

Abstract
Aims
Heart failure with reduced ejection fraction (HFrEF) is a disease with high mortality and morbidity. Recent positive inotropic drug developments focused on cardiac myofilaments, that is, direct activators of the myosin molecule and Ca2+ sensitizers for patients with advanced HFrEF. Omecamtiv mecarbil (OM) is the first direct myosin activator with promising results in clinical studies. Here, we aimed to elucidate the cellular mechanisms of the positive inotropic effect of OM in a comparative in vitro investigation where Ca2+‐sensitizing positive inotropic agents with distinct mechanisms of action [EMD 53998 (EMD), which also docks on the myosin molecule, and levosimendan (Levo), which binds to troponin C] were included.
Methods
Enzymatically isolated canine cardiomyocytes with intact cell membranes were loaded with Fura‐2AM, a Ca2+‐sensitive, ratiometric, fluorescent dye. Changes in sarcomere length (SL) and intracellular Ca2+ concentration were recorded in parallel at room temperature, whereas cardiomyocyte contractions were evoked by field stimulation at 0.1 Hz in the presence of different OM, EMD, or Levo concentrations.
Results
SL was reduced by about 23% or 9% in the presence of 1 μM OM or 1 μM EMD in the absence of electrical stimulation, whereas 1 μM Levo had no effect on resting SL. Fractional sarcomere shortening was increased by 1 μM EMD or 1 μM Levo to about 152%, but only to about 128% in the presence of 0.03 μM OM. At higher OM concentrations, no significant increase in fractional sarcomere shortening could be recorded. Contraction durations largely increased, whereas the kinetics of contractions and relaxations decreased with increasing OM concentrations. One‐micromole EMD or 1 μM Levo had no effects on contraction durations. One‐micromole Levo, but not 1 μM EMD, accelerated the kinetics of cardiomyocyte contractions and relaxations. Ca2+ transient amplitudes were unaffected by all treatments.
Conclusions
Our data revealed major distinctions between the cellular effects of myofilament targeted agents (OM, EMD, or Levo) depending on their target proteins and binding sites, although they were compatible with the involvement of Ca2+‐sensitizing mechanisms for all three drugs. Significant part of the cardiotonic effect of OM relates to the prolongation of systolic contraction in combination with its Ca2+‐sensitizing effect.
Keywords: Omecamtiv mecarbil; Heart failure with reduced ejection fraction; Myosin activators; Positive inotropy, diastolic dysfunction
Introduction
Heart failure (HF) with reduced ejection fraction (HFrEF) is a complex clinical syndrome resulting from structural and functional impairments of the myocardium and involves contractile dysfunction of left ventricular (LV) cardiomyocytes. Cardiotonic agents are frequently considered for the therapy of acute and chronic HF to counter cardiac pump function. Traditional β‐mimetic positive inotropic drugs [i.e. β‐receptor agonists and inhibitors of the phosphodiesterase (PDE) III isoenzyme] are effective but potentially harmful. These Ca2+‐mobilizing agents augment the amplitude of intracellular Ca2+ transients during systoles; therefore, cardiomyocyte contractions and relaxations become larger and faster. Unfortunately, safety concerns limit the applicability of β‐mimetic drugs. This is because stimulation of the β‐adrenergic cascade increases the oxygen demand of cardiomyocytes that is often hard to tolerate by the failing heart.
In addition to the Ca2+ sensitizers, recently, a new group of positive inotropic drugs have been developed for the therapy of HFrEF, which directly modulate the thick myofilaments of cardiac sarcomeres (rather than increasing the intracellular Ca2+ transient): the selective cardiac myosin activators. 1 , 2 The central concept behind drug developments of these kinds relates to the augmentation of cardiac contractile state without increasing myocardial energy demand. Nevertheless, the relationships between intracellular Ca2+ concentration and sarcomeric responses in the presence myofilament targeted agents have not been fully elucidated yet.
A phase II clinical trial on omecamtiv mecarbil (OM) demonstrated a significant increase in LV ejection fraction (EF). 3 , 4 Moreover, a large‐scale phase III clinical trial has recently revealed lower incidence of a composite endpoint of HF event or death from cardiovascular (CV) causes in patients with chronic HFrEF receiving OM. 5 , 6 Nevertheless, slight increases in circulating troponin levels were also occasionally observed in patients on OM, and this raised awareness to possible side effects of the drug. Similarly to OM, a previous drug candidate, the thiadiazinone derivative EMD 53998 (EMD), also exerts positive inotropic effects through a molecular interaction with the myosin motor protein. 7 , 8 , 9 In contrast to OM and EMD, the frequently used inodilator, levosimendan (Levo), is thought to increase myocardial contractility through myofilament Ca2+ sensitization due to its binding to the thin filament protein, troponin C [10]. Of note, the cardiac effects of EMD and Levo might be complicated by the inhibition of the PDE III isoenzyme. 10 , 11
In the present study, we aimed to compare intracellular Ca2+ trasients with parameters describing cardiomyocyte contractions and relaxations in the presence and absence of increasing OM concentrations, and these effects were contrasted to those recorded in the presence of supramaximal EMD or Levo concentrations. Our results shed new light on the complex relationships between the extents and kinetics of systolic and diastolic sarcomere length (SL) changes upon OM administrations in a framework where Ca2+ sensitization and myosin activation appear to be tightly coupled to each other.
Materials and methods
Animals
Adult mongrel dogs of either sex were used here according to a protocol approved by the local Animal Care Committee (2/2020/DEMÁB). All animals received human care in compliance with the ‘Principles of Laboratory Animal Care’, formulated by the National Society for Medical Research and the Guide for the Care and Use of Laboratory Animals, prepared by the Institute of Laboratory Animal Resources and published by the National Institutes of Health (NIH Publication No. 86‐23, Revised 1996).
