, Ogretmen B. Ceramide induced mitophagy and tumor suppression. Biochim Biophys Acta. 1853; 2015: 2834–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yan‐Ting Zhou PG, Karim A, Shimabukuro M, Baetens MHD, Orci L, Unger RH. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A. 2000; 97: 1784–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001; 107: 813–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chavez JA, Summers SA. A ceramide‐centric view of insulin resistance. Cell Metab. 2012; 15: 585–594. [DOI] [PubMed] [Google Scholar]
- 74. Di Paola M, Cocco T, Lorusso M. Ceramide interaction with the respiratory chain of heart mitochondria. Biochemistry. 2000; 39: 6660–6668. [DOI] [PubMed] [Google Scholar]
- 75. Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, Dogra S, Öhman MK, Takeda K, Sugii S, Pewzner‐Jung Y, Futerman AH, Summers SA. CerS2 haploinsufficiency inhibits β‐oxidation and confers susceptibility to diet‐induced steatohepatitis and insulin resistance. Cell Metab. 2014; 20: 687–695. [DOI] [PubMed] [Google Scholar]
- 76. Kogot‐Levin A, Saada A. Ceramide and the mitochondrial respiratory chain. Biochimie. 2014; 100: 88–94. [DOI] [PubMed] [Google Scholar]
- 77. Roszczyc‐Owsiejczuk K, Zabielski P. Sphingolipids as a culprit of mitochondrial dysfunction in insulin resistance and type 2 diabetes. Front Endocrinol (Lausanne). 2021; 12: 635175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, Ramshesh VK, Peterson YK, Lemasters JJ, Szulc ZM, Bielawski J, Ogretmen B. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012; 8: 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Spassieva SD, Mullen TD, Townsend DM, Obeid LM. Disruption of ceramide synthesis by CerS2 down‐regulation leads to autophagy and the unfolded protein response. Biochem J. 2009; 424: 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Preuss C, Jelenik T, Bodis K, Mussig K, Burkart V, Szendroedi J, Roden M, Markgraf DF. A new targeted lipidomics approach reveals lipid droplets in liver, muscle and heart as a repository for diacylglycerol and ceramide species in non‐alcoholic fatty liver. Cell. 2019; 8: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liu Q, Chen X, Macdonnell SM, Kranias EG, Lorenz JN, Leitges M, Houser SR, Molkentin JD. Protein kinase Cα, but not PKCβ or PKCγ, regulates contractility and heart failure susceptibility: implications for ruboxistaurin as a novel therapeutic approach. Circ Res. 2009; 105: 194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010; 106: 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Simon JN, Chowdhury SA, Warren CM, Sadayappan S, Wieczorek DF, Solaro RJ, Wolska BM. Ceramide‐mediated depression in cardiomyocyte contractility through PKC activation and modulation of myofilament protein phosphorylation. Basic Res Cardiol. 2014; 109: 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jia G, Whaley‐Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia‐ and insulin‐resistance‐induced heart disease. Diabetologia. 2018; 61: 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wilson AJ, Gill EK, Abudalo RA, Edgar KS, Watson CJ, Grieve DJ. Reactive oxygen species signalling in the diabetic heart: emerging prospect for therapeutic targeting. Heart. 2018; 104: 293–299. [DOI] [PubMed] [Google Scholar]
- 86. Kaludercic N, Di Lisa F. Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Front Cardiovasc Med. 2020; 7: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bugger H, Abel ED. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci (Lond). 2008; 114: 195–210. [DOI] [PubMed] [Google Scholar]
- 88. Galloway CA, Yoon Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid Redox Signal. 2015; 22: 1545–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Aimo A, Castiglione V, Borrelli C, Saccaro LF, Franzini M, Masi S, Emdin M, Giannoni A. Oxidative stress and inflammation in the evolution of heart failure: from pathophysiology to therapeutic strategies. Eur J Prev Cardiol. 2020; 27: 494–510. [DOI] [PubMed] [Google Scholar]
