

Abstract
INTRODUCTION:
The nuclear pore complex (NPC) is the molecular conduit in the nuclear membrane of eukaryotic cells that regulates import and export of biomolecules between the nucleus and the cytosol, with vertebrate NPCs ~110 to 125 MDa in molecular mass and ~120 nm in diameter. NPCs are organized into four main rings: the cytoplasmic ring (CR) at the cytosolic side, the inner ring and the luminal ring on the plane of the nuclear membrane, and the nuclear ring facing the nucleus. Each ring possesses an approximate eightfold symmetry and is composed of multiple copies of different nucleoporins. NPCs have been implicated in numerous biological processes, and their dysfunctions are associated with a growing number of serious human diseases. However, despite pioneering studies from many groups over the past two decades, we still lack a full understanding of NPCs’ organization, dynamics, and complexity.
RATIONALE:
We used the Xenopus laevis oocyte as a model system for the structural characterization because each oocyte possesses a large number of NPC particles that can be visualized on native nuclear membranes without the aid of detergent extraction. We used single-particle cryo–electron microscopy (cryo-EM) analysis on data collected at different stage tilt angles for three-dimensional reconstruction and structure prediction with AlphaFold for model building.
RESULTS:
We reconstructed the CR map of X. laevis NPC at 6.9 and 6.7 Å resolutions for the full CR protomer and a core region, respectively, and predicted the structures of the individual nucleoporins using AlphaFold because no high-resolution models of X. laevis Nups were available. For any ambiguous subunit interactions, we also predicted complex structures, which further guided model fitting of the CR protomer. We placed the nucleoporin or complex structures into the CR density to obtain an almost full CR atomic model, composed of the inner and outer Y-complexes, two copies of Nup205, two copies of the Nup214-Nup88-Nup62 complex, one Nup155, and five copies of Nup358. In particular, we predicted the largest protein in the NPC, Nup358, as having an S-shaped globular domain, a coiled-coil domain, and a largely disordered C-terminal region containing phenylalanine-glycine (FG) repeats previously shown to form a gel-like condensate phase for selective cargo passage. Four of the Nup358 copies clamp around the inner and outer Y-complexes to stabilize the CR, and the fifth Nup358 situates in the center of the cluster of clamps. AlphaFold also predicted a homo-oligomeric, likely specifically pentameric, coiled-coil structure of Nup358 that may provide the avidity for Nup358 recruitment to the NPC and for lowering the threshold for Nup358 condensation in NPC biogenesis.
CONCLUSION:
Our studies offer an example of integrative cryo-EM and structure prediction as a general approach for attaining more precise models of megadalton protein complexes from medium-resolution density maps. The more accurate and almost complete model of the CR presented here expands our understanding of the molecular interactions in the NPC and represents a substantial step forward toward the molecular architecture of a full NPC, with implications for NPC function, bio-genesis, and regulation.
Graphical Abstract
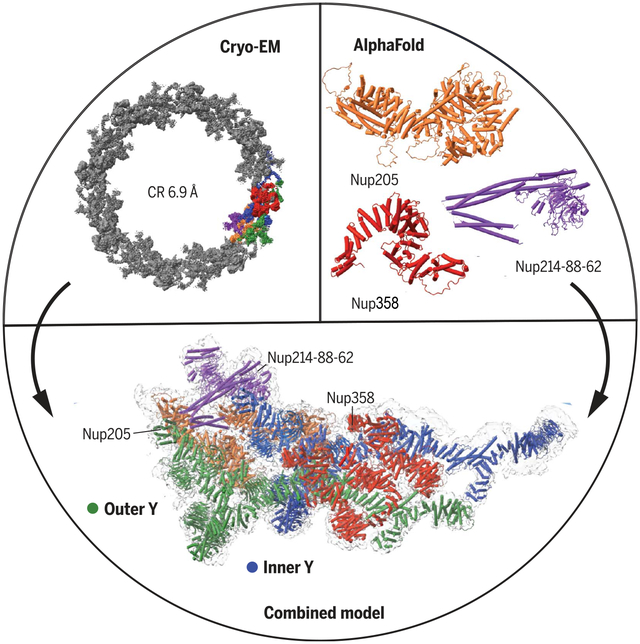
Cryo-EM structure of the cytoplasmatic ring of the nuclear pore complex from X. leavis. The 6.9 Å map was generated with single-particle cryo-EM, and the model was built with AlphaFold structure prediction. The secondary structural elements guided EM map fitting, resulting in an almost complete model of the complex. The approach allowed the identification of five copies of Nup358 and a second copy of the trimeric Nup214-Nup88-Nup62 complex.
The nuclear pore complex (NPC) is the conduit for bidirectional cargo traffic between the cytoplasm and the nucleus. We determined a near-complete structure of the cytoplasmic ring of the NPC from Xenopus oocytes using single-particle cryo–electron microscopy and AlphaFold prediction. Structures of nucleoporins were predicted with AlphaFold and fit into the medium-resolution map by using the prominent secondary structural density as a guide. Certain molecular interactions were further built or confirmed by complex prediction by using AlphaFold. We identified the binding modes of five copies of Nup358, the largest NPC subunit with Phe-Gly repeats for cargo transport, and predicted it to contain a coiled-coil domain that may provide avidity to assist its role as a nucleation center for NPC formation under certain conditions.
The nuclear pore complex (NPC) regulates nucleocytoplasmic passage of biomolecules and has been implicated in numerous biological processes, with their dysfunctions associated with a growing number of diseases (1–6). An NPC is composed of multiple copies of more than 30 nucleoporins (Nups) with structural elements of stacked a-helical repeats and/or β-propellers, about a third of which also contain phenylalanin-eglycine (FG) repeat sequences for selective transport of cargoes (7–10). The approximately eightfold symmetric NPC can be divided into the cytoplasmic ring (CR) at the cytosolic side, the inner ring (IR) and the luminal ring (LR) on the plane of the nuclear membrane, and the nuclear ring (NR) facing the nucleus (Fig. 1A) (3, 4, 11–13). Tremendous progress has been made toward unveiling the architecture of this enormous molecular machine (11–20). Here, we present the cryo–electron microscopy (cryo-EM) structure of the CR from Xenopus laevis oocytes.
Fig. 1. Cryo-EM map of the X. laevis NPC.
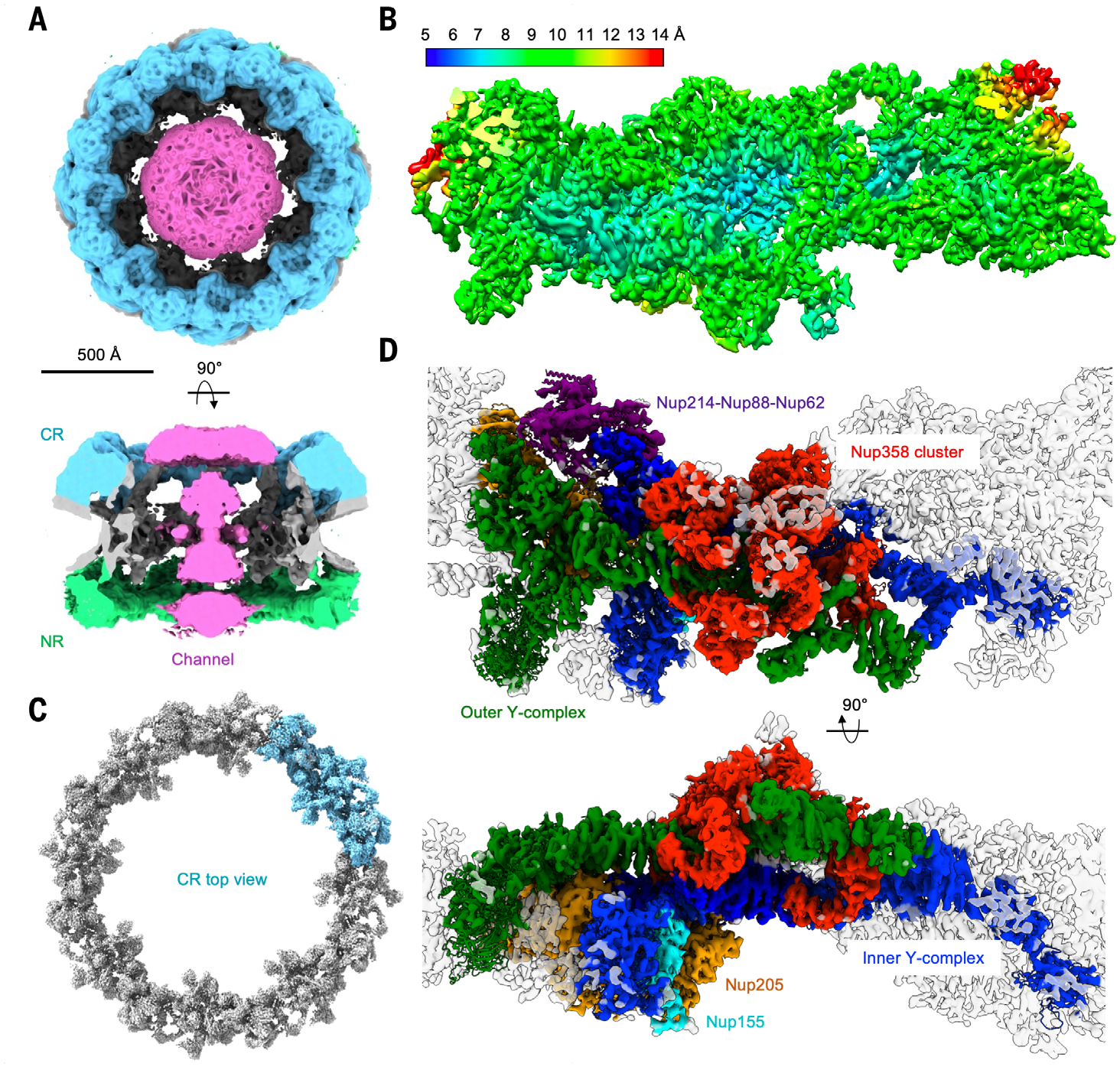
(A) Cryo-EM density of the X. laevis NPC (contour level, 3.0 σ) in top and side views, shown with CR in cyan, NR in green, IR and membrane region in gray, and the channel density in magenta. The map is eightfold symmetrized and at 19.8 Å resolution. (B) Cryo-EM density of a CR protomer at 6.9 Å resolution colored by local resolution. (C) Cryo-EM density of the X. laevis NPC CR ring (top view; contour level, 9.5 σ) composed from the 6.9 Å CR protomer map by assuming the eightfold symmetry. One of the CR protomers is shown in cyan. (D) Cryo-EM density (contour level, 4.5 σ) of a CR protomer superimposed with the final model in two orientations and colored by their model colors, with inner Y-complex in blue, outer Y-complex in green, Nup205 in orange, Nup214-Nup88-Nup62 complex in purple, Nup358 in red, and Nup155 in cyan.