Isolation of canine LV cardiomyocytes
Canine cardiomyocytes were studied since their electrophysiological properties are similar to those of humans. Cardiomyocytes were isolated from the mid‐myocardial region of the LV as described previously. 12 , 13 Briefly, hearts were isolated from anaesthetized (ketamine‐HCl 10 mg/kg, Richter Gedeon, Hungary, xylazine‐HCl 1 mg/kg, Eurovet Animal Health BV, The Netherlands) adult mongrel dogs (N = 6) weighed 10.6 ± 2.1 kg (7–15 kg) and aged 15.1 ± 4.2 months (10.9–23.9 months). Thereafter, the hearts were subjected to a perfusion system where the left anterior descending coronary artery was cannulated. Single cardiomyocytes were obtained by enzymatic dispersion technique by using a Ca2+‐free Joklik solution (Minimum Essential Medium Eagle, Joklik Modification; Sigma‐Aldrich Co., St. Louis, MO, USA) for 5 min, followed by 30‐min perfusion with Joklik solution containing 1 mg/mL collagenase (Type II, Worthington Biochemical Co., Lakewood, NJ, USA) and 0.2% bovine serum albumin (Fraction V, Sigma) in the presence of 50 μM Ca2+. Then cardiomyocytes were grounded, and normal Ca2+ concentration was restored progressively. Cardiomyocytes were stored at 15°C until the measurements.
Experimental set‐up
Cardiomyocytes were loaded with 5 μM Fura‐2AM Ca2+‐sensitive ratiometric fluorescent dye for 30 min in the presence of Pluronic F‐127 (25 mg/mL) to avoid early elimination of the dye from the intracellular space. Twenty‐five milligrams of Pluronic F‐127 was dissolved in 1 mL DMSO, and this solvent was used to make a Fura‐2AM stock solution. Cells were then incubated for 30 min to allow the intracellular esterases to relase Fura‐2. Cardiomyocytes were placed in a chamber on the stage of an inverted microscope (Nikon TS‐100). The final volume of the chamber filled with Tyrode solution (containing 144 mM NaCl, 5.6 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 5 mM HEPES, and 11 mM dextrose, pH = 7.4) was 1 mL. After sedimentation, a rod‐shaped cardiomyocyte with clear striation and acceptable contraction upon field‐stimulation was selected for further experiments. Field stimulation was performed at 0.1 Hz (Experimenta Setup, MDE, Heidelberg). Alternating excitation, wavelengths of 340 and 380 nm were used to monitor the fluorescence signals of Ca2+‐bound and Ca2+‐free Fura‐2 dye, respectively. Fluorescent emission was detected above 510 nm in case of both wavelengths, and traces were digitized at 120 Hz using the FeliX Software (Ratiomaster RM‐50 system, Horiba, New Brunswick, NJ, USA). 14
Determination of effects on resting SLs
The experimental protocol was the following: Cardiomyocytes were paced at 0.1 Hz for at least 2–3 min to achieve a steady state at the beginning of each experiment. The resting (unstimulated) SL was continuously measured after this initial conditioning using a high‐speed camera. OM (with final concentrations of 0.03, 0.1, 0.3, or 1 μM), Levo (1 μM), or EMD (1 μM) was added for 5–8 mins to the experimental chamber.
Determination of contractile parameters and Ca2+ transients
Contractile responses were monitored at 0.1 Hz pacing rates. Multiple parameters of cardiomyocyte contractions, relaxations, and intracellular Ca2+ transients were assessed. Resting SL of the cardiomyocytes was measured using a high‐speed camera. Fractional sarcomere shortening (FS) was calculated using the following equation: FS = (diastolic SL − systolic SL/diastolic SL) * 100. Duration of contraction (DC; s) was defined as the time period from the beginning till the end of the contraction of the cardiomyocyte. Rates (μm/s) of contraction and relaxation were determined by linear fits to the apparently linear phases of contractions and relaxations, respectively. The resting Ca2+ level was estimated by the Fura‐2 ratio (fluorescent intensity ratio at 340 and 380 nm excitations) at baseline (before cardiomyocyte stimulation). The amplitude of Ca2+ transients was defined as the difference between the peak levels of Ca2+ transients and resting Ca2+ levels (340/380 nm ratio). The rate of Ca2+ transient increase was considered as the slope of a line fitted by linear regression to the ascending phases of Ca2+ transients (1/s). The Ca2+ transient decay kinetics was fitted to a single exponential and described by its rate constant (K; 1/s), which was calculated from the following equation: Y = (Y0 − plateau)*exp(− K * X) + plateau, where X = time, Y starts at Y0 and decays (in one phase) down to plateau level, Y0 and plateau are in the same units as Y, and K is expressed in the reciprocal of X‐axis units.
Data analysis and statistics
Results were evaluated and graphs were created in the GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA, USA). The number of experiments in each group varied between 6 and 10 from six different hearts. Background fluorescence intensity levels were obtained at the end of the measurements, on a region without cardiomyocytes and were manually subtracted the fluorescence intensities for background correction. Values were evaluated for normality (Kolmogorov–Smirnov normality test) and were then evaluated by paired t‐tests, ordinary one‐way ANOVA or Kruskal–Wallis test with multiple comparisons as appropriate. Group descriptions are given as mean ± SEM values. Statistical significance was accepted at P < 0.05.
Results
OM and EMD, but not Levo, reduce cardiomyocyte resting SL
A dose‐dependent decrease in the resting SL of cardiomyocytes was observed upon OM administrations (i.e. from the drug‐free control of 1.96 ± 0.01 μm to 1.94 ± 0.04, 1.77 ± 0.04, 1.62 ± 0.05, or 1.50 ± 0.05 μm at 0.03 μM OM, 0.1 μM OM, 0.3 μM OM, or 1 μM OM, respectively; SL changes at 0.1 μM OM concentrations and beyond were significant) (Figure 1A,D ). One‐micromole EMD also evoked a significant reduction in resting SL (i.e. to 1.83 ± 0.08 μm; Figure 1B,D ), whereas Levo did not affect resting SL (Figure 1C,D ). Intracellular resting Ca2+ concentrations remained unchanged during OM, EMD, or Levo treatments (Figure 1A,C,E ).
Figure 1.