- 90. Steinberg SF. Oxidative stress and sarcomeric proteins. Circ Res. 2013; 112: 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lorenzo O, Ramirez E, Picatoste B, Egido J, Tunon J. Alteration of energy substrates and ROS production in diabetic cardiomyopathy. Mediators Inflamm. 2013; 2013: 461967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Murray AJ, Anderson RE, Watson GC, Radda GK, Clarke K. Uncoupling proteins in human heart. Lancet. 2004; 364: 1786–1788. [DOI] [PubMed] [Google Scholar]
- 93. Boudina S, Han YH, Pei S, Tidwell TJ, Henrie B, Tuinei J, Olsen C, Sena S, Abel ED. UCP3 regulates cardiac efficiency and mitochondrial coupling in high fat‐fed mice but not in leptin‐deficient mice. Diabetes. 2012; 61: 3260–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005; 112: 2686–2695. [DOI] [PubMed] [Google Scholar]
- 95. Dludla PV, Nkambule BB, Tiano L, Louw J, Jastroch M, Mazibuko‐Mbeje SE. Uncoupling proteins as a therapeutic target to protect the diabetic heart. Pharmacol Res. 2018; 137: 11–24. [DOI] [PubMed] [Google Scholar]
- 96. Madhusudhan T, Wang H, Dong W, Ghosh S, Bock F, Thangapandi VR, Ranjan S, Wolter J, Kohli S, Shahzad K, Heidel F, Krueger M, Schwenger V, Moeller MJ, Kalinski T, Reiser J, Chavakis T, Isermann B. Defective podocyte insulin signalling through p85‐XBP1 promotes ATF6‐dependent maladaptive ER‐stress response in diabetic nephropathy. Nat Commun. 2015; 6: 6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med. 2012; 18: 59–68. [DOI] [PubMed] [Google Scholar]
- 98. Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008; 29: 42–61. [DOI] [PubMed] [Google Scholar]
- 99. Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS‐1 pancreatic β‐cell apoptosis. Endocrinology. 2006; 147: 3398–3407. [DOI] [PubMed] [Google Scholar]
- 100. Luan Y, Luan Y, Yuan RX, Feng Q, Chen X, Yang Y. Structure and function of mitochondria‐associated endoplasmic reticulum membranes (MAMs) and their role in cardiovascular diseases. Oxid Med Cell Longev. 2021; 2021: 4578809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Gao P, Yan Z, Zhu Z. Mitochondria‐associated endoplasmic reticulum membranes in cardiovascular diseases. Front Cell Dev Biol. 2020; 8: 604240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Diaz‐Juarez J, Suarez JA, Dillmann WH, Suarez J. Mitochondrial calcium handling and heart disease in diabetes mellitus. Biochim Biophys Acta Mol Basis Dis. 2021; 1867: 165984. [DOI] [PubMed] [Google Scholar]
- 103. Oliveira PJ, Seiça R, Coxito PM, Rolo AP, Palmeira CM, Santos MS, Moreno AJ. Enhanced permeability transition explains the reduced calcium uptake in cardiac mitochondria from streptozotocin‐induced diabetic rats. FEBS Lett. 2003; 554: 511–514. [DOI] [PubMed] [Google Scholar]
- 104. Pu J, Ha CW, Zhang S, Jung JP, Huh WK, Liu P. Interactomic study on interaction between lipid droplets and mitochondria. Protein Cell. 2011; 2: 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Veliova M, Petcherski A, Liesa M, Shirihai OS. The biology of lipid droplet‐bound mitochondria. Semin Cell Dev Biol. 2020; 108: 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Seferovic PM, Paulus WJ. Clinical diabetic cardiomyopathy: a two‐faced disease with restrictive and dilated phenotypes. Eur Heart J. 2015; 36: 1718–1727. [DOI] [PubMed] [Google Scholar]
- 107. Pham I, Cosson E, Nguyen MT, Banu I, Genevois I, Poignard P, Valensi P. Evidence for a specific diabetic cardiomyopathy: an observational retrospective echocardiographic study in 656 asymptomatic type 2 diabetic patients. Int J Endocrinol. 2015; 2015: 743503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mizamtsidi M, Paschou SA, Grapsa J, Vryonidou A. Diabetic cardiomyopathy: a clinical entity or a cluster of molecular heart changes? Eur J Clin Invest. 2016; 46: 947–953. [DOI] [PubMed] [Google Scholar]
- 109. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974; 34: 29–34. [DOI] [PubMed] [Google Scholar]