Structure determination
We directly spread the nuclear envelopes (NEs) of actinomycin D (ActD)–treated X. laevis oocytes (18) onto Lacey grids with carbon foil on gold support and applied the Benzonase nuclease to remove contaminating chromatin (fig. S1A). Cryo-EM data collection was conducted at different stage tilts and in counting mode by use of a K3 detector mounted on a Titan Krios microscope at 1.4 Å pixel size. Representative three-dimensional (3D) plots composed of the X and Y positions and the defocus levels (ΔZ) of the NPC particles in selected tilt images-showed the location-dependent variation of the defocus values consistent with the tilt planes (fig. S1B). Data processing performed at the bin2 pixel size (2.8 Å) gave rise to an eightfold averaged full NPC structure, subtracted CR structure, and NR structure at 19.8, 14.6, and 14.7 Å resolutions, respectively (Fig. 1A, fig. S2, and table S1). Symmetry expansion, density subtraction, and 3D classification led to CR and NR protomers at 11.1 and 15.1 Å resolutions.
Final per-particle refinement and masking resulted in maps at 6.9 and 6.7 Å resolutions for the full CR protomer and a core region, respectively (Fig. 1, B and C; fig. S2; and table S1). The Fourier shell correlation (FSC) plots and 3D FSC plots for both maps are shown (fig. S3, A to D), as well as particle orientation distributions (fig. S3, E and F). The histograms of per-angle FSC indicated fairly iso-tropic resolutions along different orientations (fig. S3, C and D). The map used for density interpretation is the 6.9 Å resolution map of the full protomer. Despite the modest 6.9 Å resolution of the full CR protomer, the secondary structures, especially helices, are apparent in the maps (Fig. 1, B and C).
Model building using AlphaFold
We used the recently implemented breakthrough algorithm for protein structure prediction (AlphaFold) (21, 22), mainly as the ColabFold notebook (22) with extended capability to predict homo- and heterocomplexes, to build a nearly complete model of the CR protomer (fig. S4), which contains the inner and outer Y-complexes, two copies of Nup205, two copies of the Nup214-Nup88-Nup62 complex, one Nup155, and five copies of Nup358 (Fig. 1D).
Because no high-resolution models of X. laevis Nups were available, the workflow first involved prediction of five independent models of individual Nups, which in almost all cases gave essentially the same structures (tables S2 and S3). For each prediction, we present the overall and per-residue pLDDT (predicted local distance difference test; 0 to 100, with 100 being the best), the pTM (predicted template modeling; 0 to 1, with 1 being the best), and the predicted alignment error (PAE) matrix (expected position error at residue x when the predicted and true structures are aligned on residue y, representing confidence of the relative positioning of each pair of residues or domains) (tables S2 and S3). We picked the top-ranked model by pLDDT for single proteins and by pTM for complexes in each case for density fitting unless otherwise noted. Whereas helical Nups used the prominent helical features in the maps for their fitting, Nups with mainly a β-propeller domain required prediction of binary complexes with contacting helical Nups to guide the fitting (table S4). Last, for any ambiguous subunit interactions, we predicted complex structures, which further guided model fitting of the CR protomer (table S4). X. laevis Nups that have a substantial region not covered by homology to structural homologs in other species include Nup107, Nup133, Nup160, Nup205, and Nup358 (tables S5 and S6 and fig. S5).
The Y-complex
The CR contains 16 copies of the Y-shaped complex (Y-complex), encircling head to tail to form the inner and outer layers of eight Y-complexes each in the ring (Fig. 1D) (23). Each Y-complex is composed of Nup160 and Nup37 (one short arm); Nup85, Nup43, and Seh1 (the other short arm); and Nup96, Sec13, Nup107, and Nup133 (the long stem) (Fig. 2A). Structural superposition revealed conformational differences between inner and outer Y-complexes at near Nup133 (Fig. 2B and Movie 1), likely because of the need to accommodate the different diameters at the inner and outer layers.
Fig. 2. Fitting of Y-complex Nups with AlphaFold.
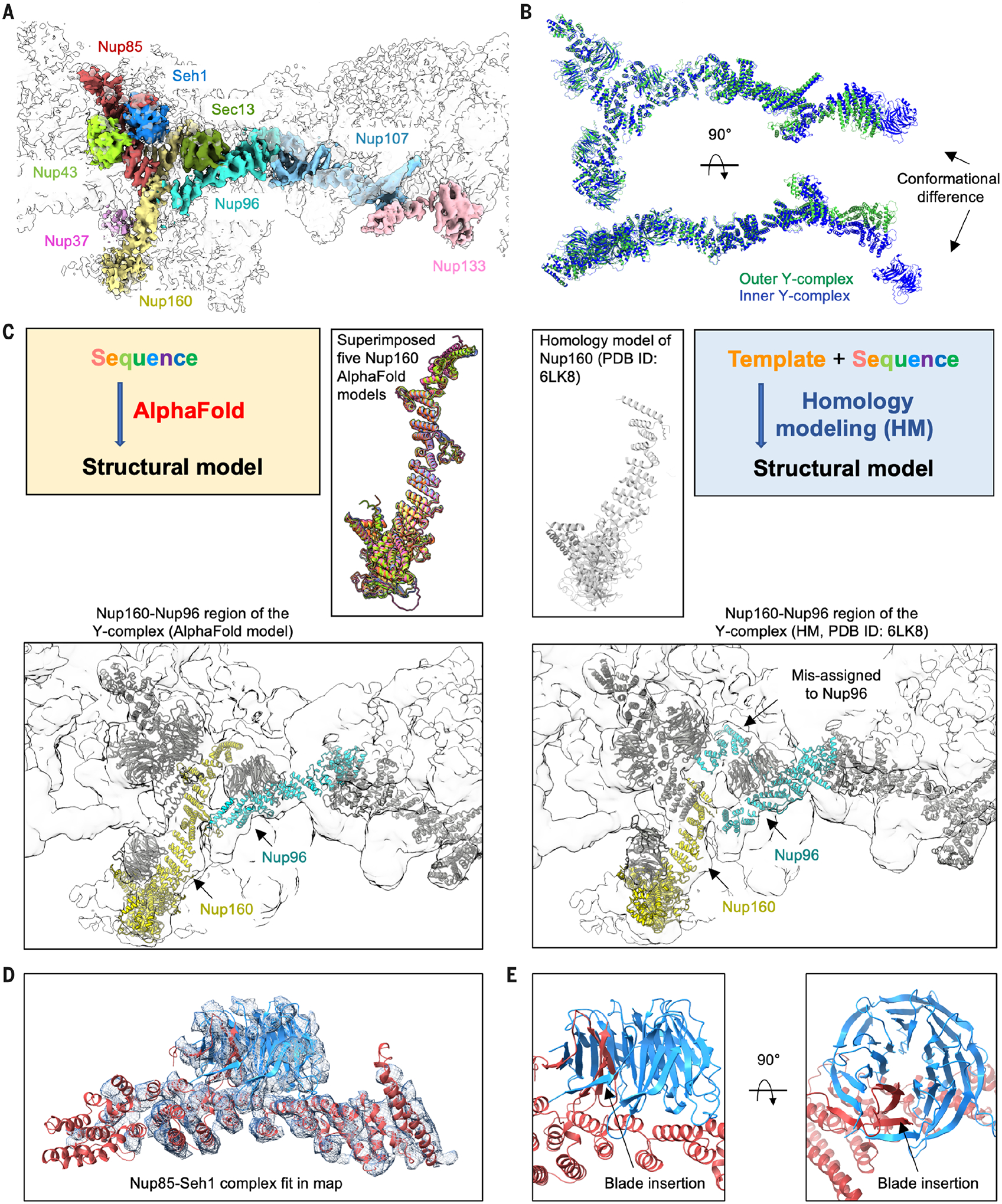
(A) Cryo-EM density (contour level, 8.0 σ) of the outer Y-complex colored by individual Nups. The β-propeller domain of Nup133 was not built because of lack of density. (B) Two views of superimposed inner Y-complex (blue) and outer Y-complex (green) by the two short arms of the Y-complexes. The distal ends of aligned Nup133 without counting the β-propeller have a distance of ~38 Å. (C) Comparison of AlphaFold prediction (left) and homology modeling (right) for Nup160. The cryo-EM density (contour level, 4.5 σ) and the positioning of Nup160 (yellow) and Nup96 (cyan) by the two predictions are shown at bottom. (D and E) AlphaFold-generated model of the Nup85-Seh1 complex fitted with (D) the cryo-EM density (contour level, 4.5 σ) and shown to highlight the inserted blade.
Movie 1. Conformational difference between inner and outer Y-complexes.
The movie shows models of the complete Y-complexes, from 90° rotation around the horizontal axis to transition between conformations of the outer and inner Y-complexes, with the main difference at Nup133. Details are reported in Fig. 2.
