

Abstract
The COVID-19 pandemic has highlighted the need for human respiratory tract-based assay platforms for efficient discovery and development of antivirals and disease-modulating therapeutics. Physiologically relevant tissue models of the lower respiratory tract, including the respiratory bronchioles and the alveolar sacs, are of high interest because they are the primary site of severe SARS-CoV-2 infection and are most affected during the terminal stage of COVID-19. Current epithelial lung models used to study respiratory viral infections include lung epithelial cells at the air-liquid interface with fibroblasts and endothelial cells, but such models do not have a perfusable microvascular network to investigate both viral infectivity and viral infection-induced thrombotic events. Using a high throughput, 64-chip microfluidic plate-based platform, we have developed two novel vascularized, lower respiratory tract multi-chip models for the alveoli and the small airway. Both models include a perfusable microvascular network consisting of human primary microvascular endothelial cells, fibroblasts and pericytes. The established biofabrication protocols also enable the formation of differentiated lung epithelial layers at the air-liquid interface on top of the vascularized tissue bed. We validated the physiologically relevant cellular composition, architecture and perfusion of the vascularized lung tissue models using fluorescence microscopy, flow cytometry, and electrical resistance measurements. These vascularized, perfusable microfluidic lung tissue on high throughput assay platforms will enable the development of respiratory viral infection and disease models for research investigation and drug discovery.
1. Introduction
The lower respiratory tract (LRT) is the primary site for many respiratory diseases, ranging from chronic obstructive pulmonary disease (COPD) to restrictive diseases such as idiopathic pulmonary fibrosis (IPF), in addition to severe respiratory diseases caused by bacterial or viral infections, including SARS-CoV-2, the causative agent of COVID-19. Regardless of the respiratory disease, the microvascular network in the pulmonary interstitium plays a crucial role in common pathologies associated with pulmonary disease, such as fibrosis and edema [1–4]. In many cases, aggravation of clinical symptoms arise due to unique role of the microvascular network in cell-cell crosstalk [5, 6]. For instance, pulmonary thrombosis appears to be common in COVID-19, and there is now emerging evidence that SARS-CoV-2 infection of the microvasculature is critical in prolonging the course and the severity of COVID-19 [7, 8]. The need for a rapid therapeutic response to the COVID-19 pandemic has highlighted the lack of clinically relevant in vitro lung tissue models that faithfully mimic the pathomechanism of respiratory diseases while enabling high-throughput screening (HTS) of existing and novel therapeutics for disease-corrective pharmacological interventions. In the case of SARS-COV-2, currently used cellular models for HTS rely on monolayer cultures such as Vero [9, 10], Huh7 [11], A549-ACE2, 293T-ACE2/TMPRSS2 [12–15], and Calu3 [16, 17]. However, these monoculture systems are not predictive of drug activity in animal models nor humans, and some cell lines such as Vero are not human respiratory epithelial cells at all [18].
Several epithelial lung models have already been established with various degrees of physiological complexity. Generally, such models are comprised of lung epithelial cells at an air-liquid interface (ALI) to induce differentiation of the basal cells to distinct lung epithelial cell types, including those that express specific membranal receptors that make them permissive to viral infection. In some engineered lung tissue models, a monolayer culture of lung epithelial cells in the ALI is co-cultured with endothelial and fibroblast cells to recapitulate the respiratory tissue more accurately. These can either be in transwell plates or microfluidic devices that incorporate fluid and air flow; some also include mechanical stretching to mimic the lung strain during breathing [19–21]. For example, the EpiAirway [22, 23] and EpiAlveolar [24] tissues are human lung tissue equivalents in a transwell plate which are (1) complex, (2) permissive of viral infections, (3) produce disease-relevant cytokines upon infection and (4) are effective drug screening assay platforms for antivirals [25].
While transwell plates are compatible with HTS, they are static cultures and do not include mechanical stretching and fluid flow, which are quite relevant to the pulmonary pathophysiology (e.g., V/Q mismatch, physiological dead space). Several microfluidic lung-on-a-chip (LOC) models have been developed to include an ALI-induced layer of differentiated lung epithelial cells, with accompanying fibroblast and endothelial cells to create a barrier-like cellular arrangement while creating aqueous flow over the cell surfaces. In most reported LOC models, small airway epithelial cells (SAEC) are typically seeded on the top and pulmonary endothelial cells underneath a porous polyester (PET) membrane to mimic the bronchiolar-capillary junction of the lung [19, 20, 26–30]. These LOC models are permissive to both influenza and SARS-CoV-2 infections, and some include a vacuum-driven, mechanical contraction to mimic the bronchioles’ tubular, mechanical stretching during inspiration/expiration. These models have also been used to model edema and asthma pathologies [20]. Although highly relevant to in vivo physiology, the described LOC have two major drawbacks when it comes to HTS application; first, are very low-throughput (e.g., singular chip) and second, are made out of polydimethylsiloxane (PDMS) [31]. PDMS absorbs highly hydrophobic compounds, which can distort any pharmacokinetics studies conducted during early drug development process. To specifically address the HTS need, Draper Laboratory, Inc. [32, 33] recently developed a 96-well microfluidic platform for the lung. Additionally, Alveolix developed a second-generation LOC that utilizes a stretchable membrane to mimic breathing on a multi-chip platform that is amenable to HTS [21].
While the aforementioned lung tissue models include physiological features that are important in homeostatic lung function, they primarily rely on a barrier model, with a monolayer of endothelial cells underneath the epithelial layer, which does not necessarily recapitulate the complex, three-dimensional (3D) network of capillaries with clear input and output parent vessels. Microvascular networks have organ-specific footprints and are heterogenous among different organs and tissues. In the lung, microvascular endothelial cells behave based on the microenvironment created by organ-specific tissues ranging from sheer stress, pH, and oxygen saturation [3, 4, 34–37]. Hence, microvasculature constitutes an indispensable role in shaping, directing, and forming the organs and their functions [38]. Because there are important morphological, physiological, and phenotypic differences between endothelial cells in monolayers versus capillaries, these distinction are critical when developing clinically relevant tissue models. Therefore, there is a critical need to develop lung tissue models that incorporate a functional, microvascular network on an HTS-compatible assay platform that allow researchers to recreate pulmonary pathophysiology and identify novel therapeutics in a more clinically predictive manner.
Here, we describe such vascularized alveolar (VAMC) and bronchiolar multi-chip (VBMC) models that are HTS-applicable. By successfully reconfiguring a commercially available microfluidic plate (figure 1b), we were able to incorporate both a complex 3D microvascular network and ALI-induced respiratory epithelium (figure 1c). To create a dynamically perfusable microvasculature underneath an ALI-induced epithelial layer, pulmonary microvascular endothelial cells, pericytes, and fibroblasts were co-cultured along with differentiated alveolar and small airway epithelial cells (figure 1d & 1e). Through the establishment of multiple orthogonal parameters to characterize the two models, we have two vascularized, perfusable respiratory multi-chip models that are amenable to multiple high-throughput instruments for screening and disease modeling purposes.
Figure 1.
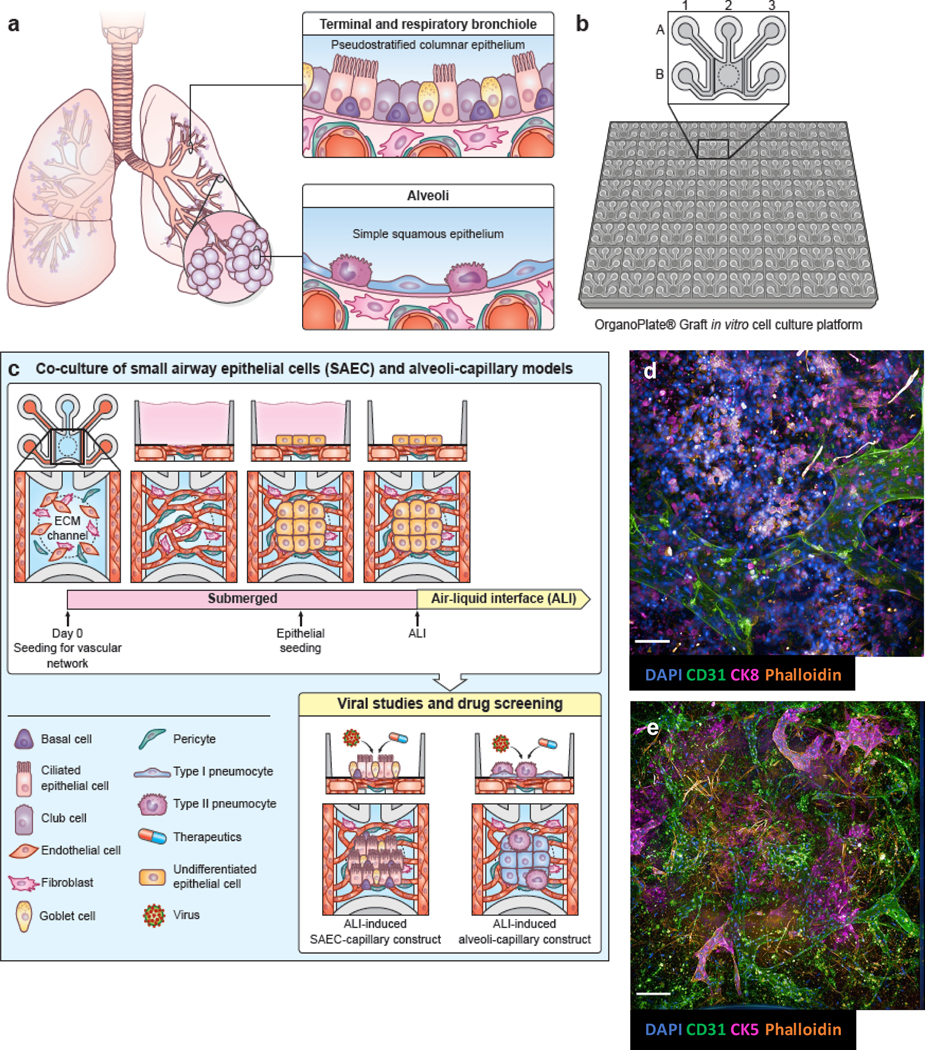
(a) anatomy of small airway and alveoli in the human lower respiratory tract. (b) schematic of 384-well, 64-chip based Mimetas Organograft® (c) timeline of co-culture of vascular cells, composed of human pulmonary microvascular endothelial cells, pericytes and fibroblasts; and epithelial cells. Epithelial cells are placed under air-liquid interface (ALI) which allows for maturation into distinct sub-population. Figure legend included. (d) maximum projection of alveolar-capillary tissue construct. (e) maximum projection of small airway-capillary tissue construct. (a), (b), and (c) by NIH Medical Arts. Scale bar = 100 μm.