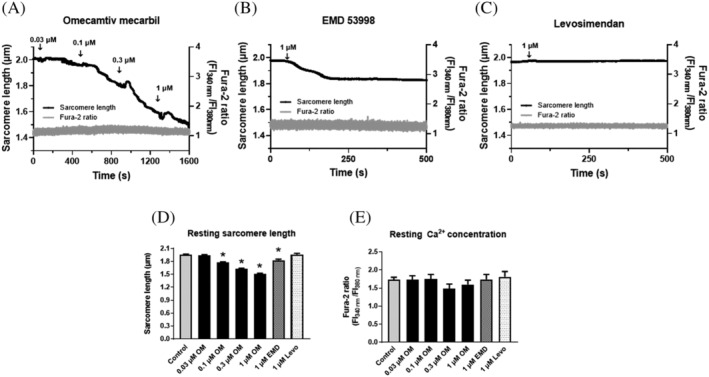
Distinct effects of OM, EMD or Levo on cardiomyocyte resting sarcomere lengths. OM (A) or EMD (B) decreased SL in resting isolated left ventricular canine cardiomyocytes. OM was added without field stimulation in a cumulative manner. EMD or Levo (C) was applied at a single concentration (1 μM). SL (black trace, left axis) and intracellular Ca2+ concentration (Fura‐2340/380 fluorescent intensity ratio, grey trace, right axis) were recorded simultaneously. Single representative examples are shown in the presence of OM, EMD, or Levo. EMD, Levo, or OM administrations are indicated by arrows. Resting SL decreased in the presence of OM (A,D) or EMD (B,D), whereas Levo did not affect SL (C,D) in the absence of Ca2+ concentration changes (E). Significant differences are indicated by asterisks when P < 0.05 vs. before treatment. EMD, EMD 53998; FI, fluorescence intensity; Levo, levosimendan; OM, omecamtiv mecarbil.
OM, EMD, and Levo differently affected cardiomyocyte contraction and relaxation kinetics
A significant decrease in the systolic peak SL was observed at 0.03, 0.01, 0.3, or 1 μM OM concentrations (1.65 ± 0.03 μm 1.53 ± 0.03 μm, 1.39 ± 0.02 μm, 1.34 ± 0.03 μm, respectively, vs. 1.74 ± 0.01 μm measured under control conditions, P < 0.05 for all; Figure 2A,G ) in field‐stimulated cardiomyocytes. Similarly to OM, EMD significantly decreased the systolic peak SL (i.e. to 1.51 ± 0.03 μm, P < 0.05; Figure 2B,G ). The effects of Levo on systolic peak SL decreases also reached the significance level (i.e. this parameter decreased to 1.63 ± 0.03 μm, P < 0.05; Figure 2C,G ). OM increased fractional sarcomere shortening at 0.03 μM (i.e. to 14.34 ± 1.64%), but not at higher drug concentrations, whereas EMD and Levo significantly increased this parameter at 1 μM (i.e. to 17.36 ± 0.98% and 17.02 ± 0.80%, respectively, from the drug‐free control level of 11.17 ± 0.55%, P < 0.05; Figure 2A–C,G,H ). The amplitudes of Ca2+ transients were not affected by any of the applied drugs (Figure 2D–F,I ).
Figure 2.
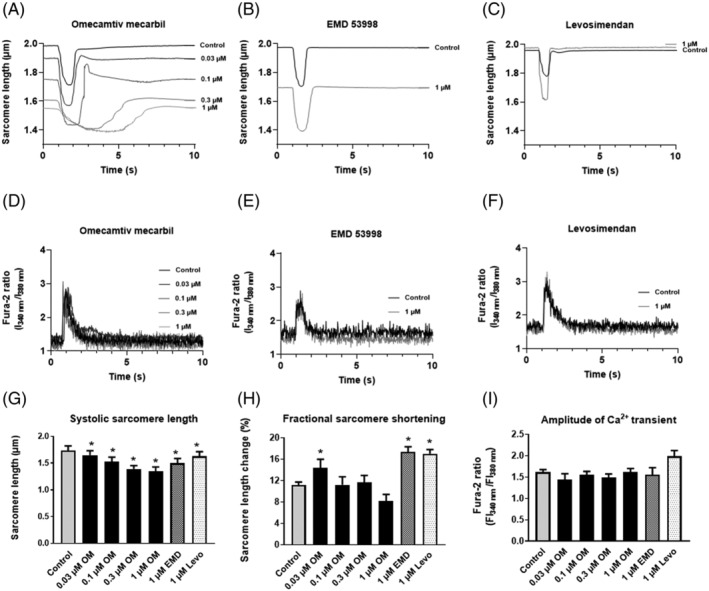
Effects of OM, EMD, and Levo on sarcomere length and intracellular Ca2+ transients during field‐stimulation. OM (A) was added to the tissue chamber in different concentrations (0.03, 0.1, 0.3, and 1 μM). Steady‐state conditions were reached within 5–8 min, and thereafter, contractile parameters were recorded. EMD (B) and Levo (C) were applied at a single concentration of 1 μM. Intracellular Ca2+ transients (D–F) were monitored before and after the treatments (at drug concentrations as indicated). Results of representative examples are shown in the presence of OM (A,D), EMD (B,E), or Levo (C,F). Changes in systolic peak SLs (G), fractional sarcomere shortenings (H), and amplitudes of Ca2+ transients (I) are given in bar graphs, where bars represent mean ± SEM. Significant differences are indicated by asterisks when P < 0.05 vs. before treatment. EMD, EMD 53998; FI, fluorescence intensity; Levo, levosimendan; OM, omecamtiv mecarbil.