The AlphaFold-generated Nup160 structure fits well with the density of the inner and outer Y-complexes (Fig. 2C, fig. S5A, and tables S2 and S3). By contrast, the published homology model of X. laevis Nup160 [Protein Data Bank (PDB) ID 6LK8] (14) misses a C-terminal region (Fig. 2C), which may have led to the incorrect assignment of its density to Nup96 (Fig. 2C and fig. S5B) (14). Thus, building full-length models with AlphaFold may not only increase the structural accuracy of the individual subunits but also help to better assign and interpret densities.
How β-propeller Nups in the Y-complex—Nup37, Nup43, Seh1, and Sec13—fit in the CR map cannot be easily discerned. We therefore predicted structures of these Nups in complex with their contacting α-helical Nups. Seh1-Nup85, Nup43-Nup85, and Sec13-Nup96 complexes were all predicted with excellent pTM and pLDDT scores and fitted the cryo-EM density as a rigid body (Fig. 2D; fig. S5, C and D; and table S4). The Seh1-Nup85 and Sec13-Nup96 complexes exhibited hybrid β-propeller structures in which an insertion blade from the interacting helical domain completes the seven-bladed propellers (Fig. 2E and fig. S5D), as also observed in previous crystal structures of the corresponding, but partial, yeast and human complexes (24–26). AlphaFold failed to predict the Nup37-Nup160 complex (fig. S5E) (27), and we instead used the crystal structure to guide the Nup37 positioning in the map.
Nup205 and the Nup214-Nup88-Nup62 complex
Two AlphaFold-generated Nup205 models, which are larger than and quite different from the homologous crystal structure (28), were each fitted well at the channel side of the two Y-complexes to act as a bridge between them (Fig. 3A; Movie 2; fig. S6, A and B; and tables S5 and S6). The outer Nup205 runs from the C-terminal part of Nup160 to Nup85, and the inner Nup205 interacts with Nup160 at its N-terminal domain but tilts away from Nup85 at its C-terminal domain because of the interaction with the neighboring Nup214-Nup88-Nup62 complex (Fig. 3, A and B).
Fig. 3. Interactions mediated by Nup205 and the Nup214-Nup88-Nup62 complex.
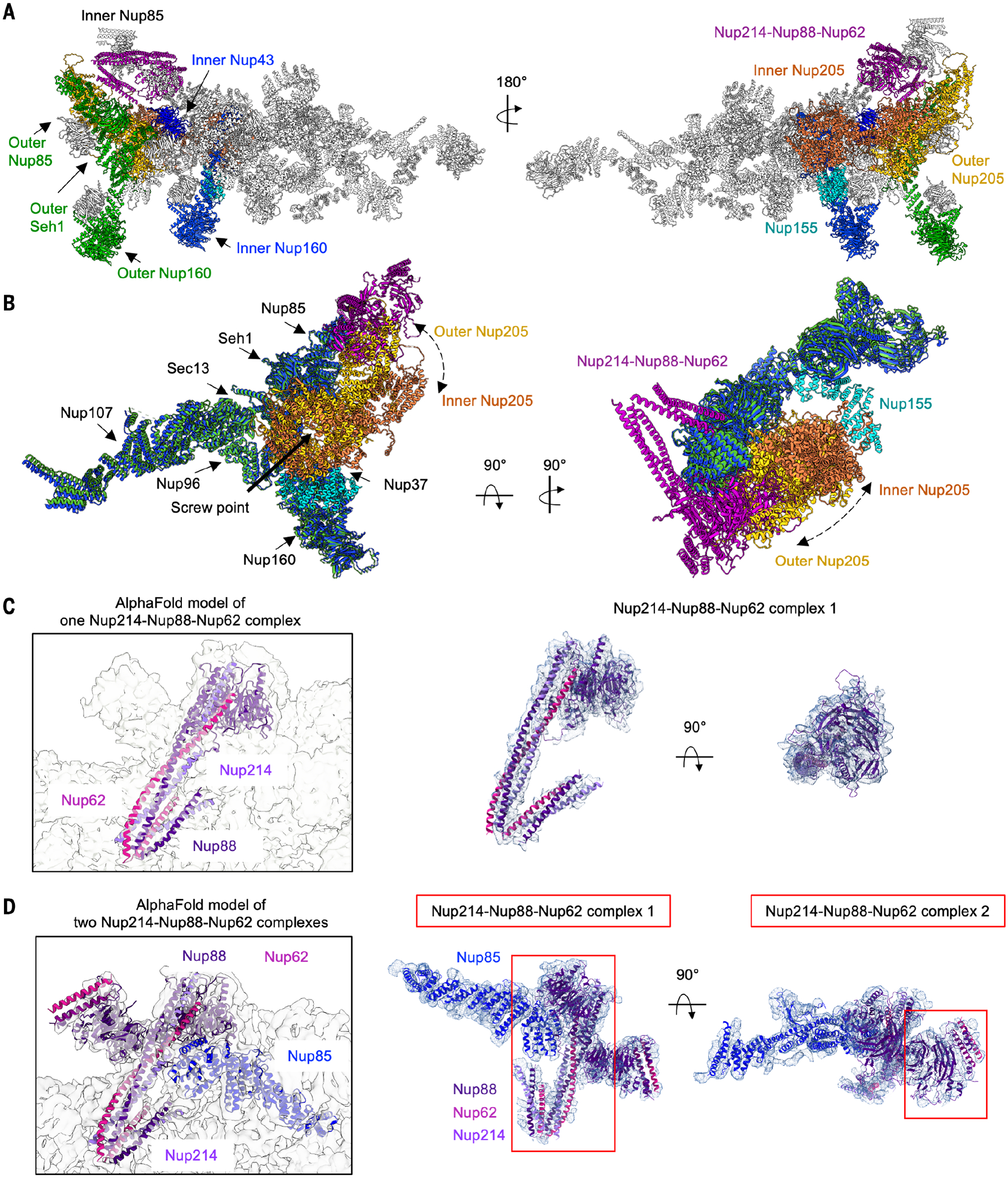
(A) Overall interactions of inner Nup205 (orange) and outer Nup205 (yellow) with Y-complexes. Outer Nup205 directly interacts with Nup160, Nup85, and Seh1 of the outer Y-complex and with Nup43 of the inner Y-complex. The inner Nup205 directly interacts with Nup160 of the inner Y-complex, C-terminal region of Nup155, and Nup88 β-propeller in the Nup214-Nup88-Nup62 complexes. The dashed arrows indicate the locational difference between inner and outer Nup205. (B) Superposition of the inner (blue) and outer (green) Y-complexes together with the bound Nup205 molecules, showing the positions of the inner (orange) and outer (yellow) Nup205 relative to Nup85 and Nup160. The N-terminal region of Nup205 binds similarly to inner and outer Nup160 molecules; the C-terminal domain binds outer Nup85 but pivots away from the inner Nup85 because of the presence of the Nup214-Nup88-Nup62 complexes. (C) Overview (left) and fitting (right) of the AlphaFold-predicted one Nup214-Nup88-Nup62 complex into the cryo-EM density map of NPC CR monomer (contour level, 4.5 σ). (D) Overview (left) and fitting (right) of the AlphaFold-predicted Nup214-Nup88-Nup62 complexes into the cryo-EM density map (contour level, 4.5 σ), with the neighboring inner Nup85. Two Nup214-Nup88-Nup62 complexes are shown.
Movie 2. Interactions formed by Nup205 and the Nup214-Nup88-Nup62 complex.
The movie highlights inner and outer Nup205 and the ternary Nup214-Nup88-Nup62 complex and their interactions. The model rotates 360° along the vertical axis and 360° along the horizontal axis. Detailed interactions are reported in Fig. 3.
We fit a prominent, flag-shaped density over inner Nup85 and extending to the outer Nup85 by generating a composite model of the Nup214-Nup88-Nup62 complex (fig. S6C). The three proteins have been previously predicted to form coiled-coil interactions (4, 29–32). According to AlphaFold, Nup88 and Nup214 also contain β-propeller domains, and complex prediction confirmed the coiled coils and agreed well with the CR map: the β-propeller of Nup88 and one end of the helical bundle as the flag base, the long helical bundle as the flagpole, and the shorter helical bundle as the banner (Fig. 3C). By contrast, the previous X. laevis CR structure presented only a polyalanine model for this complex (fig. S6D) (14). The β-propeller domain of Nup214 does not have density, likely because of a flexible linkage. A given Nup85 can only bind to either Nup205 (for outer Nup85) or the Nup214-Nup88-Nup62 complex (for inner Nup85), but not both (Fig. 3, A and D), which explains the differential modes of Nup205 interactions with the Y-complexes.
We noticed another piece of nearby density, which was previously suggested as a second Nup214-Nup88-Nup62 complex (14) and was fitted as such in a recent paper (20), which is in agreement with the expected stoichiometry from mass spectrometry data (13). Our density fit well with the flag base (Fig. 3D). However, the flag pole is largely missing. We do not know whether this is due to a partial disorder of this region or a lower occupancy of the second complex as a result of ActD treatment in our sample. The Nup88-Nup214-Nup62 complex resembles the X. laevis Nup54-Nup58-Nup62 complex anchored by Nup93 of the IR or yeast Nup49-Nup57-Nsp1 complex in its coiled-coil region (fig. S6C) (33, 34), suggesting that coiled-coil structures are frequently used building blocks in NPC assembly.
The five copies of Nup358
The largest protein in the NPC, Nup358 (also known as RANBP2, or RAN binding protein 2), is composed of a largely disordered C-terminal region with FG repeats for gel-like phase formation and selective cargo passage and with binding sites for RANGAP, RAN, and other effectors (Fig. 4A) (7, 8, 23, 35). AlphaFold predicted the Nup358 N-terminal region as having a large α-helical domain (~800 residues), a linker, and an isolated single α-helix (Fig. 4, A and B). Previously, only the structures of a small N-terminal region (~150 residues) of human and chimpanzee NUP358 were solved (36) and used for homology modeling in X. laevis NPC (fig. S7A and tables S5 and S6) (14). The Nup358 globular domain is an S-shaped structure, and we identified five copies of Nup358 in the CR map (Fig. 4C and fig. S7B), which is consistent with the previous understanding of Nup358 as one of the most abundant proteins in the NPC (Fig. 4C and fig. S7B) (4).