2. Materials & Methods
2.1. Mimetas Organograft® Plate
The Organograft® plate was purchased from Mimetas US (Mimetas, 6401–400-B). It is a 64-chip based microfluidic platform that has been designed to fit within a traditional 384-well plate (figure 1b). It was originally intended to vascularize tumor spheroids [39]; instead, we took advantage of the open access (a diameter of 1 mm) to establish a vascular network underneath in the gel chamber while allowing the respiratory epithelial cells to seed along the open access and crosstalk with the vascular cells in the gel without any impeding membrane.
2.2. Pulmonary tissue construct
Primary human pulmonary microvascular endothelial cells.
Cells (HPMEC) were obtained from Promocell (C-12281). They were cultured on coated flasks (Angioproteomie, cAP-01) before being plated for co-culture on the microfluidic platform. Prior to experiment, cells were given fresh media (Lifeline Technology, LM-0002) every other day. The cells were dissociated from the sub-culture flasks using 0.05% trypsin (ThermoFisher Scientific, 25300).
Primary human pulmonary fibroblasts.
Fibroblasts were obtained from Lifeline Technology (FC-0049). They were cultured on coated flasks (Angioproteomie, cAP-01) before being plated for co-culture on the microfluidic platform. Prior to experiment, cells were given fresh media (Lifeline Technology, LL-0001) every other day. The cells were dissociated from the sub-culture flasks using 0.05% trypsin.
Primary human placental microvascular pericytes.
Pericytes were obtained from Angioproteomie (cAP-0029). They were cultured on coated flasks (Angioproteomie, cAP-01) before being plated for co-culture on the microfluidic platform. Prior to experiment, cells were given fresh media (Angioproteomie, cAP-09) every other day. The cells were dissociated from sub-culture flasks using 0.05% trypsin.
Primary human pulmonary alveolar epithelial cells.
Alveolar epithelial cells were obtained from Cell Biologics (H-6053). The cells were cultured on coated flasks (0.1% gelatin coating solution, AATC® PCS-999–027). The cells were given fresh media (Cell Biologics, H6621) that had final concentration of 2.5 ng/ml EGF every other day. Cells were passaged using 0.25% trypsin (ThermoFisher Scientific, 25200056). To plate on the open access of the microfluidic platform, the open access well was coated for 2 min with gelatin coating solution, and then aspirated before the alveolar cells were pipetted into the well.
Primary human pulmonary small airway epithelial cells.
SAEC were obtained from Lifeline Technology (FC-0016). The epithelial cells were cultured on flasks coated with 50 ug/ml fibronectin (ThermoFisher Scientific, 33016015) dissolved in UltraPure™ DNase/RNase-Free Distilled Water (ThermoFisher Scientific, 10–977-015) for at least 1h RT or overnight. The cells were passaged once using 0.25% trypsin (Gibco, 15050065). Unlike alveolar epithelial cells, the SAEC were directly maturated on the microfluidic platform along with the microvascular network prior to ALI induction.
2.3. Gel loading, cell seeding for 3D, microfluidic respiratory model
The Organograft® plate was used to co-culture the cells for small airway- and alveoli-capillary models (figure 1b). For gel conditions that either contained cells for microvasculature or did not contain any cells, the gel was seeded first prior to cell seeding in the side lanes. For gel seeding, 3 ml of 5 mg/ml fibrinogen (Sigma, F3879–1G) was prepared by diluting 15 mg/ml stock fibrinogen with DPBS (ThermoFisher Scientific, 14190136). 100 ul of 10U thrombin was prepared in DPBS from 100U thrombin stock solution (Sigma, T6884–1KU). 3 ul of fibrin solution was injected immediately onto the inlet of the gel chamber after the addition of the thrombin into fibrinogen solution. The liquified gel was observed by naked eye to confirm the end-to-end filling of the gel chamber. The final working concentration of thrombin was 1U/ml and 4.5 mg/ml for fibrinogen when the mixed solution was injected into the inlet of the gel chamber.
For the gel condition that contained vascular cells (HPMEC, fibroblasts, and pericyte), the cells were counted by Cell Countess II (ThermoFisher Scientific, A27977), then resuspended in media solution before being centrifuged at 500 xg for 5 minutes. After supernatant removal, the fibrinogen solution was added at working volume. From this fibrinogen/cell solution, smaller working solution was taken and mixed with thrombin to inject into the inlet of gel chamber to create suspended cells in the gel for microvascular network formation. The gel was left in 37°C for 30 minutes to allow fibrin polymerization.
For the side lanes of the microfluidic chip, HPMEC were counted by Cell Countess II, then resuspended in media solution before being centrifuged at 500 xg for 5 minutes. 1 ul of HPMEC were injected into the inlets of the side lanes and were left in 37°C for 15 minutes. Once the side lanes were filled with the cells, 50 ul of media was pipetted into the wells. In the VAMC model, the media for the side lanes contained 2.5% FBS, 42.5 ng/ml recombinant human vascular endothelial growth factor (rhVEGF, R&D Systems, 298-VE), 25 ng/ml recombinant human angiopoietin-1 (rhANG1, R&D Systems, 923-AN) and the middle gel chambers and corresponding inlet wells were provided media with 2.5% FBS, 85 ng/ml rhVEGF, 100 ng/ml rhANG1. Both media contained 0.075 U/ml aprotinin (Sigma, A4529–100MG) to prevent fibrin gel degradation. In the VBMC model, the media contained 1:1 mixture of 5% FBS endothelial media and the SAEC media, and rhVEGF, rhANG1 and aprotinin concentration was similar to that of the alveolar construct except for the side lanes which contained 100 ng/ml rhANG1.
For the VAMC model, the microvascular network was allowed to develop for a week prior to placing the alveolar monolayer on top. For the VBMC model, the microvascular network was also cultured for 3 days to allow for vascular development prior to epithelial seeding. The plates are placed in 37°C for at least 1h, before being moved onto the perfusion rocker (Mimetas, MI-OFPR-1) with angle set at 12°, rocking every 8 minutes.
For flow resistance of vascular network, all chips within each plate are placed under the same sheer stress caused by the parameters of the perfusion rocker. Sheer stress was known for each chip, as it was part of the product development by the manufacturer. At the settings at which the perfusion rocker was set (12° every 8 min), peak flow rate is calculated to be 1.5 dyn/cm2. If the gel chamber contained zero or non-perfusable vessels, the flow resistance across the cross-sectional area of the microvasculature is effectively zero. However, in the vascularized alveolar and bronchiolar multi-chip models, there was evidence of perfusable vascular bed, as identified by image-based perfusion assay. The total flow of the vascular bed can be up to 50% of the flow through the parent tube (the side channels). These values were obtained by the manufacturer.
Lastly, the chips were always cultured on the perfusion rocker. It had been previously reported that the lack of active perfusion by the rocker showed remarkable difference in tube formation between perfused and non-perfused chips [40]. Since the continuous perfusion was critical in both the perfusion of the parent vessels and to create volume and pressure gradient to induce vascularization in the gel chamber, all chips were cultured on the perfusion rocker.