A progressive prolongation of contraction time was observed upon increasing OM concentrations (1.50 ± 0.33 s, 2.31 ± 0.21 s, 3.85 ± 0.15 s, and 5.89 ± 0.57 s, at 0.03, 0.1, 0.3, and 1 μM OM concentrations, respectively, vs. 0.95 ± 0.05 s in drug‐free controls), whereas EMD and Levo did not affect this parameter (Figure 3A ). The prolongation of contractile responses could be attributed to slower kinetics of both contractions (from 0.65 ± 0.05 μm/s to 0.51 ± 0.13, 0.27 ± 0.10, 0.11 ± 0.02, and 0.05 ± 0.01 μm/s) and relaxations (from 0.95 ± 0.10 μm/s to 1.12 ± 0.30, 0.29 ± 0.06, 0.13 ± 0.03, and 0.05 ± 0.01 μm/s), respectively, with increasing OM concentrations (control, 0.03 μM, 0.1 μM, 0.3 μM, and 1 μM, respectively; Figure 3B,C ). In contrast to OM and EMD, Levo increased the kinetics of both contractions (Figure 3B ) and relaxations (Figure 3C ). The durations of the Ca2+ transients (Figure 3D ), kinetics of the upstrokes of intracellular Ca2+ transients (Figure 3E ), and the kinetics of the Ca2+ transient decays (illustrated by the rate constant, K) remained unaffected by all drug treatments (Figure 3F ).
Figure 3.
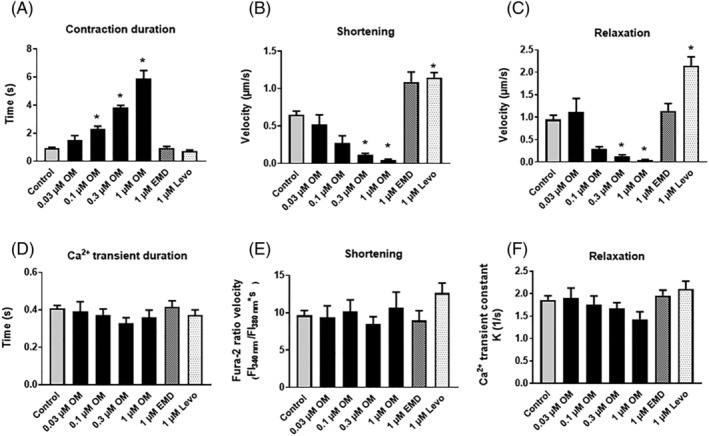
Effects of OM, EMD and Levo on the kinetics of cardiomyocyte contractions and relaxations. The durations of contractions (A) were progressively prolonged by increasing OM concentrations but remained unchanged during EMD or Levo administrations. The kinetics of contractions (B) were slowed down by higher OM concentrations (0.3 and 1 μM). Relaxation speeds of (E) were also greatly diminished by these high OM concentrations (C). Intracellular Ca2+ transient durations (D) were unaffected by all treatments. The kinetics of the upstrokes of intracellular Ca2+ transients (E) did not change during the treatments. Intracellular Ca2+ concentration transient decays (represented by the rate constant, K) (F) remained also unaltered during drug treatments. Significant differences are indicated by asterisks when P < 0.05 vs. before treatment. EMD, EMD‐53998; FI, fluorescence intensity; Levo, levosimendan; OM, omecamtiv mecarbil.
OM uniquely alters intracellular Ca2+–SL relationship
To characterize further the relationships between intracellular Ca2+ transients and contractile responses, sarcomere length was expressed as a function of intracellular Ca2+ concentration upon positive inotropic agent administrations (Figure 4 ). These Ca2+–SL relationships did not largely differ before and after 0.03 μM OM exposures (Figure 4A ). However, following 0.1, 0.3, or 1 μM OM administrations, loop diagrams markedly and progressively shifted downwards, suggestive for a Ca2+‐sensitizing effect that was present both at systolic and diastolic intracellular Ca2+ concentrations (Figure 4B–D ). The effect of 1 μM EMD on Ca2+–SL relationship was similar to that observed in the presence of 0.1 μM OM (Figure 4E ). Ca2+–SL relationship in the presence of Levo was enlarged towards shorter systolic SLs, consistent with a Ca2+‐sensitizing effect that developed during systoles but not during diastoles (Figure 4F ).
Figure 4.
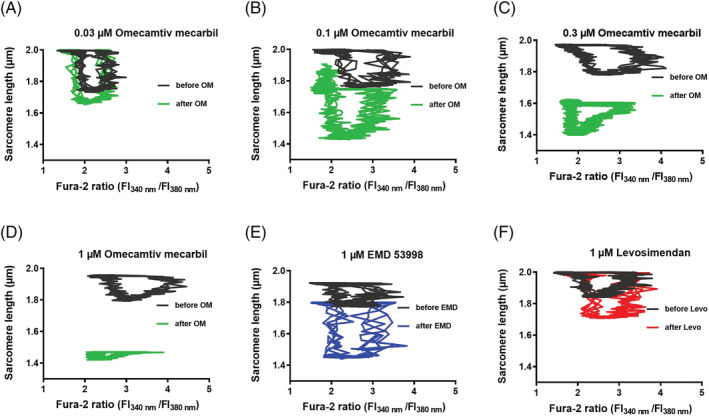
Intracellular Ca2+–sarcomere length relationships before and after OM, EMD, or Levo administrations. SL values were plotted as the function of the intracellular Ca2+ concentration during representative cardiac cycles. Responses of four consecutive cardiac cycles are shown for all panels. Traces before (grey) and after treatments (black) are shown. Upon electrical stimulation, a rapid increase in the intracellular Ca2+ concentration was seen, which was followed by contraction (evidenced by decrease in SL). Thereafter, intracellular Ca2+ returned to diastolic (low) levels, followed by relaxation (evidenced by increase in SL). Ca2+–SL relationships were progressively shifted downwards with increasing OM concentrations (A–D), suggestive for a Ca2+‐sensitizing effect during the entire cardiac cycle. The EMD‐evoked Ca2+‐sensitization was similar to that induced by 0.1 μM OM (E). Levo did not alter diastolic SL, but augmented SL decreases during systoles (F). EMD, EMD 53998; FI, fluorescence intensity; Levo, levosimendan; OM, omecamtiv mecarbil.