- 110. Movahed MR, Hashemzadeh M, Jamal MM. Diabetes mellitus is a strong, independent risk for atrial fibrillation and flutter in addition to other cardiovascular disease. Int J Cardiol. 2005; 105: 315–318. [DOI] [PubMed] [Google Scholar]
- 111. de Simone G, Devereux RB, Chinali M, Lee ET, Galloway JM, Barac A, Panza JA, Howard BV. Diabetes and incident heart failure in hypertensive and normotensive participants of the Strong Heart Study. J Hypertens. 2010; 28: 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Maisch B, Alter P, Pankuweit S. Diabetic cardiomyopathy—fact or fiction? Herz. 2011; 36: 102–115. [DOI] [PubMed] [Google Scholar]
- 113. Lorenzo‐Almorós A, Tuñón J, Orejas M, Cortés M, Egido J, Lorenzo Ó. Diagnostic approaches for diabetic cardiomyopathy. Cardiovasc Diabetol. 2017; 16: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rijzewijk LJ, van der Meer RW, Smit JWA, Diamant M, Bax JJ, Hammer S, Romijn JA, de Roos A, Lamb HJ. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol. 2008; 52: 1793–1799. [DOI] [PubMed] [Google Scholar]
- 115. Rider OJ, Apps A, Miller J, Lau JYC, Lewis AJM, Peterzan MA, Dodd MS, Lau AZ, Trumper C, Gallagher FA, Grist JT. Noninvasive in vivo assessment of cardiac metabolism in the healthy and diabetic human heart using hyperpolarized 13C MRI. Circ Res. 2020; 126: 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Bonds DE, Craven TE, Buse J, Crouse JR, Cuddihy R, Elam M, Ginsberg HN, Kirchner K, Marcovina S, Mychaleckyj JC, O'Connor PJ, Sperl‐Hillen JA. Fenofibrate‐associated changes in renal function and relationship to clinical outcomes among individuals with type 2 diabetes: the Action to Control Cardiovascular Risk in Diabetes (ACCORD) experience. Diabetologia. 2012; 55: 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J, Berria R, Ma JZ, Dwivedi S, Havranek R, Fincke C, DeFronzo R, Bannayan GA, Schenker S, Cusi K. A placebo‐controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006; 355: 2297–2307. [DOI] [PubMed] [Google Scholar]
- 118. Harrison SA, Alkhouri N, Davison BA, Sanyal A, Edwards C, Colca JR, Lee BH, Loomba R, Cusi K, Kolterman O, Cotter G, Dittrich HC. Insulin sensitizer MSDC‐0602K in non‐alcoholic steatohepatitis: a randomized, double‐blind, placebo‐controlled phase IIb study. J Hepatol. 2020; 72: 613–626. [DOI] [PubMed] [Google Scholar]
- 119. Chen Z, Vigueira PA, Chambers KT, Hall AM, Mitra MS, Qi N, McDonald WG, Colca JR, Kletzien RF, Finck BN. Insulin resistance and metabolic derangements in obese mice are ameliorated by a novel peroxisome proliferator‐activated receptor γ‐sparing thiazolidinedione. J Biol Chem. 2012; 287: 23537–23548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Tahrani AA, Barnett AH, Bailey CJ. SGLT inhibitors in management of diabetes. Lancet Diab Endocrinol. 2013; 1: 140–151. [DOI] [PubMed] [Google Scholar]
- 121. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR, CANVAS Program Collaborative Group . Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017; 377: 644–657. [DOI] [PubMed] [Google Scholar]
- 122. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015; 373: 2117–2128. [DOI] [PubMed] [Google Scholar]
- 123. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Silverman MG, Zelniker TA, Kuder JF, Murphy SA, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Ruff CT, Gause‐Nilsson IAM, Fredriksson M, Johansson PA, Langkilde AM, Sabatine MS. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019; 380: 347–357. [DOI] [PubMed] [Google Scholar]
- 124. McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Belohlavek J, Böhm M. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019; 381: 1995–2008. [DOI] [PubMed] [Google Scholar]
- 125. Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, Januzzi J, Verma S, Tsutsui H, Brueckmann M, Jamal W, Kimura K, Schnee J, Zeller C, Cotton D, Bocchi E, Böhm M, Choi DJ, Chopra V, Chuquiure E, Giannetti N, Janssens S, Zhang J, Gonzalez Juanatey JR, Kaul S, Brunner‐la Rocca HP, Merkely B, Nicholls SJ, Perrone S, Pina I, Ponikowski P, Sattar N, Senni M, Seronde MF, Spinar J, Squire I, Taddei S, Wanner C, Zannad F. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. 2020; 383: 1413–1424. [DOI] [PubMed] [Google Scholar]