Fig. 4. Nup358 interacts with the Y-complexes as clamps.
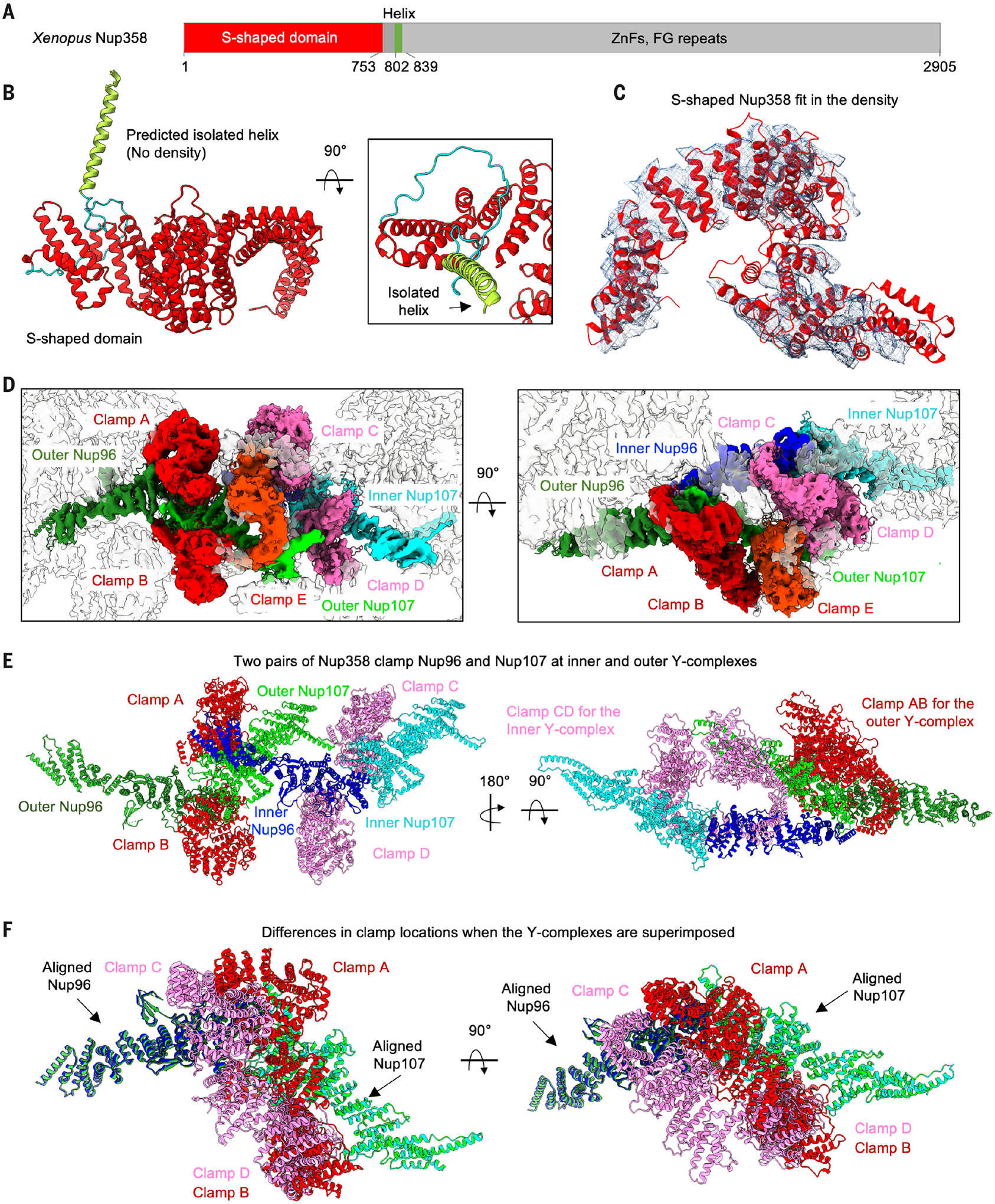
(A) Domain organization of X. laevis Nup358 and the approximate boundaries. ZnFs, zinc fingers. (B) AlphaFold-predicted structure of the N-terminal region of Nup358, showing the S-shaped globular domain, an isolated helix, and the flexible linker in between. (C) Fitting of Nup358 globular domain to the density (contour level, 8.0 σ). (D) The region of the map (contour level, 8.0 σ) containing five Nup358 molecules (labeled as clamps A to E) and two Y-complexes (Nup96-Nup107 complex), in two orientations. (E) Two Nup358 molecules each clamp around Nup96-Nup107 at the inner and outer Y-complexes. Clamps A and B (red) are for the outer Y-complex, and clamps C and D (pink) are for the inner Y-complex. The last Nup358 (clamp E, orange) contacts clamp C and Nup107 of the outer Y-complex. (F) Relative shifts in the clamp location on the two Y-complexes. The clamps B and D are similar in their location on Nup107, whereas clamps A and C have a shift in their position on Nup96.
The full model of Nup358 molecules shows that four of the copies clamp around the inner and outer Y-complexes near the junction of Nup96 and Nup107 (Fig. 4, D and E, and Movie 3), likely to stabilize the CR. In the outer Y-complex, clamp A contacts Nup96 and Nup107 with ~750 and 400 Å2 buried surface area, respectively, and clamp B contacts Nup107 with ~630 Å2 buried surface area, as calculated on the PDBePISA server (37). In the inner Y-complex, clamp C contacts Nup96 with only ~270 Å2 buried surface area, and clamp D interacts with Nup107 with ~750 Å2 buried surface area. Superposition of the inner and outer Nup96-Nup107 complexes showed that clamps B and D both contact Nup107 in a similar mode of binding, but clamps A and C are shifted significantly to account for the differences in the surface area burial (Fig. 4F). The fifth Nup358 (clamp E), situating in the center of the Nup358 cluster, contacts clamp C (~1700 Å2) and Nup107 (~600 Å2) of the outer Y-complex. Thus, the apparent weaker interaction to the Y-complex by clamp C is compensated by the additional interaction from clamp E.
Movie 3. Interactions of Nup358 with the Y-complexes.
The movie shows five copies of Nup358 and their interactions with inner and outer Nup96 and Nup107. The model zooms in to the five Nup358 clamps and then rotates 75° along the horizontal axis. Detailed interactions are reported in Fig. 4.
Homo-oligomeric Nup358
We wondered whether the predicted isolated helix (Fig. 4B) following the S-shaped domain forms a coiled-coil structure, which is however invisible because of its flexible linkage. We thus used the COILS sever (38), which predicted up to 100% coiled-coil propensity for this helix (Fig. 5A). We then used AlphaFold to predict how the helix would assemble into oligomers. We input the number of protomers as six because coiled-coil structures with more than five subunits are very rare, and six should cover almost all possibilities. AlphaFold predicted a pentameric coiled coil plus a single helix as the top-ranked model with a pTM of 0.74 and pLDDT of 82.2. This is then followed by two trimeric coiled-coil complexes with pTMs of 0.45 and 0.44, a tetramer and a dimer with a pTM of 0.57, and last, a hexameric coiled coil with a pTM of 0.39 (Fig. 5B). The pentameric coiled coil also had the highest per-residue pLDDT scores at its core region (bluest) when displayed onto the structure (Fig. 5C).
Fig. 5. Nup358 is predicted to contain an oligomeric coiled coil.