Interactions between diastolic and systolic sarcomere dynamics
To elucidate further the three different drug–target interactions, parameters of systolic responses upon OM, EMD, or Levo administrations were expressed as functions of their respective diastolic SLs. This approach verified hypothetical similarities between the OM and EMD dependent effects, in particular when interrelations between diastolic SLs and peak systolic SL (Figure 5A ) or contraction durations (Figure 5B ) were analysed. Nevertheless, diastolic SL–fractional sarcomere shortening relationships suggested distinctions between OM and EMD (Figure 5C ). Furthermore, Levo induced systolic parameters apparently did not require changes in diastolic SL, consistently with a Levo‐induced Ca2+‐sensitizer mechanism different from those evoked by OM or EMD (Figure 5A–C ).
Figure 5.
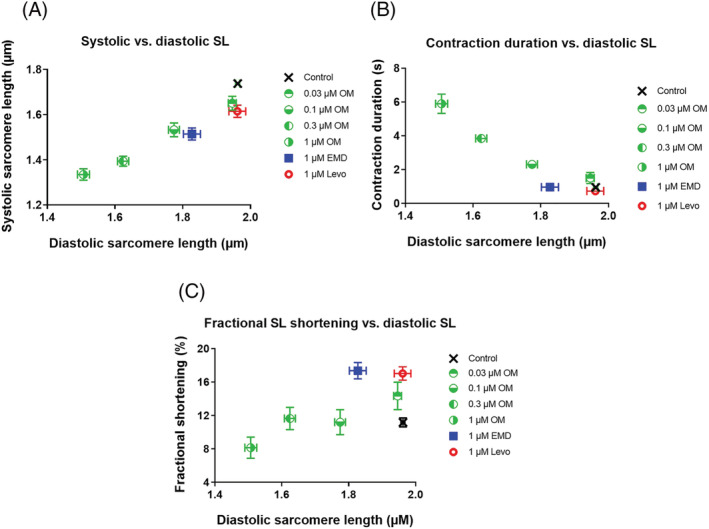
Hypothetical dependences of systolic cardiomyocyte parameters on diastolic sarcomere length. Both peak systolic and diastolic SLs decreased with increasing OM concentrations, and with EMD administration, but not after Levo application (A). Contraction durations increased with increasing OM concentrations, and with EMD administration, but not after Levo application (B). Fractional sarcomere shortening did not increase when diastolic SL decreased following OM administrations; however, it increased by EMD or Levo (C).
Discussion
In this study, we identified similarities and major differences among the contractile effects of OM, EMD, and Levo on systolic and diastolic indices of cardiomyocyte contractions of enzymatically isolated intact canine LV cardiomyocytes. Our data are consistent with an OM‐evoked Ca2+‐sensitizing effect, whereby its positive inotropic effect develops primarily by prolongation of systolic contractions rather than by major changes in fractional sarcomere shortening or augmentation of the kinetics of cardiac contractions or relaxations.
Here, we report that OM, EMD, and Levo evoke roboust changes in cardiomyocyte contractions and relaxations in the absence of significant changes in intracellular Ca2+ transients, suggestive for Ca2+ sensitization for all three agents. 2 Nevertheless, the characteristics of these Ca2+‐sensitizing mechanisms were different for OM, EMD, and Levo and hence resulted in different kinds of contractile responses. Although both Levo and EMD have inhibitory effect on PDE III, the unchanged intracellular Ca2+ levels were somewhat surprising in the presence of these agents and can be potentially explained by PDE isoforms (other than PDE III) not inhibited by these agents in canine cardiomyocytes at the employed drug concentrations. 7 , 10 , 15
OM evoked a reduction in diastolic SL of unstimulated cardiomyocytes of dog hearts at low intracellular Ca2+ concentrations. The magnitude of this decrease in SL was comparable with that observed in stimulated cardiomyocytes during diastoles, suggesting that the OM‐dependent activation of the actin–myosin interaction did not require Ca2+. These findings are also in accord with the OM‐stimulated increase of basal myosin ATPase activity of rabbit hearts 1 and isometric force production in permeabilized cardiomyocytes of rat hearts at diastolic Ca2+ levels. 15 Similarly to OM, EMD but not Levo decreased resting SL, revealing similarities for the two myosin‐binding agents and a distinct effect for the thin filament selective Levo in their Ca2+‐sensitizing effects. Here, we also show that the reductions in diastolic and systolic SLs and contraction durations are tightly coupled to the applied OM concentrations and thus illuminate different facets of the same drug‐target interaction on the myosin molecule. The reduction in diastolic SL was probably also responsible for the unchanged fractional sarcomere shortenings in the presence of high OM concentrations despite the observed reductions in peak systolic SLs. Interestingly, in an independent study, fractional cell shortening did increase by OM in rat cardiomyocytes 1 ; nevertheless, based on our results, we propose that the increase in fractional cell shortening can be limited by the OM‐evoked decrease in resting SL at higher OM concentrations. 16
The range of OM concentrations (i.e. between 0.01 and 1 μM), where reductions in resting SL were observed overlapped with that reported during its clinical administrations. 3 , 4 , 6 In the most recent clinical trial of OM (GALACTIC‐HF), a guided dose titration strategy was applied to achieve plasma concentrations of at least 200 ng/mL (0.5 μM) while avoiding concentrations >1.000 ng/mL (2.5 μM). It is important to note that OM concentrations >1200 ng/ml (3 μM) (three times higher than the maximum concentration used in our in vitro experiments) were previously reported to lead to excessive prolongation of the systole, thus limiting coronary blood flow during diastole and possibly leading to myocardial ischemia. 17 , 18 In animals and humans, the pharmacodynamic signature of OM is an increase in the systolic ejection time (SET). 3 , 4 , 6 , 19 This observation is the reflection of the drug's most significant mechanism of action, as contractile activity can be maintained by OM even when cytoplasmic Ca2+ concentration falls. Accordingly, in the first‐in‐man, dose‐escalating study, OM augmented left ventricular systolic function and induced a dose‐dependent increase in SET, stroke volume (SV), fractional shortening (FS) measured by echocardiography, and left ventricular ejection fraction (LVEF). Despite the slower kinetics of force generation in vitro, the maximal rate of LV pressure development (dP/dtmax) was shown to be unaffected by OM in vivo, 19 suggestive for distinct pharmacokinetic properties for OM in vitro and in vivo.