2.3.1. Differentiation of small airway epithelial cells on Organograft® platform
The SAECs were seeded onto the open window of the Organograft® chip (figure 1b, well B2) on 3 days in vitro (DIV) post-seeding of the vascular network on the plate. The submerged culture was kept between DIV4–7 to allow the SAEC to adhere and form a monolayer across the entire open access window. Afterwards, the culture was induced into ALI through media change from the original growth media to ALI media in all wells of the chip except for the middle gel chamber well – which was left devoid of any media. The ALI media was a 1:1 mixture of human tracheobronchial epithelial cell (HBTEC) Air-Liquid Interface Differentiation medium (Lifeline, LM-0050) and the endothelial culture media containing 5% FBS with corresponding VEGF, ANG1 and aprotinin concentrations from the previous sub-section. The ALI culture was maintained over 2 weeks and the tissues were harvested on DIV14 for endpoint analysis.
The maturity of the SAEC was established using immunofluorescence microscopy, as it had been previously reported the identification of canonical markers and TEER correlated with the differentiation of the epithelial cells [41]. For VBMC, similar routes of validation were taken, and both IF and TEER evaluation showed marked difference between immature and fully differentiated SAEC population.
2.4. Image-based perfusion
Opera Phenix (Perkin Elmer) was used to produce images of perfusion. Perfusion images were taken at single z-plane for both brightfield and Cy3 channels (ex: 554 nm, em: 568 nm). 70 kDa Texas Red Dextran (ThermoFisher Scientific, D1864) was used to observe the perfusion of the parent channels via microvascular network in the gel chamber. Columbus software (Perkin Elmer) was used for further qualitative analysis. ImageJ was used to stitch different fields in wells if images were acquired at objectives higher than 10x, i.e., 20x, 40x.
2.5. Immunofluorescence
Opera Phenix (Perkin Elmer) was used to image the canonical markers for each cellular type. Under the confocal settings, lasers were adjusted so that each channel was taken in separate sequence to minimize any signal bleed-throughs. For antibody staining, the chips were fixed with 4% PFA in DPBS from 32% PFA solution (Fisher Scientific, 50–980-495) for 20 min at rt on a rocker or overnight at 4°C. After DPBS wash, the samples were blocked and permeated with 1% bovine serum albumin (BSA, Sigma, A9418), 2% Triton X-100 (ThermoFisher Scientific, 85111) and 5% normal goat serum (ThermoFisher Scientific, 31872) for 1h at RT. For both primary and secondary antibody staining, the chips were left to rock at least overnight at RT to ensure the binding of the antibody in the 3D construct. Unless separately indicated, all primary antibodies were stained at 1:100 dilution and all secondary antibodies were stained at 1:500. Alveoli-capillary constructs were stained for caveolin-1 (BD Biosciences, 2297), ABCA3 at 1:200 (ThermoFisher Scientific, PA5–52478), and CK8 (Cell Signaling Technology, 4546). Small airway-capillary constructs were stained for α-tubulin (Thermo Fisher Scientific, MA1–80017), MUC5AC (Sigma Aldrich, M5293), and CK5 (Abcam, ab193895). For endothelial cells in the vascular network, CD31 (Dako, M0823) and vWF (Dako, GA52761–2). E-cadherin (Cell Signaling Technology, 3195) was used as tight junction staining. For phalloidin staining, 1:50 dilution was used to stain the actin filaments in the tissue constructs. 1:1000 dilution was used for DAPI to stain for the nuclei in the VAMC and VBMC models. For image analysis of wells taken with Opera Phenix, the online Columbus software was used (Perkin Elmer). IF microscopy of the E-cadherin junctions in fully vascularized alveolar and bronchiolar constructs was performed with Leica DMi8 confocal microscopy with 25x water objective (figure 6d, 6e).
2.6. Flow Cytometry
Sony SH800S Cell Sorter (Sony Biotechnology) was used to identify specific cellular population dissociated from the microfluidic chip. A 100-um sorting chip (Sony Biotechnology, LE-C3210) was used for the experiment. For sample preparation, the microfluidic chips were washed once with DPBS after media aspiration for 5 minutes. After the DPBS wash, 0.25% trypsin solution was pipetted in the chips with volume gradient (50 ul/30 ul/30 ul) across the 3 columns comprising the microfluidic chip. The plate was placed on the perfusion rocker in the incubator for 15 minutes without any perturbation. The trypsin solution was then pipetted out with multichannel pipette onto multichannel reservoir. The solution was then placed in a conical tube and diluted with FACS buffer (1 % BSA, 0.1% sodium azide) and centrifuged at 500 xg for 4 minutes. For the 3D alveoli-capillary construct, aquaporin 5 (AQP5-PE, Santa Cruz Biotechnology, sc-514022) and surfactant protein C (SpC-AF647, Santa Cruz Biotechnology, sc-518029) were used to mark for alveolar epithelial cells. For the 3D SAEC-capillary construct, AF700-conjugated MUC5AC (Novus Biologicals, NBP2–32732AF700), PE-conjugated cytokeratin 5/8 (Novus Biologicals, NBP2–47824PE), and AF488-conjugated α-tubulin (Cell Signaling Technology, 8058S) were used to identify small airway epithelial cells. To identify the vascular cells comprising the microvascular network in the gel chamber and the side lanes, FITC-conjugated vimentin (eBioscience™, 11–9897-82) and AF700-conjugated CD31 (R&D Systems, FAB3567N-100) were used. Vimentin is a general marker for any cells of mesenchymal origin and CD31 is a more specific tight junction and adhesion molecular marker for the endothelial cells.
2.7. Trans-endothelial/epithelial electrical resistance
The automated TEER measurement system (World Precision Instruments LLC, SYS-REMS) was reconfigured to measure the TEER values of each microfluidic chip in the Mimetas Organograft® plate (figure 6a). REMS-XT software was used, and the voltage setting was set at 80K. The Corning HTS Transwell-96 (World Precision Instruments LLC, REMS-96C) electrode was configured so that the tips of the probe fit in adjacent wells, which would measure the current passing through the epithelial layer and the adjacent endothelial barrier at the gel-side lane junction. The platemap settings were adjusted so that the plate was set as a 64-well plate with 8 columns and 8 rows. The resulting raw value was then calculated with the known surface area (0.03 cm2) based on the dimensions provided in manufacturer manual for the Organograft® [39].
3. Results
3.1. Vascularized alveolar and bronchiolar multi-chip model development
We aimed to assemble two vascularized LRT multi-chip models: one for the alveoli using primary human alveolar epithelial cells (VAMC), and another for the bronchiole with primary human small airway epithelial cells (VBMC). For both models, we incorporated a perfusable microvascular network underneath a layer of ALI-induced, differentiated lung epithelial cells (figure 1a).
To assemble VAMC and VBMC models, we used the Organograft® plate from Mimetas, a high throughput microfluidic platform containing 64 individual chips per plate. Each chip consists of two parent side lanes along A1-B1 and A3-B3 wells, and a gel chamber with a gel loading inlet in A2 and open access in B2 well. The epithelial cells were seeded on the open access (figure 1b). The gel fills up the phase-guided gel chamber area from A2 to B2 wells via capillary force, and the phase guides situated between the side wells and the gel chamber allows for the gel to fill up along the desired scaffold without overflowing into the side channels, as previously reported [39].
We first created the vascularized bed in the middle gel chamber (B2 well), by seeding a mixture of HPMEC, human pulmonary fibroblasts and human primary pericytes. HPMEC-only suspension was then loaded into the side lanes (A1-B1, A3-B3 wells). After a maturation period of 3–5 days to allow for the vasculature to stabilize and connect with the parent vessels along the side channels, we then added lung epithelial cells onto the exposed gel along the open access in B2 well. The alveolar epithelial cells were cultured in submersion with Cell Biologics epithelial media for 48h before being induced into ALI, while the SAEC were treated with 1:1 HBTEC and VascuLife media for 96h before being exposed to ALI (figure 1c). Perfusion between the side lanes via microvascular network was created by rocking the plate at a set angle and time interval (12°, every 8 minutes) in 37°C. Immunofluorescence microscopy markers of various cell types were used to identify the general cell populations in the chip (CD31 for endothelial cells; CK8 for alveolar epithelial cells; CK5 for SAEC) and to demonstrate the formation of a microvascular network underneath the respiratory epithelium (figure 1d & e).
Additionally, it was seen that the vascular bed went through structural re-organization when epithelial layer was added and exposed to ALI (figures 2e-g). In the xyz-view, the cross-sections of vessels of various sizes can be observed across all conditions, and the subsequent graphical visualization and quantification can be found in supplemental figure 1g. In vasculature-only condition, the vessels are noticeably merged and in some cases with enlarged lumen size (figure 2c). However, in corresponding figures 3d and 4e, the vessels have become finer and streamlined. As there were no permeable membrane separating the epithelial and the vascular network, the changes in vascular network seem to correlate with the addition of epithelial cells in the VAMC and VBMC when compared to vasculature-only condition.
Figure 2.