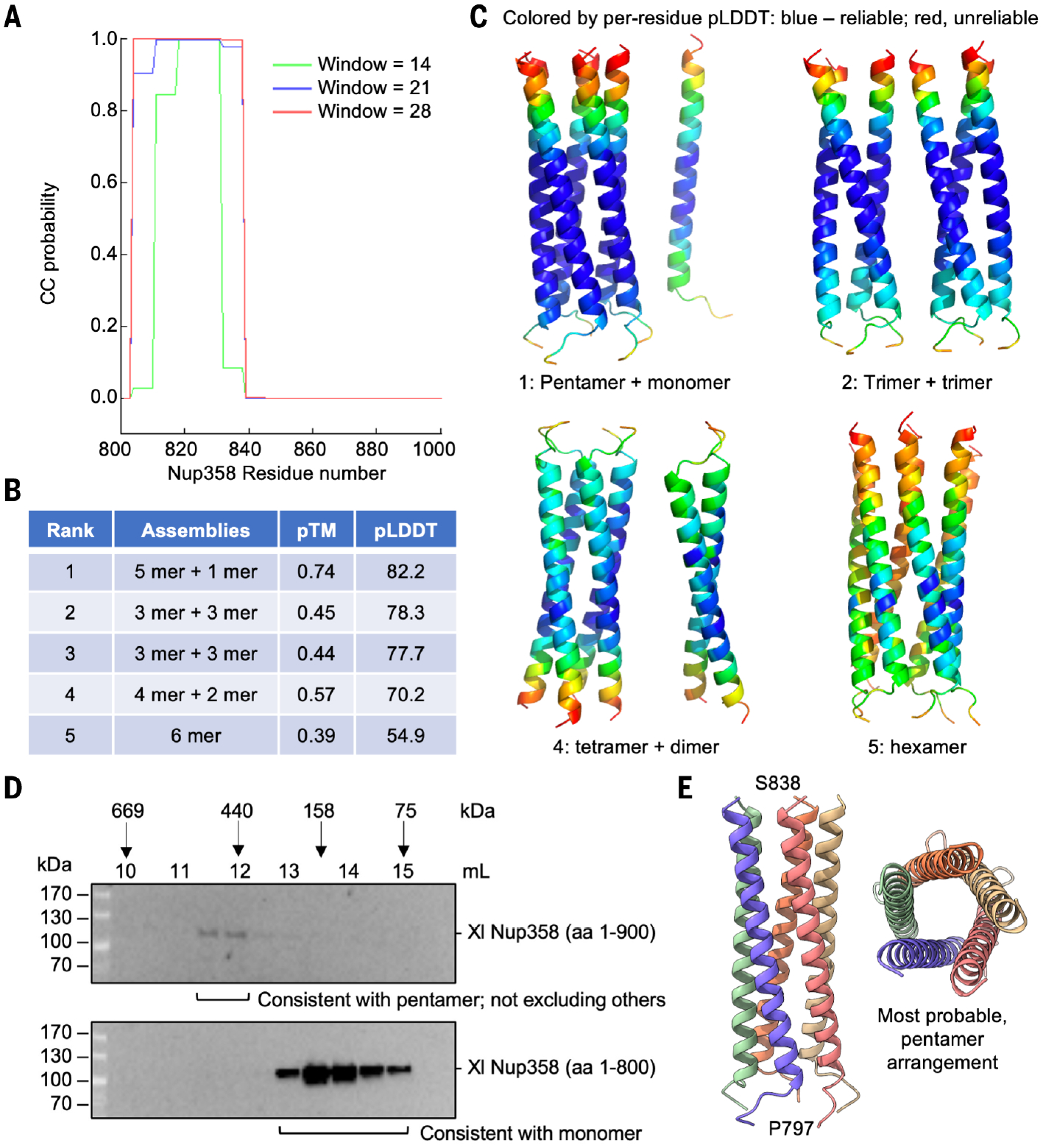
(A) Prediction of the single helix after the S-shaped globular domain for coiled-coil propensity by using a sliding window of 14, 21, or 28 residues. (B) The ranked five models of six Nup358 coiled-region protomers predicted with AlphaFold and the associated pTM and average pLDDT scores. The top model contains a pentamer and a monomer, suggesting that the pentamer is the most favorable oligomer. (C) Ribbon diagrams of four models from (B) (ranked at 1, 2, 4, and 5) and colored by per-residue pLDDT scores. A light spectrum from blue to red corresponds to highest to lowest pLDDT scores, respectively. (D) Elution fractions of X. laevis Nup358 (1 to 900, top) and Nup358 (1 to 800, bottom) from a gel filtration column. The elution positions of several standards are shown. aa, amino acid. (E) The ribbon diagram of a pentamer colored by each protomer and shown in side and top views.
To corroborate the AlphaFold prediction, we expressed and purified His-tagged X. laevis Nup358 (1 to 800, only the globular region) and Nup358 (1 to 900, with the coiled-coil region) and subjected them to gel filtration chromatography. Judging by gel filtration standards from the same column, Nup358 (1 to 800) may be consistent with a monomer, whereas Nup358 (1 to 900) may be consistent with a pentamer (Fig. 5D). A pentameric Nup358 (Fig. 5E) may help its interactions with the Y-complexes through avidity, although the potential formation of other oligomers cannot be excluded. A recent preprint reported an antiparallel tetrameric crystal structure of the coiled-coil region of human NUP358 (39), suggesting that Nup358 from different species may assume different modes of oligomerization.
A recurrent human mutation of NUP358, Thr585→Met (T585M) (equivalent to X. laevis T584M), is associated with autosomal-dominant acute necrotizing encephalopathy (ADANE) (40, 41). Thr585 is mapped to a partially buried site in direct interaction with the hydrophobic side chain of Leu450 (fig. S7C), suggesting that the mutation might affect the conformation of the structure and reduce its interaction with the Y-complexes. The dominant nature of this presumed loss-of-function mutation is consistent with the multimeric nature of Nup358 in which the mutant co-oligomerizes with the wild-type protein to reduce the avidity for its interaction with the Y-complexes.
Nup155 and unassigned densities
Previously, a cryo–electron tomography (cryo-ET) study of human NPC showed localization of NUP155, a linker Nup, in both the CR and the NR (16). The AlphaFold-predicted Nup155 structure consists of a β-propeller followed by a large helical repeat domain (Fig. 6A), in an organization similar to that of Nup160 and Nup133. The helical repeat domain fits well with the CR protomer map (Fig. 6B) and interacts with inner Nup160, burying ~750 Å2 surface area, and with inner Nup205, burying ~310 Å2 surface area (Fig. 6C). We wondered whether we masked out the density for the β-propeller during high-resolution refinement. The full CR map from a previous step of data processing (fig. S2) revealed density for a complete Nup155 (Fig. 6D). In this map, the β-propeller of Nup155, the neighboring inner and outer Nup160, and inner Nup133 situate inside a membrane region of the density (Fig. 6D). The β-propeller domains of Nup155 and Nup133have been shown to possess amembrane-anchoring domain known as amphipathic lipid packing sensor (ALPS) (42, 43), which consists of a short, disordered loop that may fold into an amphipathic helix on membrane (44).
Fig. 6. Nup155 and other membrane-anchoring domains in the CR.
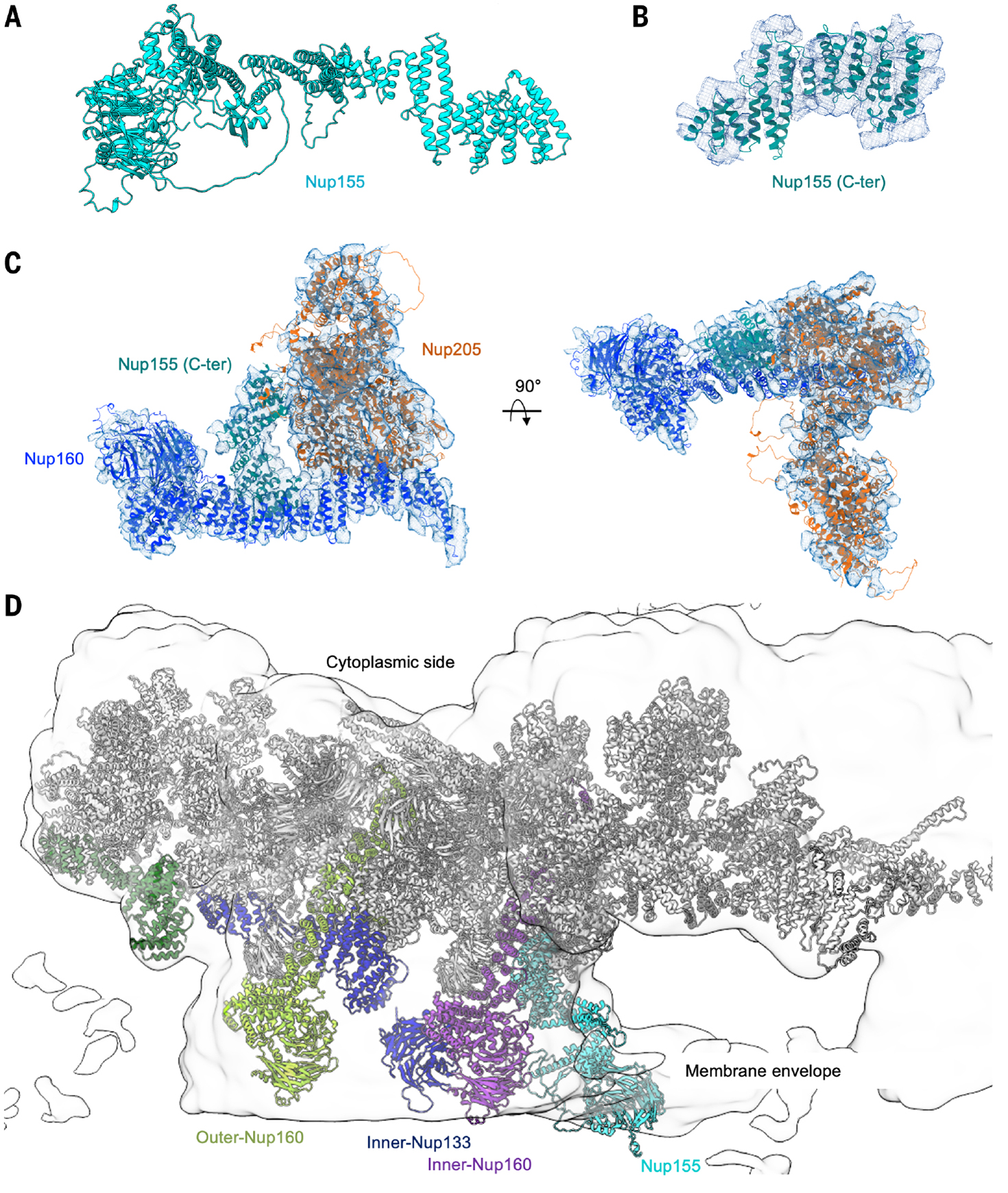
(A) AlphaFold-predicted full-length Nup155. (B) Fitting of the C-terminal region of Nup155 into the cryo-EM density (contour level, 4.5 σ). (C) Interaction of Nup155 with the neighboring inner Nup160 and Nup205 (contour level, 4.5 σ). (D) β-propeller domains of Nup155, Nup133, and Nup160 all localize to the membrane envelope region of the cryo-EM density map of NPC CR full ring at 14.6 Å resolution (contour level, 3.0 σ).
We could not assign the identity of a piece of elongated density next to inner Nup205, Nup133, and Nup107 (fig. S8A). This density was absent from a previously deposited cryo-EM map of X. laevis CR (14) but was present in the deposited cryo-ET maps of X. laevis NPC treated or not with ActD (fig. S8B) (18). Another smaller piece of unassigned density situates adjacent to Nup358, inner Nup96, and outer Nup107 (fig. S8A). The location of this density could be explained by Nup93 as suggested by a recently released paper and a preprint (20, 39). However, we were unable to properly fit Nup93 because of the weaker density.