The durations of cardiomyocyte contractions increased almost six times at high OM concentrations, implicating a prolonged activation for the contractile protein machinery due to a delay of the inactivation of the thin filaments and increased number of strongly attached cross‐bridges. 20 , 21 The increase in the half‐time of activation (t 1/2 of activation) and the decrease in the rate constant of force redevelopment (k tr , illustrating the intrinsic kinetics of the actin–myosin cross‐bridges) observed in our previous study 16 in permeabilized rat cardiomyocytes are also in line with our present observations and corroborate the decreased in vitro motilities of the myosin filaments. 22 , 23 Taken together, OM‐evoked Ca2+ sensitization may contribute to stronger, slower and prolonged cardiac contractions consistently with previous echocardiographic findings.
Of note, the OM‐evoked increase in contraction durations was not observed during EMD administrations, and this might be a consequence of their distinct binding sites on the myosin molecule. The binding site of OM on the myosin S1 domain probably resides in a cleft in the vicinity of its actin‐binding interface and of the nucleotide binding pocket. This location is supposedly ideal for allosteric modulation of both the enzymatic and mechanical properties of the cardiac myosin motor. 1 The EMD binding site is not identical with that of OM on the myosin S1 domain; nevertheless, EMD administration can also increase basal myosin ATPase activity. 9 , 24 , 25 , 26
In the present study, relaxation of intact cardiac cells was significantly attenuated, particularly at high (0.3 and 1 μM) OM concentrations, but was less affected upon EMD or Levo treatments. This is compatible with the finding of our previous study in which OM also substantially prolonged the relaxation and increased the passive stiffness of permeabilized rat myocyte‐sized preparations with a Ca2+‐independent mechanism. 16 In heart failure, abnormalities in cardiomyocyte Ca2+ cycling are mainly due to altered sarcoplasmic reticulum (SR) functions: reduced and partial cytoplasmic Ca2+ reuptake in association with abnormal SR Ca2+‐ATPase (SERCA) activity. 27 Here, we found that cardiomyocyte relaxation was impaired even at low OM concentrations (0.1–0.3 μM), whereas at high OM concentration (1 μM) major limitations in cardiomyocyte relaxation developed. Accordingly, OM‐induced diastolic dysfunction can be potentially aggravated by higher diastolic Ca2+ levels in the failing heart. An impaired diastolic performance—reflected by worsened time constant of isovolumic relaxation (τ) and the rate of the LV pressure decrease (dP/dt min )—was shown earlier after i.v. OM administration in rats with volume overload HF during OM treatments. 28 Nevertheless, OM did not impair diastolic function in healthy volunteers at plasma concentrations similar to those applied in this latter study. 17 , 18 The possibility that OM may lead to diastolic dysfunction was addressed based on the data collected in the Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure (COSMIC‐HF) trial. The post hoc analysis showed an increase in isovolumic relaxation time (IVRT) without changes in E/A ratio or E wave. Unfortunately, diastolic function was not assessed in detail in GALACTIC‐HF, which would allow further characterization of LV diastolic dysfunction upon oral administration of OM in patients with HFrEF.
Despite having no effects on the intracellular Ca2+ transients, OM, EMD and Levo differently alter Ca2+–SL relationships of cardiomyocytes. OM at high concentrations has the most prominent effect on resting SL, leading to diastolic cardiomyocyte shortening and prolonged relaxation. EMD also results in a decrease in the resting SL, but without affecting relaxation kinetics. In contrast to OM and EMD, none of these adverse effects were seen during Levo administration. We propose that the widely used clinical parameters of systolic function such as fractional shortening or ejection fraction can be misleading in case of myosin activators. Here, we showed a dramatic decrease in resting SL (which could be paralleled by the reduced left ventricular diastolic diameter in clinical studies) upon OM administrations. This factor apparently limited fractional shortening at high OM concentrations and might affect ejection fraction calculations as well. The distinct mechanism of OM action on cardiomyocyte resting SL and relaxation kinetics should raise concerns for the clinical administration of the drug and draw our attention to the importance of regular determination of serum OM levels.
Limitations
The experiments were performed in vitro in enzymatically isolated canine cardiomyocytes at room temperature and low pacing rate, and hence, caution is certainly needed when extrapolating experimental data to human hearts. As kinetics of biological processes slow down with decreasing temperatures, we opted for low‐frequency stimulation when performing experiments under steady‐state conditions. Similarly to our current findings, in one of our previous studies, 29 impaired relaxations and decreases in cardiomyocyte cell lengths were also observed at 0.5, 1, and 2 Hz pacing frequencies and 1 or 2 μM OM concentrations at 37°C. In contrast to structurally intact cardiomyocytes in vitro, potentially lower extents of myosin activations can be reached at the applied OM concentrations in vivo. It needs also to be noted that all of our experimental data were obtained in healthy animals. Nevertheless, canine cardiomyocytes are considered as relevant models for human cardiac cellular electrophysiology.
Conclusions
OM acts on cardiac muscle at resting Ca2+ concentrations, resulting in diastolic cardiomyocyte shortening; besides, it attenuates both contraction and relaxation kinetics. Our data implicate a narrow therapeutic window for OM and a risk for diastolic dysfunction at high OM concentrations.
Conflict of interest
None declared.
Funding
This work was supported by the GINOP‐2.3.2‐15‐2016‐00043 project and by the ÚNKP‐21‐3 new national excellence program of the Ministry for Innovation and Technology from the source of the national research, development, and innovation fund. The project was co‐financed by the European Union and the European Regional Development Fund. This work was also supported by project nos. TKP2020‐IKA‐04 and TKP2020‐NKA‐04 from the National Research, Development and Innovation Fund of Hungary, under the 2020‐4.1.1‐TKP2020 funding scheme. Project no. K 132623 (to AT) has been implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the K_19 funding scheme. Project no. NKFIH‐K138090 (to PPN), NKFIH‐K142764 (to NSz), and NKFIH‐FK128116 (to BH) was financed by the National Research, Development and Innovation Fund of Hungary. The research group (AT, ZP) is supported by the Hungarian Academy of Sciences.