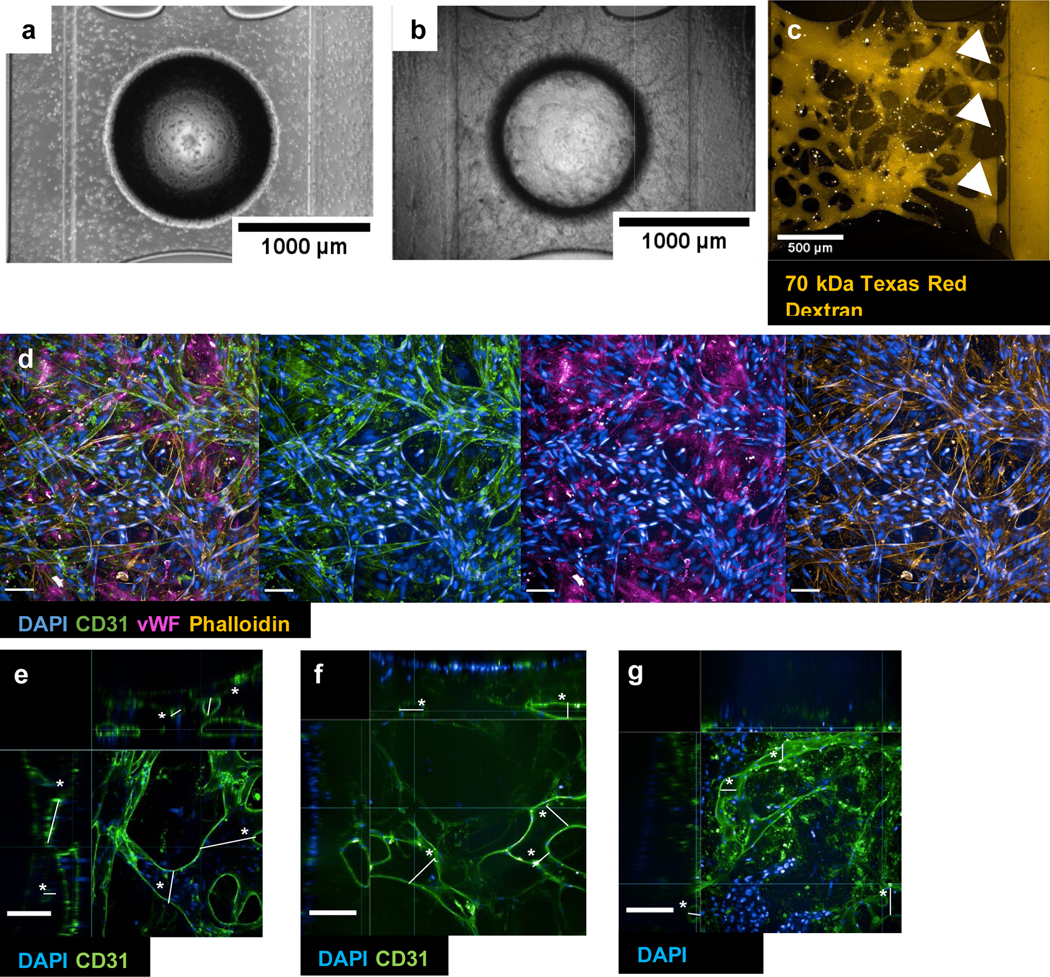
Development of microvascular network. (a) 0 days in vitro (DIV). (b) DIV14. (c) Live perfusion at DIV14 at timepoint 0. The dye was introduced into the reservoirs of the left parent channel (input channel/vessel) and the white arrows indicate the entry point from the vascular network into the output channel/vessel. (d) Immunofluorescence (IF) of vascular network underneath the open access (circular border in bright field images in (a), (b)). (e) xyz-view of vasculature-only condition. (f) xyz-view of VAMC. (g) xyz-view of VBMC (please see SI figure 1 for corresponding plot of asterisked vessels and statistics). All brightfield images taken with EVOS. Confocal taken with Opera Phenix. Scale bar = 100 μm unless indicated otherwise in individual images.
Figure 3.
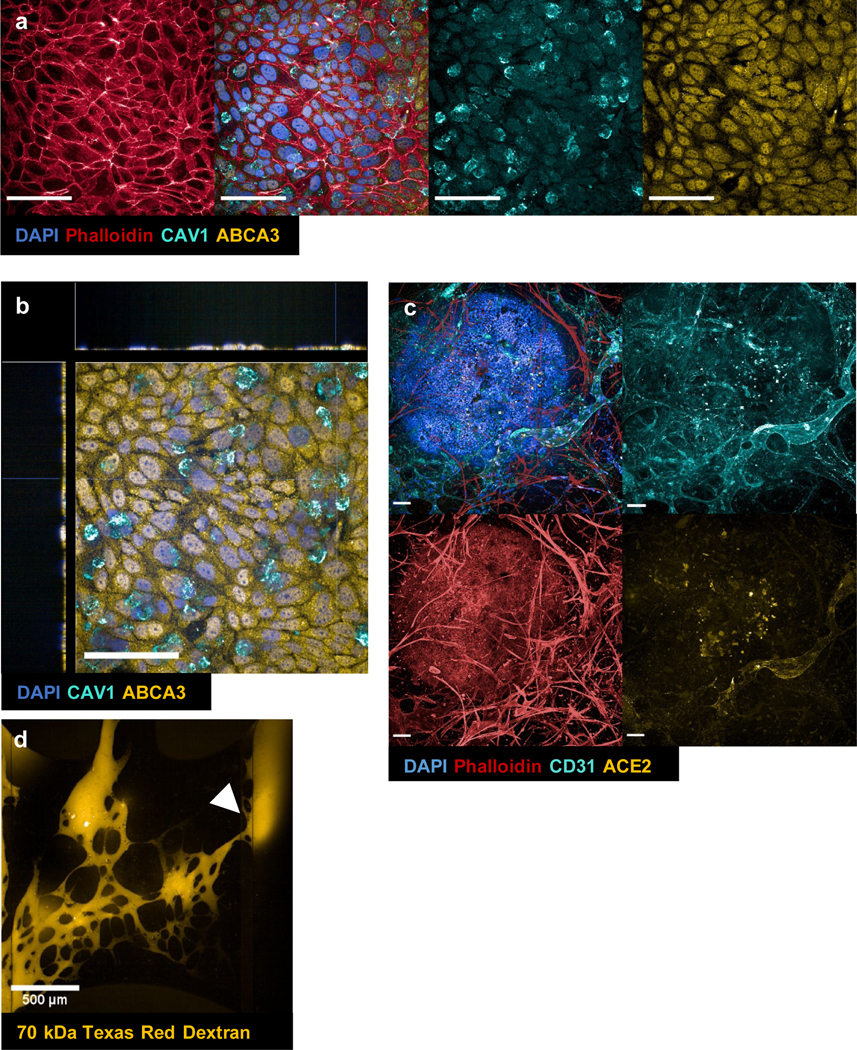
Immunofluorescence (IF) of alveolar population at the epithelial layer (a) and the epithelial-endothelial junction (c). (a) maximum projection of alveolar layer only. DAPI (blue), phalloidin (red), caveolin-1/CAV1 (cyan), ATP-binding cassette class A3/ABCA3 (yellow). (b) xyz-view of DAPI (blue), CAV1 (cyan), ABCA3 (yellow) from (a). (c) maximum projection of vascularized alveolar tissue construct (contains both epithelial and vascular layers). DAPI (blue), phalloidin (red), CD31 (cyan), ACE2 (yellow). (d) live perfusion of alveoli-capillary construct at timepoint t=0 min. The dye was introduced into the reservoirs of the left parent channel (input channel/vessel) and the white arrow indicatesthe entry point from the vascular network into the output channel/vessel. Taken with Opera Phenix. Scale bar = 100 μm unless otherwise indicated in images.
Figure 4.