Conclusion
Our nearly complete model of the CR of X. laevis NPC reveals the molecular interactions within and their biological implications. One aspect of the CR assembly that was unexpected is the observed asymmetry in the composition and mode of binding among Nups: the conformational differences between the two Y-complexes, the different binding modes of the two Nup205 molecules with the Y-complexes, the two Nup214-Nup88-Nup62 complexes side by side, and the five Nup358 complexes with contrasting binding modes. It will be interesting to know whether this asymmetry represents a basal state of the CR or is caused by ActD-mediated cargo deficiency, and whether it will be a common feature in the structures of the NR, IR, or LR. Our X. laevis NPC sample came from haploid oocytes, which may differ further from NPCs in somatic cells.
We propose that the multiple copies of Nup358 and its oligomeric coiled-coil association explain its implicated role as a key driver of NPC assembly during oogenesis in the cytosol that is different from the rapid postmitotic and the slower interphase NPC assembly (2). This process occurs on stacked membrane sheets of the endoplasmic reticulum (ER) termed annulate lamellae (AL), and Nup358 condensates from its FG repeats act as a fastener to spatially direct this NPC bio-genesis from scratch (2, 45). The additional requirement for the FG-containing Nup214 in Nup358 recruitment to the NPC (46) further suggests a role of condensation in NPC assembly. The oligomeric structure of Nup358 may lower the threshold for Nup358 condensation, thus helping to explain its nucleating role among the different Nups.
We also present an integrative approach to take advantage of the recent developments in cryo-EM technology (47, 48) and AlphaFold structure prediction (21, 22, 49), which led to a more precise modeling of the NPC. Similar approaches were also used in the structure determination of NPCs in recently published papers or preprints (19, 20, 50–52). AlphaFold prediction is in contrast to structure modeling by means of homology to deposited structures that are often partial or quite dissimilar. The goal of achieving high resolution is to obtain the best model possible; incorporating information from AlphaFold in the modeling process may be analogous to what the field did previously for stereochemical restraints (53). With the capability for complex prediction to become more routine (22, 54, 55), we anticipate that this approach will not only assist the modeling of new structures but also help to reinterpret previous medium-resolution cryo-EM maps and become a norm in structural biology.
Materials and methods
Sample preparation for cryo-EM
X. laevis has played a key role in revealing the NPC structure because each oocyte has a large number of NPC particles (11, 14, 15, 18, 56). Freshly isolated stage VI oocytes of X. laevis in the modified Barth’s saline (MBS, 10 mM HEPES at pH 7.5, 88 mM NaCl, 1 mM KCl, 0.82mM MgSO4, 0.33mM Ca(NO3)2 and 0.41 mM CaCl2) were purchased and shipped overnight from Ecocyte Bioscience US LLC. To optimize the homogeneity of the NPC sample, we incubated these oocytes with 100 mg/ml Actinomycin D (ActD) at 4°C overnight to inhibit RNA synthesis and thus RNA export for synchronization of the transport cycles (18). Each oocyte was poked at the animal pole using a sharp tweezer to result in the ejection of the nucleus, and transferred into a low salt buffer containing ActD (LSB, 10 mM HEPES at pH 7.5, 83 mM KCl, 17 mM NaCl and 7.5 μg/ml ActD). The nucleus was further washed to reduce the contaminating yolk in a new LSB solution. Two or three washed nuclei were then transferred to the surface of a freshly glow-discharged grid. The NE was poked open, spread using glass needles, incubated for 10 min in 10 μl of LSB supplemented with Benzonase Nuclease (Sigma Aldrich, E8263) to remove the contaminating chromatin, and subsequently washed twice with 10 μl of LSB. 3 ml LSB was added to the grid before blotting it for 3 to 5 s under 100% humidity at 4°C and plunged into liquid ethane using a Mark IV Vitrobot (ThermoFisher).
Negative staining EM
Nuclear membranes were applied on a freshly glow-discharged grid, using a Pelco EasyGlow, as described for cryo-EM sample preparation. Excess buffer was blotted on filter paper, and 6 μl of a 1% uranyl formate solution was applied for 30 s and blotted again on filter paper. Negatively stained samples were imaged on a Joel JEM1400 Transmission Electron Microscope at 120 keV.
Cryo-EM data collection
Screening and collection were performed at Stanford-SLAC Cryo-EM center (S2C2) with a Titan Krios electron microscope (Thermo Fisher Scientific) operating at 300 keV equipped with a K3 detector and a BioQuantum energy filter (Gatan, slit width 20 eV). Movies were collected in counting mode at a 1.4 Å pixel size (table S1). Because of the way the grids were made, most NPC particles would have a similar orientation with their eightfold axis perpendicular to a grid, and we were expected to use a series of stage tilt angles to alleviate this orientation bias for 3D reconstruction. Given the known knowledge that gold grids can minimize beam-induced movement (57), we tested a number of gold grid types with the goal of identifying one with smallest beam-induced movement that is often exaggerated at high tilt angles. These grids include Lacey carbon films on gold support, 300 mesh (Ted Pella), Quantifoil holey carbon films on gold support, R 1.2/1.3, 300 mesh (Quantifoil Micro Tools), UltrAuFoil holy gold films on gold support, R 1.2/1.3, 300 mesh (Quantifoil Micro Tools) and UltrAuFoil holy gold films on gold support overlaid with graphene (made by Wei Li Wang in the Wu lab). Lacey carbon films on gold support were shown to be the most stable and thus used for all data collection.
To alleviate the orientation bias, we initially collected datasets at stage tilts of 0°, 35°, and 45° with a total dose of 54 e/Å2 over 40 frames for 0° and 35°, and a total dose of 79.8 e/Å2 over 60 frames for 45°. An ideal tilt angle of 42° was then calculated using cryoEF (58) from a preliminary 3D reconstruction, and was used for the subsequent data collection with a total dose of 80 to 140 e/Å2 over 80 to 120 frames. SerialEM was used for fully automated data collection, with a defocus range between −1 and −3 μm.
Cryo-EM data processing
Data processing leveraged computer support from the SBgrid Consortium (59). Movies were corrected by gain reference and beam-induced motion, and summed into motion-corrected and dose weighted images using the Relion 3.08 implementation of the MotionCor2 algorithm (60, 61). The distribution of average motions per frame for each grid type at a given tilt angle was plotted using OriginLab (OriginPro 2017 Suite, OriginLab Corporation, Northampton, MA, USA) to evaluate grid-dependent drift performance.
The initial contrast transfer function (CTF) estimation of motion-corrected micrographs without dose-weighting was calculated by CTFFIND4 (62). All micrographs were manually inspected and selected based on particle uniformity and contrast, and particles were picked manually. Gctf (63) was then used to determine the per-particle defocus values (63), from which 3D plots composed of the X and Y coordinates and the CTF (Z) of the particles for selected tilt images were generated using OriginLab (OriginPro 2017 Suite, OriginLab Corporation, Northampton, MA, USA). A plane was then fit to each 3D plot of a given image (fig. S1B).
A total of 204,551 particles were manually picked, local CTF-corrected and extracted from 30,987 dose-weighted micrographs using a box size of 330 by 300 pixels at a 4× binned pixel size of 5.6 Å in RELION 3.08 (61). These particles were imported into cryoSPARC (64) to perform 2D classification, from which 124,532 good particles were selected and merged for homogeneous refinement. The published cryo-EM map of the human NPC (EMD-3103) (16) was low-pass filtered to 60 Å and used as the initial model. The homogeneous refinement with C8 symmetry resulted in a reconstruction at 22.1 Å. These reconstructed 124,532 particles were exported to RELION, 3.08 extracted again with a box size of 660 by 660 pixels and a binned pixel size of 2.8 Å, and imported back into cryoSPARC to re-perform 2D classification. 101,366 particles were selected for homogeneous refinement using the 22.1 Å map low-pass filtered to 40 Å as the initial model. The homogeneous refinement with C8 symmetry resulted in a 19.8 Å map. Particle density subtraction with the aligned 101,366 particles for separate processing of the CR or the NR was done in cryoSPARC. The new local refinement in cryoSPARC using the subtracted particles and a NR or a CR mask led to NR and CR maps at 14.7 and 14.6 Å resolutions, respectively.
The aligned 101,366 particles for the whole NPC were also exported to RELION 3.08 and ran auto-refine with local search and C8 symmetry, with the 19.8 map low-pass filtered to 30 Å as the initial model. The resolution of the auto-refined map was 19.5 Å. We then performed C8 symmetry expansion and density subtraction using a CR protomer mask, and these subtracted particles were recentered and box size re-windowed to 300 by 300 pixels, all in RELION 3.1. 3D classification using a CR protomer mask, local search with 50 iterations and K = 6 was done on these subtracted particles. A class with 333,214 particles was selected for auto-refine with a mask and local search, reaching an 11.1 Å resolution. CTF refinement accounting beam-tilt estimation, anisotropic magnification estimation and per-particles defocus estimation and the subsequent auto-refine resulted in an improved map at 9.9 Å resolution. Additional reconstructions using a tight CR protomer mask or a tight core region mask led to maps at 8.8 and 8.4 Å resolutions. These aligned 333,214 subtracted particles were also imported into cryoSPARC to perform local CTF refinement and local refinement. The final resolutions for the CR protomer and the core region were 6.9 Å and 6.7 Å, respectively. All reported resolutions were estimated based on the gold-standard FSC = 0.143 criterion (fig. S2). All final maps were corrected and sharpened by applying a negative B factor using automated procedures in RELION 3.1. Local resolution variations of cryo-EM maps were estimated using Phenix.