Ráduly, A. P. , Tóth, A. , Sárkány, F. , Horváth, B. , Szentandrássy, N. , Nánási, P. P. , Csanádi, Z. , Édes, I. , Papp, Z. , and Borbély, A. (2023) Omecamtiv mecarbil augments cardiomyocyte contractile activity both at resting and systolic Ca2+ levels. ESC Heart Failure, 10: 1326–1335. 10.1002/ehf2.14300.
Contributor Information
Zoltán Papp, Email: pappz@med.unideb.hu.
Attila Borbély, Email: borbelya@med.unideb.hu.
References
- 1. Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, Anderson RL, Sueoka SH, Lee KH, Finer JT, Sakowicz R, Baliga R, Cox DR, Garard M, Godinez G, Kawas R, Zraynack E, Lenzi D, Lu P, Muci A, Niu C, Qian X, Pierce DW, Pokrovskii M, Suehiro I, Sylvester S, Tochimoto T, Valdez C, Wang W, Katori T, Kass DA, Shen Y, Vatner SF, Morgans DJ. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science (New York, NY). 2011; 331: 1439–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Endoh M. Cardiac Ca2+ signaling and Ca2+ sensitizers. Circ J. 2008; 72: 1915–1925. [DOI] [PubMed] [Google Scholar]
- 3. Teerlink JR, Felker GM, McMurray JJV, Ponikowski P, Metra M, Filippatos GS, Ezekowitz JA, Dickstein K, Cleland JGF, Kim JB, Lei L, Knusel B, Wolff AA, Malik FI, Wasserman SM. Acute treatment with omecamtiv mecarbil to increase contractility in acute heart failure: the ATOMIC‐AHF study. J Am Coll Cardiol. 2016; 67: 1444–1455. [DOI] [PubMed] [Google Scholar]
- 4. Teerlink JR, Felker GM, McMurray JJV, Solomon SD, Adams KFJ, Cleland JGF, Ezekowitz JA, Goudev A, Macdonald P, Metra M, Mitrovic V, Ponikowski P, Serpytis P, Spinar J, Tomcsányi J, Vandekerckhove HJ, Voors AA, Monsalvo ML, Johnston J, Malik FI, Honarpour N. Chronic oral study of myosin activation to increase contractility in heart failure (COSMIC‐HF): a phase 2, pharmacokinetic, randomised, placebo‐controlled trial. Lancet. 2016; 388: 2895–2903. [DOI] [PubMed] [Google Scholar]
- 5. Teerlink JR, Diaz R, Felker GM, McMurray JJV, Metra M, Solomon SD, Legg JC, Büchele G, Varin C, Kurtz CE, Malik FI, Honarpour N. Omecamtiv mecarbil in chronic heart failure with reduced ejection fraction: rationale and design of GALACTIC‐HF. JACC Heart Fail. 2020; 8: 329–340. [DOI] [PubMed] [Google Scholar]
- 6. Teerlink JR, Diaz R, Felker GM, McMurray JJV, Metra M, Solomon SD, Adams KF, Anand I, Arias‐Mendoza A, Biering‐Sørensen T, Böhm M, Bonderman D, Cleland JGF, Corbalan R, Crespo‐Leiro MG, Dahlström U, Echeverria LE, Fang JC, Filippatos G, Fonseca C, Goncalvesova E, Goudev AR, Howlett JG, Lanfear DE, Li J, Lund M, Macdonald P, Mareev V, Momomura SI, O'Meara E, Parkhomenko A, Ponikowski P, Ramires FJA, Serpytis P, Sliwa K, Spinar J, Suter TM, Tomcsanyi J, Vandekerckhove H, Vinereanu D, Voors AA, Yilmaz MB, Zannad F, Sharpsten L, Legg JC, Varin C, Honarpour N, Abbasi SA, Malik FI, Kurtz CE. Cardiac myosin activation with omecamtiv mecarbil in systolic heart failure. N Engl J Med. 2021; 384: 105–116. [DOI] [PubMed] [Google Scholar]
- 7. Beier N, Harting J, Jonas R, Klockow M, Lues I, Haeusler G. The novel cardiotonic agent EMD 53 998 is a potent “calcium sensitizer”. J Cardiovasc Pharmacol. 1991; 18: 17–27. [DOI] [PubMed] [Google Scholar]
- 8. Dobrunz LE, Backx PH, Yue DT. Steady‐state [Ca2+]i‐force relationship in intact twitching cardiac muscle: direct evidence for modulation by isoproterenol and EMD 53998. Biophys J. 1995; 69: 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Radke MB, Taft MH, Stapel B, Hilfiker‐Kleiner D, Preller M, Manstein DJ. Small molecule‐mediated refolding and activation of myosin motor function. Elife. 2014; 3: e01603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Szilágyi S, Pollesello P, Levijoki J, Kaheinen P, Haikala H, Edes I, Papp Z. The effects of levosimendan and OR‐1896 on isolated hearts, myocyte‐sized preparations and phosphodiesterase enzymes of the guinea pig. Eur J Pharmacol. 2004; 486: 67–74. [DOI] [PubMed] [Google Scholar]
- 11. Papp Z, Édes I, Fruhwald S, De Hert SG, Salmenperä M, Leppikangas H, Mebazaa A, Landoni G, Grossini E, Caimmi P, Morelli A, Guarracino F, Schwinger RHG, Meyer S, Algotsson L, Wikström BG, Jörgensen K, Filippatos G, Parissis JT, González MJG, Parkhomenko A, Yilmaz MB, Kivikko M, Pollesello P, Follath F. Levosimendan: molecular mechanisms and clinical implications: consensus of experts on the mechanisms of action of levosimendan. Int J Cardiol. 2012; 159: 82–87. [DOI] [PubMed] [Google Scholar]
- 12. Horváth B, Váczi K, Hegyi B, Gönczi M, Dienes B, Kistamás K, Bányász T, Magyar J, Baczkó I, Varró A, Seprényi G, Csernoch L, Nánási PP, Szentandrássy N. Sarcolemmal Ca(2+)‐entry through L‐type Ca(2+) channels controls the profile of Ca(2+)‐activated Cl(−) current in canine ventricular myocytes. J Mol Cell Cardiol. 2016; 97: 125–139. [DOI] [PubMed] [Google Scholar]