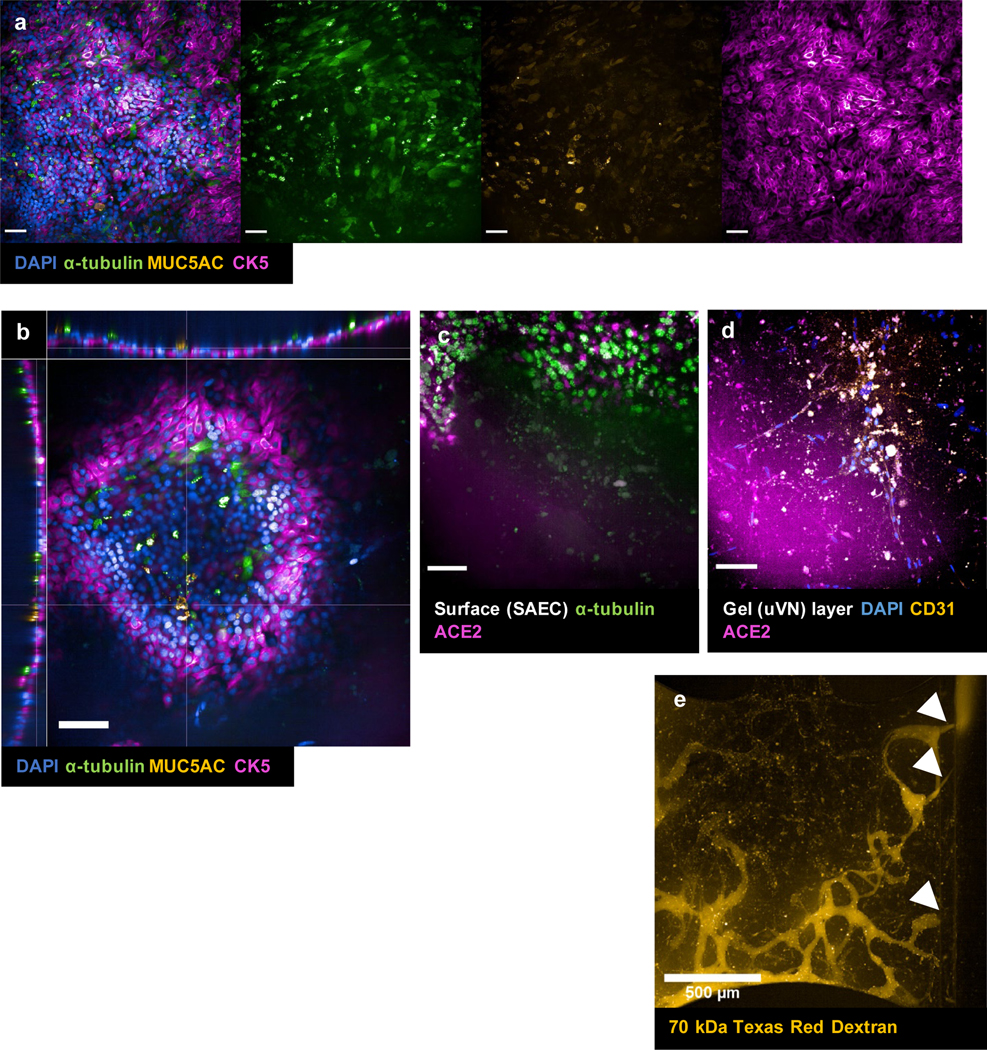
Immunofluorescence (IF) of small airway epithelial population at the epithelial level (a) and at the epithelial-endothelial junction (c), (d). (a) DAPI (blue), α-tubulin (green), MUC5AC (yellow), CK5 (magenta). (b) xyz-view of DAPI (blue), α-tubulin (green), MUC5AC (yellow), and CK5 (magenta) co-stain to show pseudostratified columnar epithelium structure of the small airway layer. (c) maximum projection of epithelial layer only. α-tubulin (green), ACE2 (magenta) along the surface/higher z-stacks of small airway epithelium. (d) maximum projection at vascular layer. DAPI (blue), CD31 (yellow), ACE2 (magenta) along vasculature layer/lower z-stacks of small airway-capillary constructs. (e) live perfusion of small airway-capillary construct at timepoint t=0 min. The dye was introduced into the reservoirs of the left parent channel (input channel/vessel) and the white arrows indicate the entry point from the vascular network into the output channel/vessel. Taken with Opera Phenix. Scale bar = 100 μm unless otherwise indicated in images.
3.1.1. Development of a perfusable vascularized bed for epithelial cell seeding
To create a perfusable microvascular network, we optimized several parameters, including media conditions, cell seeding density, and maturation time for the vascular bed and the side channels. Brightfield microscopy was used to assess the growth and stabilization of the microvasculature (figure 2a & b). By days in vitro (DIV) 14, we observed formation of microvessels when cultured in endothelial media containing 2.5% FBS, 85 ng/ml rhVEGF, 100 ng/ml rhANG1 (A2, B2 wells) and initial seeding density and ratio were (3:1:0.1 mil cells/ml) for HPMEC, fibroblasts and pericytes in the gel chamber. For the side lanes, the initial seeding density for HPMEC was 4 mil cells/ml and were cultured in endothelial media containing 2.5% FBS, 42.5 ng/ml rhVEGF, 25 ng/ml rhANG1 (A1-B3, A3-A3 wells). The network formation and connection with the parent vessels were confirmed with treatment of 70kDa Texas Red Dextran in A1-B1 media reservoirs. Fluorescence microscopy was used to observe perfusion from the input parent vessel towards the output parent vessel through the microvascular network (figure 2c). We found that the presence of microvascular network allowed for rapid perfusion of the dye, as it take 2–3 minutes from the lab hood to imaging acquisition and by the first timepoint, the dye has already reached the output parent vessel. IF evaluation of the microvascular network also showed formation of tight junctions and localization of other canonical proteins for endothelial cells, such as CD31 and vWF (figure 2d). The IF staining patterns confirmed that by DIV14, the microvascular network stabilized and uniformly spread along the height of the gel in the A2-B2 gel chamber (200–300 μm) (data not shown).
The vessel diameters were large and merged in the gel chamber prior to epithelial cell seeding and ALI induction (figure 2e). Once the alveolar and small airway epithelial cells were seeded and introduced into ALI, the vessels in the gel were noticeably smaller and refined, most noticeable during IF evaluation of CD31 and image-based perfusion assays (figures 2e-g and figures 2–4).
3.2. Validation of VAMC and VBMC models through fluorescence microscopy
For the VAMC model, we first stabilized the vascular network for one week (supplemental figure 1) prior to the seeding of alveolar cells in suspension onto open access in B2 well. 48h after the epithelial cell seeding, media from B2 was removed to expose the apical side of the epithelium to ALI (figure 1b, well B2). On DIV14, differentiation of alveolar cells to different subpopulation of epithelial cells was confirmed by immunostaining with 1) ATP-binding cassette class A3 (ABCA3) for type 2 cells; 2) caveolin-1 (CAV1) for type 1 cells; (figure 3a & b). The tissues were also stained for ACE2, as ACE2 is reported to be expressed in both alveolar cells and endothelial cells [42]. We also stained for CD31 and phalloidin to confirm the microvascular network formation (figure 3c). Compared to conditions without vascular network in the gel chamber (supplemental figure 2), vascularized alveoli constructs (figure 3d) showed immediate perfusion of the output channel/vessel even at the beginning timepoint of the perfusion assay. On DIV14, the chips were perfused with the dextran dye to confirm the integrity of the microvascular network morphology and organization in the presence of ALI-induced alveolar epithelium (figure 3d).
To biofabricate the VBMC model, we adopted the protocol described above to create the microvascular network underneath the small airway epithelium. The human primary SAEC were also seeded along the open access in B2 well (figure 1b) and were kept submerged in conditioned media (1:1 HBTEC: VascuLife media) for 96h before the media was removed to induce ALI. After 2 weeks of ALI co-culture, we confirmed the three major sub-populations of SAEC through IF staining: ciliated cells (α-tubulin+), goblet cells (MUC5AC+), and basal cells (CK5+) indicating mature SAEC culture (figure 4a). We also observed pseudostratified layers in the mature SAEC culture (figure 4b) resembling in vivo pseudostratified columnar epithelium of the H&E stains from bronchiolar biopsies [43]. Specifically, we observed the generated SAEC epithelium consist of basal cells along the basolateral surfaces towards the bottom near the gel surface, goblet cells and ciliated cells polarizing towards the top along the apical surface (figure 4b). Additionally, canonical cilia morphology was also observed on α-tubulin positive cells (figure 4a), further confirming the complete differentiation of small airway epithelium after 2 weeks of ALI treatment. Due to the thickness of the SAEC tissue, we generated two separate z-stack ranged images to capture the structure of the epithelial (figure 4c) and the underlying microvascular network layers (figure 4d). Similar to the VAMC tissue construct, we co-stained the small airway epithelial cells for ciliated cell marker α-tubulin as well as for ACE2 especially because ciliated cells have been reported to express ACE2 [12] (figure 4c). Additionally, we observed positive ACE2 staining in the epithelial layer and CD31/ACE2 co-staining in the vasculature for VBMC, just as we had for VAMC (figure 4d). We also observed positive co-localization of ACE2 with α-tubulin+ cells. Lastly, we saw dynamic perfusion in post-ALI treated SAEC-capillary constructs (figure 4c). In parallel, the formation of basement membrane in both models was also investigated through IF microscopy with collagen type III and IV. We confirmed that both ECM proteins formed in the vascular region and the basal epithelial region of VAMC and VBMC models.