Prediction of NPC subunit structures by AlphaFold
The AlphaFold structures in this study were mainly generated from the AlphaFold2 implementation in the ColabFold notebooks (49) running on Google Colaboratory (21, 22), using the default settings with Amber relaxation (msa_method=mmseqs2, homooligomer=1, pair_mode=unpaired, max_msa=512:1024, subsample_msa=True, num_relax=5, use_turbo=True, use_ptm=True, rank_by=pLDDT, num_models=5, num_samples=1, num_ensemble=1, max_recycles=3, tol=0, is_training=False, use_templates=False). The major difference of ColabFold from the native AlphaFold2 implementation is that ColabFold uses mmseqs2 (65), which the ColabFold authors suggest give equivalent results (22). For complex prediction, sequences were entered in tandem and separated by a semicolon. For coiled coil prediction, we used homooligomer=6. Due to computing memory constraints on Google Colaboratory, we sometimes split up large proteins at disordered junctions to predict each segment separately.
AlphaFold was run once with each of the 5 trained models; the five models generated were checked for consistency, and unless specified otherwise, the top-ranked model was taken in each case for density fitting. AlphaFold computes pLDDT score and pTM score to indicate the accuracy of a prediction (23). We used pLDDT for ranking single protein models and pTM for ranking protein-protein complexes, as recommended by ColabFold (22). A predicted alignment error map between pairs of residues was also calculated for each prediction, which represents confidence in domain positioning. Confidence metrics (global and per-residue pLDDT, pTM, and PAE maps) of predictions made in this work can be found in tables S2 to S4. A few larger proteins or complexes (more than 1400 residues in total length) were run on a Boston Children’s Hospital GPU cluster, by using default AlphaFold settings.
To color ribbon diagrams based on per-residue pLDDT scores (range 0 to 100, with higher being better), these scores stored at the B-factor column of the .pdb files were changed to 100-pLDDT; thus, when colored as pseudo-B-factors in Pymol (66), a light spectrum from blue to red corresponds to highest to lowest pLDDT scores.
Model fitting and building
Prior to beginning modeling, we used AlphaFold (21, 22) to generate all models of known components of the CR using the specific X. laevis sequences. An initial model of the Y-complex (PDB ID: 6LK8) (14) was fitted into the cryo-EM density using ChimeraX (67), and used as a reference for manual positioning of AlphaFold-generated subunit or complex structures into the density followed by executing the “fit in map” command to refine the replacement. Flexible loops were removed to avoid steric clash. After building the two Y-complexes, we began to model the other densities. Nup205 cryo-EM density was easily recognized behind the Y-complexes due to the large size and overall shape. Inner and outer Nup205 assume a different position due to the presence of the Nup214-Nup88-Nup62 complex in the inner Y-complex. Nup358 density was easily recognized in the presence of the generated AlphaFold model with a prominent S shape, and allowed for identification of 5 copies for each CR protomer. Nup88 density was recognized due to the β-propeller and the long a-helix. The additional density which belongs to the Nup214 β-propeller was recognized upon generation of its AlphaFold model. Building of the Nup88-Nup214-Nup62 complex was assisted by predicting the hetero-trimeric coiled coil stricture in AlphaFold, from which a composite model of the Nup88-Nup214-Nup62 complex was obtained. The final model was compared with the previous atomic model (PDB ID: 6LK8) (14). The model fitting quality was estimated for each subunit by the correlation coefficient in ChimeraX (67) and in Phenix (68). A value of correlation coefficient ranges from −1 to 1, with 1 as the perfect fit, and 0.5 to 1.0 as good fit. This modeling process using AlphaFold is reminiscent of the use of stereochemical information of amino acids and nucleic acids in the current practice of structural modeling (53) that increases model accuracy.
Nup358 expression and purification
X. laevis Nup358 constructs (residues 1–800 and 1–900) were cloned into pET21a with a C-terminal His tag. Expression was carried out in E.coli BL21 DE3. Briefly, cells were grown in terrific broth media, supplemented with 100 μg/ml of Ampicillin and 30 μg/ml of Chloramphenicol, until OD600 reached 0.6. Cells were then transferred at 4°C for 30 min before the addition of 1 mM IPTG and incubation overnight at 18°C. Cells were pelleted at 3,000 g for 20 min and resuspended in lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM TCEP, 10 mM Imidazole) supplemented with a protease inhibitor cocktail. Lysis was performed by sonication and the soluble fraction was separated by centrifugation at 40,000 g for 1 hour at 4°C. The supernatant was incubated with Ni-NTA beads pre-equilibrated with lysis buffer, and purification was performed per manufacturer’s recommendation. Eluted fractions were further separated by gel filtration chromatography with a Superdex 200 Increase 10/300 GL in gel filtration buffer (20 mM Hepes pH 7.4, 150 mM NaCl, 0.5 mM TCEP). Fractions were analyzed by Western blotting using an Anti-His antibody (Takara 631210). The Superdex 200 Increase 10/300 GL column was previously calibrated in gel filtration buffer using a high molecular weight kit from MW of 43 kDa to 669 kDa (Cytiva 28-4038-42).
Supplementary Material
ACKNOWLEDGMENTS
We thank W. Chiu for help with the design of data collection, M. Kirschner for initially offering to use oocytes from his laboratory, W. L. Wang for giving us graphene-coated UltrAuFoil holy gold films on gold support, A. N. Hayati and P. Sliz for running some AlphaFold predictions on Boston Children’s Hospital’s cluster, and H. Sharif for discussions on tilt data processing. The authors acknowledge Boston Children’s Hospital’s High-Performance Computing Resources BCH HPC Clusters Enkefalos 2 (E2) and Massachusetts Green High-Performance Computing (MGHPCC), which were made available for conducting the research reported in this publication.
Funding:
All cryo-EM data were collected at Stanford-SLAC Cryo-EM Center (S2C2) supported by the NIH Common Fund Transformative High Resolution Cryo-Electron Microscopy program (U24 GM129541). This work was also supported by the US Department of Energy, Office of Basic Energy Sciences, Nanomachine Program, under contract DE-AC02-05CH11231 (to C.B.); National Institutes of Health (NIH) grant R01GM032543 (to C.B.); and a postdoctoral fellowship from the Cancer Research Institute (to P.F.).
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability:
All data and materials reported in the main text and supplementary materials are available upon reasonable request. The electron density maps have been deposited in the Electron Microscopy Data Bank (EMDB) with accession numbers EMD-25817 and EMD-25818 for a CR protomer and a full CR ring built from the CR protomer map, respectively, and the atomic coordinates have been deposited in the Protein Data Bank with the accession number 7TDZ.
REFERENCES AND NOTES
- 1.Fernandez-Martinez J, Rout MP, One ring to rule them all? Structural and functional diversity in the nuclear pore complex. Trends Biochem. Sci 46, 595–607 (2021). doi: 10.1016/j.tibs.2021.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hampoelz B et al. , Nuclear pores assemble from nucleoporin condensates during oogenesis. Cell 179, 671–686.e17 (2019). doi: 10.1016/j.cell.2019.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glavy JS, The quest for the blueprint of the nuclear pore complex. Protein J 38, 363–376 (2019). doi: 10.1007/s10930-019-09858-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin DH, Hoelz A, The structure of the nuclear pore complex (an update). Annu. Rev. Biochem 88, 725–783 (2019). doi: 10.1146/annurev-biochem-062917-011901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sali A, From integrative structural biology to cell biology. J. Biol. Chem 296, 100743 (2021). doi: 10.1016/j.jbc.2021.100743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferreira PA, The coming-of-age of nucleocytoplasmic transport in motor neuron disease and neurodegeneration. Cell. Mol. Life Sci 76, 2247–2273 (2019). doi: 10.1007/s00018-019-03029-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frey S, Richter RP, Görlich D, FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science 314, 815–817 (2006). doi: 10.1126/science.1132516 [DOI] [PubMed] [Google Scholar]
- 8.Lemke EA, The multiple faces of disordered nucleoporins. J. Mol. Biol 428 (10 Pt A), 2011–2024 (2016). doi: 10.1016/j.jmb.2016.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Devos D et al. , Components of coated vesicles and nuclear pore complexes share a common molecular architecture. PLOS Biol 2, e380 (2004). doi: 10.1371/journal.pbio.0020380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berke IC, Boehmer T, Blobel G, Schwartz TU, Structural and functional analysis of Nup133 domains reveals modular building blocks of the nuclear pore complex. J. Cell Biol 167, 591–597 (2004). doi: 10.1083/jcb.200408109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akey CW, Interactions and structure of the nuclear pore complex revealed by cryo-electron microscopy. J. Cell Biol 109, 955–970 (1989). doi: 10.1083/jcb.109.3.955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim SJ et al. , Integrative structure and functional anatomy of a nuclear pore complex. Nature 555, 475–482 (2018). doi: 10.1038/nature26003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ori A et al. , Cell type-specific nuclear pores: A case in point for context-dependent stoichiometry of molecular machines. Mol. Syst. Biol 9, 648 (2013). doi: 10.1038/msb.2013.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang G et al. , Structure of the cytoplasmic ring of the Xenopus laevis nuclear pore complex by cryo-electron microscopy single particle analysis. Cell Res 30, 520–531 (2020). doi: 10.1038/s41422-020-0319-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y et al. , Molecular architecture of the luminal ring of the Xenopus laevis nuclear pore complex. Cell Res 30, 532–540 (2020). doi: 10.1038/s41422-020-0320-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.von Appen A et al. , In situ structural analysis of the human nuclear pore complex. Nature 526, 140–143 (2015). doi: 10.1038/nature15381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bui KH et al. , Integrated structural analysis of the human nuclear pore complex scaffold. Cell 155, 1233–1243 (2013). doi: 10.1016/j.cell.2013.10.055 [DOI] [PubMed] [Google Scholar]