- 13. Hegyi B, Horváth B, Váczi K, Gönczi M, Kistamás K, Ruzsnavszky F, Veress R, Chen‐Izu Y, Bányász T, Magyar J, Csernoch L, Nánási PP, Szentandrássy N. Ca(2+)‐activated Cl(−) current is antiarrhythmic by reducing both spatial and temporal heterogeneity of cardiac repolarization. J Mol Cell Cardiol. 2017; 109: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andrei SR, Ghosh M, Sinharoy P, Dey S, Bratz IN, Damron DS. TRPA1 ion channel stimulation enhances cardiomyocyte contractile function via a CaMKII‐dependent pathway. Channels. 2017; 11: 587–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Szilágyi S, Pollesello P, Levijoki J, Haikala H, Bak I, Tósaki A, Borbély A, Edes I, Papp Z. Two inotropes with different mechanisms of action: contractile, PDE‐inhibitory and direct myofibrillar effects of levosimendan and enoximone. J Cardiovasc Pharmacol. 2005; 46: 369–376. [DOI] [PubMed] [Google Scholar]
- 16. Nagy L, Kovács Á, Bódi B, Pásztor ET, Fülöp GÁ, Tóth A, Édes I, Papp Z. The novel cardiac myosin activator omecamtiv mecarbil increases the calcium sensitivity of force production in isolated cardiomyocytes and skeletal muscle fibres of the rat. Br J Pharmacol. 2015; 172: 4506–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cleland JGF, Teerlink JR, Senior R, Nifontov EM, Mc Murray JJV, Lang CC, Tsyrlin VA, Greenberg BH, Mayet J, Francis DP, Shaburishvili T, Monaghan M, Saltzberg M, Neyses L, Wasserman SM, Lee JH, Saikall KG, Clarke CP, Goldman JH, Wolff AA, Malik FI. The effects of the cardiac myosin activator, omecamtiv mecarbil, on cardiac function in systolic heart failure: a double‐blind, placebo‐controlled, crossover, dose‐ranging phase 2 trial. Lancet. 2011; 378: 676–683. [DOI] [PubMed] [Google Scholar]
- 18. Teerlink JR, Clarke CP, Saikali KG, Lee JH, Chen MM, Escandon RD, Elliott L, Bee R, Habibzadeh MR, Goldman JH, Schiller NB, Malik FI, Wolff AA. Dose‐dependent augmentation of cardiac systolic function with the selective cardiac myosin activator, omecamtiv mecarbil: a first‐in‐man study. Lancet. 2011; 378: 667–675. [DOI] [PubMed] [Google Scholar]
- 19. Shen Y‐T, Malik FI, Zhao X, Depre C, Dhar SK, Abarzúa P, Morganss DJ, Vatner SF. Improvement of cardiac function by a cardiac myosin activator in conscious dogs with systolic heart failure. Circ Heart Fail. 2010; 3: 522–527. [DOI] [PubMed] [Google Scholar]
- 20. Poggesi C, Tesi C, Stehle R. Sarcomeric determinants of striated muscle relaxation kinetics. Pflugers Archiv: Eur J Appl Physiol. 2005; 449: 505–517. [DOI] [PubMed] [Google Scholar]
- 21. Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000; 80: 853–924. [DOI] [PubMed] [Google Scholar]
- 22. Liu Y, White HD, Belknap B, Winkelmann DA, Forgacs E. Omecamtiv Mecarbil modulates the kinetic and motile properties of porcine β‐cardiac myosin. Biochemistry. 2015; 54: 1963–1975. [DOI] [PubMed] [Google Scholar]
- 23. Wang Y, Ajtai K, Burghardt TP. Analytical comparison of natural and pharmaceutical ventricular myosin activators. Biochemistry. 2014; 53: 5298–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ferroni C, Hano O, Ventura C, Lakatta EG, Klockow M, Spurgeon H, Capogrossi MC. A novel positive inotropic substance enhances contractility without increasing the Ca2+ transient in rat myocardium. J Mol Cell Cardiol. 1991; 23: 325–331. [DOI] [PubMed] [Google Scholar]
- 25. Solaro RJ, Gambassi G, Warshaw DM, Keller MR, Spurgeon HA, Beier N, Lakatta EG. Stereoselective actions of thiadiazinones on canine cardiac myocytes and myofilaments. Circ Res. 1993; 73: 981–990. [DOI] [PubMed] [Google Scholar]
- 26. Gambassi G, Capogrossi MC, Klockow M, Lakatta EG. Enantiomeric dissection of the effects of the inotropic agent, EMD 53998, in single cardiac myocytes. Am J Physiol. 1993; 264: H728–H738. [DOI] [PubMed] [Google Scholar]
- 27. Denniss AL, Dashwood AM, Molenaar P, Beard NA. Sarcoplasmic reticulum calcium mishandling: central tenet in heart failure? Biophys Rev. 2020; 12: 865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilson K, Guggilam A, West TA, Zhang X, Trask AJ, Cismowski MJ, de Tombe P, Sadayappan S, Lucchesi PA. Effects of a myofilament calcium sensitizer on left ventricular systolic and diastolic function in rats with volume overload heart failure. Am J Physiol Heart Circ Physiol. 2014; 307: H1605–H1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Horváth B, Szentandrássy N, Veress R, Almássy J, Magyar J, Bányász T, Tóth A, Papp Z, Nánási PP. Frequency‐dependent effects of omecamtiv mecarbil on cell shortening of isolated canine ventricular cardiomyocytes. Naunyn‐Schmiedeb Arch Pharmacol. 2017; 390: 1239–1246. [DOI] [PubMed] [Google Scholar]