3.3. Determination of cell populations in VAMC and VBMC models by flow cytometry
We also utilized flow cytometry to quantitatively characterize cell populations present in the VAMC and VBMC models. ALI-induced transwell alveoli or SAEC monoculture and submerged alveoli or SAEC monoculture were used as reference for comparison. The cell clusters for the alveolar (AQP5, Sp-C) and small airway (MUC5AC, α-tubulin) population displayed patterns dependent on the culture systems (figure 5a-f). The 2D submerged monoculture for alveolar cells showed sizeable portion with double positive +AQP5/+Sp-C (29.8%) and majority of the epithelial cells transdifferentiating to +AQP5/ (−) Sp-C (68.5%). Comparatively, for 2D alveolar mono-culture induced into ALI on a transwell platform, there was a significant shift showing +AQP5/+Sp-C (1.04%) cells with most cells showing +AQP5/ (−) Sp-C (56.1%). In the 3D alveoli-capillary construct induced into ALI, the ratios were +AQP5/+Sp-C (2.38%), +AQP5/ (−) Sp-C (39.4%) and (−) AQP5/+Sp-C (0.38%). For SAEC, the submerged monoculture showed +α-tubulin/+MUC5AC (40.8%) co-stain cells along with (−) α-tubulin/+MUC5AC signal (0.015%) and + α-tubulin/ (−) MUC5AC markers (58.9%). In ALI-induced transwell culture, +α-tubulin/+MUC5AC population increased (29.5%) while (−) α-tubulin/+MUC5AC increased (12%) and +α-tubulin/ (−) MUC5AC decreased (14.5%). The 3D small airway-capillary tissue construct showed population distribution that is in between the previous two samples: +α-tubulin/+MUC5AC (36.5%) with (−) α-tubulin/+MUC5AC (2.35%) and +α-tubulin/ (−) MUC5AC (51.1%). Non-epithelial cells were also stained for CD31 and vimentin. We were able to identify endothelial cells (6.69%), fibroblasts/pericytes (19.4%) (figure 5g). The endothelial cells, fibroblasts and pericytes were also cultured as 2D monolayer and analyzed to see the approximate localization and distribution when compared against the microvascular network in the vascularized respiratory-capillary multi-chip models (figure 5g, h). We observed the distribution of the vascular cells is similar between 2D and 3D platform, with the exception of a larger (−) CD31/ (−) vimentin population present in the stained sample (figure 5g), which are most likely the epithelial cells that were also collected during sample preparation from the 3D tissue. The population variation of sub-epithelial cell types and vascular or unstained cells can be seen changing across the different culture systems (figure 5i). Overall, the addition of the vascular network seems to facilitate both the differentiation and the stabilization of mature epithelial cell types in both models.
Figure 5.

Flow cytometric evaluation of cell types in 3D respiratory tissue construct and table with cluster frequencies. (a) bivariate plot of AQP5 and Sp-C in 3D alveoli-capillary construct. (b) bivariate plot of AQP5 and Sp-C on transwell platform, alveolar monoculture. (c) bivariate plot of AQP5 and Sp-C in submerged T-75 alveolar monoculture. (d) bivariate plot of a-tubulin and MUC5AC in 3D small airway-capillary construct. (e) bivariate plot of α-tubulin and MUC5AC on transwell platform, small airway monoculture. (f) bivariate plot of α-tubulin and MUC5AC in submerged, T-75 small airway culture. Grey = negative control; pink – stained sample. (g) bivariate plot of CD31 and vimentin in 3D respiratory construct. (h) bivariate plot of CD31 and vimentin of submerged T-75 monoculture for endothelial cells, fibroblasts and pericyte, contour plots of cell types overlaid. (i) Stacked column graph of cell populations from (a)-(f). VAMC = vascularized alveolar multi-chip; VBMC = vascularized bronchiolar multi-chip; AEC = alveolar epithelial cells; SAEC = small airway epithelial cells; TW = transwell. Blue = endothelial cells; red = fibroblasts; black = pericytes. FlowJo for compensation matrix calculation and gating analysis.
3.4. Evaluation of barrier functionality in VAMC and VBMC models
We evaluated the barrier integrity of the whole lung epithelial-endothelial junction in both respiratory models by trans-epithelial/endothelial electrical resistance (TEER) measurement at DIV14 for alveoli and at DIV21 for small airway constructs, both with and without epithelial monolayers or vasculature. As shown in figure 6a, the electrical probe was positioned so that one end would touch the open access covered by epithelial cells (well B2), and the other end of the probe over the outlet of the input parent vessel (well B1). The current then would have to travel across through the epithelial layer and across the vascular network and through the parent vessel wall lying at the gel interface to reach the other end of the probe. In conditions where there were only monolayer endothelial tubes but no vascular network and epithelial layer, the average TEER value was 43.11±Ω*cm2 (data not shown). A standard automated TEER system was taken and modified (supplemental video 4) for the Organograft® platform, and multiple wells per condition were measured for TEER evaluation.
We observed that tissues lacking epithelial monolayers (microvascular network only) exhibited the lowest TEER values in both models (12.3±6.5 Ω*cm2 in figure 6a; 51.0±9.8 Ω*cm2 in figure 6b). In the alveolar construct that contained the alveolar monolayer and the side lanes but no microvascular network, the values averaged 261.5±55.4 Ω*cm2 (figure 6a). When microvascular network was present (figure 6a) along with the alveolar monolayer, the perfusion by the μVN significantly improved the barrier functionality in the tissue construct, with average of 742.06±44.4 Ω*cm2 (p < 0.0001). In contrast, there was no significant difference in TEER value when comparing microvasculature-only conditions (51.0±9.8 Ω*cm2) and SAEC without microvasculature conditions averaging 119.19±0.33 Ω*cm2 (figure 6b). However, similar to the alveolar construct, the presence of microvasculature significantly improved the barrier function for the small airway epithelial layer – with the average of 312.26±19.6 Ω*cm2 (figure 6b, p = 0.0001).
Figure 6.
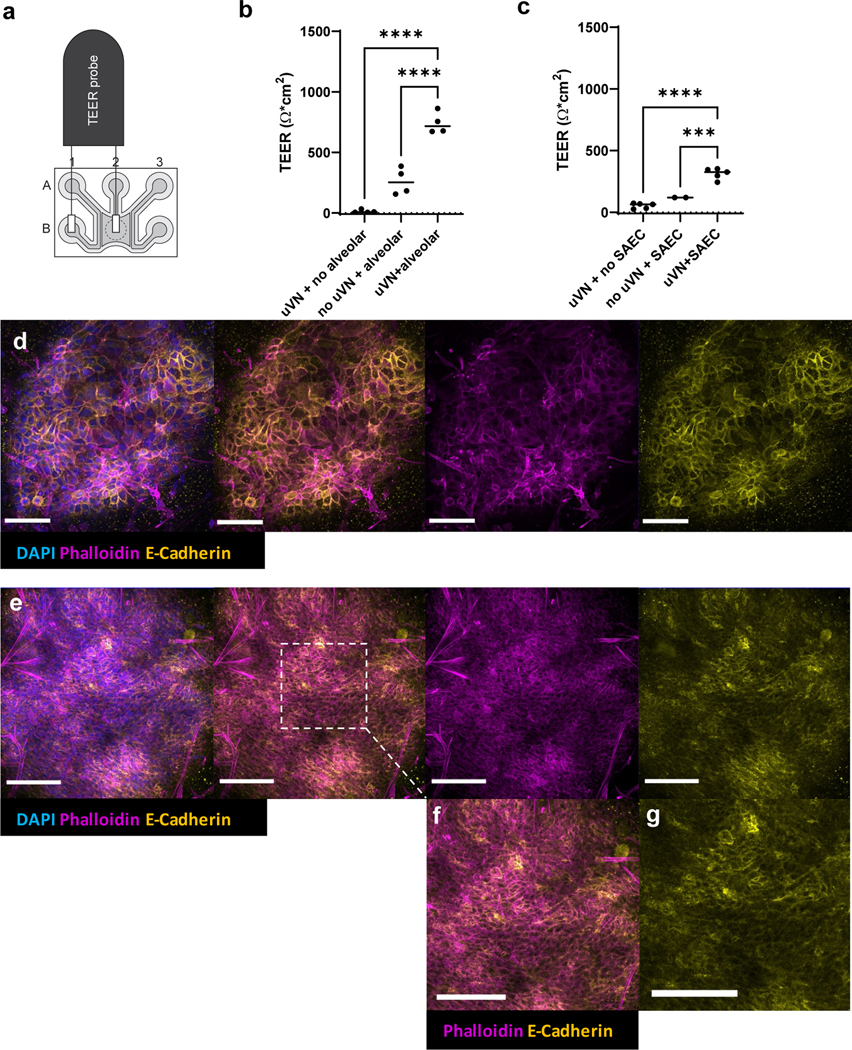
TEER quantification for respiratory models in Organograft®. (a) Schematic of the World Precision Instrument 96-transwell TEER probe positioned over single microfluidic chip. (b) TEER differences among conditions with varying cellular complexity for the alveoli-capillary construct; Ordinary one-way ANOVA, Dunnett’s multiple comparison test, **** p < 0.0001. (c) TEER differences among conditions with varying cellular complexity for the small airway-capillary construct; Ordinary one-way ANOVA, Dunnett’s multiple comparison test, ***p = 0.0001, ****p < 0.0001, μVN = microvascular network. (d) Immunofluorescence of tight junctions in VAMC. (e) Immunofluorescence of tight junctions in VBMC. (f), (g) magnification of dashed region of interest. IF taken with Leica DMi8 confocal microscopy. Scale bar = 100 μm.
4. Discussion
Current lung tissue models incorporate ALI-induced epithelial cells and also include endothelial cells underneath to the epithelial monolayer, forming a barrier interface. These lung tissue equivalents have been used to model many aspects of lung diseases like influenza and COVID-19, asthma, and ARDS by utilizing cytokine or other observable and measurable cellular changes occurring at the epithelial-endothelial junction. In such models, the endothelial cells are constructed to mainly act as barrier via a simplistic 2D monolayer or a monolayer tube [19,20]. However, these lung tissue models with monolayer endothelial tubes do not faithfully recapitulate the role of a complex, multi-dimensional microvascular network that dynamically perfuse the ALI-induced epithelium during homeostatic and disease states. Furthermore, many microfluidic LOC platforms previously described are not easily amenable to HTS and require significant manual manipulation and adjustment of the chips to acquire data using HTS instruments. To address such needs for a perfusable microvasculature in a multi-chip platform, we developed the VAMC and BVMC models that would allow: (1) formation of a complex, 3D microvascular network to support the ALI-treated epithelium with (2) the through-put to perform efficient therapeutics screening.