- 126. Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Böhm M, Brunner‐La Rocca HP, Choi DJ, Chopra V, Chuquiure‐Valenzuela E, Giannetti N. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. 2021; 385: 1451–1461. [DOI] [PubMed] [Google Scholar]
- 127. Nassif ME, Windsor SL, Borlaug BA, Kitzman DW, Shah SJ, Tang F, Khariton Y, Malik AO, Khumri T, Umpierrez G, Lamba S, Sharma K, Khan SS, Chandra L, Gordon RA, Ryan JJ, Chaudhry SP, Joseph SM, Chow CH, Kanwar MK, Pursley M, Siraj ES, Lewis GD, Clemson BS, Fong M, Kosiborod MN. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: a multicenter randomized trial. Nat Med. 2021; 27: 1954–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Bhatt DL, Szarek M, Steg PG, Cannon CP, Leiter LA, McGuire DK, Lewis JB, Riddle MC, Voors AA, Metra M, Lund LH, Komajda M, Testani JM, Wilcox CS, Ponikowski P, Lopes RD, Verma S, Lapuerta P, Pitt B, SOLOIST‐WHF Trial Investigators . Sotagliflozin in patients with diabetes and recent worsening heart failure. N Engl J Med. 2021; 384: 117–128. [DOI] [PubMed] [Google Scholar]
- 129. Zelniker TA, Braunwald E. Mechanisms of cardiorenal effects of sodium‐glucose cotransporter 2 inhibitors: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2020; 75: 422–434. [DOI] [PubMed] [Google Scholar]
- 130. Stenlöf K, Cefalu WT, Kim KA, Alba M, Usiskin K, Tong C, Canovatchel W, Meininger G. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab. 2013; 15: 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Steven S, Oelze M, Hanf A, Kröller‐Schön S, Kashani F, Roohani S, Welschof P, Kopp M, Gödtel‐Armbrust U, Xia N, Li H, Schulz E, Lackner KJ, Wojnowski L, Bottari SP, Wenzel P, Mayoux E, Münzel T, Daiber A. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017; 13: 370–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lee TM, Chang NC, Lin SZ. Dapagliflozin, a selective SGLT2 inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic Biol Med. 2017; 104: 298–310. [DOI] [PubMed] [Google Scholar]
- 133. Verma S, McMurray JJV. SGLT2 inhibitors and mechanisms of cardiovascular benefit: a state‐of‐the‐art review. Diabetologia. 2018; 61: 2108–2117. [DOI] [PubMed] [Google Scholar]
- 134. Lopaschuk GD, Verma S. Mechanisms of cardiovascular benefits of sodium glucose co‐transporter 2 (SGLT2) inhibitors: a state‐of‐the‐art review. JACC Basic Transl Sci. 2020; 5: 632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Marso SP, Daniels GH, Brown‐Frandsen K, Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, Steinberg WM, Stockner M, Zinman B, Bergenstal RM, Buse JB, LEADER Steering Committee on behalf of the LEADER Trial Investigators . Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016; 375: 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jódar E, Leiter LA, Lingvay I, Rosenstock J, Seufert J, Warren ML, Woo V, Hansen O, Holst AG, Pettersson J, Vilsbøll T. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016; 375: 1834–1844. [DOI] [PubMed] [Google Scholar]
- 137. Pfeffer MA, Claggett B, Diaz R, Dickstein K, Gerstein HC, Køber LV, Lawson FC, Ping L, Wei X, Lewis EF, Maggioni AP, McMurray J, Probstfield JL, Riddle MC, Solomon SD, Tardif JC, ELIXA Investigators . Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med. 2015; 373: 2247–2257. [DOI] [PubMed] [Google Scholar]
- 138. Holman RR, Bethel MA, Mentz RJ, Thompson VP, Lokhnygina Y, Buse JB, Chan JC, Choi J, Gustavson SM, Iqbal N, Maggioni AP, Marso SP, Öhman P, Pagidipati NJ, Poulter N, Ramachandran A, Zinman B, Hernandez AF. Effects of once‐weekly exenatide on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2017; 377: 1228–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Almutairi M, Gopal K, Greenwell AA, Young A, Gill R, Aburasayn H, Al Batran R, Chahade JJ, Gandhi M, Eaton F, Mailloux RJ, Ussher JR. The GLP‐1 receptor agonist liraglutide increases myocardial glucose oxidation rates via indirect mechanisms and mitigates experimental diabetic cardiomyopathy. Can J Cardiol. 2021; 37: 140–150. [DOI] [PubMed] [Google Scholar]