- 18.Eibauer M et al. , Structure and gating of the nuclear pore complex. Nat. Commun 6, 7532 (2015). doi: 10.1038/ncomms8532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akey CW et al. , Comprehensive structure and functional adaptations of the yeast nuclear pore complex. Cell 185, 361–378.e25 (2022). doi: 10.1016/j.cell.2021.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tai L et al. , 8 Å structure of the outer rings of the Xenopus laevis nuclear pore complex obtained by cryo-EM and AI. Protein Cell (2022). doi: 10.1007/s13238-021-00895-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jumper J et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). doi: 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mirdita M, Ovchinnikov S, Steinegger M, ColabFold - Making protein folding accessible to all. bioRxiv [Preprint] (2021). 10.1101/2021.08.15.456425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knockenhauer KE, Schwartz TU, The nuclear pore complex as a flexible and dynamic gate. Cell 164, 1162–1171 (2016). doi: 10.1016/j.cell.2016.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brohawn SG, Leksa NC, Spear ED, Rajashankar KR, Schwartz TU, Structural evidence for common ancestry of the nuclear pore complex and vesicle coats. Science 322, 1369–1373 (2008). doi: 10.1126/science.1165886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Debler EW et al. , A fence-like coat for the nuclear pore membrane. Mol. Cell 32, 815–826 (2008). doi: 10.1016/j.molcel.2008.12.001 [DOI] [PubMed] [Google Scholar]
- 26.Hsia KC, Stavropoulos P, Blobel G, Hoelz A, Architecture of a coat for the nuclear pore membrane. Cell 131, 1313–1326 (2007). doi: 10.1016/j.cell.2007.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bilokapic S, Schwartz TU, Molecular basis for Nup37 and ELY5/ELYS recruitment to the nuclear pore complex. Proc. Natl. Acad. Sci. U.S.A 109, 15241–15246 (2012). doi: 10.1073/pnas.1205151109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin DH et al. , Architecture of the symmetric core of the nuclear pore. Science 352, aaf1015 (2016). doi: 10.1126/science.aaf1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madeira F et al. , The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47 (W1), W636–W641 (2019). doi: 10.1093/nar/gkz268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bailer SM, Balduf C, Hurt E, The Nsp1p carboxy-terminal domain is organized into functionally distinct coiled-coil regions required for assembly of nucleoporin subcomplexes and nucleocytoplasmic transport. Mol. Cell. Biol 21, 7944–7955 (2001). doi: 10.1128/MCB.21.23.7944-7955.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grandi P et al. , A novel nuclear pore protein Nup82p which specifically binds to a fraction of Nsp1p. J. Cell Biol 130, 1263–1273 (1995). doi: 10.1083/jcb.130.6.1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belgareh N et al. , Functional characterization of a Nup159p-containing nuclear pore subcomplex. Mol. Biol. Cell 9, 3475–3492 (1998). doi: 10.1091/mbc.9.12.3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stuwe T et al. , Architecture of the fungal nuclear pore inner ring complex. Science 350, 56–64 (2015). doi: 10.1126/science.aac9176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chug H, Trakhanov S, Hülsmann BB, Pleiner T, Görlich D, Crystal structure of the metazoan Nup62•Nup58•Nup54 nucleoporin complex. Science 350, 106–110 (2015). doi: 10.1126/science.aac7420 [DOI] [PubMed] [Google Scholar]
- 35.Wu J, Matunis MJ, Kraemer D, Blobel G, Coutavas E, Nup358, a cytoplasmically exposed nucleoporin with peptide repeats, Ran-GTP binding sites, zinc fingers, a cyclophilin A homologous domain, and a leucine-rich region. J. Biol. Chem 270, 14209–14213 (1995). doi: 10.1074/jbc.270.23.14209 [DOI] [PubMed] [Google Scholar]
- 36.Kassube SA et al. , Crystal structure of the N-terminal domain of Nup358/RanBP2. J. Mol. Biol 423, 752–765 (2012). doi: 10.1016/j.jmb.2012.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krissinel E, Henrick K, Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 372, 774–797 (2007). doi: 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 38.Lupas A, Van Dyke M, Stock J, Predicting coiled coils from protein sequences. Science 252, 1162–1164 (1991). doi: 10.1126/science.252.5009.1162 [DOI] [PubMed] [Google Scholar]
- 39.Bley CJ et al. , Architecture of the cytoplasmic face of the nuclear pore. bioRxiv [Preprint] (2021). 10.1101/2021.10.26.465790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deshmukh P, Singh A, Khuperkar D, Joseph J, Acute necrotizing encephalopathy-linked mutations in Nup358 impair interaction of Nup358 with TNRC6/GW182 and miRNA function. Biochem. Biophys. Res. Commun 559, 230–237 (2021). doi: 10.1016/j.bbrc.2021.04.027 [DOI] [PubMed] [Google Scholar]
- 41.Shibata A, Kasai M, Hoshino A, Tanaka T, Mizuguchi M, RANBP2 mutation causing autosomal dominant acute necrotizing encephalopathy attenuates its interaction with COX11. Neurosci. Lett 763, 136173 (2021). doi: 10.1016/j.neulet.2021.136173 [DOI] [PubMed] [Google Scholar]
- 42.Kim SJ et al. , Integrative structure-function mapping of the nucleoporin Nup133 suggests a conserved mechanism for membrane anchoring of the nuclear pore complex. Mol. Cell. Proteomics 13, 2911–2926 (2014). doi: 10.1074/mcp.M114.040915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drin G et al. , A general amphipathic alpha-helical motif for sensing membrane curvature. Nat. Struct. Mol. Biol 14, 138–146 (2007). doi: 10.1038/nsmb1194 [DOI] [PubMed] [Google Scholar]
- 44.Nordeen SA, Turman DL, Schwartz TU, Yeast Nup84-Nup133 complex structure details flexibility and reveals conservation of the membrane anchoring ALPS motif. Nat. Commun 11, 6060 (2020). doi: 10.1038/s41467-020-19885-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Onischenko E et al. , Natively unfolded FG repeats stabilize the structure of the nuclear pore complex. Cell 171, 904–917.e19 (2017). doi: 10.1016/j.cell.2017.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernad R, van der Velde H, Fornerod M, Pickersgill H, Nup358/RanBP2 attaches to the nuclear pore complex via association with Nup88 and Nup214/CAN and plays a supporting role in CRM1-mediated nuclear protein export. Mol. Cell. Biol 24, 2373–2384 (2004). doi: 10.1128/MCB.24.6.2373-2384.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kühlbrandt W, Biochemistry. The resolution revolution. Science 343, 1443–1444 (2014). doi: 10.1126/science.1251652 [DOI] [PubMed] [Google Scholar]
- 48.Chiu W, Schmid MF, Pintilie GD, Lawson CL, Evolution of standardization and dissemination of cryo-EM structures and data jointly by the community, PDB, and EMDB. J. Biol. Chem 296, 100560 (2021). doi: 10.1016/j.jbc.2021.100560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tunyasuvunakool K et al. , Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596 (2021). doi: 10.1038/s41586-021-03828-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mosalaganti S et al. , Artificial intelligence reveals nuclear pore complexity. bioRxiv [Preprint] (2021). 10.1101/2021.10.26.465776. [DOI] [Google Scholar]
- 51.Huang G et al. , Cryo-EM structure of the inner ring from Xenopus laevis nuclear pore complex. bioRxiv [Preprint] (2021). 10.1101/2021.11.13.468242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tai L et al. , 8 Å structure of the nuclear ring of the Xenopus laevis nuclear pore complex solved by cryo-EM and AI. bioRxiv [Preprint] (2021). 10.1101/2021.11.10.468011. [DOI] [Google Scholar]
- 53.Hendrickson WA, Stereochemically restrained refinement of macromolecular structures. Methods Enzymol 115, 252–270 (1985). doi: 10.1016/0076-6879(85)15021-4 [DOI] [PubMed] [Google Scholar]
- 54.Evans R et al. , Protein complex prediction with AlphaFold-Multimer. bioRxiv [Preprint] (2021). 10.1101/2021.10.04.463034. [DOI] [Google Scholar]
- 55.Humphreys IR et al. , Computed structures of core eukaryotic protein complexes. Science 374, eabm4805 (2021). doi: 10.1126/science.abm4805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akey CW, Radermacher M, Architecture of the Xenopus nuclear pore complex revealed by three-dimensional cryo-electron microscopy. J. Cell Biol 122, 1–19 (1993). doi: 10.1083/jcb.122.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Russo CJ, Passmore LA, Electron microscopy: Ultrastable gold substrates for electron cryomicroscopy. Science 346, 1377–1380 (2014). doi: 10.1126/science.1259530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Naydenova K, Russo CJ, Measuring the effects of particle orientation to improve the efficiency of electron cryomicroscopy. Nat. Commun 8, 629 (2017). doi: 10.1038/s41467-017-00782-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morin A et al. , Collaboration gets the most out of software. eLife 2, e01456 (2013). doi: 10.7554/eLife.01456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng SQ et al. , MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). doi: 10.1038/nmeth.4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zivanov J et al. , New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). doi: 10.7554/eLife.42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rohou A, Grigorieff N, CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221 (2015). doi: 10.1016/j.jsb.2015.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang K, Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12 (2016). doi: 10.1016/j.jsb.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA, cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). doi: 10.1038/nmeth.4169 [DOI] [PubMed] [Google Scholar]
- 65.Steinegger M, Söding J, MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol 35, 1026–1028 (2017). doi: 10.1038/nbt.3988 [DOI] [PubMed] [Google Scholar]
- 66.Delano WL, The PyMol Molecular Graphics System (Delano Scientific, 2002). [Google Scholar]
- 67.Goddard TD et al. , UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25 (2018). doi: 10.1002/pro.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Adams PD et al. , PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 (2010). doi: 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and materials reported in the main text and supplementary materials are available upon reasonable request. The electron density maps have been deposited in the Electron Microscopy Data Bank (EMDB) with accession numbers EMD-25817 and EMD-25818 for a CR protomer and a full CR ring built from the CR protomer map, respectively, and the atomic coordinates have been deposited in the Protein Data Bank with the accession number 7TDZ.