We first developed perfusable microvasculature using primary HPMEC, fibroblasts and pericytes, which were co-cultured in the Organograft® multi-chip platform. Several parameters were optimized: (1) cell sourcing, (2) media conditions, (3) maturation and culture time for the parent vessels and the microvascular network, and (4) microvasculature morphology and physiology. For HPMEC and pulmonary fibroblasts, the cells were commercially available as primary cells from human donors. Pericytes were obtained as primary human placental pericytes, as pulmonary pericytes were not readily available. We found that culturing HPMEC, fibroblasts and pericytes in low serum (2.5% FBS) along a biochemical gradient (42.5 ng/ml rhVEGF, 25 ng/ml rhANG1 in A1-B1 and A3-B3 wells; 85 ng/ml rhVEGF, 100 ng/ml rhANG1 in A2-B2 wells) allowed for rapid angiogenesis and microvasculature formation between DIV3 and DIV5 post-seeding. In the process, we also had to assess the long-term maintenance of both the endothelial tube formation along the side lanes and the structural integrity of the microvascular network. While the vascular bed remained perfusable 2 weeks post-ALI induction, the non-physiological 90° angle at which the side channels curve caused the endothelial tubes to gradually detach from the channel walls after 3 weeks post-seeding, likely due to continuous exposure to high sheer stress and flow [44]. The parent vessels then continued to constrict and contract, and we observed decrease in the tube diameter. This was the primary reason for the timeline limit, which was maximally set to end in 3 weeks when we performed our endpoint analyses. As this may become a primary limitation in any long-term investigation of respiratory diseases, we plan to address and solve this particular co-culture issue in our future work.
Unlike SAEC, there is comparatively less research done on human alveolar epithelial differentiation and maturation that distinctly separates type I (primarily for gas exchange) and type II (stem cell-like, trans-differentiation potential into type I) alveolar cells [45–47]. Subsequently it is difficult to confidently identify type I-specific and type II-specific cells based on cellular markers. Most literature provide images of type I and II markers in separate tissue samples. By doing so, it fails to show within the same field and tissue the lack of co-localization, i.e., pure subpopulations of type I- and type II-only alveolar cells [48–51] To address this, we co-stained the alveoli-capillary construct, which did result in some co-positive cells for ATI and ATII markers. By providing data showing that the alveolar epithelial cells display of either or both types, it can provide a starting place for future work that focus specifically on differentiation of the alveolar epithelia that is directly perfused by a microvascular network. In figure 5a-c, it can be observed that the ALI-induction on 2D transwell does help with transdifferentiation of the cells into AT1-like cells (56.15%) but none that are maintained as type II. However, with the addition of microvasculature, there is a slight decrease in AQP5 expression (39.4%) while slight increase in Sp-C (0.38%).
An additional advantage of the above vascularized model may be the shortened maturation time for the SAEC in the VBMC model. While traditional ALI transwell models require an extended maturation time for SAEC to transdifferentiate [52–55], we observed that in our vascularized ALI-induced SAEC-capillary constructs, the SAEC fully matured at only 2 weeks post-ALI induction (figure 4). It may be that the presence of dynamically perfused microvessels is not only providing nutrients more efficiently, but also the lack of any physical scaffold barriers, i.e., permeable membrane, allows for direct cell-cell crosstalk and is expediting the differentiation process. Unlike the alveolar epithelia which presents in a simple monolayer (figure 3b), the small airway cells in the SAEC-capillary construct also exhibit a diverse apical landscape, with CK5-positive basal cells localizing along the basement membrane, beneath the ciliated and goblet cells in a stratified manner (figure 4b). While we only accounted for the aforementioned cell types for the small airway construct, other cell types such as clara cells, may be incorporated or characterized in future studies.
For both VAMC and VBMC models, we found that identification of cell population based on germ layers, e.g., endoderm-derived epithelial cells versus mesoderm-derived mesenchymal and endothelial cells, were possible using two distinct markers during flow cytometric analysis. CD31, a canonical marker for endothelial cells whose physiological role is to facilitate neutrophil transmigration, was used to identify the endothelial cells. Vimentin was also used as a secondary benchmark to differentiate the cell populations. The clusters clearly separated into 3 distinct populations in the 3D tissue construct (figure 5g).
While IF and flow cytometry provided us an assessment of the cellular make-up in VAMC and VBMC models, we utilized another orthogonal parameter to benchmark the tissue constructs’ physiological function. TEER was used as a measure to evaluate how modular complexity within the construct give rise to induction of barrier phenotype. Specifically, for the alveolar-capillary system, the presence of microvasculature seemed to augment the whole barrier function only minimally since alveoli consists of a simple squamous epithelium in vivo. It was the addition of the alveolar monolayer that drove the significant increase in the barrier function (figure 6b, p < 0.0001). In this case, the diffusion gradient from the side channels may be sufficient to reach the simple squamous epithelium to allow the epithelial cells to properly form tight junctions and maintain homeostasis. Once the vascular cells were seeded within the gel, the presence of vascular network just allowed for marginally improved perfusion and distribution of various supplements to efficiently reach the epithelial layer, resulting in a continued improvement in barrier function. For the SAEC-capillary construct, the vascularized SAEC conditions displayed significantly higher TEER values than that of the SAEC-only ALI conditions (figure 6c, p < 0.0001). However, SAEC-only conditions without a microvascular network showed much lower TEER values compared to the alveoli-only conditions without microvascular network. Whether this is due to the inherent functional differences between the two epithelial types (i.e., the small airway primarily conducts airflow whereas the alveoli conduct gas exchange) or whether microvasculature has a greater role in supporting the pseudostratified small airway epithelium will require further investigation for better understanding of structure and function relationship of the epithelium and capillary. To evaluate the correlation of barrier phenotype with tight junction formation, we stained with E-cadherin and phalloidin, which showed the membranal localization of the proteins in both VAMC and VBMC models. Future investigation will focus on how the permeable membrane-free VAMC and VBMC models might more closely demonstrate the human native lung tissue physiology, when compared against the current transwell culture systems and other membrane-separated multi-cellular constructs.
5. Conclusion
While there has been significant progress in developing more physiologically relevant respiratory models, this pandemic has highlighted a need for a system that is not only using simple monolayer respiratory epithelial, but also a perfusable microvascular network that is amenable for high-throughput therapeutics screening and disease modeling. We used the 64-chip Organograft® microfluidic plate to develop an ALI-induced respiratory epithelium that is perfusable by a stabilized microvascular network. Subsequently, we successfully developed small airway and alveoli models that can recapitulate key, essential epithelial-capillary interactions specifically observed from in vivo pulmonology.