- 140. Correale M, Lamacchia O, Ciccarelli M, Dattilo G, Tricarico L, Brunetti ND. Vascular and metabolic effects of SGLT2i and GLP‐1 in heart failure patients. Heart Fail Rev. 2021. 10.1007/s10741-021-10157-y [DOI] [PubMed] [Google Scholar]
- 141. Ying Y, Zhu H, Liang Z, Ma X, Li S. GLP1 protects cardiomyocytes from palmitate‐induced apoptosis via Akt/GSK3b/b‐catenin pathway. J Mol Endocrinol. 2015; 55: 245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Ding L, Zhang J. Glucagon‐like peptide‐1 activates endothelial nitric oxide synthase in human umbilical vein endothelial cells. Acta Pharmacol Sin. 2012; 33: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. dos Santos L, Salles TA, Arruda‐Junior DF, Campos LC, Pereira AC, Barreto AL, Antonio EL, Mansur AJ, Tucci PJ, Krieger JE, Girardi AC. Circulating dipeptidyl peptidase IV activity correlates with cardiac dysfunction in human and experimental heart failure. Circ Heart Fail. 2013; 6: 1029–1038. [DOI] [PubMed] [Google Scholar]
- 144. Shigeta T, Aoyama M, Bando YK, Monji A, Mitsui T, Takatsu M, Cheng XW, Okumura T, Hirashiki A, Nagata K, Murohara T. Dipeptidyl peptidase‐4 modulates left ventricular dysfunction in chronic heart failure via angiogenesis‐dependent and ‐independent actions. Circulation. 2012; 126: 1838–1851. [DOI] [PubMed] [Google Scholar]
- 145. Scheen AJ. Cardiovascular effects of new oral glucose‐lowering agents: DPP‐4 and SGLT‐2 inhibitors. Circ Res. 2018; 122: 1439–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Deacon CF. Physiology and pharmacology of DPP‐4 in glucose homeostasis and the treatment of type 2 diabetes. Front Endocrinol (Lausanne). 2019; 10: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Bethel MA, Engel SS, Green JB, Huang Z, Josse RG, Kaufman KD, Standl E, Suryawanshi S, Van de Werf F, McGuire DK, Peterson ED, Holman RR, TECOS Study Group . Assessing the safety of sitagliptin in older participants in the Trial Evaluating Cardiovascular Outcomes with Sitagliptin (TECOS). Diabetes Care. 2017; 40: 494–501. [DOI] [PubMed] [Google Scholar]
- 148. Leiter LA, Teoh H, Braunwald E, Mosenzon O, Cahn A, Kumar KM, Smahelova A, Hirshberg B, Stahre C, Frederich R, Bonnici F, Scirica BM, Bhatt DL, Raz I, SAVOR‐TIMI 53 Steering Committee and Investigators . Efficacy and safety of saxagliptin in older participants in the SAVOR‐TIMI 53 trial. Diabetes Care. 2015; 38: 1145–1153. [DOI] [PubMed] [Google Scholar]
- 149. Bando YK, Murohara T. Heart failure as a comorbidity of diabetes: role of dipeptidyl peptidase 4. J Atheroscler Thromb. 2016; 23: 147–154. [DOI] [PubMed] [Google Scholar]
- 150. White WB, Cannon CP, Heller SR, Nissen SE, Bergenstal RM, Bakris GL, Perez AT, Fleck PR, Mehta CR, Kupfer S, Wilson C, Cushman WC, Zannad F, EXAMINE Investigators . Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013; 369: 1327–1335. [DOI] [PubMed] [Google Scholar]
- 151. Li B, Zheng Z, Wei Y, Wang M, Peng J, Kang T, Huang X, Xiao J, Li Y, Li Z. Therapeutic effects of neuregulin‐1 in diabetic cardiomyopathy rats. Cardiovasc Diabetol. 2011; 10: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Huynh K, Kiriazis H, Du XJ, Love JE, Gray SP, Jandeleit‐Dahm KA, McMullen JR, Ritchie RH. Targeting the upregulation of reactive oxygen species subsequent to hyperglycemia prevents type 1 diabetic cardiomyopathy in mice. Free Radic Biol Med. 2013; 60: 307–317. [DOI] [PubMed] [Google Scholar]
- 153. Borghetti G, von Lewinski D, Eaton DM, Sourij H, Houser SR, Wallner M. Diabetic cardiomyopathy: current and future therapies. Beyond glycemic control. Front Physiol. 2018; 9: 1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Velmurugan GV, Sundaresan NR, Gupta MP, White C. Defective Nrf2‐dependent redox signalling contributes to microvascular dysfunction in type 2 diabetes. Cardiovasc Res. 2013; 100: 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]