Supplementary Material
8. Acknowledgements
This work was funded by the NIH Intramural Research Program (ZIA TR000414). We thank Lara El Touhy and Anju Singh for their advice regarding experimental setup and data analysis in flow cytometry; Hoda Zarkoob and Jiaqi Fu for guidance on experimental protocols for cell culture and immunofluorescence; and Ethan Taylor from NIH Medical Arts for the graphics in figure 1.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
9. References
- [1].Ribatti D, Nico B, Vacca A, et al. Endothelial cell heterogeneity and organ specificity. J Hematotherapy Stem Cell Res 2002. 11 81–90. [DOI] [PubMed] [Google Scholar]
- [2].Hogg JC. The traffic of polymorphonuclear leukocytes through pulmonary microvessels in health and disease. American J Roentgenology 163 769–775. [DOI] [PubMed] [Google Scholar]
- [3].Chi JT, Chang HY, Haraldsen G, et al. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci U S A 2003. 100 10623–10628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Krüger-Genge A, Blocki A, Franke RP, et al. Vascular endothelial cell biology: An update. Int J Mol Sci 2019. 20 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Probst CK, Montesi SB, Medoff BD, et al. Vascular permeability in the fibrotic lung. Eur Respir J; 2020. 56 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Renzoni EA, Walsh DA, Salmon M, et al. Interstitial vascularity in fibrosing alveolitis. Am J Respir Crit Care Med 2003. 167 438–443. [DOI] [PubMed] [Google Scholar]
- [7].Vieceli Dalla Sega F, Fortini F, Spadaro S, et al. Time course of endothelial dysfunction markers and mortality in COVID‐19 patients: A pilot study. Clin Transl Med 2021. 11 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jin Y, Ji W, Yang H, et al. Endothelial activation and dysfunction in COVID-19: from basic mechanisms to potential therapeutic approaches. Signal Transduct Target Ther 2020. 5 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ogando NS, Dalebout TJ, Zevenhoven-Dobbe JC, et al. SARS-coronavirus-2 replication in Vero E6 cells: Replication kinetics, rapid adaptation and cytopathology. J Gen Virol 2020. 101 925–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Matsuyama S, Nao N, Shirato K, et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2- expressing cells. Proc Natl Acad Sci U S A 2020. 117 7001–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mirabelli C, Wotring JW, Zhang CJ, et al. Morphological cell profiling of SARS-CoV-2 infection identifies drug Repurposing candidates for COVID-19. bioRxiv. 2020. DOI: 10.1101/2020.05.27.117184. [DOI] [PMC free article] [PubMed]
- [12].Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020. 181 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chu H, Chan JF-W, Yuen TT- T, et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: an observational study. The Lancet Microbe 2020. 1 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen H, Liu W, Liu D, et al. SARS-CoV-2 activates lung epithelia cell proinflammatory signaling and leads to immune dysregulation in COVID-19 patients by single-cell sequencing. EBioMed 2020. 70 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ma D, Chen CB, Jhanji V, et al. Expression of SARS-CoV-2 receptor ACE2 and TMPRSS2 in human primary conjunctival and pterygium cell lines and in mouse cornea. Eye 2020. 34 1212–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Park BK, Kim D, Park S, et al. Differential Signaling and Virus Production in Calu-3 Cells and Vero Cells upon SARS-CoV-2 Infection. Biomol Ther (Seoul) 2021. 29 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen F, Zhang Y, Sucgang R, et al. Meta-analysis of host transcriptional responses to SARS-CoV-2 infection reveals their manifestation in human tumors. Sci Rep 2021. 11 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ammerman NC, Beier-Sexton M, Azad AF. Growth and maintenance of vero cell lines. Curr Protoc Microbiol 2008. APPENDIX A.4E.1-A4.E.7. [DOI] [PMC free article] [PubMed]
- [19].Huh D, Matthews BD, Mammoto A, et al. Reconstituting organ-level lung functions on a chip. Science 328 1662–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Huh D, Leslie DC, Matthews BD, et al. A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Sci Transl Med. 2012. 4 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zamprogno P, Wüthrich S, Achenbach S, et al. Second-generation lung-on-a-chip with an array of stretchable alveoli made with a biological membrane. Commun Biol 2021. 4 168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gorshkov K, Chen CZ, Bostwick R, et al. The SARS-CoV-2 Cytopathic Effect Is Blocked by Lysosome Alkalizing Small Molecules. ACS Infect Dis 2021. 7 1389–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Buggele WA, Johnson KE, Horvath CM. Influenza A virus infection of human respiratory cells induces primary microRNA expression. J Biol Chem 2012. 287 31027–31040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Barosova H, Maione AG, Septiadi D, et al. Use of EpiAlveolar Lung Model to Predict Fibrotic Potential of Multiwalled Carbon Nanotubes. ACS Nano 2020. 14 3941–3956. [DOI] [PubMed] [Google Scholar]
- [25].Zarkoob H, Allué-Guardia A, Chen Y-C, et al. Modeling SARS-CoV-2 and Influenza Infections and Antiviral Treatments in Human Lung Epithelial Tissue Equivalents. bioRxiv 2021. DOI: 10.1101/2021.05.11.443693. [DOI] [PMC free article] [PubMed]
- [26].Benam KH, Mazur M, Choe Y, et al. Human lung small airway-on-a-chip protocol. Methods in Molecular Biology 2017. 1612 345–365. [DOI] [PubMed] [Google Scholar]
- [27].Li K, Yang X, Xue C, et al. Biomimetic human lung-on-a-chip for modeling disease investigation. Biomicrofluidics 2019. 13 31501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Thacker VV, Sharma K, Dhar N, et al. Rapid endotheliitis and vascular damage characterize SARS‐CoV‐2 infection in a human lung‐on‐chip model. EMBO Rep 2021. 22 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Benam KH, Villenave R, Lucchesi C, et al. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat Methods 2016. 13 151–157. [DOI] [PubMed] [Google Scholar]
- [30].Si L, Prantil-Baun R, Benam K, et al. Clinically Relevant Influenza Virus Evolution Reconstituted in a Human Lung Airway-on-a-Chip. Microbiol Spectr 2019. 9 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].van Meer BJ, de Vries H, Firth KSA, et al. Small molecule absorption by PDMS in the context of drug response bioassays. Biochem Biophys Res Commun 2017. 482 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tan K, Keegan P, Rogers M, et al. A high-throughput microfluidic microphysiological system (PREDICT-96) to recapitulate hepatocyte function in dynamic, re-circulating flow conditions. Lab Chip 2019. 19 1556–1566. [DOI] [PubMed] [Google Scholar]
- [33].Gard AL, Cain BP, Miller CR, et al. High-Throughput Human Primary Cell-Based Airway Model for Evaluating Influenza, Coronavirus, or other Respiratory Viruses in vitro. Sci Rep 2021. 11 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Aird WC. Endothelial cell heterogeneity. Cold Spring Harb Perspect Med 2012. 2 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zheng Y, Chen J, Craven M, et al. In vitro microvessels for the study of angiogenesis and thrombosis. Proc Natl Acad Sci U S A 2012. 109 9342–9347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lee S, Lim J, Yu J, et al. Engineering tumor vasculature on an injection-molded plastic array 3D culture (IMPACT) platform. Lab Chip 2019. 19 2071–2080. [DOI] [PubMed] [Google Scholar]
- [37].Phan DTT, Wang X, Craver BM, et al. A vascularized and perfused organ-on-a-chip platform for large-scale drug screening applications. Lab Chip 2017. 17 511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Augustin HG, Koh Y. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science 2017. 357 1–12. [DOI] [PubMed] [Google Scholar]
- [39].Mimetas. OrganoPlate® Graft: Our Technology. www.mimetas.com. https://www.mimetas.com/en/our-technology/ (accessed 23 May 2021).
- [40].Vormann MK, Gijzen L, Hutter S, et al. Nephrotoxicity and Kidney Transport Assessment on 3D Perfused Proximal Tubules. AAPS J 2018. 20 1–11. [DOI] [PubMed] [Google Scholar]
- [41].Bluhmki T, Bitzer S, Gindele JA, et al. Development of a miniaturized 96-Transwell air–liquid interface human small airway epithelial model. Sci Reports 2020. 10 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hamming I, Timens W, Bulthuis MLC, et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004. 203 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].SIU SOM Histology INTRO, (2009, December 14). Southern Illinois University. http://www.siumed.edu/~dking2/crr/CR007b.htm (accessed 23 May 2021). [Google Scholar]
- [44].Wang GJ, Ho KH, Hsu SH, et al. Microvessel scaffold with circular microchannels by photoresist melting. Biomed Microdevices 2007. 9 657–663. [DOI] [PubMed] [Google Scholar]
- [45].Chan MCW, Chan RWY, Yu WCL, et al. Influenza H5N1 virus infection of polarized human alveolar epithelial cells and lung microvascular endothelial cells. Respir Res 2009. 10 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Daum N, Kuehn A, Hein S, et al. Isolation, cultivation, and application of human alveolar epithelial cells. Methods Mol Biol 2012. 806 31–42. [DOI] [PubMed] [Google Scholar]
- [47].Yu WCL, Chan RWY, Wang J, et al. Viral Replication and Innate Host Responses in Primary Human Alveolar Epithelial Cells and Alveolar Macrophages Infected with Influenza H5N1 and H1N1 Viruses. J Virol 2011. 85 6844–6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hynynen K, McDannold N, Sheikov NA, et al. Local and reversible blood–brain barrier disruption by noninvasive focused ultrasound at frequencies suitable for trans-skull sonications. Neuroimage 2005. 24 12–20. [DOI] [PubMed] [Google Scholar]
- [49].Hermanns MI, Fuchs S, Bock M, et al. Primary human coculture model of alveolo-capillary unit to study mechanisms of injury to peripheral lung. Cell Tissue Res 2009. 336 91–105. [DOI] [PubMed] [Google Scholar]
- [50].Lehmann AD, Daum N, Bur M, et al. An in vitro triple cell co-culture model with primary cells mimicking the human alveolar epithelial barrier. Eur J Pharm Biopharm 2011. 77 398–406. [DOI] [PubMed] [Google Scholar]
- [51].Neuhaus W, Samwer F, Kunzmann S, et al. Lung endothelial cells strengthen, but brain endothelial cells weaken barrier properties of a human alveolar epithelium cell culture model. Differentiation 2012. 84 294–304. [DOI] [PubMed] [Google Scholar]
- [52].Staudt MR, Buro-Auriemma LJ, Walters MS, et al. Airway basal stem/progenitor cells have diminished capacity to regenerate airway epithelium in chronic obstructive pulmonary disease. American J Res Critical Care Med 2014. 190 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Cozens D, Grahame E, Sutherland E, et al. Development and optimization of a differentiated airway epithelial cell model of the bovine respiratory tract. Sci Rep; 2018. 8 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wang G, Lou HH, Salit J, et al. Characterization of an immortalized human small airway basal stem/progenitor cell line with airway region-specific differentiation capacity. Respir Res 2019. 20 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bhowmick R, Gappa-Fahlenkamp H. Cells and Culture Systems Used to Model the Small Airway Epithelium. Lung 2016. 194 419–428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.